

Abstract
Background:
Epidemiologic studies indicate that early life arsenic exposures are linked to an increased risk of cardiovascular diseases. Different oxidation and methylation states of arsenic exist in the environment and are formed in vivo via the action of arsenic ( oxidation state) methyltransferase (As3MT). Methylated arsenicals are pro-atherogenic postnatally, but pre- and perinatal effects are unclear. This is particularly important because methylated arsenicals are known to cross the placenta.
Objectives:
We tested the effects of early life exposure to inorganic and methylated arsenicals on atherosclerotic plaque formation and its composition in apolipoprotein E knock-out () mice and evaluated whether mice lacking As3MT expression were susceptible to this effect.
Methods:
We exposed or mice to inorganic or methylated arsenic in the drinking water from conception to weaning and assessed atherosclerotic plaques in the offspring at 18 wk of age. Mixed regression models were used to estimate the mean difference in each outcome relative to controls, adjusting for sex and including a random effects term to account for within-litter clustering.
Results:
Early life exposure to inorganic arsenic, and more profoundly methylated arsenicals, resulted in significantly larger plaques in the aortic arch and sinus in both sexes. Lipid levels in these plaques were higher without a substantial difference in macrophage numbers. Smooth muscle cell content was not altered, but collagen content was lower. Importantly, there were sex-specific differences in these observations, where males had higher lipids and lower collagen in the plaque, but females did not. In mice lacking As3MT, arsenic did not alter the plaque size, although the size was highly variable. In addition, control mice had significantly larger plaque size compared with control .
Conclusion:
This study shows that early life exposure to inorganic and methylated arsenicals is pro-atherogenic with sex-specific differences in plaque composition and a potential role for As3MT in mice. https://doi.org/10.1289/EHP8171
Introduction
Arsenic is a toxicant naturally found in soil, water, and air. (Mandal and Suzuki 2002; Nordstrom 2002). The World Health Organization (WHO) has set a maximum contaminant level of arsenic at for municipal drinking water (WHO 2008), but millions of people worldwide are exposed to elevated levels of arsenic (Nordstrom 2002). Arsenic increases the risk of several diseases, from cancer to cardiovascular disease (CVD) (Chen et al. 2013b; James et al. 2015; Jiang et al. 2015; Kuo et al. 2017; Mateen et al. 2017; Moon et al. 2017; Roh et al. 2017; Smith et al. 2018). It has been linked epidemiologically to increased risk for atherosclerosis, the gradual occlusion of large arteries with fibro-fatty plaques (Hsieh et al. 2011; Wang et al. 2010), which, in turn, increases the risk of mortality due to heart attack and stroke. However, we do not understand who is at risk for developing atherosclerosis following arsenic exposure.
Several studies have indicated that pregnancy is an important window for arsenic exposure (Young et al. 2018). In utero exposure to arsenic has been associated with effects in children, including decreased birth weight, potentially by decreasing gestational age and/or maternal weight gain (Kile et al. 2016; Wang et al. 2018). A number of epidemiologic studies have suggested that in utero arsenic exposure is also associated with several adverse health effects later in life, such as lung (Steinmaus et al. 2016) and bladder tumors (Smith et al. 2012; Yuan et al. 2007), diabetes (Navas-Acien et al. 2019), and coronary heart disease (Roseboom et al. 2000). Limited data have linked early life arsenic exposures to CVD. In a cross-sectional study of children in Mexico, increased carotid intima thickness, a marker for early atherosclerosis, was found to be associated with total urinary arsenic (Osorio-Yáñez et al. 2013). Although the contribution of in utero exposure could not be defined, most of the mothers lived in the same region throughout pregnancy. In a Bangladeshi cohort, arsenic exposure was associated with a greater risk of mortality from cancer or CVD in children, albeit in a sample population with a very small death rate (Rahman et al. 2013). Thus, it is unclear whether exposure to arsenic during gestation leads to increased risk of atherosclerosis in humans.
Arsenic is metabolized by a series of oxidative methylation reactions catalyzed by the enzyme arsenic (III) methyltransferase (As3MT) (Thomas 2007). The biotransformation process is a conserved mechanism that produces methylated intermediate compounds and has historically been considered a detoxification process. However, this concept has been challenged because some of the intermediate compounds of arsenic biomethylation, particularly the methylated metabolites containing trivalent arsenic, were considered more toxic than the inorganic form (Stýblo et al. 2002). For example, we have shown that in mice, in addition to inorganic arsenic, methylated forms are pro-atherogenic (Negro Silva et al. 2017). Interestingly, we also showed that arsenic-enhanced plaque formation was dependent upon As3MT expression (Negro Silva et al. 2017). This suggests that arsenic methylation is essential for the atherogenic properties of arsenic after postnatal exposure in mice. Based on the urinary profile of arsenic metabolites in epidemiologic studies, several factors may contribute to the efficiency of the biotransformation reaction, such as nutritional status (Hall et al. 2007; Howe et al. 2014; Pilsner et al. 2009), genetic background (Meza et al. 2007; Schläwicke Engström et al. 2007), gender (Lindberg et al. 2008; Vahter et al. 2002), and pregnancy (Concha et al. 1998). Indeed, pregnant women have a more efficient methylation process (Concha et al. 1998; Hall et al. 2007). Arsenic and methylated intermediates pass through the placenta, and thus the fetus has been shown to be exposed to similar concentrations of arsenic as the mother (Concha et al. 1998; Hall et al. 2007). Pregnancy and development are considered susceptible periods of exposure to toxicants (Farzan et al. 2013), but the sequela from exposure to arsenic during gestation have not been well defined.
Animal experiments have provided mechanistic insight into the underlying mechanisms of arsenic-enhanced atherosclerosis. We previously showed that postnatal exposure to low ()-to-moderate () concentrations of arsenic as sodium arsenite () (Lemaire et al. 2011; Makhani et al. 2018) or methylated arsenicals (Negro Silva et al. 2017) resulted in larger atherosclerotic plaques in 18-wk-old male mice. Our data also indicated that arsenic trioxide enhanced lipid accumulation in macrophages (Padovani et al. 2010) and increased the attachment of macrophages to the arterial endothelial cell layer in vitro (Lemaire et al. 2015). Furthermore, an in utero exposure to also increased plaque formation in mice (Srivastava et al. 2007). Microarray analysis showed that preprograming of oxidative stress and inflammatory response pathways during early life was associated with enhanced development of atherosclerosis later in life (States et al. 2012). Lipids, stress, and inflammatory pathways, which may contribute to plaque formation, were also up-regulated after arsenic exposure (as ) at high concentrations via drinking water () (States et al. 2012). However, it is unknown whether prenatal exposure to low-to-moderate arsenic concentrations are pro-atherogenic. In addition, the pro-atherogenic potential of methylated arsenicals and the impact of biotransformation remain to be elucidated.
Thus, we asked whether pre- and perinatal exposure to inorganic or methylated arsenicals would result in greater atherosclerotic plaques when measured later in life in an mouse model. Furthermore, we used our model of mice that also lack As3MT expression to determine whether arsenic-enhanced atherosclerosis following gestational exposure was dependent upon As3MT.
Material and Methods
Mice and Exposure Protocol
() mice were obtained from the Jackson Laboratory. mice (C57BL/6 background) were kindly provided by D. Thomas (U.S. Environmental Protection Agency, Research Triangle Park, NC) and used to create double-knockout (DKO) mice in our facility (Negro Silva et al. 2017). Purchased mice were acclimatized to housing conditions under a 12-h light/12-h dark cycle for at least 2 wk before experiments. All mice were fed ad libitum. Female mice were fed AIN-76A purified diet (Harlan Laboratories Inc.) containing 5% fat (by weight) with no cholesterol 1 wk prior to mating, and then parents and offspring were maintained on AIN-76A. The experimental protocol was approved by the McGill Animal Care Committee, and animals were handled in accordance with institutional guidelines. The McGill Animal Care Committee is certified by the Canadian Council on Animal Care.
Both and mice were assigned randomly by sex to mating pairs ( pairs per experimental group). mating pairs were exposed to either tap water or tap water containing arsenic in the form of ( ; Sigma-Aldrich), disodium methyl arsonate hexahydrate (; ; Chem Service), cacodylic acid (; ; Sigma-Aldrich) or monomethylarsonous acid [; ]. This is a concentration at which we have observed maximal pro-atherogenic effects postnatally (Lemaire et al. 2011; Makhani et al. 2018). This level of arsenic has been found in the drinking water in highly contaminated areas of Bangladesh (BGS 2000), Argentina (Pérez-Carrera and Fernández Cirelli 2010; Concha et al. 1998), and the United States (Neilsen et al. 2010). The synthesis of is described in the next section. Moreover, mice were exposed to either tap water or tap water containing arsenic in the form of ( ; Sigma-Aldrich). Solutions containing arsenic were prepared fresh from the powder every 2 to 3 days to minimize oxidation and hydrolytic demethylation. After confirmation of pregnancy via weight gain and vaginal plug observations in female mice, male mice were removed from the cage and pregnant females were housed separately until weaning. Exposure was from mating until 3 wk after birth. dams are sensitive to stress and environmental changes, thus to assure care of the pups, we waited until 3 wk post-birth to change to tap water. At weaning (Wk 4), both control and exposed or DKO pups were maintained for an additional 14 wk on tap water. At 18 wk of age, adult male and female offspring were euthanized by carbon dioxide asphyxiation/cervical dislocation, and subsequently, heart and aortae were collected, rinsed in phosphate-buffered saline (PBS), and fixed in 4% paraformaldehyde solution.
Synthesis of
Synthesis of was performed as/based on a previous publication (Cullen et al. 1989). The first step in the synthesis of was to prepare methylarsine oxide [] by dissolving of methylarsenate () acid sodium salt, () in of water (), Dissolution of the initial salt was promoted by gradual heating of the solution. Once dissolved, the solution was treated with sulfur dioxide (), which was bubbled through the solution. The solution quickly becomes clear (suggesting acid sensitivity), then light yellow after another 2 min. After saturating with , the solution was quickly boiled for 2 min, then cooled for 15 min. Neutralization with sodium carbonate turned the solution from light yellow to clear. The solvent was removed and was extracted with benzene. Removing the benzene in vacuo resulted in of a white solid (70% yield).
Instant hydrolysis of this tetrameric methylarsenic oxide by dissolution in water leads to stoichiometric formation of . For example, when was dissolved in water, a uniform colorless solution formed of [] in a concentration. When deuterium oxide () is used in place of water, this solution has only one peak at in the nuclear magnetic resonance spectrum at 25°C, which corresponds to the methyl signal in the hydrolyzed species , as reported previously (Petrick et al. 2001).
Plasma analyses.
Blood () was collected by cardiac puncture and plasma was obtained using collection tubes (ethylenediaminetetraacetic acid BD Vacutainer SST). Cholesterol, high- (HDL) and low-density (LDL) lipoproteins, triglycerides, and liver enzymes [aspartate aminotransferase (AST) and alanine aminotransferase (ALT)] were assessed by the Comparative Medicine and Animal Resources Center (CMARC at McGill University, Canada; Tables S1 and S2). The following products were used by CMARC: VITROS CHOL (Ortho-Clinical Diagnostics; 1669829) for cholesterol, VITROS dHDL (Ortho-Clinical Diagnostics; 6802469) for HDL, VITROS TRIG (Ortho-Clinical Diagnostics 8,329,930) for triglycerides, VITROS AST (Ortho-Clinical Diagnostics; 8433815) for AST, and VITROS ALT (Ortho-Clinical Diagnostics; 1655281) for ALT, as per the manufacturer’s protocol. These slides employed enzyme-based methods for substrate detection followed by reflectance spectrophotometry for quantification. The quantity of LDL was calculated from HDL using the Friedwald formula.
Atherosclerotic lesion characterization.
The characterization of the atherosclerotic lesions was performed as previously described (Lemaire et al. 2014, 2011; Makhani et al. 2018; Negro Silva et al. 2017). Briefly, the fixed aorta was rinsed with ultra-pure water, then cut longitudinally and stained en face with oil red O (Electronic Microscopy Sciences), which stains neutral triglycerides and lipids. Images were acquired using Infinity Capture software (Version 6.5.6) and a Lumenera camera. Percentage of lesion area of the aortic arch, as defined as the region from the first intercostal arteries to the ascending arch, was evaluated with ImageJ software (Schneider et al. 2012). Rinsed, fixed, and embedded frozen hearts were processed as previously described (Lemaire et al. 2011). Consecutive, cryosections were sliced from the aortic base throughout the aortic sinus, and three to five valve sections per animal were stained with oil red O to visualize the plaque areas and analyzed for their lipid content. We considered the atherosclerotic lesions in the sinus either as lesion area in millimeters squared or as a percentage of the total sinus area. The latter calculation uses the total sinus area to normalize the data and accounts for any bias contributed by the slicing of the heart samples. The lesion area stained with oil red O (as measured by ImageJ) was divided by the total sinus area for three to five valve sections per sample. These measurements were then averaged to obtain the final plaque size (as a percentage of aortic sinus). Aortic valves were also stained and analyzed for their collagen content (type I and III) using picrosirius red (Polysciences).
In situ immunofluorescence.
Smooth muscle cell (SMC) and macrophage content were assessed within the entire plaque area, as previously described (Lemaire et al. 2011). Briefly, slides were rinsed with PBS and blocked with 3% bovine serum albumin for 1 h. Next, they were incubated with primary antibody for 1 h at room temperature; 1:100 for monoclonal muscle cell actin [clone 1A4] (Abcam; ab7817), 1:50 for Moma-2 (Abcam; ab33451), rinsed, and incubated with fluorescently labeled secondary antibodies at room temperature for 1 h. A 1:500 dilution each of goat antimouse (Alexa 488; A11001) and a goat antirat (Alexa 488; A11006) (Invitrogen) was used to visualize muscle cell actin and macrophages respectively. Images were acquired using Infinity Capture software and a Lumenera camera. The presence of the immunofluorescent marker from three to five sections per animal was quantified using ImageJ software (Schneider et al. 2012) and expressed as a percentage of the total lesion area to define the contribution of each component to plaque independent of plaque size.
Statistical analyses.
Standard statistical analyses assume the independence of each mouse; therefore, these analyses could not be used in this in utero exposure study. The issue was that the mice in the same litter were more similar to each other than those from other mothers; this dependency violates the usual assumption of independence in standard statistical models (e.g., -tests, regression), and thus, these correlations needed to be captured in the statistical model (Haseman and Kupper 1979; Lazic and Essioux 2013). As well, assuming independence leads to an underestimation of standard errors and consequently, biased confidence intervals (CIs) and -values (Bieler and Williams 1995). The mixed-model framework (Breslow and Clayton 1993; McLean et al. 1991) accounts for these intra-litter correlations and produces appropriate inferences under certain assumptions of the model. Thus, to meet the objectives of this study, namely, to estimate the effect of arsenic on plaque size in the aortic arch and sinus and plaque contents: macrophages, lipids, collagen, and SMC, we developed simple mixed regression models in which each of the exposure groups were compared with the unexposed group for each outcome separately. The mixed-model framework accounted for the within-litter correlations by specifying that all the offspring of one dam were grouped together (a random effect was placed on the intercept for each dam). Effectively, this more complicated model is equivalent to ordinary linear regression models but accounts for the within-dam correlations, thereby providing unbiased estimates of standard errors, CIs, and -values. We used the mixed-models package [nlme package in R, version 4.0.3 (R Development Core Team)] with a compound symmetry structure corresponding to a constant correlation. We routinely verified the assumptions of normality of each outcome and used standard diagnostics to ensure that the assumptions of the model were met (especially, normality of the random effects). As described previously (Caligiuri et al. 1999; Smith et al. 2010), females have larger plaques than males, independent of arsenic exposure; thus, we included sex as a covariate in our mixed-effects model. The analyses were conducted in the open-source R program, version 4.0.3 (R Development Core Team) using the lme4 function (Bates 2014a; Pinheiro and Bates 2000) and the final model was fitted using restricted maximum likelihood (REML) on the complete data set. We then formally tested for interactions between sex and exposure groups using maximum likelihood given that likelihood ratio tests cannot be used in REML. To obtain sex-specific differences, we then fitted separate models for females and males using REML. For all the statistical analyses, we considered as statistically significant.
Results
Analysis of Atherosclerotic Plaque Size following in Utero Arsenic Exposure in an Mouse Model
We exposed mating pairs (i.e., both male and female parents) to arsenic as methylated arsenicals (, , or ), inorganic arsenic () or tap water. We continued exposure for 3 wk after birth so that subsequent litters of mice were exposed from mating to weaning, although arsenic may not be present in breast milk (Fängström et al. 2008; Islam et al. 2014). After weaning, all pups were kept on tap water until 18 wk of age, a time point at which postnatal exposure enhances lesion formation (Makhani et al. 2018). Table 1 shows the distribution of litter number, size, and sex. We did not find any differences in litter size between exposure groups but did find some differences in the proportion of males across exposure groups, confirming our decision to adjust for sex.
Table 1.
Selected characteristics of the litters from control and exposed groups.
| Genotype | Exposure | Litters ()a | of pups/litter | of pups/litter () | Proportion of males (%) |
|---|---|---|---|---|---|
| Control | 12 | 3; 5; 8; 2; 2; 3; 6; 6; 6; 3; 11; 7 | 58 | ||
| 9 | 6; 9; 5; 8; 1; 9; 5; 6; 4 | 36 | |||
| 4 | 6; 5; 4; 7 | 40 | |||
| 5 | 6; 7; 8; 3; 3 | 61 | |||
| 5 | 3; 4; 7; 5; 6 | 61 | |||
| DKO | Control | 4 | 5; 5; 4; 3 | 70 | |
| 4 | 4; 7; 6; 4 | 39 |
Note: DKO, double-knock out; , monomethylarsonous acid; , disodium methyl arsonate hexahydrate; , sodium arsenite..
These numbers represent the total number of litters exposed. However, not all litters were analyzed for all the outcomes. Outcome-specific litter numbers are presented in relevant table notes.
We assessed atherosclerotic plaque size in the aortic arch (en face) and aortic sinus after staining with oil red O. Using a mixed-effects model, we found that the effects of arsenical treatment, with respect to the control group, varied by sex for many of the outcomes, that is, there was a statistically significant interaction () between exposure groups and sex. Therefore, quantitative values for sex-wise analyses are reported in addition to litter averages for all the outcomes.
We observed that mean plaque size in the aortic arch and aortic sinus was larger in offspring of the dams exposed to either inorganic arsenic or its methylated intermediates. Figures 1 and 2 show box plots of the distributions of unadjusted results, and Table 2 shows the results adjusted for sex using the mixed models. Atherosclerotic lesions in the aortic arch were larger in the inorganic arsenic group by 2.6% [(95% CI: 0.5, 4.6%); ] and that in sinus was larger by 3.4% [(95% CI: 0.1, 6.8%); ] or by [(95% CI: , 0.05); ] compared with the control group. Surprisingly, methylated arsenical exposures were significantly associated with the enhanced size of the aortic arch lesions [ (95% CI: 7.5, 13.2%) for , 7.7% (95% CI: 5.1, 10.3%) for , and 8.4% (95% CI: 5.7, 11.0%) for )], as well as aortic sinus plaques [ (95% CI: 5.1, 14.1%) for , 3.7% (95% CI: , 8.1%) for , and 6.9% (95% CI: 2.2, 11.5%) for ], which tended to be larger than -induced plaques, particularly in the arch analyses (Figures 1 and 2 and Tables 2 and S4). For the aortic arch, we found an interaction () between sex and the exposure groups. Therefore, we analyzed each sex separately, and we observed that the mean lesion size in the aortic arch was larger in both female and male offspring from arsenical-exposed dams; the effects of were the largest in enhancing the plaques in females [ (95% CI: 9.5, 17.6%)], whereas that of were the largest in males [ (6.0, 10.8%)] Figure 1 and Tables 3 and S4). In the aortic sinus, using percentages as outcome, we did not find any interaction between sex and the exposure groups (), and, therefore, the combined analysis is sufficient. We found rather different patterns of effects when we used the absolute area of the plaque in the aortic sinus measured in millimeters squared as the metric. There was an interaction by sex for the effects of arsenical treatments compared with the control group (). Here, the effects of were the largest in enhancing plaques in the aortic sinus in females [ (95% CI: 0.03, ); ], which is consistent with our observations for aortic arch analysis. Also consistent with arch data, we found a larger effect for in enhancing the sinus plaque size in males [ (95% CI: 0.02, ); ]. Moreover, we measured circulating concentrations of lipid and liver enzymes from the plasma of both male and female mice, and no differences were observed between control and arsenical-exposed mice (Tables S1 and S2).
Figure 1.

Differences in plaque formation in the aortic arch in adult mice following early life arsenical exposure. mice were exposed to arsenicals (, , , or ) or maintained on tap water from conception to weaning. After weaning (4 wk), male and female pups were kept on tap water for an additional 14 wk. The percentage of the lesion area of the aortic arch was evaluated via en face oil red O staining. (A) Representative images. The box plots represent the distribution of unadjusted total lesion area as a percentage of total arch area, where midline, box limits, whiskers, and dots denote the median, interquartile range, minimum and maximum values, and outliers, respectively. (B) The combined data set is presented, and sex-specific data sets for (C) females and (D) are presented. The corresponding numeric data are presented in Table S4 and that adjusted by litter are presented in Tables 2 and 3. Note: , cacodylic acid; , monomethylarsonous acid; , disodium methyl arsonate hexahydrate; , sodium arsenite.
Figure 2.
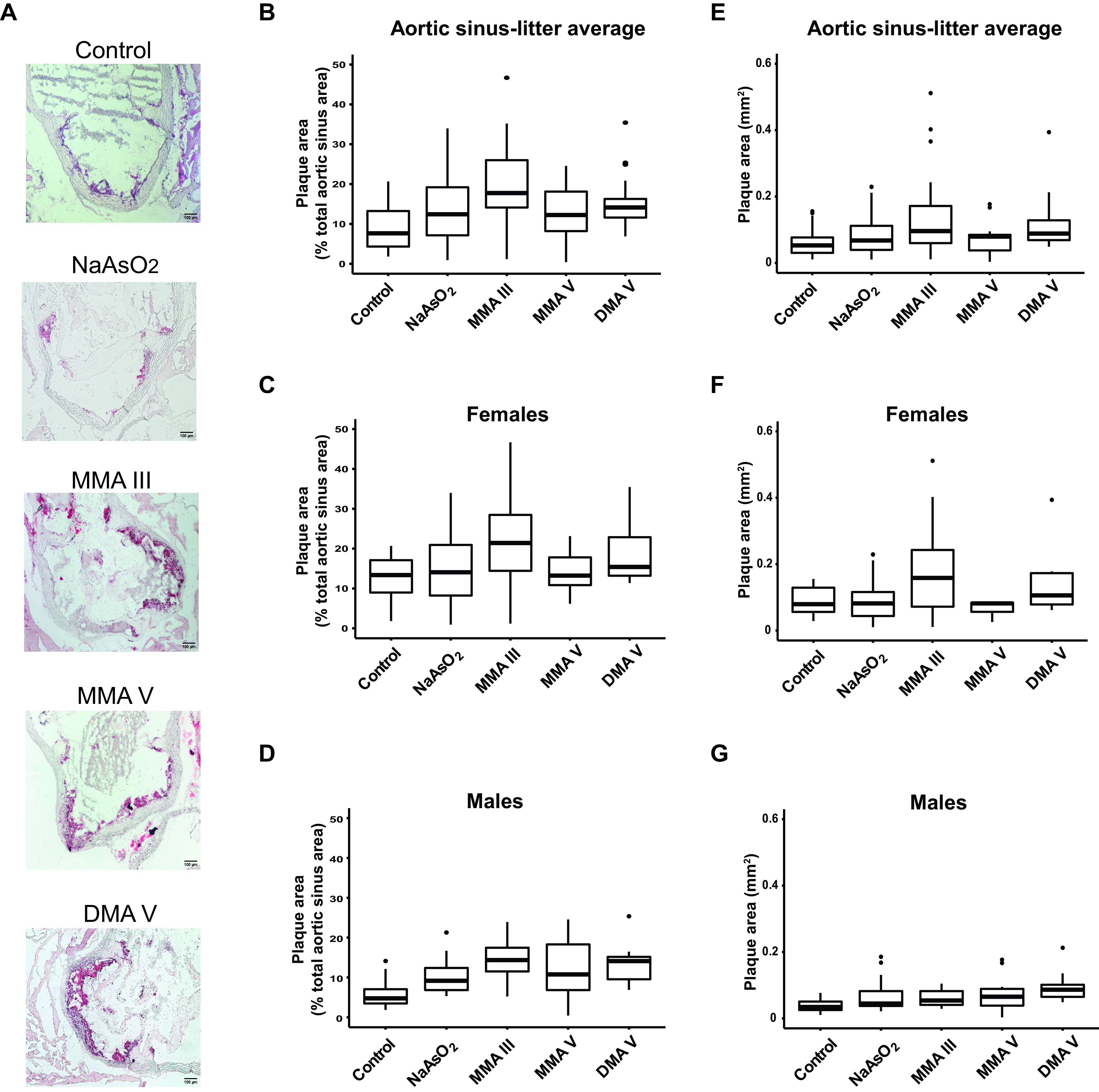
Differences in plaque formation in the aortic sinus in adult mice following early life arsenical exposure. mice were exposed as described for Figure 1 and the plaque area in the aortic sinus was assessed by oil red O staining. The percentage of the lesion area of the aortic sinus was evaluated via oil red O staining. (A) Representative images. The box plots represent the distribution of unadjusted total lesion area as a percentage of total sinus area, where midline, box limits, whiskers, and dots denote the median, interquartile range, minimum and maximum values, and outliers, respectively. (B) The combined data set is presented, and sex-specific data sets for (C) females and (D) are presented. In addition, the lesion area was measured as millimeters squared, and the unadjusted data distribution for all the litters from (E) each exposed group as well as that separated by (F) female and (G) male offspring is shown. The corresponding numeric data are presented in Table S4 and that adjusted by litter are presented in Tables 2 and 3. Note: , cacodylic acid; , monomethylarsonous acid; , disodium methyl arsonate hexahydrate; , sodium arsenite.
Table 2.
Relative differences in the plaque size in aortic arch and sinus of arsenical-treated mice relative to their control counterparts and associated 95% confidence intervals (CIs), adjusted for sex.
| Exposure groups | Lesion area | |||||
|---|---|---|---|---|---|---|
| Percentage of total archa | Percentage of aortic sinusb | Aortic sinus ()c | ||||
| Relative difference (95% CI) | -Value | Relative difference (95% CI) | -Value | Relative difference (95% CI) | -Value | |
| 2.6 (0.5, 4.6) | 0.01 | 3.4 (0.1, 6.8) | 0.04 | 0.02 (, 0.05) | 0.40 | |
| 10.4 (7.5, 13.2) | 9.6 (5.1, 14.1) | 0.07 (0.02, 0.12) | ||||
| 7.7 (5.1, 10.3) | 3.7 (, 8.1) | 0.09 | 0.01 (, 0.05) | 0.69 | ||
| 8.4 (5.7, 11.0) | 6.9 (2.2, 11.5) | 0.06 (0.01, 0.10) | 0.02 | |||
Note: Models were fitted using simple mixed regression models to control for the intra-litter correlation, and sex was included as a covariate. All exposure groups were compared with the unexposed (control) group for each outcome separately. The corresponding data distribution is depicted in Figures 1B and 2B,E. Number of litters : 12, : 9, : 4, : 5; : 5. , cacodylic acid; , monomethylarsonous acid; , disodium methyl arsonate hexahydrate; , sodium arsenite.
Percentage of oil red O-stained area over the total arch area.
Percentage of oil red O-stained lesion area over the total sinus area.
Absolute lesion area as measured by oil red O staining.
Table 3.
Differences in the plaque size in aortic arch and sinus of arsenical-treated mice relative to their control counterparts and associated 95% confidence intervals (CI), presented separately for females and males.
| Outcomes | Exposure groups | Females | Males | between males and femalesa | ||
|---|---|---|---|---|---|---|
| Relative difference (95% CI) | -Value | Relative difference (95% CI) | -Value | |||
| Lesion area (% of total arch)b | 2.9 (, 6.0) | 0.07 | 2.7 (0.7, 3.8) | 0.01 | ||
| 13.6 (9.5, 17.6) | 7.6 (4.8, 10.5) | |||||
| 8.2 (4.1, 12.3) | 7.3 (4.9, 9.6) | |||||
| 8.6 (4.4, 12.9) | 8.4 (6.0, 10.8) | |||||
| Lesion area (% of total sinus)c | 2.7 (, 8.9) | 0.38 | 4.9 (1.8, 7.9) | 0.47 | ||
| 9.3 (1.4, 17.2) | 0.02 | 8.8 (4.6, 13.1) | ||||
| 1.1 (, 9.5) | 0.80 | 5.9 (2.6, 9.3) | ||||
| 7.4 (, 16.7) | 0.11 | 7.7 (4.1, 11.2) | ||||
| Lesion area (in aortic sinus) ()d | 0.01 (, 0.06) | 0.78 | 0.03 (0, 0.06) | 0.06 | 0.02 | |
| 0.10 (0.03, 0.16) | 0.02 (, 0.06) | 0.23 | ||||
| (, 0.05) | 0.56 | 0.03 (0, 0.06) | 0.07 | |||
| 0.07 (, 0.15) | 0.08 | 0.05 (0.02, 0.09) | ||||
Note: To obtain sex-specific changes, separate models were fitted for females and males using simple mixed regression to control for the intra-litter correlation. All exposure groups were compared with the unexposed (control) group, for each outcome. , cacodylic acid; , monomethylarsonous acid; , disodium methyl arsonate hexahydrate; , sodium arsenite.
Interactions between sex and exposure groups were tested using maximum likelihood and the resulting -values are presented here. The corresponding data distribution is depicted in Figures 1C,F and 2C,D and 2F,G.
Percentage of oil red O-stained area over the total arch area; Number of litters : 12, : 9, : 4, : 5; : 5.
Percentage of oil red O-stained lesion area over the total sinus area; Number of litters : 12, : 9, : 4, : 4; : 4.
Absolute lesion area as measured by oil red O-staining; Number of litters : 12, : 9, : 4, : 4; : 4.
Analysis of Atherosclerotic Plaque Components following in Utero Arsenical Exposure in an Mouse Model
We assessed both macrophage and lipid content in the plaques following in utero and early life arsenic exposure. No statistically significant associations between macrophage content and arsenical exposure were observed, although all exposure groups showed a lower percentage of macrophages compared with the untreated control (Figure 3A–D and Tables 4 and S5). Importantly, the in utero exposure to inorganic and methylated arsenicals led to higher mean lipid content within the plaque (Figure 3E–H and Tables 4 and S5), with the -exposed group being statistically significant [ (95% CI: 4.9, 14.8%); ]. For plaque lipid content, we found an interaction () between sex and exposure groups. Therefore, analyzing each sex separately, we found a larger effect for and in males as compared with the respective unexposed controls [ (95% CI: 1.2, 12.0%); for and 13.4% (95% CI: 7.6, 19.1%); for ] (Figure 3G vs. 3H and Tables 5 and S5).
Figure 3.
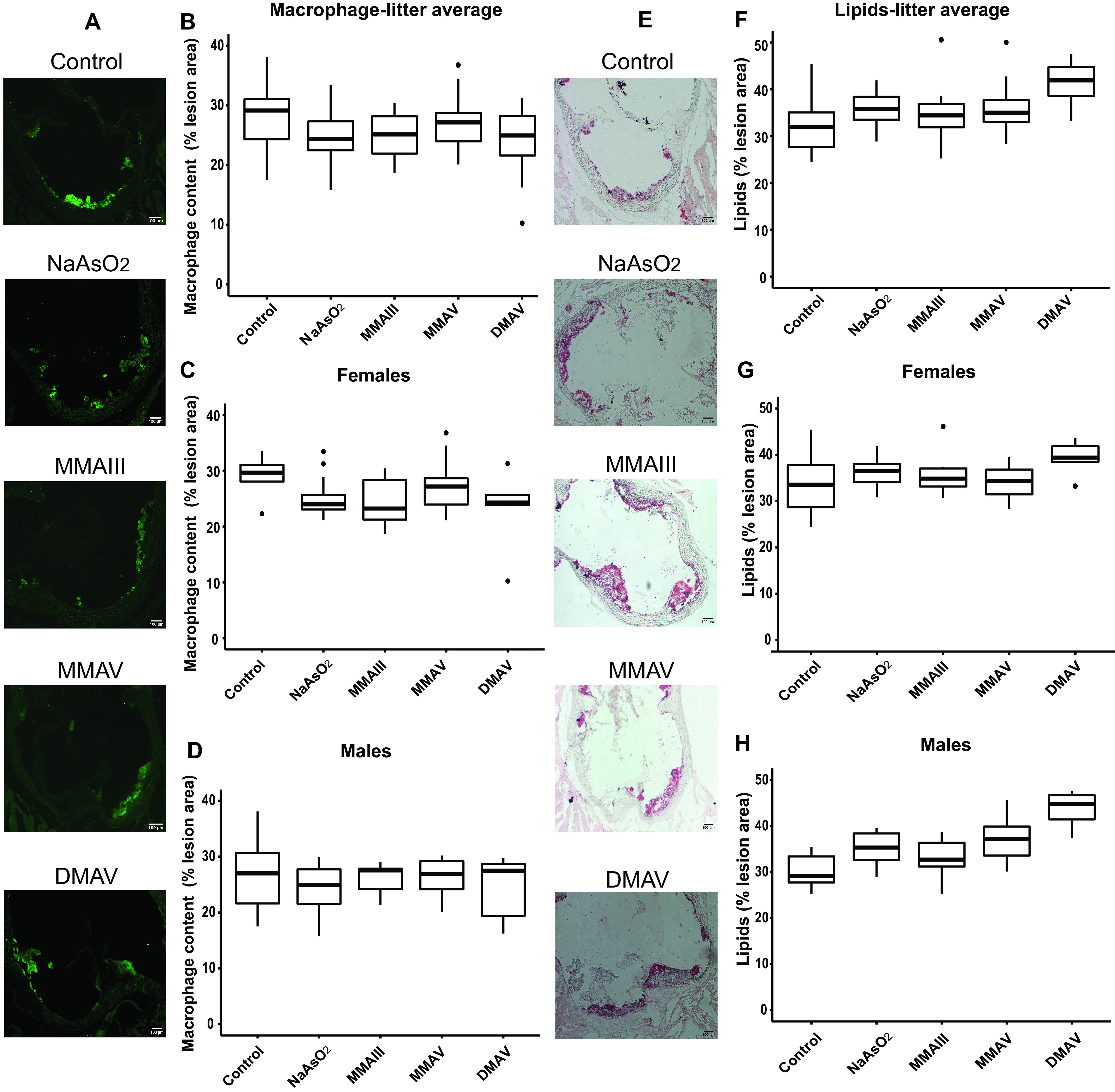
Differences in plaque macrophage and lipid content in the aortic sinus in adult mice following early life arsenical exposure. mice were exposed as described for Figure 1. (A–D) Macrophage and (E–H) lipid content were evaluated in the aortic sinus relative to the total lesion area using Moma-2 and oil red O staining, respectively. (A) and (E) show representative images. Scale bar: . (B) Box plots represent the data distribution of the unadjusted percentage of macrophages with respect to the total sinus area for all offspring in the control and arsenical-exposed groups, whereas those in (C) and (D) represent that in the female and male offspring, respectively. (E) Box plots represent the data distribution of the unadjusted percentage of lipid content with respect to the total sinus area for all offspring in the control and arsenical-exposed groups, whereas (F) and (G) represent that in the female and male offspring, respectively. The midline, box limits, whiskers, and dots denote the median, interquartile range, minimum and maximum values, and outliers, respectively. The corresponding numeric data are presented in Table S5 and that adjusted by litter are presented in Tables 4 and 5. Note: , cacodylic acid; , monomethylarsonous acid; , disodium methyl arsonate hexahydrate; , sodium arsenite.
Table 4.
Differences in the plaque components (macrophages, lipids, smooth muscle cells, and collagen) in aortic sinus of arsenical-treated mice relative to their control counterparts and associated 95% confidence intervals (CIs), adjusted for sex.
| Groups | Macrophages (% of sinus lesion area)a | Lipids (% of sinus lesion area)b | SMCs (% of sinus lesion area)c | Collagen (% of sinus lesion area)d | ||||
|---|---|---|---|---|---|---|---|---|
| Relative differences (95% CI) | -Value | Relative differences (95% CI) | -Value | Relative differences (95% CI) | -Value | Relative differences (95% CI) | -Value | |
| (, 1.2) | 0.16 | 3.8 (, 8.4) | 0.10 | (, 2.5) | 0.35 | (, 8.2) | 0.89 | |
| (, 1.8) | 0.22 | 2.7 (, 7.6) | 0.24 | (, 2.7) | 0.32 | 0.4 (, 9.3) | 0.93 | |
| (, 3.7) | 0.63 | 3.5 (, 8.1) | 0.12 | (, 3.4) | 0.43 | (, 6.9) | 0.74 | |
| (, 1.3) | 0.15 | 9.9 (4.9, 14.8) | (, 1.5) | 0.13 | (, 2.8) | 0.16 | ||
Note: Models were fitted using simple mixed regression models to control for the intra-litter correlation, and sex was included as a covariate. All exposure groups were compared with the unexposed (control) group, for each outcome separately. The corresponding data distribution is depicted in Figures 3B,F and 4B,F. , cacodylic acid; , monomethylarsonous acid; , disodium methyl arsonate hexahydrate; , sodium arsenite; , smooth muscle actin-alpha.
Percentage of Moma-2 antibody-stained area over the total lesion area in the aortic sinus as measured by the intensity of fluorescent signal at ; Number of litters : 6, : 3, : 4, : 3; : 4.
Percentage of oil red O-stained area over the total lesion area in the aortic sinus; Number of litters : 6, : 3, : 4, : 3; : 4.
Percentage of total antibody-stained area over the total lesion area in the aortic sinus as measured by the intensity of fluorescent signal at ; Number of litters : 6, : 3, : 4, : 3; : 3.
Percentage of picrosirius red-stained area over the total lesion area in the aortic sinus; Number of litters : 6, : 3, : 3, : 3; : 3.
Table 5.
Differences in the plaque components (macrophages, lipids, smooth muscle cells, and collagen) in the aortic sinus of arsenical-treated mice relative to their control counterparts and associated 95% confidence intervals (CIs), presented separately for females and males.
| Outcome | Exposure groups | Females | Males | between males and femalesa | ||
|---|---|---|---|---|---|---|
| Relative difference (95% CI) | -Value | Relative difference (95% CI) | -Value | |||
| Macrophage (% of sinus lesion area)b | (, 2.2) | 0.19 | (, 3.9) | 0.40 | 0.42 | |
| (, 1.4) | 0.12 | (, 6.0) | 0.80 | |||
| (, 4.7) | 0.61 | (, 6.0) | 0.85 | |||
| (, 0) | 0.05 | (, 4.2) | 0.50 | |||
| Lipids (% of sinus lesion area)c | 2.2 (, 11.0) | 0.59 | 5.0 (, 10.5) | 0.07 | 0.02 | |
| 2.0 (, 11.0) | 0.63 | 3.2 (, 8.8) | 0.25 | |||
| 0.7 (, 9.5) | 0.86 | 6.6 (1.2, 12.0) | 0.02 | |||
| 6.1 (, 16.1) | 0.20 | 13.4 (7.6, 19.1) | ||||
| Smooth muscle cells (% of sinus lesion area)d | 0.4 (, 5.0) | 0.84 | (, 1.3) | 0.09 | ||
| (, 3.6) | 0.61 | (, 6.7) | 0.55 | |||
| 0.2 (, 5.04) | 0.91 | (, 5.6) | 0.41 | |||
| (, 3.5) | 0.45 | (, 2.4) | 0.13 | |||
| Collagen (% of sinus lesion area)e | 8.0 (1.0, 15.0) | 0.03 | (, 0.2) | 0.05 | ||
| 6.5 (, 14.0) | 0.08 | (, 4.6) | 0.29 | |||
| 2.9 (, 10.0) | 0.37 | (, 2.8) | 0.18 | |||
| 5.9 (, 14.0) | 0.14 | (, ) | ||||
Note: To obtain sex-specific changes, separate models were fitted for females and males using simple mixed regression to control for the intra-litter correlation. All exposure groups were compared with the unexposed (control) group, for each outcome. , cacodylic acid; , monomethylarsonous acid; , disodium methyl arsonate hexahydrate; , sodium arsenite; , smooth muscle actin-alpha.
Interactions between sex and exposure groups were tested using maximum likelihood and the resulting -values are presented here. The corresponding data distribution is depicted in Figures 3C,D, 3G,H, 4C,D, and 4G,H.
Percentage of Moma-2 antibody-stained area over the total lesion area in the aortic sinus as measured by the intensity of fluorescent signal at ; Number of litters : 6, : 3, : 4, : 3; : 4.
Percentage of oil red O-stained area over the total lesion area in the aortic sinus; Number of litters : 6, : 3, : 4, : 3; : 4.
Percentage of total antibody-stained area over the total lesion area in the aortic sinus as measured by the intensity of fluorescent signal at ; Number of litters : 6, : 3, : 4, : 3; : 3.
Percentage of picrosirius red-stained area over the total lesion area in the aortic sinus; Number of litters : 6, : 3, : 3, : 3; : 3.
We also investigated collagen and SMC content using histological techniques. The mean percentage of SMCs and collagen measurement had a wide variability and did not differ between the arsenical-exposed groups and the control group (Figure 4A–D,E–H, and Tables 4 and S5). Despite not finding an overall effect, we observed an interaction between sex and exposure groups for both of these outcomes (Tables 5 and S5). When males and females were considered separately for plaque collagen content, had the largest effect on male offspring in that it resulted in profoundly lesser collagen in the exposed males [ (95% CI: , ); ], whereas in females, had the largest effect but resulted in more plaque collagen content instead [ (95% CI: 1.0, 15.0%); ] (Tables 5 and S5).
Figure 4.
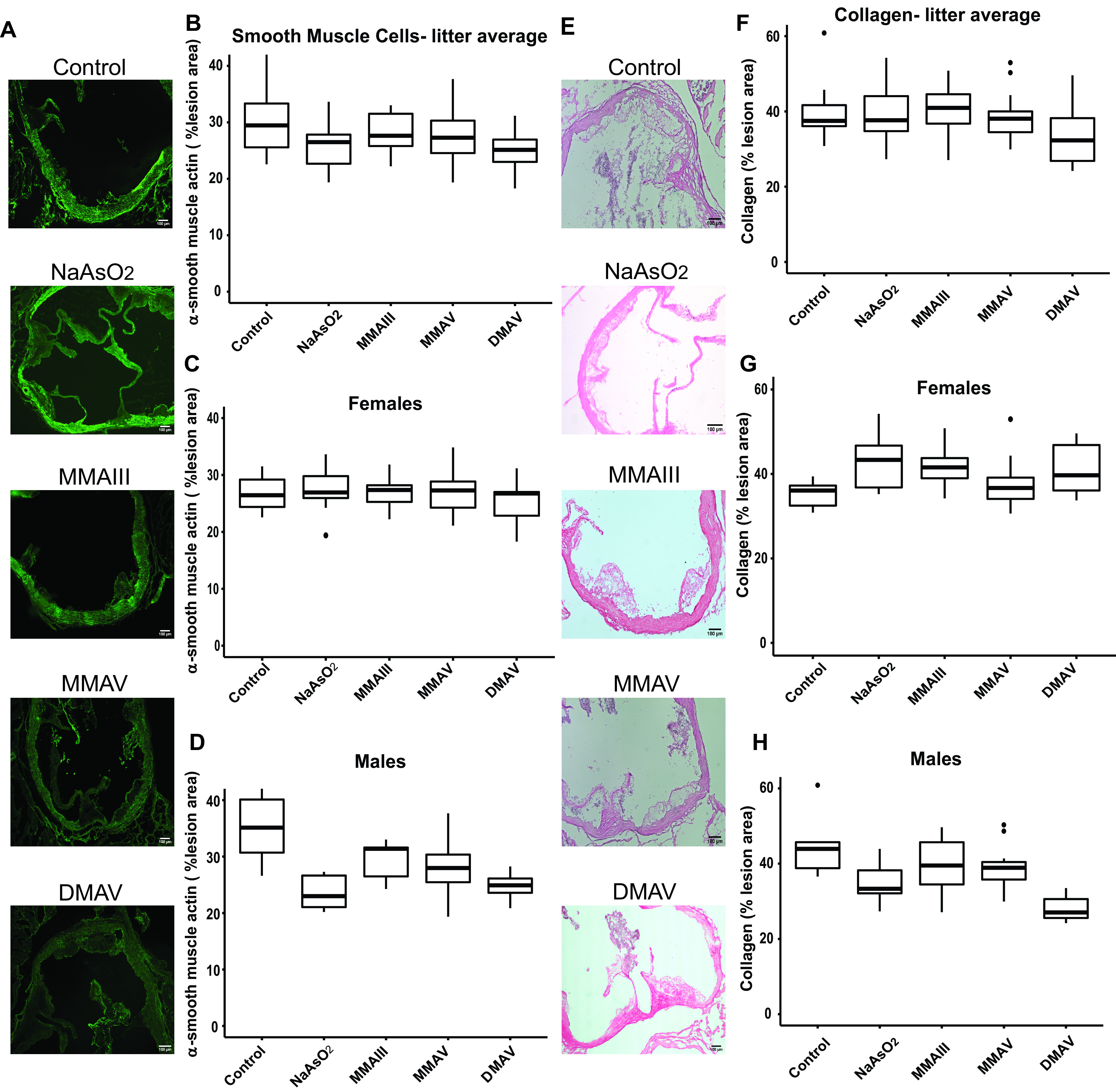
Differences in plaque SMC and collagen content in the aortic sinus in adult mice following early life arsenical exposure. mice were exposed as described for Figure 1. (A–D) SMC and (E–H) collagen content were evaluated in the aortic sinus relative to the total lesion area using muscle actin and picrosirius red staining, respectively. Scale bar: . (B) Represents the data distribution of the unadjusted percentage of SMCs with respect to the total sinus area for all offspring in the control and arsenical-exposed groups, whereas (C) and (D) represent that in the female and male offspring, respectively. (E) Represents the data distribution of the unadjusted percentage of collagen content with respect to the total sinus area for all offspring in the control and arsenical-exposed groups, whereas (F) and (G) represent that in the female and male offspring, respectively. The midline, box limits, whiskers, and dots denote the median, interquartile range, minimum and maximum values, and outliers, respectively. The corresponding numeric data are presented in Table S5 and that adjusted by litter are presented in Tables 4 and 5. Note: , cacodylic acid; , monomethylarsonous acid; , disodium methyl arsonate hexahydrate; , sodium arsenite; SMC, smooth muscle cell.
Analysis of Atherosclerotic Plaque Formation following in Utero Arsenic Exposure in an Mouse Model
We investigated the impact of the biotransformation process on atherosclerotic plaques after in utero and early life exposure. We used our DKO mice, which have proven useful to study the effects of arsenic methylation in arsenic-induced atherosclerosis (Negro Silva et al. 2017). DKO mice were exposed from mating to weaning to or tap water. Atherosclerotic plaques were assessed after 13 wk on tap water. Interestingly, did not have an effect on lesion area in the aortic arch [ (95% CI: , 7.9%)] (Figure 5 and Tables 6 and S6) or on plaque size in the aortic sinus [ (95% CI: , 3.6%) or (95% CI: , )] in DKO mice (Figures 6 and Tables 6 and S6). As well, we did not find an interaction between sex and the arsenic exposure for either of these outcomes (Table 7). Similar to mice, female DKO mice had larger plaques than male DKO mice, independent of the exposure, although there was a large variability in plaque size in the DKO cohorts (Figures 5 and 6). Of note, the basal levels of plaque in the control DKO mice were significantly higher than that of control mice (Figures 5 and 6 and Table S3).
Figure 5.
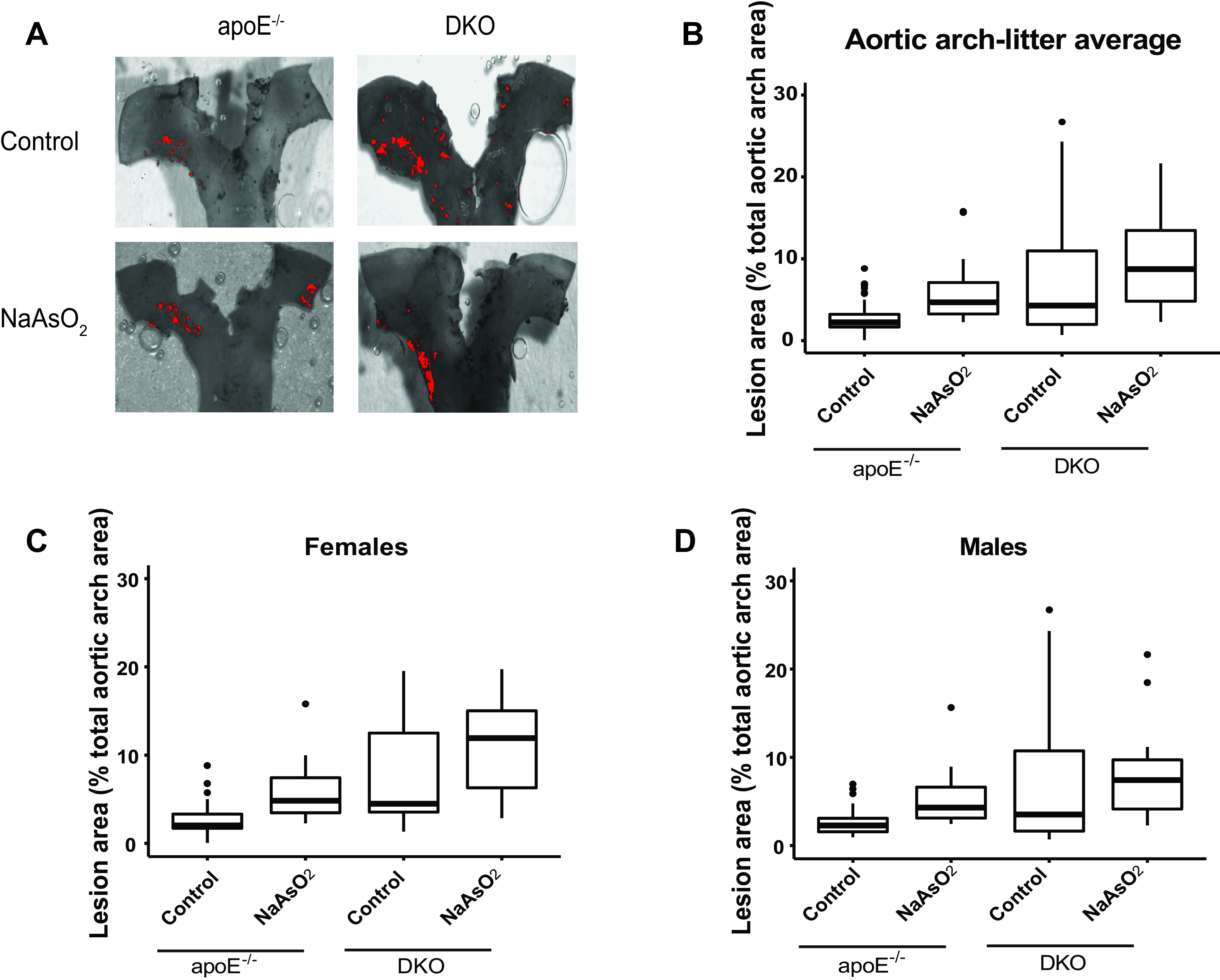
Differences in plaque formation in the aortic arch in adult DKO mice following early life arsenical exposure. or DKO mice were exposed to or maintained on tap water from conception to weaning. After weaning (4 wk), animals were kept on tap water for an additional 13 wk. The percentage of the lesion area of the aortic arch was evaluated via oil red O staining. (A) Representative images. Scale bar: . The box plots represent the distribution of unadjusted total lesion area as a percentage of total arch area. (B) The combined data set is presented, and sex-specific data sets for (C) females and (D) are presented. The midline, box limits, whiskers, and dots denote the median, interquartile range, minimum and maximum values, and outliers, respectively. The corresponding numeric data are presented in Table S6 and that adjusted by litter are presented in Tables 6 and 7. Note: DKO, double-knock out; , sodium arsenite.
Table 6.
Differences in the plaque size in the aortic arch and the sinus in the DKO mouse model in response to arsenic exposure in utero relative to their control counterpart and associated 95% confidence intervals (CIs), adjusted for sex.
| Exposure group | Lesion area | |||||
|---|---|---|---|---|---|---|
| Percentage of aortic archa | Percentage of aortic sinusb | Aortic sinus ()c | ||||
| Relative difference (95% CI) | -Value | Relative difference (95% CI) | -Value | Relative difference (95% CI) | -Value | |
| 1.3 (, 7.9) | 0.67 | (, 3.6) | 0.50 | (, 0.07) | 0.99 | |
Note: Models were fitted using simple mixed regression models to control for the intra-litter correlation, and sex was included as a covariate. All exposure groups were compared with the unexposed (control) group, for each outcome separately. The corresponding data distribution is depicted in Figures 5B, 6B and 6E. DKO, double-knock out; , sodium arsenite; Number of DKO litters : 4, : 4.
Percentage of oil red O-stained area over the total arch area.
Percentage of oil red O-stained lesion area over the total sinus area.
Absolute lesion area as measured by oil red O staining.
Figure 6.
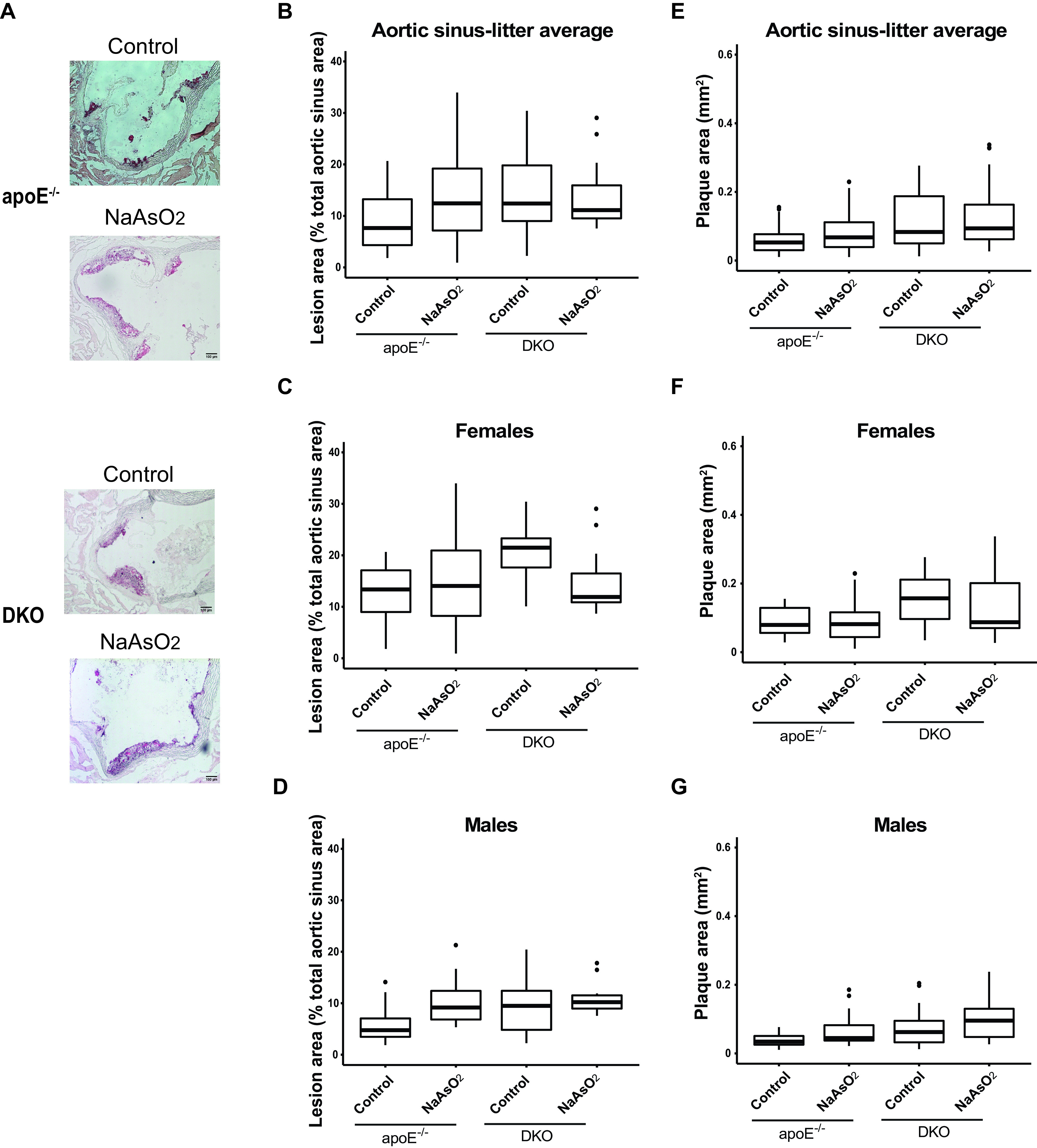
Differences in plaque formation in the aortic sinus in adult DKO mice following early life arsenical exposure. or DKO mice were exposed as described for Figure 1 and the plaque area in the aortic sinus was assessed by oil red O staining. The percentage of the lesion area of the aortic sinus was evaluated via oil red O staining. (A) Representative images. The box plots represent the distribution of unadjusted total lesion area as a percentage of the total sinus area, where midline, box limits, whiskers, and dots denote the median, interquartile range, minimum and maximum values, and outliers respectively, in the data set. (B) The combined data set is presented, and sex-specific data sets for (C) females and (D) are presented. In addition, the lesion area was also measured as millimeters squared, and the unadjusted data distribution for all the litters from (E) each exposed group as well as that separated by (F) female and (G) male offspring is shown. The corresponding numeric data are presented in Table S6 and that adjusted by litter are presented in Tables 6 and 7. Note: DKO, double-knock out; , cacodylic acid; , monomethylarsonous acid; , disodium methyl arsonate hexahydrate; , sodium arsenite.
Table 7.
Differences in the plaque size in the aortic arch and the sinus in the DKO mouse model in response to exposure in utero relative to their control counterpart and associated 95% confidence intervals (CIs), presented separately for females and males.
| Outcome | Females | Males | between males and femalesa | ||
|---|---|---|---|---|---|
| Relative differences (95% CI) | -Value | Relative differences (95% CI) | -Value | ||
| Lesion area (% of total arch area)b | 1.2 (, 9.1) | 0.73 | 1.8 (, 10.2) | 0.64 | 0.70 |
| Lesion area (% of total arch area)c | (, 3.4) | 0.24 | 1.2 (, 6.8) | 0.64 | 0.11 |
| Lesion area (in aortic sinus) ()d | 0.009 (, 0.14) | 0.87 | 0.024 (, 0.08) | 0.39 | 0.73 |
Note: To obtain sex-specific changes, separate models were fitted for females and males using simple mixed regression to control for the intra-litter correlation. All exposure groups were compared with the unexposed (control) group, for each outcome. Number of litters DKO : 4, : 4. DKO, double-knock out; , sodium arsenite.
Interactions between sex and exposure groups were tested using maximum likelihood and the resulting -values are presented here. The corresponding data distribution is depicted in Figures 5C,D, 6C,D, and 6F,G.
Percentage of oil red O-stained area over the total arch area.
Percentage of oil red O-stained lesion area over the total sinus area.
Absolute lesion area as measured by oil red O staining.
Discussion
Arsenic is an environmental toxicant to which millions of people are exposed worldwide (Naujokas et al. 2013). Previously, we and others have shown that postnatal exposure to both inorganic (Lemaire et al. 2014, 2011; Simeonova et al. 2003; Srivastava et al. 2009) and methylated arsenicals (Negro Silva et al. 2017) was pro-atherogenic in mice. Here, we provide evidence that combined pre- and perinatal exposure to moderate concentrations of these compounds also promotes atherosclerosis. These data complement other studies that have shown that early life exposure to arsenic correlated with disease later in life (Bailey and Fry 2014; Chen et al. 2019; Steinmaus et al. 2016). Importantly, these methylated arsenicals appear to be as pro-atherogenic as the parent compounds, indicating that the differences in methylation efficiency conferred by As3MT polymorphisms may not be relevant in this exposure scenario.
One criticism of previous murine transplacental models to show intergenerational effects was that high concentrations ( arsenic) were employed (Garry et al. 2015). The groundbreaking studies observing cancer outcomes following in utero exposure to arsenic in mice used 42,500 and arsenic (Waalkes et al. 2003). Arsenic exposure () during pregnancy increased the early onset of atherosclerosis in the mouse model (Srivastava et al. 2007; States et al. 2012). Our present study extends these findings to arsenic as inorganic, , and as methylated arsenicals. Thus, at least for atherosclerosis, moderate concentrations resulted in larger plaques and a similar disease phenotype as high concentrations in mice. An important consideration is that mice metabolize arsenic more quickly than humans (ATSDR 2007), thus making an extrapolation to exposed human populations difficult. Arguably, a arsenic exposure in mice approximates a exposure in humans using the human equivalent dose calculations described by the U.S. Food and Drug Administration (FDA 2005). Thus, the data presented here are relevant to low-level, more prevalent exposure groups and suggest that populations living in arsenic-endemic areas may have adverse generational health effects despite remediation of arsenic contamination from their waters.
Here, we show that a conception-to-early life exposure to arsenicals can significantly enhance atherosclerotic plaque sizes in the aortic arch and sinus in the mouse model. These effects are similar to what we observed in our postnatal exposure model (Negro Silva et al. 2017). Of note, an early life exposure to methylated arsenicals resulted in larger plaque sizes as compared with , whereas in postnatal exposures, had pro-atherogenic effects equal to or greater than methylated arsenicals (Negro Silva et al. 2017). We had observed in vitro (Padovani et al. 2010) and after a 13-wk-long postnatal exposure in vivo (Lemaire et al. 2011; Negro Silva et al. 2017) that arsenic impaired lipid handling in macrophages. Individual macrophages retained more lipids because of diminished cholesterol efflux (Lemaire et al. 2014). In the present study, we observed elevated plaque lipids without a concomitant difference in plaque macrophage contents, which is also consistent with our previous findings. SMCs and collagen are also components of the plaque and promote plaque stability (Libby et al. 2011). We previously reported that postnatal arsenical exposure decreased SMCs and collagen (Lemaire et al. 2011; Negro Silva et al. 2017), characteristic of plaques more prone to rupture (Gomez and Owens 2012; Libby et al. 2011). In the present study, we observed lower numbers of SMCs and less collagen content in treated vs. control mice in males, but not females. It is difficult to say how this would translate to humans given that the sex differences observed in mice are opposite of what is observed in humans (i.e., in mice, females have larger plaques, whereas in humans, males do). There is some literature indicating that sex is a determinant in the response to arsenic in terms of metabolic diseases. For instance, a population-based study of the residents in Central Italy exposed to low-to-medium doses of arsenic concluded that males had a higher mortality hazard ratio of arsenic-associated myocardial infarctions than females, whereas the inverse was true for diabetes (D’Ippoliti et al. 2015). This sexual dimorphism can be attributed to direct as well as indirect factors. For instance, arsenic induced sex-specific changes of the gut microbiome in mice (Chi et al. 2016) and humans (Hoen et al. 2018), which had been correlated with metabolic health (Peng et al. 2020). Moreover, epidemiologic data has suggested that women have higher arsenic methylation rates than men as measured by their urinary arsenical profile, although this effect is lost in older women (Lindberg et al. 2008). This not only suggests a role of arsenic methylation efficiency in sexually dimorphic effects of arsenic, but also implicates sex hormones as a potential factor. However, these findings are based on arsenic species measured in urine samples only. More specifically, both the gut microbiome (Chi et al. 2019) and As3MT activity (Negro Silva et al. 2017) have been linked to arsenic-induced dyslipidemia and atherosclerosis in mouse models, providing potential explanations for our observations. Furthermore, sex-specific differences were observed following in utero arsenic exposure in humans. For instance, the placentae in female fetuses had a higher expression of the arsenic transporter AQP9 (Winterbottom et al. 2017). In utero arsenic exposure led to decreased birth weight, but only in males (Xu et al. 2011). Besides, arsenic-associated changes in cognitive functions differed between sexes (Hamadani et al. 2011). Together, these data support our findings that in utero and early life arsenic exposure is linked to sex-specific differences in CVD later in life.
Our data show that mice lacking As3MT have a higher basal level of atherosclerotic plaque in the absence of drinking-water–derived arsenic exposure. This may be explained by a higher arsenic accumulation from laboratory diet in the absence of As3MT-dependent metabolism compared with As3MT wild-type mice although we did use a purified diet to minimize the amount of arsenic contributed through the food. Therefore, the impact of arsenic biotransformation on atherosclerosis is difficult to decipher in this study, as we observed slightly larger, but statistically insignificant, plaques in the aortic arch and no change in plaque size in the aortic sinus (Figures 5 and 6). Arsenic biotransformation has recently received attention in regard to its potential in arsenic pathogenesis (Chen et al. 2013a; Kuo et al. 2017). We were the first group to show that As3MT is required for arsenic-enhanced atherosclerosis following a postnatal exposure in mice (Negro Silva et al. 2017). Herein, we used our DKO model to investigate the effects of As3MT deletion on the outcomes that we had observed after prenatal exposure to inorganic arsenic in mice (Figure 1). One hypothesis is that dimethylarsenous acid () or other methylated intermediates may drive the effects. , , and were unable to induce atherosclerosis in our postnatal exposure model in DKO mice (Negro Silva et al. 2017), but it is possible that in utero exposure could be pro-atherogenic. However, these exposures were not tested in the present study. Several epidemiologic studies have correlated arsenic metabolism patterns with adverse health outcomes in at-risk populations. Recent reports from the Strong Heart Family Study showed a correlation between arsenic metabolite ratios (high DMA% and low MMA%) and the incidence of metabolic syndrome, as well as with greater waist circumference, a risk factor for CVDs, independent of arsenic exposure (Spratlen et al. 2018). Incomplete arsenic methylation (higher urinary MMA/DMA ratios) in humans was also associated with atherosclerosis in adults (Chen et al. 2013a) and with the risk of CVD (Chen et al. 2013b; Kuo et al. 2017). Interestingly, genetic variants in the 10q24.32 region near As3MT were important determinants of arsenic metabolism patterns and disease outcomes (Balakrishnan et al. 2017). For instance, distinct genetic variants in this locus were associated with arsenic metabolism efficiency as determined by urinary arsenic metabolites (Chernoff et al. 2020), as well as with the risk of skin lesions among unrelated individuals in Bangladesh (Pierce et al. 2012). Hence, the urinary arsenic profile, a surrogate for arsenic metabolism efficiency, can be an important biomarker to predict disease susceptibility associated with the As3MT locus and can have translational implications in the prevention of arsenic-associated toxicities.
In summary, our results show that methylated arsenicals were pro-atherogenic following early life exposure and that the biotransformation process was an important mechanism driving this phenotype. In addition, our data suggest that epidemiologic studies should evaluate single nucleotide polymorphisms that affect arsenic methylation efficiency in relation to in utero exposure when evaluating arsenic-exposed populations. Finally, our data further support the need to understand the mechanism by which arsenic enhances atherosclerosis in order to define appropriate interventions because, clearly, removal of arsenic is not enough to reverse damage caused by in utero exposure.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research (MOP-115000; K.K.M.) and the Heart and Stroke Foundation (G-17-00018365; K.K.M.). L.F.N.S. and K.K.M. were supported by Gordon Phillips Fellowship/McGill Faculty of Medicine and TD bank/LDI fellowship.
References
- ATSDR (Agency for Toxic Substances and Disease Registry). 2007. Toxicological Profile for Arsenic. Atlanta, GA: U.S. Department of Health and Human Services, Public Health Service. 2007. https://stacks.cdc.gov/view/cdc/11481 [accessed 16 April 2021]. [Google Scholar]
- Bailey K, Fry RC. 2014. Long-term health consequences of prenatal arsenic exposure: links to the genome and the epigenome. Rev Environ Health 29(1–2):9–12, PMID: 24552957, 10.1515/reveh-2014-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan P, Vaidya D, Franceschini N, Voruganti VS, Gribble MO, Haack K, et al. . 2017. Association of cardiometabolic genes with arsenic metabolism biomarkers in American Indian communities: the Strong Heart Family Study (SHFS). Environ Health Perspect 125(1):15–22, PMID: 27352405, 10.1289/EHP251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D, Maechler M, Bolker B, Walker S. 2014a. lme4: linear mixed-effects models using Eigen and S4. Version 1.1-7.
- Bieler G, Williams R. 1995. Cluster sampling techniques in quantal response teratology and developmental toxicity studies. Biometrics 51(2):764–776, PMID: 7662858, 10.2307/2532963. [DOI] [PubMed] [Google Scholar]
- Breslow NE, Clayton DG. 1993. Approximate inference in generalized linear mixed models. Journal of American Statistical Association 88(421):9–25, 10.1080/01621459.1993.10594284. [DOI] [Google Scholar]
- BGS (British Geological Survey). 2000. Executive summary of the main report of phase I, groundwater studies of As contamination in Bangladesh by British Geological Survey and Mott MacDonald (UK) for the Government of Bangladesh, Ministry of Local Government, Rural Development and Cooperatives DPHE and DFID (UK). http://bicn.com/acic/resources/infobank/bgs-mmi/risumm.htm [accessed 17 March 2021].
- Caligiuri G, Nicoletti A, Zhou X, Tornberg I, Hansson GK. 1999. Effects of sex and age on atherosclerosis and autoimmunity in apoE-deficient mice. Atherosclerosis 145(2):301–308, PMID: 10488957, 10.1016/S0021-9150(99)00081-7. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wu F, Liu X, Parvez F, LoIacono NJ, Gibson EA, et al. . 2019. Early life and adolescent arsenic exposure from drinking water and blood pressure in adolescence. Environ Res 178:108681, PMID: 31520830, 10.1016/j.envres.2019.108681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wu F, Liu M, Parvez F, Slavkovich V, Eunus M, et al. . 2013a. A prospective study of arsenic exposure, arsenic methylation capacity, and risk of cardiovascular disease in Bangladesh. Environ Health Perspect 121(7):832–838, PMID: 23665672, 10.1289/ehp.1205797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wu F, Parvez F, Ahmed A, Eunus M, McClintock TR, et al. . 2013b. Arsenic exposure from drinking water and QT-interval prolongation: results from the health effects of arsenic longitudinal study. Environ Health Perspect 121(4):427–432, PMID: 23384555, 10.1289/ehp.1205197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernoff M, Tong L, Demanelis K, Vander Griend D, Ahsan H, Pierce BL. 2020. Genetic determinants of reduced arsenic metabolism efficiency in the 10q24.32 region are associated with reduced as3mt expression in multiple human tissue types. Toxicol Sci 176(2):382–395, PMID: 32433756, 10.1093/toxsci/kfaa075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi L, Bian X, Gao B, Ru H, Tu P, Lu K. 2016. Sex-Specific effects of arsenic exposure on the trajectory and function of the gut microbiome. Chem Res Toxicol 29(6):949–951, PMID: 27268458, 10.1021/acs.chemrestox.6b00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi L, Lai Y, Tu P, Liu CW, Xue J, Ru H, et al. . 2019. Lipid and cholesterol homeostasis after arsenic exposure and antibiotic treatment in mice: potential role of the microbiota. Environ Health Perspect 127(9):097002, PMID: 31532247, 10.1289/EHP4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concha G, Vogler G, Lezcano D, Nermell B, Vahter M. 1998. Exposure to inorganic arsenic metabolites during early human development. Toxicol Sci 44(2):185–190, PMID: 9742656, 10.1093/toxsci/44.2.185. [DOI] [PubMed] [Google Scholar]
- Cullen WR, McBride BC, Manji H, Pickett AW, Reglinski J. 1989. The metabolism of methylarsine oxide and sulfide. Appl Organomet Chem 3(1):71–78, 10.1002/aoc.590030107. [DOI] [Google Scholar]
- D’Ippoliti D, Santelli E, De Sario M, Scortichini M, Davoli M, et al. . 2015. Arsenic in drinking water and mortality for cancer and chronic diseases in central Italy, 1990–2010. PLoS ONE 10(9):e0138182, PMID: 26383851, 10.1371/journal.pone.0138182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fängström B, Moore S, Nermell B, Kuenstl L, Goessler W, Grandér M, et al. . 2008. Breast-feeding protects against arsenic exposure in Bangladeshi infants. Environ Health Perspect 116(7):963–969, PMID: 18629322, 10.1289/ehp.11094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzan SF, Karagas MR, Chen Y. 2013. In utero and early life arsenic exposure in relation to long-term health and disease. Toxicol Appl Pharmacol 272(2):384–390, PMID: 23859881, 10.1016/j.taap.2013.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA (U.S. Food and Drug Administration). 2005. Guidance for Industry: Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/estimating-maximum-safe-starting-dose-initial-clinical-trials-therapeutics-adult-healthy-volunteers [accessed 16 April 2021].
- Garry MR, Santamaria AB, Williams AL, DeSesso JM. 2015. In utero arsenic exposure in mice and early life susceptibility to cancer. Regul Toxicol Pharmacol 73(1):378–390, PMID: 26239692, 10.1016/j.yrtph.2015.07.023. [DOI] [PubMed] [Google Scholar]
- Gomez D, Owens GK. 2012. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res 95(2):156–164, PMID: 22406749, 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M, Gamble M, Slavkovich V, Liu X, Levy D, Cheng Z, et al. . 2007. Determinants of arsenic metabolism: blood arsenic metabolites, plasma folate, cobalamin, and homocysteine concentrations in maternal–newborn pairs. Environ Health Perspect 115(10):1503–1509, PMID: 17938743, 10.1289/ehp.9906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamadani J, Tofail F, Nermell B, Gardner R, Shiraji S, Bottai M, et al. . 2011. Critical windows of exposure for arsenic-associated impairment of cognitive function in pre-school girls and boys: a population-based cohort study. Int J Epidemiol 40(6):1593–1604, PMID: 22158669, 10.1093/ije/dyr176. [DOI] [PubMed] [Google Scholar]
- Haseman JK, Kupper LL. 1979. Analysis of dichotomous response data from certain toxicological experiments. Biometrics 35(1):281–293, PMID: 574026, 10.2307/2529950. [DOI] [PubMed] [Google Scholar]
- Hoen AG, Madan JC, Li Z, Coker M, Lundgren SN, Morrison HG, et al. . 2018. Sex-specific associations of infants’ gut microbiome with arsenic exposure in a US population. Sci Rep 8(1):12627, PMID: 30135504, 10.1038/s41598-018-30581-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe CG, Niedzwiecki MM, Hall MN, Liu X, Ilievski V, Slavkovich V, et al. . 2014. Folate and cobalamin modify associations between S-adenosylmethionine and methylated arsenic metabolites in arsenic-exposed Bangladeshi adults. J Nutr 144(5):690–697, PMID: 24598884, 10.3945/jn.113.188789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YC, Lien LM, Chung WT, Hsieh FI, Hsieh PF, Wu MM, et al. . 2011. Significantly increased risk of carotid atherosclerosis with arsenic exposure and polymorphisms in arsenic metabolism genes. Environ Res 111(6):804–810, PMID: 21605854, 10.1016/j.envres.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Islam MR, Attia J, Alauddin M, McEvoy M, McElduff P, Slater C, et al. . 2014. Availability of arsenic in human milk in women and its correlation with arsenic in urine of breastfed children living in arsenic contaminated areas in Bangladesh. Environ Health 13(1):101, PMID: 25471535, 10.1186/1476-069X-13-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James KA, Byers T, Hokanson JE, Meliker JR, Zerbe GO, Marshall JA. 2015. Association between lifetime exposure to inorganic arsenic in drinking water and coronary heart disease in Colorado residents. Environ Health Perspect 123(2):128–134, PMID: 25350952, 10.1289/ehp.1307839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Liu M, Parvez F, Wang B, Wu F, Eunus M, et al. . 2015. Association between arsenic exposure from drinking water and longitudinal change in blood pressure among HEALS cohort participants. Environ Health Perspect 123(8):806–812, PMID: 25816368, 10.1289/ehp.1409004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kile ML, Cardenas A, Rodrigues E, Mazumdar M, Dobson C, Golam M, et al. . 2016. Estimating effects of arsenic exposure during pregnancy on perinatal outcomes in a Bangladeshi cohort. Epidemiology 27(2):173–181, PMID: 26583609, 10.1097/EDE.0000000000000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo CC, Moon KA, Wang SL, Silbergeld E, Navas-Acien A. 2017. The association of arsenic metabolism with cancer, cardiovascular disease, and diabetes: a systematic review of the epidemiological evidence. Environ Health Perspect 125(8):087001, PMID: 28796632, 10.1289/EHP577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazic SE, Essioux L. 2013. Improving basic and translational science by accounting for litter-to-litter variation in animal models. BMC Neurosci 14(1):37, PMID: 23522086, 10.1186/1471-2202-14-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire M, Lemarié CA, Flores Molina M, Guilbert C, Lehoux S, Mann KK. 2014. Genetic deletion of LXRα prevents arsenic-enhanced atherosclerosis, but not arsenic-altered plaque composition. Toxicol Sci 142(2):477–488, PMID: 25273567, 10.1093/toxsci/kfu197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire M, Lemarié CA, Flores Molina M, Schiffrin EL, Lehoux S, Mann KK. 2011. Exposure to moderate arsenic concentrations increases atherosclerosis in ApoE−/− mouse model. Toxicol Sci 122(1):211–221, PMID: 21512104, 10.1093/toxsci/kfr097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire M, Negro Silva LF, Lemarié CA, Bolt AM, Flores Molina M, Krohn RM, et al. . 2015. Arsenic exposure increases monocyte adhesion to the vascular endothelium, a pro-atherogenic mechanism. PLoS One 10(9):e0136592, PMID: 26332580, 10.1371/journal.pone.0136592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Hansson GK. 2011. Progress and challenges in translating the biology of atherosclerosis. Nature 473(7347):317–325, PMID: 21593864, 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- Lindberg AL, Ekström EC, Nermell B, Rahman M, Lönnerdal B, Persson LA, et al. . 2008. Gender and age differences in the metabolism of inorganic arsenic in a highly exposed population in Bangladesh. Environ Res 106(1):110–120, PMID: 17900557, 10.1016/j.envres.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Makhani K, Chiavatti C, Plourde D, Negro Silva LF, Lemaire M, Lemarié CA, et al. . 2018. Using the apolipoprotein E knock-out mouse model to define atherosclerotic plaque changes induced by low dose arsenic. Toxicol Sci 166(1):213–218, PMID: 30376133, 10.1093/toxsci/kfy201. [DOI] [PubMed] [Google Scholar]
- Mandal BK, Suzuki KT. 2002. Arsenic round the world: a review. Talanta 58(1):201–235, PMID: 18968746, 10.1016/S0039-9140(02)00268-0. [DOI] [PubMed] [Google Scholar]
- Mateen FJ, Grau-Perez M, Pollak JS, Moon KA, Howard BV, Umans JG, et al. . 2017. Chronic arsenic exposure and risk of carotid artery disease: the Strong Heart Study. Environ Res 157:127–134, PMID: 28554006, 10.1016/j.envres.2017.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean RA, Sanders WL, Stroup WW. 1991. A unified approach to mixed linear models. Am Stat 45(1):54–64, 10.1080/00031305.1991.10475767. [DOI] [Google Scholar]
- Meza M, Gandolfi AJ, Klimecki WT. 2007. Developmental and genetic modulation of arsenic biotransformation: a gene by environment interaction? Toxicol Appl Pharmacol 222(3):381–387, PMID: 17306849, 10.1016/j.taap.2006.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon KA, Oberoi S, Barchowsky A, Chen Y, Guallar E, Nachman KE, et al. . 2017. A dose-response meta-analysis of chronic arsenic exposure and incident cardiovascular disease. Int J Epidemiol 46(6):1924–1939, PMID: 29040626, 10.1093/ije/dyx202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naujokas MF, Anderson B, Ahsan H, Aposhian HV, Graziano JH, Thompson C, et al. . 2013. The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Environ Health Perspect 121(3):295–302, PMID: 23458756, 10.1289/ehp.1205875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navas-Acien A, Spratlen MJ, Abuawad A, LoIacono NJ, Bozack AK, Gamble MV. 2019. Early-life arsenic exposure, nutritional status, and adult diabetes risk. Curr Diab Rep 19(12):147, PMID: 31758285, 10.1007/s11892-019-1272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negro Silva LF, Lemaire M, Lemarié CA, Plourde D, Bolt AM, Chiavatti C, et al. . 2017. Effects of inorganic arsenic, methylated arsenicals, and arsenobetaine on atherosclerosis in the mouse model and the role of As3MT-mediated methylation. Environ Health Perspect 125(7):077001, PMID: 28728140, 10.1289/EHP806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilsen MG, Lombard PJ, Schalk LK. 2010. Assessment of Arsenic Concentrations in Domestic Well Water, by Town, in Maine, 2005–09: U.S. Geological Survey Scientific Investigations Report, 2010-5199. https://pubs.usgs.gov/sir/2010/5199/pdf/sir2010-5199_nielsen_arsenic_report_508.pdf [accessed 17 March 2021].
- Nordstrom DK. 2002. Public health. Worldwide occurrences of arsenic in ground water. Science 296(5576):2143–2145, PMID: 12077387, 10.1126/science.1072375. [DOI] [PubMed] [Google Scholar]
- Osorio-Yáñez C, Ayllon-Vergara JC, Aguilar-Madrid G, Arreola-Mendoza L, Hernández-Castellanos E, Barrera-Hernández A, et al. . 2013. Carotid intima-media thickness and plasma asymmetric dimethylarginine in Mexican children exposed to inorganic arsenic. Environ Health Perspect 121(9):1090–1096, PMID: 23757599, 10.1289/ehp.1205994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padovani AM, Flores Molina M, Mann KK. 2010. Inhibition of liver X receptor/retinoid X receptor-mediated transcription contributes to the proatherogenic effects of arsenic in macrophages in vitro. Arterioscler Thromb Vasc Biol 30(6):1228–1236, PMID: 20339114, 10.1161/ATVBAHA.110.205500. [DOI] [PubMed] [Google Scholar]
- Peng C, Xu X, Li Y, Li X, Yang X, Chen H, et al. . 2020. Sex-specific association between the gut microbiome and high-fat diet-induced metabolic disorders in mice. Biol Sex Differ 11(1):5, PMID: 31959230, 10.1186/s13293-020-0281-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Carrera A, Fernández Cirelli A. 2010. Arsenic and water quality challenges in South America. In: Water and Sustainability in Arid Regions: an Interdisciplinary Exploration of Human and Environmental Interactions. Schneier-Madanes G, Courel MF, eds. Dordrecht, Netherlands: Springer. [Google Scholar]
- Petrick JS, Jagadish B, Mash EA, Aposhian HV. 2001. Monomethylarsonous acid (MMAIII) and arsenite: LD50 in hamsters and in vitro inhibition of pyruvate dehydrogenase. Chem Res Toxicol 14(6):651–656, PMID: 11409934, 10.1021/tx000264z. [DOI] [PubMed] [Google Scholar]
- Pierce BL, Kibriya MG, Tong L, Jasmine F, Argos M, Roy S, et al. . 2012. Genome-wide association study identifies chromosome 10q24.32 variants associated with arsenic metabolism and toxicity phenotypes in Bangladesh. PLoS Genet 8(2):e1002522, PMID: 22383894, 10.1371/journal.pgen.1002522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilsner JR, Liu X, Ahsan H, Ilievski V, Slavkovich V, Levy D, et al. . 2009. Folate deficiency, hyperhomocysteinemia, low urinary creatinine, and hypomethylation of leukocyte DNA are risk factors for arsenic-induced skin lesions. Environ Health Perspect 117(2):254–260, PMID: 19270796, 10.1289/ehp.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro JC, Bates DM. 2000. Mixed-Effects Models in S and S-PLUS. New York, NY: Springer. [Google Scholar]
- Rahman M, Sohel N, Yunus M, Chowdhury ME, Hore SK, Zaman K, et al. . 2013. Increased childhood mortality and arsenic in drinking water in Matlab, Bangladesh: a population-based cohort study. PLoS One 8(1):e55014, PMID: 23383038, 10.1371/journal.pone.0055014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh T, Lynch CF, Weyer P, Wang K, Kelly KM, Ludewig G. 2017. Low-level arsenic exposure from drinking water is associated with prostate cancer in Iowa. Environ Res 159:338–343, PMID: 28841521, 10.1016/j.envres.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseboom TJ, van der Meulen JH, Osmond C, Barker DJ, Ravelli AC, Schroeder-Tanka JM, et al. . 2000. Coronary heart disease after prenatal exposure to the Dutch famine, 1944–45. Heart 84(6):595–598, PMID: 11083734, 10.1136/heart.84.6.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schläwicke Engström K, Broberg K, Concha G, Nermell B, Warholm M, Vahter M. 2007. Genetic polymorphisms influencing arsenic metabolism: evidence from Argentina. Environ Health Perspect 115(4):599–605, PMID: 17450230, 10.1289/ehp.9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH image to ImageJ: 25 years of image analysis. Nat Methods 9(7):671–675, PMID: 22930834, 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeonova PP, Hulderman T, Harki D, Luster MI. 2003. Arsenic exposure accelerates atherogenesis in apolipoprotein E-/- mice. Environ Health Perspect 111(14):1744–1748, PMID: 14594625, 10.1289/ehp.6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AH, Marshall G, Liaw J, Yuan Y, Ferreccio C, Steinmaus C. 2012. Mortality in young adults following in utero and childhood exposure to arsenic in drinking water. Environ Health Perspect 120(11):1527–1531, PMID: 22949133, 10.1289/ehp.1104867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AH, Marshall G, Roh T, Ferreccio C, Liaw J, Steinmaus C. 2018. Lung, bladder, and kidney cancer mortality 40 years after arsenic exposure reduction. J Natl Cancer Inst 110(3):241–249, PMID: 29069505, 10.1093/jnci/djx201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DD, Tan X, Tawfik O, Milne G, Stechschulte DJ, Dileepan KN. 2010. Increased aortic atherosclerotic plaque development in female apolipoprotein E-null mice is associated with elevated thromboxane A2 and decreased prostacyclin production. J Physiol Pharmacol 61(3):309–316, PMID: 20610861. [PMC free article] [PubMed] [Google Scholar]
- Spratlen MJ, Grau-Perez M, Best LG, Yracheta J, Lazo M, Vaidya D, et al. . 2018. The association of arsenic exposure and arsenic metabolism with the metabolic syndrome and its individual components: prospective evidence from the Strong Heart Family Study. Am J Epidemiol 187(8):1598–1612, PMID: 29554222, 10.1093/aje/kwy048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, D’Souza SE, Sen U, States JC. 2007. In utero arsenic exposure induces early onset of atherosclerosis in ApoE-/- mice. Reprod Toxicol 23(3):449–456, PMID: 17317095, 10.1016/j.reprotox.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Vladykovskaya EN, Haberzettl P, Sithu SD, D’Souza SE, States JC. 2009. Arsenic exacerbates atherosclerotic lesion formation and inflammation in ApoE-/- mice. Toxicol Appl Pharmacol 241(1):90–100, PMID: 19682479, 10.1016/j.taap.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- States JC, Singh AV, Knudsen TB, Rouchka EC, Ngalame NO, Arteel GE, et al. . 2012. Prenatal arsenic exposure alters gene expression in the adult liver to a proinflammatory state contributing to accelerated atherosclerosis. PLoS One 7(6):e38713, PMID: 22719926, 10.1371/journal.pone.0038713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmaus C, Ferreccio C, Acevedo J, Balmes JR, Liaw J, Troncoso P, et al. . 2016. High risks of lung disease associated with early-life and moderate lifetime arsenic exposure in northern Chile. Toxicol Appl Pharmacol 313:10–15, PMID: 27725189, 10.1016/j.taap.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stýblo M, Drobná Z, Jaspers I, Lin S, Thomas DJ. 2002. The role of biomethylation in toxicity and carcinogenicity of arsenic: a research update. Environ Health Perspect 110(suppl 5):767–771, PMID: 12426129, 10.1289/ehp.110-1241242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DJ. 2007. Molecular processes in cellular arsenic metabolism. Toxicol Appl Pharmacol 222(3):365–373, PMID: 17397889, 10.1016/j.taap.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Vahter M, Berglund M, Åkesson A, Lidén C. 2002. Metals and women’s health. Environ Res 88(3):145–155, PMID: 12051792, 10.1006/enrs.2002.4338. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Ward JM, Liu J, Diwan BA. 2003. Transplacental carcinogenicity of inorganic arsenic in the drinking water: induction of hepatic, ovarian, pulmonary, and adrenal tumors in mice. Toxicol Appl Pharmacol 186(1):7–17, PMID: 12583988, 10.1016/S0041-008X(02)00022-4. [DOI] [PubMed] [Google Scholar]
- Wang CH, Chen CL, Hsiao CK, Chiang FT, Hsu LI, Chiou HY, et al. . 2010. Arsenic-induced QT dispersion is associated with atherosclerotic diseases and predicts long-term cardiovascular mortality in subjects with previous exposure to arsenic: a 17-year follow-up study. Cardiovasc Toxicol 10(1):17–26, PMID: 19957052, 10.1007/s12012-009-9059-x. [DOI] [PubMed] [Google Scholar]
- Wang H, Li J, Zhang X, Zhu P, Hao JH, Tao FB, et al. . 2018. Maternal serum arsenic level during pregnancy is positively associated with adverse pregnant outcomes in a Chinese population. Toxicol Appl Pharmacol 356:114–119, PMID: 30075163, 10.1016/j.taap.2018.07.030. [DOI] [PubMed] [Google Scholar]
- WHO (World Health Organization). 2008. Chapter 12. Chemical fact sheets. In: Guidelines for Drinking-Water Quality [electronic resource]. 3rd ed. Geneva, Switzerland: World Health Organization, 306–308b. https://www.who.int/water_sanitation_health/dwq/GDW12rev1and2.pdf?ua=1 [accessed 16 April 2021]. [Google Scholar]
- Winterbottom EF, Koestler DC, Fei DL, Wika E, Capobianco AJ, Marsit CJ, et al. . 2017. The aquaglyceroporin AQP9 contributes to the sex-specific effects of in utero arsenic exposure on placental gene expression. Environ Health 16(1):59, PMID: 28615018, 10.1186/s12940-017-0267-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Yokoyama K, Tian Y, Piao FY, Kitamura F, Kida H, et al. . 2011. Decrease in birth weight and gestational age by arsenic among the newborn in Shanghai, China. Nihon Koshu Eisei Zasshi 58(2):89–95, PMID: 21473424. [PubMed] [Google Scholar]
- Young JL, Cai L, States JC. 2018. Impact of prenatal arsenic exposure on chronic adult diseases. Syst Biol Reprod Med 64(6):469–483, PMID: 29873257, 10.1080/19396368.2018.1480076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Marshall G, Ferreccio C, Steinmaus C, Selvin S, Liaw J, et al. . 2007. Acute myocardial infarction mortality in comparison with lung and bladder cancer mortality in arsenic-exposed region II of Chile from 1950 to 2000. Am J Epidemiol 166(12):1381–1391, PMID: 17875584, 10.1093/aje/kwm238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.