

Human papillomaviruses (HPV) have coevolved with their host by using cellular factors like bromodomain-containing protein 4 (BRD4) to control viral processes, such as genome maintenance, gene expression, and replication. Here, we show that, in addition to the C-terminal motif in BRD4, the phospho-dependent interaction domain in BRD4 interacts with E2 proteins, which enable the recruitment of BRD4S, the short isoform of BRD4, to E2.
KEYWORDS: E2, BRD4S, L1, viral gene expression, papillomavirus
ABSTRACT
The E2 protein encoded by human papillomaviruses (HPV) is a sequence-specific DNA-binding protein that recruits viral and cellular proteins. Bromodomain-containing protein 4 (BRD4) is a highly conserved interactor for E2 proteins that has been linked to E2’s functions as transcription modulator, activator of viral replication, and segregation factor for viral genomes. In addition to BRD4, a short form of BRD4 (BRD4S) is expressed from the BRD4 gene, which lacks the C-terminal domain of BRD4. E2 proteins interact with the C-terminal motif (CTM) of BRD4, but a recent study suggested that the phospho-dependent interaction domain (PDID) and the basic interaction domain (BID) in BRD4 also bind to E2. These domains are also present in BRD4S. We now find that HPV31 E2 interacts with the isolated PDID domain in living cells and also with BRD4S, which is present in detectable amounts in HPV-positive cell lines and is recruited into HPV31 E1- and E2-induced replication foci. Overexpression and knockdown experiments surprisingly indicate that BRD4S inhibits activities of E2. In line with that, the specific knockdown of BRD4S in the HPV31-positive CIN612-9E cell line induces mainly late viral transcripts. This occurs only in undifferentiated but not differentiated cells in which the productive viral replication cycle is induced. These data suggest that the BRD4S-E2 interaction is important to prevent HPV late gene expression in undifferentiated keratinocytes, which may contribute to immune evasion and HPV persistence.
IMPORTANCE Human papillomaviruses (HPV) have coevolved with their host by using cellular factors like bromodomain-containing protein 4 (BRD4) to control viral processes, such as genome maintenance, gene expression, and replication. Here, we show that, in addition to the C-terminal motif in BRD4, the phospho-dependent interaction domain in BRD4 interacts with E2 proteins, which enable the recruitment of BRD4S, the short isoform of BRD4, to E2. Knockdown and overexpression of BRD4S reveal that BRD4S is a negative regulator of E2 activities. Importantly, the knockdown of BRD4S induces mainly L1 transcripts in undifferentiated CIN612-9E cells, which maintain replicating HPV31 genomes. Our study reveals an inhibitory role of BRD4S on HPV transcription, which may serve as an immune escape mechanism by the suppression of L1 transcripts and thus contribute to the establishment of persistent HPV infections.
INTRODUCTION
Persistent infections with high-risk (HR) human papillomaviruses (HPV) such as HPV16, 18, or 31 can result in anogenital and oropharyngeal cancers (1). HPV replicate in keratinocytes and have adapted their replication cycle to the differentiation state of the cell. In undifferentiated keratinocytes, viral genome replication is limited and mainly the viral early region is transcribed, while the late promoter is silent (2). Upon differentiation, viral genomes are amplified and late viral genes are highly expressed, which finally results in the production of infectious virus (3, 4). Initial genome replication requires the E1 and E2 proteins, which form a complex that binds with high affinity to the viral origin of replication. E1 acts as the replicative helicase and also recruits host replication proteins to replicate the viral genome. Aside from recruiting E1, E2 modulates viral and cellular transcription. E2 can repress the major viral early promoter but has also transcription-activating activities. Furthermore, E2 acts as a segregation factor for viral genomes during cell division (5, 6). E2’s activities are mediated by protein-protein interactions and its sequence-specific recognition of DNA. Among the many host cell proteins reported to bind to E2 proteins, bromodomain-containing protein 4 (BRD4) stands out, as this interaction is highly conserved among all papillomaviruses tested so far (7, 8). Furthermore, the BRD4-E2 interaction has been implicated to contribute to E2’s replication, transcription modulation, and segregation activities in a virus-type-dependent manner (8). BRD4 belongs to the bromodomain and extraterminal domain (BET) protein family and acts as a coactivator of transcription. The two conserved bromodomains preferentially bind to multiacetylated peptides in histones 3 and 4 as well as transcription factors (7). The ET domain modulates transcriptional activation and chromatin remodeling by interacting with JMJD6, NSD3, CHD4, and BRG1 (9). The C-terminal motif (CTM) domain binds to the heterodimeric P-TEFB kinase, which enables it to bypass the paused state of RNA polymerase II and promotes transcriptional elongation (9). In addition, BRD4 is involved in DNA damage repair pathways. BRD4 contributes to the activation of the nonhomologous end-joining recombination pathway (10, 11) and has been implicated in the activation of a DNA damage checkpoint (12, 13). The BRD4 gene encodes two additional gene products, which are generated by alternative RNA splicing, as follows: BRD4 short (BRD4S) and BRD4 isoform B. BRD4 isoform B has been shown to insulate chromatin from DNA damage signaling (12). BRD4S acts as a corepressor for HIV-1 transcription and thus controls latency (14). Furthermore, the ratio between BRD4 and BRD4S contributes to the antileukemic effects of SPRK1 inhibition (15).
The interaction between E2 and BRD4 is mainly mediated by the CTM domain of BRD4, which is present in BRD4 but not in BRD4S or isoform B (16) (Fig. 1). However, a recent biochemical study indicated that E2 proteins are able to bind two additional domains in BRD4. While the basic residue-enriched interaction domain (BID) (amino acids [aa] 524 to 579) was identified as a universal E2 binding site, the phosphorylation-dependent interaction domain (PDID) (aa 287 to 530) only interacted with E2 proteins from HR-HPV16 and 18 in a phosphorylation-dependent manner (17). Thus, recruitment of BRD4S, which contains PDID and BID, by HR-E2 proteins might also be possible.
FIG 1.

Interaction of BRD4 and HPV31 E2 in a flow cytometry-based FRET assay. (A) Schematic depiction of BRD4 (aa 1 to 1362) and BRD4S (aa 1 to 722) and their functional domains as follows: BD1 (bromodomain 1), PDID (phosphorylation-dependent interaction domain), BD2 (bromodomain 2), NPS (N-terminal cluster of phosphorylation sites), BID (basic residue-enriched interaction domain), ET (extra terminal domain), and CTM (C-terminal motif). BRD4S contains the unique residues GPA at positions 720 to 722 compared to BRD4. Additionally, the schematic depiction of HPV31 E2 protein (aa 1 to 372) showing the transactivation domain (TAD), hinge region, and DNA-binding/dimerization domain (DBD). (B) Gating strategy for fluorescence-activated cell sorter (FACS)-FRET analysis and BFP-31 E2 domain expression and nuclear localization. (A) One experiment is shown representatively for C33A cells, which were not transfected (mock) or transfected with expression vectors for SYFP, BFP, SYFP-BFP fusion, or BRD4-SYFP and BFP-HPV31 E2. Living cells were analyzed 48 h after transfection for FRET-positive signals using MACSQuant VYB (Miltenyi Biotec). Living cells were gated for double positive cells by excitation with the 405-nm laser to measure BFP emission with the 450/50-nm band-pass filter and by excitation with the 488-nm laser to measure SYFP emission by 525/50-nm band-pass filter (lane 1). The next gate ensures that SYFP-expressing cells, which emit a YFP signal (488/525) and a FRET signal measured by 525/50-nm filter that resulted in excitation with the 405-nm laser, are excluded to avoid false-positive cell count (lane 2). The last gate includes cells that emit a BFP signal (405/450) and a FRET signal, which was generated by the energy transfer of the BFP (excited by 405-nm laser) to SYFP measured by 525/50-nm filter. (C) C33A cells were transfected with expression vectors for SYFP, BFP, SYFP-BFP fusion, or combinations of BRD4-SYFP or BRD4dCTM-SYFP and BFP-HPV31 E2 or BFP-HPV31 E2 I73L. Living cells were analyzed 48 h after transfection for FRET-positive signals using MACSQuant VYB (Miltenyi Biotec). Mean values of FRET-positive cells (%) from at least three independent experiments are shown. Error bars indicate the standard error of the mean (SEM). Statistical significance was determined by one-way analysis of variance (ANOVA) and Dunnett’s multiple-comparison test (compared to BRD4-SYFP + BFP-31 E2). **, P < 0.01; ***, P < 0.001. (D) Immunoblot of transfected C33A cells using antibodies specific for SYFP to detect BRD4 fusions or BFP to detect HPV31 E2 fusion proteins. HSP90 was used as a loading control. (E) C33A cells were transfected with combinations of BFP-HPV16 E2 or HPV31 E2 and SYFP-BRD4-PDID, SYFP-BRD4-BID, or SYFP-BRD4-CTM expression vectors, and FRET-positive cells were determined as described above. Mean values of FRET-positive cells (%) from at least three independent experiments are shown. Error bars indicate the SEM. (F) C33A cells were transfected with 500 ng of BFP-31 E2 TAD-hinge, BFP-31 E2 hinge, BFP-31 E2 hinge-DBD vectors. Whole-cell lysates were analyzed by immunoblot using an antibody specific for BFP. HSP90 was used as loading control. C33A cells transfected with 50 ng of expression vectors for BFP-31 E2 TAD-hinge, BFP-31 E2 hinge, and BFP-31 E2 hinge-DBD were analyzed by immunofluorescence with an antibody specific for BFP. DAPI was used to visualize DNA. (G) C33A cells were transfected with a total of 1 μg of expression vectors for SYFP-BRD4-PDID, -BID, -CTM, and BFP-HPV31 E2 TAD-hinge, BFP-HPV31 E2 hinge, and BFP-HPV31 E2 hinge-DBD. Mean values of FRET-positive cells (%) from three independent experiments are shown, and error bars indicate SEM.
So far, the contribution of BRD4 to the papillomavirus (PV) life cycle has been investigated without taking the role of BRD4S into account. In this study, we demonstrate that PDID and BRD4S interact with HPV31 E2 and that BRD4S inhibits E2-dependent activities. Consistent with this, BRD4S is recruited into HPV31 E1/E2-induced replication foci. Furthermore, knockdown of BRD4S by either small interfering RNA (siRNA) or CRISPR/Cas9 specifically induced late viral transcripts in undifferentiated but not in differentiated CIN612-9E cells that maintain replicating HPV31 genomes. In summary, our data suggest that the BRD4S-E2 interaction is important to limit late gene expression in undifferentiated cells, which might contribute to immune evasion by high-risk HPV and subsequently to virus persistence.
RESULTS
BRD4S interacts with HR-HPV E2.
A previous study reported the following two additional interaction sites in BRD4 for E2 proteins: BID (aa 524 to 579) was bound by all E2 proteins tested, whereas phospho-PDID (aa 287 to 530) interacted only with HR-HPV 16 and 18 E2 proteins (Fig. 1A) (7, 17). Since BID and PDID are present in BRD4S, E2 proteins may also interact with BRD4S with unknown functional consequences. We first used an in vivo protein-protein interaction assay based on the flow cytometric detection of Förster resonance energy transfer (FRET)-positive cells in which FRET signals are generated when blue fluorescent protein (BFP) fusion proteins are in close proximity (<10 nm) to super yellow fluorescent protein (SYFP) fusion proteins (18). The gating strategy is based on negative (untransfected cells, unfused BFP and SYFP) and positive (a BFP-SYFP fusion protein) controls and demonstrates that the close proximity of fluorophores in the BFP-SYFP fusion protein leads to the generation of a FRET signal resulting in 99.3% FRET-positive cells, whereas the negative control results in less than 0.5% FRET-positive cells (Fig. 1B and C). To validate the system for E2, BFP was fused to the N terminus of HPV31 E2 (BFP-31 E2) and tested in cotransfection experiments in C33A cells with SYFP fused to the C terminus of BRD4 (SYFP-BRD4) (Fig. 1B and C). As controls, an E2 mutant (E2 I73L), previously shown to reduce the interaction with the C-terminal motif (CTM) of BRD4, and a BRD4 protein, in which the CTM was deleted (BRD4 dCTM), were used (19). Cotransfection of BRD4 and HPV31 E2 resulted in 70.7% FRET-positive cells, whereas the signal decreased to 3.4 and 0.5% when coexpressing BRD4 with HPV31 E2 I73L or BRD4 dCTM with E2 (Fig. 1C), although expression of HPV31 E2 I73L and BRD4 dCTM was observed (Fig. 1D). This suggested that this assay can be used to quantitatively determine E2-BRD4 interactions in living cells. As pointed out above, BRD4 dCTM, despite the presence of the PDID and BID domains, did not result in a FRET signal with E2. Since FRET signals can only be generated when the fluorophores are in close proximity, it was possible that the two fluorophores in the context of full-length proteins are too far apart to generate a FRET signal. Therefore, we tested smaller fragments of BRD4 harboring the following described interaction domains: SYFP-BRD4 PDID (aa 287 to 530), SYFP-BRD4 BID (aa 524 to 579), and SYFP-BRD4 CTM (aa 1224 to 1362) (Fig. 1E). This revealed that the isolated PDID and CTM domains resulted in similar numbers of FRET-positive cell of 42.4% and 49.5%, respectively, whereas BID did not generate a signal (0.2%). HPV16 E2 also generated a FRET signal with PDID and CTM but not with BID (Fig. 1E), indicating that the interaction between PDID and E2 is conserved. To test which domain in HPV31 E2 is involved in the PDID interaction, the transactivation domain (TAD)-hinge, the hinge alone, or the hinge-DNA-binding domain (DBD) were tested. Expression of the BFP-E2 domain constructs was determined by Western blot analysis (Fig. 1F). Furthermore, to ensure nuclear localization, BFP-E2 domain constructs were expressed with an additional nuclear localization signal (Fig. 1F). This revealed that only the TAD-hinge, but not the hinge alone or the hinge-DBD, interacted with PDID, and the same pattern of binding was, as expected, observed for the CTM (Fig. 1G). To delineate domains in PDID involved in binding to E2, a panel of deletion mutants was tested (Fig. 2A). A complete loss of signal was observed when residues 505 to 530 were removed, and also deletion of N-terminal cluster of phosphorylation sites (NPS) significantly reduced the signal (Fig. 2A). The loss of FRET signal was specific since these constructs were expressed at similar levels as the complete PDID (Fig. 2C). Bromodomain 1 (BD1)/bromodomain 2 (BD2) and residues 505 to 530 have previously been implicated in mediating dimerization of BRD proteins (20, 21). In line with this, the coexpression of SYFP-BRD4 PDID and BFP-BRD4 PDID resulted in 31% FRET-positive cells but not the coexpression of CTM, BID, CTM and PDID, BID and PDID, or BID and CTM (Fig. 2B). However, this mainly depended on BD2 and not on NPS or residues 505 to 530 (Fig. 2B), which suggests that BRD4 proteins can dimerize via BD2 but that this is not important for binding to E2. Since PDID is part of BRD4S (Fig. 1A), we tested if BRD4S binds to E2 by coimmunoprecipitation. Flag-BRD4, -BRD4S, or -BRD4 dCTM were cotransfected with hemagglutinin (HA)-HPV31 E2, which revealed that not only BRD4 but also BRD4S interacts with E2 (Fig. 3A). Furthermore, a small amount of BRD4 dCTM could be immunoprecipitated, suggesting that the PDID in BRD4 is sufficient to mediate an interaction (Fig. 3A). Immunoblot analyses revealed that both BRD4 and BRD4S are present in normal human keratinocytes, C33A, and HPV31-positive CIN612-9E cells (Fig. 3B), suggesting that the interaction between E2 and BRD4S might be relevant for the HPV life cycle.
FIG 2.

Several domains in PDID are required for interaction with E2. (A) C33A cells were cotransfected with SYFP-BRD4 PDID, -PDIDd287-348, -PDIDdBD2, -PDIDd462-482, -PDIDdNPS, or -PDIDd505-530 and BFP-HPV31 E2 expression vectors. Mean values of FRET-positive cells (%) are derived from three independent experiments, and error bars indicate the SEM. (B) BFP-BRD4-PDID, -BID, -CTM were tested in different combinations with SYFP-BRD4-PDID, -BID, -CTM in C33A cells. Additionally, the interactions of BFP-BRD4-PDID with SYFP-BRD4-PDID, -PDIDdBD2, -PDIDdNPS, and -PDIDd505 were analyzed. Mean values of FRET-positive cells (%) are derived from three independent experiments, and error bars indicate the SEM. Statistical significance was determined by one-way ANOVA and Dunnett’s multiple-comparison test (compared to SYFP-BRD4-PDID + BFP-HPV31 E2 [A] or SYFP-BRD4-PDID + BFP-BRD4-PDID [B]); ns, P > 0.05; **, P < 0.01; ****, P < 0.0001. (C) Immunoblot analyses of C33A cells transfected with expression vectors for SYFP-BRD4-PDID, -BID, -CTM, -PDIDdBD2, -PDIDdNPS, or -PDID d505-530. Untransfected cells served as a negative control (C33A). HSP90 was used as loading control.
FIG 3.

BRD4S interacts with HPV31 E2 and is expressed in different keratinocyte cell lines. (A) C33A cells were transfected with 10 μg of expression vectors for HA-tagged HPV31 E2 and Flag-tagged BRD4, BRD4S, or BRD4dCTM proteins. Whole-cell lysates were directly analyzed (input) or precipitated with magnetic anti-Flag beads. Lysates were analyzed using α-HA and α-Flag antibodies. (B) Immunoblot analysis of endogenous BRD4S expression in whole-cell extracts of normal human keratinocytes (NHK), C33A, and CIN612-9E cells. To detect BRD4S, α-BRD4 (N-terminal) and for BRD4 detection, α-BRD4 (C-terminal) antibodies were used. Alpha-tubulin was used as a loading control.
BRD4S is recruited into E1/E2 replication foci.
To further elucidate the role of BRD4S in HPV replication, colocalization studies using indirect immunofluorescence were conducted (Fig. 4). C33A cells were transfected with vectors encoding epitope-tagged HPV31 E1, E2, BRD4, or BRD4S. HPV31 E1 and E2 alone were diffusely distributed in the nucleus, whereas BRD4 and BRD4S were located in nuclear foci (Fig. 4A) consistent with other studies (22, 23). Consistent with previous reports, coexpression of E1 and E2 proteins lead to formation of foci, which increase in size when a plasmid harboring the HPV31 upstream regulatory region (URR), which includes the viral origin of replication, is present (Fig. 4B) (23, 24). Coexpression of E2 and BRD4 led to a more structured staining pattern of E2 and to colocalization (Pearson correlation coefficient [PCC] = 0.68) (Fig. 4C and E). Coexpression of E1, E2, and BRD4 resulted in foci in which a similar degree of colocalization of E2 and BRD4 was observed (PCC = 0.69) (Fig. 4C and E). The addition of the URR reduced colocalization significantly in the absence of E1 (PCC = 0.46) but not significantly when E1 was present (PCC = 0.54) (Fig. 4C and E). Coexpression of BRD4S and E2 revealed a more diffuse expression pattern for E2 with partial BRD4S colocalization (PCC = 0.47) (Fig. 4D and E). The addition of E1 alone (PCC = 0.69), the URR alone (PCC = 0.86), or E1 and URR together (PCC = 0.72) significantly enhanced colocalization of BRD4S and E2 (Fig. 4D and E). Taken together, these data strongly indicate that not only BRD4 but also BRD4S is recruited into E1/E2 replication foci, supporting the idea that BRD4S regulates the HPV life cycle.
FIG 4.
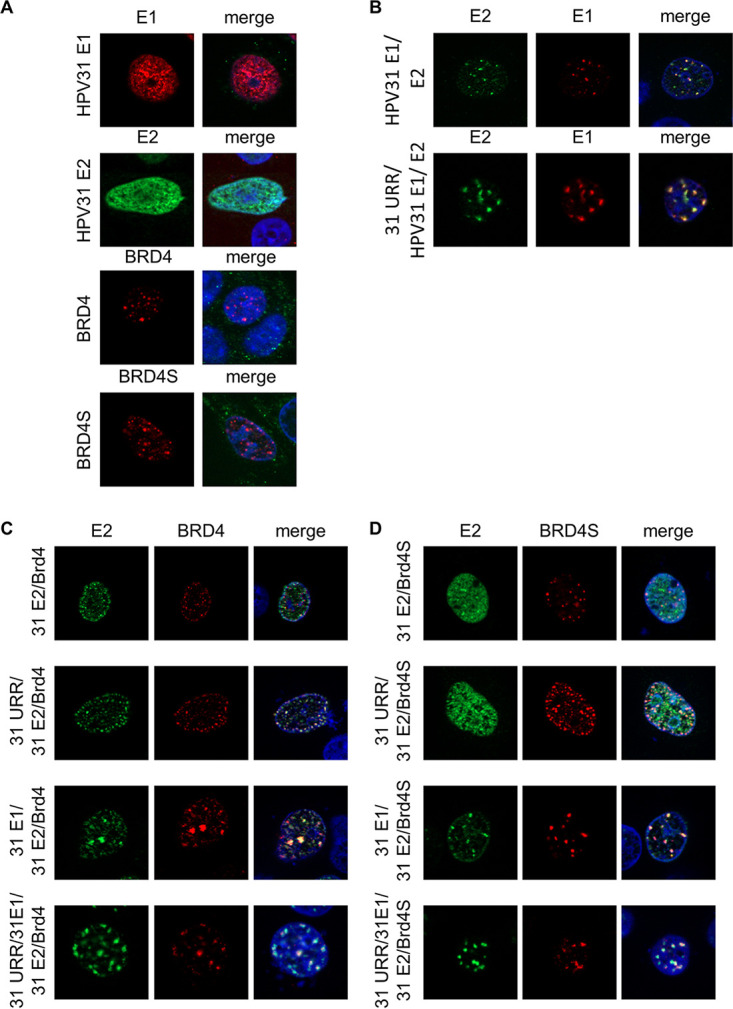
BRD4S colocalizes with E1/E2 replication foci. (A) C33A cells were transfected with expression vectors for HPV31 E1 (500 ng; pCMV neo 31 E1 – 3×Flag), HPV31 E2 (50 ng; pSX31 E2–HA), BRD4 (700 ng; pcDNA3-flag BRD4), or BRD4S (500 ng; pcDNA3-flag BRD4S) and analyzed by immunofluorescence with anti-HA or anti-Flag antibodies. DAPI was used to stain DNA. (B) C33A cells were transfected with expression vectors for HPV31 E1 (flag) and E2 (HA) and with or without 500 ng pGL31URR-Luc (31 URR) and stained with anti-HA, anti-Flag, and DAPI. (C, D) C33A cells were transfected with combinations of expression vectors for HPV31 E1 (untagged), HPV31 E2 (HA), BRD4 (flag), BRD4S (flag), or URR 31 (pGL31 URR) and stained with anti-HA, anti-Flag, and DAPI. (E) Quantification of the levels of colocalization between E2 and BRD4 or BRD4S. Data are expressed as the Pearson correlation coefficient between E2 and BRD4(S) signals. Statistical significance was determined by one-way ANOVA with Dunnett’s multiple-comparison test (ns, not significant; ***, P < 0.001; ****, P < 0.0001).
BRD4S modulates HPV31 E2 activities.
To address the functional role of BRD4S, cotransfection assays were carried out with an E2-dependent reporter plasmid. Cotransfection of increasing amounts of BRD4S with E2 strongly reduced activation of the reporter plasmid by E2 in a concentration-dependent manner (Fig. 5A). This surprisingly indicated that BRD4S limits transcriptional activation by E2. Immunoblots revealed that the expression of BRD4S increased E2 protein levels, demonstrating that the loss of transactivation by E2 in the presence of BRD4S is specific and not due to reduced E2 protein levels (Fig. 5B). Cotransfection of BRD4S also decreased the E1/E2-dependent activation of the pGL31 URR-luc plasmid in an E1/E2-dependent replication reporter assay (25) (Fig. 5c), indicating that BRD4S also inhibits replicating templates. To further validate these findings, endogenous BRD4S was depleted by small interfering BRD4S (siBRD4S), which targets the 3′ untranslated region (3′-UTR) in the exon specific for BRD4S (see Fig. 8A), and consistent with this, only BRD4S but not BRD4 RNA and protein levels were greatly diminished (Fig. 6A and B). siBRD4S increased activation of the reporter plasmid by E2 from 26-fold in the presence of a control siRNA to 78-fold, providing evidence that endogenous BRD4S inhibits E2 (Fig. 6C). Cotransfection of siBRD4S and the BRD4S expression plasmid, which does not harbor the siRNA target sequence in the 3′-UTR of BRD4S, resulted in a reduced activation by E2 (Fig. 6C), confirming that the phenotype is due to the specific knockdown of BRD4S.
FIG 5.
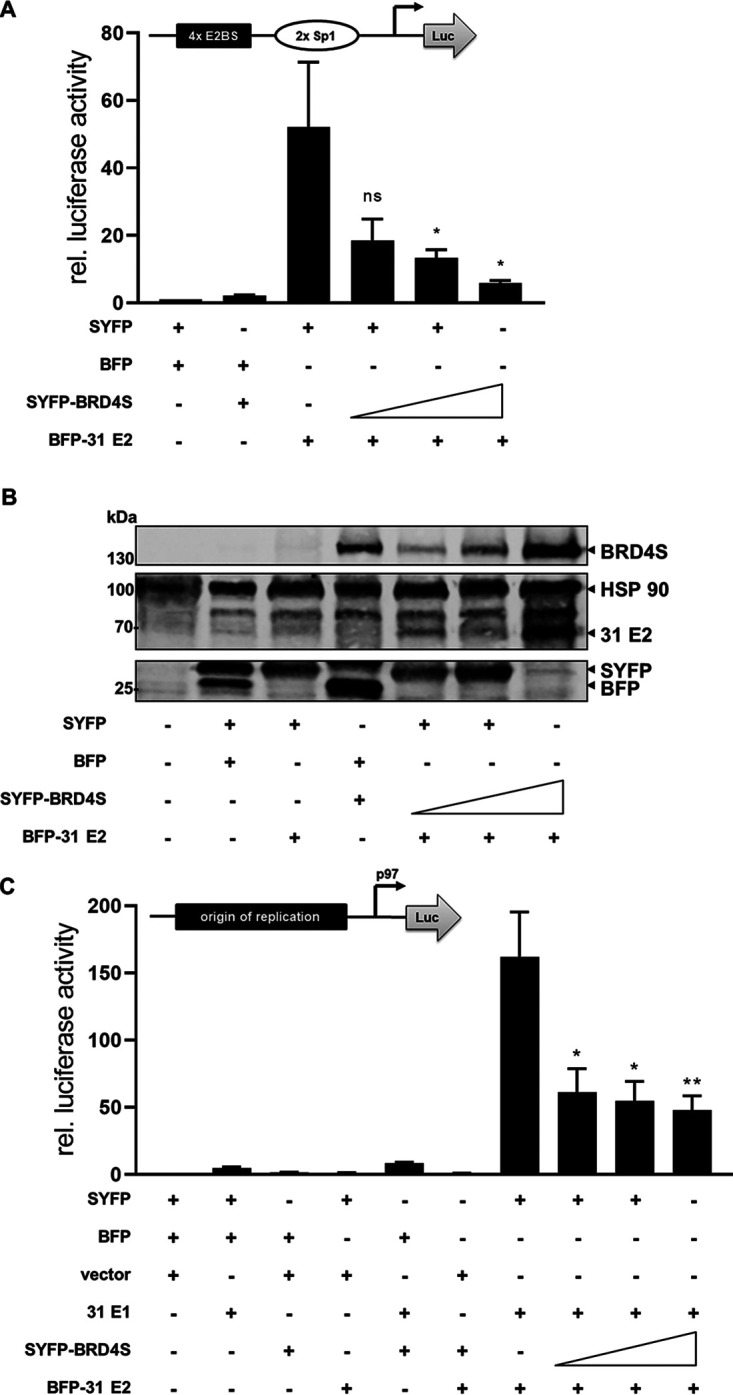
BRD4S inhibits E2 transactivation. (A) C33A cells were transfected with 0.5 ng pCMV-Gluc, 50 ng pC18-Sp1-Luc (structure shown above the graph), 10 ng of BFP-31 E2, and 25, 50, or 100 ng of SYFP-BRD4S or empty SYFP and BFP vectors. Values were calculated as the ratio of firefly to Gaussia luciferase and are shown relative to the activity of the reporter in the presence of BFP and SYFP expression vectors. Mean values are derived from at least three independent experiments, and error bars indicate the SEM. Statistical significance was determined by one-way ANOVA and Dunnett’s multiple-comparison test (compared to activity of reporter with HPV31 E2); ns, P > 0.05; *, P < 0.05. (B) Immunoblot of untransfected C33A cells or which were transfected with 100 ng of BFP-31 E2 and 250, 500, or 1,000 ng of SYFP-BRD4S or/and empty SYFP and BFP vectors using antibodies specific for SYFP to detect SYFP and BRD4S expression or BFP to detect BFP and HPV31 E2. HSP90 was used as a loading control. (C) C33A cells were transfected with 0.5 ng pCMV-Gluc, 50 ng pGL31 URR-luc (structure shown above the graph), 100 ng HPV31 E1, 10 ng of BFP-31 E2, and 25, 50, or 100 ng of SYFP-BRD4S or empty pSG5, SYFP, or BFP vectors. Values were calculated as the ratio of firefly to Gaussia luciferase and are shown relative to the activity of the reporter in the presence of pSG5-, BFP-, and SYFP expression vectors. Mean values are derived from at least three independent experiments, and error bars indicate the SEM. Statistical significance was determined by one-way ANOVA and Dunnett’s multiple-comparison test (compared to activity of reporter with HPV31 E2 and HPV31 E1); *, P < 0.05; **, P < 0.01.
FIG 6.
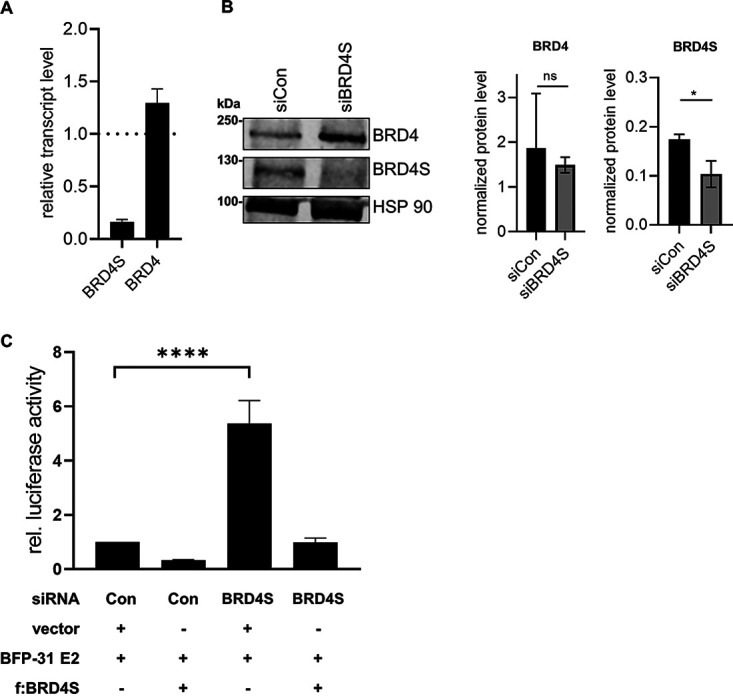
siRNA-mediated knockdown of BRD4S enhances E2-dependent reporter activity. (A) C33A cells were transfected with siRNA (90 pmol) against BRD4S, and 72 h after transfection, total RNA was analyzed for BRD4 and BRD4S expression using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a reference gene. Results are presented relative to siRNA control (=1) and represent the mean of three independent experiments. Error bars indicate the SEM. (B) Expression of BRD4S in C33A cells transfected with control siRNA (siCon) or siBRD4S was analyzed by immunoblot 72 h after transfection using α-BRD4 (N-ter) for BRD4S detection and α-BRD4 (C-ter) for BRD4 detection. HSP90 was used as reference. The graphs represent the quantification of signals for BRD4, BRD4S, and HSP90 from three independent experiments, and the error bars indicate the SEM. Statistical significance was determined by paired t test (ns, not significant; *, P < 0.05). (C) C33A cells were transfected with control siRNA (Con) or siBRD4S (15 pmol). One day later, cells were transfected with 0.5 ng pCMV-Gluc, 50 ng of pC18-Sp1-Luc, 10 ng of BFP-31 E2, and either 100 ng of Flag-tagged BRD4S or 100 ng of empty vector (pcDNA3.1). Mean values represent ratios of firefly luciferase to Gaussia luciferase activity relative to the E2-induced reporter activity from at least three independent experiments, and error bars indicate the SEM. Statistical significance was determined by one-way ANOVA and Dunnett’s multiple-comparison test; ****, P < 0.0001.
Knockdown of BRD4S induces HPV late gene transcription in undifferentiated CIN612-9E cells.
To further validate this, CIN612-9E cells, which maintain replicating HPV31 genomes, were transfected with siBRD4S. Comparable to C33A cells, siBRD4S greatly reduced BRD4S transcript and protein levels but did not reduce BRD4 transcript and only weakly reduced BRD4 protein levels (Fig. 7A). The amounts of different spliced viral RNAs were analyzed by quantitative PCR (qPCR) and revealed a 2-fold increase of E6* and E1^E4 transcripts and a significant 33-fold increase in E4^L1 transcripts, whereas E8^E2 transcripts were unchanged (Fig. 7B). Interestingly, siBRD4S did not reduce viral copy numbers, suggesting that BRD4S acts mainly at the level of transcription (Fig. 7C). CIN612-9E cells induced to differentiate by suspension in methylcellulose expressed higher levels of E1^E4 and E4^L1 transcripts than undifferentiated cells, consistent with previous reports (26, 27). Both BRD4 and BRD4S protein levels were reduced by differentiation, and siBRD4S further reduced BRD4S levels (Fig. 7A). siBRD4S did not further increase viral late gene expression compared to siControl in differentiated cells, which could be due to the differentiation-dependent decrease of BRD4S (Fig. 7D). This indicates that BRD4S mainly limits viral late transcription in undifferentiated cells. To validate these results by an alternative method, we used CRISPR/Cas9 technology to knock down BRD4S using published genomic RNAs (gRNAs) targeting the splice acceptor of the exon unique for BRD4S (Fig. 8A) (15). It turned out that a knockdown of BRD4S was only evident in CIN612-9E cells briefly after drug selection but could not be stably maintained, indicating a counter selection against the loss of BRD4S (Fig. 8B and C). Therefore, experiments were carried out immediately after drug selection using four independently generated knockdown cell lines. This revealed a 2.3-fold induction of E4^L1 transcript levels with gRNA A and a significant 2.6-fold induction with gRNA B (Fig. 8D). This correlated with the ability of gRNA B to reduce BRD4S levels more efficiently than gRNA A (Fig. 8B and C). Taken together, these data strongly suggest that BRD4S restricts mainly E4^L1 transcripts in undifferentiated CIN612-9E cells.
FIG 7.

siRNA knockdown of BRD4S increases HPV31 late gene expression. (A) CIN612-9E cells were transfected with 90 pmol of control siRNA or siBRD4S, and 72 h after transfection, whole-cell lysates from undifferentiated or differentiated cells were analyzed by immunoblot analysis using α-BRD4 (N-ter) for BRD4S detection and α-BRD4 (C-ter) for BRD4 detection. HSP90 was used as reference. (B) Total RNA of CIN612-9E cells transfected with 90 pmol of control siRNA or siBRD4S was analyzed for BRD4, BRD4S, and the HPV31 transcripts E4^L1, E1^E4, E8^E2, and E6*. As reference, gene GAPDH was used. Mean values are derived from three independent experiments and are presented relative to siRNA control (=1). (C) CIN612-9E cells transfected with 90 pmol of control siRNA or siBRD4S were differentiated the next day in 1.5% methylcellulose solution for 48 h. Total RNA was analyzed for BRD4, BRD4S, and the HPV31 transcripts E4^L1, E1^E4, E8^E2, and E6*. As reference, gene GAPDH was used. Mean values are derived from three independent experiments and are presented relative to undifferentiated siRNA control (siCon). Error bars indicate the SEM. Statistical significance was determined with a two-way ANOVA and Sidak’s multiple-comparison test; ****, P < 0.0001.
FIG 8.

CRISPR/Cas9-mediated knockdown of BRD4S increases expression of the HPV31 E4^L1 transcript in undifferentiated cells. (A) Schematic depiction of BRD4S genomic structure (intron and exon 12) with gRNA sequences gBRD4SA and gBRD4SB, which disrupt the splice acceptor site (black). Additionally, the target site of siBRD4S is shown. (B) Immunoblot analysis of CIN612-9E whole-cell extracts, stably infected with pLentiCrispr v2 (empty vector), pLentiCrispr BRD4S-ex12-SA_A (A), or pLentiCrispr BRD4S-ex12-SA_B (B). p1 shows BRD4S expression in cells harvested directly after puromycin selection, and p2 shows the same cells after they were passaged once. To detect BRD4 and BRD4S, an α-BRD4 (N-terminal) antibody was used. HSP90 was used as reference. (C) Relative BRD4S expression from CIN612-9E cells infected with lentiviral pLentiCrispr v2 vector system analyzed by qPCR. p1 summarizes four independent experiments. p2 represents the mean of two independent experiments. GAPDH was used as a reference gene. Results are presented relative to the empty pLentiCrispr v2 vector (=1). (D) Total RNA of the four independent experiments from (p1) were analyzed for the expression of BRD4, BRD4S, and the HPV31 transcripts E4^L1, E1^E4, E8^E2, and E6* using GAPDH as a reference gene. Results are presented relative to the empty pLentiCrispr v2 vector (=1). Error bars indicate the SEM. Statistical significance was determined with a two-way ANOVA and Dunnett’s multiple-comparison test; *, P < 0.05.
DISCUSSION
The interaction between PV E2 proteins and BRD4 is highly conserved and contributes to the transcription modulation, replication, and segregation activities of E2 in a virus-type-dependent manner (7, 8). A previous study reported that HR-HPV E2 interacts in addition to the CTM domain also with BID and PDID in BRD4 (17). The BRD4 gene locus generates, by alternative splicing, at least two different BRD4 proteins, BRD4 and BRD4S. As BID and PDID are also part of BRD4S, we investigated whether E2 binds to BRD4S and addressed its functional consequences. Using an in vivo protein-protein interaction assay based on the generation of FRET signals, we confirmed that PDID interacts with HPV31 and HPV16 E2 in vivo. Using domain mapping, we observed that deletion of NPS or aa 505 to 530 from PDID reduced the interaction with E2 in vivo. BD1, BD2, and aa 505 to 530 have been implicated in the dimerization of BRD4 and BRD2 proteins (20, 21). In our hands, only the deletion of BD2, but not of NPS or aa 505 to 530, significantly reduced the FRET signal of PDID with itself. This suggests that BRD4 proteins can dimerize in vivo, but this does not significantly contribute to the interaction with E2. Our data indicate that the interaction between PDID and E2 is mediated mainly by the E2 TAD-hinge domains in vivo and not as reported previously by the E2 DBD when using purified proteins (17). The hinge on its own did not give rise to a FRET signal, making it likely that the TAD domain interacts with PDID. However, attempts to test a BFP-E2 TAD construct failed due to a very low level of expression (our unpublished observation). The study by Wu et al. (17) revealed that phosphorylation of NPS modulates binding to E2. It is also known that E2 proteins can be post-transcriptionally modified which influences their activities (28–33). Thus, the discrepancies concerning the domains in E2 and BRD4 involved in binding between our data and the study by Wu et al. (17) may be due to different posttranslational modification of both proteins in the different experimental systems. Nevertheless, the data are consistent with an interaction between BRD4S and E2 in vivo and in vitro.
We now provide evidence that not only BRD4 but also BRD4S is present in detectable amounts in normal human keratinocytes (NHK), C33A, and CIN612-9E cells, which have been used for many years to explore the life cycle and replication functions of HR-HPV. Our findings indicate that the interaction between E2 and BRD4 is more complex than anticipated, as E2 does not only bind to BRD4 but also to BRD4S. We also observe that BRD4S is recruited into E1/E2 replication foci, underpinning the idea that BRD4S acts directly on the viral genome. BRD4 is required for the transcriptional activation of promoters and the repression of HR-HPV E6/E7 promoters by E2 as well as the E1/E2-dependent replication of HPV16 (7, 8). Our data show that BRD4S attenuates the transcriptional activation by E2 from nonreplicating and replicating reporter plasmids. These data suggest that BRD4S, in contrast to BRD4, is mainly an inhibitor of E2. Thus, it is likely that the ratio between BRD4 and BRD4S is crucial for HR-HPV E2 activities. How does BRD4S inhibit E2 activities? One possibility is that BRD4S is a weaker coactivator of transcription than BRD4 due to the lack of a CTM domain. In line with this, E2 still activated the reporter plasmid even at the highest amounts of coexpressed BRD4S (Fig. 5). Conversely, it has been reported that BRD4S targets repressive chromatin remodeling complexes to the HIV promoter (14). Thus, it is possible that BRD4S also recruits such complexes to E2, which could then limit transcription from E2-dependent promoters. In line with BRD4S being both an activator and an inhibitor of transcription, the specific knockdown of BRD4S by RNA interference (RNAi) in MDA-MB-231 cells upregulated 1,308 genes and downregulated 710 genes (34).
Knockdown of BRD4S revealed an increase of both spliced E6* and E1^E4 transcripts in undifferentiated CIN612-9E cells, which maintain replicating HPV31 genomes, providing further evidence that BRD4S negatively regulates HR-HPV gene expression. Interestingly, spliced E4^L1 transcripts were greatly induced, suggesting that BRD4S mainly acts as an inhibitor of late gene expression in undifferentiated cells. E2 has already been implicated in activating L1 gene expression in differentiated cells via its interaction with the CTM of BRD4 (35, 36). This has been correlated with the transcriptional control of cellular splicing factors serine/arginine-rich splicing factor (SRSF) 1, 2, and 3 by E2 in a BRD4-CTM-dependent manner (36). While we have not observed an upregulation of SRSF 1, 2, or 3 upon BRD4S knockdown in undifferentiated CIN612-9E cells (our unpublished observation), it remains possible that BRD4S and E2 not only directly control transcription of viral genes but also of cellular genes involved in HPV late mRNA splicing. It is also possible that BRD4S directly regulates splicing of late HPV transcripts since BRD4 has been implicated in regulating alternative splicing via an interaction with the splicing machinery (37). BRD4S inhibits HIV-1 transcription by facilitating a repressive nucleosome structure, which promotes HIV-1 latency (14). This could also be applicable to the HPV life cycle, as the suppression of late transcripts indicates an immune escape mechanism, which could allow the establishment of latency in basal cells of the epithelium. Thus, it is possible that the balance of BRD4 and BRD4S determines a precondition of persistence, which is a necessary requirement for cancer development.
MATERIALS AND METHODS
Recombinant plasmids.
Expression vectors pSYFP2-C1, pSYFP2-N1, and pmTagBFP-C1 used to generate different BFP- and yellow fluorescent protein (YFP)-labeled fusion proteins and the BFP-SYFP fusion protein have been previously described (38). The pmTagBFP-C1-HPV31 E2 expression plasmid was generated by inserting codon-optimized HPV31 E2 into the Kpn2I and 3′-SmaI restriction sites of pmTagBFP-C1. The I73L mutation was introduced into pmTagBFP-C1-HPV-31 E2 by site-directed mutagenesis. To obtain BFP-16 E2, HPV16 E2 was inserted into Kpn2I and 3′-BamHI restriction sites of pmTagBFP-C1. The 31E2-TAD-hinge (HPV31 nucleotide [nt] 2693 to 3298), 31E2-hinge (HPV31 nt 3299 to 3571), and 31E2-hinge-DBD (HPV31 nt 3572 to 3811) were synthesized by Thermo Fisher, including an N-terminal simian virus 40 (SV40) nuclear localization signal, and cloned into a pmTagBFP-C1 vector restricted with EcoRI and BamHI. Plasmids pcDNA3.1 f:BRD4 and pcDNA3.1 BRD4dCTM [formerly known as pcDNA3.1 f:BRD4(1-1223); kindly provided by C. M. Chiang] have been previously described (39). PCR-amplified BRD4 and BRD4dCTM containing 5′-XhoI and 3′-BamHI restriction sites were cloned into the pSYFP2-N1 vector digested with the respective enzymes. To generate the pcDNA3.1 f:BRD4S and SYFP-BRD4S expression vectors, BRD4 was amplified from pcDNA3.1 f:BRD4, adding 5′-XhoI and 3′-BamHI sites, and the C-terminal residues unique for BRD4S and then inserted into XhoI/BamHI digested pSYFP2-C1 and a modified pcDNA3.1 vector. BRD4 PDID and CTM domains have been previously described (17, 40). For cloning into the XhoI/BamHI-restricted pSYFP2-C1 vector, PDID and CTM domains were synthesized by Thermo Fisher Scientific, including an N-terminal SV40 nuclear localization signal and respective restriction sites. SYFP-BRD4 PDIDd287-348, BRD4 PDIDdBD2, BRD4 PDIDd462-482, BRD4 PDIDdNPS, and BRD4 PDIDd505-530 were generated identically. gRNA sequences for the knockdown of BRD4S, BRD4S-ex12-SA_A (gBRD4SA), and BRD4S-ex12-SA_B (gBRD4SB) described previously (15) were cloned into pLentiCRISPR v2 using BsmBI restriction sites. Plasmids pMD2.G and psPAX2 coding for lentiviral packaging elements were obtained from Addgene. The luciferase reporter plasmid pC18-Sp1-Luc has been previously described (26, 41). For luciferase-based reporter assays, the control plasmid pCMV-GLuc, coding for luciferase gene from Gaussia princeps, was used (NEB). The expression plasmids for HPV31 E1 (pCMV neo HPV31 E1-3×Flag, pCMV HPV31 E1), E2 (pSX HPV31 E2-HA), and the HPV31 URR reporter plasmid (pGL HPV31 URR-luc) have been previously described (26, 42–45).
Cell culture.
C33A and HEK293T cells were maintained in Dulbecco’s modified Eagle medium (DMEM) (Life Technologies) supplemented with 10% fetal bovine serum (FBS). CIN-612 9E cells were maintained in E-medium supplemented with 5% FBS in the presence of mitomycin C-treated NIH 3T3 J2 cells (44, 46, 47). To induce differentiation, CIN612-9E cells were suspended in E-medium/1.5% (wt/vol) methylcellulose 24 h after siRNA transfection (26). Differentiated cells were harvested 48 h later.
Flow cytometry-based FRET analysis.
To analyze protein-protein interactions in vivo via flow cytometry-based FRET analysis, 1.5 × 105 C33A cells were seeded into the wells of a 12-well plate. The next day, BFP and SYFP expression plasmids were transfected using FuGENE HD (Promega) according to manufacturer’s instructions. The amounts of BFP and SYFP expression plasmids were optimized for each plasmid combination to achieve similar expression levels of both proteins as judged by flow cytometry analyses. Two days after transfection, cells were washed with cold phosphate-buffered saline (PBS) and harvested by using 0.05% trypsin-EDTA (Life Technologies). The cells were collected by centrifugation (20 s, 18,000 × g), and the cell pellet resuspended in 400 μl PBS/1% vol/vol FBS. Flow cytometry-FRET measurements were performed with the MACSQuant VYB (Miltenyi Biotec) using 405-nm, 488-nm, and 561-nm lasers. In order to ensure the specificity of FRET signals, untransfected C33A cells or C33A cells transfected with expression plasmids for SYFP or BFP alone, BFP and SYFP together, or for a BFP-SYFP fusion protein were used to apply the previously described gating strategy (Fig. 1) (18, 38). First, cells were selected that express both SYFP and BFP (Fig. 1, lane 1). Then, a gate was set to exclude cells that exert a false-positive signal in the FRET channel due to YFP only being excited at 405 nm (Fig. 1, lane 2). Finally, the FRET signal is plotted versus BFP, and the last gate is adjusted to the cells that are coexpressing BFP and SYFP (Fig. 1, lane 3; BFP+SYFP) (18, 38). The data were evaluated with the help of MACSQuant software (Miltenyi Biotec).
Coimmunoprecipitation assays.
For coimmunoprecipitation analysis, 3 × 106 C33A cells were seeded into a 100-mm plate and transfected with 10 μg DNA the next day. Cells were lysed 48 h later in lysis buffer 50 mM Tris-HCl, pH 8.0; 0.5% (vol/vol) IGEPAL 630; 150 mM NaCl; 1 mM dithiothreitol [DTT], 1× cOmplete EDTA-free protease inhibitor cocktail (Sigma); 1× PhosSTOP EASYpack phosphatase inhibitor cocktail (Sigma). For immunoprecipitation (IP), magnetic anti-FLAG beads (Miltenyi Biotec) were used. Purification of protein-bead complexes was performed with the help of μMACS columns and μMACS separator (Miltenyi Biotec). The beads were washed with IP washing buffer (50 mM Tris-HCl (pH 8.0); 0.5% IGEPAL 630; 0 to 400 mM NaCl). Bound proteins were eluted in elution buffer (Miltenyi Biotec) heated to 95°C. The proteins were analyzed by immunoblotting.
siRNA transfection for qPCR, immunoblot analyses, and luciferase-based reporter assays.
CIN612-9E (2 × 105) or C33A (1.5 × 105) cells were transfected with 90 pmol control siRNA (siAllStar; Qiagen) or siBRD4S (UCCCAGCAAGCUAUAGCUUAA; Dharmacon) using Lipofectamine RNAiMax reagent (Thermo Fisher) according to the manufacturer’s instructions. Analyses of transcript or protein levels were carried out 72 h after transfection. For luciferase-based reporter assays, 7 × 104 C33A cells were seeded into 24-well dishes and transfected first with siRNA (15 pmol) and 24 h later with the respective plasmid vectors using FuGENE HD (Promega) according to the manufacturer’s instructions. Luciferase assays were carried out 48 h after DNA transfection.
CRISPR-mediated knockdown of BRD4S.
To achieve knockdown of BRD4S, recombinant pLentiCRISPR v2 plasmid containing gRNAs against BRD4S (pLentiCRISPR BRD4S-ex12-SA_A [gBRD4SA] and pLentiCRISPR BRD4S-ex12-SA_B [gBRD4SB] [15]) were packaged together with pMD2.G (Addgene) and psPAX2 (Addgene) in HEK293T cells, which were seeded in 60 mm with 2.5 × 106 cells followed by DNA transfection using FuGENE HD (Promega). The next day, medium was changed and 48 h after transfection, CIN612-9E cells, cultivated in 60-mm dishes, were infected with the supernatant of HEK293T cells and 5 μg/ml Polybrene. The next day, infected CIN612-9E cells were transferred to 100-mm dishes, and approximately 6 h later, cells were selected with 1 μg/ml puromycin.
Quantitative PCR.
For quantitative analysis of gene expression levels, RNA was isolated from cells using the RNeasy minikit (Qiagen), and then 1 μg of isolated RNA was transcribed using the QuantiTect reverse transcription kit (Qiagen). qPCR assays were carried out in a LightCycler 480 (Roche) using SYBR green I master mix (Roche), 25 ng of cDNA, and 0.3 μM primer pairs (Table 1) for cellular and viral genes as previously described (48).
TABLE 1.
qPCR primers
| Primer name | Sequence (source) |
|---|---|
| BRD4 2184 F | 5′-CGACTTTGAGACCCTGAAGC-3′ (15) |
| BRD4 2380 R | 5′-TCGGAGCCATCTCTGTTTC-3′ (15) |
| BRD4S 2371 R | 5′-GGCAGGACCTGTTTCGGA-3′ |
| HPV31 804 F (E1^E4) | 5′-TGTTAATGGGCTCATTTGGAA-3′ (49) |
| HPV31 3373 R (E1^E4) | 5′-GGTTTTGGAATTCGATGTGG-3′ (49) |
| HPV31 1242 F (E8^E2) | 5′-ACTTCCAGACAGCGGGTATG-3′ (49) |
| HPV31 3461 R (E8^E2) | 5′-GGTGGGTGTTTCTGTGCTCT-3′ (49) |
| HPV31 3528 F (E4^L1) | 5′-CATGCACAAACCAAACAAGG-3′ (49) |
| HPV31 5651 R (E4^L1) | 5′-GCACTGCCTGCGTGATAATA-3′ (49) |
| HPV31 E6* F | 5′-AATTGTGTCTACTGCAAAGGTGTA-3′ (49) |
| HPV31 508 R - E6* | 5′-CCAACATGCTATGCAACGTC-3′ (49) |
| GAPDH F | 5′-TGCACCACCAACTGCTTAGC-3′ |
| GAPDH R | 5′-GGCATGGACTGTGGTCATGAG-3′ |
Immunoblot analysis.
To validate the expression of BFP or SYFP fusion proteins, 3 × 105 C33A cells were seeded in 6-well dishes and transfected with 0.5 to 1 μg DNA and FuGENE HD (Promega). Cells were harvested 48 h posttransfection and lysed in 35 μl 4× RotiLoad 1 (Carl Roth) by incubating the samples for 5 min at 95°C. Protein were separated in 6% to 12% SDS-PAGE and transferred to nitrocellulose membrane (Amersham Protran; GE Healthcare) at 90 V for 90 min in 10 mM N-cyclohexyl-3-aminopropanesulfonic acid (CAPS), 10% (vol/vol) methanol, pH 10.3. Membranes were blocked for 1 h in PBS/3% bovine serum albumin (BSA). The following primary antibodies were used: DYKDDDDK Tag rabbit polyclonal antibody (Cell Signaling; number 2368S; 1:1,000), HA-Tag mouse MAb (Cell Signaling; 6E2; number 2376S; 1:1,000), BRD4 rabbit MAb (N-terminal; Thermo Fisher Scientific; 14H4L4; number 702448; 1:350), BRD4 rabbit Ab (C-terminal; Cell Signaling; E2A7X; number 13440S; 1:1,000), anti-YFP JL-8 mouse MAb (TaKaRa; number 632381; 1:1,000), anti-RFP rabbit pAb (Evrogen; number 233; 1:1,000), HSP90 mouse Ab (Santa Cruz; sc-69703; 1:2,000). Bound antibodies were detected with IRDye 680RD goat anti-mouse IgG (number 926-68070), IRDye 680RD goat anti-rabbit IgG (number 926-68071), IRDye 800CW goat anti-mouse IgG (number 926-32210), or IRDye 800CW goat anti-rabbit IgG (number 926-32211) at a 1:15,000 dilution. Antibodies were diluted in PBS/0.1% Tween 20/3% BSA, and washing steps were performed with PBS/0.1% Tween 20. Fluorescent signals were recorded with a Li-Cor Odyssey Fc (Licor).
Luciferase-based reporter assays.
A total of 7 × 104 C33A cells per well were seeded in a 24-well plate. The next day, cells were transfected with firefly luciferase reporter, Gaussia luciferase reporter, and expression plasmids using FuGENE HD (Promega) as indicated in the figure legends. Luciferase-based assays were carried out 48 h after DNA transfection (43).
Immunofluorescence analysis.
C33A cells (1 × 105) were seeded on a glass cover slip in a 6-well plate. The next day, cells were transfected with combinations of the pGL 31 URR-luc reporter and expression vectors for HPV 31 E1, E2, BRD4, or BRD4S expression plasmids. Two days after transfection, cells were washed twice with PBS, incubated with methanol-acetone (1:1) for 2 min at room temperature (RT), and then washed again with PBS. Cells were then incubated with the following primary antibodies diluted in PBS/3% BSA for 1 h at RT in a humidified chamber: HA-Tag rabbit MAb (Cell Signaling; C29F4; number 3724S; 1:16,000); DYKDDDDK Tag mouse MAb (Cell Signaling; 9A3; number 8146S; 1:1,000). Primary antibodies were detected with rabbit Alexa Fluor 488 (Thermo Fisher; A-11034), and mouse Alexa Fluor 555 (Thermo Fisher A-31570) diluted 1:2,000 in PBS/3% BSA for 1 h at RT. Cover slips were mounted and counterstained with RotiMount FluorCare 4′,6-diamidino-2-phenylindole (DAPI) (Carl Roth). Stained cells were analyzed with a Zeiss Axio Observer microscope and the appropriate filter sets in combination with a Zeiss Apotome as described previously (49). Colocalization of E2 with BRD4 or BRD4S was analyzed in images of cell nuclei that were randomly taken, and then the Pearson correlation coefficient was determined using ZEN 2 pro software (Zeiss).
ACKNOWLEDGMENT
This work was supported by a grant from the Deutsche Forschungsgemeinschaft to T.I. (IF 1/8-1).
REFERENCES
- 1.de Martel C, Plummer M, Vignat J, Franceschi S. 2017. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int J Cancer 141:664–670. 10.1002/ijc.30716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grassmann K, Rapp B, Maschek H, Petry KU, Iftner T. 1996. Identification of a differentiation-inducible promoter in the E7 open reading frame of human papillomavirus type 16 (HPV-16) in raft cultures of a new cell line containing high copy numbers of episomal HPV-16 DNA. J Virol 70:2339–2349. 10.1128/JVI.70.4.2339-2349.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kadaja M, Silla T, Ustav E, Ustav M. 2009. Papillomavirus DNA replication—from initiation to genomic instability. Virology 384:360–368. 10.1016/j.virol.2008.11.032. [DOI] [PubMed] [Google Scholar]
- 4.Stubenrauch F, Laimins LA. 1999. Human papillomavirus life cycle: active and latent phases. Semin Cancer Biol 9:379–386. 10.1006/scbi.1999.0141. [DOI] [PubMed] [Google Scholar]
- 5.Bergvall M, Melendy T, Archambault J. 2013. The E1 proteins. Virology 445:35–56. 10.1016/j.virol.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McBride AA. 2013. The papillomavirus E2 proteins. Virology 445:57–79. 10.1016/j.virol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iftner T, Haedicke-Jarboui J, Wu SY, Chiang CM. 2017. Involvement of Brd4 in different steps of the papillomavirus life cycle. Virus Res 231:76–82. 10.1016/j.virusres.2016.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McBride AA, Jang MK. 2013. Current understanding of the role of the Brd4 protein in the papillomavirus lifecycle. Viruses 5:1374–1394. 10.3390/v5061374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Y, Vakoc CR. 2017. Targeting cancer cells with BET bromodomain inhibitors. Cold Spring Harb Perspect Med 7:a026674. 10.1101/cshperspect.a026674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X, Baek G, Ramanand SG, Sharp A, Gao Y, Yuan W, Welti J, Rodrigues DN, Dolling D, Figueiredo I, Sumanasuriya S, Crespo M, Aslam A, Li R, Yin Y, Mukherjee B, Kanchwala M, Hughes AM, Halsey WS, Chiang CM, Xing C, Raj GV, Burma S, de Bono J, Mani RS. 2018. BRD4 promotes DNA repair and mediates the formation of TMPRSS2-ERG gene rearrangements in prostate cancer. Cell Rep 22:796–808. 10.1016/j.celrep.2017.12.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stanlie A, Yousif AS, Akiyama H, Honjo T, Begum NA. 2014. Chromatin reader Brd4 functions in Ig class switching as a repair complex adaptor of nonhomologous end-joining. Mol Cell 55:97–110. 10.1016/j.molcel.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 12.Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, Bryson BD, Rameseder J, Lee MJ, Blake EJ, Fydrych A, Ho R, Greenberger BA, Chen GC, Maffa A, Del Rosario AM, Root DE, Carpenter AE, Hahn WC, Sabatini DM, Chen CC, White FM, Bradner JE, Yaffe MB. 2013. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature 498:246–250. 10.1038/nature12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang J, Dulak AM, Hattersley MM, Willis BS, Nikkilä J, Wang A, Lau A, Reimer C, Zinda M, Fawell SE, Mills GB, Chen H. 2018. BRD4 facilitates replication stress-induced DNA damage response. Oncogene 37:3763–3777. 10.1038/s41388-018-0194-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conrad RJ, Fozouni P, Thomas S, Sy H, Zhang Q, Zhou MM, Ott M. 2017. The short isoform of BRD4 promotes HIV-1 latency by engaging repressive SWI/SNF chromatin-remodeling complexes. Mol Cell 67:1001–1012. 10.1016/j.molcel.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tzelepis K, De Braekeleer E, Aspris D, Barbieri I, Vijayabaskar MS, Liu WH, Gozdecka M, Metzakopian E, Toop HD, Dudek M, Robson SC, Hermida-Prado F, Yang YH, Babaei-Jadidi R, Garyfallos DA, Ponstingl H, Dias JML, Gallipoli P, Seiler M, Buonamici S, Vick B, Bannister AJ, Rad R, Prinjha RK, Marioni JC, Huntly B, Batson J, Morris JC, Pina C, Bradley A, Jeremias I, Bates DO, Yusa K, Kouzarides T, Vassiliou GS. 2018. SRPK1 maintains acute myeloid leukemia through effects on isoform usage of epigenetic regulators including BRD4. Nat Commun 9:5378. 10.1038/s41467-018-07620-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.You J. 2010. Papillomavirus interaction with cellular chromatin. Biochim Biophys Acta 1799:192–199. 10.1016/j.bbagrm.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu SY, Nin DS, Lee AY, Simanski S, Kodadek T, Chiang CM. 2016. BRD4 phosphorylation regulates HPV E2-mediated viral transcription, origin replication, and cellular MMP-9 expression. Cell Rep 16:1733–1748. 10.1016/j.celrep.2016.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banning C, Votteler J, Hoffmann D, Koppensteiner H, Warmer M, Reimer R, Kirchhoff F, Schubert U, Hauber J, Schindler M. 2010. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PLoS One 5:e9344. 10.1371/journal.pone.0009344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Senechal H, Poirier GG, Coulombe B, Laimins LA, Archambault J. 2007. Amino acid substitutions that specifically impair the transcriptional activity of papillomavirus E2 affect binding to the long isoform of Brd4. Virology 358:10–17. 10.1016/j.virol.2006.08.035. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Gutierrez P, Mundi M, Garcia-Dominguez M. 2012. Association of bromodomain BET proteins with chromatin requires dimerization through the conserved motif B. J Cell Sci 125:3671–3680. 10.1242/jcs.105841. [DOI] [PubMed] [Google Scholar]
- 21.Wang R, Li Q, Helfer CM, Jiao J, You J. 2012. Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J Biol Chem 287:10738–10752. 10.1074/jbc.M111.323493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Han X, Yu D, Gu R, Jia Y, Wang Q, Jaganathan A, Yang X, Yu M, Babault N, Zhao C, Yi H, Zhang Q, Zhou MM, Zeng L. 2020. Roles of the BRD4 short isoform in phase separation and active gene transcription. Nat Struct Mol Biol 27:333–341. 10.1038/s41594-020-0394-8. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Helfer C, Pancholi N, Bradner J, You J. 2013. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J Virol 87:3871–3884. 10.1128/JVI.03068-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sakakibara N, Chen D, Jang MK, Kang DW, Luecke HF, Wu S-Y, Chiang C-M, McBride AA. 2013. Brd4 is displaced from HPV replication factories as they expand and amplify viral DNA. PLoS Pathog 9:e1003777. 10.1371/journal.ppat.1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dreer M, Fertey J, van de Poel S, Straub E, Madlung J, Macek B, Iftner T, Stubenrauch F. 2016. Interaction of NCOR/SMRT repressor complexes with papillomavirus E8^E2C proteins inhibits viral replication. PLoS Pathog 12:e1005556. 10.1371/journal.ppat.1005556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruesch MN, Stubenrauch F, Laimins LA. 1998. Activation of papillomavirus late gene transcription and genome amplification upon differentiation in semisolid medium is coincident with expression of involucrin and transglutaminase but not keratin-10. J Virol 72:5016–5024. 10.1128/JVI.72.6.5016-5024.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson R, Laimins LA. 2006. Differentiation of HPV-containing cells using organotypic “raft” culture or methylcellulose, p 157–169. In Davy C, Doorbar J (ed), Human papillomaviruses: methods and protocols. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 28.Culleton SP, Kanginakudru S, DeSmet M, Gilson T, Xie F, Wu SY, Chiang CM, Qi G, Wang M, Androphy EJ. 2017. Phosphorylation of the bovine papillomavirus E2 protein on tyrosine regulates its transcription and replication functions. J Virol 91:e01854-16. 10.1128/JVI.01854-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DeSmet M, Jose L, Isaq N, Androphy EJ. 2019. Phosphorylation of a conserved tyrosine in the papillomavirus E2 protein regulates Brd4 binding and viral replication. J Virol 93:e01801-18. 10.1128/JVI.01801-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McBride AA, Howley PM. 1991. Bovine papillomavirus with a mutation in the E2 serine 301 phosphorylation site replicates at a high copy number. J Virol 65:6528–6534. 10.1128/JVI.65.12.6528-6534.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sekhar V, McBride AA. 2012. Phosphorylation regulates binding of the human papillomavirus type 8 E2 protein to host chromosomes. J Virol 86:10047–10058. 10.1128/JVI.01140-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas Y, Androphy EJ. 2018. Human papillomavirus replication regulation by acetylation of a conserved lysine in the E2 protein. J Virol 92:e01912-17. 10.1128/JVI.01912-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu YC, Roark AA, Bian XL, Wilson VG. 2008. Modification of papillomavirus E2 proteins by the small ubiquitin-like modifier family members (SUMOs). Virology 378:329–338. 10.1016/j.virol.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu SY, Lee CF, Lai HT, Yu CT, Lee JE, Zuo H, Tsai SY, Tsai MJ, Ge K, Wan Y, Chiang CM. 2020. Opposing functions of BRD4 isoforms in breast cancer. Mol Cell 78:1114–1132. 10.1016/j.molcel.2020.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stubenrauch F, Colbert AM, Laimins LA. 1998. Transactivation by the E2 protein of oncogenic human papillomavirus type 31 is not essential for early and late viral functions. J Virol 72:8115–8123. 10.1128/JVI.72.10.8115-8123.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klymenko T, Hernandez-Lopez H, MacDonald AI, Bodily JM, Graham SV. 2016. Human papillomavirus E2 regulates SRSF3 (SRp20) to promote capsid protein expression in infected differentiated keratinocytes. J Virol 90:5047–5058. 10.1128/JVI.03073-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uppal S, Gegonne A, Chen Q, Thompson PS, Cheng D, Mu J, Meerzaman D, Misra HS, Singer DS. 2019. The bromodomain protein 4 contributes to the regulation of alternative splicing. Cell Rep 29:2450–2460. 10.1016/j.celrep.2019.10.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider M, Yigitliler A, Stubenrauch F, Iftner T. 2018. Cottontail rabbit papillomavirus E1 and E2 proteins mutually influence their subcellular localizations. J Virol 92:e00704-18. 10.1128/JVI.00704-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee AY, Chiang CM. 2009. Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J Biol Chem 284:2778–2786. 10.1074/jbc.M805835200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. 2013. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell 49:843–857. 10.1016/j.molcel.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zobel T, Iftner T, Stubenrauch F. 2003. The papillomavirus E8^E2C protein represses DNA replication from extrachromosomal origins. Mol Cell Biol 23:8352–8362. 10.1128/mcb.23.22.8352-8362.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stubenrauch F, Straub E, Fertey J, Iftner T. 2007. The E8 repression domain can replace the E2 transactivation domain for growth inhibition of HeLa cells by papillomavirus E2 proteins. Int J Cancer 121:2284–2292. 10.1002/ijc.22907. [DOI] [PubMed] [Google Scholar]
- 43.Stubenrauch F, Zobel T, Iftner T. 2001. The E8 domain confers a novel long-distance transcriptional repression activity on the E8^E2C protein of high-risk human papillomavirus type 31. J Virol 75:4139–4149. 10.1128/JVI.75.9.4139-4149.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stubenrauch F, Hummel M, Iftner T, Laimins LA. 2000. The E8^E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J Virol 74:1178–1186. 10.1128/jvi.74.3.1178-1186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fradet-Turcotte A, Moody C, Laimins LA, Archambault J. 2010. Nuclear export of human papillomavirus type 31 E1 is regulated by Cdk2 phosphorylation and required for viral genome maintenance. J Virol 84:11747–11760. 10.1128/JVI.01445-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Frattini MG, Lim HB, Laimins LA. 1996. In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differentiation-dependent late expression. Proc Natl Acad Sci U S A 93:3062–3067. 10.1073/pnas.93.7.3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meyers C, Frattini MG, Hudson JB, Laimins LA. 1992. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 257:971–973. 10.1126/science.1323879. [DOI] [PubMed] [Google Scholar]
- 48.Straub E, Dreer M, Fertey J, Iftner T, Stubenrauch F. 2014. The viral E8^E2C repressor limits productive replication of human papillomavirus 16. J Virol 88:937–947. 10.1128/JVI.02296-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.van de Poel S, Dreer M, Velic A, Macek B, Baskaran P, Iftner T, Stubenrauch F. 2018. Identification and functional characterization of phosphorylation sites of the human papillomavirus 31 E8^E2 protein. J Virol 92:e01743-17. 10.1128/JVI.01743-17. [DOI] [PMC free article] [PubMed] [Google Scholar]