

Ubiquitin and UBL posttranslational modifications, including FAT10, are utilized and dysregulated by viruses for achievement of effective infection and virion production. The UBE1L2-FAT10 system catalyzes FAT10ylation, where one or more FAT10 molecules are covalently linked to a substrate.
KEYWORDS: FAT10, KSHV, herpesvirus, lytic replication, posttranslational modification, protein expression, ubiquitin-like protein
ABSTRACT
During Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic replication, host cell functions, including protein expression and posttranslational modification pathways, are dysregulated by KSHV to promote virus production. Here, we attempted to identify key proteins for KSHV lytic replication by profiling protein expression in the latent and lytic phases using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Proteomic analysis, immunoblotting, and quantitative PCR demonstrated that antigen F (HLA-F) adjacent transcript 10 (FAT10) and UBE1L2 (also known as ubiquitin-like modifier-activating enzyme 6 [UBA6]) were upregulated during lytic replication. FAT10 is a ubiquitin-like protein (UBL). UBE1L2 is the FAT10-activating enzyme (E1), which is essential for FAT10 modification (FAT10ylation). FAT10ylated proteins were immediately expressed after lytic induction and increased over time during lytic replication. Knockout of UBE1L2 suppressed KSHV production but not KSHV DNA synthesis. In order to isolate FAT10ylated proteins during KSHV lytic replication, we conducted immunoprecipitation using anti-FAT10 antibody and nickel-nitrilotriacetic acid (Ni-NTA) chromatography of exogenously expressed His-tagged FAT10 from cells undergoing latent or lytic replication. LC-MS/MS was performed to identify FAT10ylated proteins. We identified KSHV ORF59 and ORF61 as FAT10ylation substrates. Our study revealed that the UBE1L2-FAT10 system is upregulated during KSHV lytic replication, and it contributes to viral propagation.
IMPORTANCE Ubiquitin and UBL posttranslational modifications, including FAT10, are utilized and dysregulated by viruses for achievement of effective infection and virion production. The UBE1L2-FAT10 system catalyzes FAT10ylation, where one or more FAT10 molecules are covalently linked to a substrate. FAT10ylation is catalyzed by the sequential actions of E1 (activation enzyme), E2 (conjugation enzyme), and E3 (ligase) enzymes. The E1 enzyme for FAT10ylation is UBE1L2, which activates FAT10 and transfers it to E2/USE1. FAT10ylation regulates the cell cycle, interferon (IFN) signaling, and protein degradation; however, its primary biological function remains unknown. Here, we revealed that KSHV lytic replication induces UBE1L2 expression and production of FAT10ylated proteins, including KSHV lytic proteins. Moreover, UBE1L2 knockout suppressed virus production during the lytic cycle. This is the first report demonstrating the contribution of the UBE1L2-FAT10 system to KSHV lytic replication. Our findings provide insight into the physiological function(s) of novel posttranslational modifications in KSHV lytic replication.
INTRODUCTION
Kaposi’s sarcoma-associated herpesvirus (KSHV) is the causative agent of the endothelial cell malignancy Kaposi’s sarcoma, the B cell malignancy primary effusion lymphoma, and multicentric new Castleman’s disease (1–3). The KSHV life cycle is comprised of two phases: latent replication and lytic replication. After the primary infection, the KSHV genome is transported into the nucleus and forms circularized double-stranded DNA. During the latent infection, KSHV expresses latent genes and microRNAs to maintain the viral genome without viral production (4). Productive reactivation of KSHV is triggered by the expression of ORF50/RTA (replication and transcription activator), which is known as a “switch gene” that activates lytic replication (5, 6). ORF50/RTA expression normally occurs spontaneously in a subset of infected cells. During lytic replication, viral genomes are amplified using viral replication machinery, viral proteins are synthesized, and subsequently, progeny virions are assembled. KSHV utilizes host machineries responsible for protein expression and posttranslational modifications in order to establish an infection, promote effective virus production, and facilitate the switch from latency to lytic replication (7).
In infected cells, the ubiquitination pathway is utilized and hijacked by KSHV for the manipulation of cell proliferation, inhibition of apoptosis, and escape from immune surveillance (7). The KSHV-encoded proteins LANA (latency-associated nuclear antigen) and ORF50/RTA mediate the polyubiquitination of p53 and interferon regulatory factor 7 (IRF-7), respectively, which results in their proteasomal degradation (8, 9). Additionally, KSHV carries genes that encode the deubiquitinating enzyme (ORF64) that disassembles the polyubiquitin chains of retinoic acid-inducible gene 1 (RIG-I) and induces RIG-I stabilization (10, 11). ORF45, in its monoubiquitinated form, regulates the interaction of virions with internal lipid rafts for virion assembly and egress from infected cells (12). KSHV hijacks not only the ubiquitination pathway but also various ubiquitin-like protein (UBL) pathways, including those that generate small ubiquitin-related modifier (SUMO), interferon-stimulated gene product of 15 kDa (ISG15), and neural precursor cell expressed developmentally downregulated protein 8 (NEDD8) modifications. KSHV utilizes the SUMO, ISG15, and NEDD8 modifications as well as ubiquitination to regulate gene expression, to enhance viral replication, and to regulate both latency and lytic reactivation, respectively (13–15). Thus, mounting evidence has revealed a strong relationship between alterations in UBL modifications and regulation of the KSHV life cycle. Therefore, monitoring dynamic changes in protein expression and posttranslational modifications is imperative to understand the KSHV life cycle.
In this study, we attempted to identify novel key proteins involved in KSHV lytic replication by comparing protein expression dynamics during latent and lytic replication using liquid chromatography-tandem mass spectrometry (LC-MS/MS). We found that the levels of antigen F (HLA-F) adjacent transcript 10 (FAT10) and UBE1L2 were significantly upregulated during the KSHV lytic phase. FAT10 belongs to the UBL family and was recently discovered as a novel UBL (16). Similar to other UBLs, FAT10 modification (FAT10ylation) is catalyzed by the sequential actions of FAT10-activating enzyme (E1), FAT10-conjugating enzyme (E2), and FAT10 ligase (E3) (17). The E1 for FAT10ylation is UBE1L2 (also named ubiquitin-like modifier-activating enzyme 6 [UBA6]), which activates FAT10 and transfers the activated FAT10 to E2/USE1 (18–20). Finally, FAT10 is covalently attached to a Lys of the substrate by E3 (21, 22). The FAT10ylated protein is degraded by the 26S proteasome or stabilized via unknown mechanisms.
FAT10ylation regulates several processes, including the immune system, cell cycle, interferon (IFN) and tumor necrosis factor (TNF) signaling, and apoptosis (23, 24). The H5N1 influenza virus utilizes FAT10 for viral replication by inhibiting type I IFN (25). In human immunodeficiency virus type 1 (HIV-1) infection, Vpr, an HIV-1 accessory protein, induces the expression of FAT10 and enhances apoptosis via interacting with FAT10 (26). Thus, several viruses utilize FAT10 modifications for the establishment and maintenance of their infections. However, before this study, it was unknown whether FAT10 modifications play a role in the life cycle of KSHV. This is the first report demonstrating that the UBE1L2-FAT10 system is upregulated during the KSHV lytic cycle and contributes to viral replication.
RESULTS
KSHV lytic replication induces UBE1L2 expression and FAT10ylation.
First, we searched for critical factors involved in KSHV lytic replication by profiling differentially expressed host proteins between the lytic and latent phases of infection. In order to induce the transition from latent to lytic replication, KSHV reactivation was induced in iSLK-rKSHV.219 cells by doxycycline (Dox) and sodium butyrate (NaB) treatment for 72 h. The postreactivation (as the lytic phase) and prereactivation (as the latent phase) cell lysates were analyzed by LC-MS/MS in order to compare the protein expression patterns between the two phases. LC-MS/MS analysis revealed a total of 6,487 proteins that were expressed in iSLK-rKSHV.219 cells, and 584 proteins were upregulated more than twofold in the lytic phase relative to the latent phase. Figure 1A shows the top 10 most upregulated proteins during the lytic phase, and UBE1L2 (also known as UBA6) was the most upregulated. The full data set is shown in Table S1 in the supplemental material. The expression level of UBE1L2 increased by approximately 64-fold (26.6-fold) in the lytic phase compared to the latent phase. Next, the upregulation of UBE1L2 was validated by immunoblotting using lytic-phase-induced iSLK-rKSHV.219 (rKSHV stands for recombinant KSHV) and iVero-BAC16 (BAC stands for bacterial artificial chromosome) cells. The UBE1L2 expression levels were increased depending on the postinduction time in both iSLK-rKSHV.219 (Fig. 1B) and iVero-BAC16 cells (Fig. 1D). Figure 1C and E show the densitometric analysis of UBE1L2 expression from immunoblots shown in Fig. 1B and D, respectively. On the other hand, UBE1L2 expression in iSLK cells lacking the KSHV genome were not affected by Dox and NaB treatment even after 72 h (Fig. 1F). These results indicated that the upregulation of UBE1L2 was induced by KSHV lytic replication. UBE1L2 is the E1 enzyme of FAT10, which belongs to the UBL family (16). Therefore, we tested whether the KSHV lytic cycle affected FAT10 expression and FAT10ylation of substrate proteins. To this end, iSLK-rKSHV.219 cells were treated with Dox and NaB to induce lytic replication and were harvested at 24 to 72 h postinduction. Cell lysates were generated and subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 15% (Fig. 1G, left panel) and 6% (Fig. 1G, right panel) acrylamide gels, followed by immunoblotting with anti-FAT10 antibody. The data showed a significant increase in free FAT10 protein (18 kDa) expression and production of FAT10 conjugates (30, 45, 58, 70, 80, and 100 kDa) after lytic induction. These results indicated that the KSHV lytic cycle stimulated the UBE1L2-FAT10 system and induced the expression of FAT10 as well as UBE1L2.
FIG 1.

The expression levels of UBE1L2 and FAT10 are increased during KSHV lytic replication. (A) The upregulated cellular proteins in lytic-phase-induced iSLK-rKSHV.219 cells. The iSLK-rKSHV.219 cells were treated with (or without) Dox and NaB for 72 h and harvested. Quantitative LC-MS/MS was then used to evaluate protein expression during latent versus lytic replication. The protein expression level ranks (orders), gene names, UniProt IDs, protein names, and quantitative values of the top 10 proteins that were upregulated 72 h after lytic induction in iSLK-rKSHV.219 cells are summarized in the table. In the Log2 Fold change (Lytic/Latent) column of the table, log2-transformed fold changes were calculated from iSLK-rKSHV.219 cells during lytic infection and latent infection. In the Normalized Intensity (Lytic and Latent) columns, the values show the log2-transformed intensities after median normalization. (B to E) The upregulation of UBE1L2 (also known as UBA6) in lytic-induced Vero and SLK cells harboring the KSHV genome. In order to induce lytic replication, the iSLK-rKSHV.219 (B and C) and iSLK cells (F) were treated with Dox and NaB, and the iVero-BAC16 cells were treated with Dox (D and E). Lytic-induced cells were harvested at 24 to 72 h postinduction (hpi) followed by immunoblotting (IB) with anti-UBE1L2 antibody. UBE1L2 expression levels in lytic-induced iSLK-rKSHV.219 cells (B) and lytic-induced iVero-BAC16 cells (D). The quantification of UBE1L2 levels in lytic-induced iSLK-rKSHV.219 cells (C) and lytic-induced iVero-BAC16 cells (E) were performed by densitometric analysis using ImageJ software. The levels of UBE1L2 were normalized to the β-actin levels. Immunoblotting analyses were repeated at least three times, and representative data are shown in panels B and D. (F) The expression of UBE1L2 was not affected by Dox treatment. The iSLK cells were treated with Dox for 72 h and subjected to immunoblotting with anti-UBE1L2 antibody. (G) Production of free FAT10 and FAT10 conjugates in lytic-induced iSLK-rKSHV.219 cells. Cells were treated with Dox and NaB to induce lytic replication, and the cells were harvested at 24 to 72 hpi followed by cell lysis. Lysates (4 μg of protein/lane) were subjected to SDS-PAGE on 15% (left panel) or 6% (right panel) acrylamide gels. The proteins were transferred onto nitrocellulose membranes and blotted with anti-FAT10 antibody to monitor FAT10 expression and FAT10ylation.
KSHV lytic replication induces the transcription of FAT10 and ISG15.
Next, we tested the mRNA expression levels of ubiquitin C as well as several UBLs (FAT10, ISG15, and NEDD8) during the lytic cycle. In order to induce lytic replication, the iSLK-rKSHV.219 and iSLK cells were cultured with Dox and NaB. The cells were then harvested at 24 to 72 h postinduction, and total RNA was isolated, followed by quantitative reverse transcription-PCR (RT-qPCR) analysis. The fold changes in mRNA expression were calculated by dividing the value of the mRNA expression level found in iSLK-rKSHV.219 cells to the value in iSLK cells. As shown in Fig. 2A, the FAT10 mRNA was continuously transcribed during lytic replication and was induced by 3,000-fold 72 h after lytic induction compared to the value at 0 h. ORF59 is a KSHV early gene, whose expression is utilized as a marker for lytic replication. As expected, ISG15 expression was slightly increased after lytic induction, consistent with previous reports, which showed that ISG15 was upregulated by virus infection or lytic induction (27). Surprisingly, the UBE1L2 mRNA expression levels were not increased during the lytic cycle despite the finding that UBE1L2 protein expression was highly upregulated (Fig. 1). In addition, mRNA expression of ubiquitin C, NEDD8, and USE1 (E2 of FAT10) did not change significantly following lytic replication activation. To investigate why the protein expression of UBE1L2 was upregulated, we examined protein stabilization using a proteasome inhibitor (MG132) and a lysosome inhibitor (bafilomycin A1). Data showed that both MG132 and bafilomycin A induced UBE1L2 stabilization during the latent phase (Fig. 2B); however, UBE1L2 appeared to be more stabilized by MG132 rather than bafilomycin A1 (Fig. 2B, panels ii and v). These data suggested that UBE1L2 is regulated by both proteasomal and lysosomal degradation during latency, and the proteasome predominantly degrades UBE1L2. On the other hand, neither MG132 nor bafilomycin A1 did not significantly stabilize UBE1L2 during the lytic phase (panels ii and v). In addition, the UBE1L2 bands shifted to approximately to the upper 10 to 30 kDa during the lytic phase. These shifted UBE1L2, which is indicated by an arrowhead in Fig. 2B (panels i and iv), seem to be posttranslationally modified UBE1L2 (PTM-UBE1L2). Interestingly, PTM-UBE1L2 was stabilized by bafilomycin A1 (panels iii and vi). These data indicated the possibility that these degradation system alterations after reactivation might contribute to the upregulation of UBE1L2.
FIG 2.

Expression of FAT10 mRNA is upregulated during KSHV lytic replication in a time-dependent manner. (A) Expression of several mRNAs of interest during KSHV lytic replication. iSLK-rKSHV.219 and iSLK cells were treated with Dox and NaB to induce lytic replication. The cells were then harvested at 48 to 72 h postinduction (hpi), and total RNA was subjected to real-time RT-qPCR. The FAT10, ubiquitin C, ISG15, NEDD8, UBE1L2, USE1, and ORF59 mRNA levels were normalized to the GAPDH mRNA levels. The mRNA expression levels shown in the graphs were obtained by dividing the mRNA expression values from iSLK-rKSHV.219 cells by those from iSLK cells to exclude the effects of Dox and NaB per se. (B) The stabilization of UBE1L2 during latent and lytic phases. Nontreated iSLK-rKSHV.219 cells (latent phase) or Dox- and NaB-treated iSLK-rKSHV.219 cells (lytic phase) were cultured in medium containing 1μΜ MG132 or 1 nΜ bafilomycin A1 for 24 h. Cells were harvested and subjected to immunoblotting with anti-UBE1L2 antibody (i and iv). The quantification of UBE1L2 levels normalized with the β-tubulin in latent (left)- or lytic-induced iSLK-rKSHV.219 cells (right) and were performed using densitometric analysis using ImageJ software (ii and v). The quantification of posttranslationally modified UBE1L2 (PTM-UBE1L2) in lytic-induced iSLK-rKSHV.219 cells was performed by densitometric analysis using ImageJ software. The PTM-UBE1L2 band is indicated by an arrowhead. The levels of PTM-UBE1L2 were normalized to approximate β-tubulin levels. Immunoblotting analyses were repeated three times, and typical data are shown in panels i and iv. (C) FAT10 mRNA expression was slightly upregulated by RTA. HeLa or 293T cells were transfected with a negative-control plasmid (empty) or the expression plasmids encoding RTA and/or K-bZIP. At 48 h posttransfection, cells were collected (left and middle panels). iSLK cells were treated with Dox and NaB to induce RTA expression, and cells were cultured for 48 and 72 h (right panel). Total RNA was purified and subjected to real-time RT-qPCR for quantification of FAT10 mRNA. The FAT10 mRNA levels were normalized to the GAPDH mRNA levels. (D) Effects of RTA or K-bZIP on the mRNA expression of ubiquitin C and NEDD8. The RTA or K-bZIP expression plasmids were transfected into 293T cells. (i) Cells were harvested at 48 h posttransfection. (ii) iSLK cells were treated with Dox and NaB to induce RTA expression, and cells were cultured for 48 and 72 h, respectively. Total RNA was subjected to real-time RT-qPCR for the quantification of ubiquitin C and NEDD8 mRNA. Each mRNA level was normalized to the GAPDH mRNA level.
KSHV carries genes that encode viral transcription factors such as RTA and K-bZIP. RTA is responsible for the transition from latency to the lytic cycle and thus, induces lytic replication (6, 28). K-bZIP is expressed immediately after the initiation of reactivation (29). Both RTA and K-bZIP activate downstream gene expression, which allows for viral replication and virus production. In order to assess whether these viral transcription factors are involved in the upregulation of FAT10, we transfected RTA and/or K-bZIP expression plasmids into HeLa or 293T cells and monitored the FAT10 mRNA expression levels. In addition to those overexpressed, iSLK cells were treated with Dox and NaB to induce RTA expression, and these cells were subjected to real-time RT-qPCR for quantification of FAT10 mRNA. Consequently, RTA was deemed to enhance FAT10 transcription in both cell lines (Fig. 2C, left and middle panels). On the other hand, in RTA-expressing iSLK cells, FAT10 was not enhanced (Fig. 2C, right panel). As seen in Fig. 2C, we also evaluated the effects of RTA and K-bZIP on ubiquitin C and NEDD8 transcript levels (Fig. 2D). The overexpression of either RTA or K-bZIP appeared to suppress the ubiquitin C and NEDD8 transcript levels in 293T cells (Fig. 2D, panel i). In contrast, both ubiquitin C and NEDD8 were unaffected in RTA-expressing iSLK cells (panel ii). This might be due to the fact that other transcriptional factors, which were activated in iSLK cells, act antagonistically on RTA and K-bZIP. Taken together, these findings indicated that FAT10 mRNA expression was induced not only by RTA but also by the RTA-independent transactivators expressed during lytic replication.
UBE1L2 plays an important role in KSHV production.
Next, we attempted to unveil the role of the UBE1L2-FAT10 system in KSHV lytic replication using the CRISPR/Cas9 system to knock out UBE1L2. To this end, iSLK-rKSHV.219 cells were transduced with the CRISPR/Cas9 KO plasmid targeting UBE1L2 (or the control CRISPR/Cas9 plasmid) and incubated for 4 days followed by lytic induction with Dox and NaB. The downregulation of UBE1L2 was confirmed by immunoblotting (Fig. 3A). We analyzed the effects of the UBE1L2 knockout (KO) on intracellular KSHV DNA synthesis and extracellular KSHV production. In order to monitor KSHV DNA synthesis in UBE1L2 KO cells, the amount of KSHV DNA in CRISPR/Cas9-transduced and lytic-induced iSLK-rKSHV.219 cells were measured by real-time qPCR. The UBE1L2 KO cells exhibited slightly reduced intracellular KSHV DNA at 48 h and 72 h after lytic induction, but there was no significant difference between the UBE1L2 KO cells and control KO cells (Fig. 3B).
FIG 3.

KO of UBE1L2 in host cells reduces KSHV production but not KSHV DNA synthesis. (A) CRISPR/Cas9-mediated KO of UBE1L2 in iSLK-rKSHV.219 cells. iSLK-rKSHV.219 cells were transfected with the UBE1L2 CRISPR/Cas9 KO plasmid (KO) or control CRISPR/Cas9 plasmid (Cont) and cultured for 4 days. The UBE1L2 KO was confirmed by immunoblotting (IB) with anti-UBE1L2 antibody. The UBE1L2 and β-actin band intensities were calculated using ImageJ software, and the value of UBE1L2/β-actin in the control is presented as 1.0. (B to E) Effects of UBE1L2 KO on KSHV DNA synthesis (B), virus production (C), infectious virus production (D), and FAT10 mRNA expression (E) in iSLK-rKSHV.219 cells. In order to induce lytic replication (viral DNA synthesis and progeny virus production), the control (Cont) and UBE1L2 KO (KO) cells were treated with Dox and NaB. (B) At 48 or 72 h postinduction (hpi), the intracellular viral DNA synthesis levels were evaluated by real-time qPCR as described in Materials and Methods. (C) At 48 or 72 hpi, the extracellular KSHV production was quantitated by real-time qPCR of viral DNA derived from mature virions in the culture supernatant. (D) At 0 or 72 hpi, the culture supernatants were collected and inoculated with 293T cells for infection. At 72 h postinfection, GFP-positive cells were counted by fluorescence microscopy. (E) The expression levels of FAT10 mRNA were not affected by UBE1L2 KO. At 0 or 72 h posttreatment, cells were collected, and total RNA was subjected to real-time RT-qPCR. The FAT10 mRNA levels were normalized toward the GAPDH mRNA levels. The mRNA expression levels shown in the graphs were obtained by dividing the mRNA expression values from 72 h by those from 0 h to monitor the rate of increase. The two-tailed Student’s t test was used to indicate between-group differences. The P values are shown in each graph. N.S., not significant.
During herpesvirus assembly and egress, synthesized viral DNA is packaged by viral capsids in the nuclei, which become enveloped as they bud through the nuclear membrane. Once in the cytoplasm, tegumentation occurs and the tegumented capsids obtain their final envelope by budding into the trans-Golgi network. The progeny virus particles are then secreted to the extracellular environment (30). In order to evaluate extracellular KSHV production, we measured viral DNA in the culture medium. The KO of UBE1L2 inhibited viral production by 67% (Fig. 3C). In addition to measuring viral DNA, we also monitored the virus titers in culture medium. The culture medium was added to 293T cells, and the numbers of green fluorescent protein (GFP)-positive cells were counted to evaluate infectious virion production. As a result, the KO of UBE1L2 reduced progeny viruses released into the culture medium by approximately 50 to 60% (Fig. 3D). In addition, the expression levels of FAT10 mRNA were not changed significantly in UBE1L2 KO cells (Fig. 3E). This suggests that the mechanism of expression of FAT10 is independent of the mechanism of expression of UBE1L2. These data indicated that UBE1L2 plays a role in lytic replication, especially after viral DNA synthesis. In addition, we monitored the localization of UBE1L2 in lytic-induced iVero-BAC16 cells. UBE1L2 appeared as dots that were spread out throughout the cytoplasm before lytic induction, and after lytic induction, UBE1L2 accumulated in the peripheral nuclei (Fig. 4A). Superresolution microscopy was also performed to further examine the localization of UBE1L2. UBE1L2 colocalized with the Golgi marker 130 (GM130) (Fig. 4B). This suggests UBE1L2 acts on the Golgi apparatus during lytic replication, probably as an E1 enzyme. Taken together with Fig. 3 and 4, it is likely that UBE1L2, located in the Golgi apparatus, contributes to viral production directly or indirectly when viral particles go through Golgi apparatus.
FIG 4.
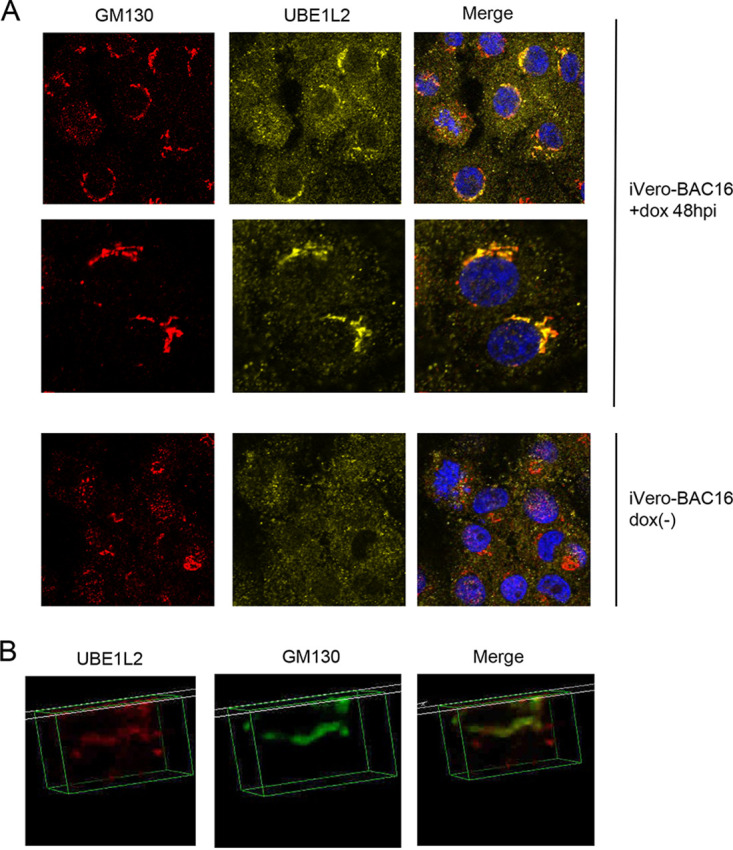
UBE1L2 is localized to the cis-Golgi during KSHV lytic replication. (A) In order to induce lytic replication, iVero-BAC16 cells were treated with doxycycline (+dox) or without doxycycline [dox(-)] for 48 h. The cells were then fixed, permeabilized, and stained with anti-GM130 antibody (red), anti-UBE1L2 antibody (yellow), and DAPI (blue). Merged images are shown on the right. (B) Lytic-replication-induced-iVero-BAC16 cells were fixed, permeabilized, and stained with anti-UBE1L2 (red) and anti-GM130 (green) antibodies followed by superresolution microscopy. The merged image is shown on the right.
Identification of FAT10ylated proteins from cells undergoing KSHV lytic replication.
FAT10 modification of substrate proteins has been reported to alter their structure and function (23, 24, 31). We found that UBE1L2 contributed to KSHV production in the lytic stage (Fig. 3) and lytic replication induced the production of FAT10ylated proteins in the host cell (Fig. 1G). Therefore, we hypothesized that these FAT10ylated proteins, especially KSHV-encoded proteins, were important for the establishment of lytic replication. Thus, we attempted to identify the proteins that were FAT10ylated during KSHV lytic replication. Figure 5A shows the experimental workflow for identifying the FAT10ylated proteins. In order to isolate FAT10ylated proteins, pulldown assays using His-tagged FAT10 (His-FAT10) as the bait protein and immunoprecipitations using anti-FAT10 antibody were conducted. His-FAT10 plasmid-transfected or untransfected iSLK-rKSHV.219 cells were treated with Dox and NaB to induce lytic replication. His-FAT10 conjugates and endogenous FAT10 conjugates were purified from cell extracts by Ni-NTA agarose and anti-FAT10 antibody-immobilized protein G beads, respectively. In order to identify the FAT10ylated proteins, FAT10 conjugates were subjected to LC-MS/MS analysis. The Ni-NTA pulldown assays captured 787 proteins, including 14 KSHV-encoded proteins, which were regarded as putative FAT10ylated substrates. Likewise, the immunoprecipitations with anti-FAT10 antibody captured 1,402 proteins, including 15 KSHV-encoded proteins, which were regarded as putative FAT10ylated substrates (the full data set is shown in Table S2 in the supplemental material). To detect FAT10ylated substrates by Ni-NTA pulldown and immunoprecipitation, radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris-HCl, 300 mM NaCl, 1% Nonidet P-40 [NP-40], 0.5% deoxycholate [DOC], and 0.1% SDS) was used for cell lysis and bead washing. Thus, we estimated that FAT10 conjugation with viral open reading frames (ORFs) was formed by covalent binding.
FIG 5.
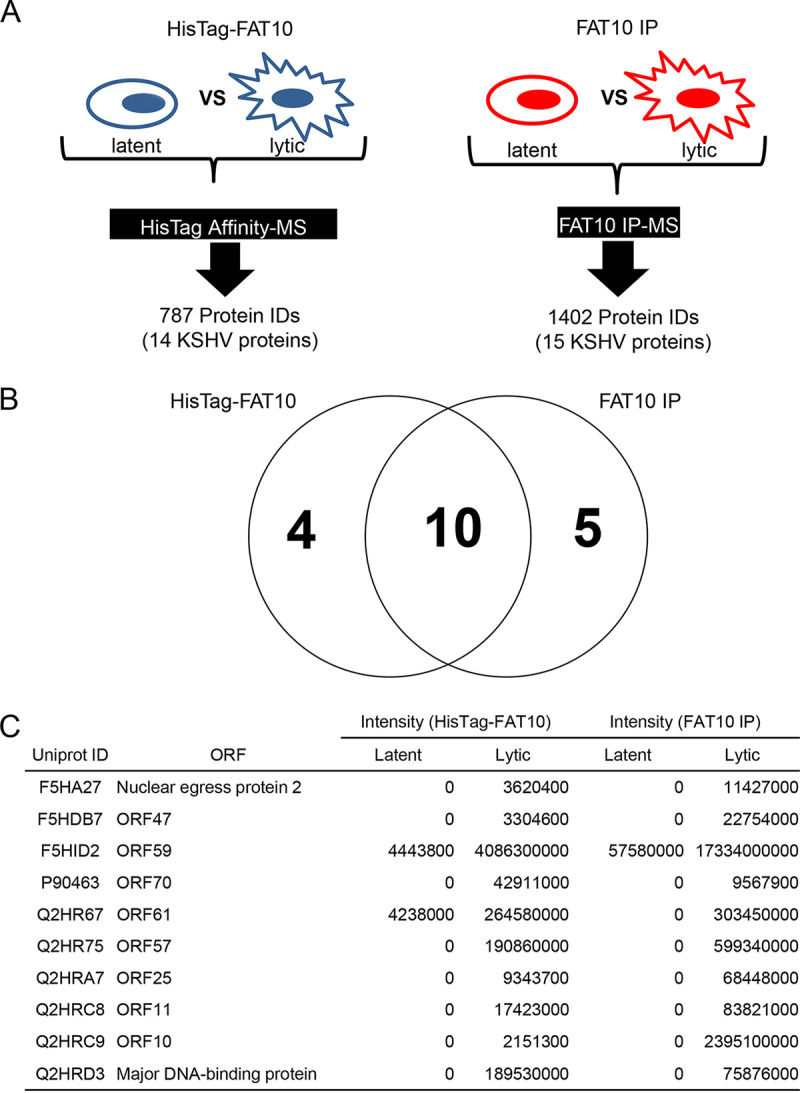
Identification of FAT10ylated proteins. (A) The proteomic workflow designed to isolate and identify proteins linked to His-tagged FAT10 or endogenous FAT10. Proteins linked to His-tagged FAT10 (HisTag-FAT10) or endogenous FAT10 were isolated by His tag affinity chromatography (HisTag Affinity) or immunoprecipitation with anti-FAT10 antibody (FAT10 IP), respectively. The isolated proteins were then identified by mass spectrometry (LC-MS/MS) analysis. (B) A Venn diagram of identified KSHV proteins captured by a Ni-NTA affinity column (HisTag FAT10) or immunoprecipitations with anti-FAT10 antibody (FAT10 IP). (C) The 10 KSHV proteins that were captured by both His tag pulldown and immunoprecipitation with anti-FAT10 antibody. The table lists the UniProt IDs, protein names, and quantitative values of FAT10ylated or His tag FAT10ylated KSHV protein expression during lytic or latent replication. The intensity columns (HisTag-FAT10 and FAT10 IP) show the observed intensities based on label-free quantification. The presence of a 0 indicates that the protein was not identified.
Our initial studies were focused on identifying FAT10ylated viral proteins. Therefore, we focused on the putative viral FAT10ylated proteins which were positive using both pulldown and immunoprecipitation methods. These comparisons revealed that 10 KSHV proteins overlapped FAT10ylated candidates from pulldown assays and those from immunoprecipitations (Fig. 5B and C). We next confirmed that ORF59 and ORF61 were covalently bound to FAT10 by pulldown analysis. Plasmids expressing His-FAT10 and either 2xStag-ORF59-3xFLAG (Stag-ORF59) or 2xStag-ORF61-3xFLAG (Stag-ORF61) were cotransfected into 293T cells, and Stag-ORF59 or Stag-ORF61 was precipitated from the cell lysates with S-protein agarose. Stag-ORF59 (42 kDa) and Stag-ORF61 (88 kDa) were detected in both total cell lysates (TCLs) and the pulldown fractions. In addition, FAT10ylated Stag-ORF59 and Stag-ORF61 migrated at approximately 60 and 100 kDa, respectively, after SDS-PAGE (Fig. 6A). Additionally, we tested other viral proteins which were listed by our MS analysis and are potentially involved in viral production, ORF11, ORF47, and nuclear egress protein 2. However, the FAT10ylation signals of these proteins were undetected (data not shown). MS analysis showed that the proportion of these proteins undergoing FAT10ylation was lower than that of ORF59 and ORF61 (Fig. 5C). Another possibility is that the expression levels of these proteins were low or unstable because of an expression vector or structural property. Due to these reasons, as well as the low sensitivity of our detection method, FAT10ylation signals of those proteins might not have been detected by our pulldown method.
FIG 6.

KSHV ORF61 and ORF59 are potential FAT10ylation substrates. (A) 293T cells were cotransfected with plasmids expressing His-FAT10 (20 kDa) and either 2xStag-ORF59-3xFLAG (Stag-ORF59; 42 kDa) or 2xStag-ORF61-3xFLAG (Stag-ORF61; 88 kDa). At 48 h posttransfection, the cells were lysed in RIPA buffer and subjected to pulldown assays (PD) using S-protein beads. The FAT10ylation of ORF61 and ORF59 was confirmed by immunoblotting (IB) using anti-His antibody. The FAT10ylated ORF59 (approximately 60 kDa) and ORF61 (approximately 110 kDa) are indicated by an asterisk. TCL, total cell lysate. (B) Effect of UBE1L2 knockout on the FAT10ylation of ORF61. 293T cells were transfected with the UBE1L2 CRISPR/Cas9 KO plasmid (UBE1L2-KO) or control CRISPR/Cas9 plasmid (control) and cultured for 4 days. KO plasmid-transfected cells were cotransfected with His-FAT10 and 2xStag-ORF61-3xFLAG plasmids. Cells were subjected to pulldown assay at 48 h posttransfection. The FAT10ylated ORF61 are both indicated by an asterisk. (C) 293T cells were cotransfected with plasmids expressing His-FAT10 and either 2xStag-ORF37-3xFLAG (Stag-ORF37; 55 kDa) or 2xStag-ORF56-3xFLAG (Stag-ORF56; 96 kDa).
These results indicated that KSHV ORF59 and ORF61 are endogenous substrates of FAT10ylation. To demonstrate the involvement of FAT10ylation on UBE1L2, we tested FAT10ylation of ORF61 using UBE1L2 knockout cells. In control cells, FAT10ylated Stag-ORF61 were detected (Fig. 6B, Control lane). On the other hand, in UBE1L2 KO cells, using an unmodified Stag-ORF61 was detected at around 88 kDa, while FAT10ylated Stag-ORF61 was not detected (UBE1L2-KO lane). Furthermore, to indicate pulldown specificity, we analyzed the FAT10ylation of ORF37 and ORF56, which were randomly chosen from KSHV ORFs that our MS analysis excluded from potential FAT10ylated candidates. As expected, we confirmed that Stag-ORF37 (55 kDa) and Stag-ORF56 (96 kDa) were not FAT10ylated (Fig. 6C). Taken together, the UBE1L2-FAT10 system mediated FAT10ylation of several KSHV lytic proteins, and UBE1L2-FAT10 was upregulated by KSHV lytic replication. We believe that these changes in FAT10ylation after lytic induction might contribute to the establishment of KSHV lytic replication, including effective viral production.
DISCUSSION
We found that FAT10 and UBE1L2 expression were induced during the KSHV lytic cycle, and the UBE1L2-FAT10 system contributed to KSHV lytic replication. In addition, pulldown experiments revealed that two KSHV proteins, ORF61 and ORF59, were conjugated to FAT10. KSHV ORF61 is commonly known as a homolog of the herpes simplex virus 1 (HSV-1) UL39 protein, which encodes the ribonucleotide reductase large subunit. KO of the Epstein-Barr virus (EBV) BORF2, another homolog of ORF61, reduced viral production by approximately 70% (32). BORF2 and ORF61 localize in the endoplasmic reticulum (ER) and mediate the relocalization of nuclear APOBEC3B in order to protect viral genomes from APOBEC3B-indued mutation (32, 33). Another group discovered by yeast two-hybrid studies that BORF2 interacted with the EBV capsid structural protein, BcLF1 (34). Therefore, FAT10 conjugation of ORF61 may be involved in progeny capsid assembly or capsid transport among the nucleus, ER, and Golgi apparatus. On the other hand, ORF59 is a multifunctional protein; ORF59 functions as a polymerase processivity factor and a double-stranded DNA-binding protein (35). ORF59 is mainly localized to the nucleus and forms viral DNA replication complexes with ORF6 and ORF9, which are essential for KSHV DNA synthesis (36, 37). Even though our data showed that KO of UBE1L2 had no effect on viral DNA synthesis (Fig. 3B), FAT10ylation of ORF59 may affect the nuclear localization of ORF59 or its recruitment to the viral DNA replication complexes.
Even though the expression of FAT10 and FAT10ylated proteins was upregulated by KSHV lytic replication (Fig. 1G), the upregulation of FAT10 protein under these same conditions was not detected in our deep proteomic analysis (Fig. 1A). Most of the UBLs, including FAT10 become covalently conjugated to their substrates immediately after substrate translation, and this may inhibit FAT10 detection by LC-MS/MS because of covalent binding of FAT10 to other proteins. In addition, it is often difficult to detect low-molecular-weight peptides, such as free FAT10, by MS analysis after LC separation of peptides. Ubiquitylome analysis, a specialized proteomic approach to identify ubiquitinated proteins, is known to be effective at overcoming these issues (38). Development of proteomic methods for identification of FAT10ylated proteins is expected and may contribute to a comprehensive understanding of the physiological function(s) of FAT10ylation.
In the current study, we found that UBE1L2 mRNA expression levels were not increased after induction of KSHV lytic replication. However, UBE1L2 protein levels were highly upregulated under these same conditions (Fig. 1A and Fig. 2A). Thus, the upregulation of UBE1L2 may be due to translational control, i.e., the induction of lytic replication upregulates UBE1L2 translation but not UBE1L2 transcription. There is a report showing that during KSHV lytic replication, translation of cellular proteins is inhibited (39). The KSHV lytic protein, ORF57, interacts with the RNA-binding protein poly(rC)-binding protein 1 (PCBP1), and this complex activates internal ribosome entry site (IRES)-mediated protein translation (39). In several viral infections, IRES-mediated protein translation allows for viral mRNA translation, even when host cell translation is inhibited. Therefore, it is likely that the translation of UBE1L2 is posttranscriptionally controlled by viral protein(s) expressed during lytic replication. UBE1L2 was regulated by both proteasomal and lysosomal degradation during latency; however, proteasome predominantly degrade UBE1L2 (Fig. 2B). Further, UBE1L2 degradation might be turned off after lytic induction, and posttranslational modification of UBE1L2 increases. These suggest that posttranslationally modified UBE1L2 might induce a new function during the lytic phase.
While most of the UBL family members possess their own E1, E2, and E3 enzymes (17, 40), the E1 and E2 of FAT10 are shared with ubiquitin (18–20). The human genome encodes only two types of E1 enzymes: UBE1 and UBE1L2 (also known as UBA6) (41). UBE1 binds and activates only ubiquitin, whereas UBE1L2 binds and activates FAT10 and ubiquitin. Activated FAT10 or ubiquitin is transferred from UBE1L2 to E2/USE1, the UBE1L2-specific E2 enzyme, that allows ubiquitin or FAT10 to bind to their substrate via E3 (21, 22). E3 then ligates FAT10 or ubiquitin to the substrate.
FAT10ylation leads to the degradation of the substrate protein by the 26S proteasome, which is independent of ubiquitin (23, 24). In addition to FAT10ylation, noncovalent interaction with FAT10 leads to change in the activity of the other protein. The deubiquitinating activity of the deubiquitinase OTUB1 is stimulated by the interaction with FAT10 (31). FAT10 can also become covalently bound to OTUB1, and this FAT10ylation induces the proteasomal degradation of OTUB1. Previous studies observed FAT10 accumulation in various tumor cells, which suggested that FAT10ylation directly or indirectly regulates the protein stability of its substrates, which contributes to the regulation of the cell cycle, DNA damage response, and apoptosis (42–44). It was also reported that interferon gamma (IFN-γ) induces FAT10 expression; however, FAT10 negatively regulates the antiviral response. For example, FAT10 noncovalently binds to RIG-I, the cytosolic viral RNA sensor for the innate immune response, and this binding inhibits RIG-I-mediated antiviral signaling pathways (45). In addition, ZNF598, E3 enzyme, catalyzes the ligation of FAT10 to RIG-I, which inhibits downstream RIG-I-mediated signaling that produces type I IFN (46).
FAT10 expression appears to enhance the pathogenesis, survival, and replication of several viruses. Zhang et al. showed that overexpression of FAT10 in human respiratory epithelial cells and mice increased H5N1 influenza virus replication by inhibiting type I IFN (25). Liu et al. also demonstrated that increased expression of FAT10 correlated with recurrence and poor prognosis in hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC) patients, which suggested that HBV-related HCC was exacerbated by the upregulation of FAT10 (43). Thus, viruses utilize the UBE1L2-FAT10 system to attenuate innate immunity responses and establish virus replication.
On the other hand, our study showed that FAT10 was covalently linked to a few KSHV proteins and the UBE1L2-FAT10 system is involved in KSHV production. Thus, we discovered a novel role of FAT10 in herpesviral infection. Since homologs of ORF59 and ORF61 are highly conserved among human herpesviruses, it is likely that these FAT10 modifications are present in other herpesvirus homologs. Our findings strongly suggest that novel UBL posttranslational modifications of KSHV proteins may be useful as therapeutic targets to treat KSHV infections. Therefore, we believe that unraveling the novel viral strategy that KSHV utilizes the FAT10 system for its lytic replication will provide insight for the development of novel KSHV treatments.
MATERIALS AND METHODS
Tetracycline/doxycycline-inducible recombinant KSHV producer cell lines.
In order to monitor KSHV lytic replication, we used the lytic replication-inducible iSLK-rKSHV.219 stable cell line (47). The iSLK-rKSHV.219 cells harbor a recombinant KSHV DNA (rKSHV.219), and this cell line was established from iSLK cells. iSLK cells are tetracycline/doxycycline-inducible (Tet-On) ORF50/RTA-expressing SLK cells (47). iSLK-rKSHV.219 cells were maintained in Dulbecco modified Eagle medium (DMEM) containing 10% fetal calf serum (FCS), 250 μg/ml G418 (Nacalai Tesque, Kyoto, Japan), 100 μg/ml hygromycin B (Wako, Osaka, Japan), and 1 μg/ml puromycin (InvivoGen, San Diego, CA, USA). iSLK cells were maintained in DMEM containing 10% FCS, 250 μg/ml G418, and 1 μg/ml puromycin. In order to induce ORF50/RTA expression, iSLK cells were treated with 8 μg/ml Dox (LKT Laboratories, St. Paul, MN, USA) and 1.5 mM NaB (Tokyo Chemical Industry, Tokyo, Japan). Another tetracycline/doxycycline-inducible recombinant KSHV producer stable cell line (iVero-BAC16) was also used in this study (48). iVero-BAC16 cells harbor the KSHV BAC16 DNA (49) and was established from iVero cells. iVero cells are tetracycline/doxycycline-inducible (Tet-On) ORF50/RTA-expressing Vero cells (48, 50). iVero-BAC16 cells were maintained in DMEM containing 10% FCS, 2.5 μg/ml puromycin, and 1000 μg/ml hygromycin B. iVero cells were maintained in DMEM containing 10% FCS and 2.5 μg/ml puromycin. In order to induce ORF50/RTA expression, iVero cells were treated with 8 μg/ml Dox. The expression of ORF50/RTA in iSLK-rKSHV.219 or iVero-BAC16 cells induces lytic replication and subsequent production of recombinant KSHV virions.
Construction of plasmids.
KSHV BAC16 was a kind gift from Jae U. Jung (University of Southern California) (49). In order to construct the 6×His-tagged FAT10 plasmid, the sense and antisense DNA oligonucleotides encoding the 6×His tag fused to FAT10 (N-terminal tagged) were synthesized based on the sequence information of the FAT10 cDNA (gene identifier [ID] 10537) obtained from Eurofins Genomics (Tokyo, Japan). After annealing the sense and antisense oligonucleotides, double-stranded DNA encoding 6×His-tagged FAT10 was cloned into pCIneo (Promega, Madison, WI, USA), generating the plasmid referred to as pCIneo-6xHis-FAT10. In order to construct the pCIneo-2xStag-ORF59-3xFLAG, pCIneo-2xStag-ORF61-3xFLAG, pCIneo-2xStag-ORF11-3xFLAG, pCIneo-2xStag-ORF47-3xFLAG, pCIneo-2xStag-ORF67-3xFLAG, pCIneo-2xStag-ORF37-3xFLAG, and pCIneo-2xStag-ORF56-3xFLAG expression plasmids, KSHV ORF59, ORF61, ORF11, ORF47, ORF67, ORF37, and ORF56 DNA were amplified by PCR from BAC16 and cloned into the pCIneo-2xS-3xFLAG (N-terminal 2xS-tag and C-terminal 3xFLAG-tag) vector. The pCIneo-2xS-3xFLAG vector was generated by inserting oligonucleotides encoding two repeats of the S-tag peptide and three repeats of the FLAG tag peptide in front of and in back of the multiple cloning site, respectively, of pCIneo (Promega, Madison, WI, USA). In order to construct the pCIneo-3xFLAG-RTA and pCIneo-3xFLAG-K-bZIP plasmids, ORF50/RTA and K-bZIP DNA were amplified by PCR from BAC16 and cloned into the pCIneo-3xFLAG (N-terminal 3xFLAG-tag) vector. The pCIneo-3xFLAG vector was generated by inserting oligonucleotides encoding three repeats of the FLAG-tag peptide in front of the multiple cloning site of pCIneo (Promega, Madison, WI, USA).
Transfection.
iSLK-rKSHV.219 cells (1 × 106) were seeded into six-well plates and transfected with 2 μg of expression plasmid utilizing ScreenFectA plus (Fujifilm, Tokyo, Japan). After incubation for 12 h, the cells were treated with 8 μg/ml Dox and 1.5 mM NaB for 48 h and harvested.
Antibodies.
Anti-UBE1L2 (catalog no. 13386) and anti-FAT10 (catalog no. 76194) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-GM130 antibody (catalog no. 610822) was purchased from BD Biosciences (Franklin Lakes, NJ, USA). Anti-His monoclonal (D291-3) and anti-S polyclonal (PM021) antibodies were obtained from Medical & Biological Laboratories (Nagoya, Japan). Secondary donkey anti-rabbit and anti-mouse IgG antibodies conjugated to Alexa Fluor 488, 594, or 647 (A-21202, A-21203, A-31571, A-21206, A-21207, and A-32795) were obtained from Molecular Probes (Eugene, OR, USA).
Preparation of samples for proteomic analysis.
For proteomic analysis of KSHV-infected cells, cell pellets were solubilized in phase-transfer buffer (12 mM sodium deoxycholate, 12 mM sodium lauroyl sarcosinate, and 50 mM ammonium bicarbonate) containing cOmplete protease inhibitor cocktail (Roche, Basel, Switzerland) and PhosSTOP phosphatase inhibitor cocktail (Roche). The protein lysates (100 μg) were reduced, alkylated, and trypsinized. After incubation at 37°C overnight, surfactants in the lysates were removed by mixing with ethyl acetate as described previously (51). The tryptic peptides were desalted with Oasis HLB (Waters, Milford, MA, USA) and fractionated into seven fractions on a C18-SCX stagetip (52). The fractions were evaporated and stored at 4°C until LC-MS/MS analysis. For the analysis of FAT10ylated proteins captured with anti-FAT10 antibody or His-FAT10ylated proteins captured with nickel-nitrilotriacetic acid (Ni-NTA), the eluents were subjected to methanol/chloroform precipitation. The protein pellets were dissolved in phase-transfer buffer containing cOmplete protease inhibitor cocktail and PhosSTOP phosphatase inhibitor cocktail. The lysates were reduced, alkylated, and trypsinized at 37°C overnight. After mixing with ethyl acetate for surfactant removal, the tryptic peptides were desalted on a C18 stagetip (53). The peptides were evaporated and stored at 4°C until LC-MS/MS analysis.
LC-MS/MS analyses.
All proteomic analyses were performed with the Q Exactive (Thermo Fisher Scientific, Waltham, MA, USA) connected to an UltiMate 3000 (Thermo Fisher Scientific, Waltham, MA, USA) and an HTC PAL autosampler (CTC Analytics, Zwingen, Switzerland). The nano-LC buffers comprised the following components: buffer A (0.1% formic acid and 2% acetonitrile) and buffer B (0.1% formic acid and 90% acetonitrile). The tryptic peptides were separated with the nano-LC in a gradient of 5 to 35% buffer B over 135 min. The flow rate of the nano-LC was 280 nl/min. The MS parameters for the Q Exactive were described in a previous phosphoproteomic study (54).
Protein identification and quantification.
The raw files were analyzed with MaxQuant 1.5.1.2 supported by the Andromeda search engine (55). For identification of human and viral proteins, a protein database from UniProt (release 2011_11, including 262 common contaminants) was combined with the amino acid sequences of 84 KSHV proteins. The enzymatic specificity was set as C terminal to Arg or Lys with the allowed cleavage at the proline bond. Two missed cleavages were tolerated. The fixed and variable modifications were specified as carbamidomethylation of cysteine residues and methionine oxidation, respectively. The protein groups and peptide levels with false discovery rates (FDRs) less than 0.01 were accepted. Proteins annotated as “Reverse Hit” or “Potential Contaminant” were omitted. The quantitative data were obtained with label-free quantification. In each sample, the log2-transformed data were normalized using median centering.
Immunoblotting.
Cells were washed with phosphate-buffered saline (PBS) and lysed with lysis buffer (20 mM Tris-HCl [pH 7.5], 300 mM NaCl, 1 mM EDTA, 0.02% sodium dodecyl sulfate (SDS), 0.5% Triton X-100, and 1 mM dithiothreitol [DTT]) containing cOmplete protease inhibitor cocktail (Roche). The cell lysates were sonicated and then centrifuged at 4°C for 15 min at 15,000 rpm. The total amount of protein contained in the supernatants were measured with a Protein Assay BCA kit (Nacalai Tesque, Kyoto, Japan), and the concentration of each sample was adjusted to 0.5 μg/μl. The proteins (4 or 10 μg) were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and were transferred onto polyvinylidene difluoride (PVDF) membranes (GE Healthcare Life Sciences, Buckinghamshire, UK). The membranes were blocked with 5% skim milk, incubated with primary antibody (1:1,000) at 4°C overnight, and then incubated with anti-mouse (GE Healthcare Life Sciences) or rabbit horseradish peroxidase (HRP)-conjugated secondary antibody (GE Healthcare Life Sciences) (1:5,000) for 2 h at room temperature. The blotted proteins were visualized with the Amersham ECL Prime Western blotting detection reagent (GE Healthcare Life Sciences).
To monitor the UBE1L2 stabilization, cells were treated with 1 μM MG132 (Peptide Institute, Osaka, Japan) or 1 nM bafilomycin A1 (AdipoGen, San Diego, CA, USA) for 24 h. Cells were lysed using a lysis buffer containing a cocktail of protease inhibitors, and cell lysates were subjected to Western blotting with anti-UBE1L2 antibody.
Quantitative reverse transcription-PCR.
Total cellular RNA was purified from cells using RNAiso Plus (TaKaRa, Osaka, Japan) and quantitated by quantitative reverse transcription-PCR (RT-qPCR). cDNA was synthesized with the ReverTra Ace qPCR RT kit (Toyobo) and then subjected to SYBR green real-time PCR using the Thunderbird qPCR Mix kit (Toyobo). The sequences of the RT-qPCR primer sets are listed in Table 1. The sequences of the primer sets designed to amplify ubiquitin C were previously reported (56). RT-qPCR with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers was also performed to serve as an internal control for input RNA levels. The relative mRNA expression levels were determined by ΔΔCT methods and normalized to GAPDH mRNA levels.
TABLE 1.
Oligonucleotide primer sequence
| Primer name | Sequence |
|---|---|
| FAT10-forward | 5′-TCTGTGTGCATGTCCGTTCC-3′ |
| FAT10-reverse | 5′-GAGGCTTCTCCGTGGCTTTA-3′ |
| UbiquitinC-forward | 5′-CACTTGGTCCTGCGCTTGA-3′ |
| UbiquitinC-reverse | 5′-TTATTGGGAATGCAACAACTTTAT-3′ |
| ISG15-forward | 5′-GCGCAGATCACCCAGAAGAT-3′ |
| ISG15-recerse | 5′-GTTCGTCGCATTTGTCCACC-3′ |
| NEDD8-forward | 5′-GCTGACCGGAAAGGAGATTGA-3′ |
| NEDD8-reverse | 5′-AACACCAGGTGAAGGACTGA-3′ |
| SUMO1-forward | 5′-TCTGACCAGGAGGCAAAACC-3′ |
| SUMO1-reverse | 5′-TGGAACACCCTGTCTTTGAC-3′ |
| ORF59-forward | 5′-GCCCACATCCACCGACTTC-3′ |
| ORF59-reverse | 5′-AGCCAGAAACCAAACCCGTT-3′ |
| UBE1L2-forward | 5′-GCAACCCTGAGGCACCTTTA-3′ |
| UBE1L2-reverse | 5′-TCCAACATTTGGCTTGCGAC-3′ |
| USE1-forward | 5′-GGACTCCCCTTGGAGGATCT-3′ |
| USE1-reverse | 5′-GCTCGAGGAATGGAAGAGGG-3′ |
| GAPDH-forward | 5′-TCGCTCTCTGCTCCTCCTGTTC-3′ |
| GAPDH-reverse | 5′-CGCCCAATACGACCAAATCC-3′ |
CRISPR/Cas9-mediated UBE1L2 KO.
The UBE1L2 CRISPR/Cas9 knockout (KO) plasmid (sc-412489) or control CRISPR/Cas9 plasmid (sc-418922) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA), and 1 μg of each plasmid was transfected into iSLK-rKSHV.219 cells or 293T cells utilizing ScreenFectA plus (Fujifilm Wako Pure Chemical Corporation, Osaka, Japan). After 4 days, the cells were treated with8 μg/ml Dox and 1.5 mM NaB. The treated cells were then incubated for 72 h to induce lytic replication.
Measurement of extracellular virus production and intracellular viral DNA synthesis.
Mature KSHV virions are released into the culture supernatant after lytic replication. In order to quantify extracellular KSHV production, qPCR was conducted on culture supernatants from KSHV-infected cells as described previously (48, 57). The UBE1L2 CRISPR/Cas9 KO plasmid-transfected iSLK-rKSHV.219 cells were cultured with 8 μg/ml of Dox for 48 h or 72 h to induce lytic replication and production of KSHV virions. Culture supernatants (300 μl) were treated with DNase I (New England Biolabs, Ipswich, MA, USA) in order to digest cellular DNA or viral DNA, which failed to be encapsidated. Viral DNA was extracted and purified from 200 μl of DNase I-treated culture supernatant using the QIAamp DNA Blood minikit (Qiagen, Hilden, Germany). The KSHV copy number was quantitated by qPCR using the KSHV-encoded ORF11-specific primer set: 5′-TTGACAACACGCACCGCAAG-3′ and 5′-AAAAATCAGCACGCTCGAGGAG-3′.
During lytic replication, the KSHV genome is amplified in host nuclei by approximately 100- to 1,000-fold. In order to quantify the intracellular KSHV DNA synthesis levels, qPCR was conducted with ORF11-specific primers described above. The UBE1L2 CRISPR/Cas9 KO plasmid-transfected and lytic replication-induced iSLK-rKSHV.219 cells were harvested at 48 h or 72 h after treatment with Dox and NaB. Total DNA, containing cellular DNA as well as newly synthesized KSHV DNA, was purified with the QIAamp DNA Blood minikit (Qiagen). Next, qPCR was conducted in order to quantitate the KSHV genome copy number, which was normalized to the total DNA concentration.
Measurement of progeny viruses contained in culture supernatant.
The UBE1L2 CRISPR/Cas9 KO plasmid-transfected iSLK-rKSHV.219 cells were cultured with 8 μg/ml of Dox for 72 h to induce lytic replication and the production of KSHV virions. The 700-μl culture supernatants were harvested. A total of 1 × 106 293T cells were plated in a 24-well plate, and 500-μl culture supernatants were inoculated. After 72 h, GFP -positive/negative cells were counted by fluorescence microscopy.
Immunofluorescence analyses.
All staining procedures were carried out at room temperature except for primary antibody incubations. For immunofluorescence experiments, cells were washed with ice-cold PBS and fixed in PBS containing 0.1% FCS and 0.01% Tween 20 (PBS-T), followed by permeabilization with 0.5% Triton X-100 in PBS for 10 min. The samples were then blocked for 1 h in 10% FCS in PBS, followed by overnight incubation at 4°C with the primary antibodies diluted in PBS-T. After the samples were washed with PBS-T, they were then incubated for 1 h with secondary antibodies conjugated to Alexa Fluor 488 or 594 (Thermo Fisher Scientific, MA, USA) diluted in PBS-T. Next, the samples were washed with PBS-T. All the primary antibodies were utilized at a 1:300 dilution, and the secondary antibodies were used at a 1:500 dilution. The slides were mounted in ProLong Diamond Antifade reagent with 4′,6′-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific) and analyzed by fluorescence confocal microscopy or a superresolution microscope. Laser scanning confocal fluorescence microscopic images were captured and processed using an LSM-810 Meta microscope (Carl Zeiss MicroImaging, Inc., Jena, Germany) with a plan-apochromat 63×/1.4-numerical-aperture oil immersion objective. Superresolution microscopic images were captured and processed with an N-SIM superresolution microscope (Nikon, Tokyo, Japan).
Purification of His-FAT10ylated proteins by Ni-NTA agarose chromatography.
The pCIneo-6xHis-FAT10 expression plasmid (15 μg) was transfected into iSLK-rKSHV.219 cells (approximately 2.5 × 107), following lytic induction. The cells were lysed in RIPA buffer (20 mM Tris-HCl [pH 7.5], 300 mM NaCl, 1% NP-40, 0.5% DOC, and 0.1% SDS) containing cOmplete protease inhibitor cocktail (Roche). Ni-NTA agarose beads (30 μl) (Qiagen) and cell lysates (5 ml) were mixed for 60 min at 4°C, and the Ni-NTA beads were washed four times with RIPA buffer. The bound His-FAT10 and His-FAT10ylated proteins were eluted with 30 μl of SDS sample buffer and separated by SDS-PAGE, followed by visualization with Coomassie brilliant blue (CBB) staining and LC-MS/MS analysis.
Immunoprecipitation.
For immunoprecipitation, lytic-induced iSLK-rKSHV.219 cells (approximately 8 × 106) were lysed in 1 ml of RIPA buffer and incubated with 10 μg of anti-FAT10 antibody (Santa Cruz Biotechnology) or an equal amount of control IgG (Santa Cruz Biotechnology) overnight at 4°C. Next, 20 μl of protein G agarose (Merck-Millipore, Darmstadt, Germany) was added to the lysates (1 ml) and incubated with mixing for 2 h at 4°C. The FAT10 antibody-immobilized protein G beads were washed four times with RIPA buffer. The immunoprecipitates were eluted with 20 μl of SDS sample buffer and separated by SDS-PAGE, followed by visualization with CBB staining and MS analysis.
Pulldown with S-protein agarose beads.
293T cells were maintained in DMEM and 10% FCS at 37°C in a humidified 5% CO2 atmosphere. 293T cells (approximately 8 × 106) were cotransfected with 2.5 μg of pCIneo-6xHis-FAT10 and 2.5 μg of pCIneo-2xS-ORF61-3xFLAG or pCIneo-2xS-ORF59-3xFLAG. After incubation for 48 h, the cells were lysed with RIPA buffer (3 ml) containing protease inhibitors. S-protein agarose beads (20 μl) (Merck-Millipore) and cell lysates (3 ml) were mixed for 60 min at 4°C, and the precipitated proteins were eluted with 20 μl of SDS sample buffer. The samples were then separated by SDS-PAGE, followed by immunoblotting. To monitor the infection of UBE1L2 KO, UBE1L2 CRISPR/Cas9 KO plasmid were introduced to 293T cells as mentioned in “CRISPR/Cas9-mediated UBE1L2 KO” and incubated for 4 days. UBE1L2-KO cells were cotransfected with 2.5 μg of pCIneo-6xHis-FAT10 and 2.5 μg of pCIneo-2xS-ORF61-3xFLAG, incubated for 48 h, and lysed with RIPA buffer (3 ml). Pulldown analysis was performed as previously described.
Supplementary Material
ACKNOWLEDGMENTS
The KSHV BAC clone, BAC16, was a kind gift from Kevin Brulois and Jae U. Jung (University of Southern California [USC], USA). We thank Gregory A. Smith (Northwestern University, USA) for the Escherichia coli strain GS1783 and Nikolaus Osterrieder (Cornell University, USA) for the pEP-KanS plasmid. Superresolution microscopy was technically supported by Nikon Solutions Co., Ltd.
A.S. was supported by grants for scientific research from the Uehara Memorial Foundation and from the JSPS for Young Scientists. M.F. was supported by a grant from the JSPS Grant-in-Aid for Scientific Research (18K06642). T.W. was supported by a grant from the JSPS Grant-in-Aid for Young Scientists (18K14910).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Nador RG, Cesarman E, Chadburn A, Dawson DB, Ansari MQ, Sald J, Knowles DM. 1996. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood 88:645–656. 10.1182/blood.V88.2.645.bloodjournal882645. [DOI] [PubMed] [Google Scholar]
- 2.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d’Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 86:1276–1280. 10.1182/blood.V86.4.1276.bloodjournal8641276. [DOI] [PubMed] [Google Scholar]
- 3.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc Natl Acad Sci U S A 93:14862–14867. 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toth Z, Brulois K, Jung JU. 2013. The chromatin landscape of Kaposi’s sarcoma-associated herpesvirus. Viruses 5:1346–1373. 10.3390/v5051346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guito J, Lukac DM. 2012. KSHV Rta promoter specification and viral reactivation. Front Microbiol 3:30. 10.3389/fmicb.2012.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F, Miller G. 1998. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc Natl Acad Sci U S A 95:10866–10871. 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashizawa A, Higashi C, Masuda K, Ohga R, Taira T, Fujimuro M. 2012. The ubiquitin system and Kaposi’s sarcoma-associated herpesvirus. Front Microbiol 3:66. 10.3389/fmicb.2012.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cai Q, Xiao B, Si H, Cervini A, Gao J, Lu J, Upadhyay SK, Verma SC, Robertson ES. 2012. Kaposi’s sarcoma herpesvirus upregulates Aurora A expression to promote p53 phosphorylation and ubiquitylation. PLoS Pathog 8:e1002566. 10.1371/journal.ppat.1002566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu J, Martin HJ, Liao G, Hayward SD. 2007. The Kaposi’s sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc. J Virol 81:10451–10459. 10.1128/JVI.00804-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzalez CM, Wang L, Damania B. 2009. Kaposi’s sarcoma-associated herpesvirus encodes a viral deubiquitinase. J Virol 83:10224–10233. 10.1128/JVI.00589-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Inn KS, Lee SH, Rathbun JY, Wong LY, Toth Z, Machida K, Ou JH, Jung JU. 2011. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J Virol 85:10899–10904. 10.1128/JVI.00690-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Zhu N, Li W, Zhu F, Wang Y, Yuan Y. 2015. Mono-ubiquitylated ORF45 mediates association of KSHV particles with internal lipid rafts for viral assembly and egress. PLoS Pathog 11:e1005332. 10.1371/journal.ppat.1005332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Izumiya Y, Ellison TJ, Yeh ET, Jung JU, Luciw PA, Kung HJ. 2005. Kaposi’s sarcoma-associated herpesvirus K-bZIP represses gene transcription via SUMO modification. J Virol 79:9912–9925. 10.1128/JVI.79.15.9912-9925.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobs SR, Stopford CM, West JA, Bennett CL, Giffin L, Damania B. 2015. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 1 interacts with a member of the interferon-stimulated gene 15 pathway. J Virol 89:11572–11583. 10.1128/JVI.01482-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hughes DJ, Wood JJ, Jackson BR, Baquero-Perez B, Whitehouse A. 2015. NEDDylation is essential for Kaposi’s sarcoma-associated herpesvirus latency and lytic reactivation and represents a novel anti-KSHV target. PLoS Pathog 11:e1004771. 10.1371/journal.ppat.1004771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu YC, Pan J, Zhang C, Fan W, Collinge M, Bender JR, Weissman SM. 1999. A MHC-encoded ubiquitin-like protein (FAT10) binds noncovalently to the spindle assembly checkpoint protein MAD2. Proc Natl Acad Sci U S A 96:4313–4318. 10.1073/pnas.96.8.4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulman BA, Harper JW. 2009. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol 10:319–331. 10.1038/nrm2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang F, Zhao B. 2019. UBA6 and its bispecific pathways for ubiquitin and FAT10. Int J Mol Sci 20:2250. 10.3390/ijms20092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiu YH, Sun Q, Chen ZJ. 2007. E1-L2 activates both ubiquitin and FAT10. Mol Cell 27:1014–1023. 10.1016/j.molcel.2007.08.020. [DOI] [PubMed] [Google Scholar]
- 20.Aichem A, Pelzer C, Lukasiak S, Kalveram B, Sheppard PW, Rani N, Schmidtke G, Groettrup M. 2010. USE1 is a bispecific conjugating enzyme for ubiquitin and FAT10, which FAT10ylates itself in cis. Nat Commun 1:13. 10.1038/ncomms1012. [DOI] [PubMed] [Google Scholar]
- 21.Gavin JM, Chen JJ, Liao H, Rollins N, Yang X, Xu Q, Ma J, Loke HK, Lingaraj T, Brownell JE, Mallender WD, Gould AE, Amidon BS, Dick LR. 2012. Mechanistic studies on activation of ubiquitin and di-ubiquitin-like protein, FAT10, by ubiquitin-like modifier activating enzyme 6, Uba6. J Biol Chem 287:15512–15522. 10.1074/jbc.M111.336198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aichem A, Catone N, Groettrup M. 2014. Investigations into the auto-FAT10ylation of the bispecific E2 conjugating enzyme UBA6-specific E2 enzyme 1. FEBS J 281:1848–1859. 10.1111/febs.12745. [DOI] [PubMed] [Google Scholar]
- 23.Pelzer C, Groettrup M. 2010. FAT10: activated by UBA6 and functioning in protein degradation. Subcell Biochem 54:238–246. 10.1007/978-1-4419-6676-6_19. [DOI] [PubMed] [Google Scholar]
- 24.Schmidtke G, Kalveram B, Groettrup M. 2009. Degradation of FAT10 by the 26S proteasome is independent of ubiquitylation but relies on NUB1L. FEBS Lett 583:591–594. 10.1016/j.febslet.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Tang J, Yang N, Liu Q, Zhang Q, Zhang Y, Li N, Zhao Y, Li S, Liu S, Zhou H, Li X, Tian M, Deng J, Xie P, Sun Y, Lu H, Zhang MQ, Jin N, Jiang C. 2016. FAT10 is critical in influenza A virus replication by inhibiting type I IFN. J Immunol 197:824–833. 10.4049/jimmunol.1501563. [DOI] [PubMed] [Google Scholar]
- 26.Snyder A, Alsauskas Z, Gong P, Rosenstiel PE, Klotman ME, Klotman PE, Ross MJ. 2009. FAT10: a novel mediator of Vpr-induced apoptosis in human immunodeficiency virus-associated nephropathy. J Virol 83:11983–11988. 10.1128/JVI.00034-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morales DJ, Lenschow DJ. 2013. The antiviral activities of ISG15. J Mol Biol 425:4995–5008. 10.1016/j.jmb.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gradoville L, Gerlach J, Grogan E, Shedd D, Nikiforow S, Metroka C, Miller G. 2000. Kaposi’s sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J Virol 74:6207–6212. 10.1128/jvi.74.13.6207-6212.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin SF, Robinson DR, Miller G, Kung HJ. 1999. Kaposi’s sarcoma-associated herpesvirus encodes a bZIP protein with homology to BZLF1 of Epstein-Barr virus. J Virol 73:1909–1917. 10.1128/JVI.73.3.1909-1917.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mettenleiter TC. 2002. Herpesvirus assembly and egress. J Virol 76:1537–1547. 10.1128/jvi.76.4.1537-1547.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bialas J, Boehm AN, Catone N, Aichem A, Groettrup M. 2019. The ubiquitin-like modifier FAT10 stimulates the activity of deubiquitylating enzyme OTUB1. J Biol Chem 294:4315–4330. 10.1074/jbc.RA118.005406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng AZ, Yockteng-Melgar J, Jarvis MC, Malik-Soni N, Borozan I, Carpenter MA, McCann JL, Ebrahimi D, Shaban NM, Marcon E, Greenblatt J, Brown WL, Frappier L, Harris RS. 2019. Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat Microbiol 4:78–88. 10.1038/s41564-018-0284-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cheng AZ, Moraes SN, Attarian C, Yockteng-Melgar J, Jarvis MC, Biolatti M, Galitska G, Dell’Oste V, Frappier L, Bierle CJ, Rice SA, Harris RS. 2019. A conserved mechanism of APOBEC3 relocalization by herpesviral ribonucleotide reductase large subunits. J Virol 93:e01539-19. 10.1128/JVI.01539-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calderwood MA, Venkatesan K, Xing L, Chase MR, Vazquez A, Holthaus AM, Ewence AE, Li N, Hirozane-Kishikawa T, Hill DE, Vidal M, Kieff E, Johannsen E. 2007. Epstein-Barr virus and virus human protein interaction maps. Proc Natl Acad Sci U S A 104:7606–7611. 10.1073/pnas.0702332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan SR, Chandran B. 2000. Characterization of human herpesvirus 8 ORF59 protein (PF-8) and mapping of the processivity and viral DNA polymerase-interacting domains. J Virol 74:10920–10929. 10.1128/jvi.74.23.10920-10929.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan SR, Bloomer C, Chandran B. 1998. Identification and characterization of human herpesvirus-8 lytic cycle-associated ORF 59 protein and the encoding cDNA by monoclonal antibody. Virology 240:118–126. 10.1006/viro.1997.8911. [DOI] [PubMed] [Google Scholar]
- 37.Wu FY, Ahn JH, Alcendor DJ, Jang WJ, Xiao J, Hayward SD, Hayward GS. 2001. Origin-independent assembly of Kaposi’s sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-K8 protein and cellular PML. J Virol 75:1487–1506. 10.1128/JVI.75.3.1487-1506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rose CM, Isasa M, Ordureau A, Prado MA, Beausoleil SA, Jedrychowski MP, Finley DJ, Harper JW, Gygi SP. 2016. Highly multiplexed quantitative mass spectrometry analysis of ubiquitylomes. Cell Syst 3:395–403.e394. 10.1016/j.cels.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nishimura K, Ueda K, Guwanan E, Sakakibara S, Do E, Osaki E, Yada K, Okuno T, Yamanishi K. 2004. A posttranscriptional regulator of Kaposi’s sarcoma-associated herpesvirus interacts with RNA-binding protein PCBP1 and controls gene expression through the IRES. Virology 325:364–378. 10.1016/j.virol.2004.04.041. [DOI] [PubMed] [Google Scholar]
- 40.Hershko A, Ciechanover A. 1998. The ubiquitin system. Annu Rev Biochem 67:425–479. 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 41.Pelzer C, Kassner I, Matentzoglu K, Singh RK, Wollscheid HP, Scheffner M, Schmidtke G, Groettrup M. 2007. UBE1L2, a novel E1 enzyme specific for ubiquitin. J Biol Chem 282:23010–23014. 10.1074/jbc.C700111200. [DOI] [PubMed] [Google Scholar]
- 42.Zhang DW, Jeang KT, Lee CG. 2006. p53 negatively regulates the expression of FAT10, a gene upregulated in various cancers. Oncogene 25:2318–2327. 10.1038/sj.onc.1209220. [DOI] [PubMed] [Google Scholar]
- 43.Liu L, Dong Z, Liang J, Cao C, Sun J, Ding Y, Wu D. 2014. As an independent prognostic factor, FAT10 promotes hepatitis B virus-related hepatocellular carcinoma progression via Akt/GSK3beta pathway. Oncogene 33:909–920. 10.1038/onc.2013.236. [DOI] [PubMed] [Google Scholar]
- 44.Aichem A, Groettrup M. 2016. The ubiquitin-like modifier FAT10 in cancer development. Int J Biochem Cell Biol 79:451–461. 10.1016/j.biocel.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen NT, Now H, Kim WJ, Kim N, Yoo JY. 2016. Ubiquitin-like modifier FAT10 attenuates RIG-I mediated antiviral signaling by segregating activated RIG-I from its signaling platform. Sci Rep 6:23377. 10.1038/srep23377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang G, Kouwaki T, Okamoto M, Oshiumi H. 2019. Attenuation of the innate immune response against viral infection due to ZNF598-promoted binding of FAT10 to RIG-I. Cell Rep 28:1961–1970.e1964. 10.1016/j.celrep.2019.07.081. [DOI] [PubMed] [Google Scholar]
- 47.Myoung J, Ganem D. 2011. Generation of a doxycycline-inducible KSHV producer cell line of endothelial origin: maintenance of tight latency with efficient reactivation upon induction. J Virol Methods 174:12–21. 10.1016/j.jviromet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nishimura M, Watanabe T, Yagi S, Yamanaka T, Fujimuro M. 2017. Kaposi’s sarcoma-associated herpesvirus ORF34 is essential for late gene expression and virus production. Sci Rep 7:329. 10.1038/s41598-017-00401-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brulois KF, Chang H, Lee AS, Ensser A, Wong LY, Toth Z, Lee SH, Lee HR, Myoung J, Ganem D, Oh TK, Kim JF, Gao SJ, Jung JU. 2012. Construction and manipulation of a new Kaposi’s sarcoma-associated herpesvirus bacterial artificial chromosome clone. J Virol 86:9708–9720. 10.1128/JVI.01019-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Campbell M, Watanabe T, Nakano K, Davis RR, Lyu Y, Tepper CG, Durbin-Johnson B, Fujimuro M, Izumiya Y. 2018. KSHV episomes reveal dynamic chromatin loop formation with domain-specific gene regulation. Nat Commun 9:49. 10.1038/s41467-017-02089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masuda T, Tomita M, Ishihama Y. 2008. Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J Proteome Res 7:731–740. 10.1021/pr700658q. [DOI] [PubMed] [Google Scholar]
- 52.Adachi J, Hashiguchi K, Nagano M, Sato M, Sato A, Fukamizu K, Ishihama Y, Tomonaga T. 2016. Improved proteome and phosphoproteome analysis on a cation exchanger by a combined acid and salt gradient. Anal Chem 88:7899–7903. 10.1021/acs.analchem.6b01232. [DOI] [PubMed] [Google Scholar]
- 53.Rappsilber J, Ishihama Y, Mann M. 2003. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75:663–670. 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 54.Abe Y, Nagano M, Tada A, Adachi J, Tomonaga T. 2017. Deep phosphotyrosine proteomics by optimization of phosphotyrosine enrichment and MS/MS parameters. J Proteome Res 16:1077–1086. 10.1021/acs.jproteome.6b00576. [DOI] [PubMed] [Google Scholar]
- 55.Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26:1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 56.Rydbirk R, Folke J, Winge K, Aznar S, Pakkenberg B, Brudek T. 2016. Assessment of brain reference genes for RT-qPCR studies in neurodegenerative diseases. Sci Rep 6:37116. 10.1038/srep37116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Watanabe T, Nakamura S, Ono T, Ui S, Yagi S, Kagawa H, Watanabe H, Ohe T, Mashino T, Fujimuro M. 2014. Pyrrolidinium fullerene induces apoptosis by activation of procaspase-9 via suppression of Akt in primary effusion lymphoma. Biochem Biophys Res Commun 451:93–100. 10.1016/j.bbrc.2014.07.068. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.