

Abstract
Securing novel, safe, and effective medicines to treat Mycobacterium tuberculosis remains an elusive goal, particularly influenced by the largely impervious Mtb envelope that limits exposure and thus efficacy of inhibitors at their cellular and periplasmic targets. The impact of physicochemical properties on pharmacokinetic parameters that govern oral absorption and exposure at sites of infection is considered alongside how these properties influence penetration of the Mtb envelope, with the likely influence of transporter proteins. The findings are discussed to benchmark current drugs and the emerging pipeline, whilst considering tactics for future rational and targeted design strategies, based around emerging data on Mtb transporters and their structures and functions.
The physicochemical properties of TB drugs are compared oral drugs and antibiotics, which support likely roles for transporters contributing to their efficacy.
Tuberculosis and the need for new drugs
Tuberculosis (TB), the disease caused by Mycobacterium tuberculosis (Mtb), is responsible for over a billion deaths over the last 200 years, making Mtb the single most important infectious agent in history.1 The latest figures from the World Health Organisation indicate 1.5 million TB fatalities per year, despite the availability of effective drugs.2 However, whilst the standard combination therapies are effective against drug-susceptible strains of TB, these medicines are over 40 years old and the successful use of the existing front-line and second-line agents has been compromised by the escalation in drug-resistant cases of TB and additional complications due to co-infection by human immunodeficiency virus (HIV).2 Therefore an urgent need to discover new TB drugs remains and the ideal next generation of anti-tubercular agents should have a shorter treatment regimen and novel mechanisms of action to ensure the effective treatment of drug-resistant TB strains and minimise interactions with co-medications. New collaborative initiatives, partly funded through philanthropic organisations,3,4 have bolstered research efforts, leading to better understanding of the disease and identifying potential new treatments undergoing clinical evaluation.5,6 The annotation of the complete Mycobacterium tuberculosis genome sequence over 20 years ago identified and delivered multiple targets for potential chemotherapeutic intervention,7 yet the outcome of many TB drug discovery programmes that screened compound libraries against these targets was the identification of potent in vitro inhibitors that failed to translate into any corresponding killing of Mtb,8 a common issue with target based drug discovery.9,10 One reason for these failures is the inability of molecules to cross the largely impenetrable Mtb cell envelope (Fig. 1), preventing many potential chemotherapeutics from reaching their required periplasmic or intracellular targets.11,12
Fig. 1. Model structure of the Mycobacterium tuberculosis cell envelope, the mycobacterial outer membrane (MOM) formed of surface lipids and mycolic acids presents an impenetrable waxy coat beyond the complex mycolyl–arabinogalactan–peptidoglycan (mAGP) layer and periplasm that exists beyond the cytoplasmic membrane. Representative porin and transmembrane proteins are indicated in blue and orange, respectively. We thank Professor Gurdyal Besra for sharing his artwork11 to enable this modified figure.

Scope of this perspective
The following sections provide commentary and comparisons of oral drugs, antibiotics, and TB drugs through their physicochemical properties, noting how these data relate to the necessary permeability of the molecules to achieve both oral exposure and antimicrobial efficacy. The likely influence of transporters on these processes is discussed in the context of differing theories on how molecules cross membranes, ahead of a section reviewing current understanding of Mtb carrier proteins.
Physicochemical properties and membrane permeability
Optimising the physicochemical properties of experimental molecules is a key aspect of modern drug discovery,13 to address the balance between activity, efficacy and minimise off-target effects.14 The principle of minimal lipophilicity15 remains a pragmatic guideline; although reducing the lipophilicity of molecules, whilst generally improving solubility, metabolic profiles,16 and off-target effects,17,18 often results in poorer membrane permeability, a key process in enabling oral exposure and accessing intracellular targets.19,20
Herein, models, based on physicochemical properties, that are used to help estimate the likelihood of achieving oral exposure with small molecules, are used to rationalise observations apparent within the data. Analyses were performed on Schultz's representative set of oral drug molecules,21 supplemented with additional TB drugs plus potential medicines in the TB pipeline. This set was annotated with GSK calculated data19 plus classification of antibiotics, nucleosides, carbohydrates, amino acids and peptides in the set. Oral agents are invariably the main goal for TB discovery programmes, given the characteristics of existing treatments and necessary supply logistics for many patients.
The Lipinski rule of 5 (Ro5, based on calculated partition coefficient, clog P > 5, molecular weight <500 and hydrogen bond donors <5/acceptors <10) was initially devised to estimate solubility and permeation to increase chances of oral exposure,22 yet became the commonly used standard in drug discovery. A stated caveat of the Ro5 recognised that transported compounds, notably are not necessarily within the proposed confines of the 90th centile thresholds, which themselves are often misinterpreted23 and misconstrued.21,24 An additional model, originating from Wellcome,25 based on lipophilicity (as log of distribution coefficient, log D7.4) versus size (calculated molar refraction, CMR) is used to predict permeability and oral exposure, based on combinations of these parameters.26 This model was designed to help understand the behaviour of more hydrophilic molecules that are either small enough to be absorbed by paracellular absorption or would have limited passive diffusion across membranes, with refinements to give predictions based on a linear discriminant function analysis, with further enhancements using chromatographic lipophilicity measurements.19 In addition to these models, there is growing empirical evidence to support the preferred physicochemical nature of compounds with high MW (the most likely measure beyond Ro5 (ref. 19 and 21), for which the AbbVie multi-parametric scoring function (AB-MPS)27 gives particularly important insight regarding a preferred log D7.4 of 3 (a value of 5 on the chromatographic scale) and minimising aromatic ring count (#Ar) and rotatable bonds (#RB). The empirically derived equation is AB-MPS = (|3 − log D7.4| + #RB + #Ar) and when AB-MPS < 15, an improved chance achieving of oral exposure was evident.27 The bilinear relationship to log D7.4 that the AB-MPS implies is consistent with observations of permeation trends noted with chromatographic methods26 and historic data.28 Finally, the property forecast index17 (PFI = Chrom log D7.4 + #Ar) is used as a benchmark quality measure, whereby PFI < 6 is a desirable target to maximise the chance of achieving good solubility and minimise developability risks29 and a clog P version of this metric was shown to be useful in predicting permeation of tuberculosis lesions.30 Together, these metrics can be used to benchmark quality and illustrate risk and likelihood of achieving reasonable pharmacokinetic exposure; there is no guarantee of success, but the chances of failure are reduced with compounds possessing better tuned physical properties.14,19
Transporters/solute carriers and oral drugs
The existence of solute carriers that act as facilitators of transmembrane passage in eukaryotic cells is well documented, yet conflicting schools of thought as to whether this is a minor31,32 or sole mechanism33 generates much debate.34 The positioning of most antimicrobial drugs on the fringes or outside of the traditional boundaries for oral drugs is suggestive of a role for carrier proteins in enabling both antimicrobial effects and oral exposure. The physicochemical make-ups of antimicrobial compounds35 – and their frequent origins from natural products – is indicative of them being transported (vide infra), which bears comparison to the hypothesis that oral drug molecules have similarities to exogenous natural products,36 shown to be the most important transporter substrates.37 A similar notion to human solute carriers evolving to facilitate the beneficial uptake of exogenous natural products37 would be to rationalise the evolution of natural antimicrobial agents with beneficial effects due to transport to the site of action. An explanation for the paucity of hits in antimicrobial screens is that corporate collections have been designed with a predominance of structures that are unlikely to achieve effective concentrations within microbes.10,38
Mapping the properties of oral drugs, antibiotics and tuberculosis drugs
The physicochemical profiles of the sets of compounds using log D7.4/CMR and Ro5 models are represented in Fig. 2 and 3, respectively. It is well-documented that anti-infective molecules possess physicochemical properties that are distinct from oral drugs,10,39,40 and these figures illustrate that most antibacterial agents and the TB drugs in particular (Fig. 2, Table 1) are outside or on the fringes of distributions produced by these models. Indeed, box plot analyses (reproduced in ESI†) illustrate significant (p < 0.05) differences in the lipophilicity and molecular weight distributions of these classes. It is noteworthy that although the majority of antibiotics in the analysis are “Lipinski compliant”, most are identified as likely candidates for transporters by their positioning in the log D7.4vs. CMR analysis due to their hydrophilic character and natural product origins.41
Fig. 2. Property distributions, using calculated Chrom log D7.4vs. CMR, of the Shultz set of representative oral drugs,21 updated from ref. 19 with the classification of antibacterials and TB drugs.

Fig. 3. Distribution of the scrutinised molecules using Lipinski MW < 500 and clog P < 5 guidelines, coloured as Fig. 2.

Physiochemical properties and descriptors for front-line and impactful TB drugs. Background colouration reflects positioning of drug with respect to i) Lipinski parameters (MW, molecular weight, in Daltons <500; clog P, calculated log partition coefficient <5; HBD hydrogen bond donors <5 and HBA acceptors <10); ii) GSK log D7.4–CMR positioning (see Fig. 4) and iii) property forecast index values PFI <6.
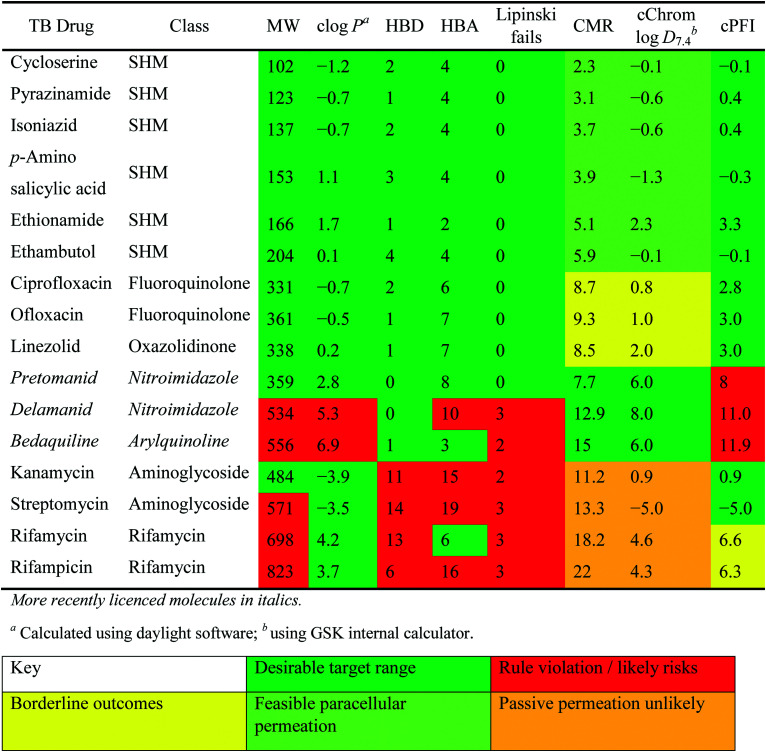
|
The lines of Chrom log D7.4 = 2.5 and CMR of 8 used in Fig. 2 are approximations of the original model bounds used to target passive permeation;25 compounds in the lower right quadrant are unlikely to be absorbed by “passive permeation”. The black diagonal line (Fig. 2) is an empirically-derived linear discriminant function line giving the best spatial separation of oral outcome predictions, the dotted lines are boundaries that indicate lesser confidence in predictive discrimination.19
Physiochemical properties of Mtb drugs
Many studies have described the physicochemical characteristics of anti-tubercular molecules, in efforts to map out the “chemical space” in which they reside and rationalise the therapeutic responses of various heterogeneous bacterial sub-populations located within diverse micro-environments,42 utilising different sources and pathways in their central carbon metabolism.43 In this context, differential expression of solute carriers is likely, such as those facilitating the import of cholesterol, of importance in mycobacterial persistence,44 which has quite distinct properties to those of carbohydrate carbon sources. These scenarios all contribute to the complexity of necessary treatment regimes. Additionally, the accumulation of drugs within the envelope, cellular and periplasmic space of Mtb will be governed by differing physical environments. It is thus logical to anticipate continued need for combination treatments of drugs with differing properties to address these physicochemical challenges.
The relatively poor state of the TB drug pipeline in 2005 was largely substantiated using properties in Ro5 calculations,45 whilst the physicochemical profile of GSK open source hit sets used the log D7.4/CMR metric to illustrate their quality as starting points for further optimisation.46,47 Given the failure of many drug discovery efforts to translate hit molecules into TB killing a better understanding of the molecular characteristics and compartmentalisation of effective clinically used TB drugs would help.30,48 The physicochemical properties of the front-line TB drugs are summarised in Table 1 where the diverse range of molecular size, lipophilicity and number of Lipinski violations is evident in the structures shown in Table 2. Importantly, whilst navigating the idiosyncrasies required to effectively kill Mtb, molecules must also have characteristics amenable to oral administration and the maintenance of effective concentrations within their pharmacokinetic and pharmacodynamic profiles. It is thus perhaps not unexpected that for effective TB treatment high drug doses are often required, which can compromise narrow safety windows and increases the risk of adverse side effects.29,49 The use of bedaquiline, which is only approved to treat multi-drug resistant TB, is a case in point where the advantages of successful TB treatment outweighs the ‘black box’ warning of cardiovascular and hepatic toxicity risks and usage limited to drug resistant cases; it is notable that all three of the most recently licenced TB medicines are in higher risk space as defined by PFI estimates, unlike more established drugs. The analysis of this set using the GSK log D7.4/CMR model reveals that TB drugs are localised at the fringes when compared to the distribution of marketed oral drugs (Fig. 2) and it is possible to include distinct sub-classifications of these front-line TB drugs in terms of their physicochemical compositions and how these relate to their human pharmacokinetics. Firstly, the three more recently licenced molecules bedaquiline, delamanid and pretomanid possess log D7.4/CMR combinations more akin to most oral medicines and dissimilar to antibiotics and other TB drugs. The natural product-derived rifampin and rifamycin are macrocycles with just two aromatic rings, few rotatable bonds and Chrom log D7.4 values close to five (on chromatographic scale) that give low AbbVie MPS scores (7.7 and 5.4 respectively) commensurate with achieving oral exposure for such high molecular weight compounds, where other antibiotics are found.27 The small hydrophilic molecules (SHMs) in the set: cycloserine, pyrazinamide, isoniazid, p-amino salicylic acid and ethionamide possess properties likely to facilitate oral exposure via paracellular absorption form the digestive tract, formerly a common paradigm for drug discovery,19 but relatively neglected in current screening-driven drug discovery. The transport mechanisms of the aminoglycosides such as streptomycin and kanamycin,50 and also oxazolidinones51 and fluoroquinolones52,53 are well-documented and these molecules all possess log D7.4–CMR profiles that are not suggestive of passive permeability. These pertinent properties of the various classifications are colour coded in Table 1 and shaded on an overlay of Fig. 2 as Fig. 4.
Structures and classifications of front-line and other significant tuberculosis drugs in Table 1.
| Small hydrophilic molecules | |||||
|---|---|---|---|---|---|
| Cycloserine | Pyrazinamide | Isoniazid | p-Amino salicylic acid | Ethionamide | Ethambutol |
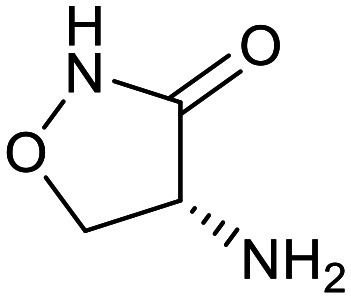
|
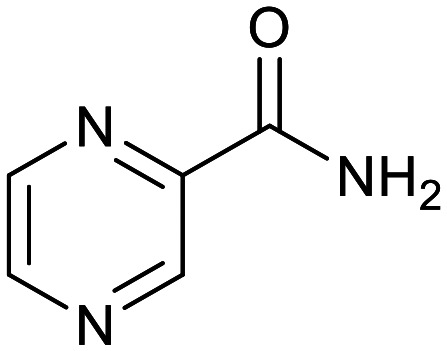
|
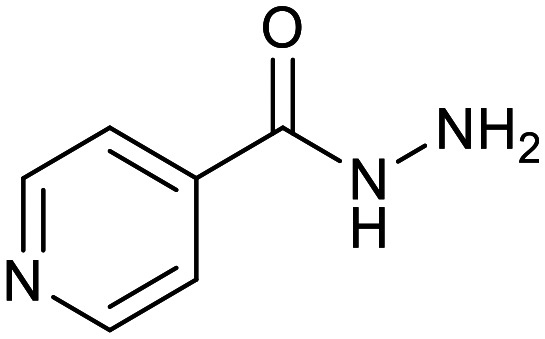
|
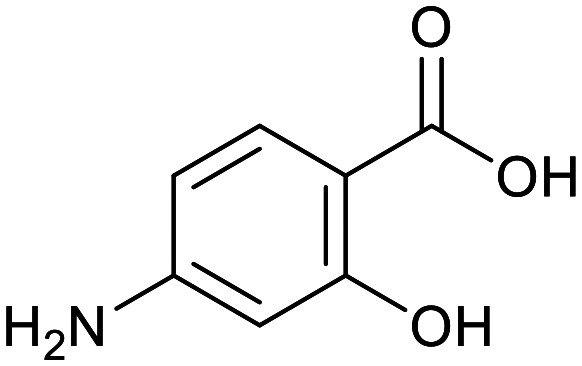
|
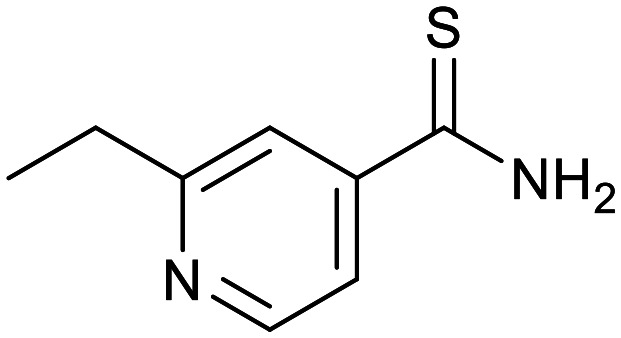
|
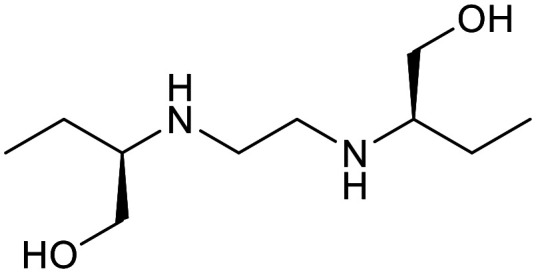
|
| Fluoroquinolones | Oxazolidinone | ||||
|---|---|---|---|---|---|
| Ciprofloxacin | Ofloxacin | Linezolid | |||
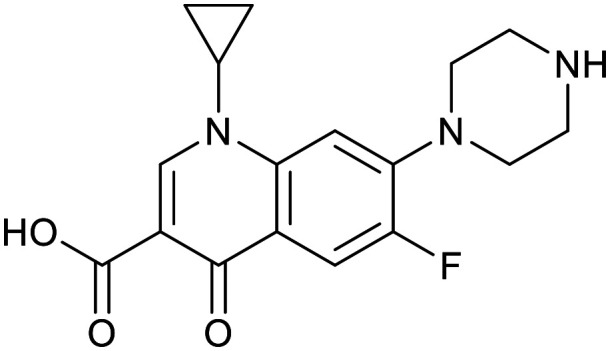
|
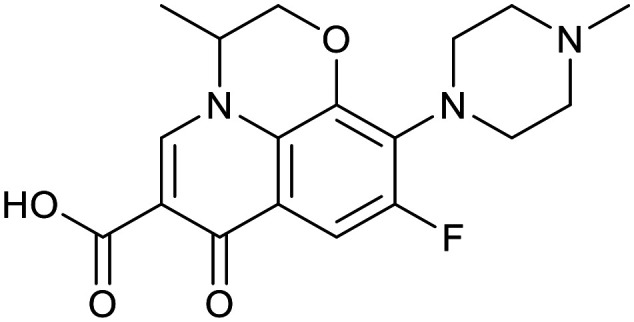
|
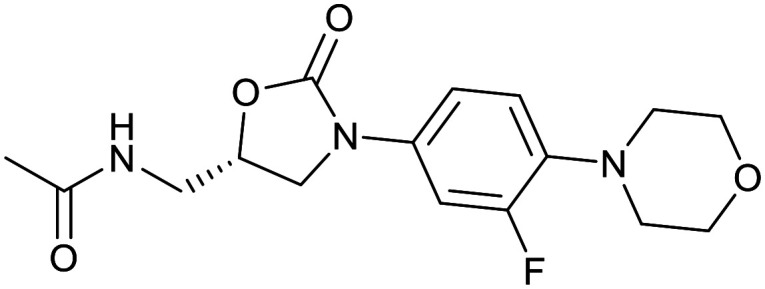
|
|||
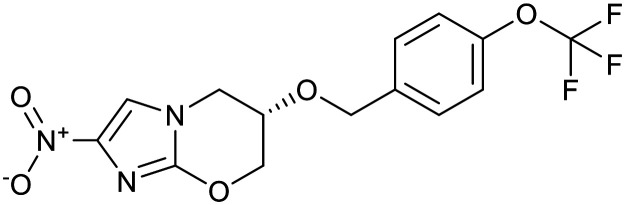
| Arylquinoline | Nitroimidazoles | ||||
|---|---|---|---|---|---|
| Bedaquiline | Pretomanid | Delamanid | |||
|
|
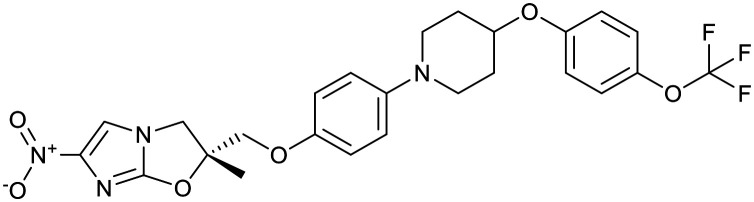
|
|||
| Aminoglycosides | |||||
|---|---|---|---|---|---|
| Kanamycin | Streptomycin | ||||
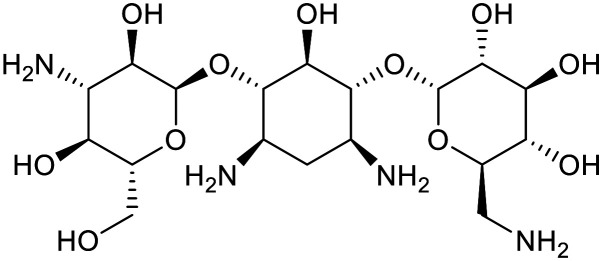
|
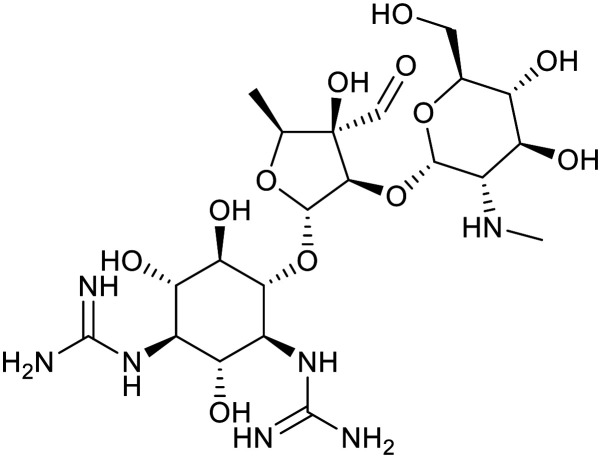
|
||||
| Rifamycins | |||||
|---|---|---|---|---|---|
| Rifamycin | Rifampicin | ||||
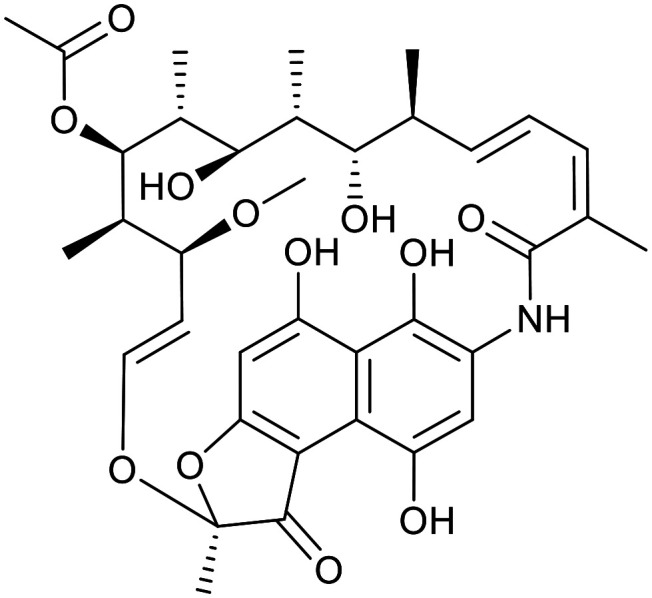
|
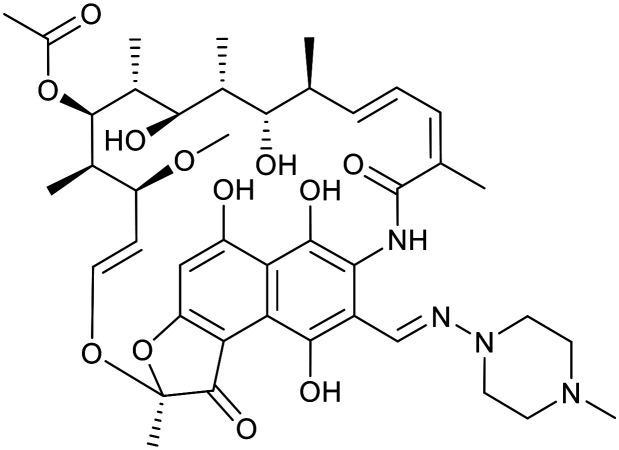
|
||||
Fig. 4. Reproduction of Fig. 2 with coloured regions highlighting likely absorption classifications.

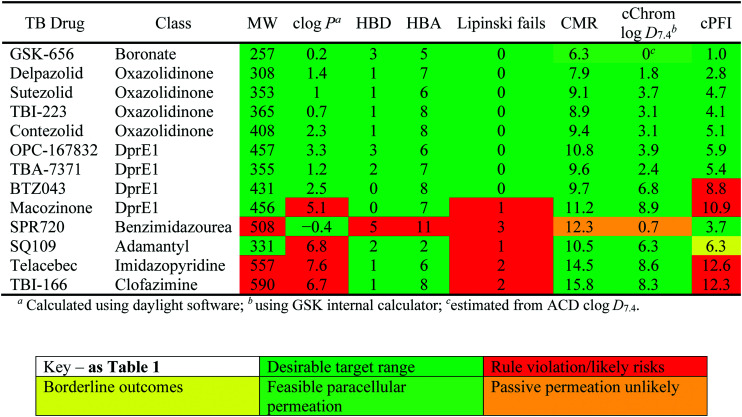
Physicochemical properties of molecules in the TB drug discovery pipeline
The physicochemical characteristics of the most advanced molecules in the TB pipeline (Table 3)5 are listed in Table 4 and analyses of these show some interesting trends and observations (Fig. 5). The shift of average properties apparent in Fig. 2, suggests that most exemplars are more similar, in terms of their log D7.4/CMR profile positioning, to oral drugs than to existing TB drugs. A notable exception is the Leucyl-tRNA synthetase inhibitor GSK-656,54 the product of structure based design that is likely to be absorbed by the paracellular mechanism, given its small size and hydrophilicity. This shift in properties could be the result of the general appreciation of optimal properties in the medicinal chemistry community – or could be reflective of their origins in high throughput screening sets. It may be also be influenced by medicinal chemistry practices in the several oxazolidinone analogues that are variations on the linezolid structure.55 It is interesting to consider that screening of company collections can identify compounds with demonstrable MICs against TB, such as the GSK open source hit set47 (Fig. 6) with properties akin to most drugs. However, such collections have few examples of small, hydrophilic molecules and likely few that mimic natural product features.38 In this open source set, the compounds were chosen based on validated antimycobacterial activity, but the targets were unknown; therefore, it is not clear how effectively activity versus these unknown targets translates into antimycobacterial activity. The imidazopyridine telacebec was optimised from a hit in a phenotypic high content screen and maintained MICs and oral exposure despite high lipophilicity.56 This could be viewed in the context of TBI-166,57 an analogue of the repurposed natural product clofazimine,58 used to treat leprosy, so may possess the potential to be transported in spite of the apparently unattractive structure in terms of high lipophilicity and high aromatic ring count. These latter compounds are the extremes of what, from a property perspective, is a pipeline with many compounds with lower risk potential as indicated by the PFI estimates. The set of compounds inhibiting decaprenylphosphoryl-β-d-ribose oxidase (DprE1) are notable for their physical make-up within the purported sweet spot for oral drugs.59 DprE1 is an essential enzyme for Mtb,60–62 as it is the only source of arabinose for the construction of cell wall arabinan structures, against which various screening efforts furnished tractable hits,8,63 with four compounds under clinical evaluation (Table 3).64 Of particular note is the extracytoplasmic localization of DprE1,65 which makes the target more accessible and thus vulnerable. Indeed, the maintenance of potent MICs with a broad range of physical characteristics within one DprE1 series was recognised, suggesting that properties alone do not control access to the target and other recognition features may facilitate penetration of the envelope.66 This latter series was identified through screening of the open source GSK set against a strain of M. bovis BCG overexpressing DprE1,67 illustrating the value of this novel combined approach to target identification and screening.
Structures of molecules currently under clinical evaluation against tuberculosis.
| Oxazolidinones | |||
|---|---|---|---|
| Delpazolid | Sutezolid | TBI-223 | Contezolid |
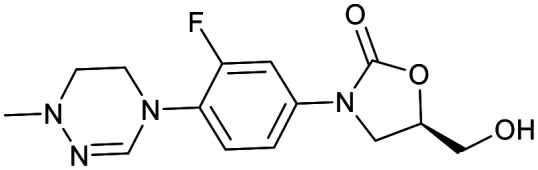
|
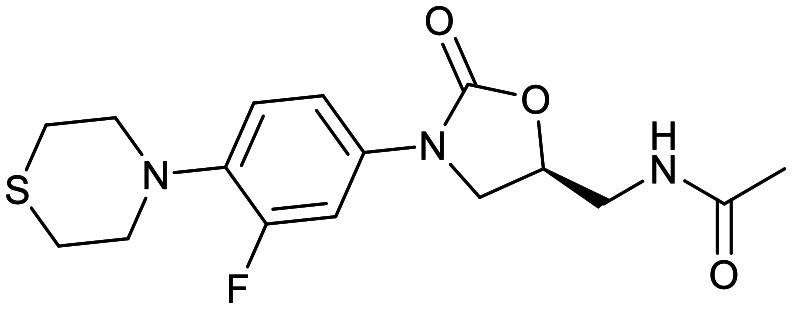
|
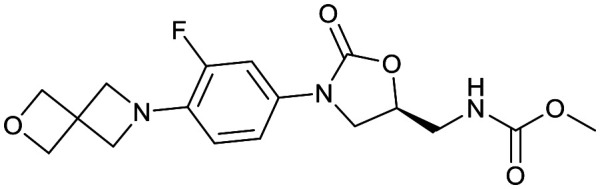
|
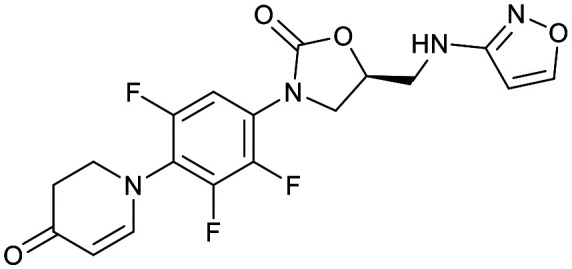
|
| DprE1 inhibitors | |||
|---|---|---|---|
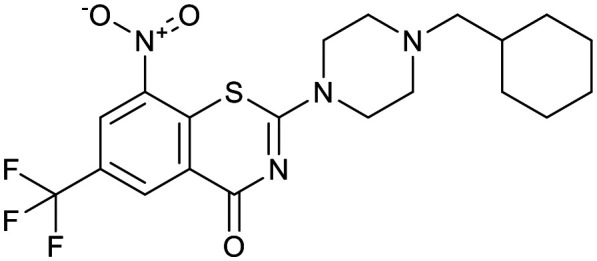
| OPC-167832 | TBA-7371 | BTZ043 | Macozinone |
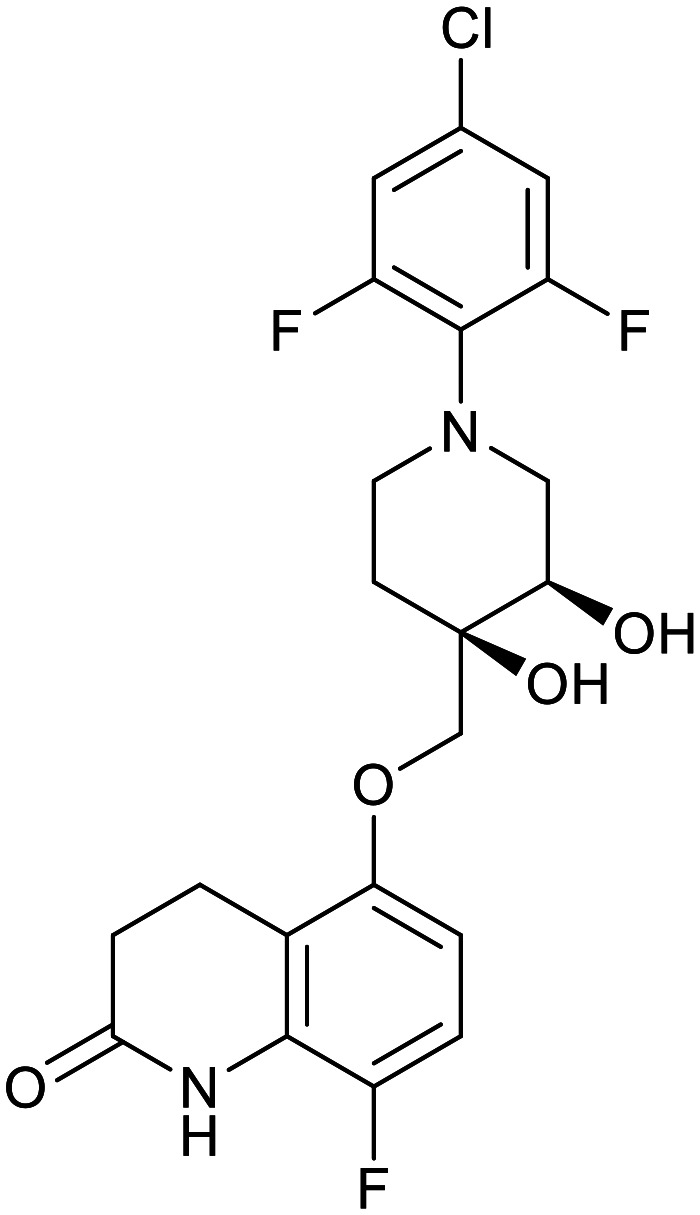
|
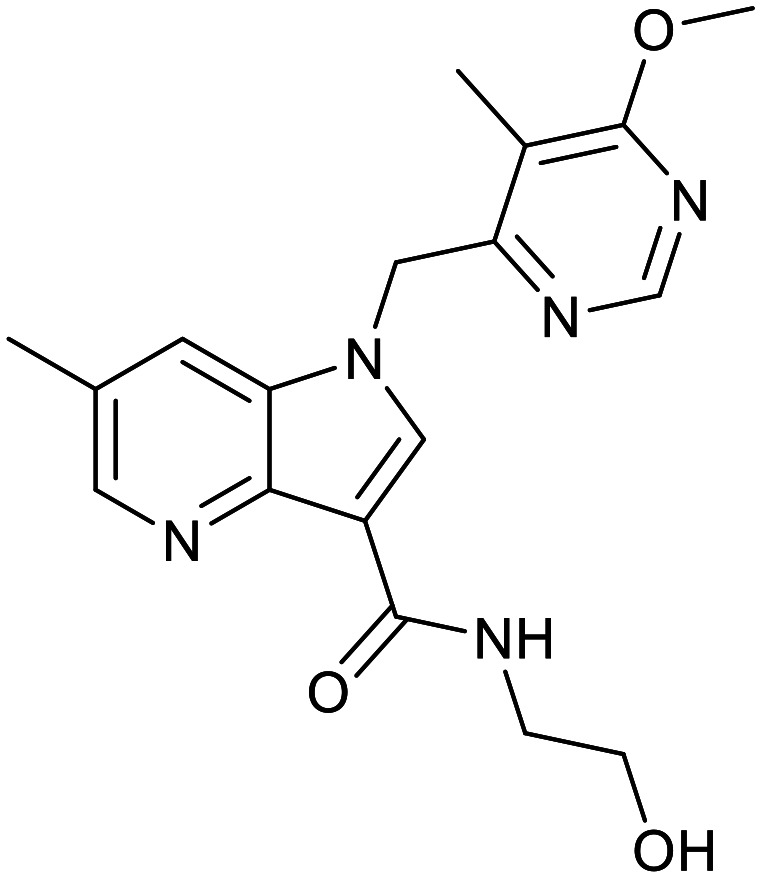
|
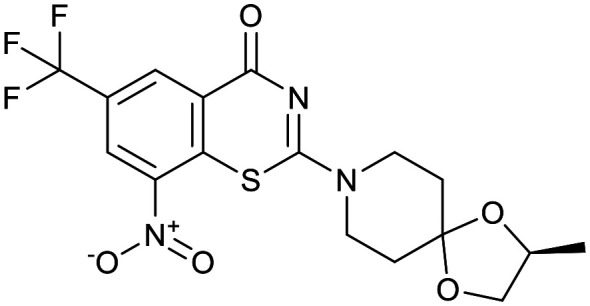
|
|
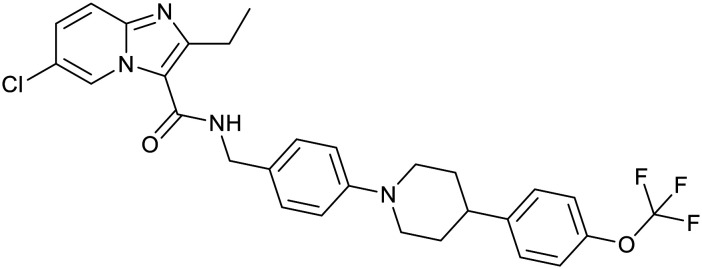
| Boronate | Benzimidazourea | Adamantyl | |
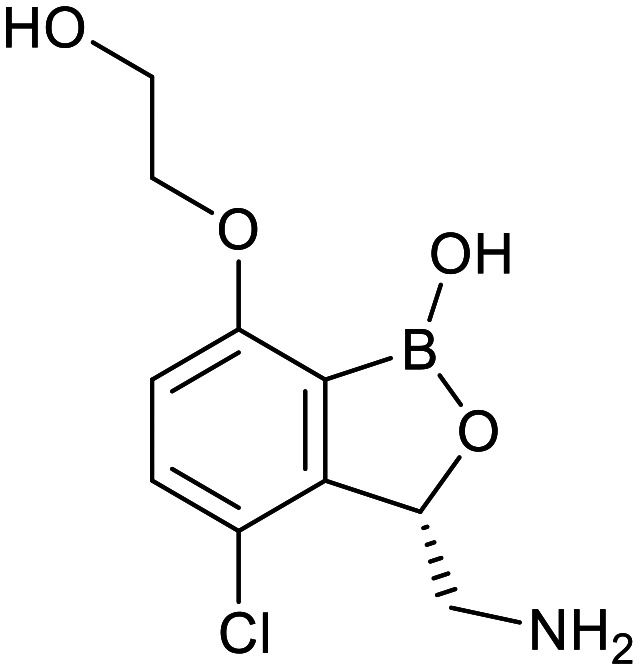
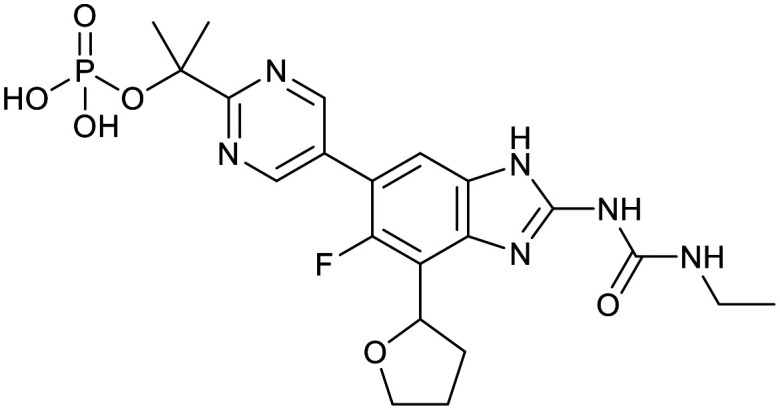
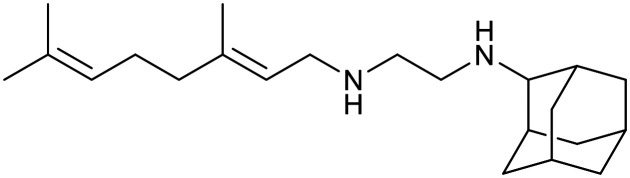
|---|---|---|---|
| GSK-656 | SPR720 | SQ109 | |
|
|
|
|
| Clofazimine | Imidazopyridine | ||
|---|---|---|---|
| TBI-166 | Telacebec | ||
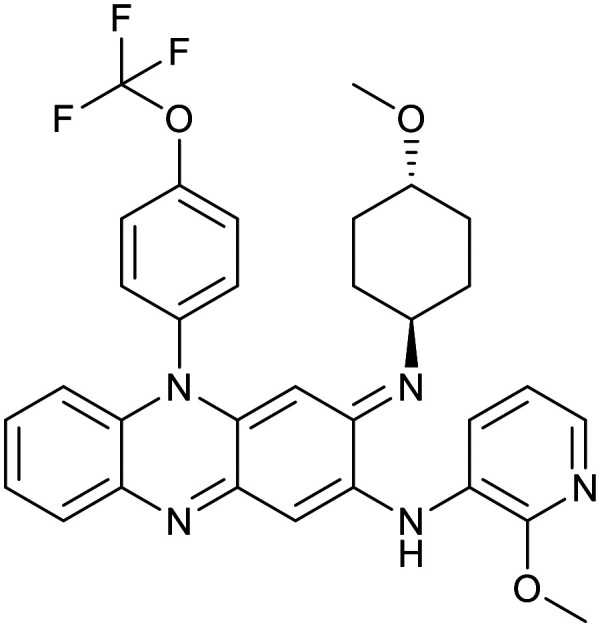
|
|
Physicochemical properties and descriptors for TB drugs in advanced development.
|
Fig. 5. Comparative distribution of Tb drugs and compounds reported in clinic development.5.

Fig. 6. cChrom log D7.4vs. CMR plot of the GSK set of 177 open source set of compounds with TB MICS versus the GSK training set of marketed drugs with human oral bioavailability greater or less than 30%.47.

Mtb transporters as a strategy
The unique lipid-rich composition of the mycobacterial envelope is a major factor that contributes to the insensitivity of Mtb to many antibiotics and it is intrinsically less permeable to chemotherapeutic agents and nutrients compared to other bacterial species.68,69 Whilst anti-tubercular agents must cross the envelope to reach their intracellular enzyme targets, the precise processes which enable this access remains elusive and the diversity of physicochemical profiles in the preceding sections is commensurate with multiple mechanisms. In principle, Mtb is able to assimilate antibiotics via several routes that may include direct diffusion of hydrophobic molecules across the mycobacterial lipid membranes or via porins and/or carrier proteins.70 Evidence for the passive diffusion of TB drugs via the “hydrophobic pathway” is supported through observations that more lipophilic derivatives of a single TB drug class were more active against Mtb69 and that perturbations in the membrane lipids, either through changes in temperature or alterations in mycolic acid content, could lead to increased uptake.68,69 However, it is difficult to reconcile the uptake of the small and hydrophilic drug molecules with passive diffusion and more lipophilic compounds would be expected to remain in this hydrophobic structure. Therefore, the notion of drug molecules crossing the mycobacterial envelope by carrier proteins is a rational conclusion and, accordingly, hijacking endogenous transport systems to facilitate the uptake of drugs represents a logical and attractive approach. Determining which endogenous and natural substrates are imported by Mtb transporters and mapping the structural recognition elements and physiochemical properties of these molecules should facilitate the identification of new anti-tubercular lead compounds. Whilst considering the role of carrier proteins facilitating the influx of compounds, the presence of efflux mechanisms is also pertinent, if beyond the scope of this review. Their mechanisms and specificities are also not yet fully understood,71,72 but physicochemical properties are likely to be amongst key recognition features. Mtb also possesses export carrier proteins such as mycobacterial membrane protein large 3 (MmpL3),64 the target of SQ109 (ref. 73) and other molecules,74–76 involved in the export of essential molecules to the periplasm,11 with studies unravelling its likely substrates utilized in cell wall biogenesis.77 Understanding the nuances and interplay of these various transport mechanisms could pave the way for targeted combination therapies and new drugs less prone to drug–drug interactions through their interference with host transporters and metabolising enzymes.78
In order for molecules/drugs to gain access to the Mtb cytoplasm there are two barriers in place to cross. First molecules must cross the waxy outer mycomembrane and then cross the inner membrane.11 Recently, the role of porins, which form channels in the outer mycomembrane, have been found to have be important in controlling the influx of nutrients, including glycerol, haem, and antibiotics.79Mtb is predicted to contain at least 37 ATP-binding cassette (ABC) transporters and 30 major facilitator permeases80 that are thought to be located in the inner membrane and have a putative role in uptake of range of diverse molecules that include phosphates, nitrites/nitrates, ions, vitamins and sugars, although the complete substrate repertoire of these importers is not known.80–84 The functional role of the transporters is emerging, and a number of different studies have found that mutant strains of Mtb that lack various importer impact on the survival of this pathogen during infection. There have been several comprehensive reviews covering the identification of Mtb transporters and the assignment of putative substrates.81–84 Herein the focus is on the recent advances in biochemical and structural characterisation of Mtb transporters that are beginning to shed light on their function and may enable drug design, both as transport inhibitors or as substrates of the more promiscuous carriers to enable passage to their targets.
Structural determination of Mtb transporters
Haem and iron transport
Iron is an essential element for the growth and survival of Mtb during infection.85,86 Whilst the host attempts to limit access to this metal, Mtb has developed effective strategies to prevent metal depletion and ensure that it can scavenge this nutrient. To do this, Mtb can assimilate iron complexed in siderophores (mycobactins)87 and through uptake of iron complexed to haem and haemoglobin.88 Two recent studies have determined the biochemical and structural basis of iron uptake.89,90 IrtAB (Rv1348–Rv1349) is an unusual ABC-transporter that was predicted to be an exporter, however instead IrtAB imports mycobactins across the inner-membrane.89 The structure of IrtAB (PDB 6TEJ) has been solved by X-ray crystallography in an inward-facing conformation at 2.7 Å and reveals insight into its mechanism of action.89 In contrast the Mtb Dpp transporter (Rv3666c–Rv3663c) is responsible for the uptake of iron complexed to heme.90 The structure of the DppA substrate binding domain (Rv3666c) has been solved to 1.3 Å (PDB 63Ed and 6E4D) and the heme binding pocket has been revealed through a combination of X-ray and modelling approaches. Both the IrtAB and Dpp transporters have been found to be important for infection in mice.91,92
Amino acids
Nitrogen metabolism is critical for the survival of Mtb during infection within the host and the assimilation of multiple amino acids, including glutamate, glutamine, aspartate, alanine, glycine, and valine, within the macrophages provide this important nitrogen source.93,94 However, despite the importance of nitrogen assimilation by this pathogen, the transporters linked to individual amino acid recognition are unresolved and to date only one crystal structure of an amino acid transporter has been determined.95 GlnH (Rv0411c) is the solute binding domain of an ABC-transporter and appears to have dual functionality in which is acts as an ABC-transporter and a signal sensor. GlnH95 recognises aspartate, glutamate and asparagine with a preference for aspartate and high-resolution crystal structures of GlnH have been solved in complex with each amino acid aspartate (PDB 6H1U), glutamate (PDB 6H2T) and asparagine (PDB 6H20).95 The GlnH co-complex structures have enabled key structural recognition elements to be determined, which map to the observed binding affinity preferences for these amino acids.95
Vitamin B12
Mtb is able to uptake exogenous vitamin B12, which is subsequently utilised as an important cofactor in a number of core biosynthetic pathways.96 The Mtb ABC-transporter (Rv1819c) responsible for the uptake of vitamin B12 is implicated to have a role in maintaining chronic infection of Mtb in mice.96 Excitingly, the structure of Rv1819c (PDB 6TQE and 6TQF) has recently been solved by single-particle cryo-electron microscopy to a resolution of 3.5 Å.97 Similar to IrtAB the structure of Rv1819c has a typical ABC-transporter exporter fold, despite having import activity. Intriguingly the Rv1819c structure reveals a very large water-filled internal cavity that has a volume that could accommodate a number of cobalamin molecules.97 Close inspection finds that this central cavity is lined with polar and negatively charged residues, which may facilitate the transport of diverse hydrophilic molecules, including cobalamin and the antimicrobial peptide bleomycin across the inner membrane of Mtb.
Carbohydrates
The sugars that are available to Mtb during infection are not well defined. Mtb is predicted to have 4 putative ABC-transporters for the uptake of carbohydrates and one transporter belonging to the major-facilitator superfamily.81 To date, functional studies of the Mtb carbohydrate importers have focused on the substrate binding domains of the ABC-transporters and three structures have been resolved. The structure of UspC, the substrate binding protein that belongs to UspABC transporter, has been determined to 1.5 Å (PDB 5K2X and 5K2Y) in an open conformation.98 Biochemical studies indicate that UspC specifically recognises amino sugars, suggesting a possible role of this importer in the recycling of peptidoglycan fragments from the Mtb cell wall.98 The structure of UgpB, belonging to the UgpABCE transporter, has been solved in both an open ligand-free conformation (PDB 4MFI, 1.5 Å)99 and a closed conformation in complex with its glycerophosphocholine substrate (PDB 6R1B 2.3 Å).100 Interestingly, a combination of biochemical and structural studies elucidated that Mtb UgpB recognises a diverse range of glycerophosphodiester substrates, indicating a role in the recycling of phospholipid metabolites, with the glycerol moiety being the main recognition element with minimal contacts with the phosphodiester head group at the entrance to the binding cavity.100
Conclusions
Given the extraordinarily complex and impenetrable Mtb cell envelope, which is major factor in the failure of TB drug discovery efforts, an exciting opportunity exists for the exploitation of Mtb transporters to deliver effective anti-tubercular agents. Increased biochemical, functional, and physiological studies have started to provide significant insights into the role of Mtb transporters and molecular mechanisms/key substrate recognition features are being established. This is key to allow the design of novel inhibitors that can inhibit or hijack the mechanisms.
Progress is being made towards new and better molecules to treat TB, evidenced by enhanced molecular quality, to meet the challenges presented by drug resistant tuberculosis cases, yet challenges remain in the shortening of therapy and treating the various stages of infection. Understating how to reach molecular targets effectively through the somewhat impenetrable Mtb cell envelope should help decrease instances of potent activity against an Mtb target not translating into an MIC. A better understanding of the properties and characteristics required to get across the cell envelope is likely to be linked to better characterisation of the expression, structure and function of transporters. An understanding of the necessary requirements for transporters may help in the development of new molecules which can be designed to be inherently recognised by these transport systems, either as substrates or inhibitors and derivatisation with recognised motifs could facilitate transport or lead to pathogen specific agents conjugated to bactericidal molecules. To employ transporters it is possible that a renewed interest in natural products101 and structures rich in natural product like motifs;38 will pay dividends, especially if the hypothesis that carriers evolved to transport necessary molecules has substance.37 In that case, screening representative natural product sets102 and designing molecules with tailored physical properties and features akin to known substrates is a logical proposition; a further inference would be the possibility that TB “drug space” is rather more limited than has been suggested.5 Such knowledge, coupled with modern computer assisted design, should enable more focussed screening and influence best practice in optimisation procedures, especially when coupled to physical properties indicative of lower developability risks. Together such practices present a more rational approach to the quest for new medicines and better prioritisation of resources; the ultimate quality of drugs may be a destination, but better starting points and waymarks on the journey14 will be pointers to success.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We gratefully acknowledged contributions from Paul Leeson, Mike Shultz, Chris Tinworth (who enabled compilation of our review sets through utilising data from their publications), and Del Besra (for helpful discussions and his artwork adapted for Fig. 1). EF is a Sir Henry Dale Fellow, jointly funded by the Wellcome Trust and Royal Society (grant number 104193/Z/14/Z and 104193/Z/14/B).
Electronic supplementary information (ESI) available: Word file with Box plot data used to perform the analyses. Excel file used to generate plots and tables in this review. See DOI: 10.1039/d0md00265h
References
- Sizemore C. F., Hafner R. and Fauci A. S., in NIH Statement on World Tuberculosis Day 2018, 2018, https://www.nih.gov/news-events/news-releases/nih-statement-world-tuberculosis-day-2018
- In Global Tuberculosis Report 2019, World Health Organization, Geneva, 2019, https://www.who.int/tb/publications/global_report/en/ [Google Scholar]
- Cole S. T. Drug Discovery Today. 2017;22:477–478. doi: 10.1016/j.drudis.2016.09.026. [DOI] [PubMed] [Google Scholar]
- De Rycker M. Horn D. Aldridge B. Amewu R. K. Barry, 3rd C. E. Buckner F. S. Cook S. Ferguson M. A. J. Gobeau N. Herrmann J. Herrling P. Hope W. Keiser J. Lafuente-Monasterio M. J. Leeson P. D. Leroy D. Manjunatha U. H. McCarthy J. Miles T. J. Mizrahi V. Moshynets O. Niles J. Overington J. P. Pottage J. Rao S. P. S. Read K. D. Ribeiro I. Silver L. L. Southern J. Spangenberg T. Sundar S. Taylor C. Van Voorhis W. White N. J. Wyllie S. Wyatt P. G. Gilbert I. H. ACS Infect. Dis. 2020;6:3–13. doi: 10.1021/acsinfecdis.9b00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V. Salina E. Reynolds R. C. Kyaw Zin P. P. Ekins S. J. Med. Chem. 2020;63(17):8917–8955. doi: 10.1021/acs.jmedchem.9b02075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mdluli K. Kaneko T. Upton A. Cold Spring Harbor Perspect. Med. 2015;5(6) doi: 10.1101/cshperspect.a021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S. T. Brosch R. Parkhill J. Garnier T. Churcher C. Harris D. Gordon S. V. Eiglmeier K. Gas S. Barry C. E. Tekaia F. Badcock K. Basham D. Brown D. Chillingworth T. Connor R. Davies R. Devlin K. Feltwell T. Gentles S. Hamlin N. Holroyd S. Hornsby T. Jagels K. Krogh A. McLean J. Moule S. Murphy L. Oliver K. Osborne J. Quail M. A. Rajandream M. A. Rogers J. Rutter S. Seeger K. Skelton J. Squares R. Squares S. Sulston J. E. Taylor K. Whitehead S. Barrell B. G. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Mikušová K. Ekins S. Drug Discovery Today. 2017;22:534–545. doi: 10.1016/j.drudis.2016.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D. Drug Discovery Today. 2007;12:1007–1012. doi: 10.1016/j.drudis.2007.10.017. [DOI] [PubMed] [Google Scholar]
- Payne D. J. Gwynn M. N. Holmes D. J. Pompliano D. L. Nat. Rev. Drug Discovery. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- Batt S. M. Minnikin D. E. Besra G. S. Biochem. J. 2020;477:1983–2006. doi: 10.1042/BCJ20200194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal-Mutalik R. Nikaido H. Proc. Natl. Acad. Sci. U. S. A. 2014;111:4958–4963. doi: 10.1073/pnas.1403078111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meanwell N. A. Chem. Res. Toxicol. 2011;24:1420–1456. doi: 10.1021/tx200211v. [DOI] [PubMed] [Google Scholar]
- Young R. J. Leeson P. D. J. Med. Chem. 2018;61:6421–6467. doi: 10.1021/acs.jmedchem.8b00180. [DOI] [PubMed] [Google Scholar]
- Hansch C. Bjorkroth J. P. Leo A. J. Pharm. Sci. 1987;76:663–687. doi: 10.1002/jps.2600760902. [DOI] [PubMed] [Google Scholar]
- Miller R. R. Madeira M. Wood H. B. Geissler W. M. Raab C. E. Martin I. J. J. Med. Chem. 2020 doi: 10.1021/acs.jmedchem.9b01813. [DOI] [PubMed] [Google Scholar]; , ASAP
- Young R. J. Green D. V. Luscombe C. N. Hill A. P. Drug Discovery Today. 2011;16:822–830. doi: 10.1016/j.drudis.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Gleeson M. P. Hersey A. Montanari D. Overington J. Nat. Rev. Drug Discovery. 2011;10:197–208. doi: 10.1038/nrd3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinworth C. P. Young R. J. J. Med. Chem. 2020;63:10091–10108. doi: 10.1021/acs.jmedchem.9b01596. [DOI] [PubMed] [Google Scholar]
- Naylor M. R. Ly A. M. Handford M. J. Ramos D. P. Pye C. R. Furukawa A. Klein V. G. Noland R. P. Edmondson Q. Turmon A. C. Hewitt W. M. Schwochert J. Townsend C. E. Kelly C. N. Blanco M. J. Lokey R. S. J. Med. Chem. 2018;61:11169–11182. doi: 10.1021/acs.jmedchem.8b01259. [DOI] [PubMed] [Google Scholar]
- Shultz M. D. J. Med. Chem. 2019;62:1701–1714. doi: 10.1021/acs.jmedchem.8b00686. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Lombardo F. Dominy B. W. Feeney P. J. Adv. Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Leeson P. D. Young R. J. ACS Med. Chem. Lett. 2015;6:722–725. doi: 10.1021/acsmedchemlett.5b00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P. D. Adv. Drug Delivery Rev. 2016;101:22–33. doi: 10.1016/j.addr.2016.01.018. [DOI] [PubMed] [Google Scholar]
- Glen R. C. Martin G. R. Hill A. P. Hyde R. M. Woollard P. M. Salmon J. A. Buckingham J. Robertson A. D. J. Med. Chem. 1995;38:3566–3580. doi: 10.1021/jm00018a016. [DOI] [PubMed] [Google Scholar]
- Young R. J., in Tactics in Contemporary Drug Design, ed. N. A. Meanwell, Springer Berlin Heidelberg, Berlin, Heidelberg, 2014, pp. 1–68 [Google Scholar]
- DeGoey D. A. Chen H. J. Cox P. B. Wendt M. D. J. Med. Chem. 2018;61:2636–2651. doi: 10.1021/acs.jmedchem.7b00717. [DOI] [PubMed] [Google Scholar]
- Sugano K. Kansy M. Artursson P. Avdeef A. Bendels S. Di L. Ecker G. F. Faller B. Fischer H. Gerebtzoff G. Lennernaes H. Senner F. Nat. Rev. Drug Discovery. 2010;9:597–614. doi: 10.1038/nrd3187. [DOI] [PubMed] [Google Scholar]
- Bayliss M. K. Butler J. Feldman P. L. Green D. V. Leeson P. D. Palovich M. R. Taylor A. J. Drug Discovery Today. 2016;21:1719–1727. doi: 10.1016/j.drudis.2016.07.007. [DOI] [PubMed] [Google Scholar]
- Sarathy J. P. Zuccotto F. Hsinpin H. Sandberg L. Via L. E. Marriner G. A. Masquelin T. Wyatt P. Ray P. Dartois V. ACS Infect. Dis. 2016;2:552–563. doi: 10.1021/acsinfecdis.6b00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L. Artursson P. Avdeef A. Ecker G. F. Faller B. Fischer H. Houston J. B. Kansy M. Kerns E. H. Kramer S. D. Lennernas H. Sugano K. Drug Discovery Today. 2012;17:905–912. doi: 10.1016/j.drudis.2012.03.015. [DOI] [PubMed] [Google Scholar]
- Smith D. Artursson P. Avdeef A. Di L. Ecker G. F. Faller B. Houston J. B. Kansy M. Kerns E. H. Kramer S. D. Lennernas H. van de Waterbeemd H. Sugano K. Testa B. Mol. Pharmaceutics. 2014;11:1727–1738. doi: 10.1021/mp400713v. [DOI] [PubMed] [Google Scholar]
- Kell D. B. Dobson P. D. Oliver S. G. Drug Discovery Today. 2011;16:704–714. doi: 10.1016/j.drudis.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Kell D. B. Oliver S. G. Front. Pharmacol. 2014;5:231. doi: 10.3389/fphar.2014.00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugumbate G. Overington J. P. Bioorg. Med. Chem. 2015;23:5218–5224. doi: 10.1016/j.bmc.2015.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hagan S. Swainston N. Handl J. Kell D. B. Metabolomics. 2015;11:323–339. doi: 10.1007/s11306-014-0733-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hagan S. Kell D. B. ADMET and DMPK. 2017;5:85–125. doi: 10.5599/admet.5.2.376. [DOI] [Google Scholar]
- Kellenberger E. Hofmann A. Quinn R. J. Nat. Prod. Rep. 2011;28:1483–1492. doi: 10.1039/C1NP00026H. [DOI] [PubMed] [Google Scholar]
- Pawlowski A. C. Johnson J. W. Wright G. D. Curr. Opin. Biotechnol. 2016;42:108–117. doi: 10.1016/j.copbio.2016.04.006. [DOI] [PubMed] [Google Scholar]
- Tommasi R. Brown D. G. Walkup G. K. Manchester J. I. Miller A. A. Nat. Rev. Drug Discovery. 2015;14:529–542. doi: 10.1038/nrd4572. [DOI] [PubMed] [Google Scholar]
- Leeson P. D. Davis A. M. J. Med. Chem. 2004;47:6338–6348. doi: 10.1021/jm049717d. [DOI] [PubMed] [Google Scholar]
- Lakshminarayana S. B. Huat T. B. Ho P. C. Manjunatha U. H. Dartois V. Dick T. Rao S. P. S. J. Antimicrob. Chemother. 2014;70:857–867. doi: 10.1093/jac/dku457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee K. Y. Carvalho L. P. S. D. Bryk R. Ehrt S. Marrero J. Park S. W. Schnappinger D. Venugopal A. Nathan C. Trends Microbiol. 2011;19:307–314. doi: 10.1016/j.tim.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey A. K. Sassetti C. M. Proc. Natl. Acad. Sci. U. S. A. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballell L. Field R. A. Duncan K. Young R. J. Antimicrob. Agents Chemother. 2005;49:2153–2163. doi: 10.1128/AAC.49.6.2153-2163.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebollo-Lopez M. J. Lelièvre J. Alvarez-Gomez D. Castro-Pichel J. Martínez-Jiménez F. Papadatos G. Kumar V. Colmenarejo G. Mugumbate G. Hurle M. Barroso V. Young R. J. Martinez-Hoyos M. González del Río R. Bates R. H. Lopez-Roman E. M. Mendoza-Losana A. Brown J. R. Alvarez-Ruiz E. Marti-Renom M. A. Overington J. P. Cammack N. Ballell L. Barros-Aguire D. PLoS One. 2015;10:e0142293. doi: 10.1371/journal.pone.0142293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballell L. Bates R. H. Young R. J. Alvarez-Gomez D. Alvarez-Ruiz E. Barroso V. Blanco D. Crespo B. Escribano J. Gonzalez R. Lozano S. Huss S. Santos-Villarejo A. Martin-Plaza J. J. Mendoza A. Rebollo-Lopez M. J. Remuinan-Blanco M. Lavandera J. L. Perez-Herran E. Gamo-Benito F. J. Garcia-Bustos J. F. Barros D. Castro J. P. Cammack N. ChemMedChem. 2013;8:313–321. doi: 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janardhan S. Ram Vivek M. Narahari Sastry G. Mol. BioSyst. 2016;12:3377–3384. doi: 10.1039/C6MB00457A. [DOI] [PubMed] [Google Scholar]
- Leeson P. D. Chem. Res. Toxicol. 2018;31:494–505. doi: 10.1021/acs.chemrestox.8b00044. [DOI] [PubMed] [Google Scholar]
- Taber H. W. Mueller J. P. Miller P. F. Arrow A. S. Microbiol. Rev. 1987;51:439–457. doi: 10.1128/MR.51.4.439-457.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbachyn M. R. Ford C. W. Angew. Chem., Int. Ed. 2003;42:2010–2023. doi: 10.1002/anie.200200528. [DOI] [PubMed] [Google Scholar]
- Vergalli J. Dumont E. Cinquin B. Maigre L. Pajovic J. Bacqué E. Mourez M. Réfrégiers M. Pagès J.-M. Sci. Rep. 2017;7:9821. doi: 10.1038/s41598-017-08775-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bounds S. J. Nakkula R. Walters J. D. Antimicrob. Agents Chemother. 2000;44:2609–2614. doi: 10.1128/AAC.44.10.2609-2614.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Hernandez V. Rock F. L. Choi W. Mak Y. S. L. Mohan M. Mao W. Zhou Y. Easom E. E. Plattner J. J. Zou W. Pérez-Herrán E. Giordano I. Mendoza-Losana A. Alemparte C. Rullas J. Angulo-Barturen I. Crouch S. Ortega F. Barros D. Alley M. R. K. J. Med. Chem. 2017;60:8011–8026. doi: 10.1021/acs.jmedchem.7b00631. [DOI] [PubMed] [Google Scholar]
- Zhang M. Sala C. Dhar N. Vocat A. Sambandamurthy V. K. Sharma S. Marriner G. Balasubramanian V. Cole S. T. Antimicrob. Agents Chemother. 2014;58:3217–3223. doi: 10.1128/AAC.02410-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pethe K. Bifani P. Jang J. Kang S. Park S. Ahn S. Jiricek J. Jung J. Jeon H. K. Cechetto J. Christophe T. Lee H. Kempf M. Jackson M. Lenaerts A. J. Pham H. Jones V. Seo M. J. Kim Y. M. Seo M. Seo J. J. Park D. Ko Y. Choi I. Kim R. Kim S. Y. Lim S. Yim S.-A. Nam J. Kang H. Kwon H. Oh C.-T. Cho Y. Jang Y. Kim J. Chua A. Tan B. H. Nanjundappa M. B. Rao S. P. S. Barnes W. S. Wintjens R. Walker J. R. Alonso S. Lee S. Kim J. Oh S. Oh T. Nehrbass U. Han S.-J. No Z. Lee J. Brodin P. Cho S.-N. Nam K. Kim J. Nat. Med. 2013;19:1157–1160. doi: 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- Xu J. Wang B. Fu L. Zhu H. Guo S. Huang H. Yin D. Zhang Y. Lu Y. Antimicrob. Agents Chemother. 2019;63:e02155-02118. doi: 10.1128/AAC.02155-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal M. Padayatchi N. Metcalfe J. Z. O'Donnell M. R. Int. J. Tuberc. Lung Dis. 2013;17:1001–1007. doi: 10.5588/ijtld.12.0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann M. M. Keseru G. M. Nat. Rev. Drug Discovery. 2012;11:355–365. doi: 10.1038/nrd3701. [DOI] [PubMed] [Google Scholar]
- Kolly G. S. Boldrin F. Sala C. Dhar N. Hartkoorn R. C. Ventura M. Serafini A. McKinney J. D. Manganelli R. Cole S. T. Mol. Microbiol. 2014;92:194–211. doi: 10.1111/mmi.12546. [DOI] [PubMed] [Google Scholar]
- Makarov V. Manina G. Mikusova K. Möllmann U. Ryabova O. Saint-Joanis B. Dhar N. Pasca M. R. Buroni S. Lucarelli A. P. Milano A. De Rossi E. Belanova M. Bobovska A. Dianiskova P. Kordulakova J. Sala C. Fullam E. Schneider P. McKinney J. D. Brodin P. Christophe T. Waddell S. Butcher P. Albrethsen J. Rosenkrands I. Brosch R. Nandi V. Bharath S. Gaonkar S. Shandil R. K. Balasubramanian V. Balganesh T. Tyagi S. Grosset J. Riccardi G. Cole S. T. Science. 2009;324:801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neres J. Pojer F. Molteni E. Chiarelli L. R. Dhar N. Boy-Röttger S. Buroni S. Fullam E. Degiacomi G. Lucarelli A. P. Read R. J. Zanoni G. Edmondson D. E. De Rossi E. Pasca M. R. McKinney J. D. Dyson P. J. Riccardi G. Mattevi A. Cole S. T. Binda C. Sci. Transl. Med. 2012;4:150ra121. doi: 10.1126/scitranslmed.3004395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikhale R. V. Barmade M. A. Murumkar P. R. Yadav M. R. J. Med. Chem. 2018;61:8563–8593. doi: 10.1021/acs.jmedchem.8b00281. [DOI] [PubMed] [Google Scholar]
- Degiacomi G. Belardinelli J. M. Pasca M. R. De Rossi E. Riccardi G. Chiarelli L. R. Appl. Sci. 2020;10:623. doi: 10.3390/app10020623. [DOI] [Google Scholar]
- Brecik M. Centárová I. Mukherjee R. Kolly G. S. Huszár S. Bobovská A. Kilacsková E. Mokošová V. Svetlíková Z. Šarkan M. Neres J. Korduláková J. Cole S. T. Mikušová K. ACS Chem. Biol. 2015;10:1631–1636. doi: 10.1021/acschembio.5b00237. [DOI] [PubMed] [Google Scholar]
- Borthwick J. A. Alemparte C. Wall I. Whitehurst B. C. Argyrou A. Burley G. de Dios-Anton P. Guijarro L. Monteiro M. C. Ortega F. Suckling C. J. Pichel J. C. Cacho M. Young R. J. J. Med. Chem. 2020;63:2557–2576. doi: 10.1021/acs.jmedchem.9b01561. [DOI] [PubMed] [Google Scholar]
- Batt S. M. Cacho Izquierdo M. Castro Pichel J. Stubbs C. J. Vela-Glez Del Peral L. Perez-Herran E. Dhar N. Mouzon B. Rees M. Hutchinson J. P. Young R. J. McKinney J. D. Barros Aguirre D. Ballell L. Besra G. S. Argyrou A. ACS Infect. Dis. 2015;1:615–626. doi: 10.1021/acsinfecdis.5b00065. [DOI] [PubMed] [Google Scholar]
- Jarlier V. Nikaido H. FEMS Microbiol. Lett. 1994;123:11–18. doi: 10.1111/j.1574-6968.1994.tb07194.x. [DOI] [PubMed] [Google Scholar]
- Brennan P. J. Nikaido H. Annu. Rev. Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- Sarathy J. P. Dartois V. Lee E. J. D. Pharmaceuticals. 2012;5:1210–1235. doi: 10.3390/ph5111210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brake L. H. M. T. Knegt G. J. D. Steenwinkel J. E. D. Dam T. J. P. V. Burger D. M. Russel F. G. M. Crevel R. V. Koenderink J. B. Aarnoutse R. E. Annu. Rev. Pharmacol. Toxicol. 2018;58:271–291. doi: 10.1146/annurev-pharmtox-010617-052438. [DOI] [PubMed] [Google Scholar]
- Szumowski J. D. Adams K. N. Edelstein P. H. Ramakrishnan L. Curr. Top. Microbiol. Immunol. 2013;374:81–108. doi: 10.1007/82_2012_300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahlan K. Wilson R. Kastrinsky D. B. Arora K. Nair V. Fischer E. Barnes S. W. Walker J. R. Alland D. Barry C. E. Boshoff H. I. Antimicrob. Agents Chemother. 2012;56:1797–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao M. McNeil M. Cook G. M. Lu X. Eur. J. Med. Chem. 2020;200:112390. doi: 10.1016/j.ejmech.2020.112390. [DOI] [PubMed] [Google Scholar]
- Poce G. Bates R. H. Alfonso S. Cocozza M. Porretta G. C. Ballell L. Rullas J. Ortega F. De Logu A. Agus E. La Rosa V. Pasca M. R. De Rossi E. Wae B. Franzblau S. G. Manetti F. Botta M. Biava M. PLoS One. 2013;8:e56980. doi: 10.1371/journal.pone.0056980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. T. Haiderer E. R. Coulson G. B. Conner K. N. Ellsworth E. Chen C. Alvarez-Cabrera N. Li W. Jackson M. Dick T. Abramovitch R. B. Antimicrob. Agents Chemother. 2019;63:e00547-00519. doi: 10.1128/AAC.00547-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C.-C. Klenotic P. A. Bolla J. R. Purdy G. E. Robinson C. V. Yu E. W. Proc. Natl. Acad. Sci. U. S. A. 2019;116:11241–11246. doi: 10.1073/pnas.1901346116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson B. Dooley K. E. Zhang Y. Back D. J. Owen A. Antimicrob. Agents Chemother. 2013;57:6366–6369. doi: 10.1128/AAC.01124-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. M. Wells R. M. Madduri A. V. R. Renfrow M. B. Ratledge C. Moody D. B. Niederweis M. Proc. Natl. Acad. Sci. U. S. A. 2014;111:1945–1950. doi: 10.1073/pnas.1311402111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P. Gu Y. Li J. Xie L. Li X. Xie J. J. Membr. Biol. 2017;250:573–585. doi: 10.1007/s00232-017-9982-x. [DOI] [PubMed] [Google Scholar]
- Niederweis M. Microbiology. 2008;154:679–692. doi: 10.1099/mic.0.2007/012872-0. [DOI] [PubMed] [Google Scholar]
- Braibant M. Gilot P. Content J. FEMS Microbiol. Rev. 2000;24:449–467. doi: 10.1111/j.1574-6976.2000.tb00550.x. [DOI] [PubMed] [Google Scholar]
- Mitra A. Speer A. Lin K. Ehrt S. Niederweis M. mBio. 2017;8:e01720-01716. doi: 10.1128/mBio.01720-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. Boshoff H. I. M. Harrison J. R. Ray P. C. Green S. R. Wyatt P. G. Barry C. E. Science. 2020;367:1147–1151. doi: 10.1126/science.aav5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boelaert J. R. Vandecasteele S. J. Appelberg R. Gordeuk V. R. J. Infect. Dis. 2007;195:1745–1753. doi: 10.1086/518040. [DOI] [PubMed] [Google Scholar]
- Kurthkoti K. Amin H. Marakalala M. J. Ghanny S. Subbian S. Sakatos A. Livny J. Fortune S. M. Berney M. Rodriguez G. M. mBio. 2017;8:e01092-01017. doi: 10.1128/mBio.01092-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow G. A. Bacteriol. Rev. 1970;34:99–125. doi: 10.1128/BR.34.2.99-125.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. M. Niederweis M. J. Bacteriol. 2011;193:1767–1770. doi: 10.1128/JB.01312-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold F. M. Weber M. S. Gonda I. Gallenito M. J. Adenau S. Egloff P. Zimmermann I. Hutter C. A. J. Hürlimann L. M. Peters E. E. Piel J. Meloni G. Medalia O. Seeger M. A. Nature. 2020;580:413–417. doi: 10.1038/s41586-020-2136-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A. Ko Y.-H. Cingolani G. Niederweis M. Nat. Commun. 2019;10:4260. doi: 10.1038/s41467-019-12109-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores-Valdez M. A. Morris R. P. Laval F. Daffé M. Schoolnik G. K. FASEB J. 2009;23:4091–4104. doi: 10.1096/fj.09-132407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez G. M. Smith I. J. Bacteriol. 2006;188:424–430. doi: 10.1128/JB.188.2.424-430.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borah K. Beyß M. Theorell A. Wu H. Basu P. Mendum T. A. Nöh K. Beste D. J. V. McFadden J. Cell Rep. 2019;29:3580–3591. doi: 10.1016/j.celrep.2019.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]; .e3584
- Agapova A. Serafini A. Petridis M. Hunt D. M. Garza-Garcia A. Sohaskey C. D. de Carvalho L. P. S. eLife. 2019;8:e41129. doi: 10.7554/eLife.41129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya N. Nkumama I. N. Newland-Smith Z. Lin L.-Y. Yin W. Cullen R. E. Griffiths J. S. Jarvis A. R. Price M. J. Chong P. Y. Wallis R. O'Hare H. M. mBio. 2018;9:e00931-00918. doi: 10.1128/mBio.00931-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinath K. Venclovas Č. Ioerger T. R. Sacchettini J. C. McKinney J. D. Mizrahi V. Warner D. F. Open Biol. 2013;3:120175. doi: 10.1098/rsob.120175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rempel S. Gati C. Nijland M. Thangaratnarajah C. Karyolaimos A. de Gier J. W. Guskov A. Slotboom D. J. Nature. 2020;580:409–412. doi: 10.1038/s41586-020-2072-8. [DOI] [PubMed] [Google Scholar]
- Fullam E. Prokes I. Fütterer K. Besra G. S. Open Biol. 2016;6:160105. doi: 10.1098/rsob.160105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D. Zhang Q. Zheng Q. Zhou H. Jin J. Zhou W. Bartlam M. Rao Z. FEBS J. 2014;281:331–341. doi: 10.1111/febs.12600. [DOI] [PubMed] [Google Scholar]
- Fenn J. S. Nepravishta R. Guy C. S. Harrison J. Angulo J. Cameron A. D. Fullam E. ACS Chem. Biol. 2019;14:1879–1887. doi: 10.1021/acschembio.9b00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan D. Nagalingam G. Payne R. Triccas J. A. Int. J. Infect. Dis. 2017;56:212–220. doi: 10.1016/j.ijid.2016.12.024. [DOI] [PubMed] [Google Scholar]
- O'Hagan S. Kell D. B. Pharmaceutical Frontiers. 2019;1:e190005. doi: 10.20900/pf20190005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.