

Abstract
There is a diverse array of influenza viruses which circulate between different species, reassort and drift over time. Current seasonal influenza vaccines are ineffective in controlling these viruses. We have developed a novel universal vaccine which elicits robust T cell responses and protection against diverse influenza viruses in mouse and human models. Vaccine mediated protection was dependent on influenza-specific CD4+ T cells, whereby depletion of CD4+ T cells at either vaccination or challenge time points significantly reduced survival in mice. Vaccine memory CD4+ T cells were needed for early antibody production and CD8+ T cell recall responses. Furthermore, influenza-specific CD4+ T cells from vaccination manifested primarily Tfh and Th1 profiles with anti-viral cytokine production. The vaccine boosted H5-specific T cells from human PBMCs, specifically CD4+ and CD8+ T effector memory type, ensuring the vaccine was truly universal for its future application. These findings have implications for the development and optimization of T cell activating vaccines for universal immunity against influenza.
Keywords: Influenza virus, T cells, Universal vaccine, IL-15, Vaccinia
1. Introduction
Influenza viruses are highly diverse and circulate within animal reservoirs with at least 18 different HA subtypes. Some of these viruses can cross-species to cause sporadic zoonotic outbreaks and may reassort their genomes to give rise to pandemic influenza viruses. Furthermore, seasonal epidemics are attributed to antigenic drift of the surface HA protein resulting in antigenic changes that require frequent updating of vaccine formulations to match circulating strains. This requires a worldwide effort of surveillance and prediction of the dominant strain for the upcoming season and generation of a vaccine candidate. Furthermore, this prediction may be incorrect or delayed, leading to mismatch between the vaccine virus and circulating strains resulting in poor vaccine efficacy, as occurred with H3N2 vaccines in the northern hemisphere during 2014 [1]. Current vaccination is dependent upon eliciting antibodies that match virus neutralising epitopes in the HA protein for protection. However the HA is a highly diverse protein that can undergo change in these dominant antigenic epitopes, resulting in “antigenic drift”. The limitations with conventional vaccine strategies have led to calls for alternative approaches to influenza vaccines, specifically “universal” influenza vaccines [2]. We explore novel vaccination strategies that utilise cross-reactive T cells to lead to a universal influenza vaccine.
T cells are critical for effective viral clearance during influenza infection, and can reduce symptom severity, duration of illness and viral shedding [3,4]. Importantly, T cells can provide heterosubtypic immunity, whereby T cells are able to recognise different influenza virus subtypes and even unrelated viruses due to conserved peptide homology [5–7]. Heterosubtypic T cell immunity is especially important when there is no protective antibody response, which can occur during seasonal influenza epidemics caused by an antigenically drifted virus, a zoonotic outbreak or pandemic. Thus, T cells can provide essential immunological protection from influenza infection.
Cytotoxic CD8+ T cells recognize viral peptides in the context of major histocompatibility complex (MHC) class I which is expressed on the surface of all cells providing immune surveillance of host cells. CD8+ T cells mediate the direct lysis of virally-infected cells and produce anti-viral cytokines, effectively reducing viral load and the duration of virus shedding. Helper CD4+ T cells recognize viral peptides within the context of MHC class II antigens which are only expressed by professional antigen presenting cells, including B cells, macrophages, dendritic cells and CD4+ T cells. In addition, it has been proposed that MHCII is also expressed by type II alveolar pneumocytes (reviewed in [8]), enabling CD4+ T cells to target a minor population of infected cells in the lung tissue combined with evidence of cytotoxicity. In a transgenic mouse model, CD4+ T cells have been found to be directly cytotoxic during influenza infection [9] and there is emerging evidence of CD4+ cytolytic activity [10], and that the magnitude of memory-influenza specific CD4+ T cell correlates with reduced infection and symptom severity in a human challenge study [3].
Our vaccine uses a live vaccinia Wyeth backbone carrying 5 influenza proteins derived from H5N1 viruses and IL-15 as a molecular adjuvant. It has remarkable heterosubtypic protective effects against influenza viruses of different subtypes in mice [11,12]. The vaccine encodes full length influenza proteins from the virion surface HA, NA, matrix protein 1 and 2, and the highly conserved NP protein. As the vaccine is able to access MHC I and II processing machinery, it is able to elicit robust CD4+ and CD8+ T cell responses. Previously we have demonstrated that this protection is attributable to robust T cell immunity, especially CD4+ T cell responses, because depletion of CD4+ T cells in vaccinated mice, but not depletion of CD8+ T cells alone, led to reduced survival and increased viral loads upon lethal H7N7 infection [11].
Other vaccine approaches to stimulate influenza T cell response in humans are currently being developed and tested in phase I and IIa clinical trials, including virus-like particles, RNA replicons, DNA vaccines, viral vectors including modified vaccinia Ankara (MVA) and chimpanzee adenovirus, and the use of adjuvants (reviewed in [13]). Some of these approaches show promising results for reducing viral loads and symptom severity [14] in human challenge studies. Therefore, we have tested our universal vaccine in a mouse system to determine the mechanism of protection by vaccine-specific CD4+ T cells, and in an in vitro system to determine its ability to recruit and expand human memory T cells as it is unbiased for HLA type by encoding full length proteins. The mechanisms of T cell protection and the universality of the vaccine is assessed in this study.
2. Materials and methods
2.1. Vaccinia vaccine: Wyeth/IL-15/5flu
The vaccine construct was previously described in detail by Poon et al. [12]. Briefly, the replication competent vaccinia Wyeth strain encodes the NP, HA and NA proteins derived from A/Vietnam/1203/2004 and the M1 and M2 proteins derived from A/CK/Indonesia/PA/2003. The vaccine encodes human IL-15 cytokine as a molecular adjuvant. Mice were either vaccinated with the H5 vaccine virus (Wyeth/IL-15/5flu, termed Vacc A), a control Wyeth vaccine (influenza protein negative, termed Vacc C), or PBS.
2.2. Influenza virus challenge of vaccinated mice
Female BALB/c (H-2d), SCID (C.B-17/Icr-scid) and nude (BALB/cAnN-nu) mice (6–8 weeks of age) were primed twice three weeks apart via the subcutaneous (s.c.) route with 1 × 107 plaque forming units (pfu) in 100 μl PBS of Vacc A, Vacc C or PBS and challenged with influenza virus three weeks later. For influenza challenge, mice were anaesthetized and infected intranasally (i.n.) with 25 μl of mouse adapted H3N2 (A/Hong Kong/1/68-MA20C, 4.72 × 105TCID50, 1LD50) (gift from Earl G. Brown, University of Ottawa) or pandemic H1N1 (A/California/04/2009, 1.36 × 106TCID50, 1LD50). All animal studies were approved by the institutional animal ethics committee (CULATR, HKU). Humane endpoints were defined as weight loss >25% from starting weight and combined with symptoms (fur ruffling, reduced activity, hunching and laboured breathing).
2.3. Depletion and transfer of T cell subsets in vaccinated mice
Adoptive transfer of T cell subsets and immune serum from vaccinated mice to naïve mice was performed to determine passive immunity [15]. Splenocytes from vaccinated mice were purified by magnetic isolation for total CD3+, CD4+ and CD8+ T cells, according to manufactures instructions (R&D systems), to greater than 95% purity as confirmed by flow cytometry (Fig. 2B). At the time of H3N2 virus challenge (1LD50), 1 × 106 purified total T cells from day 21 post 2-dose vaccination BALB/c mice were transferred intra-venously (i.v.) in 100 μl PBS to naïve BALB/c mice (Fig. 2A). In addition to passive transfer of T cells, immune serum was given to naïve mice. Immune serum at 21-days post 2-dose vaccination, was harvested by cardiac puncture from Vacc A vaccinated mice, pooled (n = 20), and heat inactivated (56 °C 60mins). Immune serum was given intra-peritoneally (i.p.), 500 μl on day −4, −2, 0 and +1 of infection, for a total volume of 2 ml per mouse [16].
Fig. 2.

Adoptive T cell transfer of CD4+ or CD8+ T cells provides challenge for protection, whilst sera shows no protection. (A) Naïve BALB/c mice were given memory splenocytes from vaccinated mice purified by magnetic selection (B) for total CD3+, CD4+ or CD8+ T cells. (A) Mice were given T cells i.v., and immune serum was given i.p. 500 μl on 4 separate days. Mice were then infected with 1LD50 H3N2, lung viral load determined at day 7 (C) (n = 3), and fold reduction in viral load compared to PBS negative controls (D), and monitored for survival to day 14 (E) (n = 5). Experiment was repeated twice. (C) Data represents the individual viral loads and group mean (n = 3), (D) data represents the average viral load reduction compared to PBS negative control mice, and (E) the % survival by day 14 post infection (n = 5).
Control groups of Vacc A and Vacc C mice served as positive and negative controls respectively. Transfer of splenocytes from Vacc C immunised mice to naïve mice had no effect (data not shown), therefore for subsequent experiments PBS alone served as negative controls and only immune cell transfer from vaccinated mice was used. Mice were monitored for survival and weight loss (n = 5). Lung viral loads were determined at day 7 post infection (n = 3). Clarified lung homogenates were titered on MDCK cells by standard TCID50 assay, as previously described [11].
In further experiments, vaccinated mice were depleted of CD4+, CD8+ or both CD4+ and CD8+ T cells prior to influenza infection (Fig. 3A). Mice were treated with GK1.5 (anti-CD4) and 2.43 (anti-CD8) monoclonal antibodies (BioXCell) at 100 μg i.p 4 times before challenge (days −16, −14, −11 and −9 for depletion at challenge) or during vaccination (days −46, −44, −42 and −39 for the first vaccination and days −25, −23, −21 and −19 for the second vaccination) [11,17]. The final depletion dosage of ‘depletion at challenge’ group was 9 days before challenge to ensure minimal depletion of transferred cells (18). Depletion was confirmed in peripheral blood at day −1 at >98% compared to isotype treated mice and non-depleted mice. Magnetically purified untouched CD3+ T cells were isolated from the spleens of naïve BALB/c mice according to manufacturer’s instructions (R&D systems), and 1 × 106 cells in 100 μl PBS were transferred i.v. to selected vaccinated depleted mice. The NP147-specific CD8+ T cell response of bronchoalveolar lavage (BAL) was determined IFN-γ ICS assay, as previously described [11].
Fig. 3.
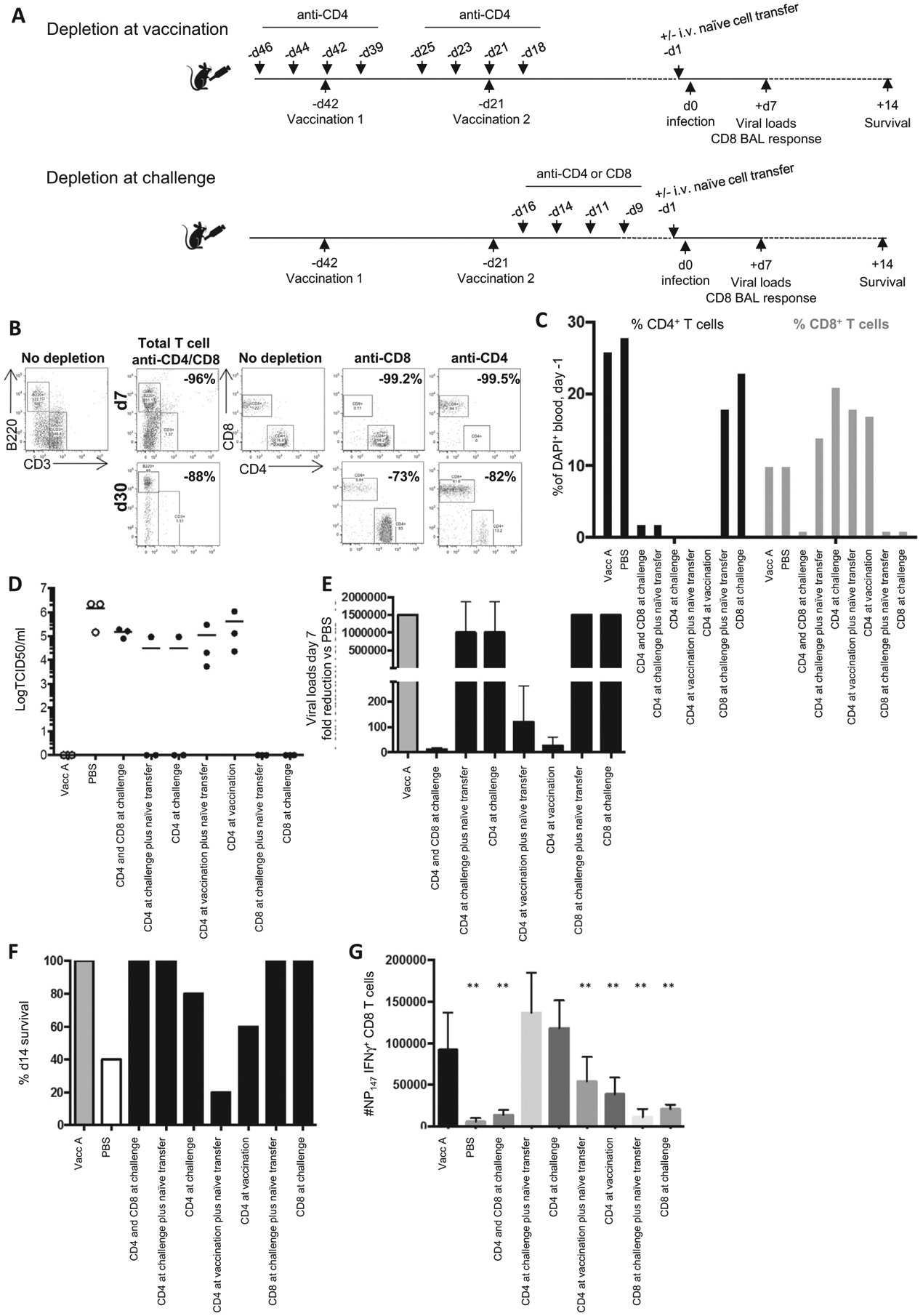
T cell depletion impairs CD4+ T cell help for memory and CD4+ T cells are needed at challenge for protection. (A) Mice were depleted for CD4+ and CD8+ at challenge, CD4+ T cells at challenge or vaccination, or CD8+ T cells at challenge. Prior to infection selected mice were given naïve splenocytes i.v. 9 days after the final depletion. (B) The efficiency of T cell depletion was assessed from the peripheral blood at day 7 and day 30 after the last dose of monoclonal antibody treatment, and % reduction compared to undepleted mice, as shown in representative FACS plots. (C) The % of CD4+ and CD8+ T cells of total DAPI+ cells in the peripheral blood was also assessed at -day 1 prior to challenge (n = 3 pooled). Mice were infected with 1LD50 H3N2, lung viral load determined at day 7 (n = 3) (D), fold reduction in viral load compared to negative controls (E), and monitored for survival day 14 (n = 5) (F). BAL NP147-specific IFN-γ+ CD8+ T cells were enumerated at day 7 post H3N2 infection (data represents n = 3, mean+/−stdev) (G), **p > 0.001 vs Vacc A controls (t-test). Experiment repeated at least twice.
2.4. Profiling of CD4 T cell subsets
A panel of antibodies was used to differentiate CD4+ T cell subsets from BAL samples of infected vaccinated mice. BAL cells were stimulated with NP55 and HA140 peptide, as previously described [11], for cytokine production following 2 h peptide stimulation in the presence of Brefeldin A for a further 4 h, for a total 6 h stimulation. The FcR was blocked prior to immune cell staining, 20 min at room temperature (anti-CD16/CD32, BD Bioscience). Cells were stained for surface markers for anti-mouse CD3-BV510, CD4-APCCy7, CD25-PerCPCy5.5, CXCR5-APC and PD1-BV605 (all Biolegend). Samples were then fixed with FoxP3 Fix/Perm buffer (eBioscience), and intracellular stain for IFN-γ-FITC, RORγt-PE, IL-17A-AF700, FoxP3-Pacific Blue, Bcl6-PETexas and IL-4-PECy7. Cells were acquired by flow cytometry on a LSR Fortessa and analysed by FlowJo software. Live lymphocyte gating, followed by CD3+ CD4+ to determine: Th2 (IL-4+), Th1 (IFN-γ+), Th17 (IL-17A+), Treg (CD25+ FoxP3+) and Tfh cells (CXCR5+).
2.5. Vaccine stimulation of human PBMC for T cell expansion
PBMCs (1 × 107) were pre-incubated with CellTrace violet (Violet Proliferation Dye 450, BD Biosciences) before generating virus/vaccine restimulated T cell lines. In parallel, 3 × 106 autologous PBMCs were pulsed with MOI4 of UV irradiated H5N2 virus (A/Eurasian Wigeon/MPF4610/07), Vacc A or Vacc C for 90 mins at 37 °C, followed by two PBS wash twice. PBMCs were coupled together and incubated for 4, 10 and 20 days in cRPMI with 10U/mL recombinant IL-2 (Roche) from day 4 onwards, with 50% culture media changes every 2 days. Restimulated PBMCs, and direct ex vivo (day 0) PBMCs were restimulated with MOI4 homologous virus or vaccine on the day of the experiment, plus BFA (BD) at 4 h post stimulation, and incubated for a further 12 h. Cells were stained with anti-human CD3-PETexas, CD4-BV605, CD8-AF700, CCR7-PE and CD45RA-APC (Biolegend). Cells were fixed with Cytofix/cytoperm buffer (BD), and stained for anti-human IFNγ-FITC (Biolegend) in Perm wash buffer (BD). Cells were assessed by flow cytometry on an LSR Fortessa and analysed on FlowJo software. IFN-γ from background controls was subtracted from each sample.
2.6. Statistics
Results represent the mean ± SD of three to five mice per group, unless indicated otherwise. Statistical significance was compared between different treatment groups using a standard unpaired, two-tailed Student’s t-test (unless indicated), #p < 0.05, *p < 0.01, ##p < 0.005, **p < 0.001. For human PBMC experiments, paired, a two-tailed Student’s t-test (two-tailed) was used for comparison of day 0 versus later time points from the same stimulation.
3. Results
A universal influenza vaccine is the “holy grail” for vaccination efforts, yet the fundamental correlates of protection beyond HAI antibodies still remains to be defined, but are essential for development of next generation vaccines. The Wyeth/IL-15/5flu vaccine has proven effective in the mouse model against a diverse array of influenza viruses including avian viruses of human health concerns (H5N1 and H7N7 and H7N9) and pandemic viruses (H1N1) [11,12]. Previously, we have shown this vaccine can elicit robust influenza-specific T cell memory responses which are sustained and recalled to the site of infection. Furthermore, depletion of total CD4+ and CD8+ T cells in vaccinated mice showed that mice lacking CD4+ T cells or CD4+ and CD8+ T cells had reduced survival and increased viral loads against HPAI H7N7 virus.
To further demonstrate T cell-dependent vaccine mediated protection, we utilised the SCID and nude mouse strains, which lack mature T plus B cells and T cells respectively. The mouse strains have a variety of differences in their background, but provide a starting point for understanding vaccine mediated protection. Vaccinated BALB/c mice which have intact T and B cells, were protected and had reduced viral loads in the lung when challenged with the H3N2 virus, whereas SCID and nude mice had elevated H3N2 virus loads (Fig. 1AB) and reduced survival (Fig. 1C). In comparison to unvaccinated controls, vaccinated nude and SCID mice had only a 4-fold reduction in lung viral load, whilst vaccinated intact BALB/c mice had over 1395 fold reduced viral loads (Fig. 1B). This minor but notable 4-fold reduction in vaccinated SCID mice may implicate the role of IL-15 meditated stimulation of innate lymphoid cells (reviewed in [19]). BALB/c vaccinated with vaccine vector control (Vacc C) mice had lower viral loads than nude and SCID mice, due to their intact immune system.
Fig. 1.
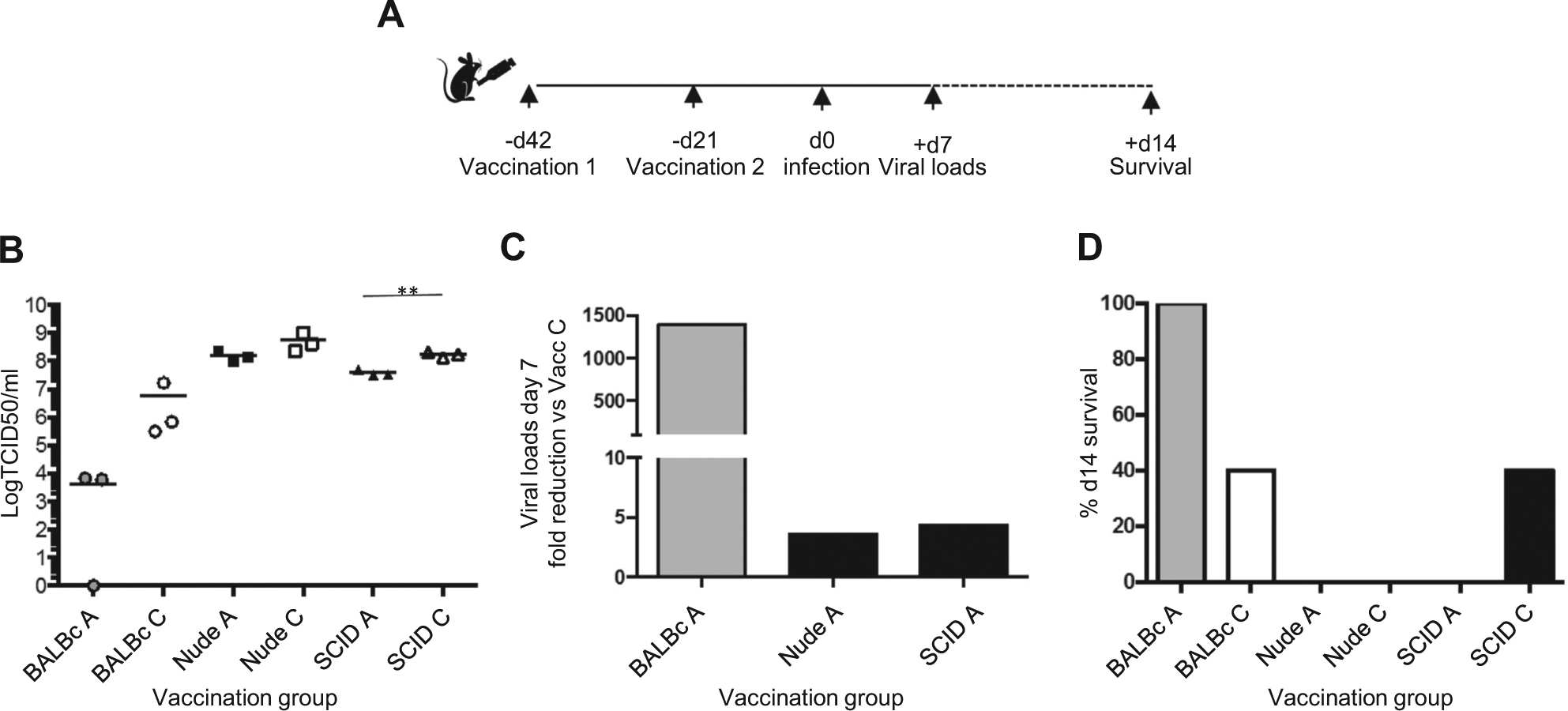
Wyeth/IL-15/5flu does not protect T cell deficient mouse strains from H3N2 infection. (A) BALB/c, nude and SCID mice were vaccinated twice s.c. with Wyeth/IL-15/5flu (Vacc A) or Wyeth (Vacc C), and challenged with 1LD50 of H3N2 virus i.n.. (B) Viral loads of day 7 post infection lungs were determined by standard TCID50 on MDCK cells, for individual mouse viral loads and group mean (n = 3). (C) Data represents the fold reduction in viral load compared to negative (Vacc C) controls of the same mouse strain (n = 3), and (D) monitored for survival day 14 (n = 5). Experiment was repeated twice.
We investigated the protective mechanisms underlying of our vaccine by passively transferring immune T cell subsets and serum to naïve mice (Fig. 2A). The purity of purified T cell population was confirmed (Fig. 2B). Transfer of purified T cells or immune serum from vaccinated BALB/c mice reduced viral loads (Fig. 2CD); immune serum by 26-fold, CD3+ T cells by 58-fold, CD4+ T cells by 88-fold and CD8+ T cells by an impressive 254-fold, whilst positive control (Vacc A) vaccinated mice had over 5800-fold reduction in viral loads by day 7 post infection (Fig. 2D). Therefore, passive transfer of individual antibody or T cell subsets from vaccinated animals cannot fully recapitulate the level of anti-viral coverage afforded by vaccination when multiple immune arms were working in concert. However, only the transfer of immune T cells, of either CD3+, CD4+ or CD8+ T cell subtype improved survival from 20% observed in negative control mice to 100% survival (Fig. 2C), whilst immune serum had no effect on viral loads or survival. Therefore, vaccine elicited memory T cell responses provide important levels of passive heterosubtypic immunity to naïve mice.
To further delineate the role of T cell immunity for vaccine-mediated protection, we depleted T cells at the time of vaccination or challenge respectively, and transferred naïve T cells to enable primary responses (Fig. 3A). Monoclonal antibody depletion was confirmed to remove total CD4+ or CD8+ T cells stably long term (Fig. 3B) and confirmed at the onset of influenza challenge (Fig. 3C). We transferred naïve splenocytes at the time of infection, coincident 9 days after the last depletion time point to ensure depletion of transferred cells would not occur [18]. Depletion of CD4+ and CD8+ T cells at the time of challenge, and CD4+ T cells at the time of vaccination, altered viral clearance to only 3–10 fold reduction in viral loads compared to unvaccinated mice (Fig. 3E). Depletion of CD4+ T cells at the time of vaccination removes their helper function reducing CD8+ T cell recall responses (Fig. 3G), and depletion at challenge negates the early antibody responses (Fig. 4).
Fig. 4.
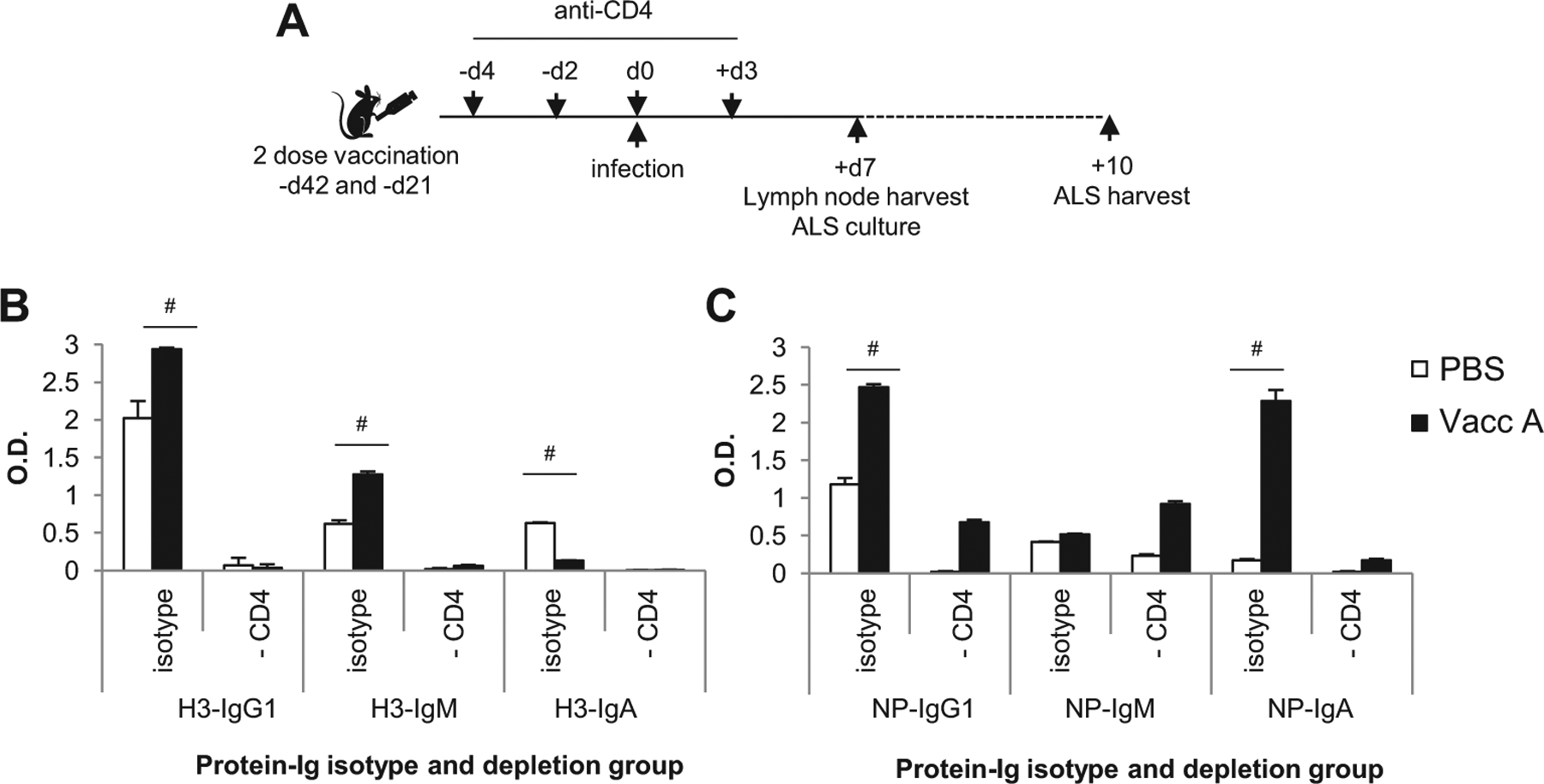
The early local antibody response from vaccinated mice is CD4+ T cell dependent. (A) Vaccinated and unvaccinated mice were depleted for CD4+ T cells, and then were infected with 1LD50 H3N2, and the local mediastinal lymph node (mLN) harvested at day 7, for a further 3 day in vitro culture to harvest antibody lymphocyte secretions (ALS). The mLN of 3 mice per group was pooled per group and 1 × 106 cells cultured. The ALS supernatant was tested in triplicate by in ELISA for H3-HA (B) and NP (C) protein, for IgG1, IgM and IgA isotype. Data represents the pooled response of triplicates, mean+/−stdev, #p > 0.05 vs PBS controls (by t-test).
Depletion of CD8+ T cells at the time of challenge, with or without the supplement of naïve T cells, had no effect on survival or viral clearance with 100% survival and an over 1.5 × 106 reduction in viral loads equivalent to positive control Vacc A mice (Fig. 3DE). Furthermore, the magnitude of the total CD8+ T cell response (Fig. 3C) and NP147 IFNγ+ CD8+ T cell response (Fig. 3G) was significantly impacted by anti-CD8 treatment, yet viral clearance was maintained (Fig. 3D). Whilst the anti-CD4 treatment groups at challenge retained their NP147 IFNγ+ CD8+ T cell response (Fig. 3G), yet maintained viral load reduction (Fig. 3E), survival (Fig. 3F) similar to fully vaccinated positive control mice.
The largest reduction in survival was observed when CD4+ T cells were depleted at vaccination time-points, due to both the disruption of NP147 IFNγ+ CD8+ T cell response (Fig. 3G) but not total CD8+ T cell recall (Fig. 3C), therefore non-specific CD8+ T cells remained, and may contribute to bystander responses. In addition, the CD4+ T cell depletion is maintained long term at 82% at 30 days post depletion in the circulation (Fig. 3BC). The anti-CD4 treatment of monoclonal GK1.5 specific for MHC-II may also target monocytes, macrophages and dendritic cells, however there was no difference in the magnitude of these responses between any treatment groups observed in the BAL (data not shown). The important of CD4+ T cells at the of vaccination for heterosubtypic protection was also confirmed by results observed from a H1N1 virus challenge model (Supplementary Fig. 1)
The local early antibody response was probed from the mediastinal lymph node at day 7 of H3N2 challenge (Fig. 4), by 3 day in vitro culture of the local lung draining lymph node, by antibody lymphocyte secretion (ALS). ELISA of the ALS was used to determine if CD4+ T cell depletion at challenge impacted the early antibody response to heterosubtypic infection, thus attributing reduced protection seen for anti-CD4 treatment partly to the antibody response. Indeed vaccinated mice mounted an early H3-HA-specific IgG1 and IgM and NP-specific IgG1 and IgA antibody response that was significantly higher than unvaccinated PBS mice (Fig. 4BC). Anti-CD4 treatment at challenge abrogated this response in both vaccinated and unvaccinated mice, with ALS at baseline levels. Therefore, anti-CD4 treatment significantly impacts the early antibody response.
Using a panel of CD4+ phenotype specific antibodies representing, Th1, Th2, Th17, Treg and Tfh phenotypes (reviewed in [20]), the CD4+ T cell profile after influenza challenge was probed in the BAL. The phenotype of CD4+ T cells elicited by the vaccine and recalled by infection were predominantly of the Th1 and Tfh phenotype (Fig. 5AB), for both the conserved NP55 peptide and heterologous HA140 peptide. Furthermore, the polyfunctional cytokine production of Th1 CD4+ T cells was determined for single, double or triple cytokine production (Fig. 5DE). Th1 IFN-γ+ was independent of Th2 IL-4+ production, and the Th2 IL-4+ response was comparable between vaccinated and unvaccinated mice (Fig. 5E). The majority of the Th1 NP55-specific CD4+ T cell response in vaccinated mice was single IFN-γ+ cytokine producing, and a partial double IFN-γ+ TNF-α+ response (Fig. 5D), whilst triple cytokine (IFN-γ+ TNF-α+ IL-2+) producing Th1 cells were rare in either vaccinated or unvaccinated mice, and therefore vaccinated CD4+ memory T cell responses are not terminally and fully differentiated and may have greater recall capacity. Therefore, it is likely that the magnitude and phenotype quality of the memory CD4+ T cells are essential for protection in the mouse model.
Fig. 5.
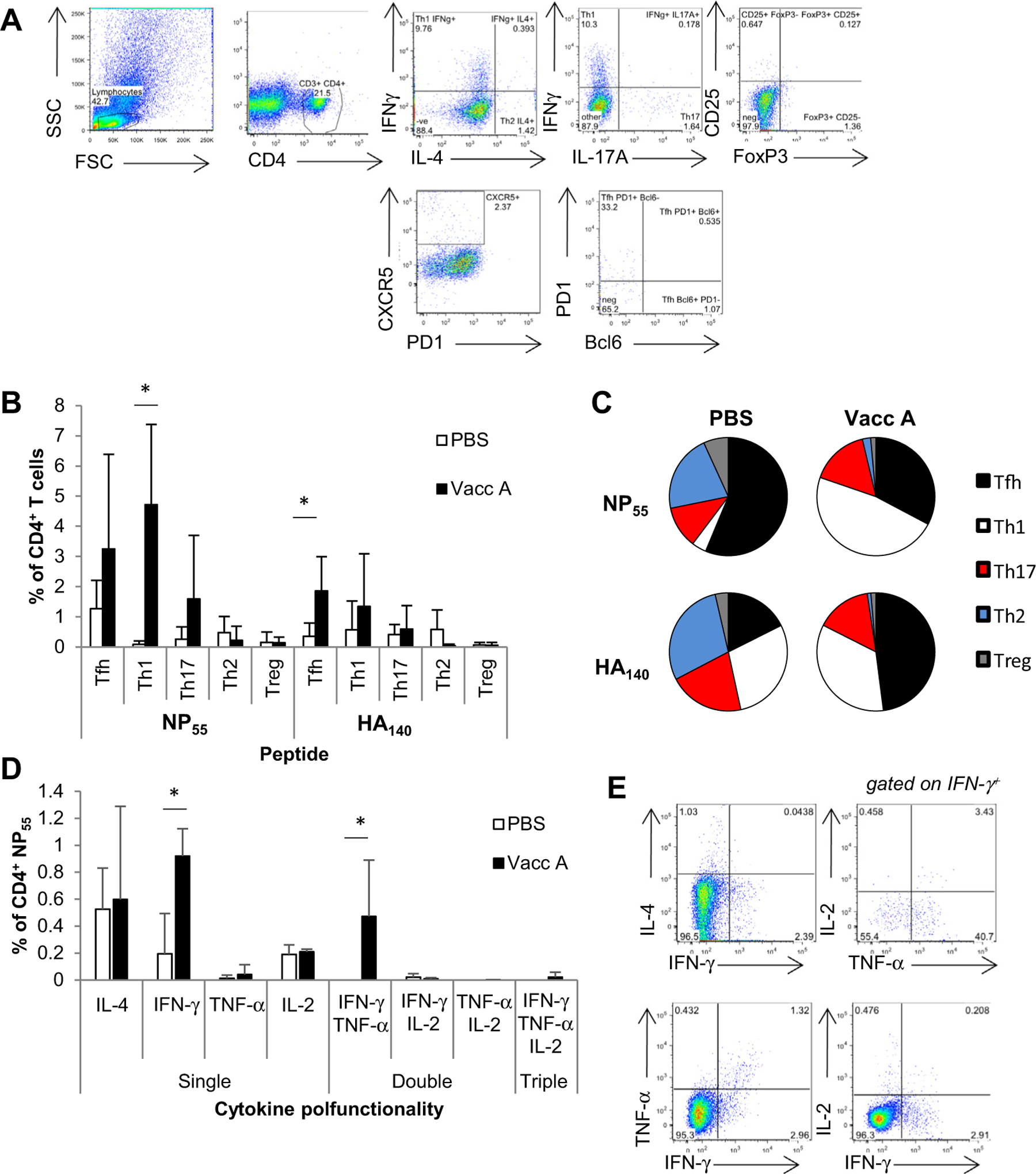
Vaccination increases Tfh and Th1 CD4+ T cell phenotype responses. BALB/c mice were vaccinated twice or received PBS, infected with H3N2 1LD50, and day 7 post infection BAL harvested. Cells were stained with a panel of antibodies representing CD4+ T cell phenotype profiles (A), stimulated with MHC-II restricted peptides, NP55 and HA140, for cytokine production of IFN-γ+ and IL-4, and analyzed by flow cytometry. (A) The CD4+ T cell panel was assessed for Tfh (CD4+ CXCR5+), Th1 (CD4+ IFN-γ+), Th2 (CD4+ IL-4+), Th17 (CD4+ IL-17A+) d and Treg (CD4+ CD25+ FoxP3+). Tfh cells were also assessed for PD1 and Bcl6 expression, and Th17 cells for RORγt which was low in the BAL, and not included in the analysis. (B) The cytokine specific and phenotype response was determined for NP55 and HA140 peptides (data represents n = 3, mean+/−stdev), and (C) their proportions. (D) Polyfunctional cytokine production was also assessed for NP55 responses in the BAL from day 7 H3N2 infection, for Th1 (IFN-γ, TNF-α and IL-2) and Th2 (IL-4) cytokines (data represents n = 3, mean+/−stdev). (E) CD4+ T cells gated for single, double and triple cytokine production. Experiment repeated at least twice, *p > 0.01 vs PBS controls (by t-test).
The Wyeth/IL-15/5flu Vacc A vaccine encodes full length H5N1-derived proteins and therefore its protection is not limited by HLA restriction profile to BALB/c mice. Indeed, C57BL6 mice have also demonstrated vaccine mediated protection [11]. To determine human T cell recruitment by the vaccine, we used in vitro restimulation protocol as a tool for preclinical development. As our vaccine is a live vector and encodes full length influenza derived proteins it does not have a host restriction, thus our vaccine could be directly translated for human use. We determined the recruitment, proliferation and activation of vaccine and H5N1-specific human T cells in an in vitro stimulation culture model (Fig. 6A), whereby direct ex vivo day 0 responses were compared with the kinetics and recruitment of influenza-specific memory at day 4, and 10 post-stimulation. CellTrace violet was used to stably and long-term label cells, and IFN-γ+ production was only seen in divided cells post stimulation (Fig. 6B). H5N2 virus restimulation recalled cross-reactive memory CD4+ and CD8+ T cells over a 10-day culture as expected at increased levels compared to the empty Vacc C vector (Fig. 6CD).
Fig. 6.
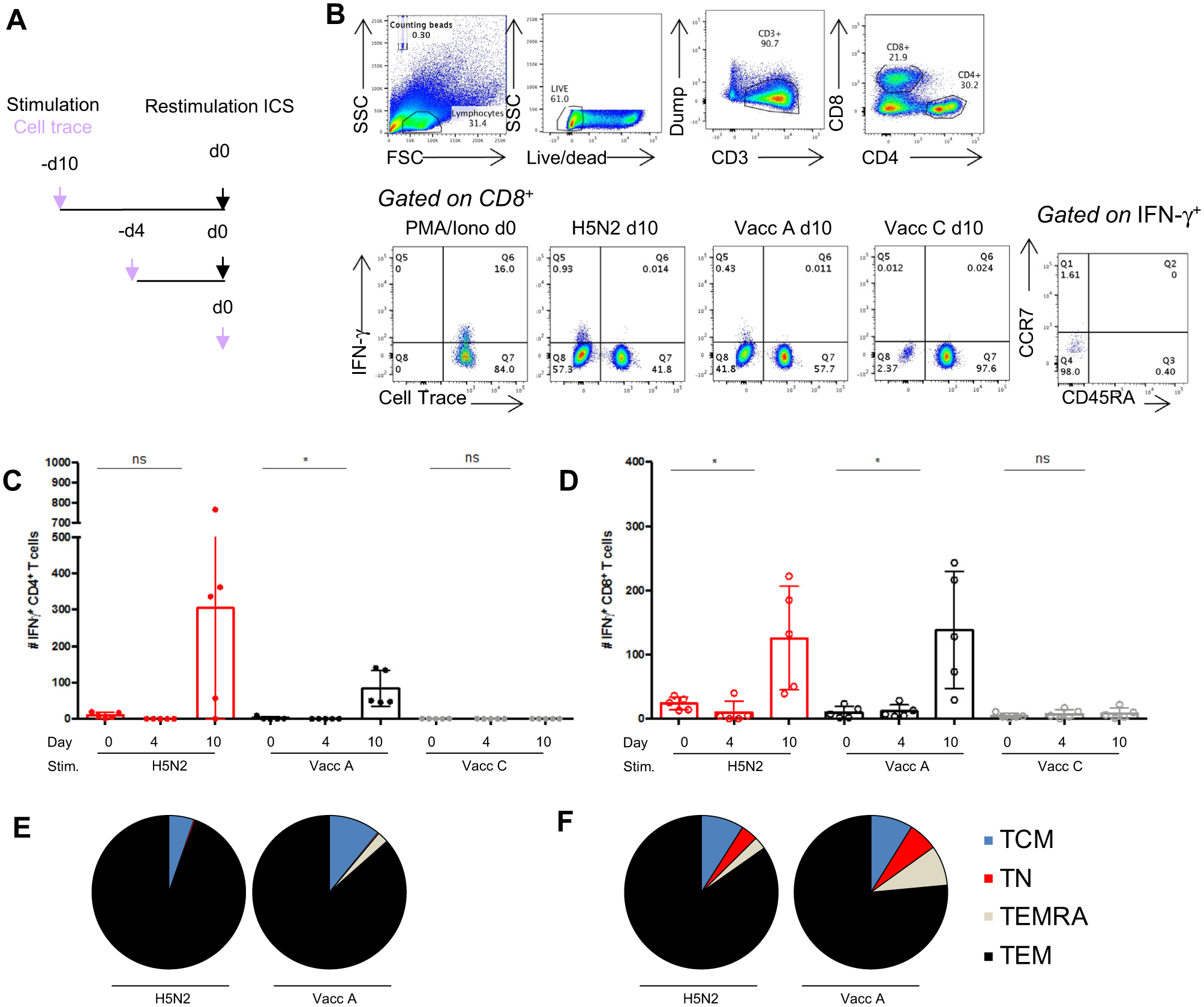
Human PBMCs restimulated with Vaccinia A proliferate, expanding influenza-specific CD4+ and CD8+ T effector memory subsets. (A) PBMCs from 5 healthy donors were expanded in vitro after stimulation with UV-inactivated H5N2, Vacc A or Vacc C viruses (MOI4) and pre-incubated with CellTrace Violet. Cells were incubated for 4 or 10 days, in the presence of IL-2 from day 4 to generate T cell lines. (A) On the day of the experiment (day 0), restimulated day 4 or 10 PBMCs and direct ex vivo day 0 PBMCs from matched donors received homologous stimulations or media stimulation (background negative control). (B) representative FACs plots show the gating strategy, live PBMCs were gated CD3+ dump−, CD4+/CD8+, cell trace and IFN-γ+, then IFN-γ+ cells for CCR7 and CD54RA for memory subsets. The # cell number of IFN-γ+ CD4+ T cells (C) and IFN-γ+ CD8+ T cells (D) (dots represents individual values (n = 5), bars represent mean+/−stdev), p > 0.01 vs day 0 responses (by t-test). The proportion memory phenotype, TEM (CCR7− CD45RA−), TEMRA (CCR7− CD45RA+), TCM (CCR7+ CD45RA−), TN (CCR7+ CD45RA+) of day 10 IFN-γ+ CD4+ (E) and CD8+ T cells (F) (mean n = 5). Experiment repeated at least twice.
The Vacc A containing H5N1-derived proteins also resulted in proliferation and dilution of total T cells for CellTrace but at levels comparable to the empty vector (data not shown), indicating the vector and culture conditions result in some cell proliferation. However, the magnitude of influenza virus-specific IFNγ+ CD4+ and CD8+ T cells was significantly increased by Vacc A stimulation compared to day 0 responses, and was at levels comparable to those caused by H5N2 virus restimulation at day 10 post-stimulation (Fig. 6CD). By contrast, the vaccine backbone, Vacc C, caused no IFN-γ+ activation of memory T cells, with minimal expansion of IFN-γ+ CD4+ and CD8+ T cell numbers at day 10 (Fig. 6CD). Furthermore, both H5N2 and Vacc A restimulation predominantly expanded the circulating T effector memory phenotype population (Fig. 6EF), which are capable of immediate cytotoxic function. Therefore, human influenza-specific T cells are recruited and expanded by the vaccine, ensuring its universality and immunogenicity.
4. Discussion
A universal influenza vaccine that provides protection against a diversity of influenza viruses including seasonal drift variants and also with ability to prearm the immune system against potential pandemic viruses has been identified as a major public health need. Our vaccine has proven effective in the mouse model against group 1 and group 2 HA viruses providing a wide breadth of heterosubtypic coverage. The vaccine encodes full length H5N1 derived HA, NA, NP, M1 and M2 proteins in a live replicating vaccinia vector, and is thus able to effectively stimulate T cell responses. We have shown these T cell responses are protective by multiple avenues. Vaccine mediated protection was T cell dependent as evident by the lack of protection in T-cell deficient SCID mice. No protection, by means of increased survival or reduced viral loads, was provided by passive transfer of high levels of immune serum [16]. Passive transfer of T cells from vaccinated mice increased survival and partially reduced viral loads, which has also been demonstrated in other heterologous influenza challenge studies [15].
The unique finding from our vaccine model is the key role of memory CD4+ T cell responses in protection. The mechanism of protection from this vaccine is not solely reliant upon cross-reactive CD8+ T cell responses, unlike other heterologous influenza challenge studies [15]. Selective depletion experiments for T cell subsets revealed an important role of vaccine-induced CD4+ T cells as the cornerstone in heterosubtypic protection, especially for establishing their helper function, augmenting local early H3-HA and NP-antibodies, CD8+ T cell memory and activation of Th1-type CD4+ T cells during challenge itself.
In this study, we conduct adoptive transfer of T cells to determine whether a particular subset of T cells from vaccinated mice alone would be sufficient for heterosubtypic protection. One should note that the cell depletion and adoptive transfer models are very different experimental approaches. The difference of results between adoptive transfer of memory T cells into naïve mice and depletion of T cell subsets from vaccinated mice, illustrates that heterosubtypic immunity against influenza is the not simple addition and subtraction for cell numbers. Adoptive transfer of CD4+ T cells did not result in reduction in viral loads like CD8+ T cells, but similarly resulted in increased survival. Therefore, a protective role of CD4+ T cells is evident beyond cytotoxic function to mediate increased protection from heterologous influenza infection, this could be attributed to mediating an earlier antibody response, bystander CD8+ T cell recruitment or cytokine production. Additional immune parameters may be present in mice vaccinated and depleted of CD8+ T cells, illustrating the connection of the immune system with multiple immune arms able to mediate heterosubtypic protection, especially CD4+ T cells.
Wyeth/IL-15/5flu is a universal vaccine, not only in terms of its remarkable breadth of anti-influenza heterosubtypic activity, but also in cross-species reactivity. The MHC genes are the most polymorphic regions in the human genome with over 8600 MHC-I and 3000 MHC-II proteins in the human population [21], which can be broken down into HLA supertypes [5] and utilised by mosaic anti-influenza vaccine strategies [22]. The vaccinia vector of Wyeth/IL-15/5flu easily encodes full-length influenza proteins and therefore has no MHC bias, a cornerstone of self-selection of immunogenic peptides. Therefore, the vaccine can be applied to different species and without ethnic bias which may occur with epitope vaccines [7]. Recently, several health authorities have specifically called for further testing of universal vaccines in human models [2,23,24]. To determine the immunogenicity of our vaccine for human T cells as pre-clinical development and extend our findings beyond mouse models, we used in vitro restimulation of healthy human PBMCs to test for expansion of T cells by the vaccine. In addition, we conducted T cell phenotyping on the expanded population. In vitro stimulation of human PBMCs with the Wyeth/IL-15/5flu, H5N2 or empty vaccine, demonstrated that the vaccine recruits cross-reactive memory influenza virus-specific T cell responses. The stimulating effect of this recombinant vaccine to human T cells can be as effectively as the one induced by influenza virus. A similar vaccine design in phase I clinical trials, a non-replicative vaccinia encoding 2 influenza proteins, MVA-NP + M1 is also able to recruit and amplify T cell responses in vivo [14,25], and is a promising vaccine candidate for wider application. Therefore T cell activating vaccines using live vaccinia viral vectors are becoming a foreseeable reality for human use.
Universal vaccination utilises conserved epitopes, which are typically located in the functionally conserved HA-stem and internal proteins (reviewed in [13]). There is emerging data on the role of non-neutralizing HA-stem antibodies providing limited heterosubtypic protection [26,27]. However, in a mouse model with comparable virus challenge doses and time-points, the Wyeth/IL-15/5flu vaccine resulted in viral clearance by day 7 and higher rates of survival against diverse influenza subtypes [11], whereas a HA-stem protein vaccine had no effect on peak viral loads or earlier viral clearance, and only provided partial cross-type protection [26]. In addition other vaccines which aim for universal immunity against influenza, have not demonstrated such breadth of coverage by a single vaccine regime against both group 1 and group 2 HA viruses and protection from lethal high dose infections. For example, the chimeric HA vaccine approach requires 3 prime boost doses using different routes and viral vectors [28,29]. Therefore, a T cell activating vaccine has a significantly wider breadth of reactivity and greater effect on reducing viral loads in the lung than a HA-stem antibody based vaccine. Similarly, the HA minus signal peptide vaccine, which is designed to stimulate T cell immunity, has only demonstrated group 1 HA protection [30]. Whilst other vaccines are also worthy candidates, our vaccine has a very good breadth of coverage against both group 1 and group 2 viruses by utilising heterosubtypic T cell immunity. There is the potential application of a combined vaccine approach to utilise both universal antibodies and T cells to provide a one-two punch to make universal vaccine mediated protection a reality.
Supplementary Material
Grant support
This project was supported by Health Medical Research Fund (project: 13121142, SAV), National Institutes of Health (NIAID contract HHSN272201400006C, LLMP), Theme-based research grant (Grant T11-705/14N), and the Intramural research program of the National Cancer Institute NIH. Wyeth/IL-15/5flu is incorporated in a patent (LPP, TAW) filed by the Office of Technology Transfer, NIH, US Department of Health and Human Services.
Footnotes
Conflicts of interest
Wyeth/IL-15/5flu is incorporated in a patent (LPP, TAW) filed by the Office of Technology Transfer, NIH, US Department of Health and Human Services. There are no other conflicts to declare.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.vaccine.2018.06.007.
References
- [1].Vestergaard LS, Nielsen J, Krause TG, Espenhain L, Tersago K, Bustos Sierra N, et al. Excess all-cause and influenza-attributable mortality in Europe, December 2016 to February 2017. Euro Surveill 2017;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Paules CI, Marston HD, Eisinger RW, Baltimore D, Fauci AS. The pathway to a universal influenza vaccine. Immunity 2017;47:599–603. [DOI] [PubMed] [Google Scholar]
- [3].Wilkinson TM, Li CK, Chui CS, Huang AK, Perkins M, Liebner JC, et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med 2012;18:274–80. [DOI] [PubMed] [Google Scholar]
- [4].Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, Carman W, et al. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat Med 2013;19:1305–12. [DOI] [PubMed] [Google Scholar]
- [5].Assarsson E, Bui HH, Sidney J, Zhang Q, Glenn J, Oseroff C, et al. Immunomic analysis of the repertoire of T-cell specificities for influenza A virus in humans. J Virol 2008;82:12241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee LY, Ha do LA, Simmons C, de Jong MD, Chau NV, Schumacher R, et al. Memory T cells established by seasonal human influenza A infection cross-react with avian influenza A (H5N1) in healthy individuals. J Clin Invest 2008;118:3478–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Quinones-Parra S, Grant E, Loh L, Nguyen TH, Campbell KA, Tong SY, et al. Preexisting CD8+ T-cell immunity to the H7N9 influenza A virus varies across ethnicities. Proc Natl Acad Sci U S A 2014;111:1049–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kim TS, Sun J, Braciale TJ. T cell responses during influenza infection: getting and keeping control. Trends Immunol 2011;32:225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brown DM, Dilzer AM, Meents DL, Swain SL. CD4 T cell-mediated protection from lethal influenza: perforin and antibody-mediated mechanisms give a one-two punch. J Immunol 2006;177:2888–98. [DOI] [PubMed] [Google Scholar]
- [10].Hildemann SK, Eberlein J, Davenport B, Nguyen TT, Victorino F, Homann D. High efficiency of antiviral CD4(+) killer T cells. PLoS One 2013;8:e60420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Valkenburg SA, Li OT, Mak PW, Mok CK, Nicholls JM, Guan Y, et al. IL-15 adjuvanted multivalent vaccinia-based universal influenza vaccine requires CD4+ T cells for heterosubtypic protection. Proc Natl Acad Sci U S A 2014;111:5676–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Poon LL, Leung YH, Nicholls JM, Perera PY, Lichy JH, Yamamoto M, et al. Vaccinia virus-based multivalent H5N1 avian influenza vaccines adjuvanted with IL-15 confer sterile cross-clade protection in mice. J Immunol 2009;182:3063–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sridhar S Heterosubtypic T-cell immunity to influenza in humans: challenges for universal T-cell influenza vaccines. Front Immunol 2016;7:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lillie PJ, Berthoud TK, Powell TJ, Lambe T, Mullarkey C, Spencer AJ, et al. Preliminary assessment of the efficacy of a T-cell-based influenza vaccine, MVA-NP+M1, in humans. Clin Infect Dis 2012;55:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hillaire ML, van Trierum SE, Kreijtz JH, Bodewes R, Geelhoed-Mieras MM, Nieuwkoop NJ, et al. Cross-protective immunity against influenza pH1N1 2009 viruses induced by seasonal influenza A (H3N2) virus is mediated by virus-specific T-cells. J Gen Virol 2011;92:2339–49. [DOI] [PubMed] [Google Scholar]
- [16].LaMere MW, Lam HT, Moquin A, Haynes L, Lund FE, Randall TD, et al. Contributions of antinucleoprotein IgG to heterosubtypic immunity against influenza virus. J Immunol 2011;186:4331–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Guo H, Santiago F, Lambert K, Takimoto T, Topham DJ. T cell-mediated protection against lethal 2009 pandemic H1N1 influenza virus infection in a mouse model. J Virol 2011;85:448–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature 1998;393:480–3. [DOI] [PubMed] [Google Scholar]
- [19].Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol 2013;13:145–9. [DOI] [PubMed] [Google Scholar]
- [20].Luckheeram RV, Zhou R, Verma AD, Xia B. CD4+T Cells: differentiation and functions. Clin Dev Immunol 2012:925135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].EMBL. https://www.ebi.ac.uk/ipd/imgt/hla/stats.html.
- [22].Stoloff GA, Caparros-Wanderley W. Synthetic multi-epitope peptides identified in silico induce protective immunity against multiple influenza serotypes. Eur J Immunol 2007;37:2441–9. [DOI] [PubMed] [Google Scholar]
- [23].Berthoud TK, Hamill M, Lillie PJ, Hwenda L, Collins KA, Ewer KJ, et al. Potent CD8+ T-cell immunogenicity in humans of a novel heterosubtypic influenza A vaccine, MVA-NP+M1. Clin Infect Dis 2011;52:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Valkenburg SA, Mallajosyula VV, Li OT, Chin AW, Carnell G, Temperton N, et al. Stalking influenza by vaccination with pre-fusion headless HA mini-stem. Sci Rep 2016;6:22666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Nachbagauer R, Krammer F. Universal influenza virus vaccines and therapeutic antibodies. Clin Microbiol Infect 2017;23:222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Krammer F, Hai R, Yondola M, Tan GS, Leyva-Grado VH, Ryder AB, et al. Assessment of influenza virus hemagglutinin stalk-based immunity in ferrets. J Virol 2014;88:3432–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nachbagauer R, Miller MS, Hai R, Ryder AB, Rose JK, Palese P, et al. Hemagglutinin Stalk Immunity Reduces Influenza Virus Replication and Transmission in Ferrets. J Virol 2015;90:3268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Baz M, Boonnak K, Paskel M, Santos C, Powell T, Townsend A, et al. Nonreplicating influenza A virus vaccines confer broad protection against lethal challenge. MBio 2015;6:e01487–e1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.