

Abstract
Endothelial cells play a major role in inflammatory responses to infection and sterile injury. Endothelial cells express Toll-like receptor 4 (TLR4) and are activated by LPS to express inflammatory cytokines/chemokines, and to undergo functional changes, including increased permeability. The extracellular signal-regulated kinase 1/2 (ERK1/2) mediates pro-inflammatory signaling in monocytes and macrophages, but the role of ERK1/2 in LPS-induced activation of microvascular endothelial cells has not been defined. We therefore studied the role of ERK1/2 in LPS-induced inflammatory activation and permeability of primary human lung microvascular endothelial cells (HMVEC). Inhibition of ERK1/2 augmented LPS-induced IL-6 and vascular cell adhesion protein (VCAM-1) production by HMVEC. ERK1/2 siRNA knockdown also augmented IL-6 production by LPS-treated HMVEC. Conversely, ERK1/2 inhibition abrogated permeability and restored cell-cell junctions of LPS-treated HMVEC. Consistent with the previously described pro-inflammatory role for ERK1/2 in leukocytes, inhibition of ERK1/2 reduced LPS-induced cytokine/chemokine production by primary human monocytes. Our study identifies a complex role for ERK1/2 in TLR4-activation of HMVEC, independent of myeloid differentiation primary response gene (MyD88) and TIR domain-containing adaptor inducing IFN-β (TRIF) signaling pathways. The activation of ERK1/2 limits LPS-induced IL-6 production by HMVEC, while at the same time promoting HMVEC permeability. Conversely, ERK1/2 activation promotes IL-6 production by human monocytes. Our results suggest that ERK1/2 may play an important role in the nuanced regulation of endothelial cell inflammation and vascular permeability in sepsis and injury.
Keywords: LPS, TLR4, acute inflammation, endothelial activation, NFκB
INTRODUCTION
The average human adult is estimated to contain in excess of 1 trillion endothelial cells (2, 7, 24, 53) as compared to 20–50 billion peripheral blood mononuclear cells and 1–4 billion circulating monocytes (40). Endothelial cells line the vast network of blood and lymphatic vessels of all tissues, and they dynamically regulate inflammation, leukocyte trafficking, coagulation, and the vascular barrier (4, 13, 22, 26, 38). Sepsis and tissue injury cause endothelial cell activation at the site of injury, as well as at remote sites. During these acute inflammatory processes, dysfunction of microvascular endothelial cells leads to vascular leak and coagulopathy, which contribute to shock, organ failure, and reduced survival (28, 31).
Endothelial cells have not traditionally been viewed as immune cells, but they are key players in the host’s immune responses in sepsis (9, 31, 55, 57). Endothelial cells express innate immune receptors, including Toll-like receptors (TLRs), NOD-like receptors and RIG-I like receptors (28). Furthermore, the direct activation of endothelial TLRs by damage- and pathogen-associated molecular proteins (DAMPs and PAMPs, respectively) increases the expression of cytokines, chemokines and adhesion molecules, promotes neutrophil-endothelial adhesion, and induces endothelial permeability (28).
TLR-dependent signaling is mediated by nuclear factor κB (NFκB) and the family of mitogen-activated protein kinases (MAPKs), including p38-MAPK, JNK, ERK1/2 and ERK5 (12, 28, 50, 51). Notably, while endothelial cells and leukocytes express TLRs and share many of the same intracellular signaling intermediaries, there are differences in inflammatory signaling between endothelial cells and leukocytes. For example, direct activation of endothelial or monocyte TLR2 or TLR4 strongly upregulates IL-6 and IL-8 production. In contrast, activation of endothelial cell TLR2 and TLR4 does not upregulate IL-1β and TNFα production, whereas these cytokines are strongly upregulated in monocytes and macrophages activated with TLR2 and TLR4 agonists (23, 37, 52). While mapping out endothelial TLR2 signaling pathways, we observed that in human umbilical vein endothelial cells (HUVEC), inhibition of MEK1, the upstream kinase of ERK1/2, augmented TLR2-induced production of IL-6, but not IL-8 (51). This effect of MEK1 inhibition on TLR2-dependent activation of HUVEC was different from the pro-inflammatory role that MEK1 is known to play in leukocytes (51). Our results suggested to us that ERK1/2 activation plays an important role in regulating TLR-dependent activation of endothelial cells.
In the current report, we tested the hypothesis that ERK1/2 activation negatively regulates TLR4-dependent activation of primary human lung microvascular endothelial cells (HMVEC). We assessed the role of ERK1/2 in LPS-induced cytokine/chemokine production, permeability, and expression of endothelial tight junction proteins.
MATERIALS AND METHODS
Primary human endothelial cells and monocytes
HMVEC (Promocell) from male and female cadavers were cultured in endothelial growth media (EGM-2 Basal Medium, Lonza) supplemented with Microvascular Endothelial Cell Growth Medium supplements (EGM-2 MV, Lonza) as described (51). Human CD14+ monocytes from healthy male and female donors (Lonza) were thawed and cultured in RPMI supplemented with 10% heat-inactivated fetal calf serum (FCS), L-glutamine, and antibiotics and used immediately for experiments. Cells were incubated at 37°C under humidified 5% CO2.
Inflammatory agonist treatment
HMVEC were added to 48-well plates (3×105 cells/well) and grown to confluence. Human monocytes were added to 48-well plates (2×105 cells/well) and used immediately. HMVEC and monocytes were pre-incubated for 1 hour with vehicle (0.01% DMSO) or ERK1/2 inhibitor (SCH772984, 1μM, SelleckChem). Then ultrapure LPS (0.01 μg/ml, List Laboratories) was added to the wells in the continued presence or absence of ERK1/2 inhibitor (1μM). Cells were cultured for another 6 hours and supernatants were collected. Cytokines were quantified in culture supernatants of using Duoset ELISA kits (R&D systems).
Lipopolysaccharide (LPS)
We utilized ultrapure LPS from E. coli O111:B4 bacteria (Ultrapure LPS, List Laboratories, Lot 4219A1) for all experiments. The LPS was tested for purity by colloidal gold staining, according to manufacturer’s instructions (Bio-Rad), and by immunoblotting for three outer membrane proteins (murein lipoprotein [MLP], peptidoglycan-associated lipoprotein [PAL], and outer membrane protein A [OmpA]), which are common contaminants of purified LPS (16, 17). The LPS contained no protein bands by gold staining, which can detect levels as low as 1 ng of protein (Supplemental Figure S1A). Similarly, none of the outer membrane proteins were detected in the LPS by immunoblotting (Figure S1B–D) (22, 23). Lysates of whole E. coli O111:B4 bacteria, which contain multiple proteins including MLP, PAL and OmpA, served as positive controls for gold stains and immunoblots.
Immunoblots
Immunoblots were performed as previously described (51). Primary antibodies used were p44/42-MAPK (ERK1/2; 1:1000; 4695; Cell Signaling), actin (0.1 mg/ml, A2066, Sigma), phospho-NF-κB p65 (1:1000; 3033; Cell Signaling), anti-murein lipoprotein (MLP), peptidoglycan-associated lipoprotein (PAL), and outer membrane protein A (OmpA). MLP, PAL, and OmpA antibodies were made as previously described and used at concentration of 1 μg/ml (17). Total protein concentrations of cell lysates were measured using the RCDC protein assay kit (Bio-Rad). Samples were separated by SDS-PAGE, transferred to PVDF membrane (Pall Corp), and blocked with 3% BSA in TBST (1 hour, room temperature). They were then incubated overnight at 4°C with primary antibodies, washed and incubated with suitable secondary antibodies conjugated to peroxidase (Jackson ImmunoResearch) and developed with SuperSignal West Dura Extended Duration Substrate (Thermo Scientific). Signal was detected with ChemiDocMP Imaging System (BioRad). Target protein expression was normalized using densitometry software to quantify band intensity with Image Lab Software (BioRad).
Transfection of HMVEC with siRNAs
ERK1-specific (L-003592-00), ERK2-specific (L-003555-00), or non-targeting (D-001810-10) ON-TARGETplus SMARTpool siRNA (Dharmacon) were used following the manufacturer’s instructions for transfecting HMVECs with DharmaFECT 4 Transfection Reagent (T-2004-01). We placed siRNA (5 μM) in serum-free medium, and diluted DharmaFECT transfection reagent in a separate tube, incubated both tubes for 5 minutes, and then combined the contents of both tubes and incubated the reagents together for another 20 minutes. HMVEC were grown to 80–90% confluence in a T-75 flask, and then the medium was replaced with antibiotic- and serum-free medium containing the transfection reaction. After 24 hours the transfection reagent media was removed, and cells were then treated with LPS (1 μg/ml) in complete media for 24 hours, and cytokines were quantified in culture supernatants.
Electric Cell-substrate Impedance Sensing (ECIS)
Transendothelial resistance (TER) was measured using the ECISZeta (Applied Biophysics) (44). HMVEC were placed in 96-well electrodes (40,000 cells/well) and allowed to adhere. Resistance (ohm), impedance (ohm), and capacitance (nanofarad) were measured at frequent intervals, using a frequency of 4000 Hz, which was determined to be the optimal frequency for endothelial cells. When cells reached a stable level of resistance, LPS (1 μg/ml) in the presence of ERK1/2 inhibitor (1μM), MyD88 inhibitor (10μM, Aobious), TRIF inhibitor (10μM, Novus), or vehicle was added to the wells, and TER was measured at frequent intervals through 15 hours. To account for differences in resistance of each well, data was normalized to the resistance of the individual well immediately before the addition of the agonist.
Immunofluorescence microscopy
HMVEC were grown to confluence on Lab Tek II chamber slides (Nunc) that had been pre-coated with collagen, and then treated for 2, 6, and 24 hours with LPS (1 μg/ml) in the presence of ERK1/2 inhibitor or vehicle. Cells were then fixed with 4% paraformaldehyde (15 minutes, 37°C) and permeabilized with 0.5% Triton X-100 in PBS (15 minutes, room temperature). After blocking (1% BSA in PBS, 30 minutes, room temperature), cells were incubated with either anti-ZO1-AlexaFluor594 (Invitrogen, 24 hours, 4°C), or anti-VE-cadherin (Santa Cruz Biotechnologies) (24 hours, 4°C) followed by AlexaFluor488-tagged secondary antibody (60 minutes, room temperature). Nuclei were counterstained with 4’6-diamidino-2-phenylindole (DAPI). Slides were visualized using fluorescent microscopy (Zeiss AxioImager D1), and images were obtained with 40X objective using AxioVision SE64 Rel 4.9.1 Software.
Statistics
Data was analyzed using GraphPad Prism version 7.00 (GraphPad Software, Inc). Results are expressed as mean ± SD. With the exception of ECIS, results were analyzed using non-parametric-based biostatistics. Mann-Whitney tests were used to compare 2 groups. P < 0.05 was considered to be statistically significant. Data from ECIS experiments were analyzed by quantifying area under the curve from baseline = 1 with addition of negative peaks and graphed as means ± SD. One-way analysis of variance (ANOVA) followed by the Sidak’s multiple comparison was then used to establish significance for multiple comparisons. P < 0.05 was considered to be statistically significant. Experiments were repeated at least twice.
RESULTS
ERK1/2 inhibitor augments LPS-induced IL-6 and VCAM-1 production, but not IL-8 or PAI-1 by HMVEC.
HMVEC were pre-incubated for 1 hour with vehicle or ERK1/2 inhibitor (SCH772984; 1 μM), and then LPS (10 ng/ml) was added in the continued presence of ERK1/2 inhibitor or vehicle. At 6 hours, levels of IL-6 and VCAM-1 were significantly higher in the supernatants of LPS-activated HMVEC treated with the ERK1/2 inhibitor (Figure 1A and 1B). In contrast, the inhibitor had no effect on the LPS-induced upregulation of IL-8 and plasminogen activator inhibitor-1 (PAI-1) in culture supernatants (Figure 1C and 1D).
Figure 1: ERK1/2 inhibitor augments LPS-induced production of IL-6 and VCAM-1, but not IL-8 or PAI-1 by HMVEC.
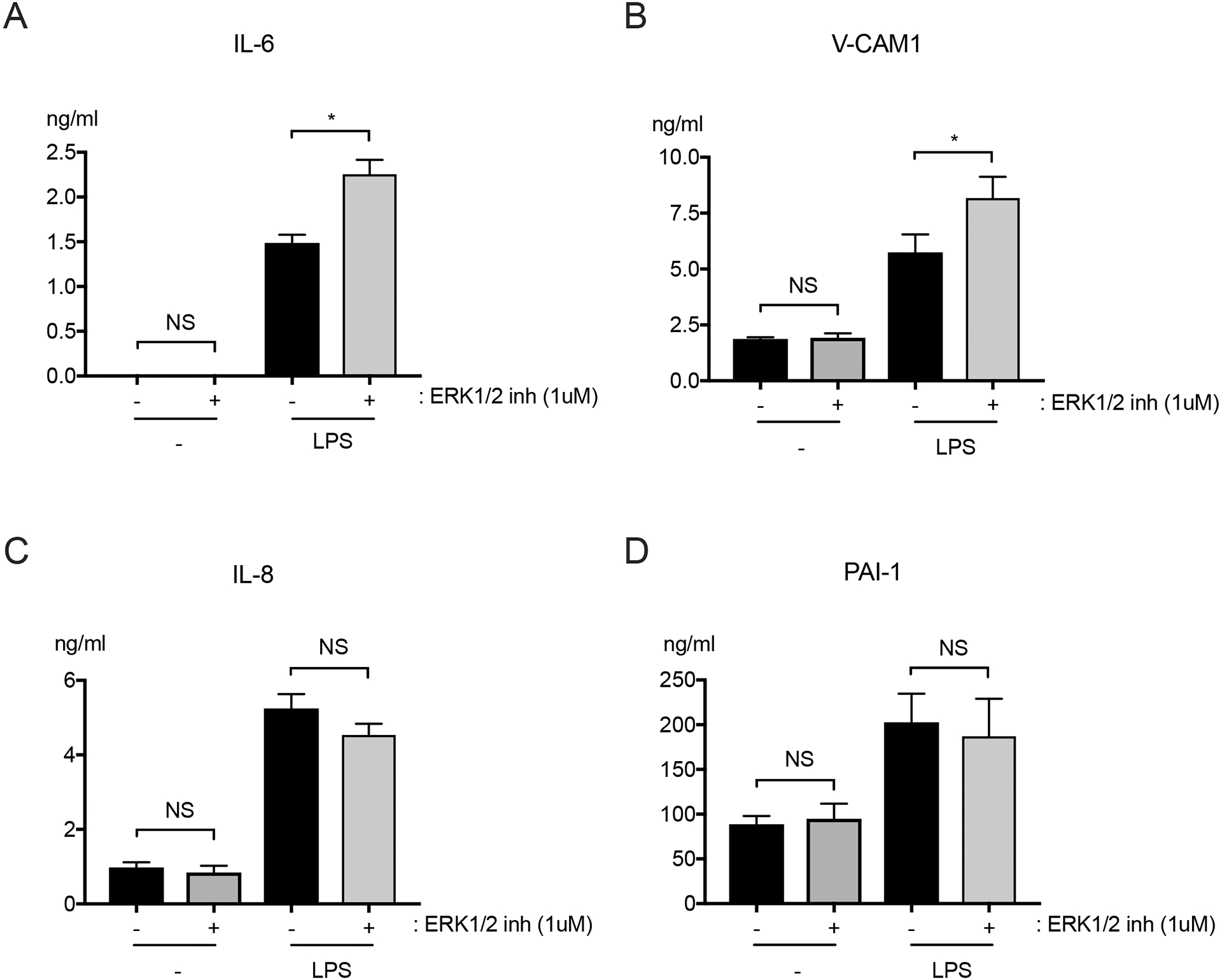
HMVEC were pre-treated with ERK1/2 inhibitor (SCH772984; 1μM), or vehicle for 1 hour, and then with LPS (10 ng/ml) for another 6 hours in the continued presence of ERK1/2 inhibitor or vehicle. Levels of (A) IL-6, (B) VCAM-1, (C) IL-8, and (D) PAI-1 were quantified in culture supernatants. n = 4; *p≤0.05; NS, not significant.
Effects of siRNA knockdown of Erk1 and Erk2 in HMVEC on LPS-induced IL-6 production.
We performed siRNA knockdown to define the individual roles of the ERK1 and ERK2 in LPS-induced activation of HMVEC. Immunoblots confirmed successful knockdown of each protein in HMVEC (Figure 2A). We observed that HMVEC that had undergone siRNA knockdown of ERK2 had significantly augmented LPS-induced IL-6 production as compared with HMVEC transfected with non-targeted siRNA. In contrast, there were not significant differences in LPS-induced cytokine production between HMVEC with ERK1 siRNA knockdown and control cells. However, combined treatment with ERK1 and ERK2 siRNAs further augmented LPS-induced IL-6 production in HMVEC as compared with individual knockdown of either protein (Figure 2B).
Figure 2: siRNA knockdown of ERK1/2 in HMVEC augments LPS-induced IL-6 production.
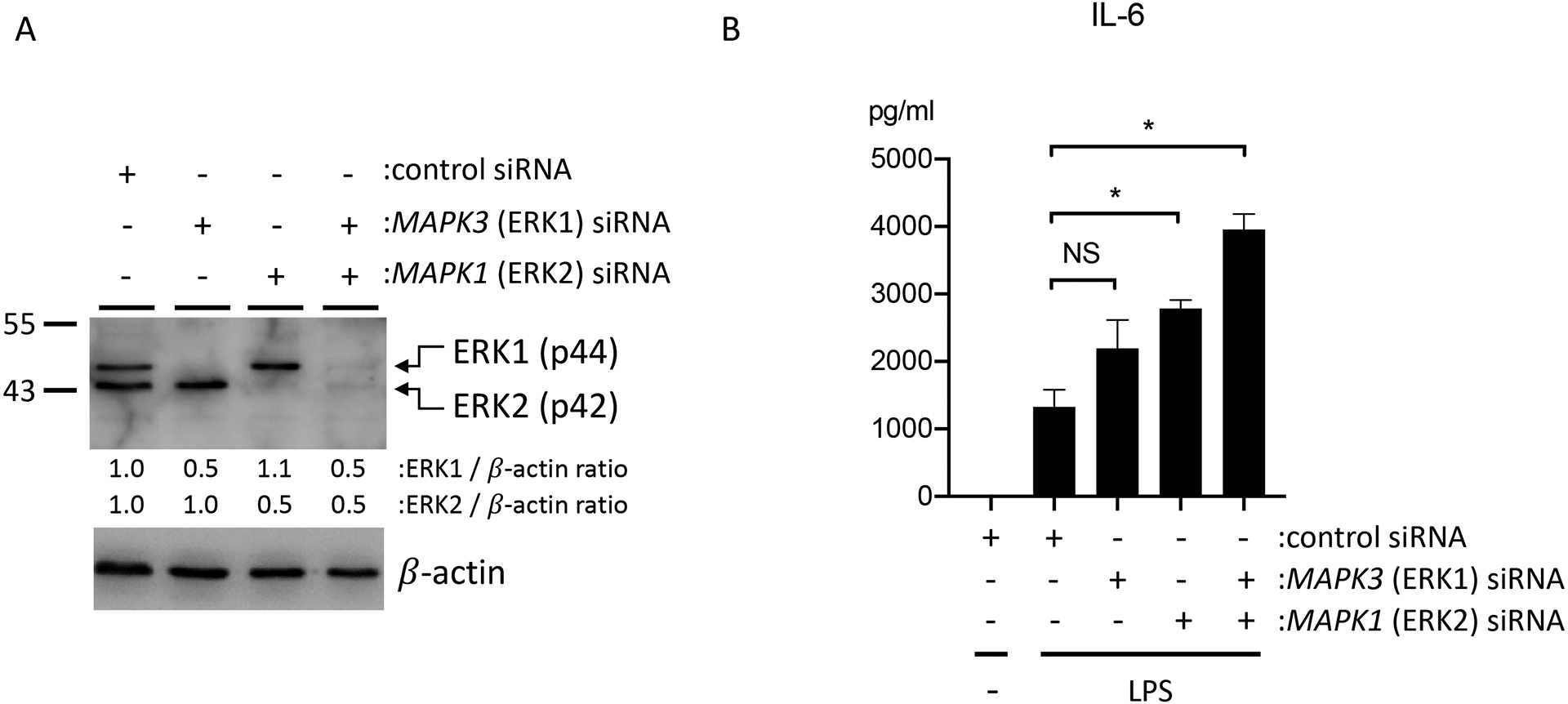
HMVEC were transfected with siRNAs for ERK1, ERK2, or both ERK1 and ERK2 for 24 hours, and then treated with LPS (1 μg/ml). (A) Expression of total ERK1/2 after siRNA transfection by western blotting. Relative density of ERK1/2 was compared to that of β-actin. (B) IL-6 levels in supernatants of LPS-treated HMVEC that had undergone siRNA knockdown of ERK1, ERK2, or both ERK1 and ERK2. n = 4; *p≤0.05; NS, not significant.
ERK1/2 inhibition decreases LPS-induced NFκB activation.
We assessed for NFκB activation HMVEC by immunoblotting cell lysates for phosphorylated NFκB. Treatment with LPS increased phosphorylated NFκB within 30 minutes. The presence of ERK1/2 inhibitor suppressed the LPS-induced phosphorylation of NFκB (Figure 3A–3B).
Figure 3: ERK1/2 inhibitor suppresses LPS-induced NFκB activation in HMVEC.
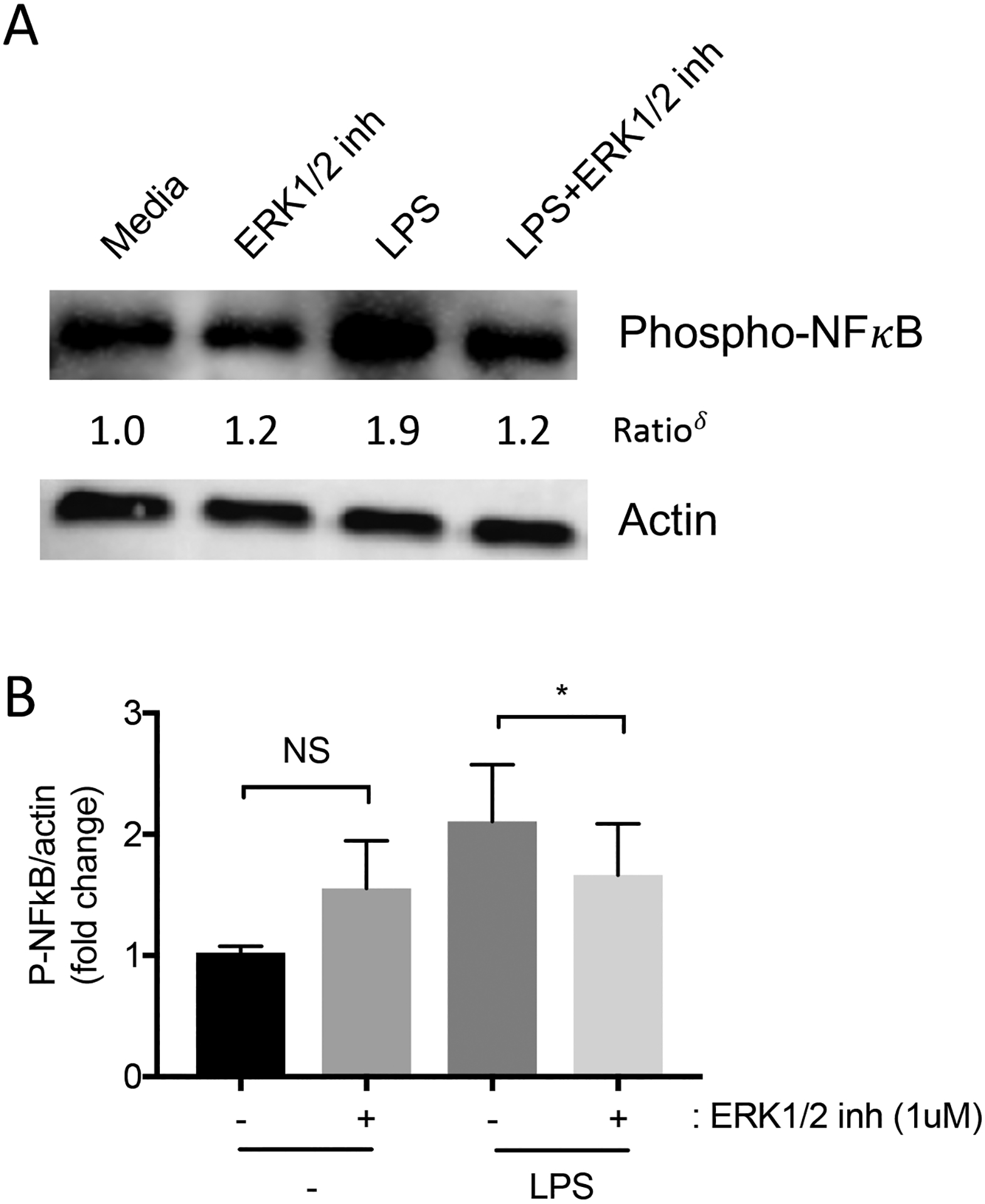
HMVEC were pre-treated for 1 hour with ERK1/2 inhibitor (1μM) or vehicle, and then with LPS (10 ng/ml) in the continued presence of ERK1/2 inhibitor for 30 minutes. (A) Representative images of phospho-NF-κB and actin were detected by immunoblots (δ: normalized density of phospho-NF-κBtreatment/normalized density of phospho-NF-κBmedia). (B) Bar graphs showing the relative abundance of phospho-NF-κB after normalization to actin. n=4; *p ≤ 0.05; NS, not significant.
Effects of ERK1/2 inhibitor on LPS-induced activation of primary human monocytes.
We tested the effects of ERK1/2 inhibition on primary human monocytes from healthy male and female donors in order to confirm that we were able to reproduce results of prior studies supporting a pro-inflammatory role for ERK1/2 in human monocytes. Monocytes were pre-treated with vehicle or with ERK1/2 inhibitor (1 μM), and then stimulated with LPS (0.01 μg/ml) for 6 hours. Consistent with previous studies showing that ERK1/2 plays a pro-inflammatory role in monocytes, the inhibition of ERK1/2 led to decreased IL-6, TNFα, IL-8, and IL1β levels in culture supernatants of LPS-treated monocytes (Figure 4A–4D).
Figure 4: ERK1/2 inhibitor decreased LPS-induced IL-6, TNFα, IL-8, and IL1β production by human monocytes.
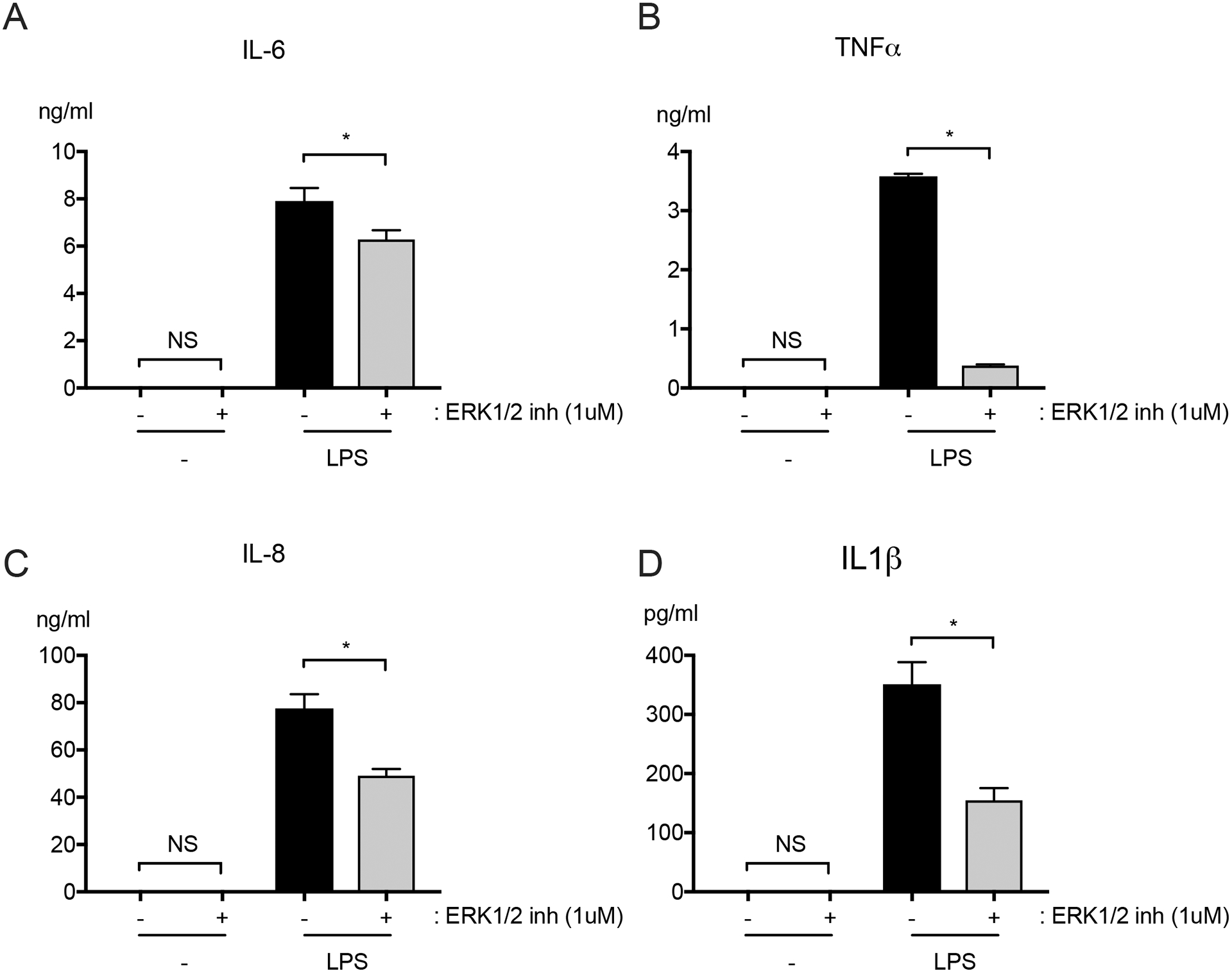
Primary human CD14+ monocytes were pretreated for 1 hour with ERK1/2 inhibitor (1 μM) and then stimulated with LPS (1 μg/ml) for 6 hours in the continued presence of ERK1/2 inhibitor or vehicle. Levels of (A) IL-6, (B) TNFα, (C) IL-8 and (D) IL1β were quantified in culture supernatants. n = 6; *p≤0.05; NS, not significant.
ERK1/2 inhibitor promotes stability of LPS-treated HMVEC monolayers.
Because ERK1/2 inhibition augments LPS-induced IL-6 production by HMVEC, we hypothesized that treatment with ERK1/2 inhibitor would exacerbate LPS-induced loosening of endothelial tight junctions and increase permeability. We treated HMVEC with LPS (1 μg/ml) in the presence and absence of ERK1/2 inhibitor, and quantified TER as a surrogate for permeability (44). Contrary to our hypothesis, treatment with ERK1/2 inhibitor abrogated LPS-induced TER (Figure 5A–5B). This suggests that ERK1/2 activation contributes to TLR4-dependent endothelial permeability. To further study the role of ERK1/2 in LPS-induced endothelial permeability, we performed fluorescence microscopy at 2, 6 and 24 hours to visualize cell-cell adhesion and tight junction proteins, including zonula occludens (ZO-1) and vascular endothelial (VE)-cadherin. Representative images following a 6-hour stimulation are shown in Figure 5B. At 6 hours, but not at 2 or 24 hours, we observed that ERK1/2 inhibitor reduced LPS-induced breakdown of cell-cell junctions and interendothelial gaps (white arrows; Figure 5C).
Figure 5: ERK1/2 inhibitor promotes stability of LPS-activated HMVEC monolayers.
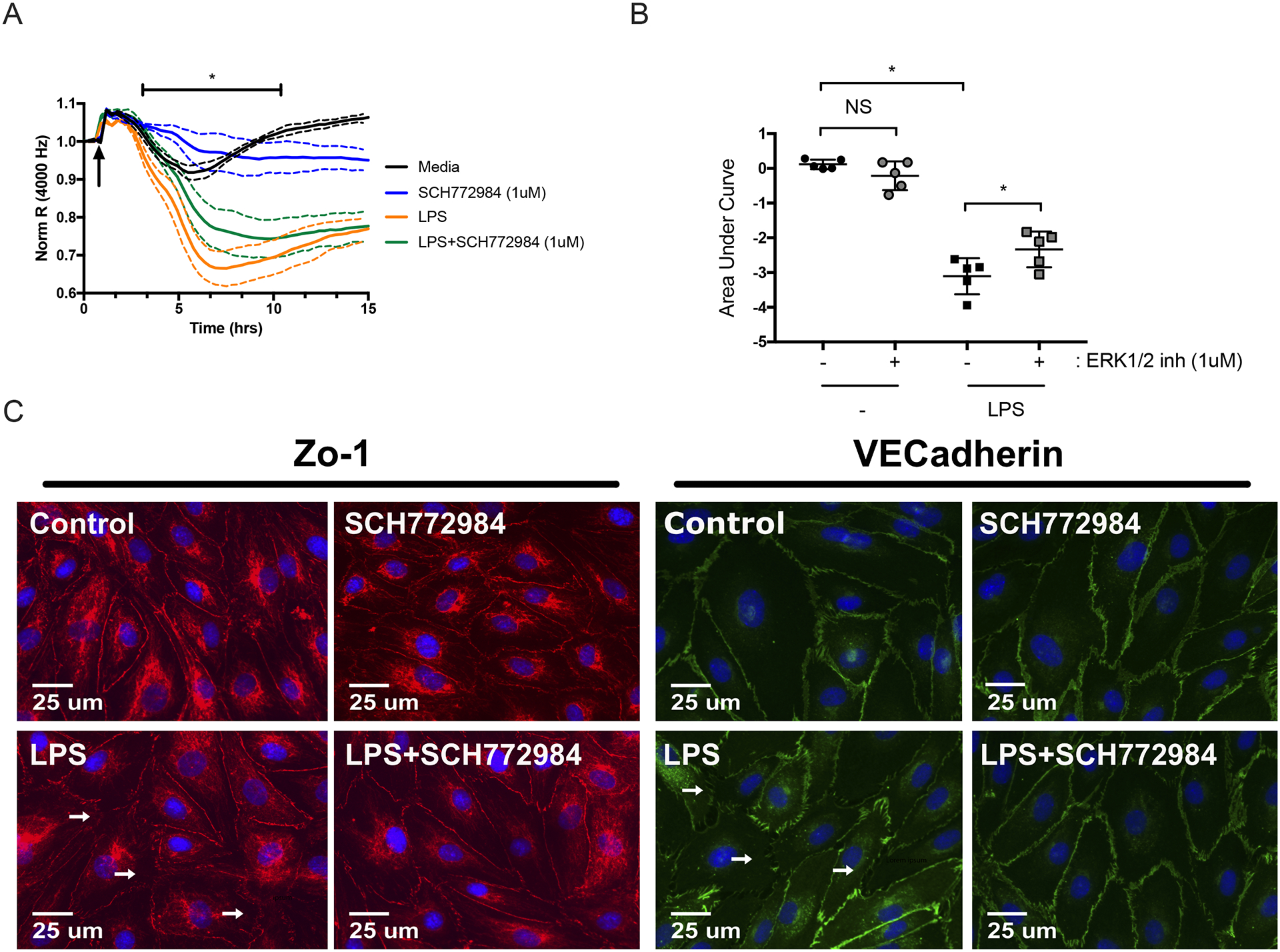
(A) ECIS was used to assess the effects of ERK1/2 inhibitor on LPS-induced HMVEC permeability. Cells were treated with LPS (1 μg/ml) in the continued presence of ERK1/2 inhibitor (1 μM) or vehicle. Data was normalized to the resistance immediately before the addition of LPS (arrow). (B) Area under the curve was then quantified from baseline = 1 with addition of negative peaks and graphed as means ± SD. Data was analyzed by one-way analysis of variance (ANOVA). n = 5; *p≤0.05; NS, not significant. (C) Representative fluorescence microscopy images of HMVEC treated with LPS (1 μg/ml) in the presence and absence of ERK1/2 inhibitor. Images were taken at 40x magnification. ZO-1(red), VEcadherin (green), or nuclei (blue). White arrows indicate examples of disrupted cellular junctions. Scale bar = 25 μm.
To begin to define the signaling pathways responsible for the disconnect in the apparent role of ERK1/2 in LPS-induced IL-6 production versus permeability, we investigated the roles of MyD88 and TRIF, two proximal adaptor proteins responsible for activation of a number of intermediary signaling molecules (10, 45). In the presence of the MyD88 inhibitor or TRIF inhibitor, TLR4-dependent IL-6 production and permeability were unaffected (S2A–S2F).
DISCUSSION
Our study identifies a novel and complex role for ERK1/2 in TLR4-dependent activation of HMVEC which differs substantially from the pro-inflammatory role of ERK1/2 in TLR4-dependent activation of leukocytes (14, 49). Furthermore, despite our finding the ERK1/2 inhibitor augments LPS-induced IL-6 production by HMVEC, we have found that the ERK1/2 inhibitor reduces LPS-induced permeability of HMVEC. This points to divergent roles of ERK1/2 in endothelial inflammatory activation and vascular leak.
The vascular endothelium plays a substantial role in the innate immune response, and regulates systemic inflammation, coagulation and vascular permeability. During sepsis, the upregulation of a number of biomarkers correlate with worse prognosis and increased organ dysfunction, including, but not limited to IL-6, IL-8, VCAM-1 and PAI-1 (5, 18, 33, 34, 41). Our data show that ERK1/2 inhibition augments upregulation of IL-6 and VCAM-1, but not IL-8 or the anti-fibrinolytic coagulation intermediary, PAI-1. This suggests that despite having a role in reducing TLR4-dependent IL-6 and adhesion molecule expression of VCAM-1 by LPS-activated endothelial cells, ERK1/2 may not play a predominant role in TLR4-dependent coagulopathy in HMVEC. This is contrary to the pro-inflammatory role of ERK1/2 in TLR4-dependent activation of leukocytes that we confirmed in LPS-activated primary human monocytes with reduced production of IL-6, TNFα, IL-8 and IL1β in the presence of the ERK1/2 inhibitor.
Because NF-κB has previously been shown to mediate LPS-induced cytokine production and adhesion molecule expression by endothelial cells (6, 11, 42, 47), we hypothesized that ERK1/2 might restrain inflammatory activation of HMVEC by modulating NF-κB activation. However, we observed that ERK1/2 inhibition reduced LPS-induced activation of NF-κB in HMVEC, despite the augmented production of IL-6 and VCAM-1. This finding has led us to speculate that in endothelial cells, non-NF-κB pathways may mediate LPS-induced upregulation of IL-6 and VCAM-1.
The differences in the role of ERK1/2 in inflammatory activation of endothelial cells and leukocytes could potentially be exploited therapeutically in patients with sepsis and other inflammatory processes that are driven by TLR4 activation, such as ischemia reperfusion injury (36). As endothelial activation and dysfunction are believed to contribute to the development of organ injury and failure in sepsis (5, 21, 34, 43), it is conceivable that augmentation of the ERK1/2 activity specifically in endothelial cells might reduce organ injury by limiting endothelial inflammatory responses while still preserving the ability of leukocyte populations to combat infection. This concept is supported by reports that ERK1/2 is critical for monocyte and macrophage development (1, 29, 39).
A perplexing aspect of our data is that ERK1/2 inhibition augmented IL-6 and VCAM-1 production but reduced permeability of HMVEC treated with LPS. Our permeability results are consistent with other reports that ERK1/2 activation increases permeability and alters cell-cell junctions in HUVEC and bovine lung artery endothelial cells (27, 46). The effects of ERK1/2 inhibition on reducing LPS-induced permeability were paralleled by preservation of adherens junction and tight junction proteins, specifically ZO-1 and VE-Cadherin, in the presence of the ERK1/2 inhibitor. Disruption of these same endothelial tight junction and adhesion proteins has been shown to cause endothelial hyperpermeability in many other inflammatory conditions, including burn injury and acute lung injury too (19, 20, 32). Thus our results on the role of ERK1/2 in TLR4-dependent endothelial permeability, and on tight junction and adhesion proteins, may have mechanistic and therapeutic relevance not only in sepsis, but also to these other inflammatory processes.
Upon LPS recognition, TLR4 intracellular signaling is divided into two signaling pathways based on the adapter proteins, MyD88 and TRIF (30, 48). We thought that it was possible that TRIF-dependent versus MyD88-dependent signaling might account for differences in the apparent role of ERK1/2 in TLR4-dependent induction of IL-6 versus permeability. However, our data using TRIF and MyD88 inhibitors suggest that neither of these proximal adapter proteins are critical for TLR4-dependent IL-6 production in HMVEC, and that ERK1/2’s effects on permeability appear to be independent of both of these adaptor proteins. Future studies will address potential alternative TLR4-dependent signaling mechanisms in human endothelial cells and further delineate proximal and distal ERK1/2 signaling pathways. It is also plausible that the downregulation of NF-κB in presence of ERK1/2 inhibitor we demonstrated can explain this preservation of the endothelial monolayer with ERK1/2 inhibition as it has been shown that inhibition of NF-κB reverses endothelial leakage in septic mice (56).
While the details of the mechanism remain to be elucidated, this disconnect between the effects of ERK1/2 in endothelial inflammation and permeability challenges the paradigm that LPS-induced inflammation per se leads to increased vascular permeability. Other studies support this notion in which signaling pathways that modulate cytokine production are separate from those that affect permeability. Interleukin signaling is an example and while an NF-κB-dependent pathway leads to transcriptional inflammatory activation in endothelial cells, a more proximal pathway separate from that is responsible for vascular barrier disruption (58). The differential effects of ERK1/2 on cytokine production and permeability may facilitate leukocyte migration and movement across the endothelial barrier by loosening cell-cell junctions, while also limiting generalized endothelial cell production of pro-inflammatory mediators. Finally, it is possible that ERK1 and ERK2 each have different functions. Our ERK1/2 inhibitor, while specific, inhibits both ERK1 and ERK2. While these proteins are generally described together and are approximately 80% homologous, there appears to be significant differences between ERK1 and ERK2 as ERK2 deficiency is embryonically lethal in mice (15). Consistent with the possibility that ERK1 and ERK2 play different roles, we found that siRNA knockdown of ERK2, but not ERK1, significantly augmented LPS-induced IL-6 production by HMVEC.
During sepsis, widespread microvascular endotheliopathy leads to vascular leak, inflammation, and coagulopathy, and promotes sepsis-induced organ failure (25). However, the precise mechanisms of sepsis-induced endotheliopathy, and the role of endothelial inflammatory pathways in driving organ injury and failure in sepsis and injury remain elusive. ERK1/2 plays key roles in numerous cellular processes including cell adhesion, differentiation, and proliferation, and in pro-inflammatory signaling in diseases such as asthma, vascular disease, ischemia-reperfusion and rheumatoid arthritis (3, 8, 35, 54). Our study has uncovered a substantial difference in the role of ERK1/2 activation in TLR4-dependent cytokine responses of HMVEC versus leukocytes. Our data suggest that ERK1/2 activity restrains LPS-induced production of IL-6 in HMVEC, in contrast to the known pro-inflammatory role of ERK1/2 in monocytes and macrophages. Paradoxically, however, ERK1/2 activity also promotes LPS-induced HMVEC permeability. Our study focused on lung microvascular endothelial cells because they are centrally involved in lung injury and respiratory failure in sepsis. Further studies are needed to determine the role of ERK1/2 in LPS-induced activation and permeability of microvascular endothelial cell from other organs, to determine the functional relevance of endothelial ERK1/2 activation in sepsis and organ injury, and to define the differences in ERK1/2-dependent signaling in endothelial cells versus leukocytes.
Supplementary Material
Figure S1: LPS purity
Gold stain of 1 μg ultrapure LPS (UP LPS, List Laboratories, Lot 4219A1) and 1 μg of lysate from E.Coli O111:B4. Immunoblot for (B) OmpA (35 kDa), (C) MLP (7 kDa), and (D) PAL (18 kDa).
Figure S2: HMVEC LPS-induced IL-6 production and permeability is independent of MyD88 and TRIF.
HMVEC were pre-treated with MyD88 inhibitor (10 μM), TRIF inhibitor (10 μM), or vehicle for 6 hours, and then with LPS (1 μg/ml) for another 15 hours in the continued presence of the inhibitors or vehicle. (A) IL-6 levels were quantified in cell supernatants. ECIS tracing of normalized resistance to assess the effects of MyD88 inhibitor (B) and TRIF inhibitor (D) on LPS-induced HMVEC permeability are shown. Raw resistance was normalized to the resistance of the corresponding well immediately before the addition of LPS (arrow). (C, E) Area under the curve was then quantified from baseline = 1 with addition of negative peaks and graphed as means ± SD. Data was analyzed by one-way analysis of variance (ANOVA). n = 6; *p≤0.05; NS, not significant.
ACKNOWLEDGEMENTS
We would like to acknowledge Angela Balolong (UC Berkeley) for her assistance in experiments.
FUNDING SOURCE:
This work was supported by a T32 Training Grant (T32GM008440-20, JH/EW), the International Anesthesia Research Society Frontiers in Anesthesia Research Award (JH), and the UCSF Department of Anesthesia and Preoperative Care (JH).
Footnotes
CONFLICTS OF INTEREST: The authors declare that there are no conflicts of interest regarding the publication of this article.
REFERENCES
- 1.Achuthan A, Aslam ASM, Nguyen Q, Lam P, Fleetwood AJ, Frye AT, et al. Glucocorticoids promote apoptosis of proinflammatory monocytes by inhibiting ERK activity. Cell Death & Disease 9(3):267–13, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aird WC. Spatial and temporal dynamics of the endothelium. Journal of Thrombosis and Haemostasis 3(7):1392–1406, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Alam R, Gorska MM. Mitogen-activated protein kinase signaling and ERK1/2 bistability in asthma. Clinical & Experimental Allergy 41(2):149–159, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alcock J, Brainard AH. Hemostatic containment – An evolutionary hypothesis of injury by innate immune cells. Medical Hypotheses 71(6):960–968, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Amalakuhan B, Habib SA, Mangat M, Reyes LF, Rodriguez AH, Hinojosa CA, et al. Endothelial adhesion molecules and multiple organ failure in patients with severe sepsis. Cytokine 88:267–273, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anand AR, Bradley R, Ganju RK. LPS-induced MCP-1 expression in human microvascular endothelial cells is mediated by the tyrosine kinase, Pyk2 via the p38 MAPK/NF-κB-dependent pathway. Molecular Immunology 46(5):962–968, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bianconi E, Piovesan A, Facchin F, Beraudi A, Casadei R, Frabetti F, et al. An estimation of the number of cells in the human body. Annals of Human Biology 40(6):463–471, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Dai L, Wu H, Zhang Z, Wang W, Fu J, Deng R, et al. Novel anti-inflammatory target of geniposide: Inhibiting ItgB1/Ras-ERK1/2 signal pathway via the miRNA-124a in rheumatoid arthritis synovial fibroblasts. International Immunopharmacology 65:284–94, 2018. [DOI] [PubMed] [Google Scholar]
- 9.Danese S, Dejana E, Fiocchi C. Immune Regulation by Microvascular Endothelial Cells: Directing Innate and Adaptive Immunity, Coagulation, and Inflammation. The Journal of Immunology 178(10):6017–6022, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Dauphinee SM, Karsan A. Lipopolysaccharide signaling in endothelial cells. Laboratory Investigation 86 (1):9–22, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Dayang E, Plantinga J, Ter Ellen B, van Meurs M, Molema G, Moser J. Identification of LPS-Activated Endothelial Subpopulations With Distinct Inflammatory Phenotypes and Regulatory Signaling Mechanisms. Frontiers in Immunology 10:1169, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doyle SL, O’Neill LAJ. Toll-like receptors: From the discovery of NFκB to new insights into transcriptional regulations in innate immunity. Biochemical Pharmacology 72 (9):1102–1113, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Garcia JG, Siflinger-Birnboim A, Bizios R, Del Vecchio PJ, Fenton 2 JW, Malik AB. Thrombin-induced increase in albumin permeability across the endothelium. Journal of Cellular Physiology 128(1):96–104, 1986. [DOI] [PubMed] [Google Scholar]
- 14.Guha M, O’Connell MA, Pawlinski R, Hollis A, McGovern P, Yan SF, et al. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor alpha expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood 98(5):1429–1439, 2001. [DOI] [PubMed] [Google Scholar]
- 15.Hatano N, Mori Y, Oh-hora M, Kosugi A, Fujikawa T, Nakai N, et al. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes to Cells 8 (11):847–856, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Hellman J, Tehan MM, Warren HS. Murein lipoprotein, peptidoglycan-associated lipoprotein, and outer membrane protein a are present in purified rough and smooth lipopolysaccharides. The Journal of Infectious Diseases 188 (2):286, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Hellman J, Loiselle PM, Tehan MM, Allaire JE, Boyle LA, Kurnick JT, et al. Outer membrane protein A (OmpA), peptidoglycan-associated lipoprotein (PAL), and murein lipoprotein (MLP) are released in experimental Gram-negative sepsis. Journal of Endotoxin Research 7(1):69–72, 2001. [PubMed] [Google Scholar]
- 18.Hoshino K, Kitamura T, Nakamura Y, Irie Y, Matsumoto N, Kawano Y, et al. Usefulness of plasminogen activator inhibitor-1 as a predictive marker of mortality in sepsis. Journal of Intensive Care 5(1):42, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang Q, Xu W, Ustinova E, Wu M, Childs E, Hunter F, et al. Myosin light chain kinase-dependent microvascular hyperpermeability in thermal injury. Shock 20(4):363–368, 2003. [DOI] [PubMed] [Google Scholar]
- 20.Huang Q, Zhao M, Zhao K. Alteration of vascular permeability in burn injury. MedicalExpress 1(2):62–76, 2014. [Google Scholar]
- 21.Iba T, Kidokoro A, Fukunaga M, Sugiyama K, Sawada T, Kato H. Association between the severity of sepsis and the changes in the hemostatic molecular markers and vascular endothelial damage markers. Shock 23(1):25–29, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Iba T, Kidokoro A, Yagi Y: The role of the endothelium in changes in procoagulant activity in sepsis. J Am Coll Surg 187(3):321–329, 1998. [DOI] [PubMed] [Google Scholar]
- 23.Imaizumi T, Itaya H, Fujita K, Kudoh D, Kudoh S, Mori K, et al. Expression of Tumor Necrosis Factor-α in Cultured Human Endothelial Cells Stimulated With Lipopolysaccharide or Interleukin-1α. Arteriosclerosis, Thrombosis, and Vascular Biology: Journal of the American Heart Association 20 (2):410, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Jaffe EA. Cell biology of endothelial cells. Human Pathology 18 (3):234–239, 1987. [DOI] [PubMed] [Google Scholar]
- 25.Joffre J, Hellman J, Ince C, Ait-Oufella H. Endothelial Responses in Sepsis. Am J Respir Crit Care Med 2020. [DOI] [PubMed] [Google Scholar]
- 26.Joyce D, Nelson D, Grinnell B. Leukocyte and endothelial cell interactions in sepsis: Relevance of the protein C pathway. Crit Care Medicine 32(5 Suppl):S280–S286, 2004. [DOI] [PubMed] [Google Scholar]
- 27.Kevil CG, Oshima T, Alexander B, Coe LL, Alexander JS. H2O2-mediated permeability: Role of MAPK and occludin. American Journal of Physiology 48 (1):C21, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Khakpour S, Wilhelmsen K, Hellman J: Vascular endothelial cell Toll-like receptor pathways in sepsis. Innate Immun 21(8):827–846, 2015. [DOI] [PubMed] [Google Scholar]
- 29.Lendemans S, Rani M, Selbach C, Kreuzfelder E, Schade FU, Flohé S. GM-CSF priming of human monocytes is dependent on ERK1/2 activation. Journal of Endotoxin Research 12(1):10–20, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Lu Y, Yeh W, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine 42(2):145–151, 2008. [DOI] [PubMed] [Google Scholar]
- 31.Mai J, Virtue A, Shen J, Wang H, Yang X. An evolving new paradigm: endothelial cells - conditional innate immune cells. Journal of Hematology & Oncology 6(1):61, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mammoto A, Mammoto T, Kanapathipillai M, Wing Yung C, Jiang E, Jiang A, et al. Control of lung vascular permeability and endotoxin-induced pulmonary oedema by changes in extracellular matrix mechanics. Nat Commun 4:1759, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Mikacenic C, Price BL, Harju-Baker S, O’Mahony DS, Robinson-Cohen C, Radella F, et al. A Two-Biomarker Model Predicts Mortality in the Critically Ill with Sepsis. Am J Respir Crit Care Med 196(8):1004–1011, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mikacenic C, Hahn WO, Price BL, Harju-Baker S, Katz R, Kain KC, et al. Biomarkers of Endothelial Activation Are Associated with Poor Outcome in Critical Illness. PloS ONE 10(10):e0141251, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okada M, Yamane M, Yamamoto S, Otani S, Miyoshi K, Sugimoto S, et al. SPRED2 deficiency may lead to lung ischemia–reperfusion injury via ERK1/2 signaling pathway activation. Surg Today 48(12):1089–1095, 2018. [DOI] [PubMed] [Google Scholar]
- 36.Prakash A, Mesa KR, Wilhelmsen K, Xu F, Dodd-o JM, Hellman J. Alveolar Macrophages and Toll-like Receptor 4 Mediate Ventilated Lung Ischemia Reperfusion Injury in Mice. Anesthesiology 117(4):822–835, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ranta V, Orpana A, Carpen O, Turpeinen U, Ylikorkala O, Viinikka L. Human vascular endothelial cells produce tumor necrosis factor-a in response to proinflammatory cytokine stimulation. Crit Care Medicine 27(10):2184–187, 1999. [DOI] [PubMed] [Google Scholar]
- 38.Repo H, Harlan JM. Mechanisms and Consequences of Phagocyte Adhesion to Endothelium. Annals of Medicine 31(3):156–165, 1999. [DOI] [PubMed] [Google Scholar]
- 39.Richardson ET, Shukla S, Nagy N, Boom WH, Beck RC, Zhou L, et al. ERK Signaling Is Essential for Macrophage Development: e0140064. PLoS ONE 10(10): 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robbins CS, Swirski FK. The multiple roles of monocyte subsets in steady state and inflammation. Cellular and Molecular Life Sciences : CMLS 67(16):2685–2693, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rondina MT, Carlisle M, Fraughton T, Brown SM, Miller 3, Russell R, Harris ES, et al. Platelet-Monocyte Aggregate Formation and Mortality Risk in Older Patients With Severe Sepsis and Septic Shock. The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences 70(2):225–231, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawa Y, Ueki T, Hata M, Iwasawa K, Tsuruga E, Kojima H, et al. LPS-induced IL-6, IL-8, VCAM-1, and ICAM-1 Expression in Human Lymphatic Endothelium. Journal of Histochemistry and Cytochemistry 56(2):97–109, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skibsted S, Jones AE, Puskarich MA, Arnold R, Sherwin R, Trzeciak S, et al. Biomarkers of Endothelial Cell Activation in Early Sepsis. Shock 39(5):427–432, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szulcek R, Bogaard HJ, van Nieuw Amerongen, Geerten P. Electric Cell-substrate Impedance Sensing for the Quantification of Endothelial Proliferation, Barrier Function, and Motility. Journal of Visualized Experiments (85):51300, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Takeda K, Akira S. Toll-like receptor signaling. Nature Reviews Immunology 4 (7):499–511, 2004. [DOI] [PubMed] [Google Scholar]
- 46.Verin AD, Liu F, Bogatcheva N, Borbiev T, Hershenson MB, Wang P, et al. Role of Ras-dependent ERK activation in phorbol ester-induced endothelial cell barrier dysfunction. AJP - Lung Cellular and Molecular Physiology 279(2):360–L370, 2000. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Wu J, Guo X, Huang X, Huang Q. RAGE Plays a Role in LPS-Induced NF-κB Activation and Endothelial Hyperpermeability. Sensors 17(4):722, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang W, Wang W, Deng M, Deng M, Liu X, Liu X, et al. TLR4 Activation Induces Nontolerant Inflammatory Response in Endothelial Cells. Inflammation 34(6):509–518, 2011. [DOI] [PubMed] [Google Scholar]
- 49.Weiss T, Shalit I, Blau H, Werber S, Halperin D, Levitov A, et al. Anti-Inflammatory Effects of Moxifloxacin on Activated Human Monocytic Cells: Inhibition of NF- B and Mitogen-Activated Protein Kinase Activation and of Synthesis of Proinflammatory Cytokines. Antimicrobial Agents and Chemotherapy 48(6):1974–1982, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilhelmsen K, Xu F, Farrar K, Tran A, Khakpour S, Sundar S, et al. Extracellular signal-regulated kinase 5 promotes acute cellular and systemic inflammation. Science Signaling 8 (391):ra86, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilhelmsen K, Mesa KR, Lucero J, Xu F, Hellman J. ERK5 protein promotes, whereas MEK1 protein differentially regulates, the Toll-like receptor 2 protein-dependent activation of human endothelial cells and monocytes. The Journal of Biological Chemistry 287(32):26478–26494, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wilhelmsen K, Mesa KR, Prakash A, Xu F, Hellman J. Activation of endothelial TLR2 by bacterial lipoprotein upregulates proteins specific for the neutrophil response. Innate Immunity 18(4):602–616, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolinsky H A Proposal Linking Clearance of Circulating Lipoproteins to Tissue Metabolic Activity as a Basis for Understanding Atherogenesis. Circulation Research 47(3):301–311, 1980. [DOI] [PubMed] [Google Scholar]
- 54.Wu Y, Wang F, Fan L, Zhang W, Wang T, Du Y, et al. Baicalin alleviates atherosclerosis by relieving oxidative stress and inflammatory responses via inactivating the κBNF- and p38 MAPK signaling pathways. Biomedicine & Pharmacotherapy 97:1673–1679, 2018. [DOI] [PubMed] [Google Scholar]
- 55.Xiao L, Liu Y, Wang N. New paradigms in inflammatory signaling in vascular endothelial cells. Am J Physiol Heart Circ Physiol 306(3):H317–25. 2014. [DOI] [PubMed] [Google Scholar]
- 56.Xu H, Ye X, Steinberg H, Liu SF. Selective blockade of endothelial NF‐κB pathway differentially affects systemic inflammation and multiple organ dysfunction and injury in septic mice. The Journal of Pathology 220(4):490–498, 2010. [DOI] [PubMed] [Google Scholar]
- 57.Young M Endothelial cells in the eyes of an immunologist. Cancer Immunol Immunother 61(10):1609–1616, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu W, London NR, Gibson CC, Davis CT, Tong Z, Sorensen LK, et al. Interleukin receptor activates a MYD88–ARNO–ARF6 cascade to disrupt vascular stability. Nature 492(7428):252–255, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: LPS purity
Gold stain of 1 μg ultrapure LPS (UP LPS, List Laboratories, Lot 4219A1) and 1 μg of lysate from E.Coli O111:B4. Immunoblot for (B) OmpA (35 kDa), (C) MLP (7 kDa), and (D) PAL (18 kDa).
Figure S2: HMVEC LPS-induced IL-6 production and permeability is independent of MyD88 and TRIF.
HMVEC were pre-treated with MyD88 inhibitor (10 μM), TRIF inhibitor (10 μM), or vehicle for 6 hours, and then with LPS (1 μg/ml) for another 15 hours in the continued presence of the inhibitors or vehicle. (A) IL-6 levels were quantified in cell supernatants. ECIS tracing of normalized resistance to assess the effects of MyD88 inhibitor (B) and TRIF inhibitor (D) on LPS-induced HMVEC permeability are shown. Raw resistance was normalized to the resistance of the corresponding well immediately before the addition of LPS (arrow). (C, E) Area under the curve was then quantified from baseline = 1 with addition of negative peaks and graphed as means ± SD. Data was analyzed by one-way analysis of variance (ANOVA). n = 6; *p≤0.05; NS, not significant.