

Abstract
More than 48 kinase inhibitors (KIs) have been approved by Food and Drug Administration. However, drug-resistance (DR) eventually occurs, and secondary mutations have been found in the previously targeted primary-mutated cancer cells. Cancer and drug research communities recognize the importance of the kinase domain (KD) mutations for kinasopathies. So far, a systematic investigation of kinase mutations on DR hotspots has not been done yet. In this study, we systematically investigated four types of representative mutation hotspots (gatekeeper, G-loop, αC-helix and A-loop) associated with DR in 538 human protein kinases using large-scale cancer data sets (TCGA, ICGC, COSMIC and GDSC). Our results revealed 358 kinases harboring 3318 mutations that covered 702 drug resistance hotspot residues. Among them, 197 kinases had multiple genetic variants on each residue. We further computationally assessed and validated the epidermal growth factor receptor mutations on protein structure and drug-binding efficacy. This is the first study to provide a landscape view of DR-associated mutation hotspots in kinase’s secondary structures, and its knowledge will help the development of effective next-generation KIs for better precision medicine.
Keywords: mutation hotspot, kinase inhibitor, gatekeeper, G-loop, αC-helix, A-loop
Introduction
Kinase activation is a commonly identified phenomenon in tumor cells through multiple mechanisms such as point mutation, amplification, gene fusion and amplification or fusion of kinase ligands [1]. To molecularly target the activated kinase in cancer patients, more than 250 kinase inhibitors (KIs) are currently undergoing clinical trials, and 48 have been approved for patient treatment by Food and Drug Administration (FDA), such as imatinib, gefitinib, sorafinib, erlotinib, dasatinib and crizotinib [2–5]. Even though these targeted therapies have greatly improved patient survival in several cancer types, resistance to these agents always occur due to diverse critical mutations [6]. With the development of next-generation sequencing technology, several somatic point mutations that induce drug-resistance (DR) to commonly used KIs have been reported, and genome-wide investigation of mutation gain or loss during DR in cancer cells has been conducted [7]. One representative example is the T315I gatekeeper mutation in ABL proto-oncogene 1, non-receptor tyrosine kinase (ABL1), which is resistant to imatinib, dasatinib and nilotinib. This mutation is known to be the cause for ~20% of resistant or relapsed chronic myeloid leukemia (CML) patients [8]. Another example is the T790M gatekeeper mutation in epidermal growth factor receptor (EGFR) [9]. T790M occurs concurrently with another primary EGFR sensitizing mutation (L858R) and has been reported to be associated with a decreased sensitivity to EGFR tyrosine KIs (TKIs) [10]. Approximately 60% of patients developing DR have this gatekeeper mutation in non-small cell lung cancer (NSCLC) [11]. KIT proto-oncogene receptor tyrosine kinase (KIT) has also shown resistance to imatinib and sunitinib when it has the T670I gatekeeper mutation in gastrointestinal stromal tumors (GIST) [12]. BCR-ABL E255K/V and Y257C are the representative G-loop mutations. These mutations disrupt an electrostatic triad required for imatinib-binding, causing imatinib-resistance to the cells [13].
To overcome DR, next-generation KIs have been developed including anti-kinase antibodies. Thus far, several studies have reported the roles of kinase domain (KD) mutations in DR. Many factors can contribute to resistance to KIs such as the target-cell extrinsic mechanisms and tumor microenvironment. And pharmacogenomic factors like gene polymorphisms can cause considerable variations in drug efficacy and toxicity. Most DR mutations arise in protein regions involved in drug interactions such as hydrophobic pocket 1/2 (HP1/2), adenine-region, type 2/3 allosteric site and G-loop. Alternatively, those mutations occur in the regions of the transitions between active and inactive kinase conformations such as G-loop, A-loop and αC-helix. To cause DR, a mutation must impair drug binding or the involved conformational changes more than ATP-binding and catalysis. Consequently, directly ATP-interacting residues of hinge or ATP-phosphate binding region are infrequently involved [13]. To date, the identified mechanisms of KI-resistant mutations arise in the four KD regions such as gatekeeper, G-loop, αC-helix and A-loop loci, but a systematic annotation for these protein substructures has not been done for somatic mutations. Since there are many millions of somatic mutations in the cancer genomes, there would be hotspot residues (i.e. the nucleotide positions with a high mutation frequency) in these regions [14–16]. Recently, Catalogue of Somatic Mutations in Cancer (COSMIC) database added the DR information for 26 drugs that target 15 kinases [17]. Additionally, the Wellcome Trust Sanger Institute screened drug response in 1001 cancer cell lines that have somatic mutation profiles after treatment with 265 anti-cancer drugs [18] including KIs. Through these studies, cancer and drug research communities have recognized the importance of KD mutations for kinasopathies and have called for systematic investigation of kinase mutation candidates associated with drug response. However, despite the exponential growth of cancer and drug data, and many bioinformatics approaches for studying KI targetable sites using genome-wide cancer mutation data [19–23], systematic investigation of the kinase mutations on DR hotspots in protein structure has not been performed yet.
Here, we first determined the loci of four protein structures in 538 human protein kinases using the annotations from the representative protein databases. By integrating somatic mutations from the large cancer genomic data sets with the four types of kinase structures, we identified 358 kinases harboring 3318 mutations that covered 702 KD hotspot residues. Specifically, there were 73, 147, 181 and 301 kinases that had drug resistance hotspot mutations in four types of structures, gatekeeper, G-loop, αC-helix and A-loop, respectively. Furthermore, we found 197 kinases that had multiple variants at one hotspot residue; some of them are well-studied mutations but others are novel. We found 645 out of 671 DR hotspot mutations in tyrosine kinases (TK) led to a decrease in the stability of their protein structure. In validation analysis, we found the evidence of inducing DR by comparing IC50 value between the cell-line with L858R primary mutation only and the one with additional EGFR T790M gatekeeper mutation. We further confirmed our findings by calculating the free energy of binding between KIs and EGFR wild type versus mutants in four DR hotspots. Overall, this is the first systematic investigation of DR hotspot mutations in the human kinome. This study will help cancer and drug research communities for the better development of next-generation KIs in the emerging cancer precision medicine.
Results
Four types of KD mutation hotspots in humans—gatekeeper, G-loop, αC-helix and A-loop
To induce DR, a mutation must impair drug binding or be involved in conformational changes. Indeed, most DR mutations arise in protein regions involved in drug interactions or in the transition between active and inactive kinase conformations [13]. To date, there are four models are known that kinase mutations may influence on the interaction between kinase and KIs: Gatekeeper, G-loop, αC-helix and A-loop (Figure 1) [13]. Specifically, we have studied and identified the locations of each of these four categories in the human kinase proteins as following. Gatekeeper residue is a single amino acid located near the protein-drug binding site. In wild-type kinases, gatekeeper residues have small side chains that sterically accommodate drugs. These small side chains can be mutated into bulky side chains that impede drug-protein binding [13]. This confers drug resistance. G-loop, which is also named glycine-rich loop, and P-loop (phosphorylation loop) have a conserved consensus motif GxGxxG, where G represents glycine, and x can be any amino acid [24]. The N-lobe of KD contains a five-stranded β-sheet (β1–β5). Of these, G-loop lies between the β1 and β2 strands [25, 26]. G-loop mutation can cause clinical resistance to type 2 KIs (T2KIs) by destabilizing the inactive conformation, stabilizing the active conformation and/or removing direct drug interactions [13]. KIs bind to the inactive conformational protein kinases. αC-helix, also called C-helix and αC, is a single α-helix located in the N-lobe of KD between β3 and β4 strands [13]. αC-helix is usually the first α-helix in the kinases domain (from N-terminus), but in some kinases such as AGC kinases, a short αB-helix may precede αC-helix [27]. Mutations at this hotspot may cause DR by destabilizing the inactive conformation of the KD. A-loop, which called activation loop or T-loop, is located in the C-lobe. A-loop contains a phosphorylation site, which upon phosphorylation, induces a conformational change on the loop, and then allows a substrate to bind. The activation loop and P + 1 loop constitute the activation segment that runs from the DFG (Asp-Phe-Gly) motif to the APE (Ala-Pro-Glu) motif [28]. A-loop mutations may have indirect effects that disfavor drug binding by increasing entropy or destabilizing the inactive conformation [29]. We searched the hotspot locus information of 538 human kinases from UniProt, KSD and NCBI databases. After removing duplicated kinases and filtering out the kinases without specific locus information of these categories, we obtained 396 unique human kinases. Among them, 348, 172, 231 and 312 kinases had Gatekeeper, G-loop, αC-helix and A-loop locus information, respectively. The detailed information is summarized in Supplementary Table S1.
Figure 1.
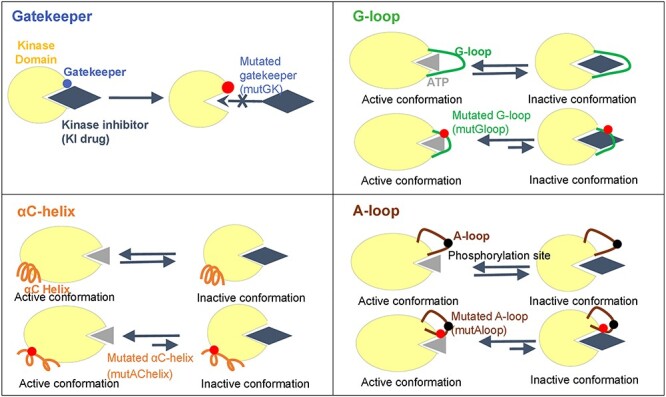
Four drug resistance models related to KD mutation hotspots.
358 human kinases had mutations on KD mutation hotspot residues
Figure 2 summarizes overall pipeline and our identification of the KD hotspot somatic mutations in humans. Figure 2A shows the overall distribution of kinases with hotspot loci among the four hotspot categories plus the ATP-binding sites (Supplementary Table S1). Overlapping these hotspot loci with four largest cancer mutation data sets resulted in 358 kinases having 3318 hotspot mutations (Figure 2B and Supplementary Table S2). These four sets were The Cancer Genome Atlas (TCGA), COSMIC, International Cancer Genome Consortium (ICGC) and Genomics of Drug Sensitivity in Cancer (GDSC), which had 308, 291, 316 and 175 kinases with mutations on KD hotspots, respectively. Since the length of hotspot regions varied, the number of kinases with mutations that occurred at KD hotspot categories was ordered by 301 (A-loop), 181 (G-loop), 147 (αC-helix) and 73 (Gatekeeper). Gatekeeper usually covers only one amino acid, so the number of mutations was the smallest among the four categories. The average length of hotspot regions of G-loop, αC-helix and A-loop was 8.6, 11.4 and 19.1 amino acids with standard deviations of 2.10, 7.12 and 4.80, respectively.
Figure 2.
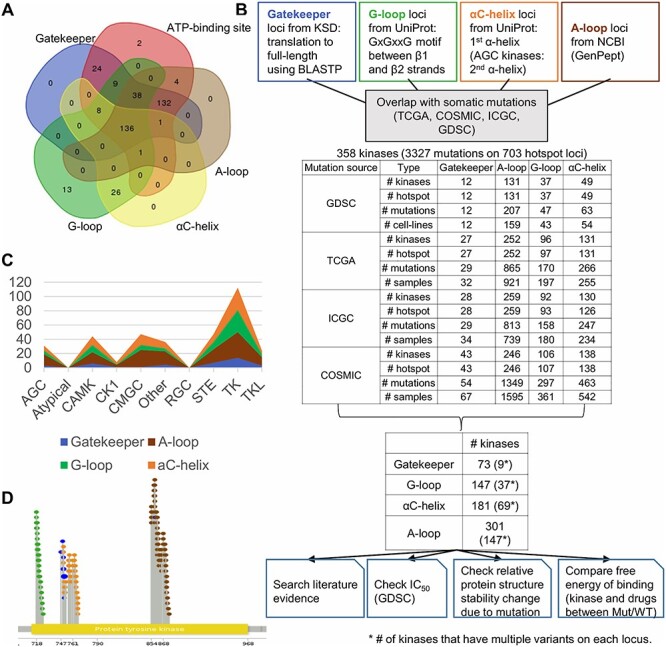
Kinase hotspot mutation statistics and the flowchart of annotation of kinase hotspot mutations. (A) Overlapping KD mutation hotspots among five structures or sites. (B) Flowchart for kinase hotspot mutation annotation. (C) Distribution of mutated kinases among four groups. TK group has the largest number of kinases harboring KD hotspot mutations. (D) Mutation hotspots on the KD sequence of EGFR. The yellow bar labels the KD of the partial sequence of EGFR. The green, blue, orange and brown colored lollipops present the mutations overlapped with G-loop, Gatekeeper, αC-helix and A-loop category residues, respectively.
Next, we checked how the kinases with KD hotspot mutations were distributed among the kinase groups. The Human Kinome database defined 10 groups of kinases: (1) the PKA, PKG, PKC families (AGC), (2) Atypical, (3) calcium/calmodulin-dependent protein kinase (CAMK), (4) casein kinase 1 (CK1), (5) containing CDK, MAPK, GSK3, CLK families (CMGC), (6) Other, (7) receptor guanylate cyclases (RGC), (8) homologs of yeast Sterile 7, Sterile 11, Sterile 20 kinases (STE), (9) TK and (10) TK-like (TKL) [30]. TK-related groups (TK and TKL groups) have been most studied; as expected, they have the highest frequency (39.0%, 136/349) of mutated hotspot loci among the 10 groups (Figure 2C). We demonstrated the hotspot mutations across the full-length protein sequence using EGFR as an example (Figure 2D). Overall, our work provided a landscape view of the KD hotspot mutations in thousands of human cancer genomes.
197 human kinases had multiple variants on individual KD hotspot residues
To assess the clinical potential [31, 32], we specifically examined hotspot residues with multiple mutations in four cancer datasets. Some examples are ABL1 (T315A/I/L/V/N), ALK (L1196M/Q), BRAF (G469A/E/R/S/V/X/del), EGFR (T790A/M), ERBB2 (V773A/M), FGFR4 (V550L/M), FLT3 (F691/L) and KIT (T670E/I/S). Through this filtering, we found 197 kinases had multiple variants on individual hotspot residues. Tables 1–4 summarize the mutations in each of the four KD hotspot categories.
Table 1.
Gatekeeper residues with multiple variants
| Kinase | Gatekeeper mutations |
|---|---|
| ABL1 | T315A/I/L/V/N |
| ALK | L1196M/Q |
| BMPR1B | T279A/I |
| EGFR | T790A/M |
| FGFR4 | V550L/M |
| FLT3 | F691/L |
| KIT | T670E/I/S |
| MAP2K7 | M196I/T |
| SYK | M448I/L |
Note: Among all 73 kinases that harbor Gatekeeper mutations, nine of them had multiple variants on each Gatekeeper residue.
Table 4.
A-loop residues with multiple variants
| Kinase | Mutations | Kinase | Mutations |
|---|---|---|---|
| ABL1 | H396P/R | MAP3K10 | A264E/T |
| ACVR1B | R379*/X | MAP3K12 | K267E/N |
| ACVR1C | P361L/S, S350*/L/X | MAP3K2 | R507Q/W |
| ACVRL1 | H355P/R | MAP3K9 | R299L/W |
| AKT1 | M306T/L | MAP4K1 | R169C/H//QT?, Y177*/X |
| ALK | G1269A/E, D1270E/G, R1275*/L/Q/X | MAPK1 | P176S/T, R191C/H |
| AMHR2 | E380K/X, Q373*/X | MAPK10 | D207A/N, G215D/S |
| AURKC | P192L/S | MAPK12 | G173D/S, W190*/L |
| BMPR1B | R376C/H | MAPK3 | R189Q/W |
| BMPR2 | M356I/T | MAPK6 | W196C/L |
| BTK | R544M/W | MAPK7 | R205C/P |
| CAMKV | G182R/V/W | MAPK8 | R189C/H/L/P |
| CDC42BPG | R219C/H | MAST1 | E552*/K |
| CDK12 | R882L/Q/W | MAST2 | G655R/V |
| CDK13 | I875S/V | MELK | A173S/T |
| CDK19 | R178*/X, L190*/X | MERTK | Q756*/X, R758H/S |
| CDK7 | A159D/T | MET | Y1230C/H |
| CDK8 | D173G/N/V, R178*/Q/X | MINK1 | P192L/S |
| P186L/S, L190F/V | MKNK2 | A254T/V | |
| CDKL2 | R149*/Q/X | MYLK | A1609S/T |
| CHEK1 | R156L/W, R160C/H | MYLK3 | R662K/M, R666*/X |
| CHEK2 | S372C/F | MYO3A | R181C/H/L/S, T184A/I |
| CLK2 | D327H/N | MYO3B | R185H/L |
| CSK | D332G/N, Q343K/L | NEK7 | R184L/W |
| CSNK1A1L | H172Q/Y, R186*/Q | NIM1K | P234L/T |
| CSNK1D | P166H/L | NLK | R287K/T, R295C/H |
| CSNK2A2 | E181D/Q | NTRK1 | D679H/N |
| DAPK3 | G171A/W | NTRK3 | S701F/T, V704D/F, R735C/H |
| DCLK3 | C514R/Y, F511C/L | NUAK1 | N201D/I, K207R/T |
| DDR1 | R798C/L | PAK4 | PR471*/X, P479L/S |
| DDR2 | Q744*/X, R752C/H | PAK5 | E596G/Q, V604F/I |
| DMPK | G233S/V | PDGFRB | R849*/G/Q, R853L/Q/W |
| DYRK1A | Q323*/H, S311F/Y | PHKG1 | F169C/V |
| DYRK2 | R378C/H/L | PIM1 | R296Q/X |
| EGFR | T854A/I/P/S, D855G/N, F856L/S/Y, G857E/R/V, | PIM2 | Y194N/S |
| L858A/G/K/M/P/Q/R/V/W, | PKN1 | E764D/K | |
| A859D/T/_L883 > V, K860E/I, | PKN3 | G704*/R, P723Q/T | |
| L861E/F/G/K/P/Q/R/V, | PRKAA2 | G159E/R/V | |
| L862P/Q/R/V, G863D/S/V, | PRKACA | V192/A, W197G/L/R/S | |
| A864E/T/V, E866D/G/K/Q/V, | PRKACB | W197G/R/S | |
| E868D/G/K/V, H870N/R/Y, | PRKACG | R195C/H | |
| A871E/G/T/V, E872*/G/K/X, | PRKCB | E490D/K | |
| G873E/Q | PRKCG | P508H/L/S, R513H/L | |
| EIF2AK1 | L495M/V, T488A/K | PRKCQ | D533E/G/H, G541R/V |
| EPHA2 | D757H/N | PRPF4B | G836R/V/W |
| EPHA3 | G766E/R, P775A/S, R769C/G/H/S | PTK2B | R586C/H |
| EPHA5 | R823Q/W, A832S/V, G837E/R, | RAF1 | R495C/H |
| I840L/N, P841L/S | RET | S891A/L/X, S904L/Y, R912Q/W | |
| EPHA7 | R781*/Q, R801K/M/S, T793I/S, | ROCK2 | D255E/V |
| G795S/V, G796E/R | ROR1 | R657C/H | |
| EPHB1 | D762E/N | ROR2 | A643P/T, G650E/V |
| ERBB2 | L869Q/R | ROS1 | V2125A/I, R2126G/L/Q/W |
| ERBB4 | G870E/R, E872K/V, E874*/K/X, | RPS6KA2 | R562C/H, A563T/V, G556C/S, M569I/V |
| G879*/E, G880E/R, M882I/R | RPS6KA3 | E217*/K | |
| FGFR1 | R646Q/W, K656*/E/M | RPS6KA4 | T188M/K, R193Q/W |
| FGFR2 | G646*/E, A648T/V, K659E/M/N | RPS6KA6 | D565A/N, G574E/R/V, G577R/X |
| FLT1 | R1060*/X | RPS6KB1 | E258*/X |
| FLT4 | R1060Q/W, Y1068*/N, R1070C/H | RPS6KC1 | S957C/F |
| FYN | G410E/R/V | SGK1 | T239N/P |
| GRK1 | G345*/X | SIK1 | P188L/R |
| GRK5 | E338*/X, R345Q/W | SIK2 | F166L/V |
| ICK | R148*/Q | STK11 | D194H/N/V/Y, L195M/P, |
| IGF1R | R1158*/X | E199*/D/K/Q/X, G215D/S, S216F/P | |
| INSRR | R1138L/Q, R1157C/H/S | STK17A | R221*/X |
| ITK | L508M/Q | STK25 | G177D/S |
| JAK2 | E1012*/X | STK3 | Q170*/X, D173V/Y, R178C/H |
| JAK3 | G987C/S | TEC | G509*/E |
| KDR | D1052G/H/N, G1063E/R, R1066H/C | TGFBR2 | R403C/H, R423*/_Y424 > S |
| KIT | C809*/G/R/X, L813I/P, A814S/T, | TIE1 | G999C/D/S, R1002Q/W, P1016H/S |
| R815_D816ins?/insVI, | TNIK | R180*/Q | |
| D816?/A/E/F/G/H/I/N/V/Y/>GP/>VVA, | TNK2 | R275*/Q, Q279*/X | |
| I817F/L/T/V, K818R/_D820 > N, | TNNI3K | G608E/R, E618D/K, K623Q/R, | |
| N819S/Y, D820A/E/G/H/N/V/Y, | Q624*/L, R629C/G | ||
| S821F/Y/_N822 > GY, | TSSK1B | R160C/H, R163L/W, R168*/Q/X | |
| N822D/H/I/K/S/T/, Y823C/D/H/N, | TSSK2 | R168C/H | |
| V825A/D/I, R830*/L | TTBK1 | R204C/H | |
| LCK | R387C/L, L388F/I, E393G/K, A396T/V | TTBK2 | R191C/G |
| LMTK2 | R307*/G | TTK | G666E/R, T675A/K |
| LTK | R678C/L/P, D681/Y, R682Q/W | TXK | F428S/Y, S407*/X |
| MAP2K3 | F209C/V | TYK2 | E1050G/K, E1053K/_Y1054 > DH, |
| MAP2K4 | G249D/R/V, S251I/N, S257/F/Y, | R1058C/H | |
| G265C/D/V | TYRO3 | R687H/S | |
| MAP2K5 | N320H/T | VRK1 | R203Q/W, G208R/V, K211Q/T, E212*/G |
| MAP2K7 | S271C/T | WEE1 | G462C/D |
| MAP3K1 | A1414/V | WEE2 | G395E/X, R398C/H |
Note: Among the 301 kinases that had A-loop mutations, 147 had multiple variants on an A-loop residue.
Mutated gatekeeper loci
Gatekeeper refers to the amino acid residue at the entrance to the hinge region of the ATP binding cleft. This residue controls the kinase sensitivity for small molecule inhibitors by allowing the inhibitor to access the binding pocket. If the gatekeeper residue is mutated, the bulky side chain may block the binding of inhibitor. In this study, we identified 81 gatekeeper mutations in 73 kinases. Interestingly, we found nine kinases had multiple variants on each hotspot locus (Table 1). It is well known that T315 gatekeeper mutation of ABL1 provided a structural rationale for the vulnerability of TKIs such as imatinib and second-generation ABL1 TKIs (dasatinib, nilotinib and bosutinib) [33]. The murine interleukin-3-dependent pro-B (Ba/F3) cell lines express EML4-ALK with L1196M gatekeeper mutation. These cells were resistant to crizotinib, whereas the cells with primary EML4-ALK were successfully inhibited by crizotinib [34]. The TKI resistance mechanisms via the secondary mutation T790M of EGFR has been well-studied for gatekeeper induced drug resistance [35]. Inhibitor ponatinib or PD173074 could not inhibit the kinase activity of FGFR4 harboring V550E/L gatekeeper mutation [36]. Fms-like TK 3 (FLT3) is one of the most frequently mutated genes in acute myeloid leukemia (AML). With F691 gatekeeper mutation, the drug response to KI gilteritinib decreased in AML patients [37]. A secondary mutation T670I conferred imatinib resistance to the cells with a primary mutation on KIT in GIST [38]. Our results had nine candidates, six of them (66.7%) are known gatekeeper mutations towards DR, while the other three (BMPR1B T279, MAP2K7 M196 and SYK M448) are novel candidates, which warrants future investigation.
Mutated G-loop loci
The glycine-rich G-loop controls ATP binding and phosphate transfer in protein kinases. Mutations limiting the flexibility of the G-loop may affect ATP binding and drug sensitivity [39]. For this category, we found 462 mutations in 147 kinases, among which, 37 kinases had multiple variants on individual hotspot residues (Table 2). For ABL1, we identified six G-loop mutated residues. Five of them (L248, G250, Q252, Y253 and E255) were discovered as the imatinib-resistant mutations from previous studies [40]. G251 residue was chosen for studying the structural effect of three KIs and the results showed different effect [41]. G469A G-loop mutation increased kinase activity of BRAF [42]. G719X G-loop mutation is one of the EGFR-TKI-sensitizing mutations in NSCLC [43].
Table 2.
G-loop residues with multiple variants
| Kinase | G-loop mutations | Kinase | G-loop mutations |
|---|---|---|---|
| ABL1 | L248R/V/_K274del, G250E/R, | IKBKB | R31*/X |
| G251C/D/E, Q252E/H/K/M/P/R, | KDR | R842C/L | |
| Y253C/F, E255D/K/L/V | MAP2K1 | G80S/D | |
| BMPR2 | R211*/P/Q/X, R213*/X, Y214*/X | MAP2K4 | R110*/P/X/Q |
| BRAF | G464E/R/V, G466A/E/R/V/X | MAP4K4 | G37*/X |
| S467F/L, F468C/L/S | MARK2 | G62C/S | |
| G469A/del/E/R/S/V/X, V471A/I/F | MAPK9 | G35E/R | |
| CDK13 | I711L/V | MYLK4 | R116C/H, Q119*/R |
| CDK2 | G13C/D/S | NTRK2 | E546*/K/V |
| CLK2 | R176*/X | PRKACA | S54F/Y |
| CSNK1A1 | S27C/F | PRKCB | S352N/G, F353L/E |
| DAPK1 | A25P/V | ROCK2 | G104R/V |
| DDR1 | E618K/Q | ROS1 | Y1960*/H |
| EGFR | G719A/C/D/S/V, S720C/F/P/T | SGK1 | L113F/P |
| G721A/D/S/V/W, F723L/S | STK11 | G56GS?/V/W, Y60*/I?, K62*/X | |
| G724D/S, T725A/M | STK26 | S34*/X | |
| EPHA2 | G620E/R | STK4 | V44A/I |
| EPHA3 | G633E/R | TGFBR1 | G212D/V, R215*/G/X/P |
| EPHA7 | I639F/T, G642A/E | TNIK | G34R/V |
| EPHA8 | G647E/R | TNK2 | G135D/S |
| EPHB4 | E625K/Q | VRK2 | G38E/R |
| FLT3 | G617E/V, S618L/_G619ins21 | ||
| A620_F621ins35/39/58/66 | |||
| G622R/_K623ins31 |
Note: Among the 147 kinases that harbor G-loop mutations, 37 had multiple variants on a G-loop residue.
Mutated αC-helix loci
A conserved salt bridge is formed between Lys and Glu, which coordinates the α- and β-phosphate groups of the ATP molecule, and drives the phosphate transfer to the substrate. Here, the conserved Glu residue is located on the αC-helix [44]. Therefore, disruption of the salt bridge by a mutation could affect the kinase activity. Our analysis revealed 704 αC-helix mutations in 181 kinases. Among them, 69 kinases had multiple variants on a specific residue (Table 3). Several cases have been reported for their effects on hindering the drug response. Ceccon et al. reported that E1167 mutation on the salt bridge of ALK stabilized the active conformation the ALK, reducing the chance of binding with the KIs [45]. When combined with a drug-sensitive L858R mutation, the D761Y mutation modestly reduced the sensitivity of mutant EGFR to TKIs in both surrogate kinase and cell-viability assays [46]. V773A αC-helix mutation of ERBB2 decreased the sensitivity to lapatinib [47]. M2001 residue of ROS proto-oncogene 1, receptor TK (ROS1) provided additional hydrophobic interactions between cabozantinib and ROS1 [48]. The G571 mutation may cause a conformational change so that the adjacent Y570 is no longer accessible and phosphorylated, resulting in a constitutively active kinase JAK2 [49].
Table 3.
αC-helix residues with multiple variants
| Kinase | Mutations | Kinase | Mutations |
|---|---|---|---|
| ABL1 | E282K/G/V, V289A/F/M | GRK4 | I223K/T |
| AKT1 | V185A/D | GSG2 | S539F/P |
| AKT2 | A190G/T | IKBKB | R53Q/W |
| ALK | M1166R/>?, E1167A/K, I1170N/S/T, I1171N/S/T | IRAK4 | Q232*/X |
| AURKB | Q112*/H | ITK | M411L/V |
| BRAF | TAPTP488/K, K499N/Q | JAK1 | R629K/_D630del, Q644*/H |
| BUB1 | P828R/S | JAK2 | G571R/S, E890*/D, D894E/G |
| CAMK2B | R66H/C | JAK3 | R870W/Q |
| CAMK4 | L93F/I/R | KDR | R880*/Q/X |
| CDK5 | R50L/Q/W | KIT | R634Q/W, L637F/H/P, K642E/N/Q, |
| CDK8 | R71*/G | Y646H/D, L647F/P | |
| CDKL3 | E50*/X | LYN | E290D/K |
| CDKL5 | R59*/Q | MAP2K2 | Q114*/H |
| CDK12 | R773C/H | MAP2K4 | E141*/K, Q142/L, L146F/P, D148/Y |
| CLK2 | E213A/Q | MAP2K7 | R162H/C |
| CSNK1A1 | E60*/K | MAPK13 | Y70C/H |
| CSNK2A1 | R80C/H | MAPKAPK3 | K80*/N/X, Q90E/P |
| CSNK2A2 | K77M/N | MARK1 | R106*/X |
| DAPK1 | R58C/H | MARK2 | V106A/L |
| DYRK1A | R205*/X | MASTL | S120F/P |
| DYRK2 | R274Q/W | MERTK | C640F/X |
| EGFR | E758D/G/K, E749_E758 > QP, | NEK7 | R76C/L/H |
| A750_E758del/delATSPKANKE/P, | NTRK3 | D584E/N, E590D/K | |
| A750_I759 > GS/PT, | PAK1 | Q304*/X | |
| T751_E758 > A/del/delTSPKANKE, | PDGFRA | L641I/V | |
| T751_I759 > AC/del/>N/>NKA/>REA/>S, | PHKG2 | R75Q/W, R82C/H | |
| S752_I759del/delSPKANKEI, | PRKAA2 | R53C/H | |
| N756S/Y/_L760del, K757M/N/R, | PRKCB | V378E/M | |
| I759N/V, D761G/N/Y, D761_E762insEAFQ, | PRKCH | D394G/V | |
| A763D/V, Y764S/_V765insHH, | PRKCI | E288*/Q | |
| V765G/M/_M766insMAS, | ROS1 | M2001T/L | |
| A767V/AAWT/_S768insSVG/_S768insYVM | PTK2B | E474K/Q | |
| EIF2AK2 | E303K/V | ROCK1 | K109E/I |
| EIF2AK3 | R633L/Q/W | RPS6KA3 | R110*/Q/X, E463K/V |
| EPHA5 | R718C/H, D720E/H/Y | STK11 | K84*/X, R86G/X |
| EPHA7 | D678E/N, S684I/R | STK17B | H79N/R |
| EPHA8 | A685G/T/V | STK3 | S72F/P |
| EPHB1 | R663L/Q/W | STK32A | E61*/X |
| ERBB2 | D769H/N/Y, V773A/M | TNNI3K | R508*/8Q, L513F/I/P |
| FLT3 | K663N/Q/R | ZAP70 | T380K/M, E386K/Q, Q388H/L |
Note: Among the 181 kinases that harbor αC-helix mutations, 69 had multiple variants on a αC- helix residue.
Mutated A-loop loci
Protein kinases are themselves controlled by the phosphorylation of activation loop [50]. When an A-loop is mutated, it may influence the control of the kinase activity. Our analysis revealed 2077 A-loop mutations in 301 kinases. About 48.8% (147/301) had multiple variants on each residue (Table 4). For example, when the cells had both T315I gatekeeper and H396R A-loop mutations on ABL1, they exhibited high-level rebastinib resistance, having IC50 value of 464.6–955.3 nM versus <100 nM to native ABL1 cells [51]. Furthermore, MET Y1230 mutants acquired crizotinib resistance in NSCLC [52]. Although this KD hotspot category had the largest number of candidate mutations, most of these mutations have not been studied for DR.
Candidate DR mutations in TK decreases the protein structure stability
To assess the effect of each mutation on the protein structure, we calculated the relative protein structure stability change of each of the 671 mutations in all the 44 kinases within TK group. For each mutation, we computed the value of energy change (ΔΔG) using MUPro software [53]. Among the 671 mutations, only 26 (3.9%) resulted in a positive value of energy change (Figure 3). We hypothesized that majority of DR hotspot mutations in TK group might decrease the protein structure stability. The decreased protein stability by KD hotspot mutations might lead to decreased affinity to KIs and, thus, cause decreased sensitivity to KIs. We specifically studied the details of four representative TKs (EGFR, ABL1, ALK and KIT) (Figure 4).
Figure 3.
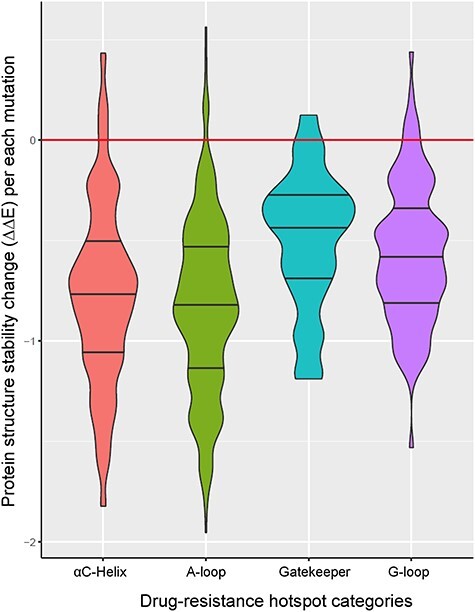
The relative protein structure stability change by a KD mutation in TKs. Almost all the hotspot mutations in TKs were predicted to decrease the protein structure stability.
Figure 4.
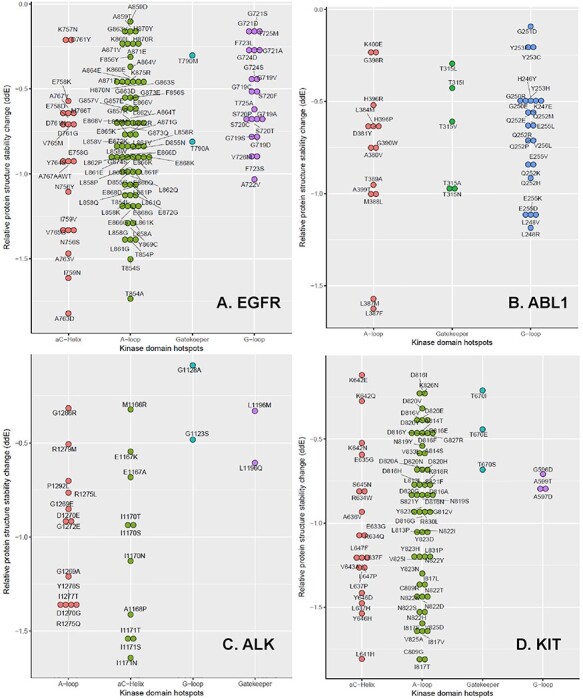
The relative protein structure stability change by a KD mutation in four TKs. These plots are drawn for the mutations that decreased the relative protein structure stability of EGFR (A), ABL1 (B), ALK (C) and KIT (D). Note there is no αC-helix for ABL1.
EGFR
Classical activating mutations like exon 19 deletion and L858R substitution account for ~45% and ~40% of the NSCLC cases with EGFR mutations, respectively [54]. These are known as good response to EGFR-inhibitors [55]. The secondary mutation T790M is known to explain ~50% of NSCLC patients who developed KI drug resistance [10]. In this study, we identified 253 cancer cells with hotspot mutations of EGFR. Their mutations covered 46 hotspot residues: 1 Gatekeeper, 8 G-loop, 16 αC-helix and 21 A-loop residues. Among them, 36 residues had multiple variants on each residue: one Gatekeeper site (T790), six G-loop sites (G719, S720, G721, F723, G724 and T725), 12 αC-helix sites (A750, T751, S752, N756, K757, E758, I759, D761, A763, Y764, V765 and A767) and 17 A-loop sites (T854, D855, F856, G857, L858, A859, K860, L861, L862, G863, A864, E866, E868, H870, A871, E872 and G873). Figure 4A shows the mutations that decreased the relative protein structure stability of EGFR. Here, T790M mutation is a canonical secondary mutation leading to DR [56]. In our results, seven COSMIC, two TCGA and one GDSC samples had T790M mutation. G719 accounts for ~4% of all EGFR activating mutations [57]. S720-mutated patients showed poor response to EGFR KIs and short overall survival (OS) in advanced NSCLC [58]. G721 mutation has been reported as potentially causing DR for erlotinib, gefitinib and lapatinib [59]. D761Y mutation modestly reduced the sensitivity of mutant EGFR to TKIs [46]. A767 mutation on EGFR exon 20 is known to be a resistant mutation [60]. M766, T854 and A859 were known as erlotinib resistant mutations [61, 62]. L858R is the highest prevalence mutation in EGFR and accounts for ~41% of all EGFR activating mutations [57]. The studies of patients with EGFR L861Q mutations showed inconsistent drug response. OS of the patients with EGFR L861Q mutations and treated with gefitinib was significantly shorter than that of other common mutation patients. However, L861Q showed better sensitivity to afatinib and progression-free survival was notably improved [63]. Figure 4A visualizes all the residues known as resistant or sensitive to EGFR KIs.
ABL1
Mutations in the KDs of BCR-ABL1 are attributed to the acquired imatinib resistance in CML patients. In particular, T315I is one of the most studied mutations that induces resistance to imatinib, nilotinib and dasatinib [64]. From our result, ABL1 has 46 mutations on the 28 residues of the KD hotspots: 1 Gatekeeper, 10 G-loop, 3 αC-helix and 14 A-loop residues. 1 Gatekeeper, 9 G-loop and 11 A-loop residues decreased the protein stability as shown in Figure 4B. Furthermore, one Gatekeeper (T315), six G-loop (L248, G250, G251, Q252, Y253 and E255), two αC-helix (E282 and V289) and one A-loop (H396) residues had multiple variants on each residue. Y253H, E255K/V, T315I/ F217L, F359V/C, Q252 are known as the imatinib-resistant BCR-ABL1 mutations [65–68]. On the other hand, L248, Y253, E255, F359 and H396 have been reported with a high response rate to dasatinib [51]. With these accumulated drug resistance reports to ABL inhibitors, the European LeukemiaNet recommends on mutation identification of the patients before changing to the treatment of another TKI or to another therapy, if the patient reaches a suboptimal response or fails to respond to imatinib [69, 70].
ALK
In our results, ALK had 19 residues with KD hotspot mutations. Eight residues had multiple variants on each site: 1 Gatekeeper site (L1196), 4 αC-helix sites (M1166, E1167, I1170 and I1171) and 3 A-loop sites (G1269, D1270 and R1275). Among these, 1 Gatekeeper, 2 G-loop, 5 αC-helix and 10 A-loop residues decreased the protein stability as shown in Figure 4C. cells with L1196M in EML4-ALK markedly reduced the sensitivity to crizotinib [71]. L1196Q and I1171N are known as conferring resistance to crizotinib in Ba/F3 cells expressing NPM-ALK [45]. ALK I1171 mutation also confers resistance to alectinib in NSCLC [72]. SNU-2535 cells derived from a NSCLC patient who harbored the G1269 mutation were resistant to crizotinib [73]. NIH 3T3 cells that had R1275Q A-loop variant were significantly more sensitive to PF-06463822 and crizotinib [74]. ALK has an ‘YxxxYY’ auto phosphorylation motif, where Y represents tyrosine and x can be any amino acid, in the A-loop and in ALK fusions. The Y1278 in ALK is the first residue in the motif to be phosphorylated. Hallberg and Palmer reported that initial activation of ALK might be mediated by the regulation of Y1278 phosphorylation and releasing ALK from inactive conformational restraints [75]. If this residue is mutated, it may affect ALK activation.
KIT
We found 38 residues having KD hotspot mutations in KIT. Among them, 20 had multiple variants on each residue: 1 Gatekeeper (T670), 5 αC-helix (R634, L637, K642, Y646 and L647) and 14 A-loop (C809, L813, A814, D816, I817, N819, D820, S821, N822, Y823, V825, K826, A829 and R830) residues. There were 1 Gatekeeper, 3 G-Loop, 11 αC-helix and 18 A-loop residues decreased the protein stability as shown in Figure 4D. In KIT, DR residue T670, which is analogous to T315 in BCR-ABL, is well studied. The bulky substituent through mutations in T670 obstructs binding of imatinib, nilotinib and dasatinib, thus conferring resistance [38]. In our study, we found six COSMIC cell lines that had mutations on these residues, four of which were derived from GIST. R815 residue corresponds to R405 in ABL1b. This residue involves in the salt bridges with E305 in the SFK-like inactive conformation and in inactive SFKs. Thus, deletion of R815 in KIT might destabilize inactive conformations and confer DR [13]. D816 mutation in A-loop leads to kinase resistance to imatinib by strongly favoring the active conformation of the KD [76]. D820 mutations were identified in three sunitinib resistant patients of GIST [77]. On the contrary, D816 and N822 mutations are common activating mutations for KIT kinase activity [38]. Lastly, Y823D is known to stabilize the active or destabilize the inactive conformation, reducing TKI binding [12].
IC50 evidence in drug resistance by KD hotspot mutations
The GDSC data provide a systematic measure of drug response (265 drugs) to many somatic mutations in 1001 cancer cell lines. Since the matched wild type samples are not available in this data set, we could not compare the drug sensitivity between mutant and wild type cells. Alternatively, we compared the IC50 values of the cell lines with the primary mutation only with that of the cell lines with both the primary and secondary mutations. This analytical strategy is useful for assessing drug sensitivity or resistance induced by a secondary mutation. We illustrated here using EGFR as an example. The primary mutation, L858R, was in both NSCLC H3255 and NCI-H1975 cell lines, while NCI-H1975 had additional T790M gatekeeper mutation. Gefitinib was reported to have a drastic increase of ATP affinity. L858R/T790M double mutant in NCI-H1975 could retain 18.6 nM affinity for gefitinib, as compared with Kd = 1.1 nM for the L858R mutant only [56]. As shown in Figure 5, the difference of IC50 values was largest between H3255 and NCI-H1975 cell lines with the treatment of gefitinib. Approximately 50% of EGFR-mutant NSCLC patients who developed resistance to gefitinib or erlotinib had this T790M mutation. Considering this strong clinical outcome, a noninvasive detection method of this mutation has been reported for easy diagnosis and exact therapy [78]. Cetuximab is an anti-EGFR monoclonal antibody. Combination of afatinib and cetuximab has been reported as overcoming T790M-mediated resistance in preclinical study [79]. The treatment of cetuximab alone still suffers from DR by T790M as shown in the first bar of Figure 5. CUDC-101 is a small molecule, anticancer agent that targets multiple proteins such as histone deacetylase (HDAC), EGFR and erb-b2 receptor TK 2 (HER2). Due to this multifunction of CUDC-101, it has been reported that a combination of EGFR inhibitors with CUDC-101 might help overcome DR [80]. However, the treatment with CUDC-101 alone may still suffer from DR as shown in the second bar. EKB-569 (pelitinib) showed its effectiveness in lung cancer-bearing T790M mutation in the previous study [81]. Consistently, in our result, this drug showed the smallest difference of IC50 compared to other drugs. This means that pelitinib may act on the cells harboring the secondary mutation. One limitation of this study is that the GDSC cell lines included only a small number of DR mutations. When larger pharmacological data sets for this purpose are available, we will include them for further examination and validation of our candidates.
Figure 5.
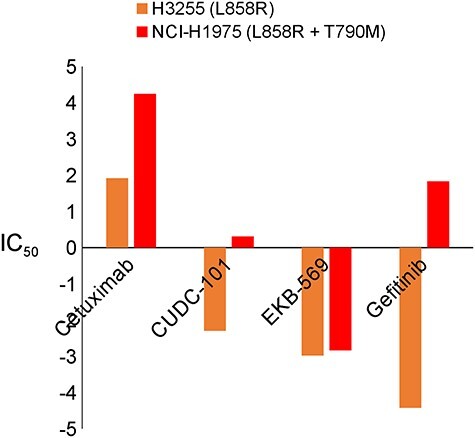
IC50 values between cells with primary only mutated and both primary and secondary mutated EGFR. IC50 value of EGFR with secondary mutation T790M shows the evidence of drug resistance.
Comparison of relative free energy of binding between wild-type and mutant EGFR
To characterize the changes on binding affinity and conformation due to mutations, we performed molecular dynamics (MD) simulation followed by structure analysis and binding free energy calculation for two drugs, afatinib and gefitinib, that bind to EGFR. The starting conformations of both complexes came from a crystal structure of EGFR L858R mutant (PDB code: 4LQM [82]). The ligand-binding poses were determined by applying the corresponding transformation matrixes generated by the least-square fitting of the secondary structures of the receptor. The root-mean-square deviation (RMSD) values are 0.321 and 0.267 Å for EGFR/afatinib (PDB code: 4G5J [83]) and EGFR/gefitinib (PDB code: 2ITY [84]), respectively. The aligned structures are shown in Figure 6. Computational mutagenesis for the four mutations of EGFR (G719S, D761Y, T790M and L861Q) was manually performed and the missing coordinates were added with the Leap module of the AMBER software package [85]. When there is no experimental structure available for a drug in complex with EGFR, one can perform molecular docking simulations to predict the possible binding modes, which can be evaluated by MD simulations and binding free energy calculations. We used the data from these MD simulations to predict the binding free energies of these two drugs for EGFR and its mutants. MM-PBSA (molecular mechanics–Poisson-Boltzmann surface area) binding free energy calculations and MM-GBSA (mechanics-generalized Born surface area) binding free energy decompositions [86–89] were performed for the collected snapshots after the MD trajectories were stabilized (see Methods). Both drugs are potent inhibitors of EGFR and the measured ki values are 0.5 [90] and 0.4 nM [91] for afatinib and gefitinib, respectively. The absolute values of different MM-PBSA energy terms are listed in Table 5. The absolute MM-PBSA binding free energies calculated with an internal dielectric constant of 1.0 are ~2–3 kcal/mol underestimated for both drugs (Table 5), the mutation effect on the binding affinities is overall reasonably predicted. For afatinib, which formed a covalent bond with the receptor with CYS797, the T790M single mutation significantly increases the binding affinity, while the L858R mutation decreases the binding affinity. In contrast, for gefitinib, which does not form any covalent bond with the receptor, both the L858R and T790M mutations increase the binding affinity. For both drugs, the double mutation (L858R/T790M) significantly decreases the binding affinities. The pattern of the G-loop mutation, G719S, is different between the two drugs due to the same fact that afatinib covalently binds to the receptor but gefitinib not. The G719S mutation significantly increases the binding affinity of afatinib, but decreases that of gefitinib. The double mutation (L858R/G719S) decreases the binding affinity for both drugs as expected (Supplementary Figures S1 and S2).
Figure 6.
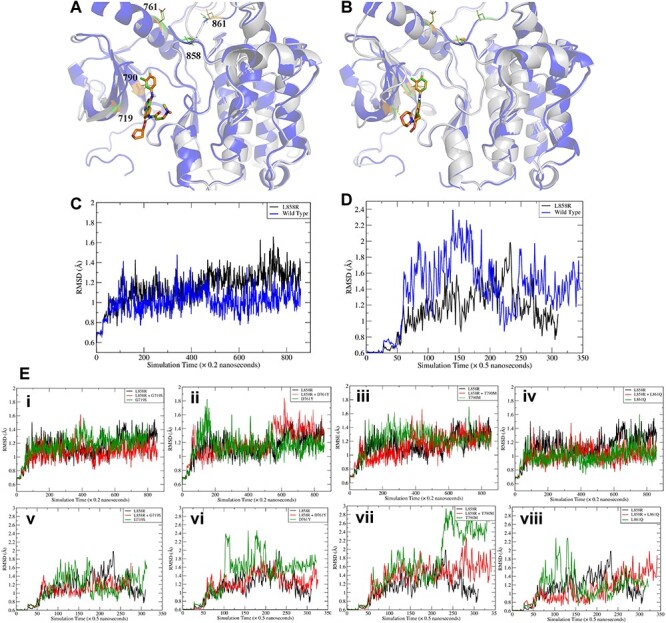
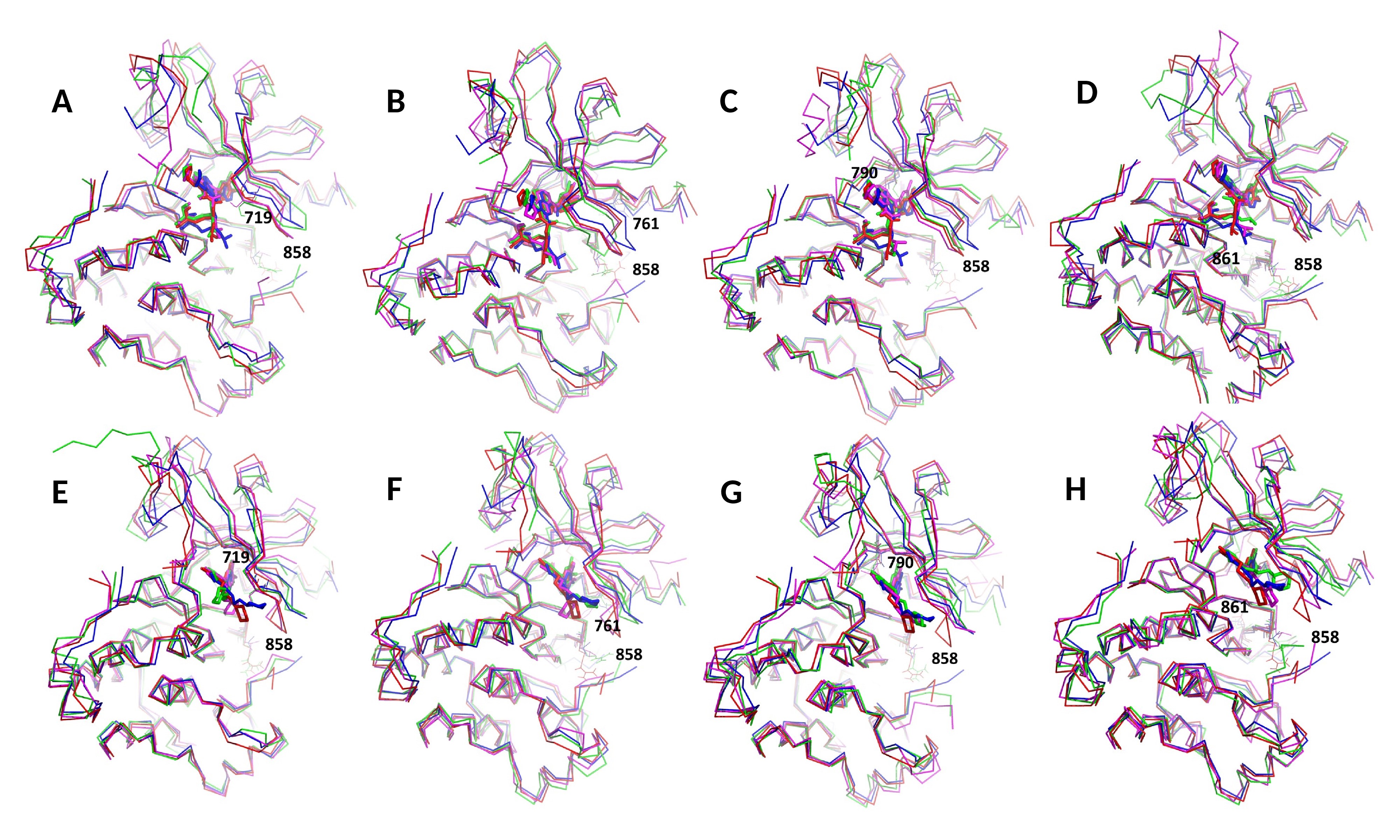
Comparisons of ligand-bound crystal structures and RMSD of wild type EGFR versus the mutant. (A) Comparisons of ligand bound crystal structures of the L858R mutant and wild type EGFR with afatinib (PDB code 2ITY) and (B) gefitinib (PDB code 4G5J). The PDB code of the L858R mutant crystal structure is 4LQM. (C-D) Comparisons of RMSD of the L858R mutant and wild type EGFR with afatinib (C) and gefitinib (D). (E) RMSD of main chain atoms of EGFR compared to a wild type crystal structure (PDB 2ITY for afatinib and 4G5J for gefitinib). (i) and (v): G719S; (ii) and (vi): D761Y; (iii) and (vii): T790M; (iv) and (viii): L861Q. i-iv are for afatinib and v-viii are for gefitinib. It is shown that all double mutants have comparable RMSDs (red curves, all ~1.2 Å for afatinib and ~1.4 for gefitinib). T790M mutants have relatively large RMSDs for both drugs.
Table 5.
Individual energy terms of MM-PBSA binding free energies of afatinib and gefitinib binding to EGFR and its mutants
| AA change | ΔEVDW ± SD | ΔEeel ± SD |
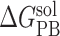 ± SD ± SD |
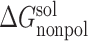 ± SD ± SD |
−TΔS ± SD | ΔGbind ± SD | ΔΔGbind |
|---|---|---|---|---|---|---|---|
| Afatinib | |||||||
| Wild type | −61.40 ± 0.10 | −48.15 ± 0.18 | 73.63 ± 0.27 | −4.98 ± 0.00 | −26.44 ± 0.04 | −10.82 ± 0.11 | 0.00 |
| L858R | −57.30 ± 0.07 | −39.64 ± 0.11 | 63.08 ± 0.35 | −4.86 ± 0.01 | −25.08 ± 0.05 | −9.82 ± 0.15 | 1.00 |
| T790M | −61.81 ± 0.12 | −51.89 ± 0.04 | 75.89 ± 0.24 | −4.89 ± 0.00 | −26.59 ± 0.04 | −12.52 ± 0.04 | -1.70 |
| L858R/T790M | −56.20 ± 0.19 | −44.91 ± 0.51 | 67.44 ± 0.63 | −4.81 ± 0.01 | −25.08 ± 0.01 | −9.70 ± 0.09 | 1.12 |
| G719S | −61.60 ± 0.07 | −49.44 ± 0.13 | 72.29 ± 0.17 | −5.08 ± 0.00 | −27.02 ± 0.01 | −13.41 ± 0.06 | -2.53 |
| L858R/G719S | −59.91 ± 0.17 | −44.81 ± 0.42 | 69.70 ± 0.73 | −5.03 ± 0.01 | −26.18 ± 0.02 | −9.95 ± 0.42 | 0.87 |
| D761Y | −59.70 ± 0.15 | −43.60 ± 0.32 | 68.53 ± 0.33 | −5.00 ± 0.01 | −25.79 ± 0.07 | −10.18 ± 0.16 | 0.64 |
| L858R/D761Y | −60.82 ± 0.21 | −43.61 ± 0.62 | 68.08 ± 0.41 | −4.95 ± 0.01 | −26.34 ± 0.05 | −11.18 ± 0.29 | -0.36 |
| L861Q | −57.05 ± 0.19 | −45.41 ± 0.19 | 68.56 ± 0.21 | −4.85 ± 0.01 | −25.25 ± 0.05 | −9.62 ± 0.02 | 1.20 |
| L858R/L861Q | −57.84 ± 0.41 | −44.37 ± 0.22 | 69.58 ± 0.27 | −4.93 ± 0.00 | −25.50 ± 0.00 | −7.71 ± 0.31 | 3.11 |
| Gefitinib | |||||||
| Wild type | −55.40 ± 0.14 | −13.78 ± 0.18 | 40.40 ± 0.43 | −4.52 ± 0.01 | −24.05 ± 0.02 | −9.25 ± 0.36 | 0.00 |
| L858R | −55.89 ± 0.24 | −13.48 ± 0.22 | 38.70 ± 0.04 | −4.62 ± 0.01 | −24.38 ± 0.07 | −10.92 ± 0.21 | -1.67 |
| T790M | −57.62 ± 0.24 | −15.56 ± 0.49 | 41.91 ± 0.35 | −4.45 ± 0.01 | −24.84 ± 0.07 | −10.88 ± 0.05 | -1.63 |
| L858R/T790M | −52.81 ± 0.29 | −11.59 ± 0.34 | 36.72 ± 0.28 | −4.28 ± 0.01 | −23.32 ± 0.03 | −8.64 ± 0.20 | 0.61 |
| G719S | −54.02 ± 0.10 | −23.07 ± 0.18 | 50.34 ± 0.55 | −4.33 ± 0.01 | −23.75 ± 0.03 | −7.33 ± 0.51 | 1.92 |
| L858R/G719S | −53.62 ± 0.41 | −12.49 ± 0.33 | 39.51 ± 0.18 | −4.40 ± 0.01 | −23.72 ± 0.08 | −7.29 ± 0.26 | 1.96 |
| D761Y | −54.15 ± 0.13 | −15.11 ± 0.25 | 41.75 ± 0.34 | −4.57 ± 0.01 | −23.78 ± 0.03 | −8.30 ± 0.28 | 0.95 |
| L858R/D761Y | −54.48 ± 0.31 | −17.72 ± 0.15 | 45.08 ± 0.60 | −4.39 ± 0.01 | −23.85 ± 0.08 | −7.66 ± 0.19 | 1.59 |
| L861Q | −51.40 ± 0.22 | −17.36 ± 0.17 | 42.07 ± 0.41 | −4.31 ± 0.01 | −22.94 ± 0.06 | −8.06 ± 0.18 | 1.19 |
| L858R/L861Q | −52.43 ± 0.04 | −13.57 ± 0.31 | 38.82 ± 0.43 | −4.23 ± 0.00 | −23.24 ± 0.05 | −8.18 ± 0.74 | 1.07 |
Note: All energy terms are in kcal/mol. The relative binding free energy of a mutant, ΔΔGbind, is calculated by subtracting the MM-PBSA binding free energy of the wild type from that of the mutant. The following is the detailed description of energy terms listed in the table. ΔEVDW: van der Waals energy. ΔEeel: electrostatic energy. 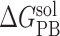 : the polar part of the solvation free energy.
: the polar part of the solvation free energy. 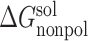 : the nonpolar part of the solvation free energy. −TΔS: the entropic contribution to the binding free energy. ΔGbind, the binding free energy, which is the sum of the above individual energy terms. ΔΔGbind is the relative binding free energy compared to the wild type. SD: standard deviation.
: the nonpolar part of the solvation free energy. −TΔS: the entropic contribution to the binding free energy. ΔGbind, the binding free energy, which is the sum of the above individual energy terms. ΔΔGbind is the relative binding free energy compared to the wild type. SD: standard deviation.
For the αC-helix (D761Y) and A-Loop (L861Q) mutations, the ligand-binding affinities of both drugs are much less affected, because the two residues have no direct contact with the ligands. As shown in Table 5, the relative binding free energies, ΔΔGbind, are rather small for both the D761Y and L861Q single mutations and the L858R/D761Y and L858R/L861Q double mutations, except for Afatinib binding to L858R/D761Y for which the binding affinity is very low. We believed that allosteric regulation plays a more important role in explaining the drug resistance of the two mutations as detailed below. Overall, the prediction performance is acceptable given the fact that MM-PBSA is inferior to a theoretically more rigorous method like free-energy perturbation and thermodynamic integration [92, 93]. It is encouraging that the MM-PBSA method can correctly predict that the additional gatekeeper and G-loop mutations can significantly decrease the binding potency of the L858R mutant for both drugs. It is demonstrated by Figure 6E that the main chain RMSDs of the secondary structures of EGFR/afatinib are ~1.2 Å, smaller than those (~1.4 Å) for EGFR/gefitinib. EGFR/afatinib is more stable than EGFR/gefitinib, which is reasonable because afatinib is covalently bonded to the receptor but gefitinib not. This observation is particularly true for the T790M and D761Y mutants. Representative MD structures of both the wild types and mutants are shown in Supplementary Figure S3.
To investigate the mutation effects of D761Y and L861Q, we performed correlation analysis using a method developed by Kong and Karplus [94]. All the calculated residue–residue correlations are normalized. For afatinib, the mean residue–residue correlations are 0.0292, 0.0298 for L858R/D761Y and L858R/L861Q, respectively, while the corrections for gefitinib are 0.0277 and 0.0288 for the two double mutants. The result suggests that afatinib has larger impact than gefitinib on the signaling pathways. The key signaling pathways linking D761 and L861 to the key residues for protein–ligand bindings were also identified by network analysis. As shown in Figure 7, hot spots residues of protein–ligand binding, such as LYS745, are linked by residues 761 and 861 through one or several bridging residues. Overall, the mutation on D761 has stronger effect on protein–ligand binding than L861, since the interaction pathways between them are shorter and have high correlation. The conclusion that afatinib has stronger impact than gefitinib on the signaling pathways is further confirmed by the interaction pathways shown in Figure 7, where afatinib has shorter and more highly correlated interaction pathways than gefitinib. Furthermore, we inspected the trajectories for L858R/D761Y and L858R/L861Q, respectively. As shown in Figure 7Bi, D761Y mutation may bring an opportunity on forming strong molecular interactions between Y761 and K860, that is, a hydrogen bond or cation-π interaction. As a result, the conformation of the αC-helix could be obviously changed and the space of the binding site for the drugs may be consequently changed, leading to a shift in the binding mode of the drug. Similar phenomenon is observed in the mutation system of L858R/L861Q, as shown in Figure 7Bii, a hydrogen bond between Q861 and Y764 is observed. However, when comparing the conformations between these different mutations, it seems the conformational change caused by the D761Y is larger than L861Q, which is consistent with the correlation analysis, as the average correlation is smaller for D761Y than L861Q for both drugs.
Figure 7.
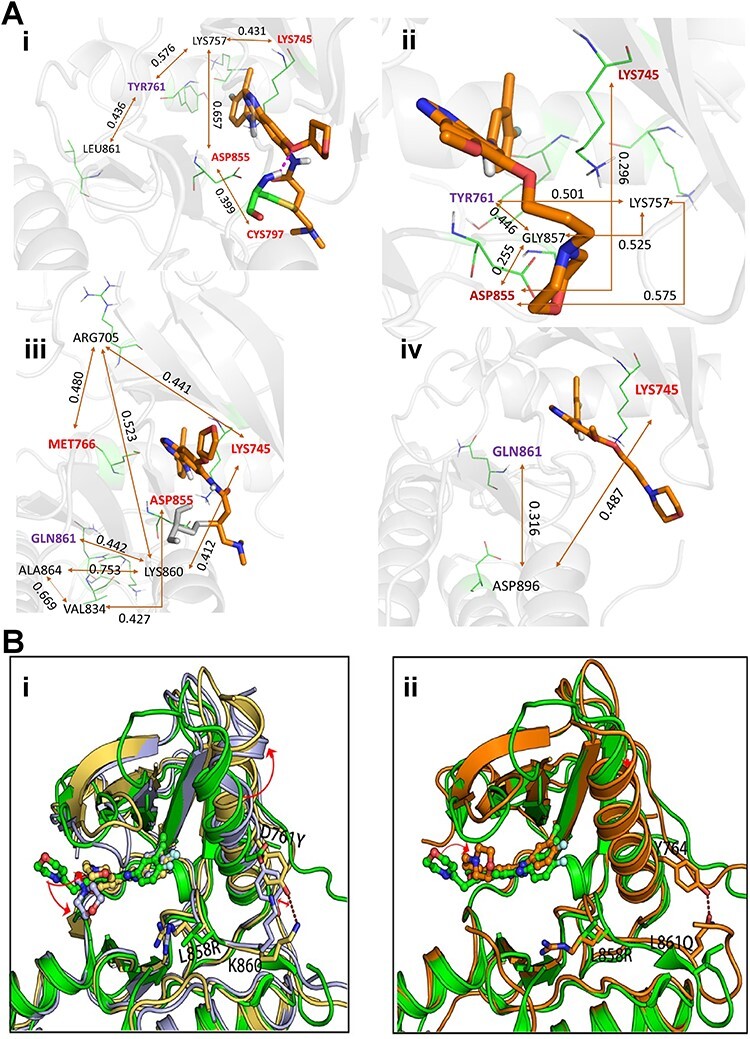
Identified interaction pathways and comparison of conformational changes due to L858R/D761Y and L858R/L861Q. (A) Interaction pathways identified by analyzing the correlations of the collected MD snapshots using Kong and Karplus’s method. (i) and (ii) L858R/D761Y; (iii) and (iv) L858R/L861Q. i and iii are for afatinib. ii and iv are for gefitinib. The number adhered to the double-head arrow indicates the correlation (ranged from 0 to 1) between two residues. The key residues which have strong contact with the ligands are labeled in red. The origin of the pathway, TYR761 or GLN861, is colored in purple. (B) Comparison of conformational changes caused by mutation L858R/D761Y (i) or L858R/L861Q (ii). Green cartoon is the experimentally obtained binding mode of gefitinib to wide type of EGFR (PDB code: 2ITY), the other colored cartoons are the binding modes of gefitinib to mutant L858R/D761Y or mutant L858R/L861Q obtained from MD simulations described in main text. Red dashed lines indicate hydrogen bonds.
Discussion
In this study, we systematically collected and curated four main protein substructures that are related to drug response and resistance and then identified 3318 mutations in 358 human kinases in these substructures as the potential candidates of association with DR. These candidates were pinpointed by our analytical framework (Figure 2B) using four large cancer genomics datasets: TCGA, ICGC, COSMIC and GDSC. Our analytical strategy could find known and novel mutations in KD hotspots (Figure 4). Among the 358 kinase candidates, we selected more reliable kinases (n = 197) with multiple variants at the same hotspot residue, based on the assumption that these kinases are common in high mutation frequency in cancer. Indeed, many known mutations with DR were included in these lists. These mutations serve as the candidates that induce different drug response, either resistant or more sensitive response, using protein structure analysis methods or cell-based assays in near future. Note that we found a feature that multiple mutations occur on a specific site, especially at gatekeeper residues (Table 1). This is commonly observed in oncogenic regions, as such mutations (e.g. EGFTR T790 site; BRAF V600 and Q61 sites) may have gained some advantages on driving cellular signaling, leading to abnormal cell growth [95].
This study has several limitations, such as incomplete curation of the protein substructure sequences, the overlap of the somatic mutation data in the three data sets (TCGA, ICGC and COSMIC), and the limited validation by simulations and molecular docking. However, these limitations do not impact much our analysis. For example, we used the union of the somatic mutations in three datasets, so overlap will not affect our results. In summary, our result is still first comprehensive and unique genomic/clinical resource for drug resistance hotspot mutations in four critical protein substructures of the human kinome. This study will be helpful for the development of personalized medicine and the design of next-generation KIs.
We attempted to find evidence that shows the drug response of different mutations from the public chemical reaction data sets like GDSC. We found the evidence of EGFR’s T790M gatekeeper mutation in DR by comparing the drug sensitivity values between the cell line with primary mutation (L858R) and the cell line with both of primary and secondary mutations (L858R + T790M). We identified the consistent result across four KIs and the result explained the most well-known case of the kinase mutation inducing DR. Through the computational mutagenesis and molecular modeling approaches, we identified the effects of four different hotpot mutations of EGFR (G719S, D761Y, T790M and L861Q) on kinase–ligand interaction. Our simulation study confirmed the working mechanism of afatinib again for T790M gatekeeper and G719S G-loop mutations, which have direct interaction with drugs. From the correlation and trajectories studies on αC-helix D761Y and the A-Loop L861Q mutations, which do not have direct contact with the ligand, we revealed evidence that these mutations may bring an opportunity of forming strong molecular interactions in the proximal regions, which may further affect the kinas–ligand binding affinity by differentiating the space of binding sites. In future work, we may expand the selection of candidate DR mutations for computational validation. For example, a web server has been recently built for evaluating drug resistance mutations in kinases by molecular docking. Using this online service, we may obtain better candidate information from the assessment of the effects of drug resistance mutations on kinase–ligand interactions. In addition, we will continue on the curation of the protein substructures as well as other functional annotations, such as the transformation of accessible chromatin, 3D nucleome and 4D nucleome, and then map the somatic mutations to them for identifying critical functional sites. Finally, we may find other types of mutations such as splicing variants and investigate their features in DR in cancer.
Materials and methods
Annotations of four types of protein substructures
We collected the information of four DR hotspots in human kinases from three resources: Kinase Sequence Database (KSD, July 2016) [12], the Universal Protein Resource (UniProt, June 2017) [96] and the Protein database of the National Center for Biotechnology Information (NCBI, June 2017) [97]. Using these data sets, we arranged the following information: hotspot residue, gene symbol, RefSeq mRNA accession, RefSeq protein accession, UniProt accession and sequence for 538 unique human kinases. The specific detail is provided below.
Collection of gatekeeper loci in human kinases. KSD contains gatekeeper and ATP-binding sites for 946 partial KD sequences, not the full-length amino-acid sequence. Since this information is not based on full-length protein sequence, we ran Basic Local Alignment Search Tool for Protein (BLASTP) to convert these amino acid residue loci into the full protein sequences [98]. From the hits of BLASTP for all 946 partial protein sequences, we extracted those with 100% identity hits only. In total, we obtained 348 kinases having gatekeeper residue information (Supplementary Table S1).
Collection of G-loop and αC-helix in human kinases. Through the literature review, we found that the G-loop lies between the β1 and β2 strands and contains a consensus GxGxxG motif [99]. After downloading the gff format file describing the β strand loci for all reviewed human kinases from UniProt, we first searched the locations of the first two β strands (β1 and β2) within the KD. Because some kinases have the first β strand located before the starting point of the KD, we allowed a ± 2 amino acids [2] window in the search. For kinases that have at least two β strands in the KD, we searched for the motif ‘GxGxxG’ in the middle of the β1 and β2 strands. For the kinases that do not have complete secondary structure information on UniProt, we checked the first 22 residues in the KD to look for the G-loop motif with ±2 AA window, since we found that the G-loop motif usually occurs in the first 20 amino acids in the KD. If a kinase had more than one KD, we searched for the G-loop (and αC-helix too) in all of the KD separately. This analysis resulted in 175 kinases having the G-loop residue loci information.
The αC-helix is defined as the first α-helix in the KD from the N-terminus and is located between β3 and β4 strands [27, 100]. However, in special kinase groups like the AGC kinase, which has 16 protein kinase families including protein kinase A, G and C families (PKA, PKC, PKG), the aC-helix is the second helix following a short αB-helix [101]. To determine the location of αC-helix, we adopted a similar strategy to that we used for the G-loop loci. We first determined the range of the KD and all the α-helix loci inside the domain for each kinase based on the full-length protein sequences using the gff format data from UniProt. We labeled the first α-helix as the αC-helix except for AGC kinases, which the second α-helix was labeled. Since the KD usually starts with a β strand, we did not check if the first α-helix begins outside the KD. We obtained αC-helix loci in 245 kinases.
Collection of A-loop in human kinases. We considered the kinases that were described as ‘Reviewed human kinase’ and whose KD locus information is defined from UniProt. As a result, 499 unique kinases were selected. For these validated kinases, we searched for A-loop information from GenPept from NCBI Protein. Using batch Entrez available at the NCBI database, we retrieved the A-loop loci information of 367 kinases.
Overlap of hotspot loci with point somatic mutations in cancer
We downloaded somatic point mutation data from four databases: TCGA, ICGC, COSMIC and GDSC. We overlapped the kinase mutation hotspot loci with non-synonymous single nucleotide variations (nsSNVs) from these mutation data sets. The UniProt accessions across all kinases from the hotspot loci table were mapped to all RefSeq mRNA accessions using the BioMart data mining tool of Ensembl [102]. We selected the mutations whose Refseq mRNA accessions were matched. We kept the mutations when the amino acid sequence of the original residue at the mutation site matched the one at the full-length amino acid sequence. Among 3327 nsSNVs, we obtained 86, 462, 705 and 2077 nsSNVs in Gatekeeper, G-loop, αC-helix and A-loop, respectively.
Relative stability change of protein structure by a hotspot mutation
We calculated the relative stability change of protein structure by a DR hotspot mutation using MuPro1.1, a computational tool that predicts the protein stability changes for single-site mutation using support vector machine [53]. A score of energy change (ΔΔG) less than 0 means that the mutation would decrease protein stability. The smaller the score is, the more confidence the prediction has. A score larger than 0 means that the mutation would increase the protein stability. The larger the score is, the more confidence the prediction has.
Drug sensitivity scores of cell lines harboring DR hotspot mutations
We downloaded the cell line information including somatic point mutations, treated drug name and drug sensitivity scores (IC50) from the Database Genomics of Drug Sensitivity in Cancer (GDSC) [18]. To search the kinases that have the DR hotspot mutations and IC50 values for the KIs, we overlapped the nsSNVs from GDSC with our hotspot loci. Since the GDSC used the cell lines from COSMIC, there is no matched normal samples in this data set. Therefore, we adopted the following comparison strategy. If there is a kinase with the primary mutation in multiple cell lines, some of which had a secondary mutation, we considered secondary mutations may confer DR, and compared their IC50 values with those cell lines without the secondary mutation.
Molecular dynamics simulation and free energy calculation
We studied a total of 20 EGFR complexes with either afatinib or gefitinib resides in the binding pocket, including the wild type, five single mutants (G719S, D761Y, T790M, L858R, L861Q) and four double mutants (L858R/G719S, L8585R/D761Y, L858R/T790M and L858R/L861Q). All 20 MD systems consist of one copy of EGFR, one copy of afatinib or gefitinib, 16 265 TIP3P [103] water molecules, 50 Cl− and a number of Na+ to neutralize the MD systems. For the force field parameters, the partial atomic charges of two ligands were derived by using the RESP [104] program to fit the HF/6-31G* electrostatic potentials generated using the GAUSSIAN 16 software package [105]. The other force field parameters came from GAFF [106] and the AMBER FF14SB [107] force field was used to model proteins. The residue topologies for ligands were prepared using the Antechamber module [108]. MD simulation was performed to produce isothermal–isobaric ensembles using the pmemd.cuda program in AMBER 18 [85]. The Particle Mesh Ewald (PME) method [109] was used to calculate the full electrostatic energy of a unit cell in a macroscopic lattice of repeating images. All bonds were constrained using the SHAKE algorithm [110] in both the minimization and MD simulation stages. For each MD simulation, the system was first relaxed to remove any possible steric clashes by a set of 10 000-step minimizations with the main chain atoms restrained using the harmonic restraint force constants decreased from 20 to 10, 5 and 1 kcal/mol/Å2, progressively. After that, the system was further relaxed by a 10 000-step minimization without any constraints or restraints. There were three phases for the subsequent MD simulations: the relaxation phase, the equilibrium phase and the sampling phase. In the relaxation phase, the simulation system was heated up progressively from 50 to 250 K at steps of 50 K. At each temperature, a 1-ns MD simulation was run without any restraints or constraints. In the following equilibrium phase, the system was equilibrated at 298 K, 1 bar for 10 ns. Finally, a 150-ns MD simulation was performed at 298 K, 1 bar to produce NTP (constant temperature and pressure) ensembles. In total, 1500 snapshots were recorded from the last simulation. A total of 300 snapshots were evenly selected for the MM-PBSA binding free energy calculation. The following are the additional settings for constant pressure MD simulations performed in this work: temperature was regulated using Langevin dynamics [111] with a collision frequency of 5 ps−1; pressure was regulated using the isotropic position scaling algorithm with the pressure relaxation time set to 1.0 ps; integration of the equations of motion was conducted at a time step of 1 fs for the relaxation phase and 2 fs for the equilibrium and sampling phases.
MM-PB/SA energy calculation and correlation analysis
For each MD snapshot, the molecular mechanical (MM) energy (EMM) and the MM-PB/SA solvation free energy were calculated without further minimization [86–89, 112, 113]. Key parameters controlling the MM-PB/SA analyses were listed as follows: external dielectric constant, ~80; internal dielectric constant, ~4; and the surface tension for estimating the nonpolar solvation energy, ~0.054. The Parse radii [114] was used in the MM-PB/SA solvation calculation using the Delphi package (http://compbio.clemson.edu/delphi). The entropic term was estimated using a method described elsewhere [115]. We conducted residue–residue correlation analysis for L858R/D761Y and L585R/L861Q for both afatinib and gefitinib using the method in Kong and Karplus [94]. Instead of only applying electrostatic and van der Waals energies in the analysis, the solvent effect was also considered by using a Generalized Born model [116]. For each MD system, 6000 MD snapshots were recorded for correlation analysis and the residue-residue interaction energies were calculated using the Sander program in AMBER18.
Competing Interests
The authors declare that they have no competing interests.
Key Points
We systematically collected and curated four main protein substructures that are related to drug response and resistance.
We identified 3318 somatic mutations in 358 kinases which formed 702 drug resistance hotspots from the four large cancer genomics datasets (TCGA, ICGC, COSMIC and GDSC).
We identified more reliable candidate mutations in 197 kinases that had multiple variants at the same hotspot residue.
We reported new evidence that the C-helix D761Y and the A-Loop L861Q mutations in EGFR may form strong molecular interactions in the proximal regions and further affect the kinase–ligand binding affinity.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank the Kinase Sequence Database for making the gatekeeper loci information available, Dr Choel Kim and Mrs Liying Qin in Baylor College of Medicine for the valuable discussion on the hotspot loci, and Drs Fang Bai, Feixiong Cheng and Junfei Zhao in the lab for their valuable help.
Pora Kim is an assistant professor in the School of Biomedical Informatics, The University of Texas Health Science Center at Houston. Her research interest includes bioinformatics and cancer genomics.
Hanyang Li was a student in Department of Bioengineering, Rice University and a visiting student in the School of Biomedical Informatics, The University of Texas Health Science Center at Houston. His research interest includes computational biology and machine learning.
Junmei Wang is an associate professor in Department of Pharmaceutical Sciences and Computational Chemical Genomic Screening Center, School of Pharmacy, University of Pittsburg. His research interest includes computer modeling and simulation of protein–ligand interactions and other biological events, pharmacometrics and computational systems pharmacology.
Zhongming Zhao holds chair professor for Precision Health and is the founding director of the Center for Precision Health, School of Biomedical Informatics, The University of Texas Health Science Center at Houston. He directs the Bioinformatics and Systems Medicine Laboratory and UTHealth Cancer Genomics Core. His research interest includes translational bioinformatics, integrative genomics and methodology development.
Funding
This work was partially supported by National Institutes of Health grants (R01LM012806 and P30DA035778A1) and Cancer Prevention and Research Institute of Texas (CPRIT RP180734 and RP170668). The funders had no role in the study design, data collection, and analysis, decision to publish, or preparation of the manuscript. Computational support from the Center for Research Computing of University of Pittsburgh is acknowledged.
References
- 1. Gross S, Rahal R, Stransky N, et al. Targeting cancer with kinase inhibitors. J Clin Invest 2015;125:1780–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uitdehaag JC, Verkaar F, Alwan H, et al. A guide to picking the most selective kinase inhibitor tool compounds for pharmacological validation of drug targets. Br J Pharmacol 2012;166:858–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Klaeger S, Heinzlmeir S, Wilhelm M, et al. The target landscape of clinical kinase drugs. Science 2017;358:eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheng F, Jia P, Wang Q, et al. Quantitative network mapping of the human kinome interactome reveals new clues for rational kinase inhibitor discovery and individualized cancer therapy. Oncotarget 2014;5:3697–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roskoski R, Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol Res 2019;144:19–50. [DOI] [PubMed] [Google Scholar]
- 6. Ahronian LG, Corcoran RB. Strategies for monitoring and combating resistance to combination kinase inhibitors for cancer therapy. Genome Med 2017;9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jia P, Jin H, Meador CB, et al. Next-generation sequencing of paired tyrosine kinase inhibitor-sensitive and -resistant EGFR mutant lung cancer cell lines identifies spectrum of DNA changes associated with drug resistance. Genome Res 2013;23:1434–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miller GD, Bruno BJ, Lim CS. Resistant mutations in CML and Ph(+)ALL - role of ponatinib. Biol Theory 2014;8:243–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Inukai M, Toyooka S, Ito S, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res 2006;66:7854–8. [DOI] [PubMed] [Google Scholar]
- 10. Wu JY, Yu CJ, Chang YC, et al. Effectiveness of tyrosine kinase inhibitors on "uncommon" epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin Cancer Res 2011;17:3812–21. [DOI] [PubMed] [Google Scholar]
- 11. Ko B, Paucar D, Halmos B. EGFR T790M: revealing the secrets of a gatekeeper. Lung Cancer (Auckl) 2017;8:147–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gajiwala KS, Wu JC, Christensen J, et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci U S A 2009;106:1542–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Barouch-Bentov R, Sauer K. Mechanisms of drug resistance in kinases. Expert Opin Investig Drugs 2011;20:153–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rogozin IB, Pavlov YI. Theoretical analysis of mutation hotspots and their DNA sequence context specificity. Mutat Res 2003;544:65–85. [DOI] [PubMed] [Google Scholar]
- 15. Jia P, Wang Q, Chen Q, et al. MSEA: detection and quantification of mutation hotspots through mutation set enrichment analysis. Genome Biol 2014;15:489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miller ML, Reznik E, Gauthier NP, et al. Pan-cancer analysis of mutation hotspots in protein domains. Cell Syst 2015;1:197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Forbes SA, Beare D, Boutselakis H, et al. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 2017;45:D777–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iorio F, Knijnenburg TA, Vis DJ, et al. A landscape of Pharmacogenomic interactions in cancer. Cell 2016;166:740–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vuong H, Cheng F, Lin CC, et al. Functional consequences of somatic mutations in cancer using protein pocket-based prioritization approach. Genome Med 2014;6:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng F, Liu C, Lin CC, et al. A gene gravity model for the evolution of cancer genomes: a study of 3,000 cancer genomes across 9 cancer types. PLoS Comput Biol 2015;11:e1004497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao J, Cheng F, Wang Y, et al. Systematic prioritization of druggable mutations in approximately 5000 genomes across 16 cancer types using a structural genomics-based approach. Mol Cell Proteomics 2016;15:642–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen Q, Cheng F, Song H, et al. Proteome-scale investigation of protein allosteric regulation perturbed by somatic mutations in 7,000 cancer genomes. Am J Hum Genet 2017;100:5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim P, Jia P, Zhao Z. Kinase impact assessment in the landscape of fusion genes that retain kinase domains: a pan-cancer study. Brief Bioinform 2018;19:450–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chmielecki J, Peifer M, Jia P, et al. Targeted next-generation sequencing of DNA regions proximal to a conserved GXGXXG signaling motif enables systematic discovery of tyrosine kinase fusions in cancer. Nucleic Acids Res 2010;38:6985–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McClendon CL, Kornev AP, Gilson MK, et al. Dynamic architecture of a protein kinase. Proc Natl Acad Sci U S A 2014;111:E4623–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bartova I, Otyepka M, Kriz Z, et al. Activation and inhibition of cyclin-dependent kinase-2 by phosphorylation; a molecular dynamics study reveals the functional importance of the glycine-rich loop. Protein Sci 2004;13:1449–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kornev AP, Taylor SS. Defining the conserved internal architecture of a protein kinase. Biochim Biophys Acta 1804;2010:440–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Scheeff ED, Eswaran J, Bunkoczi G, et al. Structure of the pseudokinase VRK3 reveals a degraded catalytic site, a highly conserved kinase fold, and a putative regulatory binding site. Structure 2009;17:128–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aleksandrov A, Simonson T. Molecular dynamics simulations show that conformational selection governs the binding preferences of imatinib for several tyrosine kinases. J Biol Chem 2010;285:13807–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Manning G, Whyte DB, Martinez R, et al. The protein kinase complement of the human genome. Science 2002;298:1912–34. [DOI] [PubMed] [Google Scholar]
- 31. Bertrand D, Drissler S, Chia BK, et al. ConsensusDriver improves upon individual algorithms for predicting driver alterations in different cancer types and individual patients. Cancer Res 2018;78:290–301. [DOI] [PubMed] [Google Scholar]
- 32. Cheng F, Zhao J, Zhao Z. Advances in computational approaches for prioritizing driver mutations and significantly mutated genes in cancer genomes. Brief Bioinform 2016;17:642–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lasater EA, Massi ES, Stecula A, et al. Novel TKI-resistant BCR-ABL1 gatekeeper residue mutations retain in vitro sensitivity to axitinib. Leukemia 2016;30:1405–9. [DOI] [PubMed] [Google Scholar]
- 34. Toyokawa G, Seto T. Updated evidence on the mechanisms of resistance to ALK inhibitors and strategies to overcome such resistance: clinical and preclinical data. Oncol Res Treat 2015;38:291–8. [DOI] [PubMed] [Google Scholar]
- 35. Morgillo F, Della Corte CM, Fasano M, et al. Mechanisms of resistance to EGFR-targeted drugs: lung cancer. ESMO Open 2016;1:e000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim DH, Kwak Y, Kim ND, et al. Antitumor effects and molecular mechanisms of ponatinib on endometrial cancer cells harboring activating FGFR2 mutations. Cancer Biol Ther 2016;17:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mori M, Kaneko N, Ueno Y, et al. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Invest New Drugs 2017;35:556–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ashman LK, Griffith R. Therapeutic targeting of c-KIT in cancer. Expert Opin Investig Drugs 2013;22:103–15. [DOI] [PubMed] [Google Scholar]
- 39. Barouch-Bentov R, Che J, Lee CC, et al. A conserved salt bridge in the G loop of multiple protein kinases is important for catalysis and for in vivo Lyn function. Mol Cell 2009;33:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Khorashad JS, Kelley TW, Szankasi P, et al. BCR-ABL1 compound mutations in tyrosine kinase inhibitor-resistant CML: frequency and clonal relationships. Blood 2013;121:489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vajpai N, Strauss A, Fendrich G, et al. Solution conformations and dynamics of ABL kinase-inhibitor complexes determined by NMR substantiate the different binding modes of imatinib/nilotinib and dasatinib. J Biol Chem 2008;283:18292–302. [DOI] [PubMed] [Google Scholar]
- 42. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54. [DOI] [PubMed] [Google Scholar]
- 43. Kim EY, Cho EN, Park HS, et al. Compound EGFR mutation is frequently detected with co-mutations of actionable genes and associated with poor clinical outcome in lung adenocarcinoma. Cancer Biol Ther 2016;17:237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Palmieri L, Rastelli G. alphaC helix displacement as a general approach for allosteric modulation of protein kinases. Drug Discov Today 2013;18:407–14. [DOI] [PubMed] [Google Scholar]
- 45. Ceccon M, Mologni L, Bisson W, et al. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol Cancer Res 2013;11:122–32. [DOI] [PubMed] [Google Scholar]
- 46. Balak MN, Gong Y, Riely GJ, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res 2006;12:6494–501. [DOI] [PubMed] [Google Scholar]
- 47. Kancha RK, von Bubnoff N, Bartosch N, et al. Differential sensitivity of ERBB2 kinase domain mutations towards lapatinib. PLoS One 2011;6:e26760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Davare MA, Vellore NA, Wagner JP, et al. Structural insight into selectivity and resistance profiles of ROS1 tyrosine kinase inhibitors. Proc Natl Acad Sci U S A 2015;112:E5381–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ma W, Kantarjian H, Zhang X, et al. Mutation profile of JAK2 transcripts in patients with chronic myeloproliferative neoplasias. J Mol Diagn 2009;11:49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ruff EF, Muretta JM, Thompson AR, et al. A dynamic mechanism for allosteric activation of aurora kinase a by activation loop phosphorylation. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zabriskie MS, Eide CA, Tantravahi SK, et al. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell 2014;26:428–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schrock AB, Lai A, Ali SM, et al. Mutation of MET Y1230 as an acquired mechanism of crizotinib resistance in NSCLC with MET exon 14 skipping. J Thorac Oncol 2017;12:e89–90. [DOI] [PubMed] [Google Scholar]
- 53. Cheng J, Randall A, Baldi P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins 2006;62:1125–32. [DOI] [PubMed] [Google Scholar]
- 54. Stewart EL, Tan SZ, Liu G, et al. Known and putative mechanisms of resistance to EGFR targeted therapies in NSCLC patients with EGFR mutations-a review. Transl Lung Cancer Res 2015;4:67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer 2010;10:760–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A 2008;105:2070–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gazdar AF. Activating and resistance mutations of EGFR in non-small-cell lung cancer: role in clinical response to EGFR tyrosine kinase inhibitors. Oncogene 2009;28(Suppl 1):S24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Klughammer B, Brugger W, Cappuzzo F, et al. Examining treatment outcomes with Erlotinib in patients with advanced non-small cell lung cancer whose tumors harbor uncommon EGFR mutations. J Thorac Oncol 2016;11:545–55. [DOI] [PubMed] [Google Scholar]
- 59. Ai X, Sun Y, Wang H, et al. A systematic profile of clinical inhibitors responsive to EGFR somatic amino acid mutations in lung cancer: implication for the molecular mechanism of drug resistance and sensitivity. Amino Acids 2014;46:1635–48. [DOI] [PubMed] [Google Scholar]
- 60. Sequist LV, Waltman BA, Dias-Santagata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 2011;3:75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Avizienyte E, Ward RA, Garner AP. Comparison of the EGFR resistance mutation profiles generated by EGFR-targeted tyrosine kinase inhibitors and the impact of drug combinations. Biochem J 2008;415:197–206. [DOI] [PubMed] [Google Scholar]
- 62. Bean J, Riely GJ, Balak M, et al. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res 2008;14:7519–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Karachaliou N, Molina-Vila MA, Rosell R. The impact of rare EGFR mutations on the treatment response of patients with non-small cell lung cancer. Expert Rev Respir Med 2015;9:241–4. [DOI] [PubMed] [Google Scholar]
- 64. O'Hare T, Eide CA, Deininger MW. Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007;110:2242–9. [DOI] [PubMed] [Google Scholar]
- 65. Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2002;2:117–25. [DOI] [PubMed] [Google Scholar]
- 66. Blasiak J, Hoser G, Bialkowska-Warzecha J, et al. Mitochondrial mutagenesis in BCR-ABL1-expressing cells sensitive and resistant to imatinib. Acta Biochim Pol 2016;63:365–70. [DOI] [PubMed] [Google Scholar]
- 67. Parker WT, Lawrence RM, Ho M, et al. Sensitive detection of BCR-ABL1 mutations in patients with chronic myeloid leukemia after imatinib resistance is predictive of outcome during subsequent therapy. J Clin Oncol 2011;29:4250–9. [DOI] [PubMed] [Google Scholar]
- 68. Parker WT, Ho M, Scott HS, et al. Poor response to second-line kinase inhibitors in chronic myeloid leukemia patients with multiple low-level mutations, irrespective of their resistance profile. Blood 2012;119:2234–8. [DOI] [PubMed] [Google Scholar]
- 69. Bitencourt R, Zalcberg I, Louro ID. Imatinib resistance: a review of alternative inhibitors in chronic myeloid leukemia. Rev Bras Hematol Hemoter 2011;33:470–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Baccarani M, Cortes J, Pane F, et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol 2009;27:6041–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010;363:1734–9. [DOI] [PubMed] [Google Scholar]
- 72. Ou SH, Milliken JC, Azada MC, et al. ALK F1174V mutation confers sensitivity while ALK I1171 mutation confers resistance to alectinib. The importance of serial biopsy post progression. Lung Cancer (Auckl) 2016;91:70–2. [DOI] [PubMed] [Google Scholar]
- 73. Kim S, Kim TM, Kim DW, et al. Heterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancer. J Thorac Oncol 2013;8:415–22. [DOI] [PubMed] [Google Scholar]
- 74. Infarinato NR, Park JH, Krytska K, et al. The ALK/ROS1 inhibitor PF-06463922 overcomes primary resistance to crizotinib in ALK-driven neuroblastoma. Cancer Discov 2016;6:96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hallberg B, Palmer RH. The role of the ALK receptor in cancer biology. Ann Oncol 2016;27(Suppl 3):iii4–iii15. [DOI] [PubMed] [Google Scholar]
- 76. Frost MJ, Ferrao PT, Hughes TP, et al. Juxtamembrane mutant V560GKit is more sensitive to Imatinib (STI571) compared with wild-type c-kit whereas the kinase domain mutant D816VKit is resistant. Mol Cancer Ther 2002;1:1115–24. [PubMed] [Google Scholar]
- 77. Guo T, Hajdu M, Agaram NP, et al. Mechanisms of sunitinib resistance in gastrointestinal stromal tumors harboring KITAY502-3ins mutation: an in vitro mutagenesis screen for drug resistance. Clin Cancer Res 2009;15:6862–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kuang Y, Rogers A, Yeap BY, et al. Noninvasive detection of EGFR T790M in gefitinib or erlotinib resistant non-small cell lung cancer. Clin Cancer Res 2009;15:2630–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Janjigian YY, Smit EF, Groen HJ, et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov 2014;4:1036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang J, Pursell NW, Samson ME, et al. Potential advantages of CUDC-101, a multitargeted HDAC, EGFR, and HER2 inhibitor, in treating drug resistance and preventing cancer cell migration and invasion. Mol Cancer Ther 2013;12:925–36. [DOI] [PubMed] [Google Scholar]
- 81. Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A 2005;102:7665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yasuda H, Park E, Yun CH, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med 2013;5:216ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Solca F, Dahl G, Zoephel A, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther 2012;343:342–50. [DOI] [PubMed] [Google Scholar]
- 84. Yun CH, Boggon TJ, Li Y, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007;11:217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Case DA, Ben-Shalom IY, Brozell SR, et al. AMBER 2018. San Francisco: University of California, 2018. [Google Scholar]
- 86. Wang J, Morin P, Wang W, et al. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of Efavirenz by docking and MM-PBSA. J Am Chem Soc 2001;123:5221–30. [DOI] [PubMed] [Google Scholar]
- 87. Kuhn B, Gerber P, Schulz-Gasch T, et al. Validation and use of the MM-PBSA approach for drug discovery. J Med Chem 2005;48:4040–8. [DOI] [PubMed] [Google Scholar]
- 88. Hou T, Wang J, Li Y, et al. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 2011;51:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hou T, Wang J, Li Y, et al. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized born surface area methods. II. The accuracy of ranking poses generated from docking. J Comput Chem 2011;32:866–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Eskens FA, Mom CH, Planting AS, et al. A phase I dose escalation study of BIBW 2992, an irreversible dual inhibitor of epidermal growth factor receptor 1 (EGFR) and 2 (HER2) tyrosine kinase in a 2-week on, 2-week off schedule in patients with advanced solid tumours. Br J Cancer 2008;98:80–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res 2004;64:6652–9. [DOI] [PubMed] [Google Scholar]
- 92. Wang L, Wu Y, Deng Y, et al. Accurate and reliable prediction of relative ligand binding potency in prospective drug discovery by way of a modern free-energy calculation protocol and force field. J Am Chem Soc 2015;137:2695–703. [DOI] [PubMed] [Google Scholar]
- 93. Wang J, Hou T. Recent advances on aqueous solubility prediction. Comb Chem High Throughput Screen 2011;14:328–38. [DOI] [PubMed] [Google Scholar]
- 94. Kong Y, Karplus M. Signaling pathways of PDZ2 domain: a molecular dynamics interaction correlation analysis. Proteins 2009;74:145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Xia J, Jia P, Hutchinson KE, et al. A meta-analysis of somatic mutations from next generation sequencing of 241 melanomas: a road map for the study of genes with potential clinical relevance. Mol Cancer Ther 2014;13:1918–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. UniProt Consortium T . UniProt: the universal protein knowledgebase. Nucleic Acids Res 2018;46:2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Coordinators NR . Database resources of the National Center for biotechnology information. Nucleic Acids Res 2016;44:D7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Boratyn GM, Camacho C, Cooper PS, et al. BLAST: a more efficient report with usability improvements. Nucleic Acids Res 2013;41:W29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Fabbro D, Cowan-Jacob SW, Moebitz H. Ten things you should know about protein kinases: IUPHAR review 14. Br J Pharmacol 2015;172:2675–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Taylor SS, Keshwani MM, Steichen JM, et al. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos Trans R Soc Lond B Biol Sci 2012;367:2517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 2010;11:9–22. [DOI] [PubMed] [Google Scholar]
- 102. Zerbino DR, Achuthan P, Akanni W, et al. Ensembl 2018. Nucleic Acids Res 2018;46:D754–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983;79:926. [Google Scholar]
- 104. Bayly CI, Cieplak P, Cornell WD, et al. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges- the RESP model. J Phys Chem 1993;97:10269–80. [Google Scholar]
- 105. Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 16, Revision B.01. Wallingford CT: Gaussian, Inc., 2016. [Google Scholar]
- 106. Wang JM, Wolf RM, Caldwell JW, et al. Development and testing of a general amber force field. J Comput Chem 2004;25:1157–74. [DOI] [PubMed] [Google Scholar]
- 107. Maier JA, Martinez C, Kasavajhala K, et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 2015;11:3696–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Wang J, Wang W, Kollman PA, et al. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 2006;25:247–60. [DOI] [PubMed] [Google Scholar]
- 109. Darden T, Perera L, Li L, et al. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure 1999;7:R55–60. [DOI] [PubMed] [Google Scholar]
- 110. Miyamoto S, Kollman PA. Settle - an analytical version of the shake and rattle algorithm for rigid water models. J Comput Chem 1992;13:952–62. [Google Scholar]
- 111. Larini L, Mannella R, Leporini D. Langevin stabilization of molecular-dynamics simulations of polymers by means of quasisymplectic algorithms. J Chem Phy 2007;126:104101. [DOI] [PubMed] [Google Scholar]
- 112. Page CS, Bates PA. Can MM-PBSA calculations predict the specificities of protein kinase inhibitors? J Comput Chem 2006;27:1990–2007. [DOI] [PubMed] [Google Scholar]
- 113. Kongsted J, Ryde U. An improved method to predict the entropy term with the MM/PBSA approach. J Comput Aided Mol Des 2009;23:63–71. [DOI] [PubMed] [Google Scholar]
- 114. Sitkoff D, Sharp KA, Honig B. Accurate calculation of hydration free energies using macroscopic solvent models. J Phys Chem 1994;98:1978–88. [Google Scholar]
- 115. Wang J, Hou T. Develop and test a solvent accessible surface area-based model in conformational entropy calculations. J Chem Inf Model 2012;52:1199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hawkins GD, Cramer C, Truhlar DG. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J Phys Chem 1996;100:19824–39. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.