

Abstract
RNA helicase A (RHA) as a member of DExH‐box subgroup of helicase superfamily II, participates in diverse biological processes involved in RNA metabolism in organisms, and these RNA‐mediated biological processes rely on RNA structure conversion. However, how RHA regulate the RNA structure conversion was still unknown. In order to unveil the mechanism of RNA structure conversion mediated by RHA, single molecule fluorescence resonance energy transfer was adopted to in our assay, and substrates RNA were from internal ribosome entry site of foot‐and‐mouth disease virus genome. We first found that the RNA structure conversion by RHA against thermodynamic equilibrium in vitro, and the process of dsRNA YZ converted to dsRNA XY through a tripartite intermediate state. In addition, the rate of the RNA structure conversion and the distribution of dsRNA YZ and XY were affected by ATP concentrations. Our study provides real‐time insight into ATP‐dependent RHA‐assisted RNA structure conversion at the single molecule level, the mechanism displayed by RHA may help in understand how RHA contributes to many biological functions, and the basic mechanistic features illustrated in our work also underlay more complex protein‐assisted RNA structure conversions.
Keywords: RNA helicase A, RNA structure conversion, single molecule fluorescence resonance energy transfer, thermodynamic equilibrium
1. INTRODUCTION
In all organisms, RNA mediates essential cellular processes, such as, transcription, 1 pre‐mRNA splicing, 2 , 3 ribosome biogenesis, 4 RNA transportation, 5 , 6 and RNA‐induced silencing complex formation, 7 , 8 , 9 which all relying on RNA structure conversion in accurate and timely fashions. 10 , 11 It is recently reported that some helicases promote RNA structure conversion. 12 , 13 , 14 , 15 , 16 , 17 The Neurospora crassa CYT‐19 mediates ATP‐dependent unfolding of both native and misfolding RNAs, and changes RNA distributions from equilibrium to kinetic control. 18 The Saccharomyces cerevisiae Ded1p disassembles and rearranges RNA structures, and facilitates the formation of less stable RNA structures from more stable ones against thermodynamic equilibrium. 15 Though it is known that helicases promote RNA structure conversion, still relatively little is understood of the dynamic mechanism of RNA structure conversion by RHA.
In order to unveil the mechanism of RNA structure conversion mediated by RHA, smFRET is mainly adopted to investigate how RHA coordinates RNA structure conversion. However, relevant reactions are needed to be quantitatively analyzed in the study of RNA structure conversions usually occur within large RNA‐protein complexes, which is difficult to control in vitro, but the investigation of protein and RNA folding indicates defined and tractable model systems can be used for the elucidation of mechanism of complex conformational rearrangements. 19 , 20
The Homo sapiens RHA, also named as DHX9, is a RNA helicase from DExH‐box family. 21 RHA is originally purified and characterized in Hela cells, and found to be located in nucleus and cytoplasm and shuttled back and forth between these two positions. 22 , 23 , 24 RHA consisted seven domains, 25 , 26 and it has been reported to unwind duplex RNA in a 3′ to 5′ direction. 27 Moreover, RHA also shows activity to unwind intra‐molecular triplex, RNA containing R‐loops and G‐quadruplexes to maintain the genomic stability. 28 , 29 , 30 RHA is shown to be an attractive ''prey'' for viruses, as there is ample evidence that it is required for the replication of several different viruses, such as, foot‐and‐mouth disease virus (FMDV), 31 human immunodeficiency virus type 1 (HIV‐I), 1 , 32 , 33 hepatitis C virus (HCV), 34 , 35 and influenza virus. 36
FMDV is a typical representative of small RNA virus family, which causes acute contact infectious diseases of cloven‐hoofed animals and causes great economic losses to the aquaculture industry. In FMDV genome, the 3′ UTR contains two stem–loops, and the S fragment interacts with each of the stem–loops. In addition, the internal ribosome entry site (IRES) also interacts with the 3′ UTR and the 3′ UTR stimulates IRES activity. S fragment and IRES are two different elements present at the 5′ end, interacted with the 3′ UTR in a dose‐dependent manner. 37 RHA plays an essential role in the replication of FMDV through ribonucleoprotein formation and interacts with the S fragment at the 5′ end of the genome. 31 In other virus such as HIV, RHA increases transcription of the HIV genome through specific binding to stem‐loop structures known as transcriptional activating regions. 32 , 38 Thus, we speculated RHA may interaction with IRES by the role of bridge with S fragment, and IRES is probably a substrate for RHA.
The IRES of FMDV was comprised of five major domains, and the sequences of strands X and Y in our work were from the domain 4 of FMDV IRES, and the strand Z was a complementary strand of strand Y, X, and Y existed as double‐stranded RNA structure in the secondary structure of IRES. 39 , 40 During viral replication of FMDV, the genomic RNA not only directs the synthesis of the viral polyprotein but also serves as template for RNA synthesis. 31 Genome copying occurs via a complementary negative‐sense RNA template and the formation of two replicating positive‐sense strands. Thus, complementary strands to Y generated in viral gene replication, indicated the strand Z existed in cells and adopted a dsRNA structure with Y.
Moreover, RHA has interaction with IRES, which can affect virus replication by affecting translation through the association of many protein factors. Thus, the RNA sequences of substrates used in our assays are selected from IRES. These three RNA strands can constitute less stable (XY) and stable (YZ) duplexes. Using smFRET, we not only probe the dynamic RNA structure conversion in real‐time to determine the pathway, but also demonstrate the key role of ATP hydrolysis in RNA structure conversion against thermodynamic equilibrium.
2. RESULTS
2.1. RNA substrate design
In order to investigate the RHA‐assisted RNA structure conversion, a simple system was designed with three ssRNA: ssRNA Y could form dsRNA XY with ssRNA X, or form dsRNA YZ with ssRNA Z as shown in Figure 1(a). In order to use the model to study the RHA‐assisted RNA structure conversion, the RNA substrate design should satisfy two requirements. One is that both dsRNA XY and YZ could be generated via annealing assay in vitro, and each substrate should have a partial duplex configuration with a ssRNA tail for efficient unwinding by RHA with ATP participation. The other is that dsRNA XY should have lower thermodynamic stability compared to dsRNA YZ.
FIGURE 1.
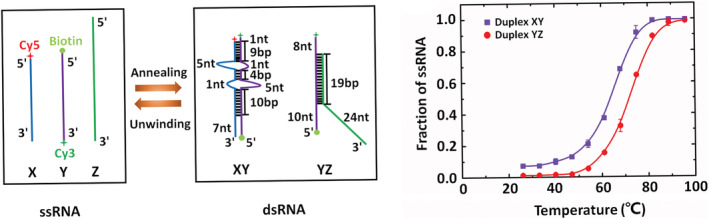
RNA substrate design. (a) Scheme of the RNA substrates. dsRNA XY can be formed from ssRNA X (blue) and Y (purple), and dsRNA YZ can be formed from ssRNA Y (purple) and Z (green) both via annealing assay in vitro; besides, dsRNA XY and YZ can be unwound by RHA. Numbers represent formed basepairs (bp) and unpaired nucleotides (nt). The 5′ end of ssRNA X was labeled with Cy5 (red asterisk) as an acceptor, and the 3′ end of ssRNA Y was labeled with Cy3 (green asterisk) as a donor. (b) Thermal melting curves of dsRNA XY and YZ. The fitted curves represent the smooth trends, and the melting temperature of dsRNA YZ is 72 ± 0.5°C, while that of dsRNA XY is 64 ± 0.5°C, revealing that dsRNA XY has a lower thermodynamic stability than dsRNA YZ
RHA can catalytically unwind a partial dsRNA with a single‐stranded tail at its 3′ end in a 3′ to 5′ direction, and the length of the 3′ tail governed the rate of duplex displacement. 27 In our experiments, dsRNA with different lengths of 3′ tail were used in unwinding assay, and the results indicated RHA unwound the dsRNA with a 3′ tail no less than 6 nt, as shown in Figure S3. Thus, the substrates used in RNA structure conversion reaction were designed to have long enough 3′ tails, as shown in Figure 1. The 3′ tail is required for the unwinding activity of RHA, and RHA has been reported to participate in diverse cellular processes involving RNA, including transcription, 41 translation, 42 RNA interference pathway, 7 innate immune response 43 and replication of a number of viruses. 31 , 35 , 36 , 44
In our design in Figure 1(a), dsRNA YZ had 19 complementary base‐pairs, and dsRNA XY had two internal loops, both had partial duplex structures with ssRNA tails. Moreover, the thermodynamic stability of both dsRNA XY and YZ was evaluated according to the melting temperatures (T m) extracted from the fitted thermal melting curves shown in Figure 1(b). The melting temperature of dsRNA YZ was 72 ± 0.5°C, which was higher than that of dsRNA XY as 64 ± 0.5°C, revealing that dsRNA YZ had higher thermodynamic stability than dsRNA XY. The RNA substrate design could satisfy both two requirements, therefore, it was next used for studying the RHA‐assisted RNA structure conversion.
2.2. Detecting RHA‐assisted RNA structure conversion using native PAGE
Native PAGE experiments were first implemented to detect the RHA‐assisted RNA structure conversion. The substrates consisted of Cy3‐labeled dsRNA YZ and Cy5‐labeled ssRNA X, and the PAGE experiments were performed in three conditions: RNA only, RNA with 100 nM RHA, and RNA with 100 nM RHA and various concentrations of ATP. The Cy5 fluorescence signal was collected for RNA structure conversion analysis. The size markers including dsRNA XY and ssRNA X, shown in left of Figure 2(a), were generated using the incomplete RNA annealing assay: ssRNA X was mixed with complementary ssRNA Y in the annealing buffer and heated at 95°C for 5 min, and slowly cooled down to the room temperature for 7 h. Moreover, in the RNA structure conversion assay, 4 nM pre‐formed dsRNA YZ and 8 nM ssRNA X were incubated with/without RHA and ATP at 26°C for different reaction time, the reaction products were equally resolved in native PAGE, and the fluorescence bands were quantified using fluorescence gel imaging scanner. Note that all the PAGE bands in Figure 2(a) were obtained in the same batch of experiments, and totally three batches of experiments were implemented.
FIGURE 2.
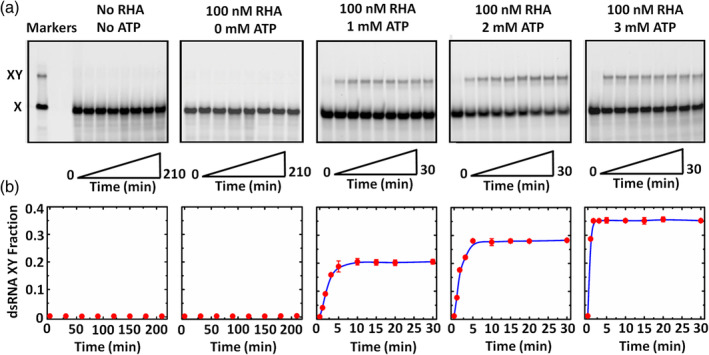
Detecting RHA‐assisted RNA structure conversion using PAGE. (a) PAGE results in different conditions. The reactions were performed at 26°C with 4 nM pre‐formed dsRNA YZ and 8 nM ssRNA X described in Materials and Methods. The reaction periods in the conditions without RHA or ATP and with RHA but without ATP were both from 0 to 210 min; and those in the conditions with RHA and ATP were all from 0 to 30 min. The reactions were stopped at indicated time, and the products were resolved in 12% native PAGE, and in addition, dsRNA XY and ssRNA X were detected using a fluorescence gel imaging scanner. Moreover, the left markers in (a) are the size markers of dsRNA XY and ssRNA X. (b) Relation between the dsRNA XY fraction and the reaction time extracted from (a). The dots represent the measured data from three repeated experiments, and the error bars represent the standard deviations. The curves were fitted according to the homogenous first order rate law
In the condition without RHA or ATP, there was almost no RNA structure conversion with a rather long reaction time up to 210 min, since the fluorescence signal of dsRNA XY could not be detected. When RHA was added but without introducing ATP, still no RNA structure conversion was observed in 210 min. By introducing both RHA and ATP, there was obvious RNA structure conversion since the fluorescence signal of dsRNA XY could be detected. Since RHA could not unwind dsRNA with nonhydrolyzabel ATP analogs (ADPNP, ADP‐BeFx, and ADP‐AlF4, data were not shown here), ATP was used in our study. Additionally, the RNA structure conversion speed was estimated according to the dsRNA XY fraction quantification in Figure 2(b), and the dsRNA XY fraction (XY%) is defined as XY% = XY/(X + XY), in which X and XY are their corresponding fluorescence intensities. With ATP concentrations of 1, 2, and 3 mM, the corresponding XY% were 21.07%, 28.45%, and 35.69%, and the time reaching steady states was ~5, ~3, and ~ 2 min obtained via fitting according to the homogenous first order rate law. The results illustrated that higher ATP concentration promoted the RHA‐assisted RNA structure conversion with higher conversion rate and faster conversion speed.
The above PAGE experiments not only verified that the RNA structure conversion relied on both RHA and ATP, but also provided details on the conversion speeds and rates; however, it still could hardly reflect the temporal dynamic process during RNA structure conversion. In order to study its dynamic process, smFRET was next adopted to deeply understand the RHA‐assisted RNA structure conversion.
2.3. Design and verification on smFRET assay
Before smFRET detection, the FRET signal in the RHA‐assisted RNA structure conversion was first verified using ensemble FRET as shown in Figure 3(a), in which, the 5′ end of ssRNA X was labelled with Cy5 as an acceptor, while the 3′ end of ssRNA Y in dsRNA YZ was labelled with Cy3 as a donor. Therefore, the FRET signal should occur when dsRNA YZ was converted to dsRNA XY, since the distance between Cy5 and Cy3 was less than 80 Å. In this verification, 100 nM Cy3‐labeled dsRNA YZ and 40 nM Cy5‐labeled ssRNA X were incubated at 26°C for 10 min in 220 μl reaction buffer. Different reaction systems, RNA only, RNA with 100 nM RHA, RNA with 100 nM RHA, and 1/2/3 mM ATP, were measured, respectively. According to the fluorescence intensities measured by the microplate reader shown in Figure 3(b), when Cy3‐labeled dsRNA YZ was incubated with Cy5‐labeled ssRNA X, no FRET signal was observed, revealing that no RNA structure conversion occurred; and when only introducing RHA, there was still no obvious RNA structure conversion; but only when both RHA and ATP were introduced, distinct FRET peaks at ~675 nm with the full width at half maximum (FWHM) of ~30 nm proved that RNA structure conversion occurred. Moreover, the FRET peak intensity increased by introducing more ATP. The ensemble FRET results not only proved that smFRET could be used for dynamically probing the RNA structure conversion, but also were consistent with the PAGE results that the RNA structure conversion only occurred in RHA assistance with the involvement of ATP.
FIGURE 3.
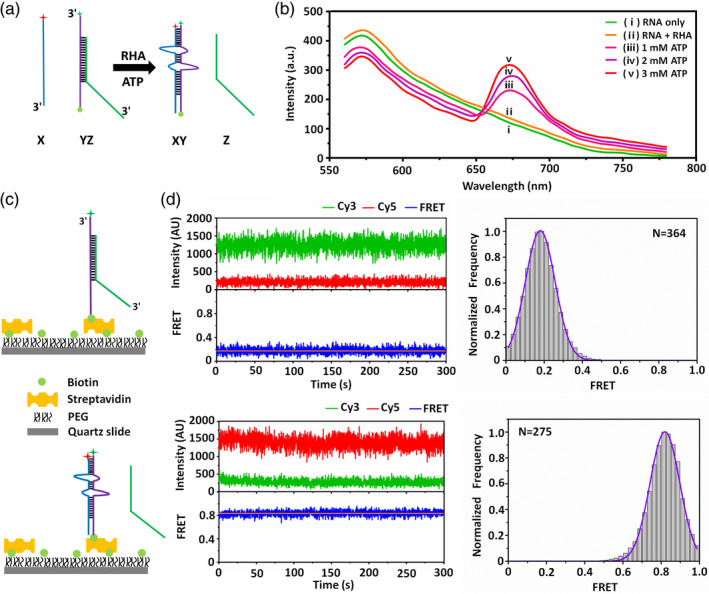
Design and verification on smFRET. (a) Strategy of the ensemble FRET assay. dsRNA YZ was converted to dsRNA XY, and the FRET signal was generated due to a distance sufficiently close between Cy3 and Cy5. (b) Ensemble FRET spectra in the presence of (i) RNA, (ii) RNA with RHA, RNA with RHA and different concentrations of ATP as (iii) 1, (iv) 2, and (v) 3 mM. Pre‐formed dsRNA YZ was incubated with ssRNA X at 26°C for 10 min. ssRNA Y and X were labeled with Cy3 (as donor) and Cy5 (as acceptor), respectively. The fluorescence signal with the peak at 575 nm was the FRET signal. (c) Strategy of the smFRET assay. dsRNA YZ was immobilized on a PEG‐coated sample chamber surface via the connection between the biotin and the streptavidin. Biotinylated ssRNA Y was labeled with Cy3 (green asterisk). (d) Representative time traces of dsRNA XY and YZ, as well as the statistical smFRET efficiency histograms. The smFRET efficiency of dsRNA XY is ~0.83 statistically analyzed from 275 molecules; and that of dsRNA YZ is ~0.17 statistically analyzed from 364 molecules
Figure 3(c) shows the smFRET design on the RHA‐assisted RNA structure conversion. Labelled with Cy3 at the 3′ end and biotin at the 5′ end of ssRNA Y, dsRNA YZ was immobilized on the PEG‐coated sample chamber surfaces through the connection between biotin and streptavidin. The adopted PEG‐coated sample chamber could significantly reduce the non‐specific binding of proteins to the sample chamber surfaces. 45 , 46 , 47 , 48 , 49 , 50 , 51 With the introduction of Cy5‐labeled ssRNA X accompanied with RHA and ATP, the RNA structure conversion occurred as dsRNA YZ started to convert to dsRNA XY, generating the smFRET signal. Via collecting temporal fluorescence signals of both donor (Cy3) and acceptor (Cy5), the dynamics during the RHA‐assisted RNA structure conversion could be probed. Before smFRET detection on the RHA‐assisted RNA structure conversion, the smFRET efficiencies of dsRNA XY and YZ were previously measured: Figure 3(d) displays the representative time traces of dsRNA XY and YZ with the temporal resolution of 100 ms, as well as the statistical data collected from multiple targets. The smFRET efficiency reached ~0.83 corresponding to the dsRNA XY condition. While in the dsRNA YZ condition, the smFRET efficiency should be 0 since dsRNA YZ just had the donor fluorophore of Cy3. However, a rather low smFRET efficiency as ~0.17 still appeared mainly due to the fluorescence leakage of TIRFM system. As samples never comprised 100% photoactive donor and acceptor dyes, the donor‐only and acceptor‐only populations were selected from the measurements and used to determine the leakage and direct excitation, uncorrected smFRET efficiency of ~0.15 due to leakage often occurred, 52 , 53 which was close to that in our work. According to the above verifications, it is proved that smFRET could be used to probe the dynamic RNA structure conversion.
2.4. Detecting RHA‐assisted RNA structure conversion using smFRET
The dynamic RNA structure conversion was first probed using smFRET in two conditions without RHA or ATP and with 100 nM RHA but without ATP, and Figure 4(a),(b) show their representative time traces (more time traces are shown in Figure S5) and smFRET efficiency histograms, respectively. In these conditions, there was only one state corresponding to low smFRET efficiency of ~0.17, proving that there was no RNA structure conversion.
FIGURE 4.
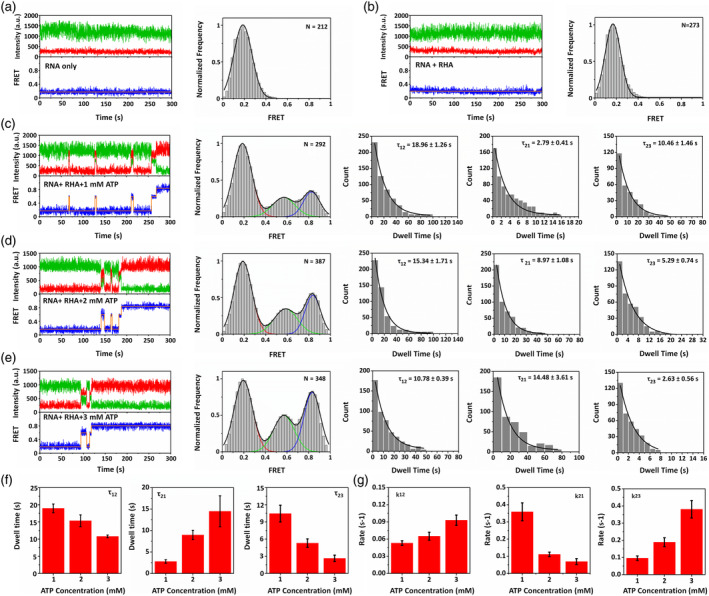
Detecting RNA‐assisted RNA structure conversion from dsRNA YZ to XY through an intermediate state using smFRET. Representative time trace and smFRET efficiency histogram in the condition (a) without RHA or ATP and (b) with 100 nM RHA but without ATP. Representative time trace, smFRET efficiency histogram, dwell time of τ12, τ21, and τ23 in the conditions with 100 nM RHA and (c) 1, (d) 2, and (e) 3 mM ATP. Blue curves in dwell time figures were fitted according to the single exponential decay function. (f) Dwell time of τ12, τ21, and τ23 and (g) transition rates of k12, k21, and k23 corresponding to ATP of 1, 2, and 3 mM
Figure 4(c), (d), (e) reveal the results corresponding to the conditions with both 100 nM RHA and ATP, but different ATP concentrations as 1, 2, and 3 mM, respectively. According to their representative time traces (more time traces are shown in Figure S4) and smFRET efficiency histograms, there were obvious three states: low smFRET efficiency of ~0.17 as State 1 indicating dsRNA YZ, high smFRET efficiency of ~0.83 as State 3 indicating dsRNA XY, and middle smFRET efficiency of ~0.58 as State 2 indicating an intermediate structure. The smFRET results show that the RNA structure conversion experienced an intermediate state from dsRNA YZ to dsRNA XY, which can hardly be unveiled using ensemble FRET or PAGE experiments.
Moreover, in order to acquire the kinetics of RNA structure conversion, the dwell time (τ) in different state transitions was analyzed. According to the time traces, dsRNA YZ converted to dsRNA XY through the intermediate state during RNA structure conversion. Since dsRNA XY was a relatively stable structure, almost no dsRNA XY transformed to other states, thus τ32 and τ31 were not considered here. Therefore, only τ12, τ21, and τ23 were analyzed in Figures 4(c)‐(e), representing the state transition from dsRNA YZ to intermediate structure, that from intermediate structure to dsRNA YZ, and that from intermediate structure to dsRNA XY, respectively. When the introduced ATP concentration was 1 mM, dsRNA YZ converted to dsRNA XY through the intermediate structure, and τ12, τ21, and τ23 are 18.96 ± 1.26 s, 2.79 ± 0.41 s, and 10.46 ± 1.46 s, respectively. When the introduced ATP concentration was 2 mM, dsRNA YZ converted to dsRNA XY through the intermediate structure with τ12, τ21 and τ23 of 15.34 ± 1.71 s, 8.97 ± 1.08 s, and 5.29 ± 0.74 s, respectively. When the introduced ATP concentration was 3 mM, dsRNA YZ converted to dsRNA XY through the intermediate structure with τ12, τ21, and τ23 of 10.78 ± 0.39 s, 14.48 ± 3.61 s, and 2.63 ± 0.56 s, respectively.
According to Figures 4(a)‐(e) as well as the dwell time analysis in Figure 4(f), RNA structure conversion only occurred with both RHA and ATP, and with higher introduced ATP concentrations, k12 and k23 obviously increased while only k21 decreased as shown in Figure 4(g), revealing that higher ATP concentration promoted the transition from stable dsRNA YZ to less stable dsRNA XY, which was consistent with the results from native PAGE.
3. DISCUSSION
RNA structure conversion is also known as conformation switch, and it always contains functional RNA folding, which can dynamically regulate many biological processes. 19 , 54 The RNA structures converting to each other are usually mutually exclusive, and one RNA structure is disrupted for the formation of another one. DEAD‐box protein Ded1 has been reported to promote loops generation in two mutually exclusive RNA structure conversions using a tripartite model, and Ded1‐assisted RNA structure conversions were found for the first time in vitro. 15 In addition, the DEAD‐box protein CYT‐19, as a general RNA chaperone, allows RNA refold and modulate the distribution of native and misfolded RNAs from equilibrium to kinetic control. The functions of RNAs depending on forming multiple conformations may rely on proteins to increase the populations of less stable conformations. 18 Proteins have functions in RNA structure rearrangement and RNA‐protein interaction, and these functions are the premise of regulating RNA structure conversion, as well as connect with the abilities of proteins to hydrolyze ATP and unwind RNA duplexes. 13 , 14 , 15 Moreover, several helicases have been shown recently not only to unwind RNA duplexes, but also to facilitate RNA duplexes formation. 16 , 55 However, whether the proteins of DExH‐box family can promote RNA structure conversions is still not known.
RHA, as a typical member of the DExH‐box family, is different from Ded1 in unwinding patterns. Ded1 exists helicase activity in ''local strand separation'' mechanism, it promotes the unwinding initiation by directly binding to the double‐stranded region of the substrate via opening a limited number of base pairs. 56 In contrast, RHA exists helicase activity in ''directional translocation'' mechanism, it utilizes ATP to unwind duplexes through translocating rapidly, possessively, and directionally from 3′ to 5′. 27 The mechanism of Ded1‐assisted RNA structure conversion has been investigated in recent years, but whether and how RHA modulate RNA structure conversion is still needed to be investigated.
In our study, we used a model to investigate the real‐time RNA structure conversion by RHA at the single molecule level, the RNA sequences of substrates used in our assays were selected from the IRES of FMDV genome, and we speculated RHA remodelled IRES‐related structures and played a coordinating role in IRES‐mediated protein translation. Despite its simplicity, the model could recapitulate essential aspects of much more complex physiological structure conversions, and the basic mechanistic features elucidated in our work also underlay more complex protein‐assisted RNA structure conversions.
Furthermore, we revealed the distinct thermodynamic stabilities of dsRNA YZ and XY by the determination of T m value, and the results illustrated that dsRNA YZ was more stable than dsRNA XY. As we known, the distribution of different RNA structures might be affected by the thermodynamic stability and structural characteristics of structures in protein‐assisted RNA structure conversion. Nevertheless, our study indicated the distribution of dsRNA XY and YZ in RHA‐assisted RNA structure conversion against thermodynamic equilibrium, and this phenomenon was similar with Ded1 and CYT‐19. 15 , 18
We found that dsRNA YZ converted to dsRNA XY through an intermediate state in an ATP‐dependent manner, and we speculated the intermediate structure was a tripartite specie (Figure 5). The acceleration of strand annealing by a protein might rely on the stabilization of inherently unstable tripartite species that formed during the rate limiting steps of duplex formation. 57 , 58 , 59 However, the structure conversion might also proceed through other tripartite complexes but not detected in our measurements, and how exactly the transition between tripartite intermediate structure and final RNA structure should be further investigated.
FIGURE 5.
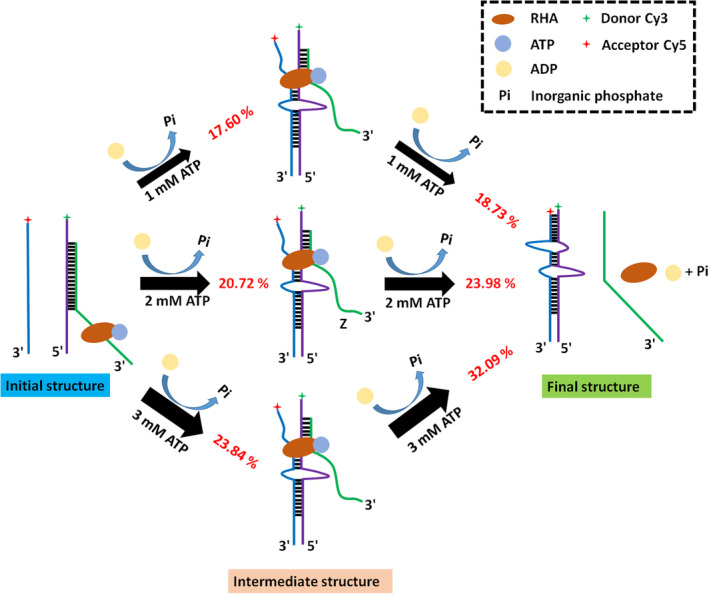
Scheme of the RHA‐assisted RNA structure conversion in an ATP‐dependent manner. The initial structure converted into final structure through an intermediate structure. The thicker arrows indicate the faster speeds of structure conversion, the thinner arrow indicate the slower speeds of structure conversion. And the red percentages are the proportion of the immediate structure and dsRNA XY generated
The annealing activity of RHA was necessary for the RNA structure conversion in our study, and other recent studies also found that RHA had annealing activity. For example, in the investigation of the interaction between RHA and oncogenic transcription factor EWS‐FLI1, RHA could facilitate the annealing of complementary single strand RNA to yield double strand RNA in a time‐dependent fashion. 17 In addition, single molecule fluorescence assays were also used to find that the unwinding process of dsRNA by RHA consisted of discrete sub‐steps including binding, activation, unwinding, stalling and reactivation. The unwinding process was repeated by a single RHA molecule without dissociation, and repetitive RNA unwinding by RHA facilitates RNA annealing. 60 The annealing activity of RHA was also related to virus replication, and RHA promoted the annealing of HIV‐1 RNA and tRNALys3, which is the primer of the HIV‐1 viral RNA. 61 , 62
Here, we found RHA promoted the annealing between strand X and Y to form dsRNA XY in the RNA structure conversion, and the formation of dsRNA XY relied on the unwinding of pre‐formed dsRNA YZ, the fluctuations of non‐steady state of RNA rearrangement, the generation and stabilization of dsRNA XY. This is the first time to observe the RHA‐assisted RNA structure conversion against thermodynamic equilibrium at the single molecule level in vitro, and the higher concentrations of ATP promoted RNA structure conversion in faster speed. The mechanism of RHA‐assisted RNA structure conversion is significant to understand the functions of RHA in many biological processes, especially virus replication, so it also lays the foundation for the further research of antiviral drugs.
4. MATERIALS AND METHODS
4.1. Protein expression
The bac‐to‐bac baculovirus expression system was used for recombinant RHA production. Firstly, SF9 cells (Invitrogen) were cultured in the Grace insect culture medium (Gibco) containing 10% fetal bovine serum (Gibco) at 27°C using a constant temperature and humidity incubator (Thermo Fisher). Then, in protein expression, the RHA gene (Thermo Fisher) was cloned into the pFastBac dual donor plasmid, and the recombinant pFastBac‐RHA‐His vector containing a full‐length RHA gene sequence and a 6 × His tag was transformed into the MAX Efficiency DH10Bac competent E. coli (Takara, Japan) to generate a recombinant bacmid. Next, the recombinant bacmid DNA was transfected into the SF9 insect cells to generate the recombinant baculovirus using Cellfectin II reagent (Invitrogen) when the cell density was ~2.5 × 106 cells/ml and the cell viability was higher than 95%. Finally, the recombinant protein was expressed by amplifying the baculovirus in the infected SF9 insect cells, and the SF9 cells were harvested 72 h post‐infection.
4.2. Protein purification
Firstly, the harvested cells were washed with the phosphate buffer saline (10 mM NaH2PO4, pH 7.4, 140 mM NaCl, 3 mM KCl), homogenized in 2 ml hypotonic Buffer A (25 mM HEPES‐KOH pH 7.8, 10 mM NaCl, 10 mM Na2S2O5, 5 mM MgCl2, 7 mM β‐mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride and 0.1% (v/v) Aprotinin (Sigma)), and incubated with 2 ml high‐salt Buffer B (25 mM HEPES‐KOH pH 7.8, 1 M NaCl, 10 mM Na2S2O5, 5 mM MgCl2, 1.6 mM imidazole, 7 mM β‐mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride, 0.1% (v/v) Aprotinin, and 30% glycerol) for 30 min. Then, the ultrasonic broken cells were centrifuged at 14,000 rpm for 20 min using centrifuge (Eppendorf, Germany) to obtain the cell supernatant, and the supernatant was incubated with Ni2+‐NTA‐agarose (Qiagen, Germany) using a rotating apparatus (Thermo Fisher) for 2 h. The recombinant protein was purified by a series of wash steps with 0.8, 5, 20, 40, 80, 300 mM imidazole in Buffer C (25 mM HEPES‐KOH pH 7.8, 0.5 M NaCl, 10 mM Na2S2O5, 5 mM MgCl2, 7 mM β‐mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride, 0.1% (v/v) Aprotinin, and 15% glycerol). Afterwards, the protein was mainly eluted with 80 and 300 mM imidazole in Buffer C and centrifuged by an ultrafiltration centrifugal tube (Millipore) with Buffer D (25 mM HEPES‐KOH pH 7.8, 0.1 M NaCl, 10% glycerol and 1 mM DTT). Next, the protein was diluted by 10 times with Buffer E (12.5 mM HEPES‐KOH pH 7.8, 5% glycerol) and incubated with Capto DEAE Sepharose (GE Healthcare) using a rotating apparatus (Thermo Fisher) for 2 h. The further purification was implemented by a series of wash steps with 0.05, 0.15, 0.25, 0.35, 0.45, and 0.55 M NaCl in Buffer E, and the protein was eluted with 0.25, 0.35, and 0.45 M NaCl in Buffer E. All the above steps were implemented at 4°C. The molecular weight of RHA was 140 KDa, and the protein purity was assessed by SDS‐PAGE and Coomassie (Figure S1). After the protein was centrifuged using ultrafiltration centrifugal tube (Millipore), the protein concentration was determined by Bradford Coomassie protein assay kit (Sigma) and the protein was aliquot and stored at −80°C. Purified RHA was verified to be active before experiments (Figure S2).
4.3. Substrate preparation
Three kinds of single‐stranded RNA (ssRNA, Dharmacon) were used: the sequences of ssRNA X and Y were from the IRES of FMDV, and ssRNA Z was a complementary strand of strand Y. They constituted to two distinct RNA conformations as double‐stranded RNA (dsRNA) XY and YZ. Fluorophores Cy3 (as donor) and Cy5 (as acceptor) were attached to the 3′ end of ssRNA Y and the 5′ end of ssRNA X, respectively. Furthermore, biotin was attached to the 5′ end of ssRNA Y in order to immobilize it on the sample chamber in smFRET assay. Efficiencies of all the labelling reactions were proved higher than 80% according to RNA extinction coefficients and fluorophores measured by UV–vis spectroscopy (Shimadzu, Japan). The purification of RNA was implemented by native polyacrylamide gel electrophoresis (PAGE). The sequences of ssRNA X, Y, and Z are listed.
ssRNA X: 5′‐Cy5‐UCAGGCGUAACUCAAAGCCCCUUCUCGGACAAGCAC‐3′.
ssRNA Y: 5′‐biotin‐CUCGGGAUCCGAGAAGGAAAAAGCUUCUACGCCUGAA‐Cy3‐3′.
ssRNA Z: 5′‐UAGAAGCUUUUUCCUUCUCUCACAAUAACAGCAUCAAUGACAU‐3′.
Additionally, to generate dsRNA YZ, ssRNA Y was first mixed with complementary ssRNA Z in the annealing buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl) without ATP, then heated at 95°C for 5 min, and finally cooled down slowly to the room temperature for 7 h. The annealing procedures of ssRNA X and Y for dsRNA XY generation were the same as those of ssRNA Y and Z.
4.4. Thermal melting curve determination
Melting temperature (T m) of dsRNA was measured in the reaction buffer containing 30 mM Tris–HCl, 0.1 mg/ml BSA, 1% glycerol, 2 mM DTT, 1 unit/μl RNase inhibitor, and 3 mM MgCl2. Firstly, 16 nM dsRNA (XY or YZ) was incubated from 26 to 96°C in a temperature calibrated PCR machine (Bio‐Rad) for 5 min. Then, the reaction was transferred to the stop buffer containing 50 mM EDTA•Na2, 1% SDS, and 10% glycerol in ice. Next, 10 μl aliquot was added to 12% native PAGE in order to separate ssRNA and dsRNA. Finally, the unwound fraction of dsRNA could be determined to generate the thermal melting curve.
4.5. RNA structure conversion detection using native PAGE
The RNA structure conversion contains the unwinding reaction of dsRNA and the annealing reaction of ssRNA. PAGE based RNA structure conversion detection was performed in 110 μl reaction buffer (30 mM Tris–HCl, 0.1 mg/ml BSA, 1% glycerol, 2 mM DTT, 1 unit/μl RNase inhibitor and 3 mM MgCl2). Before reaction, pre‐formed 4 nM dsRNA YZ was pre‐incubated with 8 nM ssRNA X at 26°C for 5 min. The reaction was initiated by adding 100 nM RHA and a certain concentration of ATP. Here, the concentrations of ATP used in native PAGE were 0, 1, 2, and 3 mM. Specified time points were taken by adding 10 μl stop buffer containing 50 mM EDTA•Na2, 1% SDS, and 10% glycerol. Products as dsRNA and ssRNA were resolved using 12% native PAGE, and the gels were visualized via fluorescence gel imaging (Typhoon, GE Healthcare) according to the Cy5 fluorescence intensity. The band intensities of the reaction products were extracted and the data points were fitted according to the homogenous first order rate law.
4.6. RNA structure conversion detection using ensemble FRET
100 nM Cy3‐labeled dsRNA YZ and 40 nM Cy5‐labeled ssRNA X were incubated at 26°C for 10 min in 220 μl reaction buffer (30 mM Tris–HCl, 0.1 mg/ml BSA, 1% glycerol, 2 mM DTT, 1 unit/μl RNase inhibitor and 3 mM MgCl2). Three different reaction systems, RNA only, RNA with 100 nM RHA, and RNA with 100 nM RHA and a certain concentration of ATP, were detected using a microplate spectrophotometer (Tecan, Switzerland). Cy3 was excited at a wavelength of 545 nm and the fluorescence emission was measured from 560 to 780 nm. The concentrations of ATP used in the assays based on ensemble FRET were 0, 1, 2, and 3 mM.
4.7. RNA structure conversion detection using smFRET
The kinetics of RNA structure conversion was probed by smFRET using total internal reflection fluorescence microscopy (TIRFM) based on a commercial inverted fluorescence microscope (Olympus IX81, Japan). In the TIRFM system, a semiconductor laser (Coherent) with the wavelength of 532 nm and the power of 24 mW was used for excitation, a 100× oil immersion micro‐objective (Olympus, Japan) was adopted for fluorescence signal collection, the donor and acceptor fluorescence signals were split by a dual‐channel imaging system (DV2, Photometrics), and an EMCCD camera (Evolve 512, Photometrics) was used for simultaneously recording fluorescence images in both the donor and acceptor channels. In the measurements, the exposure time of the EMCCD was fixed as 100 ms with the gain of 300, and the fluorescence images were recorded at a rate of 10 fps. As shown in Figure S4, our homemade sample adding device consisted of a sample stage, a sample chamber, a sample injector and a poly‐tetrafluoroethylene tube. Due to the substrate dsRNA YZ was immobilized on the sample chamber in advance, and fluorescence images could be collected when adding samples using the sample adding device, we could observe the RNA structure conversion in real time. After fluorescence image collection, smFRET data were analyzed by vbFRET. 63
In sample preparation, 50 pM Cy3‐labeled dsRNA YZ was immobilized on the surfaces of sample chambers coated with a mixture of 99% mPEG (m‐PEG‐5000, Laysan Bio, Inc.) and 1% biotin‐PEG (biotin‐PEG‐5000, Laysan Bio. Inc.). The biotinylated samples were immobilized through streptavidin (Pierce) to the biotinylated PEG, and the unbound samples were removed by washing with T50 buffer (10 mM Tris–HCl pH = 8.0, 50 mM NaCl). Reactions were performed at a constant temperature of 26°C in the reaction buffer (100 nM RHA, 30 mM Tris–HCl, 0.1 mg/ml BSA, 1% glycerol, 2 mM DTT, 1 unit/μl RNase inhibitor, 3 mM MgCl2, 100 pM ssRNA X and a certain concentration of ATP). The concentrations of ATP used in the smFRET to investigate RNA structure conversion were 0, 1, 2, and 3 mM. Besides, 5% (v/v) D‐glucose (Sigma) and 1 mM 6‐hydroxy‐2,5,7,8‐tetramethylchroman‐2‐carboxylicacid (Trolox) were also added together with an oxygen scavenging system consisting of 0.04 mg/ml catalase (Sigma) and 1 mg/ml glucose oxidase (Sigma) to reduce the fluorescence quenching.
CONFLICT OF INTEREST
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AUTHOR CONTRIBUTIONS
Chengcheng Wu: Investigation; visualization; writing‐original draft. Yanke Shan: Investigation; validation; visualization; writing‐original draft. Shouyu Wang: Software; supervision; writing‐review & editing. Fei Liu: Conceptualization; funding acquisition; supervision; writing‐review & editing.
Supporting information
Figure S1 Purification of RHA. (A) RHA was purified by Ni2+‐NTA‐agarose and stained with Coomassie Blue. Lane 1–2: eluted by elution buffer with 0.8 mM imidazole; lane 3: eluted by elution buffer with 5 mM imidazole; lane 4: flow through; lane 5–6: eluted by elution buffer with 20 mM imidazole; lane 7–8: eluted by elution buffer with 40 mM imidazole; lane 9–14: eluted by elution buffer with 80 mM imidazole; lane 15–18: eluted by elution buffer with 300 mM imidazole. (B) RHA was purified by Capto DEAE Sepharose and stained with Coomassie Blue. Lane 1–3: eluted by elution buffer with 0.05 M NaCl; lane 4–6: eluted by elution buffer with 0.15 M NaCl; lane 7: flow through; lane 8–14: eluted by elution buffer with 0.25 M NaCl; lane 15–29: eluted by elution buffer with 0.35 M NaCl; lane 30–31: eluted by elution buffer with 0.45 M NaCl. M: Protein marker 170 KDa. The red arrow indicated the bands of RHA fractions.
Figure S2. Verification on RHA activity. The unwinding assays were carried out at 26°C for 30 min, and RNA denaturation at 95°C for 5 min in lane 4 was used as an experimental control, and the bands in lane 1 illustrated that RHA had good activity.
Figure S3. Helicase activity of RHA for the different types of RNA duplexes. The 3′ tail of the dsRNA used in assays were 0 nt (lane 2), 5 nt (lane 3), 6 nt (lane 4), 7 nt (lane 5) and 10 nt (lane 6), respectively. RNA denaturation at 95°C for 5 min in lane1 was used as an experimental control, and the results illustrated that RHA unwound dsRNA with a 3′ tail no less than 6 nt.
Figure S4. A homemade sample adding device. The sample adding device consisted of a sample stage, a sample chamber, a sample injector and a poly‐tetrafluoroethylene tube that reduced the non‐specific protein adsorption.
Figure S5. Representative time traces of RHA‐assisted RNA structure conversion in different conditions as (A) without RHA or ATP, (B) with 100 nM RHA but without ATP, with 100 nM RHA and (C) 1 mM, (D) 2 mM, (E) 3 mM ATP. In these figures, the Cy3 fluorescence intensity is in green, the Cy5 fluorescence intensity is in red, and the smFRET efficiency is in blue.
ACKNOWLEDGEMENT
This work was supported by the National Natural Science Foundation of China [31870154 to F.L.]; the National Key Research and Development Program [2015BAD12B01 to F.L., 2018YFD0500100 to F.L.]; Jiangsu Key Research and Development Program [BE2018709 to F.L.] and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Wu C, Shan Y, Wang S, Liu F. Dynamically probing ATP‐dependent RNA helicase A‐assisted RNA structure conversion using single molecule fluorescence resonance energy transfer. Protein Science. 2021;30:1157–1168. 10.1002/pro.4081
Funding information Jiangsu Key Research and Development Program, Grant/Award Number: BE2018709; National Key Research and Development Program, Grant/Award Numbers: 2015BAD12B01, 2018YFD0500100; National Natural Science Foundation of China, Grant/Award Number: 31870154; Priority Academic Program Development of Jiangsu Higher Education Institutions
REFERENCES
- 1. Fujii R, Okamoto M, Aratani S, et al. A role of RNA helicase A in cis‐acting transactivation response element‐mediated transcriptional regulation of human immunodeficiency virus type 1. J Biol Chem. 2001;276:5445–5451. [DOI] [PubMed] [Google Scholar]
- 2. Cordin O, Hahn D, Beggs JD. Structure, function and regulation of spliceosomal RNA helicases. Curr Opin Cell Biol. 2012;24:431–438. [DOI] [PubMed] [Google Scholar]
- 3. Lee Y, Rio DC. Mechanisms and regulation of alternative pre‐mRNA splicing. Annu Rev Biochem. 2015;84:291–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bohnsack MT, Martin R, Granneman S, Ruprecht M, Schleiff E, Tollervey D. Prp43 bound at different sites on the pre‐rRNA performs distinct functions in ribosome synthesis. Mol Cell. 2009;36:583–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakielny S, Fischer U, Michael WM, Dreyfuss G. RNA transport. Annu Rev Neurosci. 1997;20:269–301. [DOI] [PubMed] [Google Scholar]
- 6. Cole CN, Scarcelli JJ. Transport of messenger RNA from the nucleus to the cytoplasm. Curr Opin Cell Biol. 2006;18:299–306. [DOI] [PubMed] [Google Scholar]
- 7. Robb GB, Rana TM. RNA helicase A interacts with RISC in human cells and functions in RISC loading. Mol Cell. 2007;26:523–537. [DOI] [PubMed] [Google Scholar]
- 8. Shimada Y, Mohn F, Bühler M. The RNA‐induced transcriptional silencing complex targets chromatin exclusively via interacting with nascent transcripts. Genes Dev. 2016;30:2571–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hessam S, Sand M, Skrygan M, Bechara FG. The microRNA effector RNA‐induced silencing complex in hidradenitis suppurativa: A significant dysregulation within active inflammatory lesions. Arch Dermatol Res. 2017;309:557–565. [DOI] [PubMed] [Google Scholar]
- 10. Schroeder R, Barta A, Semrad K. Strategies for RNA folding and assembly. Nat Rev Mol Cell Biol. 2004;5:908–919. [DOI] [PubMed] [Google Scholar]
- 11. Staley JP, Guthrie C. Mechanical devices of the spliceosome: Motors, clocks, springs, and things. Cell. 1998;92:315–326. [DOI] [PubMed] [Google Scholar]
- 12. Bhaskaran H, Russell R. Kinetic redistribution of native and misfold RNAs by a DEAD‐box chaperone. Nature. 2007;449:1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rocak S, Linder P. DEAD‐box proteins: The driving forces behind RNA metabolism. Nat Rev Mol Cell Biol. 2004;5:232–241. [DOI] [PubMed] [Google Scholar]
- 14. Bowers HA, Maroney PA, Fairman ME, et al. Discriminatory RNP remodeling by the DEADbox protein DED1. RNA. 2006;12:903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang Q, Fairman ME, Jankowsky E. DEAD‐box‐protein‐assisted RNA structure conversion towards and against thermodynamic equilibrium values. J Mol Biol. 2007;368:1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cordin O, Banroques J, Tanner NK, Linder P. The DEAD‐box protein family of RNA helicases. Gene. 2006;367:17–37. [DOI] [PubMed] [Google Scholar]
- 17. Erkizan HV, Schneider JA, Sajwan K, et al. RNA helicase A activity is inhibited by oncogenic transcription factor EWS‐FLI1. Nucleic Acids Res. 2015;43:1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhaskaran H, Russell R. Kinetic redistribution of native and misfolded RNAs by a DEAD‐box chaperone. Nature. 2007;449:1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lilley DM. Structure, folding and mechanisms of ribozymes. Curr Opin Struct Biol. 2005;15:313–323. [DOI] [PubMed] [Google Scholar]
- 20. Thirumalai D, Hyeon C. RNA and protein folding: Common themes and variations. Biochemistry. 2005;44:4957–4970. [DOI] [PubMed] [Google Scholar]
- 21. Tang H, McDonald D, Middlesworth T, Hope TJ, Wong‐Staal T. The carboxyl terminus of RNA helicase A contains a bidirectional nuclear transport domain. Mol Cell Biol. 1999;19:3540–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aratani S, Oishi T, Fujita H, et al. The nuclear import of RNA helicase A is mediated by importin‐alpha3. Biochem Biophys Res Commun. 2006;340:125–133. [DOI] [PubMed] [Google Scholar]
- 23. Fujita H, Ohshima T, Oishi T, et al. Relevance of nuclear localization and functions of RNA helicase A. Int J Mol Med. 2005;15:555–560. [PubMed] [Google Scholar]
- 24. Fuchsova B, Hozak P. The localization of nuclear DNA helicase II in different nuclear compartments is linked to transcription. Exp Cell Res. 2002;279:260–270. [DOI] [PubMed] [Google Scholar]
- 25. Zhang S, Grosse F. Domain structure of human nuclear DNA helicase II (RNA helicase A). J Biol Chem. 1997;272:11487–11494. [DOI] [PubMed] [Google Scholar]
- 26. Ranji A, Shkriabai N, Kvaratskhelia M, Musier‐Forsyth K, Boris‐Lawrie K. Features of double‐stranded RNA‐binding domains of RNA helicase A are necessary for selective recognition and translation of complex mRNAs. J Biol Chem. 2011;286:5328–5337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee CG, Hurwitz J. A new RNA helicase isolated from HeLa cells that catalytically translocates in the 3′ to 5′ direction. J Biol Chem. 1992;267:4398–4407. [PubMed] [Google Scholar]
- 28. Jain A, Bacolla A, Del Mundo IM, Zhao J, Wang G, Vasquez KM. DHX9 helicase is involved in preventing genomic instability induced by alternatively structured DNA in human cells. Nucleic Acids Res. 2013;41:10345–10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jain A, Bacolla A, Chakraborty P, Grosse F, Vasquez KM. Human DHX9 helicase unwinds triple‐helical DNA structures. Biochemistry. 2010;49:6992–6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chakraborty P, Grosse F. WRN helicase unwinds Okazaki fragment‐like hybrids in a reaction stimulated by the human DHX9 helicase. Nucleic Acids Res. 2010;38:4722–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lawrence P, Rieder E. Identification of RNA helicase A as a new host factor in the replication cycle of foot‐and‐mouth disease virus. J Virol. 2009;83:11356–11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li J, Tang H, Mullen TM, et al. A role for RNA helicase A in post‐transcriptional regulation of HIV type 1. Proc Natl Acad Sci U S A. 1999;96:709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bolinger C, Sharma A, Singh D, Yu L, Boris‐Lawrie K. RNA helicase A modulates translation of HIV‐1 and infectivity of progeny virions. Nucleic Acids Res. 2010;38:1686–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. He QS, Tang H, Zhang J, Truong K, Wong‐Staal F, Zhou D. Comparisons of RNAi approaches for validation of human RNA helicase A as an essential factor in hepatitis C virus replication. J Virol Methods. 2008;154:216–219. [DOI] [PubMed] [Google Scholar]
- 35. Isken O, Baroth M, Grassmann CW, et al. Nuclear factors are involved in hepatitis C virus RNA replication. RNA. 2007;13:1675–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin L, Li Y, Pyo HM, et al. Identification of RNA helicase A as a cellular factor that interacts with influenza a virus NS1 protein and its role in the virus life cycle. J Virol. 2012;86:1942–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Serrano P, Miguel Pulido MR, Sáiz M, Martínez‐Salas E. The 3′ end of the foot‐and‐mouth disease virus genome establishes two distinct long‐range RNA–RNA interactions with the 5′ end region. J Gen Virol. 2006;87:3013–3022. [DOI] [PubMed] [Google Scholar]
- 38. Reddy TR, Tang H, Xu W, Wong‐Staal F. Sam68, RNA helicase A and Tap cooperate in the post‐transcriptional regulation of human immunodeficiency virus and type D retroviral mRNA. Oncogene. 2000;19:3570–3575. [DOI] [PubMed] [Google Scholar]
- 39. Filbin ME, Kieft JS. Toward a structural understanding of IRES RNA function. Curr Opin Struct Biol. 2009;19:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carrillo C, Tulman ER, Delhon G, et al. Comparative genomics of foot‐and‐mouth disease virus. J Virol. 2005;79:6487–6504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fuller‐Pace FV. DExD/H box RNA helicases: Multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 2006;34:4206–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hartman TR, Qian S, Bolinger C, Fernandez S, Schoenberg DR, Boris‐Lawrie K. RNA helicase A is necessary for translation of selected messenger RNAs. Nat Struct Mol Biol. 2006;13:509–516. [DOI] [PubMed] [Google Scholar]
- 43. Zhang Z, Yuan B, Lu N, Facchinetti V, Liu YJ. DHX9 pairs with IPS‐1 to sense double‐stranded RNA in myeloid dendritic cells. J Immunol. 2011;187:4501–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jeang KT, Yedavalli V. Role of RNA helicases in HIV‐1 replication. Nucleic Acids Res. 2006;34:4198–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhao Y, Terry DS, Shi L, et al. Substrate‐modulated gating dynamics in a Na+‐coupled neurotransmitter transporter homologue. Nature. 2011;474:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Heng J, Zhao Y, Liu M, et al. Substrate‐bound structure of the E. coli multidrug resistance transporter MdfA. Cell Res. 2015;25:1060–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lamichhane R, Solem A, Black W, Rueda D. Single‐molecule FRET of protein‐nucleic acid and protein‐protein complexes: Surface passivation and immobilization. Methods. 2010;52:192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Blanchard SC, Kim HD, Gonzalez RL Jr, Puglisi JD, Chu S. tRNA dynamics on the ribosome during translation. Proc Natl Acad Sci U S A. 2004;101:12893–12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Charles PT, Stubbs VR, Soto CM, Martin BD, White BJ, Taitt CR. Reduction of non‐specific protein adsorption using poly(ethylene) glycol (PEG) modified polyacrylate hydrogels in immunoassays for staphylococcal enterotoxin B detection. Sensors. 2009;9:645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jain A, Liu R, Ramani B, et al. Probing cellular protein complexes using single‐molecule pull‐down. Nature. 2011;473:484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jain A, Liu R, Xiang YK, Ha T. Single‐molecule pull‐down for studying protein interactions. Nat Protoc. 2012;7:445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ha T, Enderle T, Ogletree DF, Chemla DS, Selvin PR, Weiss S. Probing the interaction between two single molecules: Fluorescence resonance energy transfer between a single donor and a single acceptor. Proc Natl Acad Sci U S A. 1996;93:6264–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ha T. Single‐molecule fluorescence resonance energy transfer. Methods. 2001;25:78–86. [DOI] [PubMed] [Google Scholar]
- 54. Russell R, Zhuang X, Babcock HP, et al. Exploring the folding landscape of a structured RNA. Proc Natl Acad Sci U S A. 2002;99:155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang Q, Jankowsky E. ATP‐ and ADP‐dependent modulation of RNA unwinding and strand annealing activities by the DEAD‐box protein DED1. Biochemistry. 2005;44:13591–13601. [DOI] [PubMed] [Google Scholar]
- 56. Yang Q, Del Campo M, Lambowitz AM, Jankowsky E. DEAD‐box proteins unwind duplexes by local strand separation. Mol Cell. 2007;28:253–263. [DOI] [PubMed] [Google Scholar]
- 57. Eguchi Y, Tomizawa J. Complex formed by complementary RNA stem‐loops and its stabilization by a protein: Function of CoIE1 Rom protein. Cell. 1990;60:199–209. [DOI] [PubMed] [Google Scholar]
- 58. Schroeder R, Grossberger R, Pichler A, Waldsich C. RNA folding in vivo . Curr Opin Struct Biol. 2002;12:296–300. [DOI] [PubMed] [Google Scholar]
- 59. Eckardt S, Romby P, Sczakiel G. Implications of RNA structure on the annealing of a potent antisense RNA directed against the human immunodeficiency virus type 1. Biochemistry. 1997;36:12711–12721. [DOI] [PubMed] [Google Scholar]
- 60. Koh HR, Xing L, Kleiman L, Myong S. Repetitive RNA unwinding by RHA facilitates RNA annealing. Nucleic Acids Res. 2014;42:8556–8564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xing L, Liang C, Kleiman L. Coordinate roles of Gag and RNA helicase A in promoting the annealing of tRNALys3 to HIV‐1 RNA. J Virol. 2011;85:1847–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xing L, Niu M, Kleiman L. In vitro and in vivo analysis of the interaction between RNA helicase A and HIV‐1 RNA. J Virol. 2012;86:13272–13280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Roy R, Hohng S, Ha T. A practical guide to single molecule FRET. Nat Methods. 2008;5:507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Purification of RHA. (A) RHA was purified by Ni2+‐NTA‐agarose and stained with Coomassie Blue. Lane 1–2: eluted by elution buffer with 0.8 mM imidazole; lane 3: eluted by elution buffer with 5 mM imidazole; lane 4: flow through; lane 5–6: eluted by elution buffer with 20 mM imidazole; lane 7–8: eluted by elution buffer with 40 mM imidazole; lane 9–14: eluted by elution buffer with 80 mM imidazole; lane 15–18: eluted by elution buffer with 300 mM imidazole. (B) RHA was purified by Capto DEAE Sepharose and stained with Coomassie Blue. Lane 1–3: eluted by elution buffer with 0.05 M NaCl; lane 4–6: eluted by elution buffer with 0.15 M NaCl; lane 7: flow through; lane 8–14: eluted by elution buffer with 0.25 M NaCl; lane 15–29: eluted by elution buffer with 0.35 M NaCl; lane 30–31: eluted by elution buffer with 0.45 M NaCl. M: Protein marker 170 KDa. The red arrow indicated the bands of RHA fractions.
Figure S2. Verification on RHA activity. The unwinding assays were carried out at 26°C for 30 min, and RNA denaturation at 95°C for 5 min in lane 4 was used as an experimental control, and the bands in lane 1 illustrated that RHA had good activity.
Figure S3. Helicase activity of RHA for the different types of RNA duplexes. The 3′ tail of the dsRNA used in assays were 0 nt (lane 2), 5 nt (lane 3), 6 nt (lane 4), 7 nt (lane 5) and 10 nt (lane 6), respectively. RNA denaturation at 95°C for 5 min in lane1 was used as an experimental control, and the results illustrated that RHA unwound dsRNA with a 3′ tail no less than 6 nt.
Figure S4. A homemade sample adding device. The sample adding device consisted of a sample stage, a sample chamber, a sample injector and a poly‐tetrafluoroethylene tube that reduced the non‐specific protein adsorption.
Figure S5. Representative time traces of RHA‐assisted RNA structure conversion in different conditions as (A) without RHA or ATP, (B) with 100 nM RHA but without ATP, with 100 nM RHA and (C) 1 mM, (D) 2 mM, (E) 3 mM ATP. In these figures, the Cy3 fluorescence intensity is in green, the Cy5 fluorescence intensity is in red, and the smFRET efficiency is in blue.