

Abstract
Neem (Azadirachta indica) is a very popular traditional medicinal plant used since ancient times to treat numerous ailments. MicroRNAs (miRNAs) are highly conserved, non-coding, short RNA molecules that play important regulatory roles in plant development and metabolism. In this study, deploying a high stringent genome-wide computational-based approach and following a set of strict filtering norms a total of 44 potential conserved neem miRNAs belonging to 21 families and their corresponding 48 potential target transcripts were identified. Important targets include Squamosa promoter binding protein-like proteins, NAC, Scarecrow proteins, Auxin response factor, and F-box proteins. A biological network has also been developed to understand the miRNA-mediated gene regulation using the minimum free energy (MFE) values of the miRNA-target interaction. Moreover, six selected miRNAs were reported to be involved in secondary metabolism in other plant species (miR156a, miR156l, miR160, miR164, miR171, miR395) were validated by qPCR and their tissue-specific differential expression pattern was observed in leaves and stem. Except for ain-miR395, all the other miRNAs were found overexpressed in the stem as compared to leaves. To the best of our knowledge, this is the first report of neem miRNAs and we believe the finding of the present study will be useful for the functional genomic study of medicinal plants.
Supplementary Information
The online version contains supplementary material available at 10.1007/s13205-021-02839-z.
Keywords: Neem (Azadirachta indica), Medicinal plant, Micrornas, Secondary metabolites, QRT-PCR
Introduction
Azadirachta indica, commonly known as neem or nimba (Family: Meliaceae) is a large evergreen tree native to the Indian sub-continent which grows widely in tropical and subtropical climates worldwide (Subapriya and Nagini 2005; Tiwari et al. 2014). Neem has broad-spectrum medicinal properties including anti-inflammatory, immunomodulatory, antihyperglycemic, antimalarial, antiulcer, antimicrobial, antifungal, antioxidant, and anticarcinogenic (Bhambhani et al. 2017; Álvarez-Caballero and Coy-Barrera 2019; Cesa et al. 2019; Elgorashi and McGaw 2019). Due to its incredible medicinal value, in the year 2012 United Nations declared the neem tree as “Tree of the twenty-first century” (http://www.aajmag.ca/the-tree-of-the-21st-century/). Approximately 250 active compounds have been extracted from different tissues of the neem tree so far (Sarah et al. 2019). Compounds of interest obtained from neem are mainly secondary metabolites such as diterpenoids, triterpenoids, flavonoids, steroids, etc. (Sarah et al. 2019). Moreover, in recent years there is a growing interest to unravel the genome and transcriptome of neem to comprehend the molecular mechanism behind the secondary metabolites production (Krishnan et al. 2012; Kuravadi et al. 2015), and among these approaches, non-coding RNAs, more specifically microRNAs (miRNAs) are considered the prime research hotspot (Wu et al. 2020).
MicroRNAs (miRNAs) are non-coding, highly conserved, 20–24 nucleotides long RNA molecules that play crucial roles in post-transcriptional gene regulation, repressing gene expression due to high complementary binding sites in target mRNAs, either by triggering transcriptional repression or via targeting mRNA degradation (Paul et al. 2011; Wu et al. 2020). MicroRNA biogenesis in plants starts when miRNA genes are transcribed by RNA polymerase II into long primary transcripts (pri-miRNAs), which are later trimmed by ribonuclease III-like Dicer (DCL1) enzyme, thus producing miRNA precursors (pre-miRNAs) with a characteristic stem-loop structure (Wu et al. 2012; Budak and Akpinar 2015). Instantaneously, DCL1 further recognizes and cleaves the pre-miRNA hairpin loop and produces short double-stranded RNAs (dsRNAs) or duplexes, and eventually, one strand of the duplex known as guide strand acts as mature miRNA (Paul et al. 2011; Yang and Li 2012). Plant microRNAs have been implicated in several metabolic and biological pathways such as tissue development and differentiation, biotic and abiotic stress responses, phytohormone signaling, and secondary metabolite production (Naya et al. 2014; Gupta et al. 2017; Kundu et al. 2017; Paul 2017; Sharma et al. 2019; Paul et al. 2020).
The recent development of next-generation sequencing technology has led to the identification of several conserved as well as novel miRNAs in a number of non-model plant species (Wu et al. 2012; Paul et al. 2014; Kundu et al. 2017), however, the overall procedure is expensive, time-consuming, and requires high technical expertise. Nevertheless, the highly conserved nature of miRNAs has simplified the process of characterization of potential miRNAs in new plant species through homologs identification (Dai et al. 2011). Still, computational approaches require comprehensive reference miRNA databases, and they usually follow strict and high-confidence pipelines (Alptekin et al. 2017). For instance, to increase the precision of the prediction as well as to avoid the false positive outputs during in silico miRNA prediction, several parameters such as length, MFE, GC content, and the minimum free energy index (MFEI) of the pre-miRNAs should be studied. Nonetheless, experimental validation of the predicted miRNAs is greatly suggested (Zhang et al. 2006b; Sharma et al. 2019).
Even though miRNAs play numerous regulatory roles and participate in a wide range of secondary metabolite biosynthetic pathways in plants, no scientific initiative has been taken so far to study neem miRNAs, hence, this study aimed to employ the published neem draft genome sequence (GenBank accession AMWY00000000) (Kuravadi et al. 2015) to identify and characterize miRNAs and their respective targets, as well as to study their expression pattern in leaves and stem to gain a better understanding of the physiological role of miRNAs in neem. Here, for the first time, we report a total of 44 miRNAs belonging to 21 families and 48 corresponding targets in A. indica. Among the predicted miRNAs, six selected miRNAs involved in secondary metabolism were validated by qPCR.
Materials and methods
Computational identification of neem miRNA
For the computational prediction of potential neem miRNAs, a reference set of plant miRNAs consisting of a total of 1430 known mature miRNA sequences from Glycine max, Arabidopsis thaliana, and Citrus sinensis were retrieved from the miRbase database (Kozomara et al. 2019) (http://www.mirbase.org/cgi-bin/browse.pl). Subsequently, each miRNA sequence was individually searched (BLASTn) against the neem genome and sequences displaying exact match were chosen manually. The potential precursor sequences of approximately 400 nt (200 nt upstream and 200 nt downstream to the BLAST hit region) were mined and sequences coded for proteins were discarded. Secondary structures of the precursors were generated using the mfold web server (http://unafold.rna.albany.edu/?q=mfold/RNA-Folding-Form). Since the stable secondary structure is considered as one of the main characters of an authentic miRNA precursor, some previously demonstrated strict filtering criteria (Paul et al. 2011) were applied in this study during secondary structure prediction such as: (i) the pre-miRNA must have a stem-loop structure containing the mature miRNA sequence within one arm; (ii) possible mature miRNA should not be located in the terminal loop of the hairpin structure, (iii) mature miRNA should have less than nine mismatches with the opposite miRNA* sequence, and (iv) the potential stem-loop candidate should have minimum negative MFE or ΔG (− kcal/mol) and higher MFEI. The formula for calculating MFEI is as follows:
The detailed methodology, described previously by other authors (Paul et al. 2011; Roy et al. 2020) was illustrated graphically in Fig. 1 with minor modifications.
Fig. 1.
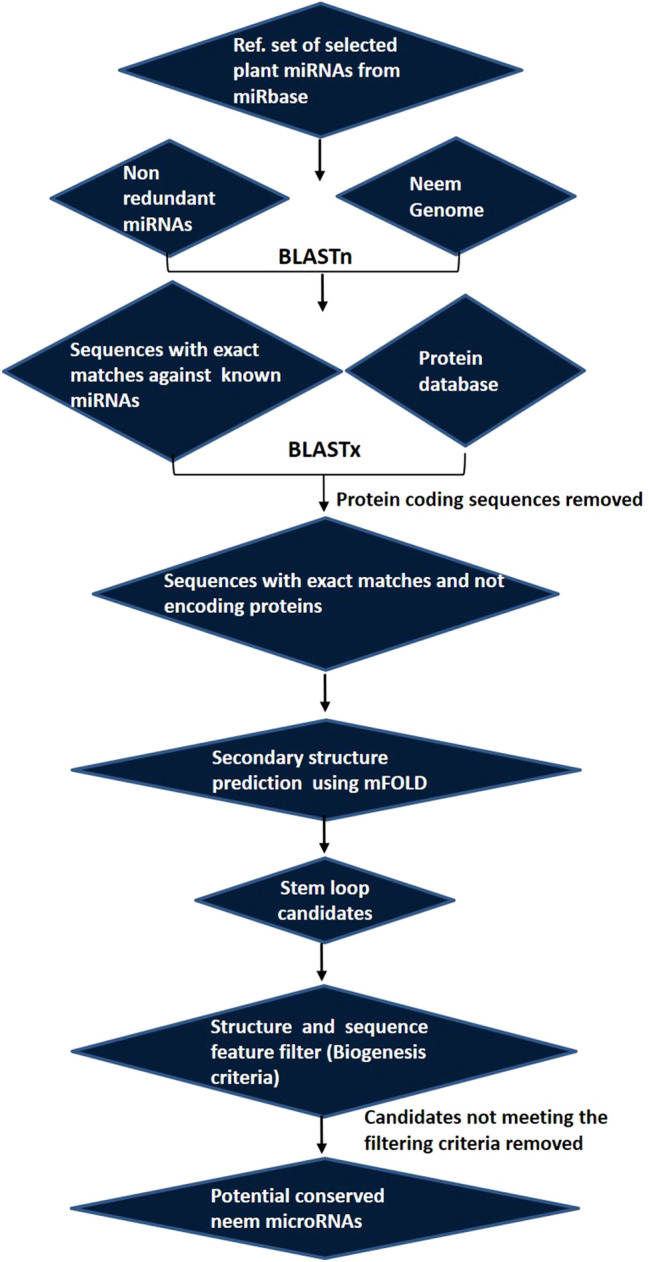
The graphical workflow of neem miRNA prediction procedure
Phylogenetic and conservation analysis of the potential neem miRNAs and their precursors
For phylogenetic analysis, ortholog precursor sequences of nine selected plant species were retrieved from the miRBase database, including model and non-model plants, as well as relevant agricultural crops: Citrus sinensis, Manihot esculenta, Arabidopsis thaliana, Glycine max, Prunus persica, Arabidopsis lyrate, Malus domestica, Cucumis melo, and Populus trichocarpa. Multiple sequence alignment (MSA) and phylogenetic tree construction were carried out employing MEGA X software. The phylogenetic tree was generated by the maximum likelihood statistical method based on the Tamura-Nei model with 1000 boot-strapped replicates. Conservation analysis for the nucleotide composition of the identified neem pre-miRNA orthologs was performed by the WebLogo tool (https://weblogo.berkeley.edu/logo.cgi) and a synteny map was generated using potential neem miRNA precursors and the well-annotated sweet orange (Citrus sinensis) genome, the phylogenetically closest species to neem (Darzentas 2010; Krishnan et al. 2012).
Prediction of A. indica miRNA targets, functional annotation, and pathway analysis
To predict the potential A. indica miRNA targets the Plant Small RNA Target Analysis Server (psRNATarget) (Dai et al. 2018) was employed in this study. The selection parameters were adjusted as follows: maximum expectation value of 2.5, translation inhibition ranges of 9–11 nt, number of top targets of 10, the penalty for G:U pair of 0.5, and number of mismatches allowed in the seed region of 1.5. Due to the unavailability of the neem protein database in the psRNATarget server, the target transcript search was performed against the transcript database of sweet orange (Citrus sinensis). The QuickGO analysis toolkit (https://www.ebi.ac.uk/QuickGO/) was used in this study to obtain the GO annotations of the potential target transcripts and to know their co-regulation a biological network was generated using the MFE values of the miRNA-target interaction; the biological network of the miRNAs and their targets was visualized by Cytoscape 3.2 (https://cytoscape.org/release_notes_3_2_0.html). Finally, the metabolic pathways regulated by the potential neem miRNAs have been investigated through KAAS (KEGG Automatic Annotation Server) (http://www.genome.jp/tools/kaas/) with the Bi-directional Best Hit (BBH) method (Panda et al. 2014).
Collection of plant materials, RNA extraction, and tissue-specific miRNA expression analysis
For experimental work, healthy young leaves and stem samples of A. indica were collected from a local nursery, immediately frozen in liquid N2, and stored at − 80 °C until further analysis. Small RNA (< 200 nt) was isolated from tissue samples using the mirVana™ miRNA Isolation Kit (Thermo Scientific, Wilmington, USA) according to the manufacturer’s instructions and pooled separately for each type of sample. The quality and quantity of RNA samples were measured with Nanodrop One (Thermo Scientific, Wilmington, USA), and 1 µg of RNA for individual samples was subsequently polyadenylated (using modified oligo dT primer) as well as reverse transcribed using mRQ Buffer and enzyme provided with Mir-X miRNA First-Stand Synthesis kit (Takara, Tokyo, Japan). Prior to the qRT-PCR experiment, a No-RT (-RT) control PCR was performed to monitor any genomic DNA contamination. The qRT-PCR experiment was performed by Step One Real-Time PCR System (Applied biosystems, Carlsbad, CA) and Mir-X miRNA TB Green qRT-PCR kit (Takara, Tokyo, Japan) using the entire predicted miRNA sequence as a forward primer and the adapter-specific mRQ3′ primer provided with the kit as the reverse primer. Each reaction was made in 12.5 µl volume containing 1X SYBR Advantage Premix, 1X ROX dye, 0.2 µM each of forward and reverse primers as indicated above, and 2 µl of the first-strand cDNA. Six miRNAs potentially involved in secondary metabolism (ain-miR156a, ain-miR156l, ain-miR160a-5p, ain-miR164, ain-miR171b, and ain-miR395) were selected for the qRT-PCR experiment and the program used is as follows: initial denaturation at 95 °C for 10 s followed by 45 cycles of denaturation at 95 °C for 5 s and annealing at 60 °C for 20 s, and finally a dissociation curve 95 °C for 30 s, 55 °C for 20 s and 95 °C for 20 s. Results were analyzed by ΔΔCt method, where the Ct value obtained after each reaction was normalized to the Ct value of U6 snRNA (U6 snRNA primer was provided with the kit) whose expression was consistent across the conditions. Subsequently, to see the differential expression pattern of selected miRNAs in leaves and stem tissues the expression level of ain-miR156a was set as 1 (control), and the expression level of all other miRNAs was quantified relative to it, as reported previously (Pirrò et al. 2016). qRT-PCR data were assessed using inferential statistics which includes student t-test and one-way ANOVA. In this study, all the qPCR experiments were carried out with two pooled biological replicates and three technical replicates as previously reported in many studies (Ho et al. 2016; Lee et al. 2017).
Results and discussion
Characterization of miRNAs in A. indica
The majority of mature miRNAs in plants are evolutionary highly conserved among species. This conserved nature of plant miRNAs has significantly enhanced the identification of potential conserved miRNAs in new plant species via a bioinformatic approach. In this study using high stringent filtering norms a total of 44 potential conserved miRNAs belonging to 21 families were predicted computationally in A. indica. Most of the identified miRNAs are 21 nt long while the predicted pre-miRNAs/precursors displayed great size variability ranging between 65 and 192 nt, with an average of 101 nt (Table 1). Regarding the miRNA location, 56.82% of the putative miRNAs were found located in the 3′ arm of the stem-loop precursors, while the remaining 43.18% was located in the 5′ arm. It is well established that low MFE values of stem-loop precursors attain more stable predictions (Bonnet et al. 2004). In this study, the MFE values of the precursors were found quite low ranging from − 27.60 to − 92.60 with an average of − 46.16 while the MFEI values fluctuated between 0.76 and 1.4 with an average of 1.07, excluding the possibility of being other small RNAs. Out of the 44 predicted neem miRNAs, 50% start with Uracil (U) at the 5′ extreme, in agreement with previous reports, which play a role in miRNA biogenesis (Unver and Budak 2009). A microRNA family is a group of miRNAs that derive from the common ancestor and have similar physiological functions. Among the neem miRNA families, the maximum number of members was found in the family miR156 and miR166 containing five members each. The predicted stem-loop precursors of neem miRNA with higher MFEI values (top 10) are shown in Fig. 2.
Table 1.
Identified potential miRNAs in neem (Azadirachta indica)
| Putative miRNA | miRNA sequence 5′ 3′ | LM (nt) | Position | LP (nt) | G + C content (%) | MFE/ ΔG (kcal/mol) | MFEI |
|---|---|---|---|---|---|---|---|
| ain-miR156a-5p | UGACAGAAGAGAGUGAGCAC | 20 | 5′ | 82 | 48.78 | − 45.40 | 1.14 |
| ain-miR156g | ACAGAAGAUAGAGAGCACAG | 20 | 5′ | 80 | 38.75 | − 40.30 | 1.30 |
| ain-miR156j-3p | AUCCAACGAAGCAGGAGCUGC | 21 | 3′ | 85 | 44.71 | − 35.50 | 0.93 |
| ain-miR156l | UUGACAGAAGAUAGAGAGCAC | 21 | 5′ | 86 | 38.37 | − 45.60 | 1.38 |
| ain-miR157d | UGACAGAAGAUAGAGAGCAC | 20 | 5′ | 84 | 39.29 | − 44.90 | 1.36 |
| ain-miR159c-3p | CUUGGACUGAAGGGAGCUCCC | 21 | 3′ | 168 | 49.70 | − 92.60 | 1.11 |
| ain-miR159e-3p | UUUGGAUUGAAGGGAGCUCUA | 21 | 3′ | 178 | 44.94 | − 81.80 | 1.02 |
| ain-miR159f-3p | AUUGGAGUGAAGGGAGCUCCA | 21 | 3′ | 173 | 50.85 | − 83.70 | 0.95 |
| ain-miR160a-3p | GCGUAUGAGGAGCCAUGCAUA | 21 | 3′ | 83 | 53.01 | − 51.10 | 1.16 |
| ain-miR160a-5p | GCCUGGCUCCCUGUAUGCCAU | 21 | 5′ | 78 | 56.41 | − 40.80 | 0.93 |
| ain-miR160c-3p | GCGUGCGAGGAGCCAUGCAUG | 21 | 3′ | 84 | 53.57 | − 44.50 | 0.99 |
| ain-miR160f | UGCCUGGCUCCCUGUAUGCCA | 21 | 5′ | 80 | 58.75 | − 46.30 | 0.99 |
| ain-miR164i | UGGAGAAGCAGGGCACGUGCA | 21 | 5′ | 73 | 42.47 | − 35.80 | 1.15 |
| ain-miR166a-3p | UCGGACCAGGCUUCAUUCCCCC | 22 | 3′ | 164 | 36.59 | − 52.10 | 0.87 |
| ain-miR166c-5p | GGGAAUGUUGUCUGGUUCGAG | 21 | 5′ | 90 | 45.56 | − 44.80 | 1.09 |
| ain-miR166d | UCGGACCAGGCUUCAUUCCCC | 21 | 3′ | 118 | 40.68 | − 41.70 | 0.87 |
| ain-miR166l | GGAAUGUUGUCUGGCUCGAGG | 21 | 5′ | 158 | 34.81 | − 47.30 | 0.86 |
| ain-miR166m | CGGACCAGGCUUCAUUCCCC | 20 | 3′ | 116 | 41.38 | − 40.70 | 0.85 |
| ain-miR167a | UGAAGCUGCCAGCAUGAUCUA | 21 | 5′ | 78 | 46.15 | − 41.80 | 1.16 |
| ain-miR169a-5p | CAGCCAAGGAUGACUUGCCGA | 21 | 5′ | 74 | 41.89 | − 31.10 | 1.00 |
| ain-miR169c | CAGCCAAGGAUGACUUGCCGG | 21 | 5′ | 152 | 36.18 | − 43.90 | 0.80 |
| ain-miR169h | UAGCCAAGGAUGACUUGCCUG | 21 | 3′ | 192 | 40.10 | − 58.60 | 0.76 |
| ain-miR171b-3p | CGAGCCGAAUCAAUAUCACUC | 21 | 3′ | 76 | 34.21 | − 28.50 | 1.10 |
| ain-miR171c-3p | UUGAGCCGUGCCAAUAUCACG | 21 | 3′ | 83 | 50.60 | − 46.60 | 1.11 |
| ain-miR171j-3p | UGAUUGAGCCGUGCCAAUAUC | 21 | 3′ | 77 | 48.05 | − 44.60 | 1.21 |
| ain-miR171k-3p | UUGAGCCGCGCCAAUAUCACU | 21 | 3′ | 90 | 41.11 | − 38.70 | 1.05 |
| ain-miR172a | AGAAUCUUGAUGAUGCUGCAU | 21 | 3′ | 120 | 38.33 | − 56.10 | 1.22 |
| ain-miR172d-3p | AGAAUCUUGAUGAUGCUGCAG | 21 | 3′ | 124 | 42.74 | − 53.40 | 1.01 |
| ain-miR172d-5p | GCGGCAUCAUCAAGAUUCACA | 21 | 5′ | 115 | 46.09 | − 53.80 | 1.02 |
| ain-miR172i-3p | GGAAUCUUGAUGAUGCUGCAU | 21 | 3′ | 120 | 45.00 | − 56.60 | 1.05 |
| ain-miR319k | UUGGACUGAAGGGAGCUCCCU | 21 | 3′ | 168 | 49.40 | − 89.30 | 1.08 |
| ain-miR390a-3p | CGCUAUCCAUCCUGAGUUUCA | 21 | 3′ | 80 | 47.50 | − 38.50 | 1.01 |
| ain-miR390g | AAGCUCAGGAGGGAUAGCGCC | 21 | 5′ | 75 | 48.00 | − 38.40 | 1.07 |
| ain-miR394b-5p | UUGGCAUUCUGUCCACCUCC | 20 | 5′ | 75 | 44.00 | − 27.60 | 0.84 |
| ain-miR395d | CUGAAGUGUUUGGGGGAACUC | 21 | 3′ | 65 | 52.31 | − 38.70 | 1.14 |
| ain-miR396b-3p | GCUCAAGAAAGCUGUGGGAAA | 21 | 3′ | 86 | 40.70 | − 39.20 | 1.12 |
| ain-miR396k-5p | UUCCACAGCUUUCUUGAACUU | 21 | 5′ | 86 | 40.70 | − 39.20 | 1.12 |
| ain-miR397b-5p | UCAUUGAGUGCAGCGUUGAUG | 21 | 5′ | 72 | 45.83 | − 34.60 | 1.05 |
| ain-miR399f-5p | GGGCAAUUCUCCUUUGGCAGA | 21 | 5′ | 68 | 47.06 | − 42.40 | 1.32 |
| ain-miR403-3p | UUAGAUUCACGCACAAACUCG | 21 | 3′ | 89 | 41.57 | − 35.70 | 0.96 |
| ain-miR408d | UGCACUGCCUCUUCCCUGGC | 20 | 3′ | 82 | 52.43 | − 39.90 | 0.93 |
| ain-miR827 | UUAGAUGACCAUCAACAAACA | 21 | 3′ | 76 | 28.95 | − 29.20 | 1.33 |
| ain-miR2111-5p | UAAUCUGCAUCCUGAGGUUUG | 21 | 5′ | 68 | 39.71 | − 37.70 | 1.40 |
| ain-miR2275a-3p | UUUAGUUUCCUCCAAUAUCUUA | 22 | 3′ | 72 | 31.94 | − 32.20 | 1.40 |
LM Length of mature miRNAs, LP length of precursor
Fig. 2.

Secondary structures (stem-loops) of predicted neem miRNA precursors (Top 10 structures with higher MFEI values are shown). Mature miRNAs are highlighted with red font
Conservation and phylogenetic analysis of predicted neem miRNAs
In this study, randomly selected neem miRNA precursors ain-miR390, ain-miR396, and ain-mir403 displayed distinct clusters along with their respective orthologs in the phylogenetic tree (Fig. 3), as well as showed high sequence conservation (Fig. 4), indicating that not only the mature miRNAs but also their precursors are evolutionarily highly conserved in plants (Zhang et al. 2006a). Interestingly, it was previously noticed that the loop region of the miR159, miR319, and miR394 families from various plant species shows a very high amount of conservation indicating their origin from a common ancestor (Dezulian et al. 2005), while numerous complete precursor sequences from miR166 family displayed high sequence identity in soybean (Li et al. 2017). Moreover, the widespread distribution of putative neem miRNA orthologs in C. sinensis genome as revealed by synteny mapping confirming their sequence conservation nature across species (Fig. 5). A conserved synteny map can provide the possibility of translational applications to economically relevant species (Bellato et al. 2019). Altogether, the comparative genomics approach allows extrapolating knowledge between related species as well as demonstrating their evolutionary connection (Jain and Das 2016).
Fig. 3.

Phylogenetic tree showing the evolutionary conservation of ain-miR390, ain-miR396, and ain-miR403 precursor candidates (highlighted with red boxes) as they form distinct clusters along with their respective orthologs. The tree was constructed using the maximum likelihood method based on the Tamura–Nei model with 1000 boot-strapped replicates
Fig. 4.

Nucleotide conservation analysis using WebLogo tool for a ain-miR390 and b ain-miR396 and their orthologs in nine plant species. The height of the stacks represents the level of nucleotide conservation at a certain position. Invariant nucleotides show a single letter, while variable nucleotides display the most common substitutions
Fig. 5.

Comparative synteny map of 44 potential neem miRNAs. Here all the miRNAs are physically mapped against chromosomes 1–9 of well-annotated phylogenetically close species sweet orange (Citrus sinensis). The lines represent the matches, confirming their sequence conservation nature across species
Predicted targets for identified neem miRNAs and their functional analysis
Plant miRNAs were previously thought to primarily target transcription factors, but subsequent research has revealed that miRNA also targets functional genes in plants involved in a variety of physiological processes such as root and stem development, leaf morphogenesis, signal transduction, and stress response (Paul 2017; Sharma et al. 2019, 2020; Paul et al. 2020). Plant miRNAs bind to their target transcripts through perfect or near-perfect complementary sites, unlike animal miRNAs (Bartel, 2004), and based on this mechanism a homology search-based approach was applied to predict putative targets in neem using the psRNAtarget server. MiRNAs function as a negative regulator of gene expression by promoting the cleavage of target mRNAs or repressing their translation and that seems to be the crucial mode of gene regulation. A total of 48 putative target transcripts of the potential neem miRNAs were identified in this study (Supplementary Table 1). To better understand the role of miRNAs, the function of the target gene set was analyzed using the GO term. GO analysis in this study showed that identified targets are involved in a variety of molecular, biological, and cellular processes (Fig. 6). It is well established that transcription factors have critical roles in the regulation of diverse developmental processes in plants (Ramachandran et al. 1994). In this work we found some significant target transcription factors including Squamosa promoter binding protein-like protein (SPLs) (target of ain-miR156) that are involved in leaf, flower, and fruit development (Liu et al. 2017; Shinde et al. 2020); NAC transcription factor (target of ain-miR164) plays a vital role in disease resistance mechanism; APETALA2 (target of ain-miR172), has shown to control flowering time and floral organ identity (Zhou et al. 2010); Auxin response factors (target of ain-miR160), involved in hormone signaling as well as participate in leaf and stem development (Varkonyi-Gasic et al. 2010); and F-box proteins (target of ain-miR394) involved in plant vegetative and reproductive growth and development (Parry and Estelle 2006). Nevertheless, previous studies demonstrated that microRNAs can regulate the secondary metabolic pathways in plants by controlling several target proteins including transcription factors (Gupta et al. 2017). In this current report, few neem miRNA target proteins such as SPLs, NAC domain-containing protein, Scarecrow proteins (target of ain-miR171), F-box proteins, Acetyl-CoA C-acyltransferase (target of ain-miR395), and Auxin response factors have been identified to have a potential role to regulate plant bioactive compound biosynthesis. SPLs are reported to have a role in anthocyanin and sesquiterpene biosynthesis as well as to regulate the metabolic flux during the flavonoid biosynthetic pathway (Gupta et al. 2017; Liu et al. 2017), while a significant role of scarecrow proteins during anthocyanin and flavonoid biosynthesis in the strawberry was noticed (Pillet et al. 2015). (Feder et al. 2015) found that the accumulation of naringenin chalcone, a yellow flavonoid pigment, in the fruit rind of muskmelons (Cucumis melo) is negatively correlated with F-box protein (CmKFB) expression. Besides functioning as a transcription factor, the NAC protein family has also been reported to regulate flavonoid biosynthesis under high light (Morishita et al. 2009). Acetyl-CoA C-Acetyltransferase was found to act as a core enzyme during terpenoid biosynthesis in Ginkgo biloba (Chen et al. 2017). Likewise, the auxin response factor might play an important role in anthocyanin biosynthesis (Sun et al. 2017), and FAD-binding and BBE domain-containing protein (target of ain-miR408) possibly participate in the biosynthesis of benzylisoquinoline alkaloids (Boke et al. 2015).
Fig. 6.

GO enrichment analysis of putative target transcripts regulated by identified neem miRNAs represented as GO terms for biological processes (green), molecular functions (blue), and cellular components (pink)
Our pathway exploration of neem miRNA target genes was enriched by KEGG analysis using KASS. KEGG pathway analysis revealed that the potential neem miRNA targets are involved in a total of 41 highly diversified biochemical pathways. “Metabolic pathways” was the most significantly enriched, followed by “biosynthesis of secondary metabolites” and ¨plant hormone signal transduction¨ (Fig. 7). Interestingly, this analysis also showed that putative target genes of the neem miRNAs are not only involved in plant metabolism but also various human diseases including tuberculosis, viral carcinogenesis, and Epstein-Barr virus infection. Since the discovery of the ability of plant-derived miRNAs in the cross-kingdom gene regulation (Samad et al. 2020), new findings contribute to the emerging discussion of the potential applications of miRNAs in therapeutic purposes. Nonetheless, co-regulation of several target genes was documented by gene network analysis (Fig. 8).
Fig. 7.

KEGG pathways mapped using the KAAS tool among the predicted targets of the identified miRNAs in neem. Axis x corresponds to biochemical pathways and axis y column refers to the number of associated miRNA targets
Fig. 8.

MFE based network interaction of potential neem miRNAs and their corresponding targets. Co-regulation of multiple targets is observed for several of the identified miRNAs-
Tissue-specific expression analysis of A. indica miRNAs
The selected secondary metabolism-related neem miRNAs ain-miR156a, ain-miR156l, ain-miR160, ain-miR164, ain-miR171, and ain-miR395 were successfully validated and they showed a distinct expression pattern between leaves and stem. The tissue-specific expression pattern of miRNAs between leaves and stem has been reported previously in various plant species including opium poppy, common grapes, and sweet sorghum (Mica et al. 2009; Yu et al. 2015; Boke et al. 2015). It is well established that some miRNAs are only expressed in certain tissues or developmental stages and this temporal and spatial expression may be involved in complex gene regulation of the growth and development of plants (Mao et al. 2012; Zhang et al. 2019).
A differential expression pattern of ain-miR156l, ain-miR160, ain-miR164, and ain-miR171 were noticed in the two sampled neem tissues when compared against the ain-miR156a (Fig. 9). In leaf, miR164 was found the most abundant, while in stem tissue it was miR160. A similar kind of expression pattern was reported in the economically important plant Larix olgensis where miR160 was significantly overexpressed in the stems of young plants (Zhang et al. 2019). As various experiments have shown miRNAs are not only involved in specific tissue development but also are key in the transformation of plant development stages and secondary metabolites accumulation (Li et al. 2021), so further research is required to decipher the complex regulatory network of miRNAs in neem.
Fig. 9.

Quantitative RT-PCR analysis of mature miRNAs in A. indica leaf and stem. The relative expression levels of five selected miRNAs involved in secondary metabolism were calculated by the CT method and normalized against the expression of miR156-a, which was set as 1. U6 was used as an endogenous control. Significant differences at P < 0.05 (One-Way ANOVA) are indicated with an asterisk
In summary, the characterization of miRNAs and their targets is a crucial step towards initiating an investigation relevant to miRNA in a non-model plant species. Since this is the first miRNA report from high-value medicinal plant neem we believe this can be advantageous to comprehend their possible role in regulating the biosynthesis of secondary metabolites of interest. Nevertheless, further studies are important to elucidate this complex regulatory network
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 (DOCX 19 KB). Putative targets of predicted ain-miRNAs
Acknowledgements
The research facility provided by the Departamento Regional de Bioingenieria, Queretaro is thankfully acknowledged
Authors Contributions
Conceptualization, experimental design, data analysis, writing, editing SP.; Experimental procedures, data collection and analysis, writing, PRP.; Bioinformatics and statistical analysis, critical writing, reviewing ASr, PI.-AB., SR, ASh; Project administration ASh. All authors have read and agreed to the published version of the manuscript.
Declarations
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Sujay Paul and Paula Reyes-Pérez contributed equally to this work.
Contributor Information
Sujay Paul, Email: spaul@tec.mx.
Ashutosh Sharma, Email: asharma@tec.mx.
References
- Alptekin B, Akpinar BA, Budak H. A comprehensive prescription for plant miRNA identification. Front Plant Sci. 2017;7:2058. doi: 10.3389/fpls.2016.02058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez-Caballero JM, Coy-Barrera E. Chemical and antifungal variability of several accessions of azadirachta indica A. Juss. from six locations across the colombian caribbean coast: identification of antifungal azadirone limonoids. Plants. 2019;8:555. doi: 10.3390/plants8120555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bellato M, De Marchi D, Gualtieri C, et al. A bioinformatics approach to explore MicroRNAs as tools to bridge pathways between plants and animals is DNA damage response (DDR) a potential target process? Front Plant Sci. 2019;10:1535. doi: 10.3389/fpls.2019.01535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhambhani S, Lakhwani D, Gupta P, et al. Transcriptome and metabolite analyses in Azadirachtaindica: identification of genes involved in biosynthesis of bioactive triterpenoids. Sci Rep. 2017;7:5043. doi: 10.1038/s41598-017-05291-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boke H, Ozhuner E, Turktas M, et al. Regulation of the alkaloid biosynthesis by miRNA in opium poppy. Plant Biotechnol J. 2015;13:409–420. doi: 10.1111/pbi.12346. [DOI] [PubMed] [Google Scholar]
- Bonnet E, Wuyts J, Rouzé P, Van de Peer Y. Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics. 2004;20:2911–2917. doi: 10.1093/bioinformatics/bth374. [DOI] [PubMed] [Google Scholar]
- Budak H, Akpinar BA. Plant miRNAs: biogenesis, organization and origins. Funct Integr Genomics. 2015;15:523–531. doi: 10.1007/s10142-015-0451-2. [DOI] [PubMed] [Google Scholar]
- Cesa S, Sisto F, Zengin G, et al. Phytochemical analyses and pharmacological screening of neem oil. South Afr J Bot. 2019;120:331–337. doi: 10.1016/j.sajb.2018.10.019. [DOI] [Google Scholar]
- Chen Q, Yan J, Meng X, et al. Molecular cloning, characterization, and functional analysis of acetyl-CoA C-acetyltransferase and mevalonate kinase genes involved in terpene trilactone biosynthesis from ginkgobiloba. Molecules. 2017;22:74. doi: 10.3390/molecules22010074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Zhuang Z, Zhao PX. Computational analysis of miRNA targets in plants: current status and challenges. Brief Bioinform. 2011;12:115–121. doi: 10.1093/bib/bbq065. [DOI] [PubMed] [Google Scholar]
- Dai X, Zhuang Z, Zhao PX. psRNATarget: a plant small RNA target analysis server (2017 release) Nucleic Acids Res. 2018;46:W49–W54. doi: 10.1093/nar/gky316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzentas N. Circoletto visualizing sequence similarity with circos. Bioinformatics. 2010;26:2620–2621. doi: 10.1093/bioinformatics/btq484. [DOI] [PubMed] [Google Scholar]
- Dezulian T, Palatnik JF, Huson D, Weigel D. Conservation and divergence of microRNA families in plants. Genome Biol. 2005;6:1–25. doi: 10.1186/gb-2005-6-11-p13. [DOI] [Google Scholar]
- Elgorashi EE, McGaw LJ. African plants with in vitro anti-inflammatory activities: a review. South Afr J Bot. 2019;126:142–169. doi: 10.1016/j.sajb.2019.06.034. [DOI] [Google Scholar]
- Feder A, Burger J, Gao S, et al. A kelch domain-containing f-box coding gene negatively regulates flavonoid accumulation in muskmelon. Plant Physiol. 2015;169:1714–1726. doi: 10.1104/pp.15.01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta OP, Karkute SG, Banerjee S, et al. Contemporary understanding of miRNA-based regulation of secondary metabolites biosynthesis in plants. Front Plant Sci. 2017;8:374. doi: 10.3389/fpls.2017.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho C-L, Tan Y-C, Yeoh K-A, et al. De novo transcriptome analyses of host-fungal interactions in oil palm Elaeis guineensis Jacq. BMC Genom. 2016;17:66. doi: 10.1186/s12864-016-2368-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain A, Das S. Synteny and comparative analysis of miRNA retention, conservation, and structure across Brassicaceae reveals lineage- and sub-genome-specific changes. Funct Integr Genomics. 2016;16:253–268. doi: 10.1007/s10142-016-0484-1. [DOI] [PubMed] [Google Scholar]
- Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019;47:D155–D162. doi: 10.1093/nar/gky1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan NM, Pattnaik S, Jain P, et al. A draft of the genome and four transcriptomes of a medicinal and pesticidal angiosperm Azadirachtaindica. BMC Genomics. 2012;13:464. doi: 10.1186/1471-2164-13-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu A, Paul S, Dey A, Pal A. High throughput sequencing reveals modulation of microRNAs in Vignamungo upon Mungbeanyellowmosaic India virus inoculation highlighting stress regulation. Plant Sci. 2017;257:96–105. doi: 10.1016/j.plantsci.2017.01.016. [DOI] [PubMed] [Google Scholar]
- Kuravadi NA, Yenagi V, Rangiah K, et al. Comprehensive analyses of genomes, transcriptomes and metabolites of neem tree. PeerJ. 2015;3:e1066. doi: 10.7717/peerj.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W-K, Namasivayam P, Ong Abdullah J, Ho C-L. Transcriptome profiling of sulfate deprivation responses in two agarophytes Gracilariachangii and Gracilariasalicornia (Rhodophyta) Sci Rep. 2017;7:46563. doi: 10.1038/srep46563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Xin X, Li J, Cui Y, Hou Y, Zhai L, Wang X, Fu Y, Liu R, Bian S. Conservation and diversification of the miR166 family in soybean and potential roles of newly identified miR166s. BMC Plant Biol. 2017;17:1–18. doi: 10.1186/s12870-016-0951-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lin Q, Yan M, et al. Relationship between secondary metabolism and miRNA for important flavor compounds in different tissues of tea plant (Camelliasinensis) As revealed by genome-wide miRNA analysis. J Agric Food Chem. 2021;69:2001–2012. doi: 10.1021/acs.jafc.0c07440. [DOI] [PubMed] [Google Scholar]
- Liu M-Y, Wu X-M, Long J-M, Guo W-W. Genomic characterization of miR156 and Squamosa promoter binding protein-like genes in sweet orange (Citrussinensis) Plant Cell Tissue Organ Cult. 2017;130:103–116. doi: 10.1007/s11240-017-1207-6. [DOI] [Google Scholar]
- Mao W, Li Z, Xia X, et al. A combined approach of high-throughput sequencing and degradome analysis reveals tissue specific expression of micrornas and their targets in cucumber. PLoS ONE. 2012;7:e33040. doi: 10.1371/journal.pone.0033040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mica E, Piccolo V, Delledonne M, et al. High throughput approaches reveal splicing of primary microRNA transcripts and tissue specific expression of mature microRNAs in vitis vinifera. BMC Genomics. 2009;10:558. doi: 10.1186/1471-2164-10-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita T, Kojima Y, Maruta T, et al. arabidopsis NAC transcription factor, ANAC078, regulates flavonoid biosynthesis under high-light. Plant Cell Physiol. 2009;50:2210–2222. doi: 10.1093/pcp/pcp159. [DOI] [PubMed] [Google Scholar]
- Naya L, Paul S, Valdés-López O, et al. Regulation of copper homeostasis and biotic interactions by microRNA 398b in common bean. PLoS ONE. 2014;9:e84416. doi: 10.1371/journal.pone.0084416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda D, Dehury B, Sahu J, et al. Computational identification and characterization of conserved miRNAs and their target genes in garlic (Alliumsativum L.) expressed sequence tags. Gene. 2014;537:333–342. doi: 10.1016/j.gene.2014.01.010. [DOI] [PubMed] [Google Scholar]
- Parry G, Estelle M. Auxin receptors: a new role for F-box proteins. Curr Opin Cell Biol. 2006;18:152–156. doi: 10.1016/j.ceb.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Paul S. Identification and characterization of microRNAs and their targets in high-altitude stress-adaptive plant maca (Lepidiummeyenii Walp) 3 Biotech. 2017;7:103. doi: 10.1007/s13205-017-0734-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, Kundu A, Pal A. Identification and validation of conserved microRNAs along with their differential expression in roots of Vigna unguiculata grown under salt stress. Plant Cell Tissue Organ Cult. 2011;105:233–242. doi: 10.1007/s11240-010-9857-7. [DOI] [Google Scholar]
- Paul S, Kundu A, Pal A. Identification and expression profiling of Vignamungo microRNAs from leaf small RNA transcriptome by deep sequencing. J Integr Plant Biol. 2014;56:15–23. doi: 10.1111/jipb.12115. [DOI] [PubMed] [Google Scholar]
- Paul S, de la Fuente-Jiménez JL, Manriquez CG, Sharma A. Identification, characterization and expression analysis of passion fruit (Passiflora edulis) microRNAs. 3 Biotech. 2020;10:25. doi: 10.1007/s13205-019-2000-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillet J, Yu H-W, Chambers AH, et al. Identification of candidate flavonoid pathway genes using transcriptome correlation network analysis in ripe strawberry (Fragaria × ananassa) fruits. J Exp Bot. 2015;66:4455–4467. doi: 10.1093/jxb/erv205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirrò S, Zanella L, Kenzo M, et al. MicroRNA from Moringaoleifera: identification by high throughput sequencing and their potential contribution to plant medicinal value. PLoS ONE. 2016;11:e0149495. doi: 10.1371/journal.pone.0149495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Hiratsuka K, Chua N-H. Transcription factors in plant growth and development. Curr Opin Genet Dev. 1994;4:642–646. doi: 10.1016/0959-437X(94)90129-Q. [DOI] [PubMed] [Google Scholar]
- Roy S, Nath D, Paul P, Chakraborty S. Computational identification of conserved microRNAs and functional annotation of their target genes in Citruslimon identification of microRNA in Citruslimon. South Afr J Bot. 2020;130:109–116. doi: 10.1016/j.sajb.2019.12.009. [DOI] [Google Scholar]
- Samad AFA, Kamaroddin MF, Sajad M. Cross-kingdom regulation by plant microRNAs provides novel insight into gene regulation. Adv Nutr. 2020;12:197–211. doi: 10.1093/advances/nmaa095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarah R, Tabassum B, Idrees N, Hussain MK. Natural bio-active compounds. Berlin: Springer; 2019. Bio-active compounds isolated from neem tree and their applications; pp. 509–528. [Google Scholar]
- Sharma A, Bejerano PIA, Maldonado IC, et al. Genome-wide computational prediction and experimental validation of quinoa (Chenopodiumquinoa) microRNAs. Can J Plant Sci. 2019;99:666–675. doi: 10.1139/cjps-2018-0296. [DOI] [Google Scholar]
- Shinde H, Dudhate A, Anand L, et al. Small RNA sequencing reveals the role of pearl millet miRNAs and their targets in salinity stress responses. South Afr J Bot. 2020;132:395–402. doi: 10.1016/j.sajb.2020.06.011. [DOI] [Google Scholar]
- Subapriya R, Nagini S. Medicinal properties of neem leaves: a review. Curr Med Chem Agents. 2005;5:149–156. doi: 10.2174/1568011053174828. [DOI] [PubMed] [Google Scholar]
- Sun Y, Qiu Y, Duan M, et al. Identification of anthocyanin biosynthesis related microRNAs in a distinctive Chinese radish (Raphanussativus L.) by high-throughput sequencing. Mol Genet Genomics. 2017;292:215–229. doi: 10.1007/s00438-016-1268-y. [DOI] [PubMed] [Google Scholar]
- Tiwari R, Verma AK, Chakraborty S, et al. Neem (Azadirachtaindica) and its potential for safeguarding health of animals and humans: a review. J Biol Sci. 2014;14:110. doi: 10.3923/jbs.2014.110.123. [DOI] [Google Scholar]
- Unver T, Budak H. Conserved microRNAs and their targets in model grass species Brachypodiumdistachyon. Planta. 2009;230:659–669. doi: 10.1007/s00425-009-0974-7. [DOI] [PubMed] [Google Scholar]
- Varkonyi-Gasic E, Gould N, Sandanayaka M, et al. Characterisation of microRNAs from apple (Malusdomestica’RoyalGala’) vascular tissue and phloem sap. BMC Plant Biol. 2010;10:159. doi: 10.1186/1471-2229-10-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Wang M, Ma Y, et al. High-Throughput sequencing and characterization of the small RNA transcriptome reveal features of novel and conserved microRNAs in panax ginseng. PLoS ONE. 2012;7:e44385. doi: 10.1371/journal.pone.0044385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Liu S, Qi H, et al. Research progress on plant long non-coding RNA. Plants. 2020;9:408. doi: 10.3390/plants9040408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Li L. Analyzing the microRNA transcriptome in plants using deep sequencing data. Biology Basel. 2012;1:297–310. doi: 10.3390/biology1020297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Cong L, Zhu Z, et al. Identification of differentially expressed microRNA in the stems and leaves during sugar accumulation in sweet sorghum. Gene. 2015;571:221–230. doi: 10.1016/j.gene.2015.06.056. [DOI] [PubMed] [Google Scholar]
- Zhang B, Pan X, Wang Q, et al. Computational identification of microRNAs and their targets. Comput Biol Chem. 2006;30:395–407. doi: 10.1016/j.compbiolchem.2006.08.006. [DOI] [PubMed] [Google Scholar]
- Zhang B, Pan X, Cobb GP, Anderson TA. Plant microRNA: a small regulatory molecule with big impact. Dev Biol. 2006;289:3–16. doi: 10.1016/j.ydbio.2005.10.036. [DOI] [PubMed] [Google Scholar]
- Zhang S, Yan S, Zhao J, et al. Identification of miRNAs and their target genes in larix olgensis and verified of differential expression miRNAs. BMC Plant Biol. 2019;19:247. doi: 10.1186/s12870-019-1853-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Liu Y, Liu Z, et al. Genome-wide identification and analysis of drought-responsive microRNAs in Oryzasativa. J Exp Bot. 2010;61:4157–4168. doi: 10.1093/jxb/erq237. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file1 (DOCX 19 KB). Putative targets of predicted ain-miRNAs