

Abstract
Chemical library screening approaches that focus exclusively on catalytic events may overlook unique effects of protein–protein interactions that can be exploited for development of specific inhibitors. Phosphotyrosyl (pTyr) residues embedded in peptide motifs comprise minimal recognition elements that determine the substrate specificity of protein tyrosine phosphatases (PTPases). We incorporated aminooxy-containing amino acid residues into a 7-residue epidermal growth factor receptor (EGFR) derived phosphotyrosine-containing peptide and subjected the peptides to solution-phase oxime diversification by reacting with aldehyde-bearing druglike functionalities. The pTyr residue remained unmodified. The resulting derivatized peptide library was printed in microarrays on nitrocellulose-coated glass surfaces for assessment of PTPase catalytic activity or on gold monolayers for analysis of kinetic interactions by surface plasmon resonance (SPR). Focusing on amino acid positions and chemical features, we first analyzed dephosphorylation of the peptide pTyr residues within the microarrayed library by the human dual-specificity phosphatases (DUSP) DUSP14 and DUSP22, as well as by PTPases from poxviruses (VH1) and Yersinia pestis (YopH). In order to identify the highest affinity oxime motifs, the binding interactions of the most active derivatized phosphopeptides were examined by SPR using noncatalytic PTPase mutants. On the basis of high-affinity oxime fragments identified by the two-step catalytic and SPR-based microarray screens, low-molecular-weight nonphosphate-containing peptides were designed to inhibit PTP catalysis at low micromolar concentrations.
Keywords: aminooxy phosphopeptide, catalytic assay, fragment-based drug design, kinetic assay, microarray, oxime-containing phosphopeptide, protein tyrosine phosphatase, substrate affinity screening, surface plasmon resonance
Graphical Abstract

INTRODUCTION
Protein tyrosine phosphatases (PTPases) dephosphorylate tyrosine residues within proteins and work in concert with protein tyrosine kinases to regulate signal transduction pathways. Because of the critical involvement of signal transduction pathways in regulating pathological processes in cancer and infectious diseases,1,2 PTPases have emerged as important targets for the development of therapeutic inhibitors. Despite growing successes in the development of small molecules that inhibit kinases, modulators of PTPases have proven to be far more challenging, and no inhibitor has yet achieved clinical approval.3 Effective PTPase targeting by small-molecule inhibitors is complicated by the relatively smooth protein surfaces and, in the case of dual-specificity PTPases (DUSPs), shallow catalytic pockets that accommodate phosphotyrosine (pTyr), phosphoserine (pSer), and phosphothreonine (pThr) residues. In addition, because recognition and binding of PTPase substrates involves a network of hydrogen and ionic bonds within a highly conserved catalytic pocket ((H/V)C(X5)R(S/T)), preferred ligands are typically negatively charged, which results in limited specificity, cell permeability, and bioavailability. An alternative approach to increase the likelihood of finding druggable features of PTPases is to exploit interactions with protein features that extend beyond the catalytic pocket. Synthetic peptide substrates have previously been used as display platforms for nonhydrolyzable pTyr-mimicking residues,4 which led to the identification of the difluorophosphonomethyl-aryl moiety as a starting point for the design of small-molecule inhibitors by converting a good substrate into a high-affinity inhibitor.4,5 In the approach described herein, we used a high-affinity peptide substrate as a scaffold for presenting microarrayed libraries of druglike fragments to identify motifs for inhibitor design.6–9 The goal of this work was to devise a method for developing inhibitors of protein–protein interactions (PPIs) that are intrinsic to PTPase catalytic specificities. In order to investigate substrate binding interactions in proximity to the PTPase catalytic cleft, we sequentially examined modified amino acid residues adjacent to the unaltered the pTyr residue of the peptide. A diversified library was created by incorporating 300 different druglike fragments at six different positions on the EGFR-derived peptide “VDADEpYL”. Detailed interactions with the substrate library were examined using PTPases from smallpox virus (Variola major H1 (VH1)), the plague bacillus (Yersinia outer protein H (YopH)), and the human enzymes DUSP14 and DUSP22. In order to identify fragments that could be useful in inhibitor design, primary results were obtained from catalytic assays with the microarrayed library, and kinetic binding data were obtained from the microarrayed library by using a biosensor assay. As proof of principle, a high-affinity oxime fragment identified by the two-step catalytic and surface plasmon resonance (SPR)-based microarray screens was employed to design low-molecular weight, non-phosphate-containing peptides, which were able to inhibit PTP catalysis at low micromolar concentrations.
RESULTS AND DISCUSSION
Methodology.
A flowchart depicting the general approach used is presented in Figure 1. We synthesized a library comprised of 1800 distinct oxime-modified phosphopeptides based on the EGFR-derived heptapeptide sequence (biotinlinker-VDADEpYL-NH2, 5), which includes the autophosphorylation site Tyr 992.10 The EGFR-derived pTyr-containing peptide is a reliable substrate for many PTPases.11,12 Along with 5, the library was deposited by inkjet printing of microarrays on either a nitrocellulose-coated or a gold-coated glass slide for use in a fluorescent-based catalytic assay or SPR based kinetic assay, respectively. The percent dephosphorylation (in comparison to the parent peptide 5) and the binding interaction of each library component with PTPases was assessed with VH1, DUSP14, DUSP22, and YopH. Combining the data from both experiments allowed us to identify fragments that may have the greatest utility as building blocks for the design of potential inhibitors. Active, wild-type PTPases were used for all catalytic assays, while catalytically inactive mutant enzymes were used in SPR binding-interaction studies to avoid dephosphorylation of the pTyr residue. These PTPase substrate-trapping mutants, which do not possess measurable catalytic activities but still bind tightly to pTyr substrates,13 can be used to identify physiological substrates in vivo and in vitro,14 as well as to elucidate binding modes by crystallography.15
Figure 1.

Flowchart depicting the methodology used in this study.
Synthetic Discussion.
Synthesis of Aminooxy-Containing Amino Acid Reagent 4.
The preparation of aminooxy-containing peptides 6a–f (Figure 2) by standard Fmoc-based solid-phase protocols entailed use of the orthogonally protected amino acid 4 (Scheme 1). The synthesis of 4 started from compound 1, which was obtained from Cbz-(Asp)-OBn by literature procedures.16 Reaction of 1 with N-hydroxyphthalimide under Mitsunobu coupling conditions gave the adduct 2 in good yield. Similar Mitsunobu coupling on the carboxymethyl ester variant of 1 had been reported to give low yields and cyclized byproducts.17 This necessitated a two-step protocol that involved conversion of the side-chain hydroxyl to a mesyl ester followed by nucleophilic displacement with N-hydroxyphthalimide.17 Starting from 1, a similar two-step approach has been reported to prepare 2.16 In the current work, treatment of 2 with methylhydrazine removed the phthalimide group to provide the free aminooxy group, which was derivatized with Boc anhydride to give the globally protected analogue 3 (Scheme 1). Hydrogenolytic removal of the N-Cbz and O-Bn groups, followed by treatment with Fmoc succinimidyl carbonate (Fmoc-OSu) and NaHCO3 in aqueous dioxane, gave the desired N-Fmoc-protected 4 in good yield. The synthesis of 4 has been previously reported by a slightly different protocol.17,18 Reagent 4 was employed in solid-phase peptide synthesis (SPPS), using standard Fmoc procedures to prepare the biotinylated aminooxy-containing peptides 6a–f (Figure 2).
Figure 2.

Structures of the parent peptide 5 and the biotinylated aminooxy-containing peptides 6a–f.
Scheme 1. Synthesis of the Orthogonally Protected Amino Acid Reagent 4.

Synthesis of a Library of Biotinylated Oxime-Containing Peptides.
Peptides and proteins undergo clean oxime-forming reactions rapidly in near-quantitative yield without the need of side-chain protection. The resulting products are highly stable.19–21 We have previously prepared libraries of oxime-diversified peptides by incorporating aminooxy-containing residues within parent peptides and then reacting the peptides in parallel with aldehydes in DMSO in the presence of acetic acid. The oxime-forming reactions were nearly quantitative, and products of >90% purity were routinely obtained. These products are stable and can be subjected directly to biological evaluation without the need for purification.22–25 In our current work we employed a collection of 300 aldehydes (structures are shown in Tables S1–S3 in the Supporting Information) that were reacted with the biotinylated aminooxy-containing peptides 6a–f. The optimized reaction conditions for oxime ligation used DMSO as solvent and acetic acid as catalyst at room temperature overnight, with 1/1/5 molar concentrations of aminooxy/RCHO/acetic acid (Scheme 2).18,19 This resulted in the formation of a library of 1800 distinct oxime-containing phosphopeptides at final concentrations of 100 μM. In light of well-established precedence, the libraries were used without purification and the solutions were used directly to prepare microarrays on nitrocellulose slides26 for screening as described below.
Scheme 2. Synthesis of Oxime-Containing Peptide Librarya.

aThe aldehydes are given in Tables S1–S3 in the Supporting Information.
Synthesis of Side-Chain-Modified Peptides 11a,b and 12.
Oxime-containing peptides that displayed the aldehydes w33 and w202 (Table S2 and Figure S3 in the Supporting Information) were selected for further examination from the high-affinity substrates observed in the microarray assays. The original oxime-aminooxy oxygens of w33 and w202 were replaced by methylene groups, while the remainder of the oxime functionality was replaced by structurally homologous amides. This resulted in the design of peptides 11a (w33) and 11b (w202) as the monosubstituted congeners and peptide 12 as a disubstituted congener that combined features of peptides w33 and w202 (Figure 3). An essential feature of 11a,b and 12 was the inclusion of ornithine residues as homologous variants of the aminooxy-containing residue 4. By employing 4-methyltrityl (Mtt) side-chain amine protection of the ornithine residues in combination with Rink amide MBHA resin, it was possible to remove the Mtt protection by treatment with a low concentration of TFA (1%) that preserved peptide attachment to the resin. Amidation of the ornithine side chain was then achieved using the appropriate carboxylic acid prior to Fmoc removal and peptide chain extension. Cleavage of the completed peptide from the resin was then accomplished using 95% TFA. Although the 3,5-dichloropyridine-4-carboxylic acid needed to prepare the ornithine amide Zzz in peptides 11b and 12 (Figure 3) was commercially available (Aldrich), it was necessary to synthesize the 4-(4-(trifluoromethyl)pyridine-2-yl)benzoic acid (10, Scheme 3), which was required to prepare the ornithine amide Yyy in peptides 11a and 12 (Figure 3).
Figure 3.

Structures of side-chain-modified substrates based on peptide 5.
Scheme 3. Synthesis of Reagent 10.

The synthesis of 10 was accomplished in two steps by initial Suzuki coupling of 2-bromo-4-(trifluoromethyl)pyridine (7) and (4-(methoxycarbonyl)phenyl)boronic acid (8) to yield the methyl ester 9, followed by hydrolysis of the ester under alkaline conditions (Scheme 3).
Fluorometric Array.
The PTPase activity of each enzyme was measured against the peptide libraries using a fluorescence-based microarray assay as a primary screening tool. The unmodified EFGR-derived heptapeptide 5 served as a baseline to compare the effect of each oxime-containing peptide within the library. The level of dephosphorylation was assessed by using an antibody that detects phosphotyrosine within the substrate peptide and a fluorescently labeled secondary antibody (antimouse IgG). Figure 4 shows a representative slide of the oxime-phoshopeptide library and controls that were spotted in triplicate and treated with PTPase under fixed conditions. The magnified section compares the difference between a buffer-treated slide (no PTPase) versus a PTPase-treated slide. Each spot represents an individual phosphopeptide, and dephosphorylation is observed by a decrease in fluorescence signal (Figure 4) in comparison to the corresponding spot on the non-enzyme-treated slide. The results from each PTPase are depicted in the heat map diagram of Figure S2 in the Supporting Information, which shows dephosphorylation values for individual components of the entire oxime-containing phosphopeptide library, peptide 5, and peptides 6a–f.
Figure 4.

Representative slide spotted with the oxime-containing phosphopeptide library and treated with PTPase or buffer solution. After probing with an Alexafluor647-conjugated detection antibody, the fluorescent signal from each spot is measured. Strong signals are colored red and white and signify a greater amount of pTyr remaining, while weaker signals are light red and black and signify less pTyr remaining. The magnified images show the contrast between the buffer-treated and enzyme-treated slides.
Figure 5 shows the change in dephosphorylation for the oxime-containing phosphopeptides on treatment with each PTPase (averaged for each spot), in comparison to 5 (the baseline). The peptides are grouped according to the position of each amino acid residue relative to pTyr. Protein concentrations and incubation times were determined from test experiments of the PTPases with 5 and 6a–f, in which approximately 40% dephosphorylation was obtained (data not shown). This was done in order to ascertain whether fragments particular to individual oxime-containing peptides enhanced or reduced the dephosphorylaiton of pTyr residues in comparison to the parent peptide 5. When they were tested against the entire peptide library, the PTPase activities of each enzyme with peptide 5 as substrate were as follows: VH1, 24%; DUSP14, 28%; DUSP22, 6%; YopH, 96%. This apparent change in dephosphorylation may be a result of scaling up enzyme solutions for the larger peptide library, slight errors in measuring enzyme concentration (by UV/vis), and/or changes in the specific activity of the enzyme over time. Because we were primarily interested in fragments associated with enhancing dephosphorylation for use in the design of inhibitors, the dephosphorylation levels corresponding to peptide 5 against VH1, DUSP14, and DUSP22 were acceptable. Although the high activity of YopH across the entire library was not unexpected,27 this made it difficult to control cleavage conditions for YopH in this assay. As seen in Figure 5, large differences from the baseline (peptide 5) for YopH are generally only apparent for decreases in percent dephosphorylation. This inhibitory effect is most notable with the oxime-containing peptides from 6d,e, in which the respective Asp and Glu residues have been modified. This is consistent with findings from other studies showing that these two residues are crucial for substrate–enzyme binding interactions.28 The data set for YopH remains biased toward decreases in dephosphorylation, due to the high amount of dephosphorylation observed with the parent peptide 5. For this reason, YopH was used as a positive control in subsequent binding experiments and analyses. In Figure 5, VH1 demonstrates varying degrees of activity toward the entire peptide library, with the exception of those in group 6f, in which the Leu residue C-proximal to the pTyr was modified.
Figure 5.

Effect of residue position of the peptide on enzymatic cleavage of pTyr: change in percent dephosphorylation in comparison to parent peptide 5 for each oxime-containing peptide, grouped according to the amino acid residue that was modified in 6a–f.
Most of the peptides in 6f appear to have an enhancing effect on VH1 PTPase activity. From a drug design viewpoint, it may be advantageous to target areas on the protein that are reachable from this residue, keeping in mind the more polar oxime handle that replaced the bulkier alkyl chain of the Leu. In contrast, DUSP14 appears to be less sensitive to residue positioning, with the exception of the C-terminal Leu, as seen in Figure 5. Nearly all modifications to residues in the N-terminal direction of pTyr produce an increase in dephosphorylation. Modification of either of the Asp residues (groups 6b or 6d) appears to enhance DUSP22 activity (Figure 5), indicating that negative chemical functionalities are disfavored at these positions for this PTPase.
The fragments for each oxime-containing phosphopeptide were grouped into 11 categories according to chemical structure and functional group. For each of the 11 structural categories, the median difference in percent dephosphorylation between each oxime-phosphopeptide and 5 are depicted graphically in Figure 6. Differences in the abilities of the functional groups to affect the PTPase dephosphorylation of the pTyr-containing peptides are readily apparent. For clarity, the effects of each fragment have also been arranged according to residue proximity to the pTyr residue and by PTPase. Bubble sizes represent the standard deviations of the difference in percent dephosphorylation for the fragment-containing peptides associated with that category (i.e., a smaller bubble signifies a tighter agreement between the fragments in that category). The red dotted lines intersect the x axis at zero to better visualize effects that groups have on the ability of the PTPase to dephosphorylate the phosphopeptide (bubbles above the line signify a median increase in percent dephosphorylation in comparison to 5, while bubbles below the line signify a median decrease within the group). Because the fragment library was chosen on the basis of druglike properties, many of the members possess qualities of multiple functional/structural groups. Therefore, a high degree of overlap is not surprising. However, a number of notable features can be discerned from these graphs. For example, in the case of VH1 (Figure 6B), fragments containing nitrile groups seem to be particularly disfavored at the Val (−5) and Asp (−2) positions. On the basis of these results, it would be beneficial to avoid nitrile groups in the design of VH1 inhibitors, at least in the chemical space associated with the areas on the protein that interact with these Val and Asp residues. In addition, although there is a high degree of overlap among many of the bubbles in the graph for DUSP22 (Figure 6D), there is a clear differentiation between the phosphate mimetics and zwitterions versus the remaining groups at the Glu (−1) position. In the design of inhibitors of DUSP22, it may be beneficial to place functional groups that possess phosphate-like qualities in close proximity. The large differences observed between each PTPase suggest that the enzymes may be selectively targeted using specifically designed small molecules and the results could serve as a baseline for further structure–activity relationship studies.
Figure 6.

Effect of functional group/chemical structure on enzymatic cleavage of pTyr: change in percent dephosphorylation in comparison to parent peptide 5 for each oxime-containing peptide, grouped according to functional group/chemical structure as named in the legend.
PTPase Kinetic Interactions.
The binding interactions between a select number of oxime-containing phosphopeptides and PTPases were calculated using an SPR-based biosensor that permitted continuous measurement in real time. For these kinetic assays, a subset of peptides was chosen according to their range of activities observed in the catalytic assay. A total of 45 oxime-containing phosphopeptides were selected in order to accommodate controls and allow for replicate spots. The chosen peptides include 6 peptides that showed little to no dephosphorylation, 5 peptides that showed specific activity toward a particular enzyme, and 17 peptides that showed the highest levels of dephosphorylation (representing at least 1 peptide from each of the 6a–f groups so that each residue position was included). The biotinylated peptides and controls were mixed with NeutrAvidin, and each mixture was spotted onto a gold-coated SPR slide that was derivatized with a self-assembled monolayer of carboxyl-terminated alkanethiols followed by 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide (EDC)/N-hydroxysuccinimide (NHS) coupling chemistry. Kinetic binding studies were then performed for each PTPase, with anti-pTyr antibody and histidine-tagged maltose binding protein (His-MBP) acting as positive and negative binding controls, respectively. Figure 7 shows representative sensorgrams from the kinetic assays for the control proteins, VH1 C110S, and DUSP14 C111S. For binding interactions between peptide 5 and the positive control proteins, anti-pTyr mouse antibody and YopH, noticeable binding curves were observed, although the slope of each curve was different. The on-rate (ka) and off-rate (kd) for the antibody were slower than for YopH, indicating a slower but stronger complex formation. The binding curves between VH1 C110S and peptides 5 and 6f-w202 (the nomenclature indicates the parent peptide with the associated oxime group as shown in Tables S1–S3 in the Supporting Information) as well as the negative control, PEG (a 12-chain poly(ethylene glycol) (PEG) with a terminal ethanolamide) are shown in Figure 7B. As depicted, negligible binding was observed between the PTPase and PEG, indicating little nonspecific binding. The higher binding affinity for VH1 and peptide 6f-w202 (KD = 73 ± 27 nM) versus peptide 5 (KD = 96 ± 21 nM) was consistent with the higher level of dephosphorylation in the catalytic assay (43% and 24% for peptides 6f-w202 and 5, respectively). Binding curves for DUSP14 C111S and peptides 5, 6f-w99, and 6f-w202 are shown in Figure 7C, with the associated KD values for each peptide being as follows: 5, 1400 ± 1300 nM; 6f-w202, 640 ± 150 nM; 6f-w99, 94 ± 94 nM. Binding curves for 5 and 6f-w99 are atypical and probably are not best fit to a Langmuir binding curve (also demonstrated by the high standard deviation values for both). However, the trends in measured binding affinities are consistent with data from the corresponding catalytic assays, in which dephosphorylation values of 28%, 87%, and 99% were observed, respectively. The high binding affinity, and likely also the potency, of peptide 6f-w99 can be attributed to the slow dissociation rate, which indicates a stable complex with the PTPase. Under the conditions used for the other PTPases, binding interactions were not observed between DUSP22 and the phosphopeptides, including the unmodified parent peptide. Figure 8 presents the kinetic data, ka (association rate or on-rate) and kd (dissociation or off-rate), as they relate to the catalytic data (percent dephosphorylation), for VH1 and DUSP14 with a subset of the oxime-containing phosphopeptide library. From a drug design perspective, peptides that reside at the top of the graph in the upper and lower left quadrants include fragments with ideal inhibitor qualities of high catalytic activity and a long residence time (slow off-rate).
Figure 7.

Biomolecular interactions of phosphatases and phosphopeptide substrates: representative sensorgrams depicting the kinetic interaction between each PTPase or control protein and the parent peptide 5, negative control PEG, or oxime-containing peptides 6f-w202 and 6f-w99.
Figure 8.

Depiction of catalytic and kinetic affinity: mapping of the on-rate (ka) and off-rate (kd) of each ligand–enzyme interaction with respect to catalytic activity (% dephosphorylation). Labels are included for select oxime-containing peptides.
Substrate Enhancement and PTPase Inhibitors.
Peptide 12 was synthesized in order to test whether multiple fragments that increased dephosphorylation and binding activity (in comparison to 5) could be combined to generate an additive effect (Figure 3). Using ornithine amides to stabilize the fragment bonds, peptide 12 merges two fragments (w33 and w202) that showed moderate to high affinity for VH1 and DUSP14 in both assays. The dually functionalized phosphopeptide 12 was spotted onto a nitrocellulose slide, along with the monosubstituted peptides (11a,b), the original oxime-containing phosphopeptides 6d-w33 and 6f-w202, and the parent peptide 5. The catalytic assay was performed in the same manner as with the oxime-containing library described previously. The percent dephosphorylation results are presented in Figure 9 as fold change relative to the percent dephosphorylation of 5. Peptides modified with 4-(4-(trifluoromethyl)pyridin-2-yl)benzoic acid (w33) presented the highest activities, especially with VH1, while the ornithine amides reduced substrate activity levels. However, there was no clear enhancement of catalytic activity toward the bisubstituted peptide in comparison with the monosubstituted peptide. Though they were not further tested, it is conceivable that other fragment combinations besides w33 and w202 may result in increased PTPase activity. Since the fragment motifs were envisioned to afford enhanced interactions with regions proximal to but outside the catalytic cleft, we wondered whether reasonable affinity could be retained in the absence of the pTyr residue. In order to examine this hypothesis, peptides 13–15 were synthesized (Scheme 4) by tethering two w33 fragments in the absence of a pTyr residue. In order to enhance the hydrolytic stability of the oxime-ether moiety, the original oxime aminooxy oxygen was replaced by a methylene group, while the remainder of the oxime functionality was replaced by a structurally homologous amide (compound 10, Scheme 3). The best inhibitory potency (Figure 10) was achieved by 14 against VH1 (IC50 = 4 μM), while the same compound exhibited the poorest inhibitory potency against DUSP14. Both VH1 and DUSP14 were inhibited equally by 13 (IC50 = 38 and 31 μM, respectively), while inhibition by 15 was greater for VH1 than for DUSP14.
Figure 9.
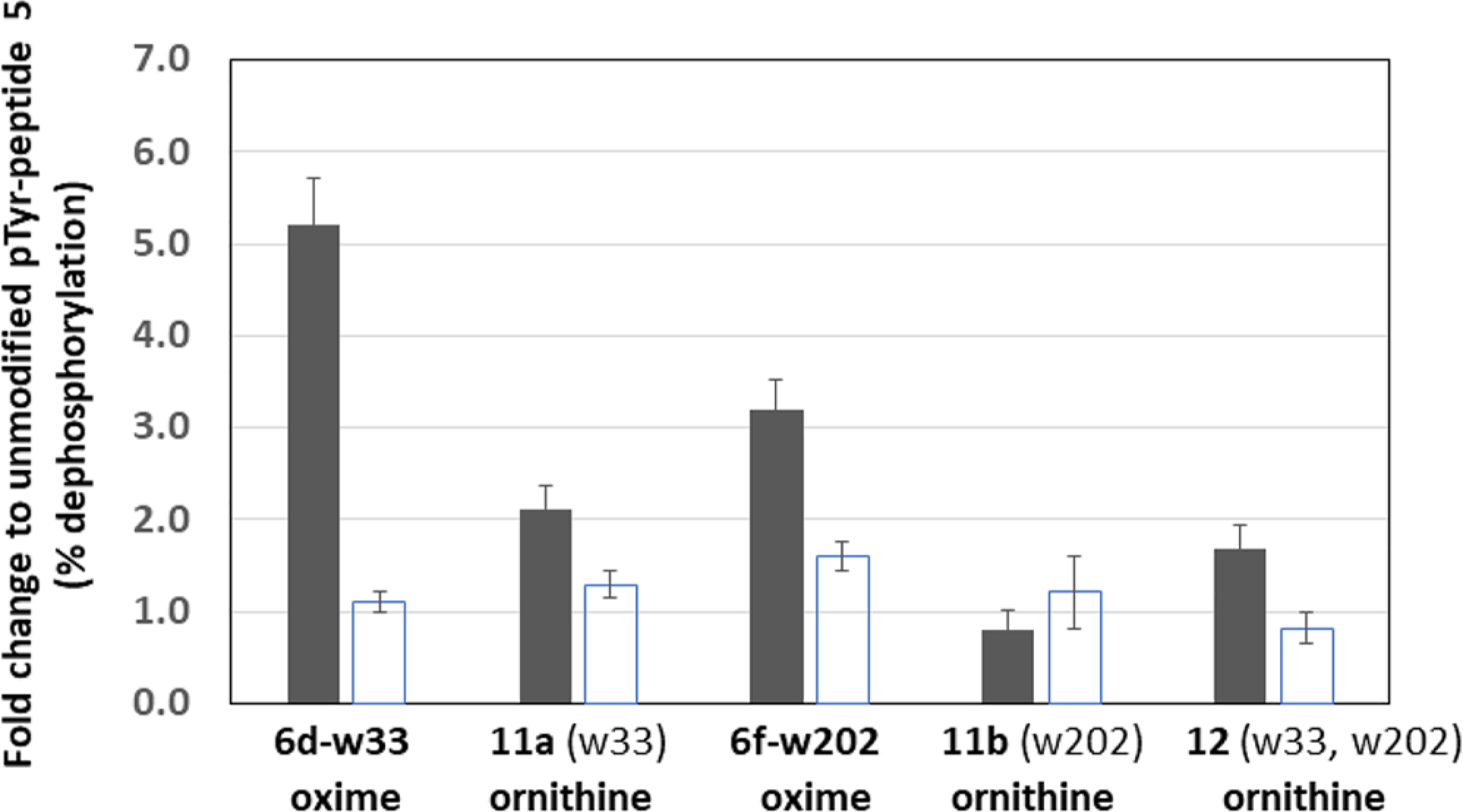
Enhanced peptide substrates. Peptides substituted with fragments w33 and w202 were examined as oxime or ornithine amides in a PTPase assay with VH1 (dark bars) and DUSP14 (open bars). Fragments w33 and w202 merged onto a single peptide backbone (12) were compared to monosubstituted peptides (11a,b). Data ± SD are presented as substrate activity (fold change) in comparison to the unmodified peptide 5.
Scheme 4. Synthesis of Peptides 13–15a.

aReagents and conditions: (a) 20% piperidine/NMP (room temperature, 15 min); (b) HATU, HOAt, DIEA, NMP (room temperature, 3 h); (c) 1% TFA, DCM (room temperature, 15 min); (d) TFA/TIPS/H2O (95/2.5/2.5) (room temperature, 1 h). Tpb = 4-(4trifluoromethyl)pyridin-2-yl)benzoic acid (10).
Figure 10.

Transformation of high-affinity substrates to inhibitors by substitution of two fragments onto a single peptide scaffold. Side chains of peptides 13–15 were replaced with a hydrolytically stable mimic of the oxime group from 6d-w33 and tested in a PTPase assay with peptide 5 as the substrate. Potencies for inhibition of peptide 5 dephosphorylation (IC50 ± SD) are shown.
CONCLUSION
Combinatorial synthetic efforts that targeted key substrate interactions with the catalytic site were previously reported to develop potent PTPase inhibitors.29,30 In the study described here, diverse chemical fragments were introduced into all residues of a short peptide substrate as the basis of a combinatorial screening approach. We synthesized a tethered fragment library of 1800 unique oxime phosphopeptide derivatives of a high-affinity heptapeptide scaffold that is widely employed as a general PTPase substrate. The entire oxime-containing peptide library was spotted onto microarray slides and evaluated against a panel of PTPases. This allowed us to measure a total of 7200 different catalytic interactions in a single assay. The results were analyzed according to amino acid position on the peptide chain, as well as by chemical structure or functional group of each oxime-derived fragment. The potencies of select oxime-containing peptides were confirmed by a microarray-based kinetic assay that simultaneously measured the association and dissociation rates of up to 200 peptide-enzyme complexes in real time. By combining the results from the catalytic and the kinetic assays, we identified multiple fragments that were preferred in different locations on the peptide. We removed the pTyr residue to form tethered bivalent constructs 13–15 and found that these exhibited micromolar PTPase inhibitory potencies. We rationalized that this reflected enhanced non-pTyr-dependent interactions with regions proximal to, but outside, the catalytic cleft. Collectively, these results suggest that chemical fragments identified by the described peptide-display method can be used as building blocks for designing nonpeptidyl inhibitors of PTPases.
EXPERIMENTAL SECTION
Synthesis.
General Considerations.
The following reagents are commercially available from Sigma-Aldrich: 2-bromo-4-(trifluoromethyl)pyridine (7) (cat. no. 661139) and (4-(methoxycarbonyl)phenyl)boronic acid (8) (cat. no. 594539). The following reagents are available from Chem-Impex, International: Fmoc-Orn(Mtt)-OH (cat. no. 03729); Fmoc-L-Tyr(HPO3Bzl)-OH (cat. no. 03746); Fmoc-Glu(OtBu)-OH (cat. no. 02413); Fmoc-Asp(OtBu)-OH (cat. no. 00494); Fmoc-Ala-OH (cat. no. 02369); Fmoc-Val-OH (cat. no. 02470), and (+) biotinyl-6-aminohexanoic acid (cat. no. 14003). Preparative high-pressure liquid chromatography (HPLC) was conducted using a Waters Prep LC4000 system having photodiode array detection and Phenomenex C18 columns (250 mm × 21.2 mm, 10 μm particle size, 110 Å pore size) at a flow rate of 10 mL/min. Binary solvent systems were employed consisting of A = 0.1% aqueous TFA and B = 0.1% TFA in CH3CN. Purified samples were lyophilized to provide final products as white powders. The mass spectra of the final peptides were measured by electrospray ionization-mass spectra (ESI-MS) using an Agilent 1200 HPLC system equipped with a diode-array detector set to measure UV absorption at 220 nm. Chromatography was carried out on a narrow-bore (100 mm × 2.1 mm), small-particle (3.5 μm) Zorbax Rapid-Resolution reversed-phase C18 column coupled with a C18 guard column of the same bonded phase (12.5 mm × 2.1 mm) eluted with a linear gradient of MeOH/H2O at a flow rate of 300 μL/min. Solvent A consisted of 5% CH3OH/H2O containing 0.1% CH3COOH, and solvent B consisted of 90% CH3OH/H2O containing 0.1% CH3COOH, employed in the following gradient: isocratic A for 3 min; linear gradient of B to 100% B in 9 min; isocratic B for 5 min; linear reset to 100% A in 5 min; isocratic A for 3 min to equilibrate.
(S)-Benzyl-2-(((benzyloxy)carbonyl)amino)-4-(((tert-butoxycarbonyl)amino)oxy)butanoate (3).
To a solution of 2 (2.96 g, 6.1 mmol) in CH2Cl2 (10 mL) at 0 °C was added methylhydrazine (0.80 mL, 0.70 mmol), and the reaction mixture was stirred at 0 °C (2 h). The mixture was filtered, and the filtrate was concentrated and dried under high vacuum. The resulting crude material was dissolved in THF (10 mL), and to this were added triethylamine (1.22 mL, 0.89 mmol) and Boc2O (1.9 g, 1.9 mmol); the solution was stirred at room temperature (overnight). The reaction mixture was diluted with EtOAc (20 mL), washed with H2O (2 × 20 mL) and brine (20 mL), and dried (MgSO4). The crude material was purified using silica gel CombiFash (Hex/EtOAc, 2/1) to give product 3 as a colorless oil (1.86 g, 4.1 mmol, 72%). 1H NMR (400 MHz, CDCl3): δ 7.23–7.31 (m, 10H), 5.12 (m, 2H), 5.08 (m, 2H), 4.52 (m, 1H), 3.81–3.92 (m, 2H), 2.09 (m, 2H), 1.42 (s, 9H). 13C NMR (400 MHz, CDCl3): δ 171.87 (1C), 156.89 (1C), 156.14 (1C), 136.36 (1C), 135.27 (1C), 128.52 (2C), 128.40 (2C), 128.35 (1C), 128.24 (2C), 127.97 (1C), 127.85 (2C), 85.10 (1C), 72.76 (1C), 67.17 (1C), 66.78 (1C), 51.72 (1C), 30.25 (1C), 28.11 (3C). ESI-MS (m/z): calcd for C24H30N2O7 + Na, 481.2; found, 481.2 [M + Na]+.
(S)-2-((((9H-fluoren-9-yl)methoxy)carbonyl)amino)-4-(((tert-butoxycarbonyl)amino)oxy)butanoic Acid (4).
A mixture of 3 (1.86 g, 4.1 mmol) and 10% Pd·C (0.19 g) was dissolved in MeOH (5 mL) and stirred under H2 (overnight). The mixture was passed through Celite, and the solvent was removed under reduced pressure. The crude material was dissolved in dioxane (10 mL) and H2O (10 mL), FmocOSu (1.64 g, 8.2 mmol) and NaHCO3 (0.68 g, 4.9 mmol) were added, and the reaction mixture was stirred at room temperature (overnight). The reaction mixture was washed with Et2O (10 mL), and the remaining aqueous layer was acidified with 1 N HCl to pH 3–4 and extracted with EtOAc (3 × 20 mL). The organic phase was dried (MgSO4) and filtered, and the solvent was evaporated. The crude product was purified using silica gel CombiFlash (MeOH/CH2Cl2 0–85%) and lyophilized from CH3CN/H2O (1/1 with 0.1% TFA) to provide product 4 as a white solid (1.65 g, 3.6 mmol, 88%). 1H NMR (400 MHz, CDCl3): δ 7.72 (d, J = 7.2 Hz, 2H), 7.62 (d, J = 7.2 Hz, 2H), 7.41 (s, 1H), 7.36 (t, J = 7.2 Hz, 2H), 7.27 (td, J = 1.2, 7.2 Hz, 2H), 4.57 (m, 1H), 4.36 (m, 2H), 4.21 (m, 1H), 3.96 (m, 1H), 2.14 (m, 1H), 1.46 (s, 9H). 13C NMR (400 MHz, CDCl3): δ 174.86 (1C), 157.78 (1C), 156.69 (1C), 143.87 (1C), 143.73 (1C), 141.23 (2C), 127.64 (2C), 127.04 (1C), 127.02 (1C), 125.22 (2C), 119.88 (2C), 82.71 (1C), 73.24 (1C), 67.26 (1C), 51.79 (1C), 47.08 (1C), 29.85 (1C), 28.11 (3C). ESI-MS (m/z): calcd for C24H28N2O7 + Na, 479.2; found, 479.2 [M + Na]+.
Methyl 4-(4-(Trifluoromethyl)pyridin-2-yl)benzoate (9).
To a solution of 2-bromo-4-(trifluoromethyl)pyridine (7) (0.547 mL, 4.42 mmol) in a solution of toluene/EtOH/H2O (5/1/5, 26.4 mL) were added Na2CO3 (4.22 g, 39.8 mmol), Pd(PPh3)4 (0.511 g, 0.442 mmol), and (4-(methoxycarbonyl)-phenyl)boronic acid (8) (1.194 g, 6.64 mmol), and the mixture was stirred at 110 °C (overnight). The mixture was cooled to room temperature, diluted with EtOAc (200 mL), and washed with H2O (50 mL) and brine (2 × 50 mL) and then dried (Na2SO4), filtered, and concentrated. The resulting residue was purified by silica gel CombiFlash to provide 9 as a white solid (805 mg, 65% yield). 1H NMR (400 MHz, CDCl3): δ 8.83 (d, J = 5.0 Hz, 1H), 8.11 (d, J = 8.4 Hz, 2H), 8.05 (d, J = 8.4 Hz, 2H), 7.91 (d, J = 0.6 Hz, 1H), 7.46–7.41 (m, 1H), 3.90 (s, 3H). DUIS-MS (m/z): 282 (MH+).
4-(4-(Trifluoromethyl)pyridin-2-yl)benzoic Acid (10).
To a solution of 9 (805 mg, 2.86 mmol) in THF/H2O (2/1, 9 mL) was added LiOH·H2O (240 mg, 5.7 mmol), and the mixture was stirred at room temperature (overnight). The mixture was acidified with ethereal HCl (3 mL, 2.0 M in Et2O) and concentrated, and the resulting residue was suspended in MeOH (3 mL), collected by filtration, washed (MeOH), and dried to provide 10 as a white solid (757 mg, 99% yield). 1H NMR (400 MHz, DMSO-d6): δ 13.04 (brs, 1H), 8.93 (d, J = 5.0 Hz, 1H), 8.34 (s, 1H), 8.31–8.22 (m, 2H), 8.03 (dd, J = 8.2, 1.3 Hz, 2H), 7.74 (d, J = 5.0 Hz, 1H). ESI-MS (m/z): 268.1 (MH+).
Synthesis of Peptides 5 and 6a–f.
Peptides were synthesized on Rink amide MBHA resin (Novabiochem, cat. no. 01–64-0037) by standard Fmoc solid-phase protocols using active ester coupling methodology in N,N-dimethylformamide (DMF). In summary, coupling was achieved by reacting each residue, amino acid, or acid (5.0 equiv based on resin loading), 1-hydroxybenzotriazole (HOBt) (5.0 equiv), and N,N’-diisopropylcarbodiimide (DIC) (5.0 equiv) (single couple, 2 h). Peptides 6a–f were prepared using reagent 4 where appropriate. The final resin was washed with DMF, MeOH, CH2Cl2, and Et2O and then dried under vacuum (overnight). Peptides were cleaved from the resin by treatment with TFA/triisopropylsilane (TIPS)/H2O (95/2.5/2.5, 5 mL, 4 h). The resin was removed by filtration, the peptide was precipitated in cold Et2O, and the precipitate was centrifuged and washed with Et2O. The resulting white solid was dissolved in 50% aqueous CH3CN (4 mL) and purified by reverse-phase preparative HPLC (analytical HPLCs of purified peptides are shown in Figure S1 in the Supporting Information).
Peptide 5. ESI-MS (m/z): calcd for C52H80N11O20PS − H, 1240.4; found, 1240.3 [M − H]−.
Peptide 6a. ESI-MS (m/z): calcd for C51H79N12O21PS + Na, 1281.5; found, 1281.3 [M + Na]+.
Peptide 6b. ESI-MS (m/z): calcd for C52H83N12O19PS+ H, 1243.5; found, 1243.3 [M + H]+.
Peptide 6c. ESI-MS (m/z): calcd for C53H83N12O21PS+ H, 1287.5; found, 1287.4 [M + H]+.
Peptide 6d. ESI-MS (m/z): calcd for C52H83N12O19PS + Na, 1265.5; found, 1265.4 [M + Na]+.
Peptide 6e. ESI-MS (m/z): calcd for C59H87N12O19PS + Na, 1251.5; found, 1251.4 [M + Na]+.
Peptide 6f. ESI-MS (m/z): calcd for C50H77N12O21PS + Na, 1268.5; found, 1268.2 [M + Na]+.
Synthesis of Peptides 11a,b and 12.
For peptides 11a,b and 12 active ester coupling was employed using N-Fmoc-protected amino acid (4 equiv), HATU (3.8 equiv), and DIEA (8.0 equiv) in N-methyl-2-pyrrolidinone (NMP). Coupling of Fmoc-L-Orn(Mtt)–OH was achieved using Fmoc-L-Orn(Mtt)-OH (2.0 equiv), HATU (1.9 equiv), and DIEA (4.0 equiv) in NMP by double coupling. After the completion of coupling of Fmoc-L-Orn(Mtt)-OH, the resin was then subjected to a global Mtt-deprotection by DCM/TFA/TIPS 95/1/4 (10 min × 2). The resulting resins were washed with 10% DIEA in NMP and were subsequently coupled with 3,5-dichloropyridine-4-carboxylic acid or 4-(4-(trifluoromethyl)-pridin-2-yl)benzoic acid (10). Peptide chains were then elongated using standard SPPS. The completed resins were treated with TFA/TIPS/H2O cocktail (95/2.5/2.5) for 3 h and then filtered. Cold Et2O was added to the filtrates, and the resulting precipitates were washed with cold Et2O (50 mL × 3). The crude product was dissolved in DMSO and purified by reverse-phase preparative HPLC (Analytical HPLCs of purified peptides are shown in Figure S1 in the Supporting Information).
Peptide 11a. Preparative HPLC conditions: linear gradient elution (79.9/20/0.1 H2O/acetonitrile/TFA to 29.9/70/0.1 H2O/acetonitrile/TFA over 30 min). ESI-MS (m/z): calcd for C66H91F3N13O19PS − H, 1488.6; found, 1488.4 [M − H]−.
Peptide 11b. Preparative HPLC conditions: linear gradient elution (79.9/20/0.1 H2O/acetonitrile/TFA to 54.9/45/0.1 H2O/acetonitrile/TFA over 30 min). ESI-MS (m/z): calcd for C57H80Cl2N13O21PS − H, 1414.4; found 1414.3, [M − H]−.
Peptide 12. Preparative HPLC conditions: linear gradient elution (69.9/30/0.1 H2O/acetonitrile/TFA to 39.9/60/0.1 H2O/acetonitrile/TFA over 30 min). ESI-MS (m/z): calcd for C71H91Cl2F3N15O20PS − H, 1662.5; found, 1662.3 [M − H]−.
Synthesis of Peptides 13–15.
Peptides 13–15 were synthesized on Rink amide MBHA resin by standard Fmoc solid-phase protocols using the active ester coupling methodology as described above, except that N-methyl-2-pyrrolidone (NMP) was used in place of DMF. Initial coupling was achieved on Fmoc-deprotected resin with a solution of Fmoc-L-Orn(Mtt)-OH (5.0 equiv), HATU (5.0 equiv), HOAt (5.0 equiv), and DIEA (10 equiv) in NMP (5 mL). The mixture was shaken for 3 h, and the solvent was then drained. The resin was washed with NMP (5 mL × 3, in 5 min/wash, drained after each wash) and iPrOH (5 mL × 3, in 5 min/wash, drained after each wash). The above sequence was repeated for the coupling of Fmoc-L-Gly-OH (5.0 equiv) (for peptides 14 and 15), Fmoc-L-Orn(Mtt)-OH (5.0 equiv), and acetic anhydride in pyridine (1/9 v/v, 5 equiv). After the completion of the peptide backbone, the resin was then subjected to a global Mtt-deprotection in a wash sequence of 1% TFA in CH2Cl2 (5 mL × 3, in 5 min/wash, drained after each wash), DIEA (5 mL × 3, in 5 min/wash, drained after each wash), and NMP (5 mL × 3, in 5 min/wash, drained after each wash). The resin was then reacted with a solution of 4-(4-(trifluoromethyl)pyridin-2-yl)benzoic acid (10, 10 equiv), HATU (5.0 equiv), HOAt (5.0 equiv), and DIEA (10 equiv) in NMP (5 mL). After the mixture was shaken (3 h), the solvent was drained. The resin was washed with NMP (5 mL × 3, in 5 min/wash, drained after each wash) and iPrOH (5 mL × 3, in 5 min/wash, drained after each wash). The final product was cleaved from the resin by shaking in a TFA/TIPS/H2O cocktail (95/2.5/2.5) (7.5 mL, 30 min). Another 7.5 mL portion of the cocktail was diluted with TFA (7.5 mL) to make a 15 mL solution. This solution was used to wash the resin twice (7.5 mL/wash, in 15 min/wash). The filtrate was collected, combined, and concentrated. Addition of cold Et2O caused precipitation of a white solid. The mixture was centrifuged and decanted. The white precipitate was collected and washed with cold Et2O (5 mL × 2). The crude product was dissolved in CH3CN and purified by reverse-phase preparative HPLC.
Peptide 13 (Ac-OtOt-NH2): 33 mg, 81% yield. ESI-MS (m/z): calcd for C38H37F6N7O5, 785.3; found, 786.3 [M + H]+, 808.2 [M + Na]+, 393.7 [M + 2H]+.
Peptide 14 (Ac-OtGOt-NH2): 26 mg, 59% yield. ESI-MS (m/z): calcd for C40H40F6N8O6, 842.3; found, 843.3 [M + H]+, 865.3 [M + Na]+, 422.2 [M + 2H]+.
Peptide 15 (Ac-OtGGOt-NH2): 16 mg, 34% yield. ESI-MS (m/z): calcd for C42H43F6N9O7, 899.3; found, 900.2 [M + H]+, 922.2 [M + Na]+, 450.7 [M + 2H]+.
Biological Studies.
Materials.
Single-pad and 16-pad nitrocellulose-coated FAST slides were purchased from KeraFAST, Inc. (Boston, MA, USA). Gold SPRi slides and index matching oil were purchased from Horiba Scientific (Edison, NJ, USA). NeutrAvidin, biotin-LC-NHS, biotin-PEG12-NHS, EDC, and NHS were purchased from Thermo Scientific (Rockford, IL, USA). Flexchip-blocking buffer was purchased from GE Healthcare Life Sciences (Piscataway, NJ, USA). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise mentioned.
Proteins.
Monoclonal anti-pTyr mouse antibody was purchased from Cell Signaling Technology, Inc. (#9411, Danvers, MA, USA). Alexafluor-conjugated goat antimouse antibody was purchased from Life Technologies, Inc. (Grand Island, NY, USA). The PTPase domain of YopH (residues 164–468) was expressed in Escherichia coli and purified as described previously,31 as were the Variola major H1 (VH1)32 and human DUSP14 dual specificity phosphatases.33 The catalytic domain of human DUSP22 was expressed and purified using a methodology previously described.34 The substrate trapping mutants of DUSP14 (C111S), DUSP22 (C88S), VH1 (C110S), and YopH (C403A/D356A) were constructed with a QuikChange site-directed mutagenesis kit (Agilent Technologies), following the manufacturer’s instructions. The nucleotide sequences of all expression vectors were confirmed experimentally.
Generation of pTyr-Peptide Library Arrays on Nitrocellulose Substrates.
The aminooxy-containing pTyr-peptides 6a–f, oxime-containing pTyr-peptides, and 5 (100 μM) were mixed with NeutrAvidin (33 μM) in citrate-buffered saline (CBS; 10 mM citrate buffer and 100 mM NaCl, pH 6.2) containing 50% glycerol overnight at 4 °C. The NeutrAvidin–peptide mixture was spotted identically onto eight FAST-slide nitrocellulose slides in triplicate using an inkjet ArrayJet microarrayer (Edinburgh, Scotland), and the slides were desiccated overnight under vacuum (25 mmHg) at 22 °C.
Catalytic Activity Assay.
Protein concentrations were determined by UV/vis using a NanoDrop 2000 instrument (Thermo Fischer Scientific, Waltham, MA, USA). Each slide was blocked with 1x flexchip blocking buffer for 1 h at 22 °C. Individual slides were treated with a PTPase diluted in CBS (pH 6.2) containing 0.05% Tween-20 and 1 mM DTT as follows: YopH (1 μg/mL, 1 min), VH1 (50 μg/mL, 15 min), DUSP14 (50 μg/mL, 15 min), and DUSP22 (100 μg/mL, 5 min). Two slides were treated with buffer only. Following the incubation periods, the PTPase and buffer solutions were discarded and the slides were washed with 1x TBS-T (4 × 5 mL, 3 min). The slides were then incubated with anti-pTyr mouse antibody (1/2000, 5 mL) overnight at 4 °C followed by a wash step. The primary antibody was probed with an Alexafluor647-conjugated-conjugated goat antimouse antibody (1/2500, 5 mL) for 1 h at 22 °C. The slides were then washed with 1x TBS-T (4 × 5 mL, 3 min) followed by one wash with distilled H2O and then air-dried. Each slide was scanned at 635 nm using a GenePix Microarray Scanner 4400A (Molecular Devices, Sunnyvale, CA, USA) with the PMT gain set to 400 and the power setting at 10.
Generation of pTyr-Peptide Subset Arrays on Au Substrates.
Self-assembled monolayers (SAMs) of 11-mercaptoundecanoic acid (11-MUA) were formed onto four gold slides by submerging each clean slide in a 2.5 mM ethanolic solution of 11-MUA for 20 h at 22 °C and rinsing with fresh EtOH. NeutrAvidin was then immobilized onto each surface via typical amine-coupling chemistry: the slides were submerged in 5 mL of an aqueous mixture of EDC (0.2 M) and NHS (0.05 M) for 10 min at 22 °C, followed by 5 mL of NeutrAvidin (200 μg/mL) in 10 mM sodium acetate (pH 5.0) for 2 h at 22 °C, and then 5 mL of 1 M ethanolamine-HCl (pH 8.5) for 10 min at 22 °C. The slides were rinsed with distilled H2O and then dried under a gentle stream of nitrogen. The oxime-containing pTyr-peptides, 6a–f, and 5 (100 μM in CBS, pH 6.2, with 20% glycerol), as well as controls (biotin-LC-NHS and biotin-PEG12 pretreated with 1 M ethanolamine-HCl pH 8.5) were arrayed onto the NeutrAvidin-coated gold slides in triplicate using an inkjet ArrayJet microarrayer, and the slides were desiccated overnight under vacuum (25 Torr) at 22 °C. Spotted slides were stored at 4 °C in an airtight container before use.
Kinetic Assay.
The SPRi-Plex II instrument (Horiba Scientific, Edison, NJ, USA) was cleaned and calibrated before each experiment as per the manufacturer’s instructions. Kinetic experiments were performed at 25 °C in 1x CBS-P (pH 6.2) containing 1 mM DTT and 1x flexchip-blocking buffer. Injections of anti-pTyr mAb (1/2000) were made before and after injection cycles of PTPases to confirm reproducibility. Serially diluted catalytically inactive mutant PTPases (DUSP14 C111S, DUSP22 C88S, VH1 C110S) were injected for 4 min at 50 μL/min, followed by 6 min of dissociation in buffer and then complete dissociation in 10 mM glycine-HCl pH 1.5 (2 min). PTPase concentrations were 0.05, 0.1, 0.5, 1, 2, and 5 μM. Experiments were repeated for DUSP22 C88S at concentrations of 0.1, 0.5, 1, 2.5, 5, 10, 20, and 40 μM. YopH C403A/D356A (1 μM) and His6-MBP (1 μM) were also injected as positive and negative controls, respectively. Sensorgrams were subtracted from reference spots and fitted to a Langmuir binding curve using the ScrubberGen 2.0 software (Horiba Scientific, Edison, NJ, USA).
PTPase Peptide Substrate and Inhibitor Assays.
Oxime-functionalized pTyr-containing peptides 6d-w33 and 6f-w202, aminooxy-functionalized pTyr-containing peptides 6d,f and EGFR pTyr-containing peptides 5, 11a,b, and 12 (100 μM each) were mixed with NeutrAvidin (33 μM) in CBS containing 40% glycerol for 30 min at 24 °C. Six replicates of each NeutrAvidin–peptide mixture were spotted in 16 identical blocks on a 16-pad nitrocellulose FAST slide using an ArrayJet microarraye,r and the slide was desiccated overnight under vacuum (25 mmHg) at 22 °C. The slide was blocked with 1x flexchip blocking buffer (5 mL) for 1 h at 22 °C. Using a 16-well gasket, each block was treated for 10 min with 25 μg/mL (80 μL) of VH1, DUSP14, VH1(C110S), or DUSP14-(C111S), 6 ng/mL of either YopH or YopH (C403A/D356A), or buffer only. PTPase dilution buffer consisted of CBS (pH 6.2) containing 0.05% Tween-20 and 1 mM DTT. The PTPase and buffer solutions were discarded, and the slides were washed with 1x TBS-T (3 × 5 mL, 5 min). The slides were then incubated with anti-pTyr mouse antibody (1/1000, 5 mL) for 1 h at 24 °C followed by a wash step. The primary antibody was probed with an Alexafluor 647-conjugated goat antimouse antibody (1/2500, 5 mL) for 1 h at 24 °C. The slides were then washed three times with 1x TBS-T followed by one time with distilled H2O and then air-dried. Each slide was scanned at 635 nm using a GenePix Microarray Scanner 4400A (Molecular Devices, Sunnyvale, CA, USA) with the PMT gain set to 400 and the power setting at 10. The peptide 5 microarray, assembled as above, was used to evaluate the inhibitory activity of fragment w33 substituted peptides 13–15. VH1 and DUSP14 were incubated (30 min, 24 °C) with dilutions (0–200 μM) of 13–15, and PTPase activity was assessed with the peptide 5 microarray (15 min, 24 °C) as above. IC50 values for inhibitors were determined with peptide 5 concentrations of 178 μM and inhibition curves that were fitted by nonlinear regression methods using ORIGIN 9.0 (OriginLab, Northampton, MA).
Supplementary Material
ACKNOWLEDGMENTS
This project was supported in part by federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract HHSN261200800001E and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by appointment of M.H. and B.M.Z. to the Research Participation Program for the U.S. Army Medical Research and Materiel Command, administered through an agreement between the U.S. Department of Energy and the USAMRMC. This work was supported in part by a JSPS Research Fellowship for Japanese Biomedical and Behavioral Researchers at NIH (K.T.). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services or the Department of Defense, nor does the mention of trade names, commercial products or organizations imply endorsement by the U.S. Government.
ABBREVIATIONS USED
- DUSP
dual-specificity phosphatase
- pTyr
phosphotyrosine
- PTPase
protein tyrosine phosphatase
- SPR
surface plasmon resonance
- VH1
Variola major H1
- YopH
Yersinia outer protein H
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscombs-ci.8b00122.
Structures of aldehydes used to make the oxime libraries, analytical HPLCs of synthetic peptides, and additional figures illustrating a heat map of catalytic data and representative sensorgrams (PDF)
REFERENCES
- (1).Alonso A; Sasin J; Bottini N; Friedberg I; Osterman A; Godzik A; Hunter T; Dixon J; Mustelin T Protein Tyrosine Phosphatases in the Human Genome. Cell 2004, 117 (6), 699–711. [DOI] [PubMed] [Google Scholar]
- (2).Tonks NK Protein tyrosine phosphatases - from housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013, 280 (2), 346–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Tautz L; Mustelin T Strategies for developing protein tyrosine phosphatase inhibitors. Methods 2007, 42, 250–260. [DOI] [PubMed] [Google Scholar]
- (4).Gao Y; Wu L; Luo JH; Yang D; Zhang Z-Y; Burke TR Jr. Examination of novel non-phosphorous-containing phosphotyrosyl mimetics against Protein Tyrosine Phosphatase 1B and demonstration of differential affinities toward Grb2 SH2 domains. Bioorg. Med. Chem. Lett 2000, 10, 923–927. [DOI] [PubMed] [Google Scholar]
- (5).Wood WJ; Patterson AW; Tsuruoka H; Jain RK; Ellman JA Substrate activity screening: a fragent-based method for the rapid idenitification of nonpeptidic protease inhibitors. J. Am. Chem. Soc 2005, 127 (44), 15521–15527. [DOI] [PubMed] [Google Scholar]
- (6).Erlanson DA; Wells JA; Braisted AC Tethering: Fragment-Based Drug Discovery. Annu. Rev. Biophys. Biomol. Struct 2004, 33, 199–223. [DOI] [PubMed] [Google Scholar]
- (7).Vetter SW; Keng Y-F; Lawrence DS; Zhang Z-Y Assessment of Protein-tyrosine Phosphatase 1B substrate specificity using ″Inverse Alanine Screening″. J. Biol. Chem 2000, 275, 2265–2268. [DOI] [PubMed] [Google Scholar]
- (8).Köhn M; Gutierrez-Rodrguez M; Jonkheijm P; Wetzel S; Wacker R; Schroeder H; Prinz H; Niemeyer CM; Breinbauer R; Szedlaczek SE; Waldmann H A microarray strategy for mapping the substrate specificity of protein tyrosine phosphatases. Angew. Chem., Int. Ed 2007, 46, 7700–7703. [DOI] [PubMed] [Google Scholar]
- (9).Mitra S; Barrios AM Identifying selective protein tyrosine phosphatase substrates and inhibitors from a fluorogenic, combinatorial peptide library. ChemBioChem 2008, 9, 1216–1219. [DOI] [PubMed] [Google Scholar]
- (10).Rotin D; Margolis B; Mohammadi M; Daly RJ; Daum G; Li N; Fischer EH; Burgess WH; Ullrich A; Schlessinger J SH2 domains prevent tyrosine dephosphorylation of the EGF receptor: identification of Try992 as the high-affinity binding site for SH2 domains of phospholipase C gamma. EMBO J. 1992, 11 (2), 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Beresford N; Patel S; Armstrong J; Balázs S; Fordham-Skelton AP; Tabernero L MptpB, a virulence factor from Mycobacterium tuberculosis, exhibits triple-specificity phosphatase activity. Biochem. J 2007, 406, 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang Z-Y; Maclean D; McNamara DJ; Sawyer TK; Dixon JE Protein tyrosine phosphatase substrate specificity: size and phosphotyrosine position requirements in peptide substrates. Biochemistry 1994, 33, 2285–2290. [DOI] [PubMed] [Google Scholar]
- (13).Flint AJ; Tiganis T; Barford D; Tonks NK Development of ″substrate-trapping″ mutants to idenitfy physiological substrates of protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 1680–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Blanchetot C; Chagnon M; Dubé N; Hallé M; Tremblay ML Substrate-trapping techniques in the idenitification of cellular PTP targets. Methods 2005, 35 (1), 44–53. [DOI] [PubMed] [Google Scholar]
- (15).Ivanov M; Stuckey JA; Schubert HL; Saper MA; Bliska JB Two substrate-targeting sites in the Yersinia protein tyrosine phosphatase co-operate to promote bacterial virulence. Mol. Microbiol 2005, 55 (5), 1346–1356. [DOI] [PubMed] [Google Scholar]
- (16).Haney CM; Loch MT; Horne WS Promoting peptide α–helix formation with dynamic covalent oxime side-chain cross-links. Chem. Commun 2011, 47, 10915–10917. [DOI] [PubMed] [Google Scholar]
- (17).Liu F; Thomas J; Burke TR Jr. Synthesis of homologous series of sidechain-extended orthogonally protected aminooxy-containing amino acids. Synthesis 2008, 2008, 2432–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Liu F; Stephen AG; Waheed AA; Aman MJ; Freed EO; Fisher RJ; Burke TR Jr. SAR by oxime-containing peptide libraries: application to Tsg101 ligand optimization. ChemBioChem 2008, 9, 2000–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Shao J; Tam JP Unprotected Peptides as Building Blocks for the Synthesis of Peptide Dendrimers with Oxime, Hydrazone, and Thiazolidine Linkages. J. Am. Chem. Soc 1995, 117 (14), 3893–3899. [Google Scholar]
- (20).Rose K; Zeng W; Regamey P-O; Chernushevich IV; Standing KG; Gaertner HF Natural Peptides as Building Blocks for the Synthesis of Large Protein-like Molecules with Hydrazone and Oxime Linkages. Bioconjugate Chem. 1996, 7 (5), 552–556. [DOI] [PubMed] [Google Scholar]
- (21).Kalia J; Raines RT Hydrolytic stability of hydrazones and oximes. Angew. Chem., Int. Ed 2008, 47 (39), 7523–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Liu F; Stephen AG; Waheed AA; Aman MJ; Freed EO; Fisher RJ; Burke TR Jr. SAR by oxime-containing peptide libraries: application to Tsg101 ligand optimization. ChemBioChem 2008, 9 (12), 2000–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Liu F; Park J-E; Qian W-J; Lim D; Scharow A; Berg T; Yaffe MB; Lee KS; Burke TR Identification of High Affinity Polo-like Kinase 1 (Plk1) Polo-box Domain Binding Peptides Using Oxime-Based Diversification. ACS Chem. Biol 2012, 7 (5), 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bahta M; Liu F; Kim S-E; Stephen AG; Fisher RJ; Burke TR Jr Oxime-based linker libraries as a general approach for the rapid generation and screening of multidentate inhibitors. Nat. Protoc 2012, 7, 686–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhao XZ; Hymel D; Burke RT Application of oxime-diversification to optimize ligand interactions within a cryptic pocket of the polo-like kinase 1 polo-box domain. Bioorg. Med. Chem. Lett 2016, 26, 5009–5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Stillman BA; Tonkinson JL FAST slides: a novel surface for microarrays. BioTechniques 2000, 29 (3), 630–635. [DOI] [PubMed] [Google Scholar]
- (27).de la Puerta ML; Trinidad AG; Rodríguez M; Bogetz J; Sánchez Crespo M; Mustelin T; Alonso A; Bayón Y Characterization of new substrates targeted by Yersinia tyrosine phosphatase YopH. PLoS One 2009, 4 (2), No. e4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhang Z-Y; Thieme-Sefler AM; Maclean D; McNamara DJ; Dobrusin EM; Sawyer TK; Dixon JE Substrate specificty of the protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U. S. A 1993, 90, 4446–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhang S; Chen L; Luo Y; Gunawan A; Lawrence DS; Zhang Z-Y Acquisition of a potent and selective TC-PTP inhibitor via a stepwise fluorophore-tagged combinatorial synthesis and screening strategy. J. Am. Chem. Soc 2009, 131, 13072–13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang S; Liu S; Tao R; Wei D; Chen L; Shen W; Yu ZH; Wang L; Jones DR; Dong XC; Zhang Z-Y A highly selective and potent PTP-MEG2 inhibitor with therapeutic potential for type 2 diabetes. J. Am. Chem. Soc 2012, 134, 18116–18124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhang Z-Y; Clemens JC; Schubert HL; Stuckey JA; Fischer MW; Hume DM; Saper MA; Dixon JE Expression, purification, and physiochemical characterisation of a recombinant Yersinia protein tyrosine phosphatase. J. Biol. Chem 1992, 267, 23759–23766. [PubMed] [Google Scholar]
- (32).Tropea JE.; Phan J.; Waugh DS. Overproduction, purification, and biochemical characterisation of the dual specificity H1 protein phosphatase encoded by Variola Major Virus. Protein Expression Purif. 2006, 50, 31–36. [DOI] [PubMed] [Google Scholar]
- (33).Lountos GT; Tropea JE; Cherry S; Waugh DS Overproduction, purification, and structure determination of human dual specificity phosphatase 14. Acta Crystallogr., Sect. D: Biol. Crystallogr 2009, 65, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tropea JE; Cherry S; Nallamsetty S; Bignon C; Waugh DS A generic method for the production of recombinant proteins in Escherichia coli using a dual hexahistidine-maltose-binding protein affinity; Humana Press: Totowa, NJ, 2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.