

Summary
Autophagy plays an important role in lipid breakdown, mitochondrial turnover, and mitochondrial function during brown adipose tissue (BAT) activation by thyroid hormone, but its role in BAT during adaptive thermogenesis remains controversial. Here, we examined BAT from mice exposed to 72 h of cold challenge as well as primary brown adipocytes treated with norepinephrine and found increased autophagy as well as increased β-oxidation, mitophagy, mitochondrial turnover, and mitochondrial activity. To further understand the role of autophagy of BAT in vivo, we generated BAT-specific Atg5 knockout (Atg5cKO) mice and exposed them to cold for 72 h. Interestingly, BAT-specific Atg5cKO mice were unable to maintain body temperature after chronic cold exposure and displayed deranged mitochondrial morphology and reactive oxygen species damage in their BAT. Our findings demonstrate the critical role of autophagy in adaptive thermogenesis, fatty acid metabolism, and mitochondrial function in BAT during chronic cold exposure.
Subject areas: Endocrine System Physiology, Molecular Physiology, Cell Biology
Graphical abstract
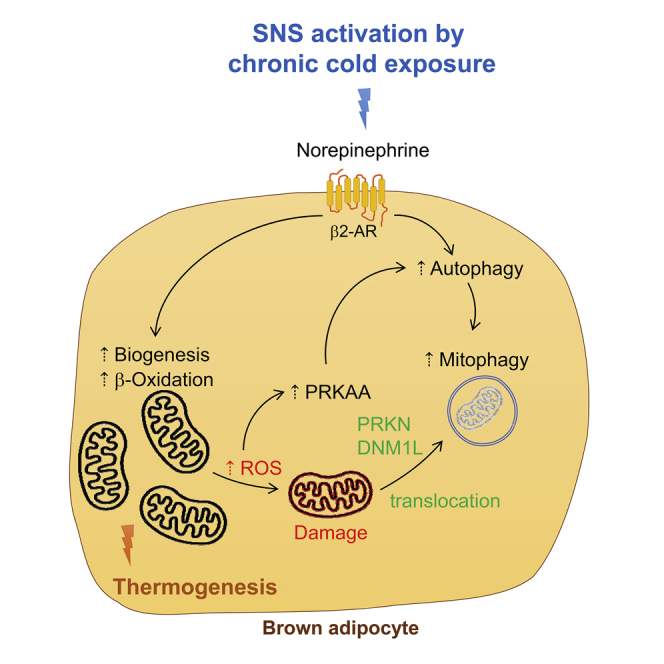
Highlights
-
•
Chronic cold stress stimulates β-oxidation in BAT to increase thermogenesis
-
•
Chronic cold stress stimulates mitophagy in BAT
-
•
Autophagy is critical for mitochondrial quality control to sustain thermogenesis
Endocrine System Physiology ; Molecular Physiology ; Cell Biology
Introduction
Adaptive thermogenesis is an essential survival mechanism for homeotherms that occurs whenever the ambient temperature falls below thermoneutrality (Silva, 2003). Brown adipose tissue (BAT) is a specialized tissue for generating adaptive thermogenesis (Lowell and Spiegelman, 2000; Stock, 1989) with high oxidative capacity. During sustained cold exposure, BAT undergoes remodeling to increase its thermogenic potential. Chronic cold exposure causes increased mitochondrial activity in BAT with induction of β-oxidation of fatty acids, electron transport activity, and uncoupling protein 1 (UCP1) expression to generate heat (Cannon and Nedergaard, 2004; Blondin et al., 2014, 2017). However, this increased mitochondrial activity also increases reactive oxygen species (ROS) (Barja De Quiroga et al., 1991; Buzadzic et al., 1999) that can lead to mitochondrial damage and impaired thermogenesis (Lettieri-Barbato, 2019).
To prevent accumulation of oxidative damage, BAT utilizes autophagy to remove and recycle damaged organelles such as mitochondria (mitophagy). BAT-specific knockdown of autophagy led to mitochondrial damage in BAT and impaired thermogenesis by thyroid hormone (T3) (Yau et al., 2019). However, the role of autophagy in BAT during cold exposure is not well understood as there are conflicting results in the literature (Martinez-Lopez et al., 2016) (Cairo et al., 2016; Lu et al., 2018). Martinez-Lopez et al. found that lipophagy was acutely upregulated after 1-h cold exposure to facilitate lipid utilization (Martinez-Lopez et al., 2016). Cairo et al. showed that autophagy was inhibited after 24 h of cold exposure and was associated with reduced clearance of UCP1 to facilitate heat generation (Cairo et al., 2016). On the other hand, Lu et al. observed increased mitophagy after 7 days of cold exposure (Lu et al., 2018). To better understand the regulation of autophagy during adaptive thermogenesis and autophagy's role in lipid metabolism and mitochondrial activity in BAT during cold stress, we examined autophagy in BAT from mice exposed to chronic cold exposure for 72 h and in primary brown adipocytes treated with norepinephrine (NE). Autophagy in BAT was highly stimulated after chronic cold exposure and in brown adipocytes after NE stimulation. This induction of autophagy was accompanied by increased autophagy gene transcription, β-oxidation of fatty acids, mitophagy, and mitochondrial turnover. BAT-specific Atg5 KO mice also were unable to maintain body temperature during cold exposure and showed evidence of ROS damage in BAT. Our findings demonstrate the critical role of autophagy for thermogenesis in BAT during chronic cold exposure and its impact on lipid metabolism and mitochondrial function.
Results
Chronic cold challenge induces autophagy in BAT
To examine the effect of cold exposure on autophagy in BAT, we housed C57BL/6J mice at 4°C for 72 h and at 30°C, the thermoneutral (TN) temperature for mice. Mice then were sacrificed and BAT harvested for analyses. After 72 h of cold exposure, there was a marked increase in MAP1LC3BII/microtubule-associated protein 1 light chain 3 beta II and decrease in SQSTM1 protein expression as well as increased MAP1LC3BI to MAP1LC3BII conversion suggesting there was an increase in autophagy after chronic cold exposure (Figure 1A).
Figure 1.

Chronic cold exposure increases autophagy in BAT and primary brown adipocytes
(A) Representative immunoblots and densitometry analysis of MAP1LC3BII, SQSTM1, and MAP1LC3BII:MAP1LC3BI ratio in BAT of mice subjected to 72 h of cold exposure.
(B) Immunoblots and densitometry analysis of PRKAA and MTOR pathway in BAT during cold exposure.
(C) mRNA expression of autophagy genes in the BAT of mice exposed to cold. Values are means ± SEM for 5 mice in each group.
(D) Autophagy flux analysis in primary brown adipocytes. Cells were treated with or without NE for 72 h, and bafilomycin (Baf) was added 6 h before harvest.
(E) Confocal microscopic images and quantification of red puncta in brown adipocyte cell line transfected with RFP-eGFP-MAP1LC3B plasmid and incubated with or without 1 μM NE for 72 h. Scale bar, 20 μm. Quantification of images (at least 10 transfected cells per sample in 3 different fields) was done using ImageJ software.
(F) mRNA expression of autophagy genes in primary brown adipocytes treated with 1 μM NE for 72 h. Statistical significance shows as ∗p < 0.05.
PRKAA/AMPK activation stimulates autophagy by phosphorylating the autophagosome initiator protein, Unc-51 Like Autophagy Activating Kinase 1 (ULK1) at serine 555 (S555) (Egan et al., 2011; Mao and Klionsky, 2011). In contrast, MTOR activation inhibits autophagy (Kim and Guan, 2015). We observed an increase in phosphorylation of PRKAA, its downstream target ACC, as well as ULK1 at S555 but no change in phosphorylation of MTOR or its downstream targets RPS6KB1 and EIF4EBP1, demonstrating that PRKAA was driving the increase in autophagy (Figure 1B). At the transcriptional level, there was induction of Map1lc3b, Sqstm1, Atg5, Atg7, and Ulk1 mRNA levels at 72 h suggesting that the increase in autophagy was associated with an increase in autophagy gene expression at 72 h (Figure 1C). Consistent with BAT activation, the mRNA levels of Ucp1 and Dio2 were also up-regulated after 72 h of cold exposure (Figure 1C).
Long-term NE treatment induces autophagy in primary brown adipocytes
Cold exposure activates adrenergic signaling in BAT to initiate thermogenesis (Silva, 2003). We next examined the direct effect of adrenergic stimulation on BAT autophagy by treating primary brown adipocytes with NE. We observed that when cells were treated with NE for 72 h, they showed increased MAP1LC3BII and reduced SQSTM1 expression that was consistent with increased autophagy flux (Figure 1D). When we used bafilomycin (Baf), to block late autophagy, there was a greater increase in MAP1LC3BII after Baf addition in the NE-treated cells versus untreated control cells (0.83 versus 0.54), showing that NE increased autophagic flux (Figure 1D). We further examined autophagic flux by transfecting a tandem fluorescent Map1lc3b (RFP-eGFP-MAP1LC3B) plasmid expressing MAP1LC3B with the green and red fluorescent domains into a brown adipocyte mouse mBAP-9 cell line. Both green and red signals are emitted when the fusion protein is within an autophagosome. However, the green signal is quenched and only red fluorescence is emitted in the acidic environment of the fused autolysosome. In cells incubated with NE for a period of 72 h, there was increased number of red puncta indicating they had higher autophagic flux than control cells (Figure 1E).
Similar to our in vivo observations after cold exposure, we observed induction of autophagy gene expression after 72 h of NE treatment in primary brown adipocytes (Figure 1F). We also detected upregulation of Ucp1 and Dio2 gene expression after 72 h of NE treatment. These results strongly suggested that autophagy was activated after chronic BAT stimulation.
Chronic cold exposure increases β-oxidation of fatty acids in BAT
To examine mitochondrial activity in BAT after chronic cold exposure, we next performed analyses of acylcarnitine and tricarboxylic acid (TCA) intermediates in BAT from mice exposed to cold temperature for 72 h. Chronic cold exposure significantly increased short-, medium-, and long-chain acylcarnitine species (Figures 2A–2C) suggesting there was increased mitochondrial β-oxidation. The level of acetyl-CoA (C2 carnitine) was only mildly upregulated, most likely due to rapid oxidation of acetyl-CoA via the TCA cycle because TCA cycle intermediates also were increased (Figure 2D). Additionally, we found that phosphorylated adipose triglyceride lipase (PNPLA2/ATGL) and hormone-sensitive lipase (LIPE/HSL) were both decreased, whereas CPT1α and MCAD1 protein expression were increased (Figure 2E). These findings highlighted that autophagy, rather than lipase activity, may be the major mechanism for mobilizing triglycerides from fat droplets and hydrolyzing them to free fatty acids for mitochondrial β-oxidation. Taken together, these results showed that chronic cold exposure stimulated CPT1α and MCAD protein expression in BAT to increase fatty acid β-oxidation in vivo.
Figure 2.

Chronic cold exposure induces lipophagy and β-oxidation in BAT
(A–D) Metabolomic profiling of (A) short-, (B) medium-, and (C) long-chain acylcarnitines species and (D) level of TCA cycle intermediates in BAT.
(E) Immunoblot and densitometry analysis of lipases and β-oxidation markers in BAT. Values are means ± SEM for 5 mice in each group. Statistical significance shows as ∗p < 0.05.
Chronic cold exposure increases mitophagy and mitochondrial turnover in BAT
We also examined mitophagy in BAT from mice after 72 h of cold exposure and observed increased amounts of autophagosomes, some of which contained mitochondrial remnants by electron microscopy (Figure 3A). We then analyzed the mitochondrial fraction of BAT and found that both MAP1LC3BII and SQSTM1 proteins were recruited to the mitochondrial fraction of cold-treated mice (Figure 3B). PRKN/E3 ubiquitin-protein ligase parkin and PINK1/PTEN-induced kinase 1 coordinate to activate selective mitophagy of damaged mitochondria (Wei et al., 2015; Jin and Youle, 2012; Bingol and Sheng, 2016). Dynamin-related protein 1 (DNM1L/DRP1) is responsible for mitochondrial fission, which facilitates PRKN-mediated mitophagy (Burman et al., 2017). Of note, DNM1L and PRKN levels were significantly higher in the mitochondrial fraction of BAT from mice exposed to 72 h of cold challenge than those from control mice (Figure 3B) and suggest that mitochondrial fission and mitophagy were increased by chronic cold exposure.
Figure 3.

Chronic cold exposure induces mitophagy and mitochondrial turnover in BAT
(A) Representative electron microscopic images showing BAT of mice housed at TN (30°C) or in cold (4°C) for 72 h. White box shows an autophagosome containing mitochondria. Scale bar, 0.2 μm.
(B) Immunoblots and densitometry of autophagy proteins in mitochondrial fraction of BAT.
(C) Confocal microscopic images and quantification of red puncta in brown adipocyte cell line transfected with mito-RFP-EGFP plasmid and treated with or without 1 μM NE for 72 h. Scale bar, 20 μm. Quantification of images (at least 10 transfected cells per sample in 3 different fields) was done using ImageJ software.
(D) Immunoblots and densitometry of mitochondrial proteins in BAT.
(E) Confocal microscopic images showing brown adipocyte cell line transfected with pMitoTimer plasmid and treated with or without 1 μM NE for 72 h. Bafilomycin (Baf) was added 6 h before harvest. Scale bar, 20 μm.
(F) Seahorse analysis of oxygen consumption rate (OCR) for primary brown adipocytes treated with 1 μM NE for 72 h. For the NE-treated group, 1 μM NE was added to the assay media 1 h before OCR measurement.
(G) Representative immunoblots and densitometry showing mitochondrial protein expression in primary brown adipocytes treated with 1 μM NE for 72 h. Statistical significance shows as ∗p < 0.05, ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001.
We next transfected mBAP-9 brown adipocytes with a tandem-tagged mito-RFP-EGFP plasmid, which encoded a fusion protein containing a mitochondrial targeting signal with green and red fluorescent proteins, with and without NE for 72 h. Similar to MAP1LC3B-RFP-EGFP, only red fluorescence is detected after mitochondria are localized within autolysosomes (mitophagy). NE treatment markedly increased the number of red puncta (Figure 3C) and demonstrated that β-adrenergic stimulation of brown adipocytes increased mitophagy.
Cold exposure for 72 h significantly increased the expression of mitochondrial proteins such as UCP1, VDAC, TOMM20, and COX4I1 as well as the master regulator for mitochondrial biogenesis, PCG1α (Figure 3D). These findings suggested that there was significant mitochondrial biogenesis that led to an overall net gain of mitochondrial level despite active mitophagy. To further examine mitochondrial turnover, we transfected mBAP-9 cells with a pMitoTimer plasmid (Laker et al., 2014), a fluorescent protein containing a DsRed mutation that causes a time-dependent transition from green fluorescence to a more stable red conformation over 48 h (Terskikh et al., 2000) to monitor the synthesis, maturation, and degradation of mitochondria. In the control cells, we detected both red and green signals, resulting in a yellow color in the overlay (Figure 3E). However, treating cells with NE for 72 h reduced the red signal and increased the green fluorescent signal indicating removal of mature mitochondria and synthesis of new mitochondria under this condition (Figure 3E). Moreover, when autophagy was inhibited by Baf, NE treatment did not reduce the red signal, suggesting that mitochondrial clearance was blocked by decreased mitophagy. Taken together, these findings suggested that there was both degradation of mature mitochondria by mitophagy and synthesis of new mitochondria that led to a net increase in the total amount of mitochondria. We next examined the effect of this increased mitochondrial turnover on mitochondrial respiration by Seahorse oximetry by treating primary brown adipocytes with NE for 72 h. These cells showed significantly higher basal oxygen consumption rate (OCR) as well as maximum respiratory capacity than untreated cells (Figure 3F). Consistent with increased mitochondrial activity, NE-treated cells also showed increased amounts of mitochondrial proteins (Figure 3G). Collectively, these findings showed that β-adrenergic stimulation increased mitophagy, mitochondria number, and mitochondrial turnover to stimulate mitochondrial respiration.
Autophagy is essential for BAT thermogenesis during chronic cold exposure
To determine whether autophagy was absolutely required for thermogenesis during chronic cold challenge, we generated BAT-specific Atg5 conditional knockout mice (Atg5cKO) by injecting Ucp1 promoter-driven Cre-expressing adeno-associated virus (AAV) into Atg5flox/flox mice and exposed them to either TN conditions or cold challenge for 72 h. After harvesting BAT, we confirmed that Atg5 expression was decreased in Atg5cKO mice but they did not exhibit any decrease in autophagy at TN (Figure 4A). During chronic cold exposure, BAT from control mice increased expression of Atg5, MAP1LC3BII, and SQSTM1 protein as well as MAP1LC3BII: MAP1LC3BI ratio consistent with high autophagic flux (Figure 4A). In contrast, BAT from Atg5cKO mice showed no change in MAP1LC3BII, increased SQSTM1, and reduced MAP1LC3BII:MAP1LC3BI ratio in response to cold exposure, suggesting there was decreased autophagy flux (Figure 4A). When we examined body temperature, wild-type (WT) and Atg5cKO mice maintained body temperature at TN and WT mice maintained body temperature after 72-h cold exposure (Figure 4B). In contrast, Atg5cKO mice were unable to sustain their body temperature after cold exposure (Figure 4B). These findings clearly demonstrated that autophagy in BAT was necessary for thermogenesis in vivo during chronic cold exposure.
Figure 4.

Autophagy in BAT is required for thermogenesis in chronic cold exposure
(A) Representative blots and densitometry of autophagy proteins in BAT of control and Atg5cKO mice housed at TN (30°C) or in cold (4°C) for 72 h.
(B) Body temperature of control and Atg5cKO mice housed at TN or in cold for 72 h.
(C) Electron microscopic images showing damaged mitochondria in cold-exposed Atg5cKO mice. Scale bars: 2 μm (10,000×) and 0.5 μm (25,000×).
(D) Immunoblots and densitometry showing increased level of protein carbonyls in cold-exposed Atg5cKO mice.
(E) Representative immunoblots and densitometry showing mitochondrial proteins.
(F) Immunoblots showing autophagy proteins in mitochondrial fraction of BAT.
(G) Representative immunoblots and densitometry of proteins involved in lipolysis and PRKAA pathway in BAT.
(H) Seahorse analysis of OCR in primary brown adipocytes transfected with control or Atg5 siRNA for 48 h before analysis. Statistical significance shows as ∗p < 0.05 and ∗∗∗∗p < 0.0001.
See also Figure S1.
We next examined the effects of blocking autophagy in BAT on mitochondria after 72-h cold exposure. Using electron microscopy, we found that BAT mitochondria from WT mice had an elongated oval shape at TN. Upon cold challenge, these mitochondria became more round but maintained intact membranes and normal cristae structure. Interestingly, when Atg5cKO mice were exposed to chronic cold challenge, BAT mitochondria became swollen and contained distorted cristae (Figure 4C). Additionally, protein carbonyl formation due to ROS generated from mitochondria was markedly increased in Atg5cKO mice during chronic cold exposure but only mildly upregulated in WT mice (Figure 4D). Accordingly, we observed increases of mitochondrial proteins (COX4I1, TOM20, and VDAC but not UCP1) in Atg5cKO mice exposed to chronic exposure due to increased accumulation of mitochondria when autophagy was inhibited by Atg5 KO (Figure 4E). These increases were unlikely due to increased mitochondrial biogenesis after KO because the level of PPARG coactivator 1 alpha (PPARGC1A/PGC1α) protein was not further induced in Atg5cKO mice exposed to chronic exposure.
We next analyzed the BAT mitochondrial fractions from WT and Atg5cKO mice exposed to chronic cold challenge (Figure 4F). We observed decreased ULK1 accumulation in BAT mitochondria from Atg5cKO mice, which in turn led to less MAP1LC3BII recruitment to BAT mitochondria than WT mice. There also was increased PRKN recruitment due to increased mitochondrial membrane damage and decreased mitophagy. Additionally, there was increased SQSTM1 with decreased LC3B recruitment indicative of decreased mitophagy. This decrease in mitophagy most likely led to the morphological changes observed in mitochondria, ROS damage, and insufficient thermogenesis observed in the BAT from Atg5cKO mice (Figures 4A–4D).
We next examined the effects of chronic cold exposure on the cell signaling of BAT from Atg5cKO mice by measuring phosphorylation of proteins (PNPLA2 and ACACA) involved in lipid metabolism in BAT (Figure 4G). PNPLA2 protein phosphorylation was higher in Atg5cKO mice than control mice during cold exposure, suggesting there was a compensatory increase in lipase activity when lipophagy was inhibited. Of note, the BAT expression of Ucp1 mRNA as well as sqstm1, Map1lc3b, Pink1, and Prkn were similar in WT and Atg5cKO mice after chronic cold exposure (Figure S1). Phosphorylated PRKAA protein was increased in both WT and Atg5cKO mice during chronic cold exposure; however, it was higher in Atg5cKO mice (Figure 4E), and was most likely due to lower intracellular ATP, and higher AMP, levels in the Atg5cKO mice owing to impaired mitochondrial function (Hinchy et al., 2018). These data were also consistent with the higher ROS damage found in Atg5cKO mice because mitochondrial ROS can directly promote PRKAA activation (Rabinovitch et al., 2017). Significantly, the increase in PRKAA did not further increase phosphorylation of ACACA and ULK1, two PRKAA targets, suggesting that inhibition of fatty acid synthesis and ULK1 activation already was maximal in both groups of mice.
Finally, to investigate the effect of autophagy loss on mitochondrial function during chronic cold exposure, we used Seahorse oximetry to measure OCR in primary brown adipocytes transfected with Atg5 small interfering RNA (siRNA) in the presence of NE (Figure 4G). We found that primary brown adipocytes with Atg5 knockdown showed significantly less basal OCR, maximal respiratory capacity, and acute NE-stimulated OCR (Figure S1B) than control cells despite having higher amounts of mitochondrial proteins (Figure S1C). These data strongly suggested that impaired autophagy in brown adipocytes led to mitochondrial dysfunction and impaired thermogenic response to NE.
Discussion
In the present study, we report that BAT thermogenesis in response to chronic cold exposure required autophagy. This finding was consistent with earlier reports suggesting there was increased autophagy during long-term cold exposure (Martinez-Lopez et al., 2016; Lu et al., 2018). However, it contrasted with an earlier report of decreased autophagy in BAT after 24 h of cold exposure (Cairo et al., 2016). It is possible that these differences may be due to temporal effects of cold exposure on autophagy, with the increase in autophagy during chronic exposure attributable to the transcriptional induction of autophagy genes that we observed after 72 h of cold exposure (Figures 1C and 1E). Our findings also were consistent with our previous study showing that T3 induced autophagy in BAT during activated thermogenesis at room temperature (Yau et al., 2019). Taken together with our findings, induction of autophagy appears to occur during both chronic cold exposure and pharmacological stimulation of BAT.
BAT has been reported to undergo metabolic adaptations such as lipogenesis (Christoffolete et al., 2004), mitochondrial biogenesis (Murholm et al., 2009; Yu et al., 2015; Gospodarska et al., 2015), mitophagy (Lu et al., 2018), and UCP-1 induction during chronic cold exposure. Here, we found that chronic cold stress led to increased fatty acid β-oxidation, mitochondrial turnover, respiration, and thermogenesis in addition to autophagy. We found that chronic cold stress also mildly increased the level of protein carbonyls (Figure 4D) indicative of increased ROS generated by active mitochondria. This increased protein oxidation coincided with the marked increase in mitochondrial activity that occurs in BAT during cold exposure. As ROS directly activates PRKAA (Rabinovitch et al., 2017), it likely plays an important role in stimulating autophagy in BAT during chronic cold exposure to promote mitophagy. We also observed PRKAA activation during chronic cold exposure, which can stimulate autophagy as well as increase transcription of autophagy genes (Sukumaran et al., 2020). This increase in transcription would lead to the synthesis and replenishment of autophagy proteins that are necessary for sustained induction of autophagy.
We found that induction of autophagy/mitophagy was important for removal of damaged mitochondria in BAT after chronic cold exposure or in primary brown adipocytes after NE stimulation (Figures 3C and 3E). These processes were accompanied by recruitment of PRKN and DNM1L protein to BAT mitochondria after chronic cold exposure (Figure 3D). These proteins are critical for mitophagy and mitochondrial fission, respectively. There also was increased mitochondrial turnover, mitochondrial biogenesis, total mitochondrial protein, and mitochondrial activity after cold exposure or NE stimulation (Figures 3C–3F). When autophagy was blocked by inhibiting Atg5 expression in BAT during cold exposure or NE stimulation of brown adipocytes, both mitochondrial integrity and activity were compromised and were accompanied by a corresponding increase in protein carbonyls reflecting oxidative damage (Figures 4B, 4C, 4D, and 4H). There also was accumulation of PRKN and SQSTM1, and decreased recruitment of ULK1 and MAP1LC3BII in the mitochondrial fraction of BAT, consistent with impaired mitophagy after Atg5 KO. Additionally, autophagy per se did not appear to affect mitochondrial biogenesis directly because we observed similar levels of PPARGC1A protein in both control and Atg5cKO mice after 72 h of cold exposure. Therefore, it is unlikely that the defective thermogenesis in Atg5cKO mice was due to reduced synthesis of healthy mitochondria in BAT. Instead, it is more likely that accumulation of damaged mitochondria from impaired mitophagy increased oxidative stress and damage, which in turn further reduced mitochondrial function. It was reported that mice that lacked all thermogenic capacity, such as the Ucp1 KO mice, had severely reduced body temperature of ∼30°C after 3 h of cold (4°C) exposure (Shabalina et al., 2010; Meyer et al., 2010). Interestingly, the Atg5cKO mice still were able to maintain their body temperature at ∼35.5°C after 72 h of cold exposure suggesting that they still possessed some functional mitochondria activity despite impaired mitophagy. These findings suggested that impaired mitophagy reduced, but did not totally abolish, adaptive thermogenesis during chronic cold exposure. Further studies using indirect calorimetry to compare Atg5 KO and Ucp1 KO mice under similar cold stress conditions would be useful for analyzing the degree of thermogenic impairment specifically due to loss of autophagy versus complete loss of Ucp1.
Chronic cold exposure increased short-, medium-, and long-chain acylcarnitines as well as increased Cpt1a and Mcad mRNA expression in BAT (Figure 2). These findings strongly suggested there was increased acylcarnitine flux and β-oxidation of fatty acids. Interestingly, pPNPLA2 and pLIPE also were down-regulated in BAT during chronic cold exposure (Figure 2E) suggesting that enzymatic hydrolysis of triglycerides was not a major mechanism for generating fatty acid substrates for β-oxidation. Our findings were consistent with a previous study that showed that acute cold exposure markedly stimulated BAT lipolysis to provide fuel for β-oxidation and generate heat, which then led to rapid depletion of lipid droplets after 12 h (Christoffolete et al., 2004). Moreover, lipophagy contributed to this process by breaking down lipid droplets stored in BAT (Martinez-Lopez et al., 2016; Zhang et al., 2020). Somewhat surprisingly, we did not observe autophagosomes-containing lipid droplets in the electron micrographs of BAT from mice chronically exposed to cold temperature (Figure 3A). It is possible that there was rapid lipophagy of fat droplets that was difficult to detect by electron microscopy or perhaps direct utilization of fatty acids generated by white adipose tissue lipolysis did not require incorporation into fat droplets after uptake by BAT during chronic cold exposure. However, it is noteworthy that phosphorylation of PRKAA and PNPLA2 were increased in BAT from Atg5 KO mice after chronic cold exposure. Thus it appears that compensatory lipase activity was stimulated when autophagy (presumably lipophagy) was inhibited (Figure 4G).
Besides its effects on lipid metabolism and mitochondrial quality control during chronic cold stimulation, autophagy also may have effects on other cellular processes in BAT. Cold acclimation induced hyperplasia of BAT with significant proliferation of preadipocytes and vascular endothelial cells (Klingenspor, 2003; Bukowiecki et al., 1986; Fukano et al., 2016). In this connection, autophagy was found to regulate both brown adipocyte differentiation (Fu et al., 2019; Martinez-Lopez et al., 2013) and angiogenesis (Hassanpour et al., 2018; Du et al., 2012). Chronic cold exposure also induced the conversion of competent WAT into brown-like fat cells that have thermogenic potential (browning) (Herz and Kiefer, 2019; Wu et al., 2013; Lin et al., 2015). Further studies are needed to better understand the role of autophagy in these chronic processes induced by long-term cold exposure.
In summary, our study demonstrated that chronic cold exposure and/or NE stimulation induced autophagy, β-oxidation of fatty acids, and mitochondrial turnover and activity in BAT and primary brown adipocytes. Autophagy played a critical role in maintaining mitochondrial integrity and function that is necessary for thermogenesis during chronic cold exposure in vivo. Mitophagy was induced in BAT during chronic cold exposure, and its loss led to impaired thermogenesis in vivo and decreased respiration in primary brown adipocytes. BAT activity has been negatively associated with obesity and diabetes in man (Cypess and Kahn, 2010; Yoneshiro et al., 2019). Thus, our results support the notion that pharmacological modulation of autophagy in BAT to mimic the thermogenic effects observed during chronic cold exposure may be a potential approach to improve BAT activity for the treatment of metabolic diseases such as obesity and diabetes.
Limitations of the study
It is possible that chronic cold exposure has effects on BAT that are independent of sympathetic activation. However, we were unable to perform BAT denervation to examine cold-induced autophagy with impaired sympathetic nerve activation. It is also possible that lipophagy plays a role in facilitating fatty acid oxidation in chronic cold exposure to sustain thermogenesis. However we did not show evidence of lipophagy because this is beyond the scope of this study.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Paul Michael Yen, MD (paul.yen@duke-nus.edu.sg).
Materials availability
This study did not generate new reagents. All reagents are commercially available.
Data and code availability
This study did not generate new dataset or code.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
The research was funded by National Medical Research Council in Singapore (NMRC) (CSASI19may-0002) (P.M.Y.), NMRC/CIRG/1457/2016 (P.M.Y.), MOH-000319 (MOH-OFIRG19may-0002) (B.K.S.) and the Citrin Foundation (W.W.Y).
The author would like to thank Dr. Rohit A. Sinha, Dr. Madhulika Tripathi, Dr. Eveline Bruinstroop, Dr. Ronny Lesmana, Dr. Kenji Ohba, Mr. Sherwin Xie, Ms. Andrea Lim, Ms. Jiapei Ho, and Dr. Ufuk Degirmenci for their helpful advice, technical support, and reagents. We are thankful to Prof. Jean-Paul Kovalik and Prof. Jianhong Ching for performing metabolic profiling (Metabolomics@ Duke-NUS). We would also like to thank Prof. T. Yoshimori (Osaka University, Osaka, Japan) and Dr. Andreas Till (Institute of Clinical Molecular Biology Christian-Albrechts-University of Kiel; Kiel, Germany) for gifting plasmids.
Author contributions
Conceptualization, W.W.Y., K.A.W., and P.M.Y.; methodology, W.W.Y., K.A.W., and P.M.Y.; investigation, K.A.W., W.W.Y., N.K.T., B.K.S., J.Z., and Y.W.; writing − original draft, K.A.W. and W.W.Y.; writing − review & editing, W.W.Y., K.A.W., B.K.S., and P.M.Y.; funding acquisition, P.M.Y. and B.K.S.; resources, B.-H.B. and P.M.Y.; supervision, P.M.Y.
Declaration of interests
The authors declare no competing interests.
Published: May 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102434.
Supplemental information
References
- Barja De Quiroga G., Lopez-Torres M., Perez-Campo R., Abelenda M., Paz Nava M., Puerta M.L. Effect of cold acclimation on GSH, antioxidant enzymes and lipid peroxidation in brown adipose tissue. Biochem. J. 1991;277(Pt 1):289–292. doi: 10.1042/bj2770289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B., Sheng M. Mechanisms of mitophagy: PINK1, parkin, USP30 and beyond. Free Radic. Biol. Med. 2016;100:210–222. doi: 10.1016/j.freeradbiomed.2016.04.015. [DOI] [PubMed] [Google Scholar]
- Blondin D.P., Daoud A., Taylor T., Tingelstad H.C., Bezaire V., Richard D., Carpentier A.C., Taylor A.W., Harper M.E., Aguer C., Haman F. Four-week cold acclimation in adult humans shifts uncoupling thermogenesis from skeletal muscles to brown adipose tissue. J. Physiol. 2017;595:2099–2113. doi: 10.1113/JP273395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondin D.P., Labbe S.M., Tingelstad H.C., Noll C., Kunach M., Phoenix S., Guerin B., Turcotte E.E., Carpentier A.C., Richard D., Haman F. Increased brown adipose tissue oxidative capacity in cold-acclimated humans. J. Clin. Endocrinol. Metab. 2014;99:E438–E446. doi: 10.1210/jc.2013-3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukowiecki L.J., Geloen A., Collet A.J. Proliferation and differentiation of brown adipocytes from interstitial cells during cold acclimation. Am. J. Physiol. 1986;250:C880–C887. doi: 10.1152/ajpcell.1986.250.6.C880. [DOI] [PubMed] [Google Scholar]
- Burman J.L., Pickles S., Wang C., Sekine S., Vargas J.N.S., Zhang Z., Youle A.M., Nezich C.L., Wu X., Hammer J.A., Youle R.J. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017;216:3231–3247. doi: 10.1083/jcb.201612106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzadzic B., Korac B., Petrovic V.M. The effect of adaptation to cold and re-adaptation to room temperature on the level of glutathione in rat tissues. J. Therm. Biol. 1999;24:373–377. [Google Scholar]
- Cairo M., Villarroya J., Cereijo R., Campderros L., Giralt M., Villarroya F. Thermogenic activation represses autophagy in brown adipose tissue. Int. J. Obes. (Lond) 2016;40:1591–1599. doi: 10.1038/ijo.2016.115. [DOI] [PubMed] [Google Scholar]
- Cannon B., Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol. Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- Christoffolete M.A., Linardi C.C., De Jesus L., Ebina K.N., Carvalho S.D., Ribeiro M.O., Rabelo R., Curcio C., Martins L., Kimura E.T., Bianco A.C. Mice with targeted disruption of the Dio2 gene have cold-induced overexpression of the uncoupling protein 1 gene but fail to increase brown adipose tissue lipogenesis and adaptive thermogenesis. Diabetes. 2004;53:577–584. doi: 10.2337/diabetes.53.3.577. [DOI] [PubMed] [Google Scholar]
- Cypess A.M., Kahn C.R. Brown fat as a therapy for obesity and diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2010;17:143–149. doi: 10.1097/MED.0b013e328337a81f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J., Teng R.J., Guan T., Eis A., Kaul S., Konduri G.G., Shi Y. Role of autophagy in angiogenesis in aortic endothelial cells. Am. J. Physiol. Cell Physiol. 2012;302:C383–C391. doi: 10.1152/ajpcell.00164.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D.F., Shackelford D.B., Mihaylova M.M., Gelino S., Kohnz R.A., Mair W., Vasquez D.S., Joshi A., Gwinn D.M., Taylor R. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X., Jin L., Han L., Yuan Y., Mu Q., Wang H., Yang J., Ning G., Zhou D., Zhang Z. miR-129-5p inhibits adipogenesis through autophagy and may Be a potential biomarker for obesity. Int. J. Endocrinol. 2019;2019:5069578. doi: 10.1155/2019/5069578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukano K., Okamatsu-Ogura Y., Tsubota A., Nio-Kobayashi J., Kimura K. Cold exposure induces proliferation of mature Brown adipocyte in a ss3-adrenergic receptor-mediated pathway. PLoS One. 2016;11:e0166579. doi: 10.1371/journal.pone.0166579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gospodarska E., Nowialis P., Kozak L.P. Mitochondrial turnover: a phenotype distinguishing brown adipocytes from interscapular brown adipose tissue and white adipose tissue. J. Biol. Chem. 2015;290:8243–8255. doi: 10.1074/jbc.M115.637785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassanpour M., Rezabakhsh A., Pezeshkian M., Rahbarghazi R., Nouri M. Distinct role of autophagy on angiogenesis: highlights on the effect of autophagy in endothelial lineage and progenitor cells. Stem Cell Res. Ther. 2018;9:305. doi: 10.1186/s13287-018-1060-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz C.T., Kiefer F.W. Adipose tissue browning in mice and humans. J. Endocrinol. 2019;241:R97–R109. [PubMed] [Google Scholar]
- Hinchy E.C., Gruszczyk A.V., Willows R., Navaratnam N., Hall A.R., Bates G., Bright T.P., Krieg T., Carling D., Murphy M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018;293:17208–17217. doi: 10.1074/jbc.RA118.002579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.M., Youle R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012;125:795–799. doi: 10.1242/jcs.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.C., Guan K.L. mTOR: a pharmacologic target for autophagy regulation. J. Clin. Invest. 2015;125:25–32. doi: 10.1172/JCI73939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenspor M. Cold-induced recruitment of brown adipose tissue thermogenesis. Exp. Physiol. 2003;88:141–148. doi: 10.1113/eph8802508. [DOI] [PubMed] [Google Scholar]
- Laker R.C., Xu P., Ryall K.A., Sujkowski A., Kenwood B.M., Chain K.H., Zhang M., Royal M.A., Hoehn K.L., Driscoll M. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J. Biol. Chem. 2014;289:12005–12015. doi: 10.1074/jbc.M113.530527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettieri-Barbato D. Redox control of non-shivering thermogenesis. Mol. Metab. 2019;25:11–19. doi: 10.1016/j.molmet.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.Z., Martagon A.J., Cimini S.L., Gonzalez D.D., Tinkey D.W., Biter A., Baxter J.D., Webb P., Gustafsson J.A., Hartig S.M., Phillips K.J. Pharmacological activation of thyroid hormone receptors elicits a functional conversion of white to Brown fat. Cell Rep. 2015;13:1528–1537. doi: 10.1016/j.celrep.2015.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowell B.B., Spiegelman B.M. Towards a molecular understanding of adaptive thermogenesis. Nature. 2000;404:652–660. doi: 10.1038/35007527. [DOI] [PubMed] [Google Scholar]
- Lu Y., Fujioka H., Joshi D., Li Q., Sangwung P., Hsieh P., Zhu J., Torio J., Sweet D., Wang L. Mitophagy is required for brown adipose tissue mitochondrial homeostasis during cold challenge. Sci. Rep. 2018;8:8251. doi: 10.1038/s41598-018-26394-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao K., Klionsky D.J. AMPK activates autophagy by phosphorylating ULK1. Circ. Res. 2011;108:787–788. doi: 10.1161/RES.0b013e3182194c29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lopez N., Athonvarangkul D., Sahu S., Coletto L., Zong H., Bastie C.C., Pessin J.E., Schwartz G.J., Singh R. Autophagy in Myf5+ progenitors regulates energy and glucose homeostasis through control of brown fat and skeletal muscle development. EMBO Rep. 2013;14:795–803. doi: 10.1038/embor.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Lopez N., Garcia-Macia M., Sahu S., Athonvarangkul D., Liebling E., Merlo P., Cecconi F., Schwartz G.J., Singh R. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the Brown adipose tissue and liver. Cell Metab. 2016;23:113–127. doi: 10.1016/j.cmet.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C.W., Willershauser M., Jastroch M., Rourke B.C., Fromme T., Oelkrug R., Heldmaier G., Klingenspor M. Adaptive thermogenesis and thermal conductance in wild-type and UCP1-KO mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010;299:R1396–R1406. doi: 10.1152/ajpregu.00021.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murholm M., Dixen K., Qvortrup K., Hansen L.H., Amri E.Z., Madsen L., Barbatelli G., Quistorff B., Hansen J.B. Dynamic regulation of genes involved in mitochondrial DNA replication and transcription during mouse brown fat cell differentiation and recruitment. PLoS One. 2009;4:e8458. doi: 10.1371/journal.pone.0008458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch R.C., Samborska B., Faubert B., Ma E.H., Gravel S.P., Andrzejewski S., Raissi T.C., Pause A., St-Pierre J., Jones R.G. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 2017;21:1–9. doi: 10.1016/j.celrep.2017.09.026. [DOI] [PubMed] [Google Scholar]
- Shabalina I.G., Hoeks J., Kramarova T.V., Schrauwen P., Cannon B., Nedergaard J. Cold tolerance of UCP1-ablated mice: a skeletal muscle mitochondria switch toward lipid oxidation with marked UCP3 up-regulation not associated with increased basal, fatty acid- or ROS-induced uncoupling or enhanced GDP effects. Biochim. Biophys. Acta. 2010;1797:968–980. doi: 10.1016/j.bbabio.2010.02.033. [DOI] [PubMed] [Google Scholar]
- Silva J.E. The thermogenic effect of thyroid hormone and its clinical implications. Ann. Intern. Med. 2003;139:205–213. [PubMed] [Google Scholar]
- Stock M.J. Thermogenesis and brown fat: relevance to human obesity. Infusionstherapie. 1989;16:282–284. doi: 10.1159/000222402. [DOI] [PubMed] [Google Scholar]
- Sukumaran A., Choi K., Dasgupta B. Insight on transcriptional regulation of the energy sensing AMPK and biosynthetic mTOR pathway genes. Front. Cell Dev. Biol. 2020;8:671. doi: 10.3389/fcell.2020.00671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terskikh A., Fradkov A., Ermakova G., Zaraisky A., Tan P., Kajava A.V., Zhao X., Lukyanov S., Matz M., Kim S. "Fluorescent timer": protein that changes color with time. Science. 2000;290:1585–1588. doi: 10.1126/science.290.5496.1585. [DOI] [PubMed] [Google Scholar]
- Wei H., Liu L., Chen Q. Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochim. Biophys. Acta. 2015;1853:2784–2790. doi: 10.1016/j.bbamcr.2015.03.013. [DOI] [PubMed] [Google Scholar]
- Wu J., Cohen P., Spiegelman B.M. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev. 2013;27:234–250. doi: 10.1101/gad.211649.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau W.W., Singh B.K., Lesmana R., Zhou J., Sinha R.A., Wong K.A., Wu Y., Bay B.H., Sugii S., Sun L., Yen P.M. Thyroid hormone (T3) stimulates brown adipose tissue activation via mitochondrial biogenesis and MTOR-mediated mitophagy. Autophagy. 2019;15:131–150. doi: 10.1080/15548627.2018.1511263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneshiro T., Wang Q., Tajima K., Matsushita M., Maki H., Igarashi K., Dai Z., White P.J., Mcgarrah R.W., Ilkayeva O.R. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature. 2019;572:614–619. doi: 10.1038/s41586-019-1503-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Zhang S., Cui L., Wang W., Na H., Zhu X., Li L., Xu G., Yang F., Christian M., Liu P. Lipid droplet remodeling and interaction with mitochondria in mouse brown adipose tissue during cold treatment. Biochim. Biophys. Acta. 2015;1853:918–928. doi: 10.1016/j.bbamcr.2015.01.020. [DOI] [PubMed] [Google Scholar]
- Zhang X., Wu D., Wang C., Luo Y., Ding X., Yang X., Silva F., Arenas S., Weaver J.M., Mandell M. Sustained activation of autophagy suppresses adipocyte maturation via a lipolysis-dependent mechanism. Autophagy. 2020;16:1668–1682. doi: 10.1080/15548627.2019.1703355. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new dataset or code.