

Abstract
Over 90% of pancreatic ductal adenocarcinomas (PDAC) express mesothelin (MSLN). Overexpression or knockdown of MSLN has been implicated in PDAC aggressiveness. This activity has been ascribed to MSLN-induced activation of MAPK or NF-κB signaling pathways and to interaction of MSLN with its only known binding partner, MUC16. Here, we used CRISPR/Cas9 gene editing to delete MSLN from PDAC, then restored expression of wild-type (WT) or Y318A mutant MSLN by viral transduction. We found that MSLN KO cells grew in culture and as subcutaneous tumors in mouse xenografts at the same rate as WT cells but formed intraperitoneal metastases poorly. Complementation with WT MSLN restored intraperitoneal growth, whereas complementation with Y318A mutant MSLN, which does not bind MUC16, was ineffective at enhancing growth in both MUC16(+) and MUC16(−) models. Restoration of WT MSLN did enhance growth but did not affect cell-to-cell binding, cell viability in suspension or signaling pathways previously identified as contributing to the protumorigenic effect of MSLN. RNA deep sequencing of tumor cells identified no changes in transcriptional profile that could explain the observed phenotype. Furthermore, no histologic changes in tumor cell proliferation or morphology were observed in mature tumors. Examination of nascent MSLN KO tumors revealed decreased microvascular density as intraperitoneal tumors were forming, followed by decreased proliferation, which resolved by 2 weeks postimplantation. These data support a model whereby MSLN expression by tumor cells contributes to metastatic colonization.
Implications:
MSLN confers a growth advantage to tumor cells during colonization of peritoneal metastasis. Therapeutic blockade of MSLN might limit peritoneal spread.
Introduction
Mesothelin (MSLN) is a cell surface differentiation antigen with normal expression largely limited to the mesothelial cells of the pleura, pericardium, and peritoneum. Many solid tumors also express MSLN, including mesothelioma, pancreatic and biliary carcinomas, ovarian, lung, thymic, and gastric cancers (1). Multiple studies have shown that >90% of pancreatic ductal adenocarcinomas (PDAC) express MSLN, and three-quarters of patient tumors express MSLN in at least 25% of cancer cells (2–4). Among patients with early-stage PDAC able to undergo potentially curative surgical resection, those with the highest intensity of MSLN expression have >12-fold risk of dying from their tumor within one year of their surgery compared with patients that do not express MSLN (5). Preclinical studies have shown that incubating PDAC cells with exogenous MSLN can increase their invasiveness (6), and that overexpression of MSLN in human PDAC cell lines increases cell growth (7), migration, and invasion in culture. It also increases tumor growth in mouse models, while knockdown retards these processes (8).
MSLN is synthesized as a 71-kDa precursor protein (9). The precursor protein is cleaved by the intracellular protease furin to produce a 31 kD N-terminal fragment called megakaryocyte-potentiating factor (MPF) and mature 40-kDa MSLN that is the C-terminal fragment. The mature glycosylated MSLN is expressed on the cell surface through a glycosylphosphatidylinositol-linkage but can also be cleaved by ADAM metallopeptidase domain 17 enzyme (TACE/ADAM17; ref. 9). This shed form is detected in the serum of some patients (10, 11). The physiologic function of MSLN remains unknown; MSLN knockout mice have no observable phenotype (12). The protein has no conserved domains and shares homology only with stereocillin and otoancorin, two proteins of the inner ear important for mechanoreception of sound stimulation (13). MSLN has no intracellular domain for signaling and a single binding partner has been identified, mucin 16 (MUC16; ref. 14). Although current data suggest that MUC16 binding constitutes one means by which MSLN affects tumorigenicity (6), it remains unclear whether all MSLN activity requires MUC16 binding or if MSLN also acts through other mechanisms such as activation of NF-κB (15) or MAPK pathways (6).
MSLN has recently become a popular target for anticancer therapeutics due to the strong differential expression between normal and tumor cells and lack of expression in the parenchyma of vital organs (9, 16, 17). An array of anti-MSLN therapeutics is currently being tested in clinical trials, including vaccines, antibody-based biologics, and engineered cell therapies (1, 18). All are designed to use MSLN as an address for delivery of the therapeutic moiety, without consideration to whether they disrupt protumorigenic MSLN signaling. There is no published data demonstrating whether these therapeutics interfere with MSLN activity, as the mechanism and phenotype of that activity is poorly defined. Here, we have used CRISPR-Cas9 to genetically eliminate expression of MSLN. We identified a deletion phenotype, and then used this model to help us understand the mechanism of MSLN action and the signaling pathway(s) responsible for the protumorigenic phenotype.
Materials and Methods
Cell culture and reagents
PK-1 and KLM1 cells were the gift of Ira Pastan and MIA PaCa-2 cells were provided by Perwez Hussain (both NCI, Bethesda, MD). Identity was confirmed by short tandem repeat analysis. Cell culture, cell growth assays, and generation of stable cell lines are described in Supplementary Methods. LMB-100 immunotoxin was manufactured by Roche and provided for these studies through a Collaborative Research and Development Agreement. The LMB-74 immunotoxin was produced as described previously (19).
MSLN KO line generation
Design of gRNAs for CRISPR-Cas9 and vector cloning is described in Supplementary Methods. Cas9 and gRNA-containing vectors were nucleofected into KLM1 cells using solution V with a Nucleofector 2b device (Amaxa). The top 5% GFP(+) population was sorted with a MoFlo Astrios sorter (Beckman Coulter). Expanded single-cell clones that lacked MSLN expression were identified by flow cytometry. The pmaxGFP vector (Amaxa), lacking gRNA and Cas9 endonuclease inserts, was used to generate a Mock cell line control as described above. Final products were analyzed for GFP expression to confirm lack of plasmid integration into the genome.
Flow cytometry
Samples were analyzed on a BD FACSCalibur or Canto II as described in Supplementary Methods. For examining noncovalent back-binding of shed MSLN in conditioned medium to intact cells, KO#2 cells detached as described above were incubated for 1 hour with conditioned medium from the cell types of interest, then washed before antibody labeling. Conditioned medium was generated by growing an equal number of the indicated cell type for 24 hours in fresh medium before harvest. MSLN concentration in conditioned medium was assessed by ELISA to confirm similar concentration between comparators.
ELISA
Medium was collected from cultured cells after indicated time, then cleared of debris by centrifugation. Tumor lysates were harvested as described above. MSLN was detected using DMSLNO Kit (R&D Systems). For MPF ELISA, streptavidin-coated SD GOLD 96-well plate (MesoScale Discovery, L15SA-1) was used with biotinylated anti-MPF capture antibody (MPF49) and sulfo-tagged anti-MPF detection antibody (MPF25; from Liang Cao, NCI, Bethesda, MD; ref. 20). Signal was detected using read buffer (MesoScale Discovery, R92TC-3) and luminescence measured with a QuickPlex SQ 120 (Meso-Scale Discovery).
Reverse-phase protein arrays
Proteome Profiler Human Phospho-Kinase Array (ARY003B), Human Protease Array (ARY021B), and Human Angiogenesis Array (ARY007) kits were purchased from R&D Systems. Changes in protein expression were evaluated as per the manufacturer’s instructions.
qPCR
Total RNA was extracted using RNeasy Mini Kit (Qiagen). iScript cDNA Synthesis Kit (Bio-Rad) was used for reverse transcription. Primers were obtained from IDT Technologies (Supplementary Table S1). Real-time qPCR was performed using the iTaq Universal SYBR Green Supermix (Bio-Rad) on a LightCycler 480 instrument (Roche). Gene expression levels were normalized to housekeeping gene HPRT.
Mouse tumor experiments
All animal experiments were performed in accordance with NIH guidelines and approved by the NCI Animal Care and Use Committee. Female 6- to 10-week-old athymic nude mice (Charles River Laboratories) were inoculated either subcutaneously with 3 × 106 cells in 4.0 mg/mL Matrigel (Corning) or intraperitoneally with 3 × 106 cells in RPMI1640 lacking additives. Subcutaneous tumors were measured in two dimensions by digital calipers and tumor volume was calculated using the formula: 0.4 × width2 × length. Cohorts of mice bearing intraperitoneal tumors were euthanized when the first mouse became moribund (~40 days after inoculation) unless indicated otherwise, and then all visible tumor was dissected from the abdominal cavity and weighed. For orthotopic injection, anesthetized mice (7–10-week-old) were inoculated into the pancreas with 2.5 × 105 cells in 4% methylcellulose through laparotomy incision. After 5 to 6 weeks, mice were euthanized, and the pancreas weighed.
Histologic analyses
Samples were sent to Pathology/Histotechnology Laboratory Core facility for all histologic studies. Analysis and quantification were performed by a trained veterinary pathologist affiliated with the Core.
Bioluminescence imaging
d-Luciferin (Luc; Sigma, 2.5 mg per mouse) was injected intraperitoneally and allowed to perfuse for 5 to 10 minutes. Mice were anesthetized under isoflurane at 2% to 5% or euthanized with CO2 inhalation and imaged in an IVIS spectrum and charged-coupled device optical imaging system with Living Image Software (PerkinElmer). All bioluminescence imaging (BLI)–positive regions were collected for pathologic analysis.
RNA-deep sequencing
Total RNA from triplicate samples was assessed by the Sequencing Facility-Illumina-Center for Cancer using a HiSeq 2500 Next Generation Sequencing instrument. See Supplementary Methods for details of data analysis.
Statistical analysis
GraphPad Prism and Microsoft Excel were used for all graphing, statistical calculations, and curve fitting. Data are presented as mean ± SD. Comparisons of >2 groups were assessed by ANOVA. For post hoc analysis and two group comparisons, Mann–Whitney nonparametric test or two-tailed Student t test with Welch correction were used. All experiments were confirmed by repeat. Data are presented in composite form (as noted) when appropriate (N.S., no statistically significant difference).
Results
PK-1 is a pancreatic cancer cell line derived from a patient liver metastasis. The KLM1 pancreatic cancer cell line was produced by using intraportal injection to seed PK-1 cells into the liver, harvesting the tumor that formed, growing out the cells, and repeating the process a second time. This produced a more aggressive cell line adapted for metastatic growth (21). Immunoblot of protein lysates from PK-1 and KLM1 showed increased expression of MSLN protein in KLM1 (Fig. 1A), suggesting that MSLN may contribute to the enhanced metastatic potential of this cell line. Therefore, we used KLM1 as an initial model to investigate the role of MSLN in tumor growth and spread.
Figure 1.
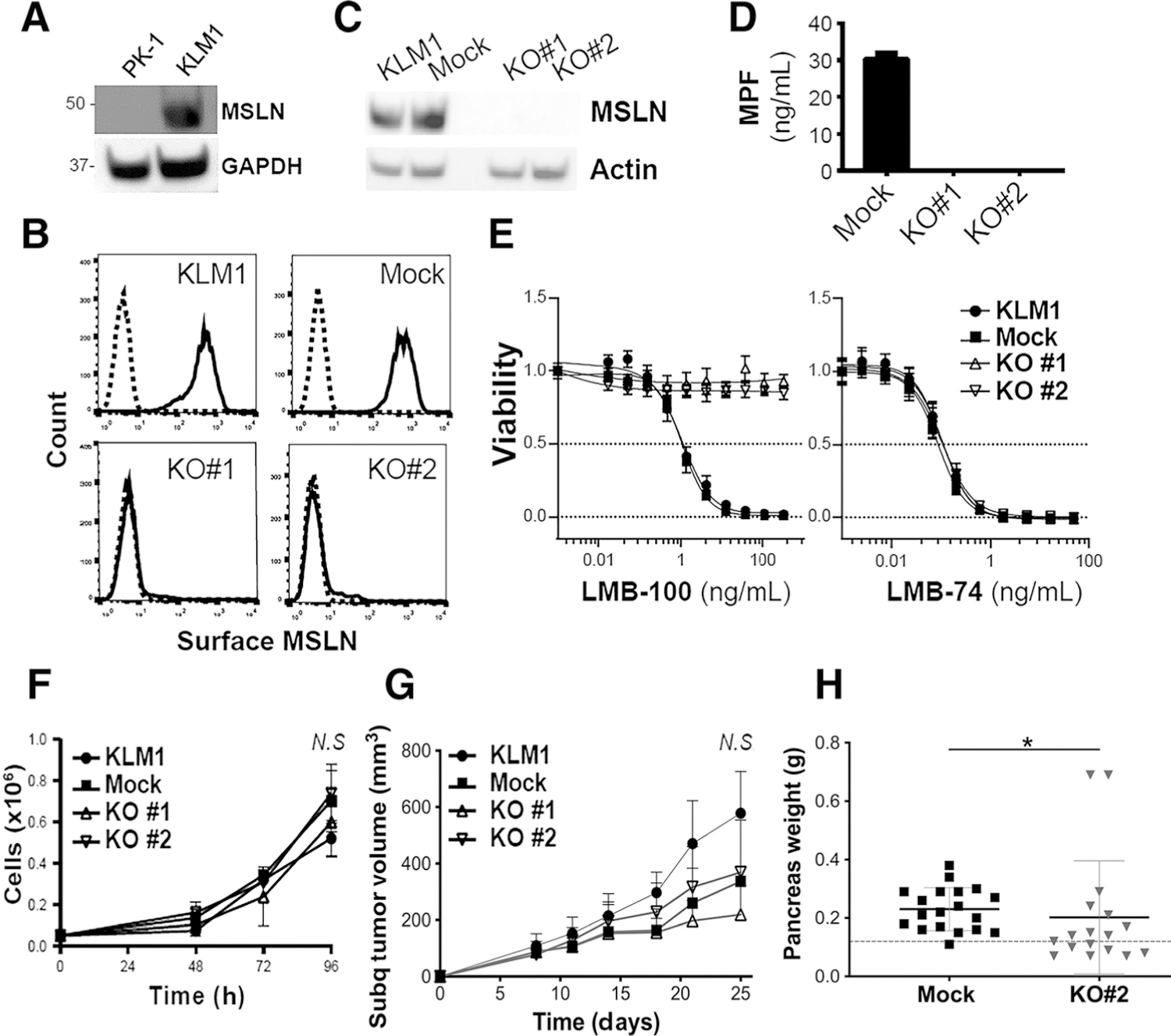
Synthesis and validation of MSLN KO cells and effect on tumor growth. A, Immunoblot of cell culture lysates showing increased expression of MSLN in KLM1 compared with PK1. GAPDH was used as loading control. B, Surface expression of MSLN as measured by flow cytometry on parental KLM1, Mock, KO#1 and KO#2 cell lines. Solid line shows cells stained with anti-MSLN primary antibody and dotted line shows stained with isotype control. C, Immunoblot of cell culture lysates for MSLN (40 kDa). Actin was used as loading control (D). Average MPF concentration in the media of cells cultured for 72 hours was measured by ELISA. Data are the results of triplicate readings from a representative experiment. E, Parental KLM1, Mock, KO#1 and KO#2 were treated with a concentration gradient of the MSLN-targeting immunotoxin, LMB-100 (left), or the transferrin receptor-targeting immunotoxin LMB-74 (right). Representative experiment shows n = 6 replicates per data point. F, In vitro growth rates of cell lines. Biological triplicates from a representative experiment are shown. Cell numbers at 96 hours were not statistically different between groups as determined by ANOVA (N.S.). G, Nude mice were inoculated subcutaneously into the flank with equal numbers of Mock, KO#1 or KO#2 cells and then tumor volume was serially measured by digital calipers over 25 days (average shown for n = 10 mice per group). No statistically significant difference seen by ANOVA (N.S.). H, Nude mice were inoculated orthotopically into the pancreas with equal numbers of Mock or KO#2 cells (Mock n = 19, KO#2 n = 17 over three separate experiments). Mice were euthanized after 5 to 6 weeks. Pancreata were collected and weighed. Dotted line indicates average normal pancreas weight of nude mice (0.12 g; *, P < 0.05; Mann–Whitney nonparametric test).
To generate the KLM1 KO cell line, a gRNA was designed to target the human MSLN gene 12 bp after the translational start site in exon 3 (Supplementary Fig. S1). Sequence alignment of the three human MSLN transcript variants showed that the target sequence was identical across all transcripts. KLM1 cells were transfected with an expression vector coding for this gRNA, the Cas9 enzyme and GFP. No surface MSLN expression was detected in two clones, called KO#1 and KO#2, whereas surface MSLN expression of the empty vector–transfected cell line (Mock) resembled parental KLM1 (Fig. 1B). Loss of MSLN expression was confirmed by immunoblot of cultured cell lysates (Fig. 1C). Loss of MPF expression in the KO lines was confirmed by ELISA using conditioned media from each cell line (Fig. 1D). To further demonstrate the loss of MSLN with a functional assay, we treated these cell lines with LMB-100, an MSLN-targeted recombinant immunotoxin that specifically kills cells expressing surface MSLN, or LMB-74, an identically structured immunotoxin that targets the transferrin receptor (19). All four cell lines were sensitive to LMB-74; however, the MSLN KO cell lines were completely resistant to the cytotoxic effects of LMB-100 (Fig. 1E). Mock cells were similarly sensitive to LMB-100 as parental KLM1, with IC50 in the picomolar range. Together, these data demonstrate successful deletion of MSLN from the KO cell lines.
To determine whether loss of MSLN affects the growth rate of tumor cells, proliferation of KO and Mock cells was assessed. MSLN loss did not affect the growth of the KO cell lines in culture (Fig. 1F), nor when cells were grown as subcutaneous xenografts in nude mice (Fig. 1G). MSLN loss did result in a small but statistically significant decrease in the orthotopic tumor growth of KO#2 cells (Fig. 1H). Intraperitoneal metastases were noted in mice bearing orthotopic tumors (data not shown), but the rates were not documented because it was most likely that these deposits were due to leakage artifact during pancreas inoculation with tumor cell solution.
To examine rates of metastatic colonization, we performed intrasplenic, tail vein, and intraperitoneal inoculations of tumor cells. Despite the origin of the KLM1 cell line, no liver or lung metastases formed after intrasplenic or tail vein inoculations. When these cell lines were inoculated intraperitoneally, a statistically significant decrease in intraperitoneal tumor burden was observed for MSLN KO cells compared with Mock or parental KLM1 (Fig. 2A). When KO lines grew, visible tumors were always located at the inoculation site. These data demonstrate that MSLN expression is important for growth of some pancreatic cancers in the intra-abdominal cavity. Examination of formalin-fixed tumor samples revealed no differences in the gross morphology of the KO tumors (hematoxylin and eosin), proliferation as measured by Ki-67 staining, composition of cancer-associated fibroblasts as measured by staining for smooth muscle actin (SMA), area of collagen deposition as measured by Masson Trichrome, nor microvessel density as measured by vascular endothelial marker CD31 (Fig. 2B). To examine whether MSLN KO delays intraperitoneal tumor growth versus completely preventing intraperitoneal take, KO tumors were grown for 3 additional weeks beyond the initial 6-week experimental endpoint (Fig. 2C). With this additional time, KO tumors grew to the same size as the Mock tumors grown for 3 weeks less (Fig. 2D). Furthermore, MSLN KO did not affect tropism of tumor deposits; KO cells grown for 9 weeks established tumor deposits just as widely throughout the intraperitoneal cavity as Mock cells (Fig. 2E). To confirm that loss of MSLN expression is retained throughout the in vivo experiment in 9-week-old MSLN KO intraperitoneal tumors, we performed immunoblot on these tumor cell lysates. Continued loss of expression of MSLN protein was observed KLM1 (Fig. 2F). Together, these data indicate that MSLN deletion delays pancreatic tumor cell growth in the intraperitoneal cavity.
Figure 2.
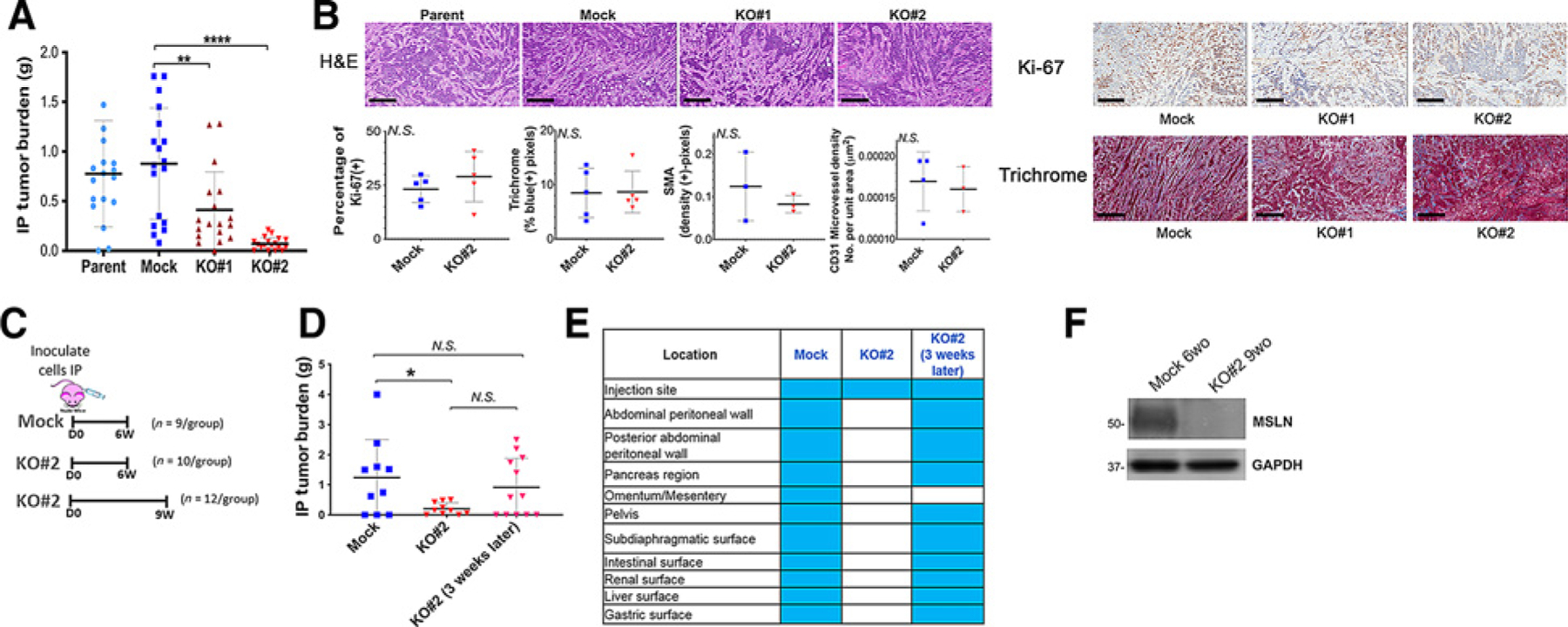
Effect of MSLN KO on intraperitoneal metastasis model. A, Nude mice were inoculated intraperitoneally with 3 × 106 parental KLM1, Mock, KO#1, or KO#2 cells. Mice were euthanized after approximately 6 weeks and all visible tumor in the abdominal cavity was harvested and weighed. Shown are composite results from two experiments (n = 18 for Mock, KO#1 and KO#2; n = 17 for Parent). **, P < 0.01, ****, P < 0.0001 (Mann–Whitney non-parametric test; B), Representative histology stainings of tumors (n = 3–6 mice per group) from (A); scale bars, 300 μm. Samples were analyzed by a consultant veterinary pathologist who found no differences (tumors from n = 3–5 mice obtained from two independent experiments). Quantitation is shown below for staining of Ki-67, SMA (a marker of cancer-associated fibroblasts), collagen on Masson Trichrome and CD31 (a vascular endothelial marker). N.S. = not significant. C, Schema of inoculations for D. D, Experiment in A was repeated but some KO tumors were grown for 3 additional weeks beyond original endpoint (n = 10–12/ group over 3 independent experiments). *, P < 0.05, N.S. = not significant (Mann–Whitney nonparametric test. E, Location of tumor deposits found in animals from D. F, Lysates from harvested KLM1 KO#2 intraperitoneal tumors grown for 9 weeks (9wo) were immunoblotted with anti-MSLN antibody to demonstrate loss of expression of MSLN in vivo. KLM1 Mock intraperitoneal tumors grown for 6 weeks (9wo) were used as positive control for MSLN expression. GAPDH was used as loading control.
Because no histologic changes were observed in end-stage tumors, we hypothesized that MSLN must confer a growth advantage at an earlier period of tumor development. To test this hypothesis, we followed growth and establishment of tumors over time (Fig. 3A). Co-expression of Luc/GFP allowed for early localization and tracking of tumor deposits in the intraperitoneal cavity by BLI (Supplementary Fig. S2). Intraperitoneal tumors were harvested 3 to 14 days postintraperitoneal injection and histologic features were examined to determine which step(s) of the peritoneal carcinomatosis cascade are impaired by MSLN loss. By day 3, MSLN KO tumors had statistically significant decrease in microvascular density as measured by CD31 staining (Fig. 3B). A statistically significant decrease in proliferation and invasion and a more marked decline in microvascular density were observed by 7 days after tumor cell inoculation (Fig. 3B–D). The differences in microvascular density and proliferation resolved by 14 days (Fig. 3B and C) demonstrating that MSLN provides a growth advantage to tumor cells during metastatic seeding. MSLN has no intracellular domain and a single known binding partner, MUC16. MUC16 binds to the N-terminus of cell surface MSLN and mutation of tyrosine-318 of MSLN to alanine (Y318A) has been reported to ablate this binding (22). To confirm that slowed intraperitoneal growth in KO cells is specifically due to MSLN loss and also to determine whether the MSLN/MUC16 interaction contributes to pancreatic tumor intraperitoneal growth, KO#2 cells were transduced with the full-length cDNAs encoding wild-type (WT) or Y318A MSLN (KO#2+WT or KO#2+Y318A, respectively; Fig. 4A). As expected, transduction with WT or Y318A MSLN restored expression of WT MPF (Supplementary Fig. S3A). In addition, KO#2+WT and KO#2+Y318A strongly expressed MSLN protein on the cell surface (Supplementary Fig. S3B). We found that shed WT MSLN in the conditioned medium of KO#2+WT cells bound to KO#2 cells, whereas shed Y318A MSLN did not (Supplementary Fig. S3C), indicating that this mutation does ablate MSLN association with MUC16 on the tumor cell surface. Expression of WT or Y318A MSLN in KO#2 cells did not change the growth rate of these cells in culture as compared with KO#2 or Mock (Fig. 4B). However, restoration of WT MSLN did rescue intraperitoneal growth: KO#2+WT grew just as well in the intraperitoneal cavity as Mock cells (Fig. 4C). Conversely, complementation of Y318A MSLN could not rescue the MSLN KO phenotype: KO#2+Y318A cells grew intraperitoneal tumors just as poorly as KO#2 cells (Fig. 4C). These data conclusively demonstrate that WT MSLN replacement is both necessary and sufficient to reverse the KO phenotype and that the retardation of intraperitoneal growth in KO#2 cells is specific to MSLN loss.
Figure 3.
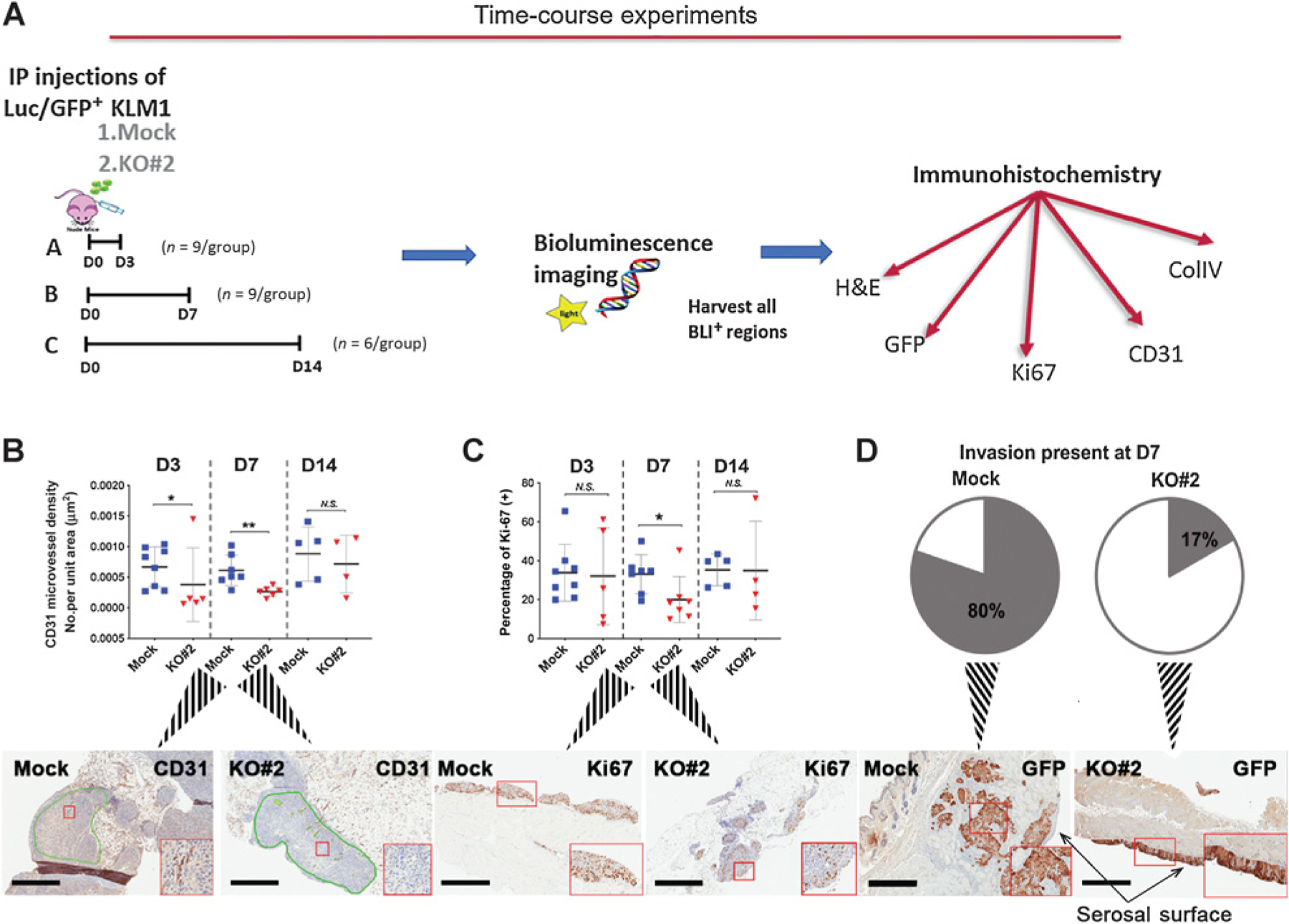
Role of MSLN in establishment of intraperitoneal tumors. A, Schema of time-course experiments. A total of 3 × 106 Luc/GFP cells were inoculated into the mouse intraperitoneal cavity. (n = 4–8/ group over three independent experiments; B–D) Histologic analyses of intraperitoneal tissues containing tumors of (B) microvessel density at 3 (D3), 7 (D7), and 14 (D14) days, green lines show tumor regions selected for pathological analysis (C) proliferation;*, P < 0.05; **, P < 0.01, N.S. = not significant (Mann–Whitney nonparametric test) and (D) invasion of tumor tissue at 7 days after intraperitoneal tumor cell inoculation. 4/5 (80%) Mock and 1/6 (17%) KO#2 tumor tissues showed invasion. Insets (B–D) show magnification of region in red box; scale bars, 600 μm.
Figure 4.
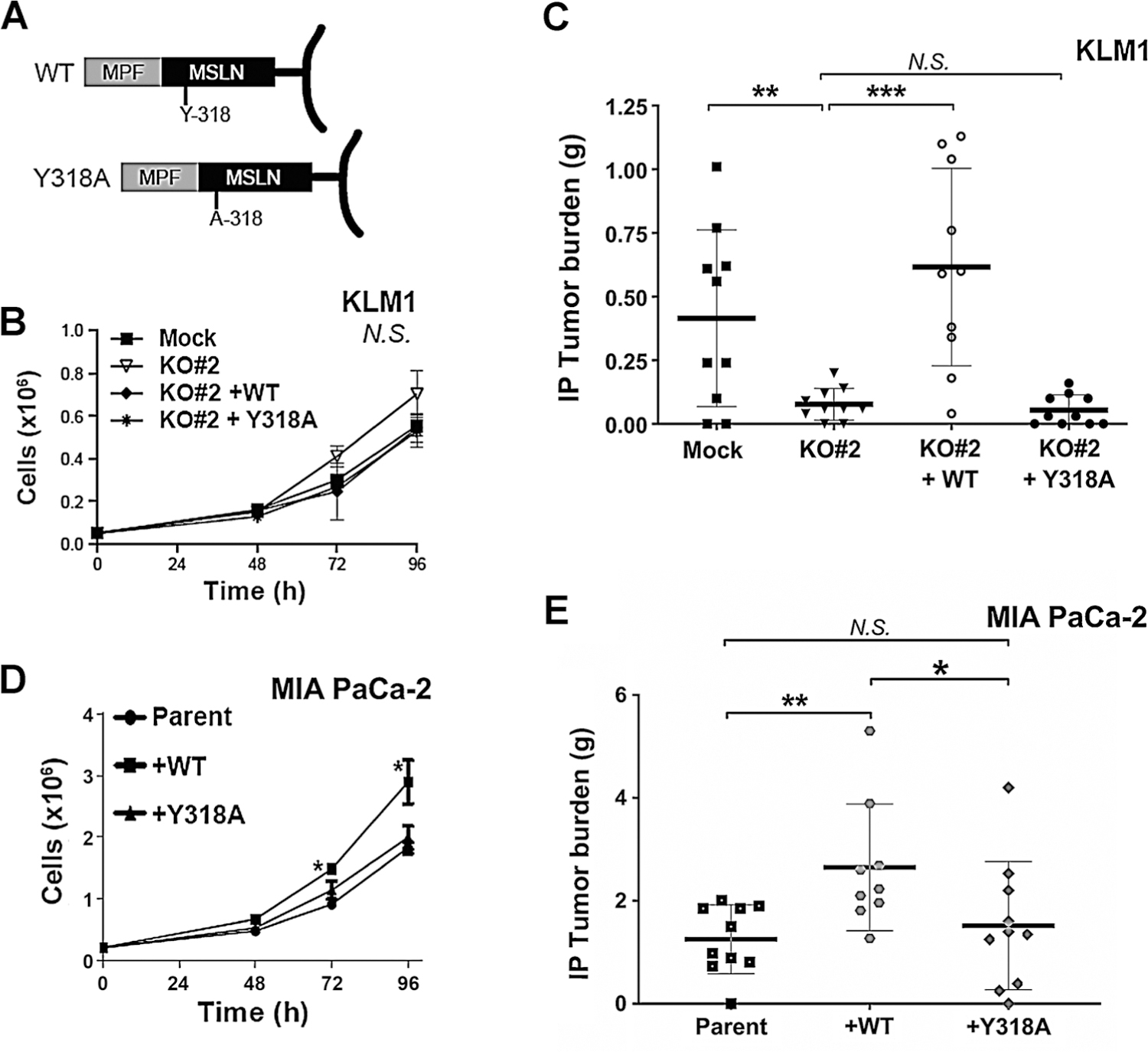
Expression of WT but not Y318A MSLN enhances tumor cell growth. A, Schema of the engineered constructs. B, Growth rate of KLM1 cell lines (Mock, KO#2, and the transduced cell lines KO#2+WT or KO#2+Y318A) in culture was measured by counting triplicate wells on the indicated days. There was no statistically significant difference in cell count at 96 hours as determined by ANOVA (N.S.). C, Nude mice were inoculated into the abdominal cavity with 3 × 106 cells of the indicated cell types. Mice were euthanized after approximately 6 weeks. All visible tumor in the abdominal cavity was harvested and weighed. Figure shows average for 10 mice per group; **, P < 0.01; ***, P < 0.001, N.S. not significant (Mann–Whitney nonparametric test). D, Growth rate of parent MIA PaCa-2 cells and those transduced with WT or Y318A mutant MSLN was measured by counting triplicate wells on the indicated days. A statistically significant increase in growth was found in +WT cells compared with parent and +Y318A (*, P < 0.05 on post hoc comparison). E, Intraperitoneal growth of MIA PaCa-2–derived cells was assessed as in C. Figure shows average for 9–10 mice per group; *, P < 0.05 and **, P < 0.01, N.S. = not significant (Mann–Whitney nonparametric test).
To corroborate the importance of Y-318 in MSLN activity, we performed additional experiments using MIA PaCa-2 cells (23). It has been previously reported that stable overexpression of WT MSLN in MIA PaCa-2 cells, which express little or no endogenous MSLN, increases cell proliferation and migration in cell culture and enhances tumor growth in mouse xenograft models (8, 24, 25). We hypothesized that overexpression of WT MSLN, but not Y318A mutant, in MIA PaCa-2 cells would enhance their growth in culture. Therefore, we transduced MIA PaCa-2 parent cells with the full-length cDNAs encoding WT or Y318A MSLN to produce MIA PaCa-2+WT or MIA PaCa-2+Y318A, respectively. Robust overexpression of MSLN protein was observed in both MIA PaCa-2+WT and MIA PaCa-2+Y318A transduced cell lines (Supplementary Fig. S3D) and conditioned medium (Supplementary Fig. S3E). MSLN was also detected on the surface of these cells (Supplementary Fig. S3F). Proliferation of the two cell lines in culture was compared with parent MIA PaCa-2 cells. We found that addition of WT MSLN enhanced cell growth as seen previously (8, 25); however, cells overexpressing Y318A MSLN grew at the same rate as parent cells that lacked MSLN (Fig. 4D). Intraperitoneal tumor progression of these cell lines corroborated with their growth in culture; addition of WT MSLN enhanced intraperitoneal tumor growth, whereas addition of Y318A MSLN did not (Fig. 4E). MIA PaCa-2 intraperitoneal tumors overexpressing WT or mutant MSLN continued to express the respective transgene throughout the in vivo experiment (Supplementary Fig. S3G) and showed no differences in cellular morphology compared with the parent (Supplementary Fig. S3H). These data confirm that Y318A point mutation is sufficient to eliminate the protumorigenic phenotype of MSLN.
Several different mechanisms might explain why WT MSLN promotes intraperitoneal growth whereas the Y318A MSLN cannot. First, to confirm that transgene expression is retained throughout the in vivo experiment in KLM1 intraperitoneal tumors, we performed immunoblot on KLM1 tumor cell lysates to assess MSLN expression. Continued robust overexpression of both WT and Y318A MSLN proteins was observed (Fig. 5A). Next, we investigated whether MSLN expression affected the ability of tumor cells to grow and survive in low adherence conditions as would be present following intraperitoneal inoculation, but not under other conditions, such as subcutaneous growth or growth on tissue culture plastic. Equal numbers of KLM1-derived cells were plated into ultra-low adherence wells. A colorimetric assay was used to measure viability in this assay rather than counting cells (as in all previous experiments) because the cells rapidly adhered to each other to make three-dimensional clumps under the low adherence conditions. This clumping effect was much reduced in KO#2 cells: Clumps were smaller, appearing as a starry sky of small granules after crystal violet staining rather than plump worm-like aggregates (Fig. 5B). However, because complementation with either WT or Y318A MSLN restored clumping, the clumping phenotype could not be associated with the impaired tumor growth phenotype. In addition, colorimetric assay showed no difference in the viability of the four cell lines after 96 hours of incubation under low adherence conditions (Fig. 5C).
Figure 5.
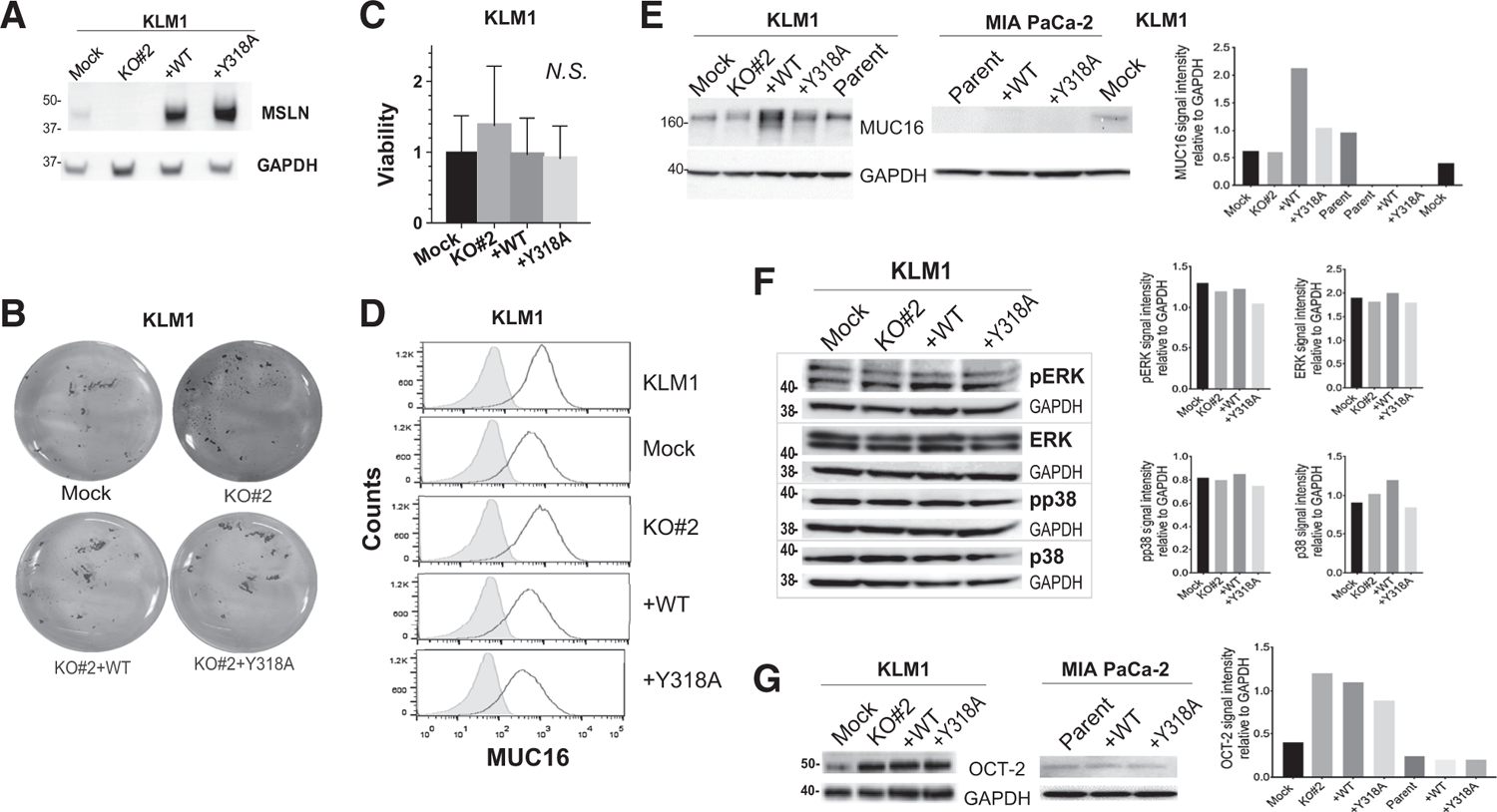
Identifying a mechanism for MSLN tumorigenicity. A, Lysates from harvested KLM1 intraperitoneal tumors were immunoblotted with anti-MSLN antibody to demonstrate continued expression of the transgene in vivo. B and C, Tumor cells were plated at equal numbers onto ultra-low adherence plates. B, After 24 hours, the cells were stained with crystal violet and photographed. C, Viability was measured by colorimetric assay after 72 hours. N.S. = no significant difference. D, Cell membrane expression of MUC16 was measured by flow cytometry (black outline). Shaded peak shows control where primary antibody was omitted. Immunoblots were performed to examine: E, Total MUC16 expression and associated quantitation (F) total and phosphorylated ERK and p38, and (G) total OCT-2 expression and associated quantitation in KLM1 and/or MIA PaCa-2 cultured cell lysates. GAPDH was used as loading control in A and E–G.
Overexpression of MUC16 has previously been shown to correlate with pancreatic cancer cell survival and tumorigenicity, whereas knockdown impairs viability and migration of pancreatic cancer cells (6, 26, 27). We hypothesized that overexpression of MSLN might alter the expression or distribution of MUC16 in pancreatic cancer cells and therefore induce the observed growth phenotypes. In KLM1, we found that surface expression of MUC16 was similar regardless of MSLN expression status (Fig. 5D); however, KO#2 WT cells had higher total MUC16 expression than the other cell types (Fig. 5E). In contrast, MIA PaCa-2 cells expressed no MUC16 regardless of their MSLN expression status (Fig. 5E), as shown previously by others at both mRNA and protein level (26). These data indicate that MUC16 is unlikely to be responsible for the enhanced proliferation of the +WT cells.
Because MSLN loss suppressed angiogenesis during peritoneal metastasis seeding, we investigated changes in abundance of common angiogenesis-related proteins using reverse-phase protein array (RPPA) in Mock, KO#2, KO#2 +WT and KO#2 +Y318A cells. Changes in abundance of plasminogen activator inhibitor-1 (PAI-1) were observed on RPPA (Supplementary Fig. S4A); however, no specific bands were detected on dedicated immunoblot (Supplementary Fig. S4B). To identify other signaling pathways activated by MSLN expression, we used additional RPPAs of signaling phosphoprotein kinases and cellular proteases. Phosphorylation of the MAPK proteins p38 and ERK has previously been associated with MSLN signaling through MUC16-dependent and -independent processes, as well as with changes in matrix metalloprotease 7 (MMP-7; ref. 6). Non-specific changes in pp38 expression that did not correlate with phenotype were observed on RPPA (Supplementary Fig. S4C), but no changes in pERK (Supplementary Fig. S4C) or MMP abundance (Supplementary Fig. S4D) were seen. Dedicated immunoblot confirmed that no consistent changes in phosphorylated or total p38 or ERK occurred in response to genetic manipulations of MSLN expression (Fig. 5F). Changes in the abundance of Cathepsin B that did correspond to phenotype were seen on protease RPPA (Supplementary Fig. S4D). Follow-up with dedicated immunoblot could not confirm this observation (Supplementary Fig. S4E). Despite screening the cancer cells for molecular mediators of MSLN action using three different RPPAs assessing abundance of 135 different potential protein mediators, we were unable to identify any changes that correlated with MSLN expression or intraperitoneal growth phenotype.
Others have shown that MSLN overexpression in MIA PaCa-2 cells causes constitutive activation of the NF-κB signaling axis to promote tumor cell growth. Activated NF-κB then augments transcription of the homeobox transcription factor OCT-2, which enhances repression of miR-198, a tumor suppressor. Because the MSLN coding sequence (CDS) is targeted by miR-198, the reduced expression of miR-198 further enhances MSLN levels (15). Importantly, none of the three putative mIR-198–binding sites in the MSLN CDS overlap the point mutation that produces the mutant Y318A MSLN (Supplementary Fig. S5A). To assess the involvement of the NF-κB signaling pathway in our models, we compared expression of NF-κB downstream target OCT-2 in KLM1 and MIA PaCa-2 cell lines lacking or overexpressing MSLN. Intraperitoneal growth phenotype did not correlate with change in protein expression of OCT-2 in either cell line (Fig. 5G). We did see a small increase in OCT-2 expression in KLM1 KO#2, +WT and +Y318A compared with Mock, which is best explained as a clonal artifact of KO#2 and derivatives. Supporting this, we observed no difference in phosphorylation of NF-κB subunit p65 with changes in MSLN expression, further demonstrating that MSLN expression does not activate NF-κB signaling (Supplementary Fig. S5B). Our data do not support a role for NF-κB–mediated induction of OCT-2 as a mechanism for the protumorigenic phenotype induced by MSLN expression in contrast with a previous report (15).
Because previously reported mechanisms for MSLN action could not adequately account for our phenotypic data, we performed an unbiased screen to identify new targets by comparing mRNA expression patterns in MSLN WT and KOs in both cells grown in culture and in intraperitoneal tumors tissues using RNA deep sequencing. Data have been deposited in NCBI’s Gene Expression Omnibus (GEO) with a GEO Series accession number (GSE)131664. Thirteen curated genes were identified to have at least a 1.5-fold change in expression with P < 0.05 in KO#1 and KO#2 cell lines and tumor tissues as compared with Mock (Fig. 6A). Simple qPCR was used to validate these hits by assessing the pattern of expression in new samples taken from Mock, KO#2, KO#2+WT and KO#2+Y318A cells grown in culture. For a target to qualify as a true hit, mRNA expression must match to the intraperitoneal growth phenotype of each cell type (Supplementary Fig. S6A). Protein phosphatase 1 regulatory inhibitor subunit 1B (PPP1R1B), was the only target of the 13 hits from RNA deep sequencing that passed qPCR validation in the 4 cell lines (Fig. 6B; Supplementary Fig. S6B). Further assessment by immunoblot showed that PPP1R1B was not expressed at the protein level, ruling out this candidate (Fig. 6C). These data indicate that MSLN does not act at the transcriptional level to enhance tumor growth.
Figure 6.
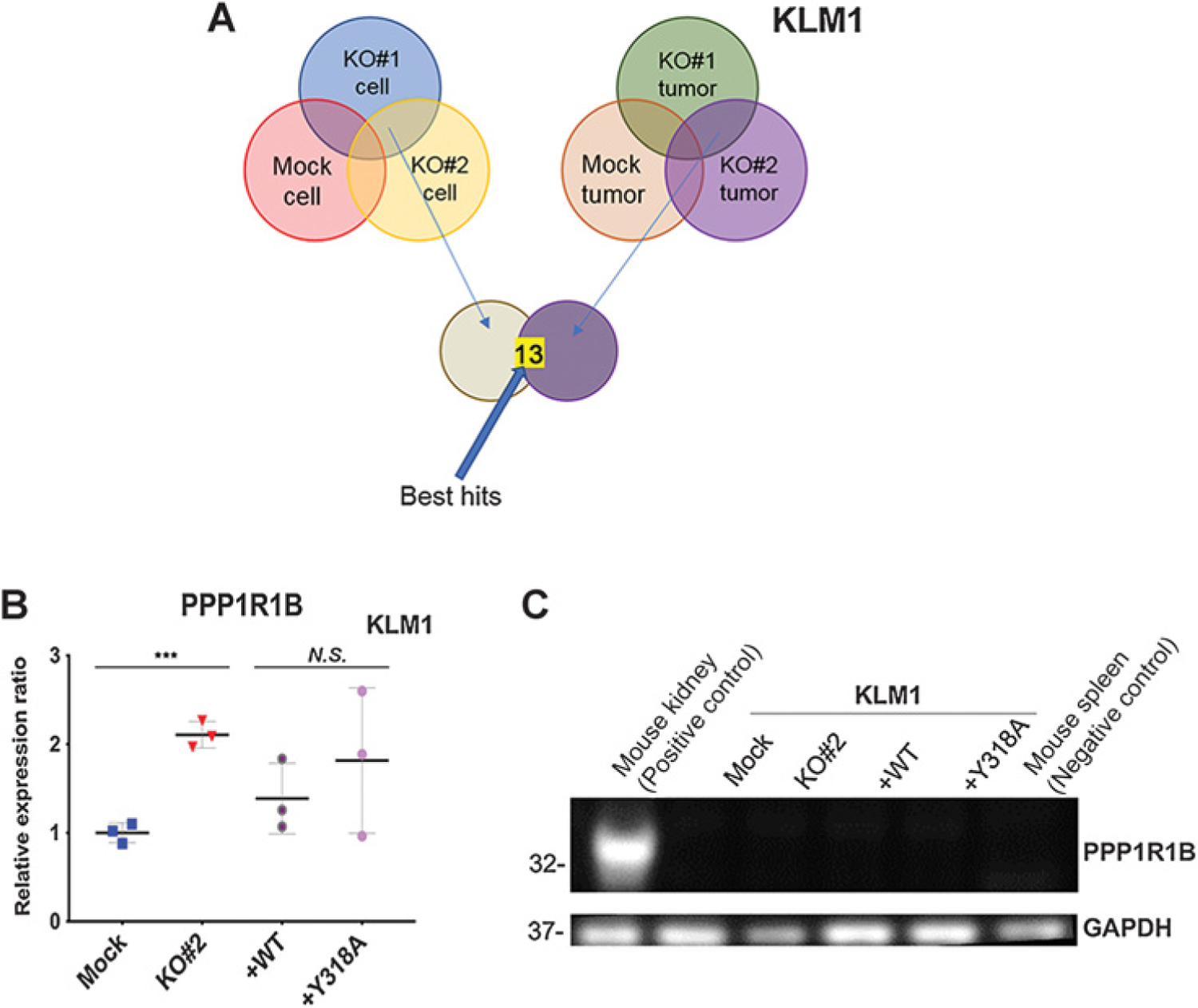
Identifying a novel mediator for MSLN activity using RNAseq and qPCR. A, Analysis schema for RNAseq transcriptional profiling. Thirteen genes were identified to be differentially expressed with at least a 1.5-fold change in expression and P < 0.05 in both KO#1 and KO#2 cell lines and tumor tissues as compared with Mock. B, qPCR showing PPP1R1B relative gene expression in KLM1 cell lines ***, P < 0.001, N.S. = not significant (Student t test with Welch correction). C, Immunoblot showing PPP1R1B expression in KLM1-cultured cell lysates. GAPDH was used as loading control.
Discussion
Here, we conclusively demonstrate for the first time that MSLN plays a role in peritoneal carcinomatosis of pancreatic cancer by positively regulating angiogenesis, proliferation, and invasion during metastatic colonization. We also show that a single point mutation at Y318 is sufficient to disrupt this activity. No previously proposed mechanism of MSLN tumorigenicity, nor transcriptional changes, account for these phenomena.
We have identified a robust and specific in vivo phenotype, impairment of intraperitoneal growth, which is associated with MSLN KO. This phenotype is not an artifact of biased clonal selection. First, both KLM1 KO clones demonstrated the same intraperitoneal phenotype. Second, the KLM1 KO cells grew similarly in cell culture and as subcutaneous xenografts to those expressing MSLN, ruling out that cells with a generalized enhancement of growth were selected. Third, by restoring MSLN expression to these cells and reversing the phenotype, we validated that poor intra-abdominal growth was specific to MSLN loss. Of interest, although we significantly overexpressed MSLN in KO#2+WT cells compared with Mock, further promotion of KLM1 intraperitoneal tumor growth was not observed. We suspect that the very high baseline MSLN expression in the Mock and parent cell lines already saturates the pathway for MSLN effect so that further increase in MSLN production no longer increases growth. Nevertheless, the strong intraperitoneal growth phenotype in cells expressing WT MSLN enabled us to unambiguously determine that Y318 is critical to the protumorigenic activity of MSLN and then to use this point mutant MSLN to assess the robustness of previously reported mechanisms of MSLN protumorigenicity.
Our data show that MSLN confers this growth advantage to pancreatic tumor cells within the first 7 days of metastatic colonization by increasing microvasculature, proliferation, and invasion. We do not see a later sustained protumor effect of MSLN expression. We would therefore expect that therapeutic blockade of MSLN could not shrink or cure established peritoneal metastases but might be effective in inhibiting peritoneal spread of PDAC. Usage of lymphocyte-deficient mice in our study could potentially limit our conclusions as published studies have shown that presence of lymphocytes that recognize MSLN correlates with improved patient outcomes (28). Consistent with our results, Mizukami and colleagues (29) recently used the clinical anti-MSLN mAb amatuximab in immunocompetent mice to show that blockade of MSLN reduced the number of gross intraperitoneal tumor masses but did not prevent peritoneal dissemination. MSLN KO or blockade can delay intraperitoneal tumor growth but does not prevent it in the presence or absence of intact immune signaling.
Peritoneal carcinomatosis is the second most common distribution of metastasis after hepatic metastasis in PDAC and confers an extremely poor prognosis (30, 31). A recent population-based study of patients newly diagnosed between 1995 and 2009 found that patients presenting with peritoneal carcinomatosis had a median survival of 6 weeks compared with 8 to 12 weeks for patients diagnosed with metastatic disease confined to the liver (31). Peritoneal metastasis is thought to occur when cells exfoliate from the primary tumor into the intra-abdominal cavity. This is followed by invasion of the free-floating cancer cells into peritoneal surfaces. This process is distinct from the hematogenous dissemination required for hepatic or pulmonary spread (32), and cells optimized for these processes have different gene expression profiles (33, 34). There are currently no studies examining a link between high MSLN expression and peritoneal carcinomatosis in pancreatic cancer. However, in patients with stage III gastric cancer who underwent potentially curative resection, higher tumor MSLN expression was predictive of peritoneal recurrence (35). It is also intriguing that other malignancies known to commonly seed the peritoneum coincidentally have high frequencies of tumor MSLN expression, including mesothelioma, ovarian, and gastric cancer (17, 35). Although 9% to 16% of patients with pancreatic cancer present with ascites or obvious peritoneal carcinomatosis at the time of diagnosis, many others will later develop it as their disease progresses (36). Available treatments for peritoneal carcinomatosis are entirely palliative and largely inadequate at reducing patient discomfort from ascites, anorexia, early satiety, abdominal pain, and bowel obstruction. Limiting or preventing peritoneal spread of disease would be a significant clinical advance. Our data show that deletion of MSLN delays intraperitoneal tumor growth by impairing metastatic colonization.
Previous studies have shown that forced overexpression of MSLN increases pancreatic cancer cell proliferation, migration in wound healing and invasion assays, and tumor volume in mouse xenografts (8). Multiple molecular mechanisms for MSLN protumorigenic activity have previously been explored. Rump and colleagues (37) first suggested a link between MSLN and the peritoneal metastasis of ovarian cancer when they identified MUC16 (CA125) on ovarian cancer cells as a binding partner for MSLN, and then demonstrated that this receptor could mediate heterotypic adhesion of cancer cells to MSLN-expressing cells like the mesothelial cells that line peritoneal surfaces. The loss of tumor cell MSLN in our study would not be expected to affect heterotypic adhesion because MSLN on the serosal surface remains intact as well as MUC16 levels in the tumor cell membrane. This initial study was followed up by Gubbels and colleagues (14) who showed that MUC16 N-linked glycans were critical for this binding, although MSLN does not contain any classic carbohydrate recognition domains like a typical lectin. They also provided evidence that MSLN/MUC16 interactions may facilitate homotypic cell–cell binding to create cell clusters, a configuration that others have shown to have higher metastatic potential than individual shed tumor cells (38). As MSLN and MUC16 are frequently coexpressed by tumor cells (37, 39), we considered enhanced homotypic binding one possible explanation for our results. However, our data under low adherence cell culture conditions showed that only MSLN KO and not Y318A point mutation inhibited tumor cell clustering, whereas both were associated with poor cancer growth, indicating that there must be an additional or alternative mechanism to explain MSLN activity.
Others have suggested that MSLN signals through MUC16 by means of a ligand–receptor interaction. In contrast with MSLN, MUC16 is a very large (22,152 amino acids), heavily glycosylated transmembrane protein that includes a cytoplasmic tail domain with proven intracellular signaling function (40). Binding of soluble, recombinant MSLN to membrane-bound MUC16 has been extensively studied: It occurs with affinity in the nanomolar range (14), requires a minimum 64 amino acid binding fragments consisting of the N-terminus of mature MSLN, and can be partially decreased by mutation of W321 of MSLN or completely abolished by mutation of Y318 (22). As soluble, shed MSLN contains the minimum binding region for MUC16 interaction (11), it is also expected to bind cell surface MUC16. Binding of recombinant MSLN to MUC16 on pancreatic cancer cells has been shown to upregulate MMP-7 (6). Consistent with binding of MSLN to MUC16 playing a role in MSLN tumorigenicity, the complementation of Y318A-mutant MSLN could not restore intraperitoneal growth of our KO cells. However, MIA PaCa-2 cells lack MUC16 expression yet Y318A mutation also ablates the enhanced cell growth phenotype conferred by WT MSLN in this model system. In addition, Chen and colleagues (6) have previously reported that cell surface interaction of MSLN and MUC16 induces phosphorylation of p38 in a MUC16-dependent fashion to activate a MAPK-dependent signaling pathway. This leads to upregulation of MMP-7 synthesis, thereby promoting migration and invasion. Furthermore, MSLN can still upregulate MMP-7 in cells lacking MUC16 via activation of an ERK1/2 MAPK-dependent pathway. We saw no evidence for activation of either p38 or ERK1/2, nor for upregulation of MMP-7 in our model system. Although we cannot rule out that the MAPK pathway, MMPs or MUC16 signaling may play a protumorigenic role in some pancreatic cancers, another mechanism must be responsible for the protumorigenic behavior of MSLN in our system.
Others proposed that MSLN and miR-198, a tumor-suppressing miRNA, are involved in an intricate reciprocal regulatory loop mediated by NF-κB (15). Specifically, MSLN overexpression triggers nuclear translocation of NF-κB by an undiscovered mechanism. This induces expression the NF-κB target gene OCT-2 that is the proposed end-mediator of MSLN-triggered NF-κB signaling. OCT-2 is a POU family homeobox octamer transcription factor that represses miR-198 expression, such that MSLN overexpression results in decreased miR-198 and increased tumor growth. Although MIA-PaCa2 were used in the original studies investigating this pathway, we identified no changes in OCT-2 levels upon KO or overexpression of MSLN in MIA-PaCa2 and confirmed this with KLM1. OCT-2–mediated repression of mIR-198 is not responsible for MSLN protumorigenic activity.
Previously delineated posttranslational pathways for MSLN activity cannot explain the activity we observed. Moreover, RNA seq analysis demonstrated that phenotypic intraperitoneal growth changes caused by MSLN deletion are not regulated at the transcriptional level. Our screen identified a surprisingly small number of potential targets that were up- or downregulated in response to MSLN loss despite the profound phenotypic changes we observed in intraperitoneal tumor growth. However, even these changes in gene expression did not convey to total protein abundance for some of these potential targets and failed to correspond to phenotype in KO+Y318A and KO+WT cells for all others. Only a few scenarios can account for this finding. First, that protumorigenic MSLN signaling occurs at the posttranslational level through a molecular mechanism that remains undefined. Alternatively, MSLN expression does not affect signaling pathways within the cancer cell, but instead MSLN acts as a paracrine signal to nontumor cells that are specific to the intraperitoneal cavity. This latter possibility better explains the compartment specificity of the phenotype observed and warrants further investigation.
In summary, we have uncovered the pathophysiologic mechanism underlying MSLN-induced protumorigenic effects. We show that MSLN expression stimulates intraperitoneal growth of pancreatic cancer by positively regulating angiogenesis, proliferation, and invasion during metastatic colonization. This activity requires the presence of Y-318. The protumorigenic activity of MSLN is not accounted for by any previously reported molecular mechanism, including MUC16 binding, activation of NF-κB or the MAPK signaling pathways, nor by upregulation of MMPs. Change in MSLN expression does not substantively alter the cancer cell transcriptional program, modify phosphorylation status of the major protein kinase signaling molecules, nor reflect in abundance of cellular proteases or protein mediators of angiogenesis. This raises the possibility that MSLN may function through a paracrine signaling mechanism targeting receptors located on an unidentified cell type (or types) present in the intraperitoneal cavity.
Supplementary Material
Acknowledgments
The authors would like to thank Liang Cao and Ira Pastan (both NCI) for providing necessary reagents to complete our experiments. The authors also thank Elijah Edmondson (NCI) for pathological analysis, David Venzon (NCI) for providing valuable advice on statistics, as well as Cynthia Hurlbert for helping with article editing. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (Project No. ZIA BC 011652).
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Hassan R, Thomas A, Alewine C, Le DT, Jaffee EM, Pastan I. Mesothelin immunotherapy for cancer: ready for prime time? J Clin Oncol 2016;34:4171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res 2001;7:3862–8. [PubMed] [Google Scholar]
- 3.Hassan R, Laszik ZG, Lerner M, Raffeld M, Postier R, Brackett D. Mesothelin is overexpressed in pancreaticobiliary adenocarcinomas but not in normal pancreas and chronic pancreatitis. Am J Clin Pathol 2005;124:838–45. [PubMed] [Google Scholar]
- 4.Scales SJ, Gupta N, Pacheco G, Firestein R, French DM, Koeppen H, et al. An antimesothelin-monomethyl auristatin e conjugate with potent antitumor activity in ovarian, pancreatic, and mesothelioma models. Mol Cancer Ther 2014;13: 2630–40. [DOI] [PubMed] [Google Scholar]
- 5.Winter JM, Tang LH, Klimstra DS, Brennan MF, Brody JR, Rocha FG, et al. A novel survival-based tissue microarray of pancreatic cancer validates MUC1 and mesothelin as biomarkers. PLoS ONE 2012;7:e40157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen SH, Hung WC, Wang P, Paul C, Konstantopoulos K. Mesothelin binding to CA125/MUC16 promotes pancreatic cancer cell motility and invasion via MMP-7 activation. Sci Rep 2013;3:1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bharadwaj U, Li M, Chen C, Yao Q. Mesothelin-induced pancreatic cancer cell proliferation involves alteration of cyclin E via activation of signal transducer and activator of transcription protein 3. Mol Cancer Res 2008;6:1755–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li M, Bharadwaj U, Zhang R, Zhang S, Mu H, Fisher WE, et al. Mesothelin is a malignant factor and therapeutic vaccine target for pancreatic cancer. Mol Cancer Ther 2008;7:286–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang K, Pastan I. Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc Natl Acad Sci U S A 1996;93:136–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hassan R, Remaley AT, Sampson ML, Zhang J, Cox DD, Pingpank J, et al. Detection and quantitation of serum mesothelin, a tumor marker for patients with mesothelioma and ovarian cancer. Clin Cancer Res 2006;12:447–53. [DOI] [PubMed] [Google Scholar]
- 11.Ho M, Onda M, Wang QC, Hassan R, Pastan I, Lively MO. Mesothelin is shed from tumor cells. Cancer Epidemiol Biomarkers Prev 2006;15:1751. [DOI] [PubMed] [Google Scholar]
- 12.Bera TK, Pastan I. Mesothelin is not required for normal mouse development or reproduction. Mol Cell Biol 2000;20:2902–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sathyanarayana BK, Hahn Y, Patankar MS, Pastan I, Lee B. Mesothelin, stereocilin, and otoancorin are predicted to have superhelical structures with ARM-type repeats. BMC Struct Biol 2009;9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gubbels JA, Belisle J, Onda M, Rancourt C, Migneault M, Ho M, et al. Mesothelin-MUC16 binding is a high affinity, N-glycan dependent interaction that facilitates peritoneal metastasis of ovarian tumors. Mol Cancer 2006;5:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marin-Muller C, Li D, Bharadwaj U, Li M, Chen C, Hodges SE, et al. A tumorigenic factor interactome connected through tumor suppressor micro-RNA-198 in human pancreatic cancer. Clin Cancer Res 2013;19:5901–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov 2016;6:133–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lv J, Li P. Mesothelin as a biomarker for targeted therapy. Biomark Res 2019;7:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen H, Cuevas C, et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer Cell 2015; 28:638–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolyvas E, Rudloff M, Poruchynsky M, Landsman R, Hollevoet K, Venzon D, et al. Mesothelin-targeted immunotoxin RG7787 has synergistic antitumor activity when combined with taxanes. Oncotarget 2017;8:9189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onda M, Nagata S, Ho M, Bera TK, Hassan R, Alexander RH, et al. Megakaryocyte potentiation factor cleaved from mesothelin precursor is a useful tumor marker in the serum of patients with mesothelioma. Clin Cancer Res 2006;12: 4225–31. [DOI] [PubMed] [Google Scholar]
- 21.Kimura Y, Kobari M, Yusa T, Sunamura M, Kimura M, Shimamura H, et al. Establishment of an experimental liver metastasis model by intraportal injection of a newly derived human pancreatic cancer cell line (KLM-1). Int J Pancreatol 1996;20:43–50. [DOI] [PubMed] [Google Scholar]
- 22.Kaneko O, Gong L, Zhang J, Hansen JK, Hassan R, Lee B, et al. A binding domain on mesothelin for CA125/MUC16. J Biol Chem 2009;284:3739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yunis AA, Arimura GK, Russin DJ. Human pancreatic carcinoma (MIA PaCa-2) in continuous culture: sensitivity to asparaginase. Int J Cancer 1977; 19:128–35. [DOI] [PubMed] [Google Scholar]
- 24.Hucl T, Brody JR, Gallmeier E, Iacobuzio-Donahue CA, Farrance IK, Kern SE. High cancer-specific expression of mesothelin (MSLN) is attributable to an upstream enhancer containing a transcription enhancer factor dependent MCAT motif. Cancer Res 2007;67:9055–65. [DOI] [PubMed] [Google Scholar]
- 25.Zheng C, Jia W, Tang Y, Zhao H, Jiang Y, Sun S. Mesothelin regulates growth and apoptosis in pancreatic cancer cells through p53-dependent and -independent signal pathway. J Exp Clin Cancer Res 2012;31:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haridas D, Chakraborty S, Ponnusamy MP, Lakshmanan I, Rachagani S, Cruz E, et al. Pathobiological implications of MUC16 expression in pancreatic cancer. PLoS ONE 2011;6:e26839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muniyan S, Haridas D, Chugh S, Rachagani S, Lakshmanan I, Gupta S, et al. MUC16 contributes to the metastasis of pancreatic ductal adenocarcinoma through focal adhesion mediated signaling mechanism. Genes Cancer 2016;7: 110–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soares K, Rucki A, Kim V, Foley K, Solt S, Wolfgang C, et al. TGF-β blockade depletes T regulatory cells from metastatic pancreatic tumors in a vaccine dependent manner. Oncotarget 2015;6:43005–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mizukami T, Kamachi H, Fujii Y, Matsuzawa F, Einama T, Kawamata F, et al. The anti-mesothelin monoclonal antibody amatuximab enhances the antitumor effect of gemcitabine against mesothelin-high expressing pancreatic cancer cells in a peritoneal metastasis mouse model. Oncotarget 2018;9: 33844–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yachida S, Iacobuzio-Donahue CA. The pathology and genetics of metastatic pancreatic cancer. Arch Pathol Lab Med 2009;133:413–22. [DOI] [PubMed] [Google Scholar]
- 31.Thomassen I, Lemmens VE, Nienhuijs SW, Luyer MD, Klaver YL, de Hingh IH. Incidence, prognosis, and possible treatment strategies of peritoneal carcinomatosis of pancreatic origin: a population-based study. Pancreas 2013;42:72–5. [DOI] [PubMed] [Google Scholar]
- 32.Mura G, Verdelli B. The features of peritoneal metastases from gastric cancer. J Cancer Metastasis Treat 2016;2:365–74. [Google Scholar]
- 33.Nishimori H, Yasoshima T, Hata F, Denno R, Yanai Y, Nomura H, et al. A novel nude mouse model of liver metastasis and peritoneal dissemination from the same human pancreatic cancer line. Pancreas 2002;24:242–50. [DOI] [PubMed] [Google Scholar]
- 34.Nishimori H, Yasoshima T, Denno R, Shishido T, Hata F, Honma T, et al. A new peritoneal dissemination model established from the human pancreatic cancer cell line. Pancreas 2001;22:348–56. [DOI] [PubMed] [Google Scholar]
- 35.Shin SJ, Park S, Kim MH, Nam CM, Kim H, Choi YY, et al. Mesothelin expression is a predictive factor for peritoneal recurrence in curatively resected stage III gastric cancer. Oncologist 2019;24:e1108–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zervos EE, Osborne D, Boe BA, Luzardo G, Goldin SB, Rosemurgy AS. Prognostic significance of new onset ascites in patients with pancreatic cancer. World J Surg Oncol 2006;4:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rump A, Morikawa Y, Tanaka M, Minami S, Umesaki N, Takeuchi M, et al. Binding of ovarian cancer antigen CA125/MUC16 to mesothelin mediates cell adhesion. J Biol Chem 2004;279:9190–8. [DOI] [PubMed] [Google Scholar]
- 38.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014;158:1110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimizu A, Hirono S, Tani M, Kawai M, Okada K, Miyazawa M, et al. Coexpression of MUC16 and mesothelin is related to the invasion process in pancreatic ductal adenocarcinoma. Cancer Sci 2012;103:739–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Das S, Batra SK. Understanding the unique attributes of MUC16 (CA125): potential implications in targeted therapy. Cancer Res 2015; 75:4669–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.