

Abstract
Host immunity has an essential role in the clinical management of cancers. Therefore, it is advantageous to choose therapies that can promote tumor cell death and concurrently boost host immunity. The dynamic tumor microenvironment (TME) determines whether an antineoplastic drug will elicit favorable or disparaging immune responses from tumor-infiltrating lymphocytes (TILs). CD8+ T cells are one of the primary tumor-infiltrating immune cells that deliver antitumor responses. Here, we review the influence of various factors in the TME on CD8+ T cell exhaustion and survival, and possible strategies for restoring CD8+ T cell effector function through immunotherapy.
Keywords: tumor-infiltrating lymphocytes, CD8+ T cell, tumor microenvironment, immunotherapy, chemotherapeutic agents
Teaser:
Cancer therapies influence tumor microenvironment (TME) plasticity and alter CD8+ T cell-mediated antitumor responses. Therefore, an understanding of TME plasticity and its impact on the immune system is crucial for designing a successful cancer therapy.
Introduction
The proper understanding of host immunity and its role in cancer is crucial for successful cancer therapy. Considering the enormous importance of the immune system in cancer therapy, several immunomodulatory drugs and immunotherapeutic agents have been used to treat various malignancies. Typical examples of immunomodulatory agents are cytokines, including interferons (IFNs), interleukin-2 (IL-2), IL-11, granulocyte-macrophage colony-stimulating factor (GM-CSF), and erythropoietin [1]. The classical example of immunomodulatory drugs in clinical use are thalidomide, lenalidomide, and pomalidomide [2]. These three drugs induce the cellular release of IL-2 [2]. Similar to immunomodulating drugs, the US Food and Drug Administration (FDA) has approved immune checkpoint inhibitors that can target T cell co-inhibitory molecules, including cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), programmed cell death protein (PD-1), and programmed death-ligand 1 (PD-L1). These immunotherapeutics have proven efficacy in enhancing T cell-mediated antitumor effects in various malignancies, including melanoma, nonsmall cell lung cancer (NSCLC), and other cancers [3]. CTLA-4 and PD-1 regulate both peripheral and tumor-infiltrating T cells. In the peripheral immune system, CTLA-4 regulates the proliferation of T cells in lymph nodes during the early immune response. However, PD-1 works during the later phase of immune response and inhibits T cell function [4]. CTLA-4 is expressed mainly on regulatory T cells (Tregs); within the tumors, however, PD-1 is primarily expressed on exhausted T cells [3]. Several tumors express PD-L1 and interact with T cells expressing PD-1; this interaction promotes compromised T cell effector function [5].
Tumor cells favor creating a protumorigenic local environment, also known as the TME. The TME encompasses lymphatic and blood vessels, extracellular matrix (ECM), stromal cells (e.g., fibroblasts), lymphocytes, neutrophils, natural killer (NK) cells, NK-T cells, tumor-associated macrophages (TAMs), RNAs, secreted proteins, and small organelles [6]. Based on the presence of immune cells, TMEs are classified as inflamed, immunological ignorant, or immune desert [7]. Based on the presence and absence of T cell infiltrates, the inflamed TME can be divided into T cell-inflamed versus non-T cell-inflamed TME. T cell-inflamed TME contains T cells and its subsets and chemokines [8]. It also contains macrophages, B cells, and plasma cells, whereas the non-T cell-inflamed TME consists mainly of macrophages [9]. By contrast, immunological-ignorant TME lacks activation markers on T cells. Immunological ignorance indicates a state in which adaptive immune system components are unable to recognize or respond to pathogens or tumor antigens [10]. The immune-desert TME is the immune-neglected region within the tumor that lacks functional effector T cells [11].
The reprogramming of TME residents directly or indirectly affects the immune and stromal cells, which consequently regulates the growth and survival of cancer cells [12]. The TME can be considered a combat zone in which host immune and non-immune cells interact with tumor cells, and several soluble mediators, including ILs, are released. Tumor cell plasticity induces polarization of the TME in either an immunosuppressive or inflammatory direction. The immunosuppressive landscape of TME attenuates the antitumor immunity of the host, leading to tumor progression and cancer cell growth [13]. The importance of the TME is exemplified in neuroblastoma, which primarily affects children. Although the MYCN oncogene is mutated in many patients, MYCN-targeted therapy fails to influence neuroblastoma. However, MYCN mutation is not solely responsible for neuroblastoma malignancies; the TME also contributes substantially to neuroblastoma tumorigenesis and, thus, strategies to target both tumor cells and TME should be considered [14,15]. Similar to MYCN, c-MYC is another proto-oncogene with an important role in regulating the immune response in the TME [16]. c-MYC impedes cancer immunotherapy and, therefore, the Myc inhibitor, MYCi361 and its improved analog, MYCi975, can sensitize tumors to anti-PD1 immunotherapy [17]. In contrast to c-MYC, loss of phosphatase and tensin homolog (PTEN) in the tumor is associated with an immunosuppressive TME [18]. Similar to the loss of PTEN, the loss of liver kinase B1 (LKB1), a serine/threonine kinase, has also been reported to regulate TME and causes dysregulation of cytokine and chemokine secretion, including CCL2, and increased recruitment of protumorigenic macrophages [19].
The immunotherapeutic approach to induce tumor cell death by tumor-infiltrating T cells, targeting specific antigens expressed on tumor cells, is an emerging area to treat different malignancies. Recent studies showed that the exhaustion and functional impairments of T cells in TME are defining features of many cancers. Therefore, a thorough understanding of the intrinsic properties if T cells, such as their survival and effector functions, will be fundamental for restoring an antitumor immune response [20]. The presence of immune cells, especially T cells within TME, determines whether the tumor is hot (T cell inflamed) or cold (T cell non-inflamed) [8]. Hot tumors express chemokines, T cell markers, and IFN I signatures that regulate overall tumor immunity [8]. The importance of TIL in tumor immunity is illustrated by metastatic melanoma, in which T cell infiltration is considered as a prognostic biomarker in response to therapy. However, the analysis of intertumoral CD8+ T cell counts showed heterogeneity for synchronous and metachronous metastases. In synchronous tumors, CD8+ T cell density was comparable; however, in metachronous tumors, CD8+ T cell density was variable [21]. These observations suggest that heterogeneity in CD8+ T cell infiltration into tumors governs differential immunity.
Tumor immunity is a multistep process. During the initial stage, tumor cells with oncogenic mutation(s) express specific antigens that distinguish cancer cells from healthy cells and help immune cells (i.e., antigen-presenting cells; APCs) recognize the specific tumor antigens. APCs present tumor antigens to T cells in the lymph nodes, and this interaction between APCs and T cells results in T cell priming and activation. The activated T cells migrate and infiltrate into the tumor via the circulation. The tumor-infiltrating T cells recognize specific antigen(s) on cancer cells and eventually destroy them. The dying tumor cells further release antigen(s), which ensures the tumor immunity cycle [22]. Several factors, including cytokines, kinases, and cellular metabolites, regulate the survival and effector function of tumor-infiltrating T cells. In this review, we discuss various TME-derived factors that influence tumor-infiltrating CD8+ T cell dysfunction and consider different strategies that can help to restore their function and survival to further enhance immunotherapy efficacy.
Infiltration of CD8+ T cells in solid tumors
Crosstalk between the peripheral immune system and TME is crucial for the antitumor responses of effector CD8+ T cells. Therefore, the proper understanding of peripheral and tumor-infiltrating T cells is crucial from a therapeutic perspective. The priming, expansion, and migration of T cells specific to tumor antigens occur in the local peripheral lymphoid organ, especially in tumor-draining lymph nodes (dLN). dLN-resident T cells are activated by soluble antigens of the tumor, tumor fragments, and apoptotic tumor cells. Dendritic cells (DCs) can also carry tumor antigenic peptides from the tumor to dLN [23]. During the late stages of cancer, CD8+ T cells in lymphoid organs of tumor-bearing mice and patients lose their effector function, ultimately diminishing the antitumor response [24]. TILs are one of the integral and essential components of the TME. Tumor-infiltrating mononuclear immune cells constitute a significant fraction of TILs and serve as a predictive indicator of drug efficacy in several cancers. TILs have an essential role in creating either pro- or anti-tumorigenic TME, affecting tumor progression and therapy resistance [25]. T lymphocytes are significant components of TILs, of which CD4+, CD8+ and Tregs are frequently observed in various cancer. CD8+ T cells have a crucial role in host antitumor immune responses in conjunction with CD4+ T cells. CD8+ T cells release granzyme B, perforin, and IFN-γ, and act as cytotoxic cells that ultimately kill cancer cells. Analyses of Cancer Genome Atlas (TCGA) data sets using Oncomine Platform (Thermo Fisher, Ann Arbor, MI, USA) for CD8 gene expression in malignancies such as brain, breast, colorectal (CRC), and ovarian tumors, indicated that CD8 gene expression is downregulated in the brain, CRC, and ovarian cancer, but unaltered in breast cancer (Figure 1a–d). However, analyses in melanoma and lung cancers using TCGA data sets suggested no major difference in CD8A copy number between normal and malignant tissues (Figure 1e,f). The TCGA data sets indicate heterogeneity of CD8 gene expression in different malignancies. Tumor-residing CD8+ T cells (TILs) are mainly memory T cells and, therefore, are called tissue-resident memory (TRM) T cells. TRM cells are characterized by the high expression of surface markers CD103 and/or CD49a integrins, and T cell activation marker, CD69 [26]. Mechanisms of antigen-specific CD8+ T cell activation, expansion, and migration into the tumor are presented in Figure 2.
Figure 1.
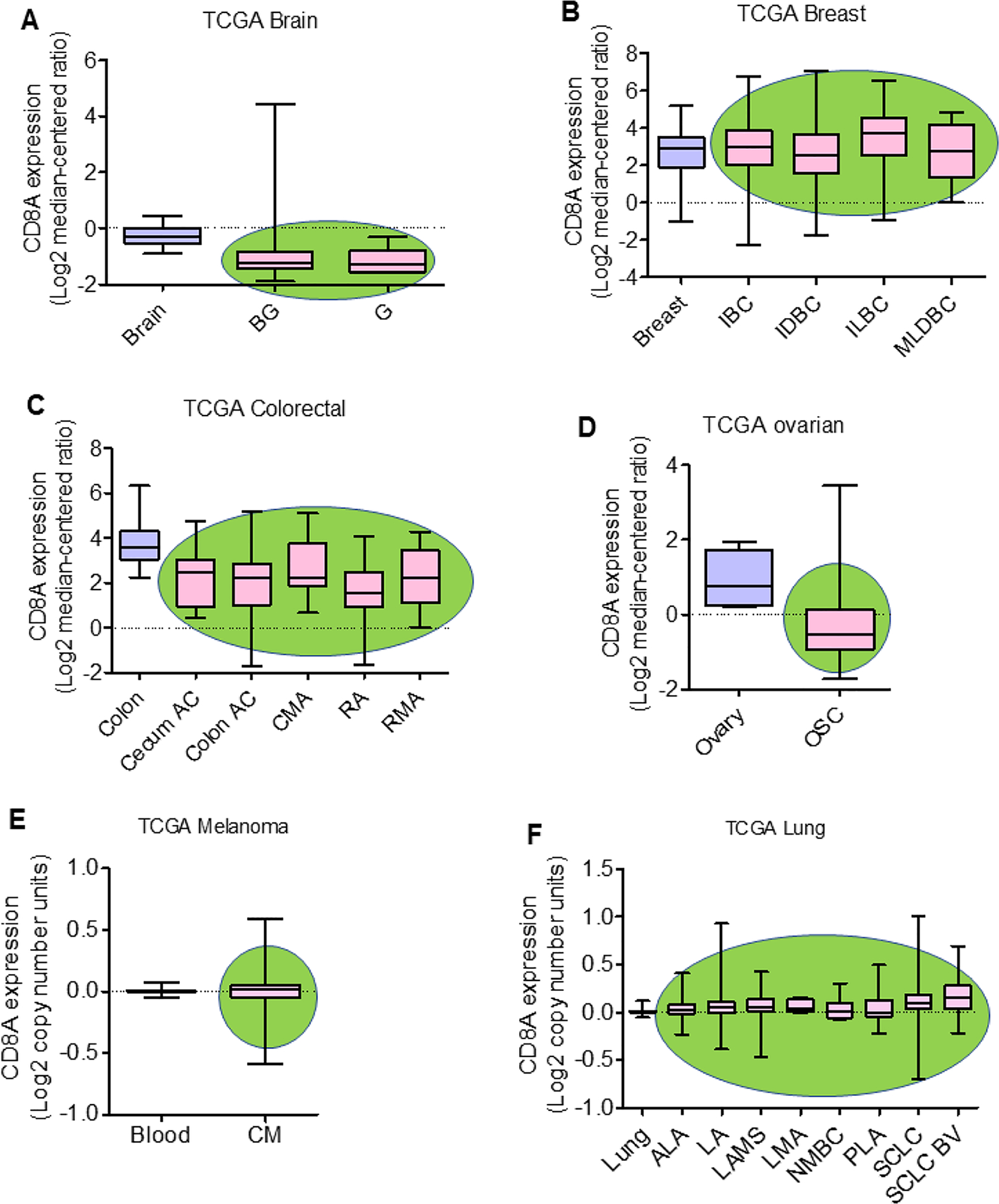
CD8A expression in solid malignancies. CD8 gene (i.e., CD8A) expression in brain, breast, colorectal, and ovarian cancers, and copy number in melanoma and lung cancer were analyzed by using The Cancer Genome Atlas (TCGA) with the Oncomine™ Platform (Thermo Fisher, Ann Arbor, MI, USA) on May 11, 2020 and July 11, 2020, respectively. CD8A gene expression in (a) brain (N=10), brain glioblastoma (BG; N=542) and glioblastoma (G; N=5); (b) breast (N=61), invasive breast carcinoma (IBC; N=76), invasive ductal breast carcinoma (IDBC;N=389), invasive lobular breast carcinoma (ILBC; N=36), and mixed lobular and ductal breast carcinoma (MLDBC; N=7); (c) colon (N=19), cecum adenocarcinoma (cecum AC; N=24), colon adenocarcinoma (colon AC; N=102), colon mucinous adenocarcinoma (CMA; N=20), rectal adenocarcinoma (RA; N=60) and rectal mucinous adenocarcinoma (RMA; N=6); (d) in ovary (N=8) and ovarian serous cystadenocarcinoma (OSC; N=586). CD8A copy number in (e) blood (N=312) and cutaneous melanoma (CM; N=58); and (f) lung (N=396), acinar lung adenocarcinoma (ALA; N=13), lung adenocarcinoma (LA; N=331), lung adenocarcinoma, mixed subtype (LAMS; N=92), lung mucinous adenocarcinoma (LMA; N=9), non-mucinous bronchioloalveolar carcinoma (NMBC; N=18), papillary lung adenocarcinoma (PLA; N=18), squamous cell lung carcinoma (SCLC; N=471), and squamous cell lung carcinoma, basaloid variant, (SCLC BV; N=17). The green oval background in the graph represents tumor tissues.
Figure 2.
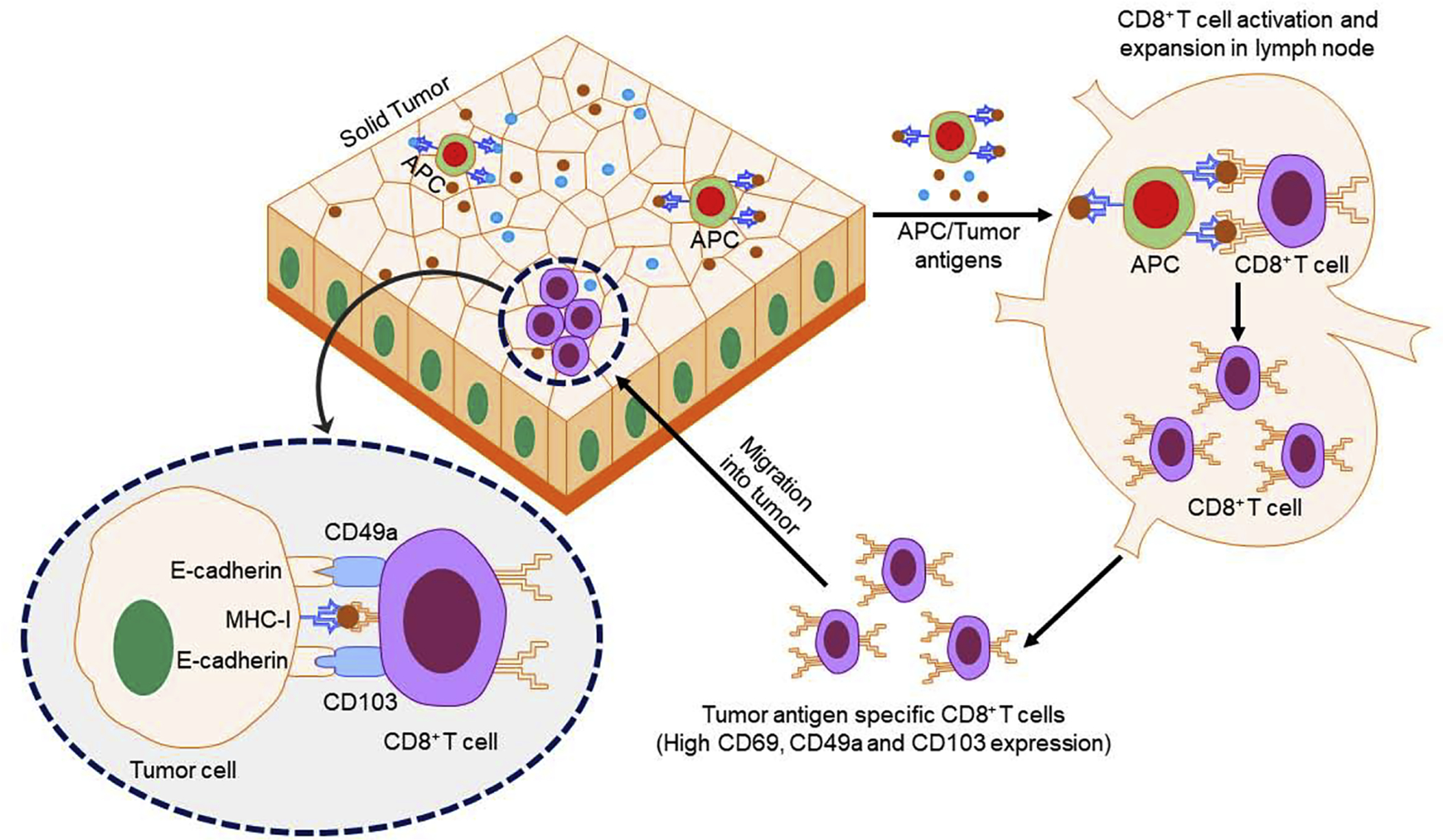
Schematic diagram for CD8+ T cell infiltration into tumors. Tumor antigens (TAs) are released by tumors and recognized by antigen-presenting cells (APCs), such as dendritic cells (DCs), in the lymph nodes. In other modes, tumor-infiltrating APCs loaded with TAs travel to the lymph nodes. The APCs process and present TA to CD8+ T cells, resulting in CD8+ T cell activation and generation of TA-specific CD8+ T cells. These TA-specific CD8+ T cells express CD49a and CD103 on their surfaces and bind with E-cadherin on tumor cells. The TA-specific CD8+ T cells recognize antigens present on tumor cells and induce tumor cell death. Abbreviation: MHC-I, major histocompatibility complex class I.
Apart from TRM T cells, there are several other phenotypes of CD8+ T cells in the TME. Notably, the stem-like CD8+ T cell phenotype is considered to be important for antitumor responses [6]. Stem-like CD8+ T cells differentiate into effector cytotoxic CD8+ T cells and localize in the tumor niche, where APC populations are abundant [6]. However, there are some malignancies in which APCs within tumors are minimal and correspond to reduced CD8+ T cell infiltration, leading to immune escape by tumors. In the absence of adequate APCs, the activation of CD8+ T cells is considerably compromised.
Interestingly, tumor-infiltrating CD8+ T cells can also serve as a bystander activation where T cells activated for a specific antigen can also respond against unrelated antigen(s) [27]. For example, in CRC and lung cancer, tumor-infiltrating CD8+ T cells show reactivity with tumor antigens as well as unrelated antigens, such as Epstein–Barr virus (EBV), human cytomegalovirus (HCMV), or influenza virus [28]. The presence of bystander CD8+ T cells in a tumor might prevent its immune escape. The latter is a protumor phenomenon that leads to tumor cell growth and survival, and failure to respond to the antitumor responses triggered by CD8+ T cells. The decreased infiltration of CD8+ T cells is also regulated by immune suppressive mechanisms, mediated via indoleamine-2, 3-dioxygenase (IDO), PD-L1/B7-H1, and FoxP3+ Tregs [29]. Cancer radiotherapy also affects the tumor infiltration of CD8+ T cells. Radiotherapy causes blood vessel damage in the TME, which permits infiltration of TAMs, myeloid-derived suppressor cells (MDSCs), and Tregs that ultimately attenuate CD8+ T cell infiltration into the tumor, inducing immunosuppression [30]. Tumor-infiltrating CD8+ T cells are prognostically and predictively important in breast malignancies [31]. The predictive value of TILs is particularly relevant in triple-negative breast cancers (TNBCs) and human epidermal growth factor receptor 2 (HER2+) breast cancers [32]. TNBCs are lymphocyte-predominant breast cancers, showing clusters of lymphocyte infiltrates [33]. Interestingly, T cell infiltration into TNBC tumors is associated with overall survival [34]. However, in HER2+ breast cancer, lymphocyte infiltration is not directly correlated with survival [35]. Similar to TNBCs, increased TILs and decreased Tregs correlate with improved survival in patients with ovarian cancer [36]. Unlike TNBC, pancreatic ductal adenocarcinoma (PDAC) lacks a defined tumor infiltration of CD8+ T cells; however, PDAC TME is flooded with inflammatory cells. In PDAC, the presence of inflammatory infiltrates in TME accelerates tumor growth and metastasis and suppresses the effector function of cytotoxic CD8+ T cells [37]. These examples collectively suggest that tumor-infiltrating CD8+ T cells have an essential role in tumor regression and overall survival. However, the heterogeneous infiltration of CD8+ T cells into tumors remains unclear in different tumors. This distribution could be regulated by several factors, including cytokine milieu in TME, the status of protein kinases, and alteration of metabolic status.
Cytokines and its role in CD8+ T cell infiltration into tumor
Cytokines are a group of small secretory proteins made by various types of cell, including immune and tumor cells. Cytokines are crucial regulators of TME plasticity and have a significant role in CD8+ T cell infiltration and effector function (Table 1). IL-2 regulates CD8+ T cell activation and proliferation and induces CD8+ T cell-mediated anti-tumor responses [38]. It also synergizes with IL-12 and IFN-γ in the augmenting cytotoxic function of tumor-infiltrating CD8+ T cells [39]. In contrast to these cytokines, several other cytokines, including IL-10 and IL-35, suppress the antitumor function of tumor-infiltrating CD8+ T cells [40]. IL-10 is an immunosuppressive cytokine that affects CD8+ T cell tumor reactivation mainly via tempering IFN-γ production [41]. IL-10 and IL-35 are released by Tregs and upregulate co-inhibitory proteins, such as PD-1, PD-L1, and CTLA-4 expression on tumor-infiltrating CD8+ T cells. T cell co-inhibitory molecules promote dysfunction in T cell activation, expansion, effector function, and survival. The combined network of IL-10 and IL-35 co-inhibitory proteins suppresses the effector function and induces exhaustion in tumor-infiltrating CD8+ T cells [40]. In addition to releasing IL-10 and IL-35, Tregs also interact with CD11c+ DCs and diminish the activation and expansion of CD8+ T cells in the tumor. Recent studies showed that depletion of Tregs in PDAC increases the infiltration of IFN-γ-producing cytotoxic CD8+ T cells into the tumor [42]. Another essential cytokine, transforming growth factor-beta (TGF-β), is also reported to have roles in tumor growth/survival and host immunity. TGF-β induces immunosuppression in the host and helps the tumor escape from the immune system [43]. Thus, it has been reported that TGF-β attenuation promotes CD8+ T cell-mediated antitumor responses [43]. Similarly, a study in a murine model of PDAC showed that adoptive transfer of TGFβ-insensitive CD8+ T cells induced tumor regression [44]. IL-4 and IL-12 similarly affect CD8+ T cell effector function adversely by influencing the expansion of CD8+ Tregs with Foxp3−IL-10+ phenotype [45]. Tumor necrosis factor alpha (TNF-α) released by cancer and stromal cells is reported to induce apoptosis in tumor-infiltrating CD8+ T cells [46].
Table 1.
Effect of TME cytokines on tumor-infiltrating CD8+ T cells
| Cytokine | Source in TME | Effect on CD8+ T cells (TILs) | Refs |
|---|---|---|---|
| IL-2 | Intratumoral injection of IL-2 | Increases CD8+ T cell infiltration | [38] |
| IL-10 | Tregs | Increases CD8+ T cell exhaustion | [40] |
| IL-15 | Tumor and stroma | Increases CD8+ T cell infiltration | [47] |
| IL-17 | Helper T cells | Increases CD8+ T cell effector function | [49] |
| IL-18 | Tumor cells | Increases CD8+ T cell cytotoxicity | [50] |
| IL-35 | Tregs | Decreases CD8+ T cell effector function | [40] |
| TNF-α | Cancer and stromal cells | Induces CD8+ T cell exhaustion | [46] |
Alongside immunosuppressive cytokines, in TME, anti-inflammatory cytokines are present that help to restore cytotoxic CD8+ T cell function. The pleiotropic cytokine, IL-15, is secreted by various immune and non-immune cell types and is associated with inflammation and defensive immunity [47]. IL-15 induces NK, NK-T, and cytotoxic CD8+ T cells, and is considered a potential candidate for cancer immunotherapy [48]. The role of IL-15 in promoting antitumor responses has been shown in ovarian cancer. Ovarian tumor antigen loaded on cytokine-matured human DCs, along with a combination of IL-15 and a p38 inhibitor, induced CD4+Th17-mediated responses and CD8+ T cell cytotoxicity [49]. Th17 helper cells are a subset of CD4+ T cells that release a proinflammatory cytokine, IL-17, which also increases antitumor immunity. In an ovarian cancer model, IL-17 in TME induced an antitumor immune responses [49]. IL-18, also known as IFN-γ-inducing factor (IGIF), is an antitumor cytokine and promotes T cell activation [50]. In syngeneic and humanized tumor models, blockade of ectoenzyme CD39 induced the release of active IL-18 and prompted the expansion of tumor-infiltrating CD8+ effector T cells [51]. Therefore, there is growing interest in identifying cytokines in the TME to understand their functions in regulating tumor-infiltrating CD8+ T cells and that could be used to alter TME dynamics to restore the antitumor efficacy of CD8+ T cells.
Small-molecular-weight cytokines, also known as chemokines, have a regulatory role in T cell migration. Effector T cell migration is a crucial step required for antitumor response. Chemokines regulate the infiltration of T cells in the TME. The association between T cell infiltration and chemokine receptor CXCR3 and ligands CXCL9, CXCL10, and CXCL11 in recruiting CD8+ T cells, Th1 cells, and NK cells has been reported in tumors [52]. Chemokines expressed in melanomas include CCL2, CCL3, CCL4, CCL5, CCL19, CCL21, CXCL9, CXCL10, CXCL11, and CXCL13, and are associated with infiltration of lymphocytes [53]. Interestingly, CD8β transcript expression is tightly associated with the higher expression of CCL2, CCL4, CCL5, CCL19, CCL21, CXCL9, CXCL10, CXCL11, CXCL13, and XCL2 in lymphocytes infiltrating melanoma tumors [53]. Given the crucial role of cytokines/chemokines in regulating antitumor responses of CD8+ T cells, cytokine-based therapy has been used in various cancers. One of the first cytokine-based therapies in cancer was IL-2-based therapy. Systemic IL-2 treatment has been used to induce an anticancer immune response in metastatic melanoma and renal cancer; however, systemic IL-2 (i.e., aldesleukin) treatment is associated with toxicity [54]. However, the low-dose IL-2-derivative PEGylated IL-2 (i.e., NKTR-214) showed promising antitumor efficacy with acceptable toxicity [54]. These emerging results suggest a promising therapeutic future for cytokine-based cancer therapy to improve T cell-mediated antitumor effects.
Immunotherapeutics to potentiate tumor-infiltrating CD8+ T cells
Significant research has focused on finding immunotherapeutic approaches to increase and restore functions of tumor-infiltrating CD8+ T cells for cancer therapy. CD8+ T cell dysfunction is relatively common in solid tumors and, therefore, several approaches have been suggested to increase CD8+ T cell infiltration and restore its functions. Some of the methods tested include the therapeutic use of checkpoint inhibitor (CPI) antibodies, T cell co-stimulatory molecules, agonistic antibodies, chimeric antigen receptor (CAR)-T cells, TCR-transduced T cells, and TIL-based cancer therapy (Figure 3).
Figure 3.
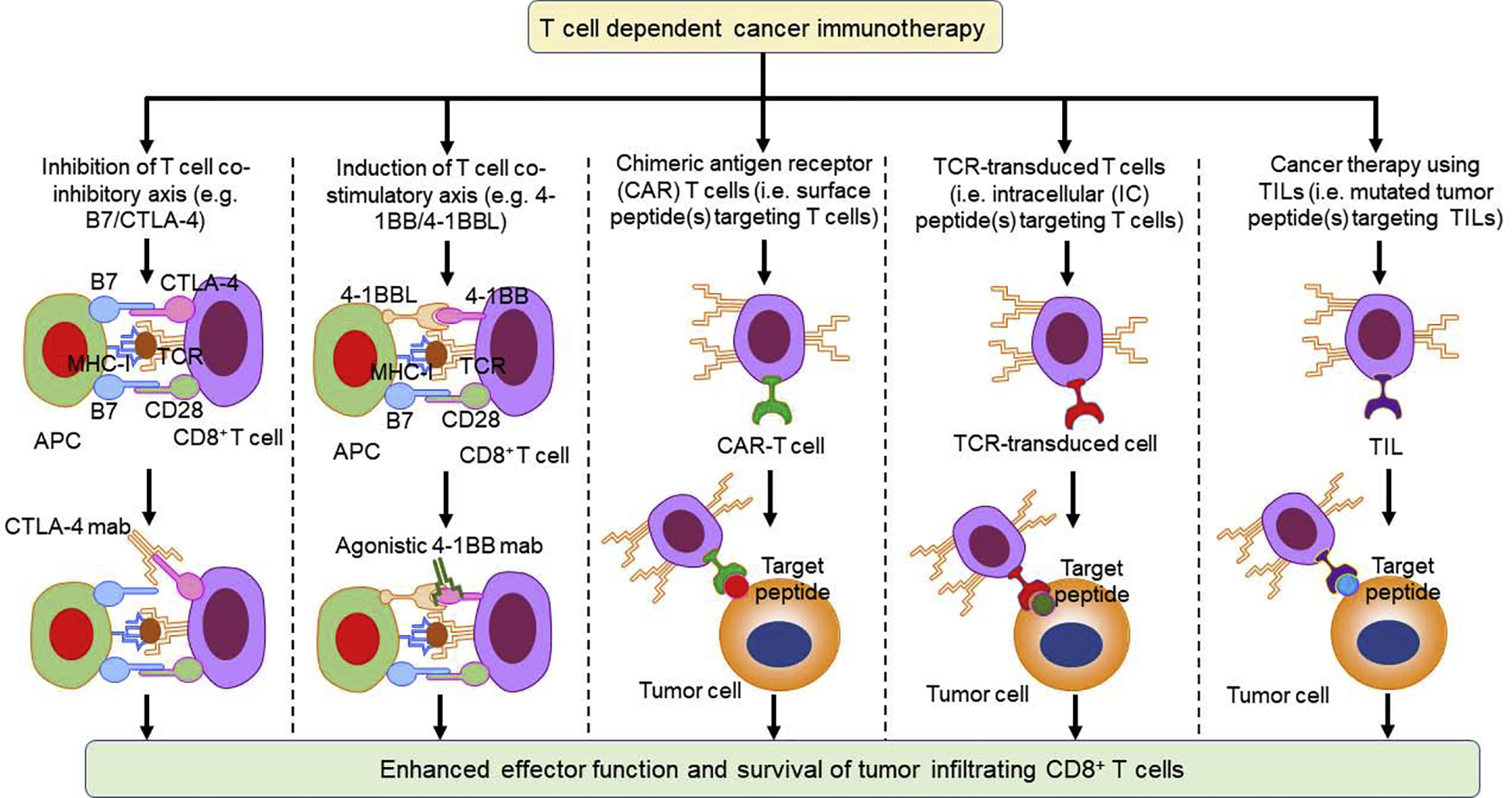
Hypothetical T cell-dependent cancer therapy to enhance CD8+ T cell infiltration and effector function. Various approaches can improve CD8+ T cell infiltration and effector function, including inhibition of T cell co-inhibitory axis (e.g., B7/CTLA-4) by using CTLA-4 blocking antibodies, induction of T cell costimulatory axis (e.g., 4-1BB/4-1BBL) by using a 4-1BB agonistic antibodies, and using chimeric antigen receptor (CAR) T cells, T cell receptor (TCR)-transduced T cells, and tumor-infiltrating T cells (TILs). Abbreviation: MHC-I, major histocompatibility complex class I.
Targeting the T cell co-inhibitory axis for cancer therapy
The expression of T cell co-inhibitory molecules (PD1, PD-L1, and CTLA-4) is reported to attenuate T cell activation, functions, and survival. Importantly, blockade of these co-inhibitory molecules reprograms T cell functions in several cancers [55]. However, such blockade has shown limited benefit against certain types of hematological and solid cancer.
The T cell co-inhibitory molecules, CTLA-4, and PD1, are crucial regulators of central and peripheral tolerance. T cell progenitors originate from bone marrow and undergo development in the thymus, where immature T cells proliferate and generate a range of TCRs because of the recombination of TCR gene segments [56]. The T cells that can bind to self-antigens are deleted to prevent autoreactivity through a process termed ‘central tolerance’. Although this is useful to prevent autoimmune reactivity, it is not suitable for the anticancer response [57]. CTLA-4 is involved significantly in central tolerance and, during T cell development, CTLA-4 regulates effector and regulatory T cells [58]. One of the mechanisms by which CTLA-4 regulates T cell tolerance is by increasing the T cell activation threshold. The minimum immune response resulting from the increased activation threshold of T cells against self-peptide (s) is favorable for host immunity [58]. However, CTLA-4 function is crucial for T cell homeostasis and, therefore, optimal expression of CTLA-4 is important for satisfactory therapeutic outcomes. Genetic loss of CTLA-4 induces uncontrolled T cell proliferation, leading to health complications and even death [59].
Another T cell co-inhibitory axis, PD1/PD-L1, is also associated with the regulation of central tolerance. This axis regulates the transition of thymocytes from the thymic double-negative (DN) to double-positive phase (CD4+CD8+) of T cells. This transition is promoted by loss of PD-1, suggesting that PD-1 is involved in the regulation of the T cell repertoire [60]. Naïve T cells released from the thymus to the peripheral, spleen, and lymphoid organs interact with APCs, presenting foreign antigenic peptide(s) or tumor antigen(s). Sometimes, naïve T cells react against self-peptides because some TCRs recognize the self-protein [61]. To prevent such T cell autoreactivity, several co-inhibitory molecules inhibit T cell activation, known as peripheral tolerance [62]. These co-inhibitory molecules, including CTLA-4 and PD-1, regulate T cell activation in different stages. CTLA-4 and PD-1 differentially regulate CD4+ and CD8+ T cell phenotypes, respectively [59]. CTLA-4 regulates T cell activation at early stages in lymphoid tissues, whereas PD-1 regulates T cell activation at late activation stages in peripheral tissue [63]. One of the mechanisms of peripheral immune tolerance is reported to be via the interaction between PD-L1 on APCs and PD-1 on T cells that attenuate the T cell immune response against self-antigen(s) [64]. Other peripheral tolerance mechanisms include T cell exhaustion and an increase in Treg cell population [65]. Treatment with an anti-CTLA-4 antibody in a tumor model showed activation of effector T cell function, promoted Treg depletion, and enhanced the antitumor immune response and tumor regression [66,67]. Although CTLA-4 depletes Tregs in TME, anti-CTLA-4 is not associated with Treg depletion in the peripheral system. The peripheral depletion of Tregs is also associated with the presence of the Fc receptor on host cells. Therefore, the efficacy of anti-CTLA-4 antibody-based immunotherapy in inflamed tumors is potentially dependent on Fc gamma receptor (FcγR) binding [68]. Considering the differential regulatory role of PD-1 and CTLA-4 in immune tolerance, their combined blockade is suggested to provide a significant antitumor response by increasing the effector:suppressor immune cell ratio [69].
The blockade of the PD-1/PD-L1 T cell co-inhibitory axis has been targeted to enhance CD8+ T cell infiltration into tumors and restore antitumor immune responses [70]. PD-1 differentially affects CD8+ T cell effector function in different cancer types [71–74]. For example, in certain breast cancer subtypes, regardless of PD-1 expression on CD8+ T cells, the effector function is unaltered [74]. By contrast, in melanoma, the presence of PD-1 on CD8+ T cells decreases the T cell effector function [75,76]. Similarly, blockade of another T cell co-inhibitory molecule, CTLA-4, effectively promotes tumor regression by restoring the cytotoxic function of tumor-infiltrating CD8+ T cells in multiple cancers [77,78]. In CT26 colon cancer and ID8-VEGF ovarian cancer animal models, PD-1 demonstrated a regulatory role in Foxp3+ Treg and CD8+ T cell function, whereas increased CTLA-4 expression was associated with dysfunction of CD8+ T effector cells. CD8+ T cell dysfunctions, especially their proliferation and the property to release cytokines, were severely hampered in PD-1+CTLA-4+CD8+ T cells compared with either PD-1+CD8+ or CTLA4+CD8+ T cells. These results suggest that combined overexpression of PD-1 and CTLA-4 on CD8+ T cells adversely impacts CD8+ T cell-mediated antitumor immune responses during cancer therapy [78]. The members of the innate immune system, DCs and NK cells, and members of the adaptive immune system, T helper cells (CD4+ T cell subset), facilitate the priming of CD8+ T cells for a tumor antigen. PD-1 and CTLA-4 restrict CD8+ T cell priming and, thus, increase the exhaustion of tumor-infiltrating CD8+ T cells [79]. In addition, PD-1 and CTLA-4 can crosstalk with other proteins to render CD8+ T cells dysfunctional. Vascular endothelial growth factor (VEGF) and semaphorin receptor, neuropilin-1 (Nrp-1), are overexpressed on tumor-infiltrating CD8+ T cells and, together with PD-1 or CTLA-4, adversely affect T cell functions [80]. In human lung cancer, Nrp-1 is present on CD8+PD-1+ cells (i.e., Nrp1+PD-1+CD8+ T cells) and interacts with semaphorin 3A on tumor cells. The interaction of Nrp-1 with semaphorin 3A resulted in attenuation of CD8+ T cell migration and cytotoxicity. In the B16F10 melanoma model, CD8+ T cells, expressing Nrp1 and PD-1 (i.e., Nrp1+PD-1+CD8+) are present in the tumor and exhibit an exhausted state, as evidenced by elevated expression of LAG-3, Tim-3, and CTLA-4 [80].
The infiltration of CD8+ T cells into tumors is also regulated by WNT/β-catenin signaling. In human metastatic melanomas, upregulation of intrinsic WNT/β-catenin signaling is associated with the absence of a T cell gene signature [81]. Similarly, in the autochthonous mouse melanoma model, increased tumor-intrinsic WNT/β-catenin signaling induced T cell depletion and resistance towards anti-PD-1 or anti-CTLA-4 therapy [81].
The blockade of PD-1 has shown limited efficacy in the clinic for some cancers types. Therefore, fully understanding the molecular mechanisms mediated by checkpoint inhibitors will allow new strategies to be developed to restore CD8+ T cell functions. Thus, other pharmacological agents have been used to increase the efficacy of checkpoint inhibitors. For example, PD-1 blockade alone in CT26 colon cancer, TSA mammary adenocarcinoma, and MCA38 colon cancer mouse models did not show significant therapeutic benefit. However, the combined treatment of PD-1 and intratumoral injection of the highly interferogenic toll-like receptor 9 (TLR9) agonist, SD101, showed measurable antitumor responses by intratumoral CD8+ T cells [82]. In a pancreatic cancer mouse model, TGF-β in the TME impeded the efficacy of combined treatment of gemcitabine and anti-PD-1. However, this combined treatment increased intratumoral cytotoxic CD8+ T cells and reduced tumor burden when TGF-β was blocked [83]. The US Food and Drug Administration (FDA) has approved inhibitory antibodies against CTLA-4 (i.e., Ipilimumab), PD1 (i.e., nivolumab, pembrolizumab, and cemiplimab), and PD-L1 (i.e., atezolizumab, avelumab, and durvalumab). Table 2 provides examples of clinical trials for solid tumors, targeting T cell co-inhibitory or co-stimulatory proteins.
Table 2.
Co-stimulatory/co-inhibitory molecule-based immunotherapy to increase tumor-infiltrating CD8+ T cellsa
| Target | Immunotherapeutics | ClinicalTrials.gov identifier | Cancer | Refs |
|---|---|---|---|---|
| PD-1 | Nivolumab, pembrolizumab, cemiplimab | NCT00730639, NCT02142738, NCT04154943 | Advanced or recurrent malignancies, metastatic NSCLC, skin cancer stage II-IV, cutaneous squamous cell carcinoma | [71,72] |
| PDL-1 | Atezolizumab, avelumab | NCT02031458, NCT01772004 | Advanced or metastatic NSCLC, metastatic or locally advanced solid tumors | [73,74] |
| CTLA-4 + PD-1 | Ipilimumab + nivolumab | NCT02970981 | Melanoma | |
| CD70 | ARGX-110 | NCT02759250, NCT01813539 | Nasopharyngeal carcinoma, advanced malignancies | [113] |
| ICOS + PD-1 | GSK3359609+Pembrolizumab | NCT04128696 | Recurrent or metastatic head and neck squamous cell carcinoma | [125] |
| GITR | INCAGN01876 | NCT03126110 | Advanced or metastatic malignancies | |
| OX40 | PF-04518600 | NCT02315066 | Advanced or metastatic carcinoma | |
| 4-1BB | PF-05082566, BMS-663513 | NCT01307267, NCT01471210 | Solid tumors or B cell lymphomas; advanced and/or metastatic solid tumors and relapsed/refractory B cell nonHodgkin’s lymphoma | [114,115] |
| CD39 | IPH5201 | NCT04261075 | Advanced solid tumors | |
| CD73 | MEDI9447 | NCT03611556 | Metastatic pancreatic cancer |
Abbreviation: CD, cluster of differentiation.
T cell exhaustion is the dysfunction or physical elimination of T cells in response to a specific antigen(s) during chronic diseases, including viral infection and cancer [84]. T cells undergoing exhaustion lose their proliferation capacity and effector functions, and their memory recall is compromised [85]. Exhausted T cells are characterized by increased expression of co-inhibitory receptors, including PD-1, CTLA-4, lymphocyte activation gene-3 (LAG3; CD223), T cell immunoglobulin and mucin domain-3 (TIM-3), CD39, CD96, CD160, T cell immunoreceptor with Ig and ITIM domains (TIGIT), 2B4 (CD244), and B and T lymphocyte attenuator (BTLA) [85]. The therapeutic use of immune checkpoint inhibitors of PD-1 and other molecules might be able to reinvigorate exhausted T cells and provide clinical benefit to patients with various malignancies (Table 3) [86–107]. Unfortunately, outcomes of PD1-based checkpoint blockade are not effective for all patients, and T cell exhaustion is not always reversed in patients with advanced malignancies [108]. Recently, tumor-reactive TILs have been reported that have hallmarks of exhausted and memory cells along with the expression of PD-1 and T cell factor 1 (Tcf1). PD1+TCf1+ TILs are associated with immunotherapy-induced proliferation [109]. In some patients with cancer, the expression of PD-1 is higher on activated and functional CD8+ T cells. Therefore, anti-PD1 antibody-based immunotherapy is not effective in these patients to reinvigorate exhausted T cells [110]. Concurrently, a study in syngeneic mouse tumor models showed that PD1 blockade-based immunotherapy could not recover dysfunctional PD-1+CD38+CD8+ T cells [111]. The failure of checkpoint blockade-based immunotherapy to reinvigorate exhausted T cells could be associated with the intrinsic and extrinsic mechanisms of resistance. The mechanism of T cell exhaustion is different in different cancers. For example, a study in melanoma (B16F10), breast (E0771), and lung carcinoma (LLC) cell lines suggested that each tumor has a characteristic and distinct TIL exhaustion signature [88]. As an advance in the field of T cell exhaustion and reinvigoration, a physiologically relevant high-throughput assay to identify the modulators of T cell exhaustion has been reported that can be used to screen small molecules that can reverse T cell exhaustion in the lymphocytic choriomeningitis virus model (LCMV) [112]. It is believed that a rationalized approach using a combination of small-molecule and checkpoint inhibitors could be used to reinvigorate exhausted CD8+ T cells in cancer immunotherapy.
Table 3.
Drug targets that can revert CD8+ T cell exhaustion in cancera
| Target | Current development stage | Cancer types where these inhibitory receptors are expressed on CD8+ T cells | Refs |
|---|---|---|---|
| PD1 | In clinical use | Melanoma and RCC, NSCLC, glioblastoma | [86–88] |
| CTLA-4 | In clinical use | Ovarian cancer | [89] |
| LAG-3 | In clinical trails | Fibrosarcoma, melanoma, colorectal adenocarcinoma, glioblastoma | [88,90] |
| TIM-3 | In clinical trails | RCC, acute myelogenous leukemia, gastric cancer | [91–93] |
| TIGIT | In clinical trails | Follicular lymphoma, colon carcinoma, lung cancer, glioblastoma | [88,94–96] |
| BTLA | In clinical trails | Metastatic melanoma, hepatocellular carcinoma | [97,98] |
| CD39 | In clinical trails | Glioblastoma | [88] |
| VISTA | In clinical trials | Squamous cell carcinoma, human ovarian and endometrial cancer, colon cancer | [99–101] |
| 2B4 | Preclinical stage | Pancreatic adenocarcinoma, lung cancer | [102,103] |
| CD96 | Preclinical stage | Colon carcinoma, fibrosarcoma, melanoma, lung adenocarcinoma | [104,105] |
| CD160 | Preclinical stage | Pancreatic cancer, lung cancer | [96,107] |
Abbreviations: CD, cluster of differentiation; LAG-3, lymphocyte-activation gene 3; TIM-3, T-cell immunoglobulin and mucin domain-3; VISTA, V-type immunoglobulin domain-containing suppressor of T cell activation.
Targeting the T cell co-stimulatory axis in cancer therapy
In addition to clinical trials with anti-PD-1 and anti-CTLA-4 agents, other trials targeting T cell co-stimulatory proteins are ongoing (Table 2). Examples of T cell co-stimulatory molecules that can be targeted to restore T cell functions are 4-1BB, OX40, CD40, CD27, CD70, inducible T-cell co-stimulator (ICOS), and glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR) [113–115]. The activation of 4-1BB, a TNFR family member, increases the cytotoxicity of CD8+ T cells via the induction of IFN-γ [116]. Clinical trials with the agonistic monoclonal antibodies (mAbs) utomilumab (PF-05082566) and urelumab (BMS-663513) that activate 4-1BB are ongoing for multiple cancers [117]. Although clinical trials have shown robust anti-tumor responses, considerable toxicity has also been observed because of FcγR interactions with nonspecific targets. To reduce/overcome the unacceptable toxicity of 4-1BB agonistic mAbs, an Fc-free tumor-targeted 4-1BB-agonistic antibody was used that delivered better therapeutic efficacy [118]. OX40 is another T cell co-stimulatory molecule on the surface of activated T cells. The interaction between OX40 and its ligand, OX40L, promotes T cell expansion and Treg suppression to increase antitumor responses [119]. Given the central role of OX40 in the regulation of CD8+ T cell effector function, an anti-OX40 agonistic mAb is under clinical trials for cancer. 4-1BB and OX40 agonistic mAbs have also been demonstrated to restore MEK inhibitor-induced T cell dysfunction [120]. Another T cell co-stimulatory axis, CD40L-CD40, has also been reported in supporting MEK inhibitor-induced cancer cell death in animal models. In the mutant KRas-driven pancreatic cancer animal model, a combination of a MEK inhibitor and an agonistic anti-CD40 mAb showed robust antitumor responses [121]. The CD27-CD70 axis is also important in T cell activation, and increased expression of CD70 and CD27 is reported both in hematological and solid cancers [113,122]. In papillary thyroid cancer, CD70 expression was detected on tumor cells, whereas the expression of the CD70 receptor, CD27, was mainly on intratumoral lymphocytes [123]. GITR is another co-stimulatory molecule that takes part in T cell activation. The combined inhibition of PD-1 and GITR synergistically increased T cell tumor infiltration and durable anti-tumor responses by CD8+ T cells [124]. ICOS is an additional co-stimulatory molecule present on activated T cells and has also been targeted in cancer [125]. Interestingly, the combined use of ICOS aptamer and CTLA-4 inhibition demonstrated strong antitumor responses by cytotoxic T cells [126]. Given the importance of T cell co-stimulatory molecules in cancer immunotherapy, current onco-immunology research has focused on finding novel therapeutics to trigger T cell co-stimulatory pathways.
Several preclinical studies and clinical trials are underway to test the therapeutic efficacy of co-inhibitory or costimulatory immune receptors. However, the FDA has so far approved only anti-PD1/PD-L1 and anti-CTLA-4 antibody-based cancer immunotherapy for clinical use in some advanced malignancies. Most of the immunotherapeutic agents that target co-stimulatory molecules are not yet clinically approved. There could be several reasons for the delayed use of agonistic antibodies in clinics, including traditional bivalent antibody format and co-stimulatory molecules on Tregs [127]. In the traditional bivalent antibody format, differential Fc gamma receptors (FcγR) are a major concern. FcγR regulates antibody-induced activation of effector T cells and antitumor response [127]. As an example, to achieve the antitumor activity of the 4-1BB agonistic antibody requires FcγR-dependent Treg depletion and FcγR-independent 4-1BB agonism [128]. One of the primary reasons for the failure of agonistic antibodies against co-stimulatory molecules is because Tregs express several co-stimulatory molecules [128]. Conceivably, co-stimulatory agonistic antibodies might prove an effective therapeutic option in cancer; however, a detailed mechanistic understanding will be necessary for their clinical application.
CAR-T cell therapy in cancer
The adoptive transfer of engineered T cells has shown clinical benefits to some patients with cancer. In hematological cancer, the adoptive transfer of cytotoxic lymphocytes targeting tumor cells has been successful [129]. TCRs can be manipulated to improve their survival and effector function. Therefore, CAR-T cells bearing specificity against tumor antigens have been developed and have shown promising results in hematological cancers. However, CAR-T cells to treat solid cancers are still in early stages, with no success thus far. The primary reason for their failure against solid tumor is because the immunosuppressive environment within TME favors tumor growth by promoting CD8+ T cell dysfunction. CAR-T cells in TME are pushed to become nonfunctional and express T cell co-inhibitory surface molecules [130,131]. Therefore, attempts are also being made to improve the adoptive transfer of TILs that target neoantigens in various malignancies [132].
The other reasons for the failure of CAR-T cell therapy in solid tumors are thought to include desmoplastic components (i.e., fibrous or connective tissue), which prevent CAR-T cells from entering tumors. It has been reported that mild hypothermia abolishes desmoplastic integrity in tumors. The inclusion of mild hyperthermia and CAR-T cell therapy resulted in increased T cell infiltration into human melanoma tumors [133]. Similar to mild hypothermia, cytokines and chemotherapeutic agents have also been used to improve CAR-T cell infiltration of various tumors. For example, combined treatment of IL-12 with the chemotherapeutic agent doxorubicin enhanced the infiltration of CD8+ T cells into tumors, leading to robust anticancer effects [134]. Another approach to improve the efficacy of CAR-T cell therapy in cancer could be the combined use of CAR-T cells along with blockade of T cell co-inhibitory molecules. Several clinical trials for the application of CAR-T cells alone or in a combination with CPI are ongoing in solid tumors. Table 4 provides examples of clinical trials of CAR-T cells against solid tumors. In several current CAR-T clinical trials, adverse effects have been reported because of adoptive transfer. Therefore, new approaches to using CAR-T cell-based immunotherapy with reduced adverse effects will be essential.
Table 4.
Clinical trials of CAR-T cell therapy in cancer
| Target | ClinicalTrials.gov identifier | Cancer |
|---|---|---|
| TM4SF1 and EpCAM | NCT04151186 | Epithelial-derived solid tumors |
| c-Met/PD-L1 | NCT03672305 | Primary hepatocellular carcinoma |
| GPC3-T2 | NCT03198546 | Hepatocellular carcinoma |
| CTLA-4, PD-1, and MUC1 | NCT03179007 | Advanced solid tumor |
| NKG2D | NCT04270461 | Solid tumors |
| PD-1 and mesothelin | NCT03747965 | Solid tumors |
| CEA | NCT03682744 | Adenocarcinoma, peritoneal metastases, or malignant ascites |
| B7–H3 | NCT04185038 | Diffuse intrinsic pontine glioma/diffuse midline glioma and recurrent or refractory pediatric central nervous system tumors |
| MUC1 | NCT03525782 | NSCLC |
| EGFR | NCT03618381 | Recurrent/refractory solid tumors |
Abbreviations: CD, cluster of differentiation; CEA, carcinoembryonic antigen; EpCAM, epithelial cell adhesion molecule; TM4SF1, transmembrane 4 L six family member 1; c-Met, tyrosine-protein kinase Met or hepatocyte growth factor receptor (HGFR); GPC3, glypican 3; MUC1, Mucin 1; NKG2D, natural killer group 2 member D.
Cancer therapy using TCR-transduced T cells
CAR-T cell therapy targets surface molecules of immune cells and cannot target intracellular peptide(s) presented by major histocompatibility complexes (MHCs). Therefore, to target intracellular peptides presented by MHCs, TCR-transduced T cells have been developed [135]. These cells are generated from patient T cells and are designed to encode the TCRαβ protein, which targets tumor or virus-infected cells [136]. The large-scale production of TCR-transduced cells is achieved using semiautomated devices and modular systems [136]. TCR-transduced T cells showed prolonged survival (i.e., >18 months) and activation even after the secondary encounter with antigen(s) [137]. Several tumor antigens have been used to generate TCR-transduced T cells, including trophoblast glycoprotein (TPBG) in renal cancer [138], placenta-specific 1 (PLAC1) in breast cancer [139], nucleophosmin 1 (NPM1) in acute myeloid leukemia (AML) [140], NY-ESO-1 in synovial cell sarcoma, in patients with melanoma and common epithelial tumors [141], and MAGE-A4 in esophageal cancer [142], and effectively demonstrated antitumor responses in preclinical studies. Diffuse intrinsic pontine glioma is a lethal cancer prevalent in children. In >70% of diffuse intrinsic pontine glioma cases, an amino acid substitution from lysine (K) to methionine (M) at position 27 of histone 3 variant 3 (H3.3) has been reported. Interestingly, TCR-transduced T cells against synthetic peptides with H3.3K27M mutation were proposed to treat HLA-A2+H3.3K27M+ glioma cells [143]. Clinical trials are underway to validate the therapeutic efficacy of TCR-transduced T cell-based cancer therapy in patients with various malignancies, including metastatic synovial cell sarcoma, melanoma, and esophageal cancer [141,142]. Taken together, TCR-transduced T cell therapy is a powerful therapeutic tool with the potential to improve T cell-mediated antitumor responses.
Cancer therapy using TILs
CAR and TCR-transduced T cell-based cancer immunotherapies are useful to target surface and intracellular antigens; however, an adoptive transfer of TILs has been proposed that can specifically target tumor antigens [144]. In TIL-dependent cancer therapy, the initial step involves the identification of mutated protein in the tumor. The mutated protein is then inserted into autologous APCs, such as DCs. Further steps include co-culture of autologous TILs with antigen-loaded APCs and selection of antigen-specific TILs. Their expansion follows the selection of antigen-specific TILs. The expanded T cells are used for transfusion in the donor patients [132]. The efficacy of TIL-based immunotherapy has been appreciated in various cancers, including metastatic melanoma and metastatic human papillomavirus (HPV)-associated carcinomas, ovarian and breast cancers [145–148]. Several clinical trials are underway to establish the antitumor efficacy of TIL-based cancer immunotherapy alone or in combination with other anticancer agents in patients with different malignancies, including metastatic head and neck squamous cell carcinoma (IDNCT03083873), cervical carcinoma (IDNCT03108495), ovarian cancer, anaplastic thyroid cancer, osteosarcoma, or other bone and soft tissue sarcomas (IDNCT03449108). Taken together, TIL-based cancer therapy has the potential to treat the advanced stage of various undruggable solid malignancies.
Emerging modulators of CD8+ T cell function
Role of protein kinases in CD8+ T cell infiltration of tumors
The CD8+ T cell effector function is regulated by various factors, such as antigen receptors, co-stimulatory molecules, cytokines, and protein kinases [149]. Interestingly, several upstream MAPKs have been reported to be mutated or upregulated in multiple cancers [77,150,151]. However, attempts to inhibit upstream and downstream MAPKs have not been successful because of ensuing drug resistance and their impact on suppressing the immune compartment. The primary reason for the failure of MAPK inhibitors is a lack of understanding of the effects of MAPK inhibition on CD8+ T cell function and survival. Several examples suggest that understanding the roles of protein kinases in T cell biology will help strategize augmenting antitumor responses, perhaps with an additional agent(s). As an example, lymphocyte function-associated antigen-1 (LFA-1) mediates T cell activation via ERK1/2 signaling upon TCR engagement [152]. Furthermore, intercellular adhesion molecule-1 (ICAM-1) ligation with LFA-1 increases ERK1/2 signaling to enhance CD8+ T cell activation [152]. Jun N-terminal kinase (JNK), a MAPK, has a paradoxical role in the regulation of CD4+ and CD8+ T cells [153]. A study in vivo using B16, a melanoma cell line, and EL-4, a lymphoma cell line, showed that JNK1-knockout mice were susceptible to tumor growth, specifically because of CD8+ T cell dysfunction [153]. Mutations in the BRAF proto-oncogene, a serine/threonine kinase, has an essential role in oncogenesis. A study showed that melanoma cells treated short term (3–7 days) with a BRAF inhibitor were efficiently recognized by autologous CD8+ T cells (TILs). However, melanoma cells treated long term (14–21 days) with BRAF inhibitor were weakly recognized by autologous CD8+ T cells (TILs). These results suggest that BRAF inhibition differentially modulates tumor antigen expression and mediates tumor resistance, perhaps by decreasing functional CD8+ T cells [154]. Another study using BRAF and MEK inhibitors (dabrafenib and trametinib, respectively) showed a differential impact on CD8+ T cells [77]. Dabrafenib treatment in human activated T cells (in vitro), induced ERK phosphorylation (i.e., activation) and did not suppress CD8+ T cell function. However, trametinib treatment decreased ERK phosphorylation, T cell proliferation, and cytokine expression [77]. However, combining trametinib with checkpoint inhibitors for PD-1 or CTLA-4 increased CD8+ T cell infiltration and promoted a robust antitumor response in a CT26 tumor model [77]. T cell activation is also reported to induce TCR-ZAP-70 protein tyrosine kinase-mediated downstream signaling, which regulates T cell motility, adhesion, and cytokine production. Rasal1, a GTPase-activating protein, binds with the TCR-ZAP-70 binding protein and inhibits the Ras-ERK pathway, T cell activation, and antitumor responses [155]. Rasal1 is expressed on activated T cells, and its knockdown induced CD8+ T cell infiltration in B16 melanoma and EL-4 lymphoma tumors [155]. Upon TCR-mediated activation, tumor-infiltrating CD8+ T cells showed reduced expression of ERK and the protein tyrosine kinase, ITK. However, in splenic CD8+ T cells, there were no such alterations in ERK and ITK kinase expression, suggesting that ERK and ITK kinases are differentially regulated in peripheral and tumor-infiltrating CD8+ T cells [156]. Sorafenib, a multikinase inhibitor and first-line therapy for hepatocellular carcinoma (HCC), can provide limited benefit to patients with HCC. The success of sorafenib treatment was associated with the activation status of ERK in HCC [157]. The analyses of human and mouse HCC samples treated with sorafenib demonstrated two heterogeneous phenotypes of active ERK and PD-1 within the same tumor. The first phenotype was pERKlowPD-1+, where tumor infiltration of CD8+ T cells was very high and these CD8+ T cells had higher expression of PD-1. The second phenotype, pERKhighPD-1−, had low expression of PD1. Interestingly, the pERKlowPD-1+ phenotype showed decreased overall and disease-free survival compared with the pERKhighPD-1− phenotype. Importantly, the pERKhighPD-1− phenotype had CD8+ T cells with low expression of PD-1, suggesting that the status of PD-1 on tumor-infiltrating CD8+ T cells is a deciding factor for antitumor responses [158]. MAP2K (or MEK) is an upstream regulator of MAPK. The targeted inhibition of MEK induced tumor suppression and the increased lifespan and infiltration of CD8+ T cells; however, MEK inhibition attenuated T cell priming in lymph nodes [151]. In a CT26 tumor model in BALB/c mice, combined therapy of anti-PD-L1 with a MEK inhibitor showed robust and durable antitumor efficacy, including complete remission of tumors in some mice [151]. Another MAP3K family member, Mixed Lineage Kinase-3 (MLK3), has a paradoxical role in the regulation of cancer cell death [159] and inhibits T cell activation and cytotoxicity [160]. Using a murine breast cancer cell line, 4T1 xenograft model in Balb/c mice, inhibition of MLK3 was shown to induce tumor infiltration of cytotoxic CD8+ T cells [160]. MLK3 inhibition also increases CD70 expression on CD8+ T cells, associated with increased apoptosis of these cells [161]. Interestingly, in a TNBC in vivo model, combined treatment with a MLK3 pharmacological inhibitor and CD70 antagonist promoted CD8+ T cell survival, tumor infiltration, and antitumor efficacy [161]. MAP4K4 is also an upstream regulator of MAPKs. The upregulated expression of MAP4K4 inhibited LFA-1 during T cell activation, whereas ablation of MAP4K4 increased LFA-1 expression on CD8+ T cells. The increased LFA-1 expression on T cells was associated with increased T cell interaction with APCs, which increased the CD8+ T cell effector function [162].
The receptor tyrosine kinase (RTK) receptor family member, epidermal growth factor receptor (EGFR), is overexpressed in epithelial cancer cells. A study carried out to understand how EGFR tyrosine kinase inhibitors (TKIs) influence significant MHC-I in NSCLC cells showed that inhibition of EGFR in NSCLC increased the gene and surface protein expression of MHC-I. T cells recognize tumor antigens presented by the MHC-I molecule on APCs and deliver an antitumor response. The ERK-MEK inhibitor, trametinib, was able to increase MHC-I, whereas the phosphatidylinositol 3-kinase inhibitor buparlisib was incapable of inducing MHC-I expression in NSCLC. These results suggest that MEK-ERK inhibition induces EGFR-activated MHC-I expression [163].
The Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathway regulates a range of cytokines and growth factors. However, JAK2 inhibitors in TNBC did not show any efficacy, perhaps because the TNBC cells acquired platelet-derived growth factor receptor beta (PDGFRβ)-mediated drug resistance. To overcome failure of the JAK2 inhibitor as a single agent, triple combination therapy with PDGFRβ, JAK2, and MEK1/2 inhibitors increased tumor-infiltrating CD8+ T cells significantly [164]. The JAK/STAT pathway is attenuated by cytokine-inducible SH2-containing protein (CISH) [165]. Downregulation in JAK/STAT signaling induced TCR-mediated activation of tumor-infiltrating CD8+ T cells [165,166]. Interestingly, induction of CISH is involved in granulocyte-macrophage colony-stimulating factor (GM-CSF)-mediated DC development, and DC-mediated cytotoxic T cell activation [165]. Therefore, it is clear that protein kinases have a crucial role in CD8+ T cell tumor infiltration, survival, and antitumor responses. Hence, a proper understanding of protein kinases could lead to the management of different types of cancer.
Metabolic factors regulating CD8+ T cell infiltration of tumors
Metabolism is the sum of the biochemical reactions within living cells that provide the necessary energy for vital cellular processes. Cellular metabolism allows T cells to acquire and utilize energy necessary for their survival, proliferation, and function. The alteration of metabolism in the TME significantly affects TIL survival and antitumor responses. Similarly, the survival and effector function of tumor-infiltrating CD8+ T cells is directly associated with metabolic factors in the TME [167–169]. In activated T cells, metabolic reprogramming induces glycolytic flux and lactate, lipid, and protein production [170]. However, impaired glucose metabolism is a major issue that adversely affects tumor-infiltrating CD8+ T cells. Effector T cells require a healthy glucose metabolism; however, lack of necessary nutrients and other factors impairs glucose metabolism in tumor-infiltrating CD8+ T cells. In clear cell renal cell carcinoma (ccRCC), tumor-infiltrating CD8+ T cells are abundant. However, they are deficient in their effector functions because of perturbed metabolism. Further analyses suggested that tumor-infiltrating CD8+ T cells in ccRCC were unable to uptake glucose for glycolysis. As a result, the mitochondria in CD8+ T cells were fragmented, hyperpolarized, and unable to generate robust reactive oxygen species (ROS) because of inhibition of mitochondrial superoxide dismutase 2 (SOD2) [171]. In human and mouse melanomas, insufficiency in glycolytic metabolism and oxidative phosphorylation (OXPHOS) in tumor-infiltrating CD8+ T cells is also reported [168].
Rapidly proliferating tumor cells consume most of the nutrients and, therefore, creates a nutrient-insufficient environment for tumor-infiltrating CD8+ T cells. In the pro-TME, infiltrating CD8+ T cells also lack or have dysfunctional metabolic enzyme activities. For example, in melanoma, tumor-infiltrating CD8+ T cells had decreased activity of the glycolytic pathway enzymes, ENOLASE 1 (i.e., alpha-enolase) and phosphoenolpyruvate (PEP) [168]. ENOLASE 1 is an upstream enzyme responsible for PEP production in the glycolytic pathway. Furthermore, PEP is a substrate for pyruvate synthesis in glycolysis. Therefore, when ENOLASE1 activity is low, the supply of pyruvate increases CD8+ T cell effector function via the upregulation of glycolysis. Pyruvate is involved in glycolysis and OXPHOS reactions; therefore, the supply of pyruvate increases the function of tumor-infiltrating CD8+ T cells [168]. The active metabolism of PEP promotes calcium ion (Ca2+)-nuclear factor of activated T cells 1 (NFATc1) signaling to induce T cell effector function [172]. Molecular studies in a melanoma model showed that PEP decreases sarco/ER Ca2+-ATPase activity to promote Ca2+-NFATc1 signaling in T cells [172]. Therapeutically, phosphoenolpyruvate carboxykinase 1 has been targeted to increase phosphoenolpyruvate in T cells to increase CD8+ T cell effector function [172]. The aberrant increase in the glycolytic gate-keeping enzyme, pyruvate dehydrogenase kinase 1 (PDK1), was reported to have a protumor role and to adversely regulate the survival and effector functions of tumor-infiltrating CD8+ T cells [169]. In ovarian cancer, PDK1 regulates PD-L1 through activation of JNK activity. The overexpression of PDK1 can enhance the PD-1–PD-L1 axis in CD8+ T cells and attenuate its effector function [169]. Given the crucial role of glucose metabolism in tumor-infiltrating CD8+ T cell function, several direct or indirect approaches have been proposed to restore normal glucose metabolism in tumor-infiltrating CD8+ T cells. As an example, acetate, a monocarboxylic acid anion, has been suggested to overcome restricted glucose metabolism in tumor-infiltrating CD8+ T cells [173]. The positive influence of acetate treatment has also been reported in acetyl-coenzyme A (acetyl-CoA) metabolism, lipogenesis, and protein acetylation [173,174].
Amino acids have a crucial role in T cell differentiation and functions. For example, glutamine metabolism, an α-amino acid, has a regulatory role in successful adoptive immunotherapy [175]. A study using a tumor-inoculated mouse model showed that the adoptive transfer of CD8+ T cells with attenuated glutamine metabolism promoted tumor regression and overall survival. The attenuation of glutamine metabolism induced CD8+ T cell proliferation and survival via downregulation of PD-1 expression on CD8+ T cells [175]. Another α-amino acid, L-arginine, improves T cell metabolism, tumor infiltration, and antitumor immune responses [176,177]. In a study where ovalbumin-expressing B16 melanoma cells were orthotopically implanted in C57BL/6 mice, adoptive transfer of L-arginine-treated OT-1 T cells (specific for ovalbumin antigen) significantly induced antitumor responses and increased the survival of mice [177]. In general, DCs are involved in antitumor responses; however, in a study of the BALB/NeuT mammary carcinoma model, a phenotype (i.e., MHC II+/CD11b+/CD11chigh) of tumor-infiltrating DCs (i.e., TIDCs) decided the suppression of CD8+ T cell-mediated antitumor responses [176]. Interestingly, arginase metabolism by L-arginine regulates TIDC-mediated CD8+ T cell function and survival [176]. Apart from glutamine and L-arginine, several other amino acids are also involved in regulating tumor-infiltrating CD8+ T cell functions. Therefore, a thorough assessment of these amino acids within TME could help strategies for effective cancer immunotherapy.
Fatty acid catabolism has a significant role in tumor-infiltrating CD8+ T cell effector function and T cell-mediated antitumor immune responses [178]. In a mouse model of melanoma, because of concurrent hypoglycemic and hypoxic conditions within tumors, the expression of peroxisome proliferator-activated receptor-α (PPAR-α) and fatty acid catabolism increased in tumor-infiltrating CD8+ T cells [178]. To survive in a hypoxic TME, tumor-infiltrating CD8+ T cells adapt to increase hypoxia-inducible factor (HIF) transcription factors, specifically HIF-1α, but not HIF-2α [179]. The nucleoside, adenosine, is a component of DNA and RNA. Interestingly, its accumulation is involved in tumor growth and progression and impairs the effector function of CD8+ T cells. Further analysis demonstrated that adenosine attenuates central memory T cell generation via the A2A receptor-mediated pathway in peripheral and tumor-infiltrating T cells [180]. Besides the metabolic factors discussed earlier, additional metabolic factors regulate tumor-infiltrating CD8+ T cell survival and function. A detailed understanding of all these metabolic factors could help to restore the antitumor functions of tumor-infiltrating CD8+ T cells.
Toll-like receptor agonists in cancer immunotherapy
TLRs are involved in the regulation of immune activation upon pathogenic encounter. TLRs can act as natural immune modulators and, therefore, its potential could be exploited in cancer immunotherapy [181]. TLRs recognize pathogen-associated molecular patterns (PAMPs), which are expressed by pathogens and also associated with endogenous damage-associated molecular patterns (DAMPs), which are released from dying and stressed cells [182]. Given the immune-stimulatory nature of TLRs, TLR agonists have been implicated in cancer immunotherapy to boost host immunity against tumors [183]. TLR agonists are reported to promote immune cell activation in TME and to attenuate tolerance and immune-inhibitory signaling [184]. The role of several TLRs, including TLR1/2, TLR3, TLR4, TLR5, TLR7, TLR8, and TLR9 ligands, are well established in enhancing CD8+ T cell antitumor efficacy [185–190]. TLR1/2 and 7 decrease PD1 expression on CD8+ T cells, leading to increased antitumor efficacy and tumor suppression [185]. The regulatory function of TLR1/2 has been reported in 4-1BB-mediated activation of CD8+ T cells and antitumor responses [191]. The agonist of TLR1/2, diprovocim, improved antitumor CD8+ T cell efficacy during anti-PD-L1-based cancer immunotherapy [192]. In a murine model of leukemia, small-molecule agonist 23 (SMU-Z1) induced TLR2 via association with TLR1, which promoted the proliferation of CD8+ T cells [193]. This TLR2 agonist was also associated with the human DC phenotype CD141+, required to induce antitumor effects by CD8+ T cells [194]. TLR3 agonists have also been reported to improve CD8+ T cell effector function along with antibody-mediated cancer immunotherapy [186].
Irreversible T cell exhaustion is associated with the failure of checkpoint inhibitors. Targeting the CD40–TLR4 axis restored T cell effector function in a preclinical cancer model [195]. TLR4 agonists and other cancer therapeutics have been reported to successfully promote CD8+ T cell antitumor efficacy [187,196]. CBLB502, an agonist of TLR5, promoted CD8+ T cell-mediated antitumor responses in graft-versus-tumor models [197]. In colon and mammary metastatic preclinical in vivo models, TLR5 agonists showed organ-specific immuno-adjuvant function independent of tumor antigens [188]. The intratumoral treatment with a TLR7 agonist promoted M1 macrophages in TME and induced CD8+ T cell effector function by increasing IFN-γ expression [198]. An agonist of TLR7/8 (i.e., MEDI9197) promoted localized effects, which led to enhanced CD8+ T cell effector function and antitumor effects [199]. The bispecific agonist of TLR7/8 has been reported in melanoma, bladder, and RCC tumor models. The TLR7/8 bispecific agonist increased DC activation and expansion in lymph nodes, leading to enhanced priming and expansion of cytotoxic CD8+ T cells [200]. TLR9 has been reported to regulate the accumulation, maturation, and lymph node migration of antigen-loaded tumor DCs, which ultimately expand cytotoxic CD8+ T cells, leading to the enhanced killing of tumor cells [201]. Together, these results suggest TLR agonists as potential therapeutic tools to enhance anticancer efficacy in cancer immunotherapy.
STING in cancer immunotherapy
The stimulator of interferon genes (STING) is an intracellular receptor of the endoplasmic reticulum (ER), which is crucial for DNA-mediated immunological reactions [202]. It is also a cytosolic DNA-sensing pathway. The DNA sensor in the cytosol is cyclic GMP-AMP Synthase (cGAS), which activates the innate immune system mainly via IFN-1 expression [203]. STING-induced IFN-1 production promotes DC activation, which ultimately activates CD8+ T cells against tumor antigens [204]. The differential expression of STING has been reported in CRC. Higher STING expression is observed in early stages of cancer and is associated with higher tumor infiltration of CD8+ T cells. The higher expression of STING in CRC is also associated with increased overall survival in patients with cancer. By contrast, lower STING expression is associated with lower infiltration and lower survival of patients [205]. Cyclic dinucleotides (CDNs) are STING agonists that could induce an immune response [206]. In an in vivo study, colon cancer tumors treated with a STING agonist showed a decreased tumor burden and increased expression of ICOS and IFNγ on CD8+ T cells [205]. STING also modulates chemotherapeutic drug efficacy. For example, in BRCA-deficient models of TNBC, the efficacy of PARP-1 inhibitors was dependent on STING-mediated CD8+ T cell antitumor effects [207]. The combined efficacy of STING-based immunotherapy with VEGFR2, PD-1, and CTLA-4 blockade has shown promising outcomes in cancer therapy by improving CD8+IFN-γ+ T cell population and function [208].
Apart from immunotherapeutic agents, STING agonists and other chemotherapeutic drugs have shown promising results. For example, in a preclinical in vivo tumor model, combined treatment with a STAT3 inhibitor and STING agonist promoted CD8+ T cell infiltration, decreased Tregs and MDSCs in TME, leading to an antitumor response [209]. CDNs have some limitations, including barriers to drug delivery and their rapid clearance; therefore, an improved version of CDNs has also been reported. STING-activating nanoparticles (STING-NPs), along with immune checkpoint inhibitors, increase STING signaling in TME and sentinel lymph nodes, leading to enhanced immunogenic and tumoricidal microenvironments [206]. The emerging and promising role of STING agonists might have a promising future in improving CD8+ T cell effector functions, leading to a robust antitumor response.
Concluding remarks
In summary, cancer-induced immunosuppressive environments in the host and TME are key reasons for the failure of numerous antineoplastic therapeutics and, subsequently, drug resistance. Current approaches to cancer treatment are based on treating tumor cells to induce cell death. However, the discovery of checkpoint inhibitors and their efficacies have forced us to consider tumor immunity for a successful treatment plan. Emerging trends suggest that targeting tumor and immune systems have promising therapeutic outcomes, especially in hematological cancer; however, the results of several ongoing clinical trials in patients with solid malignancies are still anticipated. Chemotherapeutic agent-induced TME plasticity is crucial for CD8+ T cell-mediated antitumor responses and, thus, a clear understanding of the drug-induced alterations in TME will be essential to overcome drug resistance. Therefore, basic knowledge of antineoplastic therapeutic-induced changes in cytokines/chemokines, protein kinases, and metabolic factors in the TME will be vital to restoring CD8+ T cell-mediated antitumor responses.
Highlights.
Current cancer therapy targets both the tumor and the immune system.
Tumor microenvironment (TME) regulates the anti-tumor immune responses.
Tumor-infiltrating CD8+ T cells are crucial for tumor cell killing.
Cytokines, kinases, and metabolic factors regulate TME and CD8+ T cell functions.
Tumor infiltration and functions of CD8+ T cells could be restored by immunotherapy.
Acknowledgments
The authors acknowledge Enrico Benedetti for providing access to departmental resources and financial support, Arnav Rana for editing the manuscript, and funding support from the Department of Surgery, Veterans Affairs Career Scientist Award (BX004855), and National Cancer Institute Award (CA 216410) to A.R. B.R. is supported by NCI Award (CA 219764) and Veterans Affairs Merit Award (BX003296).
Author biographies

Sandeep Kumar
Sandeep Kumar has a broad background in immunology and cancer biology, with specific training and expertise in T cell-mediated antitumor responses. During his postdoctoral training at Roswell Park Cancer Institute, Dr Kumar explored role of cytokines/chemokines associated with apoptosis resistance in cancer cells. Currently, Dr Kumar is exploring the role of various MAPK upstream and downstream family members in T cell biology and the implications of these findings for immunotherapy in breast and pancreatic cancers.

Basabi Rana
Basabi Rana received her PhD in biochemistry from the University of Calcutta (now Kolkata), and postdoctoral training from Boston University School of Medicine. She has worked extensively on elucidating the signaling mechanisms that govern WNT/β-catenin signaling and on the effect of TRAIL-PPARγ ligand combination therapies to ameliorate resistance. Current research in her laboratory focuses on understanding the signaling mechanisms of therapy resistance and in designing strategies to overcome these, with specific emphasis on sorafenib resistance in hepatocellular carcinoma.

Ajay Rana
Ajay Rana completed his PhD in biochemistry at the Indian Institute of Chemical Biology and his postdoctoral training at the Harvard Medical School in cell signaling and cancer biology. Dr Rana is well recognized in the cell signaling and breast cancer fields. The current focus of his laboratory is to delineate MAPKs and other related proteins in breast, pancreatic, and liver cancer, with particular emphasis on identifying mechanism-based targeted therapies.

Sunil K. Singh
Sunil Singh received his PhD in life sciences from Jawaharlal Nehru University, New Delhi, India. Currently, he is studying the regulatory function of MAPK upstream regulators in pancreatic and breast malignancies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vacchelli E et al. (2016) Trial Watch–immunostimulation with cytokines in cancer therapy. Oncoimmunology 5, e1115942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kronke J et al. (2014) Lenalidomide induces degradation of IKZF1 and IKZF3. Oncoimmunology 3, e941742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei SC et al. (2017) Distinct cellular mechanisms underlie anti-CTLA-4 and Anti-PD-1 checkpoint blockade. Cell 170, 1120–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buchbinder EI and Desai A (2016) CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol 39, 98–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seidel JA et al. (2018) Anti-PD-1 and Anti-CTLA–4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol 8, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jansen CS et al. (2019) An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 576, 465–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Son SM et al. (2020) Distinct tumor immune microenvironments in primary and metastatic lesions in gastric cancer patients. Sci Rep 10, 14293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trujillo JA et al. (2018) T cell-inflamed versus non-T cell-inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res 6, 990–1000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gajewski TF et al. (2017) Cancer immunotherapy targets based on understanding the T cell-inflamed versus non-T cell-inflamed tumor microenvironment. Adv Exp Med Biol 1036, 19–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans RA et al. (2016) Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight 1, e88328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen DS and Mellman I (2017) Elements of cancer immunity and the cancer-immune set point. Nature 541 (7637), 321–330 [DOI] [PubMed] [Google Scholar]
- 12.Lambrechts D et al. (2018) Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med 24, 1277–1289 [DOI] [PubMed] [Google Scholar]
- 13.Pearce OMT et al. (2018) Deconstruction of a metastatic tumor microenvironment reveals a common matrix response in human cancers. Cancer Discov 8, 304–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valentijn LJ et al. (2012) Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc Natl Acad Sci U S A 109 (47), 19190–19195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Relation T et al. (2018) Intratumoral delivery of interferongamma-secreting mesenchymal stromal cells repolarizes tumor-associated macrophages and suppresses neuroblastoma proliferation in vivo. Stem Cells 36, 915–924 [DOI] [PubMed] [Google Scholar]
- 16.Casey SC et al. (2018) The MYC oncogene is a global regulator of the immune response. Blood 131, 2007–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han H et al. (2019) Small-molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer Cell 36, 483–497 e415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y et al. (2018) PTEN loss promotes intratumoral androgen synthesis and tumor microenvironment remodeling via aberrant activation of RUNX2 in castration-resistant prostate cancer. Clin Cancer Res 24, 834–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pena CG et al. (2015) LKB1 loss promotes endometrial cancer progression via CCL2-dependent macrophage recruitment. J Clin Invest 125, 4063–4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rooney MS et al. (2015) Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160 (1–2), 48–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obeid JM et al. (2017) The heterogeneity of tumor-infiltrating CD8+ T cells in metastatic melanoma distorts their quantification: how to manage heterogeneity? Melanoma Res 27, 211–217 [DOI] [PubMed] [Google Scholar]
- 22.Chen DS and Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10 [DOI] [PubMed] [Google Scholar]
- 23.Lu B and Finn OJ (2008) T-cell death and cancer immune tolerance. Cell Death Differ 15, 70–79 [DOI] [PubMed] [Google Scholar]
- 24.Thompson ED et al. (2010) Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. J Exp Med 207, 1791–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finotello F and Trajanoski Z (2018) Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol Immunother 67, 1031–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corgnac S et al. (2018) The emerging role of CD8(+) tissue resident memory T (TRM) cells in antitumor immunity: a unique functional contribution of the CD103 integrin. Front Immunol 9, 1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ehl S et al. (1997) Bystander activation of cytotoxic T cells: studies on the mechanism and evaluation of in vivo significance in a transgenic mouse model. J Exp Med 185, 1241–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simoni Y et al. (2018) Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557 (7706), 575–579 [DOI] [PubMed] [Google Scholar]
- 29.Spranger S et al. (2013) Up-regulation of PD–L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 5 (200), 200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jarosz–Biej M et al. (2019) Tumor microenvironment as a ‘game changer’ in cancer radiotherapy. Int J Mol Sci 20, 3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mahmoud SM et al. (2011) Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol 29, 1949–1955 [DOI] [PubMed] [Google Scholar]
- 32.Salgado R et al. (2015) The evaluation of tumor-infiltrating lymphocytes (TILs) in breast cancer: recommendations by an International TILs Working Group 2014. Ann Oncol 26, 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X et al. (2019) Infiltration of CD8(+) T cells into tumor cell clusters in triple-negative breast cancer. Proc Natl Acad Sci U S A 116, 3678–3687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Byun KD et al. (2018) T-cell immunoglobulin mucin 3 expression on tumor infiltrating lymphocytes as a positive prognosticator in triple-negative breast cancer. J Breast Cancer 21, 406–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee KH et al. (2018) The prognostic and predictive value of tumor-infiltrating lymphocytes and hematologic parameters in patients with breast cancer. BMC Cancer 18, 938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Toker A et al. (2018) Regulatory T cells in ovarian cancer are characterized by a highly activated phenotype distinct from that in melanoma. Clin Cancer Res 24, 5685–5696 [DOI] [PubMed] [Google Scholar]
- 37.Komura T et al. (2015) Inflammatory features of pancreatic cancer highlighted by monocytes/macrophages and CD4+ T cells with clinical impact. Cancer Sci 106, 672–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jackaman C et al. (2003) IL-2 intratumoral immunotherapy enhances CD8+ T cells that mediate destruction of tumor cells and tumor-associated vasculature: a novel mechanism for IL-2. J Immunol 171, 5051–5063 [DOI] [PubMed] [Google Scholar]
- 39.Bhat P et al. (2017) Interferon-gamma derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death Dis 8, e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sawant DV et al. (2019) Adaptive plasticity of IL-10(+) and IL-35(+) Treg cells cooperatively promotes tumor T cell exhaustion. Nat Immunol 20, 724–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiao J et al. (2019) Targeting tumors with IL-10 prevents dendritic cell-mediated CD8(+) T cell apoptosis. Cancer Cell 35, 901–915 e904 [DOI] [PubMed] [Google Scholar]
- 42.Jang JE et al. (2017) Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep 20, 558–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas DA and Massague J (2005) TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380 [DOI] [PubMed] [Google Scholar]
- 44.Principe DR et al. (2019) TGFbeta blockade augments PD-1 inhibition to promote T-cell-mediated regression of pancreatic cancer. Mol Cancer Ther 18, 613–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Noble A et al. (2016) IL-12 and IL-4 activate a CD39-dependent intrinsic peripheral tolerance mechanism in CD8(+) T cells. Eur J Immunol 46, 1438–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bertrand F et al. (2015) Blocking tumor necrosis factor alpha enhances CD8 T-cell-dependent immunity in experimental melanoma. Cancer Res 75, 2619–2628 [DOI] [PubMed] [Google Scholar]
- 47.Santana Carrero RM et al. (2019) IL-15 is a component of the inflammatory milieu in the tumor microenvironment promoting antitumor responses. Proc Natl Acad Sci U S A 116, 599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doedens AL et al. (2016) Molecular programming of tumor-infiltrating CD8+ T cells and IL15 resistance. Cancer Immunol Res 4, 799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cannon MJ et al. (2013) Modulation of p38 MAPK signaling enhances dendritic cell activation of human CD4+ Th17 responses to ovarian tumor antigen. Cancer Immunol Immunother 62, 839–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Timperi E et al. (2017) IL-18 receptor marks functional CD8(+) T cells in non–small cell lung cancer. Oncoimmunology 6, e1328337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li XY et al. (2019) Targeting CD39 in cancer reveals an extracellular ATP- and inflammasome-driven tumor immunity. Cancer Discov 9, 1754–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stoll G et al. (2018) Impact of chemotactic factors and receptors on the cancer immune infiltrate: a bioinformatics study revealing homogeneity and heterogeneity among patient cohorts. Oncoimmunology 7, e1484980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harlin H et al. (2009) Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 69, 3077–3085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Charych DH et al. (2016) NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clin Cancer Res 22, 680–690 [DOI] [PubMed] [Google Scholar]
- 55.Philip M et al. (2017) Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545 (7655), 452–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jung D and Alt FW (2004) Unraveling V(D)J recombination; insights into gene regulation. Cell 116, 299–311 [DOI] [PubMed] [Google Scholar]
- 57.Su MA and Anderson MS (2014) Breaking through the central tolerance ceiling to unleash anticancer immune responses. Oncoimmunology 3, e950169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Verhagen J et al. (2013) CTLA-4 controls the thymic development of both conventional and regulatory T cells through modulation of the TCR repertoire. Proc Natl Acad Sci U S A 110, E221–E230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wei SC et al. (2019) Negative co-stimulation constrains T cell differentiation by imposing boundaries on possible cell states. Immunity 50, 1084–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nishimura H et al. (2000) Facilitation of beta selection and modification of positive selection in the thymus of PD-1-deficient mice. J Exp Med 191, 891–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hogquist KA and Jameson SC (2014) The self-obsession of T cells: how TCR signaling thresholds affect fate ‘decisions’ and effector function. Nat Immunol 15, 815–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nurieva RI et al. (2011) Molecular mechanisms of T-cell tolerance. Immunol Rev 241, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Waldman AD et al. (2020) A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 20, 651–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Okazaki T and Honjo T (2006) The PD-1-PD-L pathway in immunological tolerance. Trends Immunol 27, 195–201 [DOI] [PubMed] [Google Scholar]
- 65.Wherry EJ (2011) T cell exhaustion. Nat Immunol 12, 492–499 [DOI] [PubMed] [Google Scholar]
- 66.Simpson TR et al. (2013) Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 210, 1695–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ha D et al. (2019) Differential control of human Treg and effector T cells in tumor immunity by Fc-engineered anti-CTLA-4 antibody. Proc Natl Acad Sci U S A 116, 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Arce Vargas F et al. (2018) Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell 33, 649–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wei SC et al. (2019) Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc Natl Acad Sci U S A 116 (45), 22699–22709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang M et al. (2019) PD-1 blockade augments humoral immunity through ICOS-mediated CD4(+) T cell instruction. Int Immunopharmacol 66, 127–138 [DOI] [PubMed] [Google Scholar]
- 71.Topalian SL et al. (2019) Five-year survival and correlates among patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer treated with nivolumab. JAMA Oncol 381(21), 2020–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Vugt MJH et al. (2019) Immunogenicity of pembrolizumab in patients with advanced tumors. J Immunother Cancer 7, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Seetharamu N et al. (2017) New PD-L1 inhibitors in non-small cell lung cancer - impact of atezolizumab. Lung Cancer (Auckl) 8, 67–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dirix LY et al. (2018) Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN Solid Tumor study. Breast Cancer Res Treat 167, 671–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Egelston CA et al. (2018) Human breast tumor-infiltrating CD8(+) T cells retain polyfunctionality despite PD-1 expression. Nat Commun 9, 4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ahmadzadeh M et al. (2009) Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 114, 1537–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu L et al. (2015) The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res 21, 1639–1651 [DOI] [PubMed] [Google Scholar]
- 78.Duraiswamy J et al. (2013) Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res 73, 3591–3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Farhood B et al. (2019) CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol 234, 8509–8521 [DOI] [PubMed] [Google Scholar]
- 80.Leclerc M et al. (2019) Regulation of antitumour CD8 T-cell immunity and checkpoint blockade immunotherapy by Neuropilin-1. Nat Commun 10, 3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spranger S et al. (2015) Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523 (7559), 231–235 [DOI] [PubMed] [Google Scholar]
- 82.Wang S et al. (2016) Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc Natl Acad Sci U S A 113 (46), E7240–E7249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Principe DR et al. (2020) Long-term gemcitabine treatment reshapes the pancreatic tumor microenvironment and sensitizes murine carcinoma to combination immunotherapy. Cancer Res 80(15), 3101–3115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Saeidi A et al. (2018) T-cell exhaustion in chronic infections: reversing the state of exhaustion and reinvigorating optimal protective immune responses. Front Immunol 9, 2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McLane LM et al. (2019) CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol 37, 457–495 [DOI] [PubMed] [Google Scholar]
- 86.Topalian SL et al. (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366 (26), 2443–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gettinger S et al. (2016) Nivolumab monotherapy for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol 34 (25), 2980–2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Woroniecka K et al. (2018) T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res 24, 4175–4186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Friese C et al. (2020) CTLA-4 blockade boosts the expansion of tumor-reactive CD8(+) tumor–infiltrating lymphocytes in ovarian cancer. Sci Rep 10, 3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Woo SR et al. (2012) Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 72, 917–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cai C et al. (2016) Tim-3 expression represents dysfunctional tumor infiltrating T cells in renal cell carcinoma. World J Urol 34, 561–567 [DOI] [PubMed] [Google Scholar]
- 92.Zhou Q et al. (2011) Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood 117, 4501–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Takano S et al. (2016) An increased number of PD-1+ and Tim-3+ CD8+ T cells is involved in immune evasion in gastric cancer. Surg Today 46, 1341–1347 [DOI] [PubMed] [Google Scholar]
- 94.Yang ZZ et al. (2020) TIGIT expression is associated with T-cell suppression and exhaustion and predicts clinical outcome and anti-PD-1 response in follicular lymphoma. Clin Cancer Res 26, 5217–5231 [DOI] [PubMed] [Google Scholar]
- 95.Johnston RJ et al. (2014) The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 26, 923–937 [DOI] [PubMed] [Google Scholar]
- 96.Anestakis D et al. (2020) Carboplatin chemoresistance is associated with CD11b(+)/Ly6C(+) myeloid release and upregulation of TIGIT and LAG3/CD160 exhausted T cells. Mol Immunol 118, 99–109 [DOI] [PubMed] [Google Scholar]
- 97.Ritthipichai K et al. (2017) Multifaceted role of BTLA in the control of CD8(+) T-cell fate after antigen encounter. Clin Cancer Res 23, 6151–6164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Simpson LL (1986) A preclinical evaluation of aminopyridines as putative therapeutic agents in the treatment of botulism. Infect Immun 52, 858–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kondo Y et al. (2016) Differential contribution of three immune checkpoint (VISTA, CTLA-4, PD-1) pathways to antitumor responses against squamous cell carcinoma. Oral Oncol 57, 54–60 [DOI] [PubMed] [Google Scholar]
- 100.Mulati K et al. (2019) VISTA expressed in tumour cells regulates T cell function. Br J Cancer 120, 115–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Deng J et al. (2019) Hypoxia-induced VISTA promotes the suppressive function of myeloid-derived suppressor cells in the tumor microenvironment. Cancer Immunol Res 7, 1079–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mittal R et al. (2014) Phenotypic T cell exhaustion in a murine model of bacterial infection in the setting of pre-existing malignancy. PLoS ONE 9, e93523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen CW et al. (2019) 2B4 but not PD-1 blockade improves mortality in septic animals with preexisting malignancy. JCI Insight 4, e127867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mittal D et al. (2019) CD96 is an immune checkpoint that regulates CD8(+) T-cell antitumor function. Cancer Immunol Res 7, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang H et al. (2020) Direct interaction between CD155 and CD96 promotes immunosuppression in lung adenocarcinoma. Cell Mol Immunol. Published online September 11, 2020. 10.1038/s41423-020-00538-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Miller BC et al. (2019) Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol 20, 326–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu S et al. (2020) CD160 expression on CD8(+) T cells is associated with active effector responses but limited activation potential in pancreatic cancer. Cancer Immunol Immunother 69, 789–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kim JM and Chen DS (2016) Immune escape to PD-L1/PD-1 blockade: seven steps to success (or failure). Ann Oncol 27, 1492–1504 [DOI] [PubMed] [Google Scholar]
- 109.Siddiqui I et al. (2019) Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity 50, 195–211 [DOI] [PubMed] [Google Scholar]
- 110.Fourcade J et al. (2010) Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med 207, 2175–2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Verma V et al. (2019) PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat Immunol 20, 1231–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Marro BS et al. (2019) Discovery of small molecules for the reversal of T cell exhaustion. Cell Rep 29, 3293–3302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Silence K et al. (2014) ARGX-110, a highly potent antibody targeting CD70, eliminates tumors via both enhanced ADCC and immune checkpoint blockade. MAbs 6, 523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gopal AK et al. (2020) First-in-human study of utomilumab, a 4-1BB/CD137 agonist, in combination with rituximab in patients with follicular and other CD20(+) non-Hodgkin lymphomas. Clin Cancer Res 26, 2524–2534 [DOI] [PubMed] [Google Scholar]
- 115.Timmerman J et al. (2020) Urelumab alone or in combination with rituximab in patients with relapsed or refractory B-cell lymphoma. Am J Hematol 95, 510–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ye Q et al. (2014) CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin Cancer Res 20, 44–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chin SM et al. (2018) Structure of the 4-1BB/4-1BBL complex and distinct binding and functional properties of utomilumab and urelumab. Nat Commun 9, 4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Compte M et al. (2018) A tumor-targeted trimeric 4-1BB-agonistic antibody induces potent anti-tumor immunity without systemic toxicity. Nat Commun 9, 4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Deng J et al. (2019) OX40 (CD134) and OX40 ligand, important immune checkpoints in cancer. Onco Targets Ther 12, 7347–7353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dushyanthen S et al. (2017) Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer. Nat Commun 8, 606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Baumann D et al. (2020) Proimmunogenic impact of MEK inhibition synergizes with agonist anti–CD40 immunostimulatory antibodies in tumor therapy. Nat Commun 11, 2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Inaguma S et al. (2020) CD70 expression correlates with a worse prognosis in malignant pleural mesothelioma patients via immune evasion and enhanced invasiveness. J Pathol 250, 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zwaenepoel K et al. (2017) CD70 and PD-L1 in anaplastic thyroid cancer - promising targets for immunotherapy. Histopathology 71, 357–365 [DOI] [PubMed] [Google Scholar]
- 124.Wang B et al. (2018) Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8(+) T cell dysfunction and maintain memory phenotype. Sci Immunol 3 (29), eaat7061. [DOI] [PubMed] [Google Scholar]
- 125.Kaplon H et al. (2020) Antibodies to watch in 2020. MAbs 12, 1703531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Soldevilla MM et al. (2019) ICOS costimulation at the tumor site in combination with CTLA-4 blockade therapy elicits strong tumor immunity. Mol Ther 27, 1878–1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Teige I et al. (2019) Targeting the antibody checkpoints to enhance cancer immunotherapy–focus on FcgammaRIIB. Front Immunol 10, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Buchan SL et al. (2018) Antibodies to costimulatory receptor 4-1BB enhance anti-tumor immunity via T regulatory cell depletion and promotion of CD8 T cell effector function. Immunity 49, 958–970 [DOI] [PubMed] [Google Scholar]
- 129.Bhat P et al. (2017) Interferon-gamma derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death & Disease 8 (6), e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Watanabe K et al. (2018) Expanding the therapeutic window for CAR T cell therapy in solid tumors: the knowns and unknowns of CAR T cell biology. Front Immunol 9, 2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yamamoto TN et al. (2019) Developing neoantigen-targeted T cell-based treatments for solid tumors. Nat Med 25, 1488–1499 [DOI] [PubMed] [Google Scholar]
- 132.Yossef R et al. (2018) Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 3, e122467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen Q et al. (2019) Photothermal therapy promotes tumor infiltration and antitumor activity of CAR T cells. Adv Mater 31, e1900192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hu J et al. (2018) T-cell homing therapy for reducing regulatory T cells and preserving effector T-cell function in large solid tumors. Clin Cancer Res 24, 2920–2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xu Y et al. (2018) A novel antibody-TCR (AbTCR) platform combines Fab-based antigen recognition with gamma/delta-TCR signaling to facilitate T-cell cytotoxicity with low cytokine release. Cell Discov 4, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jin J et al. (2018) Enhanced clinical-scale manufacturing of TCR transduced T-cells using closed culture system modules. J Transl Med 16, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Coccoris M et al. (2008) Long-term functionality of TCR-transduced T cells in vivo. J Immunol 180, 6536–6543 [DOI] [PubMed] [Google Scholar]
- 138.Xu Y et al. (2019) Preclinical development of T-cell receptor-engineered T-cell therapy targeting the 5T4 tumor antigen on renal cell carcinoma. Cancer Immunol Immunother 68, 1979–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Li Q et al. (2018) PLAC1-specific TCR-engineered T cells mediate antigen-specific antitumor effects in breast cancer. Oncol Lett 15, 5924–5932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.van der Lee DI et al. (2019) Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J Clin Invest 129, 774–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Robbins PF et al. (2011) Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29, 917–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kageyama S et al. (2015) Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res 21, 2268–2277 [DOI] [PubMed] [Google Scholar]
- 143.Chheda ZS et al. (2018) Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med 215, 141–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dafni U et al. (2019) Efficacy of adoptive therapy with tumor-infiltrating lymphocytes and recombinant interleukin-2 in advanced cutaneous melanoma: a systematic review and meta-analysis. Ann Oncol 30, 1902–1913 [DOI] [PubMed] [Google Scholar]
- 145.Andersen R et al. (2015) Tumor infiltrating lymphocyte therapy for ovarian cancer and renal cell carcinoma. Hum Vaccin Immunother 11, 2790–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Stevanovic S et al. (2019) A Phase II study of tumor-infiltrating lymphocyte therapy for human papillomavirus-associated epithelial cancers. Clin Cancer Res 25, 1486–1493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Santoiemma PP and Powell DJ Jr. (2015) Tumor infiltrating lymphocytes in ovarian cancer. Cancer Biol Ther 16, 807–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dieci MV et al. (2018) Update on tumor-infiltrating lymphocytes (TILs) in breast cancer, including recommendations to assess TILs in residual disease after neoadjuvant therapy and in carcinoma in situ: a report of the International Immuno-Oncology Biomarker Working Group on Breast Cancer. Semin Cancer Biol 52 (Pt 2), 16–25 [DOI] [PubMed] [Google Scholar]
- 149.Finlay D and Cantrell D (2011) The coordination of T-cell function by serine/threonine kinases. Cold Spring Harb Perspect Biol 3, a002261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Jin X et al. (2016) The p38 MAPK inhibitor BIRB796 enhances the antitumor effects of VX680 in cervical cancer. Cancer Biol Ther 17, 566–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ebert PJR et al. (2016) MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 44, 609–621 [DOI] [PubMed] [Google Scholar]
- 152.Li D et al. (2009) LFA-1 regulates CD8+ T cell activation via T cell receptor-mediated and LFA-1-mediated Erk1/2 signal pathways. J Biol Chem 284 (31), 21001–21010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gao Y et al. (2005) JNK1 is essential for CD8+ T cell-mediated tumor immune surveillance. J Immunol 175, 5783–5789 [DOI] [PubMed] [Google Scholar]
- 154.Pieper N et al. (2018) Evolution of melanoma cross-resistance to CD8(+) T cells and MAPK inhibition in the course of BRAFi treatment. Oncoimmunology 7, e1450127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Thaker YR et al. (2019) GTPase-activating protein Rasal1 associates with ZAP-70 of the TCR and negatively regulates T-cell tumor immunity. Nat Commun 10, 4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.di Bari MG et al. (2009) TGF-beta modulates the functionality of tumor-infiltrating CD8+ T cells through effects on TCR signaling and Spred1 expression. Cancer Immunol Immunother 58, 1809–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Liang Y et al. (2017) Phosphorylated ERK is a potential prognostic biomarker for Sorafenib response in hepatocellular carcinoma. Cancer Med 6, 2787–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Chen J et al. (2016) Sorafenib-resistant hepatocellular carcinoma stratified by phosphorylated ERK activates PD-1 immune checkpoint. Oncotarget 7 (27), 41274–41284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mishra et al. (2010) Mixed lineage kinase-3/JNK1 axis promotes migration of human gastric cancer cells following gastrin stimulation. Mol Endocrinol 24, 598–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kumar S et al. (2020) Mixed lineage kinase 3 inhibition induces T cell activation and cytotoxicity. Proc Natl Acad Sci U S A 117, 7961–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kumar S et al. (2020) Rationalized inhibition of mixed lineage kinase 3 and CD70 enhances life span and antitumor efficacy of CD8(+) T cells. J Immunother Cancer 8, e000494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Esen E et al. (2020) MAP4K4 negatively regulates CD8 T cell-mediated antitumor and antiviral immunity. Sci Immunol 5 (45), eaay2245. [DOI] [PubMed] [Google Scholar]
- 163.Watanabe S et al. (2019) Mutational activation of the epidermal growth factor receptor down-regulates major histocompatibility complex class I expression via the extracellular signal-regulated kinase in non-small cell lung cancer. Cancer Sci 110, 52–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kalimutho M et al. (2019) Blockade of PDGFRbeta circumvents resistance to MEK-JAK inhibition via intratumoral CD8(+) T-cells infiltration in triple-negative breast cancer. J Exp Clin Cancer Res 38, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Miah MA and Bae YS (2013) Regulation of DC development and DC-mediated T-cell immunity via CISH. Oncoimmunology 2, e23404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Palmer DC et al. (2015) Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J Exp Med 212, 2095–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bengsch B et al. (2016) Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity 45, 358–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gemta LF et al. (2019) Impaired enolase 1 glycolytic activity restrains effector functions of tumor–infiltrating CD8(+) T cells. Sci Immunol 4 (31), eaap9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Wang JJ et al. (2019) Aberrant upregulation of PDK1 in ovarian cancer cells impairs CD8(+) T cell function and survival through elevation of PD-L1. Oncoimmunology 8, e1659092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Maciolek JA et al. (2014) Metabolism of activated T lymphocytes. Curr Opin Immunol 27, 60–74 [DOI] [PubMed] [Google Scholar]
- 171.Siska PJ et al. (2017) Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight 2, e93411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ho PC et al. (2015) Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Liu X et al. (2018) Acetate production from glucose and coupling to mitochondrial metabolism in mammals. Cell 175, 502–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Qiu J et al. (2019) Acetate promotes T cell effector function during glucose restriction. Cell Rep 27, 2063–2074 e2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Nabe S et al. (2018) Reinforce the antitumor activity of CD8(+) T cells via glutamine restriction. Cancer Sci 109, 3737–3750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Norian LA et al. (2009) Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L-arginine metabolism. Cancer Res 69, 3086–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Geiger R et al. (2016) L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 167, 829–842 e813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zhang Y et al. (2017) Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell 32, 377–391 e379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Palazon A et al. (2017) An HIF-1alpha/VEGF-A axis in cytotoxic T cells regulates tumor progression. Cancer Cell 32, 669–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mastelic-Gavillet B et al. (2019) Adenosine mediates functional and metabolic suppression of peripheral and tumor–infiltrating CD8(+) T cells. J Immunother Cancer 7, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Adams S (2009) Toll-like receptor agonists in cancer therapy. Immunotherapy 1, 949–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kaczanowska S et al. (2013) TLR agonists: our best frenemy in cancer immunotherapy. J Leukoc Biol 93, 847–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lu H et al. (2012) VTX-2337 is a novel TLR8 agonist that activates NK cells and augments ADCC. Clin Cancer Res 18, 499–509 [DOI] [PubMed] [Google Scholar]
- 184.Prins RM et al. (2006) The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol 176, 157–164 [DOI] [PubMed] [Google Scholar]
- 185.Zahm CD et al. (2018) TLR stimulation during T-cell activation lowers PD-1 expression on CD8(+) T cells. Cancer Immunol Res 6, 1364–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ming Lim C et al. (2013) TLR3 agonists improve the immunostimulatory potential of cetuximab against EGFR(+) head and neck cancer cells. Oncoimmunology 2, e24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Coelho-Dos-Reis JG et al. (2016) Co-administration of alpha-GalCer analog and TLR4 agonist induces robust CD8(+) T-cell responses to PyCS protein and WT-1 antigen and activates memory-like effector NKT cells. Clin Immunol 168, 6–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Brackett CM et al. (2016) Toll-like receptor-5 agonist, entolimod, suppresses metastasis and induces immunity by stimulating an NK-dendritic-CD8+ T-cell axis. Proc Natl Acad Sci U S A 113, E874–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Shayan G et al. (2018) Phase Ib study of immune biomarker modulation with neoadjuvant cetuximab and TLR8 stimulation in head and neck cancer to overcome suppressive myeloid signals. Clin Cancer Res 24, 62–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kuai R et al. (2018) Dual TLR agonist nanodiscs as a strong adjuvant system for vaccines and immunotherapy. J Control Release 282, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Joseph AM et al. (2016) Cross-talk between 4-1BB and TLR1-TLR2 signaling in CD8+ T cells regulates TLR2’s costimulatory effects. Cancer Immunol Res 4, 708–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wang Y et al. (2018) Adjuvant effect of the novel TLR1/TLR2 agonist Diprovocim synergizes with anti-PD-L1 to eliminate melanoma in mice. Proc Natl Acad Sci U S A 115 (37), E8698–E8706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Cen X et al. (2019) TLR1/2 Specific small-molecule agonist suppresses leukemia cancer cell growth by stimulating cytotoxic T lymphocytes. Adv Sci (Weinh) 6, 1802042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Pearson FE et al. (2018) Activation of human CD141(+) and CD1c(+) dendritic cells in vivo with combined TLR3 and TLR7/8 ligation. Immunol Cell Biol 96, 390–400 [DOI] [PubMed] [Google Scholar]
- 195.Khalil DN et al. (2019) In situ vaccination with defined factors overcomes T cell exhaustion in distant tumors. J Clin Invest 129, 3435–3447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Tsukamoto H et al. (2019) An agonistic anti-Toll-like receptor 4 monoclonal antibody as an effective adjuvant for cancer immunotherapy. Immunology 158, 136–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ding X et al. (2012) A TLR5 agonist enhances CD8(+) T cell-mediated graft-versus-tumor effect without exacerbating graft-versus-host disease. J Immunol 189, 4719–4727 [DOI] [PubMed] [Google Scholar]
- 198.Sato–Kaneko F et al. (2017) Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight 2, e93397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mullins SR et al. (2019) Intratumoral immunotherapy with TLR7/8 agonist MEDI9197 modulates the tumor microenvironment leading to enhanced activity when combined with other immunotherapies. J Immunother Cancer 7, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kim H et al. (2018) Polymeric nanoparticles encapsulating novel TLR7/8 agonists as immunostimulatory adjuvants for enhanced cancer immunotherapy. Biomaterials 164, 38–53 [DOI] [PubMed] [Google Scholar]
- 201.Kang TH et al. (2019) TLR9 acts as a sensor for tumor-released DNA to modulate anti–tumor immunity after chemotherapy. J Immunother Cancer 7, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ishikawa H and Barber GN (2008) STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455 (7213), 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wu J et al. (2013) Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339 (6121), 826–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Flood BA et al. (2019) STING pathway agonism as a cancer therapeutic. Immunol Rev 290, 24–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Chon HJ et al. (2019) STING signaling is a potential immunotherapeutic target in colorectal cancer. J Cancer 10, 4932–4938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Shae D et al. (2019) Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol 14, 269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Pantelidou C et al. (2019) PARP inhibitor efficacy depends on CD8(+) T-cell recruitment via intratumoral STING pathway activation in BRCA-deficient models of triple-negative breast cancer. Cancer Discov 9, 722–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yang H et al. (2019) STING activation reprograms tumor vasculatures and synergizes with VEGFR2 blockade. J Clin Invest 129, 4350–4364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Pei J et al. (2019) STAT3 inhibition enhances CDN-induced STING signaling and antitumor immunity. Cancer Lett 450, 110–122 [DOI] [PubMed] [Google Scholar]