

Abstract
Protein kinase C α (PKCα) is a ubiquitously expressed member of the PKC family of serine/threonine kinases with diverse functions in normal and neoplastic cells. Early studies identified anti-proliferative and differentiation-inducing functions for PKCα in some normal tissues (e.g., regenerating epithelia) and pro-proliferative effects in others (e.g., cells of the hematopoietic system, smooth muscle cells). Additional well documented roles of PKCα signaling in normal cells include regulation of the cytoskeleton, cell adhesion, and cell migration, and PKCα can function as a survival factor in many contexts. While a majority of tumors lose expression of PKCα, others display aberrant overexpression of the enzyme. Although cancer-related mutations of PKCα are uncommon, rare examples of driver mutations have been detected in certain cancer types (e. g., chordoid gliomas). Here we review the role PKCα in various cancers, describe mechanisms by which PKCα affects cancer-related cell functions, and discuss how the diverse functions of PKCα contribute to tumor suppressive and tumor promoting activities of the enzyme. We end the discussion by addressing mutations and expression of PKCα in tumors and the clinical relevance of these findings.
Keywords: Protein kinase C (PKC), tumor suppression, tumor promotion, migration, metastasis, survival, chemoresistance
1. Introduction
Protein kinase C α (PKCα), which is encoded by the PRKCA gene, is a founding member (Parker et al., 1986) of a family of 11 serine/threonine kinases collectively termed protein kinase C (PKC). PKC was initially isolated as a protease-activated kinase from rat brain (Inoue et al., 1977; Takai et al., 1977). However, with cloning of the enzyme (Coussens et al., 1986; Parker et al., 1986), it became apparent that PKC is a family of isozymes that show strong homology in their C-terminal kinase domains, but differ in their regulatory domain and in their cofactor requirements (Newton, 2001, 2018). Along with PKCβI, PKCβII, and PKCγ, PKCα is a member of the conventional or classical subfamily (cPKCs), which are dependent on phosphatidylserine, calcium and diacylglycerol for activity. The other PKC subfamilies are the novel PKCs, PKCδ, PKCε, PKCη and PKCθ, which are calcium-independent, and the atypical PKCs, PKCι\λ and PKCζ, which require neither calcium nor DAG for activity.
Physiological activation of PKCα and the other classical and novel PKCs involves generation of diacylglycerol (DAG), primarily by phospholipase C-mediated cleavage of membrane phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) downstream of tyrosine kinase and G protein-coupled receptors (Becker and Hannun, 2005; Newton, 2018). Hydrolysis of PI(4,5)P2 also generates inositol 1,4,5-trisphosphate (IP3) which leads to increased cytosolic Ca2+, an essential cofactor for the conventional PKCs (Newton, 2018). Under resting conditions, PKCα is maintained in an inactive conformation in the cytosol. DAG recruits PKCα to the membrane, and membrane association activates the enzyme by inducing a conformational change that releases an autoinhibitory N-terminal pseudosubstrate domain from the active site. PKCα signaling is terminated by depletion of membrane DAG by the actions of diacylglycerol kinases and lipases. DAG metabolism results in release of the enzyme from the membrane into the cytosol and restoration of the inactive conformation. Thus, membrane association is the activating event for PKCα and its activity in cells can be monitored by its translocation from the cytosol to the membrane. Several pharmacological agonists can interact with the DAG binding site and activate classical and novel PKCs, including tumor promoting phorbol esters, such as phorbol 12-myristate 13-acetate (PMA, also known as 12-O-tetradecanoylphorbol 13-acetate (TPA)) and bryostatins (Newton, 2018).
In addition to membrane binding, activation of PKCα is dependent on phosphorylation at three sites located in the activation loop (Thr497), turn motif (Thr638), and hydrophobic motif (Ser657) of the enzyme (Keranen et al., 1995; Newton, 2018). Phosphorylation both stabilizes the protein and primes it for activation. However, unlike other kinases such as AKT and ERK, phosphorylation at these sites is part of the initial processing of the enzyme and is not indicative of activation per se. Despite excellent reviews by the Newton group (Newton, 2001, 2018), there is considerable confusion regarding the role of these phosphorylations, and multiple reports equate priming site phosphorylation with activation of PKCα. Part of this confusion likely arises from 32PO4 incorporation studies indicating that PKCα is phosphorylated upon activation (Lee et al., 1996; Mitchell et al., 1989; Ng et al., 1999b); however, this phosphorylation occurs on Thr250 and Ser260 (Ng et al., 1999b) and not at priming sites. Thus, priming site phosphorylation cannot be used as a marker of PKCα activation, with translocation of the primed protein to the membrane and substrate phosphorylation providing the best indication of its activation.
Signal termination of PKCα can occur through diacylglycerol metabolism or through agonist-induced downregulation, a key aspect of PKC biology. Early studies, mainly using overexpressed protein and PMA or bryostatin, led to a model where prolonged activation results in caveolar internalization, priming site dephosphorylation, and subsequent proteasomal degradation of PKCα (Hansra et al., 1999; Lee et al., 1996; Prevostel et al., 2000). However, studies in multiple cell types by our group and others have identified at least two distinct pathways of endogenous PKCα downregulation, with the prevailing earlier model likely reflecting a combination of these mechanisms (Leontieva and Black, 2004; Lum et al., 2013a; Lum et al., 2013b; Prevostel et al., 2000). PMA can fully activate endogenous PKCα without leading to dephosphorylation of the enzyme (Lee et al., 1996; Leontieva and Black, 2004; Lum et al., 2013a); rather, prolonged PMA treatment elicits proteasomal degradation of the fully phosphorylated active enzyme at the plasma membrane (Lum et al., 2013a; Lum et al., 2013b). This is also the predominant mechanism of downregulation elicited by bryostatin. However, bryostatin also triggers clathrin-independent internalization of endogenous PKCα to a perinuclear compartment where the fully primed enzyme undergoes delayed lysosomal degradation (Leontieva and Black, 2004; Lum et al., 2013b), a mechanism that is consistent with extensive evidence for a role of PKCα in regulation of membrane trafficking in cells (Alvi et al., 2007). A small proportion of the internalized enzyme undergoes complete desphosphorylation and rapid proteasomal degradation in the perinuclear region (Leontieva and Black, 2004; Lum et al., 2013b). While PMA induces downregulation of endogenous PKCα only at the plasma membrane, it can also channel exogenously overexpressed protein to the perinuclear/lysosomal degradation pathway (Lum et al., 2013b), providing an explanation for the PMA-induced dephosphorylation of PKCα observed in previous studies (Prevostel et al., 2000). Surprisingly, our work also determined that PKCα (but not PKCδ and PKCε) is highly resistant to downregulation following prolonged diacylglycerol-mediated activation (Lum et al., 2016). This sustained activation of PKCα in the presence of its physiological activator is in keeping with the prolonged activation/membrane association of PKCα seen in growth-arrested, differentiated cells in epithelial tissues such as intestine (Frey et al., 1997; Saxon et al., 1994; Verstovsek et al., 1998), skin (Tibudan et al., 2002), and endometrium (Hsu et al., 2018) (see Figure 1).
Figure 1.

Immunohistochemical analysis of PKCα distribution in normal epithelial tissues.
A. Mouse small intestine. PKCα (brown staining) is diffusely distributed in the cytoplasm of proliferating crypt cells (arrowheads), indicating that it is inactive in these cells. However, the enzyme becomes activated/membrane-associated coincident with growth arrest at the crypt/villus junction, a pattern that is maintained in post-mitotic and differentiating cells of the villus (arrow). B. Murine vaginal squamous epithelium. PKCα is inactive/cytoplasmic in proliferating basal cells, but is activated/membrane-associated in the non-proliferative suprabasal cells. C. Mouse endometrium. Ki67 positive (dark nuclear staining in upper right panel) proliferating cells (arrows) express low levels of cytoplasmic staining for PKCα (upper left panel, arrows), whereas non-proliferative Ki67 negative cells show robust membrane staining for PKCα (arrowheads). The lower panel is a higher magnification image of non-proliferating cells showing membrane association of PKCα.
The realization that PKCs are the major cellular receptors for tumor promoting phorbol esters led to the idea that these enzymes act as oncogenes; however, subsequent studies have revealed that the role of these kinases in cancer is considerably more complex, with individual PKC isozymes having diverse and often opposing effects dependent on context (Black and Black, 2012; Black, 2001). In this review, we outline the role and regulation of PKCα in various cancer types, describe mechanisms by which PKCα affects cancer-related cell functions, and address the clinical relevance of these findings. We have concentrated on studies where the involvement of PKCα has been confirmed through knockdown, genetic deletion, overexpression and/or use of inhibitors. Inevitably, this approach will miss some functions of PKCα where studies have not identified the specific isozyme mediating “effects of PKC”.
2. PKCα signaling in regulation of cell proliferation, differentiation, and tumorigenesis
The involvement of PKCα in regulation of cell proliferation, differentiation and tumorigenesis has been investigated in normal cells and tissues and in multiple tumor types. Together, these studies highlight the complexity and context-dependence of the effects of PKCα on these processes, with opposite effects seen in different systems and even within the same tissue and tumor type. While PKCα signaling promotes cell cycle progression and tumorigenesis in some contexts, it can mediate growth arrest, differentiation, and tumor suppression in others. This complexity suggests a function of PKCα as a molecular sensor that can promote or repress cell proliferation and differentiation in response to specific environmental cues. In this vein, it has been suggested that the responses induced by PKCα are not an intrinsic property of the kinase, but instead reflect dynamic interactions of the kinase with cell type-specific factors such as anchoring proteins, modulators, and substrates (Nakashima, 2002). In the following sections, we discuss current understanding of the contribution of PKCα to these processes in different tissue types.
2.1. Roles in normal tissues
As discussed in more detail in the sections below, PKCα signaling has pro-proliferative functions in some normal tissues and growth inhibitory/differentiation-inducing effects in others. We note that in-depth studies of PKCα function in normal tissues are limited, a caveat that should be kept in mind in synthesizing currently available data. The most detailed analysis of growth regulatory roles of PKCα in normal cells comes from studies in the hematopoietic system and in regenerating epithelial tissues, including the intestinal epithelium, the epidermis, and the endometrial epithelium. While PKCα appears to have both pro- and anti-proliferative/differentiation-inducing effects in cells of the immune system (see Section 2.2), the enzyme predominantly regulates anti-proliferative and tumor suppressive signaling in regenerating epithelia (see Section 2.3). Additional support for pro-proliferative effects of the kinase in normal tissues comes from studies in muscle cells, Schwann cells, and osteoblasts. Foegh and colleagues showed that a single dose of antisense oligonucleotide targeting PKCα caused long-lasting (at least 4 days) growth inhibition of vascular smooth muscle cells (Leszczynski et al., 1996), a finding that was subsequently confirmed by others (Jiang et al., 2017; Okazaki et al., 2000; Shaikh et al., 2013). Consistent with high expression of PKCα in myometrial cells, the kinase is required for endothelin-1-induced proliferation of these cells (Eude et al., 2002; Tertrin-Clary et al., 1999). PKCα activation enhances the proliferation of Schwann cells following peripheral nerve injury, through an ERK-dependent mechanism (Li et al., 2020), and promotes the proliferation of primary human osteoblasts (Lampasso et al., 2002), while suppressing osteoblastic differentiation (Nakura et al., 2011). There is little information on the involvement of PKCα signaling in regulation of cell proliferation and differentiation in the normal mammary gland, liver, and lung. However, extensive evidence supports a tumor promoting role of the kinase in breast cancer and hepatocellular carcinoma, while results are mixed for lung cancer.
2.2. The hematopoietic system and hematological malignancies
PKCα signaling has been linked to regulation of both cell proliferation and differentiation in normal hematopoietic cells. An early study using transgenic mice overexpressing wild-type PKCα in thymocytes pointed to a pro-proliferative role in these cells (Iwamoto et al., 1992). Although no proliferative effects were observed in vivo, isolated PKCα-overexpressing thymocytes showed markedly enhanced proliferation and IL-2 production in response to a weak anti-CD3 antibody stimulus, in association with membrane association/activation of the overexpressed enzyme. Studies in PKCα knockout mice revealed a marked reduction in T cell proliferative responses, while B cell proliferation was unaffected (Pfeifhofer et al., 2006). PKCα also appears to cooperate with PKCθ to regulate T cell proliferation: while mice lacking either PKCα or PKCθ showed only a mild activation defect in a graft-versus-host model, double PKCα/PKCθ knockout mice had a severe defect in alloreactive T cell proliferation (Gruber et al., 2009). In other studies, PKCα was identified as a major effector of cytokine (stem cell factor and erythropoietin)-induced proliferative signaling in erythroid progenitor cells (Haslauer et al., 1999). In contrast, expression of constitutively active PKCα provided a signal for differentiation of primary hematopoietic progenitor cells into macrophages, mimicking the effects of macrophage colony-stimulating factor (Pierce et al., 1998). Furthermore, overexpression of PKCα, but not PKCβII, ε, ζ, or η, induced differentiation of a mouse myeloid progenitor cell line into mature macrophages in response to PMA stimulation (Mischak et al., 1993).
Several groups have addressed the role of PKCα in hematological malignancy. Interestingly, in all cases, the effects of PKCα were associated with growth inhibition and tumor suppression. Using mouse models, the Michie laboratory showed that subversion of PKCα signaling in hematopoietic progenitor cells by stable expression of kinase dead PKCα (PKCα K368R) results in the development of B cell chronic lymphocytic leukemia (CLL)-like disease, with PKCα-deficient cells phenotypically resembling human B-CLL cells (Nakagawa et al., 2006). Follow-up studies revealed that PKCα deficiency promotes lineage plasticity in B-committed precursors by perturbing critical B-lineage transcription factors, pointing to downregulation of PKCα expression/activity as a mechanism for lineage transdifferentiation in B-cell malignancy (Nakagawa et al., 2012). Further evidence for a suppressive role of the enzyme was provided by a series of studies by the Fields group. Their work determined that PKCα promotes cytostasis and megakaryocytic differentiation of K562 human erythroleukemia cells (Hocevar et al., 1992; Murray et al., 1993) and that the effect is mediated by isozyme-specific sequences within the catalytic domain (Walker et al., 1995). Studies by the Schwende group (Dieter and Schwende, 2000) demonstrated the ability of PKCα to promote the differentiation of THP-1 monocyte-like cells (derived from a childhood case of acute monocytic leukemia) into macrophage-like cells, with antisense-induced deficiency of the enzyme enhancing the proliferation of these cells. Additional studies indicated that sustained activation of PKCα is required for differentiation of HL-60 promyelocytic leukemia cells into macrophages (Aihara et al., 1991). Consistent with a tumor suppressive role of PKCα in patients, a recent study identified loss of PKCα as an indicator of relapse and very poor outcome in pediatric T-cell acute lymphoblastic leukemia (T-ALL) patients (Milani et al., 2014).
2.3. Regenerating epithelial tissues
2.3.1. The role of PKCα signaling in maintenance of intestinal homeostasis and in the development of intestinal/colon cancer
The contribution of PKCα signaling to maintenance of intestinal epithelial homeostasis has been extensively studied by our group and others. Using a combined morphological and biochemical approach, we demonstrated that (a) PKCα is cytosolic/inactive in proliferating cells of intestinal and colonic crypts of mice, rats, and humans, and (b) the enzyme undergoes plasma membrane association, indicative of kinase activation, coincident with cell growth arrest in the mid to upper crypt region (Frey et al., 2000; Frey et al., 1997; Hao et al., 2011; Saxon et al., 1994; Verstovsek et al., 1998), a finding that was confirmed by others (Jiang et al., 1995) (Figure 1A). Follow-up studies revealed that PKCα activation triggers a coordinated program of molecular events leading to intestinal epithelial cell cycle withdrawal into G0 (Frey et al., 2000). PKCα induces the expression of the cyclin-dependent kinase (CDK) inhibitors p21Cip1 and p27Kip1 and rapidly downregulates D-type cyclins, thereby inhibiting the activity of all major G1/S cyclin/CDK complexes and inducing changes in the pocket proteins, p107, pRb, and p130, that drive cells to exit the cell cycle (Frey et al., 2000). This program requires sustained PKCα activity and prolonged stimulation of the ERK/MAP pathway (Clark et al., 2004). PKCα downregulates cyclin D1 via two mechanisms: blockade of cyclin D1 translation initiation by PP2A-mediated hypophosphorylation/activation of the translational repressor, 4E-BP1, and inhibition of cyclin D1 transcription (Guan et al., 2007; Hizli et al., 2006; Pysz et al., 2014; Pysz et al., 2009). Studies by our group further showed that PKCα suppresses the expression of members of the Inhibitor of DNA Binding family, Id1, Id2, Id3, and Id4, in intestinal epithelial cells via an ERK-dependent mechanism (Hao et al., 2011), a function that has been noted in other systems (Akakura et al., 2010; Hill et al., 2014). Since Id1 has mitogenic functions, this finding further supports the growth-suppressive activity of PKCα in intestinal tissue.
Extensive evidence supports a tumor suppressive role for PKCα in the intestine and colon. The enzyme is commonly lost in colorectal cancer (Kahl-Rainer et al., 1994; Kahl-Rainer et al., 1996; Suga et al., 1998; Verstovsek et al., 1998) and in murine models of intestinal neoplasia (Klein, I. K. et al., 2000; Oster and Leitges, 2006) (Figure 2A). PKCα deficiency is seen in aberrant crypt foci and pre-malignant adenomas (Gökmen-Polar et al., 2001; Kahl-Rainer et al., 1996; Klein, Irene K. et al., 2000; Oster and Leitges, 2006; Pysz et al., 2009), indicating that expression of the enzyme is suppressed early during tumor development (Figure 2A). Restoration of PKCα expression/activity markedly suppresses anchorage-independent growth of human colon cancer cell lines in vitro and tumor formation in vivo, independent of the status of the APC/β-catenin pathway and other known genetic alterations in colon tumors (Batlle et al., 1998; Pysz et al., 2009). In contrast, PKCα deficiency increases cell proliferation, decreases differentiation, and enhances the transformed phenotype of colon cancer cells (Abraham et al., 1998; Scaglione-Sewell et al., 1998). Strong support for the tumor suppressive effects of PKCα comes from studies by the Leitges group using PKCα knockout mice and the ApcMin/+ mouse model of intestinal neoplasia (Oster and Leitges, 2006). PKCα deficiency enhanced crypt cell mitotic index and promoted the development of spontaneous intestinal tumors even in wild-type mice (i.e., in the absence of the Min mutation). In ApcMin/+ mice, loss of PKCα led to an increased number of tumors in the intestine, with lesions displaying a more aggressive histopathological phenotype characterized by invasion into the muscle layers, central necrotic areas, and less differentiated structure. Consistent with the development of more advanced disease, PKCα-deficient ApcMin/+ mice died significantly earlier than their PKCα-expressing littermates. Mechanistic analysis pointed to a role of PKCα in negative regulation of the epidermal growth factor receptor (EGFR) pathway (see Section 2.8.1), while excluding a direct effect of PKCα activity on WNT/APC signaling in this model. Studies by Oh and colleagues, on the other hand, indicate that PKCα signaling can inhibit the WNT/β-catenin pathway by phosphorylating N-terminal residues (Ser33/Ser37/Thr41) in β-catenin, marking it for degradation (Gwak et al., 2006; Gwak et al., 2009). The same group identified a small molecule activator of PKCα, CGK062, that promotes PKCα-mediated phosphorylation of β-catenin at Ser33/Ser37 and downregulation of the protein, thereby suppressing cell proliferation (Gwak et al., 2012). Another study determined that PKCα-mediated phosphorylation of the orphan nuclear receptor, RORα, at Ser35 blocks β-catenin transcriptional activity in colon cancer cells (Lee et al., 2010). Additional studies are needed to unravel the molecular mechanisms underlying the tumor suppressive effects of PKCα in intestinal cells. Nonetheless, the tumor suppressive effects of PKCα are relevant to human disease since loss of PKCα is an early event in colon tumorigenesis and is associated with advanced disease and poor prognosis in patients (Chen et al., 2016; Kahl-Rainer et al., 1994; Kahl-Rainer et al., 1996; Suga et al., 1998; Verstovsek et al., 1998).
Figure 2.
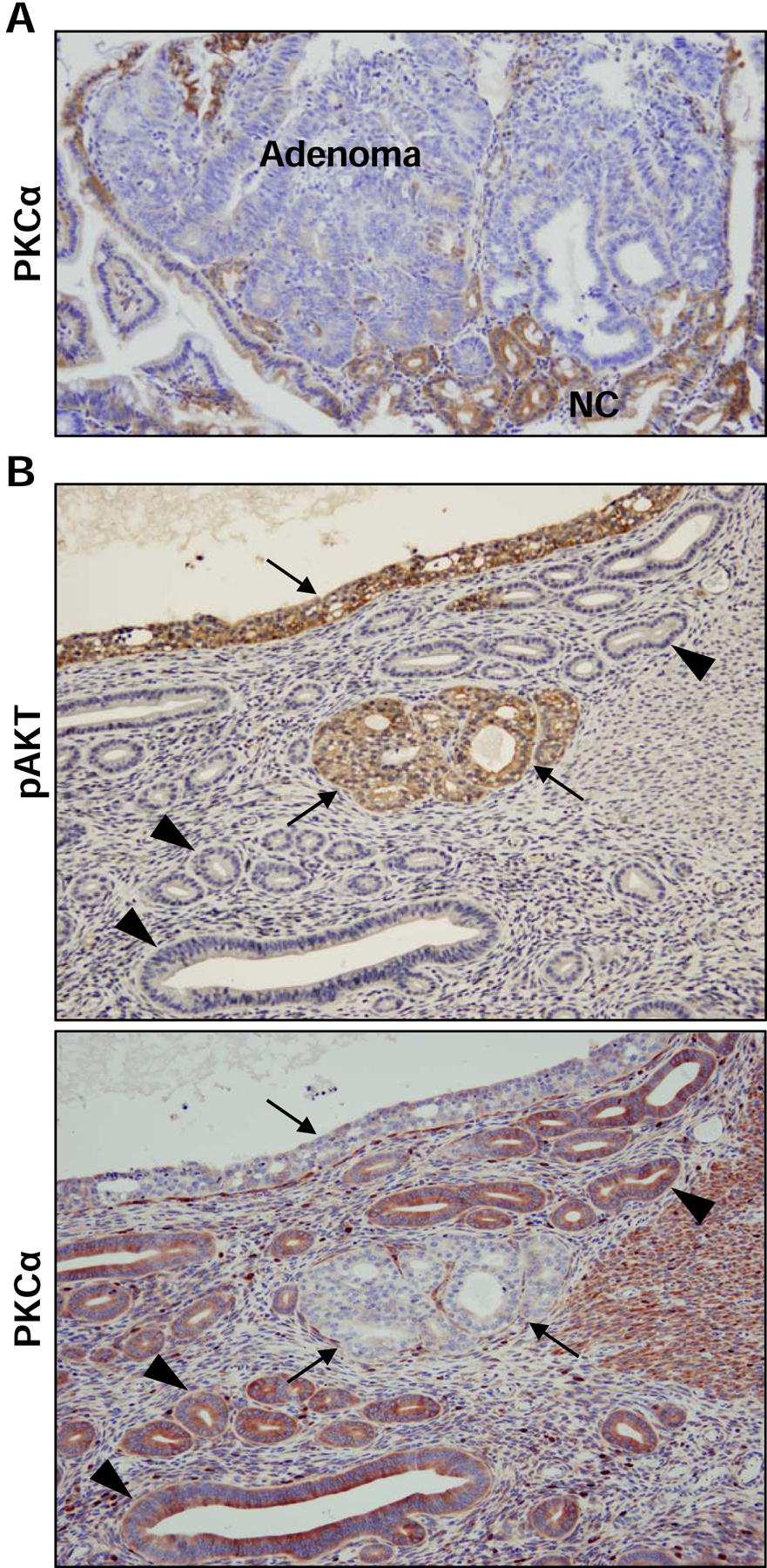
Immunohistochemical analysis of PKCα expression in epithelial tumors.
A. APCmin/+ mouse intestine. Note the lack of PKCα staining in adenoma tissue which contrasts with the robust staining of the surrounding normal epithelium. NC: normal crypts. B. PTEN 4−5/+mouse endometrium. Upper panel: Note the robust phospho-AKT (pAKT) staining that is characteristic of hyperplastic tumors (arrows) but not normal epithelium (arrowheads) in this model of endometrial cancer. Lower panel: While PKCα is clearly detected in the phospho-AKT negative normal epithelium (arrowheads), it is uniformly absent from phospho-AKT positive hyperplastic tumors (arrows).
2.3.2. Functions in the epidermis and skin carcinogenesis
Cell proliferation is restricted to the basal layer in the normal epidermis. Terminal differentiation involves detachment of keratinocytes from the basement membrane, migration into the suprabasal compartment, irreversible exit from the cell cycle, and expression of differentiation-specific genes. A role for PKCα in growth arrest and differentiation of normal epidermal cells has been reported by several groups. Early studies by Yuspa and colleagues (Denning et al., 1995) pointed to PKCα as a major player in the induction of differentiation markers during Ca2+-induced keratinocyte differentiation, a role that was confirmed by the Bilke group using PKCα-specific antisense oligonucleotides (Yang et al., 2003). Consistent with this role, immunohistochemical analysis by the Denning laboratory demonstrated PKCα membrane translocation/activation and substrate phosphorylation in suprabasal keratinocytes of the human epidermis, in association with differentiation-associated growth arrest (Bollag, 2009; Tibudan et al., 2002), a pattern also seen in the murine vaginal squamous epithelium (Figure 1B) and reminiscent of that seen for PKCα in intestinal crypts (Section 2.3.1). Additional studies by this group confirmed a role for PKCα in triggering irreversible cell cycle withdrawal in keratinocytes through coordinated changes in cell cycle regulatory molecules (Jerome-Morais et al., 2009; Tibudan et al., 2002). As in intestinal cells, these changes included induction of the CDK inhibitors p21Cip1 and p27Kip1 and activation of pocket proteins. A growth inhibitory role is further supported by PKCα knockdown studies in human keratinocyte organotypic raft cultures, which resulted in increased basal and suprabasal cell proliferation, decreased differentiation and reduced epidermal stratification (Bollag, 2009; Jerome-Morais et al., 2009). Interestingly, TGFβ-induced growth inhibition of normal human keratinocytes appears to be mediated by PKCα via a mechanism that involves phosphorylation of the EF-hand Ca2+-binding protein, S100C/A11, for p21Cip1 induction (Sakaguchi et al., 2004).
Although the preponderance of evidence points to a tumor suppressive role for PKCα in the skin, the role of the enzyme in skin carcinogenesis remains unclear. PKCα is downregulated in papillomas arising in wild-type mice subjected to the two-stage protocol of skin carcinogenesis, which involves a single, sub-carcinogenic application of the chemical initiator mutagen 7,12-dimethylbenz[a]anthracene (DMBA) followed by repeated applications of TPA (Hara et al., 2005). PKCα knockout mice show enhanced papilloma formation in this model and are also significantly more susceptible to skin tumor development by repeated DMBA treatment (Hara et al., 2012), pointing to a key role of PKCα loss in skin carcinogenesis. On the other hand, studies with targeted overexpression of PKCα in the epidermis have given inconsistent results. One group found that PKCα overexpression enhances CXCR2-dependent tumor formation in response to DMBA/TPA treatment (Cataisson et al., 2016; Cataisson et al., 2009), while others have found no effect on tumor development (Jansen et al., 2001; Wang and Smart, 1999). Overexpression of PKCα in the mouse epidermis enhances cytokine signaling (Cataisson et al., 2003; Cataisson et al., 2009; Wang and Smart, 1999), cell differentiation (Palazzo et al., 2017) and apoptosis (Cataisson et al., 2003), and the balance of these effects under different experimental conditions may explain the discrepancies between these studies. Furthermore, consistent with loss of PKCα in papillomas of wild-type mice, expression of the PKCα transgene was suppressed in tumors that formed in the mutant mice (Wang and Smart, 1999), providing a possible explanation for the lack of phenotype in some PKCα overexpression models. Additional studies will be required to resolve these inconsistent results. Interestingly, PKCα expression is downregulated in patient samples of basal cell carcinoma and PKCα deficiency may be associated with increased Gli1 transcriptional activity in these lesions (Neill et al., 2003).
2.3.3. The role of PKCα in endometrial epithelial cells and endometrial cancer
Immunohistochemical analysis by our group showed that PKCα is expressed in the proliferative and secretory endometrial epithelium during the human menstrual cycle and that PKCα expression and activation are dynamically regulated in the murine uterine epithelium during the estrus cycle (Hsu et al., 2018). Levels are generally low in metestrus and diestrus and the enzyme is maintained in an inactive state during these phases. However, coincident with progesterone-induced growth arrest and differentiation during proestrus/estrus, PKCα expression levels markedly increase and the enzyme is robustly activated, as indicated by strong plasma membrane association (Figure 1C). Thus, PKCα activation appears to be positioned to mediate anti-proliferative signaling in the normal murine endometrial epithelium.
Analysis of the effects of PKCα in endometrial cancer cells in vitro has given conflicting results (Haughian and Bradford, 2009; Haughian et al., 2009; Hsu et al., 2018; Thorne et al., 2013; Wu et al., 2005). A series of studies by the Bradford group supports a pro-proliferative role of PKCα in these cells. PKCα knockdown in several endometrial cancer cell lines resulted in reduced growth, formation of fewer colonies in soft agar, and impaired xenograft tumor formation in mice (Haughian and Bradford, 2009; Haughian et al., 2009). Constitutively active PKCα, in turn, potentiated estrogen receptor-dependent transcription and increased proliferation of Ishikawa cells (Thorne et al., 2013). However, another study in Ishikawa cells (Wu et al., 2005) indicated that PKCα signaling does not play a significant role and studies in RL95–2 endometrial cancer cells have linked PKCα to retinoic acid-induced differentiation (Carter et al., 1998). Furthermore, work by our group showed potent suppression of anchorage-independent growth by PKCα in a large panel of endometrial cancer cell lines (Hsu et al., 2018). While additional studies are required to resolve these conflicting results, anti-proliferative effects of PKCα are consistent with our finding that PKCα signaling is tumor suppressive in the endometrium (Hsu et al., 2018). Loss of PKCα is an early event in mutant PTEN-driven endometrial tumorigenesis in mice and PKCα deficiency accelerated tumor formation in this model (Hsu et al., 2018) (Figure 2B). Our data point to disruption of a PKCα→PP2A-family phosphatase⊣AKT signaling axis as a key step in endometrial tumor initiation, contributing to robust, unopposed AKT activation and promoting transformation of endometrial cells. This finding is consistent with evidence from Leitges and colleagues that knockout of PKCα enhances PI3K-AKT signaling in mice (Leitges et al., 2002). PKCα is also lost at both the mRNA and protein levels in early stage human endometrial cancers, with loss increasing with more advanced stage (Hsu et al., 2018). PKCα deficiency predicts worse patient outcome, supporting the clinical relevance of its tumor suppressive effects in this tissue (Hsu et al., 2018).
2.4. PKCα signaling in the lung
Limited evidence supports a pro-proliferative role of PKCα signaling in lung cancer cells. PKCα expression is upregulated in many human non-small cell lung cancer (NSCLC) cell lines (Clark et al., 2003; Salama et al., 2019) and some lung tumor tissues (Wang et al., 2013) compared with primary human lung epithelial cells or adjacent normal lung tissue, respectively. In addition, loss of PKCα in A549 NSCLC cells by treatment with PKCα-specific siRNA or transfection of miR203, a direct regulator of PKCα expression, suppressed cell proliferation (Wang et al., 2013), while overexpression of the kinase had the opposite effect (Wang et al., 2013). PKCα silencing experiments further revealed that NSCLC cells expressing mutant EGFR rely on PKCα for activation of the mTORC1 signaling pathway and to facilitate growth under serum-free conditions (Salama et al., 2019).
In contrast to these findings, other studies support an anti-proliferative, tumor suppressive role of PKCα in the lung. The Kazanietz group demonstrated that S-phase-specific activation of endogenous PKCα in NSCLC cells results in p21Cip1 upregulation, G2/M cell cycle arrest, and cell senescence (Oliva et al., 2008). The link between PKCα signaling and induction of senescence observed in this study has been confirmed by others (Akakura et al., 2010). Analysis of PKCα in gene expression datasets by Fields and colleagues revealed significant downregulation of the enzyme in the three major subtypes of human NSCLC (adenocarcinoma, squamous cell, and large cell carcinoma), with PKCα loss increasing with tumor stage (Hill et al., 2014). Immunohistochemical analysis of patient tissues and lung adenomas arising in the LSL-Kras murine lung cancer model confirmed that loss of PKCα is a frequent event in NSCLC (Hill et al., 2014). Furthermore, genetic deletion of PKCα in three murine lung adenocarcinoma models (LSL-Kras, LA2-Kras, and urethane exposure) significantly increased tumor number, size, and aggressiveness, while promoting progression from adenoma to carcinoma and reducing mouse survival (Hill et al., 2014). Consistent with the findings of Kazanietz et al. (Oliva et al., 2008), PKCα deficiency also resulted in bypass of oncogene-induced senescence. Loss of PKCα expanded the tumor-initiating bronchio-alveolar stem cell population, both in vitro and in vivo, an effect that was attributed to relief from a PKCα→p38MAPK⊣TGFβ signaling axis that suppresses Id protein expression, inhibits cell proliferation, and regulates KRAS-induced senescence. The basis for the opposing activities of PKCα in lung cancer cells remains to be defined in future studies and may reflect inherent differences in the lung cancer subtypes analyzed (e.g., tumors with wild-type or activating mutations of EGFR).
2.5. The mammary epithelium and breast cancer cells
Limited studies in normal and non-transformed mammary epithelial cells provide conflicting data on the role of PKCα in regulation of cell growth and differentiation in mammary tissue. Elevated levels of PKCα in COMMA-1D mouse mammary epithelial cells resulted in anchorage-independent growth in vitro and tumorigenesis in vivo (Zeng et al., 2002). In contrast, overexpression of PKCα in non-tumorigenic MCF10A human mammary epithelial cells suppressed cell proliferation, with cells accumulating in G1 phase of the cell cycle likely as a result of increased p27Kip1 expression (Sun and Rotenberg, 1999). It should be noted, however, that the validity of MCF10A cells as a model of normal mammary epithelial cells has been questioned (Qu et al., 2015). Thus, in depth studies of the expression, subcellular distribution and function of PKCα in normal mammary epithelium are required to resolve these conflicting findings and determine the role of PKCα in normal mammary biology.
Analysis of PKCα function in breast cancer cells provides a somewhat clearer picture. One of the earliest reports of pro-proliferative effects of PKCα in any system resulted from analysis of breast cancer cells by the Parker group (Ways et al., 1995). Ectopic overexpression of PKCα in PKCα-low MCF7 breast cancer cells enhanced their proliferative rate and tumorigenic properties in vitro and in nude mice. Notably, PKCα overexpression reduced estrogen receptor levels, indicating that PKCα may regulate the switch from ER-positive to ER-negative status in breast cancer. Tonetti et al. confirmed these intriguing results in a series of compelling studies. Stable expression of PKCα in hormone-dependent T47D breast cancer cells resulted in reduced ER function, increased proliferation rate, and hormone-independent growth that could not be inhibited by the antiestrogen tamoxifen (Chisamore et al., 2001; Tonetti et al., 2000; Tonetti et al., 2003). PKCα may engage an AP-1-Notch-4 signaling pathway to promote ER-independent, tamoxifen-resistant proliferation of breast cancer cells (Yun et al., 2013). A caveat of these findings, discussed by the authors of both reports, is that overexpression of PKCα altered the levels of several other members of the PKC family, with marked upregulation of PKCβ observed by both groups. However, a positive role for PKCα signaling in breast cancer cell proliferation was demonstrated more recently by the Larsson group through PKCα knockdown experiments in MDA-MB-231 cells (Lonne et al., 2010). These investigators also confirmed the association between PKCα expression and ER negativity/increased proliferative activity of breast cancer cells, as well as poor survival of breast cancer patients. Interestingly, PKCα can be upregulated and activated by ErbB2 in breast cancer cells through a Src-dependent mechanism, and ErbB2 overexpression correlates with PKCα membrane localization in breast tumors (Tan et al., 2006). A recent study by the Weinberg group identified a platelet-derived growth factor receptor (PDGFR)-PKCα-FRA1 signaling network that is activated specifically in breast cancer stem cells, and appears to play a key role in breast cancer initiation (Tam et al., 2013). However, analysis of the role of PKCα in all-trans-retinoic acid (ATRA)-induced inhibition of breast cancer cell growth has yielded conflicting results. Consistent with a growth promoting role, studies in ER-negative, retinoic acid receptor-positive SKBR-3 breast cancer cells pointed to suppression of a PKCα-ERK signaling axis as a key event in ATRA-induced G1 arrest (Nakagawa et al., 2003). In contrast, studies by the Talmage group demonstrated a requirement for PKCα expression and activity in retinoic acid-induced growth arrest of ER-positive T47D cells (Cho and Talmage, 2001; Cho et al., 1997), pointing to a potential role for ER-signaling in modulating the tumorigenic effects of PKCα.
2.6. Glioma cells
Consistent with high expression of PKCα protein in gliomas relative to astrocytes and low grade astrocytomas (Baltuch et al., 1995; Mandil et al., 2001), several studies support a pro-proliferative role of the enzyme in glioma. Using PKCα-specific antisense oligonucleotide, the Yong group showed that PKCα deficiency reduces the proliferation rate of rat C6 glioma cells (Baltuch et al., 1995). Follow-up studies by the same group determined that PKCα signaling is necessary and sufficient for PMA-induced cell cycle progression in several human glioma cell lines (Besson and Yong, 2000). Antisense experiments pointed to an unexpected role of p21Cip1 in facilitating cell cycle progression and hyperproliferation mediated by PKCα in these cells (Besson and Yong, 2000). In keeping with these findings, PKCα overexpression in the human U87 glioma cell line enhanced cell proliferation via a mechanism involving the regulatory domain of the kinase (Mandil et al., 2001). Interestingly, the Parker group reported that PKCα protein but not catalytic activity is required for U87MG glioma cell proliferation and that the fully phosphorylated, folded form of the kinase confers activity-independent effects (Cameron et al., 2008). Analysis of the expression of lncRNAs in human glioma cells identified TCONS_00020456 as an oncosuppressor that targets SMAD2/PKCα signaling to inhibit glioma cell proliferation in vitro and tumor progression in vivo (Tang et al., 2020). While these studies support a pro-proliferative role of PKCα signaling in glioma, it is becoming increasingly evident that glioma cell lines may not be optimal models for studying the disease (Lenting et al., 2017). In this regard, two recent studies (Goode et al., 2018; Rosenberg et al., 2018) involving genomic profiling of patient samples identified a novel mutation in PRKCA (D463H) as a hallmark of chordoid gliomas, with characteristics of oncogenic, gain-of-function mutations rather than inactivating alterations. While expression of wild-type or kinase-dead D463A PKCα in immortalized human astrocytes, the cells of origin of choroid gliomas, failed to affect proliferation, the D463H mutant protein was shown to drive MEK-dependent, anchorage-independent growth of these cells. D463H PKCα is less stable than the wild type enzyme (with a half-life of 4.5 h vs 17 h, respectively), and displays a defect in membrane anchorage (Rosenberg et al., 2018). The authors suggest that neomorphic functions of the mutant kinase may reflect altered substrate specificity. It is also possible that the mutation abolishes kinase activity entirely and that the oncogenic properties of the mutant protein involve novel kinase-independent mechanisms. Additional studies using relevant preclinical models are required to better understand the role of PKCα signaling in glioma.
2.7. Other tumor types
Evidence for the involvement of PKCα in regulation of cell proliferation and tumorigenesis in the liver, pancreas, and melanocytes/melanoma will be summarized briefly here as these tissues have been less extensively studied. A pro-proliferative role for PKCα in hepatocellular carcinoma is supported by some studies. Antisense inhibition of PKCα expression in poorly differentiated human hepatocellular carcinoma cells reduced cell growth, in association with p21Cip1 induction and cyclin D1 downregulation (Wu et al., 2008), and stable knockdown of PKCα using siRNA technology slowed the proliferation of SK-Hep-1 cells (Hsieh et al., 2007). In contrast, the finding that PKCα inhibited the growth of HepG2 cells points to an anti-proliferative role in hepatocellular carcinoma. PKCα-mediated growth inhibition in these cells involved RAS- and RAF-independent activation of MEK/ERK (Wen-Sheng, 2006) and ERK-dependent induction of miR-101, which targets two subunits of the PRC2 methyltransferase complex, enhancer of zeste homolog 2 (EZH2) and embryonic early development (EED), reducing methylation of histone 3 lysine 27 to regulate gene expression (Chiang et al., 2010).
Conflicting results have also been reported in pancreatic cancer. Consistent with frequent upregulation of PKCα expression in pancreatic tumors, some studies support a pro-proliferative role of PKCα in pancreatic cancer cells. Inhibition of PKCα expression in AR4–2J cells using antisense mRNA markedly reduced cell growth, and the extent of growth inhibition paralleled the reduction in PKCα immunoreactivity (Zhang et al., 1997). Conversely, overexpression of PKCα enhanced the tumorigenicity of HPAC cells in mice, while decreasing survival (Denham et al., 1998). However, an anti-proliferative role is indicated by the G1 phase arrest induced by activation of endogenous PKCα or microinjection of recombinant PKCα in DanG pancreatic cancer cells, an effect that involves p21Cip1 induction, inhibition of cdk2 activity, and hypophosphorylation of pRb (Detjen et al., 2000).
Studies by several groups point to a role of PKCα in melanoma cell growth arrest and differentiation. In early studies, Niles and colleagues showed that overexpression of PKCα (2–4 fold) in B16 mouse melanoma cells resulted in longer doubling times, diminished anchorage-independent growth, and increased melanin production (Gruber et al., 1992). PKCα overexpressing clones were further shown to produce smaller tumors with longer latency in mice. The same group determined that PKCα mRNA and protein are upregulated 6–8 fold during retinoic acid induced growth arrest and differentiation of melanoma cells (Niles and Loewy, 1989), an effect that was confirmed by others (Oka et al., 1993), and that PKCα is required for the differentiation program in these cells (Gruber et al., 1992). Follow-up studies excluded a requirement for PKCα enzyme activity in the effect, suggesting that PKCα works through a nonenzymatic protein-protein mechanism to mediate the actions of retinoic acid in B16 melanoma cells (Niles, 2003). PKCα has also been shown to play a critical role in mannosylerythritol lipid-induced differentiation of melanoma B16 cells (Zhao et al., 2001). Consistent with these findings, PKCα deficiency enhances DNA synthesis in A-375 human melanoma cells, while increased levels of the enzyme have anti-proliferative effects in these cells (Krasagakis et al., 2002; Krasagakis et al., 2004).
2.8. Underlying mechanisms
2.8.1. Modulation of growth factor receptor signaling
Extensive evidence supports the ability of PKCα to modulate signaling from growth factor receptors, a function that is thought to contribute directly to the cancer-related effects of the enzyme. Almost all these studies point to an inhibitory role of PKCα on growth factor activity. The best characterized targets of the kinase are members of the epidermal growth factor receptor family, including the EGFR and ErbB2. Early studies determined that phorbol esters can downmodulate EGFR signaling in non-transformed cells as well as cancer cells (Friedman et al., 1984), an effect that has been implicated in both negative feedback following activation by EGF (Chen et al., 1996; Welsh et al., 1991) and in transmodulation of EGFR signaling by multiple G-protein coupled receptors, including receptors for prostaglandin F2α, angiotensin II, vasopressin, carbachol, bombesin, and luteinizing hormone releasing hormone (LHRH) receptors (de Asua and Goin, 1992; Santiskulvong and Rozengurt, 2007; Yates et al., 2005).
Sequencing data and mutational analysis have led to a consensus that the effects of PKC activation are mediated by phosphorylation of Thr654 in the juxtamembrane region of EGFR (Bao et al., 2000; Countaway et al., 1990; Davis and Czech, 1985; Hunter et al., 1984; Lin, 1986). While studies that directly address the role of PKCα in phosphorylation of Thr654 have been limited, siRNA-mediated knockdown experiments have pointed to PKCα as the major isoform that phosphorylates this site (Koese et al., 2013; Macdonald-Obermann and Pike, 2009; Morrison et al., 1993; Santiskulvong and Rozengurt, 2007) (although PKD has also been implicated in Thr654 phosphorylation in some circumstances (Bagowski et al., 1999; Kluba et al., 2015) such as following PDGFR activation).
PKC-mediated phosphorylation of EGFR reduces high affinity binding of EGF and the autophosphorylation activity of the receptor, thereby inhibiting activation of downstream effectors. These effects that can be seen both in cells and with purified kinases in vitro (Bao et al., 2000; Downward et al., 1985; Hunter et al., 1984; Lund et al., 1990). Mutation of Thr654 to alanine inhibited the effects of PKC (Lund et al., 1990), while mutation to glutamate reduced kinase activity of EGFR in response to EGF, confirming the involvement of Thr654 (Morrison et al., 1993). However, it should be noted that these mutations did not totally block the effects of PKC agonists (Lund et al., 1990; Morrison et al., 1993), indicating that other factors are also involved. Since high affinity binding of EGF and EGFR activation require receptor dimerization (Bjorkelund et al., 2013; Ozcan et al., 2006), these effects may be explained by the finding that PKC activation or mutation of Thr654 to glutamate inhibits ligand-induced EGFR dimerization (Kluba et al., 2015). This idea is supported by evidence that the juxtamembrane domain containing Thr654 is involved in the link between receptor dimerization and ligand affinity (Macdonald-Obermann and Pike, 2009).
Additional effects of PKCα-mediated phosphorylation include disruption of ligand-dependent EGFR trafficking and degradation in the endolysosomal compartment (Bakker et al., 2017). These effects are consistent with extensive evidence for the involvement of PKCα signaling in regulation of membrane trafficking, including endocytosis (Alvi et al., 2007; Elnakat et al., 2009; Jeong et al., 2019; Ranganathan et al., 2004), endocytic recycling (Alvi et al., 2007; Bailey et al., 2014; Hellberg et al., 2009; Idkowiak-Baldys et al., 2006), and exocytosis (Tzeng et al., 2017). Studies in B82 cells, A431 cells and normal human fibroblasts expressing wild-type Thr654 or Ala654 mutant EGFR (Koese et al., 2013; Lund et al., 1990) indicate that, in addition to inhibition of its tyrosine kinase activity, Thr654 phosphorylation can prevent internalization and degradation of EGFR. The retention of EGFR at the plasma membrane appears to reflect a direct effect of Thr654 phosphorylation on trafficking rather than inhibition of the tyrosine kinase activity of the receptor, since it was seen with an EGFR truncation mutant lacking enzymatic activity (Koese et al., 2013; Lund et al., 1990). In contrast, other studies using EGFR expressing CHO and HEK293 cells did not observe an effect of PKC agonists on ligand-induced receptor internalization; instead, PKC activation inhibited subsequent degradation of the EGFR through Thr654-dependent redirection of the internalized receptor away from late endosomes and into a recycling compartment (Bao et al., 2000; Liu et al., 2013). This effect involves a caveolin- and ganglioside-containing sub-compartment of recycling endosomes, termed the pericentrion (El-Osta et al., 2011; Idkowiak-Baldys et al., 2006), that localizes to the perinuclear region in a PKC- and phospholipase C-dependent manner (Liu et al., 2013). Wild-type EGFR, but not a Thr654 to alanine mutant, is redirected to this compartment and thereby sequestered away from ligands, remaining inactive (Liu et al., 2013). The differences between these studies likely reflect differential engagement of interaction partners, substrates and downstream targets of PKCα in these cells. For example, studies in A431 squamous cell carcinoma cells, head-and-neck cancer cells and breast cancer cells have determined that annexin A6 acts as a scaffold to recruit PKCα to the EGFR and promote Thr654 phosphorylation, resulting in a reduction in ligand-induced EGFR auto-phosphorylation, internalization and degradation (Koese et al., 2013). This effect likely contributes to the tumor suppressive effects of annexin A6 in several cancer types (Qi et al., 2015). On the other hand, a caveolin-1 and ganglioside GM3-dependent complex consisting of EGFR, CD82 and PKCα promotes internalization of the receptor without triggering its degradation (Wang, X.Q. et al., 2007), pointing to its redirection to recycling endosomes/the pericentrion. Thr654 phosphorylation also inhibits binding of calmodulin to the juxtamembrane region of EGFR (Aifa et al., 2006), an interaction that has been implicated in regulation of EGFR activity and trafficking though early endosomes (Li et al., 2012; Tebar et al., 2002). There is also evidence that PKCα can promote the degradation of EGFR by destabilizing the de-ubiquitinase, ubiquitin-specific protease 8 (USP8), via inhibition of AKT activity (Cai et al., 2010).
In addition to the EGFR, PKCα has been implicated in regulation of other growth factor receptors, particularly by promoting trafficking to a perinuclear recycling compartment. ErbB2 has a threonine in its juxatamembrane domain (Thr686) that is analogous to Thr654 in EGFR. PKC agonist treatment induces phosphorylation at Thr686 of ErbB2, reduces tyrosine phosphorylation and activity of the receptor and, as seen with the EGFR in some studies, increases its internalization and trafficking to a perinuclear recycling compartment without triggering its degradation (Bailey et al., 2014; Ouyang et al., 1998; Ouyang et al., 1996). Although the involvement of PKCα in Thr686 phosphorylation of ErbB2 has not been tested directly, its role is supported by the ability of (a) constitutively active PKCα to trigger internalization of ErbB2 in SKBR3 cells (Jeong et al., 2019), and (b) PKCα-knockdown to block trafficking of ErbB2 to a perinuclear recycling compartment in response to PKC-agonist treatment or HSP90 inhibition. PKCα also regulates trafficking of cMet (the hepatocyte growth factor (HGF) receptor) to a perinuclear compartment without affecting its initial internalization or its activity (Kermorgant et al., 2004). However, the situation for the PDGFβ receptor is somewhat different: here PKCα also redirects the receptor to a recycling compartment but, in this case, the result is rapid recycling rather than perinuclear sequestration (Hellberg et al., 2009). PKCα functions in a negative feedback loop to regulate the RET tyrosine kinase (Andreozzi et al., 2003). RET binds to and activates PKCα and PKCα, in turn, phosphorylates and downregulates RET, inhibiting mitogenic signaling.
PKCα can also regulate growth factor signaling by acting downstream of growth factor receptors. For example, PKCα regulates insulin action by inhibiting the activation of MEK-ERK and PI3K-AKT signaling downstream of the insulin receptor. Blockade of PI3K-AKT activation involves PKCα binding to insulin receptor substrate 1 (IRS1) and inhibition of its phosphorylation (Caruso et al., 1999; Oriente et al., 2005), an effect that may be mediated by direct phosphorylation of Ser24 to prevent phosphoinositide binding of the IRS1 PH domain (Nawaratne et al., 2006). PKCα-mediated inhibition of PI3K-AKT signaling has been observed in multiple cell types, including intestinal epithelial cells (Guan et al., 2007), endometrial cancer cells (Hsu et al., 2018), prostate cancer cells (Tanaka et al., 2003), and head-and-neck squamous cell carcinoma (HNSCC) cells (Hoshino et al., 2012), and the effect appears to be accomplished by several mechanisms. In intestinal and HNSCC cells, PKCα suppresses the catalytic activity of PI3K (Guan et al., 2007; Hoshino et al., 2012); in prostate cancer cells, on the other hand, PKCα reduces AKT phosphorylation without affecting PI3K activity (Tanaka et al., 2003). The exact mechanism by which PKCα reduces PI3K activity remains to be elucidated: while the effect has been linked to phosphorylation of Ser361/652 in the p84α regulatory subunit of PI3K in HNSCC cells (Hoshino et al., 2012), in vitro analysis has suggested a role for direct phosphorylation of the catalytic subunit (Sipeki et al., 2006). Analysis in endometrial and prostate cancer cells indicates that PP2A-dependent, PHLPP1/2-independent dephosphorylation of AKT downstream of PI3K can also be involved (Hsu et al., 2018; Tanaka et al., 2003). As discussed above, inhibition of AKT by PKCα in HeLa cells destabilized the de-ubiquitinase, USP8, leading to enhanced ubiquitination and degradation of EGFR (Cai et al., 2010). Consistent with the reported growth inhibitory effects of sustained ERK-MAPK signaling in several systems, PKCα can also activate the MEK-ERK pathway to inhibit growth factor receptor signaling, as observed in intestinal epithelial cells (Clark et al., 2004), colon cancer cells (Hao et al., 2011; Kaur et al., 2018; Pysz et al., 2014), and hepatocellular carcinoma cells (Wen-Sheng and Jun-Ming, 2005), with dominant effects of the PKCα-ERK module over growth promoting signaling from serum growth factors (Clark et al., 2004; Pysz et al., 2014). As noted in section 2.3, PKCα has also been shown to regulate WNT signaling by phosphorylating N-terminal serine residues in β-catenin to mark it for degradation (Gwak et al., 2006; Gwak et al., 2009) or by phosphorylation of the orphan nuclear receptor, RORα, at Ser35 to block β-catenin transcriptional activity (Lee et al., 2010). PKCα may also engage an AP1-Notch-4 signaling pathway to promote ER-independent proliferation of breast cancer cells (Yun et al., 2013).
2.8.2. Cell cycle specific effects
Although anti- and pro-proliferative effects of PKCα have been well documented in many cancer types, relatively few studies have investigated specific effects of the kinase on the cell cycle. Available evidence points to D-type cyclins and the CDK inhibitory proteins, p21Cip1 and p27Kip1, as important targets of PKCα activity (Black and Black, 2012; Black, 2001). As discussed above, our analysis in intestinal epithelial and colorectal cancer cells delineated a program of cell cycle withdrawal triggered by PKCα that involves rapid downregulation of D-type cyclins and induction of p21Cip1 and p27Kip1, inhibition of G1/S cyclin/CDK complex activity, and changes in the pocket proteins, p107, pRb, and p130, characteristic of G1 arrest and cell cycle withdrawal (Frey et al., 2000; Frey et al., 1997; Pysz et al., 2009). Delayed progression through G2/M was also noted. Similar effects on D-type cyclins and/or CDK inhibitors were reported in keratinocytes (Jerome-Morais et al., 2009; Tibudan et al., 2002) and DanG pancreatic cancer cells (Detjen et al., 2000). In NSCLC cells, p21Cip1 was induced when PKCα was activated in S-phase, but not G1, and led to G2/M arrest and a senescence phenotype (Oliva et al., 2008). PKCα overexpression in non-tumorigenic MCF10A human mammary epithelial cells inhibited G1-S phase transit as a result of increased p27Kip1 expression (Sun and Rotenberg, 1999). Interestingly, induction of p27Kip1 by PKCα in intestinal epithelial cells, keratinocytes, and pancreatic cancer cells was delayed relative to upregulation of p21Cip1 (Detjen et al., 2000; Frey et al., 1997; Jerome-Morais et al., 2009; Tibudan et al., 2002), suggesting that the prolonged activation of the enzyme seen in differentiated epithelial tissues may help maintain their growth arrested state by supporting the expression of p27Kip1 (Figure 1).
Regulation of CDK inhibitory proteins and D-type cyclins also plays a role in growth-promoting functions of PKCα signaling. Antisense-mediated PKCα knockdown in poorly differentiated human hepatocellular carcinoma cells induced G1 arrest in association with p21Cip1 induction and cyclin D1 downregulation (Wu et al., 2008). Interestingly, in glioma cells, PKCα upregulated p21Cip1 without affecting cyclin D1 levels (Besson and Yong, 2000), and increased levels of p21Cip1 were required to promote assembly of cyclin D-CDK4/6 complexes and phosphorylation/inactivation of pRb (Besson and Yong, 2000). This pro-proliferative role of p21Cip1 points to the importance of cyclin D downregulation for the growth arrest induced by PKCα in cells where it also induces p21Cip1.
PKCα uses multiple mechanisms to regulate cell cycle proteins. In hepatocellular carcinoma cells, PKCα appears to promote p21Cip1 upregulation by a p53-dependent mechanism (Wu et al., 2008). In contrast, PKCα induces p21Cip1 expression in p53 mutant colon (Pysz et al., 2009) and pancreatic (Detjen et al., 2000) cancer cells and the upregulation of p21Cip1 in intestinal epithelial cells and colon cancer cells is mediated by MEK-ERK signaling (Clark et al., 2004). PKCα signaling also regulates cyclin D1 levels by two different mechanisms in intestinal epithelial and colon cancer cells. The enzyme inhibits cyclin D1 translation through PP2A-mediated activation of the translational repressor 4E-BP1 (Guan et al., 2007; Hizli et al., 2006; Pysz et al., 2009) and represses cyclin D1 transcription, likely through a MEK-ERK dependent mechanism (Clark et al., 2004; Guan et al., 2007; Pysz et al., 2009).
3. Role of PKCα in migration and metastasis
PKCα signaling has been implicated in cell motility, migration, epithelial-to-mesenchymal transition (EMT), invasion, and metastasis in many tumor types. In contrast to the complexity of PKCα function in regulation of cell proliferation, differentiation, and tumorigenesis (Section 2), the kinase generally plays a positive role in migratory processes and the metastatic program. These effects likely reflect the well documented functions of PKCα in regulation of the cytoskeleton, adhesion, motility and migration in normal cells. PKCα localizes at focal adhesions, filopodia, and lamellipodia in vitro and interacts with and/or phosphorylates a number of proteins that are associated with these structures and with cell migration, including syndecan-4 (Koo et al., 2006; Lim et al., 2003; Oh et al., 1997), Rho guanidine dissociation inhibitor 1 (RhoGDI1) (Dovas et al., 2010), p190RhoGAP (Lévay et al., 2009),β1 integrin (Ng et al., 1999a), formin-like 2 (FMNL2) (Kitzing et al., 2010), filamin A (Urra et al., 2018; Zhou et al., 2010), vinculin (Ziegler et al., 2002), and fascin (Anilkumar et al., 2003).
PKCα activity markedly increases the motility and migration of colon cancer cells (Hu et al., 2013; Masur et al., 2001), especially in cells with low E-cadherin expression. The enzyme also has pro-migratory effects in melanoma cells (Byers et al., 2010; Sullivan et al., 2000), breast cancer cells (Parsons et al., 2002; Pham et al., 2017), lung cancer cells (O’Neill et al., 2011; Wang et al., 2013), and kidney cancer cells among others. A role for PKCα in EMT has been described in multiple systems (Kyuno et al., 2013; Llorens et al., 2019; Qi et al., 2014). PKCα activity regulates the expression and stability of transcription factors that play key roles in regulation of the EMT program, including ZEB1 (Llorens et al., 2019), ZEB2 (Abera and Kazanietz, 2015), Snail (Abera and Kazanietz, 2015; Kyuno et al., 2013), and Twist1 (Abera and Kazanietz, 2015; Tedja et al., 2019), with phosphorylation of Twist1 at ser144 by PKCα preventing its ubiquitination and degradation (Tedja et al., 2019). Notably, a recent study by the Weinberg group identified a PKCα-FRA1 signaling axis as a gatekeeper of the EMT program in TNBC (Tam et al., 2013). PKCα signaling can also promote tumor cell invasion, as shown in colon cancer cells (Batlle et al., 1998), hepatocellular carcinoma cells (Hsieh et al., 2007), pancreatic cancer cells (Kyuno et al., 2013), endometrial cancer cells (Haughian and Bradford, 2009), melanoma cells (Putnam et al., 2009), and glioblastoma cells (Lin et al., 2010). Upregulation and activation of PKCα downstream of ErbB2 is critical for breast cancer cell invasion (Tan et al., 2006) and induction of matrix metalloprotease (MMP)-9 expression by PKCα is required for glioblastoma invasion (Lin et al., 2010). A recent study in NSCLC cells (Cooke et al., 2019) using RNA interference to silence individual PKC isozymes demonstrated the ability of PKCα, but not PKCδ or PKCε, to potently induce the expression of the extracellular matrix proteases MMP1, MMP9, and MMP10 (Cooke et al., 2019).
The involvement of PKCα in metastasis has been best characterized in breast cancer. PKCα downregulation by microRNA-200b in triple negative breast cancer cells suppressed metastasis in an orthotopic mouse xenograft tumor model (Humphries et al., 2014). Similarly, pharmacological inhibition of PKCα by in vivo administration of the PKCα selective inhibitory peptide, αV5–3, markedly reduced intravasation and lung seeding of mammary cancer cells by suppressing MMP-9 and NF-κB activity and cell surface expression of the chemokine receptor CXCR4 (Kim et al., 2011). Interestingly, PKCα inhibition had no effect on the primary tumor, indicating that PKCα signaling does not play a role in proliferation and survival per se in this model. In human melanoma cells, inhibition of PKCα expression with a phosphorothioate antisense oligonucleotide (ISIS-3521, also known as aprinocarsen) suppressed metastatic potential in athymic mice (Dennis et al., 1998) and the metastasis suppressor KiSS1 reduces human ovarian cancer metastasis by inhibiting the activity of PKCα (Jiang et al., 2005).
A major mechanism by which PKCα promotes cell migration is through coordinated activation of Rho-GTPases, particularly RhoA and Rac1. Multiple steps in cell migration are regulated by these GTPases, including formation of actin-rich lamellipodia and invadipodia, focal adhesion assembly at the leading edge, and secretion of MMPs (Clayton and Ridley, 2020). Regulation of RhoA and Rac1 by PKCα is mediated by the interaction of its catalytic domain (but not that of PKCδ or PKCε (Lim et al., 2003)) with the cytoplasmic tail of syndecan-4, a ubiquitously expressed heparin sulfate proteoglycan that has been implicated in regulation of cellular migration, mechanotransduction, endocytosis, and proliferation (Elfenbein and Simons, 2013). Syndecan-4 acts as a receptor for heparin binding extracellular proteins including growth factors, such as FGF2, VEGF and PGDF, and extracellular matrix components, such as fibronectin and vitronectin (Elfenbein and Simons, 2013). Following engagement by heparin binding proteins, syndecan-4 undergoes PI(4,5)P2-dependent oligomerization, which results in direct activation of PKCα (Koo et al., 2006; Lim et al., 2003; Oh et al., 1997). This activation differs from the canonical PKCα activation in that it is independent of DAG and Ca2+, but is dependent on PI(4,5)P2 and syndecan-4 oligomerization (Horowitz et al., 1999; Horowitz and Simons, 1998). Interestingly, PKCδ may negatively regulate this pathway through phosphorylation of syndecan-4 on Ser183, which inhibits PI(4,5)P2 binding and oligomerization, thus preventing PKCα activation (Murakami et al., 2002).
As with other GTPases, RhoGTPases act as molecular switches which are active in the GTP bound state and inactive when bound to GDP (Etienne-Manneville and Hall, 2002; Parri and Chiarugi, 2010). Both the intrinsic GTPase activity of these proteins and the dissociation of GDP are slow; thus, their activity is primarily regulated by proteins that modulate GTPase activity and GDP/GTP exchange. These proteins are activated by a number of guanine nucleotide exchange factors (GEFs) that enhance the dissociation of GDP and thus enable binding of GTP. Conversely, RhoGTPases are negatively regulated by GTPase activating proteins (GAPs), which enhance their intrinsic GTPase activity, and by guanidine dissociation inhibitors (GDIs), which sequester RhoGTPases away from the membrane while inhibiting GDP/GTP exchange (Etienne-Manneville and Hall, 2002; Parri and Chiarugi, 2010). In the unstimulated state, syndecan-4 suppresses Rho-GTPase activity by forming a complex with synectin and RhoGDI1 (RhoGDIα), which enhances the interaction of Rho-GTPases with RhoGDI1 (Elfenbein et al., 2009). Following syndecan-4 oligomerization, PKCα phosphorylates RhoGDI1 at Ser34, which releases RhoA (Dovas et al., 2010), and ser96, which releases RhoG and Rac1 (Dovas et al., 2010; Elfenbein et al., 2009; Knezevic et al., 2007). Activation of Rac1 following its release is also dependent on RhoG which forms a Rac-specific GEF by complexing with ELMO and Dock180 (Elfenbein et al., 2009; Katoh et al., 2006). Thus, while syndecan-4 restricts Rho-GTPase activity in the unstimulated state, its ability to stimulate PKCα following oligomerization allows for selective activation of RhoA and Rac1 at sites of matrix and/or growth factor binding to support directional migration (Bass et al., 2007).
In addition to promoting the spatial activation of Rho-GTPases at the leading edge, PKCα helps to regulate the phasic activation of Rac1 and RhoA during adhesion and migration through p190RhoGAP. PKCα phosphorylates p190RhoGAP at Ser1221 and Thr1226 (Lévay et al., 2009), blocking its interaction with acidic phospholipids and increasing its activity towards RhoA, while decreasing its activity towards Rac1 (Lévay et al., 2009). This supports the initial inhibition of RhoA upon matrix engagement that is required for proper adhesion formation and membrane protrusion at the leading edge (Bidaud-Meynard et al., 2017; Lévay et al., 2009).
The effects of PKCα on cell motility, migration and invasion are also mediated by its ability to regulate integrin recycling, an essential event in migration and metastasis (Ng et al., 1999a). PKCα activation by syndecan-4 or PMA upregulates β1 integrin and induces dynamin- and caveolin-dependent internalization of α5β1 integrin (Bass et al., 2011). As with activation of Rac1, the ability of PKCα to promote integrin endocytosis involves downstream activation of RhoG (Bass et al., 2011); however, it also involves direct interaction between PKCα and β1 integrin (Ng et al., 1999a) and phosphorylation of FMNL2 (Kitzing et al., 2010). The interaction of PKCα with the cytoplasmic tail of β1 integrin is mediated by the V3 variable domain of the enzyme (Parsons et al., 2002) and is thus distinct from its interaction with syndecan-4, which is mediated by the catalytic domain (Lim et al., 2003). Preventing the interaction between PKCα and β1 integrin with a blocking peptide or inhibiting PKCα-induced β1 integrin internalization blocks migration of breast cancer cells (Ng et al., 1999a; Parsons et al., 2002).
FMNL2 belongs to a group of Diaphanous (mDia)-related formins that regulate actin polymerization and are intimately involved in migration control (Bogdan et al., 2013; Breitsprecher and Goode, 2013). Knockdown of FMNL2 reduces PKCα-induced internalization of α2β1 and α5β1 integrins and cell migration/invasion, pointing to a role for FMNL2 in mediating the effects of PKCα on cell migration, invasion and metastasis (Wang et al., 2015; Zhong et al., 2018). FMNL2 is initially targeted to lamellipodia and filopodia by RhoC and CDC42 (Block et al., 2012; Kitzing et al., 2010), where it interacts directly with PKCα (Wang et al., 2015). PKCα activates FMNL2 through phosphorylation of Ser1072, promoting FMNL2 binding to the cytoplasmic tail of α integrin and actin-dependent internalization of FMNL2/α integrin/β1 integrin complexes (Wang et al., 2015).
PKCα can also drive lamellipodia protrusion at the leading edge and cell migration through effects on the actin cytoskeleton. PKCα interacts with filamin A (Tigges et al., 2003; Urra et al., 2018), a crosslinker of polymerized actin with crucial roles in adhesion and migration (Feng and Walsh, 2004; Zhou et al., 2010). Phosphorylation of filamin A on Ser2152 by PKCα protects filamin A from degradation and enhances cytoskeletal remodeling and cell migration (Urra et al., 2018; Zhou et al., 2010). Another mechanism by which PKCα can enhance migration is through interaction with Disks-Large 1 (DLG1, SAP97) (O’Neill et al., 2011), a member of the Scribble/LGL/DLG polarity complex that regulates cell polarity and directed migration (Marziali et al., 2019; Saito et al., 2018). DLG1 is a substrate for PKCα and these proteins colocalize at the leading edge in migrating cells (O’Neill et al., 2011). PKCα contains a PDZ-ligand at its C-terminus that mediates direct binding to the third PDZ-domain of DLG1. Deletion of the PDZ-ligand or knockdown of DGL1 reduces the ability of PKCα to promote migration, confirming the role of this interaction in the pro-migratory effects of PKCα (O’Neill et al., 2011). Although the precise mechanisms by which the PKCα-DLG1 interaction affects migration remains to be determined, it likely reflects, at least in part, the role of DLG1 in establishing front rear polarity in migrating cells (Marziali et al., 2019; O’Neill et al., 2011).
Other less well characterized mechanisms for regulation of migration by PKCα include inhibition of secretion of the metallopeptidase inhibitor TIMP1 through Rab37 phosphorylation (Tzeng et al., 2017), suppression of p120catenin expression through FOXC2 (Pham et al., 2017), and activation of p38 MAPK (Hsieh et al., 2007).
As noted for PKCα-mediated regulation of cell proliferation and tumorigenesis, effects of the enzyme on migration in cancer cells can be context-dependent. Consistent with a role in regulation of invasion and metastasis, a computational network analysis of published data identified PKCα as one of the top ten signaling hubs in control of focal adhesions and invadopodia (Hoshino et al., 2012). However, follow-up studies in HNSCC cells, which retain high levels of PKCα, indicated that the effects of the kinase on the invasive phenotype are dependent on the status of the PI3K/PTEN signaling axis. In PI3K/PTEN wild-type cells, PKCα knockdown reduced invadopodia formation, indicating that PKCα enhances their invasive phenotype. However, in cells with activating mutations in PI3K or loss of PTEN, PKCα knockdown enhanced invadopodia formation and extracellular matrix degradation, pointing to an inhibitory role in invasion in this context. Mechanistic analysis revealed a PKCα-mediated negative feedback loop that dampened PI3K activity. In keeping with these findings, the PI3K-high/PKCα-low signaling state was found to predict the most invasive phenotype in HNSCC (Hoshino et al., 2012).
A role of PKCα in promoting metastasis appears at odds with its tumor suppressive properties in multiple cancer types and the fact that its expression is commonly lost in advanced metastatic disease (Sections 2 and 5). Similarly, the PKCα target DLG1 can act as a tumor suppressor and is frequently lost in invasive carcinomas (Marziali et al., 2019). Thus, a positive contribution of PKCα to invasion and metastasis is likely limited to tumor types where the kinase does not have a tumor suppressive role, or to tumors where downstream tumor suppressive signaling has been inactivated. Notably, RhoGTPases that are downstream of PKCα in migration signaling are commonly overexpressed in cancer (Jansen et al., 2018), indicating that tumors have alternate mechanisms for upregulation of these pathways in the absence of PKCα activity.
4. The role of PKCα in cell survival and drug resistance
Multiple studies point to a protective role of PKCα in the response of tumor cells to chemotherapy. The ability of PKCα overexpression to confer drug resistance has been observed with a variety of chemotherapeutic agents in a broad range of cancer types. For example, increased levels of the kinase protected acute lymphocytic leukemia (ALL) cells from the antitumor effects of etoposide and cytosine arabinoside (araC) (Jiffar et al., 2004; Ruvolo et al., 1998), RT4 bladder cancer cells from the effects of doxorubicin (Kong et al., 2005), and C6 glioma cells from tamoxifen treatment (Tian et al., 2009). Conversely, reducing PKCα expression using antisense RNA, ribozyme or RNAi technology sensitized cancer cells to a variety of chemotherapeutic agents, including vincristine and prednisone in ALL (Lei et al., 2016), cisplatin and taxol in ovarian cancer (Zhao et al., 2012), erlotinib in lung cancer (Abera and Kazanietz, 2015), mitomycin-C and 5-fluorouracil in MKN-45 gastric cancer (Jiang et al., 2004), and cisplatin in prostate (Villar et al., 2009) and cervical cancer cells (Mohanty et al., 2005). The clinical relevance of these findings is highlighted by evidence that PKCα expression correlates with resistance to antiestrogen therapy in breast cancer patients (Assender et al., 2007).
A major mechanism underlying PKCα-mediated drug resistance appears to involve regulation of apoptosis, particularly through BCL2 family proteins. Even in the absence of cytotoxic treatments, reduced expression of PKCα leads to an increase in spontaneous apoptosis in some cell lines, including COS cells (Whelan and Parker, 1998), HepG2 hepatocellular carcinoma cells (Zhu, B.H. et al., 2005), T98G and U87 glioblastoma cells (Leirdal and Sioud, 1999; Mandil et al., 2001), and MKN-45 gastric cancer cells (Jiang et al., 2004). PKCα can phosphorylate BCL2 on Ser70 in vitro (Ruvolo et al., 1998), a modification that has been linked to enhanced anti-apoptotic activity (Ruvolo et al., 2001). In cells, PKCα can associate with mitochondrial membranes (Ruvolo et al., 1998; Wang, W.L. et al., 2007) and the enzyme induces phosphorylation of BLC2 in murine fibroblasts (Wang, W.L. et al., 2007), HL60 promyelocytic leukemia cells (Jiffar et al., 2004), REH pre-B ALL cells (Ruvolo et al., 1998), LNCaP prostate cancer cells (Villar et al., 2009), and U87MG glioblastoma cells (Joniova et al., 2014). In addition to direct phosphorylation, PKCα-mediated inhibition of mitochondrial PP2A appears to contribute to enhanced BCL2 phosphorylation and activity (Jiffar et al., 2004; Ruvolo et al., 1998). PKCα also supports the expression of BCL-xL in U87MG and T98G glioblastoma cells (Leirdal and Sioud, 1999) and in rat hepatic epithelial cells (Hsieh et al., 2003) (but not keratinocytes (Jost et al., 2001)), indicating that the kinase affects multiple BCL2 family members. Mitochondrial PKCα can also act through modulation of pro-apoptotic BH3 proteins since PKCα-mediated inhibition of BAD, rather than activation of BCL2, is involved in blocking mitochondrial permeability-transition (MPT)-dependent necrosis induced by peroxynitrite in U937 lymphoma cells (Cerioni et al., 2006). Additional mechanisms that have been associated with the anti-apoptotic effects of PKCα include downregulation of the apoptosis mediators FEM1b and Apaf-1 in Rack1-overexpressing Jurkat and CCRF-CEM T-cell ALL cells (Lei et al., 2016), induction of nuclear translocation of NFκB in T24 and 5637 bladder cancer cells (Zheng et al., 2017), cytoplasmic localization of p53 in M21 melanoma cells (Smith et al., 2012), and upregulation of Dicer in T24 and 5637 bladder cancer cells (Jiang et al., 2015).
Although less common, PKCα can also have a pro-apoptotic role in cancer. For example, in renal tubular cells, the cells of origin for renal clear cell carcinoma (Santiago et al., 2006), polychlorinated biphenyls (PCBs) activate PKCα, in association with downregulation of BCL2 and activation of caspase-3. In hormone-dependent LNCaP cells, PMA leads to prolonged association of PKCα with non-nuclear membranes and induction of apoptosis (Powell et al., 1996) and PKCα mediates apoptosis induced by DAG-lactones (Garcia-Bermejo et al., 2002). Interestingly, the pro-apoptotic effects of PKCα in prostate cancer appear to be cell-type specific since ribozyme-mediated knockdown of PKCα sensitizes hormone-independent DU145 cells to cisplatin-induced apoptosis (Orlandi et al., 2003).
Multiple studies have implicated PKCα in drug resistance through regulation of P-glycoprotein/MDR1 (ABCB1) and related ATP binding cassette (ABC) efflux pumps (Mayati et al., 2017). A major consideration in interpreting these studies is that many have relied on the use of the classical PKCα inhibitor Gö6976 to attribute the effects to PKC α. Gö6976, and general PKC inhibitors such as bisindolylmaleimides, are inhibitors of ABC pumps (Budworth et al., 1996; Robey et al., 2007), complicating interpretation of the findings. Nonetheless, it would appear that PKCα can reduce cellular drug accumulation by enhancing the activity of efflux pumps. Serines 661, 667 and 671 within the linker region of P-glycoprotein are phosphorylated by PKCs both in vitro and in cells (Chambers, 1998; Idriss et al., 2000), with Ser671 identified as a PKCα target (Ahmad et al., 1994). While mutational analysis has revealed that phosphorylation of serines 661, 667 and 671 is not necessary for P-glycoprotein to confer multidrug resistance (Germann et al., 1996; Goodfellow et al., 1996), mutation of these sites to alanine negatively impacts the interaction of P-glycoprotein with verapamil, vinblastine and rhodamine 123 (Szabo et al., 1997), and alanine mutation of Ser671 abolished the ability of PKCα to enhance verapamil-induced ATPase activity of P-glycoprotein (Ahmad et al., 1994). In addition to regulation of P-glycoprotein activity, PKCα also appears to regulate its expression, likely through modulation of its transcription. PKCα overexpression upregulates P-glycoprotein in MCF-7 breast cancer cells and increases ABCB1 promoter activity (Gill et al., 2001). Conversely, the increased sensitivity of OV1228 ovarian cancer cells upon siRNA-mediated knockdown of PKCα is linked to downregulation of P-glycoprotein at both the mRNA and protein levels (Zhao et al., 2012). PKCα likely affects other ABC proteins since it can phosphorylate MRP2 (ABCC2) and cooperates with PKA to modulate its internalization in rat liver cells (Crocenzi et al., 2008; Wimmer et al., 2008). The ability of PKCα to phosphorylate RLIP76 (Singhal et al., 2005), a multifunctional ATP-dependent non-ABC transporter (Vatsyayan et al., 2010), and increase its ability to transport doxorubicin in lung cancer cells (Singhal et al., 2005), indicates that PKCα can affect drug resistance through multiple types of efflux pumps.
5. PKCα in tumors
5.1. PKCα mutations in cancer
Mutations in PKCα have been detected in multiple cancer types (Antal et al., 2015; Bridge et al., 2013; Goode et al., 2018) and several mutant forms have been assessed for activity using a FRET-based reporter (CKAR) that measures the ability of the enzyme to phosphorylate a designed PKC-selective substrate peptide (RFRRFQTLKIKAKA) in cells (Antal et al., 2015; Violin et al., 2003). The tested mutations either had no effect or were deleterious to kinase activity against the reporter (Antal et al., 2015). Consistent with the previously demonstrated tumor suppressive activity of PKCα in many cancers (described in Section 2 above), no activating mutations were detected in this study (Antal et al., 2015). The effects of several of the mutations could be attributed to alterations in known PKCα regulatory mechanisms, such as inhibition of priming phosphorylation or mutations in the C1A and C1B domain affecting responses to the various agonists tested (Antal et al., 2015). In the case of several other mutations, however, it remains to be determined if the observed effects are due to loss of kinase activity per se or to altered substrate specificity affecting phosphorylation of the target peptide (see below).
Although multiple cancer-related mutations in PKCα have been detected (Antal et al., 2015; Bridge et al., 2013; Goode et al., 2018), they are surprisingly rare given the role of this isozyme as a tumor suppressor in many tissues (Section 2). To date, analysis of publicly available databases using cBioPortal reveals mutations in only 767 out of a total of 47,497 samples, or 1.6% (755 of 45,089 patients, or 1.7%). Furthermore, despite multiple reports documenting loss of PKCα expression in tumors (e.g., PKCα deficiency occurs in up to 60% of colon and endometrial cancers; see below), the majority of observed genetic changes in PKCα involve amplifications (67%) rather than deletions (3%), nonsense/frameshift mutations that would lead to a truncated protein (5%), or fusions (3%) (see Section 5.2 below for further discussion of PKCα amplification.) Missense mutations, which account for 22% of PKCα genetic changes, are scattered throughout the protein with no obvious hotspots or association with any particular protein domains. Furthermore, genetic changes in PKCα are predominantly heterozygous and are associated with tumors with high overall mutational burden (Antal et al., 2015; Goode et al., 2018). Thus, although it is clear that tumor-associated mutations in PKCα can affect its kinase activity (Antal et al., 2015), in most cases, they are more likely to be passenger mutations rather than drivers of tumorigenesis.
While most genetic alterations of PKCα in tumors are likely to be passenger mutations, some mutations are characteristic of individual tumor types, arguing for a potential driver role. Three studies examining chromosomal rearrangements in papillary glioneuronal tumors, rare grade 1 mixed neuronal-glial cancers, have identified t(9;7) rearrangements in 9 out of 10 cases. These rearrangements fuse N-terminal sequences of solute carrier family 44 choline transporter member 1 (SLC44A1) to the C-terminal kinase domain of PKCα (Bridge et al., 2013; Pages et al., 2015). Although this mutation is likely to lead to a constitutively active kinase with altered subcellular targeting, and the presence of the fusion protein has been associated with increased ERK activation (Pages et al., 2015), studies directly examining its effects on kinase activity/selectivity or on cell transformation are yet to be reported. Fusions containing the PKCα kinase domain have also been detected with IGF2BP3 and TANC2 in squamous lung tumors (Stransky et al., 2014), with ATP2B4 and RNF1 in pigmented epithelioid melanocytomas (Cohen et al., 2017), and with KIRREL in a fibrous histiocytoma (Walther et al., 2015). However, since these fusions have only been identified in single patients and are, thus, not characteristic of the tumor type, their potential role in tumorigenesis remains to be determined.
A D463H PKCα mutation has been identified as a driver mutation in chordoid gliomas, a rare slow growing, well circumscribed brain tumor. Analysis of 28 samples in two independent studies identified a heterozygous D463H mutation in PKCα that is likely to occur in 100% of these tumors (Goode et al., 2018; Rosenberg et al., 2018). The mutation is clonal within these tumors and is the only consistent genetic change seen in this cancer type, suggesting that it is an early initiating event in chordoid glioma tumorigenesis. Analysis of the effects of the D463H mutation support its tumorigenic effects; the mutant enhances anchorage-independent growth of immortalized astrocytes and is linked to activation of key oncogenic signaling pathways mediated by EIF2, RAS and AKT (Goode et al., 2018; Rosenberg et al., 2018). Notably, the ability to enhance anchorage-independent growth and ERK phosphorylation was unique to D463H PKCα, and was not seen with expression of either the wild-type or kinase-dead D463A protein (Goode et al., 2018). The D463H mutation converts a highly conserved aspartic acid in the kinase active site into a histidine. D463 lies at a critical position in the kinase domain, acting as a proton acceptor during ATP hydrolysis (Goode et al., 2018; Rosenberg et al., 2018). While histidine at this position is still compatible with kinase activity (Goode et al., 2018; Rosenberg et al., 2018), its presence significantly alters the hydrogen bonds available for molecular recognition and likely affects the substrate specificity of PKCα (Rosenberg et al., 2018). Heterozygous missense mutations clustering at a mutational hotspot in a crucial functional domain of a molecule are usually gain-of-function (Goode et al., 2018). Thus, the D463H PKCα likely acts as a novel oncogene (rather than an inactive tumor suppressor) that drives tumor formation in chordoid gliomas through aberrant substrate phosphorylation or through kinase-independent functions (Goode et al., 2018; Rosenberg et al., 2018).
The only other cancer-related mutation that has been extensively characterized is a rare D294G mutation that has been detected in select pituitary, thyroid and breast tumors (Alvaro et al., 1993; Prevostel et al., 1998; Vallentin et al., 2001) and is associated with an invasive phenotype (Alvaro et al., 1993). This mutation, which occurs in the V3 hinge region of PKCα, does not affect the intrinsic activity of the kinase, but alters its targeting to membrane regions (Prevostel et al., 1998; Quittau-Prevostel et al., 2004), substrate specificity (Prevostel et al., 1998; Zhu, Y. et al., 2005), and ability to transmit tumor suppressive signaling (Zhu, Y. et al., 2005).
Overall, the data indicate that, in the case of PKCα, genetic changes play only a minor role in the transformed phenotype of common cancers. However, mutation of PKCα appears to play a pivotal role in a few rare cancers. In these cases, the mutations do not simply modulate the normal functions of PKCα. Instead, they alter the targeting and specificity of the protein, introducing novel (as yet unidentified) functions that are specific to those individual cancer types.
5.2. Aberrant expression of PKCα in tumors
Analysis of PKCα expression in publicly available databases supports a role of the kinase as a tumor suppressor in a majority of cancer types. The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) shows significant downregulation of PKCα mRNA in bladder urothelial carcinoma, breast invasive carcinoma, cervical squamous cell carcinoma, colon adenocarcinoma, glioblastoma multiforme, chromophobe renal carcinoma, lung squamous cell carcinoma, prostate adenocarcinoma, rectal adenocarcinoma, renal clear cell carcinoma, renal papillary cell carcinoma, thyroid carcinoma, and uterine corpus endometrial carcinoma (Figure 3). Clinical Proteomic Tumor Analysis Consortium (CPTAC) data (https://proteomics.cancer.gov/data-portal), currently available for seven tumor types, indicate that PKCα protein is downregulated in breast, colon, ovarian, renal clear cell, and uterine corpus cancer. In contrast, some tumor types show upregulation of PKCα mRNA, including cholangiocarcinoma, head-and-neck squamous cell carcinoma, and liver hepatocellular carcinoma. Marginal upregulation of PKCα mRNA is seen in lung adenocarcinoma, although CPTAC data indicate significant downregulation of PKCα protein in these tumors (also see Section 2.4).
Figure 3.
Expression of PKCα in normal and tumor tissue from TCGA.
Graph was generated from TCGA data by the UALCAN portal (Chandrashekar et al., 2017). BLCA: Bladder urothelial carcinoma; BRCA: Breast invasive carcinoma; CESC: Cervical squamous cell carcinoma; CHOL: Cholangiocarcinoma; COAD: Colon adenocarcinoma; ESCA: Esophageal carcinoma; GMB: Glioblastoma multiforme; HNSC: Head and Neck squamous cell carcinoma; KICH: Kidney Chromophobe; KIRC: Kidney renal clear cell carcinoma; KIRP: Kidney renal papillary cell carcinoma; LIHC: Liver hepatocellular carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; PAAD: Pancreatic adenocarcinoma; PRAD: Prostate adenocarcinoma; PCPG: Pheochromocytoma and Paraganglioma; READ: Rectum adenocarcinoma; SARC: Sarcoma; SKCM: Skin cutaneous melanoma; THCA: Thyroid carcinoma; THYM: Thymoma; STAD: Stomach adenocarcinoma; UCEC: Uterine corpus endometrial carcinoma.
Given the general downregulation of PKCα seen in these datasets, it is surprising that the PRKCA gene is commonly amplified in tumors, with the prevalence of amplification reaching 8–25% in breast cancer and 22% in bladder cancer (www.cBioportal.org). However, no correlation between copy number and mRNA expression was seen for PKCα in the TCGA database, arguing that PKCα is not the target of amplification of the 17q region. Possible direct targets for amplification in this region based on their oncogenic properties include SOX9, RPS6KB1, PPM1D, BCAS3 and DDX5 (Ferrari et al., 2016) and more distantly HER2/ErbB2.
Although few studies have compared PKCα expression in normal and tumor tissues, available data generally support findings from the large TCGA and CPTAC datasets. For example, loss of PKCα mRNA and/or protein has been reported to be a recurrent event in human colon cancer (Kahl-Rainer et al., 1994; Kahl-Rainer et al., 1996; Suga et al., 1998; Verstovsek et al., 1998), NSCLC (Hill et al., 2014), endometrial cancer (Hsu et al., 2018), and renal clear cell carcinoma (Zhan et al., 2017), with animal models of intestinal, NSCL and endometrial cancer supporting the generality of the findings (Hill et al., 2014; Hsu et al., 2018; Klein, I. K. et al., 2000; Oster and Leitges, 2006; Pysz et al., 2009). PKCα expression is also lost in human basal cell carcinoma (Neill et al., 2003) and pediatric T-ALL (Milani et al., 2014), and is reduced in CLL cells (Abrams et al., 2006). Loss of PKCα can occur early in tumor development (e.g.,(Hill et al., 2014; Hsu et al., 2018; Kahl-Rainer et al., 1996; Klein, I. K. et al., 2000; Oster and Leitges, 2006; Pysz et al., 2009)) and has been associated with advanced disease and poor patient survival in colon, endometrial, and lung cancer, as well as pediatric T-ALL (Hill et al., 2014; Hsu et al., 2018; Milani et al., 2014; Suga et al., 1998). Similarly, published reports support the upregulation of PKCα in hepatocellular and head-and-neck squamous cell carcinoma (Martinez-Gimeno et al., 1995; Tsai et al., 2005; Wu et al., 2007; Zhen-jin et al., 2011), and a correlation between PKCα levels and disease stage (Zhen-jin et al., 2011) and poor patient survival (Cohen et al., 2009; Wu et al., 2007) was also noted in these studies. However, published reports for bladder urothelial cancer have not supported the downregulation of PKCα seen in TCGA datasets. While one study reported no change in PKCα expression in tumors at different stages (Langzam et al., 2001), others have shown that PKCα is upregulated in this tumor type and that its expression correlates with recurrence, increased invasion and tumor progression (Aaltonen et al., 2006; Du et al., 2014; Jiang et al., 2015; Kong et al., 2005; Zheng et al., 2017). Whether this discrepancy reflects a preponderance of tumors with PRKCA amplification in the samples examined remains to be determined.
Analysis of PKCα expression in breast cancer has provided conflicting results. Several studies have reported that the kinase is upregulated in breast tumors relative to normal breast tissue, and high expression has been linked to estrogen/progesterone receptor negative and triple negative status, tamoxifen resistance, advanced stage and poor survival (Assender et al., 2007; Hsu et al., 2014; Lahn et al., 2004; Lonne et al., 2010; Pham et al., 2017; Tonetti et al., 2012; Tonetti et al., 2003). However, other studies have indicated that PKCα is downregulated in breast tumors (Ainsworth et al., 2004; Kerfoot et al., 2004), and that expression is lower in more advanced disease (Ainsworth et al., 2004). Interestingly, in ErbB2 (HER2) positive breast cancer, high PKCα expression is associated with better recurrence-free survival, possibly resulting from the ability of PKCα to attenuate Notch signaling and promote sensitivity to anti-ErbB2 therapy (Pandya et al., 2016) and/or PKCα-induced internalization of the receptor (Bailey et al., 2014; Ouyang et al., 1998; Ouyang et al., 1996). Although the basis for the discrepancy between these studies remains to be determined, it likely reflects the heterogeneity of the disease and differential expression of PKCα in the breast cancer subtypes analyzed in these reports. In this regard, analysis of over 320 breast tumors representative of all histological sub-types by the Larsson group revealed that the vast majority of these cancers (72%) are PKCα negative and that smaller subgroups with higher PKCα levels display more aggressive clinicopathological features (Lonne et al., 2010). Similar conclusions were reported by the Tonetti group, with approximately 20% of samples analyzed displaying PKCα positivity in association with more aggressive disease (Pham and A., 2016).
Analysis of PKCα expression in intestinal (Oster and Leitges, 2006), endometrial (Hsu et al., 2018), and kidney (Zhan et al., 2017) cancer revealed a correlation between PKCα mRNA and protein in these tissues, indicating that PKCα levels are controlled primarily at the level of mRNA accumulation. These findings are consistent with extensive TCGA data demonstrating alterations in PKCα mRNA levels in tumors (see above). Little is known regarding the mechanism(s) underlying altered PKCα mRNA expression in tumors. Studies in breast cancer cells revealed that ErbB2 activation can upregulate PKCα protein; however, effects on PKCα mRNA were not evaluated (Tan et al., 2006). PKCα expression is upregulated by the transcription factors Ets-like protein-1 (Elk-1) and myeloid zinc finger-1 (MZF-1) in human hepatocellular carcinoma cells (Hsieh et al., 2006) and by insulin-like growth factor-2 mRNA-binding protein 1 (IGF2BP1) in melanoma cells (Ye et al., 2017). The SOX9 transcription factor represses the expression of PKCα in proliferating intestinal epithelial cells both in vitro and in vivo via a DNA binding-independent mechanism (Dupasquier et al., 2009). Since SOX9 is upregulated by WNT-APC signaling, it has been proposed that this transcription factor may mediate repression of PKCα in colorectal cancers with constitutive activation of the WNT-APC pathway. However, additional mechanisms are likely involved since PKCα is broadly lost in intestinal and colon cancers with and without alterations in WNT/APC signaling and expression of the protein is retained in a proportion of APC and β-catenin mutant tumors (Pysz et al., 2009). MicroRNAs that target PKCα have been shown to regulate PKCα expression levels in various tumor types. PKCα was identified as a direct target of microRNA-200b and microRNA-216a in TNBC cells (Humphries et al., 2014) and MCF-7 cells (Cui et al., 2019), respectively. Since both of these microRNAs are profoundly downregulated in breast cancer tissues (Humphries et al., 2014; Xie et al., 2019), their loss may account for increased levels of PKCα in some breast cancers. In additional examples, PKCα is targeted by microRNA-483–3p to promote resistance of ovarian cancer cells to platinum compounds, and by microRNA-203 to inhibit cell proliferation and migration in lung cancer cells (Wang et al., 2013).
6. PKCα as a biomarker and therapeutic target
6.1. PKCα as a diagnostic and prognostic biomarker
The prognostic value of aberrant PKCα expression has been reported by multiple groups in a broad spectrum of tumor types. Depending on the tumor, and likely reflecting the role of the kinase in individual cancers (Section 2), both loss and upregulation of the kinase can be predictive of outcome. In recent studies, loss of PKCα defined a new sub-type of pediatric T-ALL with extremely poor outcome (Milani et al., 2014). PKCα deficiency is also associated with advanced disease and poor prognosis in colon and endometrial cancer (Chen et al., 2016; Hsu et al., 2018; Kahl-Rainer et al., 1994; Kahl-Rainer et al., 1996; Suga et al., 1998; Verstovsek et al., 1998) and with higher stage in NSCLC (Hill et al., 2014). On the other hand, immunohistochemical analysis of over 320 breast tumors, representative of all histological subtypes in proportions corresponding to incidence, linked PKCα positivity (observed in 28% of all tumors analyzed) to lack of estrogen and progesterone receptor expression, higher histological grade, and increased proliferation rate. PKCα positivity predicted worse disease outcome and significantly poorer 10-year survival in patients, and multivariate analysis pointed to PKCα as an independent prognostic factor in breast cancer (Lonne et al., 2010). These findings have been confirmed by others (Hsu et al., 2014). Tonetti and colleagues further demonstrated that PKCα positively correlates with the aggressive TNBC form of breast cancer in both African American and Caucasian patients, and with high grade breast tumors in African-American patients (Tonetti et al., 2012). Elevated PKCα expression is also associated with poor prognosis in patients with hepatocellular carcinoma (Ye et al., 2017) and gastric cancer (Lin et al., 2008; Lin et al., 2013), and expression of phosphorylated PKCα in the context of BCL2 phosphorylation predicted poor survival in acute myeloid leukemia patients (Kurinna et al., 2006). Analysis of 117 melanoma samples also linked overexpression of PKCα with poorer overall patient survival, and enhanced expression of the kinase in the context of BRAFV600E or NRAS mutations was associated with an 11-fold increased risk of death from the disease (Mahapatra et al., 2019).
Consistent with the role of PKCα in cell survival and chemoresistance (Section 4), PKCα expression can also be predictive of drug response. For example, increased levels of the kinase are predictive of endocrine resistance (tamoxifen treatment failure) and duration of response in breast cancer patients (Assender et al., 2007; Frankel et al., 2007; Lonne et al., 2010; Tonetti et al., 2003). Limited evidence further supports a role for PKCα as a diagnostic marker. The ability of serum samples from lung cancer patients, but not healthy controls, to phosphorylate a PKCα-specific peptide substrate suggests that serum PKCα may serve as a biomarker for detection of some lung tumors (Kang et al., 2013). Similarly, an SLC44A1-PRKCA fusion has been identified as a specific characteristic of papillary glioneuronal tumors with high diagnostic value for this rare tumor type (Pages et al., 2015).
6.2. PKCα as a therapeutic target
The concept of PKC isozymes as tumor promoters led to an interest in targeting these kinases for therapy. UCN-01 (7-hydroxystaurosporine), developed as an inhibitor of classical PKCs, has antitumor activity in preclinical models and has shown some efficacy in early stage clinical trials (Tse et al., 2007). However, UCN-01 is now recognized to be a broad-range kinase inhibitor. Additional UCN-01 targets include multiple CDKs (Akiyama et al., 1997), GSK3β (Leclerc et al., 2001), CHK1 (Busby et al., 2000; Graves et al., 2000) and PDK1 (Sato et al., 2002), and its antitumor activity is thought to reflect activity toward these targets rather than PKCs (Graves et al., 2000; Signore et al., 2014; Wang et al., 1995).
The only drug to enter clinical trials that specifically targets PKCα is aprinocarsen (LY900003, ISIS 3521), a phosphorothiote antisense oligonucleotide that targets the 3’ UTR of PKCα mRNA (Lahn et al., 2003). Preclinical studies showed that aprinocarsen effectively reduces PKCα expression and reduces tumor growth in mouse xenograft models of pancreatic, breast, prostate and NSCLC cell lines that express high levels of the protein (Denham et al., 1998; Dennis et al., 1998; Lahn et al., 2003; Shen et al., 1999; Yazaki et al., 1996). Aprinocarsen was well tolerated in patients, with the most consistent side effects being fatigue and thrombocytopenia (Advani et al., 2005; Nemunaitis et al., 1999; Paz-Ares et al., 2006; Rao et al., 2004; Ritch et al., 2006; Yuen et al., 1999), the latter likely reflecting the role of PKCα in megakaryocytic differentiation (Hocevar et al., 1992; Murray et al., 1993). However, the drug failed to show clinical efficacy in phase II trials for astrocytoma (Grossman et al., 2005), breast (Roychowdhury and Lahn, 2003), colorectal (Cripps et al., 2002), ovarian (Advani et al., 2004) and hormone-refractory prostate (Tolcher et al., 2002) cancers. While moderate activity was reported for NSCLC in combination with gemcitabine and carboplation (Ritch et al., 2006), a subsequent phase III trial determined that aprinocarsen has no efficacy in this disease (Paz-Ares et al., 2006). A phase II study indicated activity in low-grade non-Hodgkin’s leukemia (Rao et al., 2004); however, only one phase III trial has reported results and aprinocarsen was withdrawn in 2005 (Kelland, 2006). In hindsight, the failure of aprinocarsen in many of these studies is unsurprising given the growth inhibitory/tumor suppressive activity of PKCα in leukemia cells as well as colorectal, ovarian, and possibly NSCL cancer cells (see Section 2). Furthermore, the enzyme is downregulated in colorectal, NSCL (squamous and adenocarcinoma), prostate, and ovarian cancer (Section 5.2). Thus, based on our growing knowledge, therapeutic exploitation of PKCα signaling in many cancers will require interventions that enhance downstream tumor suppressive signaling, either through inhibition of target oncogenic factors or activation of tumor suppressors.
7. Conclusions
Although the diversity of PKCα cellular functions that impinge on tumorigenesis complicates the definition of the roles of PKCα in cancer, several themes are emerging. Studies in normal tissues indicate that the kinase is generally growth inhibitory/differentiation-inducing in regenerating epithelia, with suppressive effects on growth factor receptor signaling and cell cycle progression through G1/S and G2/M phases being major underlying mechanisms (Figure 4). Consistent with the fact that most cancers arise from epithelial tissues, this activity is mirrored in a predominantly tumor suppressive role of the enzyme, supported by frequent downregulation of PKCα mRNA and protein in most cancer types (Figure 3). The importance of PKCα deficiency in tumors is further indicated by its reported prognostic value in pediatric T-ALL, colon, endometrial, and NSCLC cancer, as well as by the finding that PKCα knockout or inhibition leads to enhanced tumorigenesis in all murine cancer models reported to date (i.e., models of intestinal, skin, endometrial, and lung cancer as well as B-CLL). Although PKCα is growth inhibitory in the majority of normal tissues, it also has physiological pro-proliferative effects, most notably in the hematopoietic system. Similarly, important tumor promoting effects are seen in some cancer types, especially in glioma and breast tumors, likely due to the well documented ability of PKCα to mediate cell survival, migration, EMT and metastasis. Although inactivating mutations in PKCα are not commonly seen in tumors, gain-of-function driving mutations have been noted in rare glioma subtypes. In some tumor types such as breast cancer, both tumor promoting and tumor suppressive effects of PKCα have been observed. The opposing effects of PKCα in breast cancers may reflect unique signaling cues from the tumor microenvironment as well as specific cellular changes within individual tumors that differentially affect the ability of PKCα to mediate its anti-proliferative, survival and migratory effects.
Figure 4.
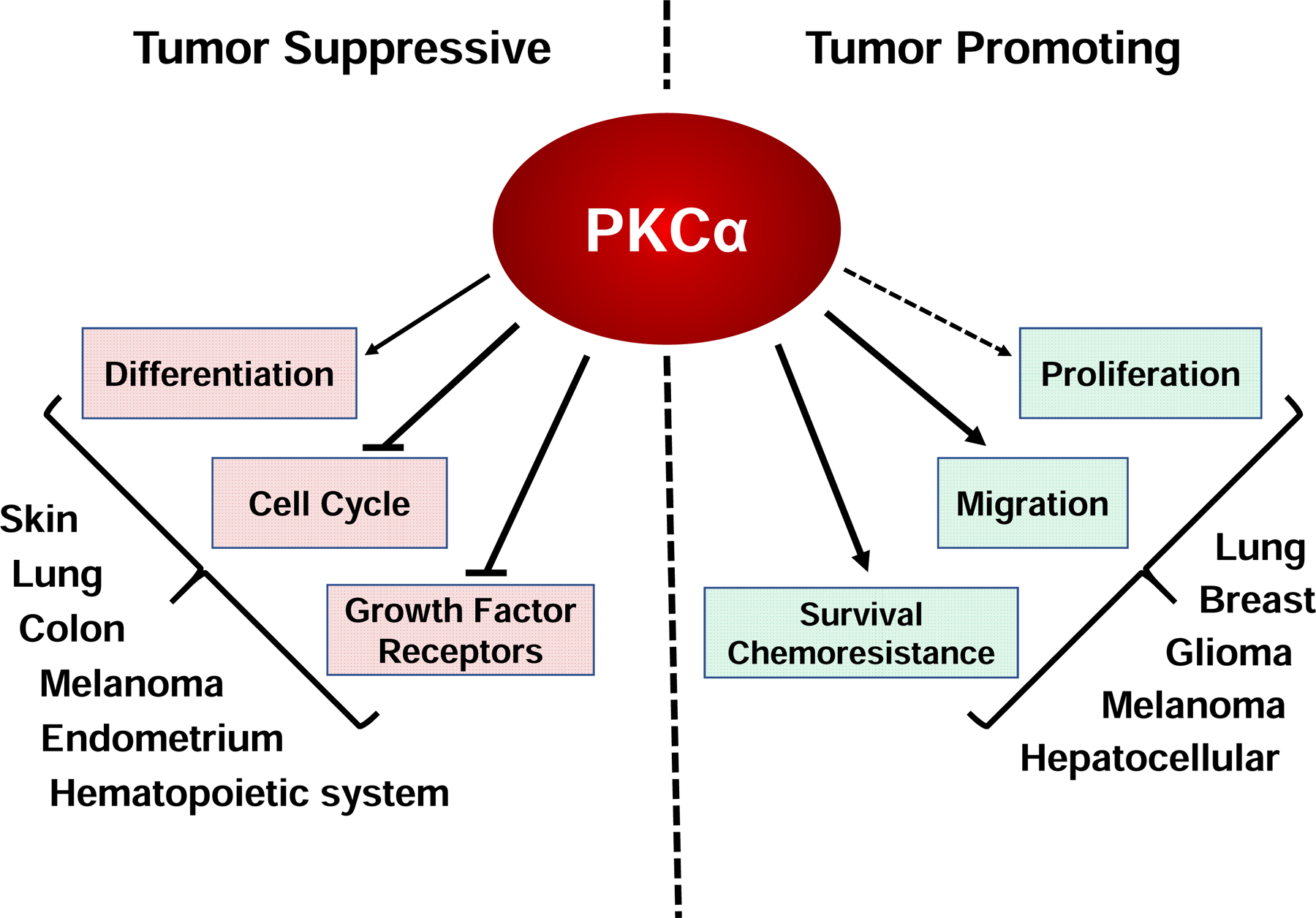
Overview of the role of PKCα in cancer.
In line with its pleotropic effects, PKCα appears to regulate both tumor initiation and progression and, depending on context, loss or upregulation of PKCα can promote these processes. Downregulation of the enzyme in human precancerous lesions, such as colonic adenomas, and in early stage human cancers of many types points to a role in initiation. Involvement in cancer initiation is further supported by the ability of PKCα knockout to increase intestinal adenoma formation in APCMin/+mice and skin papilloma formation in response to DMBA/TPA, accelerate the formation of hyperplastic endometrial lesions in the context of inactivating PTEN mutation, and expand the tumor-initiating bronchio-alveolar stem cell population in models of NSCLC. A role of PKCα deficiency in tumor progression is indicated by the correlation between loss of PKCα expression and more advanced disease in multiple human cancers, and by the ability of PKCα knockout to promote the development of invasive tumors in murine models of intestinal cancer and NSCLC. On the other hand, retention or upregulation of PKCα expression can also promote cancer initiation and progression. PKCα expression and activity in breast cancer stem cells has been shown to promote breast cancer initiation. Consistent with enhanced expression of the enzyme in the aggressive triple negative form of breast cancer, aberrant PKCα activity appears to regulate the switch from ER-positive to ER-negative status, promote hormone-independent, tamoxifen-resistant proliferation, and enhance EMT, invasion and metastasis of breast cancer cells.
Despite its clear role in tumorigenesis, therapeutic exploitation of PKCα remains elusive. Not surprisingly given its emergence as a predominantly tumor suppressive factor that is downregulated in many tumors, direct targeting of PKCα activity has proved unsuccessful in the clinic. Furthermore, its tumor suppressive roles in many healthy tissues would argue against the advisability of direct targeting of PKCα activity even for therapy of cancers where it has oncogenic roles. Thus, exploitation of the therapeutic potential of targeting PKCα signaling will require a more comprehensive understanding of the pathways that mediate its tumor suppressive and tumor promoting functions, with additional studies needed to identify downstream effectors that can be targeted for cancer treatment. Approaches that restore PKCα expression in tumors may also be of benefit in many cancer types.
Acknowledgements
We apologize to those whose work could not be cited due to space limitations. We would also like to thank our laboratory members and colleagues for their contribution to understanding the complex functions of PKCα in cancer. Work in our laboratory was supported by National Institutes of Health R01 grants CA054807, CA191894, DK54909, DK60632, pilot funds from the Fred & Pamela Buffett Cancer Center, and the Cancer Center Core Grant P30 CA036727.
Abbreviations
- ALL
acute lymphoblastic leukemia
- ABC
ATP binding cassette
- CDK
cyclin-dependent kinase
- CLL
chronic lymphocytic leukemia
- DAG
diacylglycerol
- DLG1
Disks-Large 1
- DMBA
7,12-dimethylbenz[a]anthracene
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- EMT
epithelial-to-mesenchymal transition
- GAP
GTPase activating protein
- GDI
guanidine dissociation inhibitor
- GEF
guanine nucleotide exchange factor
- HNSCC
head-and-neck squamous cell carcinoma
- Id
Inhibitor of DNA Binding
- lncRNA
long non-coding RNA
- IRS1
insulin receptor substrate 1
- NSCLC
non-small cell lung cancer
- PDGFR
platelet-derived growth factor receptor
- PI(4,5)P2
phosphatidylinositol 4,5-bisphosphate
- PKC
protein kinase C
- PMA
phorbol 12-myristate 13-acetate
- PP2A
protein phosphatase 2A
- PTEN
Phosphatase And Tensin Homolog
- pRb
retinoblastoma protein
- RhoGDI1
Rho guanidine dissociation inhibitor 1
- TPA
12-O-tetradecanoylphorbol-13-acetate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interest
None.
Declaration of Competing Interest
The authors have no conflicts of interest to declare.
References
- Aaltonen V, Koivunen J, Laato M, Peltonen J, 2006. Heterogeneity of cellular proliferation within transitional cell carcinoma: correlation of protein kinase C alpha/betaI expression and activity. J Histochem Cytochem 54(7), 795–806. [DOI] [PubMed] [Google Scholar]
- Abera MB, Kazanietz MG, 2015. Protein kinase Cα mediates erlotinib resistance in lung cancer cells. Mol Pharmacol 87(5), 832–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham C, Scaglione-Sewell B, Skarosi SF, Qin W, Bissonnette M, Brasitus TA, 1998. Protein kinase C α modulates growth and differentiation in Caco-2 cells. Gastroenterology 114(3), 503–509. [DOI] [PubMed] [Google Scholar]
- Abrams ST, Lakum T, Lin K, Jones GM, Treweeke AT, Farahani M, Hughes M, Zuzel M, Slupsky JR, 2006. B-cell receptor signaling in chronic lymphocytic leukemia cells is regulated by overexpressed active protein kinase CβII. Blood 109(3), 1193–1201. [DOI] [PubMed] [Google Scholar]
- Advani R, Lum BL, Fisher GA, Halsey J, Geary RS, Holmlund JT, Kwoh TJ, Dorr FA, Sikic BI, 2005. A phase I trial of aprinocarsen (ISIS 3521/LY900003), an antisense inhibitor of protein kinase C-α administered as a 24-hour weekly infusion schedule in patients with advanced cancer. Invest New Drugs 23(5), 467–477. [DOI] [PubMed] [Google Scholar]
- Advani R, Peethambaram P, Lum BL, Fisher GA, Hartmann L, Long HJ, Halsey J, Holmlund JT, Dorr A, Sikic BI, 2004. A Phase II trial of aprinocarsen, an antisense oligonucleotide inhibitor of protein kinase C α, administered as a 21-day infusion to patients with advanced ovarian carcinoma. Cancer 100(2), 321–326. [DOI] [PubMed] [Google Scholar]
- Ahmad S, Safa AR, Glazer RI, 1994. Modulation of P-glycoprotein by protein kinase C α in a baculovirus expression system. Biochemistry 33(34), 10313–10318. [DOI] [PubMed] [Google Scholar]
- Aifa S, Frikha F, Miled N, Johansen K, Lundstrom I, Svensson SP, 2006. Phosphorylation of Thr654 but not Thr669 within the juxtamembrane domain of the EGF receptor inhibits calmodulin binding. Biochem Biophys Res Commun 347(2), 381–387. [DOI] [PubMed] [Google Scholar]
- Aihara H, Asaoka Y, Yoshida K, Nishizuka Y, 1991. Sustained activation of protein kinase C is essential to HL-60 cell differentiation to macrophage. Proc Natl Acad Sci U S A 88(24), 11062–11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainsworth PD, Winstanley JHR, Pearson JM, Bishop HM, Garrod DR, 2004. Protein kinase C alpha expression in normal breast, ductal carcinoma in situ and invasive ductal carcinoma. Eur J Cancer 40(15), 2269–2273. [DOI] [PubMed] [Google Scholar]
- Akakura S, Nochajski P, Gao L, Sotomayor P, Matsui S, Gelman IH, 2010. Rb-dependent cellular senescence, multinucleation and susceptibility to oncogenic transformation through PKC scaffolding by SSeCKS/AKAP12. Cell Cycle 9(23), 4656–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama T, Yoshida T, Tsujita T, Shimizu M, Mizukami T, Okabe M, Akinaga S, 1997. G1 Phase Accumulation Induced by UCN-01 Is Associated with Dephosphorylation of Rb and CDK2 Proteins as well as Induction of CDK Inhibitor p21/Cip1/WAF1/Sdi1 in p53-mutated Human Epidermoid Carcinoma A431 Cells. Cancer Research 57(8), 1495–1501. [PubMed] [Google Scholar]
- Alvaro V, Lévy L, Dubray C, Roche A, Peillon F, Querat B, Joubert D, 1993. Invasive human pituitary tumors express a point-mutated alpha-protein kinase-C. J Clin Endocrinol Metab 77(5), 1125–1129. [DOI] [PubMed] [Google Scholar]
- Alvi F, Idkowiak-Baldys J, Baldys A, Raymond JR, Hannun YA, 2007. Regulation of membrane trafficking and endocytosis by protein kinase C: emerging role of the pericentrion, a novel protein kinase C-dependent subset of recycling endosomes. Cell Mol Life Sci 64(3), 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreozzi F, Melillo RM, Carlomagno F, Oriente F, Miele C, Fiory F, Santopietro S, Castellone MD, Beguinot F, Santoro M, Formisano P, 2003. Protein kinase Cα activation by RET: evidence for a negative feedback mechanism controlling RET tyrosine kinase. Oncogene 22(19), 2942–2949. [DOI] [PubMed] [Google Scholar]
- Anilkumar N, Parsons M, Monk R, Ng T, Adams JC, 2003. Interaction of fascin and protein kinase Cαa: a novel intersection in cell adhesion and motility. EMBO J 22(20), 5390–5402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter EW, Gallegos LL, Miller CJ, Furnari FB, Hunter T, Brognard J, Newton AC, 2015. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell 160(3), 489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assender JW, Gee JM, Lewis I, Ellis IO, Robertson JF, Nicholson RI, 2007. Protein kinase C isoform expression as a predictor of disease outcome on endocrine therapy in breast cancer. J Clin Pathol 60(11), 1216–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagowski CP, Stein-Gerlach M, Choidas A, Ullrich A, 1999. Cell-type specific phosphorylation of threonines T654 and T669 by PKD defines the signal capacity of the EGF receptor. EMBO J 18(20), 5567–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TA, Luan H, Tom E, Bielecki TA, Mohapatra B, Ahmad G, George M, Kelly DL, Natarajan A, Raja SM, Band V, Band H, 2014. A kinase inhibitor screen reveals protein kinase C-dependent endocytic recycling of ErbB2 in breast cancer cells. The Journal of biological chemistry 289(44), 30443–30458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker J, Spits M, Neefjes J, Berlin I, 2017. The EGFR odyssey - from activation to destruction in space and time. J Cell Sci 130(24), 4087–4096. [DOI] [PubMed] [Google Scholar]
- Baltuch GH, Dooley NP, Rostworowski KM, Villemure JG, Yong VW, 1995. Protein kinase C isoform α overexpression in C6 glioma cells and its role in cell proliferation. J Neurooncol 24(3), 241–250. [DOI] [PubMed] [Google Scholar]
- Bao J, Alroy I, Waterman H, Schejter ED, Brodie C, Gruenberg J, Yarden Y, 2000. Threonine phosphorylation diverts internalized epidermal growth factor receptors from a degradative pathway to the recycling endosome. The Journal of biological chemistry 275(34), 26178–26186. [DOI] [PubMed] [Google Scholar]
- Bass MD, Roach KA, Morgan MR, Mostafavi-Pour Z, Schoen T, Muramatsu T, Mayer U, Ballestrem C, Spatz JP, Humphries MJ, 2007. Syndecan-4-dependent Rac1 regulation determines directional migration in response to the extracellular matrix. J Cell Biol 177(3), 527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass MD, Williamson RC, Nunan RD, Humphries JD, Byron A, Morgan MR, Martin P, Humphries MJ, 2011. A syndecan-4 hair trigger initiates wound healing through caveolin-and RhoG-regulated integrin endocytosis. Dev Cell 21(4), 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlle E, Verdu J, Dominguez D, del Mont Llosas M, Diaz V, Loukili N, Paciucci R, Alameda F, de Herreros AG, 1998. Protein kinase C-α activity inversely modulates invasion and growth of intestinal cells. The Journal of biological chemistry 273(24), 15091–15098. [DOI] [PubMed] [Google Scholar]
- Becker KP, Hannun YA, 2005. Protein kinase C and phospholipase D: intimate interactions in intracellular signaling. Cell Mol Life Sci 62(13), 1448–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson A, Yong VW, 2000. Involvement of p21Waf1/Cip1 in protein kinase C alpha-induced cell cycle progression. Mol Cell Biol 20(13), 4580–4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidaud-Meynard A, Biname F, Lagree V, Moreau V, 2017. Regulation of Rho GTPase activity at the leading edge of migrating cells by p190RhoGAP. Small GTPases 10(2), 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorkelund H, Gedda L, Malmqvist M, Andersson K, 2013. Resolving the EGF-EGFR interaction characteristics through a multiple-temperature, multiple-inhibitor, real-time interaction analysis approach. Mol Clin Oncol 1(2), 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black AR, Black JD, 2012. Protein kinase C signaling and cell cycle regulation. Front Immunol 3, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JD, 2001. Protein kinase C isozymes in colon carcinogenesis: guilt by omission. Gastroenterology 120(7), 1868–1872. [DOI] [PubMed] [Google Scholar]
- Block J, Breitsprecher D, Kuhn S, Winterhoff M, Kage F, Geffers R, Duwe P, Rohn JL, Baum B, Brakebusch C, Geyer M, Stradal TE, Faix J, Rottner K, 2012. FMNL2 drives actin-based protrusion and migration downstream of Cdc42. Curr Biol 22(11), 1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan S, Schultz J, Grosshans J, 2013. Formin’ cellular structures: Physiological roles of Diaphanous (Dia) in actin dynamics. Commun Integr Biol 6(6), e27634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag WB, 2009. Protein kinase Cα puts the hand cuffs on epidermal keratinocyte proliferation. J Invest Dermatol 129(10), 2330–2332. [DOI] [PubMed] [Google Scholar]
- Breitsprecher D, Goode BL, 2013. Formins at a glance. J Cell Sci 126(Pt 1), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge JA, Liu XQ, Sumegi J, Nelson M, Reyes C, Bruch LA, Rosenblum M, Puccioni MJ, Bowdino BS, McComb RD, 2013. Identification of a novel, recurrent SLC44A1-PRKCA fusion in papillary glioneuronal tumor. Brain Pathol 23(2), 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budworth J, Davies R, Malkhandi J, Gant TW, Ferry DR, Gescher A, 1996. Comparison of staurosporine and four analogues: their effects on growth, rhodamine 123 retention and binding to P-glycoprotein in multidrug-resistant MCF-7/Adr cells. Br J Cancer 73(9), 1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busby EC, Leistritz DF, Abraham RT, Karnitz LM, Sarkaria JN, 2000. The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res 60(8), 2108–2112. [PubMed] [Google Scholar]
- Byers HR, Boissel SJ, Tu C, Park HY, 2010. RNAi-mediated knockdown of protein kinase C-alpha inhibits cell migration in MM-RU human metastatic melanoma cell line. Melanoma Res 20(3), 171–178. [DOI] [PubMed] [Google Scholar]
- Cai J, Crotty TM, Reichert E, Carraway KL 3rd, Stafforini DM, Topham MK, 2010. Diacylglycerol kinase δ and protein kinase Cα modulate epidermal growth factor receptor abundance and degradation through ubiquitin-specific protease 8. The Journal of biological chemistry 285(10), 6952–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron AJ, Procyk KJ, Leitges M, Parker PJ, 2008. PKC alpha protein but not kinase activity is critical for glioma cell proliferation and survival. Int J Cancer 123(4), 769–779. [DOI] [PubMed] [Google Scholar]
- Carter CA, Parham GP, Chambers T, 1998. Cytoskeletal reorganization induced by retinoic acid treatment of human endometrial adenocarcinoma (RL95–2) cells is correlated with alterations in protein kinase C-α. Pathobiology 66(6), 284–292. [DOI] [PubMed] [Google Scholar]
- Caruso M, Miele C, Oriente F, Maitan A, Bifulco G, Andreozzi F, Condorelli G, Formisano P, Beguinot F, 1999. In L6 skeletal muscle cells, glucose induces cytosolic translocation of protein kinase C-α and trans-activates the insulin receptor kinase. The Journal of biological chemistry 274(40), 28637–28644. [DOI] [PubMed] [Google Scholar]
- Cataisson C, Joseloff E, Murillas R, Wang A, Atwell C, Torgerson S, Gerdes M, Subleski J, Gao JL, Murphy PM, Wiltrout RH, Vinson C, Yuspa SH, 2003. Activation of cutaneous protein kinase Cα induces keratinocyte apoptosis and intraepidermal inflammation by independent signaling pathways. J Immunol 171(5), 2703–2713. [DOI] [PubMed] [Google Scholar]
- Cataisson C, Michalowski AM, Shibuya K, Ryscavage A, Klosterman M, Wright L, Dubois W, Liu F, Zhuang A, Rodrigues KB, Hoover S, Dwyer J, Simpson MR, Merlino G, Yuspa SH, 2016. MET signaling in keratinocytes activates EGFR and initiates squamous carcinogenesis. Sci Signal 9(433), ra62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataisson C, Ohman R, Patel G, Pearson A, Tsien M, Jay S, Wright L, Hennings H, Yuspa SH, 2009. Inducible cutaneous inflammation reveals a protumorigenic role for keratinocyte CXCR2 in skin carcinogenesis. Cancer Res 69(1), 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerioni L, Palomba L, Brune B, Cantoni O, 2006. Peroxynitrite-induced mitochondrial translocation of PKCα causes U937 cell survival. Biochem Biophys Res Commun 339(1), 126–131. [DOI] [PubMed] [Google Scholar]
- Chambers TC, 1998. [24] Identification of phosphorylation sites in human MDR1 P-glycoprotein, ABC Transporters: Biochemical, Cellular, and Molecular Aspects. Academic Press, pp. 328–342. [DOI] [PubMed] [Google Scholar]
- Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce-Rodriguez I, Chakravarthi B, Varambally S, 2017. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 19(8), 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Xie H, Wells A, 1996. Mitogenic signaling from the egf receptor is attenuated by a phospholipase C-gamma/protein kinase C feedback mechanism. Mol Biol Cell 7(6), 871–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang Y, Zhang Y, Wan Y, 2016. Low expression of PKCα and high expression of KRAS predict poor prognosis in patients with colorectal cancer. Oncol Lett 12(3), 1655–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CW, Huang Y, Leong KW, Chen LC, Chen HC, Chen SJ, Chou CK, 2010. PKCα mediated induction of miR-101 in human hepatoma HepG2 cells. J Biomed Sci 17, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisamore MJ, Ahmed Y, Bentrem DJ, Jordan VC, Tonetti DA, 2001. Novel antitumor effect of estradiol in athymic mice injected with a T47D breast cancer cell line overexpressing protein kinase Cα. Clin Cancer Res 7(10), 3156–3165. [PubMed] [Google Scholar]
- Cho Y, Talmage DA, 2001. Protein kinase Cα expression confers retinoic acid sensitivity on MDA-MB-231 human breast cancer cells. Exp Cell Res 269(1), 97–108. [DOI] [PubMed] [Google Scholar]
- Cho Y, Tighe AP, Talmage DA, 1997. Retinoic acid induced growth arrest of human breast carcinoma cells requires protein kinase Cα expression and activity. J Cell Physiol 172(3), 306–313. [DOI] [PubMed] [Google Scholar]
- Clark AS, West KA, Blumberg PM, Dennis PA, 2003. Altered protein kinase C (PKC) isoforms in non-small cell lung cancer cells: PKCδ promotes cellular survival and chemotherapeutic resistance. Cancer Res 63(4), 780–786. [PubMed] [Google Scholar]
- Clark JA, Black AR, Leontieva OV, Frey MR, Pysz MA, Kunneva L, Woloszynska-Read A, Roy D, Black JD, 2004. Involvement of the ERK signaling cascade in protein kinase C-mediated cell cycle arrest in intestinal epithelial cells. The Journal of biological chemistry 279(10), 9233–9247. [DOI] [PubMed] [Google Scholar]
- Clayton NS, Ridley AJ, 2020. Targeting Rho GTPase Signaling Networks in Cancer. Front Cell Dev Biol 8(222), 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen EEW, Zhu H, Lingen MW, Martin LE, Kuo WL, Choi EA, Kocherginsky M, Parker JS, Chung CH, Rosner MR, 2009. A feed-forward loop involving protein kinase Cα and microRNAs regulates tumor cell cycle. Cancer Res 69(1), 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JN, Joseph NM, North JP, Onodera C, Zembowicz A, LeBoit PE, 2017. Genomic Analysis of Pigmented Epithelioid Melanocytomas Reveals Recurrent Alterations in PRKAR1A, and PRKCA Genes. Am J Surg Pathol 41(10), 1333–1346. [DOI] [PubMed] [Google Scholar]
- Cooke M, Casado-Medrano V, Ann J, Lee J, Blumberg PM, Abba MC, Kazanietz MG, 2019. Differential Regulation of Gene Expression in Lung Cancer Cells by Diacyglycerol-Lactones and a Phorbol Ester Via Selective Activation of Protein Kinase C Isozymes. Sci Rep 9(1), 6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Countaway JL, McQuilkin P, Girones N, Davis RJ, 1990. Multisite phosphorylation of the epidermal growth factor receptor. Use of site-directed mutagenesis to examine the role of serine/threonine phosphorylation. The Journal of biological chemistry 265(6), 3407–3416. [PubMed] [Google Scholar]
- Coussens L, Parker PJ, Rhee L, Yang-Feng TL, Chen E, Waterfield MD, Francke U, Ullrich A, 1986. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science (New York, N.Y.) 233(4766), 859–866. [DOI] [PubMed] [Google Scholar]
- Cripps MC, Figueredo AT, Oza AM, Taylor MJ, Fields AL, Holmlund JT, McIntosh LW, Geary RS, Eisenhauer EA, 2002. Phase II randomized study of ISIS 3521 and ISIS 5132 in patients with locally advanced or metastatic colorectal cancer: a National Cancer Institute of Canada clinical trials group study. Clin Cancer Res 8(7), 2188–2192. [PubMed] [Google Scholar]
- Crocenzi FA, Sanchez Pozzi EJ, Ruiz ML, Zucchetti AE, Roma MG, Mottino AD, Vore M, 2008. Ca(2+)-dependent protein kinase C isoforms are critical to estradiol 17β-D-glucuronide-induced cholestasis in the rat. Hepatology 48(6), 1885–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Wang J, Liu S, Qu D, Jin H, Zhu L, Yang J, Zhang J, Li Q, Zhang Y, Yao Y, 2019. miR-216a promotes breast cancer cell apoptosis by targeting PKCα. Fundam Clin Pharmacol 33(4), 397–404. [DOI] [PubMed] [Google Scholar]
- Davis RJ, Czech MP, 1985. Tumor-promoting phorbol diesters cause the phosphorylation of epidermal growth factor receptors in normal human fibroblasts at threonine-654. Proc Natl Acad Sci U S A 82(7), 1974–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Asua LJ, Goin M, 1992. Prostaglandin F2α decreases the affinity of epidermal growth factor receptors in Swiss mouse 3T3 cells via protein kinase C activation. FEBS Letters 299(3), 235–238. [DOI] [PubMed] [Google Scholar]
- Denham DW, Franz MG, Denham W, Zervos EE, Gower WR Jr., Rosemurgy AS, Norman J, 1998. Directed antisense therapy confirms the role of protein kinase C-α in the tumorigenicity of pancreatic cancer. Surgery 124(2), 218–223; discussion 223–214. [PubMed] [Google Scholar]
- Denning MF, Dlugosz AA, Williams EK, Szallasi Z, Blumberg PM, Yuspa SH, 1995. Specific protein kinase C isozymes mediate the induction of keratinocyte differentiation markers by calcium. Cell Growth Differ 6(2), 149–157. [PubMed] [Google Scholar]
- Dennis JU, Dean NM, Bennett CF, Griffith JW, Lang CM, Welch DR, 1998. Human melanoma metastasis is inhibited following ex vivo treatment with an antisense oligonucleotide to protein kinase C-α. Cancer Lett 128(1), 65–70. [DOI] [PubMed] [Google Scholar]
- Detjen KM, Brembeck FH, Welzel M, Kaiser A, Haller H, Wiedenmann B, Rosewicz S, 2000. Activation of protein kinase Cα inhibits growth of pancreatic cancer cells via p21cip-mediated G1 arrest. J Cell Sci 113 ( Pt 17), 3025–3035. [DOI] [PubMed] [Google Scholar]
- Dieter P, Schwende H, 2000. Protein kinase C-α and -β play antagonistic roles in the differentiation process of THP-1 cells. Cell Signal 12(5), 297–302. [DOI] [PubMed] [Google Scholar]
- Dovas A, Choi Y, Yoneda A, Multhaupt HA, Kwon SH, Kang D, Oh ES, Couchman JR, 2010. Serine 34 phosphorylation of rho guanine dissociation inhibitor (RhoGDIα) links signaling from conventional protein kinase C to RhoGTPase in cell adhesion. The Journal of biological chemistry 285(30), 23296–23308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J, Waterfield MD, Parker PJ, 1985. Autophosphorylation and protein kinase C phosphorylation of the epidermal growth factor receptor. Effect on tyrosine kinase activity and ligand binding affinity. The Journal of biological chemistry 260(27), 14538–14546. [PubMed] [Google Scholar]
- Du HF, Ou LP, Yang X, Song XD, Fan YR, Tan B, Luo CL, Wu XH, 2014. A new PKCα/β/TBX3/E-cadherin pathway is involved in PLCε-regulated invasion and migration in human bladder cancer cells. Cell Signal 26(3), 580–593. [DOI] [PubMed] [Google Scholar]
- Dupasquier S, Abdel-Samad R, Glazer RI, Bastide P, Jay P, Joubert D, Cavailles V, Blache P, Quittau-Prevostel C, 2009. A new mechanism of SOX9 action to regulate PKCα expression in the intestine epithelium. J Cell Sci 122(Pt 13), 2191–2196. [DOI] [PubMed] [Google Scholar]
- El-Osta MA, Idkowiak-Baldys J, Hannun YA, 2011. Delayed phosphorylation of classical protein kinase C (PKC) substrates requires PKC internalization and formation of the pericentrion in a phospholipase D (PLD)-dependent manner. The Journal of biological chemistry 286(22), 19340–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfenbein A, Rhodes JM, Meller J, Schwartz MA, Matsuda M, Simons M, 2009. Suppression of RhoG activity is mediated by a syndecan 4-synectin-RhoGDI1 complex and is reversed by PKCα in a Rac1 activation pathway. J Cell Biol 186(1), 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfenbein A, Simons M, 2013. Syndecan-4 signaling at a glance. J Cell Sci 126(Pt 17), 3799–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elnakat H, Gonit M, Salazar MD, Zhang J, Basrur V, Gunning W, Kamen B, Ratnam M, 2009. Regulation of folate receptor internalization by protein kinase C α. Biochemistry 48(34), 8249–8260. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A, 2002. Rho GTPases in cell biology. Nature 420(6916), 629–635. [DOI] [PubMed] [Google Scholar]
- Eude I, Dallot E, Ferre F, Breuiller-Fouche M, 2002. Protein kinase Cα is required for endothelin-1-induced proliferation of human myometrial cells. Biol Reprod 66(1), 44–49. [DOI] [PubMed] [Google Scholar]
- Feng Y, Walsh CA, 2004. The many faces of filamin: a versatile molecular scaffold for cell motility and signalling. Nat Cell Biol 6(11), 1034–1038. [DOI] [PubMed] [Google Scholar]
- Ferrari A, Vincent-Salomon A, Pivot X, Sertier AS, Thomas E, Tonon L, Boyault S, Mulugeta E, Treilleux I, MacGrogan G, Arnould L, Kielbassa J, Le Texier V, Blanche H, Deleuze JF, Jacquemier J, Mathieu MC, Penault-Llorca F, Bibeau F, Mariani O, Mannina C, Pierga JY, Tredan O, Bachelot T, Bonnefoi H, Romieu G, Fumoleau P, Delaloge S, Rios M, Ferrero JM, Tarpin C, Bouteille C, Calvo F, Gut IG, Gut M, Martin S, Nik-Zainal S, Stratton MR, Pauporte I, Saintigny P, Birnbaum D, Viari A, Thomas G, 2016. A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Nat Commun 7(1), 12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel LB, Lykkesfeldt AE, Hansen JB, Stenvang J, 2007. Protein Kinase C α is a marker for antiestrogen resistance and is involved in the growth of tamoxifen resistant human breast cancer cells. Breast Cancer Res Treat 104(2), 165–179. [DOI] [PubMed] [Google Scholar]
- Frey MR, Clark JA, Leontieva O, Uronis JM, Black AR, Black JD, 2000. Protein kinase C signaling mediates a program of cell cycle withdrawal in the intestinal epithelium. J Cell Biol 151(4), 763–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey MR, Saxon ML, Zhao X, Rollins A, Evans SS, Black JD, 1997. Protein kinase C isozyme-mediated cell cycle arrest involves induction of p21waf1/cip1 and p27kip1 and hypophosphorylation of the retinoblastoma protein in intestinal epithelial cells. The Journal of biological chemistry 272(14), 9424–9435. [DOI] [PubMed] [Google Scholar]
- Friedman B, Frackelton AR Jr., Ross AH, Connors JM, Fujiki H, Sugimura T, Rosner MR, 1984. Tumor promoters block tyrosine-specific phosphorylation of the epidermal growth factor receptor. Proc Natl Acad Sci U S A 81(10), 3034–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bermejo ML, Leskow FC, Fujii T, Wang Q, Blumberg PM, Ohba M, Kuroki T, Han KC, Lee J, Marquez VE, Kazanietz MG, 2002. Diacylglycerol (DAG)-lactones, a new class of protein kinase C (PKC) agonists, induce apoptosis in LNCaP prostate cancer cells by selective activation of PKCα. The Journal of biological chemistry 277(1), 645–655. [DOI] [PubMed] [Google Scholar]
- Germann UA, Chambers TC, Ambudkar SV, Licht T, Cardarelli CO, Pastan I, Gottesman MM, 1996. Characterization of phosphorylation-defective mutants of human P-glycoprotein expressed in mammalian cells. The Journal of biological chemistry 271(3), 1708–1716. [DOI] [PubMed] [Google Scholar]
- Gill PK, Gescher A, Gant TW, 2001. Regulation of MDR1 promoter activity in human breast carcinoma cells by protein kinase C isozymes α and θ. Eur J Biochem 268(15), 4151–4157. [DOI] [PubMed] [Google Scholar]
- Gökmen-Polar Y, Murray NR, Velasco MA, Gatalica Z, Fields AP, 2001. Elevated Protein Kinase C βII Is an Early Promotive Event in Colon Carcinogenesis. Cancer Research 61(4), 1375–1381. [PubMed] [Google Scholar]
- Goode B, Mondal G, Hyun M, Ruiz DG, Lin YH, Van Ziffle J, Joseph NM, Onodera C, Talevich E, Grenert JP, Hewedi IH, Snuderl M, Brat DJ, Kleinschmidt-DeMasters BK, Rodriguez FJ, Louis DN, Yong WH, Lopes MB, Rosenblum MK, Butowski N, Tihan T, Bollen AW, Phillips JJ, Wiita AP, Yeh I, Jacobson MP, Bastian BC, Perry A, Solomon DA, 2018. A recurrent kinase domain mutation in PRKCA defines chordoid glioma of the third ventricle. Nat Commun 9(1), 810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow HR, Sardini A, Ruetz S, Callaghan R, Gros P, McNaughton PA, Higgins CF, 1996. Protein kinase C-mediated phosphorylation does not regulate drug transport by the human multidrug resistance P-glycoprotein. The Journal of biological chemistry 271(23), 13668–13674. [DOI] [PubMed] [Google Scholar]
- Graves PR, Yu L, Schwarz JK, Gales J, Sausville EA, O’Connor PM, Piwnica-Worms H, 2000. The Chk1 protein kinase and the Cdc25C regulatory pathways are targets of the anticancer agent UCN-01. The Journal of biological chemistry 275(8), 5600–5605. [DOI] [PubMed] [Google Scholar]
- Grossman SA, Alavi JB, Supko JG, Carson KA, Priet R, Dorr FA, Grundy JS, Holmlund JT, 2005. Efficacy and toxicity of the antisense oligonucleotide aprinocarsen directed against protein kinase C-α delivered as a 21-day continuous intravenous infusion in patients with recurrent high-grade astrocytomas. Neuro Oncol 7(1), 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber JR, Ohno S, Niles RM, 1992. Increased expression of protein kinase C α plays a key role in retinoic acid-induced melanoma differentiation. The Journal of biological chemistry 267(19), 13356–13360. [PubMed] [Google Scholar]
- Gruber T, Hermann-Kleiter N, Pfeifhofer-Obermair C, Lutz-Nicoladoni C, Thuille N, Letschka T, Barsig J, Baudler M, Li J, Metzler B, Nusslein-Hildesheim B, Wagner J, Leitges M, Baier G, 2009. PKCθ cooperates with PKCα in alloimmune responses of T cells in vivo. Mol Immunol 46(10), 2071–2079. [DOI] [PubMed] [Google Scholar]
- Guan L, Song K, Pysz MA, Curry KJ, Hizli AA, Danielpour D, Black AR, Black JD, 2007. Protein kinase C-mediated down-regulation of cyclin D1 involves activation of the translational repressor 4E-BP1 via a phosphoinositide 3-kinase/Akt-independent, protein phosphatase 2A-dependent mechanism in intestinal epithelial cells. The Journal of biological chemistry 282(19), 14213–14225. [DOI] [PubMed] [Google Scholar]
- Gwak J, Cho M, Gong SJ, Won J, Kim DE, Kim EY, Lee SS, Kim M, Kim TK, Shin JG, Oh S, 2006. Protein-kinase-C-mediated β-catenin phosphorylation negatively regulates the Wnt/ β-catenin pathway. J Cell Sci 119(Pt 22), 4702–4709. [DOI] [PubMed] [Google Scholar]
- Gwak J, Jung SJ, Kang DI, Kim EY, Kim DE, Chung YH, Shin JG, Oh S, 2009. Stimulation of protein kinase C-α suppresses colon cancer cell proliferation by down-regulation of β-catenin. J Cell Mol Med 13(8B), 2171–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwak J, Lee JH, Chung YH, Song GY, Oh S, 2012. Small molecule-based promotion of PKα-mediated β-catenin degradation suppresses the proliferation of CRT-positive cancer cells. PLoS One 7(10), e46697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansra G, Garcia-Paramio P, Prevostel C, Whelan RD, Bornancin F, Parker PJ, 1999. Multisite dephosphorylation and desensitization of conventional protein kinase C isotypes. Biochem J 342 ( Pt 2)(Pt 2), 337–344. [PMC free article] [PubMed] [Google Scholar]
- Hao F, Pysz MA, Curry KJ, Haas KN, Seedhouse SJ, Black AR, Black JD, 2011. Protein kinase Cα signaling regulates inhibitor of DNA binding 1 in the intestinal epithelium. The Journal of biological chemistry 286(20), 18104–18117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, Matsumura S, Hakuno F, Takahashi S, Chida K, 2012. PKCα suppresses 7,12-dimethylbenz[a]anthracene-induced skin tumor formation. Anticancer Res 32(8), 3097–3101. [PubMed] [Google Scholar]
- Hara T, Saito Y, Hirai T, Nakamura K, Nakao K, Katsuki M, Chida K, 2005. Deficiency of protein kinase Cα in mice results in impairment of epidermal hyperplasia and enhancement of tumor formation in two-stage skin carcinogenesis. Cancer Res 65(16), 7356–7362. [DOI] [PubMed] [Google Scholar]
- Haslauer M, Baltensperger K, Porzig H, 1999. Erythropoietin- and stem cell factor-induced DNA synthesis in normal human erythroid progenitor cells requires activation of protein kinase Cα and is strongly inhibited by thrombin. Blood 94(1), 114–126. [PubMed] [Google Scholar]
- Haughian JM, Bradford AP, 2009. Protein kinase C alpha (PKCα) regulates growth and invasion of endometrial cancer cells. J Cell Physiol 220(1), 112–118. [DOI] [PubMed] [Google Scholar]
- Haughian JM, Reno EM, Thorne AM, Bradford AP, 2009. Protein kinase C alpha-dependent signaling mediates endometrial cancer cell growth and tumorigenesis. Int J Cancer 125(11), 2556–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellberg C, Schmees C, Karlsson S, Ahgren A, Heldin CH, 2009. Activation of protein kinase C α is necessary for sorting the PDGF β-receptor to Rab4a-dependent recycling. Mol Biol Cell 20(12), 2856–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill KS, Erdogan E, Khoor A, Walsh MP, Leitges M, Murray NR, Fields AP, 2014. Protein kinase Cα suppresses Kras-mediated lung tumor formation through activation of a p38 MAPK-TGFβ signaling axis. Oncogene 33(16), 2134–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hizli AA, Black AR, Pysz MA, Black JD, 2006. Protein kinase C α signaling inhibits cyclin D1 translation in intestinal epithelial cells. The Journal of biological chemistry 281(21), 14596–14603. [DOI] [PubMed] [Google Scholar]
- Hocevar BA, Morrow DM, Tykocinski ML, Fields AP, 1992. Protein kinase C isotypes in human erythroleukemia cell proliferation and differentiation. J Cell Sci 101 ( Pt 3), 671–679. [DOI] [PubMed] [Google Scholar]
- Horowitz A, Murakami M, Gao Y, Simons M, 1999. Phosphatidylinositol-4,5-bisphosphate mediates the interaction of syndecan-4 with protein kinase C. Biochemistry 38(48), 15871–15877. [DOI] [PubMed] [Google Scholar]
- Horowitz A, Simons M, 1998. Phosphorylation of the cytoplasmic tail of syndecan-4 regulates activation of protein kinase Cα. The Journal of biological chemistry 273(40), 25548–25551. [DOI] [PubMed] [Google Scholar]
- Hoshino D, Jourquin J, Emmons SW, Miller T, Goldgof M, Costello K, Tyson DR, Brown B, Lu Y, Prasad NK, Zhang B, Mills GB, Yarbrough WG, Quaranta V, Seiki M, Weaver AM, 2012. Network Analysis of the Focal Adhesion to Invadopodia Transition Identifies a PI3K-PKCα Invasive Signaling Axis. Science Signaling 5(241), ra66–ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YC, Jao HC, Yang RC, Hsu HK, Hsu C, 2003. Suppression of protein kinase Cα triggers apoptosis through down-regulation of Bcl-xL in a rat hepatic epithelial cell line. Shock 19(6), 582–587. [DOI] [PubMed] [Google Scholar]
- Hsieh YH, Wu TT, Huang CY, Hsieh YS, Hwang JM, Liu JY, 2007. p38 mitogen-activated protein kinase pathway is involved in protein kinase Calpha-regulated invasion in human hepatocellular carcinoma cells. Cancer Res 67(9), 4320–4327. [DOI] [PubMed] [Google Scholar]
- Hsieh YH, Wu TT, Tsai JH, Huang CY, Hsieh YS, Liu JY, 2006. PKCα expression regulated by Elk-1 and MZF-1 in human HCC cells. Biochem Biophys Res Commun 339(1), 217–225. [DOI] [PubMed] [Google Scholar]
- Hsu AH, Lum MA, Shim KS, Frederick PJ, Morrison CD, Chen B, Lele SM, Sheinin YM, Daikoku T, Dey SK, Leone G, Black AR, Black JD, 2018. Crosstalk between PKCα and PI3K/AKT Signaling Is Tumor Suppressive in the Endometrium. Cell Rep 24(3), 655–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YH, Yao J, Chan LC, Wu TJ, Hsu JL, Fang YF, Wei Y, Wu Y, Huang WC, Liu CL, Chang YC, Wang MY, Li CW, Shen J, Chen MK, Sahin AA, Sood A, Mills GB, Yu D, Hortobagyi GN, Hung MC, 2014. Definition of PKC-α, CDK6, and MET as therapeutic targets in triple-negative breast cancer. Cancer Res 74(17), 4822–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Xia L, Zhou H, Wu B, Mu Y, Wu Y, Yan J, 2013. TF/FVIIa/PAR2 promotes cell proliferation and migration via PKCα and ERK-dependent c-Jun/AP-1 pathway in colon cancer cell line SW620. Tumour Biol 34(5), 2573–2581. [DOI] [PubMed] [Google Scholar]
- Humphries B, Wang Z, Oom AL, Fisher T, Tan D, Cui Y, Jiang Y, Yang C, 2014. MicroRNA-200b targets protein kinase Cα and suppresses triple-negative breast cancer metastasis. Carcinogenesis 35(10), 2254–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter T, Ling N, Cooper JA, 1984. Protein kinase C phosphorylation of the EGF receptor at a threonine residue close to the cytoplasmic face of the plasma membrane. Nature 311(5985), 480–483. [DOI] [PubMed] [Google Scholar]
- Idkowiak-Baldys J, Becker KP, Kitatani K, Hannun YA, 2006. Dynamic sequestration of the recycling compartment by classical protein kinase C. The Journal of biological chemistry 281(31), 22321–22331. [DOI] [PubMed] [Google Scholar]
- Idriss H, Urquidi V, Basavappa S, 2000. Selective modulation of P-glycoprotein’s ATPase and anion efflux regulation activities with PKC α and PKC ɛ in Sf9 cells. Cancer Chemother Pharmacol 46(4), 287–292. [DOI] [PubMed] [Google Scholar]
- Inoue M, Kishimoto A, Takai Y, Nishizuka Y, 1977. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. II. Proenzyme and its activation by calcium-dependent protease from rat brain. The Journal of biological chemistry 252(21), 7610–7616. [PubMed] [Google Scholar]
- Iwamoto T, Hagiwara M, Hidaka H, Isomura T, Kioussis D, Nakashima I, 1992. Accelerated proliferation and interleukin-2 production of thymocytes by stimulation of soluble anti-CD3 monoclonal antibody in transgenic mice carrying a rabbit protein kinase Cɛ. The Journal of biological chemistry 267(26), 18644–18648. [PubMed] [Google Scholar]
- Jansen AP, Dreckschmidt NE, Verwiebe EG, Wheeler DL, Oberley TD, Verma AK, 2001. Relation of the induction of epidermal ornithine decarboxylase and hyperplasia to the different skin tumor-promotion susceptibilities of protein kinase Cα, -δ and -ϵ transgenic mice. Int J Cancer 93(5), 635–643. [DOI] [PubMed] [Google Scholar]
- Jansen S, Gosens R, Wieland T, Schmidt M, 2018. Paving the Rho in cancer metastasis: Rho GTPases and beyond. Pharmacol Ther 183, 1–21. [DOI] [PubMed] [Google Scholar]
- Jeong J, Choi J, Kim W, Dann P, Takyar F, Gefter JV, Friedman PA, Wysolmerski JJ, 2019. Inhibition of ezrin causes PKCα-mediated internalization of erbb2/HER2 tyrosine kinase in breast cancer cells. The Journal of biological chemistry 294(3), 887–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerome-Morais A, Rahn HR, Tibudan SS, Denning MF, 2009. Role for protein kinase C-α in keratinocyte growth arrest. J Invest Dermatol 129(10), 2365–2375. [DOI] [PubMed] [Google Scholar]
- Jiang R, Shi Y, Zeng C, Yu W, Zhang A, Du Y, 2017. Protein kinase Cα stimulates hypoxiainduced pulmonary artery smooth muscle cell proliferation in rats through activating the extracellular signalregulated kinase 1/2 pathway. Mol Med Rep 16(5), 6814–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang XH, Tu SP, Cui JT, Lin MC, Xia HH, Wong WM, Chan AO, Yuen MF, Jiang SH, Lam SK, Kung HF, Soh JW, Weinstein IB, Wong BC, 2004. Antisense targeting protein kinase C α and β1 inhibits gastric carcinogenesis. Cancer Res 64(16), 5787–5794. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Berk M, Singh LS, Tan H, Yin L, Powell CT, Xu Y, 2005. KiSS1 suppresses metastasis in human ovarian cancer via inhibition of protein kinase C alpha. Clin Exp Metastasis 22(5), 369–376. [DOI] [PubMed] [Google Scholar]
- Jiang YH, Aukema HM, Davidson LA, Lupton JR, Chapkin RS, 1995. Localization of protein kinase C isozymes in rat colon. Cell Growth Differ 6(11), 1381–1386. [PubMed] [Google Scholar]
- Jiang Z, Kong C, Zhang Z, Zhu Y, Zhang Y, Chen X, 2015. Reduction of protein kinase C α (PKC-α) promote apoptosis via down-regulation of Dicer in bladder cancer. J Cell Mol Med 19(5), 1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiffar T, Kurinna S, Suck G, Carlson-Bremer D, Ricciardi MR, Konopleva M, Andreeff M, Ruvolo PP, 2004. PKC α mediates chemoresistance in acute lymphoblastic leukemia through effects on Bcl2 phosphorylation. Leukemia 18(3), 505–512. [DOI] [PubMed] [Google Scholar]
- Joniova J, Misuth M, Sureau F, Miskovsky P, Nadova Z, 2014. Effect of PKCα expression on Bcl-2 phosphorylation and cell death by hypericin. Apoptosis 19(12), 1779–1792. [DOI] [PubMed] [Google Scholar]
- Jost M, Huggett TM, Kari C, Boise LH, Rodeck U, 2001. Epidermal growth factor receptor-dependent control of keratinocyte survival and Bcl-xL expression through a MEK-dependent pathway. The Journal of biological chemistry 276(9), 6320–6326. [DOI] [PubMed] [Google Scholar]
- Kahl-Rainer P, Karner-Hanusch J, Weiss W, Marian B, 1994. Five of six protein kinase C isoenzymes present in normal mucosa show reduced protein levels during tumor development in the human colon. Carcinogenesis 15(4), 779–782. [DOI] [PubMed] [Google Scholar]
- Kahl-Rainer P, Sedivy R, Marian B, 1996. Protein kinase C tissue localization in human colonic tumors suggests a role for adenoma growth control. Gastroenterology 110(6), 1753–1759. [DOI] [PubMed] [Google Scholar]
- Kang JH, Mori T, Kitazaki H, Niidome T, Takayama K, Nakanishi Y, Katayama Y, 2013. Kinase activity of protein kinase Cα in serum as a diagnostic biomarker of human lung cancer. Anticancer Res 33(2), 485–488. [PubMed] [Google Scholar]
- Katoh H, Hiramoto K, Negishi M, 2006. Activation of Rac1 by RhoG regulates cell migration. J Cell Sci 119(Pt 1), 56–65. [DOI] [PubMed] [Google Scholar]
- Kaur N, Lewis R, Black A, Black J, 2018. Abstract 3466: Growth inhibitory MEK-ERK signaling in the intestinal epithelium. Cancer Research 78(13 Supplement), 3466–3466. [Google Scholar]
- Kelland L, 2006. Discontinued drugs in 2005: oncology drugs. Expert Opin Investig Drugs 15(11), 1309–1318. [DOI] [PubMed] [Google Scholar]
- Keranen LM, Dutil EM, Newton AC, 1995. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr Biol 5(12), 1394–1403. [DOI] [PubMed] [Google Scholar]
- Kerfoot C, Huang W, Rotenberg SA, 2004. Immunohistochemical analysis of advanced human breast carcinomas reveals downregulation of protein kinase C α. J Histochem Cytochem 52(3), 419–422. [DOI] [PubMed] [Google Scholar]
- Kermorgant S, Zicha D, Parker PJ, 2004. PKC controls HGF-dependent c-Met traffic, signalling and cell migration. EMBO J 23(19), 3721–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Thorne SH, Sun L, Huang B, Mochly-Rosen D, 2011. Sustained inhibition of PKCα reduces intravasation and lung seeding during mammary tumor metastasis in an in vivo mouse model. Oncogene 30(3), 323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzing TM, Wang Y, Pertz O, Copeland JW, Grosse R, 2010. Formin-like 2 drives amoeboid invasive cell motility downstream of RhoC. Oncogene 29(16), 2441–2448. [DOI] [PubMed] [Google Scholar]
- Klein IK, Ritland SR, Burgart LJ, Ziesmer SC, Roche PC, Gendler SJ, Karnes WE, 2000. Adenoma-specific Alterations of Protein Kinase C Isozyme Expression in ApcMIN Mice. Cancer Research 60(8), 2077–2080. [PubMed] [Google Scholar]
- Klein IK, Ritland SR, Burgart LJ, Ziesmer SC, Roche PC, Gendler SJ, Karnes WE Jr., 2000. Adenoma-specific alterations of protein kinase C isozyme expression in Apc(MIN) mice. Cancer Res 60(8), 2077–2080. [PubMed] [Google Scholar]
- Kluba M, Engelborghs Y, Hofkens J, Mizuno H, 2015. Inhibition of Receptor Dimerization as a Novel Negative Feedback Mechanism of EGFR Signaling. PLoS One 10(10), e0139971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knezevic N, Roy A, Timblin B, Konstantoulaki M, Sharma T, Malik AB, Mehta D, 2007. GDI-1 phosphorylation switch at serine 96 induces RhoA activation and increased endothelial permeability. Mol Cell Biol 27(18), 6323–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koese M, Rentero C, Kota BP, Hoque M, Cairns R, Wood P, Vila de Muga S, Reverter M, Alvarez-Guaita A, Monastyrskaya K, Hughes WE, Swarbrick A, Tebar F, Daly RJ, Enrich C, Grewal T, 2013. Annexin A6 is a scaffold for PKCα to promote EGFR inactivation. Oncogene 32(23), 2858–2872. [DOI] [PubMed] [Google Scholar]
- Kong C, Zhu Y, Liu D, Yu M, Li S, Li Z, Sun Z, Liu G, 2005. Role of protein kinase C-alpha in superficial bladder carcinoma recurrence. Urology 65(6), 1228–1232. [DOI] [PubMed] [Google Scholar]
- Koo BK, Jung YS, Shin J, Han I, Mortier E, Zimmermann P, Whiteford JR, Couchman JR, Oh ES, Lee W, 2006. Structural basis of syndecan-4 phosphorylation as a molecular switch to regulate signaling. J Mol Biol 355(4), 651–663. [DOI] [PubMed] [Google Scholar]
- Krasagakis K, Fimmel S, Genten D, Eberle J, Quas P, Ziegler W, Haller H, Orfanos CE, 2002. Lack of protein kinase C (PKC)-β and low PKC-α, -δ, -ε, and -ζ isozyme levels in proliferating human melanoma cells. Int J Oncol 20(4), 865–871. [PubMed] [Google Scholar]
- Krasagakis K, Lindschau C, Fimmel S, Eberle J, Quass P, Haller H, Orfanos CE, 2004. Proliferation of human melanoma cells is under tight control of protein kinase C alpha. J Cell Physiol 199(3), 381–387. [DOI] [PubMed] [Google Scholar]
- Kurinna S, Konopleva M, Palla SL, Chen W, Kornblau S, Contractor R, Deng X, May WS, Andreeff M, Ruvolo PP, 2006. Bcl2 phosphorylation and active PKC α are associated with poor survival in AML. Leukemia 20(7), 1316–1319. [DOI] [PubMed] [Google Scholar]
- Kyuno D, Kojima T, Yamaguchi H, Ito T, Kimura Y, Imamura M, Takasawa A, Murata M, Tanaka S, Hirata K, Sawada N, 2013. Protein kinase Cα inhibitor protects against downregulation of claudin-1 during epithelial-mesenchymal transition of pancreatic cancer. Carcinogenesis 34(6), 1232–1243. [DOI] [PubMed] [Google Scholar]
- Lahn M, Kohler G, Sundell K, Su C, Li S, Paterson BM, Bumol TF, 2004. Protein kinase C alpha expression in breast and ovarian cancer. Oncology 67(1), 1–10. [DOI] [PubMed] [Google Scholar]
- Lahn M, Sundell K, Moore S, 2003. Targeting protein kinase C-alpha (PKC-α) in cancer with the phosphorothioate antisense oligonucleotide aprinocarsen. Ann N Y Acad Sci 1002(1), 263–270. [DOI] [PubMed] [Google Scholar]
- Lampasso JD, Marzec N, Margarone J 3rd, Dziak R, 2002. Role of protein kinase C α in primary human osteoblast proliferation. J Bone Miner Res 17(11), 1968–1976. [DOI] [PubMed] [Google Scholar]
- Langzam L, Koren R, Gal R, Kugel V, Paz A, Farkas A, Sampson SR, 2001. Patterns of protein kinase C isoenzyme expression in transitional cell carcinoma of bladder. Relation to degree of malignancy. Am J Clin Pathol 116(3), 377–385. [DOI] [PubMed] [Google Scholar]
- Leclerc S, Garnier M, Hoessel R, Marko D, Bibb JA, Snyder GL, Greengard P, Biernat J, Wu YZ, Mandelkow EM, Eisenbrand G, Meijer L, 2001. Indirubins inhibit glycogen synthase kinase-3β and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? The Journal of biological chemistry 276(1), 251–260. [DOI] [PubMed] [Google Scholar]
- Lee HW, Smith L, Pettit GR, Bingham Smith J, 1996. Dephosphorylation of activated protein kinase C contributes to downregulation by bryostatin. Am J Physiol 271(1 Pt 1), C304–311. [DOI] [PubMed] [Google Scholar]
- Lee JM, Kim IS, Kim H, Lee JS, Kim K, Yim HY, Jeong J, Kim JH, Kim JY, Lee H, Seo SB, Kim H, Rosenfeld MG, Kim KI, Baek SH, 2010. RORalpha attenuates Wnt/β-catenin signaling by PKCα-dependent phosphorylation in colon cancer. Mol Cell 37(2), 183–195. [DOI] [PubMed] [Google Scholar]
- Lei J, Li Q, Gao Y, Zhao L, Liu Y, 2016. Increased PKCα activity by Rack1 overexpression is responsible for chemotherapy resistance in T-cell acute lymphoblastic leukemia-derived cell line. Sci Rep 6(1), 33717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leirdal M, Sioud M, 1999. Ribozyme inhibition of the protein kinase Cα triggers apoptosis in glioma cells. Br J Cancer 80(10), 1558–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitges M, Plomann M, Standaert ML, Bandyopadhyay G, Sajan MP, Kanoh Y, Farese RV, 2002. Knockout of PKCα enhances insulin signaling through PI3K. Mol Endocrinol 16(4), 847–858. [DOI] [PubMed] [Google Scholar]
- Lenting K, Verhaak R, Ter Laan M, Wesseling P, Leenders W, 2017. Glioma: experimental models and reality. Acta Neuropathol 133(2), 263–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontieva OV, Black JD, 2004. Identification of two distinct pathways of protein kinase Cα down-regulation in intestinal epithelial cells. The Journal of biological chemistry 279(7), 5788–5801. [DOI] [PubMed] [Google Scholar]
- Leszczynski D, Joenvaara S, Foegh ML, 1996. Protein kinase C-α regulates proliferation but not apoptosis in rat coronary vascular smooth muscle cells. Life Sci 58(7), 599–606. [DOI] [PubMed] [Google Scholar]
- Lévay M, Settleman J, Ligeti E, 2009. Regulation of the substrate preference of p190RhoGAP by protein kinase C-mediated phosphorylation of a phospholipid binding site. Biochemistry 48(36), 8615–8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Panina S, Kaur A, Ruano MJ, Sanchez-Gonzalez P, la Cour JM, Stephan A, Olesen UH, Berchtold MW, Villalobo A, 2012. Regulation of the ligand-dependent activation of the epidermal growth factor receptor by calmodulin. The Journal of biological chemistry 287(5), 3273–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang Z, Wang J, Du S, Yao D, Cao R, Cui S, 2020. Protein Kinase Cα Promotes Proliferation and Migration of Schwann Cells by Activating ERK Signaling Pathway. Neuroscience 433, 94–107. [DOI] [PubMed] [Google Scholar]
- Lim ST, Longley RL, Couchman JR, Woods A, 2003. Direct binding of syndecan-4 cytoplasmic domain to the catalytic domain of protein kinase Cα (PKCα) increases focal adhesion localization of PKC alpha. The Journal of biological chemistry 278(16), 13795–13802. [DOI] [PubMed] [Google Scholar]
- Lin C, 1986. Protein kinase C phosphorylation at Thr 654 of the unoccupied EGF receptor and EGF binding regulate functional receptor loss by independent mechanisms. Cell 44(6), 839–848. [DOI] [PubMed] [Google Scholar]
- Lin CW, Shen SC, Chien CC, Yang LY, Shia LT, Chen YC, 2010. 12-O-tetradecanoylphorbol-13-acetate-induced invasion/migration of glioblastoma cells through activating PKCα/ERK/NF-κB-dependent MMP-9 expression. J Cell Physiol 225(2), 472–481. [DOI] [PubMed] [Google Scholar]
- Lin KY, Fang CL, Uen YH, Chang CC, Lou HY, Hsieh CR, Tiong C, Pan S, Chen SH, 2008. Overexpression of protein kinase Cα mRNA may be an independent prognostic marker for gastric carcinoma. J Surg Oncol 97(6), 538–543. [DOI] [PubMed] [Google Scholar]
- Lin SC, Chen WY, Lin KY, Chen SH, Chang CC, Lin SE, Fang CL, 2013. Clinicopathological correlation and prognostic significance of protein kinase Cα overexpression in human gastric carcinoma. PLoS One 8(2), e56675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Idkowiak-Baldys J, Roddy PL, Baldys A, Raymond J, Clarke CJ, Hannun YA, 2013. Sustained activation of protein kinase C induces delayed phosphorylation and regulates the fate of epidermal growth factor receptor. PLoS One 8(11), e80721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens MC, Rossi FA, Garcia IA, Cooke M, Abba MC, Lopez-Haber C, Barrio-Real L, Vaglienti MV, Rossi M, Bocco JL, Kazanietz MG, Soria G, 2019. PKCα Modulates Epithelial-to-Mesenchymal Transition and Invasiveness of Breast Cancer Cells Through ZEB1. Front Oncol 9(1323), 1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonne GK, Cornmark L, Zahirovic IO, Landberg G, Jirstrom K, Larsson C, 2010. PKCα expression is a marker for breast cancer aggressiveness. Mol Cancer 9(1), 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum MA, Balaburski GM, Murphy ME, Black AR, Black JD, 2013a. Heat shock proteins regulate activation-induced proteasomal degradation of the mature phosphorylated form of protein kinase C. The Journal of biological chemistry 288(38), 27112–27127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum MA, Barger CJ, Hsu AH, Leontieva OV, Black AR, Black JD, 2016. Protein Kinase Cα (PKCα) Is Resistant to Long Term Desensitization/Down-regulation by Prolonged Diacylglycerol Stimulation. The Journal of biological chemistry 291(12), 6331–6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum MA, Pundt KE, Paluch BE, Black AR, Black JD, 2013b. Agonist-induced down-regulation of endogenous protein kinase C α through an endolysosomal mechanism. The Journal of biological chemistry 288(18), 13093–13109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund KA, Lazar CS, Chen WS, Walsh BJ, Welsh JB, Herbst JJ, Walton GM, Rosenfeld MG, Gill GN, Wiley HS, 1990. Phosphorylation of the epidermal growth factor receptor at threonine 654 inhibits ligand-induced internalization and down-regulation. The Journal of biological chemistry 265(33), 20517–20523. [PubMed] [Google Scholar]
- Macdonald-Obermann JL, Pike LJ, 2009. The intracellular juxtamembrane domain of the epidermal growth factor (EGF) receptor is responsible for the allosteric regulation of EGF binding. The Journal of biological chemistry 284(20), 13570–13576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahapatra L, Andruska N, Mao C, Gruber SB, Johnson TM, Fullen DR, Raskin L, Shapiro DJ, 2019. Protein kinase C-α is upregulated by IMP1 in melanoma and is linked to poor survival. Melanoma Res 29(5), 539–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandil R, Ashkenazi E, Blass M, Kronfeld I, Kazimirsky G, Rosenthal G, Umansky F, Lorenzo PS, Blumberg PM, Brodie C, 2001. Protein kinase Cα and protein kinase Cδ play opposite roles in the proliferation and apoptosis of glioma cells. Cancer Res 61(11), 4612–4619. [PubMed] [Google Scholar]
- Martinez-Gimeno C, Diaz-Meco MT, Dominguez I, Moscat J, 1995. Alterations in levels of different protein kinase C isotypes and their influence on behavior of squamous cell carcinoma of the oral cavity: ɛPKC, a novel prognostic factor for relapse and survival. Head Neck 17(6), 516–525. [DOI] [PubMed] [Google Scholar]
- Marziali F, Dizanzo MP, Cavatorta AL, Gardiol D, 2019. Differential expression of DLG1 as a common trait in different human diseases: an encouraging issue in molecular pathology. Biol Chem 400(6), 699–710. [DOI] [PubMed] [Google Scholar]
- Masur K, Lang K, Niggemann B, Zanker KS, Entschladen F, 2001. High PKC α and low E-cadherin expression contribute to high migratory activity of colon carcinoma cells. Mol Biol Cell 12(7), 1973–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayati A, Moreau A, Le Vee M, Stieger B, Denizot C, Parmentier Y, Fardel O, 2017. Protein Kinases C-Mediated Regulations of Drug Transporter Activity, Localization and Expression. Int J Mol Sci 18(4), 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani G, Rebora P, Accordi B, Galla L, Bresolin S, Cazzaniga G, Buldini B, Mura R, Ladogana S, Giraldi E, Conter V, Te Kronnie G, Valsecchi MG, Basso G, 2014. Low PKCδ expression within the MRD-HR stratum defines a new subgroup of childhood T-ALL with very poor outcome. Oncotarget 5(14), 5234–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischak H, Pierce JH, Goodnight J, Kazanietz MG, Blumberg PM, Mushinski JF, 1993. Phorbol ester-induced myeloid differentiation is mediated by protein kinase C-α and -δ and not by protein kinase CβII, -ε, -ζ, and -η. The Journal of biological chemistry 268(27), 20110–20115. [PubMed] [Google Scholar]
- Mitchell FE, Marais RM, Parker PJ, 1989. The phosphorylation of protein kinase C as a potential measure of activation. Biochem J 261(1), 131–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty S, Huang J, Basu A, 2005. Enhancement of cisplatin sensitivity of cisplatin-resistant human cervical carcinoma cells by bryostatin 1. Clin Cancer Res 11(18), 6730–6737. [DOI] [PubMed] [Google Scholar]
- Morrison P, Takishima K, Rosner MR, 1993. Role of threonine residues in regulation of the epidermal growth factor receptor by protein kinase C and mitogen-activated protein kinase. The Journal of biological chemistry 268(21), 15536–15543. [PubMed] [Google Scholar]
- Murakami M, Horowitz A, Tang S, Ware JA, Simons M, 2002. Protein kinase C (PKC) δ regulates PKCα activity in a Syndecan-4-dependent manner. The Journal of biological chemistry 277(23), 20367–20371. [DOI] [PubMed] [Google Scholar]
- Murray NR, Baumgardner GP, Burns DJ, Fields AP, 1993. Protein kinase C isotypes in human erythroleukemia (K562) cell proliferation and differentiation. Evidence that βII protein kinase C is required for proliferation. The Journal of biological chemistry 268(21), 15847–15853. [PubMed] [Google Scholar]
- Nakagawa R, Soh JW, Michie AM, 2006. Subversion of protein kinase C α signaling in hematopoietic progenitor cells results in the generation of a B-cell chronic lymphocytic leukemia-like population in vivo. Cancer Res 66(1), 527–534. [DOI] [PubMed] [Google Scholar]
- Nakagawa R, Vukovic M, Cosimo E, Michie AM, 2012. Modulation of PKC-α promotes lineage reprogramming of committed B lymphocytes. Eur J Immunol 42(4), 1005–1015. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Fujii T, Yokoyama G, Kazanietz MG, Yamana H, Shirouzu K, 2003. Cell growth inhibition by all-trans retinoic acid in SKBR-3 breast cancer cells: involvement of protein kinase Cα and extracellular signal-regulated kinase mitogen-activated protein kinase. Mol Carcinog 38(3), 106–116. [DOI] [PubMed] [Google Scholar]
- Nakashima S, 2002. Protein kinase Cα(PKCα): regulation and biological function. J Biochem 132(5), 669–675. [DOI] [PubMed] [Google Scholar]
- Nakura A, Higuchi C, Yoshida K, Yoshikawa H, 2011. PKCα suppresses osteoblastic differentiation. Bone 48(3), 476–484. [DOI] [PubMed] [Google Scholar]
- Nawaratne R, Gray A, Jorgensen CH, Downes CP, Siddle K, Sethi JK, 2006. Regulation of insulin receptor substrate 1 pleckstrin homology domain by protein kinase C: role of serine 24 phosphorylation. Mol Endocrinol 20(8), 1838–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neill GW, Ghali LR, Green JL, Ikram MS, Philpott MP, Quinn AG, 2003. Loss of Protein Kinase Cα Expression May Enhance the Tumorigenic Potential of Gli1 in Basal Cell Carcinoma. Cancer Research 63(15), 4692–4697. [PubMed] [Google Scholar]
- Nemunaitis J, Holmlund JT, Kraynak M, Richards D, Bruce J, Ognoskie N, Kwoh TJ, Geary R, Dorr A, Von Hoff D, Eckhardt SG, 1999. Phase I evaluation of ISIS 3521, an antisense oligodeoxynucleotide to protein kinase C-alpha, in patients with advanced cancer. J Clin Oncol 17(11), 3586–3595. [DOI] [PubMed] [Google Scholar]
- Newton AC, 2001. Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem Rev 101(8), 2353–2364. [DOI] [PubMed] [Google Scholar]
- Newton AC, 2018. Protein kinase C: perfectly balanced. Crit Rev Biochem Mol Biol 53(2), 208–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T, Shima D, Squire A, Bastiaens PI, Gschmeissner S, Humphries MJ, Parker PJ, 1999a. PKCα regulates β1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J 18(14), 3909–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng T, Squire A, Hansra G, Bornancin F, Prevostel C, Hanby A, Harris W, Barnes D, Schmidt S, Mellor H, Bastiaens PI, Parker PJ, 1999b. Imaging protein kinase Cα activation in cells. Science (New York, N.Y.) 283(5410), 2085–2089. [DOI] [PubMed] [Google Scholar]
- Niles RM, 2003. Vitamin A (retinoids) regulation of mouse melanoma growth and differentiation. J Nutr 133(1), 282S–286S. [DOI] [PubMed] [Google Scholar]
- Niles RM, Loewy BP, 1989. Induction of protein kinase C in mouse melanoma cells by retinoic acid. Cancer Res 49(16), 4483–4487. [PubMed] [Google Scholar]
- O’Neill AK, Gallegos LL, Justilien V, Garcia EL, Leitges M, Fields AP, Hall RA, Newton AC, 2011. Protein kinase Cα promotes cell migration through a PDZ-dependent interaction with its novel substrate discs large homolog 1 (DLG1). The Journal of biological chemistry 286(50), 43559–43568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh ES, Woods A, Couchman JR, 1997. Syndecan-4 proteoglycan regulates the distribution and activity of protein kinase C. The Journal of biological chemistry 272(13), 8133–8136. [DOI] [PubMed] [Google Scholar]
- Oka M, Ogita K, Saito N, Mishima Y, 1993. Selective increase of the α subspecies of protein kinase C and inhibition of melanogenesis induced by retinoic acid in melanoma cells. J Invest Dermatol 100(2 Suppl), 204S–208S. [PubMed] [Google Scholar]
- Okazaki J, Mawatari K, Liu B, Kent KC, 2000. The effect of protein kinase C and its alpha subtype on human vascular smooth muscle cell proliferation, migration and fibronectin production. Surgery 128(2), 192–197. [DOI] [PubMed] [Google Scholar]
- Oliva JL, Caino MC, Senderowicz AM, Kazanietz MG, 2008. S-Phase-specific activation of PKCα induces senescence in non-small cell lung cancer cells. The Journal of biological chemistry 283(9), 5466–5476. [DOI] [PubMed] [Google Scholar]
- Oriente F, Andreozzi F, Romano C, Perruolo G, Perfetti A, Fiory F, Miele C, Beguinot F, Formisano P, 2005. Protein kinase C-α regulates insulin action and degradation by interacting with insulin receptor substrate-1 and 14–3-3ϵ. The Journal of biological chemistry 280(49), 40642–40649. [DOI] [PubMed] [Google Scholar]
- Orlandi L, Binda M, Folini M, Bearzatto A, Villa R, Daidone MG, Zaffaroni N, 2003. Ribozyme-mediated inhibition of PKCα sensitizes androgen-independent human prostate cancer cells to cisplatin-induced apoptosis. Prostate 54(2), 133–143. [DOI] [PubMed] [Google Scholar]
- Oster H, Leitges M, 2006. Protein kinase Cα but not PKCζ suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res 66(14), 6955–6963. [DOI] [PubMed] [Google Scholar]
- Ouyang X, Gulliford T, Epstein RJ, 1998. The duration of phorbol-inducible ErbB2 tyrosine dephosphorylation parallels that of receptor endocytosis rather than threonine-686 phosphorylation: implications for the physiological role of protein kinase C in growth factor receptor signalling. Carcinogenesis 19(11), 2013–2019. [DOI] [PubMed] [Google Scholar]
- Ouyang X, Gulliford T, Zhang H, Huang GC, Epstein R, 1996. Human cancer cells exhibit protein kinase C-dependent c-erbB-2 transmodulation that correlates with phosphatase sensitivity and kinase activity. The Journal of biological chemistry 271(36), 21786–21792. [DOI] [PubMed] [Google Scholar]
- Ozcan F, Klein P, Lemmon MA, Lax I, Schlessinger J, 2006. On the nature of low- and high-affinity EGF receptors on living cells. Proc Natl Acad Sci U S A 103(15), 5735–5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pages M, Lacroix L, Tauziede-Espariat A, Castel D, Daudigeos-Dubus E, Ridola V, Gilles S, Fina F, Andreiuolo F, Polivka M, Lechapt-Zalcman E, Puget S, Boddaert N, Liu XQ, Bridge JA, Grill J, Chretien F, Varlet P, 2015. Papillary glioneuronal tumors: histological and molecular characteristics and diagnostic value of SLC44A1-PRKCA fusion. Acta Neuropathol Commun 3(1), 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo E, Kellett MD, Cataisson C, Bible PW, Bhattacharya S, Sun HW, Gormley AC, Yuspa SH, Morasso MI, 2017. A novel DLX3-PKC integrated signaling network drives keratinocyte differentiation. Cell Death Differ 24(4), 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya K, Wyatt D, Gallagher B, Shah D, Baker A, Bloodworth J, Zlobin A, Pannuti A, Green A, Ellis IO, Filipovic A, Sagert J, Rana A, Albain KS, Miele L, Denning MF, Osipo C, 2016. PKCζ Attenuates Jagged-1-Mediated Notch Signaling in ErbB-2-Positive Breast Cancer to Reverse Trastuzumab Resistance. Clin Cancer Res 22(1), 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker PJ, Coussens L, Totty N, Rhee L, Young S, Chen E, Stabel S, Waterfield MD, Ullrich A, 1986. The complete primary structure of protein kinase C--the major phorbol ester receptor. Science (New York, N.Y.) 233(4766), 853–859. [DOI] [PubMed] [Google Scholar]
- Parri M, Chiarugi P, 2010. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal 8(1), 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M, Keppler MD, Kline A, Messent A, Humphries MJ, Gilchrist R, Hart IR, Quittau-Prevostel C, Hughes WE, Parker PJ, Ng T, 2002. Site-directed perturbation of protein kinase C- integrin interaction blocks carcinoma cell chemotaxis. Mol Cell Biol 22(16), 5897–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz-Ares L, Douillard JY, Koralewski P, Manegold C, Smit EF, Reyes JM, Chang GC, John WJ, Peterson PM, Obasaju CK, Lahn M, Gandara DR, 2006. Phase III study of gemcitabine and cisplatin with or without aprinocarsen, a protein kinase C-alpha antisense oligonucleotide, in patients with advanced-stage non-small-cell lung cancer. J Clin Oncol 24(9), 1428–1434. [DOI] [PubMed] [Google Scholar]
- Pfeifhofer C, Gruber T, Letschka T, Thuille N, Lutz-Nicoladoni C, Hermann-Kleiter N, Braun U, Leitges M, Baier G, 2006. Defective IgG2a/2b class switching in PKCα−/− mice. J Immunol 176(10), 6004–6011. [DOI] [PubMed] [Google Scholar]
- Pham TND, A., T.D., 2016. Protein Kinase C Alpha in Breast Cancer: A Focus on Endocrine Resistant and Triple Negative Breast Cancer. J Cancer Biol Res 4(1), 1076. [Google Scholar]
- Pham TND, Perez White BE, Zhao H, Mortazavi F, Tonetti DA, 2017. Protein kinase C α enhances migration of breast cancer cells through FOXC2-mediated repression of p120-catenin. BMC Cancer 17(1), 832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce A, Heyworth CM, Nicholls SE, Spooncer E, Dexter TM, Lord JM, Owen-Lynch PJ, Wark G, Whetton AD, 1998. An activated protein kinase C α gives a differentiation signal for hematopoietic progenitor cells and mimicks macrophage colony-stimulating factor-stimulated signaling events. J Cell Biol 140(6), 1511–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell CT, Brittis NJ, Stec D, Hug H, Heston WD, Fair WR, 1996. Persistent membrane translocation of protein kinase C α during 12–0-tetradecanoylphorbol-13-acetate-induced apoptosis of LNCaP human prostate cancer cells. Cell Growth Differ 7(4), 419–428. [PubMed] [Google Scholar]
- Prevostel C, Alice V, Joubert D, Parker PJ, 2000. Protein kinase Cα actively downregulates through caveolae-dependent traffic to an endosomal compartment. J Cell Sci 113 ( Pt 14), 2575–2584. [DOI] [PubMed] [Google Scholar]
- Prevostel C, Alvaro V, Vallentin A, Martin A, Jaken S, Joubert D, 1998. Selective loss of substrate recognition induced by the tumour-associated D294G point mutation in protein kinase Cα. Biochem J 334 ( Pt 2)(Pt 2), 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam AJ, Schulz VV, Freiter EM, Bill HM, Miranti CK, 2009. Src, PKCα, and PKCδ are required for αvβ3 integrin-mediated metastatic melanoma invasion. Cell Commun Signal 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pysz MA, Hao F, Hizli AA, Lum MA, Swetzig WM, Black AR, Black JD, 2014. Differential regulation of cyclin D1 expression by protein kinase C α and ϵ signaling in intestinal epithelial cells. The Journal of biological chemistry 289(32), 22268–22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pysz MA, Leontieva OV, Bateman NW, Uronis JM, Curry KJ, Threadgill DW, Janssen KP, Robine S, Velcich A, Augenlicht LH, Black AR, Black JD, 2009. PKCα tumor suppression in the intestine is associated with transcriptional and translational inhibition of cyclin D1. Exp Cell Res 315(8), 1415–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Liu S, Guo C, Wang J, Greenaway FT, Sun MZ, 2015. Role of annexin A6 in cancer. Oncol Lett 10(4), 1947–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Sun B, Zhao X, Du J, Gu Q, Liu Y, Cheng R, Dong X, 2014. Wnt5a promotes vasculogenic mimicry and epithelial-mesenchymal transition via protein kinase Cα in epithelial ovarian cancer. Oncol Rep 32(2), 771–779. [DOI] [PubMed] [Google Scholar]
- Qu Y, Han B, Yu Y, Yao W, Bose S, Karlan BY, Giuliano AE, Cui X, 2015. Evaluation of MCF10A as a Reliable Model for Normal Human Mammary Epithelial Cells. PLoS One 10(7), e0131285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quittau-Prevostel C, Delaunay N, Collazos A, Vallentin A, Joubert D, 2004. Targeting of PKCα and ϵ in the pituitary: a highly regulated mechanism involving a GD(E)E motif of the V3 region. J Cell Sci 117(Pt 1), 63–72. [DOI] [PubMed] [Google Scholar]
- Ranganathan S, Liu CX, Migliorini MM, Von Arnim CA, Peltan ID, Mikhailenko I, Hyman BT, Strickland DK, 2004. Serine and threonine phosphorylation of the low density lipoprotein receptor-related protein by protein kinase Cα regulates endocytosis and association with adaptor molecules. The Journal of biological chemistry 279(39), 40536–40544. [DOI] [PubMed] [Google Scholar]
- Rao S, Watkins D, Cunningham D, Dunlop D, Johnson P, Selby P, Hancock BW, Fegan C, Culligan D, Schey S, Morris TCM, Lissitchkov T, Oliver JW, Holmlund JT, 2004. Phase II study of ISIS 3521, an antisense oligodeoxynucleotide to protein kinase C alpha, in patients with previously treated low-grade non-Hodgkin’s lymphoma. Ann Oncol 15(9), 1413–1418. [DOI] [PubMed] [Google Scholar]
- Ritch P, Rudin CM, Bitran JD, Edelman MJ, Makalinao A, Irwin D, Lilenbaum R, Peterson P, John WJ, 2006. Phase II study of PKC-α antisense oligonucleotide aprinocarsen in combination with gemcitabine and carboplatin in patients with advanced non-small cell lung cancer. Lung Cancer 52(2), 173–180. [DOI] [PubMed] [Google Scholar]
- Robey RW, Shukla S, Steadman K, Obrzut T, Finley EM, Ambudkar SV, Bates SE, 2007. Inhibition of ABCG2-mediated transport by protein kinase inhibitors with a bisindolylmaleimide or indolocarbazole structure. Mol Cancer Ther 6(6), 1877–1885. [DOI] [PubMed] [Google Scholar]
- Rosenberg S, Simeonova I, Bielle F, Verreault M, Bance B, Le Roux I, Daniau M, Nadaradjane A, Gleize V, Paris S, Marie Y, Giry M, Polivka M, Figarella-Branger D, Aubriot-Lorton MH, Villa C, Vasiljevic A, Lechapt-Zalcman E, Kalamarides M, Sharif A, Mokhtari K, Pagnotta SM, Iavarone A, Lasorella A, Huillard E, Sanson M, 2018. A recurrent point mutation in PRKCA is a hallmark of chordoid gliomas. Nat Commun 9(1), 2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychowdhury D, Lahn M, 2003. Antisense therapy directed to protein kinase C-alpha (Affinitak, LY900003/ISIS 3521): Potential role in breast cancer. Seminars in Oncology 30(2), 30–33. [DOI] [PubMed] [Google Scholar]
- Ruvolo PP, Deng X, Carr BK, May WS, 1998. A functional role for mitochondrial protein kinase Cα in Bcl2 phosphorylation and suppression of apoptosis. The Journal of biological chemistry 273(39), 25436–25442. [DOI] [PubMed] [Google Scholar]
- Ruvolo PP, Deng X, May WS, 2001. Phosphorylation of Bcl2 and regulation of apoptosis. Leukemia 15(4), 515–522. [DOI] [PubMed] [Google Scholar]
- Saito Y, Desai RR, Muthuswamy SK, 2018. Reinterpreting polarity and cancer: The changing landscape from tumor suppression to tumor promotion. Biochim Biophys Acta Rev Cancer 1869(2), 103–116. [DOI] [PubMed] [Google Scholar]
- Sakaguchi M, Miyazaki M, Sonegawa H, Kashiwagi M, Ohba M, Kuroki T, Namba M, Huh NH, 2004. PKCα mediates TGFβ-induced growth inhibition of human keratinocytes via phosphorylation of S100C/A11. J Cell Biol 164(7), 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama MF, Liu M, Clarke CJ, Espaillat MP, Haley JD, Jin T, Wang D, Obeid LM, Hannun YA, 2019. PKCα is required for Akt-mTORC1 activation in non-small cell lung carcinoma (NSCLC) with EGFR mutation. Oncogene 38(48), 7311–7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago MF, Perez-Reyes PL, Lopez-Aparicio P, Recio MN, Perez-Albarsanz MA, 2006. Differential effects of PCBs on the induction of apoptosis machinery and PKCα translocation in rat renal tubular cell cultures. Toxicol Lett 163(2), 91–100. [DOI] [PubMed] [Google Scholar]
- Santiskulvong C, Rozengurt E, 2007. Protein kinase Cα mediates feedback inhibition of EGF receptor transactivation induced by Gq-coupled receptor agonists. Cell Signal 19(6), 1348–1357. [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T, 2002. Interference with PDK1-Akt survival signaling pathway by UCN-01 (7-hydroxystaurosporine). Oncogene 21(11), 1727–1738. [DOI] [PubMed] [Google Scholar]
- Saxon ML, Zhao X, Black JD, 1994. Activation of protein kinase C isozymes is associated with post-mitotic events in intestinal epithelial cells in situ. J Cell Biol 126(3), 747–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglione-Sewell B, Abraham C, Bissonnette M, Skarosi SF, Hart J, Davidson NO, Wali RK, Davis BH, Sitrin M, Brasitus TA, 1998. Decreased PKC-α expression increases cellular proliferation, decreases differentiation, and enhances the transformed phenotype of CaCo-2 cells. Cancer Res 58(5), 1074–1081. [PubMed] [Google Scholar]
- Shaikh S, Sarkar J, Pramanik A, Karmakar K, Chakraborti S, 2013. Effect of m-calpain in PKCα-mediated proliferation of pulmonary artery smooth muscle cells by low dose of ouabain. Indian J Biochem Biophys 50(5), 419–427. [PubMed] [Google Scholar]
- Shen L, Dean NM, Glazer RI, 1999. Induction of p53-dependent, insulin-like growth factor-binding protein-3-mediated apoptosis in glioblastoma multiforme cells by a protein kinase Cα antisense oligonucleotide. Mol Pharmacol 55(2), 396–402. [DOI] [PubMed] [Google Scholar]
- Signore M, Pelacchi F, di Martino S, Runci D, Biffoni M, Giannetti S, Morgante L, De Majo M, Petricoin EF, Stancato L, Larocca LM, De Maria R, Pallini R, Ricci-Vitiani L, 2014. Combined PDK1 and CHK1 inhibition is required to kill glioblastoma stem-like cells in vitro and in vivo. Cell Death Dis 5(5), e1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal SS, Yadav S, Singhal J, Drake K, Awasthi YC, Awasthi S, 2005. The role of PKCα and RLIP76 in transport-mediated doxorubicin-resistance in lung cancer. FEBS Lett 579(21), 4635–4641. [DOI] [PubMed] [Google Scholar]
- Sipeki S, Bander E, Parker PJ, Farago A, 2006. PKCα reduces the lipid kinase activity of the p110α/p85α PI3K through the phosphorylation of the catalytic subunit. Biochem Biophys Res Commun 339(1), 122–125. [DOI] [PubMed] [Google Scholar]
- Smith SD, Enge M, Bao W, Thullberg M, Costa TD, Olofsson H, Gashi B, Selivanova G, Stromblad S, 2012. Protein kinase Cα (PKCα) regulates p53 localization and melanoma cell survival downstream of integrin alphav in three-dimensional collagen and in vivo. The Journal of biological chemistry 287(35), 29336–29347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C, 2014. The landscape of kinase fusions in cancer. Nat Commun 5(1), 4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga K, Sugimoto I, Ito H, Hashimoto E, 1998. Down-regulation of protein kinase C-α detected in human colorectal cancer. Biochem Mol Biol Int 44(3), 523–528. [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Stone M, Marshall JF, Uberall F, Rotenberg SA, 2000. Photo-induced inactivation of protein kinase Cα by dequalinium inhibits motility of murine melanoma cells. Mol Pharmacol 58(4), 729–737. [DOI] [PubMed] [Google Scholar]
- Sun XG, Rotenberg SA, 1999. Overexpression of protein kinase Cα in MCF-10A human breast cells engenders dramatic alterations in morphology, proliferation, and motility. Cell Growth Differ 10(5), 343–352. [PubMed] [Google Scholar]
- Szabo K, Bakos E, Welker E, Muller M, Goodfellow HR, Higgins CF, Varadi A, Sarkadi B, 1997. Phosphorylation site mutations in the human multidrug transporter modulate its drug-stimulated ATPase activity. The Journal of biological chemistry 272(37), 23165–23171. [DOI] [PubMed] [Google Scholar]
- Takai Y, Kishimoto A, Inoue M, Nishizuka Y, 1977. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. I. Purification and characterization of an active enzyme from bovine cerebellum. The Journal of biological chemistry 252(21), 7603–7609. [PubMed] [Google Scholar]
- Tam WL, Lu H, Buikhuisen J, Soh BS, Lim E, Reinhardt F, Wu ZJ, Krall JA, Bierie B, Guo W, Chen X, Liu XS, Brown M, Lim B, Weinberg RA, 2013. Protein kinase C α is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 24(3), 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Li P, Sun M, Yin G, Yu D, 2006. Upregulation and activation of PKCα by ErbB2 through Src promotes breast cancer cell invasion that can be blocked by combined treatment with PKC alpha and Src inhibitors. Oncogene 25(23), 3286–3295. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Gavrielides MV, Mitsuuchi Y, Fujii T, Kazanietz MG, 2003. Protein kinase C promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK and inhibition of the Akt survival pathway. The Journal of biological chemistry 278(36), 33753–33762. [DOI] [PubMed] [Google Scholar]
- Tang C, Wang Y, Zhang L, Wang J, Wang W, Han X, Mu C, Gao D, 2020. Identification of novel LncRNA targeting Smad2/PKCα signal pathway to negatively regulate malignant progression of glioblastoma. J Cell Physiol 235(4), 3835–3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebar F, Villalonga P, Sorkina T, Agell N, Sorkin A, Enrich C, 2002. Calmodulin regulates intracellular trafficking of epidermal growth factor receptor and the MAPK signaling pathway. Mol Biol Cell 13(6), 2057–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedja R, Roberts CM, Alvero AB, Cardenas C, Yang-Hartwich Y, Spadinger S, Pitruzzello M, Yin G, Glackin CA, Mor G, 2019. Protein kinase Cα-mediated phosphorylation of Twist1 at Ser-144 prevents Twist1 ubiquitination and stabilizes it. The Journal of biological chemistry 294(13), 5082–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tertrin-Clary C, Eude I, Fournier T, Paris B, Breuiller-Fouche M, Ferre F, 1999. Contribution of protein kinase C to ET-1-induced proliferation in human myometrial cells. Am J Physiol 276(3), E503–511. [DOI] [PubMed] [Google Scholar]
- Thorne AM, Jackson TA, Willis VC, Bradford AP, 2013. Protein Kinase C α Modulates Estrogen-Receptor-Dependent Transcription and Proliferation in Endometrial Cancer Cells. Obstet Gynecol Int 2013, 537479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F, Wu H, Li Z, Wang N, Huang J, Li C, Xie F, 2009. Activated PKCα/ERK1/2 signaling inhibits tamoxifen-induced apoptosis in C6 cells. Cancer Invest 27(7), 802–808. [DOI] [PubMed] [Google Scholar]
- Tibudan SS, Wang Y, Denning MF, 2002. Activation of protein kinase C triggers irreversible cell cycle withdrawal in human keratinocytes. J Invest Dermatol 119(6), 1282–1289. [DOI] [PubMed] [Google Scholar]
- Tigges U, Koch B, Wissing J, Jockusch BM, Ziegler WH, 2003. The F-actin cross-linking and focal adhesion protein filamin A is a ligand and in vivo substrate for protein kinase Cα. The Journal of biological chemistry 278(26), 23561–23569. [DOI] [PubMed] [Google Scholar]
- Tolcher AW, Reyno L, Venner PM, Ernst SD, Moore M, Geary RS, Chi K, Hall S, Walsh W, Dorr A, Eisenhauer E, 2002. A randomized phase II and pharmacokinetic study of the antisense oligonucleotides ISIS 3521 and ISIS 5132 in patients with hormone-refractory prostate cancer. Clin Cancer Res 8(8), 2530–2535. [PubMed] [Google Scholar]
- Tonetti DA, Chisamore MJ, Grdina W, Schurz H, Jordan VC, 2000. Stable transfection of protein kinase C alpha cDNA in hormone-dependent breast cancer cell lines. Br J Cancer 83(6), 782–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonetti DA, Gao W, Escarzaga D, Walters K, Szafran A, Coon JS, 2012. PKCα and ERβ Are Associated with Triple-Negative Breast Cancers in African American and Caucasian Patients. Int J Breast Cancer 2012, 740353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonetti DA, Morrow M, Kidwai N, Gupta A, Badve S, 2003. Elevated protein kinase C alpha expression may be predictive of tamoxifen treatment failure. Br J Cancer 88(9), 1400–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai JH, Tsai MT, Su WW, Chen YL, Wu TT, Hsieh YS, Huang CY, Yeh KT, Liu JY, 2005. Expression of protein kinase C alpha in biopsies and surgical specimens of human hepatocellular carcinoma. Chin J Physiol 48(3), 139–143. [PubMed] [Google Scholar]
- Tse AN, Carvajal R, Schwartz GK, 2007. Targeting checkpoint kinase 1 in cancer therapeutics. Clin Cancer Res 13(7), 1955–1960. [DOI] [PubMed] [Google Scholar]
- Tzeng HT, Li TH, Tang YA, Tsai CH, Frank Lu PJ, Lai WW, Chiang CW, Wang YC, 2017. Phosphorylation of Rab37 by protein kinase C alpha inhibits the exocytosis function and metastasis suppression activity of Rab37. Oncotarget 8(65), 108556–108570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urra H, Henriquez DR, Canovas J, Villarroel-Campos D, Carreras-Sureda A, Pulgar E, Molina E, Hazari YM, Limia CM, Alvarez-Rojas S, Figueroa R, Vidal RL, Rodriguez DA, Rivera CA, Court FA, Couve A, Qi L, Chevet E, Akai R, Iwawaki T, Concha ML, Glavic A, Gonzalez-Billault C, Hetz C, 2018. IRE1α governs cytoskeleton remodelling and cell migration through a direct interaction with filamin A. Nat Cell Biol 20(8), 942–953. [DOI] [PubMed] [Google Scholar]
- Vallentin A, Lo TC, Joubert D, 2001. A single point mutation in the V3 region affects protein kinase Cα targeting and accumulation at cell-cell contacts. Mol Cell Biol 21(10), 3351–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatsyayan R, Lelsani PC, Awasthi S, Singhal SS, 2010. RLIP76: a versatile transporter and an emerging target for cancer therapy. Biochem Pharmacol 79(12), 1699–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstovsek G, Byrd A, Frey MR, Petrelli NJ, Black JD, 1998. Colonocyte differentiation is associated with increased expression and altered distribution of protein kinase C isozymes. Gastroenterology 115(1), 75–85. [DOI] [PubMed] [Google Scholar]
- Villar J, Quadri HS, Song I, Tomita Y, Tirado OM, Notario V, 2009. PCPH/ENTPD5 expression confers to prostate cancer cells resistance against cisplatin-induced apoptosis through protein kinase Cα-mediated Bcl-2 stabilization. Cancer Res 69(1), 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, Zhang J, Tsien RY, Newton AC, 2003. A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase C. J Cell Biol 161(5), 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SD, Murray NR, Burns DJ, Fields AP, 1995. Protein kinase C chimeras: catalytic domains of α and βII protein kinase C contain determinants for isotype-specific function. Proc Natl Acad Sci U S A 92(20), 9156–9160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther C, Hofvander J, Nilsson J, Magnusson L, Domanski HA, Gisselsson D, Tayebwa J, Doyle LA, Fletcher CD, Mertens F, 2015. Gene fusion detection in formalin-fixed paraffin-embedded benign fibrous histiocytomas using fluorescence in situ hybridization and RNA sequencing. Lab Invest 95(9), 1071–1076. [DOI] [PubMed] [Google Scholar]
- Wang C, Wang X, Liang H, Wang T, Yan X, Cao M, Wang N, Zhang S, Zen K, Zhang C, Chen X, 2013. miR-203 inhibits cell proliferation and migration of lung cancer cells by targeting PKCα. PLoS One 8(9), e73985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HQ, Smart RC, 1999. Overexpression of protein kinase C-α in the epidermis of transgenic mice results in striking alterations in phorbol ester-induced inflammation and COX-2, MIP-2 and TNF-α expression but not tumor promotion. Journal of Cell Science 112(20), 3497–3506. [DOI] [PubMed] [Google Scholar]
- Wang Q, Worland P, Clark J, Carlson B, Sausville E, 1995. Apoptosis in 7-hydroxystaurosporine-treated T lymphoblasts correlates with activation of cyclin-dependent kinases 1 and 2 [published erratum appears in Cell Growth Differ 1995 Oct;6(10):1339]. Cell Growth Differ 6(8), 927–936. [PubMed] [Google Scholar]
- Wang WL, Yeh SF, Huang EY, Lu YL, Wang CF, Huang CY, Lin WJ, 2007. Mitochondrial anchoring of PKCα by PICK1 confers resistance to etoposide-induced apoptosis. Apoptosis 12(10), 1857–1871. [DOI] [PubMed] [Google Scholar]
- Wang XQ, Yan Q, Sun P, Liu JW, Go L, McDaniel SM, Paller AS, 2007. Suppression of epidermal growth factor receptor signaling by protein kinase C-α activation requires CD82, caveolin-1, and ganglioside. Cancer Res 67(20), 9986–9995. [DOI] [PubMed] [Google Scholar]
- Wang Y, Arjonen A, Pouwels J, Ta H, Pausch P, Bange G, Engel U, Pan X, Fackler OT, Ivaska J, Grosse R, 2015. Formin-like 2 Promotes β1-Integrin Trafficking and Invasive Motility Downstream of PKCα. Dev Cell 34(4), 475–483. [DOI] [PubMed] [Google Scholar]
- Ways DK, Kukoly CA, deVente J, Hooker JL, Bryant WO, Posekany KJ, Fletcher DJ, Cook PP, Parker PJ, 1995. MCF-7 breast cancer cells transfected with protein kinase C-alpha exhibit altered expression of other protein kinase C isoforms and display a more aggressive neoplastic phenotype. J Clin Invest 95(4), 1906–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JB, Gill GN, Rosenfeld MG, Wells A, 1991. A negative feedback loop attenuates EGF-induced morphological changes. J Cell Biol 114(3), 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen-Sheng W, 2006. Protein kinase C α trigger Ras and Raf-independent MEK/ERK activation for TPA-induced growth inhibition of human hepatoma cell HepG2. Cancer Lett 239(1), 27–35. [DOI] [PubMed] [Google Scholar]
- Wen-Sheng W, Jun-Ming H, 2005. Activation of protein kinase C alpha is required for TPA-triggered ERK (MAPK) signaling and growth inhibition of human hepatoma cell HepG2. J Biomed Sci 12(2), 289–296. [DOI] [PubMed] [Google Scholar]
- Whelan RD, Parker PJ, 1998. Loss of protein kinase C function induces an apoptotic response. Oncogene 16(15), 1939–1944. [DOI] [PubMed] [Google Scholar]
- Wimmer R, Hohenester S, Pusl T, Denk GU, Rust C, Beuers U, 2008. Tauroursodeoxycholic acid exerts anticholestatic effects by a cooperative cPKCα-/PKA-dependent mechanism in rat liver. Gut 57(10), 1448–1454. [DOI] [PubMed] [Google Scholar]
- Wu H, Chen Y, Liang J, Shi B, Wu G, Zhang Y, Wang D, Li R, Yi X, Zhang H, Sun L, Shang Y, 2005. Hypomethylation-linked activation of PAX2 mediates tamoxifen-stimulated endometrial carcinogenesis. Nature 438(7070), 981–987. [DOI] [PubMed] [Google Scholar]
- Wu TT, Hsieh YH, Hsieh YS, Liu JY, 2008. Reduction of PKCα decreases cell proliferation, migration, and invasion of human malignant hepatocellular carcinoma. J Cell Biochem 103(1), 9–20. [DOI] [PubMed] [Google Scholar]
- Wu TT, Hsieh YH, Wu CC, Hsieh YS, Huang CY, Liu JY, 2007. Overexpression of protein kinase Cα mRNA in human hepatocellular carcinoma: a potential marker of disease prognosis. Clin Chim Acta 382(1–2), 54–58. [DOI] [PubMed] [Google Scholar]
- Xie Q, Wang S, Zhao Y, Zhang Z, Qin C, Yang X, 2019. MicroRNA-216a suppresses the proliferation and migration of human breast cancer cells via the Wnt/β-catenin signaling pathway. Oncol Rep 41(5), 2647–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LC, Ng DC, Bikle DD, 2003. Role of protein kinase C α in calcium induced keratinocyte differentiation: defective regulation in squamous cell carcinoma. J Cell Physiol 195(2), 249–259. [DOI] [PubMed] [Google Scholar]
- Yates C, Wells A, Turner T, 2005. Luteinising hormone-releasing hormone analogue reverses the cell adhesion profile of EGFR overexpressing DU-145 human prostate carcinoma subline. Br J Cancer 92(2), 366–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazaki T, Ahmad S, Chahlavi A, Zylber-Katz E, Dean NM, Rabkin SD, Martuza RL, Glazer RI, 1996. Treatment of glioblastoma U-87 by systemic administration of an antisense protein kinase C-α phosphorothioate oligodeoxynucleotide. Mol Pharmacol 50(2), 236–242. [PubMed] [Google Scholar]
- Ye JC, Hsu LS, Tsai JH, Yang HL, Hsiao MW, Hwang JM, Lee CJ, Liu JY, 2017. MZF-1/Elk-1/PKCα is Associated with Poor Prognosis in Patients with Hepatocellular Carcinoma. J Cancer 8(15), 3028–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen AR, Halsey J, Fisher GA, Holmlund JT, Geary RS, Kwoh TJ, Dorr A, Sikic BI, 1999. Phase I study of an antisense oligonucleotide to protein kinase C-α (ISIS 3521/CGP 64128A) in patients with cancer. Clin Cancer Res 5(11), 3357–3363. [PubMed] [Google Scholar]
- Yun J, Pannuti A, Espinoza I, Zhu H, Hicks C, Zhu X, Caskey M, Rizzo P, D’Souza G, Backus K, Denning MF, Coon J, Sun M, Bresnick EH, Osipo C, Wu J, Strack PR, Tonetti DA, Miele L, 2013. Crosstalk between PKCα and Notch-4 in endocrine-resistant breast cancer cells. Oncogenesis 2, e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Xu H, Glazer RI, 2002. Transformation of mammary epithelial cells by 3-phosphoinositide-dependent protein kinase-1 (PDK1) is associated with the induction of protein kinase Cα. Cancer Res 62(12), 3538–3543. [PubMed] [Google Scholar]
- Zhan B, Kong C, Zhang Z, Dong X, Zhang N, 2017. Inhibition of PKCα reduces the ability of migration of kidney cancer cells but has no impact on cell apoptosis. Exp Ther Med 13(5), 2473–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Wen J, Aletta JM, Rubin RP, 1997. Inhibition of expression of PKC-α by antisense mRNA is associated with diminished cell growth and inhibition of amylase secretion by AR4–2J cells. Exp Cell Res 233(1), 225–231. [DOI] [PubMed] [Google Scholar]
- Zhao LJ, Xu H, Qu JW, Zhao WZ, Zhao YB, Wang JH, 2012. Modulation of drug resistance in ovarian cancer cells by inhibition of protein kinase C-alpha (PKC-α) with small interference RNA (siRNA) agents. Asian Pac J Cancer Prev 13(8), 3631–3636. [DOI] [PubMed] [Google Scholar]
- Zhao X, Murata T, Ohno S, Day N, Song J, Nomura N, Nakahara T, Yokoyama KK, 2001. Protein kinase Cα plays a critical role in mannosylerythritol lipid-induced differentiation of melanoma B16 cells. The Journal of biological chemistry 276(43), 39903–39910. [DOI] [PubMed] [Google Scholar]
- Zhen-jin Z, Peng L, Fa-yu L, Liyan S, Chang-fu S, 2011. PKCα take part in CCR7/NF-κB autocrine signaling loop in CCR7-positive squamous cell carcinoma of head and neck. Mol Cell Biochem 357(1–2), 181–187. [DOI] [PubMed] [Google Scholar]
- Zheng J, Kong C, Yang X, Cui X, Lin X, Zhang Z, 2017. Protein kinase C-α(PKCα) modulates cell apoptosis by stimulating nuclear translocation of NF-kappa-B p65 in urothelial cell carcinoma of the bladder. BMC Cancer 17(1), 432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Wang K, Xu H, Kong F, 2018. Silencing Formin-like 2 inhibits growth and metastasis of gastric cancer cells through suppressing internalization of integrins. Cancer Cell Int 18(1), 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou AX, Hartwig JH, Akyurek LM, 2010. Filamins in cell signaling, transcription and organ development. Trends Cell Biol 20(2), 113–123. [DOI] [PubMed] [Google Scholar]
- Zhu BH, Yao ZX, Luo SJ, Jiang LM, Xiao JW, Liu SC, Liu JB, Sun JM, Pei ZY, 2005. Effects of antisense oligonucleotides of PKC-α on proliferation and apoptosis of HepG2 in vitro. Hepatobiliary Pancreat Dis Int 4(1), 75–79. [PubMed] [Google Scholar]
- Zhu Y, Dong Q, Tan BJ, Lim WG, Zhou S, Duan W, 2005. The PKCα-D294G mutant found in pituitary and thyroid tumors fails to transduce extracellular signals. Cancer Res 65(11), 4520–4524. [DOI] [PubMed] [Google Scholar]
- Ziegler WH, Tigges U, Zieseniss A, Jockusch BM, 2002. A lipid-regulated docking site on vinculin for protein kinase C. The Journal of biological chemistry 277(9), 7396–7404. [DOI] [PubMed] [Google Scholar]