

Abstract
Although immune checkpoint inhibitors (ICIs), such as anti-programmed cell death protein 1 (PD-1), can deliver durable anti-tumor effects, most patients with cancer fail to respond. Recent studies suggest that ICI efficacy correlates with a higher load of tumor-specific neoantigens and development of vitiligo in patients with melanoma. Here, we report that patients with low melanoma neoantigen burdens who responded to ICIs had tumors with higher expression of pigmentation-related genes. Moreover, expansion of peripheral blood CD8+ T cell populations specific for melanocyte antigens was observed only in patients who responded to anti-PD-1 therapy, suggesting that ICIs can promote breakdown of tolerance toward tumor-lineage self-antigens. In a mouse model of poorly immunogenic melanomas, skewing of epitope recognition toward wild type melanocyte antigens was associated with markedly improved anti-PD-1 efficacy in two independent approaches: introduction of neoantigens by ultraviolet (UV) B radiation mutagenesis, or the therapeutic combination of ablative fractional photothermolysis plus imiquimod. Complete responses against UV mutation-bearing tumors after anti-PD-1 resulted in protection from subsequent engraftment of melanomas lacking any shared neoantigens, as well as pancreatic adenocarcinomas forcibly overexpressing melanocyte-lineage antigens. Our data demonstrate that somatic mutations are sufficient to provoke strong anti-tumor responses after checkpoint blockade, but long-term responses are not restricted to these putative neoantigens. Epitope skewing toward T cell recognition of wild type tumor-lineage self-antigens represents a common pathway for successful response to ICIs, which can be evoked in neoantigen-deficient tumors by combination therapy with ablative fractional photothermolysis and imiquimod.
One Sentence Summary:
Neoantigens sensitize melanomas to checkpoint blockade and trigger epitope skewing to self-antigens, a process mimicked by combination immunotherapy.
Introduction
Clinical responses to immune checkpoint inhibitors (ICIs) targeting the cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) (1) and programmed cell death-1 (PD-1) pathways (2, 3) achieve impressive and durable clinical benefit in a subset of patients with cancer. T cell infiltration and spontaneous tumor inflammation (4–7) and higher putative neoantigen loads are associated with enhanced efficacy of ICI in melanoma, non-small cell lung cancer, and colorectal cancer (8–10). However, the proportion of nonsynonymous mutations encoding neoantigens for which specific T cells can be detected is low in many tumor types, and recurrent classes of neoantigens associated with response to ICIs are not found in all melanoma cohorts (11). Moreover, numbers of mutations and putative neoantigens do not predict clinical benefit for individuals, as excellent responses to checkpoint blockade are still observed in some patients with low neoantigen burdens.
In patients with melanoma, ICI treatment is often associated with development of vitiligo, a depigmentation disorder resulting from autoimmune destruction of melanocytes. Vitiligo appears after anti-PD-1 therapy in one of four patients with melanoma (12), but not other cancers, suggesting that it is a melanocyte-lineage-specific adverse event. A recent prospective study reported that development of vitiligo among patients with melanoma is associated with higher rates of objective responses to anti-PD-1 therapy (12). Vitiligo likely arises from immune responses against melanocyte-lineage antigens shared by melanocytes and melanomas rather than neoantigens, but it is unclear whether autoimmunity against these wild type melanocyte antigens contributes to melanoma clearance, or whether vitiligo development is simply a correlating adverse effect.
Here, we identify a functional role of self-reactive T cells recognizing wild type melanocyte-lineage antigens in successful immunity against neoantigen-bearing melanomas after anti-PD-1 therapy. In poorly immunogenic melanomas and pancreatic ductal adenocarcinomas with few neoantigens, response to checkpoint blockade can be rescued by a combinatorial therapeutic strategy that promotes epitope skewing toward wild type tumor-lineage antigens. These results establish a functional role for tumor lineage-specific autoimmunity in responses to checkpoint blockade. They also provide a rationale for optimization of cancer therapies that enhance expression, presentation, or epitope skewing toward wild type tumor-lineage self-antigens.
Results
Recognition of wild type melanocyte antigens following immune checkpoint blockade in patients with melanoma
To understand determinants of response in patients without large neoantigen burdens, we first interrogated a dataset of pre-treatment melanoma biopsies from patients receiving the CTLA-4 inhibitor ipilimumab (11). Gene set enrichment analysis (GSEA) (13) of RNA-sequencing profiles of patients with comparably low neoantigen loads [10-100 predicted neoantigens with ≤50 nM binding affinity (11, 14) or 100-1000 predicted neoantigens with ≤500 nM binding affinity for human leukocyte antigen (HLA) class I molecules (15)] revealed that the top Gene Ontology (GO) biological process gene sets enriched in ipilimumab responders compared with non-responders included multiple pigmentation-related gene sets (data file S1 and fig. S1A). Moreover, the GO:0043473 pigmentation gene set was enriched in low neoantigen ipilimumab responders compared to non-responders (Fig. 1A) as well as nivolumab or pembrolizumab responders from two additional cohorts of patients with melanoma (Fig. 1, B and C) (14, 15). A caveat of these analyses is that they are limited by having used thresholds to define low neoantigen subsets with comparable neoantigen burdens between responders and non-responders (fig. S1, B and C).
Fig. 1. Anti-PD-1 therapy increases recognition of wild type melanocyte antigens in patients with melanoma.
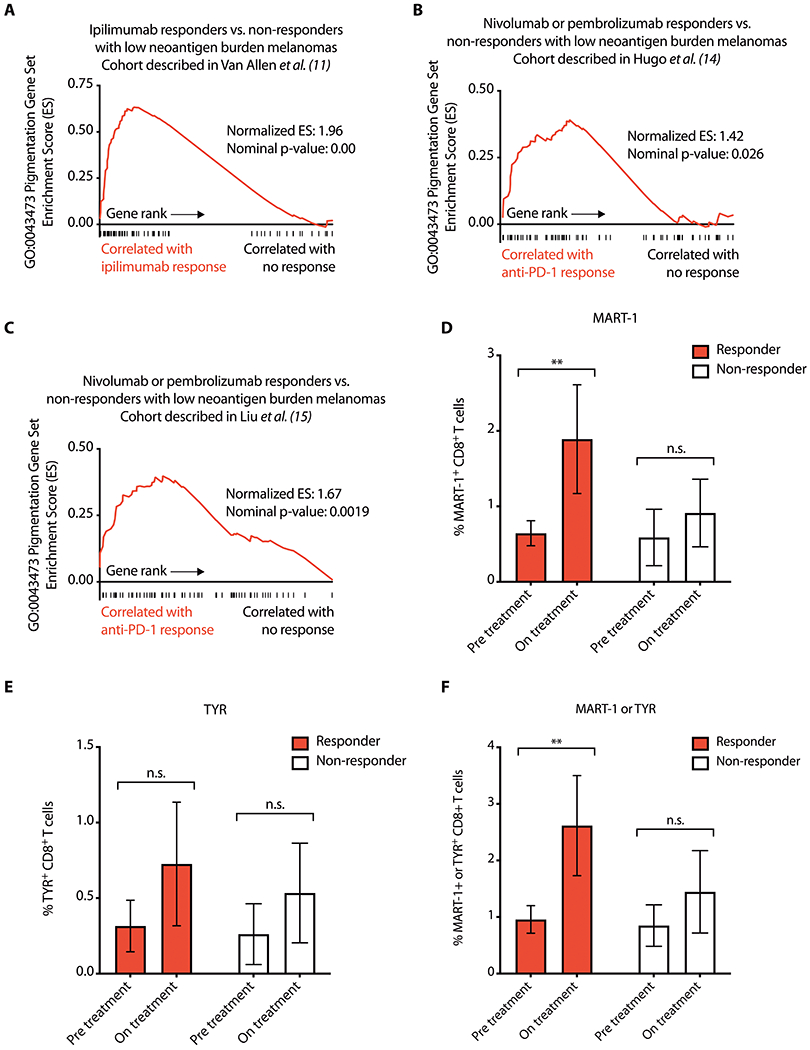
(A) GSEA plot showing enrichment of the pigmentation gene set GO:0043473 in ipilimumab responders among patients with melanoma in the Van Allen et al. dataset (11) with low neoantigen burdens. (B and C) GSEA plot showing enrichment of the pigmentation gene set GO:0043473 in nivolumab or pembrolizumab responders among patients with melanoma with low neoantigen burdens in the Hugo et al. dataset (14) (B) or Liu et al. dataset (15) (C). (D to F) Frequencies of HLA-A*0201:MART-1 (ELAGIGILTV) dextramer+ (D), HLA-A*0201:TYR (YMDGTMSQV) dextramer+ (E), or combined HLA-A*0201:MART-1 and HLA-A*0201:TYR dextramer+ (F) CD8+ T cells in peripheral blood mononuclear cells from 18 patients with metastatic melanoma treated with anti-PD-1. Data are shown as mean ± SEM (n=13 responders, n=5 non-responders). **p<0.01, n.s. not significant using a Wilcoxon paired t-test. ES: enrichment score.
The GO:0043473 pigmentation gene set includes many melanocyte differentiation antigens that are shared by melanoma cells and normal melanocytes and have been implicated as immune targets in the pathogenesis of vitiligo (16). We hypothesized that development of immune activity against wild type melanocyte antigens contributes to efficacy of anti-CTLA-4 and anti-PD-1 therapies, and thus higher expression of tumor-lineage self-antigens in melanoma may compensate for low neoantigen burden.
To test this hypothesis, we assessed antigen-specific CD8+ T cells in pre-treatment and on anti-PD-1 therapy peripheral blood samples from HLA-A02+ patients with melanoma, including 13 responders and 5 non-responders (data file S1). We measured the proportion of circulating CD8+ T cells specific for the melanocyte antigens melanoma antigen recognized by T cells 1 (MART-1) and tyrosinase (TYR) using human leukocyte antigen (HLA) class 1 dextramers. Anti-PD-1 treatment was associated with expansion of MART-1-specific CD8+ T cells in responders but not non-responders (p = 0.0046, Fig. 1D and fig. S1D). No significant increase in TYR-specific CD8+ T cells was observed in our small cohort (Fig. 1E). When the proportion of CD8+ T cells specific to MART-1 and TYR were combined, we also found an increase on anti-PD-1 treatment in responders (p = 0.0081) but not in non-responders (Fig. 1F). Although vitiligo is a striking and visible adverse event associated with immunotherapies, immune responses against melanocyte antigens that fail to reach the threshold for clinical vitiligo development may still constitute clinically meaningful activity against melanoma cells. In this cohort, anti-PD-1 therapy led to an expansion of MART-1-specific CD8+ T cells in responders, but only 5 of 13 responders developed vitiligo (data file S1).
Ultraviolet B radiation-associated somatic mutations enhance response to checkpoint blockade
To study the role of wild type self-antigen recognition in the response to checkpoint blockade and the contribution of tumor-specific neoantigens in vivo, we used a mouse melanoma model. We derived a stable cell line (D4M.3A.3, “parental”) from a single cell clone of the poorly immunogenic D4M.3A melanoma cell line (17) established from a Tyr::CreER;BrafCA;Ptenlox/lox mouse (18) fully backcrossed to the C57BL/6 background. Exome sequencing revealed that, compared to the wild type C57BL/6 reference genome, this parental D4M.3A.3 cell line contains 1.78 mutations/Mb, far fewer than the 46.86 mutations/Mb present in the B16-F10 mouse melanoma cell line (data file S1). To mimic the mutagenic effects of sun exposure, we subjected our parental D4M.3A.3 melanoma line to ultraviolet B (UVB) irradiation in vitro and isolated single cell “UV clones”. Two clones, D3UV2 (“UV2”) and D3UV3 (“UV3”), had the same in vitro growth kinetics (fig. S2A and B) and expression of PD-1, programmed cell death 1 ligand 1 (PD-L1), and major histocompatibility complex (MHC) class I and II following stimulation with interferon-γ (IFNγ) (fig. S2C). There was no difference in pigment gene expression between UV2 and the parental D4M.3A.3 cell line as determined by GSEA (fig. S2D). Compared to the parental D4M.3A.3 cell line, exome sequencing of UV2 and UV3 revealed an additional 61.08 and 58.78 mutations/Mb, respectively (data file S1), which is within the 0.1 to 100/Mb range of somatic mutation rates reported in human melanomas (19). As expected, most mutations resulted from C>T transitions associated with UVB mutagenesis and occurred at a 2:1 ratio of nonsynonymous to synonymous events (fig. S2E and data file S1).
In vivo growth kinetics of melanoma grafts generated by subcutaneous flank inoculation of 1 × 106 parental D4M.3A.3 or UVB-mutagenized cells in 0.1 ml of phosphate buffered saline (PBS) were comparable in immunodeficient nonobese diabetic (NOD)/severe combined immunodeficient (SCID)/γchainnull (NSG) mice (Fig. 2A). However, implantation of melanoma cells into immunocompetent syngeneic C57BL/6 hosts resulted in slower tumor growth of UV2 and UV3 tumors than parental D4M.3A.3 melanomas, resulting in longer survival (p = 0.002 for UV2; p = 0.002 for UV3, Fig. 2B and C). Anti-PD-1 treatment of C57BL/6 mice was initiated on day 8 after tumor cell inoculation. Compared to parental D4M.3A.3 melanomas, survival of mice with UV clone tumors was improved by anti-PD-1 (p = 0.022 for UV2; p = 0.009 for UV3), with stable complete clearance of 20 to 60% of these tumors versus 0% of parental D4M.3A.3 melanomas (Fig. 2, B and C). The UV2 melanoma cell line was selected for further analysis because of its greater immunogenicity and response rate to anti-PD-1 treatment (Fig. 2B).
Fig. 2. UVB-induced mutations enhance anti-tumor immunity and recognition of wild type melanocyte antigens after PD-1 blockade.
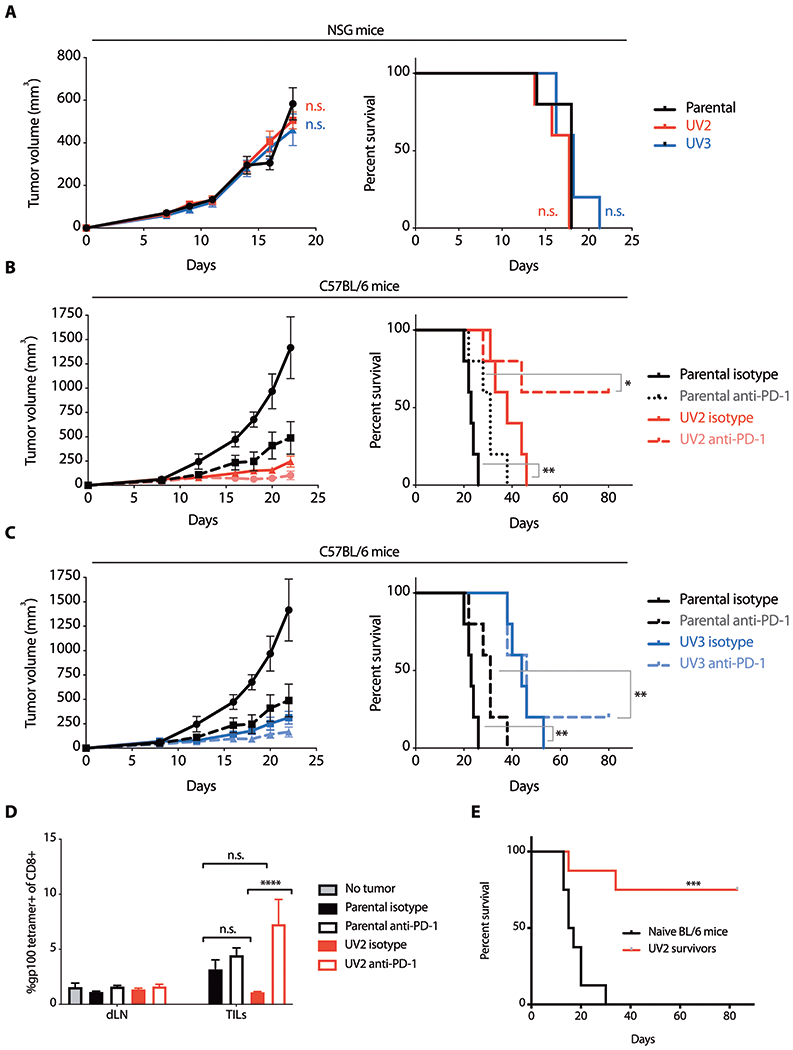
(A) Growth of parental D4M.3A.3 and UVB-mutagenized UV2 and UV3 melanomas in NSG mice and corresponding survival. Data are shown as mean tumor size ± SEM (n=5 per group). (B and C) Parental D4M.3A.3 and UV2 (B) or UV3 (C) melanoma growth in C57BL/6 mice and corresponding survival after treatment with anti-PD-1 or isotype-matched control antibody on days 8, 10, 12, 14, and 16 after tumor cell inoculation. Data are shown as mean tumor size ± SEM (n=5 per group). (D) Tumor-infiltrating CD8+ T cells isolated from parental D4M.3A.3 or UV2 melanomas (TILs) or CD8+ T cells isolated from draining lymph nodes (dLNs) harvested 5 days after initiation of anti-PD-1 therapy or isotype control were evaluated for binding to gp100:H-2Db tetramer (n=3 mice for no tumor control, n=8 mice for all other groups). Data are shown as mean ± SEM. (E) Survival of UV2 melanoma-bearing mice with complete responses to anti-PD-1 and age-matched naïve C57BL/6 mice after rechallenge with subcutaneous parental D4M.3A.3 melanoma cell inoculation (n=8 mice per group). *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; n.s., no significant difference for statistical analyses conducted by two-way analysis of variance (ANOVA) with Tukey’s post hoc test [A (left panel) and D] or log-rank test [A (right panel), B, C, and E].
Somatic mutations increase melanoma inflammation and epitope skewing to wild type melanocyte antigens after checkpoint blockade
To further investigate responses against wild type melanocyte antigens, we measured gp100:H-2Db tetramer staining in tumor-infiltrating CD8+ T cells. Glycoprotein 100 (gp100), also known as premelanosome protein PMEL, is a melanocyte and melanoma antigen enriched in melanosomes. In low mutational load parental D4M.3A.3 melanomas, anti-PD-1 treatment did not measurably affect the proportion of CD8+ T cells recognizing gp100 antigen (Fig. 2D). In contrast, anti-PD-1 treatment of mice bearing UV2 melanomas increased the gp100-specific CD8+ T cell population (p < 0.0001, Fig. 2D), similar to findings in human circulating CD8+ T cells (Fig. 1, D to F). Parental D4M.3A.3 and UV2 melanoma grafts exhibited comparable expression of gp100 mRNA (fig. S3A). We next sought to evaluate the functional importance of shared melanocyte-lineage antigens as T cell targets in long-term anti-tumor immunity. To do this, mice with complete UV2 melanoma regressions after anti-PD-1 treatment were rechallenged with inoculation of 1 x 105 parental D4M.3A.3 melanoma cells in 0.1 ml PBS on the opposite flank (Fig. 2E). When challenged over 3 months after treatment, six of eight UV2 melanoma survivors rejected parental D4M.3A.3 melanomas, although initial anti-PD-1-mediated tumor regression in these mice was dependent on the presence of UVB-induced mutations (Fig. 2B). Thus, UVB-induced mutations were sufficient to provoke an initial anti-melanoma immune response (Fig. 2, B and C). However, responses to a secondary tumor challenge were not restricted to these same putative neoantigens.
GSEA of RNA-sequencing data from bulk tumors (data file S1) revealed strong enrichment of multiple immune-associated gene sets in UV2 melanomas compared with parental D4M.3A.3 melanomas, extending across innate and adaptive immunity (Fig. 3A and data file S1). Immunohistochemical analysis revealed higher numbers of tumor-infiltrating T cells in UV2 than in parental D4M.3A.3 melanomas (p < 0.0001, Fig. 3B and C). By flow cytometry, UV2 tumors contained more CD8+ T cells (p = 0.008, Fig. 3D) and had correspondingly greater immune cytolytic activity (fig. S3B) (20). However, this was accompanied by evidence of impaired CD8+ T cell responses, such as greater numbers of CD4+FOXP3+ T regulatory (Treg) cells (p = 0.003) (Fig. 3D). In addition, transcriptional analysis of UV2 tumors revealed increased expression of inhibitory receptors and molecules, including interleukin-10 (IL-10), programmed cell death 1 ligand 2 (PD-L2), C-C motif chemokine 22 (CCL22), and lymphocyte-activation gene 3 (LAG3) (fig. S3C). Treatment of UV2 tumors with anti-PD-1 increased the proportion of CD8+ T cells positive for Ki67 (p < 0.0001, Fig. 3E). In contrast, in parental D4M.3A.3 melanomas anti-PD-1 treatment did not alter the CD8:Treg ratio or the proportion of GzmB+ CD8+ T cells and had only a small effect on the proportion of CD8+ T cells expressing Ki67 (p = 0.016; Fig. 3E).
Fig. 3. UVB irradiation promotes recruitment of tumor-infiltrating immune cells and is associated with T cell dysfunction that is reversed by PD-1 blockade.
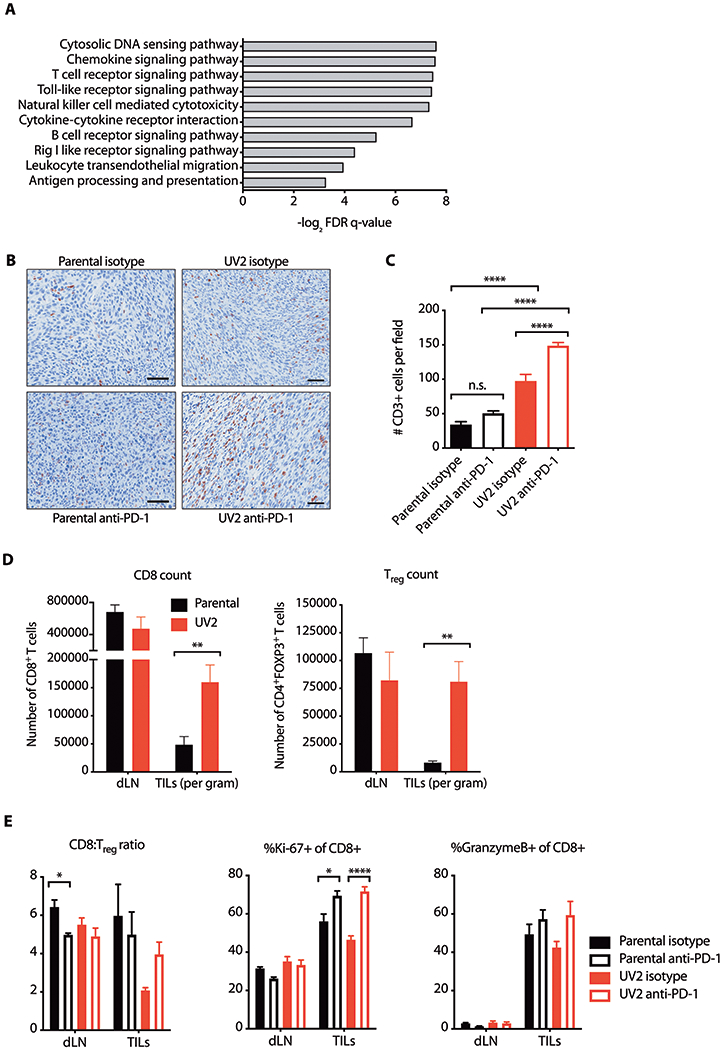
(A) GSEA of RNA-sequencing data from bulk tumor grafts in C57BL/6 hosts. Representative top-scoring KEGG gene sets enriched in UV2 compared with parental D4M.3A.3 melanomas with nominal p values<0.01 are shown. FDR, false discovery rate. (B and C) CD3+ expression in parental D4M.3A.3 and UV2 melanomas 5 days after initiation of therapy was evaluated by immunohistochemistry. (B) Representative fields shown. Scale bar: 50 μm. (C) CD3+ expression quantified in nine randomly selected intratumoral fields from three mice per group. Data are presented as mean ± SEM. ****p<0.0001; n.s. not significant as evaluated by one-way ANOVA with Tukey’s post hoc test. (D) Tumor infiltrating lymphocytes (TILs) or cells isolated from draining lymph nodes (dLNs) harvested 5 days after anti-PD-1 therapy initiation were characterized by flow cytometry. Numbers of CD8+ and Treg (CD4+FOXP3+) T cells per gram are shown as mean ± SEM (n=12 pooled to 6 per group). **p<0.01 as evaluated by two-tailed t-test. (E) TILs or cells isolated from draining lymph nodes (dLNs) harvested 5 days after anti-PD-1 therapy initiation were characterized by flow cytometry. Ratios of CD8+ T cells to Treg cells, and proportions of CD8+ T cells positive for Ki67 or granzyme B are shown as means ± SEM (n=12 pooled to 6 per group). *p<0.05; ****p<0.0001 as evaluated by one-way ANOVA with Tukey’s post hoc test.
Rescuing response to checkpoint inhibition in cancers with low mutational burdens
Our data demonstrate that implantation of the parental D4M.3A.3 melanoma line in vivo provides a stringent in vivo model of neoantigen-deficient, poorly inflamed, immunotherapy-resistant tumors. To extend these findings to patients with melanoma, we hypothesized that stimulating inflammation in a localized tumor lesion could improve broad systemic responses against wild type melanocyte antigens after immune checkpoint blockade. To this end, we used two local treatments: ablative fractional photothermolysis (aFP) (21) and topical imiquimod, a Toll-like receptor 7 (TLR7) agonist used in the treatment of basal cell and squamous cell carcinomas, actinic keratoses, and lentigo maligna melanoma (22, 23). Higher expression of TLR7 is associated with longer survival in patients with melanoma (fig. S4A).
Mice with bilateral flank parental D4M.3A.3 melanomas were treated with indicated combinations of imiquimod, aFP, and anti-PD-1 starting on day 6 after tumor inoculation (Fig. 4, A and B, and fig. S4B). Treatments of aFP and topical imiquimod were applied to only one tumor per mouse, whereas anti-PD-1 treatment was administered systemically. The parameters of aFP were adjusted to ablate only ~2.5 to 5% of subcutaneous tumor volumes, thereby aiming to enhance inflammation without eliminating tumor-infiltrating immune cell populations. Complete response rates, with complete regression of both tumors, improved from 0% with any single agent therapy to 10% with any combination of two treatments, to 50% after the triple combination of imiquimod+aFP+anti-PD-1 (p = 0.0003, Fig. 4, A and B, and fig. S4B). Combinatorial benefit of triple therapy with anti-CTLA-4 instead of anti-PD-1 (imiquimod+aFP+anti-CTLA-4) was also observed, with complete responses in 25% of mice (p = 0.007; fig. S4C). Virtually identical responses were observed in tumors on both mouse flanks despite only unilateral imiquimod+aFP treatments (fig. S4D), suggesting that local administration of imiquimod and aFP mediates an abscopal effect against mutation-deficient contralateral melanomas when combined with checkpoint inhibition. Vitiligo was observed in only 1 of 24 evaluable mice with complete responses against melanoma (fig. S4E). Furthermore, no gross toxicities such as rashes, hair loss, poor diet, diarrhea or colitis, sudden death, or failure to thrive were observed.
Fig. 4. Imiquimod and aFP improve responses of poorly immunogenic melanoma and pancreatic ductal adenocarcinoma to checkpoint blockade.
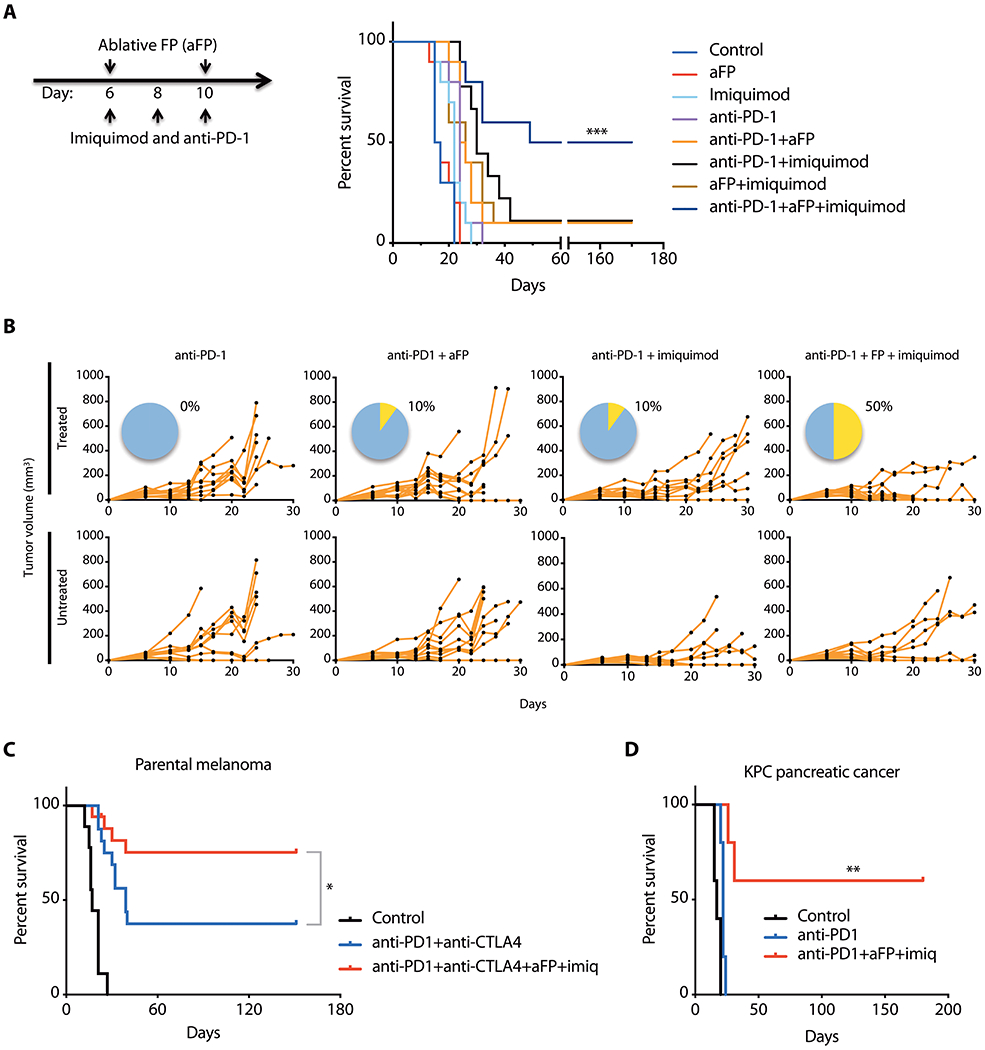
(A) Survival of C57BL/6 mice after parental D4M.3A.3 melanoma inoculation (day 0) and combination treatments using anti-PD-1, ablative fractional photothermolysis (aFP), and imiquimod administered on the days indicated (n=10 mice per group). ***p<0.001 comparing triple therapy to anti-PD-1 using a log-rank test. (B) Spider plots show growth of individual tumors in C57BL/6 mice on treated and untreated flanks after therapy with aFP and/or imiquimod administered to the treated tumor only. Pie charts show percent complete responses with regression of tumors on both flanks (yellow). (C) Survival of C57BL/6 mice after parental D4M.3A.3 melanoma inoculation and treatment with isotype-matched antibodies (n=9), anti-PD-1+anti-CTLA-4 (n=16), or quadruple therapy using anti-PD-1+anti-CTLA-4+aFP+imiquimod (n=17). *p<0.05 using a log-rank test. (D) Triple therapy induces tumor regression in a mouse model of poorly immunogenic pancreatic ductal adenocarcinoma. Survival of C57BL/6 mice after subcutaneous inoculation of KPC pancreatic cancer cells and treatment with isotype-matched antibody, anti-PD-1, or triple therapy using anti-PD-1+aFP+imiquimod (n=5 per group). **p<0.01 comparing triple therapy to anti-PD-1 using a log-rank test.
Responses to anti-PD-1+anti-CTLA-4 in mice with parental D4M.3A.3 melanomas were superior to either alone (p < 0.0001, Fig. 4C). Addition of imiquimod plus aFP further increased the complete bilateral complete response rate to 75% (p = 0.043; Fig. 4C and fig. S4F). We additionally tested imiquimod+aFP+anti-PD-1 triple therapy as a treatment for pancreatic ductal adenocarcinoma using the transplantable syngeneic genetically-engineered KPC mouse model (KrasLSL.G12D;p53R172H;Pdx1::Cre) (24). Whereas anti-PD-1 monotherapy initiated on day 6 after tumor implantation provided no benefit, triple therapy induced bilateral pancreatic tumor regressions with durable complete responses in 60% of mice (p = 0.002; Fig. 4D and fig. S4G). Last, when the brand of imiquimod used in our initial experiments was no longer available for purchase, we tested a different brand/formulation that similarly demonstrated efficacy of triple therapy (fig. S4H).
Combination immunotherapy confers long-term immunity against shared tumor-lineage antigens
RNA-sequencing was performed to investigate mechanisms underlying triple therapy responses against low mutational burden tumors. In parental D4M.3A.3 melanomas treated with imiquimod+aFP+anti-PD-1 compared to anti-PD-1 alone, GSEA identified enrichment of several immune-related KEGG gene sets (Fig. 5A and data file S1). In parental D4M.3A.3 melanomas, addition of imiquimod and aFP to checkpoint inhibitor antibodies substantially increased intratumoral CD3+ T cell density (p < 0.0001) and the CD8:Treg ratio (p < 0.0001) compared to isotype-control antibodies or anti-PD-1, respectively (Fig. 5B, C, and E). Imiquimod alone (p < 0.0001) or with anti-PD-1 (p = 0.029) or aFP (p = 0.0005) expanded the granzyme B+ fraction of CD8+ tumor infiltrating lymphocytes (TILs) (Fig. 5D and E). In draining lymph nodes (dLNs), PD-L2 expression on CD11c+ dendritic cells (DCs) was reduced by imiquimod, suggesting that imiquimod makes DCs less suppressive (p < 0.0001, Fig. 5D). Similar changes were observed in both directly treated and untreated contralateral tumors and dLNs, indicating that local imiquimod has broad immune effects (fig. S5A and B). These data suggest that imiquimod enhances antigen presentation and activates the T cell compartment independently of anti-PD-1, but anti-PD-1 is needed to increase T cell infiltration, proliferation, and effector activity in tumors. Depletion of CD8+ T cells abrogated triple therapy-mediated parental D4M.3A.3 melanoma regression and survival (p < 0.0001; Fig. 5F and fig. S5C). Tumor rejection was also dependent on Batf3 expression (p = 0.0005; Fig. 5G and fig. S5D), suggesting that triple therapy efficacy requires cross-priming of anti-tumor CD8+ T cells (25–28).
Fig. 5. Addition of imiquimod and aFP enhances the frequency and function of tumor-infiltrating T cells after checkpoint blockade.
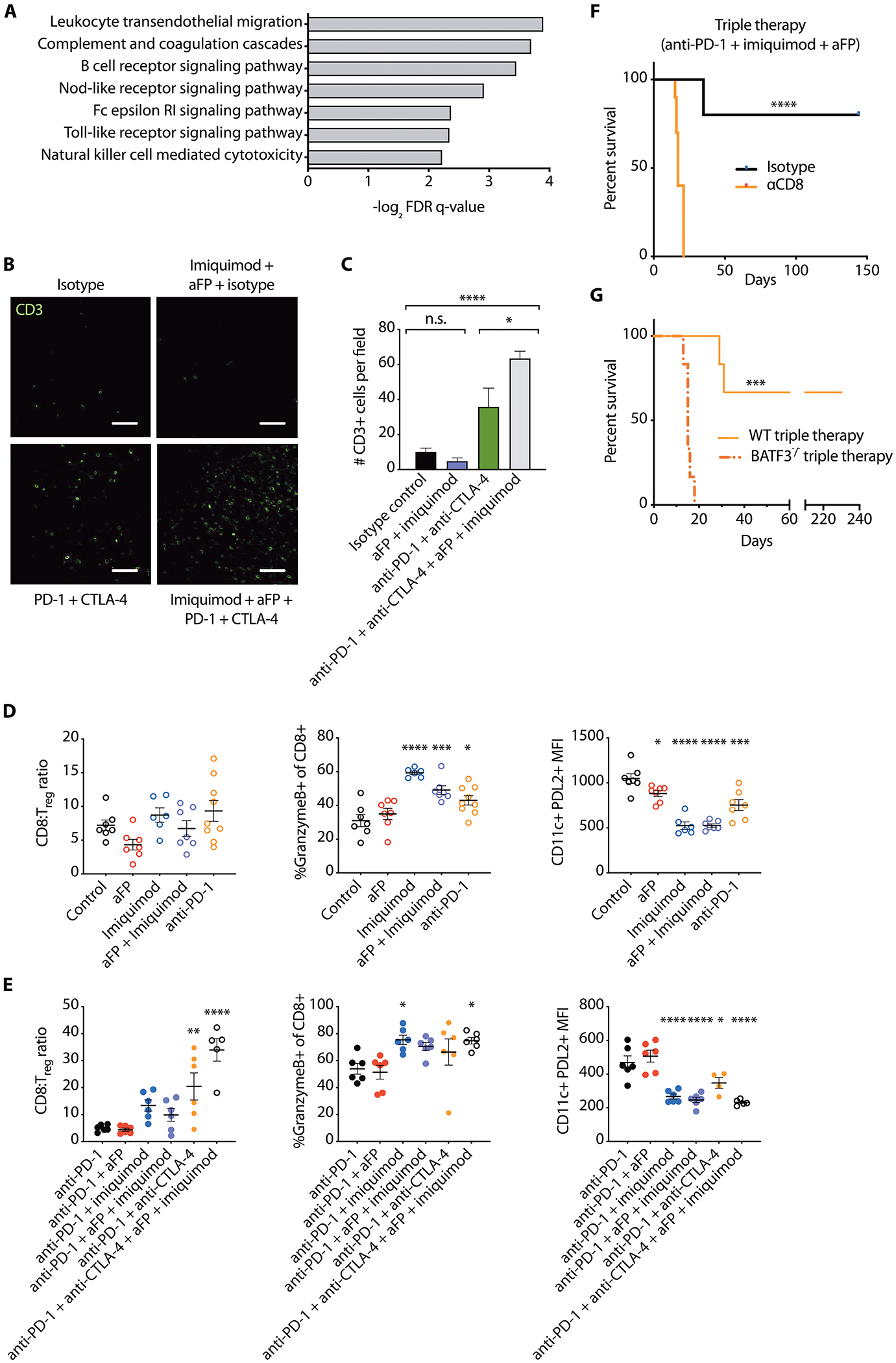
(A) Representative top-scoring KEGG gene sets enriched in bulk parental D4M.3A.3 melanomas in C57BL/6 mice treated with triple therapy (anti-PD-1+aFP+imiquimod) compared to anti-PD-1 monotherapy with nominal p values<0.01. FDR, false discovery rate. (B) Representative fields of immunofluorescence microscopy and (C) quantification of six randomly selected fields reveal CD3+ cells in parental melanomas 5 days after initiation of therapy. Scale bar: 100 μm. Data are presented as mean ± SEM. *p<0.05; ****p<0.0001; n.s. not significant by one-way ANOVA with Tukey’s post hoc test. (D and E) Immune infiltrates in contralateral (untreated flank) tumors and dLNs 5 days after initiation of indicated treatments were characterized by flow cytometry. Ratios of CD8+ T cells to Tregs (CD4+FoxP3+) and proportion of CD8+ T cells that are positive for granzyme B in tumors are shown, as well as expression of PD-L2+ on CD11c+ DCs in dLNs. Data are shown as mean ± SEM; in (D) n≥=6 mice per group; in (E) n=12 pooled to 6 per group. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 compared with isotype control (D), or anti-PD-1 (E) by one-way ANOVA with Dunnett’s post hoc test. (F) Survival of mice receiving anti-CD8 depleting antibodies or isotype-matched control antibodies after inoculation of parental D4M.3A.3 melanoma cells and triple therapy. n=10 mice per group. ****p<0.0001 by log-rank test. (G) Survival of C57BL/6 and BATF3−/− mice following parental melanoma inoculation (day 0) and triple therapy. n=6 mice per group. ***p<0.001 by log-rank test. WT, wild type.
Of note, GSEA revealed that the same GO pigmentation gene set enriched in low neoantigen patient ipilimumab or anti-PD-1 responders compared with non-responders (Fig. 1, A to C) is enriched in parental D4M.3A.3 mouse melanomas after triple therapy (Fig. 6A), but not after anti-PD-1 monotherapy. Further, protein expression of gp100 and another well-known melanocyte gene, tyrosinase related protein-2 (TRP2), was shown by immunofluorescence in tumor samples isolated from mice treated with triple therapy but not with anti-PD-1 monotherapy in parental D4M.3A.3 melanomas (Fig. 6B and fig. S6, A and B). Multiple pathways including cytokine signaling (29) and stress responses such as induction of p53 (30) have been reported to regulate melanocyte antigen expression. p53 Hallmark and KEGG pathways are also enriched after triple therapy in parental D4M.3A.3 melanomas (data file S1) and may respond to intratumoral inflammation and thermal injury by aFP.
Fig. 6. Combination immunotherapy promotes recognition and long-term immunity against wild type tumor-lineage self-antigens.
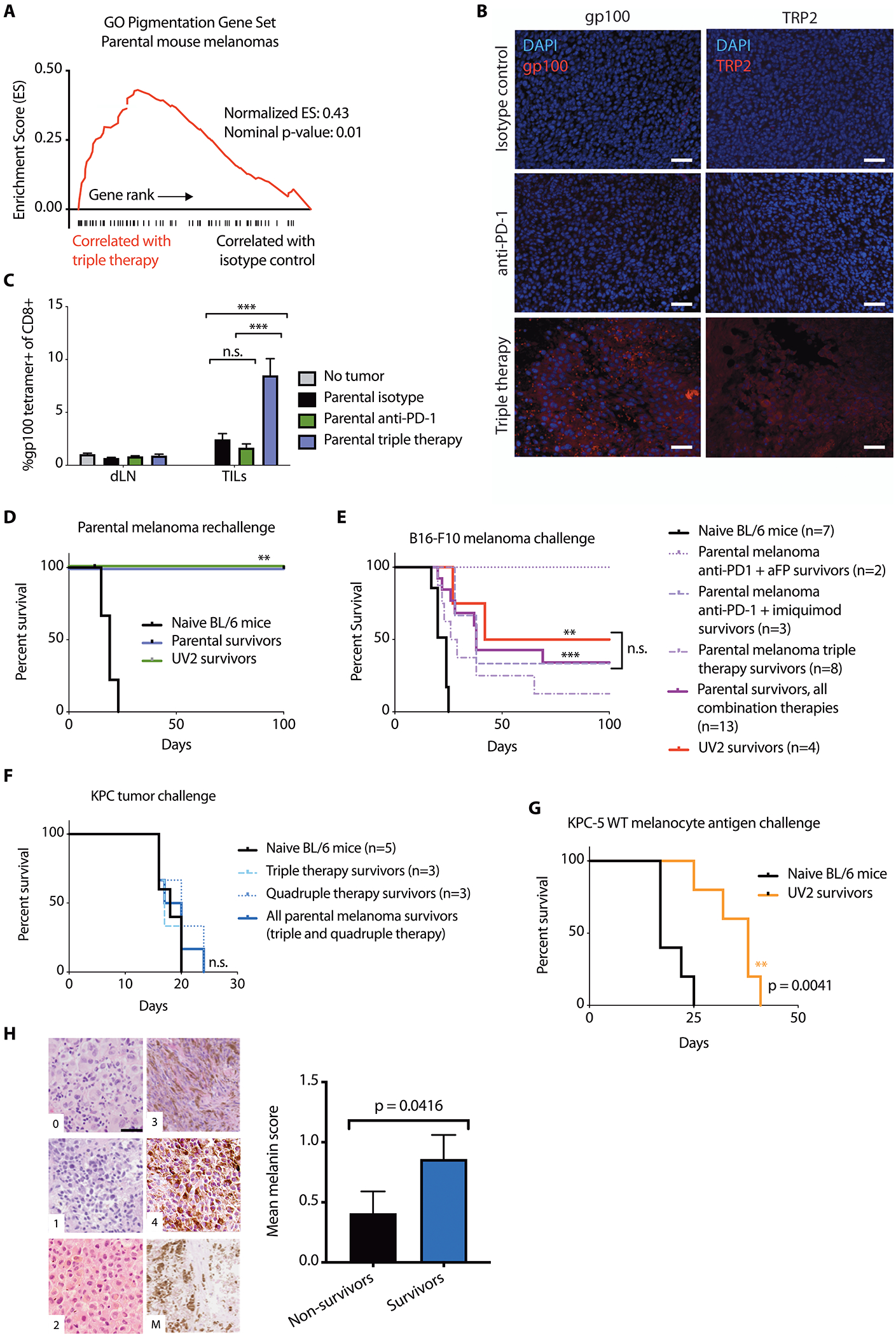
(A) GSEA plot showing enrichment of pigmentation gene set GO:0043473 in triple therapy (anti-PD-1+aFP+imiquimod)-treated mouse parental D4M.3A.3 melanomas. (B) Immunofluorescence imaging reveals expression of gp100 (left) and TRP2 (right) after anti-PD-1+aFP+imiquimod triple therapy in mouse parental melanomas 5 days after initiation of therapy. Scale bar: 50 μm. (C) CD8+ T cells from treated flank parental melanomas (TILs) and from draining lymph nodes (dLNs) 5 days after initiation of therapy were evaluated for binding to gp100:H-2Db tetramer (n=4 mice for no tumor control, n=8 for all other groups). Data are shown as means ± SEM. (D) Survival of parental (n=3) or UV2 (n=3) melanoma-bearing mice with complete responses to triple therapy or naïve mice (n=10), after challenge with 1 × 105 parental melanoma cells. (E) Survival of UV2 melanoma-bearing mice with complete responses to anti-PD-1+anti-CTLA-4 (n=4), or parental melanoma-bearing mice with complete responses to triple therapy (n=8), anti-PD-1+aFP (n=2), anti-PD-1+imiquimod (n=3), or naïve mice (n=7), after challenge with 1 x 105 B16-F10 melanoma cell inoculation. (F) Survival of parental melanoma-bearing mice with complete responses to triple therapy (n=3) or quadruple therapy (n=3), or naïve mice (n=5), after subcutaneous challenge with 1 × 105 KPC cells. (G) Survival of UV2 melanoma-bearing mice with complete responses to anti-PD-1 (n=5), or naïve mice (n=5) after challenge with subcutaneous inoculation of 1.5 × 105 KPC-5 wild type melanocyte antigen combo cells. (H) Tumor samples from patients with melanoma treated with anti-PD-1 were scored in a blinded manner based on melanin content in hematoxylin and eosin sections. Representative images (left) depict cytoplasmic melanin scoring using published parameters (37). Scale bar: 20 μm. Patient melanin scores were stratified by survival status at 2 years after starting anti-PD-1 (right). For patients with multiple tumor blocks available (from multiple masses or time points), the mean score was calculated per patient. Data are shown as mean average melanin score ± SEM, n=22 non-survivors and 27 survivors at 2-year follow up. **p<0.01; ***p<0.001; n.s., no significant difference for statistical analyses conducted by one-way ANOVA with Tukey’s post hoc test (C), log-rank test (D to G), or Mann-Whitney test (H).
To test whether upregulated wild type melanocyte antigens might be targeted by T cells after triple therapy, we analyzed mouse parental D4M.3A.3 melanomas for melanocyte antigen recognition by tumor-infiltrating CD8+ T cells using gp100:H-2Db tetramer staining. Triple therapy produced a strong increase in the frequency of gp100-tetramer-positive CD8+ TILs (p < 0.001) compared with either no treatment or anti-PD-1 treatment alone (Fig. 6C). This larger population of gp100-specific CD8+ TILs in triple therapy-treated tumors retains the same functionality as gp100-specific CD8+ TILs from isotype or anti-PD-1-treated tumors, as demonstrated by comparable expression of Ki-67, granzyme B, CD44, and PD-1 (fig. S6C).
These results demonstrate that, in poorly immunogenic melanomas treated with anti-PD-1, either a high somatic mutation load or addition of imiquimod and aFP leads to measurable expansion of CD8+ T cell populations capable of recognizing wild type melanocyte antigens.
To evaluate long-term immunological anti-tumor memory, mice exhibiting complete regression of parental D4M.3A.3 melanomas after triple therapy were rechallenged with a second melanoma inoculation. Both UV2 melanoma (high mutational load) and parental D4M.3A.3 melanoma (low mutational load) survivors after triple therapy were resistant to rechallenge with parental D4M.3A.3 melanoma cells (p = 0.001, Fig. 6D). In addition, 38% of parental D4M.3A.3 melanoma survivors after combination immunotherapy (p = 0.0003) and 50% of UV2 melanoma survivors after anti-PD-1+anti-CTLA-4 therapy (p = 0.004) were protected against unrelated syngeneic B16-F10 mouse melanoma (Fig. 6E). Whole-exome sequencing revealed that B16-F10 shares no amino acid-altering mutations with the parental D4M.3A.3 or UV2 melanoma cell lines (data file S1). In contrast, parental D4M.3A.3 and UV2 melanoma survivors had no immunity against KPC pancreatic tumors (Fig. 6F and fig. S6D).
We next investigated whether expression of wild type melanocyte antigens is sufficient for productive memory responses in UV2 melanoma survivors. Mice with complete UV2 melanoma regression after anti-PD-1 monotherapy were subsequently challenged with inoculation of KPC pancreatic tumor cells overexpressing five wild type melanocyte antigens: gp100, Tyr, tyrosinase-related protein 1 (Tyrp1), melanoregulin (Mreg), and dopachrome tautomerase (Dct). Compared to naïve mice, UV2 melanoma survivors displayed longer survival after KPC-5 wild-type melanocyte antigen challenge (p = 0.004; Fig. 6G).
In a cohort of 49 patients with melanoma treated with anti-PD-1, we observed that long-term, sustained anti-tumor immune responses were associated with a more highly differentiated state with greater pigment production (Fig. 6H and fig. S6E). Increased melanogenesis results from upregulation of pigment-producing enzymatic machinery, including multiple wild type melanocyte antigens (31). Histologic sections of melanoma samples were scored for melanin content (Fig. 6H). Melanomas from survivors at 2-year follow up after initiation of anti-PD-1 therapy had higher melanin scores than tumors from non-survivors (p = 0.0416; Fig. 6H and fig. S6E). Together, these data suggest that immune targeting of wild type melanocyte-lineage antigens may be particularly important for durable responses to checkpoint blockade therapy in patients with melanoma.
Discussion
Our results demonstrate two mechanisms by which ICI responses can be enhanced: the presence of neoantigens or induction of local inflammation using aFP plus imiquimod. Furthermore, in our mouse models, introduction of putative neoantigens from somatic UVB-induced mutations was sufficient to provoke a stronger anti-melanoma response after checkpoint blockade. These results support evidence of enhanced ICI efficacy in patients with higher predicted neoantigen loads (8–10, 14) and in a recently reported UVB-induced mouse melanoma model (32). We observed that anti-PD-1 leads to expansion of gp100 tetramer-recognizing CD8+ TILs in mutation-rich, but not matched mutation-poor, melanomas, and this skewing of immune responses against wild type melanocyte-lineage antigens correlates with treatment efficacy.
A functional role for epitope skewing from neoantigens or melanoma-specific antigens to antigens shared by normal melanocytes is established by the resistance of UV2 anti-PD-1 complete responders to challenge with parental D4M.3A.3 melanomas sharing no putative neoantigens. Moreover, in UV2 melanoma survivors subsequently challenged with pancreatic tumor grafts, overexpression of just five wild type melanocyte antigens in pancreatic cancer cells was sufficient to confer a survival benefit. These data suggest that neoantigens, which can themselves be targets of anti-tumor activity, may also contribute to breaking tolerance toward wild type melanocyte self-antigens in the context of anti-PD-1 treatment.
For poorly immunogenic cancers bearing low mutational burdens, the aFP+imiquimod+immune checkpoint combination facilitates an abscopal effect, with destruction of both directly-treated and contralateral established tumors in mouse models of melanoma and pancreatic ductal adenocarcinoma with low putative neoantigen burdens. CD8+ T cells and Batf3 expression were necessary for combination therapy efficacy, and the addition of imiquimod and aFP to checkpoint inhibitor treatment promoted tumor inflammation, increased the tumor infiltrating CD8:Treg ratio, and enhanced antigen presentation.
Combination therapy mediated changes in gene expression associated with immune signaling and pigmentation. As observed with UV2 melanomas, addition of aFP and imiquimod overcomes tolerance toward the melanocyte self-antigen gp100 in parental D4M.3A.3 melanomas, but without introduction of more mutations. Published T cell receptor (TCR) sequencing of B16 mouse melanomas suggests that gp100-specific T cell clones, which are commonly found at low frequencies before immunotherapy, can be expanded by checkpoint blockade (33). Additional studies will help determine how checkpoint blockade affects priming versus amplification of T cell clones specific for other wild type melanocyte antigens. AFP produces microscopic channels of thermal injury, releasing denatured tumor and wild type antigens, with sparing of immune cells in intact interspersed tissue (34). T cell escape from self-tolerance after triple therapy is likely mediated by a combination of this local inflammation with TLR7 agonism by imiquimod-induced DC maturation.
Our in vivo studies revealed an association between recognition of wild type melanocyte antigens and long-term melanocyte-lineage-specific immunity. Parental D4M.3A.3 melanoma triple therapy complete responders and UV2 melanoma anti-PD-1 complete responders were resistant to subsequent challenge with B16-F10 melanomas that share no protein-coding mutations; however they demonstrated no evidence of protective immunity against pancreatic ductal adenocarcinoma tumors, which are of another cellular lineage. These combination therapy and epitope skewing findings have important implications for immunotherapy in patients with cancer, where low mutational burdens remain an obstacle to achieving durable clinical benefit.
In 18 patients with melanoma studied by dextramer staining of peripheral blood mononuclear cells, we observed increases in the proportion of MART-1-specific circulating CD8+ T cells after initiation of anti-PD-1 therapy in anti-PD-1 responders, but not in non-responders. Additional studies of larger groups of patients as well as use of immunopeptidomics will be helpful in determining whether T cell populations specific for MART-1, TYR, and other wild type melanocyte antigens could be useful biomarkers in monitoring responses to immune checkpoint blockade. We also found that higher melanoma melanin content, which reflects higher expression of wild type melanocyte antigens involved in pigment production, was correlated with 2-year survival after anti-PD-1 treatment. Collectively, these results support a potential functional role of T cell activity against wild type melanocyte antigens in the response to ICIs in melanoma, despite the additional key role of tumor-restricted neoantigens in at least a subset of patients.
Limitations of this study include the prospect that purposeful induction of epitope skewing, through treatments such as the combination therapies described here, may trigger autoimmune adverse events despite lack of apparent gross toxicities in our preclinical models. Our mouse experiments were conducted using syngeneic subcutaneous tumor grafts with clonal mutations rather than genetically engineered mouse models, and the effects of tumor heterogeneity on epitope skewing remains to be addressed. In addition, our observation of gp100-directed CD8+ TIL induction is likely an underestimate of the breadth of self-antigen-directed T cell responses. Although we provide evidence of epitope skewing and long-term immunity against several wild type melanocyte antigens, further studies are required to identify a comprehensive set of melanocyte antigens targeted by the immune response after combination immunotherapy. We did not observe changes in proliferation or activation markers expressed on gp100-specific CD8+ TILs; however further evaluation of inhibitory markers may help to more comprehensively characterize the functionality of this self-antigen directed population. It will also be of interest to investigate potential modulatory effects of other immune cell types including CD4+ T cells, Tregs, and macrophages through depletion and other experiments.
It is possible that immunity against wild type melanocyte antigens occurs in patients with melanoma and contributes to immunotherapy efficacy even in individuals without overt vitiligo. Many patients with melanoma who respond to anti-PD-1 never develop vitiligo, and in our cohort of responders, checkpoint blockade increased the frequency of melanocyte-specific CD8+ T cells regardless of whether vitiligo appeared. In our experiments, vitiligo was rarely observed among mice with complete responses against melanoma, further suggesting that a therapeutic window exists in which combination immunotherapies can drive responses against tumor-lineage self-antigens and have clinical benefit without dangerous toxicities involving autoimmune destruction of the organ of tumor origin. Strategies such as this may safely achieve efficacy in poorly inflamed cancers and offer potential promise to the many patients whose cancers are currently refractory to checkpoint inhibitors.
Materials and Methods
Study design.
This was a preclinical study of determinants of ICI response in melanomas with low neoantigen loads. We hypothesized that expression and recognition of tumor-lineage self-antigens renders tumors more responsive to ICIs. Patient melanoma RNA-sequencing data were analyzed by GSEA, melanoma differentiation and pigment pathway activation were determined by melanin scoring of histologic sections, and circulating MART-1 and TYR-specific CD8+ T cell populations were quantified by dextramer staining of peripheral blood mononuclear cells isolated from patients with melanoma. We also used mouse models of melanoma and pancreatic ductal adenocarcinoma to investigate effects of UVB-associated neoantigen introduction or combination of aFP+imiquimod+ICI treatment on tumor growth and survival. Mouse melanomas, TILs, and dLNs were characterized by flow cytometry immunophenotyping and gp100 tetramer staining, immunohistochemistry and immunofluorescence for CD3 and melanocyte antigens, and GSEA of tumor RNA-sequencing.
Informed consent was obtained and serial tumor and blood samples from patients with melanoma were collected under institutional review board protocol DFCI 11-181. All patients underwent standard of care follow-up at Massachusetts General Hospital, consisting of radiographic assessment every 12 weeks, clinical evaluation by the involved oncology team, and assessment by the treating medical oncologist in which physical exam and laboratory values were analyzed. Treatment response was classified on the basis of a combination of radiographic and physical exam findings. Blood was separated into plasma and peripheral blood mononuclear cells at the time of phlebotomy. Experiments using patient blood samples or tumors were blinded to responder status.
For in vivo mouse experiments, mice were randomly assigned to each treatment group. Measurements after treatment were not blinded. Sample sizes were selected on the basis of previous experience and published experiments. The number of samples and replicates for each experiment are listed in accompanying figure legends. No outliers were excluded from any experiments or analyses reported in this manuscript.
In vivo mouse studies.
Eight-week-old female C57BL/6 and NSG mice were obtained from The Jackson Laboratory. To minimize variation in pathogen exposure in our experiments, all mice were obtained from the same mouse facility at the same age and housed together in groups of up to five mice per cage. Mice were housed in solid-bottomed cages with chipped-wood and wood-shaving bedding that was changed as needed (minimum weekly) and provided ad libitum access to water and feed. Melanoma or KPC cells were inoculated subcutaneously at the flanks at a concentration of 1 x 106 cells in 0.1 ml phosphate buffered saline (PBS) per site. Blocking antibodies were administered intraperitoneally at a dose of 200 μg in 0.1 ml of PBS per mouse. For UV clone experiments, antibodies were administered on days 8, 10, 12, 14, and 16 after tumor cell inoculation. Anti-PD-1 [29F.1A12 (35)] was provided by the Freeman laboratory, and isotype-matched antibodies (2A3) were acquired from BioXCell. For combination therapy experiments, treatments were started when tumor volumes were approximately 50 mm3 (day 6 or 8 after tumor cell inoculation): anti-PD-1 (29F.1A12) or isotype-matched (2A3) and anti-CTLA-4 (9D9) or isotype-matched (MPC-11) were administered on days 6, 8, and 10; on days 8, 10, and 12 (triple therapy); or on days 8, 10, and 12 (quadruple therapy). Left flank tumors were treated with 5% imiquimod or vehicle lotion concurrently with antibody treatments, and aFP using a CO2 laser (UltraPulse DeepFX, Lumenis, Yokneam, Israel) on the first and last day of antibody treatment. Strides Pharma 5% imiquimod cream (10 μl per tumor) was used for most experiments. After Strides Pharma imiquimod became unavailable for purchase, Perrigo 5% imiquimod cream (5 μl per tumor) was used. For aFP, a 5 mm x 5 mm scanning pattern with 100 mJ energy per pulse, 5% nominal surface coverage, and 120 μm nominal spot size was applied. For aFP dosimetry, 100 mJ energy per pulse penetrates to ~2.5 mm depth below the skin surface. Assuming a 50 mm3 tumor extends from about 0.3 mm from the skin surface (estimate based on (36)), a 5×5 mm aFP pattern reaches between 2.5% of tumor volume (for larger tumors measuring between 2.2 to 4.7 mm in depth) and 5% of tumor volume (for smaller tumors measuring less than 2.2 mm in depth). For CD8 depletion, rat anti-mouse CD8a (clone 2.43) or isotype-matched (LTF-2) antibody was administered every 3 days for the duration of the experiment, starting 6 days before tumor inoculation. Tumor volume was calculated from caliper measurements as length x (width2/2). For experiments evaluating survival, mice were euthanized when tumors reached a maximum length or width of 20 mm or 10 mm (corresponding to an estimated maximum tumor volume of 4000 mm3 or 500 mm3) in experiments with one or two tumors per mouse, respectively, or earlier in cases of severe tumor ulceration or failure to thrive as determined by veterinary staff at the Massachusetts General Center for Comparative Medicine. For most rechallenge experiments, mice with previous complete responses of their melanomas to immunotherapy were inoculated with 1 × 105 cells in 0.1 ml of PBS at one flank and euthanized when the resulting tumors reached a maximum length or width of 10 mm (corresponding to an estimated maximum tumor volume of 500 mm3) or for severe tumor ulceration or failure to thrive. For rechallenge experiments with KPC-5 wild type melanocyte antigen cells, 1.5 × 105 cells in 0.1 ml of PBS were inoculated at one flank. All studies and procedures involving animal subjects were performed in accordance with policies and protocols approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital.
Statistical analysis.
Statistical analyses were performed using GraphPad Prism. For two-way comparisons, significance was determined by two-tailed Student’s t-tests, or Mann-Whitney t-tests for unpaired datasets that did not pass the Shapiro-Wilk normality test, or Wilcoxon’s matched-pairs signed rank tests for paired datasets that did not pass the Shapiro-Wilk normality test. For multiple comparisons, significance was determined by one- or two-way analysis of variance (ANOVA) with Tukey’s method for multiple comparisons of every mean with every other mean, or Dunnett’s method for multiple comparisons of every mean to a control mean. Kaplan-Meier analyses were conducted using the log-rank (Mantel-Cox) test. p values less than 0.05 were considered statistically significant.
Supplementary Material
Fig. S1. Ipilimumab, nivolumab, and pembrolizumab responders and non-responders with low melanoma neoantigen loads.
Fig. S2. Characterization of UV2 and UV3 melanoma cell lines.
Fig. S3. RNA-sequencing reveals enhanced cytotoxic activity and upregulation of T cell dysfunction markers in UV2 melanomas compared to parental D4M.3A.3 melanomas.
Fig. S4. Addition of imiquimod and aFP to immune checkpoint blockade induces an abscopal effect in mice bearing melanomas or pancreatic ductal adenocarcinomas.
Fig. S5. Combination immunotherapy improves T cell responses and is associated with markers of increased DC infiltration and function.
Fig. S6. Melanoma treatment with combination immunotherapy promotes melanocyte antigen expression and immune elimination of melanomas but not pancreatic tumors.
Data file S1. Supplementary tables.
Data file S2. Raw data.
Materials and Methods
Acknowledgments:
We thank members of the Fisher, Manstein, Sharpe, and Freeman laboratories for assistance and advice, with special thanks to C. T. Powell for help with manuscript preparation and T. Utne for help with animal studies. We gratefully acknowledge M. Bosenberg (Yale) and M. McMahon (University of Utah), who originally generated the BRAF/PTEN murine tumor model, and S. K. Dougan (Dana-Farber Cancer Institute) for the KPC cell line. We also thank the Dana-Farber Cancer Institute and Harvard Cancer Center for the use of the Specialized Histopathology Core.
Funding: This work was supported by the NIH under grants T32 GM007753 to J.A.L. and M.Y.S., T32CA207021 to M.W.L, F30 ES025626 to J.A.L., 5P01 CA163222 to D.E.F., K.T.F., and L.A.G., 5R01 AR043369 to D.E.F., 5R01CA222871 to D.E.F., 5R01AR072304 to D.E.F., P50CA101942 to A.H.S. and G.J.F., P01 AI056299 to A.H.S. and G.J.F., P50CA206963 to G.J.F., NCI-1RO1CA155010-02 to C.J.W., an NSF Graduate Research Fellowship to V.R.J., a Cancer Research Institute CLIP Award to N.H., a Leukemia and Lymphoma Society Scholar Award to C.J.W., the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation to D.E.F., K.T.F., L.A.G., and N.H., the Evergrande Center for Immunologic Diseases to A.H.S., Cancer Research UK under grant C20510/A13031 to D.J.A., the Mildred Scheel Grant of the German Cancer Society to E.R., the Filling the Gap Grant of the University of Zurich, Switzerland to E.R., and an Early Postdoc Mobility fellowship (no. P2ZHP3_178022) from the Swiss National Science Foundation to M.M. Dana-Farber and the Harvard Cancer Center are supported in part by an NCI Cancer Center Support Grant NIH 5 P30 CA06516. A fractional CO2 laser was provided as an unrestricted gift by Lumenis Ltd., Yokneam, Israel.
Competing interests: G.J.F. and A.H.S. have patents/pending royalties on the PD-1 pathway from Bristol-Myers-Squibb, Roche, Merck, EMD-Serono, Boehringer-Ingelheim, AstraZeneca, Dako, Leica, Mayo Clinic, and Novartis. G.J.F. has served on advisory boards for Roche, Bristol-Myers-Squibb, Xios, Origimed, Triursus, iTeos, NextPoint, IgM, Jubilant, and GV20. G.J.F. has equity in NextPoint, Triursus, Xios, iTeos, IgM, and GV20. A.H.S. is on the scientific advisory boards for Surface Oncology, Sqz Biotech, Elstar Therapeutics, Elpiscience, Selecta and Monopteros, consults for Novartis, and has research funding from Merck, Novartis, Roche, Ipsen, and Quark Ventures. A.H.S. has patents/pending royalties from Roche and Novartis on intellectual property on the PD-1 pathway. C.J.W. and N.H. hold equity in BioNTech. V.R.J. is currently employed by BioNTech US, Inc. L.A.G. is a consultant for Foundation Medicine, Novartis, and Boehringer Ingelheim; an equity holder in Foundation Medicine; and a member of the Scientific Advisory Board at Warp Drive. L.A.G. receives sponsored research support from Novartis. D.W.M. is a Scientific Advisor for Qu Biologics, Inc. (Vancouver BC Canada). G.M.B. has a sponsored research agreement with Takeda Oncology, Olink Proteomics, and Palleon Pharmaceuticals; has served on a scientific advisory board for Nektar Therapeutics and Novartis; and has been a lecturer for Novartis and Takeda Oncology. D.E.F. has a financial interest in Soltego, a company developing salt inducible kinase inhibitors for topical skin-darkening treatments that might be used for a broad set of human applications. The interests of D.E.F. were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. A provisional patent application has been filed (J.A.L., D.E.F., M.K., and D.M.) based on this work (“Intratumoral Immunization and Immune Checkpoint Blockade Combinatorial Therapy for Treatment of Metastatic Melanoma”) application number US2016/060321. All other authors declare that they have no competing interests.
Footnotes
Data and materials availability: All data associated with this study are in the paper or Supplementary Materials. Mouse cell line whole-exome sequencing data are available in the European Nucleotide Archive (accession numbers ERP001691 and ERP010158). Mouse tumor RNA-sequencing data are available at Gene Expression Omnibus (accession number GSE159344).
References
- 1.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJM, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ, Improved survival with ipilimumab in patients with metastatic melanoma, N. Engl. J. Med 363, 711–723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M, Safety, activity, and immune correlates of anti-PD-1 antibody in cancer, N. Engl. J. Med 366, 2443–2454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, Elassaiss-Schaap J, Algazi A, Matéus C, Boasberg P, Tumeh PC, Chmielowski B, Ebbinghaus SW, Li XN, Kang SP, Ribas A, Safety and Tumor Responses with Lambrolizumab (Anti-PD-1) in Melanoma, N. Engl. J. Med 369, 134–144 (2013), doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, Murphy KM, Gajewski TF, Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells, J. Exp. Med 208, 2005–2016 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, Murphy KM, Schreiber RD, Type I interferon is selectively required by dendritic cells for immune rejection of tumors, J. Exp. Med 208, 1989–2003 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF, Up-Regulation of PD-L1, IDO, and Tregs in the Melanoma Tumor Microenvironment Is Driven by CD8+ T Cells, Sci. Transl. Med 5, 200ra116–200ra116 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, Jansen P, Gaffal E, Steitz J, Tolba R, Kalinke U, Limmer A, Jönsson G, Hölzel M, Tüting T, Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 4, 674–687 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, Li Y, Elipenahli C, Liu C, Harbison CT, Wang L, Ribas A, Wolchok JD, Chan TA, Genetic basis for clinical response to CTLA-4 blockade in melanoma, N. Engl. J. Med 371, 2189–2199 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA, Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer, Science 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA Jr., PD-1 Blockade in Tumors with Mismatch-Repair Deficiency, N. Engl. J. Med 372, 2509–2520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM, Utikal J, Hassel JC, Weide B, Kaehler KC, Loquai C, Mohr P, Gutzmer R, Dummer R, Gabriel S, Wu CJ, Schadendorf D, Garraway LA, Genomic correlates of response to CTLA-4 blockade in metastatic melanoma, Science 350, 207–211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hua C, Boussemart L, Matéus C, Routier E, Boutros C, Cazenave H, Viollet R, Thomas M, Roy S, Benannoune N, Tomasic G, Soria J-C, Champiat S, Texier M, Lanoy E, Robert C, Association of Vitiligo With Tumor Response in Patients With Metastatic Melanoma Treated With Pembrolizumab. JAMA Dermatol. 152, 45–51 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles, Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, Kelley MC, Sosman JA, Johnson DB, Ribas A, Lo RS, Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma, Cell 165, 35–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Amon L, Zimmer L, Gutzmer R, Satzger I, Loquai C, Grabbe S, Vokes N, Margolis CA, Conway J, He MX, Elmarakeby H, Dietlein F, Miao D, Tracy A, Gogas H, Goldinger SM, Utikal J, Blank CU, Rauschenberg R, von Bubnoff D, Krackhardt A, Weide B, Haferkamp S, Kiecker F, Izar B, Garraway L, Regev A, Flaherty K, Paschen A, Van Allen EM, Schadendorf D, Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma, Nat Med 25, 1916–1927 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van den Boorn JG, Konijnenberg D, Dellemijn TAM, van der Veen JPW, Bos JD, Melief CJM, Vyth-Dreese FA, Luiten RM, Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients, J. Invest. Dermatol 129, 2220–2232 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Jenkins MH, Steinberg SM, Alexander MP, Fisher JL, Ernstoff MS, Turk MJ, Mullins DW, Brinckerhoff CE, Multiple murine BRafV600E melanoma cell lines with sensitivity to PLX4032, Pigment Cell Melanoma Res (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, You MJ, DePinho RA, McMahon M, Bosenberg M, Braf(V600E) cooperates with Pten loss to induce metastatic melanoma, Nat Genet 41, 544–552 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortés ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, Lin P, Lichtenstein L, Heiman DI, Fennell T, Imielinski M, Hernandez B, Hodis E, Baca S, Dulak AM, Lohr J, Landau D-A, Wu CJ, Melendez-Zajgla J, Hidalgo-Miranda A, Koren A, McCarroll SA, Mora J, Lee RS, Crompton B, Onofrio R, Parkin M, Winckler W, Ardlie K, Gabriel SB, Roberts CWM, Biegel JA, Stegmaier K, Bass AJ, Garraway LA, Meyerson M, Golub TR, Gordenin DA, Sunyaev S, Lander ES, Getz G, Mutational heterogeneity in cancer and the search for new cancer-associated genes, Nature 499, 214–218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N, Molecular and genetic properties of tumors associated with local immune cytolytic activity, Cell 160, 48–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manstein D, Herron GS, Sink RK, Tanner H, Anderson R, Fractional photothermolysis: a new concept for cutaneous remodeling using microscopic patterns of thermal injury, Lasers Surg. Med 34, 426–438 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Naylor MF, Crowson N, Kuwahara R, Teague K, Garcia C, Mackinnis C, Haque R, Odom C, Jankey C, Cornelison RL, Treatment of lentigo maligna with topical imiquimod, Br. J. Dermatol 149 Suppl 66, 66–70 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Swetter SM, Chen FW, Kim DD, Egbert BM, Imiquimod 5% cream as primary or adjuvant therapy for melanoma in situ, lentigo maligna type, Journal of the American Academy of Dermatology 72, 1047–1053 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA, Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice, Cancer cell 7, 469–483 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM, Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity, Science 322, 1097–1100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edelson BT, KC W, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG, Bhattacharya D, Stappenbeck TS, Holtzman MJ, Sung S-SJ, Murphy TL, Hildner K, Murphy KM, Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells, J. Exp. Med 207, 823–836 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuertes MB, Kacha AK, Kline J, Woo S-R, Kranz DM, Murphy KM, Gajewski TF, Host type I IFN signals are required for antitumor CD8 +T cell responses through CD8α +dendritic cells, J. Exp. Med 208, 2005–2016 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, Barczak A, Rosenblum MD, Daud A, Barber DL, Amigorena S, van’t Veer LJ, Sperling AI, Wolf DM, Krummel MF, Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity, Cancer cell 26, 638–652 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fu C, Chen J, Lu J, Yi L, Tong X, Kang L, Pei S, Ouyang Y, Jiang L, Ding Y, Zhao X, Li S, Yang Y, Huang J, Zeng Q, Roles of inflammation factors in melanogenesis (Review), Mol Med Rep 21, 1421–1430 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui R, Widlund HR, Feige E, Lin JY, Wilensky DL, Igras VE, D’Orazio J, Fung CY, Schanbacher CF, Granter SR, Central role of p53 in the suntan response and pathologic hyperpigmentation, Cell 128, 853–864 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Lo JA, Fisher DE, The melanoma revolution: From UV carcinogenesis to a new era in therapeutics, Science 346, 945–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Perry CJ, Meeth K, Thakral D, Damsky W, Micevic G, Kaech S, Blenman K, Bosenberg M, UV-induced somatic mutations elicit a functional T cell response in the YUMMER1.7 Mouse Melanoma Model, Pigment Cell Melanoma Res (2017), doi: 10.1111/pcmr.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuehm LM, Wolf K, Zahour J, DiPaolo RJ, Teague RM, Checkpoint blockade immunotherapy enhances the frequency and effector function of murine tumor-infiltrating T cells but does not alter TCRβ diversity, Cancer Immunol. Immunother 68, 1095–1106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawakubo M, Demehri S, Manstein D, Fractional laser exposure induces neutrophil infiltration (N1 phenotype) into the tumor and stimulates systemic anti-tumor immune response, PLoS ONE 12, e0184852 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, Greenfield EA, Liang SC, Sharpe AH, Lichtman AH, Freeman GJ, Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis, Eur. J. Immunol 33, 3117–3126 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Hansen LS, Coggle JE, Wells J, Charles MW, The influence of the hair cycle on the thickness of mouse skin, Anat Rec 210, 569–573 (1984). [DOI] [PubMed] [Google Scholar]
- 37.Viros A, Fridlyand J, Bauer J, Lasithiotakis K, Garbe C, Pinkel D, Bastian BC, Improving melanoma classification by integrating genetic and morphologic features, PLoS Med. 5, e120 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD, A Versatile Viral System for Expression and Depletion of Proteins in Mammalian Cells, PLoS ONE 4, e6529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM, arXiv (2013). [Google Scholar]
- 40.Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, Furlotte NA, Eskin E, Nellåker C, Whitley H, Cleak J, Janowitz D, Hernandez-Pliego P, Edwards A, Belgard TG, Oliver PL, McIntyre RE, Bhomra A, Nicod J, Gan X, Yuan W, van der Weyden L, Steward CA, Bala S, Stalker J, Mott R, Durbin R, Jackson IJ, Czechanski A, Guerra-Assunção JA, Donahue LR, Reinholdt LG, Payseur BA, Ponting CP, Birney E, Flint J, Adams DJ, Mouse genomic variation and its effect on phenotypes and gene regulation, Nature 477, 289–294 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM, CellProfiler: image analysis software for identifying and quantifying cell phenotypes, Genome Biol 7, R100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng PF, Dummer R, Levesque MP, Data mining The Cancer Genome Atlas in the era of precision cancer medicine, Swiss Med Wkly 145, w14183 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2, Nat Meth 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li B, Dewey CN, RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome, BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Supek F, Bošnjak M, Škunca N, Šmuc T, REVIGO summarizes and visualizes long lists of gene ontology terms, PLoS ONE 6, e21800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Ipilimumab, nivolumab, and pembrolizumab responders and non-responders with low melanoma neoantigen loads.
Fig. S2. Characterization of UV2 and UV3 melanoma cell lines.
Fig. S3. RNA-sequencing reveals enhanced cytotoxic activity and upregulation of T cell dysfunction markers in UV2 melanomas compared to parental D4M.3A.3 melanomas.
Fig. S4. Addition of imiquimod and aFP to immune checkpoint blockade induces an abscopal effect in mice bearing melanomas or pancreatic ductal adenocarcinomas.
Fig. S5. Combination immunotherapy improves T cell responses and is associated with markers of increased DC infiltration and function.
Fig. S6. Melanoma treatment with combination immunotherapy promotes melanocyte antigen expression and immune elimination of melanomas but not pancreatic tumors.
Data file S1. Supplementary tables.
Data file S2. Raw data.
Materials and Methods