

Influenza A virus (IAV) infection poses a global health threat and is among the most common contagious pathogens to cause severe respiratory infections in humans and animals. ISGs play a key role in host defense against IAV infection.
KEYWORDS: IFI35, influenza virus, NS1, RIG-I, ubiquitination, antagonism
ABSTRACT
Interferon-stimulated genes (ISGs) create multiple lines of defense against viral infection. Here, we show that interferon-induced protein 35 (IFI35) inhibits swine (H3N2) influenza virus replication by directly interacting with the viral protein NS1. IFI35 binds more preferentially to the effector domain of NS1 (amino acids 128 to 207 [128-207aa]) than to the viral RNA sensor RIG-I. This promotes mutual antagonism between IFI35 and NS1 and frees RIG-I from IFI35-mediated K48-linked ubiquitination and degradation. However, IFI35 does not interact with the NS1 encoded by avian (H7N9) influenza virus, resulting in IFI35 playing an opposite virus-enabling role during highly pathogenic H7N9 virus infection. Notably, replacing the 128-207aa region of H7N9 NS1 with the corresponding region of H3N2 NS1 results in the chimeric NS1 acquiring the ability to bind to and mutually antagonize IFI35. IFI35-deficient mice accordingly exhibit more resistance to lethal H7N9 infection than exhibited by their wild-type control. Our data uncover a novel mechanism by which IFI35 regulates RIG-I-mediated antiviral immunity through mutual antagonism with influenza virus protein NS1.
IMPORTANCE Influenza A virus (IAV) infection poses a global health threat and is among the most common contagious pathogens to cause severe respiratory infections in humans and animals. ISGs play a key role in host defense against IAV infection. In line with others, we show IFI35-mediated ubiquitination of RIG-I to be involved in innate immunity. Moreover, we define a novel role of IFI35 in regulating the type I interferon (IFN) pathway during IAV infection. We found that IFI35 regulates RIG-I-mediated antiviral signaling by interacting with IAV NS1. H3N2 NS1, but notably not H7N9 NS1, interacts with IFI35 and efficiently suppresses IFI35-dependent ubiquitination of RIG-I. IFI35 deficiency protected mice from H7N9 virus infection. Therefore, manipulation of IFI35-NS1 provides a new approach for the development of anti-IAV treatments.
INTRODUCTION
Innate immune responses serve as the first line of antiviral defense and are essential for immune surveillance against viral infection. Acute respiratory viral infections, such as those caused by novel coronavirus (SARS-CoV-2) and highly pathogenic avian H7N9 influenza virus, usually evade host innate immunity by suppressing production of host type I interferons (IFNs) at an early stage (1–3). Type I IFNs have been proven to be important components of early immune response against influenza A virus (IAV) infection (4–8). IAV nonstructural protein NS1 is widely regarded as the common factor utilized by virus to inhibit host type I IFN production (9–11). Various strategies that may account for NS1 inhibition of type I IFN production have been documented. These include sequestration of double-stranded RNA (dsRNA), interaction with retinoic acid-inducible gene 1 (RIG-I), inhibition of downstream processes such as the activation of IFN regulatory factor 3 (IRF3), and inhibition of the activities of IFN-inducible enzymes such as double-stranded RNA-dependent protein kinase (PKR) and oligoadenylate synthase protein (OAS) (9, 12–14). Among these strategies, inhibition of type I IFNs by directing the binding of NS1 to RIG-I is employed by specific influenza virus strains (14–16). This is further supported by the fact that some highly pathogenic IAV strains encoding NS1 exhibit more efficiency in inhibiting type I IFN production than do weakly pathogenic IAV strains encoding NS1 (17, 18). However, the molecular mechanisms behind these observed differences in NS1-mediated antagonism of type I IFNs during either weakly or highly pathogenic IAV strains remain elusive.
Our previous work using subcellular proteomics identified the host factor interferon inducible protein IFI35 (also known as IFP35) upregulated in the cytosol and nucleus of swine influenza virus (SIV) H3N2-infected human lung epithelial cell line A549 (19). IFI35 is a 35-kDa protein and was originally identified as an interferon-induced protein through differential screening of the cDNA library of HeLa cells (20). It contains an N-terminal leucine zipper (L-zip), followed by two tandem N-myc-interacting (NMI) domains (NIDs) (21). IFI35 can translocate from the cytoplasm to the nucleus in a variety of interferon-stimulated cells, including fibroblasts, monocytes, macrophages, and epithelial cells (22). To date, IFI35 has been mainly reported to play roles in regulating production of innate immunity type I IFNs and proinflammatory cytokines (22–26). Notably, IFI35 has either anti- or provirus roles dependent upon specific viral infection (27–30). For example, in bovine foamy virus (BFV) infections, IFI35 can efficiently inhibit BFV replication via interaction with the homologous regulatory protein of prototype FV, arresting viral replication and repressing viral transcription (29). However, during vesicular stomatitis virus (VSV) infection, IFI35 was reported to support VSV replication through negatively regulating RIG-I activation (27). Additionally, IFI35 has been shown to induce production of proinflammatory cytokines (e.g., interleukin 12p40 [IL-12p40] and IL-12p80) following highly pathogenic H5N1 virus infection (31). All these studies have revealed critical and distinct roles of IFI35 in various viral infections. However, whether IFI35 interacts directly with IAV viral proteins or it plays a role in regulating type I IFN production during IAV infection remains largely unknown.
In the present study, we examined the ability of IFI35 to interact with NS1 encoded by swine (H3N2) and avian (H7N9/2013) influenza A virus, as well as the effect of IFI35-NS1 interaction on regulating the RIG-I-mediated type I IFN responses. We found that IFI35 interacts with NS1-H3N2 but not with NS1-H7N9. The IFI35-NS1 interaction results in IFI35 and NS1 mutually antagonizing each other, subsequently regulating RIG-I-mediated antivirus responses. Mechanistically, IFI35 is more preferentially associated with NS1 than with RIG-I, leading to reduced K48-linked ubiquitination and degradation of RIG-I. Our work provides a novel mechanism by which highly pathogenic H7N9 virus, with mutant IFI35 binding sites in NS1, evades RIG-I-mediated innate immune surveillance and antivirus immunity.
RESULTS
IFI35 interacts with swine influenza virus protein NS1.
In SIV H3N2-infected A549 cells, IFI35 can translocate from the cytoplasm into the nucleus, where the main subcellular site for viral protein NS1 is located (19). This phenotype prompted us to test whether IFI35 can interact with NS1. First, we examined the subcellular distribution of IFI35 upon influenza virus infection at different time points. In mock-infected cells, IFI35 was evenly distributed in the whole-cell cytosol (Fig. 1A), as has been previously reported (27). At 6 h postinfection (hpi), IFI35 was present predominately in the cytoplasm and colocalized with NS1 protein to form punctate bodies (Fig. 1A). However, at later stages after infection, both IFI35 and NS1 were predominately colocalized in the nucleus (Fig. 1A). This result implies that IFI35 may interact with NS1 during either early or late viral infection. To further validate this, we transfected 293T cells with Flag-tagged NS1 and Myc-tagged IFI35 plasmid, either alone or in combination, for 24 h. NS1 and IFI35 transfected alone resulted in predominately nuclear and cytosolic localization, respectively (Fig. 1B). However, 293T cells cotransfected with NS1 and IFI35 at 24 h led to a marked increase of NS1 cytoplasmic localization, and a strong colocalization of NS1 with IFI35 was found in the cytoplasm (Fig. 1B). This colocalization was not dependent on the time course of transfection, the various tagged expression plasmids, or the transfected cell types (Fig. 1C). We then addressed the NS1-IFI35 interaction in the reciprocal coimmunoprecipitation (co-IP) study in 293T cells after plasmid cotransfection (Fig. 1D). Expression vectors Flag-NS1and Myc-IFI35 were transfected into the 293T cells, the cell lysate was subjected to immunoprecipitation using anti-Flag antibody or anti-Myc antibody at 36 h posttransfection, and the coimmunoprecipitated proteins were detected by Western blotting. The results showed that NS1 was associated with the IFI35 protein (Fig. 1D). In addition, The IFI35-NS1 interaction was confirmed when Escherichia coli bacteria expressed this recombinant protein-protein direct interaction through an in vitro GST pulldown assay. His-tagged IFI35 was only pulled down in the presence of glutathione S-transferase (GST)-tagged NS1 (Fig. 1E). Collectively, these data clearly demonstrate the direct interaction between IFI35 and SIV H3N2 viral protein NS1.
FIG 1.

IFI35 interacts with SIV NS1-H3N2. (A) IFI35 colocalizes with NS1-H3N2 in A549 cells after infection with SIV H3N2 at 6 h or 24 h. A549 cells were transfected with Myc-IFI35 plasmid and then mock infected or infected with H3N2 at an MOI of 1. Cells were fixed and stained with mouse anti-NS1 Mab followed by FITC-conjugated IgG (green) and with anti-rabbit poly PAb, followed by Alexa Fluor 546-conjugated goat anti-rabbit IgG (red). The nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; blue) and analyzed by confocal microscopy. Mock-infected cells served as a control. (B) 293T cells were transfected with Flag-NS1 alone, Myc-IFI35 alone, or Flag-NS1 together with Myc-IFI35 at 24 h, and cells were fixed and stained with mouse anti-Flag Mab (green), rabbit anti-Myc pAb (red), and DAPI (nucleus; blue). More than 100 cells expressing NS1 alone or NS1 together with IFI35 were counted, and the percentage of cells with cytoplasmic localization of NS1 is shown (lower left). (C) 293T cells were cotransfected with Flag-NS1 and Myc-IFI35 at 6 h or 12 h, and A549 cells were cotransfected with HA-NS1 and GFP-IFI35 at 24 h. The cells were fixed, permeabilized, and stained for Flag-NS1 (green), Myc-IFI35 (red), and HA-NS1 (red). (D) Reciprocal coimmunoprecipitation (co-IP) of Flag-NS1 and Myc-IFI35 in 293T cells. 293T cells were cotransfected with Flag-NS1 and Myc-IFI35. The cell lysates were immunoprecipitated and immunoblotted (IB) with specific antibodies, as indicated. LC, light chain. (E) Bacterially expressed GST or GST-NS1 protein was immobilized on glutathione-Sepharose beads and then incubated with bacterially expressed His-NS1. His-tagged proteins in the GST pulldown assay were examined by IB with anti-IFI35 antibody. The bacterially expressed GST, GST-NS1, and His-IFI35 proteins before incubation were used as input and then IB with the indicated antibodies. Data are representative of at least three independent experiments.
NS1 effector domain interacts with IFI35 NIDs.
The IAV NS1 protein is comprised of two major domains: the N-terminal RNA binding domain (RBD; amino acids [aa] 1 to 73) and the C-terminal effector domain (ED; amino acid 85 to the C terminus) (32). To determine which of these two domains binds IFI35, we designed and constructed the following truncated plasmids: NS1-1-73aa (amino acids 1 to 73), NS1-74-207aa (amino acids 74 to 207), and NS1-128-230aa (amino acids 128 to 230) (Fig. 2A). In order to detect their subcellular location, 293T cells were transfected with these deletion plasmids alone or in combination with IFI35 plasmid for 24 h. As expected, NS1-1-73aa diffused in the nucleus, but it did not colocalize with IFI35 in the nucleus or cytoplasm (Fig. 2B). In contrast, NS1-74-207aa was mainly dispersed in the cytoplasm and was localized together with IFI35 and formed clustered distributions in the cytoplasm of cotransfected cells (Fig. 2B and C). NS1-128-230aa appeared only with many aggregations in the nucleus; however, it clustered with IFI35 in the cytoplasm (Fig. 2B and C). This led us to assume that the ED, but not the RBD, of NS1 can interact with IFI35. To test this hypothesis, we conducted a reciprocal co-IP experiment and confirmed that the ED, but not the RBD, is the interacting domain of NS1 for binding to IFI35 (Fig. 2D). The GST pulldown assay further identified that 128-207aa of NS1 was responsible for the NS1-IFI35 interaction (Fig. 2E). In an attempt to map the region of IFI35 responsible for the interaction with NS1, we designed several internal deletions of IFI35 (Fig. 2F). 293T cells were transfected to overexpress GFP-tagged IFI35 and its mutants lacking various domains together with Flag-tagged NS1. Deletion of the NIDs completely abolished the binding of IFI35 to NS1, whereas IFI35 with any other mutant domains had a binding affinity similar to that of full-length IFI35 (Fig. 2G). This result confirmed earlier studies suggesting that the NIDs were the main interacting domains of IFI35 (24, 25). Taken together, these data indicate that the effector domain of NS1 interacts with the NIDs of IFI35.
FIG 2.

Mapping of the domains of interaction between IFI35 and NS1-H3N2. (A) Schematic representation of the deletion mutants of NS1-H3N2. (B and C) 293T cells were transfected with Flag-NS1 deletion mutant plasmids, comprising amino acids 1 to 73 (1-73aa), 74 to 207 (74-207aa), and 128 to 230 (128-230aa), alone (B) or with Flag-NS1 deletion plasmids together with Myc-IFI35 (C) at 24 h, and the cells were fixed and stained with mouse anti-Flag MAb (green), rabbit anti-Myc PAb (red), and DAPI (blue). (D) Reciprocal co-IP of Flag-NS1 or its deletion mutant plasmids and Myc-IFI35 in 293T cells. 293T cells were cotransfected with Flag-NS1 or its deletion mutant plasmids and Myc-IFI35. The cell lysates were IP and immunoblotted (IB) with specific antibodies, as indicated. LC, light chain. (E) Whole-cell lysates (WCLs) of 293T cells transfected with Flag-NS1 or Myc-NS1(128-207aa) together with Flag-GST vector and Flag-GST-IFI35 were subjected to GST pulldown followed by IB with anti-Flag or anti-GST (upper panels). WCLs were assessed by IB with the indicated antibodies (lower panels). (F) Schematic representation of the deletion mutants of IFI35. (G) Co-IP analysis of the IFI35 interaction domains with NS1-H3N2. 293T cells were cotransfected with Flag-NS1 plasmids and GFP-tagged IFI35 or its deletion mutants. The cell lysates were IP and IB with specific antibodies, as indicated. HC, heavy chain. Data are representative of at least three independent experiments.
Interaction of IFI35 with NS1 is not conserved in various IAV strains.
It is well established that the structure and function of IAV NS1 exhibit strain-specific differences (33–35). To investigate whether the interaction of IFI35 with NS1 applied to various IAV strains, 293T cells were singly transfected with NS1 from the H7N9 IAV strain. As anticipated, NS1 consistently localized primarily in the nucleus with a minor cytoplasmic component across different virus strains (Fig. 1B and 3A). However, in IFI35 plasmid-cotransfected cells, NS1 from the H3N2 type strain colocalized with IFI35 in the cytoplasm (Fig. 1B). In contrast, coexpression of IFI35 that did not colocalize with NS1-H7N9 had only a minor effect on NS1 subcellular distribution (Fig. 3A). IFI35 colocalized with NS1 appeared in H3N2-infected cells (Fig. 1A), but this colocalization was not observed in H7N9-infected cells (Fig. 3B). Further co-IP assays showed that NS1 from H3N2, but not from H7N9, interacted with IFI35 in infected cells (Fig. 3C). These results collectively demonstrate that the interaction of IFI35 with NS1 is viral strain specific.
FIG 3.
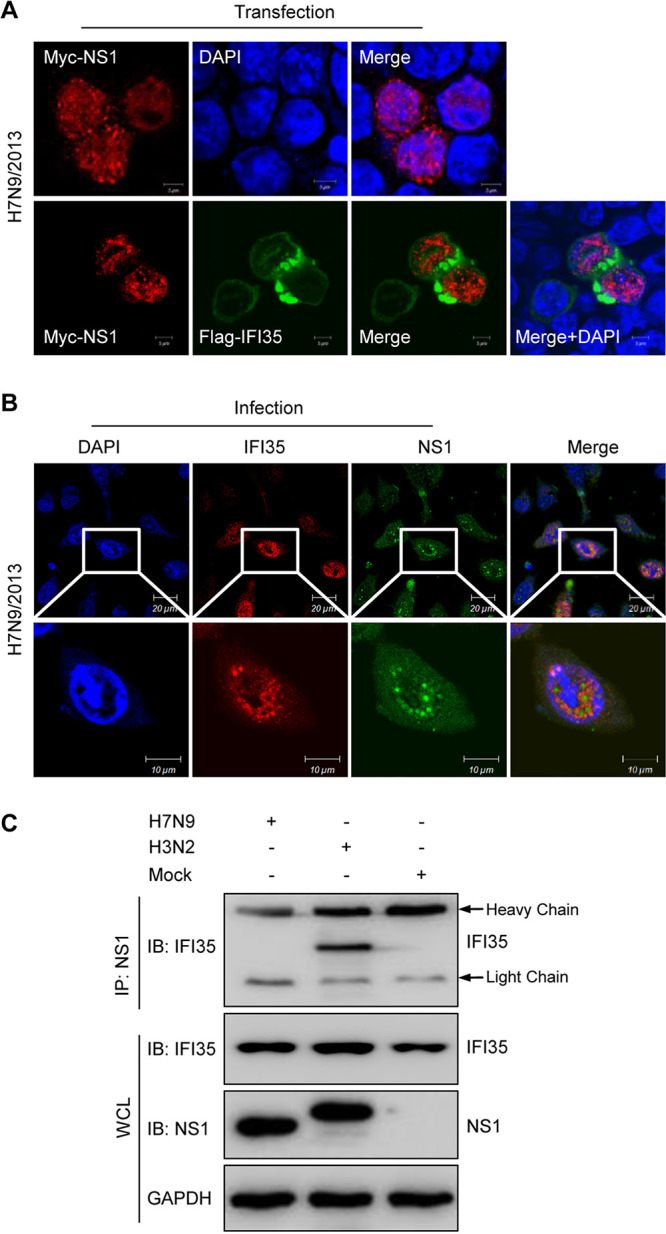
IFI35 interacts with NS1 in a virus strain-specific manner. (A) Localization of H7N9-NS1 and IFI35 in HEK293T cells was determined by confocal microscopy. HEK293T cells were transfected with Myc-NS1 plasmid alone or together with Flag-IFI35 at 24 h. Cells were fixed and stained with anti-rabbit PAb followed by Alexa Fluor 546-conjugated goat anti-rabbit IgG (red) or with mouse anti-Flag Mab followed by FITC-conjugated IgG (green), The nuclei were stained with DAPI (blue) and analyzed by confocal microscopy. (B) Localization of IFI35 and NS1-H7N9 protein in A549 cells 24 h after H7N9 infection. A549 cells were transfected with Myc-IFI35 plasmid and then infected with H7N9 at an MOI of 1. Cells were fixed and stained with mouse anti-NS1 Mab followed by FITC-conjugated IgG (green) and with anti-Myc PAb followed by Alexa Fluor 546-conjugated goat anti-rabbit IgG (red), The nuclei were stained with DAPI (blue) and analyzed by confocal microscopy. (C) Co-IP analysis of the NS1-IAV interaction with IFI35 after IAV infection at 24 h. Whole-cell lysates (WCLs) of A549 cells were mock infected or infected with H3N2 or H7N9 at an MOI of 1 and then subjected to co-IP, followed by IB with anti-NS1 or anti-IFI35 (upper panels). WCL was assessed by IB with the indicated antibodies (lower panels). Data are representative of at least three independent experiments.
IFI35 functions in opposite roles in an IAV strain-specific manner.
Influenza viral protein NS1 is a well-known antagonist to IFN production (10, 11). Thus, we sought to determine whether the NS1-IFI35 interaction affects the NS1 antagonization of virus-triggered type I IFN signaling pathways during IAV infection. Quantitative real-time PCR (qRT-PCR) analysis also indicated that transcripts of IFNB1, ISG15, and ISG56 were significantly upregulated in IFI35-overexpressing cells infected with H3N2 virus (Fig. 4A and C). Conversely, overexpression of IFI35 substantially decreased IFNB1, ISG15, and ISG56 expression in H7N9-infected cells (Fig. 4C). These data suggest that IFI35 plays opposing roles in response to different IAV strain infections. To further understand the endogenous role of IFI35 during IAV infection, we employed short hairpin RNA (shRNA)-mediated knockdown of IFI35 expression. Western blotting confirmed the knockdown efficiency of shRNAs (Fig. 4B). H3N2-infected IFI35 knockdown cells (ShIFI35-#520) had a significant decrease in mRNA levels of IFNB1, ISG15, and ISG56 in comparison to negative-control shRNA (shNC)-expressing cells (Fig. 4D). In contrast, in H7N9-infected cells, knockdown of IFI35 significantly elevated transcripts of IFNB1, ISG15, and ISG56 (Fig. 4D). These results prompted us to assess how gain or loss of IFI35 may influence virus replication. Strikingly, IFI35 functions as a negative regulator for suppressing replication of H3N2 virus but positively promotes H7N9 virus replication (Fig. 4E and F). These findings reveal that IFI35 has opposing functions in regulating IAV replication through controlling IFN antiviral signaling in a strain-specific manner.
FIG 4.

IFI35 regulates IAV-triggered signaling in a strain-specific manner. (A) A549 cells were transfected with Myc-IFI35 plasmid for 36 h and then left uninfected or infected with SIV H3N2 (MOI = 1) for 18 h. Cell lysates were analyzed by Western blotting and probed with antibodies as indicated. GAPDH expression was used as a loading control. (B) A549 cells were transfected with negative-control shRNA (shN.C.), positive-control shGAPDH (used as a positive control), or IFI35-specific shRNA (shIFI35-#930 and shIFI35-#520) as indicated. At 48 h posttransfection, Western blot analysis was used to determine the expression of IFI35 and GAPDH. (C) A549 cells were transfected with Myc-empty vector (vector) or Myc-IFI35 plasmid for 36 h and then mock infected or infected with H3N2 or H7N9 (MOI = 1) for 18 h. Total RNA was extracted from samples prepared and treated with DNase I. The expression of IFNB1, ISG15, ISG56, and GAPDH (control) was analyzed by quantitative real-time PCR (qRT-PCR). (D) A549 cells were transfected with the indicated shRNA for 36 h and then cells were left uninfected or were infected with H3N2 or H7N9 (MOI = 1). After 18 h, total RNA was analyzed using qRT-PCR to determine expression levels of IFNB1, ISG15, ISG56, and GAPDH. (E and F) H3N2 or H7N9 virus genome copies (M1 mRNA) were quantified by quantitative real-time PCR in A549 cells treated as described for panels C and D. Values are means ± SD of three independent experiments. Student's t test; *, P < 0.05; **, P < 0.01; ***, P < 0.005.
NID2 of IFI35 interacts with RIG-I and promotes RIG-I degradation through K48-linked ubiquitination.
IFI35 has been proven to interact with RIG-I (27). GST pulldown studies in transfected cells consistently showed that Flag-RIG-I can be pulled down with Flag-GST-IFI35 but not with Flag-GST vector, indicating a physical interaction between IFI35 and RIG-I (Fig. 5A). Next, we mapped which domain of IFI35 is responsible for the IFI35-RIG-I interaction. Co-IP experiments demonstrated that IFI35 without its N-terminal leucine zipper domain (IFI35-ΔL-zip) and/or NID1 (IFI35-ΔNID1) retained the ability to bind to RIG-I, while IFI35 without NID2 (IFI35-ΔNID2) did not (Fig. 5B). These results demonstrate that the interaction between IFI35 and RIG-I is specific and depends on the presence of NID2. Further ubiquitination assays showed that NIDs, but not the L-zip domain, mediated K48-linked polyubiquitination of RIG-I (Fig. 5C). Therefore, we conclude that IFI35 negatively regulates RIG-I stability through NID2 binding to RIG-I. RIG-I contains N-terminal CARDs, a middle helicase domain, and a C-terminal domain (CTD) (36–38). We designed several internal deletions of RIG-I (Fig. 5D). Domain mapping experiments indicated that the helicase domains and CTD, but not CARDs of RIG-I, could independently interact with the IFI35 (Fig. 5E and F). Next, we questioned if the region of RIG-I might be ubiquitinated by IFI35. Ubiquitination experiments indicated that overexpression of IFI35 markedly increased K48-linked polyubiquitination of RIG-I-CARDs (Fig. 5G). Taken together, these data demonstrate that NIDs of IFI35 are responsible for the interaction and ubiquitination of CARDs of RIG-I.
FIG 5.

IFI35 interacts with RIG-I. (A) WCLs of 293T cells transfected with Flag-RIG-I together with Flag-GST vector or Flag-GST-IFI35 were subjected to GST pulldown followed by IB with anti-Flag (upper panels). WCL was assessed by IB with the indicated antibodies (lower panels). (B) NID2 of IFI35 interacts with RIG-I. Co-IP analysis of the IFI35 interaction domains with RIG-I. 293T cells were cotransfected with Flag-RIG-I plasmids and GFP-tagged IFI35 or its deletion mutants. Cell lysates were IP and IB with specific antibodies, as indicated. (C) NIDs of IFI35 mediated K48-linked ubiquitination of RIG-I. 293T cells were cotransfected with HA-Ub-K48, Flag-RIG-I plasmids, and GFP vector or GFP-tagged IFI35 deletion mutants, and cells were treated with MG132 (10 μM) for 12 h. The cell lysates were IP and IB with the indicated antibodies. GAPDH expression was used as a loading control. (D) Schematic representation of the deletion mutants of RIG-I. (E) 293T cells were cotransfected with a fixed Myc-IFI35 plasmid, GFP-RIG-I plasmid, or its truncation mutants. Cell lysates were IP and IB with specific antibodies (upper panels). WCL was assessed by IB with the indicated antibodies (lower panels). (F) 293T cells were transfected with RIG-I deletion mutant plasmids, together with Myc-IFI35 at 24 h, and cells were fixed and stained for GFP (green), Myc-IFI35 (red), and DAPI (blue). (G) 293T cells were cotransfected with a fixed GFP-RIG-I-CARDs plasmid, Myc-IFI35 plasmid, and HA-K48-Ub plasmid. The cell lysates were treated with MG132 and then IP with GFP and IB with specific antibodies (upper panels). WCL was assessed by IB with the indicated antibodies (lower panels). Data are representative of at least three independent experiments.
Interaction of IFI35 with NS1 has mutual antagonism in regulating RIG-I activity.
Our observation that both viral proteins NS1-H3N2 and RIG-I can interact with IFI35, and that both bind the same NID2 of IFI35, led us to examine whether NS1-H3N2 could compete with RIG-I for binding to IFI35. To test this hypothesis, fixed amounts of plasmids expressing Flag-GST-IFI35 and Flag-RIG-I, plus increasing amounts of a plasmid encoding Flag-tagged NS1-H3N2, were cotransfected into 293T cells. IFI35 efficiently pulls down RIG-I; however, the amount of RIG-I that bound to IFI35 gradually decreased as the amount of NS1-H3N2 protein increased (Fig. 6A). These results suggest that NS1-H3N2 physically sequesters the RIG-I-IFI35 interaction in a dose-dependent manner. It is well established that IAV protein NS1 is the main IFN antagonist (14, 39). It has been documented that interaction with RIG-I is a main strategy employed by PR8 NS1 to inhibit type I IFN production (14). NS1-H3N2 can successfully pull down IFI35 but failed to pull down RIG-I in cotransfected cells through GST pulldown assays (Fig. 6B). This result further supports that NS1-H3N2 has higher affinity to IFI35 than RIG-I and that NS1-H3N2 is probably employed to alter the mechanism to inhibit RIG-I signaling induction but not to directly bind to RIG-I.
FIG 6.

IFI35 regulates RIG-I activation by mutual antagonism with NS1. (A) NS1-H3N2 disrupts IFI35-RIG-I interaction. 293T cells transfected with Flag-RIG-I together with Flag-GST vector, or Flag-GST-IFI35 and/or increased amounts of Flag-NS1, were subjected to GST pulldown followed by IB with anti-Flag or anti-GST (upper panels). WCL was assessed by IB with the indicated antibodies (lower panels). GAPDH expression was used as a loading control. (B) NS1-H3N2 does not directly interact with RIG-I. 293T cells transfected with Flag-RIG-I or Myc-IFI35 (used as a positive control) together with Flag-GST vector and/or Flag-GST-NS1 were subjected to GST pulldown followed by IB with the indicated antibodies (upper panels). WCL was assessed by IB with the indicated antibodies (lower panels). GAPDH expression was used as a loading control. (C) 293T cells were transfected with Flag-empty vector (Flag-vector), Flag-RIG-I, or Flag-RIG-I and Myc-IFI35 together with increasing amounts of Flag-NS1. In addition, IFN-β-Luc reporter and pRL-TK were transfected. Luciferase assays were performed 24 h after transfection. (D) 293T cells were transfected with Flag-vector, Flag-RIG-I, or Flag-RIG-I and Flag-NS1 together with increasing amounts of Myc-IFI35. In addition, IFN-β-Luc reporter and pRL-TK were transfected. Luciferase assays were performed 24 h after transfection. (E) 293T cells were cotransfected with a fixed amount of both viral Flag-RIG-I plasmid (2 μg) and Myc-IFI35 plasmid (2 μg) with increasing amounts of NS1-expressing plasmid (from 0.5, 1, to 2 μg). One set of cells was treated with dimethyl sulfoxide (DMSO) for 12 h (upper panels), and the other set of cells was treated with MG132 (10 μM) for 12 h (lower panels). Cell lysates were analyzed by IB with the indicated antibodies. GAPDH expression was detected as a loading control. (F) NS1-H3N2 overexpression decreased K48-linked ubiquitination of RIG-I. Flag-RIG-I was coexpressed with HA-Ub-K48, Myc-IFI35, and increasing amounts of NS1 in 293T cells. Forty-eight hours after transfection, the cells were treated with DMSO or MG132 for 12 h. Subsequently, one set of cells treated with DMSO was analyzed by IB with the indicated antibodies (lower panels). The other set of cells treated with MG132 was used for co-IP and IB with the indicated antibodies (upper panels). Data are representative of at least three independent experiments.
As with NS1, IFI35 has been shown to be negatively regulated by RIG-I-mediated IFN induction (27), which prompted us to test whether NS1, when preferentially bound to IFI35, may mutually inhibit each other, which then subsequently influences RIG-I activation. Consistent with previous results, IFI35 markedly suppressed the RIG-I-induced IFN-β promoter activation, but increasing amounts of NS1-H3N2 were able to overcome the IFI35-mediated inhibition of RIG-I activation (Fig. 6C). On the other hand, transfection with NS1-H3N2 alone markedly suppressed RIG-I-induced IFN-β promoter activation but when cotransfected with increasing amounts of IFI35 was able to overcome the NS1-mediated inhibition of RIG-I activation (Fig. 6D). These results indicate that IFI35-NS1 mutual antagonism regulates RIG-I activation. Since IFI35 negatively regulated RIG-I activation mainly through promotion of K48-linked ubiquitination and degradation of RIG-I (14), we then examined whether IFI35-mediated degradation of RIG-I can be blocked by NS1. We transfected 293T cells with a fixed amount of Flag-RIG-I and Myc-IFI35, along with increasing amounts of NS1-expressing plasmid. Flag-RIG-I expression was analyzed by Western blotting after 36 h in the whole-cell lysates. We observed a remarkable loss of Flag-RIG-I in the presence of IFI35; however, the levels of Flag-RIG-I reduction were reversed and a dose-dependent accumulation appeared along with increasing amounts of NS1-H3N2 (Fig. 6E). The levels of Flag-RIG-I did not fluctuate when cells were treated with the proteasome inhibitor MG132 (Fig. 6E). Moreover, we observed that NS1-H3N2 inhibited IFI35 promoted the K48-linked ubiquitination of RIG-I in a dose-dependent manner (Fig. 6F). Collectively, these results demonstrate that NS1-H3N2 specifically disrupts the IFI35-RIG-I interaction, which leads to attenuation of IFI35-mediated RIG-I K48-linked ubiquitination and degradation.
IFI35 binding region 128-207aa of NS1 is required for immune antagonism.
Since the 128-207aa region of NS1-H3N2 is responsible for the NS1-IFI35 interaction, we then sought to investigate whether replacing 128-207aa of NS1-H7N9 with 128-207aa of NS1-H3N2 (named NS1-73) (Fig. 7A) could enable the chimeric molecule NS1-73 to interact with IFI35. As expected, immunoprecipitation experiments showed that NS1-73 efficiently bound to IFI35, while NS1-H7N9 did not (Fig. 7B). Confocal microscopy further confirmed the colocalization of IFI35 and NS1-73 in transfected cells (Fig. 7C). In order to analyze whether IFI35-mediated degradation of RIG-I is affected by the IFI35 binding site 128-207aa of NS1, we transfected 293T cells with a fixed amount of Flag-RIG-I and Myc-IFI35, along with NS1-H3N2, NS1-H7N9, or NS1-73, and analyzed them by a Western blotting assay. As anticipated, the amount of Flag-RIG-I was dramatically reduced in the presence of IFI35. However, adding NS1-H3N2 or NS1-73, but not NS1-H7N9, markedly reversed the reduction of RIG-I (Fig. 7D), suggesting that NS1-73 functions similarly to NS1-H3N2 in blocking IFI35-mediated RIG-I degradation. In addition, NS1-73-inhibited IFI35 promoted the K48-linked ubiquitination of RIG-I (Fig. 7E). To further explore whether NS1-73 directly targets IFI35 to influence RIG-I signal transduction, we questioned if overexpression of NS1-73 can overcome the RIG-I inhibition by IFI35. IFI35 markedly suppressed the RIG-I-induced IFN-β promoter activation, while NS1-73, but not NS1-H7N9, was able to overcome the IFI35-mediated inhibition of RIG-I (Fig. 7F). Taken together, these results indicate that the IFI35 binding site 128-207aa in NS1 is required for mutual antagonism between NS1 with IFI35.
FIG 7.

IFI35 binding region 128-207aa of NS1-H3N2 is required for immune antagonism. (A) Schematic representation of the replacement region 128-207aa of NS1-H7N9 with the corresponding region of NS1-H3N2. (B) Chimeric NS1 protein NS1-73 interacts with IFI35. 293T cells were transfected with 3×Flag-empty vector (3×Flag-vector), 3×Flag-NS1-H7N9, 3×Flag-NS1-73, or 3×Flag-NS1-H3N2 together with Myc-IFI35 and were subjected to IP followed by IB with anti-Flag or anti-Myc (upper panels). WCL was assessed by IB with the indicated antibodies (lower panels). (C) Localization of NS1-73 and IFI35 in 293T cells was determined by confocal microscopy. 293T cells were transfected with 3×Flag-NS1-73 plasmid alone or together with Myc-IFI35 at 24 h. Cells were fixed and stained with rabbit anti-Myc PAb followed by FITC-conjugated anti-rabbit IgG (green) and with mouse anti-Flag MAb followed by Alexa Fluor 546-conjugated goat anti-mouse IgG (red), The nuclei were stained with DAPI (blue) and analyzed by confocal microscopy. (D) 293T cells were cotransfected with a fixed amount of both Flag-RIG-I plasmid (1 μg) and Myc-IFI35 plasmid (1 μg) with NS1-H7N9 (1 μg), NS1-73 (1 μg), or NS1-H3N2 (1 μg). The histogram represents the ratio of normalized RIG-I protein intensity band to that of control group (transfected with Flag-RIG-I alone) (upper panel). Cell lysates were analyzed by IB with the indicated antibodies. Actin expression was detected as a loading control (lower panel). (E) NS1-73 overexpression decreased K48-linked ubiquitination of RIG-I. Flag-RIG-I was coexpressed with HA-Ub-K48, Myc-IFI35, and NS1-H7N9 or NS1-73 in 293T cells. At 48 h after transfection, the cells were treated with MG132 for 12 h. Subsequently, one set of cells was analyzed by IB with the indicated antibodies (lower panels). The other set of cells was used for co-IP and IB with the indicated antibodies (upper panels). (F) 293T cells were transfected with Flag-vector, Flag-RIG-I, or Myc-IFI35 together with NS1-H7N9 or NS1-73. In addition, IFN-β-Luc reporter and pRL-TK were transfected. Luciferase assays were performed 24 h after transfection. Data are representative of at least three independent experiments.
IFI35 deficiency protects mice from lethal H7N9 virus.
To further investigate the roles of IFI35 in IAV infection in vivo, we generated IFI35-deficient (Ifi35−/−) mice using the Talen approach, which caused a ∼1-kb chromosomal deletion at the IFI35 locus (exons 4 to 7, 3′ untranslated region [UTR]) in the mouse genome (Fig. 8A). Successful deletion of Ifi35 was verified by PCR (Fig. 8B). We then intranasally infected Ifi35+/+ and Ifi35−/− mice with H7N9 and monitored their weight loss and survival for 2 weeks. In the H7N9-infected setting, Ifi35−/− mice exhibited less body weight loss, as well as a lower level of morbidity, than their littermate wild-type control mice (Fig. 8C and D). Accordingly, in comparison with the wild-type control, Ifi35−/− mice exhibited lower titers of H7N9 in the lung after infection at day 2 (Fig. 8E). Notably, histopathological analysis of the lungs of Ifi35−/− mice showed less immune cell infiltration and less tissue damage than that of the wild-type control after H7N9 virus infection at day 2 (Fig. 8F). Immunofluorescence staining of the viral nuclear protein NP indicated that the expression of viral proteins was significantly lower in the lungs of Ifi35−/− mice than in the lungs of Ifi35+/+ mice infected with H7N9 (Fig. 8G). Taken together, these results indicate that IFI35 deficiency protected mice from H7N9 virus infection by affecting viral replication and IAV-driven inflammation at the acute phase.
FIG 8.

IFI35 deficiency protects mice from lethal H7N9 virus. (A and B) Generation and identification of IFI35-deficient mice. (A) Schematic representation of IFI35 knockout strategy. Exons 4 to 7 (E4 to E7) of the IFI35 gene were deleted, which results in a frameshift mutation and loss of IFI35 expression. (B) Genotyping confirms the Ifi35 wild-type, heterogeneous, and knockout mice. Lane M, molecular weight markers. (C) Body weights of wild-type (WT) Ifi35+/+ and IFI35 knockout (KO) Ifi35−/− mice were measured for 14 days after intranasal administration of influenza virus H7N9. Values are the means of results of at least three independent experiments ± standard error of the mean (SEM). n = 8 to 14 per group. (D) The survival frequency of H7N9 virus-infected Ifi35+/+ and Ifi35−/− mice was monitored for 14 days. Data represent at least three independent experiments; n = 8 to 14 per group. Kaplan-Meier survival curves were analyzed by the Mantel-Cox log rank test. (E) Viral titers of H7N9 virus-infected lungs of Ifi35+/+ and Ifi35−/− mice at day 2 after infection. Data represent two independent experiments; n = 3 to 5 per group. Virus titers are depicted in TCID50/ml. (F) Histology of lung tissue of Ifi35+/+ and Ifi35−/− mice infected with H7N9 virus for the indicated days. Pathology was examined by H&E staining. (G) Representative confocal microscopy images of lung sections of Ifi35+/+ and Ifi35−/− mice infected with H7N9 virus for the indicated days. Tissues were fixed and stained with mouse MAb antiviral protein NP, followed by Alexa Fluor 546-conjugated goat anti-mouse IgG (red), and the nuclei were stained with DAPI (blue). Data are representative of at least three independent experiments. Differences between the experimental and control groups (C and E) were determined by two-way ANOVA tests. P values of <0.05 (*) were considered statistically significant.
DISCUSSION
In this study, we demonstrate that influenza viral protein NS1 binds to IFI35 in a strain-specific manner. NS1-H3N2, but not NS1-H7N9, interacts with IFI35. The interaction between NS1 and IFI35 mutually inhibits each other from regulating RIG-I-mediated innate antiviral response. Mechanistically, our serious interaction studies showed that NS1-H3N2 directly and preferentially interacted with IFI35 and impaired IFI35-mediated ubiquitination and degradation of RIG-I. Lastly, in a mouse model, IFI35-deficient mice were more resistant to lethal virus H7N9 infection than their wild-type (WT) counterparts. These findings reveal the previously unrecognized opposing roles of IFI35 in regulating replication of various influenza virus strains.
Viruses have evolved complicated mechanisms to evade the host innate immune response. For example, virus-encoded IFN antagonists inhibit host innate antiviral responses by targeting IFN gene expression or IFN-induced host effector molecules. Multifunctional nonstructural protein NS1 of influenza virus has been regarded as a major antagonist to host IFN induction. NS1 prevents virally induced type I IFN production primarily by suppressing RIG-I signal transduction and/or inhibiting the activation of transcription factor IRF3 (40). However, the differences in the sequences of the NS1 account for many of the functional differences between the NS1 proteins of different virus strains (12). One such functional difference is evident in the mechanisms by which NS1 employs distinct strategies to block antiviral IFN induction (40, 41). Specifically, several lines of evidence suggest that NS1-PR8 may mediate its pretranscriptional block on IFN-β induction by forming a complex with RIG-I (42, 43). However, direct binding of NS1-PR8 to RIG-I has yet to be demonstrated, and it is not clear if coprecipitation of RIG-I is a feature exhibited by all influenza A virus NS1 proteins. In this study, we did not detect a direct interaction between NS1-H3N2 and RIG-I through GST pulldown. Thus, the weak ability of NS1-H3N2 to bind to RIG-I could partially account for the high activation of IFN transcription during H3N2 virus infection. Given that NS1 can indeed markedly repress RIG-I-mediated activation of IFN-β promoter in vitro, further investigations are required to figure out the distinct mechanisms employed by strain-specific NS1 for suppressing RIG-I activation.
Previous studies suggest that the host innate immune response against influenza virus infection relies heavily on various ISGs, the key antiviral components induced by IFNs (44, 45). Several ISGs, including Mx proteins (46), IFN-inducible transmembrane (IFITM) proteins (47), and viperin (48), have been shown to function in limiting influenza virus infection and replication. Consistently, our data showed that IFI35, as one ISG, restricted H3N2 virus replication through positive regulation of RIG-I-mediated IFN induction. Mechanistically, IFI35 was found to directly bind NS1-H3N2 and to function as an antiviral factor. Our detailed biochemical analysis further revealed that the NID of IFI35 associates with the ED of NS1. To date, only IFI35 and N-myc-interacting protein (Nmi) have been shown to contain the NID (22, 24). The precise structure and biological function of the NIDs have not been revealed; however, it has been suggested that NIDs mediate the homo- and heterodimerization of IFI35 and NMI and are required for the protein-protein interaction in cell signal transduction (24). Here, we show novel data that NIDs of IFI35 could not only interact with the viral protein NS1 but also bind with the host protein RIG-I. Further studies have revealed that NS1 could preferably bind to IFI35 and disrupt the IFI35-RIG-I interaction. Our results further identified that NIDs are required for IFI35-mediated RIG-I ubiquitination. We therefore postulate that the IFI35-NS1 interaction not only disrupts the IFI35-RIG-I interaction but also potentially blocks NID-mediated RIG-I degradation. Given that the pivotal role of RIG-I in the response to influenza virus infection has also been assessed in many studies (49, 50), our results show the significance of the IFI35-NS1 interaction enhancement of host RIG-I activation during early virus infection. Obviously, the IFI35-NS1 interaction facilitates host immune surveillance rather than virus infection. Ubiquitination of RIG-I was shown to be the major posttranslational modification to negatively control RIG-I activation (51). Several E3 ubiquitin ligases like RNF125 (52) and c-Cbl ubiquitin ligase (53), which target RIG-I for K48-linked ubiquitination, have been identified. IFI35 was reported to downregulate RIG-I by promoting RIG-I K48-linked ubiquitination and degradation (27). Due to the lack of E3 ligase domains, IFI35 probably works as an adaptor by recruiting E3 ligase for RIG-I ubiquitination. Which type of E3 ligase is recruited by IFI35 to ubiquitinate RIG-I warrants further investigation. Furthermore, we found that the IFI35-mediated K48-linked ubiquitination of RIG-I was blocked by NS1-H3N2 in a dose-dependent manner but was not inhibited by NS1-H7N9. The prior interaction of IFI35 and NS1 not only blocked IFI35-mediated RIG-I ubiquitination and degradation but also simultaneously impaired the NS1-initiated IFN antagonist processes. We identified 128-207aa of NS1 as the region of the ED that determines the NS1-IFI35 interaction. However, we do not identify the crucial single amino acid residue in the region that is responsible for binding to IFI35, since the amino acid identity within this region is not consistent among various subtypes, H1, H3, H5, H7, and H9 (data not shown). It will be intriguing to address whether the de novo motif and/or precise specific tertiary structure in this region exists and are specifically responsible for the NS1-IFI35 interaction.
IFI35 has been proven to promote or inhibit the type I IFN response to virus infection (27–29). In line with this, we demonstrate the opposing roles of IFI35 in regulating the RIG-I–type I IFN axis under different influenza virus strain infections. Specifically, IFI35 works as an antivirus factor to suppress H3N2 infection, while it functions as a provirus factor to promote H7N9 virus infection. Our study proposes that the key factor that determines whether IFI35 works as a “friend” or “foe” for influenza virus is the interaction of IFI35 with viral protein NS1. During H3N2 infection, IFI35 attenuates viral replication through direct and strong binding to NS1, which is supported by the notion that IFI35 plays an antivirus role by interacting with viral protein (27, 29). In contrast, IFI35 does not interact with NS1-H7N9 and probably facilitates virus immune evasion at an early stage through blocking RIG-I-mediated immune surveillance. In this regard, the IFI35 binding site’s mutation of NS1-H7N9 at least partially explains the lower type I IFN production at the early stage and the greater pathology of H7N9 infection than of H3N2 infection. In addition, IFI35 has been shown to play multiple roles in promoting inflammatory cytokine and chemokine expression in various models. First, extracellular IFI35 associates with Nmi, forming a damage-associated molecular pattern (DAMP), which activates macrophages to release proinflammatory cytokines (23). IFI35 promotes chemokine CCL5 and CXCL10 expression and exacerbates the pathogenesis of glomerular inflammation (54). IFI35 has recently been suggested to increase IL-12p40 expression under highly pathogenic H5N1 infection (31). Based on these findings, we proposed the Ifi35−/− mice are resistant to lethal H7N9 infection due to at least two reasons. First, higher type I IFN production at an early stage facilitates host innate immune surveillance. Second, lower proinflammatory responses in Ifi35−/− mice might reduce immunopathology caused by excessive inflammation. Ifi35−/− mice also exhibited resistance to highly pathogenic H5N1 virus infection (31).
In summary, our study demonstrates a previously unrecognized role of IFI35 in regulating RIG-I-mediated innate antiviral responses during influenza virus infection. IFI35 functions as either a positive or negative regulator of antiviral signaling, which is determined by the interaction with viral protein NS1. Our findings present novel insights into the role of IFI35-NS1 in the regulation of RIG-I-mediated antiviral signaling.
MATERIALS AND METHODS
Reagents and antibodies.
Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA), TRIzol reagent (Invitrogen, Grand Island, NY), RNeasy minikit (Qiagen, GmbH, Hilden, Germany), SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA), SYBR Premix Ex Taq (perfect real time; TaKaRa), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (catalog no. 10236276001; Roche, Mannheim, Germany), and Protein A/G PLUS-agarose (Santa Cruz, Dallas, TX) were obtained from the indicated sources. The dual-luciferase reporter assay system was from Promega. Mouse monoclonal antibodies (MAbs) to NP and NS1 proteins of IAV were produced in our laboratory. Anti-IFI35 MAb (ab55332; Abcam), mouse anti-GAPDH MAb (ab9485; Abcam), rabbit anti-beta actin polyclonal antibody (PAb) (ab8227; Abcam), mouse anti-Flag M2 MAb (F1804; Sigma), rabbit anti-Myc PAb (R1208-1; Huaan Biological Technology), mouse anti-Myc MAb (ab56; Abcam), rabbit anti-GFP PAb (2956; CST), mouse anti-GFP (4B10) Mab (2955; CST), mouse anti-GST MAb (M0807-1; Huaan Biological Technology), mouse anti-HA MAb (Sigma-Aldrich, Oakville, Canada), and horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG and HRP-conjugated goat anti-rabbit IgG (Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD) were obtained from the indicated sources.
Ifi35 knockout mouse generation and virus infection.
Four- to 8-week-old C57BL/6J female and male mice were purchased from the Zhejiang experimental animal center. The Ifi35−/− deficient mice were produced by Talen from C57BL/6J mice. The genotyping of Ifi35+/+ and Ifi35−/− mice was performed with the following primers: forward primer 5′-TGCCTGAAGGTTCTGCTCTGGTCAC-3′ and reverse primer 5′-GGCCTTGCCAAAGAAGATCTCCAGC-3′ for Ifi35+/+ mice; forward primer 5′-TGCCTGAAGGTTCTGCTCTGGTCAC-3′ and reverse primer 5′-AGCCAGTGTCAAGGTCTGCGTTCTG-3′ for Ifi35−/− mice. The newly constructed Ifi35−/− mice were maintained as heterozygotes by backcrossing with WT C57BL/6J mice for at least four generations. All of the mice were bred and maintained in a specific-pathogen-free animal facility at Zhejiang University. Ifi35+/+ and Ifi35−/− mice used in this study were littermates of the same sex on a C57BL/6J background at the ages indicated in the specific experiments. Animal care and experimental procedures were in accordance with the Animal Research Committee guidelines of Zhejiang University. All animals were handled according to protocols reviewed and approved by the Institutional Animal Care and Use Committee (Zhejiang University). For virus infection, the mice were intranasally infected with H7N9 virus in 50 μl phosphate-buffered saline (PBS) at 104.8 50% tissue culture infectious doses (TCID50) per mouse. Survival and weight loss were monitored daily until 14 days postinfection (dpi). IAV infection was conducted in a biosafety level 3 facility. Inoculated animals were assessed daily. Those with severe morbidity (greater than 30% weight loss plus severe clinical impairment) were humanely euthanized according to our approved protocol.
Cell culture and virus infection.
Human lung epithelial A549 cells, human embryonic kidneys (HEK) 293T cells, and Madin-Darby canine kidney (MDCK) cells (American Type Culture Collection, Manassas, VA) were routinely cultured at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies Gibco, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Strains A/HangZhou/1/2013 (H7N9) and A/swine/Guangdong/04 (H3N2) were stored in our laboratory (19, 55, 56). For virus infection, cells were inoculated with IAV at the indicated multiplicity of infection (MOI). After adsorption for 1 h at 37°C, the cells were washed with phosphate-buffered saline (PBS) and cultured in DMEM containing 2 μg/ml TPCK (L-1-tosylamido-2-phenylethyl chloromethyl ketone)-treated trypsin. All experiments with virus infection were carried out at a biosafety level 3 laboratory.
Virus titration.
In this study, virus titers were determined by TCID50 assay on MDCK cells using the Reed and Muench method. For in vivo experiments, lungs of virus-infected mice were collected, weighed, and homogenized in cold PBS. After centrifugation at 1,000 × g for 5 min at 4°C, the supernatants were stored at −80°C or were followed by virus titration. The supernatants were serially diluted 10-fold with DMEM and adsorbed onto confluent MDCK cells for 1.5 h at 37°C. The inoculum was removed, and cells were washed three times with PBS and cultured with DMEM containing 2% FBS with 1 μg/ml TPCK-treated trypsin. The assay was carried out in eight parallel experiments. After 72 h of incubation, the cells were then fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100 in PBS, and stained with anti-NP mouse monoclonal antibodies, followed by fluorescein isothiocyanate (FITC)-conjugated IgG. Virus titers were defined as the number of cells positive for the anti-NP signals. For in vitro experiments, the IAV genome copies were determined by detecting the M1 gene of IAV by using quantitative real-time PCR (qRT-PCR). All experiments with virus infection were carried out at a biosafety level 3 laboratory.
Plasmid construction and transfection.
Standard molecular biology procedures were performed for all plasmid constructions. The full-length cDNA encoding human IFI35 and viral protein NS1 of H3N2 were amplified from mRNA of H3N2 virus-infected A549 cells by RT-PCR and cloned into the following vectors: pCMV-N-Myc (Clontech, Palo Alto, CA), pCMV-N-Flag (Clontech), pCMV-N-HA (Clontech), pCI-neo (Promega, Madison, WI), pEGFP-C3 (Clontech), pGE×4T-1 (GE Healthcare Biosciences, Piscataway, NJ), p3×Flag-CMV-10 (Sigma), and pET-28a (Novagen, Madison, WI). To produce various truncated forms of the plasmids for IFI35, RIG-I, and NS1, indicated fragments of DNA were amplified from Flag-IFI35, GFP-RIG-I, and Flag-NS1, respectively. Several deletion forms of IFI35 and NS1 plasmids were produced and designed as GFP-IFI35-L-zip (IFI35-1-80aa), GFP-IFI35-NID1 (IFI35-81-170aa), GFP-IFI35-NID2 (IFI35-177-288aa), GFP-IFI35-ΔL-zip, GFP-IFI35-ΔNID1, GFP-IFI35-ΔNID2, Flag-NS1-1-73aa, Flag-NS1-74-207aa, Flag-NS1-128-230aa, and Myc-NS1-128-207aa. Flag-GST-IFI35 and Flag-GST-NS1 plasmids were constructed by fusion of the expressing GST with wild-type full-length IFI35 and NS1, respectively. The deletions of RIG-I fragments were amplified and constructed into pEGFP-C3 vector and named GFP-RIG-I-CARDs, GFP-RIG-I-Helix, GFP-RIG-I-CTD. This protein configuration allows simultaneous detection of the wild-type IFI35, RIG-I, or NS1 in cells using three different antibodies, anti-Flag, anti-Myc, anti-GST, and anti-GFP, plus anti-IFI35 or anti-NS1 antibody, for immunostaining or immunoprecipitation. Reporter plasmid constructions were performed as previously reported with some modifications (57). Reporter plasmid pGluc-IFNβ, which carries the Gaussia luciferase gene under the control of human IFN-β promoter, was created by cloning the human IFN-β promoter (nucleotides [nt] −125 to +19) into pGluc-basic vector (New England BioLabs, Ipswich, MA). Another reporter plasmid, pISRE-Luc, was obtained from Agilent Technologies. Flag-tagged RIG-I expression plasmid was kindly provided by Danying Chen (College of Life Sciences, Peking University). Hemagglutinin (HA)-tagged Ub-K48 plasmid was kindly provided by Zongping Xia (Institute of Life Sciences, Zhejiang University). Various influenza virus NS1-expressing plasmids were constructed by amplifying the NS1 genes from the corresponding virus strains. NS1-H3N2 was cloned from A/swine/Guangdong/04 (H3N2), and NS1-H7N9 was cloned from A/HangZhou/1/2013 (H7N9). For chimeric NS1-expressing plasmid construction, the nucleic acids coding for 128-207aa of NS1-H7N9 were replaced by the corresponding region of NS1-H3N2, and the chimeric nucleic acid gene was synthesized directly and then constructed into the p3×Flag-CMV-10 vector. All constructs were validated by restriction enzyme digestion and cDNA sequencing. Primer sequences are available upon request. All constructions were transfected into cells using Lipofectamine 2000 reagent according to the manufacturer’s recommendations for transient expression.
qRT-PCR analysis.
At 24 h after the A549 monolayers were transfected with overexpression or knockdown IFI35 plasmid, they were infected with IAV for the indicated times. After this, the cells were washed 3 times with ice-cold PBS and then lysed with TRIzol reagent. Total cellular RNA was extracted using the RNeasy minikit according to the manufacturer’s protocol. RNA concentrations were measured using a spectrophotometer (260 nm/280 nm). Then, the RNA was reverse-transcribed to cDNA using 200 U SuperScript III reverse transcriptase. These cDNAs were used for quantitative real-time PCR (qRT-PCR), which was performed by using the 7500 real-time PCR system (Applied Biosystems, Foster City, CA) with a total volume of 20 μl containing 100 ng of cDNA template, 1× SYBR Premix Ex Taq, and 200 nM each primer. The primer pair sequences for qRT-PCR are available upon request. Parallel mock-infected A549 was used for controls.
shRNA knockdown.
Short hairpin RNAs (shRNAs) were used to knock down human IFI35 expression. Annealed oligonucleotides containing the 21-nt shRNA were cloned into the pGPU6/Neo-shRNA (Clontech) according to the manufacturer’s protocol. The sequence of IFI35 shRNA (shIFI35-#520) is 5′-GCACACGATCAACATGGAGGATTCAAGAGATCCTCCATGTTGATCGTGTGTGCTT-3′. The sequence of IFI35 shRNA (shIFI35-#930) is 5’GTGCTCAACATTCCTGATATATCTTTCAAGAGAGATATCAGGAATGTTGAGCACTT-3′, and an shRNA plasmid targeting the GAPDH (glyceraldehyde 3-phosphate dehydrogenase) (shGAPDH) and an shLUC plasmid targeting the firefly luciferase reporter gene (shNC) were used as negative controls. For shRNA knockdown, A549 cells in 6-well plates were transfected with 2 μg of shNC, shIFI35-#520, shIFI35-#930, and shGAPDH plasmid using Lipofectamine 2000. Cells were replaced with fresh medium 6 h posttransfection and cultured for 48 h in a CO2 incubator at 37°C. The knockdown efficiency was examined by Western blotting.
Confocal immunofluorescence assay.
Coverslip-adhered A549 or 293T cell monolayers were transfected with the indicated mammalian expression vector or infected with IAV for the indicated times. Treated cells were washed twice with PBS-Tween 20 (PBST), fixed with cold acetone-methanol (1/1) for 20 min at −20°C, and then allowed to air dry. The fixed cells were incubated with primary antibodies overnight. Cells were then incubated with FITC-conjugated secondary antibodies (KPL) or Alexa Fluor 546-conjugated secondary antibodies (Invitrogen) at 37°C for 1 h. Cellular nuclei were stained with 10 μg/ml DAPI for 5 min. For in vivo lung tissues, the PR8- or H7N9-infected mice were sacrificed at day 2 postinfection, and lungs were perfused with cold PBS and incubated in 4% paraformaldehyde for 48 h. Lungs were washed for 15 min in serial washes of PBS, 30% ethanol, and 50% ethanol and then stored in 70% ethanol at 4°C until processing for paraffin embedding, sectioning, and staining with anti-influenza virus NP mouse MAb, followed by Alexa Fluor 546-conjugated IgG, and the nuclei were stained with 10 μg/ml DAPI for 5 min. The triply stained cells and tissues were washed three times with PBS and subsequently viewed with an LSM780 laser scanning confocal microscope (Zeiss, Oberkochen, Germany). Images were acquired and analyzed using Zen 2012 (Zeiss).
Immunoblotting and co-IP.
Samples were prepared and treated as described above, and cell monolayers were washed with PBS and lysed with lysis buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, and an anti-protease Halt protease inhibitor single-use cocktail (Promega, Madison, WI). The protein concentrations were determined by use of a bicinchoninic acid (BCA) analysis kit. Protein fractions (20 μg) were used to load an SDS-PAGE gel to perform a Western blot assay. The coimmunoprecipitation (co-IP) assay was conducted in 293T or A549 cells. Briefly, the 293T cells, transfected alone or cotransfected with the indicated plasmids, were lysed with NP-40 lysis buffer after 36 to 48 h. For A549, the mock-infected or IAV-infected cells were lysed after 24 h. The supernatant was incubated with the indicated antibodies at 4°C for 2 h. Immune complexes were precipitated by incubation with Protein A/G PLUS-agarose (Santa Cruz, Dallas, TX) at 4°C for 2 h. After three stringent washes in NP-40 lysis buffer, immunoprecipitated proteins were analyzed by Western blotting.
GST pulldown.
The glutathione S-transferase (GST) pulldown assay was conducted in vivo and in vitro. For the in vivo assay, 293T cells were cotransfected with the indicated plasmids for 48 h, the cell lysate was bound to glutathione-Sepharose beads (GE Healthcare) at 4°C for 2 h, the beads were washed 3 times with PBS, and the samples were analyzed by Western blotting with the indicated antibodies. The GST pulldown in vitro assay was performed by following the protocol of the GST protein interaction pulldown kit (catalog no. 21516; Thermo, Rockford, IL). The GST or GST-NS1 and His-IFI35 fusion proteins expressed by E. coli and immobilized on glutathione-Sepharose beads were incubated at 4°C for 8 h. The beads were washed extensively and boiled in SDS loading buffer, and the precipitated proteins were subjected to SDS-PAGE and detected by immunoblotting with mouse anti-GST MAb and mouse anti-His MAb.
Luciferase assay.
293T cells were transfected with the pGLuc-IFN-β or pGLuc-ISRE promoter luciferase reporter plasmid (0.5 μg per well) together with the indicated plasmids. After 24 or 48 h, the cells were lysed with passive lysis buffer, and the luciferase activity in the lysates was determined with the dual-luciferase reporter assay system. The Renilla luciferase construct pRL-TK (0.05 μg per well) was simultaneously transfected as an internal control.
In vivo ubiquitination assay.
293T cells were transiently cotransfected with the indicated plasmids. At 24 h posttransfection, cells were treated with 10 μM MG132 where indicated. Samples were harvested 24 h posttransfection and lysed using a 1% NP-40 lysis buffer supplemented with 0.1% protease inhibitor cocktail and the deubiquitinase inhibitor N-ethylmaleimide. Samples were boiled for 10 min in 1% SDS and diluted 10 times in lysis buffer. Lysed proteins were then immunoprecipitated overnight at 4°C with MAb Flag for 2 h and incubated with Protein A/G PLUS-Agarose for 2 h at room temperature. Immunoprecipitated protein was washed 4 times with supplemented lysis buffer and denatured in 2% SDS-loading dye, followed by SDS-PAGE analysis and then transfer to a nitrocellulose membrane. Polyubiquitination was detected by immunoblotting with a monoclonal anti-HA antibody.
Lung tissue histology.
The trachea and lungs from control or virus-infected mice at the indicated postinfection times were dissected, fixed in 10% formalin, embedded in paraffin, sectioned, stained with hematoxylin and eosin (H&E) solution, and visualized by light microscopy for histologic changes.
Statistical analysis.
GraphPad Prism 6 software was used for data analysis using Student's t test for the statistical analysis of two independent treatments. Kaplan-Meier survival curves were analyzed by the Mantel-Cox log rank test. Differences in weight loss and viral titer were analyzed by two-way analysis of variance (ANOVA) tests. For all tests, P values of <0.05 (*) were considered statistically significant, and P values of <0.01 (**) and <0.001 (***) were considered highly significant.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (grant no. 31402209), the National Postdoctoral Program for Innovative Talents (grant no. 2015T80625), and the China Postdoctoral Science Foundation (grant no. 2014M551757).
We thank all members of the MOA Key Laboratory of Animal Virology for kindly providing helpful insights and generous support.
REFERENCES
- 1.Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, tenOever BR. 2020. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181:1036–1045.e9. 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park A, Iwasaki A. 2020. Type I and type III interferons—induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe 27:870–878. 10.1016/j.chom.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arilahti V, Makela SM, Tynell J, Julkunen I, Osterlund P. 2014. Novel avian influenza A (H7N9) virus induces impaired interferon responses in human dendritic cells. PLoS One 9:e96350. 10.1371/journal.pone.0096350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solorzano A, Webby RJ, Lager KM, Janke BH, Garcia-Sastre A, Richt JA. 2005. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J Virol 79:7535–7543. 10.1128/JVI.79.12.7535-7543.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beilharz MW, Cummins JM, Bennett AL. 2007. Protection from lethal influenza virus challenge by oral type 1 interferon. Biochem Biophys Res Commun 355:740–744. 10.1016/j.bbrc.2007.02.019. [DOI] [PubMed] [Google Scholar]
- 6.Arimori Y, Nakamura R, Yamada H, Shibata K, Maeda N, Kase T, Yoshikai Y. 2013. Type I interferon limits influenza virus-induced acute lung injury by regulation of excessive inflammation in mice. Antiviral Res 99:230–237. 10.1016/j.antiviral.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Ludwig S, Wolff T. 2009. Influenza A virus TRIMs the type I interferon response. Cell Host Microbe 5:420–421. 10.1016/j.chom.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Garcia-Sastre A, Biron CA. 2006. Type 1 interferons and the virus-host relationship: a lesson in detente. Science 312:879–882. 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 9.Hale BG, Randall RE, Ortin J, Jackson D. 2008. The multifunctional NS1 protein of influenza A viruses. J Gen Virol 89:2359–2376. 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- 10.Haye K, Burmakina S, Moran T, Garcia-Sastre A, Fernandez-Sesma A. 2009. The NS1 protein of a human influenza virus inhibits type I interferon production and the induction of antiviral responses in primary human dendritic and respiratory epithelial cells. J Virol 83:6849–6862. 10.1128/JVI.02323-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marc D. 2014. Influenza virus non-structural protein NS1: interferon antagonism and beyond. J Gen Virol 95:2594–2611. 10.1099/vir.0.069542-0. [DOI] [PubMed] [Google Scholar]
- 12.Krug RM. 2015. Functions of the influenza A virus NS1 protein in antiviral defense. Curr Opin Virol 12:1–6. 10.1016/j.coviro.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Min JY, Krug RM. 2006. The primary function of RNA binding by the influenza A virus NS1 protein in infected cells: Inhibiting the 2'-5' oligo (A) synthetase/RNase L pathway. Proc Natl Acad Sci U S A 103:7100–7105. 10.1073/pnas.0602184103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M, Jr, Sastre A. 2007. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol 81:514–524. 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Opitz B, Rejaibi A, Dauber B, Eckhard J, Vinzing M, Schmeck B, Hippenstiel S, Suttorp N, Wolff T. 2007. IFN beta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell Microbiol 9:930–938. 10.1111/j.1462-5822.2006.00841.x. [DOI] [PubMed] [Google Scholar]
- 16.Guo Z, Chen LM, Zeng H, Gomez JA, Plowden J, Fujita T, Katz JM, Donis RO, Sambhara S. 2007. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am J Respir Cell Mol Biol 36:263–269. 10.1165/rcmb.2006-0283RC. [DOI] [PubMed] [Google Scholar]
- 17.Geiss GK, Salvatore M, Tumpey TM, Carter VS, Wang X, Basler CF, Taubenberger JK, Bumgarner RE, Palese P, Katze MG, Garcia-Sastre A. 2002. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc Natl Acad Sci U S A 99:10736–10741. 10.1073/pnas.112338099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forbes NE, Ping J, Dankar SK, Jia JJ, Selman M, Keleta L, Zhou Y, Brown EG. 2012. Multifunctional adaptive NS1 mutations are selected upon human influenza virus evolution in the mouse. PLoS One 7:e31839. 10.1371/journal.pone.0031839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu XP, Wang SY, Yu Y, Zhang JY, Sun ZY, Yan Y, Zhou JY. 2013. Subcellular proteomic analysis of human host cells infected with H3N2 swine influenza virus. Proteomics 13:3309–3326. 10.1002/pmic.201300180. [DOI] [PubMed] [Google Scholar]
- 20.Bange FC, Vogel U, Flohr T, Kiekenbeck M, Denecke B, Bottger EC. 1994. I 35 is an interferon-induced leucine-zipper protein that undergoes interferon-regulated cellular redistribution. J Biol Chem 269:1091–1098. 10.1016/S0021-9258(17)42225-3. [DOI] [PubMed] [Google Scholar]
- 21.Chen J, Shpall RL, Meyerdierks A, Hagemeier M, Bottger EC, Naumovski L. 2000. Interferon-inducible Myc/STAT-interacting protein Nmi associates with IFP 35 into a high molecular mass complex and inhibits proteasome-mediated degradation of IFP 35. J Biol Chem 275:36278–36284. 10.1074/jbc.M006975200. [DOI] [PubMed] [Google Scholar]
- 22.Zhou X, Liao J, Meyerdierks A, Feng L, Naumovski L, Bottger EC, Omary MB. 2000. Interferon-alpha induces nmi-IFP35 heterodimeric complex formation that is affected by the phosphorylation of IFP35. J Biol Chem 275:21364–21371. 10.1074/jbc.M003177200. [DOI] [PubMed] [Google Scholar]
- 23.Xiahou Z, Wang X, Shen J, Zhu X, Xu F, Hu R, Guo D, Li H, Tian Y, Liu Y, Liang H. 2017. NMI and IFP35 serve as proinflammatory DAMPs during cellular infection and injury. Nat Commun 8:950. 10.1038/s41467-017-00930-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang L, Tang Y, Tie Y, Tian C, Wang J, Dong Y, Sun Z, He F. 2007. The PH domain containing protein CKIP-1 binds to IFP35 and Nmi and is involved in cytokine signaling. Cell Signal 19:932–944. 10.1016/j.cellsig.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 25.Wang X, Johansen LM, Tae HJ, Taparowsky EJ. 1996. IFP 35 forms complexes with B-ATF, a member of the AP1 family of transcription factors. Biochem Biophys Res Commun 229:316–322. 10.1006/bbrc.1996.1799. [DOI] [PubMed] [Google Scholar]
- 26.Das A, Dinh PX, Pattnaik AK. 2015. Trim21 regulates Nmi-IFI35 complex-mediated inhibition of innate antiviral response. Virology 485:383–392. 10.1016/j.virol.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 27.Das A, Dinh PX, Panda D, Pattnaik AK. 2014. Interferon-inducible protein IFI35 negatively regulates RIG-I antiviral signaling and supports vesicular stomatitis virus replication. J Virol 88:3103–3113. 10.1128/JVI.03202-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng W, Li X, Wang J, Li X, Cao H, Wang Y, Zeng Q, Zheng SJ. 2014. A critical role of interferon-induced protein IFP35 in the type I interferon response in cells induced by foot-and-mouth disease virus (FMDV) protein 2C. Arch Virol 159:2925–2935. 10.1007/s00705-014-2147-7. [DOI] [PubMed] [Google Scholar]
- 29.Tan J, Qiao W, Wang J, Xu F, Li Y, Zhou J, Chen Q, Geng Y. 2008. IFP35 is involved in the antiviral function of interferon by association with the viral Tas transactivator of bovine foamy virus. J Virol 82:4275–4283. 10.1128/JVI.02249-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu X, Yang W, Liu R, Geng Y, Qiao W, Tan J. 2014. N-Myc interactor inhibits prototype foamy virus by sequestering viral Tas protein in the cytoplasm. J Virol 88:7036–7044. 10.1128/JVI.00799-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gounder AP, Yokoyama CC, Jarjour NN, Bricker TL, Edelson BT, Boon ACM. 2018. Interferon induced protein 35 exacerbates H5N1 influenza disease through the expression of IL-12p40 homodimer. PLoS Pathog 14:e1007001. 10.1371/journal.ppat.1007001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Qi X, Lu C. 2012. Mutations in the C-terminal tail of NS1 protein facilitate the replication of classical swine H1N1 influenza A virus in mice. Folia Microbiol (Praha) 57:169–175. 10.1007/s12223-012-0110-0. [DOI] [PubMed] [Google Scholar]
- 33.Mitra S, Kumar D, Hu L, Sankaran B, Moosa MM, Rice AP, Ferreon JC, Ferreon ACM, Prasad BVV. 2019. Influenza A virus protein NS1 exhibits strain-independent conformational plasticity. J Virol 93:e00917-19. 10.1128/JVI.00917-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carrillo B, Choi JM, Bornholdt ZA, Sankaran B, Rice AP, Prasad BV. 2014. The influenza A virus protein NS1 displays structural polymorphism. J Virol 88:4113–4122. 10.1128/JVI.03692-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajsbaum R, Albrecht RA, Wang MK, Maharaj NP, Versteeg GA, Nistal-Villan E, Garcia-Sastre A, Gack MU. 2012. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog 8:e1003059. 10.1371/journal.ppat.1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrage F, Dutta K, Nistal-Villan E, Patel JR, Sanchez-Aparicio MT, De Ioannes P, Buku A, Aseguinolaza GG, Garcia-Sastre A, Aggarwal AK. 2012. Structure and dynamics of the second CARD of human RIG-I provide mechanistic insights into regulation of RIG-I activation. Structure 20:2048–2061. 10.1016/j.str.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, Gerlier D, Cusack S. 2011. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 147:423–435. 10.1016/j.cell.2011.09.039. [DOI] [PubMed] [Google Scholar]
- 38.Peisley A, Wu B, Xu H, Chen ZJ, Hur S. 2014. Structural basis for ubiquitin-mediated antiviral signal activation by RIG-I. Nature 509:110–114. 10.1038/nature13140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayman A, Comely S, Lackenby A, Hartgroves LCS, Goodbourn S, McCauley JW, Barclay WS. 2007. NS1 proteins of avian influenza A viruses can act as antagonists of the human alpha/beta interferon response. J Virol 81:2318–2327. 10.1128/JVI.01856-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, Garcia-Sastre A. 2000. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol 74:7989–7996. 10.1128/jvi.74.17.7989-7996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X, Li M, Zheng H, Muster T, Palese P, Beg AA, Garcia-Sastre A. 2000. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J Virol 74:11566–11573. 10.1128/jvi.74.24.11566-11573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steidle S, Martinez-Sobrido L, Mordstein M, Lienenklaus S, Garcia-Sastre A, Staheli P, Kochs G. 2010. Glycine 184 in nonstructural protein NS1 determines the virulence of influenza A virus strain PR8 without affecting the host interferon response. J Virol 84:12761–12770. 10.1128/JVI.00701-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kochs G, Martinez-Sobrido L, Lienenklaus S, Weiss S, Garcia-Sastre A, Staeheli P. 2009. Strong interferon-inducing capacity of a highly virulent variant of influenza A virus strain PR8 with deletions in the NS1 gene. J Gen Virol 90:2990–2994. 10.1099/vir.0.015727-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan WM, Aramini JM, Montelione GT, Krug RM. 2002. Structural basis for ubiquitin-like ISG 15 protein binding to the NS1 protein of influenza B virus: a protein-protein interaction function that is not shared by the corresponding N-terminal domain of the NS1 protein of influenza a virus. Virology 304:291–301. 10.1006/viro.2002.1663. [DOI] [PubMed] [Google Scholar]
- 45.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, Dabelic R, Manicassamy B, Aitchison JD, Aderem A, Elliott RM, Garcia-Sastre A, Racaniello V, Snijder EJ, Yokoyama WM, Diamond MS, Virgin HW, Rice CM. 2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691–695. 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Staeheli P, Haller O, Boll W, Lindenmann J, Weissmann C. 1986. Mx protein: constitutive expression in 3t3 cells transformed with cloned Mx Cdna confers selective resistance to influenza virus. Cell 44:147–158. 10.1016/0092-8674(86)90493-9. [DOI] [PubMed] [Google Scholar]
- 47.Brass AL, Huang IC, Benita Y, John SP, Krishnan MN, Feeley EM, Ryan BJ, Weyer JL, van der Weyden L, Fikrig E, Adams DJ, Xavier RJ, Farzan M, Elledge SJ. 2009. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 139:1243–1254. 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang XY, Hinson ER, Cresswell P. 2007. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe 2:96–105. 10.1016/j.chom.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 49.Weber M, Sediri H, Felgenhauer U, Binzen I, Banfer S, Jacob R, Brunotte L, Garcia-Sastre A, Schmid-Burgk JL, Schmidt T, Hornung V, Kochs G, Schwemmle M, Klenk HD, Weber F. 2015. Influenza virus adaptation PB2-627K modulates nucleocapsid inhibition by the pathogen sensor RIG-I. Cell Host Microbe 17:309–319. 10.1016/j.chom.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu GQ, Park HS, Pyo HM, Liu Q, Zhou Y. 2015. Influenza A virus panhandle structure is directly involved in RIG-I activation and interferon induction. J Virol 89:6067–6079. 10.1128/JVI.00232-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maelfait J, Beyaert R. 2012. Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol Mol Biol Rev 76:33–45. 10.1128/MMBR.05012-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arimoto KI, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. 2007. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci U S A 104:7500–7505. 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen WL, Han CF, Xie B, Hu X, Yu Q, Shi LY, Wang QQ, Li DL, Wang JL, Zheng P, Liu Y, Cao XT. 2013. Induction of Siglec-G by RNA viruses inhibits the innate immune response by promoting RIG-I degradation. Cell 152:467–478. 10.1016/j.cell.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 54.Shirai K, Shimada T, Yoshida H, Hayakari R, Matsumiya T, Tanji K, Murakami M, Tanaka H, Imaizumi T. 2017. Interferon (IFN)-induced protein 35 (IFI35) negatively regulates IFN-beta-phosphorylated STAT1-RIG-I-CXCL10/CCL5 axis in U373MG astrocytoma cells treated with polyinosinic-polycytidylic acid. Brain Res 1658:60–67. 10.1016/j.brainres.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 55.Wu XP, Wang HL, Bai L, Yu Y, Sun ZY, Yan Y, Zhou JY. 2013. Mitochondrial proteomic analysis of human host cells infected with H3N2 swine influenza virus. J Proteomics 91:136–150. 10.1016/j.jprot.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 56.Yu X, Jin T, Cui Y, Pu X, Li J, Xu J, Liu G, Jia H, Liu D, Song S, Yu Y, Xie L, Huang R, Ding H, Kou Y, Zhou Y, Wang Y, Xu X, Yin Y, Wang J, Guo C, Yang X, Hu L, Wu X, Wang H, Liu J, Zhao G, Zhou J, Pan J, Gao GF, Yang R, Wang J. 2014. Influenza H7N9 and H9N2 viruses: coexistence in poultry linked to human H7N9 infection and genome characteristics. J Virol 88:3423–3431. 10.1128/JVI.02059-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li W, Chen H, Sutton T, Obadan A, Perez DR. 2014. Interactions between the influenza A virus RNA polymerase components and retinoic acid-inducible gene I. J Virol 88:10432–10447. 10.1128/JVI.01383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]