

Abstract
While engineered chimeric antigen receptor (CAR) T cells have shown promise in detecting and eradicating cancer cells within patients, it remains difficult to identify a set of truly cancer-specific CAR-targeting cell surface antigens to prevent potentially fatal on-target off-tumor toxicity against other healthy tissues within the body. To help address this issue, we present a novel tamoxifen-gated photoactivatable split-Cre recombinase optogenetic system, called TamPA-Cre, that features high spatiotemporal control to limit CAR T cell activity to the tumor site. We created and optimized a novel genetic AND gate switch by integrating the features of tamoxifen-dependent nuclear localization and blue-light-inducible heterodimerization of Magnet protein domains (nMag, pMag) into split Cre recombinase. By fusing the cytosol-localizing mutant estrogen receptor ligand binding domain (ERT2) to the N-terminal half of split Cre(2–59aa)-nMag, the TamPA-Cre protein ERT2-CreN-nMag is physically separated from its nuclear-localized binding partner, NLS-pMag-CreC(60–343aa). Without tamoxifen to drive nuclear localization of ERT2-CreN-nMag, the typically high background of the photoactivation system was significantly suppressed. Upon blue light stimulation following tamoxifen treatment, the TamPA-Cre system exhibits sensitivity to low intensity, short durations of blue light exposure to induce robust Cre-loxP recombination efficiency. We finally demonstrate that this TamPA-Cre system can be applied to specifically control localized CAR expression and subsequently T cell activation. As such, we posit that CAR T cell activity can be confined to a solid tumor site by applying an external stimulus, with high precision of control in both space and time, such as light.
Keywords: optogenetics, immunotherapy, CAR T cell, Cre-loxP recombination, ERT2, nMag and pMag
Graphical Abstract
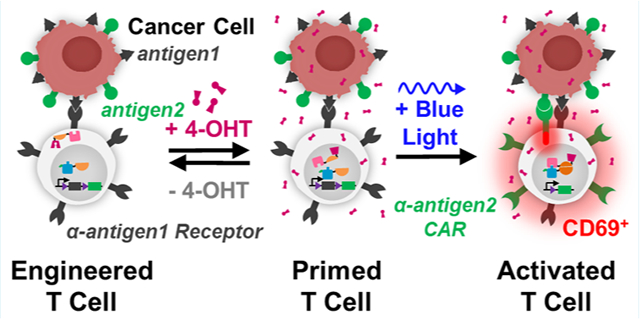
Chimeric antigen receptor (CAR) T cell therapy has emerged as a promising tool in the ongoing battle against cancer. CAR T cells targeting the tumor-associated antigen (TAA) CD19 have been successfully employed to treat several CD19+ B-cell lymphomas and leukemias.1–4 However, one fundamental risk to patient safety is on-target off-tumor toxicity, whereby CAR T cells attack unintended healthy cells which express the same tumor-associated antigen (TAA) targeted by the CAR. In the case of α-CD19 CAR T cells, on-target off-tumor toxicity results in B-cell aplasia—a condition which is uniquely survivable with immunoglobulin-replacement therapy. However, most on-target off-tumor toxicities are difficult to predict and constitute serious safety risks for patients.5–8 For example, in clinical trials, CAR T cells targeting ERBB2+ breast,9 CAIX+ renal,10 and CEACAM5+ colorectal11 cancerous tissues also attacked healthy TAA+ pulmonary, biliary epithelial, and gastrointestinal as well as pulmonary tissues, respectively, resulting in patient death, liver toxicity, and acute respiratory toxicity.
In efforts to prevent on-target off-tumor toxicity, synthetic biologists have introduced genetically encoded Boolean logic gates designed to allow CAR T cells to interpret a unique combination of TAA inputs.12 For example, splitting the CAR’s intracellular costimulatory and activation domains between two different TAA-recognizing proteins creates an AND gate by which binding to both TAAs is required to drive full CAR-mediated T cell effector function.13 Similarly, costimulatory and activation CAR domains can be reconstituted via small-molecule-driven dimerization, but failure to limit such diffusible small molecules only to the tumor site in vivo leaves healthy tissues susceptible to on-target off-tumor toxicity.14 More recently, the mechanically sensitive synNotch receptor has been used to automatically drive CAR transcription upon binding to the target cell.15 Antigen recognition can also be altered at the protein level using tumor-targeting single-chain variable fragment adaptor molecules to tune and gate CAR activity.16 However, in all of these AND-gated systems, patients are still at risk of on-target off-tumor toxicity when healthy tissues also express the same pair of antigens (Table S1).
Although such CAR T cells are capable of interpreting more complex signals in vivo, it remains challenging to identify an ideal patient-specific combination of TAAs.5–8 However, if TAA+ cancerous and healthy tissues are spatially distinct in vivo, an external stimulus with high spatiotemporal control can be used to limit CAR T cell activity to the tumor site. Light is an ideal stimulus in that it can be quickly and reversibly applied with precise control over the dosage, duration, and location of stimulation. On-target off-tumor toxicity would likewise be limited to the light-stimulated region, allowing for more flexibility in selecting TAAs to target. Therefore, CAR T cells equipped with optogenetic logic gates may have the potential to limit CAR T cell activation to the tumor site, thus protecting healthy tissues elsewhere in the body from on-target off-tumor toxicity (Figure 1).
Figure 1.
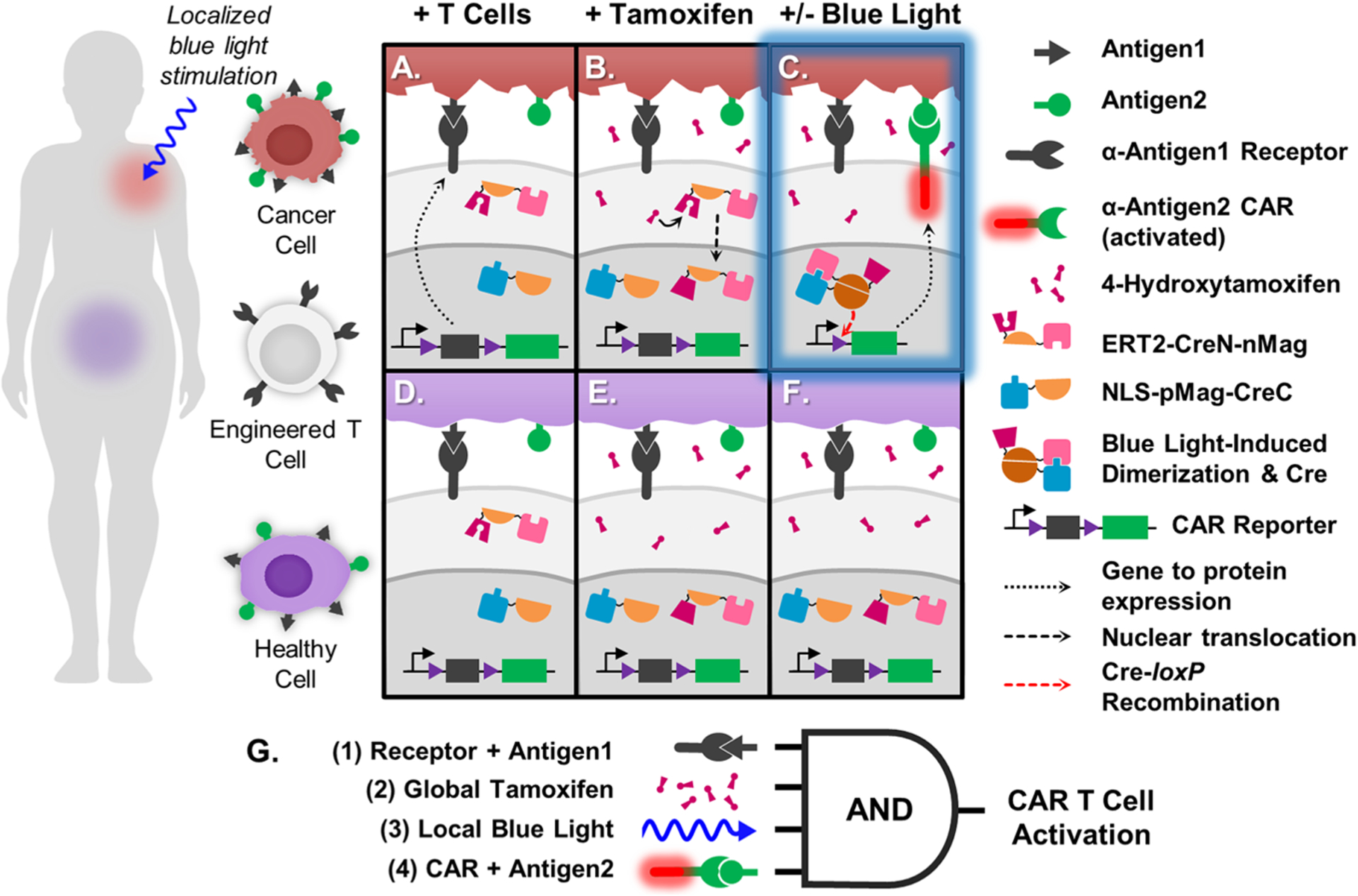
Schematic of TamPA-Cre application and molecular mechanism. (Left) Person with Antigen1+ Antigen2+ cancerous (red) and healthy (purple) tissues in separate regions of the body. Engineered T cells express TamPA-Cre (ERT2-CreN-nMag and NLS-pMag-CreC) and the CAR Reporter genetic construct, consisting of a constitutive promoter driving expression of the floxed (purple) α-Antigen1 Receptor CDS with stop codons (black), followed by α-Antigen2 CAR (green). Upon intravenous introduction, the engineered T cells bind and localize to both cancerous (A) and healthy (D) Antigen1+ cells. TamPA-Cre is inactive as its NLS-pMag-CreC and ERT2-CreN-nMag protein halves nuclear and cytosolically localized, respectively. After administration of tamoxifen, metabolite 4-OHT binds with ERT2-CreN-nMag to drive nuclear localization (B,E), priming TamPA-Cre. Next, blue light is applied to the cancerous tissue region only (C), inducing nMag-pMag heterodimerization which restores active TamPA-Cre recombinase activity within the nucleus. The floxed α-Antigen1 Receptor CDS in the CAR Reporter is excised through Cre-loxP recombination along with its stop codons, thus allowing for α-Antigen2 CAR expression. The T cell is finally activated upon CAR-mediated binding to Antigen2. T cells localized to the healthy tissue region (F) are not exposed to blue light and thus do not express α-Antigen2 CAR, effectively protecting healthy cells that express both Antigen1 and Antigen2. (G) Boolean logic representation of the AND-gated TamPA-Cre system. (1) T cells first bind to cells expressing Antigen1 via the Receptor. (2) Then, TamPA-Cre is be primed with tamoxifen before receiving localized (3) blue light stimulation in the cancerous tissue region, driving Cre-loxP recombination and CAR expression. (4) T cell activation is triggered when Antigen2 is recognized by the CAR.
A multitude of versatile optogenetic light-switches have been developed to both study and actuate a variety of cellular functions by modulating gene expression, enzyme function, protein translocation, and so on.17,18 Optogenetic systems sensitive to tissue-penetrating red, far-red, or near-infrared light (NIR), such as PhyB-PIF319 and BphP1-PpsR2,20 exhibit relatively weak light-induced responses and consist of high-molecular-weight components which can additionally limit protein expression and gene delivery. On the other hand, systems like UVB8 and Dronpa rely on violet or ultraviolet light which may damage DNA.21,22 We therefore focused on optogenetic systems responsive to 400–500 nm wavelength light. The limited tissue penetration (<1 mm) of such wavelengths has been overcome by using implantable micron-scale inorganic light emitting diodes controlled wirelessly with radio frequencies23 or NIR light,24 or by stimulating antigen-targeted lanthanide-doped upconversion nanoparticles with NIR light.25,26 NIR-wavelength two-photon illumination can also activate short-wavelength-sensitive proteins at precise locations deeper within tissues.27 As has been extensively reviewed,17,18 several genetically encoded blue-light-sensitive proteins such as AsLOV2, GIGANTEA-FKF1, VVD, Magnet, and Cry2-CIB1 have been used to transcriptionally regulate transient gene expression. While removing blue light from these systems would advantageously turn off CAR expression, these systems often suffer from leaky inducible promoters and weak light-induced gene expression. On the other hand, genome-editing blue-light-switches have been used to permanently alter gene expression,28–30 but leakiness in the optogenetic systems themselves could lead to irreversible premature CAR expression. Therefore, it was critical to design a blue-light optogenetic system that is tightly regulated to help prevent on-target off-tumor toxicity.
In this work, we introduce a new robust optogenetic split-Cre system called TamPA-Cre, which is gated by the drug tamoxifen for nuclear translocation and utilizes the blue light photoactivatable Magnet dimerizing pair, nMag-pMag, for transcriptional activation, to strictly regulate CAR expression with high spatiotemporal resolution. By gating CAR expression with both tamoxifen and blue light, the TamPA-Cre system exhibits significantly suppressed background activity compared with the blue-light-gated PA-Cre-M system.29 Furthermore, the TamPA-Cre system is sensitive to low intensity, short-duration blue light exposure like the PA-Cre-M system. We demonstrate that TamPA-Cre-induced CAR expression is highly dependent on cells receiving both tamoxifen and blue light stimulation and that TamPA-Cre can drive CAR-mediated T cell activation against specific TAA+ target cells. Additionally, we demonstrate that our TamPA-Cre system exhibits virtually no background activity under room lights, in contrast to the PA-Cre-M system. Finally, we integrated the multiantigen recognition approach with our drug-and-light-inducible system to allow for high spatiotemporal control of T cell activation. TamPA-Cre can thus be helpful in locally regulating CAR expression in T cells that might otherwise cause on-target off-tumor toxicity in healthy tissues in the body.
RESULTS AND DISCUSSION
Testing Photoactivatable Split-Cre Systems.
To develop the optogenetic circuit, we first tested the two most recently developed blue light photoactivatable split-Cre systems. The PA-Cre-2.0 system (hereafter referred to as PA-Cre-C) relies on the blue-light-induced heterodimerization between CIB1 and CRY2(L348F) domains (Arabidopsis thaliano, heterodimer half-life: 24 min) to reconstruct functional Cre recombinase from the split CreN(19–104) and CreC(106–343) components.30 The PA-Cre system (hereafter referred to as PA-Cre-M) utilizes the Neurospora crassa-derived Vivid photoreceptor mutant heterodimers negative Magnet (nMag) and positive Magnet (pMag) to reconstitute its split CreN(19–59) and CreC(60–343) components into functional Cre with blue light (heterodimer half-life: 1.8 h).29 To compare these two systems (Figure S1A–C), we developed a puromycin-selected HEK293T cell line that stably expresses the Cre-loxP deletion-based EGFP Reporter construct (Figures 2A, S1J). We also built a novel controllable blue light stimulation apparatus that uses a diffusion filter to distribute light relatively evenly across a standard cell culture plate within a mammalian cell culture incubator (Figure S2).
Figure 2.
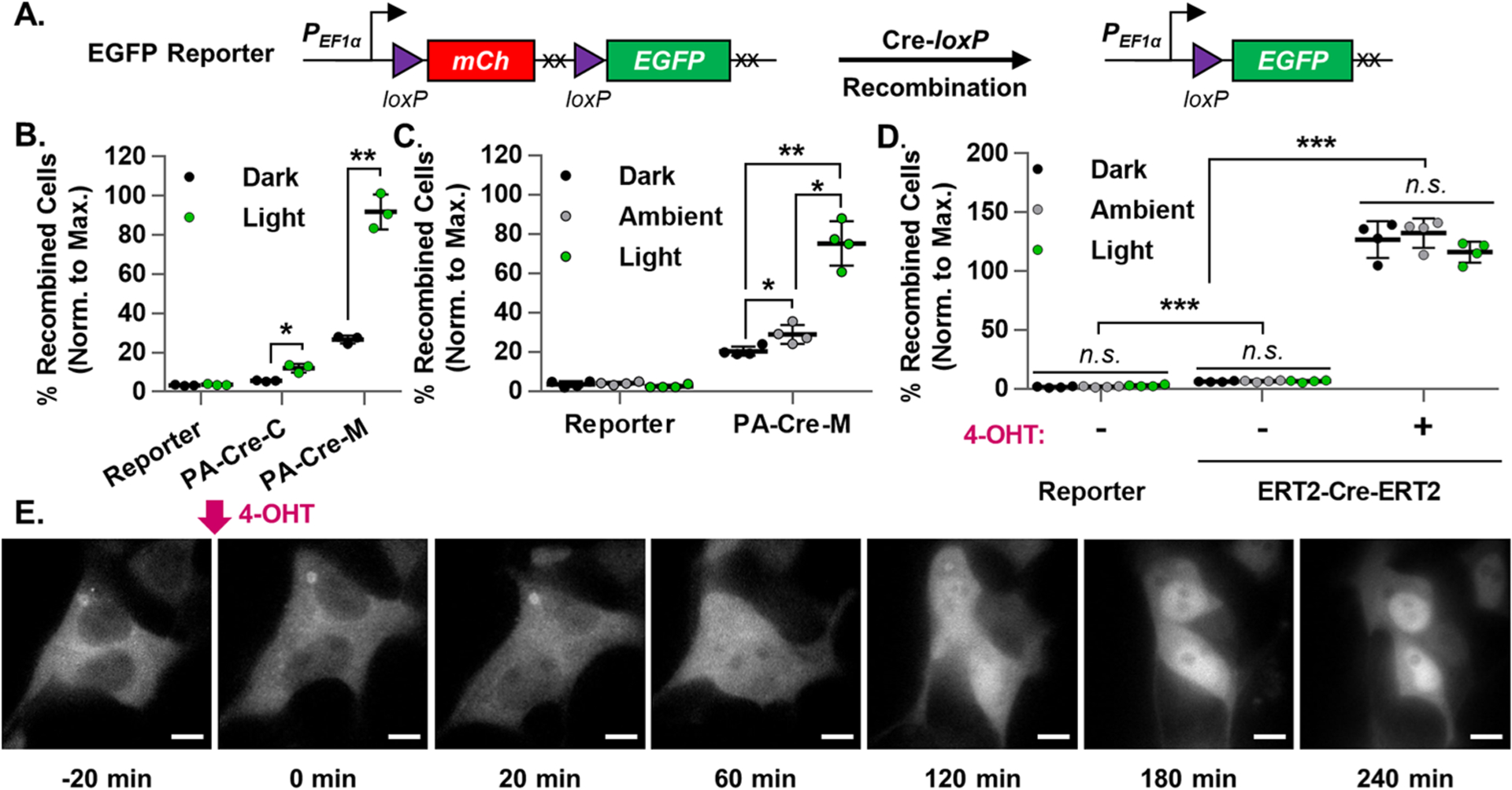
Evaluation of photoactivatable and tamoxifen-dependent Cre recombinase systems. (A) Schematic representation of the EGFP Reporter genetic construct before and after Cre-loxP recombination. The hEF1α promoter drives mCherry expression. During Cre-loxP recombination, the floxed mCherry is irreversibly excised along with its stop codons (XX), thus allowing for EGFP expression. The normalized percentage of recombined cells was calculated for EGFP Reporter HEK293T transiently transfected with the following constructs which did (Light) or did not (Ambient, Dark) receive the indicated blue light stimulation (473 ± 29 nm): (B) PA-Cre-C, PA-Cre-M, or Cre PAconstructs, (30 W/m2, 30s) (n = 3), or (C) PA-Cre-M or Cre constructs, (15 W/m2, 1s per min, 24 h) (n = 4), or (D) ERT2-Cre-ERT2 or Cre constructs, (15 W/m2, 1s per min, 24 h) (n = 4). Reporter = untransfected EGFP Reporter HEK293T cell line. Blue light stimulation started, and flow cytometry measurements were taken 24 and 72 h post-transfection, respectively. Percentage of recombined HEK293T cells (normalized to maximal recombination) = 100%*(% of EGFP+ cells)/(mean % of EGFP+ cells in corresponding Cre groups). (E) Representative time-lapse fluorescence microscopy images of HEK293T cells transiently expressing ERT2-mCherry before and after the addition of nuclear-localizing 4-OHT (500 nM) (imaged every 2 min, 100× magnification, scale bar = 10 μm, n = 6 independently measured cells).
To compare the two photoactivatable split-Cre systems, EGFP Reporter HEK293T cells were or were not (Reporter) transiently transfected with either PA-Cre-C, PA-Cre-M, or Cre constructs (Figure S1A–D). Then, cells either were (Light) or were not (Dark) briefly stimulated with a single pulse of blue light (472 ± 29 nm, 30 W/m2, 30 s). The percentage of EGFP+ recombined cells in each cell group was determined by flow cytometry 24 h later. All EGFP Reporter HEK293T cell groups were normalized to the mean EGFP+ percentage of cells in the maximally recombined corresponding Cre+ positive control groups to account for differences in transfection efficiency between experiments.
As can be seen in Figure 2B, EGFP Reporter HEK293T cells in both Light PA-Cre-C and Light PA-Cre-M groups responded to blue light stimulation with the expected increase in Cre-loxP recombination compared to their respective Dark counterparts (a 2.1- and 3.4-fold increase, respectively). The PA-Cre-M groups exhibited near maximal light-induced Cre-loxP recombination (90.8 ± 9.0%, mean ± standard deviation), whereas the Light PA-Cre-C groups reached only a fraction of that (12.1 ± 2.2%). While different blue light intensities and patterns of exposure may have improved Cre-loxP recombination levels in the Light PA-Cre-C group, we prioritized robust recombination efficiency driven by minimal blue light energy. Therefore, we decided to pursue the highly sensitive, efficient, and robust nMag-CreN59 and pMag-CreC60 heterodimerizing domains of the PA-Cre-M system in our design.
However, despite the high recombination efficiencies of the Light PA-Cre-M groups, the Dark PA-Cre-M groups suffered from high background levels of spontaneous Cre-loxP recombination (26.7 ± 2.0%), which was found to be proportional to PA-Cre-M protein expression levels (Figure S3A). Furthermore, while stimulating the PA-Cre-M system in EGFP Reporter HEK293T cells with a pulsatile light protocol intended to improve its performance (15 W/m2, 1s per min, 24 h), we found it necessary to actively protect nonblue-light-stimulated (Dark) cell groups from all light sources. Without protection, a small but significant additional percentage of cells underwent Cre-loxP recombination (Ambient: 28.9 ± 4.8%, Dark: 20.4 ± 2.4%), presumably driven by incidental exposure to the blue wavelengths of the laboratory’s ambient white room lighting while in the incubator (0.34–0.78 W/m2, Figure 2C). Taken together, these results indicated that the highly photosensitive PA-Cre-M system was susceptible to leaky Cre-loxP recombination, which we next sought to minimize without sacrificing the system’s light-driven high recombination efficiency.
Testing Cre-ERT2 Systems.
We focused on suppressing background Cre-loxP recombination in nonstimulated cells by spatially segregating one of the PA-Cre-M heterodimer split-Cre protein halves outside of the nucleus, since neither nuclear localization sequence (NLS) tagged CreN-nMag-NLS nor NLS-pMag-CreC protein halves alone were able to drive significant recombination (Figures S1C, S3B). Although this could be achieved in a number of different ways (e.g., using the AsLOV2-based blue-light-inducible nuclear localization signal system),31 it was preferable to find an orthogonally inducible, well-gated, and robust system compatible with use in vivo. The U.S. Food and Drug Administration-approved drug tamoxifen and its active metabolites have been widely used to induce nuclear translocation of proteins, including Cre, which are fused to the T2 mutant Estrogen Receptor ligand binding domain (ERT2),32–34 particularly to induce genomic changes in vivo in transgenic mouse models.35
Because tamoxifen and its active metabolites are known to be somewhat photosensitive,22 we examined whether or not relevant amounts of blue light stimulation interfered with the tamoxifen-gated ERT2-Cre-ERT2 Cre-loxP recombination system (Figures 2D, S1E).36 Applying the same pulsatile light stimulation pattern used in Figure 2C, we found that tamoxifen metabolite 4-hydroxytamoxifen (4-OHT, 500 nM) stimulated robust Cre-loxP recombination efficiency in ERT2-Cre-ERT2+ EGFP Reporter HEK293T cells, surpassing the efficiency of the Cre+ positive control group despite blue light illumination (Dark: 127.0 ± 15.6%, Ambient: 132.4 ± 12.4%, Light: 116.1 ± 9.0%). Furthermore, cell groups lacking 4-OHT stimulation exhibited minimal background recombination (Dark: 6.1 ± 0.4%, Ambient: 6.5 ± 0.7%, Light: 6.5 ± 0.8%), highlighting that 4-OHT functions independently of the tested blue light stimulation parameters.
Tamoxifen-induced nuclear import translocation dynamics in HEK293T cells were further characterized using an ERT2-mCherry fusion protein (Figure S1F). Using time-lapse fluorescence microscopy, we discovered that nuclear translocation driven by 4-OHT (500 nM) occurred on the order of hours. ERT2-mCherry protein was only clearly nuclear-localized (with a nuclear-to-cytosolic mean fluorescence intensity ratio ≥2) after approximately 3 h (Figure 2E), highlighting an effective time window for blue light stimulation to drive robust Cre-loxP recombination in ERT2-based photoactivatable construct designs.
Design and Function of TamPA-Cre.
We therefore integrated components of both the Tamoxifen-ERT2 and Photoactivatable-Cre systems to create a novel genetically encoded AND gate in which both tamoxifen and blue light stimulation are needed to drive Cre-loxP recombination (Figure 3A). In our system, named TamPA-Cre, the ERT2 domain was fused to the smaller CreN59-nMag construct (Figure S1H1) and not to pMag-CreC60 (Figure S1C2) which contains potentially competitive NLSs native to Cre. The TamPA-Cre system’s heterodimers were designed to be expressed at similar levels, like the PA-Cre-M system, with both proteins translated from a single transcript and separated by the P2A self-cleaving peptide (Figure S1B). The ERT2-CreN(2–59) sequence was fused to the N-terminus of nMag (without NLS), and fluorescent marker P2A-mCherry was added to the C-terminus of NLS-pMag-CreC60 to confirm expression. Our design also included a codon-diversified pMag coding sequence (CDS) to prevent potential recombination introduced by lentiviral gene transfer between the nearly identical nMag and pMag CDSs (Figure S4).37 Although this construct worked well for transient expression, its large single transcript size significantly impeded lentivirus production efficiency. Therefore, the ERT2-CreN-nMag and NLS-pMag-CreC components were separated into two vectors, each with a unique fluorescent marker (Figure S1H). The similarity in transcript size of ERT2-CreN-nMag and NLS-pMag-CreC (approximately 1.6 and 1.4 kb, respectively) resulted in similar protein expression levels and comparable efficiency in lentivirus production. We additionally created ERT2-CreN-nMagHigh1, which contains the mutated nMag variant nMagHigh1 (M135I/M165I) previously shown to improve light-induced heterodimerization with pMag.38 Along with NLS-pMag-CreC, this system is referred to as TamPA-Cre-nH1 (Figure S1I).
Figure 3.
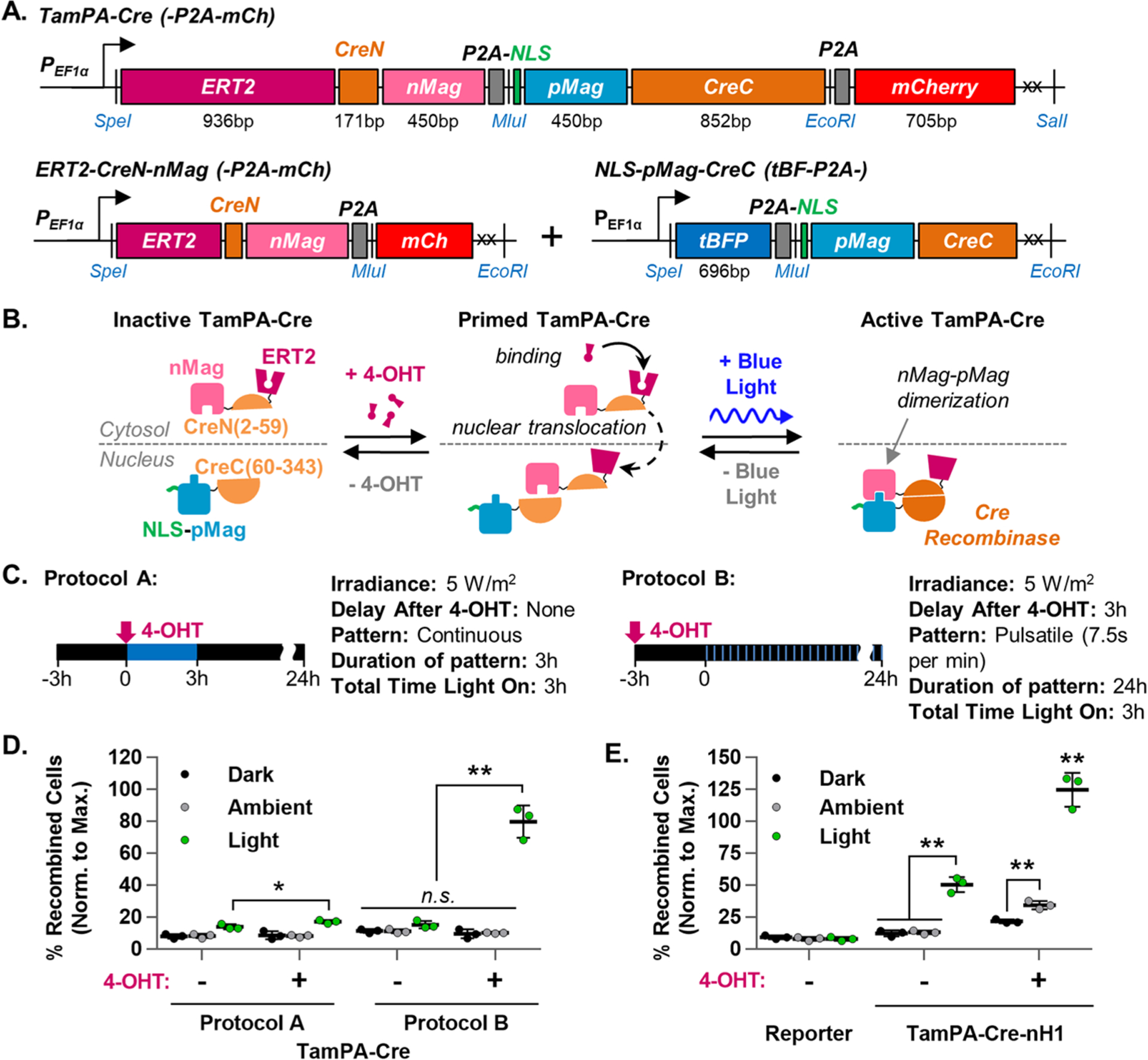
Design and optimization of the TamPA-Cre system in HEK293T cells. (A) Schematic of TamPA-Cre as a single genetic construct with codon-diversified pMag and marker P2A-mCh (top), or as two genetic constructs: ERT2-CreN-nMag with marker P2A-mCh (bottom left) and NLS-pMag-CreC with marker tBFP-P2A (bottom right). (B) Schematic depicting the mechanism of the tamoxifen- AND blue-light-gated TamPA-Cre system. (C) Schematic illustrating two different tamoxifen and blue light stimulation protocols, each providing the same total amount of blue light energy (473 ± 29 nm, not to scale). Protocol A: 3 h of continuous blue light stimulation (5 W/m2) started immediately after the addition of 4-OHT (500 nM). Protocol B: 24 h of pulsatile blue light stimulation (5 W/m2, 7.5s per min) started 3 h after 4-OHT addition. (D) The percentage of recombined TamPA-Cre+ EGFP Reporter HEK293T cells (normalized to maximal recombination) which were (Light) or were not (Ambient, Dark) subjected to protocol A or B (n = 3). (E) Recombination in TamPA-Cre-nH1+ EGFP Reporter HEK293T cells subjected (Light) or not (Ambient, Dark) to Protocol B (n = 3). Reporter = untransfected EGFP Reporter HEK293T cell line (n = 3). Blue light stimulation started and flow cytometry measurements taken 24 and 72 h post-transfection, respectively. Percentage of recombined HEK293T cells (normalized to maximal recombination) = 100%*(% of EGFP+ cells)/(mean % of EGFP+ cells in corresponding Cre groups).
The mechanism of TamPA-Cre activation is illustrated in Figure 3B. Without tamoxifen or blue light stimulation, TamPA-Cre is inactive with ERT2-CreN-nMag and NLS-pMag-CreC proteins spatially segregated to the cytosol and nucleus, respectively. This physical separation prevents the spontaneous, concentration-dependent nMag-pMag dimerization seen in the PA-Cre-M system and thus suppresses the unwanted background levels of Cre-loxP recombination (Figure S3A). Also, unlike the PA-Cre-M system, inactive TamPA-Cre is designed to be insensitive to nonspecific light stimulation from, for example, the ambient white fluorescent lighting found in a typical lab or clinical environment. To restore its light sensitivity, TamPA-Cre must be primed by 4-OHT, which drives ERT2-CreN-nMag protein nuclear translocation. With both ERT2-CreN-nMag and NLS-pMag-CreC protein halves residing in the nucleus, blue light stimulation induces nMag-pMag heterodimerization, bringing CreN and CreC protein halves together to reconstitute the Cre recombinase activity of active TamPA-Cre.
Optimizing Tamoxifen and Blue Light Stimulation.
To optimize Cre-loxP recombination, we tested the TamPA-Cre system in EGFP Reporter HEK293T cells with a variety of tamoxifen and blue light stimulation protocols. Two parameters were found to be particularly important: the light stimulation pattern (pulsatile versus continuous exposure), and the time at which light was started relative to 4-OHT addition. Drawn from the 3 h short illumination pattern shown to improve PA-Cre-M function,29 Figure 3C illustrates two different tamoxifen and light stimulation protocols that each call for a total 3 h of 5 W/m2 blue light stimulation. In Protocol A, light is applied continuously with concurrent 4-OHT addition. In Protocol B, 4-OHT is added 3 h prior to delivering light in an even pulsatile pattern over the course of 24 h (Figure 3C).
Under Protocol A, light-stimulated TamPA-Cre drove Cre-loxP recombination in only a minor percentage of EGFP Reporter HEK cells (17.3 ± 1.1%, Figure 3D). However, TamPA-Cre-mediated recombination was improved when the start of light stimulation was delayed by several hours relative to 4-OHT addition (24.6 ± 5.1%, Figure S5A). Considering that the 4-OHT-stimulated ERT2-mCh fusion protein reached significant 4-OHT-induced nuclear translocation only after approximately 3 h (Figure 2E), the poor function of TamPA-Cre under Protocol A is likely due to suboptimal nuclear ERT2-CreN-nMag concentrations.
TamPA-Cre-mediated recombination was also improved to a similar extent by administering light in a pulsatile pattern started concurrently with tamoxifen stimulation (25.4 ± 3.5%, Figure S5B). Therefore, Protocol B was created to merge the advantages of both a delay in light stimulation and a pulsatile light pattern. In EGFP Reporter HEK293T cells exposed to the same total 3 h of blue light stimulation but delivered in a pulsatile pattern (7.5 s per min, 24 h) started 3 h post-tamoxifen stimulation (Protocol B), TamPA-Cre drove robust levels of Cre-loxP recombination (79.9 ± 10.1%, Figure 3D)—on par with the performance of PA-Cre-M (84.8 ± 3.1%, Figure S5C). Moreover, the PA-Cre-M system’s high levels of nonlight-stimulated background Cre-loxP recombination (Dark: 32.9 ± 6.3%, Ambient: 45.2 ± 8.1%) were significantly suppressed by the AND gate of the TamPA-Cre system (Dark: 9.6 ± 2.8%, Ambient: 10.3 ± 0.3%). The AND gate also successfully suppressed blue light-stimulated recombination in the absence of 4-OHT (15.4 ± 2.3%).
With the AND gate working as intended, we further investigated whether the reported 13-fold increase in heterodimerization between the pMag and nMagHigh1 domains38 would additionally improve recombination efficiency as TamPA-Cre-nH1 (Figure S1I). Under Protocol B, a greater percentage of TamPA-Cre-nH1+ EGFP Reporter HEK cells did indeed undergo Cre-loxP recombination (125.0 ± 13.3%, Figure 3E), but at the cost of an increase in background recombination in nonblue-light-stimulated cell groups (Dark: 21.8 ± 1.5%, Ambient: 34.6 ± 3.2%). The AND gate of TamPA-Cre-nH1 also failed to sufficiently suppress light-induced recombination in the absence of 4-OHT (50.7 ± 5.9%). The TamPA-Cre system was thus a safer choice for us to use in reaching our goal to strictly regulate localized CAR expression in T cells to avoid on-tumor off-target toxicities.
Design and Function of the CAR Reporter.
The TamPA-Cre system was next applied to induce CAR expression in the physiologically relevant Jurkat T cell line. To demonstrate our overall concept outlined in Figure 1, CD19 and CD38 TAAs were targeted with an α-CD19 CAR and a c-myc-tagged α-CD38 Receptor, respectively (Figure S6A).39 A Jurkat T cell line stably expressing the α-CD19CAR-EGFP was verified to undergo CD19-specific CAR-mediated T cell activation, as measured by upregulated CD69 expression. Coincubation with CD19+ Toledo Target cells activated Jurkat CAR T cells (81.1 ± 0.35%), whereas coincubation with CD19− K562 Target cells did not (3.18 ± 0.09%, Figure S6B,D). Likewise, Jurkat T cells stably expressing myc-α-CD38 Receptor were not activated upon binding to CD38+ Toledo Target cells, as expected (Figure S6D).
The CAR and Receptor constructs were next integrated into several different floxed reporter designs to achieve the initial expression of the tumor-anchoring myc-α-CD38 Receptor and α-CD19 CAR expression only upon induction via TamPA-Cre-mediated Cre-loxP recombination. After testing the TamPA-Cre system with several different reporter configurations (Figure S7), the deletion-based Cre-loxP recombination CAR Reporter was selected to create the CAR Reporter Jurkat T cell line (Figure 4A, S1K). As CAR overexpression can lead to spontaneous T cell activation (Figure S6D), this cell line was transduced with a single copy (on average) of the CAR Reporter. The CAR Reporter consists of an hEF1α promoter driving the expression of floxed myc-α-CD38Receptor (with stop codons to halt translation), followed by α-CD19CAR-EGFP. The Receptor is designed to allow patient-administered CAR T cells to home in and bind to CD38+ cells, both cancerous and healthy, in vivo. When the cancerous region is locally stimulated with blue light following global tamoxifen administration, TamPA-Cre excises myc-α-CD38Receptor from the genome (along with its stop codons), allowing for rapid α-CD19CAR-EGFP expression and subsequent T cell effector functions (Figure 4B,E).
Figure 4.
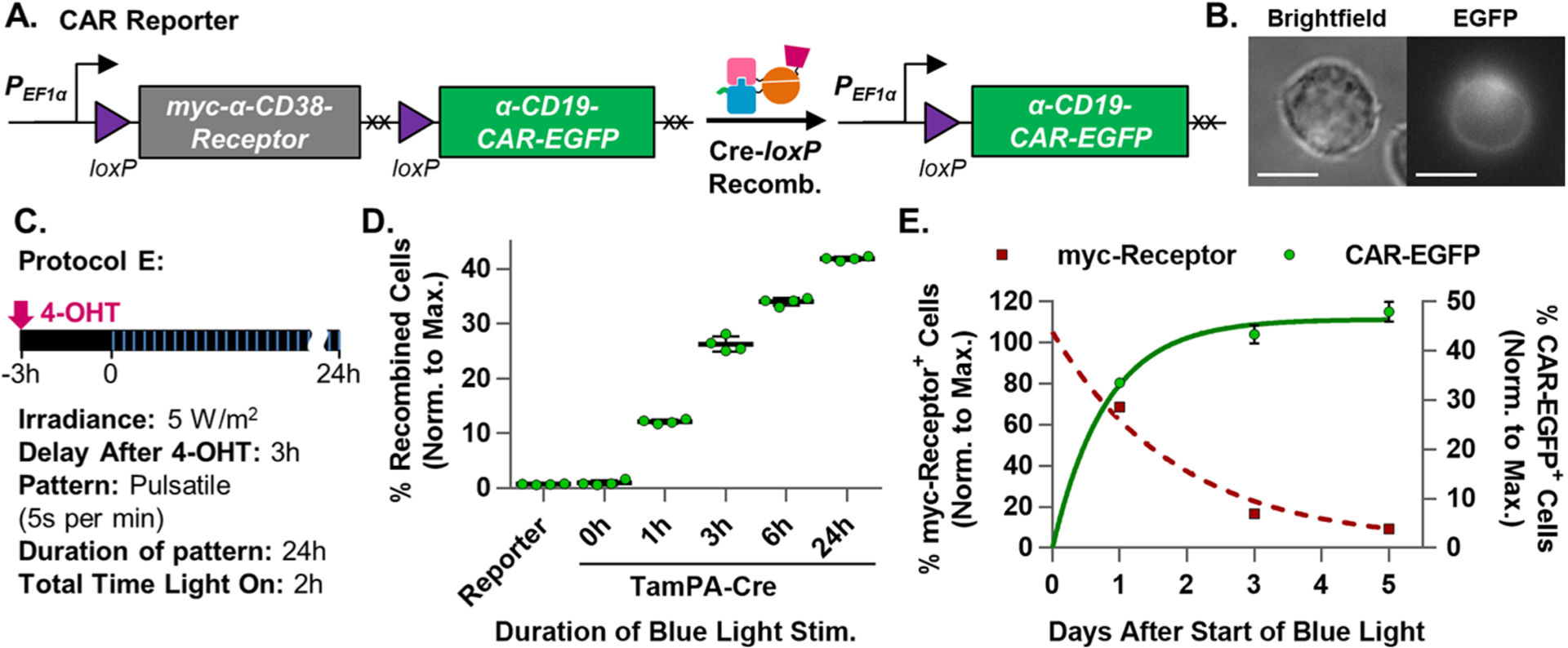
Optimization and characterization of the TamPA-Cre system in Jurkat T cells. (A) Schematic of the CAR Reporter construct before and after TamPA-Cre-mediated Cre-loxP recombination. The hEF1α promoter initially drives myc-α-CD38-Receptor expression. During Cre-loxP recombination, the floxed myc-α-CD38Receptor (with its stop codons, XX) is irreversibly excised allowing for (B) α-CD19CAR-EGFP expression (Jurkat T cell, 100×, scale bar = 10 μm). (C) Schematic illustrating tamoxifen (500 nM) and blue light stimulation (473 ± 29 nm) Protocol E: (5 W/m2, 5 s per min, 24 h) started 3 h after 4-OHT addition. (D) The percentage of recombined TamPA-Cre+ CAR Reporter Jurkat T cells (normalized to maximal recombination) exposed to Protocol E over the course of 0, 1, 3, 6, or 24 h (n = 4). (E) Normalized percentage of myc-Receptor+ and CAR-EGFP+ TamPA-Cre+ CAR Reporter Jurkat T cells stimulated by Protocol E, measured 1, 3, and 5 days after start of blue light stimulation, fitted with exponential decay and association trendlines (GraphPad, Table S2) (n = 4). Reporter = CAR Reporter Jurkat T cell line. CAR-EGFP flow cytometry measurements taken 72 h after the start of blue light stimulation. Percentage of recombined (% CAR-EGFP+) Jurkat T cells (normalized to maximal recombination) = 100%*(% of CAR-EGFP+ cells)/(initial % of CAR Reporter+ cells, measured via myc).
Both components of either the PA-Cre-M or TamPA-Cre system were transduced into CAR Reporter Jurkat T cells sequentially at high copy number and maintained via puromycin selection to create stable cell lines. CAR Reporter Jurkat T cells transduced with only one of the two TamPA-Cre components were unable to undergo Cre-loxP recombination indicating that, like PA-Cre-M, both split-Cre protein halves are necessary for function (Figure S6E). The PA-Cre-M+ and TamPA-Cre+ CAR Reporter Jurkat T lines were protected from light whenever possible during cell line development and culture to minimize potential ambient light-driven background recombination.
In order to preemptively address reported blue light phototoxicity concerns in T cells,40 we further reduced the total amount of blue light stimulation time from 3 h in Protocol B to 2 h in Protocol E (5 W/m2, 5 s per min, 24 h, Figure 4C). This reduction was possible because of the slow dissociation kinetics of the PA-Cre-M system’s nMag-pMag heterodimerized proteins (half-life: 1.8 h).38 Assuming stimulated TamPA-Cre heterodimerized proteins dissociate similarly upon the removal of light, less than 1% of active TamPA-Cre proteins will have been lost to dissociation during the 52.5 s or 55 s of darkness between the blue light pulses of Protocol B and E, respectively (Figure S8A). In essence, the pulsatile light pattern uses minimal light energy to maintain near-maximal levels of tamoxifen-primed active TamPA-Cre expression (Figure S8B). Three hours after incubation with 4-OHT, TamPA-Cre+ CAR Reporter Jurkat T cells were subjected to Protocol E’s 5 s per min pulsatile blue light stimulation pattern for 0, 1, 3, 6, or 24 h, after which the percentage of CAR-EGFP+ recombined cells in each group was determined by flow cytometry 48 h after the start of light exposure (Figure 4D). A positive control Cre+ CAR Reporter Jurkat T cell line with comparable protein expression levels could not be established because of long-term cytotoxicity. Therefore, the percentage of recombined CAR-EGFP+ cells in each CAR Reporter Jurkat T cell group was normalized to its mean maximal recombination measured from the initial percentage of myc-α-CD38 Receptor+ cells capable of undergoing recombination in each group. Cre-loxP recombination efficiency increased exponentially with the total duration of blue light stimulation and plateaued within 24 h of exposure (Figure S9, Table S2).
Following Protocol E with 24 h of blue light stimulation, the percentage of TamPA-Cre+ CAR Reporter Jurkat T cells expressing myc-α-CD38 Receptor and α-CD19 CAR-EGFP was tracked over several days (Figure 4E, Table S2). Without a means to replenish receptor proteins after the myc-α-CD38 Receptor CDS is excised during Cre-loxP recombination, the fraction of Receptor+ cells decayed exponentially over time (half-life: ~ 33.7 h), reaching a minimum of 9.3% ± 1.3% 5 days after the start of blue light stimulation. Simultaneously, the percentage of CAR+ cells increased exponentially (half-life: 13.3 h), more rapidly than the decrease in myc-α-CD38 Receptor expression. As such, engineered TamPA-Cre CAR Reporter T cells should remain anchored as locally applied blue light triggers CAR expression for tumor eradication (Figure 5A).
Figure 5.
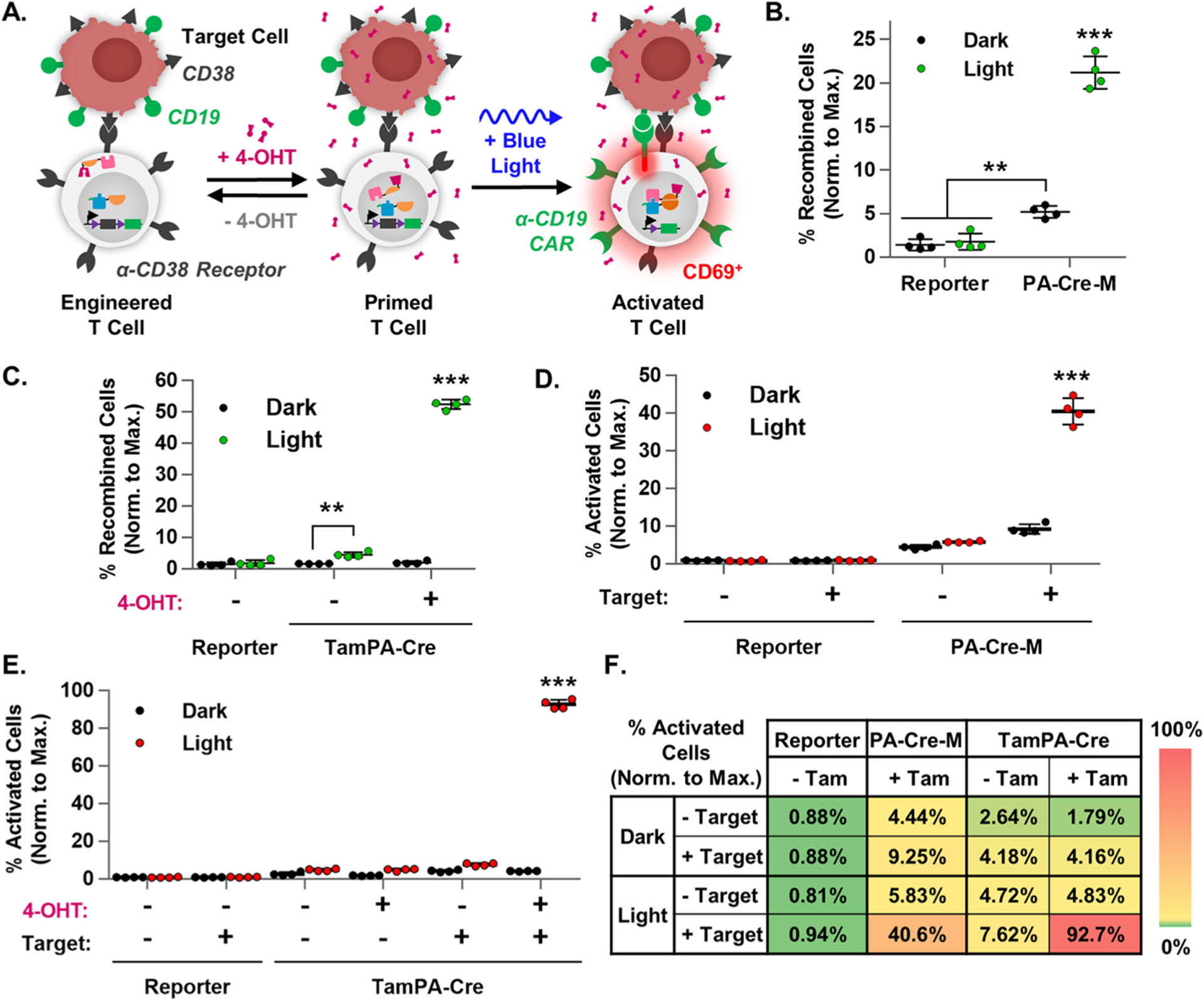
TamPA-Cre drives CAR-mediated T cell activation. (A) Schematic of the tamoxifen- and blue light-induced TamPA-Cre system in a CAR Reporter T cells driving recombination and CAR-mediated T cell activation upon binding to a TAA+ Target cells. The percentage of recombined cells (normalized to maximal recombination) in (B) PA-Cre-M+ and (C) TamPA-Cre+ CAR Reporter Jurkat T cells that did (+ 4-OHT) or did not (− 4-OHT) receive tamoxifen (500 nM) stimulation, and did (Light) or did not (Dark) receive blue light stimulation (473 ± 29 nm) as outlined in Protocol E (n = 4). The percentage of activated cells (normalized to maximal recombination) of samples from (B) and (C) that were (+ Target) or were not (− Target) coincubated with CD19+ Target cells (1:1), as reported in (D) and (E) respectively. (n = 4). (F) A heat map summary of T cell activation in Reporter, PA-Cre-M, and TamPA-Cre groups, with higher and lower efficiencies shown in red and green, respectively. Reporter = CAR Reporter Jurkat T cell line. Coincubation started and flow cytometry measurements taken 48 and 72 h after the start of blue light stimulation, respectively. Percentage of recombined Jurkat T cells (normalized to maximal recombination) = 100%*(% of CAR-EGFP+ cells)/(initial% of CAR Reporter+ cells, measured via myc). Percentage of activated Jurkat T cells (normalized to maximal recombination) = 100%*(% of CD69+ cells)/(initial % of CAR Reporter+ cells, measured via myc).
TamPA-Cre Drives CAR-Mediated T Cell Activation.
In a head-to-head comparison, CAR Reporter Jurkat T cell lines expressing TamPA-Cre, PA-Cre-M, or neither (Reporter) were (Light) or were not (Dark) subjected to tamoxifen and/or blue light stimulation in accordance with Protocol E. Two days after the start of light stimulation, each group of cells either were (+Target) or were not (−Target) cocultured for 24 h with an equal number of CD19+ Toledo Target cells. All groups were then analyzed 24 h later for the expression of α-CD19 CAR-EGFP and the early T cell activation marker CD69 via flow cytometry. The percentage of recombined CAR-EGFP+ cells and the percentage of activated CD69+ cells in each CAR Reporter Jurkat T cell group were normalized to maximal recombination calculated from the initial percentage of myc-α-CD38 Receptor+ cells capable of undergoing Cre-loxP recombination.
In a trend consistent with HEK293T experiments, light stimulation drove a significant 4.1-fold increase in the normalized percentage of recombined PA-Cre-M+ CAR Reporter Jurkat T cells (Light: 21.2 ± 1.9%, Dark: 5.2 ± 0.7%), whereas tamoxifen- and blue light-stimulated TamPA-Cre+ cells exhibited a robust 27.1-fold increase (Light + 4-OHT: 52.4 ± 1.5%, Dark + 4-OHT: 1.9 ± 0.5%, Figures 5B,C, S10A,B). Upon coculture with CD19+ Target cells, light-stimulated PA-Cre-M+ groups exhibited activation in less than half of CAR Reporter Jurkat T cells—a 4.4-fold increase compared to those without light stimulation (40.6 ± 3.5% and 9.3 ± 1.3%, respectively, Figures 5D, S10C). On the other hand, tamoxifen- and blue-light-stimulated TamPA-Cre drove CAR-mediated T cell activation in nearly all CAR Reporter Jurkat T cells cocultured with CD19+ Target cells (92.7 ± 2.4%)—a 22.3-fold increase from those groups lacking light stimulation (4.2 ± 0.1%, Figures 5E,F, S10D). Furthermore, without tamoxifen, blue light stimulation drove only a minor increase in TamPA-Cre-mediated Cre-loxP recombination and activation (4.4 ± 0.8% and 7.6 ± 0.8%, respectively), indicating that the AND gate functions as intended in Jurkat T cells.
While the suppression of partially stimulated cells was not perfect, the TamPA-Cre system’s high recombination efficiency upon complete stimulation makes it amenable to further means of suppression (e.g., lowering TamPA-Cre expression, additional gating, etc.) Moreover, exposing TamPA-Cre+ CAR Reporter Jurkat T cells to 48 h of ambient light did not drive any additional background recombination, unlike PA-Cre-M+ cells (Figure S6F). Therefore, without tamoxifen stimulation, cells expressing the TamPA-Cre system are relatively safe from background CAR expression—a requirement for practical applications. As such, with robust and well-gated tamoxifen- and blue light-inducible CAR expression and T cell activation, the TamPA-Cre system proves to be an effective tool with which to control localized CAR-mediated T cell activation
SUMMARY AND FUTURE DIRECTIONS
We have developed a novel logic-gated optogenetic split Cre system by integrating both ERT2-fusion proteins with the blue light-inducible nMag-pMag heterodimerizing domains to drive robust Cre-loxP recombination with significantly suppressed background. Only after treatment with tamoxifen is the TamPA-Cre system primed to be activated by short pulses of low intensity blue light stimulation. The tamoxifen gate helps prevent the spontaneous Cre-loxP recombination within cells prior to specific blue light stimulation—a weakness of other photoactivatable Cre-loxP systems. Applying the TamPA-Cre system to our floxed CAR-Reporter construct in Jurkat T cells, we were able to precisely induce CAR expression and antigen-specific T cell activation. With its unique high spatiotemporal control over T cell activation, the TamPA-Cre system could be used to locally induce T cell effector functions against cancer cells in vivo while avoiding on-target off-tumor toxicity in TAA+ healthy tissues.
The TamPA-Cre system also offers improved spatiotemporal control over other engineered CAR systems, like SynNotch41 and SUPRA CAR.42 Suicide switches43,44 and iCARs45 can be further integrated into our robust photoactivatable system to prevent potential on-target off-tumor toxicity caused by TamPA-Cre-activated CAR T cells leaving the stimulated region following tumor eradication. CRISPR-Cas9 technology can also help integrate large TamPA-Cre and engineered CAR T cell system designs into safe and effective loci in the genome.46 Furthermore, while the highly controllable TamPA-Cre system can replace virtually any Cre-loxP system, we foresee that it will serve as a particularly useful alternative to CRE-ERT2 systems in mouse lines where spontaneous Cre-loxP background recombination in vivo is already an established problem.47
MATERIALS AND METHODS
Cre and CAR Construct Design.
Detailed information on cloning and constructs can be found in the file Supplementary Construct Spreadsheet but is briefly explained here. pCDNA3.1-CMV (Thermo Fisher, no. V79020), pSin-EF1α (Addgene no. 16579), and pHR-PGK (Addgene no. 79125, 79130) mammalian expression vector backbones were used. Molecular cloning was done in DH5α competent cells (Thermo Fisher no. 18265017), and plasmids were isolated using miniprep (Sigma, no. PLN350) and maxiprep (Qiagen, no. 12163) kits. Constructs were created using restriction enzymes with T4 ligation (New England Biolabs) or using Gibson Assembly (NEB no. E2622L) methods. Sufficient plasmid concentration (>0.5 μg/mL) and purity (260/280 ≥ 1.8, 260/230 ≥ 2.0) were ensured for all experiments as measured by a Nanodrop 2000c Spectrophotometer (Thermo Fisher no. ND2000C).
For PA-Cre-C, both photoactivatable Cre partners CRY2-(L348F)-CreN (Addgene no. 75368) and CIBI-CreC(N1) (Addgene no. 75367) were cloned into a pSin vector separated by an IRES and preceded by the PGK promoter and mCherry marker. PA-Cre-M photoactivatable Cre partners CreN-nMag-NLS and NLS-pMag-CreC were cloned from a synthesized DNA template of the Sato Lab’s published PA-Cre-M construct (a generous gift from the Wilson Wong Lab, Boston University, MA), into pSin-EF1a with mCherry and tBFP fluorescent markers, respectively. Before both PA-Cre-M components were cloned into pSin-EF1a with an mCherry marker, the pMag sequence was codon-diversified and synthesized (Integrated DNA Technologies) in an effort to prevent unwanted sequence recombination between nMag and pMag during lentiviral production. ERT2-CreN59 was cloned from a pSin-6xUAS-CMVmin-ERT2-Cre-ERT2 template (a kind gift from the Todd Coleman Lab, UCSD) and assembled with nMag (NLS removed) in pCDNA3.1-CMV or with nMag-P2A-mCherry in pCDNA3.1 and pSin-EF1α. The full constitutively expressed Cre sequence (Addgene no. 14797) with mCherry marker was cloned into the pSin-PGK vector. Tamoxifen-dependent ERT2-Cre-ERT2 was cloned into pSin-EF1a along with a c-terminal mCherry marker to create pSin-EF1α-ERT2-Cre-ERT2–2xP2A-mCh.
CAR and receptor constructs were assembled from first-generation α-CD19 CAR (19z1) and α-PSMA chimeric costimulatory receptor (P28BB) templates (both generously donated by the Sadelain Lab, Memorial Sloan-Kettering Cancer Center, NY). The α-CD38 scFv domain was synthesized (Integrated DNA Technologies) based on U.S. patent application US20100267145A1, with IgG signaling peptide and VL-VH linker inspired by a third-generation α-CD19 CAR sequence Daofeng Liu from Baylor College of Medicine, TX kindly shared with us. The α-CD19-CAR-EGFP construct consists of a CD8 signaling peptide, α-CD19 scFv, CD28 hinge, transmembrane, and intracellular domain, 4–1BB domain, CD3-zeta domain, GGSGGT linker, and EGFP. The α-CD38-Receptor construct consists of an IgG signaling peptide, c-Myc tag, α-CD38 scFv, and a codon-diversified CD28 hinge, transmembrane, and truncated/nonfunctional (4aa) intracellular domain. CAR and receptor constructs were assembled in pCDNA3.1-CMV and transferred to pSin-EF1α vector.
EGFP and CAR Reporter constructs containing floxed components were partially assembled in the pLV-CMV-LoxP-DsRed-LoxP-EGFP (Addgene no. 65726) vector and were fully assembled in pSin-EF1α. The Lox66 site was generated via overlap extension PCR using MEA344 and 345 primers, while Lox71 was created by using a mutated reverse primer (MEA346) to change the template’s LoxP site into a Lox71-containing PCR product for subsequent assembly.
Cell Culture and Reagents.
The following cell lines were purchased from ATCC: human embryonic kidney cells (HEK293T), Jurkat T cells (Clone E6–1, TIB-152), K-562 lymphoblasts (CCL-243, CD38−/CD19− target cells), and Toledo B lymphocytes (CRL-2631, CD38+/CD19+ target cells). HEK293T cells were cultured in Dulbecco’s modified eagle medium (DMEM) (Gibco no. 11995065) supplemented with 2 mM l-glutamine, 1 mM sodium pyruvate, 100 units/ml penicillin, 100 μg/mL streptomycin (Gibco no. 15140122), and 10% (v/v) fetal bovine serum (FBS) (Thermo Fischer Scientific no. 10438026) in standard tissue-culture-treated dishes or plates. K-562, Jurkat, and Toledo cells were cultured in RPMI-1640 medium with l-glutamine, supplemented with 10% (v/v) FBS, 1× penicillin, and streptomycin in nontissue-culture-treated plates or flasks. HEK293T EGFP Reporter and Jurkat PA-Cre-M and TamPA-Cre cell lines were maintained in puromycin (Sigma no. P8833-100MG).
(Z)-4-Hydroxytamoxifen (Sigma no. H7904–5MG) was dissolved in 200-proof ethanol with heat to make 50 mM frozen stock solution aliquots (protected from light with foil). Immediately before adding 4-OHT to cells, an aliquot of stock solution was serially diluted in the appropriate fresh cell culture medium to 500 nM in experiments calling for the addition of 4-OHT.
Lentivirus Production.
Lenti-X 293T cells (Clontech Laboratories no. 632180) cultured in DMEM + 10% FBS + 1× Pen/Strep were used to produce VSV-G pseudotyped lentivirus with a second-generation lentiviral system. Genes of interest were cloned into transfer plasmid pPKm-145 (Addgene no. 90505), which was cotransfected into Lenti-X-293T cells with envelope plasmid pCMV-VSV-G (Addgene no. 8454) and packaging plasmid pCMVΔR8.2 (Addgene no.12263) at a 2:1:1 molar ratio using the ProFection Mammalian Transfection System (Promega, Madison, WI no. E1200). Viral supernatant was collected and filtered (0.22 μm) 48 h after transfection and either used directly or concentrated 100× using PEG-it Virus Precipitation Solution (System Biosciences, Palo Alto, CA) before use. Excess virus was aliquoted and stored at −80 °C for future use.
Lentiviral Transduction in Cell Lines.
The EGFP Reporter HEK293T cell line was made by lentiviral transduction. First, functional titer was determined by mCherry expression. Then, the cell line was generated from a group in which 1–20% of cells were transduced (~1–2 copied/cell) and was selected and maintained by culturing in 0.5 μg/mL puromycin.
For Jurkat T cells, unconcentrated or concentrated lentivirus supernatant was added directly to 100 000 cells/mL in culture medium and incubated 24 h, after which the virus was removed and the cells plated in fresh culture medium. After 4–5 days, lentivirus functional titration of the CAR Reporter construct in Jurkat cells was measured by AlexaFluor647-conjugated antibody immunostaining against the myc-tag (Cell Signaling no. 2233S) on constitutively expressed myc-α-CD38 Receptor. A group in which 1–20% of cells were transduced (~1–2 copies/cell) was sorted via myc-tag antibody staining using the FACSAria II Sorter at UCSD Human Embryonic Stem Cell Core Facility (La Jolla, CA). Cells were expanded in culture medium, aliquoted, and cryopreserved. Sorted CAR Reporter Jurkat cells were likewise also transduced with PA-Cre-M and TamPA-Cre constructs, with nMag and pMag constructs transduced sequentially with FACS sorting between each transduction. Unless otherwise specified, cell culture plates containing cells transduced or transfected with complete PA-Cre-M or TamPA-Cre were handled in a room illuminated by red light (HIGROW, 36 W, 660 nm) and were otherwise protected from light with aluminum foil to prevent premature recombination. Functional titration of the α-CD19CAR-EGFP construct in Jurkat cells was measured by EGFP expression, and a group in which 1–20% of cells were transduced (~1–2 copies/cell) was selected for and maintained using puromycin.
Jurkat T cell lines expressing the CAR Reporter were occasionally enriched via MACS selection using a 1:10 dilution of Biotin-conjugated antibody against the myc-tag (Jackson ImmunoResearch Inc. no. 115-066-006) to label cells and Anti-Biotin MicroBeads in the MidiMACS Separator with LS Columns (Miltenyi Biotec, Germany) to separate cells with the CAR Reporter.
Light Stimulation.
An intensity-adjustable blue light control system was made by Phillip Kyriakakis. To diffuse the light more evenly across the plate of cells, first, the blue LED (LUXEON Rebel no. LXML-PB02, 472 nm) was mounted with Dual Lock Reclosable Fasteners (3M) in the center of the bottom of a deep, polyester white-walled container with a clear PET plastic lid and synthetic rubber gasket (IKEA no. 402.574.99). Small plastic Decorating Clips (3M) were used to hold the LED wire along the inside walls of the container, keeping it out of the light path. Then, a static cling frosted window film (Beautyhero) was cut to size and placed on the inside of the lid. Closing with the lid, the container was flipped lid-side-down and centered on top of a lidless black plastic box (Hammond no. 1591ESBK) in which a cell culture plate would be placed for blue light exposure. The blue light intensity was adjusted and measured using a power meter (Newport, Model 843-R) before each experiment to ensure the plate wells farthest from the light source were receiving the intended irradiance flux density. For experiments, the LED container was placed on top of the black box holding the plate of cells immediately before placing in 37 °C, 5% CO2 cell culture incubator for the indicated blue light exposure patterns. The control box and computer were operated outside of the incubator. The spectrum of the blue LED used in this experiment was measured using the TECAN Infinite M1000 Pro, which measured a peak intensity at 473 nm with a 29 nm bandwidth. (See Figure S2 for photographs and measurement data.)
Plates of cells exposed to the Ambient light condition were placed in the frontmost position on the top shelf of a shared cell culture incubator, where the cells were intermittently exposed to approximately 0.34–0.78 W/m2 of white fluorescent light according to normal lab use. During a typical business day between 8:00 am and 8:00 pm, we estimate that the incubator door is opened approximately 100 times per day for about 5 s each. However, we purposely forwent strict control over the Ambient light condition in order to faithfully simulate light exposure in our busy shared lab environment. Therefore, we expect—and indeed find—that cells exposed to the Ambient light condition may or may not exhibit statistically significant higher recombination levels than those kept in the dark simply because of the inherently variable day-to-day use of the incubator.
HEK 293T Experiments.
With the exception of the Ambient light condition, all samples were protected from environmental light with aluminum foil immediately after transfection and throughout the duration of the experiment unless otherwise specified (i.e., during blue light stimulation). All procedures outside of the incubator were conducted in red light only to minimize any effects that ambient light from the room and biosafety hood may have on photoactivation.
EGFP Reporter HEK293T cells were plated at 100 000 cells/well in a tissue-culture-treated 24-well plate. After 24 h, cells were transfected with construct plasmid(s) using Lipofectamine 3000 (Thermo Fisher Scientific no. L3000015). Eight hours after transfection, each group was split into multiple 24-well plates (10–15% confluence/well), each plate corresponding to a unique light condition (Dark, Ambient, or Light). Dark condition plate(s) remained wrapped in foil until end-point measurements. Foil was removed from Ambient condition plate(s) at this point until end-point analysis to allow for exposure to the normal light of our shared lab environment. Twenty-four hours post-transfection, foil was temporarily removed from Light condition plates to expose them to the specified blue light condition, after which the Light condition’s plate(s) were again wrapped in foil until end-point analysis.
Jurkat Experiments.
Throughout cell line creation, culture, and experimentation, Jurkat T cells expressing the CAR Reporter along with complete PA-Cre-M or TamPA-Cre were protected from environmental light with aluminum foil at all times unless otherwise specified (e.g., Ambient light condition, blue light stimulation). All procedures outside of the incubator were conducted in red light only to minimize any effects that ambient light from the room and biosafety hood may have on photoactivation.
CAR Reporter Jurkat T cell lines with or without PA-Cre-M or TamPA-Cre were plated at 100 000 cells/well in multiple nontissue-culture-treated 24-well plates (Genesee Scientific Corp. no. 25-102), with one set of TamPA-Cre cells receiving 500 nM 4-OHT. Unless otherwise specified, one of the plates was moved into a separate cell culture incubator and stimulated with 5 W/m2 of pulsatile blue light (5s per min) for 24 h and then wrapped in foil. Forty-eight hours after blue light stimulation started, each group of Jurkat T cells was passed to a round-bottom nontissue-culture-treated 96-well plate (2 wells, 100 000 cells each), one with and one without 100 000 CD19+ Toledo target cells (200 μL/well). After a further 24 h of incubation, Jurkat T cells were immunostained with an APC-conjugated antibody against early T cell activation marker CD69 (BioLegend no. 310910) and measured via flow cytometry. Recombination (% EGFP+ cells) and activation (% CD69+ cells) measurements were normalized to the initial percentage of cells expressing the CAR Reporter, as measured by immunostaining for the myc-tag on CD38Receptor). Jurkat groups were not normalized to cells expressing Cre (as was done with HEK293T cells) because stable cell lines with constitutive Cre expressed at levels comparable to PA-Cre-M or TamPA-Cre died over time, preventing the creation of a stable cell line (data not shown).
Jurkat T cells expressing α-CD19CAR-EGFP or myc-α-CD38Receptor-EGFP were shown to bind to CD19+CD38+ Toledo target cells via a novel flow cytometry-based binding assay. Briefly, Toledo target cells were dyed with CellTracker deep red (Ex/Em: 630/650 nm, ThermoFisher), mixed 1:1 with CAR or Receptor Jurkat T cells (~150 000 cells each) in 1 mL of cell culture medium and placed in a 37 °C rotator for 30–45 min to allow for binding. Then, the mixture of cells was directly measured at slow speed by flow cytometer (BD Accuri C6) to identify the percentage of CAR+ or Receptor+ Jurkat T cells (EGFP+) that were also bound to target cells (CellTracker+).
Microscopy.
Prior to imaging, cells were plated on 35 mm no. 0 glass bottom dishes (Cell E&G, San Diego, CA) coated with either 20 μg/mL fibronectin or 10 μg/mL poly-l-Lysine for adherent and suspension cells, respectively (Sigma, St. Louis, MO). Images were acquired on a Nikon Eclipse Ti inverted microscope with a cooled charge-coupled device (CCD) camera. The following filters were used to image the indicated fluorophores: tBFP (420/40 nm excitation, 480/40 nm emission, 455 nm LP dichroic mirror), mCherry (580/10 nm excitation, 630/20 nm emission, 595 LP dichroic mirror), and EGFP (457–487 nm excitation, 502–538 nm emission, GFP filter cube set). Analysis was conducted using MetaFluor 7.8 or MetaMorph 7.8 software (Molecular Devices, San Jose, CA).
Flow Cytometry Measurements.
Fluorescent and immunostained cells were measured using a BD Accuri C6 flow cytometer equipped with 488 and 640 nm excitation lasers. EGFP expression was measured using the 488 nm excitation laser with a 533/30 nm emission filter. Cell labeling antibodies conjugated with far-red dyes were measured using the 640 nm excitation laser with a 675/25 nm emission filter. Before measuring, live cells were trypsinized (if adherent), washed three times with FACS wash buffer (filter-sterilized 0.5% BSA in PBS or autoMACS Running Buffer (Miltenyi Biotec, Germany)), then either measured directly or immunostained for 20 min at 37 °C, washed thrice, and then measured. Data was analyzed using FlowJo software (TreeStar). Samples of plain HEK293T, Jurkat, and Toledo cells were included with each experiment as negative controls for gating purposes. For data presentation, the normalized values are shown to easily compare the differences among the experimental groups
Data Representation and Statistical Analysis.
In all EGFP Reporter HEK293T cell line experiments, the percentage of cells in each experimental and negative control (Reporter) group that underwent Cre-loxP recombination (% EGFP+) was divided by the mean percentage of maximally recombined cells of the corresponding Cre+ positive control groups and then multiplied by 100% to represent the% Recombined Cells (Norm. to Max.). This was done to account for differences in transfection efficiency between experiments.
In all CAR Reporter Jurkat T cell line experiments, the percentage of cells in each experimental group that underwent either Cre-loxP recombination (% EGFP+) or CAR-mediated T cell activation (% CD69+) was divided by the initial percentage of nonrecombined CAR-Reporter+ Jurkat T cells for each group and then multiplied by 100% to represent the% Recombined or Activated Cells (Norm. to Max.), respectively. Attempts to create a Cre+ CAR Reporter Jurkat T cell line to function as a positive control resulted in cell death due to the known toxicity of constitutively expressed Cre and were therefore unable to be used as a measurement of maximal recombination efficiency. Instead, the initial percentage of nonrecombined CAR-Reporter+ Jurkat T cells—as measured by myc—with the potential to undergo recombination was used to normalize measurements for each group.
The mean ± standard deviation dot plots of n independently tested groups (≥10 000 cells/group) were assessed by the Student’s t test (two-tailed, two-sample unequal variance) to determine statistical significance between sets of different experimental conditions. P values were determined as follows: not significant (n.s.) if p > 0.05, * if p ≤ 0.05, ** if p ≤ 0.01, ***if p ≤ 0.001. Error bars represent the standard deviation.
Supplementary Material
ACKNOWLEDGMENTS
Special thanks to the laboratories of Wilson Wong (Boston University, MA), Michel Sadelain (Sloan Kettering Institute, NY), and Daofeng Liu (Baylor College of Medicine, TX) who generously provided helpful constructs and information.
Funding
This work was supported in part by grants from NIH HL121365, GM125379, GM126016, CA204704 and CA209629 (to Y.W.), the Galvanizing Engineering in Medicine program under the Institute of Engineering in Medicine and Altman Clinical and Translational Research Institute (ACTRI) at UC San Diego (to Y.W. and X.X.), the American Cancer Society Award ACS-IRG 70-002 (to X.X.), and the Integrative Bioengineering of Heart, Vessels and Blood NHLBI Training Grant 5T32HL105373-04 (to M.A.). The funding agencies had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
ABBREVIATIONS
- 4-OHT
(Z)-4-hydroxytamoxifen
- A.U.
arbitrary units
- aa
amino acids
- bp
base pairs
- CAR
chimeric antigen receptor
- CDS
coding sequence
- CRISPR
clustered regularly inter-spaced short palindromic repeats
- EGFP
enhanced green fluorescent protein
- ERT2
T2 mutant estrogen receptor ligand binding domain
- IL-2
interleukin-2
- kb
kilobase pairs
- LED
light emitting diode
- LOV2
light-oxygen-voltage-sensing domain
- mCherry
a monomeric red fluorescent protein
- NIR
near-infrared
- NLS
nuclear localization sequence
- nMag
negative magnet protein of the light-inducible Magnet system
- Norm.
normalized
- PA-Cre-C
CRY2(L348F)-CIB1-based photoactivatable Cre recombinase30
- PA-Cre-M
nMag-pMag-based photoactivatable Cre recombinase29
- pMag
positive magnet protein of the light-inducible Magnet system
- TAA
tumor-associated antigen
- Tam
tamoxifen
- TamPA-Cre
tamoxifen and photoactivatable Cre recombinase
- UCNPs
up-conversion nanoparticles
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acssynbio.9b00175.
Truth table highlighting potential on-target off-tumor toxicity risks for several engineered CAR T cell system, schematic representations of genetic constructs, design and characterization of the blue light stimulation apparatus, additional characterization of the PA-Cre-M system, diversification of the pMag CDS, optimizing TamPA-Cre recombination efficiency through different tamoxifen- and blue light-stimulation protocols, CAR-mediated T cell activation is antigen specific at low CAR expression levels, testing different Cre-loxP CAR Reporter designs, and other data as mentioned in the text (PDF)
Supplementary Construct Spreadsheet: oligonucleotide and genetic construct information (XLSX)
The authors declare the following competing financial interest(s): Y.W. is a scientific co-founder of Cell E&G Inc and Acoustic Cell Therapy LLC. However, these financial interests do not affect the design, conduct, or reporting of this research.
REFERENCES
- (1).Porter DL, Levine BL, Kalos M, Bagg A, and June CH (2011) Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med 365 (8), 725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF, Milone MC, Levine BL, and June CH (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med 368 (16), 1509–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Maude SL, Shpall EJ, and Grupp SA (2014) Chimeric antigen receptor T-cell therapy for ALL. Hematology Am. Soc. Hematol Educ Program 2014 (1), 559–64. [DOI] [PubMed] [Google Scholar]
- (4).Maude SL, Teachey DT, Porter DL, and Grupp SA (2015) CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 125 (26), 4017–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, Qu J, Wasielewska T, He Q, Fink M, Shinglot H, Youssif M, Satter M, Wang Y, Hosey J, Quintanilla H, Halton E, Bernal Y, Bouhassira DC, Arcila ME, Gonen M, Roboz GJ, Maslak P, Douer D, Frattini MG, Giralt S, Sadelain M, and Brentjens R (2014) Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med 6 (224), 224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Alonso-Camino V, Harwood SL, Alvarez-Mendez A, and Alvarez-Vallina L (2016) Efficacy and toxicity management of CAR-T-cell immunotherapy: a matter of responsiveness control or tumour-specificity? Biochem. Soc. Trans 44 (2), 406–11. [DOI] [PubMed] [Google Scholar]
- (7).Frey N, and Porter D (2019) Cytokine Release Syndrome with Chimeric Antigen Receptor T Cell Therapy. Biol. Blood Marrow Transplant 25, e123. [DOI] [PubMed] [Google Scholar]
- (8).Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, Pequignot E, Gonzalez VE, Chen F, Finklestein J, Barrett DM, Weiss SL, Fitzgerald JC, Berg RA, Aplenc R, Callahan C, Rheingold SR, Zheng Z, Rose-John S, White JC, Nazimuddin F, Wertheim G, Levine BL, June CH, Porter DL, and Grupp SA (2016) Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discovery 6 (6), 664–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, and Rosenberg SA (2010) Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther 18 (4), 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lamers C (2009) Treatment of metastatic renal cell cancer with autologous T cells genetically retargeted against carbonic anhydrase IX - first clinical experience. Hum. Gene Ther 20 (11), 1544–1545. [DOI] [PubMed] [Google Scholar]
- (11).Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, Byatte AJ, Kirillova N, Valle JW, Sharma SK, Chester KA, Westwood NB, Halford SER, Nabarro S, Wan S, Austin E, and Hawkins RE (2017) The clinical efficacy of first-generation carcinoembryonic antigen (CEA-CAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol. Immunother 66 (11), 1425–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Weinberg BH, Pham NTH, Caraballo LD, Lozanoski T, Engel A, Bhatia S, and Wong WW (2017) Large-scale design of robust genetic circuits with multiple inputs and outputs for mammalian cells. Nat. Biotechnol 35 (5), 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kloss CC, Condomines M, Cartellieri M, Bachmann M, and Sadelain M (2013) Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol 31 (1), 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wu CY, Roybal KT, Puchner EM, Onuffer J, and Lim WA (2015) Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 350 (6258), aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, and Lim WA (2016) Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164 (4), 770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Cho JH, Collins JJ, and Wong WW (2018) Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 173 (6), 1426–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ueda Y, and Sato M (2018) Induction of Signal Transduction by Using Non-Channelrhodopsin-Type Optogenetic Tools. ChemBioChem 19 (12), 1217–1231. [DOI] [PubMed] [Google Scholar]
- (18).Tan P, He L, Han G, and Zhou Y (2017) Optogenetic Immunomodulation: Shedding Light on Antitumor Immunity. Trends Biotechnol 35 (3), 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Shimizu-Sato S, Huq E, Tepperman JM, and Quail PH (2002) A light-switchable gene promoter system. Nat. Biotechnol 20 (10), 1041–4. [DOI] [PubMed] [Google Scholar]
- (20).Kaberniuk AA, Shemetov AA, and Verkhusha VV (2016) A bacterial phytochrome-based optogenetic system controllable with near-infrared light. Nat. Methods 13 (7), 591–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Huang X, Yang P, Ouyang X, Chen L, and Deng XW (2014) Photoactivated UVR8-COP1 module determines photo-morphogenic UV-B signaling output in Arabidopsis. PLoS Genet 10 (3), e1004218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Sinha DK, Neveu P, Gagey N, Aujard I, Benbrahim-Bouzidi C, Le Saux T, Rampon C, Gauron C, Goetz B, Dubruille S, Baaden M, Volovitch M, Bensimon D, Vriz S, and Jullien L (2010) Photocontrol of protein activity in cultured cells and zebrafish with one- and two-photon illumination. ChemBioChem 11 (5), 653–63. [DOI] [PubMed] [Google Scholar]
- (23).Park SI, Brenner DS, Shin G, Morgan CD, Copits BA, Chung HU, Pullen MY, Noh KN, Davidson S, Oh SJ, Yoon J, Jang KI, Samineni VK, Norman M, Grajales-Reyes JG, Vogt SK, Sundaram SS, Wilson KM, Ha JS, Xu RX, Pan TS, Kim TI, Huang YG, Montana MC, Golden JP, Bruchas MR, Gereau RW, and Rogers JA (2015) Soft, stretchable, fully implantable miniaturized optoelectronic systems for wireless optogenetics. Nat. Biotechnol 33 (12), 1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Jeong JW, McCall JG, Shin G, Zhang YH, Al-Hasani R, Kim M, Li S, Sim JY, Jang KI, Shi Y, Hong DY, Liu YH, Schmitz GP, Xia L, He ZB, Gamble P, Ray WZ, Huang YG, Bruchas MR, and Rogers JA (2015) Wireless Optofluidic Systems for Programmable In Vivo Pharmacology and Optogenetics. Cell 162 (3), 662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).He L, Zhang Y, Ma G, Tan P, Li Z, Zang S, Wu X, Jing J, Fang S, Zhou L, Wang Y, Huang Y, Hogan PG, Han G, and Zhou Y (2015) Near-infrared photoactivatable control of Ca(2+) signaling and optogenetic immunomodulation. eLife 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Yu N, Huang L, Zhou Y, Xue T, Chen Z, and Han G (2019) Near-Infrared-Light Activatable Nanoparticles for Deep-Tissue-Penetrating Wireless Optogenetics. Adv. Healthc Mater 8, 1801132. [DOI] [PubMed] [Google Scholar]
- (27).Repina NA, Rosenbloom A, Mukherjee A, Schaffer DV, and Kane RS (2017) At Light Speed: Advances in Optogenetic Systems for Regulating Cell Signaling and Behavior. Annu. Rev. Chem. Biomol. Eng 8, 13–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Nihongaki Y, Kawano F, Nakajima T, and Sato M (2015) Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol 33 (7), 755–60. [DOI] [PubMed] [Google Scholar]
- (29).Kawano F, Okazaki R, Yazawa M, and Sato M (2016) A photoactivatable Cre-loxP recombination system for optogenetic genome engineering. Nat. Chem. Biol 12 (12), 1059. [DOI] [PubMed] [Google Scholar]
- (30).Taslimi A, Zoltowski B, Miranda JG, Pathak GP, Hughes RM, and Tucker CL (2016) Optimized second-generation CRY2-CIB dimerizers and photoactivatable Cre recombinase. Nat. Chem. Biol 12 (6), 425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Niopek D, Benzinger D, Roensch J, Draebing T, Wehler P, Eils R, and Di Ventura B (2014) Engineering light-inducible nuclear localization signals for precise spatiotemporal control of protein dynamics in living cells. Nat. Commun 5, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Feil R, Wagner J, Metzger D, and Chambon P (1997) Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem. Biophys. Res. Commun 237 (3), 752–757. [DOI] [PubMed] [Google Scholar]
- (33).Hirrlinger J, Requardt RP, Winkler U, Wilhelm F, Schulze C, and Hirrlinger PG (2009) Split-CreERT2: Temporal Control of DNA Recombination Mediated by Split-Cre Protein Fragment Complementation. PLoS One 4 (12), e8354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Besser S, Wilhelm F, Winkler U, Requardt R, Schulze C, Hirrlinger PG, and Hirrlinger J (2011) Split-Cre and Split-Creert2: Versatile Genetic Coincidence Detectors for Precise Analysis of Cell Populations in Vivo. Glia 59, S100–S100. [Google Scholar]
- (35).Feil S, Valtcheva N, and Feil R (2009) Inducible Cre mice. Methods Mol. Biol 530, 343–63. [DOI] [PubMed] [Google Scholar]
- (36).Casanova E, Fehsenfeld S, Lemberger T, Shimshek DR, Sprengel R, and Mantamadiotis T (2002) ER-based double iCre fusion protein allows partial recombination in forebrain. Genesis 34 (3), 208–14. [DOI] [PubMed] [Google Scholar]
- (37).Komatsubara AT, Matsuda M, and Aoki K (2015) Quantitative analysis of recombination between YFP and CFP genes of FRET biosensors introduced by lentiviral or retroviral gene transfer. Sci. Rep 5, 13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kawano F, Suzuki H, Furuya A, and Sato M (2015) Engineered pairs of distinct photoswitches for optogenetic control of cellular proteins. Nat. Commun 6, 6256. [DOI] [PubMed] [Google Scholar]
- (39).Drent E, Groen RW, Noort WA, Themeli M, Lammerts van Bueren JJ, Parren PW, Kuball J, Sebestyen Z, Yuan H, de Bruijn J, van de Donk NW, Martens AC, Lokhorst HM, and Mutis T (2016) Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica 101 (5), 616–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Phototoxicity revisited. Nat. Methods 2018, 15 (10), 751. DOI: DOI: 10.1038/s41592-018-0170-4 [DOI] [PubMed] [Google Scholar]
- (41).Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, and Lim WA (2016) Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164 (4), 770–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Cho JH, Collins JJ, and Wong WW (2018) Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 173, 1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B, Liu H, Cruz CR, Savoldo B, Gee AP, Schindler J, Krance RA, Heslop HE, Spencer DM, Rooney CM, and Brenner MK (2011) Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N. Engl. J. Med 365 (18), 1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Gargett T, and Brown MP (2014) The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol 5, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Fedorov VD, Themeli M, and Sadelain M (2013) PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci. Transl Med 5 (215), 215ra172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gonen M, and Sadelain M (2017) Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543 (7643), 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Kristianto J, Johnson MG, Zastrow RK, Radcliff AB, and Blank RD (2017) Spontaneous recombinase activity of Cre-ERT2 in vivo. Transgenic Res 26 (3), 411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.