

Summary
Tau aggregates contribute to neurodegenerative diseases including frontotemporal dementia and Alzheimer’s disease (AD). Although RNA promotes tau aggregation in vitro, whether tau aggregates in cells contain RNA is unknown. We demonstrate in cell culture and mouse brains that both cytosolic and nuclear tau aggregates contain RNA, with enrichment for snRNAs and snoRNAs. Nuclear tau aggregates colocalize with and alter the composition, dynamics, and organization of nuclear speckles, which are membraneless organelles involved in pre-mRNA splicing. Moreover, several nuclear speckle components, including SRRM2, mislocalize to cytosolic tau aggregates in cells, mouse brains, and patient brains with AD, frontotemporal dementia (FTD), and corticobasal degeneration (CBD). Consistent with these alterations we observe the presence of tau aggregates is sufficient to alter pre-mRNA splicing. This work identifies tau alteration of nuclear speckles as a feature of tau aggregation that may contribute to the pathology of tau aggregates.
Graphical Abstract
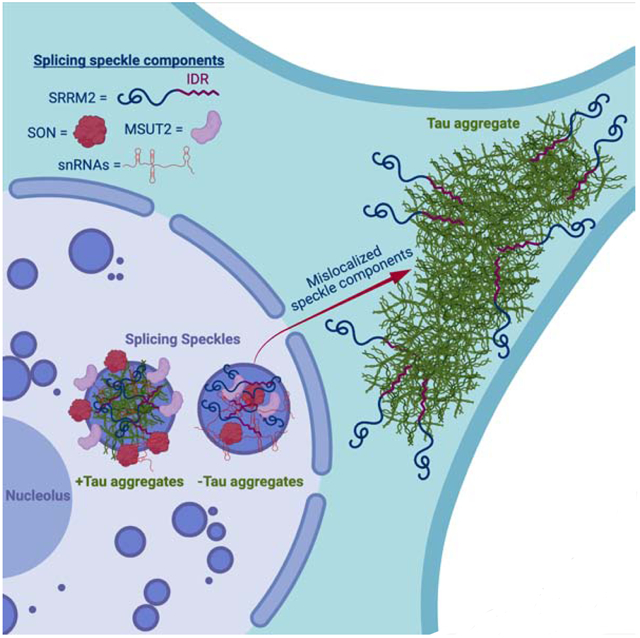
ETOC blurb
Insoluble tau aggregates are present in multiple neurodegenerative diseases, including Alzheimer’s disease. In this article, Lester et al. show that tau aggregates are enriched for snRNAs and snoRNA, alter splicing speckles, and mislocalize nuclear splicing proteins. This could help explain RNA processing defects seen in patients with tau pathology.
Introduction
Fibrillar aggregates of the microtubule associated protein tau (tau) are seen in numerous neurodegenerative diseases collectively referred to as tauopathies (Orr et al., 2017). Tauopathies have a variety of etiologies ranging from mutations in tau that promote its aggregation, such as in the inherited frontotemporal dementia with parkinsonism-17 (FTDP-17), to environmental triggers such as head trauma giving rise to chronic traumatic encephalopathy (CTE), to the incompletely understood link between beta-amyloid and tauopathy in Alzheimer’s disease (AD) (Aoyagi et al., 2019; Goedert et al., 1988; Wischik et al., 1988).
Several lines of evidence suggest that the formation and propagation of tau oligomers or aggregates is a key driver of toxicity in tauopathies. First, mutations that promote tau aggregation are causative in FTDP-17 (Goedert and Spillantini, 2000). Second, the rate of cognitive decline in AD is closely related to the rate of tau aggregate formation (Hanseeuw et al., 2019). Third, tau aggregates and tauopathy can be transmitted by inoculation in cells and mice (Aoyagi et al., 2019; Kaufman et al., 2016; Sanders et al., 2014; Woerman et al., 2016). Induction of tau aggregates in cell models also can be toxic (Sanders et al., 2014). Fourth, reduction of tau is neuroprotective in mouse models of AD (DeVos et al., 2018). Understanding how tau oligomers or aggregates form and how they induce neurotoxicity may lead to the development of therapeutics for numerous neurodegenerative diseases.
Tau is present in the human central nervous system as six splice isoforms. These isoforms differ in the number of N terminal inserts—0N, 1N, or 2N—and the number of microtubule repeat binding domains (RD)—3R or 4R (Buée et al., 2000; Park et al., 2016). The N terminal inserts have been shown to impact tau’s localization, interactions with membranes, spacing between microtubules, and signal transduction (Brandt et al., 1995; Chen et al., 1992; Lee et al., 1998; Liu and Götz, 2013). The positively charged RD has been shown to form the core of the amyloid fibrils present in the brains of patients with tauopathies and this is also where the majority of disease causing mutations are found (Buée et al., 2000; Falcon et al., 2018, 2019; Fitzpatrick et al., 2017; Goedert, 2005; Wegmann et al., 2013; Zhang et al., 2020).
Several observations suggest RNA may affect the formation of tau aggregates. First, tau binds RNA (Dinkel et al., 2015; Schröder et al., 1984; Wang et al., 2006; Zhang et al., 2017). Second, in vitro RNA promotes the conversion of soluble tau into insoluble aggregated tau, possibly because the negatively charged phosphate backbone of RNA can neutralize the positively charged RD of tau allowing for tighter packing of tau molecules and cross-β fibril formation (Ambadipudi et al., 2017; Dinkel et al., 2015; Kampers et al., 1996). Third, tau immunopurifies with a number of RNA binding proteins in both the aggregated and unaggregated states (Bai et al., 2013; Broccolini et al., 2000; Gunawardana et al., 2015; Hales et al., 2014a, 2014b; Hsieh et al., 2019; Meier et al., 2016). Fourth, tau aggregates in AD and Pick’s disease have been found to stain positive for RNA using RNA dyes (Ginsberg et al., 1997, 1998). Finally, analysis of the RNAs interacting with tau in an unaggregated state by iCLIP suggests that tau preferentially interacts with tRNAs (Zhang et al., 2017). Thus, important questions are whether pathological tau aggregates contain RNA, and if so, what is the nature of those RNAs and what are the possible physiologic or pathologic consequences of their interaction?
Herein, we investigated the RNA composition of tau aggregates in both cell culture and mouse model systems. Similar to earlier results, we found that tau aggregates form in the cytosol and the nucleus (Bukar Maina et al., 2016; Gil et al., 2017; Rady et al., 1995; Sanders et al., 2014; Ulrich et al., 2018). We found that both cytosolic and nuclear tau aggregates contain RNA and are enriched for RNAs involved in RNA splicing and modification including snRNAs and snoRNAs, as well as repetitive Alu RNAs. We also found that nuclear tau aggregates contain snRNAs and are concentrated in, and alter the composition, organization and dynamics of, splicing speckles, which are non-membranous assemblies of RNA and protein containing nascent RNA transcripts and splicing machinery (Galganski et al., 2017). Surprisingly, we discovered that the serine arginine repetitive matrix protein 2 (SRRM2), a protein component of splicing speckles, mislocalizes from nuclear splicing speckles to cytosolic tau aggregates in cellular models of tauopathy, tauopathy mouse models, and patients with AD, frontotemporal lobar degeneration (FTLD), and corticobasal degeneration (CBD). These extensive interactions of tau with splicing speckles and the splicing machinery correlate with splicing alterations seen in cells that form tau aggregates. This is notably similar to how cytosolic sequestration of RNA binding proteins such as TDP-43 and FUS in amyotrophic lateral sclerosis (ALS) can lead to alterations in nuclear RNA processing promoting neurodegeneration (Lagier-Tourenne et al., 2012; Polymenidou et al., 2011).
RESULTS
Cytosolic and nuclear tau aggregates contain RNA
To determine whether tau aggregates contain RNA, we employed a previously developed HEK293 tau biosensor cell line (Holmes et al., 2014; Sanders et al., 2014). The HEK293 biosensor cells express the 4R repeat domain (RD) of tau with the P301S mutation tagged with either cyan-fluorescent protein (CFP) or yellow fluorescent protein (YFP). Fluorescent tau aggregates can be induced in these HEK293 cells via lipofection of preformed non-fluorescent tau aggregates isolated from the brains of mice expressing 0N4R tau with the P301S mutation (P301S mice, Tg2541) (Holmes et al., 2014; Sanders et al., 2014). As previously seen (Sanders et al., 2014), we observed fluorescent tau aggregates in both the cytosol and the nucleus of the HEK293 cells following transfection of clarified brain homogenate from mice expressing P301S human tau, but not from mice expressing wild-type (WT) tau (WT mice, Tg21221), which do not develop tauopathy (Fig. 1A–B). Nuclear tau aggregates are not an artifact of the truncated K18 tau expressed in HEK293 cells since we also observed the formation of both nuclear and cytosolic tau aggregates in a tau seeding model expressing full length P301S 0N4R tau-YFP in H4 neuroglioma cells (Fig. S1A, Supp. video 2). Consistent with these fluorescent bodies being insoluble tau aggregates, fluorescence recovery after photobleaching (FRAP) revealed that both nuclear and cytosolic tau aggregates are immobile and do not recover after photobleaching (Fig. S1B, C).
Figure 1: Tau biosensor cell schematic and tau aggregates in mice contain poly(A) RNA.
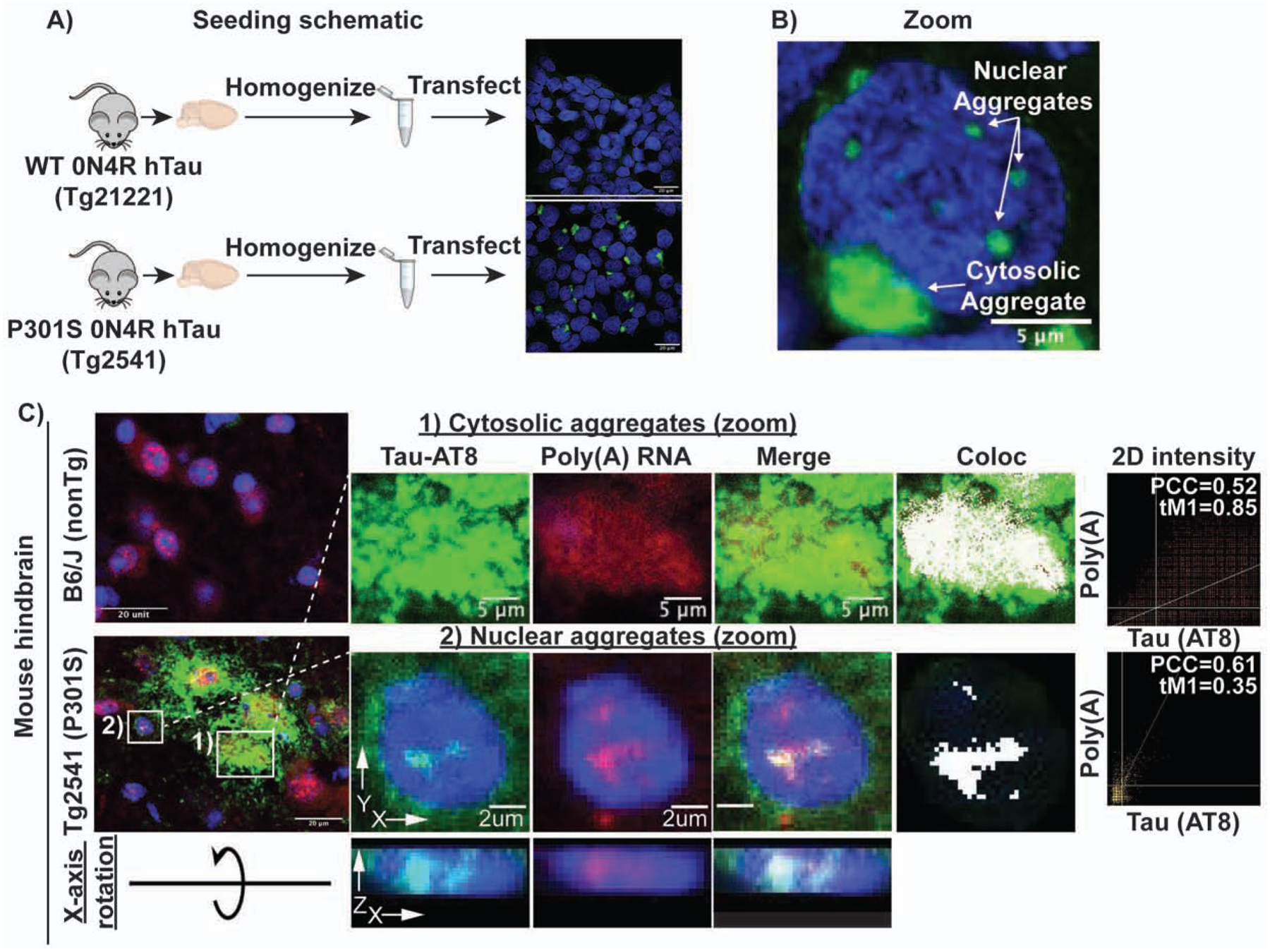
(A) Schematic showing experimental design of tau seeding in HEK293 biosensor cells. Brain homogenate from mice expressing either WT (rTg21221) or P301S (rTg2541) 0N4R tau was homogenized, clarified by successive centrifugation, and transfected into HEK293 cells expressing tau K18 (4R repeat domain) tagged with either CFP or YFP. Only cells transfected with P301S homogenate formed bright fluorescent aggregates. (B) Tau aggregates form in both the nucleus and the cytosol following transfection of P301S tau homogenate. (C) Cytosolic and nuclear tau aggregates contain poly(A) RNA in mouse brain. White pixels in Coloc image show pixels above the Costes determined thresholds in 2D intensity plots (PCC = Pearson correlation coefficient and tM1= thresholded manders colocalization (% of tau pixels above threshold that colocalize with poly(A) pixels above threshold)). X-axis rotation shows AT8 and oligo(dT) staining within the nucleus of mouse Tg2541 cells. See also Figure S1.
Using fluorescence in-situ hybridization (FISH) for poly(A) RNA we observed that cytosolic and nuclear tau aggregates showed 1.5- and 1.72-fold enrichment of poly(A)+ RNA staining, respectively (Fig. S1D). We also examined the presence of poly(A) RNA in tau aggregates in the brains of 6 month old P301S mice, in which transmissible tau aggregates start to form at 1.5 months (Holmes et al., 2014; Yoshiyama et al., 2007). Unlike in humans, where tau pathology develops in the frontal cortex, tau pathology in P301S mice tau pathology develops primarily in the hindbrain (Johnson et al., 2017). Nuclear tau and tau aggregates have been previously observed in the brains of mice and humans, however their function and role in disease is poorly understood (Bengoa-Vergniory et al., 2021; Bukar Maina et al., 2016; Gil et al., 2017; Jiang et al., 2019; Liu and Götz, 2013; Metuzals et al., 1988; Montalbano et al., 2020; Papasozomenos, 1995; Rady et al., 1995; Ulrich et al., 2018). We observed nuclear tau aggregates in the hindbrain stain strongly for poly(A) RNA (Fig. 1C, S1E, S7A, Supp. Video 1). We also observed a redistribution of poly(A) signal to overlap with the cytosolic tau aggregates in P301S mouse brains (Fig. 1C, S1E, S7A). Thus, in both mouse and cellular models of tau pathologies, tau forms cytosolic tangles and nuclear puncta that contain RNA.
Tau aggregates in HEK293 cells and mouse brains are enriched for snRNAs and snoRNAs
To determine the identity of the RNAs present in tau aggregates, we first purified tau aggregates from HEK293 tau biosensor cells using differential centrifugation and fluorescent activated particle sorting (FAPS) and then sequenced the associated RNA. By comparing the abundance of RNAs in total RNA and tau aggregates, we observed that tau aggregates contain a diverse transcriptome (Fig. 2A) and were enriched for small non-coding RNAs, particularly snoRNAs and minor snRNAs (Fig. 2B). Some mRNAs were also enriched in tau aggregates, notably mRNAs coding for voltage gated calcium channel complex, histone proteins, centrosomal proteins, and proteins involved in splicing regulation (Fig. S2A, B).
Figure 2: The RNA composition of tau aggregates in cellular and mouse tauopathy model systems.
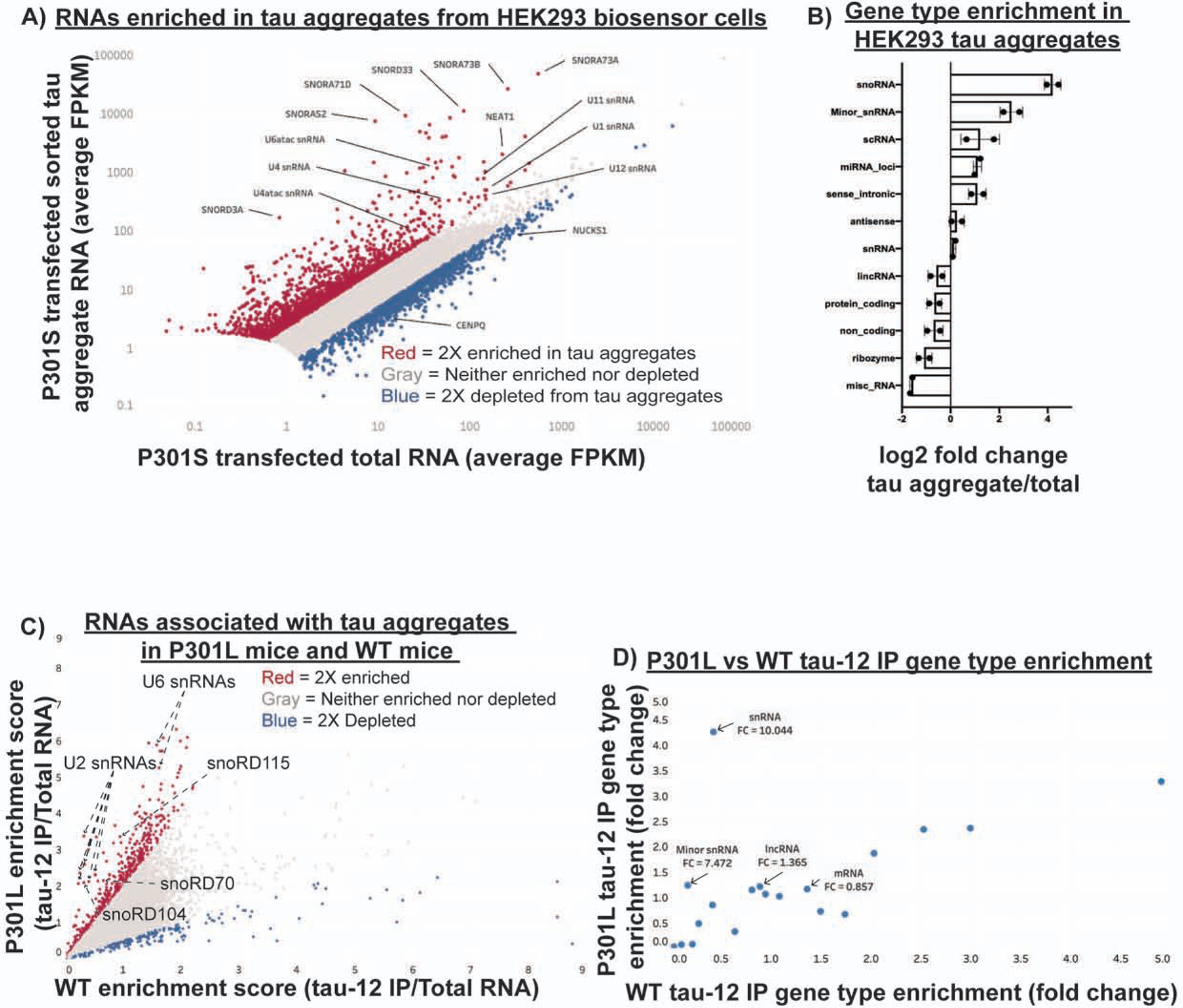
(A) Scatter plot of RNA sequencing showing average FPKM of two replicates for tau aggregate associated RNA and total RNA. Genes in red are two-fold enriched in tau aggregates and genes in blue are two-fold depleted from tau aggregates. Genes with fewer than 5 FPKM were removed from the analysis due to low coverage. (B) Fold change in the percentage of total FPKM for each gene type between the tau aggregate RNA and total RNA. Percentage of total FPKM was calculated by grouping genes using the Ensembl GRCh38.p13 biomart gene types. (C) Scatter plot of RNA sequencing data from two replicates of mouse brain tau aggregate isolation in P301L and WT mice. Enrichment scores were calculated by dividing the insoluble tau IP FPKM by the total RNA FPKM for each replicate. Genes in red are two-fold enriched and genes in blue are two-fold depleted from the P301L sarkosyl insoluble tau aggregates. (D) Gene type enrichment in the P301L and WT samples. Fold change for each gene type was calculated by dividing the percentage of total FPKM made up by each gene type in the insoluble tau-IP by the percentage of total FPKM made up by each gene type in the total RNA. The numbers below the gene type names indicate the P301L/WT enrichment. See also Figure S2.
Analysis of RNAs expressed from multicopy genes using RepEnrich (Criscione et al., 2014) showed enrichment of RNAs from the multicopy snoRNAs (U3, U17, and U8) and the multicopy snRNAs (U2 and U1) (Figure S2C–D). Consistent with this observation, U1 snRNA has previously been observed to be enriched in AD tau aggregates by PCR (Hales et al., 2014a). Additionally, this analysis showed some enrichment of tRNAs, as previously observed with non-aggregated tau (Zhang et al., 2017). We also observed enrichment of RNAs from specific types of transposable elements, namely Alu elements (Fig. S2C–D).
Tau aggregates in P301L mouse brain are also enriched for snRNAs
To investigate whether tau aggregates in mouse brains contain similar RNAs to tau aggregates identified in HEK293 cells, we fractionated mouse brains with a 1% sarkosyl extraction followed by tau immunoprecipitation (IP) using the tau-12 antibody to isolate tau aggregates as previously described (Diner et al., 2017). Western blot analysis showed that the sarkosyl extraction enriched for insoluble tau in the P301L mice (rTg4510) but not in the WT mice (rTg21221) (Fig. S2H). To identify RNAs specifically enriched in the aggregated tau fraction, we compared the enrichment of RNAs in the insoluble tau-12 IP relative to total RNAs between the WT and P301L mice (Fig. 2C).
Analysis of RNAs enriched in tau aggregates isolated from mouse brain relative to total RNA revealed an enrichment of specific RNAs, including snRNAs and snoRNAs in the P301L tau IP samples (Fig. 2C, D). This is similar to the RNA composition of tau aggregates isolated from HEK293 cells (Fig S2F). For example, we observed that snoRD115, snoRD104, snoRD70, U2 snRNA, and U6 snRNA, were enriched in the P301L insoluble tau fraction (Fig. 2C). In contrast to the HEK293 tau aggregates, only particular snoRNAs were enriched. This could be due to a variety of factors including differences in the isoforms of tau expressed in the two models (RD of tau in the HEK293 cells versus full-length 0N4R tau in the rTg4510 mice), the mutation in tau itself (P301S in the HEK293 biosensor cells and P301L in the rTg4510 mice), or differences in the RNA expression profiles of HEK293 cells and mouse neurons. The sarkosyl-insoluble fraction from both the P301L and WT mice revealed little to no enrichment of snRNAs or minor snRNAs suggesting that RNAs enriched in the tau-12 IP are interacting with aggregated tau rather than just enriched in the insoluble fraction (Fig. S2E). Similar to HEK293 cells, we observed some mRNAs enriched in tau aggregates from mouse brain (Figure S2G). Interestingly, some of the most enriched mRNAs are components of the centrosome (PCNT, Cep250, Cep164, Cep131) (Delaval and Doxsey, 2010; Graser et al., 2007), which is in agreement with previous work that has observed cytosolic tau aggregates concentrating at the centrosomes (Sanders et al., 2014; Santa-Maria et al., 2012). mRNAs coding for centrosomal proteins, such as PCNT, have previously been described to be present at the centrosome where they are locally translated and could become ensnared in tau aggregates (Sepulveda et al., 2018). Taken together, our results demonstrate that isolated pathological tau aggregates in mouse brains are enriched for similar types of RNAs as the tau aggregates in HEK293 cells including snRNAs and snoRNAs.
Enriched RNAs localize to tau aggregates by fluorescence in-situ hybridization
We used FISH to examine if RNA enriched in tau aggregates identified by RNA sequencing localized to cytosolic and/or nuclear tau aggregates. We performed FISH for two enriched snRNAs (U2 and RNU6ATAC), two enriched snoRNAs (snoRA73B and snoRD3A), two depleted mRNAs (CENPQ and NUCKS1), and for the enriched Alu family of multicopy RNAs (Fig. 3, S3). Interestingly, we observed that enrichment of specific RNAs differed with respect to the localization of tau aggregates. Specifically, snoRD3A had a 2.05-fold enrichment into cytosolic tau aggregates relative to bulk cytosol, while nuclear tau aggregates had a 1.35-fold enrichment of snoRD3A relative to bulk nucleoplasm (Fig. 3A). For U2 snRNA, cytosolic and nuclear tau aggregates had roughly the same fold enrichment over their respective compartments (1.48 and 1.42) yet the absolute intensity of U2 snRNA in nuclear tau aggregates was 1.87-fold higher than that of the cytosolic tau aggregates (Fig. 3B). In cells without tau aggregates, U2 snRNA localizes into discrete nuclear foci called splicing speckles, which are non-membranous RNA-protein assemblies containing factors involved in mRNA splicing (Kota et al., 2008; Wagner et al., 2004; Zhang et al., 2016). Nuclear colocalization of the splicing speckle associated snRNAs with tau implies that nuclear tau aggregation is occurring in splicing speckles (see below). Consistent with this observation, we also see colocalization of the enriched RNU6ATAC snRNA in nuclear tau aggregates (Fig. S3A).
Figure 3: FISH for RNAs identified by sequencing.
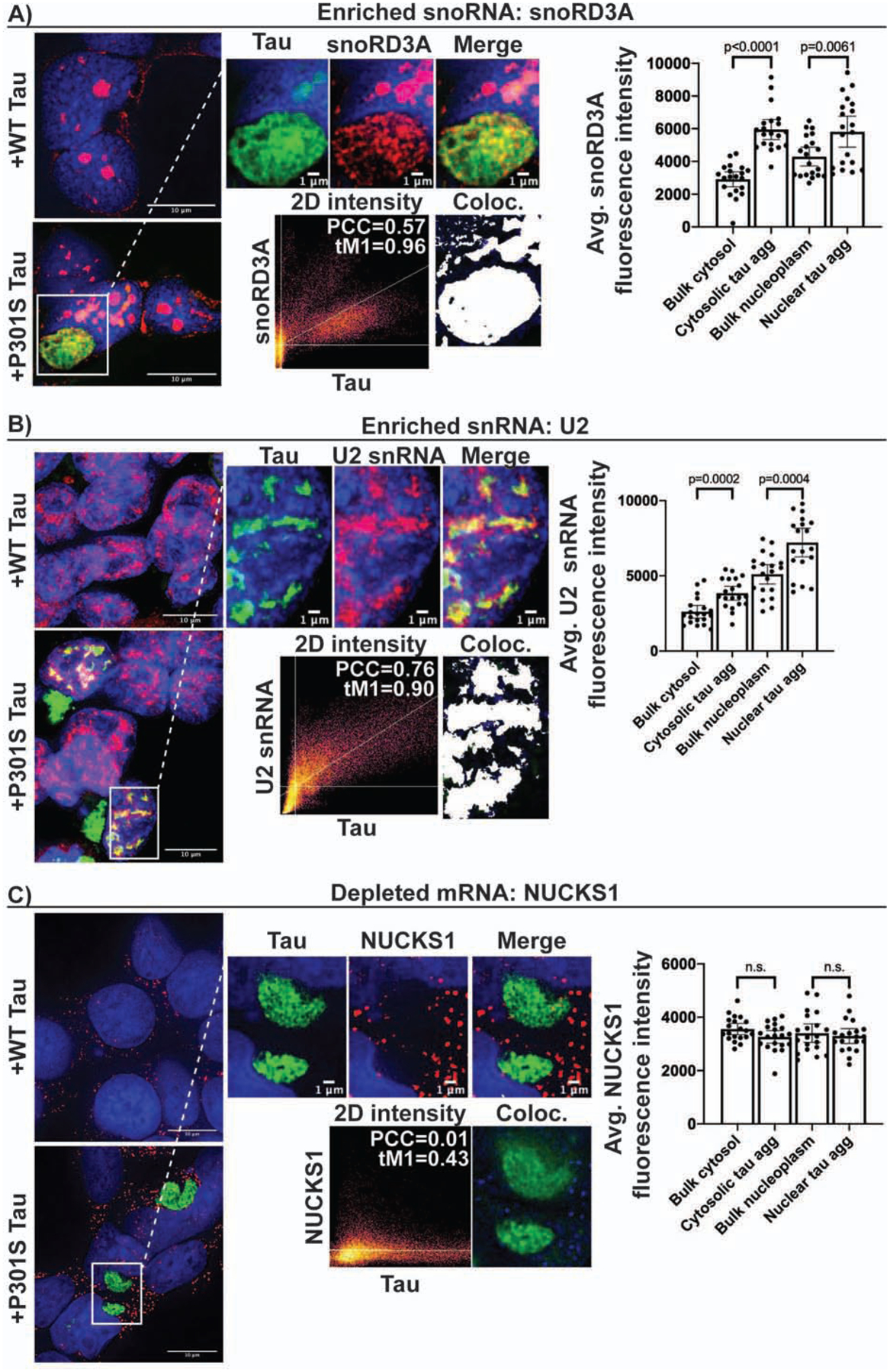
Line intensity plots and intensity quantification show enrichment of snoRD3A (A) and U2 snRNA (B) in both the nucleus and cytosol of HEK293 tau biosensor cells. We observed no enrichment of the depleted mRNA, NUCKS1 (C), into nuclear or cytosolic tau aggregates. Bar graphs show quantification of FISH fluorescence intensity within nuclear and cytosolic tau aggregates in relation to bulk cytosol and nucleoplasm (n = 20 aggregates). Data are represented as mean ± 95% confidence interval. Significance was determined using an unpaired two-tailed t-test. White pixels in Coloc image show pixels above the Costes determined thresholds in 2D intensity plots (PCC = Pearson correlation coefficient and tM1= thresholded manders colocalization (% of tau pixels above threshold that colocalize with red pixels above threshold)). See also Figure S3.
We also performed FISH for an Alu consensus sequence present in the Alu RNAs enriched from the sequencing data (Fig. S2C–D) and found that Alu signal enriched into both nuclear and cytosolic tau aggregates (Fig. S3C). Alu enrichment was greater in cytosolic aggregates (1.56-fold; p<0.0001) than in nuclear aggregates (1.23-fold; p=0.02). In the nuclear aggregates, Alu intensity was greatest on the periphery of the aggregates. The depleted mRNA NUCKS1 did not show significant intensity enrichment into either nuclear or cytosolic tau aggregates (Fig. 3C). Similarly, no positive localization correlation could be found for the depleted mRNA CENPQ and tau (Fig. S3D). Thus, tau aggregates contain, and are enriched for, a diverse set of specific RNAs.
Nuclear tau aggregates colocalize with splicing speckles in HEK293 cells
Due to the bias in both the HEK293 and mouse tau aggregate transcriptomes towards nuclear snRNAs and snoRNAs, we explored whether nuclear tau aggregates localized to the nucleolus, where snoRNAs are concentrated, or splicing speckles, which are enriched in snRNAs (Spector and Lamond, 2011).
Three lines of evidence suggest that nuclear tau aggregates colocalize with splicing speckles. First, a canonical nuclear splicing speckle marker, abcam antibody ab11826, colocalized with nuclear tau aggregates by immunofluorescence (IF). In IF, the ab11826 antibody recognizes the splicing speckle protein, SRRM2 (Ilik et al., 2020), and in agreement with this specificity, an SRRM2-halo fusion protein also localizes to nuclear tau aggregates and colocalizes with ab11826 (Fig. 4A, Fig. S4A–B). Similarly, other components of nuclear speckles co-localize with nuclear tau aggregates (see below). Second, nuclear tau aggregates are enriched for poly(A) RNA (Fig. 1C, S1D–E, Supp. Video 1) and splicing speckle associated RNAs including U2 snRNA (Fig. 3B, S2C) (Huang et al., 1994). Third, consistent with tau accumulating in splicing speckles and not with other nuclear RNA foci, nuclear tau aggregates do not colocalize with the nucleolar marker fibrillarin (Kiss, 2002) (Fig. S4C). In the brains of P301S (Tg2541) mice, we also observe pTau(S422) signal colocalizing with both SRRM2 and poly(A) in the nucleus (Fig. S7A). Thus, nuclear tau aggregates localize to SRRM2-positive nuclear splicing speckles in cell and mouse models of tauopathy.
Figure 4: Tau aggregates colocalize with splicing speckles and mislocalize SRRM2 in cellular tauopathy model systems.
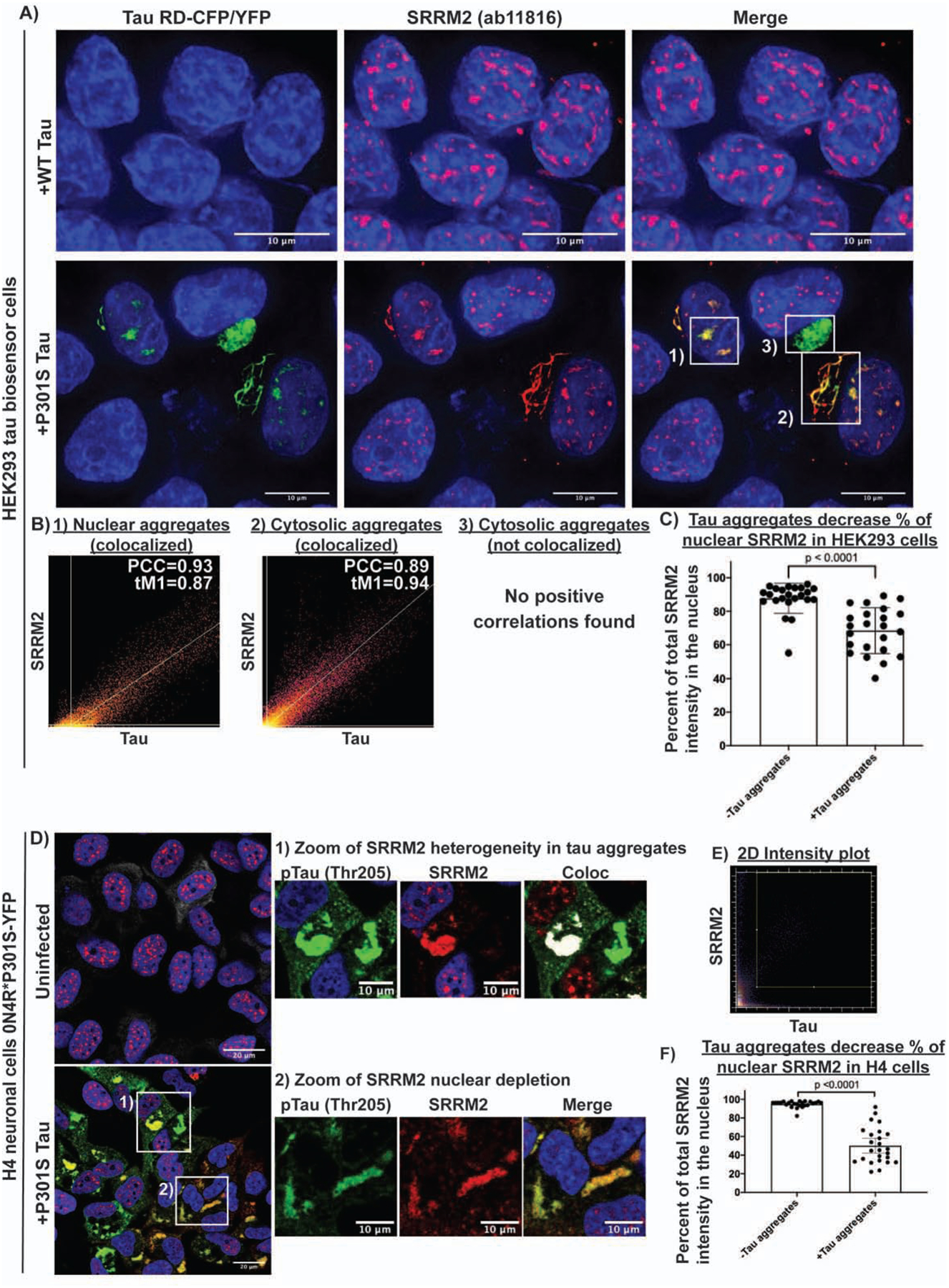
(A) Nuclear tau aggregates in HEK293 cells colocalize with SRRM2 (ab11826), a marker of splicing speckles. (B) Colocalization analysis showing the relationship between various tau aggregates and SRRM2 1) nuclear tau aggregates and splicing speckles marked by SRRM2, 2) a cytosolic tau aggregates that colocalize with cytosolic SRRM2, and 3) a cytosolic tau aggregate that does not colocalize with SRRM2. (C) Quantification of the percent of total SRRM2 intensity in the nucleus in HEK293 cells with and without tau aggregates (n = 23 cells). (D) Immunofluorescence of phospho-tau (Thr205) and SRRM2 in H4 neuroglioma cells expressing 0N4R*P301S-YFP tau +/− tau aggregates shows SRRM2 recruitment to tau aggregates is not dependent on phosphorylation at Thr205. (E) 2D intensity plot for the zoomed images showing two Thr205 positive tau aggregates, one that colocalizes with SRRM2 and one that does not colocalize with SRRM2. White pixels in Coloc image show pixels above the Costes determined thresholds in 2D intensity plot. (F) Quantification of percentage of SRRM2 in the nucleus in cells with and without tau aggregates (n = 25 cells). Images were quantified using CellProfiler. Data are represented as mean ± 95% confidence interval. Significance was determined using an unpaired two-tailed t-test. See also Figure S4.
Multiple nuclear speckle components re-localize to cytoplasmic Tau aggregates
While examining the co-localization of tau and SRRM2 in nuclear speckles in HEK293 cells, we observed that 77% of cytosolic tau aggregates also contained SRRM2 that relocalized to the cytosol (Fig 4A & S4D). Both the colocalization of nuclear tau aggregates in splicing speckles and the relocalization of SRRM2 to cytosolic tau aggregates were independent of the lipofectamine used to transfect tau into the cell models (Fig. S4E).
The accumulation of SRRM2 into cytosolic phospho-tau aggregates was sufficient to deplete nuclear SRRM2 in HEK293, and an H4 neuroglioma cell line expressing a full length 0N4R P301S tau-YFP that forms fewer nuclear tau aggregates (Fig 4C, 4F). Interestingly, in H4 cells that accumulated SRRM2 in cytosolic tau aggregates, nuclear speckles formed with no change in the intensity of the SON protein, a nuclear speckle protein that does not accumulate in cytosolic tau aggregates (Fig. S4F–G). These observations argue that cytosolic tau aggregates deplete the nucleus of SRRM2, but do not prevent the formation of nuclear speckles.
To determine if other nuclear speckle proteins also re-localized into cytosolic tau aggregates to some extent, we utilized IF to measure the average intensity enrichment of 19 other RNA binding and speckle proteins in nuclear and cytosolic tau aggregates. We observed that the SRRM2 paralogs, SRRM1 and SRRM3, did not accumulate in tau aggregates (Fig. 5A) indicating this accumulation is not shared between SRRM family members. Similarly, proteins with SR domains did not accumulate in cytosolic tau aggregates (SRSF1, SRSF2, and SRSF3) indicating that an SR domain is not sufficient for accumulation in cytoplasmic tau aggregates (Fig. 5A). Some, but not all, speckle components/splicing factors showed cytosolic tau colocalization with PNN (a known binding partner of SRRM2), SFPQ, MSUT2, DDX39B, and DYRK1A showing the strongest enrichment scores (Figure 5A, S5A) (Zimowska et al., 2003). Thus, multiple nuclear speckle proteins involved in pre-mRNA splicing mis-localize to cytosolic tau aggregates with SRRM2 and PNN being the most strongly re-localized.
Figure 5: Other proteins that localize to tau aggregates and the C-terminal region of SRRM2 is responsible for localization to tau aggregates.
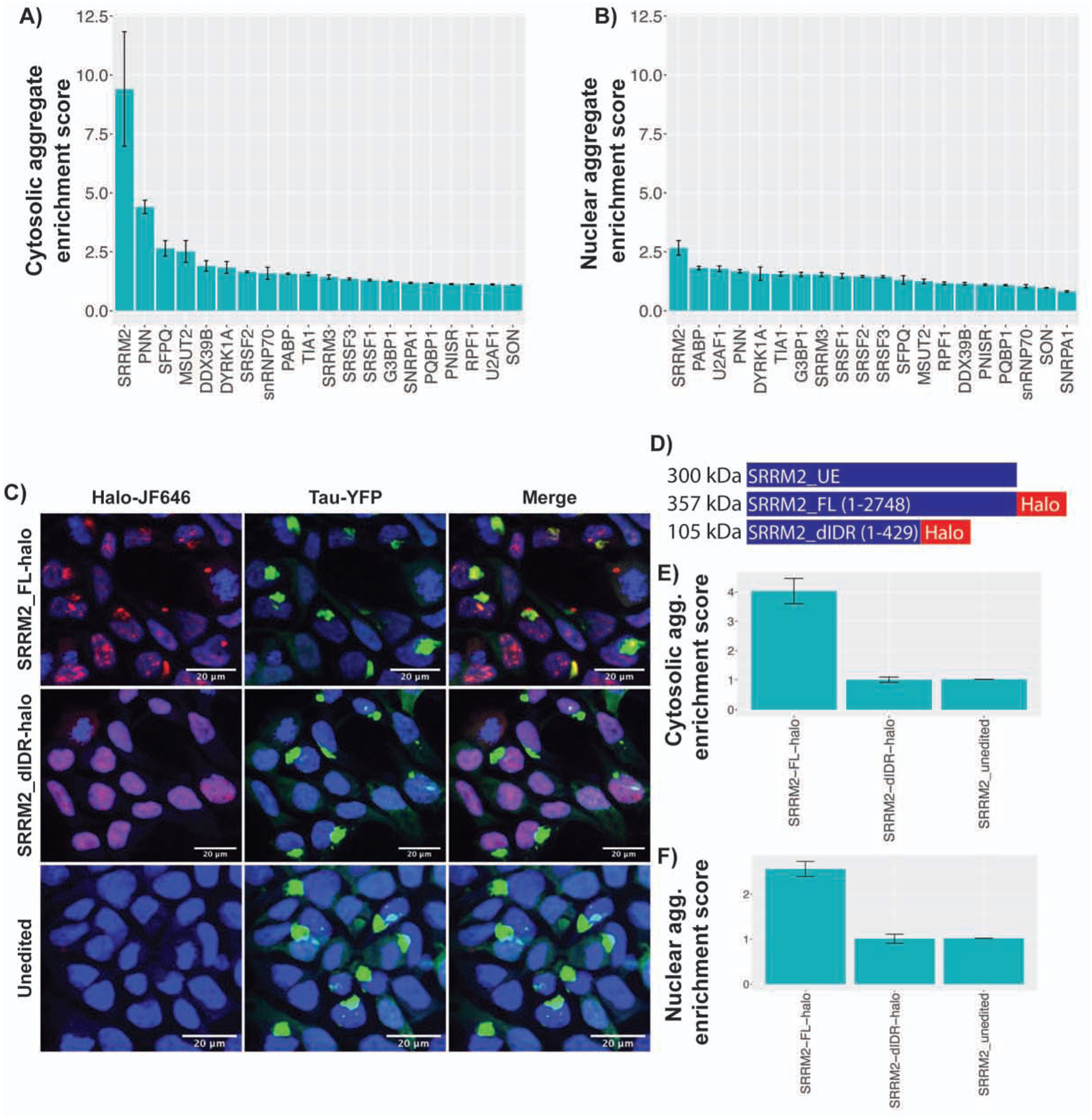
(A, B) Cytosolic and Nuclear tau aggregates enrichment scores (Median intensity within tau aggregate/median intensity within cytosol or nucleus) for 20 proteins. Ilastik was used to segment images into the following categories: nuclear tau aggregates, cytosolic tau aggregates, nucleus, cytosol, and background. Segmentation masks were fed into CellProfiler to quantify the median intensity of the interrogated protein. (C) Images of cells CRISPR edited to express endogenous FL or dIDR SRRM2 tagged with halo. (D) Schematic of SRRM2 constructs. (E, F) Cytosolic and nuclear enrichment scores for FL-Halo, dIDR-Halo, and unedited SRRM2. Data are represented as mean ± 95% confidence interval (N=25 images per condition). See also Figure S5.
Since neither SR domains nor the N-terminal conserved features of SRRM proteins appeared sufficient to recruit proteins to tau aggregates, we hypothesized that the C-terminal domain of SRRM2, which is comprised of an intrinsically disordered region (IDR) (Ilik et al., 2020), might be responsible for SRRM2 recruitment to tau aggregates. This would be consistent with the trend that intrinsically disordered regions of proteins can promote their recruitment to membraneless organelles. To test this idea, we used the CRISPaint system to create two HEK293 tau biosensor cell lines that contained a halo tag inserted into endogenous SRRM2 (Ilik et al., 2020; Schmid-Burgk et al., 2016). These two cell lines were 1) a full length SRRM2 cell line referred to as SRRM2_FL-halo (insert at aa 2708), and 2) a cell line lacking the C-terminal IDR of SRRM2 referred to as SRRM2_dIDR-halo (insert at aa 430) (Fig. 5C, S5B). We induced tau aggregation in these cells and compared the average SRRM2 halo intensity within nuclear and cytosolic tau aggregates relative to the average intensity in the bulk nucleus or cytosol respectively.
We observed that SRRM2_FL-halo was recruited to tau aggregates, but that the SRRM2_ dIDR-halo was not (Figures 5C–F). This demonstrated that SRRM2 is recruited to tau aggregates by the disordered C-terminal domain rather than the structured N-terminal domain. Since the N-terminal domain of SRRM2 is sufficient for RNA binding and interactions with the core of the spliceosome (Grainger et al., 2009; Zhang et al., 2018), this result argues that SRRM2 is not recruited to cytosolic tau aggregates by binding RNA nor the core of the spliceosome.
Tau aggregates alter the properties of nuclear speckles including pre-mRNA splicing
Since cytoplasmic tau aggregates depleted some nuclear speckle components, and tau aggregates can also form in nuclear speckles, we hypothesized that tau aggregate formation might alter the properties and function of nuclear speckles, which we examined in three experiments. First, given that nuclear speckles are highly dynamic structures (Rino et al., 2007) yet the tau aggregating in speckles was essentially static (Fig S1C), we examined if speckles with tau aggregates showed altered dynamics by performing FRAP on two tagged components of speckles, SRRM2 (Halo) and SRSF2 (mCherry). We observed that in the presence of tau aggregates, both speckle components showed an increase in the static component, and a reduced rate of recovery from FRAP (Fig 6A–B). This demonstrates that the presence of tau aggregates in nuclear speckles changes their dynamics.
Figure 6: Tau aggregation alters the dynamics and organization of splicing speckles as well as RNA splicing.
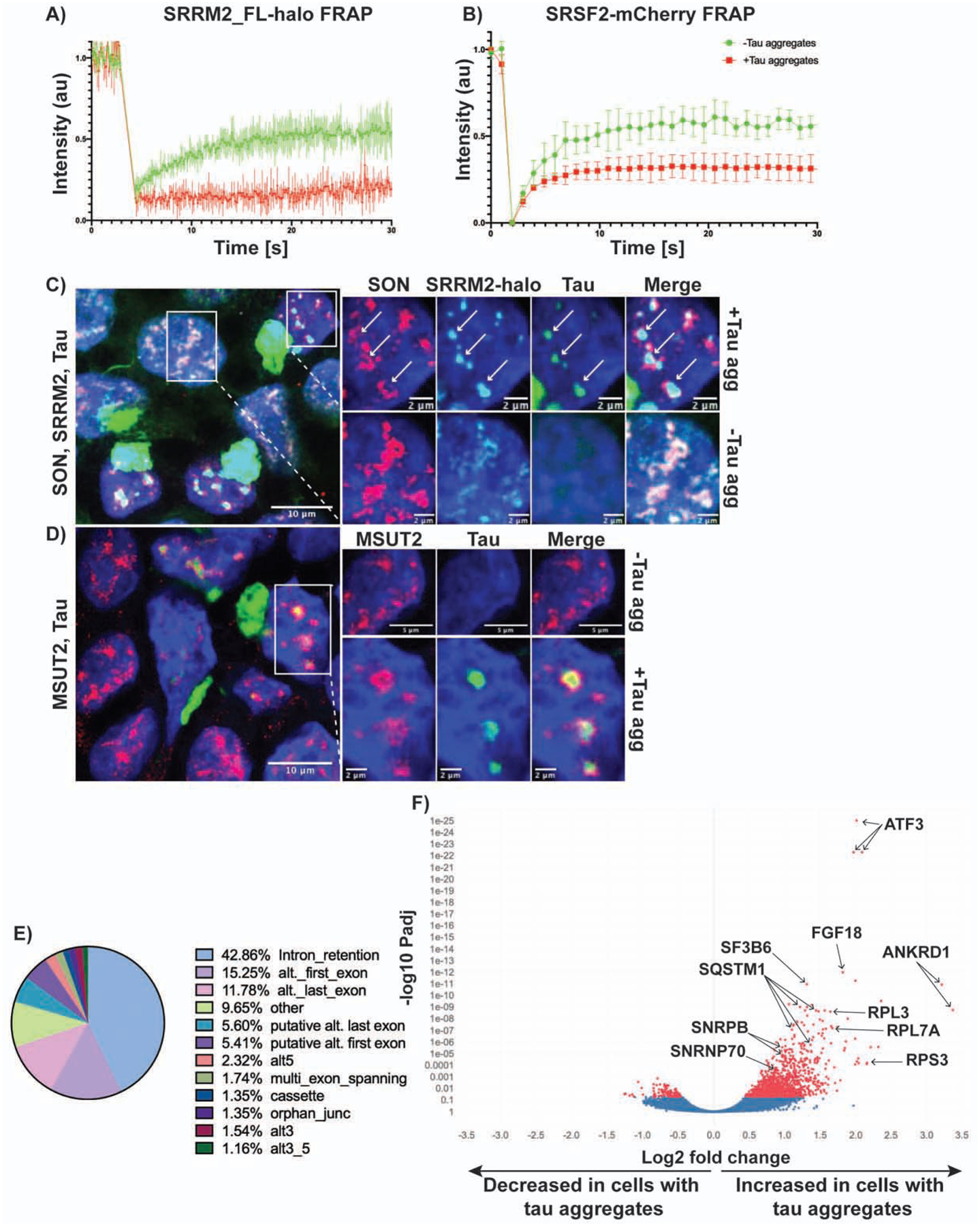
(A) FRAP of SRRM2_FL-halo and (B) SRSF2-mCherry splicing speckles with and without tau aggregates (n=5 replicates). 405 nm laser was used to photobleach and fluorescence intensity was measured using the 647 nm channel for halo-JF647 and 561 nm channel for mCherry. Data is represented as mean ± standard deviation. (C, D) Images of P301S tau transfected cells showing nuclear speckle reorganization in the presence of aggregates. SON and MSUT2 move to the periphery of speckles, while SRRM2 remains in the center of speckles with the tau aggregate. (E) MAJIQ analysis of splicing changes in cells with and without tau aggregates (n = 3 biological replicates per condition) showing that intron retention constitutes the largest percentage of local splicing variations. (F) Volcano plot showing differential intron retention in cells with tau aggregates quantified by iREAD and DEseq2 (Red = Padj < 0.05). Padj values were determined from DESeq2 using the Benjamini Hochberg method. Multiple points per gene are due to the multiple retained introns in those genes. See also Figure S6.
Second, the formation of tau aggregates in speckles alters the organization of speckle components. IF for SRRM2 and SON [a speckle protein that does not relocalize to cytosolic tau aggregates (Fig. 5A)] showed that the two proteins colocalize in speckles in the absence of tau aggregates. However, in the presence of nuclear tau aggregates, SRRM2 and tau colocalize in the center of speckles, while SON moves to the periphery and forms a ring-like structure around the aggregate (Fig. 6C). MSUT2 displayed a similar redistribution from the center of speckles to the periphery in the presence of tau aggregates. Interestingly, knockdown of MSUT2 has been shown to suppress tau toxicity in several model systems (Guthrie et al., 2011; Wheeler et al., 2019). These results suggest that the formation of tau aggregates in the nucleus disrupts the spatial organization of speckles.
Third, since nuclear speckles are thought to modulate pre-mRNA splicing (Spector and Lamond, 2011), we performed RNA-Seq on the same HEK293 cells with and without tau aggregates to determine if the presence of tau aggregates could alter splicing. We then investigated splicing patterns using two analyses: MAJIQ and iREADs (Li et al., 2020; Vaquero-Garcia et al., 2016). Using MAJIQ at a ΔPSI threshold of 0.1 and confidence threshold of 0.95, we identified 305 local splicing variations in 226 genes that are differentially spliced (Supplemental Table 4). Examination of the types of local splicing variations revealed that the largest categories were intron retention (42.86%), alternative first exons (15.25%), and alternative last exons (11.78%) (Fig. 6E, MAJIQ splicing diagrams and IGV raw read counts for one example, ATF3, are provided in Fig. S6A–B). Due to the abundance of intron retention events, we used iREAD (intron REtention Analysis and Detector) to better quantify differential intron retention between cells with and without tau aggregates. Reads that fully or partially overlap annotated introns were then used for differential expression analysis using DEseq2 (Li et al., 2020) (Love et al., 2014). We found that at a Padj < 0.05 there were 1,225 introns in 641 genes that were retained in cells with tau aggregates and 120 introns in 86 genes that were retained in cells without tau aggregates (Fig. 6F). Pre-mRNAs with retained introns in cells with tau aggregates cluster in genes affecting apoptosis and splicing associated protein (ASAP) complex, ribosome, and RNA splicing and processing (Fig. S6C). Thus, the formation of tau aggregates in cells is sufficient to induce changes in pre-mRNA splicing and is expected to have significant biological impact. Since patients with tauopathies and model systems with tau mutations show changes in pre-mRNA splicing patterns in the brain (Apicco et al., 2019; Hsieh et al., 2019; Raj et al., 2018), an alteration in splicing due to tau aggregate formation may contribute to these splicing changes (see discussion).
SRRM2 is depleted from the nucleus and relocalized to cytosolic tau neurofibrillary tangles in mouse and human tauopathies
Since SRRM2 was the most highly enriched protein identified in tau aggregates (Fig. 5A), we examined whether cytosolic tau aggregates also contain SRRM2 in mice via IF, using the ab11826 antibody on brains from WT B6/J mice, or the Tg2541 mice. In B6 control mice, SRRM2 predominantly localized to poly(A)+ nuclear splicing speckles (Fig. S7A). In contrast, we observe SRRM2 re-localized from the nucleus into cytosolic phospho-tau aggregates in the P301S-expressing Tg2541 mice (Fig. S7A). Consistent with these observations, phosphorylated SRRM2 has been previously observed to be relocalized to the cytosol in 5X FAD mouse brains using a different antibody, however the association of SRRM2 with tau aggregates was not reported (Tanaka et al., 2018). Thus, SRRM2, and potentially other speckle components, relocalize and sequestered into cytosolic tau aggregates in both cell culture models and in tauopathy mice.
To examine if SRRM2 is mis-localized in human tauopathies, we performed IF on tauopathy patient brains. We observed that in patients with the primary tauopathy CBD, SRRM2 was present in tau-containing cytosolic aggregates in the form of neuropil threads, whereas SRRM2 localized to nuclear splicing speckles in aged-matched heathy control patients (Figure 7A, patient demographics in Supplemental Table 3). Quantification of CBD and age matched control images revealed that the average nuclear SRRM2 signal was significantly lower in the CBD brains compared to the controls (Fig 7B). We also observed that SRRM2 was re-localized from the nucleus into the cytosol in the frontal cortex of multiple AD and FTLD patient brains (n=4 AD and n=4 FTLD), but not age-matched control brains (n=4, Figure 7C, S7B, patient demographics in Spplemental Table 3). These results show that cytosolic SRRM2 is a histopathological feature seen across three distinct human tauopathies.
Figure 7: SRRM2 is relocalized to cytosolic tau aggregates in human tauopathies.
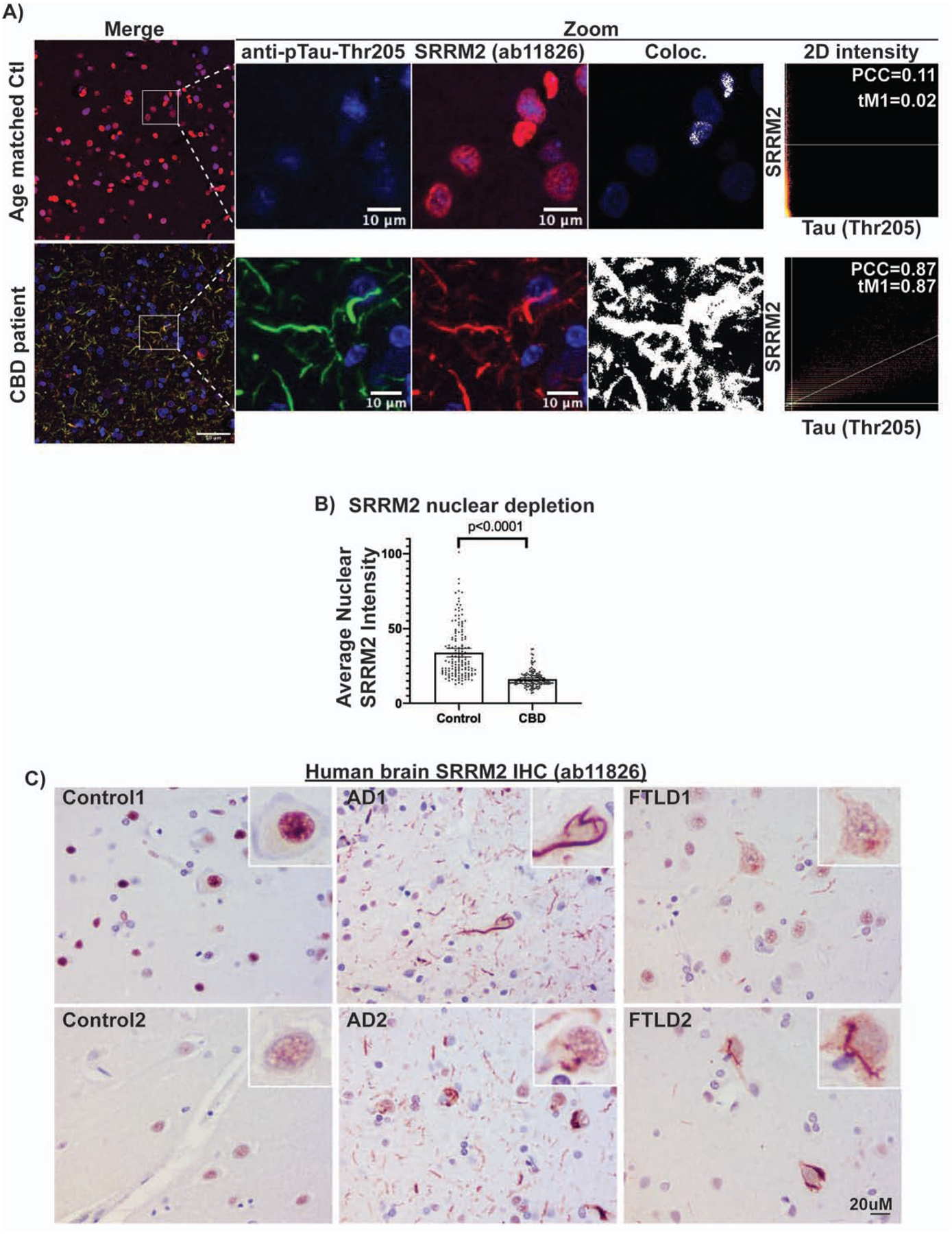
(A) IF and colocalization analysis showing hyperphosphorylated tau (pTau-Thr205, MN1020) colocalizes with SRRM2 (ab11826) in the cytosol of CBD patient brain while SRRM2 is localized to the nucleus in age matched control brain. (B) Quantification of the average nuclear SRRM2 intensity showing a significant decrease in the setting of CBD relative to age matched control. Data are represented as mean ± 95% confidence interval and p-values were determined using a Mann-Whitney test (n = 135 nuclei). Immunohistochemisty of SRRM2 in human brains showing SRRM2 redistribution to the cytosol in AD and FTD brains but retains nuclear localization in control brains. Patient demographics and additional examples can be found in Fig. S7B Supplemental Table 3. See also Figure S7.
DISCUSSION
We present several lines of evidence that both cytosolic and nuclear tau aggregates contain RNA. First, in mice and HEK293 cells, nuclear and cytosolic tau aggregates both stained positive for poly(A) RNAs, indicating the presence of mRNAs or non-coding RNAs with poly(A) tails (Fig. 1, S1D–E, S7A). Second, purification and sequencing of tau aggregates from mouse brains or HEK293 cells demonstrated the presence and enrichment of specific RNAs, most notably snRNAs and snoRNAs (Fig. 2). Third, FISH for specific RNAs in HEK293 cells validated that our sequencing identified RNAs enriched in tau aggregates (Fig. 3). Although we have not yet examined the specific RNAs present in tau aggregates in human pathologies, tau aggregates in patient brains stain positive with acridine orange, a dye with specificity for RNA (Ginsberg et al., 1997, 1998). Based on these observations, we suggest that tau aggregates generally contain RNA, and the presence of specific RNA species may alter their structure, formation, and stability. The presence of RNA in tau aggregates may explain why tau aggregates and other RNA binding proteins can co-immunoprecipitate and/or co-localize (Apicco et al., 2018; Gunawardana et al., 2015; Hales et al., 2014a, 2014b; Maziuk et al., 2018; Meier et al., 2016; Vanderweyde et al., 2016).
We observed that tau aggregates are enriched for a number of different RNA species. Most notably, we observed enrichment of specific snRNA and snoRNAs in both HEK293 reporter cells and in mouse brains, although the specific snRNA/snoRNA species can vary between model systems (Fig. 2). We also observed the enrichment of repetitive RNAs (such as tRNAs, Alu elements, and satellite RNAs) and some mRNAs coding for proteins in the centrosome and spliceosome (Fig. S2A, G). As our library preparation protocol was not specifically designed to capture tRNAs, miRNAs, or rRNAs, our analyses may underestimate the abundance of these species (Motameny et al., 2010; Xu et al., 2019). The mechanisms by which specific RNAs are enriched in tau aggregates remains to be determined but could be due to tau’s intrinsic RNA binding specificity, the structure of the tau conformers, and/or the presence of specific RNAs at sites of tau aggregation such as snRNAs in nuclear speckles or mRNAs at the centrosome (Sepulveda et al., 2018). We suggest tau aggregates in cells could be considered a representative of the growing class of RNA and protein assemblies.
We provide evidence that nuclear tau aggregates form in splicing speckles and alter their properties, composition, and function. One critical observation is that nuclear tau assemblies are observed both in HEK293 cells and P301L mice that overlap with both protein and RNA markers of nuclear speckles (Fig. 4, S4, S7). Moreover, nuclear speckles that contain tau aggregates show altered dynamics of both SRRM2 and SRSF2 (Fig. 6A) demonstrating tau aggregation has altered their material properties. Tau accumulation in speckles also changes their spatial organization with proteins partitioning into novel sub-domains of the assembly (Fig. 6C–D). Finally, since cytoplasmic tau aggregates accumulate multiple components of nuclear speckles, leading to their depletion from the nucleus, the presence of tau aggregates within cells alters the composition of nuclear speckles.
A critical question is the biological significance of the nuclear tau aggregates. In this light it is important to note we observed tau aggregates in both cell and mouse models of tauopathies demonstrating nuclear tau aggregates are not an artifact of cell line models (Fig. 1C, S1E, S7A). Moreover, we observe tau aggregates in nuclear speckles in both HEK293 cells expressing just the 4 repeat regions of tau, and in H4 neuroglioma cells expressing a full-length tau isoform (Fig. S1A) demonstrating tau accumulating in speckles is not an artifact of expressing truncated tau. Although tau is predominantly thought to be a cytosolic microtubule associated protein, numerous labs have observed aggregated and unaggregated tau in the nucleus of neuronal and non-neuronal cells (Bengoa-Vergniory et al., 2021; Bukar Maina et al., 2016; Gärtner et al., 1998; Gil et al., 2017; Jiang et al., 2019; Liu and Götz, 2013; Maj et al., 2010; Metuzals et al., 1988; Montalbano et al., 2020; Rady et al., 1995; Siano et al., 2019; Ulrich et al., 2018; Violet et al., 2014, 2015). One particularly interesting case report shows abundant nuclear tau (PHF-1, Tau-1, Tau-2, Tau-3, Tau-5 and SMI-31 positive) in the brain of a 64 year old man with pre-senile dementia and motor neuron disease whose neurologic status deteriorated rapidly prior to his death (Papasozomenos, 1995). Previous studies in HEK293 cells found that transfection of tau conformers that produced nuclear tau aggregates with a “speckled” phenotype were associated with greater cellular toxicity relative to those that only produced cytosolic tau aggregates (Sanders et al., 2014). In agreement, the suppressor of tau toxicity, MSUT2 (Guthrie et al., 2011; Wheeler et al., 2019), also localized to nuclear splicing speckles (Fig. 6D, S5A). Thus, tau’s interaction with splicing speckle components could be integral to the toxicity of tau aggregation.
A striking feature of our results is that cytoplasmic tau aggregates accumulate and mis-localize several proteins that normally accumulate in nuclear speckles (Fig. 4–6, S4–S7). The most striking of these is the SRRM2 protein, which can show an order of magnitude enrichment in cytosolic tau aggregates as compared to the bulk cytosol (Fig. 5A). Moreover, we also observed accumulation of SRRM2 in tau aggregates in mice, and in the human tauopathy, CBD (Figure 7A). Strikingly, we observed cytosolic mis-localization and/or nuclear depletion of SRRM2 in multiple tauopathies, including AD and FTLD (Fig. 7C, S7B). The consistent mis-localization of SRRM2 in cell line and animal models of tauopathy, as well as in post-mortem patient brain samples, argues the mis-localization of nuclear speckle proteins into cytosolic tau aggregates is a fundamental and consistent consequence of tau aggregation. An important issue in future work is to determine the mechanism of nuclear speckle component mis-localization.
One likely consequence of altering the composition and dynamics of nuclear speckles would be to alter pre-mRNA splicing. It is well documented that pre-mRNA splicing is altered in tauopathy patient brains, including AD patients (Hsieh et al., 2019; Raj et al., 2018). Consistent with this finding, we demonstrate that the formation of tau aggregates in HEK293 cells is sufficient to induce alterations in alternative splicing and increase the number of significantly retained introns (Fig. 6E–F, S6). Thus, tau aggregates are sufficient to alter pre-mRNA splicing, although it should be noted that splicing alterations seen in disease tissue may be complicated by additional factors including multiple cell types and neuroinflammatory responses. Interestingly, many of these retained introns triggered by tau aggregates are in RNAs that code for proteins involved in RNA processing and ribosome biogenesis (Fig. S6C). Disruptions to these processes could lead to a pathologic cascade, potentially explaining the complex alterations in ribosome function and RNA processing that have been observed in AD patients (Hsieh et al., 2019; Q et al., 2005; Raj et al., 2018). These results raise the possibility that tau aggregation per se is responsible for some of the splicing changes seen in disease tissue.
The coaggregation of RNA and proteins in tauopathies is reminiscent of pathologic RNA-protein aggregates seen in other neurodegenerative and neuromuscular diseases, such as amyotrophic lateral sclerosis and inclusion body myopathy (Ramaswami et al., 2013; Taylor et al., 2016). Thus, the sequestration of RNAs and RNA binding proteins into pathologic aggregates may represent a shared pathophysiological feature across multiple degenerative diseases affecting diverse tissue types with a common feature being depletion of critical RNA processing factors from the nucleus leading to changes in RNA processing and gene expression.
STAR METHODS TEXT
RESOURCE AVAILABILITY
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Roy Parker (Roy.Parker@Colorado.edu)
Materials Availability:
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability:
Primary sequencing data is deposited on the Gene Expression Omnibus under the accession GSE148716.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Tauopathy mouse models
Animals were maintained in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care in accord with the Guide for the Care and Use of Laboratory Animals. All procedures were approved by the University of California, San Francisco, Institutional Animal Care and Use Committee. Animals were maintained under standard environmental conditions, with a cycle of 12 hours light and 12 hours dark and free access to food and water.
For tau seeding experiments in HEK293 tau biosensor cells, the following mice were used: homozygous B6-Tg(Thy1-MAPT*P301S)2541 mice (referred to as Tg2541 or P301S tau mice in cellular seeding experiments) and FvBB6F1-Tg(Camk2a-tTa),(tetO-MAPT*wt)21221 (referred to as rTg21221 or WT tau mice in cellular seeding experiments). Mice were euthanized when the P301S tau mice developed spontaneous pathology (6–7 months). Collected brains were homogenized to 10% (wt/vol) in DPBS, aliquoted, and frozen at −80°C.
For tau isolation and sequencing of tau aggregates, the following mice were used: FvBB6F1-Tg(Camk2a-tTA)1Mmay, (tet)-tdTomato-Syp/EGFP)1.1Luo/J,(tetO-MAPT*P301L)4510 (referred to as rTg4510 or P301L mice in sequencing experiments) and FvBB6F1-Tg(Camk2a-tTa),(tetO-MAPT*wt)21221 (referred to as rTg21221 or WT tau mice in sequencing experiments). An even split of male and female mice were used and we did not observe any influence or associaition of sex on the findings.
For IF and FISH experiments, the following mice were used: homozygous B6-Tg(Thy1-MAPT*P301S)2541 mice (referred to as Tg2541 or P301S tau mice in IF and FISH experiments) and C57BL/B6 non transgenic mice (referred to as WT in IF and FISH experiments) were used as a control.
Cell culture and tau seeding of H4 biosensor cells
H4 cells (ATCC Cat# HTB-148, RRID:CVCL_1239) stably expressing the pIRESpuro3 vector (Clontech) containing a codon-optimized 0N4R MAPT gene with the P301S point mutation and tagged with YFP were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 0.2% penicillin-streptomycin, and maintained in incubators set to 37°C with 5% carbon dioxide. Cells were plated in a 12-well glass-bottomed dish at 1×105 cells/well and allowed to settle for a minimum of 2 hours prior to infection with PTA-precipitated tau prions from Tg2541 mouse brain.
Cell culture and tau seeding in HEK293 biosensor cells
HEK293 tau biosensor cells stably expressing the 4R RD of tau with the P301S mutation were purchased from ATCC (ATCC Cat# CRL-3275, RRID:CVCL_DA04) (previously described in (Holmes et al., 2014)). Cells were seeded at 2.5 × 105 cells/mL in 500uL of DMEM with 10% FBS and 0.2% penicillin-streptomycin antibiotics on PDL coated glass coverslips in a 24-well tissue culture treated plate (Corning 3526) and allowed to grow overnight in incubators set to 37°C with 5% carbon dioxide. The next day, 7ug of 1 mg/mL clarified P301L tau or WT tau mouse brain homogenate was mixed with 6uL of Lipofectamine 2000 and brought up to 100uL in PBS and allowed to sit at room temperature for 1.5 hours. The mixture was then added to 300uL of DMEM without FBS or antibiotics and mixed by pipetting. 50uL of this mixture was added to each well of a 24 well plate and allowed to incubate at 37 °C for 24 hours. Tau aggregate formation was monitored using a fluorescence microscope with a 488nm filter.
METHOD DETAILS
Clarification of brain homogenate for tau aggregate seeding in HEK293 cells
10% brain homogenate from Tg2541 or WT mice was centrifuged at 500 × g for 5 minutes, the supernatant was transferred to a new tube and centrifuged again at 1,000 × g for 5 minutes. The supernatant was again transferred to a new tube and the protein concentration was measured using bicinchoninic acid assay (BCA), and diluted in DPBS to 1 mg/mL for transfection into HEK293 tau biosensor cells.
PTA precipitation from brain homogenate for tau aggregate seeding in H4 biosensor cells
PTA precipitation of tau aggregates from mouse brain was performed as described (Woerman et al, 2016). 10% brain homogenate was incubated in final concentrations of 2% sarkosyl (Sigma, 61747) and 0.5% benzonase (Sigma, E1014–25KU) with constant agitation at 37°C for 2 hours. Sodium PTA (Sigma, P6395) was made in ultrapure MilliQ H2O and the pH was adjusted to 7.0. PTA was added to the samples to a final concentration of 2%, and samples were then incubated shaking at 37°C overnight. The samples were centrifuged at 13,200 × g at room temperature for 30 minutes, and the supernatant was removed. The resulting pellet was resuspended in 2% sarkosyl/PBS and 2% PTA. The sample was again incubated shaking at 37°C for 2 hours before a second centrifugation as above. The supernatant was again removed, and the pellet was resuspended in 1X PBS to 10% of the initial starting volume. This suspension was incubated using 1μL/well with Lipofectamine 2000 and OptiMEM at room temperature for at least 1.5 hours prior to infecting cells.
Fluorescent labeling of oligonucleotides for FISH
As previously described (Gaspar et al., 2017), DNA oligonucleotides were labeled with ddUTP-Cy5 fluorophores using terminal deoxynucleotidyl transferase (TdT). DNA oligonucleotides were designed to be antisense to the target of interested with the following specifications: 18–22 nucleotides in length and a minimum of 2 nucleotide spacing between probes. 20uM of DNA oligonucleotides were mixed with 120uM of 5-Propargylamino-ddUTP-Cy5, 10 units of TdT, and 1X TdT buffer and incubated at 37°C for 16 hours. Following incubation, oligonucleotides were precipitated in 80% ethanol with 60mM Na-acetate at −80°C for 20 minutes. The oligonucleotides were pelleted by centrifugation at 16,000g for 20 minutes at 4°C, washed with 80% ethanol 2x, air dried, and brought up in 20uL of nuclease free H20. If necessary, a further round of purification can be performed with the Zymo Oligo Clean and Concentrate spin-column kit (Zymo D4060). Labeled probe concentration was measured via NanoDrop OneC UV-Vis Spectrophotometer (Thermo Scientific 840–274200).
Tau aggregate isolation from HEK293 cells via centrifugation and flow cytometry
HEK293 biosensor cells were grown to 70–80% confluency in 245 mm square tissue culture treated dishes (Corning 07-200-599) in 50mL of DMEM (one plate per biologic replicate). 200 ug of WT or P301S tau clarified mouse brain homogenate was transfected per dish using lipofectamine 2000 and incubated at 37°C for 24 hours. Tau aggregation was monitored using the Evos M500 Imaging System with a GFP filter. Cells were harvested by scraping, centrifuged at 200 rcf, snap frozen in liquisd nitrogen, and stored at −80°C.
The cell pellet was thawed on ice for 5 minutes and resuspended in 6mL of high salt, high sucrose buffer containing RNase Inhibitors (10mM Tris-HCl pH 7.4, 0.8M NaCl, 1 mM EGTA, 10% sucrose, 0.5% NP40, Complete ultra-protease inhibitor, PhosStop Phosphatase inhibitor, 1:1500 RNasein, 1:300 Ribolock, 1:60 turbo DNAse). Cell lysate was passed through a 25 G needle 3x to homogenize and 100uL of sample was taken to extract total RNA.
Large tau complexes were pelleted by centrifugation at 21,000g for 20 minutes at 4°C, the pellet was brought up in high salt/high sucrose buffer, passed through a 27G needle, and centrifuged at 10,000g for 10 minutes at 4°C. The pellet was brought up in 1mL of DPBS and centrifuged at 500g for 5 minutes at 4°C to pellet large cellular debris. The supernatant (S3, enriched tau fraction) was taken and spotted onto a microscope slide for fluorescent imaging of tau aggregates in solution.
A BD Biosciences FACSAria Fusion flow cytometer (RRID:SCR_019309) was used to sort tau aggregates by fluorescence and size. The sheath fluid was changed to PBS, flow rate was set to 1.2, and threshold rate was set to <200 events/second. Gates were set on side scatter-H and 488 fluorescence such that WT transfected S3 fractions had <1% of particles in sorted fraction and P301S transfected S3 fractions had >30% in the sorted fraction. Roughly 1 million particles were sorted for each sample. To ensure the flow cytometer was sorting particles properly, the sorted fraction was visually inspected by fluorescence microscopy and the sorted and waste fractions were run back through the flow cytometer.
To denature tau aggregates and extract RNA, the sorted fractions were brought up in Proteinase K buffer (2M Urea, 100ug/mL proteinase K, and 3mM DTT) and incubated at room temperature for 15 minutes. Guanidine Hydrochloride was added to a final concentration of 5M and incubated at room temperature for 30 minutes. RNA was then extracted with TRIzol LS reagent (ThermoFisher 10296010). RNA concentrations were measured by QuBit RNA HS Assay kit (ThermoFisher Q328521) and Agilent 4200 TapeStation using the High Sensitivity RNA ScreenTape (Agilent 5067–5579). All samples except for the WT transfected sorted fraction yielded sufficient RNA to prepare sequencing libraries. RNA sequencing libraries were then prepared from total RNA and tau aggregate associated RNA from HEK293 biosensor cells using the Roche KAPA RNA HyperPrep Kit with RiboErase (Kapa KK8560) and sequenced on an Illumina NextSeq sequencer at the University of Colorado, Boulder BioFrontiers Sequencing Core (RRID: SCR_019308).
Isolation of tau aggregates from mouse brain
Brains were harvested from two Tg21221 (WT 0N4R human tau mouse brains) and two rTg4510 (P301L 0N4R human tau) mice and snap frozen in liquid nitrogen. Samples were thawed on ice and weighed. The brain tissue was then homogenized on ice using a dounce homogenizer and diluted to 5 mL/g in homogenization buffer with RNase inhibitors (10mM Tris-HCl pH7.4, 0.8M NaCl, 1mM EGTA, 10% sucrose, 1X Roche protease inhibitor, 1:40 promega RNasein). Aliquots were stored at −80 C.
To extract total RNA, 50uL of brain homogenate was incubated for 2 hours at room temperature in proteinase K buffer (2% SDS, 4M Urea, 10mM Tris-HCl pH4.54, 100ug/mL Proteinase K). 400uL of Urea buffer (60mM Tris-HCl pH 8.5, 8M Urea, 2% SDS) was then added and incubated for 30 minutes at room temperature. RNA was extracted from one half of this reaction using TRIzol LS solution and the other half was frozen at −80°C.
900uL of frozen brain homogenate was thawed on ice and 100uL of 10% (w/v) sarkosyl solution was added and incubated on ice for 15 minutes. Homogenate was then passed through a 25G and 27G syringe. Protein concentrations were measured by QuBit Protein Assay Kit (Thermo Fisher, Q33211) and sarkosyl buffer (50mM HEPES pH 7.2, 250 mM sucrose, 1mM EDTA, 1% w/v sarkosyl, 0.5 M NaCl) was added to reach a final concentration of 10 mg/mL. 500uL of each sample was transferred into an ultracentrifuge tube (Beckman Coulter, 349623) and centrifuged at 180,000g for 30 min at 4 deg in a Beckman Coulter Optima MAX-XP Ultracentrifuge. The supernatant (S1 fraction) was removed and stored at −80°C. The pellet was then brought up in 500uL of sarkosyl buffer and run through a 25G needle to homogenize. The sample was then centrifuged at 180,000g for 30 min at 4°C and the supernatant (S2) was removed and stored at −80°C.
For the sarkosyl insoluble RNA sequencing, P2 pellet was brought up in 100uL of proteinase K buffer and incubated at RT for 2 hours at RT. To further solubilize the sample, 400uL of urea buffer was added and incubated at RT for 30 minutes. The sample was then split in two and RNA was extracted from one half (250uL) using Trizol LS solution the other half was frozen at −80°C.
For the Tau IP, the P2 fraction was brought up in 400uL of PBS and protein concentrations were measured using the QuBit Protein Assay Kit (Thermo Fisher, Q33211). Samples were precleared with 15mg of DEPC treated (to inactivate RNAse) protein A dynabeads at room temperature for 45 minutes at RT on rotator. While preclearing, Tau12 and IgG antibodies were conjugated to 50uL (1.5mg) of protein A dynabeads for 40 minutes on rotator. Following preclear step, the sample was split into two fractions (one for the Tau12 IP and one for the IgG IP). Dynabeads with conjugated antibody were washed with PBS, brought up in 50uL of PBS and added to sample. IP was carried out on rotator at room temperature for 40 minutes. Sample was then washed 3x with PBS and 100uL of proteinase K buffer was added to the beads and incubated at RT for 2 hours. 400uL of urea buffer was added to beads and incubated for 30 minutes to further denature. Samples were then split into two 250uL fractions, one was Trizol extracted. RNA concentrations were then measured by QuBit RNA HS Assay kit (ThermoFisher Q328521) and Agilent 4200 TapeStation using the High Sensitivity RNA ScreenTape (Agilent 5067–5579). IgG IP did not pull down any RNA. RNA sequencing libraries were prepared using the Nugen Ovation SoLo RNA-Seq System, Mouse (Nugen 0501–32) and sequenced on an Illumina NovaSeq sequencer at the University of Colorado, Anschutz Genomics and Microarray Core.
Generation of Lentiviral particles
As previously described (Burke et al., 2019), HEK293T cells (T25 Flask at 80% confluence) were co-transfected with 1ug of pLenti-SRSF2-mCherry-blasticydin, 1ug of pVSV-G, 1ug of pRSV-Rev, and 1ug of pMDLg-pRRe using 16uL of lipofectamine 2000. Medium was replaced 6 hours post-transfection. Medium was then collected at 24- and 48-hours post-transfection and filter sterilized with a 0.45-um filter.
Generating SRSF2-mCherry cells via lenti-virus
HEK293 biosensor cells were seeded in a T-25 flask. When 80% confluent, the cells were incubated for 1 hour with 1mL of SRSF2-mCherry-blasticydin lentiviral particles containing 10-ug of polybrene with periodic rocking. 4mL of normal medium was then added to the flask and incubated for 24 hours. Normal medium was then aspirated and replaced with selective medium containing 10-ug/mL of Blasticidine S hydrochloride (Sigma-Aldrich). Selective medium was changed every three days. After one-week, selective medium was replaced with normal growth medium. Expression of SRSF2-mCherry was confirmed by fluorescence microscopy.
Generating halo tagged SRRM2 cells using CRISPaint
HEK293 biosensor cells were seeded in a 6 well plate. As previously described (Ilik et al., 2020; Schmid-Burgk et al., 2016) when 80% confluent, cells were transfected with 1 ug of pCRIPaint-HaloTag-PuroR plasmid (RRID:Addgene_80960)), 0.5 ug of PX458-CAS9 targeting plasmid (RRID:Addgene_48138), 0.5 ug of pCAS9-mCherry-Frame_selector plasmid (either RRID:Addgene_66939 for SRRM2_FL-Halo or RRID:Addgene_66941 for SRRM2_dIDR-Halo). After 24 hours, cells were selected using 2 ug/mL puromycin for 48 hours to enrich for edited cells. To label the halo constructs, JF646 was added to growth media at 10 nM overnight prior to cell lysis for gel analysis or fixation for imaging. CRISPR targeting guide sequences for SRRM2_FL-halo: CACCGCCATGAGACACCGCTCCTCC and AAACGGAGGAGCGGTGTCTCATGGC and SRRM2_dIDR-halo: CACCGCTGGCATGCCGAGAAACTT and AAACAAGTTTCTCGGCATGCCAGC
Immunofluorescence (IF) in HEK293 and H4 cells
Cells were fixed in 4% FPA for 10 minutes, washed 3X with DPBS, permeabilized in 0.1% Triton X-100 (Fisher BP151–100) for 5 minutes, washed 3x with PBS, and blocked with 5% bovine serum albumin (BSA) for 1 hour. Primary antibodies were diluted to desired concentration in 5% BSA and incubated overnight at 4 deg. Slides were washed 3x with DPBS and secondary antibodies were added at appropriate dilution in 5% BSA and allowed to incubate at room temperature for 1 hour. Slides were washed 2x with DPBS and then incubated in DAPI diluted in PBS (1ug/mL) for 5 minutes at RT, washed 1X with DPBS and then mounted on microscope slides with Prolong glass antifade mountant.
Fluorescence in-situ hybridization (FISH)
As previously described (Khong et al., 2017) , cells were fixed in 4% PFA for 10 minutes, washed 3x with PBS, permeabilized in 70% ethanol for 1 hour at 4 deg. Cells were then incubated in a wash buffer consisting of 2X nuclease free SSC and 10% deionized formamide (Calbiochem 4610) for 5 minutes at room temperature. The FISH probes were diluted to desired concentration in 100uL of hybridization buffer (2X nuclease-free SSC, 10% deionized formamide, 10% dextran sulfate) and spotted onto parafilm in a hybridization chamber (10cm cell culture dish lined with wet paper towels and covered with parafilm). Coverslips were then inverted onto the droplet of hybridization buffer contain the FISH probes and incubated at 37°C overnight. The slides were then transferred back to a 24 well plate and 500uL of 2X nuclease free SSC with 10% deionized formamide was added for 30 minutes at 37°C. Cells were then incubated in DPBS with 1ug/mL DAPI at room temperature for 5 minutes, washed with 2X nuclease free SSC and incubated at room temperature for 5 minutes. Coverslips were mounted on microscope slides with ProLong Glass Antifade Mountant (ThermoFisher, P36980) and allowed to cure overnight at room temperature.
minutes.
Fluorescence recovery after photobleaching
HEK293 biosensor cells were seeded in DMEM supplemented with 10% FBS and 0.2% penicillin-streptomycin at 0.25×105 cells/mL in Grenier Bio-One CELLview dishes with Glass Bottoms (Thomas Scientific, 07-000-235) and grown overnight at 37°C. The next day, clarified P301S tau brain homogenate was transfected and grown for 24 hours. A Nikon A1R Laser Scanning Confocal with environmental chamber was used to image the cells. A circular region within a tau aggregate was defined and bleached using a 405nm laser set to 100% laser power. For determining the recovery of tau within tau aggregates (Fig. S1B–C), fluorescence intensity was measured continuously for 6 minutes and 30 seconds post bleaching and normalized to an unbleached region (N=5). For determining the recovery of SRRM2_FL-halo within splicing speckles with and without tau aggregates, fluorescence intensity was measured continuously for 30 seconds and normalized to an unbleached region (n=5) (Fig. 6A). For determining the recovery of SRSF2-mCherry within splicing speckles with and without tau aggregates, fluorescence intensity was measured every 1 second for 30 seconds and normalized to an unbleached region (n=5) (Fig. 6B).
Mouse brain RNA fluorescent in situ hybridization followed by immunofluorescent staining (RNA FISH-IF)
Control (B6/J) and Tg2541 animals at approximately 6 months of age were anesthetized for whole brain collection. The mouse brains were embedded in OCT compound (Sakura, 4583) and flash-frozen in chilled isopentane. Samples were sectioned at 12μm using a cryostat and mounted on glass slides. Samples were air-dried at room temperature for 20 minutes to ensure tissue adherence to slides, then fixed in cold 4% PFA/1X PBS for 15 minutes. Samples were washed 3 times in 1X PBS for 5 minutes/wash, followed by a wash in 1X SSC for 5 minutes. Samples were transferred into 0.1X citrate buffer (Sigma, C9999) for a gentle antigen retrieval at 60°C for 1 hour 15 minutes. The slides were allowed to cool for 15 minutes, then rinsed 3 times in 1X SSC for 5 minutes/wash. Samples were then dehydrated in a graded series of ethanol washes (50%, 70%, 90% and 100%) for 3 minutes/wash and air-dried for 10 minutes. A hydrophobic barrier was drawn around the tissue and samples were blocked in a pre-hybridization buffer of 3% normal goat serum (NGS)/4X SSC at 37°C for 1 hour in a humidified chamber. Oligo(dT) probe labeled with Quasar 570 or Quasar 670 (Stellaris) was added to hybridization buffer (Stellaris, SMF-HB1–10) and incubated at 65°C for 10 minutes followed by a cooling on ice for 2 minutes. Pre-hybridization buffer was removed, and samples were then incubated in probe/hybridization buffer at 37°C overnight.
The next day, samples were washed in a dilution series of pre-hybridization buffer (twice in 4X, then once in 2X, 1X, 0.1X) at 37°C for 10 minutes/wash. Samples were then blocked in 20% NGS/1X PBST (0.1% Tween-20) at room temperature for 1 hour and incubated at room temperature overnight in primary antibodies diluted at 1:250 in 10% NGS/1X PBST.
The next day, samples were washed 3 times in 1X PBST for 10 minutes/wash, then incubated in Alexa Fluor secondary antibodies diluted at 1:500 in 10% NGS/1X PBST for 2 hours at room temperature. Samples were washed 3 times in 1X PBST for 10 minutes/wash. To quench autofluorescence, samples were incubated in 0.1% Sudan Black B in 70% ethanol for 10 minutes, then rinsed briefly in fresh 70% ethanol and transferred into 1X PBS for 5 minutes. Coverslips were mounted onto the slides using Vectashield Vibrance antifade mounting medium with DAPI (Vector Laboratories H-1800) and slides were left to dry overnight prior to being imaged with a Leica SP8 confocal microscope.
Human brain immunofluorescent staining
Human brain samples were provided by the Neurodegenerative Disease Brain Bank at the University of California, San Francisco, which receives funding support from NIH grants P01AG019724 and P50AG023501, the Consortium for Frontotemporal Dementia Research, and the Tau Consortium.
Formalin-fixed paraffin-embedded (FFPE) human brain samples from the left angular gyrus region of control individuals and patients diagnosed with corticobasal degeneration (CBD) were sliced at 8μm and mounted on glass slides. Samples were deparaffinized in a 60°C oven overnight, followed by two 10-minute xylene washes. Samples were then rehydrated in a graded series of ethanol washes (twice in 100% then once each in 90%, 70%, 50%) for 3 minutes/wash. Slides were then rinsed in cold ultrapure MilliQ H2O and transferred into 0.1X citrate buffer (Sigma, C9999) for antigen retrieval in an autoclave at 120°C for 5 minutes. Slides were allowed to cool for 15 minutes, then rinsed in 1X PBST (0.25% Triton X-100) for 15 minutes. A hydrophobic barrier was drawn around the tissue and samples were blocked in 20% normal goat serum (NGS)/1X PBST at room temperature for 1 hour in a humidified chamber. Samples were incubated at room temperature overnight in primary antibodies diluted at 1:250 in 10% NGS/1X PBST.
The next day, samples were washed 3 times in 1X PBST for 10 minutes/wash, then incubated in Alexa Fluor secondary antibodies diluted at 1:500 in 10% NGS/1X PBST for 2 hours at room temperature. Samples were washed 3 times in 1X PBST for 10 minutes/wash. To quench autofluorescence, samples were incubated in 0.1% Sudan Black B in 70% ethanol for 10 minutes, then rinsed briefly in fresh 70% ethanol and transferred into 1X PBS for 5 minutes. Samples were incubated for 10 minutes in 5μg/mL DAPI diluted in 1X PBS, then washed for 10 minutes in 1X PBS. Coverslips were mounted onto the slides using Vectashield Vibrance antifade mounting medium (Vector Laboratories H-1700) and slides were left to dry overnight prior to being imaged with a Leica SP8 confocal microscope.
Human tissue samples for immunohistochemistry
AD, FTD and non-neurologic disease control post-mortem tissue samples were obtained from the University of Pittsburgh ALS Tissue Bank, the Barrow Neurological Institute ALS Tissue Bank, and the Target ALS Human Postmortem Tissue Core. All tissues samples were collected after informed consent from the subjects or by the subjects’ next of kin, complying with all relevant ethical regulations. The protocol and consent process were approved by the University of Pittsburgh Institutional Review Board (IRB) and the Dignity Health Institutional Review Board. Clinical diagnoses were made by board certified neuropathologists. Subject demographics are listed in Supplemental Table 3.
Immunohistochemistry
Paraffin-embedded post-mortem frontal cortex tissue sections were used for this study. All sections were deparaffinized, rehydrated and antigen retrieval performed using Target Antigen Retrieval Solution, pH 9.0 (DAKO) for 20 min in a steamer. After cooling to room temperature, non-specific binding sites were blocked using Super Block (Scytek), supplemented with Avidin (Vector Labs). Primary antibodies used for immunohistochemistry were incubated overnight in Super Block with Biotin. Slides were then washed and incubated for 1 h in the appropriate biotinylated IgG secondary antibodies (1:200; Vector Labs) in Super Block. Slides were washed in PBS and immunostaining visualized using the Vectastain Elite ABC reagent (Vector Labs) and Vector Immpact NovaRED peroxidase substrate kit (Vector Labs). Slides were counterstained with hematoxylin (Sigma Aldrich) and pictures were captured using an OLYMPUS BX40 microscope equipped with a SebaCam camera.
QUANTIFICATION AND STATISTICAL ANALYSIS
Information on the statistical analysis and software used can be found in this section and in the Key Resources Table, statistical details of experiments can be found in the figure legends (including statistical tests used, value of n, what n represents, definition of center, and dispersion and precision measures). D’Agostino-Pearson was used check for normality. Normally distributed data was tested for significance using an un-paired two-tailed t-test, non-normally distributed data was analyzed using a Mann-Whitney test. All statistical analyses were performed using Prism version 9 unless otherwise mentioned.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-pTau (S202, Thr205) | Invitrogen | Cat #: MN1020, RRID:AB_223647 |
| Rabbit anti-pTau (Thr205) | Invitrogen | Cat #: 44-738G, RRID:AB_2533738 |
| Mouse anti-pTau (Ser202, Thr205) AT8 | Invitrogen | Cat #: MN1020, RRID:AB_223647 |
| Rabbit anti-pTau (Ser422) | Abcam | Cat #: ab79415, RRID:AB_1603345 |
| Mouse anti-Tau (Tau-12 clone) | Millipore | Cat #: MAB2241, RRID:AB_1977340 |
| Mouse anti-SRRM2 (SC-35) | Abcam | Cat #: ab11826, RRID:AB_298608 |
| Rabbit anti-Pinin | Thermo Scientific | Cat #: 18266-1-AP, RRID:AB_10642138 |
| Rabbit anti-SFPQ | Abcam | Cat #: ab177149, |
| Rabbit anti-ZC3H14 (MSUT2) | Sigma-Aldrich | Cat #: HPA049798, RRID:AB_2680888 |
| Rabbit anti-DDX39B | OriGene | Cat #: TA890032, |
| Rabbit anti-DYRK1A | Abcam | Cat #: ab65220, RRID:AB_1140733 |
| Rabbit anti-SRSF2 | Abcam | Cat #: ab204916, |
| Rabbit anti-SNRNP70 (U1-70K) | Abcam | Cat #: ab83306, RRID:AB_10673827 |
| Rabbit anti-PABP | Abcam | Cat #: ab21060, RRID:AB_777008 |
| Rabbit anti-TIA1 | Abcam | Cat #: ab40693, RRID:AB_2201438 |
| Rabbit anti-SRRM3 | Sigma-Aldrich | Cat #: HPA019337, RRID:AB_1848909 |
| Rabbit anti-SRSF1 (SF2) | Abcam | Cat #: ab129108, RRID:AB_11141636 |
| Mouse anti-G3BP | Abcam | Cat #: ab56574, RRID:AB_941699 |
| Rabbit anti-SNRPA1 | Millipore Sigma | Cat #: HPA045622, RRID:AB_2679394 |
| Rabbit anti-PQBP1 | Millipore Sigma | Cat #: HPA001880, RRID:AB_1079671 |
| Rabbit anti-PNISR | Millipore Sigma | Cat #: HPA038796, RRID:AB_10672652 |
| Rabbit anti-RPF1 | Sigma-Aldrich | Cat #: HPA024642, RRID:AB_1856428 |
| Rabbit anti-U2AF1 | Sigma-Aldrich | Cat #: HPA044833, RRID:AB_10960990 |
| Rabbit anti-SON | Thermo Fisher | Cat #: PA565108, RRID:AB_2662628 |
| Mouse anti-SRSF3 | Thermo Fisher | Cat #: 7B4A12, RRID:AB_2533119 |
| Rabbit anti-Fibrillarin | Invitrogen | Cat #: MA1-22000, RRID:AB_2231906 |
| Rabbit IgG isotype control antibody | ThermoFisher | Cat #: 10500C, RRID:AB_2532981 |
| Goat anti-mouse 647 | Abcam | Cat #: ab150115, RRID:AB_2687948 |
| Goat anti-rabbit 647 | Abcam | Cat #: ab150079, RRID:AB_2722623 |
| Goat anti-mouse biotinylated | Vector labs | Cat #: BP-9200, RRID:AB_2827937 |
| Bacterial and virus strains | ||
| pLenti CMV Blast | (Campeau et al., 2009) | Addgene plasmid #17451 RRID:Addgene_17451 |
| Biological samples | ||
| Human brain samples | Refer to Supplemental Table 3 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Sarkosyl | Sigma | Cat #: 61747 |
| Benzonase | Sigma | Cat #: E1014-25KU |
| Sodium PTA | Sigma | Cat #: P6395 |
| Lipofectamine 2000 | ThermoFisher | Cat #: 11668027 |
| Blasticidine S hydrochloride | Sigma | Cat #: 15205-25MG |
| Triton X-100 | Fisher | Cat #: BP151-100 |
| Deionized formamide | Calbiochem 4610 | Cat #: 4610 |
| ProLong Glass Antifade Mountant | ThermoFisher | Cat #: P36980 |
| 5-Propargylamino-ddUTP-Cy5 | JenaBioscience | Cat #: NU-1619-CY5 |
| RNasein Plus RNase Inhibitor | Promega | Cat #: N2615 |
| Terminal Deoxynucleotidyl Transferase | ThermoFisher | Cat #: EP0161 |
| cOmplete ULTRA Tablets | Sigma | Cat #: 05892791001 |
| PhosSTOP | Sigma | Cat #: 4906837001 |
| RiboLock RNase Inhibitor | ThermoFisher | Cat #: EO0382 |
| Turbo DNase | ThermoFisher | Cat #: AM2239 |
| Proteinase K | ThermoFisher | Cat #: 25-530-049 |
| Guanidine Hydrochloride | Sigma | Cat #: G3272-500G |
| TrizolLS | ThermoFisher | Cat #: 10296028 |
| Diethyl pyrocarbonate (DEPC) | Sigma | Cat #: D5758-25ML |
| 0.1X citrate buffer | Sigma | Cat #: C9999 |
| FISH Hybridization buffer | Stellaris | Cat #: SMF-HB1-10 |
| Vectashield Vibrance antifade mounting medium | Vector Laboratories | Cat #: H-1700 |
| Scytek SUPER BLOCK | Fisher | Cat #: NC9782835 |
| Avidin/Biotin blocking kit | Vector Labs | Cat #: SP-2001 |
| hematoxylin | Sigma Aldrich | Cat #: H3136 |
| Vectastain Elite ABC reagent | Vector Labs | Cat #: PK-6100 |
| Vector Immpact NovaRED peroxidase substrate kit | Vector Labs | Cat #: SK-4800 |
| Critical commercial assays | ||
| Zymo Oligo Clean and Concentrate spin-column kit | Zymo | Cat #: D4060 |
| QuBit RNA HS Assay kit | ThermoFisher | Cat #: Q328521 |
| Agilent 4200 TapeStation using the High Sensitivity RNA ScreenTape | Agilent | Cat #: 5067–5579 |
| KAPA RNA HyperPrep Kit with RiboErase | Roche | Cat #: KK8560 |
| Nugen Ovation SoLo RNA-Seq System, Mouse | Nugen | Cat #: 0501–32 |
| Dynabeads Protein A for Immunoprecipitation | ThermoFisher | Cat #: 10002D |
| Deposited data | ||
| RNA sequencing data | This paper | GSE148716 |
| Experimental models: cell lines | ||
| HEK293 tau biosensor cells | ATCC | ATCC Cat# CRL-3275, RRID:CVCL_DA04 |
| H4 cells | ATCC | ATCC Cat# HTB-148, RRID:CVCL_1239 |
| Experimental models: organisms/strains | ||
| P301S mice (Tg2541) B6-Tg(Thy1-MAPT*P301S)2541 | (Allen et al., 2002) | N/A |
| WT mice (rTg21221) FvBB6F1-Tg(Camk2a-tTa),(tetO-MAPT*wt)21221 | (Hoover et al., 2010) | N/A |
| P301L mice (rTg4510) FvBB6F1-Tg(Camk2a-tTA)1Mmay, (tet)-tdTomato-Syp/EGFP)1.1Luo/J,(tetO-MAPT*P301L)4510 | (Ramsden et al., 2005) | N/A |
| Non-transgenic mice (C57BL/6J) | Jackson Laboratory | Stock No. 000664 | B6 |
| Oligonucleotides | ||
| sgRNAs for SRRM2_FL-halo. Up: CACCGCCATGAGACACCGCTCCTCC down: AAACGGAGGAGCGGTGTCTCATGGC |
This paper | N/A |
| sgRNAs for SRRM2_dIDR-halo. Up: CACCGCTGGCATGCCGAGAAACTT down: AAACAAGTTTCTCGGCATGCCAGC |
This paper | N/A |
| Fluorescence in-situ hybridization probes | Supplemental Table 6 | N/A |
| Recombinant DNA | ||
| pCRIPaint-HaloTag-PuroR | (Schmid-Burgk et al., 2016) | Addgene plasmid #80960 RRID:Addgene_80960 |
| PX458-CAS9 | (Schmid-Burgk et al., 2016) | Addgene plasmid #48138 RRID:Addgene_48138 |
| pCAS9-mCherry-Frame_selector +0 | (Schmid-Burgk et al., 2016) | Addgene plasmid #66939 RRID:Addgene_66939 |
| pCAS9-mCherry-Frame_selector +2 | (Schmid-Burgk et al., 2016) | Addgene plasmid #66941 RRID:Addgene_66941 |
| pIRESpuro3 | Clontech | Cat: 631619 |
| Software and algorithms | ||
| CellProfiler image analysis software Version 3.1.8 | (McQuin et al., 2018) | https://cellprofiler.org/ |
| Ilastik image segmentation software Version 1.3.3 | (Berg et al., 2019) | https://www.ilastik.org/ |
| MAJIQ splicing analysis software Version 2.1 | (Vaquero-Garcia et al., 2016) | https://majiq.biociphers.org/ |
| STAR RNAseq aligner Version 2.6.0 | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| RepEnrich repetitive element analysis Version 1 | Criscione et al., 2014 | https://github.com/nskvir/RepEnrich |
| iREAD intron analysis software Version 0.8.5 | Li et al., 2020 | https://github.com/genemine/iread |
| DEseq2 Version 1.30.1 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq.html |
| Trimmomatic Version 0.36 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| FastQ Screen Version 0.12.0 | Wingett and Andrews, 2018 | https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/ |
| Prism 9 | GraphPad | https://www.graphpad.com/scientificsoftware/prism/ |
| Other | ||
| Nikon Spinning Disc Confocal microscope | Biofrontiers imaging core | RRID: SCR_018302 |
| Round cover glass, #1.5 thickness | Thomas Scientific | Cat #: 1217N79 |
| NanoDrop OneC UV-Vis Spectrophotometer | ThermoFisher | Cat #: 840–274200 |
| 245 mm square tissue culture treated dishes | Corning | Cat #: 07-200-599 |
| Evos Imaging system | Evos | Cat #: M500 |
| BD Biosciences FACSAria Fusion flow cytometer | BIofrontiers flow cytometry core | RRID:SCR_019309 |
| Illumina NextSeq | Biofrontiers sequencing core | RRID: SCR_019308 |
| Optima MAX-XP ultracentrifuge | Beckman Coulter | Cat #: 393315 |
| Ultracentrifuge tubes | Beckman Coulter | Cat #: 349623 |
Image analysis
To quantify FISH intensity within nuclear and cytosolic tau aggregates, ImageJ’s freehand selection tool was used to draw perimeters around aggregates and in regions of bulk cytosol and nucleus. The average FISH intensity was measured within the selections and used to compare enrichment of RNAs.
To quantify the percentage of SRRM2 in the cytosol of cells, CellProfiler was used. Nuclei were identified using object detection with a typical diameter between 50–200 pixels for HEK293 and H4 cells, which were then used as a mask to quantify nuclear and cytosolic SRRM2 intensity. The percentage of total SRRM2 intensity in the cytosol was calculated by dividing the cytosolic SRRM2 intensity by the sum of the nuclear and cytosolic intensities per image. Significance we determined using an unpaired two-tailed t-test.
To quantify enrichment scores of various proteins of interest (POIs) in tau aggregates, 25 images were taken in a 5×5 panel of each slide using a 40x air objective on a Nikon Spinning Disc Confocal microscope (RRID: SCR_018302). Ilastik was used to create the following segmentation masks: cytosolic tau aggregates, nuclear tau aggregates, nucleus, cytosol, and background. The RGB images and segmentation masks were fed into CellProfiler, which was used to quantify the average POI intensity within tau aggregates and the average POI intensity within the corresponding compartment. Enrichment was defined as the ratio of POI intensity within the tau aggregate to the POI intensity within the corresponding compartment.
Analysis of RNA sequencing data
Following sequencing, quality of sequencing reads were assessed using FASTQC version 0.11.5, Illumina TruSeq3 adapters and low quality reads were trimmed off using Trimmomatic version 0.36 (Bolger et al., 2014). Reads that aligned uniquely to the ribosome, yeast, or bacteria were then filtered out using FastQ Screen (Wingett and Andrews, 2018). Reads were aligned using the Spliced Transcripts Alignment to a Reference (STAR) aligner version 2.6.0 (Dobin et al., 2013) to either the Genome Reference Consortium Human Build 38 (GRCh38, acquired from NCBI) or the Genome Reference Consortium Mouse Build 38 (GRCm38, acquired from NCBI) depending on the species being analyzed. Adjusted p-values were calculated from raw read counts using DEseq2. Gene counts were used to calculate Fragments per kilobase per million read (FPKM) using transcript lengths retrieved from the Ensembl Biomart (Kinsella et al., 2011) and the following formula FPKM = (# of mapped fragments*103*106)/(transcript length in bp * total number of mapped fragments). For mouse sequencing, FPKM values were used to calculate enrichment scores for each biological replicate (n = 2) by dividing the Tau IP FPKM by the Total RNA FPKM for each replicate. Enrichment scores were then used to calculate average enrichment score for each gene and fold changes between P301L and WT mice. Gene type enrichment was determined by calculating the percentage of FPKM made up by each gene type. Repetitve elements were analyzed using a reference files acquired from repeatmasker (hg38 - Dec 2013 -RepeatMasker open-4.05 – Repeat Library 20140131) and RepEnrich (Criscione et al., 2014).
Splicing analysis: Following mapping of reads to GRCh38 using STAR, MAJIQ v2.1 was used with standard settings to quantify splicing changes. Voila was used to view results, generate splicing diagrams, and determine the relative percentage of each splicing type (only LSVs containing more than 10 reads were reported). To quantify reads mapping to introns, iREAD v0.8.5 was used along with an intron annotation file generated from ensemble v77 (Li et al., 2020). The read count output from iREAD was used as an input to DEseq2 for calling differential intron retention (Love et al., 2014).
Supplementary Material
Supplemental Video 1: 3D rotation of a mouse brain nucleus, related to Figurs 1. Nucleus is shown in blue (DAPI), AT8 phosphorylated tau is shown in green, and poly(A) RNA is shown in red.
Supplemental Video 2: 3D rotation of an H4 cells expressing 0N4R tau-YFP showing intranuclear tau aggregates, related to Figure 1. Nucleus is shown in blue (DAPI) and Tau-YFP is shown in green.
Supplemental Excel Table 1: HEK293 tau aggregate RNA sequencing data, related to Figurs 2.
Supplemental Excel Table 2: Mouse Tau aggregate RNA enrichment score, related to Figure 2.
Supplemental Excel Table 3: patient demographics for human brain samples. Related to Figure 7 and STAR Methods.
Supplemental Excel Table 4: Splicing analysis. Sheet1: MAJIQ splicing analysis of RNA seq from cells with and without tau aggregates. Sheet 2: iREADs intron retention analysis from the same experiment. Related to Figure 6.
Supplemental Excel Table 5: FISH probe sequence information. Related to Figures 1, 3 and STAR Methods.
Highlights.
Tau aggregates contain RNA and are enriched for snRNAs and snoRNAs
Nuclear tau aggregates localize to splicing speckles in model systems
Tau alters the composition, organization, and dynamics of speckles
Speckle components are mislocalized to cytoplasmic tau aggregates in patient brains
ACKNOWLEDGEMENTS
We would like to thank Joe Dragavon, Ph.D. for his assistance with microscopy at the BioFrontiers Advanced Light Microscopy Core (supported by NIST-CU 70NANB15H226), Theresa Nahreini for her assistance in the Biochemistry Cell Culture Facility at CU Boulder, and the BioFrontiers Institute Next-Gen Sequencing Core Facility, which performed the Illumina sequencing and library construction. We would also like to thank Dr. James Burke for his assistance and discussions regarding experimental design.
This work was supported by grants from the National Institutes of Health (NIH) (F30AG063468) (E.L.), (5R01GM045443-31) (R.P.) (AG002132) (S.B.P.), the Brockman Foundation (S.B.P.), the Sherman Fairchild Foundation (S.B.P.), and the Howard Hughes Medical Institute (R.P.).
DECLARATION OF INTERESTS:
Roy Parker is a founder and consultant for Faze Medicines. Stanley B. Prusiner is a member of the Scientific Advisory Boards of ViewPoint Therapeutics and New Ventures Inc., and a member of the Supervisory Board of Priavoid, none of which have contributed financial or any other support to these studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, et al. (2002). Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J. Neurosci 22, 9340–9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambadipudi S, Biernat J, Riedel D, Mandelkow E, and Zweckstetter M (2017). Liquid-liquid phase separation of the microtubule-binding repeats of the Alzheimer-related protein Tau. Nat Commun 8, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyagi A, Condello C, Stöhr J, Yue W, Rivera BM, Lee JC, Woerman AL, Halliday G, Duinen S. van, Ingelsson M, et al. (2019). Aβ and tau prion-like activities decline with longevity in the Alzheimer’s disease human brain. Science Translational Medicine 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apicco DJ, Ash PEA, Maziuk B, LeBlang C, Medalla M, Abdullatif AA, Ferragud A, Botelho E, Ballance HI, Dhawan U, et al. (2018). Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat Neurosci 21, 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apicco DJ, Zhang C, Maziuk B, Jiang L, Ballance HI, Boudeau S, Ung C, Li H, and Wolozin B (2019). Dysregulation of RNA Splicing in Tauopathies. Cell Rep 29, 4377–4388.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai B, Hales CM, Chen P-C, Gozal Y, Dammer EB, Fritz JJ, Wang X, Xia Q, Duong DM, Street C, et al. (2013). U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. PNAS 110, 16562–16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengoa-Vergniory N, Velentza-Almpani E, Silva AM, Scott C, Vargas-Caballero M, Sastre M, Wade-Martins R, and Alegre-Abarrategui J (2021). Tau-proximity ligation assay reveals extensive previously undetected pathology prior to neurofibrillary tangles in preclinical Alzheimer’s disease. Acta Neuropathologica Communications 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, Schiegg M, Ales J, Beier T, Rudy M, et al. (2019). ilastik: interactive machine learning for (bio)image analysis. Nature Methods 16, 1226–1232. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt R, Léger J, and Lee G (1995). Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol 131, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broccolini A, Engel WK, Alvarez RB, and Askanas V (2000). Paired helical filaments of inclusion-body myositis muscle contain RNA and survival motor neuron protein. Am. J. Pathol 156, 1151–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buée L, Bussière T, Buée-Scherrer V, Delacourte A, and Hof PR (2000). Tau protein isoforms, phosphorylation and role in neurodegenerative disorders11These authors contributed equally to this work. Brain Research Reviews 33, 95–130. [DOI] [PubMed] [Google Scholar]
- Bukar Maina M, Al-Hilaly YK, and Serpell LC (2016). Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JM, Moon SL, Matheny T, and Parker R (2019). RNase L Reprograms Translation by Widespread mRNA Turnover Escaped by Antiviral mRNAs. Mol. Cell 75, 1203–1217.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, and Kaufman PD (2009). A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kanai Y, Cowan NJ, and Hirokawa N (1992). Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 360, 674–677. [DOI] [PubMed] [Google Scholar]
- Criscione SW, Zhang Y, Thompson W, Sedivy JM, and Neretti N (2014). Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics 15, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaval B, and Doxsey SJ (2010). Pericentrin in cellular function and disease. J Cell Biol 188, 181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Corjuc BT, Commins C, Dujardin S, Bannon RN, Corjuc D, Moore BD, Bennett RE, Jorfi M, Gonzales JA, et al. (2018). Tau reduction in the presence of amyloid-β prevents tau pathology and neuronal death in vivo. Brain 141, 2194–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner I, Nguyen T, and Seyfried NT (2017). Enrichment of Detergent-insoluble Protein Aggregates from Human Postmortem Brain. J Vis Exp [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkel PD, Holden MR, Matin N, and Margittai M (2015). RNA Binds to Tau Fibrils and Sustains Template-Assisted Growth. Biochemistry 54, 4731–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R, Crowther RA, Ghetti B, Scheres SHW, and Goedert M (2018). Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R, Crowther RA, Newell KL, Ghetti B, Goedert M, et al. (2019). Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 568, 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, and Scheres SHW (2017). Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galganski L, Urbanek MO, and Krzyzosiak WJ (2017). Nuclear speckles: molecular organization, biological function and role in disease. Nucleic Acids Res 45, 10350–10368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gärtner U, Janke C, Holzer M, Vanmechelen E, and Arendt T (1998). Postmortem changes in the phosphorylation state of tau-protein in the rat brain. Neurobiol. Aging 19, 535–543. [DOI] [PubMed] [Google Scholar]
- Gaspar I, Wippich F, and Ephrussi A (2017). Enzymatic production of single-molecule FISH and RNA capture probes. RNA 23, 1582–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil L, Federico C, Pinedo F, Bruno F, Rebolledo AB, Montoya JJ, Olazabal IM, Ferrer I, and Saccone S (2017). Aging dependent effect of nuclear tau. Brain Research 1677, 129–137. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Crino PB, Lee VM, Eberwine JH, and Trojanowski JQ (1997). Sequestration of RNA in Alzheimer’s disease neurofibrillary tangles and senile plaques. Ann. Neurol 41, 200–209. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Galvin JE, Chiu TS, Lee VM, Masliah E, and Trojanowski JQ (1998). RNA sequestration to pathological lesions of neurodegenerative diseases. Acta Neuropathol. 96, 487–494. [DOI] [PubMed] [Google Scholar]
- Goedert M (2005). Tau gene mutations and their effects. Movement Disorders 20, S45–S52. [DOI] [PubMed] [Google Scholar]
- Goedert M, and Spillantini MG (2000). Tau mutations in frontotemporal dementia FTDP-17 and their relevance for Alzheimer’s disease. Biochim. Biophys. Acta 1502, 110–121. [DOI] [PubMed] [Google Scholar]
- Goedert M, Wischik CM, Crowther RA, Walker JE, and Klug A (1988). Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. U.S.A 85, 4051–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger RJ, Barrass JD, Jacquier A, Rain J-C, and Beggs JD (2009). Physical and genetic interactions of yeast Cwc21p, an ortholog of human SRm300/SRRM2, suggest a role at the catalytic center of the spliceosome. RNA 15, 2161–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graser S, Stierhof Y-D, Lavoie SB, Gassner OS, Lamla S, Le Clech M, and Nigg EA (2007). Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol 179, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardana CG, Mehrabian M, Wang X, Mueller I, Lubambo IB, Jonkman JEN, Wang H, and Schmitt-Ulms G (2015). The Human Tau Interactome: Binding to the Ribonucleoproteome, and Impaired Binding of the Proline-to-Leucine Mutant at Position 301 (P301L) to Chaperones and the Proteasome. Mol. Cell Proteomics 14, 3000–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie CR, Greenup L, Leverenz JB, and Kraemer BC (2011). MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum Mol Genet 20, 1989–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CM, Dammer EB, Diner I, Yi H, Seyfried NT, Gearing M, Glass JD, Montine TJ, Levey AI, and Lah JJ (2014a). Aggregates of small nuclear ribonucleic acids (snRNAs) in Alzheimer’s disease. Brain Pathol. 24, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CM, Seyfried NT, Dammer EB, Duong D, Yi H, Gearing M, Troncoso JC, Mufson EJ, Thambisetty M, Levey AI, et al. (2014b). U1 small nuclear ribonucleoproteins (snRNPs) aggregate in Alzheimer’s disease due to autosomal dominant genetic mutations and trisomy 21. Mol Neurodegener 9, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanseeuw BJ, Betensky RA, Jacobs HIL, Schultz AP, Sepulcre J, Becker JA, Cosio DMO, Farrell M, Quiroz YT, Mormino EC, et al. (2019). Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease: A Longitudinal Study. JAMA Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes BB, Furman JL, Mahan TE, Yamasaki TR, Mirbaha H, Eades WC, Belaygorod L, Cairns NJ, Holtzman DM, and Diamond MI (2014). Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. U.S.A 111, E4376–4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan L-L, et al. (2010). Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh Y-C, Guo C, Yalamanchili HK, Abreha M, Al-Ouran R, Li Y, Dammer EB, Lah JJ, Levey AI, Bennett DA, et al. (2019). Tau-Mediated Disruption of the Spliceosome Triggers Cryptic RNA Splicing and Neurodegeneration in Alzheimer’s Disease. Cell Rep 29, 301–316.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Deerinck TJ, Ellisman MH, and Spector DL (1994). In vivo analysis of the stability and transport of nuclear poly(A)+ RNA. J. Cell Biol 126, 877–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilik İA, Malszycki M, Lübke AK, Schade C, Meierhofer D, and Aktaş T (2020). SON and SRRM2 are essential for nuclear speckle formation. ELife 9, e60579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Ash PEA, Maziuk BF, Ballance HI, Boudeau S, Abdullatif AA, Orlando M, Petrucelli L, Ikezu T, and Wolozin B (2019). TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol 137, 259–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NR, Condello C, Guan S, Oehler A, Becker J, Gavidia M, Carlson GA, Giles K, and Prusiner SB (2017). Evidence for sortilin modulating regional accumulation of human tau prions in transgenic mice. PNAS 114, E11029–E11036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampers T, Friedhoff P, Biernat J, Mandelkow EM, and Mandelkow E (1996). RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 399, 344–349. [DOI] [PubMed] [Google Scholar]
- Kaufman SK, Sanders DW, Thomas TL, Ruchinskas AJ, Vaquer-Alicea J, Sharma AM, Miller TM, and Diamond MI (2016). Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 92, 796–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong A, Matheny T, Jain S, Mitchell SF, Wheeler JR, and Parker R (2017). The Stress Granule Transcriptome Reveals Principles of mRNA Accumulation in Stress Granules. Molecular Cell 68, 808–820.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella RJ, Kähäri A, Haider S, Zamora J, Proctor G, Spudich G, Almeida-King J, Staines D, Derwent P, Kerhornou A, et al. (2011). Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford) 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss T (2002). Small Nucleolar RNAs: An Abundant Group of Noncoding RNAs with Diverse Cellular Functions. Cell 109, 145–148. [DOI] [PubMed] [Google Scholar]
- Kota KP, Wagner SR, Huerta E, Underwood JM, and Nickerson JA (2008). Binding of ATP to UAP56 is necessary for mRNA export. Journal of Cell Science 121, 1526–1537. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling S-C, Liang TY, Mazur C, et al. (2012). Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci 15, 1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Newman ST, Gard DL, Band H, and Panchamoorthy G (1998). Tau interacts with src-family non-receptor tyrosine kinases. J. Cell. Sci 111 ( Pt 21), 3167–3177. [DOI] [PubMed] [Google Scholar]
- Li H-D, Funk CC, and Price ND (2020). iREAD: a tool for intron retention detection from RNA-seq data. BMC Genomics 21, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, and Götz J (2013). Profiling Murine Tau with 0N, 1N and 2N Isoform-Specific Antibodies in Brain and Peripheral Organs Reveals Distinct Subcellular Localization, with the 1N Isoform Being Enriched in the Nucleus. PLOS ONE 8, e84849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maj M, Gartner W, Ilhan A, Neziri D, Attems J, and Wagner L (2010). Expression of TAU in insulin-secreting cells and its interaction with the calcium-binding protein secretagogin. Journal of Endocrinology 205, 25–36. [DOI] [PubMed] [Google Scholar]
- Maziuk BF, Apicco DJ, Cruz AL, Jiang L, Ash PEA, da Rocha EL, Zhang C, Yu WH, Leszyk J, Abisambra JF, et al. (2018). RNA binding proteins co-localize with small tau inclusions in tauopathy. Acta Neuropathol Commun 6, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: Next-generation image processing for biology. PLOS Biology 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier S, Bell M, Lyons DN, Rodriguez-Rivera J, Ingram A, Fontaine SN, Mechas E, Chen J, Wolozin B, LeVine H, et al. (2016). Pathological Tau Promotes Neuronal Damage by Impairing Ribosomal Function and Decreasing Protein Synthesis. J. Neurosci 36, 1001–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metuzals J, Robitaille Y, Houghton S, Gauthier S, and Leblanc R (1988). Paired helical filaments and the cytoplasmic-nuclear interface in Alzheimer’s disease. J Neurocytol 17, 827–833. [DOI] [PubMed] [Google Scholar]
- Montalbano M, McAllen S, Puangmalai N, Sengupta U, Bhatt N, Johnson OD, Kharas MG, and Kayed R (2020). RNA-binding proteins Musashi and tau soluble aggregates initiate nuclear dysfunction. Nature Communications 11, 4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motameny S, Wolters S, Nürnberg P, and Schumacher B (2010). Next Generation Sequencing of miRNAs – Strategies, Resources and Methods. Genes (Basel) 1, 70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr ME, Sullivan AC, and Frost B (2017). A Brief Overview of Tauopathy: Causes, Consequences, and Therapeutic Strategies. Trends Pharmacol Sci 38, 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasozomenos SC (1995). Nuclear tau immunoreactivity in presenile dementia with motor neuron disease: a case report. Clin Neuropathol 14, 100–104. [PubMed] [Google Scholar]
- Park SA, Ahn SI, and Gallo J-M (2016). Tau mis-splicing in the pathogenesis of neurodegenerative disorders. BMB Rep 49, 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling S-C, Sun E, Wancewicz E, Mazur C, et al. (2011). Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nature Neuroscience 14, 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Q D, Wr M, Q C, F L, and Jn K (2005). Ribosome dysfunction is an early event in Alzheimer’s disease (J Neurosci). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rady RM, Zinkowski RP, and Binder LI (1995). Presence of tau in isolated nuclei from human brain. Neurobiology of Aging 16, 479–486. [DOI] [PubMed] [Google Scholar]
- Raj T, Li YI, Wong G, Humphrey J, Wang M, Ramdhani S, Wang Y-C, Ng B, Gupta I, Haroutunian V, et al. (2018). Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat Genet 50, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswami M, Taylor JP, and Parker R (2013). Altered Ribostasis: RNA-Protein Granules in Degenerative Disorders. Cell 154, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, Lewis J, Carlson G, et al. (2005). Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci 25, 10637–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rino J, Carvalho T, Braga J, Desterro JMP, Lührmann R, and Carmo-Fonseca M (2007). A Stochastic View of Spliceosome Assembly and Recycling in the Nucleus. PLoS Computational Biology 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC, et al. (2014). Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82, 1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santa-Maria I, Varghese M, Ksiezak-Reding H, Dzhun A, Wang J, and Pasinetti GM (2012). Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of Tau protein in aggresomes. J. Biol. Chem 287, 20522–20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid-Burgk JL, Höning K, Ebert TS, and Hornung V (2016). CRISPaint allows modular base-specific gene tagging using a ligase-4-dependent mechanism. Nature Communications 7, 12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder HC, Bernd A, Zahn RK, and Müller WE (1984). Binding of polyribonucleotides and polydeoxyribonucleotides to bovine brain microtubule protein: age-dependent modulation via phosphorylation of high-molecular-weight microtubule-associated proteins and tau proteins. Mech. Ageing Dev 24, 101–117. [DOI] [PubMed] [Google Scholar]
- Sepulveda G, Antkowiak M, Brust-Mascher I, Mahe K, Ou T, Castro NM, Christensen LN, Cheung L, Jiang X, Yoon D, et al. (2018). Co-translational protein targeting facilitates centrosomal recruitment of PCNT during centrosome maturation in vertebrates. ELife 7, e34959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siano G, Varisco M, Caiazza MC, Quercioli V, Mainardi M, Ippolito C, Cattaneo A, and Di Primio C (2019). Tau Modulates VGluT1 Expression. Journal of Molecular Biology 431, 873–884. [DOI] [PubMed] [Google Scholar]
- Spector DL, and Lamond AI (2011). Nuclear Speckles. Cold Spring Harb Perspect Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Kondo K, Chen X, Homma H, Tagawa K, Kerever A, Aoki S, Saito T, Saido T, Muramatsu S-I, et al. (2018). The intellectual disability gene PQBP1 rescues Alzheimer’s disease pathology. Mol Psychiatry 23, 2090–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Brown RH, and Cleveland DW (2016). Decoding ALS: from genes to mechanism. Nature 539, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich G, Salvadè A, Boersema P, Calì T, Foglieni C, Sola M, Picotti P, Papin S, and Paganetti P (2018). Phosphorylation of nuclear Tau is modulated by distinct cellular pathways. Scientific Reports 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PEA, Cook C, Lummertz da Rocha E, Jansen-West K, Frame AA, Citro A, Leszyk JD, et al. (2016). Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Reports 15, 1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquero-Garcia J, Barrera A, Gazzara MR, González-Vallinas J, Lahens NF, Hogenesch JB, Lynch KW, and Barash Y (2016). A new view of transcriptome complexity and regulation through the lens of local splicing variations. ELife 5, e11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violet M, Delattre L, Tardivel M, Sultan A, Chauderlier A, Caillierez R, Talahari S, Nesslany F, Lefebvre B, Bonnefoy E, et al. (2014). A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front Cell Neurosci 8, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violet M, Chauderlier A, Delattre L, Tardivel M, Chouala MS, Sultan A, Marciniak E, Humez S, Binder L, Kayed R, et al. (2015). Prefibrillar Tau oligomers alter the nucleic acid protective function of Tau in hippocampal neurons in vivo. Neurobiol. Dis 82, 540–551. [DOI] [PubMed] [Google Scholar]
- Wagner S, Chiosea S, Ivshina M, and Nickerson JA (2004). In vitro FRAP reveals the ATP-dependent nuclear mobilization of the exon junction complex protein SRm160. J Cell Biol 164, 843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang D, Zhao J, Qu M, Zhou X, He H, and He R (2006). The proline-rich domain and the microtubule binding domain of protein tau acting as RNA binding domains. Protein Pept. Lett 13, 679–685. [DOI] [PubMed] [Google Scholar]
- Wegmann S, Medalsy ID, Mandelkow E, and Müller DJ (2013). The fuzzy coat of pathological human Tau fibrils is a two-layered polyelectrolyte brush. Proc. Natl. Acad. Sci. U.S.A 110, E313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler JM, McMillan P, Strovas TJ, Liachko NF, Amlie-Wolf A, Kow RL, Klein RL, Szot P, Robinson L, Guthrie C, et al. (2019). Activity of the poly(A) binding protein MSUT2 determines susceptibility to pathological tau in the mammalian brain. Science Translational Medicine 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingett SW, and Andrews S (2018). FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischik CM, Novak M, Thøgersen HC, Edwards PC, Runswick MJ, Jakes R, Walker JE, Milstein C, Roth M, and Klug A (1988). Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A 85, 4506–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woerman AL, Aoyagi A, Patel S, Kazmi SA, Lobach I, Grinberg LT, McKee AC, Seeley WW, Olson SH, and Prusiner SB (2016). Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. PNAS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yao J, Wu DC, and Lambowitz AM (2019). Improved TGIRT-seq methods for comprehensive transcriptome profiling with decreased adapter dimer formation and bias correction. Scientific Reports 9, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, and Lee VM-Y (2007). Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53, 337–351. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Kota KP, Alam SG, Nickerson JA, Dickinson RB, and Lele TP (2016). Coordinated dynamics of RNA splicing speckles in the nucleus. J Cell Physiol 231, 1269–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Tarutani A, Newell KL, Murzin AG, Matsubara T, Falcon B, Vidal R, Garringer HJ, Shi Y, Ikeuchi T, et al. (2020). Novel tau filament fold in corticobasal degeneration. Nature 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lin Y, Eschmann NA, Zhou H, Rauch JN, Hernandez I, Guzman E, Kosik KS, and Han S (2017). RNA stores tau reversibly in complex coacervates. PLoS Biol 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yan C, Zhan X, Li L, Lei J, and Shi Y (2018). Structure of the human activated spliceosome in three conformational states. Cell Research 28, 307–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimowska G, Shi J, Munguba G, Jackson MR, Alpatov R, Simmons MN, Shi Y, and Sugrue SP (2003). Pinin/DRS/memA Interacts with SRp75, SRm300 and SRrp130 in Corneal Epithelial Cells. Invest. Ophthalmol. Vis. Sci 44, 4715–4723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Video 1: 3D rotation of a mouse brain nucleus, related to Figurs 1. Nucleus is shown in blue (DAPI), AT8 phosphorylated tau is shown in green, and poly(A) RNA is shown in red.
Supplemental Video 2: 3D rotation of an H4 cells expressing 0N4R tau-YFP showing intranuclear tau aggregates, related to Figure 1. Nucleus is shown in blue (DAPI) and Tau-YFP is shown in green.
Supplemental Excel Table 1: HEK293 tau aggregate RNA sequencing data, related to Figurs 2.
Supplemental Excel Table 2: Mouse Tau aggregate RNA enrichment score, related to Figure 2.
Supplemental Excel Table 3: patient demographics for human brain samples. Related to Figure 7 and STAR Methods.
Supplemental Excel Table 4: Splicing analysis. Sheet1: MAJIQ splicing analysis of RNA seq from cells with and without tau aggregates. Sheet 2: iREADs intron retention analysis from the same experiment. Related to Figure 6.
Supplemental Excel Table 5: FISH probe sequence information. Related to Figures 1, 3 and STAR Methods.
Data Availability Statement
Primary sequencing data is deposited on the Gene Expression Omnibus under the accession GSE148716.