

Abstract
The development of the next generation therapy for Alzheimer’s disease (AD) presents a huge challenge given the number of promising treatment candidates that failed in trials, despite recent advancements in understanding of genetic, pathophysiologic and clinical characteristics of the disease. This review reflects some of the most current concepts and controversies in developing disease-modifying and new symptomatic treatments. It elaborates on recent changes in the AD research strategy for broadening drug targets, and potentials of emerging non-pharmacological treatment interventions. Established and novel biomarkers are discussed, including emerging cerebrospinal fluid and plasma biomarkers reflecting tau pathology, neuroinflammation and neurodegeneration. These fluid biomarkers together with neuroimaging findings can provide innovative objective assessments of subtle changes in brain reflecting disease progression. A particular emphasis is given to neurophysiological biomarkers which are well-suited for evaluating the brain overall neural network integrity and function. Combination of multiple biomarkers, including target engagement and outcome biomarkers will empower translational studies and facilitate successful development of effective therapies.
Keywords: Alzheimer’s disease, drug development, non-pharmacological treatment, clinical trials, novel biomarkers
Graphical Abstract
1. Introduction
Worldwide increase in life expectancy has produced a dramatic rise in the prevalence, and thus impact of aging-associated diseases including Alzheimer’s disease (AD) on the biomedicine and society at large. It has been estimated that 5.8 million Americans live with AD in 2020, and that number is projected to triplicate by mid-century. Currently, it is the sixth leading cause of death in the United States and the fifth leading cause of death in patients 65 years of age and older (Alzheimer’s Association, 2020)
Clinically AD is characterized by slowly progressive neurodegeneration leading to overt cognitive decline involving loss of memory and higher executive functioning. The core pathological hallmarks of AD in the brain are accumulated extracellular plaques composed of amyloid-β (Aβ) proteins, and intracellular neurofibrillary tangles (NFTs) consisting of hyperphosphorylated tau (Fig. 1), both contributing to the damage and death of neurons (Ittner and Gotz, 2011). Recent research demonstrated that sustained neuroinflammation, impairments of mitochondrial bioenergetics and protein homeostasis have also prominent role in unfolding of AD (Kinney et al., 2018; Perez Ortiz and Swerdlow, 2019; Veitch et al., 2019), thus implying to multifactorial etiology of the disease. AD exists under two forms: sporadic AD (sAD), characterized by late onset and accounts for vast majority of AD cases, and familial AD (fAD), an early onset and dominantly inherited disease, which accounts for less than 1% of cases (Alzheimer’s Association, 2020). For sAD, age is the major risk factor, but carrying the ApoE-ε4 genetic polymorphism highly increase the likelihood of developing disease in subjects older than 65 years. The fAD has more rapid progression affecting younger subjects with highly penetrant mutations in the genes encoding for amyloid precursor protein (APP), and presenilins 1 and 2 (PS1 and PS2) that result in accumulation of Aβ and formation of plaques (Bertram and Tanzi, 2012).
Figure 1.
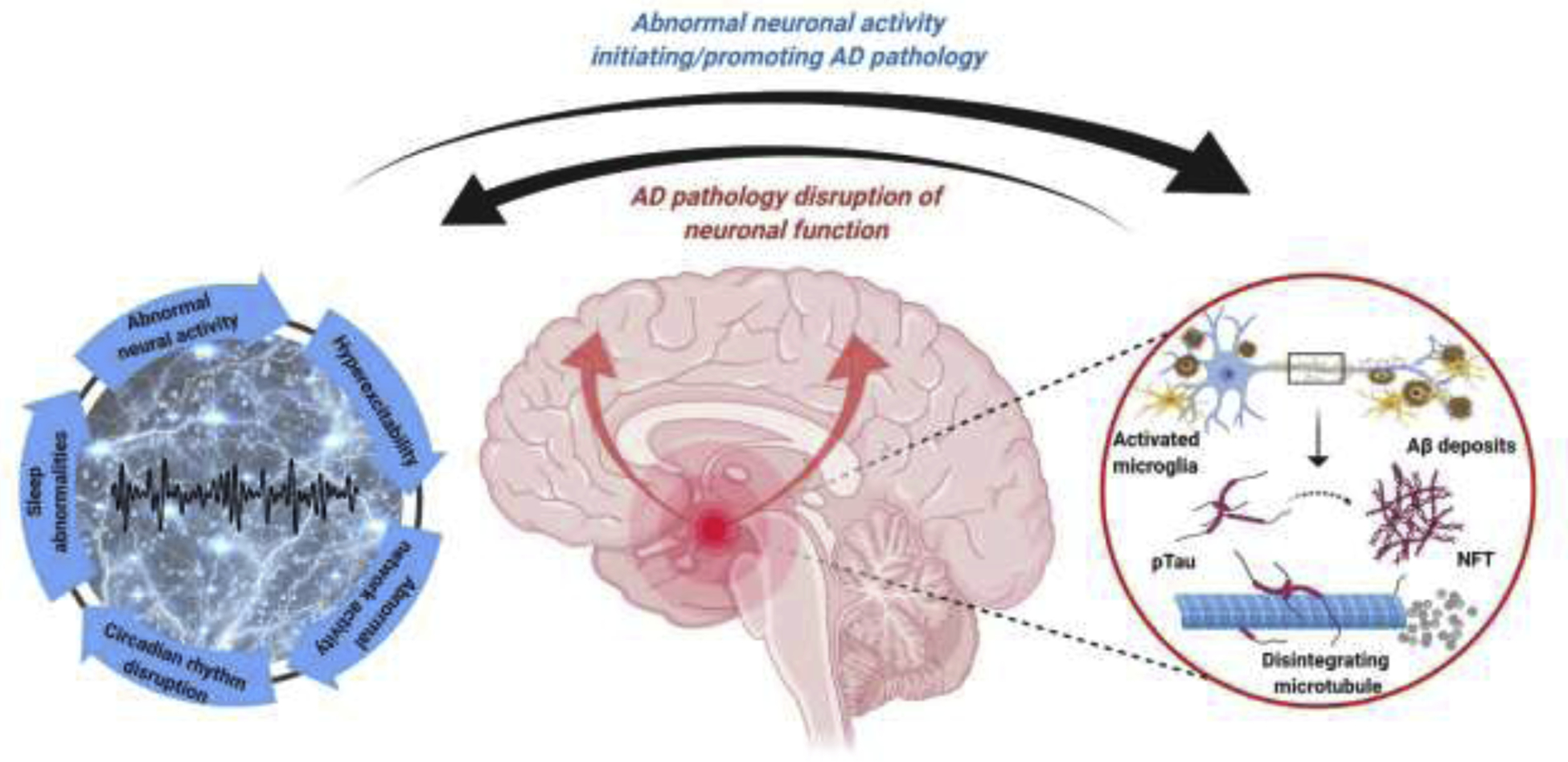
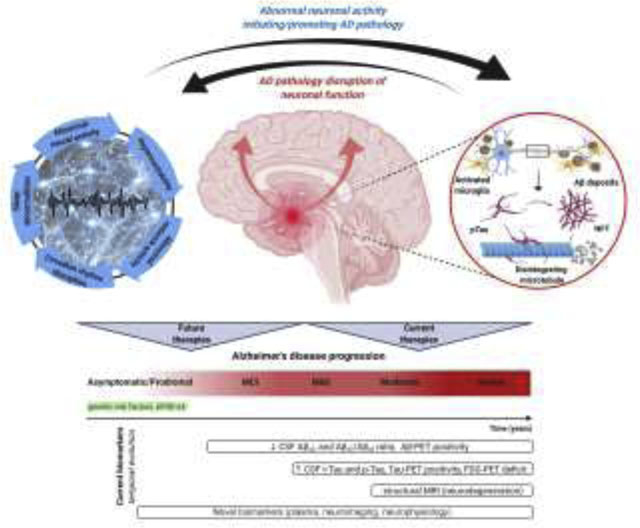
Bidirectional interaction between AD pathology and abnormal neuronal activity. Detrimental effects of AD related pathology, such as Aβ and tau oligomers are well known, resulting in abnormal neuronal activity and neuronal network function. These abnormalities lead to hyperexcitability, epileptiform activity, altered sleep/wake activity, circadian rhythm, and disruption of coherent network activities supporting cognitive functions. However, aberrant neuronal and neuronal network activities, such as sleep abnormalities can initiate and further disease progression. Therefore, a bidirectional relationship between pathological neuronal activity and AD can be proposed, indicating a vicious circle contributing to AD progression. Figure created with BioRender.com
Despite significant advances in our understanding of pathological mechanisms involved in AD, treatment options are still limited. Existing drugs provide only short-term symptomatic benefits, while the development of effective disease-modifying therapeutic interventions that would prevent, halt, or reverse AD seems to be notoriously difficult task given the large number of compounds, particularly those targeting Aβ, which failed in late-stage clinical trials (Cummings et al., 2020; Long and Holtzman, 2019). Repeated setbacks in development of AD treatments recently prompted changes in the research strategy, which increasingly begins to focus on multifaceted targets and their possible interactions rather than just on disease canonical mainstays, Aβ and hyperphosphorylated tau. At this inflection point, it is necessary to critically evaluate all potential factors that influence negative outcomes in clinical trials, from translatability of results obtained in preclinical AD models to predictive validity of currently used biomarkers for treatment efficacy and improvement in the disease state.
In this review we provide an updated outline of the current therapeutic possibilities, status of AD drug development focusing mainly on compounds being tested in clinical trials, and an overview of emerging non-pharmacological treatment interventions. We also discuss challenges and limitations in AD discovery efforts, and importance of inclusion of functional biomarkers such as neurophysiological signatures of disease, which combining with currently used fluid and imaging biomarkers would form multicomponent biomarker panel for ensuring adequate translation of preclinical to human studies and leveraging treatment development research into patients benefit.
1. Current therapeutics
The current treatment for clinically diagnosed AD patients utterly relies on pharmacological modulation of cholinergic and glutamatergic neurotransmission. There are only five drugs approved for AD by the United States Food and Drug Administration (FDA), including cholinesterase inhibitors donepezil, rivastigmine, and galantamine, a N-methyl-D-aspartate (NMDA) receptor partial antagonist memantine, and a fixed-dose combination of donepezil and memantine (Alzheimer’s Association, 2020). All these drugs, however, have limited effectiveness and they do not reverse the course of the disease, or even substantially improve cognitive dysfunction, but rather temporarily delay the deterioration of AD symptoms (Birks and Harvey, 2018; Knight et al., 2018).
Cholinergic neurons in the basal forebrain are among the earliest targets of AD pathology, and their severe loss leads to decreased cholinergic neurotransmission consequently resulting in impairment of cortical activation with cognitive and behavioral symptoms characterized by memory, sensory processing and sleep disturbances (Mesulam, 2013). Accordingly, enhancement of cholinergic neurotransmission was recognized as tenable treatment goal to ameliorate and delay these dysfunctions in AD patients. Cholinesterase inhibitors bind reversibly with and inactivate enzyme acetylcholinesterase (Nestler et al., 2015), thus inhibiting hydrolysis of neurotransmitter acetylcholine and increasing its concentrations at cholinergic synapses still unaffected by neurodegeneration. In the clinical treatment of AD, these symptomatic drugs are recommended as the first-line therapy for mild-to-moderate stage of the disease. However, systematic review of clinical data (Knight et al., 2018) showed that cholinesterase inhibitors have significant, though practically modest, effects in improving cognitive symptoms and maintaining patient functional ability over the period of 6 months to 1 year. Apart from the cholinesterase inhibitors, cholinergic drugs that directly modulate muscarinic and nicotinic acetylcholine receptors have also been considered as viable treatment options for AD, and several drug candidates are or were under development (see below).
Memantine is other FDA approved symptomatic treatment for AD in addition to the cholinesterase inhibitors. It is a low affinity and voltage-dependent antagonist of glutamatergic NMDA receptors (Nestler et al., 2015), which clinical effects likely resulted from neuronal protection of excessive NMDA activation associated with excitotoxicity. Supporting evidence suggest that memantine, at therapeutically relevant concentrations, preferentially blocks extrasynaptic NMDA receptors in condition of excessive stimulation, without disrupting synaptic NMDA receptor mediated neurotransmission (Lipton, 2005; Xia et al., 2010). Memantine is currently indicated for patients with moderate-to-severe AD as a monotherapy or in combination with cholinesterase inhibitors.
2. Potential therapeutic interventions
2.1. Drugs targeting Aβ
In the realm of AD research, amyloid cascade hypothesis (Hardy and Selkoe, 2002), has been a dominant strategy influencing considerable clinical, preclinical and drug discovery research for years. Conceptualized upon observations that increased Aβ production in neurons induces chain of neurotoxic events resulting in synaptic dysfunction and generation of tau-containing NFTs with consequent cell loss and progressive cognitive impairments, it offered an attractive framework to explain AD pathogenesis. Accordingly, pharmacologic interventions to counteract deleterious effects of Aβ by preventing its excessive production and buildups or promoting its clearance prevailed as promising approach to arrest or reverse AD. Intriguingly, even though amyloid pathology is undoubtedly involved in the trajectory of AD, none of the drug candidates targeting Aβ to modify this disease succeeded in clinical trials so far.
2.1.1. Drugs to reduce Aβ production
Aβ peptide is produced by sequential proteolytic processing of the APP by β- and γ-secretases, respectively (LaFerla et al., 2007). Inhibition of these enzymes reduces the level of Aβ in the brain and limits the deposition of amyloid plaques through the upstream interference with the amyloid cascade.
Development of β-secretase (β-site APP–cleaving enzyme 1) or BACE1 inhibitors have been considered as one of the priority disease-modifying treatment approach for AD, and several drugs have reached the late phase clinical testing, including atabecestat (JNJ-54861911), verubecestat (MK-8931), and lanabecestat (AZD3293). All these compounds were found to significantly lower the brain Aβ levels measured by positron emission tomographic (PET) imaging and in cerebrospinal fluid (CSF), clearly indicating their target engagement. However, their further clinical development was discontinued due to the unacceptable side effects or ineffectiveness to improve cognitive status in AD patients (Das and Yan, 2019). Results from the recent clinical trials of verubecestat and atabecestat surprisingly showed that both drugs have even detrimental effects on cognitive function and brain structure (Egan et al., 2019; Henley et al., 2019). On the other hand, lanabecestat which was better tolerated than other two drugs, showed convincingly lack of improvement in cognitive function of AD patients. The reasons for these negative outcomes are not yet fully revealed, but some of the potentially contributing factors include the wide substrate-binding domain of BACE1 enzyme, and propensity for non-selective blockade of both BACE1 and BACE2 isoenzymes of some of these drugs (Barao et al., 2016). There are about 45 known substrates for BACE1 enzyme, and changes in their processing induced by these drugs might cause some of the observed side effects, or even mask their cognitive benefits. In fact, interference between some of BACE1 inhibitors and enzyme’s non-amyloid substrates such as neuregulin-1, which is involved in myelination, or CHL1, the neural cell adhesion molecule close homolog of L1, involved in neurite outgrowth, might account for structural brain damage and cognitive worsening associated with their use (Ben Halima et al., 2016; Hitt et al., 2012; Zhu et al., 2018). Another possible explanation for inefficacy of these drugs is their excessive inhibition of BACE1, which in turn can compromise normal synaptic functions. For example, it has been suggested that moderate inhibition of BACE1 is sufficient to reduce the toxic effects of Aβ production while still allowing the physiological function of the enzyme at synapse, as observed in carriers of a rare APP mutation (Knopman, 2019). Non-selective BACE1/2 inhibition is additional challenging factor and some side effects reported after use of BACE1 inhibitors, like hair and skin depigmentation, could result from this property (Cebers et al., 2016). The two BACE1 inhibitors, umibecestat (CNP520) and elenbecestat (E2609), have been shown to have significantly higher selectivity to BACE1 over BACE2 enzyme comparing with formerly tested BACE1 inhibitors (Lynch et al., 2018; Neumann et al., 2018), thus lower chances to encounter targets that would lead to serious side effects. Moreover, umibecestat was shown not to produce hippocampal structural alterations as described for earlier BACE1 drugs, likely due to the more selective activity on enzyme’s subcellular domain and sparing CHL1 processing (Barao et al., 2016). However, their late-stage clinical development has been recently terminated due to inefficacy.
In the biochemical network involved in generation and accumulation of Aβ, γ-secretase is the enzyme responsible for the final step in the amyloidogenic APP processing pathway. It is a membrane-embedded protease that cleaves APP within its transmembrane domain yielding to Aβ species of different lengths, including Aβ1–40 and Aβ1–42, the most common isoforms in the amyloid plaques. The catalytic core of γ-secretase consists of presenilins (PS1 and PS2), which missense mutations are a major cause of fAD, and three accessory proteins i.e. nicastrin, anterior pharynx-defective 1 (APH1) protein, and PS enhancer 2 (Pen-2) (De Strooper, 2003). This complex enzyme, besides cleaving APP also regulates processing of series of integral membrane proteins including Notch, N-cadherin and variety of other important cellular substrates making development of γ-secretase inhibitors (GSIs) challenging similar to those for BACE1 inhibitors (Mangialasche et al., 2010). In the past decade, several GSIs progressed to clinical trials due to their high potency to lower Aβ production indicated at the preclinical level. These include: semagacestat (LY-450139), begacestat (GSI-953), and MK-0752. Unfortunately, none of these compounds were successful in clinical trials, as their use in AD patients was followed by lack of cognitive improvement and severe adverse gastrointestinal and dermatological side effects (Doody et al., 2013; Golde et al., 2013; Martone et al., 2009). Importantly, these adverse events were present at pharmacologically active concentrations and mostly account for the inherent mechanism based-toxicity, given their high affinity to block the Notch signaling pathway which is critically involved in synaptic plasticity, cell differentiation and survival (Ables et al., 2011). In an attempt to overcome toxicity issues, GSIs that are more restrictive to APP and with Notch-sparing property have been synthetized, such as avagecestat (BMS-708163) and nirogacestat (PF-3084014). Nevertheless, the effort has proven to be futile since neither lower toxicity nor increased efficacy were achieved in AD patients (Coric et al., 2015; Crump et al., 2012; Kumar et al., 2018). In addition to GSIs, γ-secretase modulators (GSMs) emerged as possibly safer agents for AD. These compounds interact with γ-secretase through the allosteric binding site without altering physiological activity of the enzyme. They were shown to reduce the deposition of pathogenic Aβ by shifting the profile of the secreted Aβ peptides toward the shorter non-amyloidogenic species (Bursavich et al., 2016). Both the first generation of GSMs, comprised of repurposed non-steroidal anti-inflammatory drugs ibuprofen, indomethacin and tarenflurbil, and second generation GSMs such as E2012, after initial promise did not show clinical benefits in AD patients (Eriksen et al., 2003; Green et al., 2009; Imbimbo, 2009; Nagy et al., 2010). Presently, it is not clear whether GSI or GSM discovery programs are completely abandoned, but in the current AD drug development pipeline there is no such drugs in clinical testing.
Experimental studies indicated that upregulation of α-secretase activity may also have therapeutic potential for AD (Lichtenthaler and Haass, 2004), since the activation of α-secretase decreases Aβ generation and concomitantly increases formation of neuroprotective soluble form of APP (APPa) by facilitating its proteolysis in non-amyloidogenic pathway (De Strooper et al., 2010). As a result development of α-secretase enhancers emerged as an innovative alternative to β- and γ-secretase inhibitors. However, even though early α-secretase enhancing compounds (Marcade et al., 2008; Snow et al., 2009) did not make significant progress in clinical studies, the rationale behind developing novel α-secretase modulators strongly recommended further efforts and thus two drug candidates, APH-1105 and ID1201, are currently in phase 2 clinical trials.
2.1.2. Drugs to facilitate Aβ clearance
Active and passive immunization against Aβ peptides has been considered as another promising therapeutic strategy to tackle AD pathology (Herline et al., 2018; van Dyck, 2018). Biochemical characterizations of toxic Aβ species that either in form of soluble oligomers, protofibrils or aggregates affect normal neuronal and synaptic functions, enable the development of both vaccines and specific antibodies with potential for their removal from the brain. Using this anti-amyloid approach considerable effects has been observed in transgenic animal models overproducing Aβ (Wilcock and Colton, 2008), yet translation of its therapeutic potential in patients seems to be difficult given the number of Aβ immunotherapy trials that failed thus far.
An early clinical study with anti-Aβ vaccine AN1792 in mild-to-moderate AD patients was stopped when some subjects developed toxicity in the form of T cell-mediated meningoencephalitis (Gilman et al., 2005). Interestingly, a recent report of 15-year postmortem neuropathological follow-up of patients involved in this trial (Nicoll et al., 2019) showed extensive and persisted plaque clearance, but this effect unfortunately did not affect progression of severe cognitive impairments and spread of tau pathology nor prolong survival time. To achieve better safety profile of anti-Aβ vaccine that stimulates an immune response against Aβ by molecular mimicry of the parts of native Aβ sequence, such as B-cell epitope but not T-cell epitope in its antigenic component, has been tailored (Schneeberger et al., 2009). Although better tolerated, this vaccine also failed to produce significant clinical effects in AD patients (Schneeberger et al., 2015). Nevertheless, active immunization strategy is still considered as potential approach for AD therapy, particularly for use in early stage of disease, or even as a preventive measure. In the recent clinical trials there are four different anti-Aβ vaccines in evaluation. In the most advanced clinical phase is investigational vaccine CAD106 (Farlow et al., 2015), which is being tested (Table 1) in cognitively unimpaired individuals who have genetic profile that place them at high risk for developing AD (elderly homozygous ApoE4 carriers). Two other vaccines, ABvac40 and UB-311, are currently in phase 2, and one vaccine, LuAF20513, is in phase 1. The latter vaccine is engineered with aim to induce production of anti-Aβ polyclonal antibodies and robust “non-self” responses of preexisting memory T-helper cells stemming from previously immunized elderly individuals against tetanus (Davtyan et al., 2013).
Table 1.
Active Phase 3 clinical trials of pharmacological treatments in AD (clinicaltrials.gov as of February 10, 2021)
| Investigational drug | Study population/treatment target | Type of treatment | Clinical trial identifier |
|---|---|---|---|
| Disease modifying treatment | |||
| Gantenerumab | prodromal to mild AD | fully human monoclonal antibody |
NCT03443973 NCT03444870 NCT02051608 NCT01224106 |
| Solanezumab | asymptomatic AD, positive amyloid PET | humanized monoclonal antibody | NCT02008357 |
| Aducanumab | prodromal to mild AD, previous participants in aducanumab clinical study | fully human monoclonal antibody | NCT04241068 |
| Lecanemab (BAN2401) | prodromal to mild AD | humanized monoclonal antibody |
NCT03887455 NCT04468659 |
| TRx0237 (Methylene blue) | MCI and probable AD, positive amyloid PET | tau aggregation inhibitor | NCT03446001 |
| AGB101 (Levetiracetam) | prodromal AD | anticonvulsant, SV2A modulator | NCT03486938 |
| BHV-4157 (Riluzole prodrug) | mild to moderate AD | glutamate release inhibitor | NCT03605667 |
| ALZT-OP1 (cromolyn+ibuprofen) | prodromal AD | mast cell stabilizer, microglial modulator, anti-inflammatory | NCT02547818 |
| Sodium Oligomannate (GV-971) | mild to moderate AD | bioactive marine-derived oligosaccharide | NCT04520412 |
| Tricaprilin | mild to moderate AD | inductor of ketosis | NCT04187547 |
| NE3107 | mild to moderate AD | sterol derivative | NCT04669028 |
| COR388 | mild to moderate AD | gingipains inhibitor | NCT03823404 |
| Preventional treatment | |||
| CAD106 | elderly homozygous ApoE4 carriers at risk of clinical AD | anti-Aβ vaccine + BACE1 inhibitor | NCT02565511 |
| Solenezumab | individuals at risk due to AD-causing mutations | monoclonal Aβ antibodies | NCT01760005 |
| Metformin | early and late MCI | antihyperglycemic agent | NCT04098666 |
| Symptomatic treatment | |||
| Brexpiprazole | agitation due to AD | dopamine D2 receptors partial agonist |
NCT03594123 NCT03724942 NCT03548584 NCT03620981 |
| AVP-786 | agitation due to AD | NMDA receptors antagonist and sigma 1 receptors agonist |
NCT03393520 NCT02446132 |
| Escitalopram | agitation due to AD | selective serotonin reuptake inhibitor | NCT03108846 |
| Mirtazapine | agitation due to AD | adrenergic α2 and serotonin 5-HT2/5-HT3 receptors antagonist | NCT03031184 |
| Nabilone | agitation due to AD | synthetic cannabinoid | NCT04516057 |
| Octohydroaminoacridine succinate | cognitive deficiencies in mild to moderate AD | acetylcholinesterase inhibitor | NCT03283059 |
| ANAVEX2–73 | cognitive deficiencies in prodromal AD | Sigma1 receptor agonist | NCT03790709 |
| Guanfacine | attention deficit in AD | adrenergic α2 receptors agonist | NCT03116126 |
| Methylphenidate | apathy in AD | dopamine and norepinephrine reuptake inhibitor | NCT02346201 |
The systemic application of anti-Aβ antibodies is another therapeutic approach to neutralize Aβ toxicity and promote plaque clearance. In contrast to active vaccination, passive immunotherapy using monoclonal antibodies has been seen as more viable strategy in directing the immune response towards particular epitope or isoform of Aβ. Also, this approach can improve treatment efficacy by preserving consistent antibody titer and enabling better control of possible adverse events. Over the past decade several monoclonal antibodies directed against Aβ such as bapineuzumab (AAB001), solanezumab (LY2062430), ponezumab (PF04360365), crenezumab (MABT5102A), gantenerumab (RG1450), aducanumab (BIIB037), donanemab (Ly3002813) and lecanemab (BAN2401) have being tested in AD patients (van Dyck, 2018), however just few of them are still in clinical evaluation (Table 1).
Bapineuzumab is a humanized IgG1 anti-Aβ monoclonal antibody corresponding to the murine clone 3D6 specific for an N-terminal Aβ epitope (residues 1–5) (Kerchner and Boxer, 2010). This was the first antibody tested in clinical trials. It binds to and clears soluble and fibrillar Aβ peptides by triggering microglial phagocytosis. Imaging analyses showed evidence of target engagement in lowering of Aβ in patients treated with bapineuzumab over placebo control, however, in two parallel phase 3 trials no significant clinical benefits on cognition were found in mild-to-moderate AD patients (Salloway et al., 2014). Moreover, high dose of bapineuzumab was associated with increased rate of symptomatic amyloid-related imaging abnormalities (ARIA) such as edema and microhemorrhages (Sperling et al., 2012), leading to the discontinuation of its further development. Solanezumab is also humanized IgG1 monoclonal antibody designed to recognize an epitope within Aβ mid-domain (residues 16–26). It increases the clearance of soluble Aβ peptides from the brain leading to dose-dependent rise in plasma and CSF Aβ levels (Farlow et al., 2012). It has a favorable safety profile, but was without marked benefits for the cognitive and functional outcomes in patients with mild-to-moderate AD in phase 3 trials (Doody et al., 2014; Honig et al., 2018). Ponezumab is a humanized IgG2 monoclonal antibody with affinity towards C-terminus (residues 30–40) that binds to soluble Aβ (La Porte et al., 2012). This antibody has safe profile expressing a lower propensity to induce immune effector response comparing to other IgG1 antibodies in the class, however, two phase 2 studies revealed clinical inefficacy, and its development was also discontinued (Burstein et al., 2013; Landen et al., 2017). Crenezumab, a humanized monoclonal IgG4 antibody with ability to bind to different Aβ species (oligomeric, fibrillar, and plaque-embedded) was designed to minimize Fc gamma receptor-mediated inflammatory activation of microglia in order to lower the risk of ARIA (Adolfsson et al., 2012). Similar to others, clinical trials with crenezumab brought to one more disappointment, since no reduction in cognitive decline of treated AD patients was demonstrated (Cummings et al., 2018; Salloway et al., 2018). Gantenerumab is the first fully human IgG1 monoclonal antibody that binds to a conformational epitopes within the N-terminus (residues 3–12) and at central region (residues 18–27) of Aβ oligomers and aggregated fibrils. In transgenic mice, gantenerumab significantly reduced amyloid plaques in the brain by recruiting microglia, but without altering Aβ plasma levels (Bohrmann et al., 2012). Even though this effect was observed in early clinical evaluation in patients with mild-to-moderate AD, subsequent phase 3 study was abruptly terminated following an interim futility analysis and an increased rate of ARIA (Ostrowitzki et al., 2017). Recently, gantenerumab has brought back in clinical evaluation in four phase 3 trials, two ongoing, for evaluating its effect on cognition in prodromal and mild AD patients, and two currently recruiting patients with early AD (Klein et al., 2019). Aducanumab, is a potent recombinant human IgG1 monoclonal antibody developed through screening libraries of memory B cells produced in cognitively healthy aged donors reacting against aggregated Aβ. This anti-Aβ antibody binds the N-terminus (residues 3–6) of the Aβ sequence and specifically targets soluble oligomers and insoluble fibrils. Initial evaluation of aducanumab convincingly showed robust, dose-dependent clearing of fibrillary amyloid plaques measured by PET imaging together with slowing of cognitive decline both in preclinical models and in mild AD patients (Sevigny et al., 2016). Later clinical assessments in two phase 3 trials were stopped early possibly due to insufficient efficacy in an interim futility analysis (Selkoe, 2019), but subsequently aducanumab development has revived after additional analysis of larger dataset showing positive results in reducing clinical decline in AD patients (Howard and Liu, 2019). Back on track of aducanumab, together with two investigational monoclonal antibodies in underway clinical trials, donanemab and lecanemab keep optimism in success of Aβ-directed immunotherapy concept after more than a decade of research futility. Donanemab, a humanized IgG1 antibody is developed from murine mE8-IgG2a which specifically targets a pyroglutamate form of Aβ (pGlu-3 Aβ) aggregated in amyloid plaques (Cynis et al., 2016). Evidence suggest that pGlu-3 Aβ modified peptide has high amyloidogenic and neurotoxic potential given its high presence in diffuse plaques and vascular amyloids that cause cerebral angiopathy in AD (Crehan et al., 2020). Preclinical study showed that donanemab reduces Aβ deposits in mice without causing ARIA (Demattos et al., 2012), recommended its further development in ongoing phase 2 trial (Irizarry et al., 2018). Recent company’s announcement hinted that compound has shown significant slowing of decline in a composite measure of cognition and daily function in subjects with early symptomatic AD, but official study report is yet to come. Lecanemab is another humanized IgG1, derived from the E22G arctic mutation in the APP that actively binds to and clears soluble Aβ protofibrils (Lannfelt et al., 2014). Driven by phase 2 data of significant plaque clearance and possible reduction in cognitive decline in patients with mild AD (Logovinsky et al., 2016), this compound has recently entered a phase 3 trial.
The passive immunotherapy using monoclonal antibodies has also been considered in primary and secondary prevention studies where it might provide better opportunity to modify disease in the protracted asymptomatic or prodromal phase of AD. Currently, solanezumab and gantenerumab are being tested in asymptomatic subjects with fAD mutations that have high risk for developing AD (Bateman et al., 2017) (Table 1), while crenezumab is in active phase 2 trial in Colombian preclinical autosomal-dominant carriers of PS1 E280A mutation (Tariot et al., 2018).
General characteristic of all failed anti-Aβ trials is that even though most investigational compounds showed robust target engagement, i.e. reducing Aβ in the brain measured by PET imaging and CSF biomarkers, they did not significantly improve performances in cognitive functions of AD patients. This dissociation demonstrates that these biomarkers are not surrogate markers for treatment efficacy in clinical trials, but more importantly question if the Aβ accumulation represents a proper target for disease modification after clinical symptoms arise. Compelling evidence showed that plasma and CSF Aβ levels are reliable indicators of pathological processes in the AD brains (Blennow and Zetterberg, 2019) and thus can serve as biomarkers for diagnostic assessment and perhaps stratification of patients in clinical trials. Similarly, imaging biomarkers facilitate accurate diagnosis and can allow tracking of distinctive changes in the brain that are characteristic for AD progression. However, the value of these biomarkers in predicting treatment benefit remains unclear, since trajectory of their changes usually does not parallel with response to treatment and clinical improvement (Penninkilampi et al., 2017), thus challenging the separation of drug from placebo in AD trials. Another frequently cited reason for lack of positive outcome in amyloid-targeted trials is probably belated initiation of the treatment when the window for disease modification has already passed and neuronal damage might advance to an irreparable stage (Jack et al., 2013; van Dyck, 2018). Therefore, demonstrating Aβ clearance in AD patients perhaps is not the ultimate marker indicating achievement of meaningful treatment benefits.
2.2. Drugs targeting tau
Disappointing clinical outcomes of drug candidates targeting accumulation of Aβ oligomers and plaque formation shifted drug discovery efforts to AD-related tau pathology. Microtubule associated protein tau (MAPT) has important cytoskeletal roles such as microtubule assembly and transport, and stabilization of neuronal axons that are impaired in AD. Tau filaments and hyperphosphorylated tau accumulated intracellularly in NFTs directly contribute to neuronal destruction and neurodegeneration. Also, it has been shown that toxic tau isoforms spread via neuronal synapses in prion-like pattern (Jucker and Walker, 2018), which theoretically could be pharmacologically blocked. Thus, multiple strategies have been proposed for reducing tau pathogenicity including stabilization of microtubules, modulation of abnormal tau phosphorylation, and inhibition or prevention of tau aggregation, seeding and spreading. Early anti-tau therapies were primarily based on inhibition of tau hyperphosphorylation and NFTs formation by modulating activity of protein kinases (i.e. glycogen-synthase-kinase-3) involved in tau metabolism. However, these attempts have been discontinued because of toxicity or lack of efficacy (Congdon and Sigurdsson, 2018). Current clinical trials include inhibitors of tau aggregation, tau antibodies, and antisense oligonucleotides against the MAPT RNA (Bittar et al., 2020; Cummings et al., 2019a). Potential of methylene blue (TRx0237) for degradation of existing NFTs and prevention of new tau aggregates associated with AD is exploring in an ongoing phase 3 trial (Table 1). Tau-targeted active and passive immunotherapies are actively being tested for safety and efficacy in early-stage AD patients. Several compounds recently advanced to phase 2 trials including vaccine AADvac-1, and monoclonal antibodies directed toward different aspects of the pathological tau such as gosuranemab (BIIB092), semorinemab (RO7105705), zagotenemab (LY3303560), and tilavonemab (ABBV-8E12) (Bittar et al., 2020; Hoskin et al., 2019). An innovative and promising approach against tau pathology in AD brain is the use of antisense oligonucleotides which have potential to safely reduce MAPT mRNA impacting post-translational modifications of tau and thereby preventing expression and seeding activity of toxic isoforms in neurons. Intrathecal delivery of such a drug IONIS-MAPTRx is currently being tested in a Phase 1/2 trial in patients with mild AD (Mignon et al., 2018).
No clinical trials have been completed up to date, therefore no conclusion can be drawn on therapeutic benefits of treatments against tau pathology. In contrast to the well-demonstrated genetic link between Aβ pathology and development of AD (see above), no such a genetic risk factor for tau pathology has been established. Based on the lack of genetic connection to AD, and the late terminal role of tau in AD, doubts have been raised about tau-related drug treatment concept.
2.3. Miscellaneous drugs with symptomatic or disease-modifying promise
While most efforts and money have been invested in preventing Aβ and tau pathologies as potential treatments for AD, there is a burgeoning interest in identifying alternative drug targets beyond these miscreant peptides that can impact the disease pathology and translate to beneficial clinical outcome. Accordingly, numerous ongoing preclinical and clinical researches in AD are focusing on modifying neurotransmission, counteracting neuroinflammation, bioenergetics, epigenetics, and protein homeostasis that contribute to AD pathophysiology at different stages.
In AD drug pipeline, several small molecules targeting neurotransmitters’ receptors in cognitive-related brain regions are in the development. This includes novel anticholinesterase inhibitor octohydroaminoacridine (Xiao et al., 2017) (Table 1), as well as cholinergic agents that directly activate muscarinic M1 or nicotinic α7 and α4β2 receptors which have shown potential to improve performance in a variety of preclinical cognitive assays and to modulate the pathogenic effects of APP/Aβ (Bertrand and Terry, 2018; Fisher, 2008; Sako et al., 2019; Stoiljkovic et al., 2015). Several selective agonists of M1 receptors including talsaclidine, MK-7622, TAK-071, HTL0018318, and AGN-242071 have being recently evaluated in clinical trials for symptomatic treatment of cognitive deficits as adjunct therapy to standard AD treatment. Unfortunately, they were all withdrawn from further development due to safety and tolerability issues, or insignificant efficacy (Voss et al., 2018). Enhancement of cholinergic transmission via nicotinic receptors has also been investigated and selective agonists of α7 (encenicline) and α4β2 (ispronicline) receptors were tested in patients with AD, but without successful due to unexpected off-target toxicity (Bertrand and Terry, 2018). Targeting serotoninergic receptors is another potential therapeutic approach to improve cognition and memory (Calhoun et al., 2017). In recent years, inhibitors of 5HT6 receptors (idalopirdine, intepirdine) were in clinical consideration for mild-to-moderate AD. These receptors are wide spread in brain regions that mediate cognition and their blockade is shown to increase concentrations of multiple neurotransmitters facilitating oscillations in neuronal networks relevant in cognition (de Jong and Mork, 2017). Although, the rationale for 5HT6 receptors antagonist use was sound, trials ended as a failure (Bennett, 2018; Khoury et al., 2018). Another 5HT6 antagonist, masupirdine (SUVN-502) also recently failed in phase 2 trial for testing its efficacy as an add-on treatment in subjects with moderate AD being on combined therapy with donepezil and memantine. Phosphodiesterase (PDE) inhibitors are also being studied as potential AD symptomatic treatment. The expression of these enzymes in the brain, and their involvement in neuroplasticity and memory consolidation proposed evaluation of several PDE targeting compounds including PDE-9, PDE-4 and PDE-3 inhibitors in AD patients (Prickaerts et al., 2017) but results did not show clinical promise so far.
Given the high prevalence of neuropsychiatric symptoms observed in AD and lack of drugs approved by FDA for this indication, several neurotransmission-based multimodal compounds that can mitigate agitation, aggression, apathy, and sleep disturbances observed in AD patients are in currently active clinical trials (Table 1). These include drugs such as serotoninergic modulators pimavanserin, escitalopram and mirtazapine, melatonin and serotonin receptors agonist piromelatine, adrenergic α2 agonist guanfacine, orexin receptor antagonists lemborexant and suvorexant, glutamate receptor antagonist riluzole, dopamine D2 receptor agonist brexpiprazole, dopamine and norepinephrine reuptake inhibitor methylphenidate, synthetic cannabinoid nabilone as well as and GABAA receptors allosteric modulators zolpidem and zoplicone which trials are recently completed (Cummings et al., 2020; b). Compounds acting through sigma receptors, chaperon proteins that modulate intracellular calcium signaling, are under investigation for therapeutic use in AD. High affinity sigma1 agonist ANAVEX2–73 (blarcamesine) which also binds to muscarinic receptors, and AVP-786 and AXS-05 that act as sigma1 agonists and NMDA antagonists are in clinical evaluations for neuropsychiatric symptoms associated with AD (Stahl, 2018). Investigational drug elayta (CT1812) is a small molecule which binds to the neuronal sigma2/PGRMC1 (progesterone receptor membrane component 1) protein and disrupts attachment of oligomeric Aβ on brain cells preventing the Aβ-induced synaptic toxicity. Compelling results in restoring cognitive deficits at preclinical studies, and good tolerance and safety profile in prior clinical study in AD patients (Grundman et al., 2019; Izzo et al., 2014) warrant it’s further testing in two ongoing phase 2 studies. Azeliragon is an inhibitor of RAGE (receptor for advanced glycation end-products) with potential to slow cognitive decline in AD (Burstein et al., 2013). Upregulation of RAGE in hippocampus has been observed in people with AD and is thought to promote neuroinflammation and brain amyloid deposition. In preclinical studies, azeliragon decreased Aβ load and improved performance on behavioral assays in transgenic mice (Walker et al., 2015). The compound failed in trial in mild AD patients with impaired glucose tolerance. Interventions to tackle AD by improving mitochondrial metabolism in AD through mild ketosis induction such those with tricaprilin, an oral formulation of caprylic triglyceride (Henderson et al., 2009), or by inhibiting activity of gingipains, proteases produced by bacteria causing periodontitis and linked to neuroinflammation, astrogliosis and cognitive impairment, with compound COR388 (Dominy et al., 2019) are also in active clinical consideration (Table 1).
Neuroinflammation has been recognized as another promising target for intervention as several studies have identified a sustained immune response in the AD (Kinney et al., 2018). Activation of the resident microglia and other immune cells in the brain has been demonstrated to exacerbate both Aβ and tau pathology. Conversely, an efficient adaptive immune response during AD might ameliorate developing pathology by modulating microglial function (Marsh et al., 2016). Thus, modifiers of innate immune activity and of microglial responses are attractive therapeutic targets for AD. DNL747 is an inhibitor of RIPK1 (receptor-interacting serine/threonine-protein kinase 1), an enzyme that mediates disease-associated microglial activation and release of pro-inflammatory cytokine in AD. The finding that genetic and pharmacological inhibition of RIPK1 induced amyloid clearance and improved memory in transgenic mice overproducing Aβ (Ofengeim et al., 2017) gave impetus to its clinical consideration in subjects with AD. Neflamapimod (VX-745) which selectively inhibits p38 mitogen-activated protein kinase alpha (p38 MAPKα), an enzyme involved in neuroinflammation and possibly Aβ toxicity (Alam, 2015) is another anti-inflammatory small molecule designed to tackle AD. A good safety profile and results of recently completed proof-of-concept study where possible improvement in episodic memory and impact on Aβ production was found in patients with early AD (Scheltens et al., 2018) supported further clinical evaluation. However, newly released data from phase 2b study showed that neflamapimod failed to significantly improve memory although it decreased levels of hyperphosphorylated tau and total tau in the CSF of treated patients compared to those given a placebo (Scheltens et al., 2019). Masitinib, a selective tyrosine kinase inhibitor is in clinical consideration for AD treatment because of its presumed potential to modulate inflammatory and neurodegenerative processes, and disrupt Aβ signaling cascade. Experimental results (Li et al., 2020) and data of cognitive improvement in AD patients in phase 2 study supported its further development and multicenter phase 3 study for its effectiveness as an add-on therapy in patients on stable dose treatment with cholinesterase inhibitors and/or memantine has recently completed, but results are yet expected to come. A current trial also evaluates NE3107, an orally bioavailable synthetic sterol derivative with anti-inflammatory property which can supposedly target multiple mechanisms of pathology in AD (Table 1). The triggering receptor expressed on myeloid cells 2 (TREM2) enhances microglial activity for Aβ uptake and modulates inflammatory signaling in AD. Moreover, mutations in the TREM2 gene have been found to increase risk for sAD similarly as observed in APOE-ε4 carriers (Gratuze et al., 2018). Also, increased levels of soluble TREM2 protein in the CSF are observed in the early symptomatic phase of AD reflecting changes of microglia activation status (Suarez-Calvet et al., 2016). These together make TREM2 an attractive target, and monoclonal TREM2 agonistic antibody AL002 heads to first-in-human study for testing safety, tolerability and its pharmacokinetic/pharmacodynamic properties (Long et al., 2019). In parallel, an antibody targeting SIGLEC-3 microglial receptor that interacts with TREM2 signaling is also in clinical development. The rationale behind this anti-SIGLEC-3 antibody named AL003 is that counteracting the function of this receptor can be more feasible way to tweak microglial activation (Timmins, 2019).
A novel strategy to harness neuroinflammation and therefore mitigate cognitive impairment in AD by remodeling the gut microbiota recently brought about approval of sodium oligomannate by China’s regulatory agency (Wang et al., 2019). Even though conditional, this approval is the first in almost two decade-lasting effort to get new AD treatment to market. The drug is currently in US clinical trial for efficacy and safety evaluation in patients with mild-to-moderate AD (Table 1).
3. Repurposed drugs
Slow pace of development of new and highly needed effective treatments for AD set off identification of repurposable drug candidates. Evidence of frequent but usually clinically silent episodes of epileptiform hyperexcitability in AD patients, which occur in early disease stage and likely contribute to cognitive deficiency (Vossel et al., 2017) propose the use of antiepileptics. Levetiracetam, acting through SV2A glycoprotein to reduce presynaptic neurotransmitter release, is considered to be good candidate for stabilization of neuronal networks hyperexcitability and improvement of AD-related cognitive symptoms (Musaeus et al., 2017), and several clinical studies currently evaluate its effects in subjects with AD including one phase 3 trial (Table 1). Daratumumab is an FDA-approved human IgG antibody that targets CD38. It is in clinical treatment of multiple myeloma, but has a broad immunomodulatory effects including role in apoptosis and immune-mediated cytotoxicity (Touzeau and Moreau, 2017). The findings of increased expression on CD8+ T-cells in the blood during early stage of AD (Sommer et al., 2017), suggesting T-cells activation in the brain and possible toxic effects, triggered clinical exploration of daratumumab in mild-to-moderate AD patients. ALZT-OP1 represents combination of two FDA-approved drugs, mast cell stabilizer cromolyn and ibuprofen. Synergistic action of these drugs was shown to promote microglia recruitment and phagocytosis of Aβ deposits (Hori et al., 2015; Zhang et al., 2018), recommending its further clinical evaluation (Table 1).
Addressing the modifiable risk factors for developing AD was another option in consideration for AD treatment. Based on the premise that insulin resistance and peripheral hyperinsulinemia contribute to AD, antidiabetics such as insulin, and thiazolidinediones, rosiglitazone and pioglitazone, were tested in clinical studies (Arnold et al., 2018). In the current AD drug pipeline, long acting formulation of metformin is being tested in MCI subjects without diabetes, for prevention of AD development (Luchsinger and Zetterberg, 2020). Notion that elevated cholesterol levels might promote disease progression triggered evaluation of statins, lipid-lowering drugs (Sparks, 2011), while neurovascular alterations well-known in AD prompted clinical testing of sartans, angiotensin II receptors blockers (Cooper et al., 2018). However, neither antidiabetics, statins nor sartans have shown efficacy on cognition or global function in AD patients so far.
4. Non-pharmacological treatments
The critical need for effective AD therapy has initiated exploration of several non-pharmacological interventions including lifestyle modification such as physical exercise, different diets, cognitive therapy (e.g. EXERT study https://www.exertstudy.org/) and neurostimulation. Currently, there are approximately 30 active clinical trials evaluating these therapeutics approaches. The preclinical and preliminary human studies testing these alternative strategies have signaled that some may have positive effects on maintaining or improving cognitive function and other aspects of well-being in people with AD, thus strengthening their further clinical development.
Lifestyle modification with regular aerobic exercising is considered as an approach with potential for delaying disease onset, slowing cognitive decline, and reducing the brain atrophy due to AD. However, a randomized multicenter trial exploring effects of aerobic fitness in patients with mild-to-moderate AD did not support an impact of regular physical training on AD-related structural and functional brain changes (Clemmensen et al., 2020; Frederiksen et al., 2018). Another attractive therapeutic concept to target attention and memory dysfunctions in AD patients is the use of personalized, interactive computer-based cognitive neurofeedback training. Although it demonstrated feasibility for use in elderly (Yeo et al., 2018), a large systemic review did not find strong evidence for meaningful benefits of this approach in improving either global cognitive functioning or performance in distinctive cognitive domains to support its certain clinical importance in AD treatment (Gates et al., 2020).
Multiple different neurostimulation techniques are in evaluation in current AD clinical trials, including deep brain stimulation, transcranial magnetic stimulation, transcranial alternating or direct current stimulation, multi-sensory gamma-range stimulation and photobiomodulation (Table 2). The underlying idea of all these approaches is that stimulating brain areas vulnerable to the developing AD pathology can modulate neural network dysfunctions and aberrant connectivity in memory circuitry and default mode network (Fig. 1) causing a clinically meaningful reduction of disease progression (Marron et al., 2018). Application of focused ultrasound stimulations (including low-intensity focused ultrasound and magnetic resonance guided focused ultrasound) are currently explored whether it can improve Aβ clearance and/or transiently open blood-brain barrier promoting CNS exposure of therapeutic agents (antibodies or other large molecule biologics) in the brain targeted regions of AD patients (Lee et al., 2019).
Table 2.
Active clinical trials of non-pharmacological neuromodulatory interventions in AD (clinicaltrials.gov as of February 10, 2021)
| Investigational technique/device | Study population | Targeted area | Clinical trial identifier |
|---|---|---|---|
| Invasive brain stimulation | |||
| Deep Brain Stimulation (DBS) - electrical, implantable device | mild AD mild to moderate AD |
Fornix Nucleus basalis of Meynert |
NCT03622905 NCT03959124 |
| Non-invasive brain stimulation | |||
| mild AD | DMN-precuneus (20 Hz) | NCT03778151 | |
| repetitive Transcranial Magnetic Stimulation (rTMS) - coil induced depolarizing magnetic field | Dorsolateral prefrontal cortex | NCT04263194 | |
| mild to moderate AD (with depression) | Dorsolateral prefrontal cortex unilateral (H1-coil) | NCT03665831 | |
| Lateral parietal areas | NCT04260724 | ||
| mild to moderate AD (with apathy) | Dorsolateral prefrontal cortex bilateral (H-coil) | NCT04562506 | |
| Dorsolateral prefrontal cortex (10 Hz) | NCT02190084 | ||
| prodromal AD (amnestic MCI) | DMN-angular gyrus (20 Hz) | NCT04045990 | |
| DMN-prespecified cortical areas | NCT04294888 | ||
| rTMS - intermittent Theta Burst (>5 Hz) Stimulation (iTBS) | MCI and mild AD | NCT04555941 | |
| Transcranial Electromagnetic Treatment (TEMT) | mild to moderate AD | MemorEM 1000 head device emitting electromagnetic waves in radiofrequency range |
NCT03927040 NCT04271163 |
| transcranial Alternating Current Stimulation (tACS) - sinusoidal low intensity electric current | mild to moderate AD | Areas with maximal tracer uptake on the amyloid PET (40 Hz) |
NCT03290326 NCT03412604 NCT03880240 NCT04515433 |
| Superior parietal cortex (40 Hz) | NCT03920826 | ||
| mild AD | Wide cortical
area (40 Hz, Nexalin device and conductive pads at forehead and mastoid
processes) Wide cortical area (4 Hz, Nexalin device and conductive pads at forehead and mastoid processes) |
NCT04088643 | |
| transcranial Direct Current Stimulation (tDCS) - direct low intensity electric current | mild
AD mild to moderate AD |
Dorsolateral prefrontal cortex |
NCT03288363 NCT04404153 |
| Sensory stimulation -GammaSense (audiovisual stimulation in gamma range) | MCI prodromal AD mild to moderate AD |
Visual and auditory brain areas (40 Hz light flickers and tone pulses) |
NCT03543878 NCT03556280 NCT03661034 |
| Photobiomodulation (PBM) - near-infrared light stimulation | mild to moderate AD | transcranial/intranasal delivery of near infrared light 40 Hz |
NCT03405662 NCT03160027 |
| subjects at risk of AD | pulses | NCT04018092 | |
| Low Intensity Focused Ultrasound Pulsation | MCI and mild AD | Hippocampus (50ms long bursts at 10 Hz) | NCT03347084 |
Deep brain stimulation (DBS) has emerged as a promising technique for treating various neuropsychiatric disorders. It involves the delivery of electrical current into specific areas of the brain through implanted electrodes. Although the mechanism of DBS is still not completely understood, preclinical findings from animal experiments and clinical observations highlight a number of possible mechanisms as to how DBS may affect AD progression and memory deterioration (Jakobs et al., 2019; Lam et al., 2020; McKinnon et al., 2019). In transgenic AD rats, chronic DBS of the fornix significantly reduces amyloid deposition in the hippocampus and cortex, inflammation, and neuronal loss (Leplus et al., 2019). Recent phase 2 clinical study have shown that targeting fornix bilaterally with DBS is safe and well tolerated procedure. It increases glucose metabolism in temporo-parietal region and decreases atrophy in the hippocampus and default mode network exerting cognitive improvement in AD patients (Lozano et al., 2016). Interestingly this study indicated greater benefits in patients aged 65 and older comparing to younger patients (Leoutsakos et al., 2018). Currently, larger phase 2b/3 trial of fornix DBS is testing efficacy of two different stimulation paradigms, low frequency (40 Hz) and high frequency (130 Hz) in older (> 65 years) mild AD patients, in whom changes in glucose PET imaging and fluid biomarkers (Aβ and tau protein) are being monitored.
Non-invasive brain stimulation methods as treatment option for improving cognitive impairments present in neurodegenerative diseases attracted great interest (Sanches et al., 2020). In recent years, potential of transcranial magnetic stimulation (TMS) of the brain to reduce cognitive symptoms in AD, particularly in the early stage of disease, has been actively explored (Dong et al., 2018; Weiler et al., 2020). Evidence suggests that patterned repetitive TMS can induce long-lasting changes in neural plasticity which are associated with improved cognition both in patients (Kumar et al., 2017; Turriziani et al., 2019; Wang et al., 2020) and rodent models of AD (Huang et al., 2017; Zhen et al., 2017). Furthermore, it has been reported that high-frequency repetitive TMS of the precuneus resulted in improvement in episodic memory together with enhancement of brain oscillations in the beta band and a modification of functional connections within the default mode network in mild AD patients (Koch et al., 2018). The meta-analysis of the effectiveness of various patterns of repetitive TMS on different cognitive domains in patients with mild cognitive impairment and AD (Chou et al., 2019) revealed that both high frequency stimulation over the left dorsolateral prefrontal cortex and low frequency stimulation over the same region at the right side significantly improved memory function. Coupling repetitive TMS of distinct brain regions (prefrontal and somatosensory associative cortices) with adaptive computerized cognitive training in AD patients presents an innovative therapeutic concept that is rationalized on advantage in the synergy between exogenous (TMS) and endogenous (cognitive engagement) concomitant stimulations (Rabey and Dobronevsky, 2016). This combinatory treatment modality is currently under active exploration in patients with MCI and mild-to-moderate AD, and several pivotal short-duration (up to 6 weeks) clinical studies already provided encouraging evidence of low- risk benefits in cognitive improvement which lasted beyond treatment interventions, and surpassed effects of the pharmacological standard of care of those patients (Bagattini et al., 2020; Sabbagh et al., 2020). Interestingly, even though paired TMS-cognitive training seems promising, recent review of one such trial (NCT02166827) by the FDA expert panel, did not support its clinical approval yet, emphasizing the need for further investigation of this treatment approach.
Other noninvasive brain stimulation methods, such as transcranial alternating current stimulation (tACS), transcranial direct current stimulation (tDCS), and transcranial electromagnetic treatment (TEMT) are also under exploration for reducing or delaying cognitive decline and ameliorating neuropsychiatric symptoms associated with AD. It is presumed that electrical current stimulations either with tACS or tDCS can potentially normalize excitatory and inhibitory balance or modulate neuronal activity in the cortex, particularly in vulnerable cortical regions related to memory, attention and executive functions (for review see Grover et al., 2021). However, the exact mechanisms underlying the effects of tACS and tDCS have not been fully established yet (e.g. see Voroslakos et al., 2018). Similarly, biophysics and mode of action of the application of pulsed transcranial infrared or near-infrared light stimulation technology known as photobiomodulation (Zomorrodi et al., 2019), have not been explored to satisfaction. Therefore, appropriately designed, multi-site randomized large-size clinical trials are necessary to further explore the full therapeutic potential of these neuromodulation techniques in AD patients (Gonsalvez et al., 2017), and confirm initial promising results before FDA consideration as valid clinical treatments.
Currently, an alternative, non-invasive brain-stimulation method GammaSense, is evaluated in MCI and AD patients. Based on recent findings showing that 40 Hz gamma frequency audio-visual sensory stimulation reverses AD-related pathologies in transgenic mice (Iaccarino et al., 2016; Martorell et al., 2019), patients are exposed to 40 Hz audio-visual sensory stimulation over a period of time. Similar long-lasting gamma-band oscillation can be also elicited using sensory stimulation in humans and mild-to-moderate AD patients (Hajós et al., 2020; Tsoneva et al., 2015; Vialatte et al., 2010), and current clinical trials investigate if similar downstream mechanisms would be present in AD patients which were observed in experimental animal studies. The therapeutic effects of long-term, at-home gamma sensory stimulation on severity of clinical symptoms and AD biomarkers, such as Aβ PET signals are being investigated in clinical trials with MCI and AD subjects (NCT03543878; NCT03556280). Although no neuromodulation treatment of AD patients has been approved by FDA up to date, the agency granted Breakthrough Device Designation for three brain stimulation methods in 2020 and 2021. These neuromodulation approaches include therapy with DBS (Functional Neuromodulation, Inc) for patients 65 years and older with mild probable AD, therapy with TEMT (NeuroEM Therapeutics, Inc) for AD patients, and therapy with gamma sensory stimulation system (Cognito Therapeutics) for cognitive and functional symptoms associated with AD.
Advances in our understanding of pathological neuronal activities together with a better insight into interactions between brain stimulation and on-going neuronal activity will further the methodology and effectiveness of neuromodulation, leading to an improved clinical therapeutic outcome. Emerging closed-loop technology represents an inventive approach based on bidirectional brain-computer interface that enables optimization of stimulation parameters through real-time decoding of neuronal activities. In this adaptive neurostimulation paradigm, neurochemical and electrophysiological disease-specific signals recorded with intracranial microelectrodes or EEG are using for precisely-timed and feedback-responsive adjusting stimulations in individual patients (Price et al., 2020), making the closed-loop method personalized and informed treatment modality with the potential for achieving better results with fewer side effects. Although there is still a relatively small number of available clinical studies using this approach, data on closed-loop DBS targeting lateral temporal cortex and fornix for improving memory encoding (Ezzyat et al., 2018; Mankin and Fried, 2020; Senova et al., 2018), as well as closed-loop acoustic stimulation during slow-wave sleep for enhancing memory consolidation (Ngo et al., 2013; Wei et al., 2020) suggest its applicability for AD treatment. Given the advantage of temporal precision for customized neurostimulation delivery with this approach, it can be possibly used for selective enhancement of reduced EEG rhythms associated with cognitive dysfunctions in AD (Babiloni et al., 2020), or for instantaneous suppression of aberrant neuronal activity such as clinically silent epileptic discharges (Vossel et al., 2017) found in some of these patients. Designing and further developing of neurostimulation methods with ability for fine-tuning stimulation parameters like closed-loop stimulations, can make neuromodulation part of precision medicine for AD therapy.
5. Functional biomarkers for assessing brain integrity and performance reflecting cognitive abilities
Functional biomarkers indicating global brain function and its subtle changes in response to therapy are underdeveloped. Many of the current clinical tests assessing cognitive function have low sensitivity and are not well suited to monitor short-term changes in patients’ everyday function in response to treatment. Currently, a number of established and novel methods are under evaluation if they can detect brain function in prodromal and early AD or MCI, including neuroimaging methods, measurements of brain blood flow and various neurophysiological techniques (Fig. 2).
Figure 2.
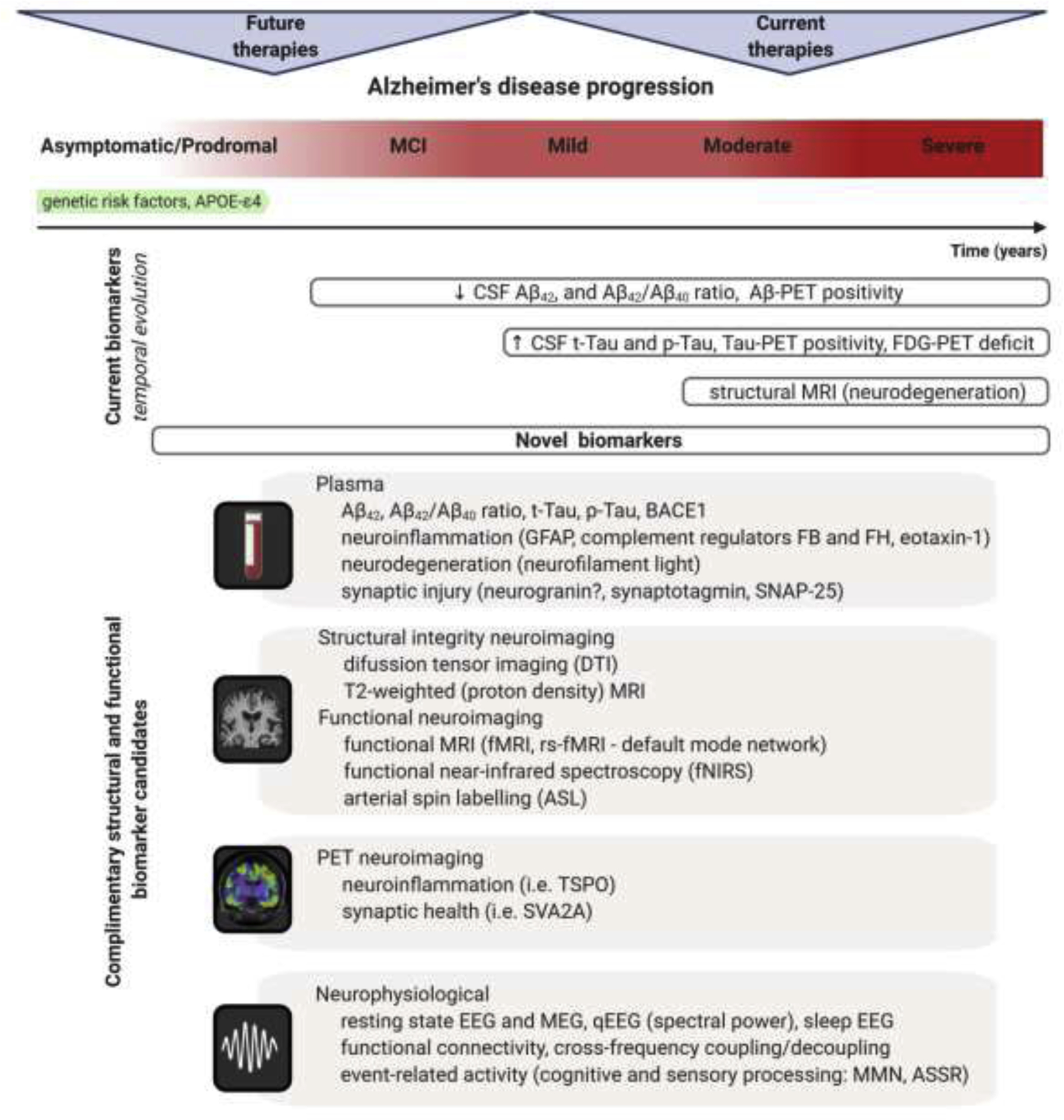
Dynamical changes of biomarkers reflecting core AD pathologies during the course of disease progression. The process of AD begins in the brain with dormant deterioration of neurons and circuits lasting years, probably decades, before the first symptoms emerge. This asymptomatic/prodromal period of the disease represents the desirable window for disease-modifying therapeutic interventions. Currently used biomarker toolbox comprising of cerebrospinal fluid (CSF) and positron emission tomography (PET) measures of Aβ and tau has high diagnostic value indicating AD pathological processes, but cannot reliably index subtle functional changes in the advent of cognitive symptoms or predict treatment response in clinical trials. Additionally, 18F-2fluoro-2-deoxy-D-glucose PET (FDG-PET), measuring neuronal activity through resting state metabolic rate of glucose in the brain, as well as magnetic resonance imaging (MRI) measuring degree of neurodegeneration are nonspecific markers and may indicate several etiologies of brain damage. Improved analytical techniques facilitate development of novel biomarkers with potential for refined and/or less invasive measuring of AD associated morphological and functional alterations through diverse transition states of the disease. Plasma Aβ and tau profiles, and set of specific markers for neuroinflammation, neurodegeneration, and synaptic injury have emerged as clinically valid measures for early diagnosis and prediction of disease progression (Hampel et al., 2020; Mila-Aloma et al., 2019; Morgan et al., 2019). Advanced neuroimaging methods demonstrate high degree of sensitivity and specificity for detection of initial pathologic and functional changes which are important for staging progression and for selecting patients for clinical trials during the prodromal phase of AD (Marquez and Yassa, 2019). Certain neurophysiological signals and applied innovative approaches for analyses are proven to be valid proxies for subtle abnormalities at neural network level and for assessment of global brain function (Babiloni et al., 2020; Stoiljkovic et al., 2019). Pertinent combination of these novel biomarkers with traditional AD biomarkers can enable more sensitive tracking of early disease-associated alterations and specifically estimate the likelihood of responding to next generation AD treatment. Abbreviations: MCI – mild cognitive impairment; GFAP – glial fibrillary acidic protein; FB – complement factor B; FH – complement factor H; SNAP-25 – synaptosomal-associated protein, 25kDa; rs-fMRI – resting state functional magnetic resonance imaging; TSPO – translocator protein; SVA2A – synaptic vesicle protein 2A; MEG – magnetoencephalography; qEEG – quantitative electroencephalography; MMN – mismatch negativity; ASSR – auditory steady-state response.
Functional magnetic resonance imaging (MRI) provides information on the brain metabolic activity by measuring blood oxygen level dependence and cerebral blood flow, detecting abnormal brain activities in MCI and AD patients, either at resting state or during cognitive challenges. Several, task-free magnetic resonance imaging (rsfMRI) reflecting default mode network (DMN) activity has shown altered connectivity in this network, including reduced connectivity between posterior cingulate cortex and precuneus in MCI or early stage of AD (Khan et al., 2020; Young et al., 2020). Task-based fMRI studies showed both hyper- and hypoactivation, which might reflect disease progression, but methodological differences between individual studies could also contribute to these findings (Young et al., 2020). This conundrum is well demonstrated by a recent report, showing that lack of established and validated analytical methods for processing complex data resulted in substantial variation in the results from 70 laboratories analyzing the exact same fMRI data (Botvinik-Nezer et al., 2020), demonstrating the challenges for integrating fMRI methods in drug discovery. Measuring cerebral glucose hypometabolism by 18F-FDG PET can detect declined activity in localized brain regions most closely associated with dementia, therefore could be also considered to discriminate in dementia due to AD, frontotemporal lobar degeneration, and dementia with Lewy bodies (Bailly et al., 2015; Moonis et al., 2020). Arterial spin labeling (ALS) MRI is an alternative method for measuring brain perfusion reflecting neuronal activity. Since ALS-MRI does not require a PET tracer, it is a clear advantage, particularly if patients are monitored longitudinally. Due to high variation in study design, such as heterogeneity of patient/control group selection, differences in disease progression and methodology applied, it is hard to draw a coherent conclusion from the available ALS data (Sierra-Marcos, 2017). Nevertheless, some overlapping patterns in cortical hypoperfusion of MCI patients have become apparent, including bilateral parietal lobes, posterior cingulate cortex, precuneus and frontal lobes compared to healthy controls (Binnewijzend et al., 2013). Disrupted frontal and long-range connectivity in the resting state has been also reported in MCI and AD patients compared to individuals with normal cognition using functional near-infrared spectroscopy (fNIRS), to the study of cerebral oxygenation (Yeung and Chan, 2020). Although these functional neuroimaging methods have high sensitivity to detect functional brain abnormalities associated with dementia, but due to their lack of diagnostic specificity they need to be supplemented by disease-relevant objective markers, such as pathological CSF or plasma Aβ/tau or Aβ/tau-PET findings in AD patients. Also, once changes in functional neuroimaging signals are well established markers of disease progression, theoretically they will be sensitive enough to detect sublime changes related to cognitive function in response to drug treatment. Nevertheless, until now, application of these neuroimaging methods provided only limited values to drug discovery due to the shortcomings in experimental design and data interpretation (Canu et al., 2018).
Recently, various neurophysiological signals have been explored in MCI and AD patients reflecting brain global brain function and integrity of neuronal circuits (Babiloni et al., 2020; Horvath et al., 2018). Although these neurophysiological markers could be associated most closely with a certain type of neurodegenerative diseases, they are mostly agnostic to the exact underlying pathology. A broad variety of neurophysiological signals have been explored in MCI and AD patients, include alterations in power distribution of spontaneous electroencephalography (EEG) or magnetoencephalography (MEG). One of the most consistent findings is a shift of EEG power distribution to lower frequencies, resulting in a widespread higher delta and theta power and reduced posterior alpha and beta power, indicating a general slowing of cortical activity in MCI and AD patients (Babiloni et al., 2020; Horvath et al., 2018; Rossini et al., 2020). Similarly, power spectral analysis of MEG has consistently demonstrated slowing of brain activity in AD patients, displaying a reduction in the frequency of the main power spectrum peak or reduction in alpha peak (Lopez-Sanz et al., 2019). Both EEG and MEG signals correlate well with the severity of neurodegeneration assessed by MRI or Aβ pathology and shifting in power spectra to lower frequencies has been suggested as an excellent marker for predicting disease progression from MCI to AD, or from mild to a more severe AD stage. Accordingly, it has been found that integrative EEG markers can predict conversion from MCI to AD with high degree (>80%) of both sensitivity and specificity (Poil et al., 2013). A longitudinal study pairing frontal EEG recording with working memory task performance demonstrated that EEG signals can predict individualized MCI risk few years before clinical diagnosis (Jiang et al., 2021). Moreover, measuring EEG microstate complexity, which has potential to give insight into altered brain dynamics underlying cognitive impairment, was shown to highly predict progression from MCI to AD in a recent small cohort study (Tait et al., 2020), providing additional evidence for validity of neurophysiological markers to index early, prodromal brain dysfunctions associated with the disease.
EEG spectral power density changes in AD patients were well recognized and subsequently some neurophysiological studies were included in AD drug development in the late 1990s. These initial, and by now mostly forgotten studies showed an excellent correlation between acute EEG changes and benefit of clinical symptoms in response to acetylcholinesterase inhibitors, even potentially predicting clinical outcome of the treatment (Almkvist et al., 2001; Knott, 2000; Lanctot et al., 2003). None of the clinical trials on BACE1 inhibitors, γ-secretase inhibitors/modulators, or Aβ antibodies included EEG or neurophysiological measurement. In light of the clinical trials showing target engagements of BACE1 inhibitors and Aβ antibodies reducing Aβ plaque loads but without improvement of cognitive function, or even accelerating cognitive and morphological decline (see above), development and validation of an objective assessment of global brain function is imperative. An addition to a validated target-engagement biomarker, it would be particularly beneficial having markers of global brain function predicting clinical outcome in trails of disease modifying therapies lasting up to two or more years. These functional biomarkers could be based and designed on neuroimaging and/or neurophysiological findings. In addition to resting EEG/MEG spectra, additional neurophysiological tests could be considered, involving neurophysiological signals of sensory processing and cognitive tasks, together with application of cutting-edge signal processing.
In summary, identification, validation and implementation of biomarkers that indicate subtle AD-related alterations during transition from normal cognitive function to early dementia, and those potentially orthogonal to Aβ and tau pathology (Whelan et al., 2019) could improve chances of success in AD drug development. Such biomarkers will increase likelihood of optimum timing and dosing, target engagement and use of endpoints that are sensitive enough to detect response to a given treatment in trial. They can be also useful for proper selection of AD patients for clinical trials and to infer individual, precision-medicine approach particularly in light of recent report (Qiu et al., 2019) of phenotypic heterogeneity among typical amyloid-positive, mild-to-moderate AD patient population.
6. Concluding remarks
There must be multiple reasons for failing of almost all clinical trials for AD therapy. Limitations due to preclinical models is one: though some drug candidates show certain signals of efficacy (or at least target engagement) in AD related transgenic animals, they turn out to be without any clinical benefits in patients. It is safe to conclude that animal models do not capture the complexity of AD pathologies, which are not exclusively related to Aβ or tau, but it includes cerebrovascular, metabolic and CNS innate immunity pathologies. Similarly, it has been pointed out that preclinical models do not faithfully mimic interactions between the main pathologies of the disease and processes underlying aging as observed in humans (Drummond and Wisniewski, 2017). Therefore, a better insight to AD pathologies based on novel findings in genomics, proteomics, and metabolomics together with a better understanding of interactions between pathological neuronal activities and disease progression will identify novel, hitherto unrecognized drug targets. Other limitations relate to timing of intervention with therapy. Since there are no foreseen treatment options to restore or reverse neuronal death and large brain volume loss in AD patients, current clinical trials are recruiting patients at earlier disease stage, or even prodromal stage. Therefore, development of novel predictive biomarkers for both diagnostics of prodromal AD and disease progression will have a profound benefit for developing disease-modifying therapies. Similarly, better functional assessment of brain function is needed, either with validated neuroimaging or neurophysiological methods. These markers should be sensitive enough to detect subtle improvements in brain function which are not easily detectable by currently used clinical cognitive assessments. Recognized surrogate markers of efficacy, reflecting global brain function would support clinical trials by providing early signals if the tested therapy would lead to clinical benefits or otherwise.
Highlights.
Development of AD disease-modifying therapies is futile thus far
Research efforts for effective AD therapy broaden targets and treatment options
Current biomarkers are not reliable predictors of effectiveness of new treatments
Advanced functional brain assessment tools can increase success in clinical trials
Acknowledgments
This work was supported by NIH grants AG067329 and AG052986 (TLH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest:
Dr. Mihály Hajós is an employee of Cognito Therapeutics, shareholder of Biogen and Pfizer. The other authors have no conflicts of interest.
References
- Ables JL, Breunig JJ, Eisch AJ, Rakic P, 2011. Not(ch) just development: Notch signalling in the adult brain. Nat Rev Neurosci 12, 269–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K, Lohmann S, Piorkowska K, Gafner V, Atwal JK, Maloney J, Chen M, Gogineni A, Weimer RM, Mortensen DL, Friesenhahn M, Ho C, Paul R, Pfeifer A, Muhs A, Watts RJ, 2012. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci 32, 9677–9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam JJ, 2015. Selective Brain-Targeted Antagonism of p38 MAPKalpha Reduces Hippocampal IL-1beta Levels and Improves Morris Water Maze Performance in Aged Rats. J Alzheimers Dis 48, 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almkvist O, Jelic V, Amberla K, Hellstrom-Lindahl E, Meurling L, Nordberg A, 2001. Responder characteristics to a single oral dose of cholinesterase inhibitor: a double-blind placebo-controlled study with tacrine in Alzheimer patients. Dement Geriatr Cogn Disord 12, 22–32. [DOI] [PubMed] [Google Scholar]
- Alzheimer’s Association, 2020. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement 16, 391–460. [DOI] [PubMed] [Google Scholar]
- Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang HY, Ahima RS, Craft S, Gandy S, Buettner C, Stoeckel LE, Holtzman DM, Nathan DM, 2018. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 14, 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiloni C, Blinowska K, Bonanni L, Cichocki A, De Haan W, Del Percio C, Dubois B, Escudero J, Fernandez A, Frisoni G, Guntekin B, Hajos M, Hampel H, Ifeachor E, Kilborn K, Kumar S, Johnsen K, Johannsson M, Jeong J, LeBeau F, Lizio R, Lopes da Silva F, Maestu F, McGeown WJ, McKeith I, Moretti DV, Nobili F, Olichney J, Onofrj M, Palop JJ, Rowan M, Stocchi F, Struzik ZM, Tanila H, Teipel S, Taylor JP, Weiergraber M, Yener G, Young-Pearse T, Drinkenburg WH, Randall F, 2020. What electrophysiology tells us about Alzheimer’s disease: a window into the synchronization and connectivity of brain neurons. Neurobiol Aging 85, 58–73. [DOI] [PubMed] [Google Scholar]
- Bagattini C, Zanni M, Barocco F, Caffarra P, Brignani D, Miniussi C, Defanti CA, 2020. Enhancing cognitive training effects in Alzheimer’s disease: rTMS as an add-on treatment. Brain Stimul 13, 1655–1664. [DOI] [PubMed] [Google Scholar]
- Bailly M, Destrieux C, Hommet C, Mondon K, Cottier JP, Beaufils E, Vierron E, Vercouillie J, Ibazizene M, Voisin T, Payoux P, Barre L, Camus V, Guilloteau D, Ribeiro MJ, 2015. Precuneus and Cingulate Cortex Atrophy and Hypometabolism in Patients with Alzheimer’s Disease and Mild Cognitive Impairment: MRI and (18)F-FDG PET Quantitative Analysis Using FreeSurfer. Biomed Res Int 2015, 583931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barao S, Moechars D, Lichtenthaler SF, De Strooper B, 2016. BACE1 Physiological Functions May Limit Its Use as Therapeutic Target for Alzheimer’s Disease. Trends Neurosci 39, 158–169. [DOI] [PubMed] [Google Scholar]
- Bateman RJ, Benzinger TL, Berry S, Clifford DB, Duggan C, Fagan AM, Fanning K, Farlow MR, Hassenstab J, McDade EM, Mills S, Paumier K, Quintana M, Salloway SP, Santacruz A, Schneider LS, Wang G, Xiong C, Network, D.-T.P.C.f.t.D.I.A., 2017. The DIAN-TU Next Generation Alzheimer’s prevention trial: Adaptive design and disease progression model. Alzheimers Dement 13, 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Halima S, Mishra S, Raja KMP, Willem M, Baici A, Simons K, Brustle O, Koch P, Haass C, Caflisch A, Rajendran L, 2016. Specific Inhibition of beta-Secretase Processing of the Alzheimer Disease Amyloid Precursor Protein. Cell Rep 14, 2127–2141. [DOI] [PubMed] [Google Scholar]
- Bennett DA, 2018. Lack of Benefit With Idalopirdine for Alzheimer Disease: Another Therapeutic Failure in a Complex Disease Process. JAMA 319, 123–125. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE, 2012. The genetics of Alzheimer’s disease. Prog Mol Biol Transl Sci 107, 79–100. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Terry AV Jr., 2018. The wonderland of neuronal nicotinic acetylcholine receptors. Biochem Pharmacol 151, 214–225. [DOI] [PubMed] [Google Scholar]
- Binnewijzend MA, Kuijer JP, Benedictus MR, van der Flier WM, Wink AM, Wattjes MP, van Berckel BN, Scheltens P, Barkhof F, 2013. Cerebral blood flow measured with 3D pseudocontinuous arterial spin-labeling MR imaging in Alzheimer disease and mild cognitive impairment: a marker for disease severity. Radiology 267, 221–230. [DOI] [PubMed] [Google Scholar]
- Birks JS, Harvey RJ, 2018. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev 6, CD001190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittar A, Bhatt N, Kayed R, 2020. Advances and considerations in AD tau-targeted immunotherapy. Neurobiol Dis 134, 104707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K, Zetterberg H, 2019. Fluid biomarker-based molecular phenotyping of Alzheimer’s disease patients in research and clinical settings. Prog Mol Biol Transl Sci 168, 3–23. [DOI] [PubMed] [Google Scholar]
- Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, Messer J, Oroszlan K, Rauchenberger R, Richter WF, Rothe C, Urban M, Bardroff M, Winter M, Nordstedt C, Loetscher H, 2012. Gantenerumab: a novel human anti-Abeta antibody demonstrates sustained cerebral amyloid-beta binding and elicits cell-mediated removal of human amyloid-beta. J Alzheimers Dis 28, 49–69. [DOI] [PubMed] [Google Scholar]
- Botvinik-Nezer R, Holzmeister F, Camerer CF, Dreber A, Huber J, Johannesson M, Kirchler M, Iwanir R, Mumford JA, Adcock RA, Avesani P, Baczkowski BM, Bajracharya A, Bakst L, Ball S, Barilari M, Bault N, Beaton D, Beitner J, Benoit RG, Berkers RMWJ, Bhanji JP, Biswal BB, Bobadilla-Suarez S, Bortolini T, Bottenhorn KL, Bowring A, Braem S, Brooks HR, Brudner EG, Calderon CB, Camilleri JA, Castrellon JJ, Cecchetti L, Cieslik EC, Cole ZJ, Collignon O, Cox RW, Cunningham WA, Czoschke S, Dadi K, Davis CP, Luca AD, Delgado MR, Demetriou L, Dennison JB, Di X, Dickie EW, Dobryakova E, Donnat CL, Dukart J, Duncan NW, Durnez J, Eed A, Eickhoff SB, Erhart A, Fontanesi L, Fricke GM, Fu S, Galván A, Gau R, Genon S, Glatard T, Glerean E, Goeman JJ, Golowin SAE, González-García C, Gorgolewski KJ, Grady CL, Green MA, Guassi Moreira JF, Guest O, Hakimi S, Hamilton JP, Hancock R, Handjaras G, Harry BB, Hawco C, Herholz P, Herman G, Heunis S, Hoffstaedter F, Hogeveen J, Holmes S, Hu C-P, Huettel SA, Hughes ME, Iacovella V, Iordan AD, Isager PM, Isik AI, Jahn A, Johnson MR, Johnstone T, Joseph MJE, Juliano AC, Kable JW, Kassinopoulos M, Koba C, Kong X-Z, Koscik TR, Kucukboyaci NE, Kuhl BA, Kupek S, Laird AR, Lamm C, Langner R, Lauharatanahirun N, Lee H, Lee S, Leemans A, Leo A, Lesage E, Li F, Li MYC, Lim PC, Lintz EN, Liphardt SW, Losecaat Vermeer AB, Love BC, Mack ML, Malpica N, Marins T, Maumet C, McDonald K, McGuire JT, Melero H, Méndez Leal AS, Meyer B, Meyer KN, Mihai G, Mitsis GD, Moll J, Nielson DM, Nilsonne G, Notter MP, Olivetti E, Onicas AI, Papale P, Patil KR, Peelle JE, Pérez A, Pischedda D, Poline J-B, Prystauka Y, Ray S, Reuter-Lorenz PA, Reynolds RC, Ricciardi E, Rieck JR, Rodriguez-Thompson AM, Romyn A, Salo T, Samanez-Larkin GR, Sanz-Morales E, Schlichting ML, Schultz DH, Shen Q, Sheridan MA, Silvers JA, Skagerlund K, Smith A, Smith DV, Sokol-Hessner P, Steinkamp SR, Tashjian SM, Thirion B, Thorp JN, Tinghög G, Tisdall L, Tompson SH, Toro-Serey C, Torre Tresols JJ, Tozzi L, Truong V, Turella L, van ‘t Veer AE, Verguts T, Vettel JM, Vijayarajah S, Vo K, Wall MB, Weeda WD, Weis S, White DJ, Wisniewski D, Xifra-Porxas A, Yearling EA, Yoon S, Yuan R, Yuen KSL, Zhang L, Zhang X, Zosky JE, Nichols TE, Poldrack RA, Schonberg T, 2020. Variability in the analysis of a single neuroimaging dataset by many teams. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursavich MG, Harrison BA, Blain JF, 2016. Gamma Secretase Modulators: New Alzheimer’s Drugs on the Horizon? J Med Chem 59, 7389–7409. [DOI] [PubMed] [Google Scholar]
- Burstein AH, Zhao Q, Ross J, Styren S, Landen JW, Ma WW, McCush F, Alvey C, Kupiec JW, Bednar MM, 2013. Safety and pharmacology of ponezumab (PF-04360365) after a single 10-minute intravenous infusion in subjects with mild to moderate Alzheimer disease. Clin Neuropharmacol 36, 8–13. [DOI] [PubMed] [Google Scholar]
- Calhoun A, Ko J, Grossberg GT, 2017. Emerging chemical therapies targeting 5-hydroxytryptamine in the treatment of Alzheimer’s disease. Expert Opin Emerg Drugs 22, 101–105. [DOI] [PubMed] [Google Scholar]
- Canu E, Sarasso E, Filippi M, Agosta F, 2018. Effects of pharmacological and nonpharmacological treatments on brain functional magnetic resonance imaging in Alzheimer’s disease and mild cognitive impairment: a critical review. Alzheimers Res Ther 10, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebers G, Lejeune T, Attalla B, Soderberg M, Alexander RC, Budd Haeberlein S, Kugler AR, Ingersoll EW, Platz S, Scott CW, 2016. Reversible and Species-Specific Depigmentation Effects of AZD3293, a BACE Inhibitor for the Treatment of Alzheimer’s Disease, Are Related to BACE2 Inhibition and Confined to Epidermis and Hair. J Prev Alzheimers Dis 3, 202–218. [DOI] [PubMed] [Google Scholar]
- Chou YH, Ton That V, Sundman M, 2019. A systematic review and meta-analysis of rTMS effects on cognitive enhancement in mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging 86, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemmensen FK, Hoffmann K, Siersma V, Sobol N, Beyer N, Andersen BB, Vogel A, Lolk A, Gottrup H, Hogh P, Waldemar G, Hasselbalch SG, Frederiksen KS, 2020. The role of physical and cognitive function in performance of activities of daily living in patients with mild-to-moderate Alzheimer’s disease - a cross-sectional study. BMC Geriatr 20, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congdon EE, Sigurdsson EM, 2018. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 14, 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper HA, Scalia R, Rizzo V, Eguchi S, 2018. Angiotensin II- and Alzheimer-Type Cardiovascular Aging. Circ Res 123, 651–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coric V, Salloway S, van Dyck CH, Dubois B, Andreasen N, Brody M, Curtis C, Soininen H, Thein S, Shiovitz T, Pilcher G, Ferris S, Colby S, Kerselaers W, Dockens R, Soares H, Kaplita S, Luo F, Pachai C, Bracoud L, Mintun M, Grill JD, Marek K, Seibyl J, Cedarbaum JM, Albright C, Feldman HH, Berman RM, 2015. Targeting Prodromal Alzheimer Disease With Avagacestat: A Randomized Clinical Trial. JAMA Neurol 72, 1324–1333. [DOI] [PubMed] [Google Scholar]
- Crehan H, Liu B, Kleinschmidt M, Rahfeld JU, Le KX, Caldarone BJ, Frost JL, Hettmann T, Hutter-Paier B, O’Nuallain B, Park MA, DiCarli MF, Lues I, Schilling S, Lemere CA, 2020. Effector function of anti-pyroglutamate-3 Abeta antibodies affects cognitive benefit, glial activation and amyloid clearance in Alzheimer’s-like mice. Alzheimers Res Ther 12, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump CJ, Castro SV, Wang F, Pozdnyakov N, Ballard TE, Sisodia SS, Bales KR, Johnson DS, Li YM, 2012. BMS-708,163 targets presenilin and lacks notch-sparing activity. Biochemistry 51, 7209–7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J, Blennow K, Johnson K, Keeley M, Bateman RJ, Molinuevo JL, Touchon J, Aisen P, Vellas B, 2019a. Anti-Tau Trials for Alzheimer’s Disease: A Report from the EU/US/CTAD Task Force. J Prev Alzheimers Dis 6, 157–163. [DOI] [PubMed] [Google Scholar]
- Cummings J, Lee G, Ritter A, Sabbagh M, Zhong K, 2020. Alzheimer’s disease drug development pipeline: 2020. Alzheimers Dement (N Y) 6, e12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J, Ritter A, Rothenberg K, 2019b. Advances in Management of Neuropsychiatric Syndromes in Neurodegenerative Diseases. Curr Psychiatry Rep 21, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL, Cohen S, van Dyck CH, Brody M, Curtis C, Cho W, Ward M, Friesenhahn M, Rabe C, Brunstein F, Quartino A, Honigberg LA, Fuji RN, Clayton D, Mortensen D, Ho C, Paul R, 2018. ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 90, e1889–e1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cynis H, Frost JL, Crehan H, Lemere CA, 2016. Immunotherapy targeting pyroglutamate-3 Abeta: prospects and challenges. Mol Neurodegener 11, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das B, Yan R, 2019. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 33, 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davtyan H, Ghochikyan A, Petrushina I, Hovakimyan A, Davtyan A, Poghosyan A, Marleau AM, Movsesyan N, Kiyatkin A, Rasool S, Larsen AK, Madsen PJ, Wegener KM, Ditlevsen DK, Cribbs DH, Pedersen LO, Agadjanyan MG, 2013. Immunogenicity, efficacy, safety, and mechanism of action of epitope vaccine (Lu AF20513) for Alzheimer’s disease: prelude to a clinical trial. J Neurosci 33, 4923–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong IEM, Mork A, 2017. Antagonism of the 5-HT6 receptor - Preclinical rationale for the treatment of Alzheimer’s disease. Neuropharmacology 125, 50–63. [DOI] [PubMed] [Google Scholar]
- De Strooper B, 2003. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 38, 9–12. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Vassar R, Golde T, 2010. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol 6, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, Hole JT, Forster BM, McDonnell PC, Liu F, Kinley RD, Jordan WH, Hutton ML, 2012. A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer’s disease mice. Neuron 76, 908–920. [DOI] [PubMed] [Google Scholar]
- Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, Nguyen M, Haditsch U, Raha D, Griffin C, Holsinger LJ, Arastu-Kapur S, Kaba S, Lee A, Ryder MI, Potempa B, Mydel P, Hellvard A, Adamowicz K, Hasturk H, Walker GD, Reynolds EC, Faull RLM, Curtis MA, Dragunow M, Potempa J, 2019. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 5, eaau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Yan L, Huang L, Guan X, Dong C, Tao H, Wang T, Qin X, Wan Q, 2018. Repetitive transcranial magnetic stimulation for the treatment of Alzheimer’s disease: A systematic review and meta-analysis of randomized controlled trials. PLoS One 13, e0205704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, Alzheimer’s Disease Cooperative Study Steering, C., Siemers E, Sethuraman G, Mohs R, Semagacestat Study, G., 2013. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369, 341–350. [DOI] [PubMed] [Google Scholar]
- Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, Siemers E, Liu-Seifert H, Mohs R, Alzheimer’s Disease Cooperative Study Steering, C., Solanezumab Study, G., 2014. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370, 311–321. [DOI] [PubMed] [Google Scholar]
- Drummond E, Wisniewski T, 2017. Alzheimer’s disease: experimental models and reality. Acta Neuropathol 133, 155–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, Tariot PN, Vellas B, van Dyck CH, Boada M, Zhang Y, Li W, Furtek C, Mahoney E, Harper Mozley L, Mo Y, Sur C, Michelson D, 2019. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N Engl J Med 380, 1408–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen JL, Sagi SA, Smith TE, Weggen S, Das P, McLendon DC, Ozols VV, Jessing KW, Zavitz KH, Koo EH, Golde TE, 2003. NSAIDs and enantiomers of flurbiprofen target gamma-secretase and lower Abeta 42 in vivo. J Clin Invest 112, 440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzyat Y, Wanda PA, Levy DF, Kadel A, Aka A, Pedisich I, Sperling MR, Sharan AD, Lega BC, Burks A, Gross RE, Inman CS, Jobst BC, Gorenstein MA, Davis KA, Worrell GA, Kucewicz MT, Stein JM, Gorniak R, Das SR, Rizzuto DS, Kahana MJ, 2018. Closed-loop stimulation of temporal cortex rescues functional networks and improves memory. Nat Commun 9, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlow M, Arnold SE, van Dyck CH, Aisen PS, Snider BJ, Porsteinsson AP, Friedrich S, Dean RA, Gonzales C, Sethuraman G, DeMattos RB, Mohs R, Paul SM, Siemers ER, 2012. Safety and biomarker effects of solanezumab in patients with Alzheimer’s disease. Alzheimers Dement 8, 261–271. [DOI] [PubMed] [Google Scholar]
- Farlow MR, Andreasen N, Riviere ME, Vostiar I, Vitaliti A, Sovago J, Caputo A, Winblad B, Graf A, 2015. Long-term treatment with active Abeta immunotherapy with CAD106 in mild Alzheimer’s disease. Alzheimers Res Ther 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher A, 2008. M1 muscarinic agonists target major hallmarks of Alzheimer’s disease--the pivotal role of brain M1 receptors. Neurodegener Dis 5, 237–240. [DOI] [PubMed] [Google Scholar]
- Frederiksen KS, Larsen CT, Hasselbalch SG, Christensen AN, Hogh P, Wermuth L, Andersen BB, Siebner HR, Garde E, 2018. A 16-Week Aerobic Exercise Intervention Does Not Affect Hippocampal Volume and Cortical Thickness in Mild to Moderate Alzheimer’s Disease. Front Aging Neurosci 10, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates NJ, Rutjes AW, Di Nisio M, Karim S, Chong LY, March E, Martinez G, Vernooij RW, 2020. Computerised cognitive training for 12 or more weeks for maintaining cognitive function in cognitively healthy people in late life. Cochrane Database Syst Rev 2, CD012277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM, Team ANS, 2005. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64, 1553–1562. [DOI] [PubMed] [Google Scholar]
- Golde TE, Koo EH, Felsenstein KM, Osborne BA, Miele L, 2013. gamma-Secretase inhibitors and modulators. Biochim Biophys Acta 1828, 2898–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsalvez I, Baror R, Fried P, Santarnecchi E, Pascual-Leone A, 2017. Therapeutic Noninvasive Brain Stimulation in Alzheimer’s Disease. Curr Alzheimer Res 14, 362–376. [DOI] [PubMed] [Google Scholar]
- Gratuze M, Leyns CEG, Holtzman DM, 2018. New insights into the role of TREM2 in Alzheimer’s disease. Mol Neurodegener 13, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Schneider LS, Amato DA, Beelen AP, Wilcock G, Swabb EA, Zavitz KH, Tarenflurbil Phase 3 Study, G., 2009. Effect of tarenflurbil on cognitive decline and activities of daily living in patients with mild Alzheimer disease: a randomized controlled trial. JAMA 302, 2557–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover S, Nguyen JA, Reinhart RMG, 2021. Synchronizing Brain Rhythms to Improve Cognition. Annu Rev Med 72, 29–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundman M, Morgan R, Lickliter JD, Schneider LS, DeKosky S, Izzo NJ, Guttendorf R, Higgin M, Pribyl J, Mozzoni K, Safferstein H, Catalano SM, 2019. A phase 1 clinical trial of the sigma-2 receptor complex allosteric antagonist CT1812, a novel therapeutic candidate for Alzheimer’s disease. Alzheimers Dement (N Y) 5, 20–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajós M, Harvey B, Mortimore H, Strozewski N, Kolin K, Boasso A, Cimenser A, Marin A, Hajós GP, Palumbo R, Spencer KM, Malchano Z, Budson A, 2020. Sensory-Evoked Steady-State Oscillations in Alzheimer’s disease Patients: Biomarker and Therapeutic Applications American Clinical Neurophysiology Society (ACNS) Annual Meeting and Courses 2020, New Orleans, Louisiana, USA [Google Scholar]
- Hampel H, Vergallo A, Caraci F, Cuello AC, Lemercier P, Vellas B, Giudici KV, Baldacci F, Hanisch B, Haberkamp M, Broich K, Nistico R, Emanuele E, Llavero F, Zugaza JL, Lucia A, Giacobini E, Lista S, Alzheimer Precision Medicine I, 2020. Future avenues for Alzheimer’s disease detection and therapy: Liquid biopsy, intracellular signaling modulation, systems pharmacology drug discovery. Neuropharmacology, 108081. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ, 2002. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC, 2009. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab (Lond) 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley D, Raghavan N, Sperling R, Aisen P, Raman R, Romano G, 2019. Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer’s Disease. N Engl J Med 380, 1483–1485. [DOI] [PubMed] [Google Scholar]
- Herline K, Drummond E, Wisniewski T, 2018. Recent advancements toward therapeutic vaccines against Alzheimer’s disease. Expert Rev Vaccines 17, 707–721. [DOI] [PubMed] [Google Scholar]
- Hitt B, Riordan SM, Kukreja L, Eimer WA, Rajapaksha TW, Vassar R, 2012. beta-Site amyloid precursor protein (APP)-cleaving enzyme 1 (BACE1)-deficient mice exhibit a close homolog of L1 (CHL1) loss-of-function phenotype involving axon guidance defects. J Biol Chem 287, 38408–38425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu-Seifert H, Case M, Dean RA, Hake A, Sundell K, Poole Hoffmann V, Carlson C, Khanna R, Mintun M, DeMattos R, Selzler KJ, Siemers E, 2018. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N Engl J Med 378, 321–330. [DOI] [PubMed] [Google Scholar]
- Hori Y, Takeda S, Cho H, Wegmann S, Shoup TM, Takahashi K, Irimia D, Elmaleh DR, Hyman BT, Hudry E, 2015. A Food and Drug Administration-approved asthma therapeutic agent impacts amyloid beta in the brain in a transgenic model of Alzheimer disease. J Biol Chem 290, 1966–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath A, Szucs A, Csukly G, Sakovics A, Stefanics G, Kamondi A, 2018. EEG and ERP biomarkers of Alzheimer’s disease: a critical review. Front Biosci (Landmark Ed) 23, 183–220. [DOI] [PubMed] [Google Scholar]
- Hoskin JL, Sabbagh MN, Al-Hasan Y, Decourt B, 2019. Tau immunotherapies for Alzheimer’s disease. Expert Opin Investig Drugs 28, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard R, Liu KY, 2019. Questions EMERGE as Biogen claims aducanumab turnaround. Nat Rev Neurol. [DOI] [PubMed] [Google Scholar]
- Huang Z, Tan T, Du Y, Chen L, Fu M, Yu Y, Zhang L, Song W, Dong Z, 2017. Low-Frequency Repetitive Transcranial Magnetic Stimulation Ameliorates Cognitive Function and Synaptic Plasticity in APP23/PS45 Mouse Model of Alzheimer’s Disease. Front Aging Neurosci 9, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino HF, Singer AC, Martorell AJ, Rudenko A, Gao F, Gillingham TZ, Mathys H, Seo J, Kritskiy O, Abdurrob F, Adaikkan C, Canter RG, Rueda R, Brown EN, Boyden ES, Tsai LH, 2016. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 540, 230–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbimbo BP, 2009. Why did tarenflurbil fail in Alzheimer’s disease? J Alzheimers Dis 17, 757–760. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Fleisher AS, Hake AM, Liu P, Shcherbinin S, DeMattos RB, Mintun MA, 2018. TRAILBLAZER-ALZ (NCT03367403): A Phase 2 disease-modification combination therapy trial targeting multiple mechanisms of action along the amyloid pathway. Alzheimers Dement 14, P1622–P1623. [Google Scholar]
- Ittner LM, Gotz J, 2011. Amyloid-beta and tau--a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci 12, 65–72. [DOI] [PubMed] [Google Scholar]
- Izzo NJ, Xu J, Zeng C, Kirk MJ, Mozzoni K, Silky C, Rehak C, Yurko R, Look G, Rishton G, Safferstein H, Cruchaga C, Goate A, Cahill MA, Arancio O, Mach RH, Craven R, Head E, LeVine H 3rd, Spires-Jones TL, Catalano SM, 2014. Alzheimer’s therapeutics targeting amyloid beta 1–42 oligomers II: Sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity. PLoS One 9, e111899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ, 2013. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 12, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobs M, Lee DJ, Lozano AM, 2019. Modifying the progression of Alzheimer’s and Parkinson’s disease with deep brain stimulation. Neuropharmacology, 107860. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Li J, Schmitt FA, Jicha GA, Munro NB, Zhao X, Smith CD, Kryscio RJ, Abner EL, 2021. Memory-Related Frontal Brainwaves Predict Transition to Mild Cognitive Impairment in Healthy Older Individuals Five Years Before Diagnosis. J Alzheimers Dis 79, 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, Walker LC, 2018. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci 21, 1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner GA, Boxer AL, 2010. Bapineuzumab. Expert Opin Biol Ther 10, 1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan W, Amad A, Giampietro V, Werden E, De Simoni S, O’Muircheartaigh J, Westman E, O’Daly O, Williams SCR, Brodtmann A, Alzheimer’s Disease Neuroimaging I, 2020. The heterogeneous functional architecture of the posteromedial cortex is associated with selective functional connectivity differences in Alzheimer’s disease. Hum Brain Mapp 41, 1557–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury R, Grysman N, Gold J, Patel K, Grossberg GT, 2018. The role of 5 HT6-receptor antagonists in Alzheimer’s disease: an update. Expert Opin Investig Drugs 27, 523–533. [DOI] [PubMed] [Google Scholar]
- Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT, 2018. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y) 4, 575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein G, Delmar P, Voyle N, Rehal S, Hofmann C, Abi-Saab D, Andjelkovic M, Ristic S, Wang G, Bateman R, Kerchner GA, Baudler M, Fontoura P, Doody R, 2019. Gantenerumab reduces amyloid-β plaques in patients with prodromal to moderate Alzheimer’s disease: a PET substudy interim analysis. Alzheimers Res Ther 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight R, Khondoker M, Magill N, Stewart R, Landau S, 2018. A Systematic Review and Meta-Analysis of the Effectiveness of Acetylcholinesterase Inhibitors and Memantine in Treating the Cognitive Symptoms of Dementia. Dement Geriatr Cogn Disord 45, 131–151. [DOI] [PubMed] [Google Scholar]
- Knopman DS, 2019. Bad news and good news in AD, and how to reconcile them. Nat Rev Neurol 15, 61–62. [DOI] [PubMed] [Google Scholar]
- Knott VJ, 2000. Quantitative EEG methods and measures in human psychopharmacological research. Hum Psychopharmacol 15, 479–498. [DOI] [PubMed] [Google Scholar]
- Koch G, Bonni S, Pellicciari MC, Casula EP, Mancini M, Esposito R, Ponzo V, Picazio S, Di Lorenzo F, Serra L, Motta C, Maiella M, Marra C, Cercignani M, Martorana A, Caltagirone C, Bozzali M, 2018. Transcranial magnetic stimulation of the precuneus enhances memory and neural activity in prodromal Alzheimer’s disease. Neuroimage 169, 302–311. [DOI] [PubMed] [Google Scholar]
- Kumar D, Ganeshpurkar A, Kumar D, Modi G, Gupta SK, Singh SK, 2018. Secretase inhibitors for the treatment of Alzheimer’s disease: Long road ahead. Eur J Med Chem 148, 436–452. [DOI] [PubMed] [Google Scholar]
- Kumar S, Zomorrodi R, Ghazala Z, Goodman MS, Blumberger DM, Cheam A, Fischer C, Daskalakis ZJ, Mulsant BH, Pollock BG, Rajji TK, 2017. Extent of Dorsolateral Prefrontal Cortex Plasticity and Its Association With Working Memory in Patients With Alzheimer Disease. JAMA Psychiatry 74, 1266–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Porte SL, Bollini SS, Lanz TA, Abdiche YN, Rusnak AS, Ho WH, Kobayashi D, Harrabi O, Pappas D, Mina EW, Milici AJ, Kawabe TT, Bales K, Lin JC, Pons J, 2012. Structural basis of C-terminal beta-amyloid peptide binding by the antibody ponezumab for the treatment of Alzheimer’s disease. J Mol Biol 421, 525–536. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S, 2007. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci 8, 499–509. [DOI] [PubMed] [Google Scholar]
- Lam J, Lee J, Liu CY, Lozano AM, Lee DJ, 2020. Deep Brain Stimulation for Alzheimer’s Disease: Tackling Circuit Dysfunction. Neuromodulation. [DOI] [PubMed] [Google Scholar]
- Lanctot KL, Herrmann N, LouLou MM, 2003. Correlates of response to acetylcholinesterase inhibitor therapy in Alzheimer’s disease. J Psychiatry Neurosci 28, 13–26. [PMC free article] [PubMed] [Google Scholar]
- Landen JW, Andreasen N, Cronenberger CL, Schwartz PF, Borjesson-Hanson A, Ostlund H, Sattler CA, Binneman B, Bednar MM, 2017. Ponezumab in mild-to-moderate Alzheimer’s disease: Randomized phase II PET-PIB study. Alzheimers Dement (N Y) 3, 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannfelt L, Moller C, Basun H, Osswald G, Sehlin D, Satlin A, Logovinsky V, Gellerfors P, 2014. Perspectives on future Alzheimer therapies: amyloid-beta protofibrils - a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res Ther 6, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EJ, Fomenko A, Lozano AM, 2019. Magnetic Resonance-Guided Focused Ultrasound : Current Status and Future Perspectives in Thermal Ablation and Blood-Brain Barrier Opening. J Korean Neurosurg Soc 62, 10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoutsakos JS, Yan H, Anderson WS, Asaad WF, Baltuch G, Burke A, Chakravarty MM, Drake KE, Foote KD, Fosdick L, Giacobbe P, Mari Z, McAndrews MP, Munro CA, Oh ES, Okun MS, Pendergrass JC, Ponce FA, Rosenberg PB, Sabbagh MN, Salloway S, Tang-Wai DF, Targum SD, Wolk D, Lozano AM, Smith GS, Lyketsos CG, 2018. Deep Brain Stimulation Targeting the Fornix for Mild Alzheimer Dementia (the ADvance Trial): A Two Year Follow-up Including Results of Delayed Activation. J Alzheimers Dis 64, 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leplus A, Lauritzen I, Melon C, Kerkerian-Le Goff L, Fontaine D, Checler F, 2019. Chronic fornix deep brain stimulation in a transgenic Alzheimer’s rat model reduces amyloid burden, inflammation, and neuronal loss. Brain Struct Funct 224, 363–372. [DOI] [PubMed] [Google Scholar]
- Li T, Martin E, Abada YS, Boucher C, Ces A, Youssef I, Fenaux G, Forand Y, Legrand A, Nachiket N, Dhenain M, Hermine O, Dubreuil P, Delarasse C, Delatour B, 2020. Effects of Chronic Masitinib Treatment in APPswe/PSEN1dE9 Transgenic Mice Modeling Alzheimer’s Disease. J Alzheimers Dis 76, 1339–1345. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler SF, Haass C, 2004. Amyloid at the cutting edge: activation of alpha-secretase prevents amyloidogenesis in an Alzheimer disease mouse model. J Clin Invest 113, 1384–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, 2005. The molecular basis of memantine action in Alzheimer’s disease and other neurologic disorders: low-affinity, uncompetitive antagonism. Curr Alzheimer Res 2, 155–165. [DOI] [PubMed] [Google Scholar]
- Logovinsky V, Satlin A, Lai R, Swanson C, Kaplow J, Osswald G, Basun H, Lannfelt L, 2016. Safety and tolerability of BAN2401--a clinical study in Alzheimer’s disease with a protofibril selective Abeta antibody. Alzheimers Res Ther 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, Tu G, Schwabe T, Rhinn H, King R, Rosenthal A, 2019. Preclinical development of TREM2 agonist antibody (AL002) in cynomolgus monkeys. Alzheimers Dement 15, P215. [Google Scholar]
- Long JM, Holtzman DM, 2019. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 179, 312–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Sanz D, Bruna R, de Frutos-Lucas J, Maestu F, 2019. Magnetoencephalography applied to the study of Alzheimer’s disease. Prog Mol Biol Transl Sci 165, 25–61. [DOI] [PubMed] [Google Scholar]
- Lozano AM, Fosdick L, Chakravarty MM, Leoutsakos JM, Munro C, Oh E, Drake KE, Lyman CH, Rosenberg PB, Anderson WS, Tang-Wai DF, Pendergrass JC, Salloway S, Asaad WF, Ponce FA, Burke A, Sabbagh M, Wolk DA, Baltuch G, Okun MS, Foote KD, McAndrews MP, Giacobbe P, Targum SD, Lyketsos CG, Smith GS, 2016. A Phase II Study of Fornix Deep Brain Stimulation in Mild Alzheimer’s Disease. J Alzheimers Dis 54, 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchsinger JA, Zetterberg H, 2020. Tracking the potential involvement of metabolic disease in Alzheimer’s disease-Biomarkers and beyond. Int Rev Neurobiol 154, 51–77. [DOI] [PubMed] [Google Scholar]
- Lynch SY, Kaplow J, Zhao J, Dhadda S, Luthman J, Albala B, 2018. Elenbecestat, E2609, a BACE inhibitor: Results from a phase 2 study in subjects with mild cognitive impairment and mild-to-moderate dementia due to Alzheimer’s disease. Alzheimers Dement 14, P1623. [Google Scholar]
- Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M, 2010. Alzheimer’s disease: clinical trials and drug development. Lancet Neurol 9, 702–716. [DOI] [PubMed] [Google Scholar]
- Mankin EA, Fried I, 2020. Modulation of Human Memory by Deep Brain Stimulation of the Entorhinal-Hippocampal Circuitry. Neuron 106, 218–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcade M, Bourdin J, Loiseau N, Peillon H, Rayer A, Drouin D, Schweighoffer F, Desire L, 2008. Etazolate, a neuroprotective drug linking GABA(A) receptor pharmacology to amyloid precursor protein processing. J Neurochem 106, 392–404. [DOI] [PubMed] [Google Scholar]
- Marquez F, Yassa MA, 2019. Neuroimaging Biomarkers for Alzheimer’s Disease. Mol Neurodegener 14, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marron EM, Viejo-Sobera R, Quintana M, Redolar-Ripoll D, Rodriguez D, Garolera M, 2018. Transcranial magnetic stimulation intervention in Alzheimer’s disease: a research proposal for a randomized controlled trial. BMC Res Notes 11, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh SE, Abud EM, Lakatos A, Karimzadeh A, Yeung ST, Davtyan H, Fote GM, Lau L, Weinger JG, Lane TE, Inlay MA, Poon WW, Blurton-Jones M, 2016. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc Natl Acad Sci U S A 113, E1316–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martone RL, Zhou H, Atchison K, Comery T, Xu JZ, Huang X, Gong X, Jin M, Kreft A, Harrison B, Mayer SC, Aschmies S, Gonzales C, Zaleska MM, Riddell DR, Wagner E, Lu P, Sun SC, Sonnenberg-Reines J, Oganesian A, Adkins K, Leach MW, Clarke DW, Huryn D, Abou-Gharbia M, Magolda R, Bard J, Frick G, Raje S, Forlow SB, Balliet C, Burczynski ME, Reinhart PH, Wan HI, Pangalos MN, Jacobsen JS, 2009. Begacestat (GSI-953): a novel, selective thiophene sulfonamide inhibitor of amyloid precursor protein gamma-secretase for the treatment of Alzheimer’s disease. J Pharmacol Exp Ther 331, 598–608. [DOI] [PubMed] [Google Scholar]
- Martorell AJ, Paulson AL, Suk HJ, Abdurrob F, Drummond GT, Guan W, Young JZ, Kim DN, Kritskiy O, Barker SJ, Mangena V, Prince SM, Brown EN, Chung K, Boyden ES, Singer AC, Tsai LH, 2019. Multi-sensory Gamma Stimulation Ameliorates Alzheimer’s-Associated Pathology and Improves Cognition. Cell 177, 256–271 e222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon C, Gros P, Lee DJ, Hamani C, Lozano AM, Kalia LV, Kalia SK, 2019. Deep brain stimulation: potential for neuroprotection. Ann Clin Transl Neurol 6, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM, 2013. Cholinergic circuitry of the human nucleus basalis and its fate in Alzheimer’s disease. J Comp Neurol 521, 4124–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignon L, Kordasiewicz H, Lane R, Smith A, Miller T, Narayanan P, Swayze E, Norris D, Fitzsimmons B, Bennett F, 2018. Design of the First-in-Human study of IONIS-MAPTRx, a Tau-lowering antisense oligonucleotide, in patients with alzheimer disease (S2. 006). AAN Enterprises. [Google Scholar]
- Mila-Aloma M, Suarez-Calvet M, Molinuevo JL, 2019. Latest advances in cerebrospinal fluid and blood biomarkers of Alzheimer’s disease. Ther Adv Neurol Disord 12, 1756286419888819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonis G, Subramaniam RM, Trofimova A, Burns J, Bykowski J, Chakraborty S, Holloway K, Ledbetter LN, Lee RK, Pannell JS, Pollock JM, Powers WJ, Roca RP, Rosenow JM, Shih RY, Utukuri PS, Corey AS, 2020. ACR Appropriateness Criteria(R) Dementia. J Am Coll Radiol 17, S100–S112. [DOI] [PubMed] [Google Scholar]
- Morgan AR, Touchard S, Leckey C, O’Hagan C, Nevado-Holgado AJ, Consortium N, Barkhof F, Bertram L, Blin O, Bos I, Dobricic V, Engelborghs S, Frisoni G, Frolich L, Gabel S, Johannsen P, Kettunen P, Kloszewska I, Legido-Quigley C, Lleo A, Martinez-Lage P, Mecocci P, Meersmans K, Molinuevo JL, Peyratout G, Popp J, Richardson J, Sala I, Scheltens P, Streffer J, Soininen H, Tainta-Cuezva M, Teunissen C, Tsolaki M, Vandenberghe R, Visser PJ, Vos S, Wahlund LO, Wallin A, Westwood S, Zetterberg H, Lovestone S, Morgan BP, Annex, N.-W.T.C.f.N.o.M.D., Alzheimer’s, D., 2019. Inflammatory biomarkers in Alzheimer’s disease plasma. Alzheimers Dement 15, 776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musaeus CS, Shafi MM, Santarnecchi E, Herman ST, Press DZ, 2017. Levetiracetam Alters Oscillatory Connectivity in Alzheimer’s Disease. J Alzheimers Dis 58, 1065–1076. [DOI] [PubMed] [Google Scholar]
- Nagy C, Schuck E, Ishibashi A, Nakatani Y, Rege B, Logovinsky V, 2010. E2012, a novel gamma-secretase modulator, decreases plasma amyloid-beta (Aβ) levels in humans. Alzheimers Dement 6, S574. [Google Scholar]
- Nestler EJ, Hyman SE, Holtzman DM, Malenka RC, 2015. Molecular Neuropharmacology: A Foundation for Clinical Neuroscience., 3rd ed. McGraw-Hill Education/Medical. [Google Scholar]
- Neumann U, Ufer M, Jacobson LH, Rouzade-Dominguez ML, Huledal G, Kolly C, Luond RM, Machauer R, Veenstra SJ, Hurth K, Rueeger H, Tintelnot-Blomley M, Staufenbiel M, Shimshek DR, Perrot L, Frieauff W, Dubost V, Schiller H, Vogg B, Beltz K, Avrameas A, Kretz S, Pezous N, Rondeau JM, Beckmann N, Hartmann A, Vormfelde S, David OJ, Galli B, Ramos R, Graf A, Lopez Lopez C, 2018. The BACE-1 inhibitor CNP520 for prevention trials in Alzheimer’s disease. EMBO Mol Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo HV, Martinetz T, Born J, Molle M, 2013. Auditory closed-loop stimulation of the sleep slow oscillation enhances memory. Neuron 78, 545–553. [DOI] [PubMed] [Google Scholar]
- Nicoll JAR, Buckland GR, Harrison CH, Page A, Harris S, Love S, Neal JW, Holmes C, Boche D, 2019. Persistent neuropathological effects 14 years following amyloid-beta immunization in Alzheimer’s disease. Brain 142, 2113–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofengeim D, Mazzitelli S, Ito Y, DeWitt JP, Mifflin L, Zou C, Das S, Adiconis X, Chen H, Zhu H, Kelliher MA, Levin JZ, Yuan J, 2017. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc Natl Acad Sci U S A 114, E8788–E8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrowitzki S, Lasser RA, Dorflinger E, Scheltens P, Barkhof F, Nikolcheva T, Ashford E, Retout S, Hofmann C, Delmar P, Klein G, Andjelkovic M, Dubois B, Boada M, Blennow K, Santarelli L, Fontoura P, Investigators SCR, 2017. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 9, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penninkilampi R, Brothers HM, Eslick GD, 2017. Safety and Efficacy of Anti-Amyloid-beta Immunotherapy in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J Neuroimmune Pharmacol 12, 194–203. [DOI] [PubMed] [Google Scholar]
- Perez Ortiz JM, Swerdlow RH, 2019. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol 176, 3489–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poil SS, de Haan W, van der Flier WM, Mansvelder HD, Scheltens P, Linkenkaer-Hansen K, 2013. Integrative EEG biomarkers predict progression to Alzheimer’s disease at the MCI stage. Front Aging Neurosci 5, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JB, Rusheen AE, Barath AS, Rojas Cabrera JM, Shin H, Chang SY, Kimble CJ, Bennet KE, Blaha CD, Lee KH, Oh Y, 2020. Clinical applications of neurochemical and electrophysiological measurements for closed-loop neurostimulation. Neurosurg Focus 49, E6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prickaerts J, Heckman PRA, Blokland A, 2017. Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin Investig Drugs 26, 1033–1048. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Jacobs DM, Messer K, Salmon DP, Feldman HH, 2019. Cognitive heterogeneity in probable Alzheimer disease: Clinical and neuropathologic features. Neurology 93, e778–e790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabey JM, Dobronevsky E, 2016. Repetitive transcranial magnetic stimulation (rTMS) combined with cognitive training is a safe and effective modality for the treatment of Alzheimer’s disease: clinical experience. J Neural Transm (Vienna) 123, 1449–1455. [DOI] [PubMed] [Google Scholar]
- Rossini PM, Di Iorio R, Vecchio F, Anfossi M, Babiloni C, Bozzali M, Bruni AC, Cappa SF, Escudero J, Fraga FJ, Giannakopoulos P, Guntekin B, Logroscino G, Marra C, Miraglia F, Panza F, Tecchio F, Pascual-Leone A, Dubois B, 2020. Early diagnosis of Alzheimer’s disease: the role of biomarkers including advanced EEG signal analysis. Report from the IFCN-sponsored panel of experts. Clin Neurophysiol 131, 1287–1310. [DOI] [PubMed] [Google Scholar]
- Sabbagh M, Sadowsky C, Tousi B, Agronin ME, Alva G, Armon C, Bernick C, Keegan AP, Karantzoulis S, Baror E, Ploznik M, Pascual-Leone A, 2020. Effects of a combined transcranial magnetic stimulation (TMS) and cognitive training intervention in patients with Alzheimer’s disease. Alzheimers Dement 16, 641–650. [DOI] [PubMed] [Google Scholar]
- Sako Y, Kurimoto E, Mandai T, Suzuki A, Tanaka M, Suzuki M, Shimizu Y, Yamada M, Kimura H, 2019. TAK-071, a novel M1 positive allosteric modulator with low cooperativity, improves cognitive function in rodents with few cholinergic side effects. Neuropsychopharmacology 44, 950–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway S, Honigberg LA, Cho W, Ward M, Friesenhahn M, Brunstein F, Quartino A, Clayton D, Mortensen D, Bittner T, Ho C, Rabe C, Schauer SP, Wildsmith KR, Fuji RN, Suliman S, Reiman EM, Chen K, Paul R, 2018. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer’s disease (BLAZE). Alzheimers Res Ther 10, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, Guenzler V, Miloslavsky M, Wang D, Lu Y, Lull J, Tudor IC, Liu E, Grundman M, Yuen E, Black R, Brashear HR, Bapineuzumab, Clinical Trial, I., 2014. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370, 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanches C, Stengel C, Godard J, Mertz J, Teichmann M, Migliaccio R, Valero-Cabre A, 2020. Past, Present, and Future of Non-invasive Brain Stimulation Approaches to Treat Cognitive Impairment in Neurodegenerative Diseases: Time for a Comprehensive Critical Review. Front Aging Neurosci 12, 578339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheltens P, Alam J, Harrison J, Blackburn K, Prins N, 2019. Efficacy and safety results of REVERSE-SD, phase-2b clinical study of the selective p38α kinase inhibitor neflamapimod in early-stage Alzheimer’s disease (AD). Clinical Trials on Alzheimer’s Disease (CTAD), San Diego, CA. [Google Scholar]
- Scheltens P, Prins N, Lammertsma A, Yaqub M, Gouw A, Wink AM, Chu HM, van Berckel BNM, Alam J, 2018. An exploratory clinical study of p38alpha kinase inhibition in Alzheimer’s disease. Ann Clin Transl Neurol 5, 464–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger A, Hendrix S, Mandler M, Ellison N, Burger V, Brunner M, Frolich L, Mimica N, Hort J, Rainer M, Imarhiagbe D, Kurz A, Peters O, Gertz HJ, Tierney L, Mattner F, Schmidt W, Dubois B, 2015. Results from a Phase II Study to Assess the Clinical and Immunological Activity of AFFITOPE(R) AD02 in Patients with Early Alzheimer’s Disease. J Prev Alzheimers Dis 2, 103–114. [DOI] [PubMed] [Google Scholar]
- Schneeberger A, Mandler M, Otawa O, Zauner W, Mattner F, Schmidt W, 2009. Development of AFFITOPE vaccines for Alzheimer’s disease (AD)--from concept to clinical testing. J Nutr Health Aging 13, 264–267. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, 2019. Alzheimer disease and aducanumab: adjusting our approach. Nat Rev Neurol 15, 365–366. [DOI] [PubMed] [Google Scholar]
- Senova S, Chaillet A, Lozano AM, 2018. Fornical Closed-Loop Stimulation for Alzheimer’s Disease. Trends Neurosci 41, 418–428. [DOI] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A, 2016. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537, 50–56. [DOI] [PubMed] [Google Scholar]
- Sierra-Marcos A, 2017. Regional Cerebral Blood Flow in Mild Cognitive Impairment and Alzheimer’s Disease Measured with Arterial Spin Labeling Magnetic Resonance Imaging. Int J Alzheimers Dis 2017, 5479597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow AD, Cummings J, Lake T, Hu Q, Esposito L, Cam J, Hudson M, Smith E, Runnels S, 2009. Exebryl-1: A novel small molecule currently in human clinical trials as a disease-modifying drug for the treatment of Alzheimer’s disease. Alzheimers Dement 5, P418. [Google Scholar]
- Sommer A, Winner B, Prots I, 2017. The Trojan horse - neuroinflammatory impact of T cells in neurodegenerative diseases. Mol Neurodegener 12, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks DL, 2011. Alzheimer disease: statins in the treatment of Alzheimer disease. Nat Rev Neurol 7, 662–663. [DOI] [PubMed] [Google Scholar]
- Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Lieberburg I, Arrighi HM, Morris KA, Lu Y, Liu E, Gregg KM, Brashear HR, Kinney GG, Black R, Grundman M, 2012. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol 11, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl SM, 2018. New hope for Alzheimer’s dementia as prospects for disease modification fade: symptomatic treatments for agitation and psychosis. CNS Spectr 23, 291–297. [DOI] [PubMed] [Google Scholar]
- Stoiljkovic M, Kelley C, Stutz B, Horvath TL, Hajos M, 2019. Altered Cortical and Hippocampal Excitability in TgF344-AD Rats Modeling Alzheimer’s Disease Pathology. Cereb Cortex 29, 2716–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoiljkovic M, Leventhal L, Chen A, Chen T, Driscoll R, Flood D, Hodgdon H, Hurst R, Nagy D, Piser T, Tang C, Townsend M, Tu Z, Bertrand D, Koenig G, Hajos M, 2015. Concentration-response relationship of the alpha7 nicotinic acetylcholine receptor agonist FRM-17874 across multiple in vitro and in vivo assays. Biochem Pharmacol 97, 576–589. [DOI] [PubMed] [Google Scholar]
- Suarez-Calvet M, Kleinberger G, Araque Caballero MA, Brendel M, Rominger A, Alcolea D, Fortea J, Lleo A, Blesa R, Gispert JD, Sanchez-Valle R, Antonell A, Rami L, Molinuevo JL, Brosseron F, Traschutz A, Heneka MT, Struyfs H, Engelborghs S, Sleegers K, Van Broeckhoven C, Zetterberg H, Nellgard B, Blennow K, Crispin A, Ewers M, Haass C, 2016. sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early-stage Alzheimer’s disease and associate with neuronal injury markers. EMBO Mol Med 8, 466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait L, Tamagnini F, Stothart G, Barvas E, Monaldini C, Frusciante R, Volpini M, Guttmann S, Coulthard E, Brown JT, Kazanina N, Goodfellow M, 2020. EEG microstate complexity for aiding early diagnosis of Alzheimer’s disease. Sci Rep 10, 17627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariot PN, Lopera F, Langbaum JB, Thomas RG, Hendrix S, Schneider LS, Rios-Romenets S, Giraldo M, Acosta N, Tobon C, Ramos C, Espinosa A, Cho W, Ward M, Clayton D, Friesenhahn M, Mackey H, Honigberg L, Sanabria Bohorquez S, Chen K, Walsh T, Langlois C, Reiman EM, Alzheimer’s Prevention I, 2018. The Alzheimer’s Prevention Initiative Autosomal-Dominant Alzheimer’s Disease Trial: A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement (N Y) 4, 150–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins P, 2019. Industry update: the latest developments in the field of therapeutic delivery, April 2019. Ther Deliv 10, 469–479. [Google Scholar]
- Touzeau C, Moreau P, 2017. Daratumumab for the treatment of multiple myeloma. Expert Opin Biol Ther 17, 887–893. [DOI] [PubMed] [Google Scholar]
- Tsoneva T, Garcia-Molina G, Desain P, 2015. Neural dynamics during repetitive visual stimulation. J Neural Eng 12, 066017. [DOI] [PubMed] [Google Scholar]
- Turriziani P, Smirni D, Mangano GR, Zappala G, Giustiniani A, Cipolotti L, Oliveri M, 2019. Low-Frequency Repetitive Transcranial Magnetic Stimulation of the Right Dorsolateral Prefrontal Cortex Enhances Recognition Memory in Alzheimer’s Disease. J Alzheimers Dis 72, 613–622. [DOI] [PubMed] [Google Scholar]
- van Dyck CH, 2018. Anti-Amyloid-beta Monoclonal Antibodies for Alzheimer’s Disease: Pitfalls and Promise. Biol Psychiatry 83, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veitch DP, Weiner MW, Aisen PS, Beckett LA, Cairns NJ, Green RC, Harvey D, Jack CR Jr., Jagust W, Morris JC, Petersen RC, Saykin AJ, Shaw LM, Toga AW, Trojanowski JQ, Alzheimer’s Disease Neuroimaging, I., 2019. Understanding disease progression and improving Alzheimer’s disease clinical trials: Recent highlights from the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement 15, 106–152. [DOI] [PubMed] [Google Scholar]
- Vialatte FB, Maurice M, Dauwels J, Cichocki A, 2010. Steady-state visually evoked potentials: focus on essential paradigms and future perspectives. Prog Neurobiol 90, 418–438. [DOI] [PubMed] [Google Scholar]
- Voroslakos M, Takeuchi Y, Brinyiczki K, Zombori T, Oliva A, Fernandez-Ruiz A, Kozak G, Kincses ZT, Ivanyi B, Buzsaki G, Berenyi A, 2018. Direct effects of transcranial electric stimulation on brain circuits in rats and humans. Nat Commun 9, 483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss T, Li J, Cummings J, Farlow M, Assaid C, Froman S, Leibensperger H, Snow-Adami L, McMahon KB, Egan M, Michelson D, 2018. Randomized, controlled, proof-of-concept trial of MK-7622 in Alzheimer’s disease. Alzheimers Dement (N Y) 4, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Tartaglia MC, Nygaard HB, Zeman AZ, Miller BL, 2017. Epileptic activity in Alzheimer’s disease: causes and clinical relevance. Lancet Neurol 16, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker D, Lue LF, Paul G, Patel A, Sabbagh MN, 2015. Receptor for advanced glycation endproduct modulators: a new therapeutic target in Alzheimer’s disease. Expert Opin Investig Drugs 24, 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Mao Z, Ling Z, Yu X, 2020. Repetitive transcranial magnetic stimulation for cognitive impairment in Alzheimer’s disease: a meta-analysis of randomized controlled trials. J Neurol 267, 791–801. [DOI] [PubMed] [Google Scholar]
- Wang X, Sun G, Feng T, Zhang J, Huang X, Wang T, Xie Z, Chu X, Yang J, Wang H, Chang S, Gong Y, Ruan L, Zhang G, Yan S, Lian W, Du C, Yang D, Zhang Q, Lin F, Liu J, Zhang H, Ge C, Xiao S, Ding J, Geng M, 2019. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res 29, 787–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Krishnan GP, Marshall L, Martinetz T, Bazhenov M, 2020. Stimulation Augments Spike Sequence Replay and Memory Consolidation during Slow-Wave Sleep. J Neurosci 40, 811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler M, Stieger KC, Long JM, Rapp PR, 2020. Transcranial Magnetic Stimulation in Alzheimer’s Disease: Are We Ready? eNeuro 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan CD, Mattsson N, Nagle MW, Vijayaraghavan S, Hyde C, Janelidze S, Stomrud E, Lee J, Fitz L, Samad TA, Ramaswamy G, Margolin RA, Malarstig A, Hansson O, 2019. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta Neuropathol Commun 7, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Colton CA, 2008. Anti-amyloid-beta immunotherapy in Alzheimer’s disease: relevance of transgenic mouse studies to clinical trials. J Alzheimers Dis 15, 555–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Chen HS, Zhang D, Lipton SA, 2010. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J Neurosci 30, 11246–11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Wang T, Ma X, Qin Y, Li X, Zhao Z, Liu X, Wang X, Xie H, Jiang Q, Sun L, Luo B, Shang L, Chen W, Bai Y, Tang M, He M, Wu L, Ma Q, Hou D, He J, 2017. Efficacy and safety of a novel acetylcholinesterase inhibitor octohydroaminoacridine in mild-to-moderate Alzheimer’s disease: a Phase II multicenter randomised controlled trial. Age Ageing 46, 767–773. [DOI] [PubMed] [Google Scholar]
- Yeo SN, Lee TS, Sng WT, Heo MQ, Bautista D, Cheung YB, Zhang HH, Wang C, Chin ZY, Feng L, Zhou J, Chong MS, Ng TP, Krishnan KR, Guan C, 2018. Effectiveness of a Personalized Brain-Computer Interface System for Cognitive Training in Healthy Elderly: A Randomized Controlled Trial. J Alzheimers Dis 66, 127–138. [DOI] [PubMed] [Google Scholar]
- Yeung MK, Chan AS, 2020. Functional near-infrared spectroscopy reveals decreased resting oxygenation levels and task-related oxygenation changes in mild cognitive impairment and dementia: A systematic review. J Psychiatr Res 124, 58–76. [DOI] [PubMed] [Google Scholar]
- Young PNE, Estarellas M, Coomans E, Srikrishna M, Beaumont H, Maass A, Venkataraman AV, Lissaman R, Jimenez D, Betts MJ, McGlinchey E, Berron D, O’Connor A, Fox NC, Pereira JB, Jagust W, Carter SF, Paterson RW, Scholl M, 2020. Imaging biomarkers in neurodegeneration: current and future practices. Alzheimers Res Ther 12, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Griciuc A, Hudry E, Wan Y, Quinti L, Ward J, Forte AM, Shen X, Ran C, Elmaleh DR, Tanzi RE, 2018. Cromolyn Reduces Levels of the Alzheimer’s Disease-Associated Amyloid beta-Protein by Promoting Microglial Phagocytosis. Sci Rep 8, 1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen J, Qian Y, Weng X, Su W, Zhang J, Cai L, Dong L, An H, Su R, Wang J, Zheng Y, Wang X, 2017. Gamma rhythm low field magnetic stimulation alleviates neuropathologic changes and rescues memory and cognitive impairments in a mouse model of Alzheimer’s disease. Alzheimers Dement (N Y) 3, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Peters F, Filser S, Herms J, 2018. Consequences of Pharmacological BACE Inhibition on Synaptic Structure and Function. Biol Psychiatry 84, 478–487. [DOI] [PubMed] [Google Scholar]
- Zomorrodi R, Loheswaran G, Pushparaj A, Lim L, 2019. Pulsed Near Infrared Transcranial and Intranasal Photobiomodulation Significantly Modulates Neural Oscillations: a pilot exploratory study. Sci Rep 9, 6309. [DOI] [PMC free article] [PubMed] [Google Scholar]