

Abstract
CD1d-restricted invariant natural killer T cells (iNKT cells) mediate strong anti-tumor immunity when stimulated by glycolipid agonists. However, attempts to develop effective iNKT cell agonists for clinical applications have been thwarted by potential problems with dose-limiting toxicity and by activation-induced iNKT cell anergy, which limits the efficacy of repeated administration. To overcome these issues, we developed a unique bispecific T cell engager (BiTE) based on covalent conjugates of soluble CD1d with photoreactive analogs of the glycolipid α-galactosylceramide. Here we characterize the in vivo activities of iNKT cell-specific BiTEs and assess their efficacy for cancer immunotherapy in mouse models using transplantable colorectal cancer or melanoma tumor lines engineered to express human Her2 as a tumor-associated antigen. Systemic administration of conjugated BiTEs stimulated multiple iNKT cell effector functions including cytokine release, secondary activation of NK cells, and induction of dendritic cell maturation and also initiated epitope spreading for tumor-specific CD8+ cytolytic T cell responses. The anti-tumor effects of iNKT cell activation with conjugated BiTEs were further enhanced by simultaneous checkpoint blockade with antibodies to CTLA-4, providing a potential approach for combination immunotherapy. Multiple injections of covalently stabilized iNKT cell-specific BiTEs activated iNKT cells without causing iNKT anergy and exhaustion, thus enabling repeated administration for effective and nontoxic cancer immunotherapy regimens.
Introduction
Invariant Natural Killer T (iNKT) cells are a conserved subset of specialized T cells that contribute to many innate and adaptive immune responses (1). Unlike conventional T cells, iNKT cells express T cell antigen receptors (TCRs) of limited diversity, and respond to specific foreign and self-glycolipid antigens presented by the MHC class I-like CD1d protein (2). The best characterized glycolipid antigens recognized by iNKT cells are synthetic forms of α-galactosylceramide (αGalCer), which activate their proliferation, cytokine production and cytotoxic functions (3). Activation of iNKT cells by αGalCer influences downstream immune responses through rapid cytokine release and a range of cell contact dependent signals. Detailed studies in mouse models show that this generates multifaceted immune responses that include Natural Killer (NK) cell activation, dendritic cell maturation, enhanced conventional T cell priming, antibody production and reversal of immunosuppressive effects from regulatory cells (4–6).
In mouse models, many structural analogues of αGalCer have strong anti-tumor activities (7), suggesting that iNKT cell directed therapies could be useful for cancer immunotherapy in humans (8, 9). Several phase I clinical trials with αGalCer have been done in patients with advanced cancers (10–12), but progress has been hampered by potential toxicities, off-target effects and lack of clear efficacy. These problems are due in part to the uptake and presentation of glycolipids by a variety of different types of antigen presenting cells, leading to complicated and unpredictable outcomes (4, 13). Furthermore, the development of long-term unresponsiveness (anergy) and depletion of iNKT cells by multiple exposures to αGalCer following injection into mice makes repeated administration ineffective (14, 15). Approaches to activating iNKT cells that could circumvent these issues are needed to advance the development of effective cancer immunotherapies.
Previous studies showed that delivery of αGalCer in complex with soluble CD1d proteins targeted to the surface of tumor cells could more precisely focus iNKT cell responses and reduce adverse effects (16, 17). Such complexes, representing a type of bispecific T cell engager (BiTE) (18), showed improvements over free glycolipid in terms of sustained iNKT functionality and improved anti-tumor efficacy in these studies. However, the reversible glycolipid binding and rapid dissociation in vivo resulted in reduced functionality of iNKT cells, and other potential problems similar to those associated with free glycolipid (19). To overcome this limitation, we developed an approach for precise covalent conjugation of CD1d with αGalCer analogues containing a photoactivatable benzophenone group to create covalently stabilized iNKT cell specific activators (20). In the current study, we have extended our analysis of the immunological effects of iNKT cell activation by soluble tumor targeted, glycolipid conjugated iNKT cell specific BiTEs, and demonstrate their substantial anti-cancer effects in mouse models. Compared to free glycolipids or noncovalent complexes of CD1d with αGalCer, we found that covalently stabilized conjugates were significantly more effective at inducing iNKT activation, maturation of dendritic cells and secondary activation of NK cells and T cells, and did so without causing iNKT cell anergy following repeated administration. Target specificity was incorporated into conjugates by genetically fusing soluble CD1d with a single chain antibody domain with specificity for a tumor associated antigen, thus facilitating direct cytotoxic effects of iNKT cells and driving sustained immune responses within tumors to improve therapeutic outcomes.
Materials and Methods
Animal models
Female C57BL/6J (B6) mice were obtained at age 6–8 weeks from Jackson Laboratories (JAX) and maintained in specific pathogen-free conditions. CD1d knockout mice with deletions in the Cd1d1/Cd1d2 loci on the B6 background (strain B6.129S6-Del(3Cd1d2-Cd1d1)1Sbp/J) were obtained from JAX (RRID: IMSR_JAX:008881), and C57BL/6 mice with a deletion of the Traj18 gene (Jα18−/− mice) were from Dr. Mitchell Kronenberg (La Jolla Institute for Allergy and Immunology). Human CD1d knock-in mice (B6.hCD1d-KI) were from Dr. Weiming Yuan (USC School of Medicine) (21). GFP expressing transgenic mice (strain C57BL/6-Tg(UBC-GFP)30Scha/J) were from JAX (RRID: IMSR_JAX:004353). All transgenic mice were bred in our facility. Female mice 10–12 weeks of age at the time of start of the experiments were used. All experiments requiring mice were conducted in compliance with institutional guidelines and under a protocol reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at Albert Einstein College of Medicine.
Glycolipid reagents
The iNKT cell α-GalCer glycolipid agonists C26:0 (KRN7000), C11:BP, 14C-labeled C11:BP and azido-C11:BP were synthesized and subsequently solubilized at 100 μM concentration in aqueous buffer (PBS + 0.1% DMSO + 0.05% Triton X-100) as described previously (13, 20).
CD1d tetramers and recombinant CD1d-ScFv fusion proteins
Murine single chain CD1d-β2m linked to AviTag™ and hexahistidine tags (MW ~57 kDa) was expressed and purified from stably transfected CHO cells as previously described (13, 22). Mouse CD1d tetramers were either prepared as described by Im et. al. (23) or obtained from the NIH Tetramer Core Facility (http://tetramer.yerkes.emory.edu/). Recombinant mouse or human CD1d fused to single chain antibody variable fragments (ScFv) against either huHER2 or human CEA were produced from transiently transfected HEK-293 cells as previously described (16, 17, 20). Glycolipid loading and covalent conjugation was performed as described previously (20) and in the supplemental methods.
Monoclonal antibodies, FACS and ELISA
Monoclonal antibody L363 (13, 24–26), was purified from supernatants of hybridoma cultures and biotinylated with EZ-Link™ Sulfo-NHS Biotinylation Kit (ThermoFisher). Effect of UV exposure on CD1d-C11:BP conjugate stability was determined by plate based dissociation assay (20). For FACS analyses, cells were stained with LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit, for UV excitation (Thermo Fisher) or Zombie NIR™ Fixable Viability Kit (Biolegend). Fluorochrome conjugated mAb were obtained from commercial suppliers (Supplemental Table 1). Multiparameter FACS analyses incorporating up to 22 parameters were carried out using 5 laser LSRII (BD Biosciences) or 4 laser Aurora (Cytek) flow cytometers. All data analysis was done using FlowJo™ software (Becton Dickinson). Tumors were harvested aseptically for preparation of single cell suspensions. Methods for isolating tumor infiltrating lymphocytes are described in the supplemental materials. Tumor infiltrating iNKT cells from all saline, complex or conjugate treated mice were gated as live, B220-, TCRβ+, αGalCer-loaded CD1d tetramer+ cells. For t-SNE analysis, 1500 total iNKT cells from each group were concatenated and run through 1000 iterations with perplexity of 30 and learning rate of 315. For serum cytokine ELISAs, capture antibodies were coated on wells of 96 well polystyrene microtiter plates (Supplemental Table 2), and wells were blocked using 5% BSA. Serum samples diluted 1:5 in PBS were added to allow binding of the cytokine of interest to the primary capture antibodies. Biotinylated secondary antibodies were then added, followed by washing and addition of Streptavidin-HRP. ELISAs were developed using Turbo-TMB substrate (Thermo Scientific) and read using a Wallac Victor plate reader. Data were analyzed using Prism 6.0 software (GraphPad). Hamster anti-CTLA-4 (Invivomab UC10–4F10, BioXcell) and purified normal hamster IgG (BioXcell) were used for in vivo CTLA4 blockade and isotype control respectively.
Cell lines
The murine colon carcinoma MC38 (RRID:CVCL_B288) and B16.F10 melanoma (RRID:CVCL_0159) tumor cell lines were obtained from the American Type Culture Collection (ATCC), and stable transfectants expressing huHER2 were produced as previously described (16). MC38 cells and transfectants were cultured in RPMI-1640 supplemented with 15% FBS. For transfectants, 1.2 μg/ml of puromycin was included in the medium. B16.F10 cells and transfectants were cultured in complete DMEM supplemented with 10% FBS, 10 mM HEPES, essential amino acids, glutamine and β-mercaptoethanol. For transfectants, 1.2 mg/ml Geneticin was added. Human iNKT clone HDD3 previously established from a random donor blood sample (27) was maintained in complete RPMI-1640 medium containing 10% decomplemented human serum at 37°C in a humidified 5% CO2 incubator. The cultures were expanded every 3–4 weeks by stimulation with phytohemagglutinin (PHA-P, Difco, Detroit, MI) in the presence of recombinant human IL-2 (250 IU/ mL; Chiron), recombinant human IL-7 (10 ng/mL; Peprotech) and irradiated (5000 Rad) allogeneic PBMCs. Cells were expanded by splitting cultures and feeding with fresh medium containing 250 IU/ml IL-2 and 10 ng/ml IL-7 every 2–3 days. Cell lines were maintained as frozen stocks at −150ºC and used in experiments within 2 weeks of thawing. Cell lines were not routinely tested for mycoplasma.
In vivo iNKT, NK and DC activation assays
Mice were injected i.v. with 0.4 nmoles (50 μg) of complexes or conjugates or an equimolar amount of free C11:BP or C26:0 (0.4 nmoles) in 0.1 ml of vehicle (PBS + 0.1% DMSO + 0.01% Tween-20). To quantitate cytokine secretion, mice were bled from the retro-orbital plexus at 2, 5, 10 and 24 hours and serum samples were prepared for quantitation of cytokines by capture ELISA. Kinetics of NK, iNKT and DC activation was determined in mice following i.v. injections of 0.4 nmoles of conjugates or complexes, or 0.4 nmoles of free glycolipid (αGalCer C26:0). Spleens were harvested at specific time points and splenocyte suspensions were stained as described in supplemental methods. NK and iNKT cells were stained for intracellular IFNγ. iNKT cells were quantitated as percentage of all live B220-, TCR-β low and CD1d tetramer+ cells. Tetramer+ iNKT cells were co-stained for relevant activation or inhibitory markers (CD69, CD28, NK1.1, ICOS, CTLA-4, LAG3, PD-1), and CD11c+ DC were co-stained for activating or inhibitory ligands (CD80, CD86, PD-L1, PD-L2) and analyzed by flow cytometry. Assessment of iNKT cell anergy induction was done in mice primed by injection i.v. with 0.4 nmoles of complexes or conjugates (once, or three times at weekly intervals), or with saline or 0.4 nmoles of free glycolipid (αGalCer C26:0). Mice from all groups were restimulated with C26:0 on day 7 after the final priming dose, bled at 12 hours to quantitate serum IFNγ and sacrificed at 72 hours to determine iNKT numbers.
Tumor cell cytotoxicity assays
Direct cytotoxicity of tumor cells by iNKT cells was determined by standard 51Cr release assay. Target cells (MC38-huHER2) labeled with 100 μCi 51Cr as Na2CrO4 (Perkin Elmer) were co-cultured with human iNKT cell clone HDD3 at 1:1 E:T ratio for 12 hours at 370C, in the presence of anti-huHER2 complexes or conjugated BiTE (0.01– 10 μg/ml). Specific lysis was determined by measurement of gamma emmisions in the supernatant. For imaging of iNKT cell mediated tumor killing in vitro, MC38-huHER2 cells were seeded in 96 well Nunclon™ Sphera™ plates (ThermoFisher) at 3000 cells/well and grown for 5 days to allow formation of 3-dimensional tumor cell spheroids. Anti-huHER2 conjugated BiTEs or complexes containing either mouse or human CD1d were added to wells for a concentration of 10 μg/ml, and control wells received saline only. After incubating for 2 hours at 37ºC, unbound ligands were removed by centrifugation of plates and gentle washing with medium, followed by addition of 1000 purified mouse iNKT cells or 1000 human iNKT clone HDD3 cells in pre-warmed medium. Plates were then incubated for 12 hours before staining for apoptosis and necrosis using Apoptosos/Necrosis Assay Kit (Abcam) following the instructions provided by the manufacturer. Confocal images of individual spheroids were acquired as Z-stacks with a Leica SP8 point scanning confocal microscope using a 10x air objective.
Mouse tumor immunotherapy models
Tumor cells harvested at mid-log phase were injected i.v. through the tail vein for the B16.F10-huHER2 melanoma model or grafted by subcutaneous injection into the thigh for the MC38-huHER2 carcinoma model. Immunotherapy agents were administered intravenously. Mice engrafted with B16.F10-huHER2 cells were sacrificed 16 days post injection Lungs were removed and digital photographs were obtained for calculation of the area of lung surface covered with melanized tumor using Image J software (RRID: SCR_003070) (https://imagej.nih.gov/ij/) (28). For animals grafted with subcutaneous MC38 cells, tumor volumes were measured on a daily basis using a digital caliper to obtain length and width. Tumor volume was calculated using the formula V = (L x W x D) x π/6, as recommended in previous studies (29, 30). After attaining a tumor volume of approximately 1 cm3, mice were monitored for up to two days, after which they were sacrificed if the tumor continued to grow as required by the approved animal research protocol. Mice were observed for up to 80 days, and animals lacking palpable tumors at the end of this period were considered complete responses.
To determine tumor antigen specific T cell responses in tumor bearing mice, IFNγ ELISPOT was performed on total T cells and CD8 depleted T cells purified from spleens. Tumor sections fixed in formalin were analyzed by histochemistry. To study distribution and kinetics of clearance of Conjugated BiTE, 14C linked conjugates were injected intravenously in tumor bearing mice. Mice were sacrificed at several time points to harvest organs. 14C radioactivity was measured in CPM using liquid scintillation counter. Azide-linked conjugates were injected in mice to study tissue localization of conjugates. Frozen sections of harvested spleens, livers, kidneys and tumor were stained with DBCO-AF647 (31) and imaged using a panoramic slide scanner (3D Histek ltd.). For detailed methods see supplemental methods.
Statistical analysis
Results are shown as means ± 1 SE. Statistical significance between the groups was determined with 1-way or 2–way ANOVA test as indicated, and Sidak post-test was applied for data involving multiple comparisons. All P values were calculated using GraphPad Prism 6.0 software (RRID: SCR_002798). Values of P < 0.05 were considered significant.
Results
Activation of iNKT cells by covalent protein-glycolipid conjugates
Our previous work analyzed a series of photoactivable analogues of αGalCer containing a benzophenone (BP) moiety appended to a fatty acyl chain of varying length. An analogue containing an 11 carbon aliphatic chain (C11:BP, Fig. S1a) was optimal for iNKT cell stimulation when covalently conjugated to soluble mouse single chain CD1d-β2 microglobulin fusion proteins (mCD1d-β2m) by UV irradiation (20). For the current study, we produced BiTEs comprised of a mouse or human CD1d-β2m fusion proteins with single chain Fv (ScFv) antibody domains specific for the tumor associated antigen HER2/neu linked to the C-termini (Supplementary Fig. 1b), as described previously (16, 20). The proteins were loaded with saturating amounts of glycolipids C11:BP or αGalCer C26:0 to prepare noncovalent protein-glycolipid complexes (Supplementary Fig. 1a). These were irradiated with UV light (λ = 365 nm, dose of ~600 mJ/cm2), activating the BP group in the case of C11:BP to form a covalent bond with the CD1d protein (20). Size exclusion chromatography showed the resulting conjugates to be predominantly monomeric (Supplementary Fig. 1b), and their covalent stabilization was validated by assessing residual binding of monoclonal antibody L363 (specific for CD1d containing a bound αGalCer ligand (24)) following mild detergent washing (Supplementary Fig. 1c).
Systemic administration of conjugated iNKT activators in vivo showed similar early activation of splenic iNKT cells for conjugates compared to the complexes and the benchmark of αGalCer injected as a free glycolipid, based on their levels of intracellular staining for IFNγ. Conjugates gave significantly stronger activation than an equivalent dose of complexes by this criterion, and responses to both complexes and conjugates were less sustained than the response to free αGalCer (Fig. 1a). A very similar pattern of response was observed for transactivation of NK cells, based on intracellular IFNγ levels in NK1.1+ CD3 negative cells (Supplementary Fig. S2a–d). Mice lacking iNKT cells due to deletion of genes encoding either CD1d (Fig. 1b) or Jα18 (Supplementary Fig. S3a) showed no IFNγ secretion to either free glycolipid or conjugates, and adoptive transfer of purified iNKT cells into CD1d−/− mice restored responses to only conjugates but not to free αGalCer This confirmed that conjugates did not require CD1d expression by antigen presenting cells to stimulate in vivo responses.
Figure 1; In vivo activities of iNKT cell specific BiTEs.
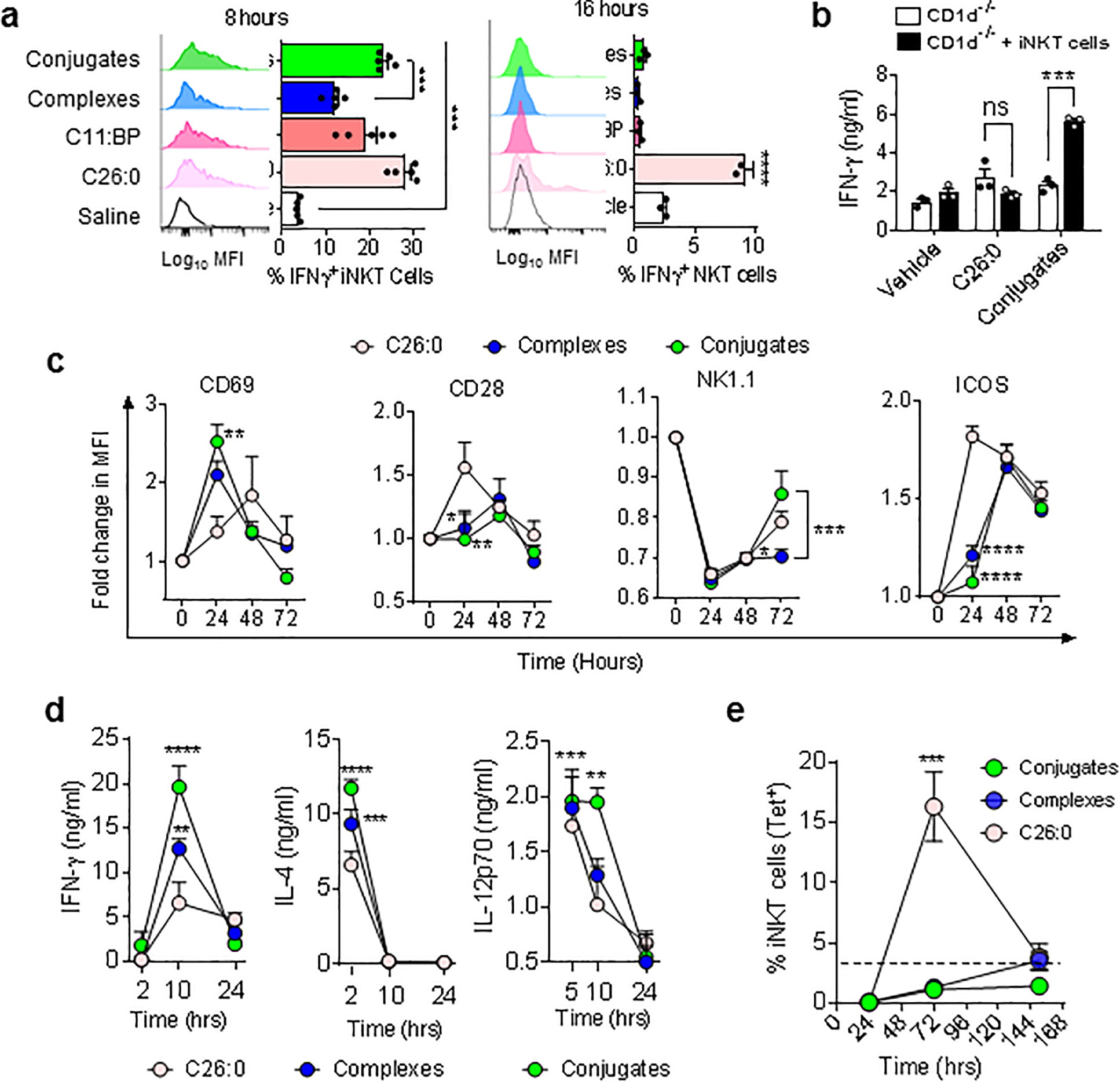
(a) Intracellular IFNγ staining of iNKT cells 8 or 16 hr post i.v. injection of WT (C57BL/6) mice with 1 nanomole of conjugates, complexes, free glycolipids (C11:BP or C26:0) or saline. (b) Serum IFNγ in CD1d−/− mice 12 hr post i.v. injection with vehicle, free glycolipid or conjugates with or without adoptive transfer of 100,000 purified iNKT cells from WT mice 16 hours earlier. (c) Surface modulation of activation related markers on splenic iNKT cells in WT mice injected once i.v. with 1 nanomole of free glycolipid (C26:0), complexes or conjugates, and sacrificed at indicated times for staining with αGalCer-loaded mCD1d tetramers and antibodies to surface molecules. Values are plotted as fold change in MFI compared to untreated controls. (d) Serum cytokine levels in WT mice following injection of free glycolipid, complexes or conjugates. (e). Expansion kinetics of iNKT cells in WT mice following a single i.v. injection of free glycolipid (C26:0), complexes or conjugates were determined by FACS analysis of splenocytes. Results are plotted as percent of total splenic TCRβ and Tetramer+ cells. Dashed line shows baseline level in saline injected animals. **P < 0.01, *** P < 0.001, ****P < 0.0001; 2-way ANOVA with Sidak multiple comparison test. All symbols or bars show means ± 1 SE for groups of 3 mice, and data are representative of at least three separate experiments.
Similar changes in levels of activation-inducible surface proteins on iNKT cells, including CD69, CD28, NK1.1 and ICOS, were observed following in vivo stimulation by complexes or conjugates (Fig. 1c). Sequential measurements of serum levels of IFNγ and other cytokines produced after iNKT cell activation, including IL-4, IL-12p70, TNFα (Fig. 1d), also showed rapid responses to complexes and conjugates, which were generally equivalent to or stronger than those stimulated by equimolar concentrations of free αGalCer C26:0. However, minimal iNKT cell expansion was observed following a single injection of complexes or conjugates, in contrast to the robust expansion at 72 hours followed by subsequent contraction of the splenic iNKT cells observed with a single injection of free αGalCer (Fig. 1e). Lack of expansion was further confirmed by selectively analyzing GFP+ iNKT cells that were adoptively transferred into wild type recipient mice prior to treatment with conjugates (Supplementary Fig. S3b). Thus, both complexes and conjugates stimulated a unique partial activation of iNKT cells in vivo, which was independent of normal antigen processing and presentation pathways requiring CD1d+ APCs. Such responses were characterized by strong transient cytokine production and faster NK transactivation, but without the proliferative expansion or subsequent contraction of iNKT cells normally associated with presentation of αGalCer (32, 33).
Reduced anergy with activation by conjugates
Induction of unresponsiveness (anergy) of iNKT cells after activation in vivo by free αGalCer is a major potential problem that limits the application of iNKT cell-directed therapies (14, 33, 34). To assess whether the partial activation observed with complexes or conjugates also caused anergy, we analyzed responses to αGalCer in mice that had previously received complexes, conjugates or free αGalCer. As expected, the response to re-stimulation with αGalCer was markedly reduced in animals that had received a single prior injection of the glycolipid, based on serum IFNγ levels (Fig. 2a) and reduced iNKT cell expansion (Fig. 2b). Responses in animals that had previously received injections of complexes also showed evidence of anergy induction, with moderate reduction of IFNγ secretion and strong decreases in iNKT cell expansion. In contrast, a single injection of conjugates induced no evidence of anergy by these criteria, and three serial injections of conjugates gave only partial loss of expansion and no suppression of IFNγ secretion by iNKT cells. Despite the marked reduction in anergy, conjugates stimulated significant increases in surface expression of multiple inhibitory checkpoint receptors including CTLA-4, Lag3 and PD-1 by iNKT cells, especially in the liver (Fig. 2c), which may have contributed to the partial reduction in iNKT cell expansion associated with multiple conjugate injections. However, the expression of ligands for CTLA-4 and PD-1 on DCs was attenuated in conjugate treated versus free αGalCer treated animals (Fig. 2d), possibly mitigating the potential inhibitory effects of these receptors following multiple conjugate stimulations.
Figure 2; Reduced anergy and exhaustion of iNKT cells following stimulation by conjugates.
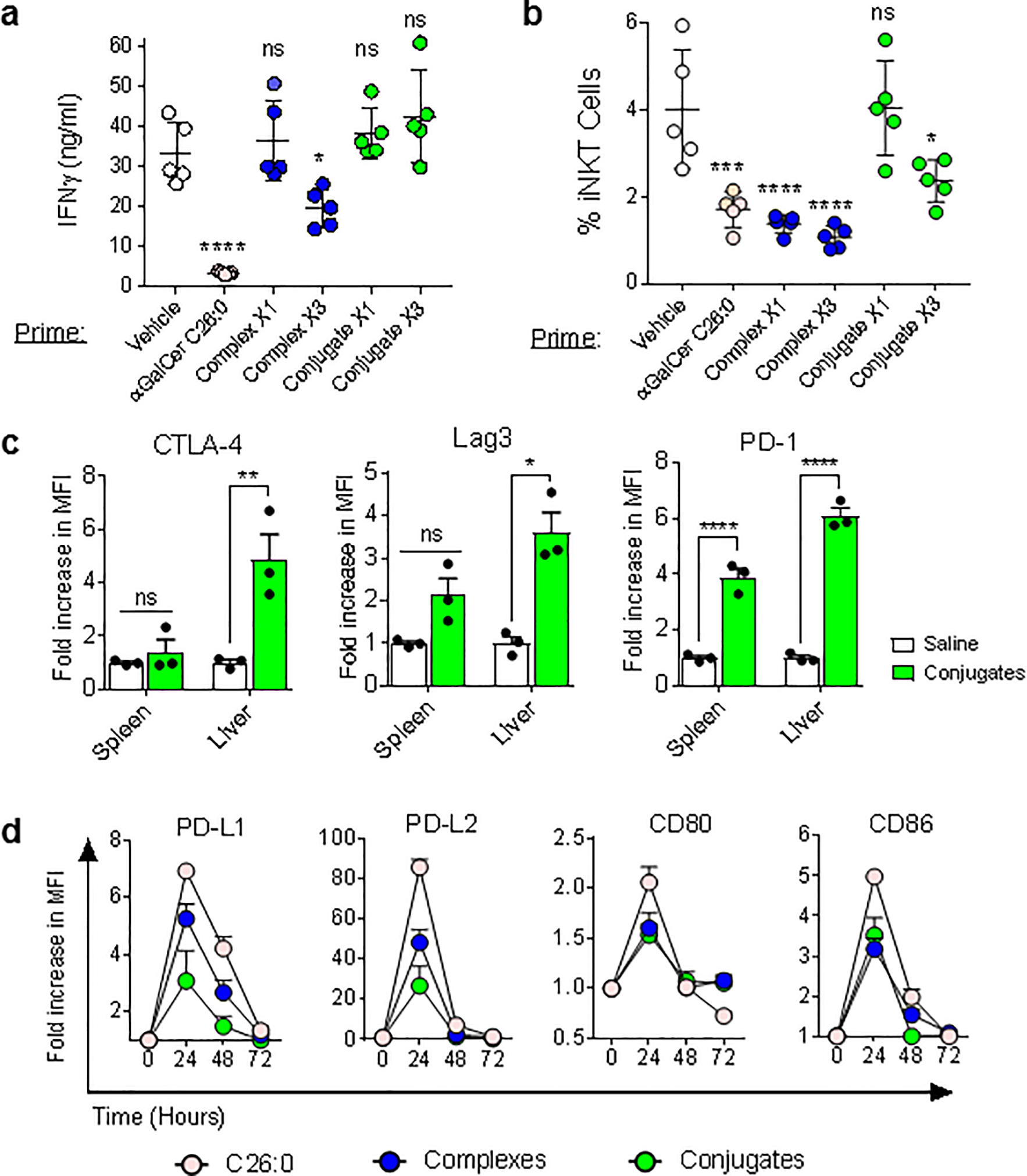
(a) Mice were primed with i.v. injections of vehicle, free glycolipid (C26:0), complexes or conjugates either once or three times at weekly intervals. One week later, all groups were re-stimulated with i.v. C26:0, and serum IFNγ was measured at 12 hours by ELISA. (b) Quantitation of iNKT cells as percentage of total T cells by tetramer staining of splenocytes harvested at 72 hours after priming and restimulation as in panel A. (c) Surface expression of inhibitory checkpoint receptors by iNKT cells was measured on spleen and liver on tetramer stained iNKT cells 48 hours after the second of two weekly injections of saline versus conjugates. (d) Upregulation of ligands for co-inhibitory receptors on spleen CD11c+ DCs after stimulation with free glycolipid, complexes or conjugates. Data are means ± 1 SE for groups of 3 – 5 mice, and are representative of at least three experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****p<0.0001, 2-way ANOVA with Sidak multiple comparisons test.
Enhanced anti-tumor activity of iNKT cell specific covalently stabilized BiTEs
Given the ability of conjugates to more strongly induce iNKT cell functions with less anergy than complexes, we directly compared iNKT cell specific BiTEs with and without covalent stabilization in mouse tumor immunotherapy models. For proof of concept studies, we incorporated the targeting capability of the anti-huHER2-specific ScFv fused to murine CD1d. In vitro studies validated that the anti-huHER2 ScFv-mCD1d fusion proteins bound specifically to tumor cells expressing huHER2, whereas similar fusion proteins containing a ScFv domain specific for human CEA or a nonspecific control ScFv did not bind detectably to huHER2 transfected cells (Supplementary Fig. S4a,b). Initial studies to assess anti-tumor effects used a mouse metastatic melanoma cells B16.F10 transfected to express huHER2. Following tumor cell injection, mice were treated on days 3 and 10 with injections of either saline, noncovalent BiTEs (complexes) or covalent BiTEs (conjugates) and sacrificed on day 16 for assessment of the extent of metastatic disease. Compared to saline treated mice, which showed extensive tumor growth with heavy melanin deposition over nearly 100% of the lung surface, animals treated with complexes showed a modest reduction in tumor burden. However, animals treated with covalently stabilized conjugates showed a substantially greater effect, with only ~5% residual tumor compared to the saline treated controls (Fig. 3a).
Figure 3; Enhancement of tumor immunotherapy with conjugates.
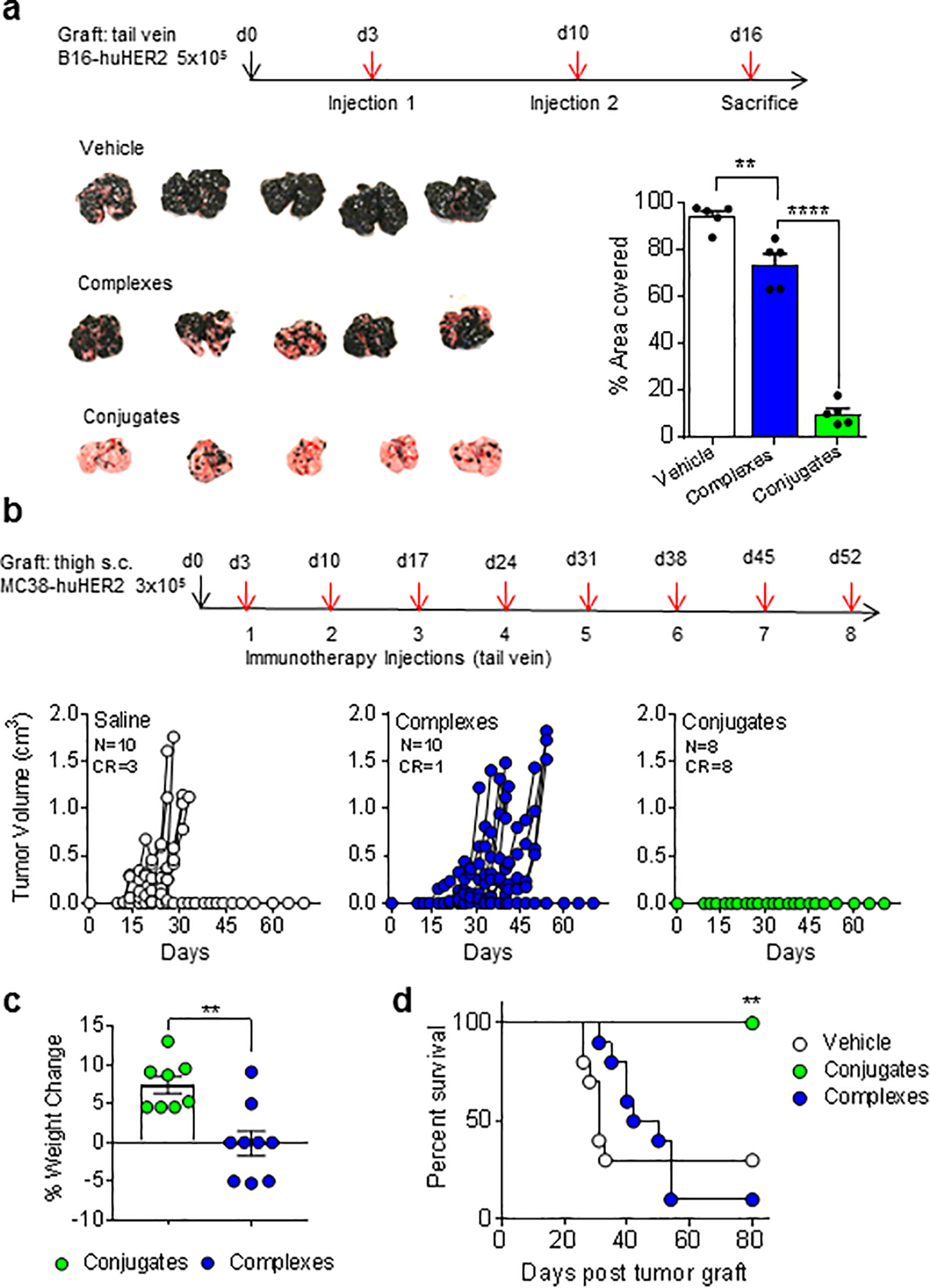
(a) Schematic for tumor injection and immunotherapy for B16.F10-huHER2 tumor model. Images of lungs harvested on day 16 from 5 mice per treatment group are shown, and extent of tumor growth was calculated as percent lung surface covered with melanized nodules. Bars show means ± 1 SE. **P < 0.01, ****p<0.0001; 2-way ANOVA with Sidak multiple comparisons test. (b) Schematic for grafting MC38-huHER2 and immunotherapy regimen. Tumor growth curves for individual mice in each treatment group are shown for groups of 8 – 10 mice. Numbers of complete responders (CR) with no detectable tumor are indicated. (c) Comparison of weight change from day of tumor graft to day 30 in mice treated with huHER2 targeted complexes or conjugates. **P < 0.01; 2-way ANOVA. (d) Survival curves for treatment groups. **P < 0.01; log rank test.
We next assessed the effects of covalent BiTEs on tumor progression in a localized subcutaneous implant model using MC38 colorectal carcinoma cells expressing huHER2. Three days after tumor initiation by subcutaneous injection of cells in the thigh, the animals received eight weekly i.v. injections of either noncovalent complexes, covalently stabilized conjugates or saline. Most saline treated animals showed rapid tumor growth, leading to death or mandatory sacrifice (i.e., following two consecutive measurements of tumor volume >1 cm3, as required by protocol) within 30 days. Animals treated with complexes showed delayed tumor growth but continued disease progression in most animals, consistent with previously published studies using this model and a similar treatment regimen (16). In contrast, covalent conjugate treated animals showed complete suppression of tumor growth in all cases (Fig. 3b) and continued to gain weight normally during treatment (Fig. 3c). A modest prolongation of survival was apparent in the complex treated animals, but substantially less than the complete survival through day 80 in the conjugate treated group (Fig. 3d). Re-challenge in these mice on day 80 with subcutaneous injection of 3 × 105 MC38-huHER2 cells showed complete resistance to re-establishment of tumors, consistent with durable anti-tumor recall immune responses.
Mechanisms of superior anti-tumor activity of covalent BiTEs
In vitro analysis showed that covalently stabilized conjugates were more potent than complexes at inducing iNKT cell mediated lysis of huHER2 expressing tumor cells in chromium release assays (Fig. 4a), possibly reflecting their greater stability. Similar results were obtained using tumor spheroids cultured with either mouse or human iNKT cells, with covalent conjugates generating greater levels of tumor cell killing and to greater depth within the tumor cell mass (Fig. 4b). To assess the importance of the specific targeting of the conjugated BiTE to huHER2, we grafted individual mice with huHER2 positive and negative MC38 cells on opposite thighs. This showed partial suppression of huHER2 negative tumors, while huHER2 positive tumors were completely suppressed (Fig. 4c). Similarly, tumors established with mixtures of huHER2 positive and negative cells showed reduced efficacy of immunotherapy, which became more pronounced as the fraction of huHER2 negative cells was increased (Supplementary Fig. S4c). This indicated that at least part of the anti-tumor activity of conjugated iNKT cell specific BiTEs was mediated by direct targeting to the tumor-associated antigen.
Figure 4; Specific cytotoxicity and tissue deposition of HER2-targeted conjugates.

(a) MC38-huHER2 cells labeled with 51Cr labeled were incubated with anti-huHER2 conjugates or complexes. Target specific killing was determined following incubation for 12 hr with human iNKT cell clone HDD3 at effector:target ratio of 1:1. (b) Spheroids of MC38-huHER2 cells were pulsed with saline or anti-huHER2 complexes or conjugates containing either murine (top) or human (bottom) CD1d. Following incubation for 12 hr with mouse spleen iNKT cells (top) or human iNKT cells (clone HDD3, bottom), spheroids were stained for markers of viability, apoptosis and necrosis. Scale bar 1 mm. (c) Mice were grafted on day 0 with 5 × 105 MC38-huHER2 cells s.c. in the right thigh and 5 × 105 huHER2 negative MC38 cells in the left thigh. Injections of saline or anti-huHER2 conjugates were given i.v. on days 3, 10 and 17, and growth of tumors on left and right thighs was followed to day 30. (d) Mice grafted with 5 × 105 MC38-huHER2 tumor cells 14 days earlier received a single i.v. injection of 1 nanomole of anti-HER2 conjugates loaded with 14C-labelled C11:BP glycolipid or 1 nanomole of 14C-labeled free C11:BP glycolipid. Individual mice were sacrificed at times shown, and 14C retention in organ or tumor tissues was determined by liquid scintillation counting. Dashed lines indicate background CPM for mice injected with saline only. *P < 0.05, **P < 0.01 ***P < 0.001, ****P < 0.0001, 2-way ANOVA with Sidak multiple comparison test.
To further evaluate this point, we used two different approaches to assess the efficacy of conjugated BiTEs to target the glycolipid ligand to the huHER2 positive tumor. Using radiolabeled 14C-C11:BP, we quantitated the delivery of conjugated BiTEs versus the free glycolipid to normal tissues and to established MC38-huHER2 tumors (Fig. 4d). This revealed higher and sustained accumulation of the glycolipid in tumor tissue compared to spleen and kidney when administered as conjugates. Liver tissue also showed relatively high levels of conjugates and free glycolipid initially, but this declined rapidly over time. A second approach using an azido modified variant of C11:BP enabled direct visualization of conjugates in tissue sections by fluorescent labelling (Supplementary Fig. S4d) (20). This confirmed the relatively high accumulation in liver and tumor tissues relative to spleen and kidney, without histologic evidence of tissue necrosis or inflammation in either tissue. Overall, these results suggested that specific targeting of conjugated BiTEs to tumors was relevant to improving anti-tumor effects by directing iNKT cell activation to the tumor site.
Analysis of TILs in conjugated BiTE treated mice
To gain further insight into the mechanisms for the superior anti-tumor effects of conjugated iNKT cell specific BiTEs, we carried out a detailed analysis of tumor infiltrating leukocytes (TILs). Tumors of MC38-huHER2 cells were first established to moderate size (0.3 cm3) in the thigh, and the animals then received two injections of saline, complexes or conjugates (Fig. 5a). Representative tumors excised from these animals were analyzed by routine histology and immunohistochemistry (Fig. 5b). This revealed dense and well delineated tumors in all three treatment groups, with reduced Ki67 staining and increased TUNEL positivity in the conjugated BiTE treated group, indicative of reduced tumor cell proliferation and increased cell death. A notable finding was the distribution of anti-CD3 staining, which was prominently confined to the margin of the tumor in saline treated tumors, but evenly distributed throughout the tumor in complex and conjugate treated tumors suggesting an effect on promoting T cell infiltration. Extraction of TILs and FACS analysis (see Supplementary Fig. S5a and b for gating strategy) revealed increased numbers of total leukocytes and T cells per gram of tumor tissue in conjugated BiTE treated animals (Fig. 5c), and phenotypic analysis showed an increase in the proportion of T cells lacking both CD4 and CD8 (i.e., double negative (DN) T cells) (Fig. 5d). FoxP3+ regulatory CD4+ T cells (Treg) were reduced in TILs from these animals (Fig. 5e). Significant effects on the composition of TILs with regard to cell populations other than T cells were also observed in animals treated with noncovalent complexes or conjugates. These included an increase in the proportion and number of NK1.1+ CD3 negative cells, most likely representing NK cells (Fig. 5f), and significant effects on myeloid lineage cells with reductions in F4/80+ macrophages and Gr-1+ neutrophils in conjugate treated tumors (Fig. 5g).
Figure 5; T cell infiltrates of tumors treated with iNKT cell specific BiTEs.
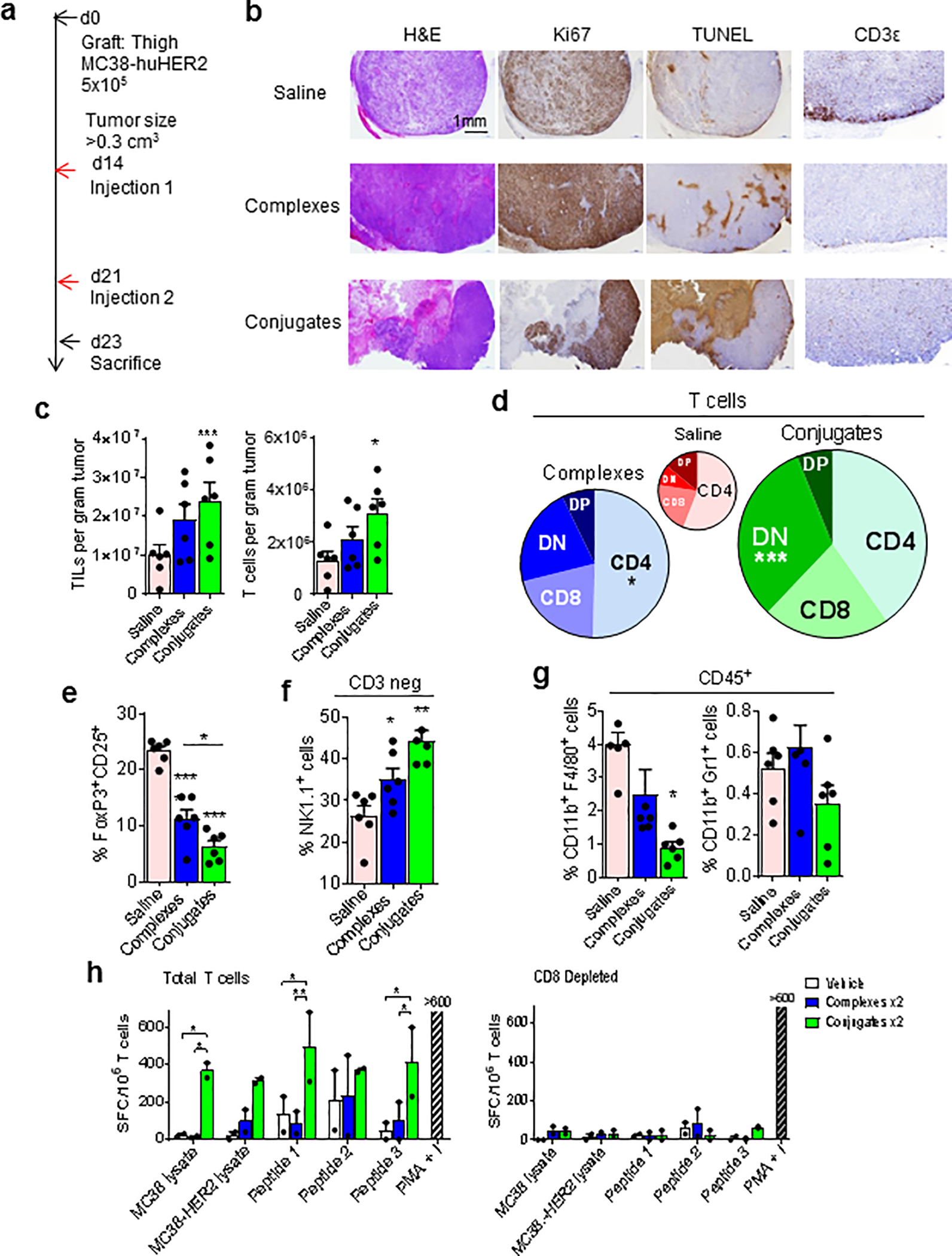
(a) Protocol for generation of tumors for TIL extraction and analysis in mice injected with saline versus huHER2-specific complexes or conjugates. (b) Immunohistochemistry of representative tumors from treated mice. (c) Quantitation of total TILs (CD45+) and T cells (CD3+) by FACS in tumors from treated mice (N = 5 mice per group). (d) Diagrams showing relative proportions of T cell subsets based on CD4 and CD8 expression, including double negative (DN) and double positive (DP) subsets. The size of each circle is proportional to the total T cell number in TILs for each treatment group. (e) Percentage of FoxP3+ CD25+ Tregs among total CD3+ cells in TILs. (f) Percentage of CD3 negative NK1.1+ cells among CD45+ TILs. (g) Myeloid subsets showing percentages of F4/80+ (macrophages) and Gr1+ (granulocytes) among total CD11b+ cells in TILs. (h) ELISPOT assay showing IFNγ spot forming cells in purified total T cells (left) and CD8 depleted T cells (right) from spleens of MC38-huHER2 tumor bearing mice injected twice with saline, complexes or conjugates and re-stimulated ex vivo with lysates of MC38, MC38-huHER2, synthetic peptides spanning three predicted MHC class I presented epitopes of huHER2, or PMA plus Ionomycin. Bars show means and range for values from two mice for each treatment group. N = 6 mice per treatment group for c – g, *P < 0.05, **P < 0.01, ***P < 0.001, 2-way ANOVA with Sidak multiple comparison test.
To functionally assess tumor specific T cell responses in TILs, we used an IFNγ ELISPOT assay with stimulation by tumor lysate or synthetic peptides from the huHER2 tumor-associated antigen (Fig. 5h). Three peptides based on sequences of predicted CD8+ T cell epitopes in huHER2 were tested, with two of these generating significant responses from T cells purified from tumors of conjugate treated mice, whereas no significant responses were observed with T cells from tumors of animals treated with saline or noncovalent complexes. T cells from conjugate treated tumors responded to lysates of MC38-huHER2 cells, and also to lysate from MC38 cells not expressing huHER2, suggesting the stimulation of epitope spreading to endogenous mouse tumor associated antigens. All of the T cell responses detected in these assays were eliminated by depletion with anti-CD8 conjugated magnetic beads, implicating CD8+ T cells as the relevant effectors.
Analysis of iNKT cells in TILs
Based on αGalCer loaded CD1d tetramer staining, iNKT cells were present at similar numbers in TILs of tumors from saline, complex or conjugate treated mice (Fig. 6a). To assess the quality of these cells in the different treatment groups, we applied an extended multicolor analysis of 13 cell surface markers (see Fig. 6d for list) and three intracellular transcription factors (FoxP3, Tbet and RORγt). Dimensional reduction of the data to identify distinct populations of tetramer+ iNKT cells was carried out using t-distributed stochastic neighbor embedding (t-SNE) (Fig 6b) (35). The concatenated data from all three treatment groups revealed up to 20 distinct phenotypic clusters based on t-SNE-1 and t-SNE-2 two dimensional plots (Fig. 6c). These clusters could be separated into four distinct groups, based on their abundance in tumors from saline, complex or conjugate treated mice (Fig. 6d). Clusters comprising Group 1 were present exclusively in saline treated mice, while Group 2 subsets predominated in complex treated and Group 3 in conjugate treated animals. Subsets in Group 4 showed approximately equal levels in both complex and conjugate treated animals. Overall, this analysis revealed a striking diversity of iNKT cells, suggesting a fine-tuning of their functions when activated by different stimuli.
Figure 6; Analysis of iNKT cells in TILs.
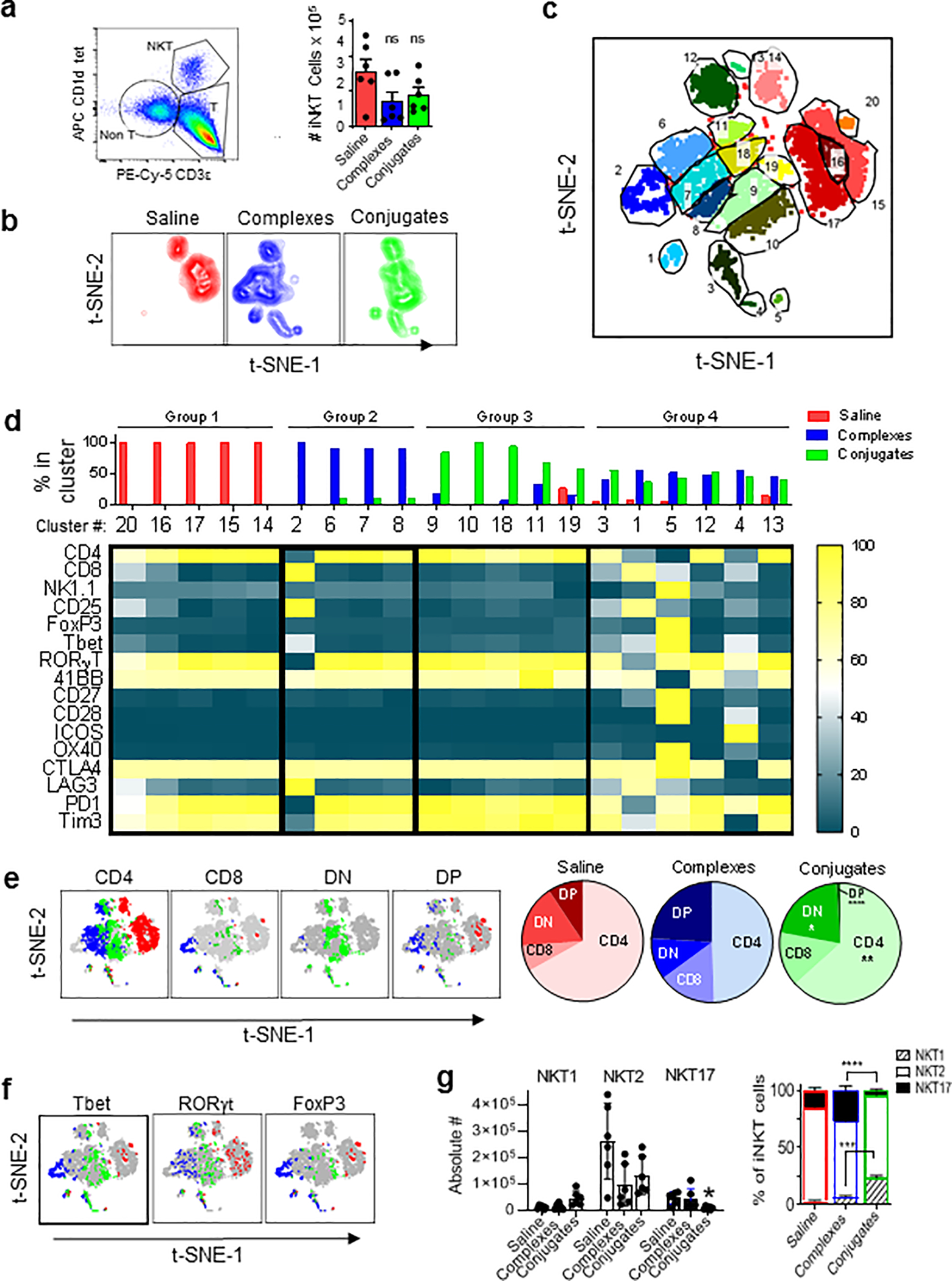
(a) Gating of iNKT cells in TILs and quantitation of absolute numbers of iNKT cells in total TILs extracted per tumor from mice with MC38-huHER2 tumors and treatments as in Fig. 5 (ns, not significant; 2-way ANOVA). (b) Two dimensional plots from t-SNE analysis of tetramer+ iNKT cells in TILs based on expression of 16 markers (listed in panel d). (c) Concatenated t-SNE analysis for all three treatment groups identifying 20 phenotypic clusters of iNKT cells. (d) Identification of four distinct groups of iNKT cells in TILs based on clustering by t-SNE. Bar graph on top shows relative abundance for each cluster in TILs of each treatment group. Heat map shows level of expression (normalized MFI) for each marker analyzed. (e) Subsets of CD4 and CD8α single positive, double positive (DP) and double negative (DN) expressing iNKT cells from t-SNE analysis mapped onto the concatenated data set (grey) in colors corresponding to each treatment group (red for saline, blue for complexes and green for conjugates). Pie charts show relative proportions of each subset by treatment group. (f) Similar to panel E, except showing distribution of signature transcription factors for functional iNKT cell subsets (i.e., classified as NKT1, NKT2 or NKT17 based on Tbet and RORγt as previously described36). (g) Bar graph on left shows absolute numbers of iNKT cells in total TILs isolated per tumor. *P < 0.05 for NKT17 cells in conjugate group compared to saline controls. Other trends observed did not reach statistical significance. Stacked bar graph on right shows subsets as percentage of total iNKT cells. Means ± 1SE for treatment groups are shown. ***P < 0.01, ****P < 0.001; 2-way ANOVA with Sidak multiple comparison test. All analyses based on data from 6 mice per group
We also evaluated the previously established iNKT cell subsets based on expression of CD4 and CD8 co-receptors, or their classification into NKT1, NKT2 and NKT17 functional subsets based on intracellular transcription factor staining (36). We observed a significant bias toward CD4 plus CD8 double positive (DP) iNKT cells in TILs from complex treated mice, whereas this subset was nearly absent in conjugate treated animals which had a greater predominance of CD4+ and double negative (DN) iNKT cells (Fig. 6e). With regard to functional subsets, all treatment groups had NKT2 cells as the majority population, and TILs from conjugated BiTE treated animals showed significantly reduced proportions and numbers of NKT17 and increased proportions of NKT1 cells (Fig. 6f and g).
Co-inhibitory receptors and checkpoint blockade in conjugated BiTE treatment
We also analyzed expression of costimulatory and co-inhibitory receptors on iNKT cells from our multiparameter analysis of TILs (Fig. 7a). Compared to saline treated mice, conjugated BiTEs gave no significant increase in expression of costimulatory receptors 4–1BB, ICOS, OX40 or CD28 on iNKT cells, although the expression was distributed in different clusters based on t-SNE analysis. With regard to co-inhibitory receptors, both complexes and conjugates induced higher expression of CTLA-4, but no change or a reduction in LAG3 (CD223), PD-1 (CD279) or Tim-3. This increase in CTLA-4 expression was consistent with the increase in this receptor observed in liver iNKT cells following treatment with conjugated BiTEs (Fig. 2c). Given these findings, we performed experiments to assess synergy between anti-CTLA4 and conjugated BiTEs in the MC38-huHER2 model using both wild type C57BL/6 mice (Supplementary Fig. S6) or human CD1d knock-in mice (Fig. 7). The latter represent a potentially more accurate model for translational studies, since they closely resemble humans with regard to numbers of iNKT cells and the strength of responses to iNKT cell activators (21, 37). Animals implanted with subcutaneous MC38-huHER2 tumors received three weekly i.v. injections of conjugated BiTEs and i.p. injections of either 100 μg of hamster anti-CTLA-4 or control normal hamster serum IgG (Fig. 7b). Under these conditions, conjugates alone were ineffective and anti-CTLA-4 alone had a small impact on tumor progression. However, strong synergy was observed with the combination of conjugates plus concurrent anti-CTLA-4 (Figs. 7c,d). A similar pattern of responses with synergy between conjugates and anti-CTLA-4 checkpoint blockade was also observed in wild type C57BL/6 mice (Supplementary Fig. S6a–d).
Figure 7; Synergistic antitumor effects of conjugates combined with checkpoint blockade.
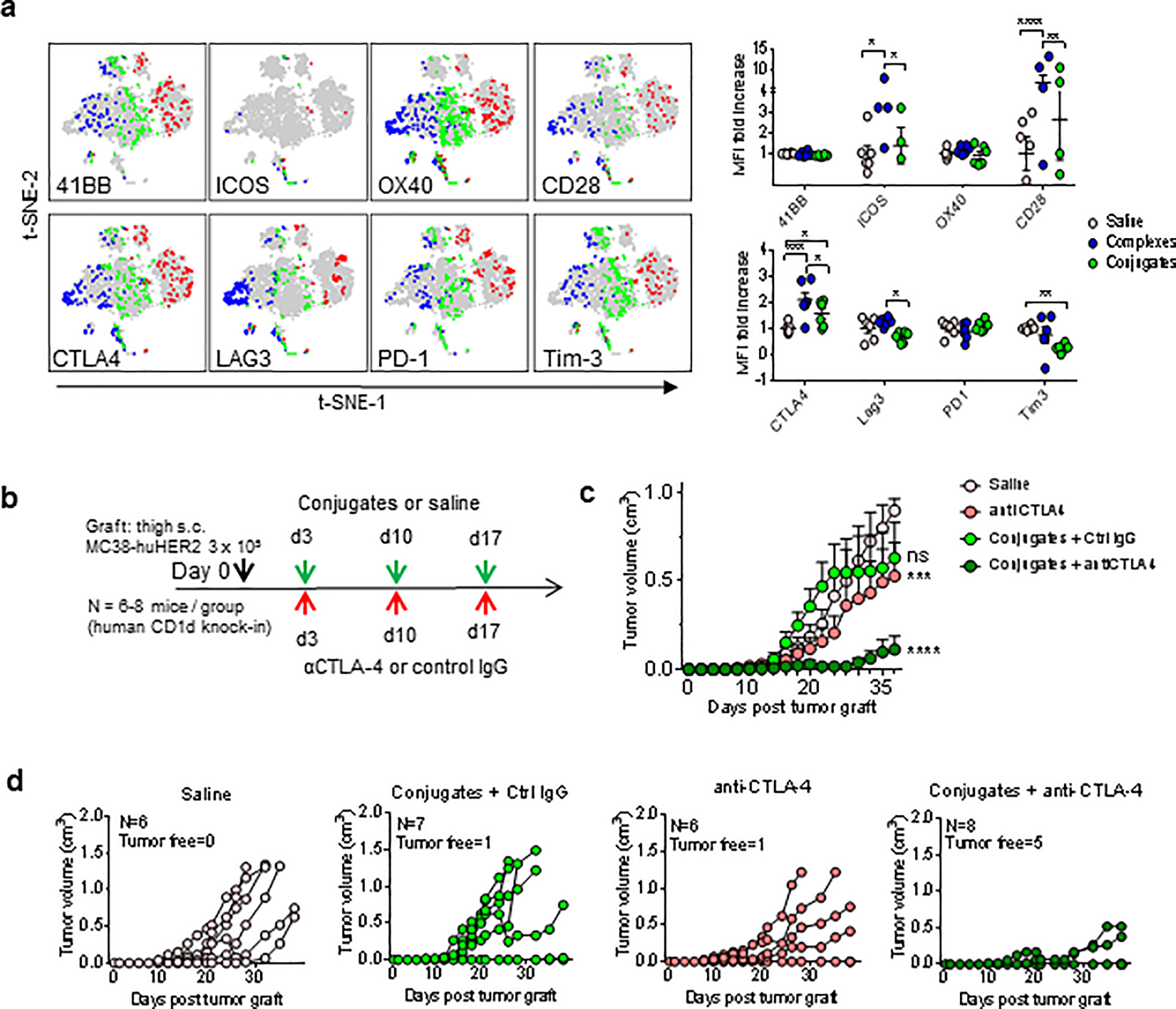
(a) Expression of costimulatory and co-inhibitory checkpoint receptors on iNKT cells in TILs from saline (red), complex (blue) or conjugate (green) treated mice. Graphs show fold increase compared to mean values for MFI in saline treated mice. (b) Scheme for combination treatment of MC38-huHER2 tumor bearing human CD1d knock-in mice with conjugates plus anti-CTLA-4 blocking antibody. For experiments using human CD1d knock-in mice, the conjugates also contained human CD1d. (c) Tumor growth shown as mean values for each treatment group (N = 6 – 8 mice per group). (d) Tumor growth curves for individual mice in each treatment group. The numbers of tumor free mice remaining at the end of observation at day 36 are indicated. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, not significant; 2-way ANOVA with Sidak multiple comparison test.
Discussion
The discovery of αGalCer as a powerful anti-tumor agent in mice (38), and its subsequent identification as a specific activator of iNKT cells (39), triggered substantial interest in the potential for iNKT cell directed cancer immunotherapy (9, 40, 41). While treatment with free αGalCer shows remarkable anti-tumor activity in mouse models, phase I clinical trials of this approach in human cancer patients have not shown clear evidence of therapeutic benefit (11, 34, 42, 43). Several problems confronting iNKT cell therapies have been identified, including depletion and dysfunction of these cells in cancer patients (44), and the tendency of strong activators such as αGalCer to induce a long-lasting anergic state (14, 32, 45, 46). The induction of anergy is a major limitation for regimens that depend on repetitive stimulation of iNKT cells to achieve strong and durable effects against established cancers. In addition, systemic administration of free glycolipid activators of iNKT cells has been linked to major toxic effects in mouse models, including severe hepatic necrosis and potentiation of septic shock (15, 47, 48). These observations indicate the need for new approaches for delivering iNKT cell activators more selectively to the tumor microenvironment while avoiding the induction of anergy.
An approach for creating more targetable iNKT cell activators is the use of soluble CD1d proteins loaded ex vivo with αGalCer (16, 17). However, this approach is compromised by the inherent instability of the glycolipid-protein complex, which is prone to rapid dissociation in vivo. To overcome this issue, we created covalent conjugates of αGalCer with soluble CD1d proteins as stable and specific bispecific iNKT cell engagers (20). The current study assessed the in vivo immune activation resulting from systemic injection of these covalently stabilized BiTEs, and their potential for anti-tumor immunotherapy. We found that conjugated BiTEs had a unique ability to optimally activate multiple immunological functions of iNKT cells while inducing only minor levels of iNKT cell expansion, and little or no hypo-responsiveness to subsequent stimulation. Compared to responses stimulated by systemic injection of free αGalCer, the iNKT cell responses to a single injection of BiTEs showed rapid and strong induction of multiple iNKT cell effector functions and upregulation of activation related molecules.
Responses to iNKT cell specific BiTEs did not require presentation by CD1d on APCs, which contrasts strongly with free αGalCer which requires presentation by CD1d+ cells, most prominently CD8α+ DEC205+ DCs in mice (4). It was also notable that activation of iNKT cell cytokine secretion appeared to occur without any specific anchoring of the BiTEs to an APC surface, as shown in mice lacking any expression of human HER2. The ability of a soluble TCR ligand to strongly stimulate cytokine production without multimerization or anchoring to an APC surface was surprising, and suggests that iNKT cells may be less dependent on costimulatory signals than conventional T cells for stimulation of effector functions. This may also be relevant to the lack of anergy following stimulation with conjugated BiTEs, as anergy of iNKT cells is potentiated when TCR stimulation is accompanied by costimulation through CD28 and ICOS, the two principle costimulatory receptors known to be active on iNKT cells (45, 46, 48). Thus, our ability to activate iNKT cells without direct presentation by APCs should bypass these costimulatory signals, explaining the lack of proliferative expansion or anergy. In addition, the ligands for multiple costimulatory and co-inhibitory receptors were less strongly upregulated on APCs by iNKT cell specific BiTEs compared to free αGalCer, which may have further reduced the induction or maintenance of anergy following iNKT cell activation.
In spite of the relatively similar effects of nonconjugated and conjugated BiTEs in activating iNKT cell responses, there were markedly superior anti-tumor responses with covalent conjugates in our experiments, or when compared to published results using similar noncovalent complexes (16, 17). This can be explained by the partial induction of hypo-responsiveness by multiple injections of complexes (16), which was attributed to the ability of glycolipid to dissociate from the CD1d protein and stimulate APC-dependent responses (19). The more stable covalently conjugated BiTEs minimized or eliminated this problem, leading to improvements in the anti-tumor immune response including recruitment of direct cytotoxicity toward tumor cells, enhanced cross-priming of CD8+ T cells and improved T cell penetration into tumors. With regard to iNKT cells within the TILs, t-SNE analysis revealed substantial heterogeneity with several distinct phenotypic clusters that distinguished noncovalent complex and conjugated BiTE treated mice. Although the functions of the different populations identified by this analysis remain to be investigated, a number of provocative findings were evident. For example, the presence of a distinctive CD8α+ subset (cluster 2) exclusively in complex treated animals could indicate a subset that is deleterious to the highly effective antitumor response induced by conjugated BiTEs. Conversely, the presence of several distinct subsets comprising Group 3 clusters may point to specific subsets or differentiation states of iNKT cells that drive anti-tumor immunity.
Our studies on combinations of iNKT cell specific conjugated BiTE injections and anti-CTLA-4 antibodies provided initial proof of concept for adding checkpoint blockade to an optimized iNKT cell activating regimen. Our choice of CTLA-4 blockade was guided in part by the increased expression of this receptor observed on iNKT cell populations in tumors of conjugated BiTE treated animals. However, given our observations of the upregulation of multiple inhibitory receptors within TILs and in other tissues of tumor bearing mice, it will be prudent to test additional combinations using other checkpoint blockade agents such as anti-PD-1/PD-L1 and anti-LAG3. Checkpoint blockade may improve responses by other cell types that are stimulated downstream of iNKT cell activation, in particular CD8+ anti-tumor cytolytic T cells. Our finding that peptide specific CD8+ T cell responses against tumor associated antigens were increased by conjugated BiTE treatment provides further incentives to explore combinations with checkpoint blockade strategies, and also for adding therapeutic tumor vaccines. While our results confirm the known direct tumoricidal activities of iNKT cells, the secondary effects of the conjugated BiTEs on enhancing tumor-specific CD8+ T cell priming as well as NK cell activation strongly suggests that these cells also contribute as terminal effectors in the potent anti-tumor effects observed in our model system. Determining the relative importance of these and other potential effector populations in animals treated with iNKT cell specific BiTEs will be a focus for future analysis, along with more detailed assessment of efficacy of our approach in models that more accurately represent clinically relevant scenarios for cancer treatment.
The production of covalently stabilized conjugated BiTEs and the validation of their immunostimulatory properties provide a promising new avenue for iNKT cell directed therapy, although several major challenges remain. One of these is the apparent need for tumor-specific targeting of the conjugates to achieve maximal therapeutic effects, which imposes limitations on the types of cancers that can be effectively treated. However, the huHER2 protein that served as the target for our preclinical studies is expressed by a substantial fraction of breast cancers, and is one of several well-established targets for clinical translation of our approach. Targets could also include normal cell surface antigens with a restricted range of expression such as CD19, which has recently been demonstrated as an effective target for αGalCer-loaded CD1d complexes in mouse models of tumor immunotherapy (49). In addition, while conjugated BiTEs were most effective in our studies when designed to specifically bind to the tumor cell surface, partial effects were observed against tumors lacking the specifically targeted tumor antigen (Fig. 4c). Further work is ongoing to assess the potential for non-targeted conjugates, especially in combination immunotherapy regimens for tumors lacking known surface targets.
Our studies have not directly compared the anti-tumor potential of iNKT cell engaging BiTEs to many other bispecific T cell engagers that may more broadly stimulate effector T cells, such as BiTEs that act through binding to CD3 to more promiscuously activate all T cells. These latter constructs represent a potent family of T cell activators, although their potential toxicities remain a major concern that may limit their efficacy (50). In contrast, the relatively low numbers of iNKT cells in normal humans, and their further reduction and loss of function in many cancer patients (11, 51), suggest that iNKT cell selective BiTEs are likely to be better tolerated, albeit less potent than more broadly acting T cell engagers. In this regard, it is encouraging that strong anti-tumor effects with iNKT cell engaging BiTEs were seen in our experiments with human CD1d knock-in mice, despite their low numbers of iNKT cells (37). These effects could potentially be amplified by combining iNKT cell selective conjugated BiTEs with strategies for increasing iNKT cell numbers and function, such as adoptive cell therapy for engineering of chimeric antigen receptor expressing iNKT cells (9, 52). These and other approaches are becoming increasingly feasible, and are likely to converge in the future to generate practical and effective methods for iNKT cell based immunotherapy of cancer.
Supplementary Material
Statement of significance:
Covalently stabilized conjugates that engage the antigen receptors of iNKT cells and target a tumor antigen activate potent anti-tumor immunity without induction of anergy or depletion of the responding iNKT cells.
Acknowledgements
We thank Dr. Amy Howell (University of Connecticut) for helpful discussions. We thank Vera DesMarais, Hillary Guzik and Andrea Briceno for expert assistance with slide scanning and imaging analyses. This work was supported by NIH grants R01 AI45889 (S. Porcelli, S. Kharkwal and N. Saavedra-Avila), U01 GM111849 (S. Porcelli and N. Saavedra-Avila), and R01 CA198095 (S. Almo). G. Besra acknowledges support from a Personal Research Chair from Mr. James Bardrick and a Royal Society Wolfson Research Merit Award. L. Carreño was supported by Fondecyt grant 1160336. Core facilities used in this study (Flow Cytometry, Histology and Comparative Pathology, and Macromolecular Therapeutics Development Facilities) were supported in part by the Albert Einstein College of Medicine Cancer Center (NIH/NCI grant P30CA13330), and used instrumentation obtained with funding from NIH Shared Instrumentation Grants 1S10OD019961-01, 1S10OD023591-01 and 1S10OD026833-01.
Footnotes
Conflict of interests: MZ and EE are employees of Vaccinex, Inc., which has proprietary interests in iNKT cell-based immunotherapeutics. SAP has previously been a paid consultant of Vaccinex, Inc. The other authors declare no potential conflicts of interest.
References
- 1.Brennan PJ, Brigl M, Brenner MB. Invariant natural killer T cells: an innate activation scheme linked to diverse effector functions. Nat Rev Immunol. 2013;13: 101–17. [DOI] [PubMed] [Google Scholar]
- 2.Rossjohn J, Pellicci DG, Patel O, Gapin L, Godfrey DI. Recognition of CD1d-restricted antigens by natural killer T cells. Nat Rev Immunol. 2012;12: 845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carreno LJ, Saavedra-Avila NA, Porcelli SA. Synthetic glycolipid activators of natural killer T cells as immunotherapeutic agents. Clin Transl Immunology. 2016;5: e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arora P, Baena A, Yu KO, Saini NK, Kharkwal SS, Goldberg MF, et al. A single subset of dendritic cells controls the cytokine bias of natural killer T cell responses to diverse glycolipid antigens. Immunity. 2014;40: 105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Godfrey DI, Le Nours J, Andrews DM, Uldrich AP, Rossjohn J. Unconventional T Cell Targets for Cancer Immunotherapy. Immunity. 2018;48: 453–73. [DOI] [PubMed] [Google Scholar]
- 6.Hong C, Lee H, Park YK, Shin J, Jung S, Kim H, et al. Regulation of secondary antigen-specific CD8(+) T-cell responses by natural killer T cells. Cancer Res. 2009;69: 4301–8. [DOI] [PubMed] [Google Scholar]
- 7.Laurent X, Bertin B, Renault N, Farce A, Speca S, Milhomme O, et al. Switching invariant natural killer T (iNKT) cell response from anticancerous to anti-inflammatory effect: molecular bases. J Med Chem. 2014;57: 5489–508. [DOI] [PubMed] [Google Scholar]
- 8.Bedard M, Salio M, Cerundolo V. Harnessing the Power of Invariant Natural Killer T Cells in Cancer Immunotherapy. Front Immunol. 2017;8: 1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wolf BJ, Choi JE, Exley MA. Novel Approaches to Exploiting Invariant NKT Cells in Cancer Immunotherapy. Front Immunol. 2018;9: 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Exley MA, Nakayama T. NKT-cell-based immunotherapies in clinical trials. Clin Immunol. 2011;140: 117–8. [DOI] [PubMed] [Google Scholar]
- 11.Nair S, Dhodapkar MV. Natural Killer T Cells in Cancer Immunotherapy. Front Immunol. 2017;8: 1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waldowska M, Bojarska-Junak A, Rolinski J. A brief review of clinical trials involving manipulation of invariant NKT cells as a promising approach in future cancer therapies. Cent Eur J Immunol. 2017;42: 181–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Im JS, Arora P, Bricard G, Molano A, Venkataswamy MM, Baine I, et al. Kinetics and cellular site of glycolipid loading control the outcome of natural killer T cell activation. Immunity. 2009;30: 888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parekh VV, Wilson MT, Olivares-Villagomez D, Singh AK, Wu L, Wang CR, et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest. 2005;115: 2572–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Singh M, Quispe-Tintaya W, Chandra D, Jahangir A, Venkataswamy MM, Ng TW, et al. Direct incorporation of the NKT-cell activator alpha-galactosylceramide into a recombinant Listeria monocytogenes improves breast cancer vaccine efficacy. Br J Cancer. 2014;111: 1945–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corgnac S, Perret R, Derre L, Zhang L, Stirnemann K, Zauderer M, et al. CD1d-antibody fusion proteins target iNKT cells to the tumor and trigger long-term therapeutic responses. Cancer Immunol Immunother. 2013;62: 747–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stirnemann K, Romero JF, Baldi L, Robert B, Cesson V, Besra GS, et al. Sustained activation and tumor targeting of NKT cells using a CD1d-anti-HER2-scFv fusion protein induce antitumor effects in mice. J Clin Invest. 2008;118: 994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Slaney CY, Wang P, Darcy PK, Kershaw MH. CARs versus BiTEs: A Comparison between T Cell-Redirection Strategies for Cancer Treatment. Cancer Discov. 2018;8: 924–34. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L, Donda A. Alpha-Galactosylceramide/CD1d-Antibody Fusion Proteins Redirect Invariant Natural Killer T Cell Immunity to Solid Tumors and Promote Prolonged Therapeutic Responses. Front Immunol. 2017;8: 1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veerapen N, Kharkwal SS, Jervis P, Bhowruth V, Besra AK, North SJ, et al. Photoactivable Glycolipid Antigens Generate Stable Conjugates with CD1d for Invariant Natural Killer T Cell Activation. Bioconjug Chem. 2018;29: 3161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wen X, Rao P, Carreno LJ, Kim S, Lawrenczyk A, Porcelli SA, et al. Human CD1d knock-in mouse model demonstrates potent antitumor potential of human CD1d-restricted invariant natural killer T cells. Proc Natl Acad Sci U S A. 2013;110: 2963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khurana A, Kronenberg M. A method for production of recombinant mCD1d protein in insect cells. J Vis Exp. 2007: 556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Im JS, Yu KO, Illarionov PA, LeClair KP, Storey JR, Kennedy MW, et al. Direct measurement of antigen binding properties of CD1 proteins using fluorescent lipid probes. J Biol Chem. 2004;279: 299–310. [DOI] [PubMed] [Google Scholar]
- 24.Yu KO, Im JS, Illarionov PA, Ndonye RM, Howell AR, Besra GS, et al. Production and characterization of monoclonal antibodies against complexes of the NKT cell ligand alpha-galactosylceramide bound to mouse CD1d. J Immunol Methods. 2007;323: 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong ZJ, Wei HM, Sun R, Tian ZG, Gao B. Isolation of murine hepatic lymphocytes using mechanical dissection for phenotypic and functional analysis of NK1.1+ cells. World J Gastroenterol. 2004;10: 1928–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arora P, Venkataswamy MM, Baena A, Bricard G, Li Q, Veerapen N, et al. A rapid fluorescence-based assay for classification of iNKT cell activating glycolipids. J Am Chem Soc. 2011;133: 5198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bricard G, Venkataswamy MM, Yu KO, Im JS, Ndonye RM, Howell AR, et al. Alpha-galactosylceramide analogs with weak agonist activity for human iNKT cells define new candidate anti-inflammatory agents. PLoS One. 2010;5: e14374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9: 671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faustino-Rocha A, Oliveira PA, Pinho-Oliveira J, Teixeira-Guedes C, Soares-Maia R, da Costa RG, et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab Anim (NY). 2013;42: 217–24. [DOI] [PubMed] [Google Scholar]
- 30.Jensen MM, Jorgensen JT, Binderup T, Kjaer A. Tumor volume in subcutaneous mouse xenografts measured by microCT is more accurate and reproducible than determined by 18F-FDG-microPET or external caliper. BMC Med Imaging. 2008;8: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeon J, Kang JA, Shim HE, Nam YR, Yoon S, Kim HR, et al. Efficient method for iodine radioisotope labeling of cyclooctyne-containing molecules using strain-promoted copper-free click reaction. Bioorg Med Chem. 2015;23: 3303–8. [DOI] [PubMed] [Google Scholar]
- 32.Wilson MT, Johansson C, Olivares-Villagomez D, Singh AK, Stanic AK, Wang CR, et al. The response of natural killer T cells to glycolipid antigens is characterized by surface receptor down-modulation and expansion. Proc Natl Acad Sci U S A. 2003;100: 10913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wingender G, Birkholz AM, Sag D, Farber E, Chitale S, Howell AR, et al. Selective Conditions Are Required for the Induction of Invariant NKT Cell Hyporesponsiveness by Antigenic Stimulation. J Immunol. 2015;195: 3838–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giaccone G, Punt CJ, Ando Y, Ruijter R, Nishi N, Peters M, et al. A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin Cancer Res. 2002;8: 3702–9. [PubMed] [Google Scholar]
- 35.Toghi Eshghi S, Au-Yeung A, Takahashi C, Bolen CR, Nyachienga MN, Lear SP, et al. Quantitative Comparison of Conventional and t-SNE-guided Gating Analyses. Front Immunol. 2019;10: 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee YJ, Wang H, Starrett GJ, Phuong V, Jameson SC, Hogquist KA. Tissue-Specific Distribution of iNKT Cells Impacts Their Cytokine Response. Immunity. 2015;43: 566–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chennamadhavuni D, Saavedra-Avila NA, Carreno LJ, Guberman-Pfeffer MJ, Arora P, Yongqing T, et al. Dual Modifications of alpha-Galactosylceramide Synergize to Promote Activation of Human Invariant Natural Killer T Cells and Stimulate Anti-tumor Immunity. Cell Chem Biol. 2018;25: 571–84 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Motoki K, Morita M, Kobayashi E, Uchida T, Akimoto K, Fukushima H, et al. Immunostimulatory and antitumor activities of monoglycosylceramides having various sugar moieties. Biol Pharm Bull. 1995;18: 1487–91. [DOI] [PubMed] [Google Scholar]
- 39.Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 1997;278: 1626–9. [DOI] [PubMed] [Google Scholar]
- 40.Krijgsman D, Hokland M, Kuppen PJK. The Role of Natural Killer T Cells in Cancer-A Phenotypical and Functional Approach. Front Immunol. 2018;9: 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Godfrey DI, Kronenberg M. Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest. 2004;114: 1379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kunii N, Horiguchi S, Motohashi S, Yamamoto H, Ueno N, Yamamoto S, et al. Combination therapy of in vitro-expanded natural killer T cells and alpha-galactosylceramide-pulsed antigen-presenting cells in patients with recurrent head and neck carcinoma. Cancer Sci. 2009;100: 1092–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ishikawa A, Motohashi S, Ishikawa E, Fuchida H, Higashino K, Otsuji M, et al. A phase I study of alpha-galactosylceramide (KRN7000)-pulsed dendritic cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. 2005;11: 1910–7. [DOI] [PubMed] [Google Scholar]
- 44.Singh AK, Shukla NK, Das SN. Altered invariant natural killer T cell subsets and its functions in patients with oral squamous cell carcinoma. Scand J Immunol. 2013;78: 468–77. [DOI] [PubMed] [Google Scholar]
- 45.Singh AK, Gaur P, Das SN. Natural killer T cell anergy, co-stimulatory molecules and immunotherapeutic interventions. Hum Immunol. 2014;75: 250–60. [DOI] [PubMed] [Google Scholar]
- 46.Iyoda T, Ushida M, Kimura Y, Minamino K, Hayuka A, Yokohata S, et al. Invariant NKT cell anergy is induced by a strong TCR-mediated signal plus co-stimulation. Int Immunol. 2010;22: 905–13. [DOI] [PubMed] [Google Scholar]
- 47.Ando T, Ito H, Ohtaki H, Kanbe A, Hirata A, Hara A, et al. Role of invariant NKT cells in lipopolysaccharide-induced lethal shock during encephalomyocarditis virus infection. Immunobiology. 2017;222: 350–7. [DOI] [PubMed] [Google Scholar]
- 48.Ito H, Koide N, Hassan F, Islam S, Tumurkhuu G, Mori I, et al. Lethal endotoxic shock using alpha-galactosylceramide sensitization as a new experimental model of septic shock. Lab Invest. 2006;86: 254–61. [DOI] [PubMed] [Google Scholar]
- 49.Das R, Guan P, Wiener SJ, Patel NP, Gohl TG, Evans E, et al. Enhancing the antitumor functions of invariant natural killer T cells using a soluble CD1d-CD19 fusion protein. Blood Adv. 2019;3: 813–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ellerman D Bispecific T-cell engagers: Towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods. 2019;154: 102–17. [DOI] [PubMed] [Google Scholar]
- 51.Molling JW, Kolgen W, van der Vliet HJ, Boomsma MF, Kruizenga H, Smorenburg CH, et al. Peripheral blood IFN-gamma-secreting Valpha24+Vbeta11+ NKT cell numbers are decreased in cancer patients independent of tumor type or tumor load. Int J Cancer. 2005;116: 87–93. [DOI] [PubMed] [Google Scholar]
- 52.Bollino D, Webb TJ. Chimeric antigen receptor-engineered natural killer and natural killer T cells for cancer immunotherapy. Transl Res. 2017;187: 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.