

Abstract
Purpose
This study investigated the neuroprotective effects of administration of ROCK inhibitor E212 on ischemic optic neuropathy.
Methods
Rats received an intravitreal injection of either E212 or PBS immediately after optic nerve infarct. The oxidative stress in the retina was detected by performing superoxide dismutase activity and CellROX assays. The integrity of retinal pigment epithelium was determined by staining of zona occludens 1. The visual function, retinal ganglion cell (RGC) density, and RGC apoptosis were determined by using flash visual-evoked potential analysis, retrograde FluoroGold labeling, and TdT-dUTP nick end-labeling assay. Macrophage infiltration was detected by staining for ED1. The protein levels of TNF-α, p-CRMP, p-AKT1, p-STAT3, and CD206 were evaluated using Western blotting.
Results
Administration of E212 resulted in a 1.23-fold increase in the superoxide dismutase activity of the retina and 2.28-fold decrease in RGC-produced reactive oxygen species as compared to the levels observed upon treatment with PBS (P < 0.05). Moreover, E212 prevented the disruption of the blood-retinal barrier (BRB) in contrast to PBS. The P1-N2 amplitude and RGC density in the E212-treated group were 1.75- and 2.05-fold higher, respectively, than those in the PBS-treated group (P < 0.05). The numbers of apoptotic RGCs and macrophages were reduced by 2.93- and 2.54-fold, respectively, in the E212-treated group compared with those in the PBS-treated group (P < 0.05). The levels of p-AKT1, p-STAT3, and CD206 were increased, whereas those of p-PTEN, p-CRMP2, and TNF-α were decreased after treatment with E212 (P < 0.05).
Conclusions
Treatment with E212 suppresses oxidative stress, BRB disruption, and neuroinflammation to protect the visual function in ischemic optic neuropathy.
Keywords: Rho kinase inhibitor, ischemic optic neuropathy, reactive oxygen species, retinal ganglion cell, blood-retinal barrier
Nonarteritic anterior ischemic optic neuropathy (NAION) is a type of acute optic neuropathy that commonly affects individuals aged ≥55 years, with an estimated annual incidence of 2.3 to 10.2 cases per 100,000 population.1–4 The clinical presentation of NAION is a sudden, isolated, and usually painless visual impairment accompanied by optic disc swelling.5 However, no effective treatment has been established thus far.
It is thought that NAION is caused by small vessel circulatory insufficiency within the optic nerve head (ONH).6 Vascular and optic disc risk factors form the two main types of risk factors for NAION. The former group of risk factors includes nocturnal systemic hypotension, impaired autoregulation of the microvascular supply, vascular occlusion, and venous insufficiency.6–10 Patients with small and crowded optic discs are at risk of developing NAION, leading to compartment compression of the ON fibers and contributing to the worsening of ON ischemia.5,11 The occurrence of ONH ischemia can induce an inflammatory reaction and optic edema, and eventually result in retinal ganglion cell (RGC) death.12–15 During ON ischemia, harmful processes (e.g., over-induction of oxidants, suppression of detoxification systems and consumption of antioxidants, and interruption the normal antioxidative process) occur in the ON and retina.16 These processes eventually lead to elevated levels of oxidative stress, causing acute ischemic optic neuropathy. Although previous studies have provided evidence that oxidative stress contributes to the degeneration of RGCs,17–19 the exact mechanism involved in this process remains unclear. Moreover, treatments for oxidative stress in patients with NAION have not yet been established.
Rho kinase (ROCK) controls several signaling pathways and biological processes.20 Abnormal activation of ROCK has been detected in diabetic nephropathy,21 cardiovascular disease,22 and central nervous system diseases, including Alzheimer's disease,23 spinal cord injury,24 stroke,25 and multiple sclerosis.26 Recently, ROCK has gained increasing importance in ocular diseases, including corneal disease, glaucoma, and ischemic retina.27 Fard et al.28 also found that activation of RhoA and microglia occurs at the site of infarction in a rodent model of AION (rAION).
ROCK inhibitors are a series of compounds that inhibit the activity of ROCK. They have been clinically approved in Japan, China, and the United States. Fasudil was clinically approved in China and Japan for the treatment of cerebral vasospasm. In addition, ripasudil and netarsudil were approved for the treatment of glaucoma in Japan and the United States, respectively.29 ROCK inhibitors are potential treatments for asthma,30 cancer,31 erectile dysfunction,32 insulin resistance,33 kidney failure,34 neuronal degeneration,35 and osteoporosis.22 Several studies have investigated its neuroprotective effects on glaucoma,36,37 Parkinson's disease,38 spinal cord injury, stroke, Alzheimer's disease, and demyelinating central nervous system diseases,35 and axonal degeneration in an ON crush model.39 Of note, a recent report suggested that K-115 (a novel ROCK inhibitor) reduces reactive oxygen species (ROS) production by attenuating the expression of NADPH oxidase 1, and eventually preventing RGC death in an ON crush model.36 Koch et al.39 demonstrated that knockdown of ROCK2 promotes RGC survival and axon outgrowth in the ON crush model. A clinical case series (n = 13) showed that an intravitreal injection of fasudil can improve visual acuity in patients with recent-onset NAION.40 However, this study was limited by the lack of a control group and the small sample size.
The novel amino-isoquinoline derivative ROCK inhibitor E212 (United States Patent Number US 10,696,638 B2; Supplementary Data S1), developed by the Industrial Technology Research Institute in Taiwan, has shown high specificity for ROCK inhibition and high selectivity toward ROCK1. The 50% inhibitory concentration (IC50) of E212 for ROCK1, ROCK2, PRKX, PRKG2, PKAC-alpha, PRKG1, PKN1, PKAC-beta, and YSK4 was 0.05, 0.35, 2.3, 2.9, 4.9, 5.5, 7.8, 8.2, and 12 µM, respectively (Supplementary Data S2). Compared with other common ROCK inhibitors, such as Y-27632 and fasudil, the IC50 of E212 was 10-fold lower. Interaction of E212 with non-kinases such as receptors, channels, and transporters was not evaluated. Therefore its role in other partial mediated activities cannot be denied. Moreover, E212 demonstrated the highest water solubility (7 mg/mL) with a good drug loading yield (2.1%) in the ophthalmic formulation. The LD50 and no-observed-effect level of E211 in the human retinal pigment epithelium (RPE) cell culture were 219 µM and 108 µM, respectively (Supplementary Data S3).
Taken together, we hypothesized that the ROCK inhibitor may have neuroprotective effects against NAION. The aim of this study was to investigate the degree of oxidative stress, inflammatory response, and RGC apoptosis in the rAION to reveal the neuroprotective effects of the novel ROCK inhibitor E212.
Methods
Animals
Adult male Wistar rats (age 7–8 weeks; weight 150–180 g) were used in this study. The rats were obtained from the breeding colony of BioLASCO Taiwan Co., Taipei, Taiwan. The animal care and experimental procedures were performed in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. All animal experiments were approved by the Institutional Animal Care and Use Committee of Tzu Chi Medical Center. All manipulation procedures were performed following the protocols used in our previous studies.41
Study Design
A total of 108 animals were used for this study. After induction of rAION in the right eye, 72 rats were randomized into two equal groups: the first group received intravitreal injection of 5 µL E212, and the second group received intravitreal injection of PBS alone in the rAION-induced right eye one hour after induction. The remaining 36 rats received sham laser treatment without the photosensitizing agent (Rose Bengal [RB]). The 33-G needles (Hamilton 7747-01 with a Gaslight syringe, IA2-1701RN 10 µL SYR; Hamilton Co., Hamilton, KS, USA) were used to perform the intravitreal injections. The remaining 36 rats received sham laser treatment without photosensitizing agents (RB) to form the sham-operated group. All animals survived until the end of the experiment without developing any complications. The rats were euthanized through insufflation of carbon dioxide. On day 1 after rAION, the level of oxidative stress in the retina was detected by performing a superoxide dismutase (SOD) activity assay, and the percentage of RGCs that produced ROS was determined by performing a CellROX assay. On day 3 after rAION, the retina and optic nerve samples were collected for immunofluorescence staining of zona occludens 1 (ZO-1) and Western blotting analysis. At week 4 after infarct, visual function was assessed by performing a flash visual-evoked potential (FVEP) analysis; the RGC density was measured by conducting retrograde FluoroGold labeling. In situ TdT-dUTP nick end-labeling (TUNEL) assays in the RGC layer and immunohistochemistry of ectodermal dysplasia 1 (ED1; biomarker of macrophage) expression in the ON were also conducted. A summary of the study is illustrated in Figure 1.
Figure 1.

Study design to investigate the role of E212 in the rAION model. The level of oxidative stress in the retina was determined at day 1 post rAION. The effect of E212 on BRB integrity was analyzed at day 3 after rAION. The neuroprotective effects of E212 were evaluated at day 28 after rAION. WB, Western blotting.
rAION Induction
The detailed induction protocol was described in our previous studies.41–43 Following the induction of general anesthesia, RB (Sigma–Aldrich Corp., St. Louis, MO, USA) was intravenously injected using a 28-gauge needle (2.5 mM RB in PBS/1 mL/kg animal weight). A laser fundus lens (Ocular Instruments, Inc., Bellevue, WA, USA) was used to focus the laser on the optic disc. Subsequently, the ONH of rats was treated with an argon laser using a 532 nm wavelength, a 500 µm spot size, and an 80 mW power (MC-500 multi-color laser; Nidek Co., Ltd., Tokyo, Japan). For each laser procedure, 12 pulses with one second duration were performed. When RB is exposed to a green laser light, it glows a bright golden color, indicating successful photodynamic therapy.44 After completing the procedure, TobraDex eye ointment (Alcon, Geneva, Switzerland) was topically administered. The rats were placed on electric heating pads (37°C) to recover. The induction protocol of sham laser treatment was the same as that of the procedure mentioned above, except for the administration of RB.
Retrograde Labeling of RGCs With FluoroGold
The FluoroGold labeling procedure was described in detail in our previous studies.45–48 To avoid overcounting the RGCs by mixing labeled RGCs with dye-engulfed macrophages and microglia, retrograde labeling of RGCs was performed one week before the rats were euthanized. The rats were anesthetized with a mixture of ketamine (40 mg/kg body weight) and xylazine (4 mg/kg body weight; Sigma-Aldrich), and placed in a stereotactic apparatus (Stoelting, Wood Dale, IL, USA). Subsequently, 2 µL of 5% FluoroGold was injected into the superior colliculus on each side using a Hamilton syringe. One week after the FluoroGold labeling was performed, the rats were euthanized, and the eyeballs were harvested. The eyeballs were fixed with paraformaldehyde for one hour. Subsequently, the retina was carefully dissected and flat mounted on a slide. The retinas were examined using a 400 epi-fluorescence microscope (Axioskop; Carl Zeiss Meditec Inc., White Plains, NY, USA) equipped with a filter set (excitation filter 350–400 nm; barrier filter 515 nm), as well as a digital camera (AxioCam MRm; Carl Zeiss Meditec Inc.,) and the AxioVision 4.0 software. We examined the retinas for RGCs at a distance between 1 mm and 3 mm from the ONH to determine the central RGC densities. At least four randomly selected areas of 62,500 µm2 each in the central (40% of the central area) regions of each retina were counted, and their corresponding averages were obtained as the mean density of RGCs per retina (n = 6 rats per group). The percentage of RGC survival was defined as the number of RGCs in each treatment group divided by the number of RGCs in the sham-operated retinas, multiplied by 100.
FVEPs
FVEPs were recorded at week 4 after infarct. The same surgery was performed in both eyes to examine the pattern of FVEPs. This procedure was approved by the Institutional Animal Care and Use Committee and described in detail in our previous studies.45–48 In brief, after induction of general anesthesia, the skin covering the skull was incised, and the brain surface was exposed using a dental drill. Two screw implants, connected to active (positive) electrodes, were fixed at the primary visual cortex region of both hemispheres. One screw implant, connected to a reference (negative) electrode, was fixed at the frontal cortex region. The ground electrode was placed on the rat's tail. A visual electrodiagnostic system (UTAS-E3000; LKC Technologies, Gaithersburg, MD, USA) was used to measure the photopic FVEPs. The settings used were as follows: background illumination off, a flash intensity of 30 cd·s/m2, single flash with a flash rate of 1.02 Hz, and a test average of 64 sweeps. To evaluate the visual function, the latency of the P1 wave, referred to as the first positive-going wavelet, and the amplitude of the P1-N2 wave in each group were compared (n = 6 rats per group).
ON and Retinal Sample Preparation
ON Preparation
After euthanasia of rats at week 4 after infarct, a segment of the ON (length 5–7 mm) was obtained and frozen at −70°C for immunohistochemistry.
Retinal Section Preparation
At week 4 after infarct, the animals were sacrificed, and eyeballs were harvested. To prepare the eyecups, the lens and vitreous body were removed from the eyeball. The remaining eyecups were fixed with 4% buffered paraformaldehyde for two hours at room temperature. The eyecups were dehydrated in 30% sucrose overnight and stored at −20°C.
In Situ TUNEL Assays
To ensure that equivalent fields were used for comparison, all frozen retinal sections were prepared using the samples cut at 1 to 2 mm distant from the ONH. TUNEL assays (DeadEnd Fluorometric TUNEL System; Promega Corporation, Madison, WI, USA) were performed to detect apoptotic cells. The TUNEL-positive cells in the RGC layer were counted in 10 high-power fields (HPF, magnification ×400). Each eye was divided into three sections, and an average measurement was obtained for comparison (n = 6 rats per group).
Immunohistochemistry
The frozen ON and retinal sections were fixed with acetone at −20°C for 30 minutes and blocked with 5% fetal bovine serum containing 1% bovine serum albumin for 15 minutes. The fixed ON section or retinal section was incubated with a primary antibody (anti-ED1 antibody: 1:50, ab31630; Abcam, Cambridge, MA, USA) or anti-ZO1 antibody (1:100, LS-B9774; Lifespan Biosciences, Seattle, WA, USA) overnight at 4°C. Subsequently, the section was incubated with the secondary antibody conjugated with fluorescein isothiocyanate (1:100 dilution; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for one hour at room temperature. For nuclear quantitation, 4,6-diamidino-2-phenylindole (DAPI, 1:1,000 dilution; Sigma-Aldrich) staining was performed. The ED1-positive cells at the ON were counted in 6 HPFs (magnification × 400), and the fluorescence intensity of ZO-1 in the RPE and ON vessel was measured using the ImageJ software (n = 6 rats per group).
In Situ Detection of ROS Production
FluoroGold labeling was performed one week before the rats were sacrificed. On day 1 after AION, the rats received an intravitreal injection of 1 µL of 50 µM CellROX Green Reagent (Life Technologies, Inc., Carlsbad, CA, USA). After two hours, the animals were sacrificed, and the eyeballs were harvested. To prepare the eyecups, the lens and vitreous body were removed. The remaining eyecups were fixed with paraformaldehyde for two hours at room temperature. The eyecups were dehydrated in increasing concentrations of sucrose. For nuclear quantitation, DAPI (1:1,000 dilution; Sigma-Aldrich) staining was performed. The CellROX-labeled and FluoroGold-positive cells in ganglion cell layer were counted in 6 HPFs (magnification × 400) at the retina (n = 6 rats per group).
SOD Activity Assay
The retinal samples were collected from each group at day 1 after rAION. These samples were homogenized in ice-cold radioimmunoprecipitation assay buffer and centrifuged at 14,000g for five minutes at 4°C. The supernatant was collected and transferred to a clean tube. The supernatant was incubated with a, SOD activity assay reaction mix (ab65354 Superoxide Dismutase Activity Assay kit [Colorimetric]; Abcam) at 37°C for 20 minutes. The optical density (OD) was measured at OD 450 nm (n = 6 rats per group).
Western Blotting
On day 3 after the occurrence of rAION, the retinal samples or ON samples from each group were collected. Western blotting was used to measure the levels of proteins in the retinas and ONs. The protein extracts were separated using a 4% to 12% NuPAGE Bis-Tris gel (Invitrogen, Carlsbad, CA, USA) and subsequently transferred to polyvinylidene difluoride membranes. After blocking with 5% nonfat milk in tris-buffered saline with Tween 20 solution (20 mM Tris-HCl [pH 7.5], 0.5 M NaCl, and 0.5% Tween-20), the membranes were reacted with either anti-p-AKT (1:1,000, antibody 8599S; Cell Signaling Technology Inc., Danvers, MA, USA), anti-phosphorylated-collapsin response mediator protein 2 (anti-p-CRMP2; 1:1,000, antibody no. 9397; Cell Signaling Technology Inc.), anti-p-signal transducer and activator of transcription 3 (anti-p-STAT3; 1:1,000, antibody no. 9145S; Cell Signaling Technology Inc.), anti-tumor necrosis factor-α (anti-TNF-α; 1:1,000, ab6671; Abcam), or anti-glyceraldehyde-3-phosphate dehydrogenase (1:1,000, antibody no. 2118S; Cell Signaling Technology Inc.) antibody overnight at 4°C. The membranes were incubated with a secondary antibody conjugated to horseradish peroxidase against the appropriate host species for one hour at room temperature. The blots were developed using an enhanced chemiluminescent substrate (Perkin-Elmer Life Science, Boston, MA, USA). An image analysis system (Amersham Biosciences, Uppsala, Sweden) was used to measure the relative intensities of the bands.
Statistical Analysis
All measurements in this study were performed in a masked fashion: the mean and standard deviations of six measurements were obtained. Statistical analysis was performed using the SPSS version 19.0 (IBM Corp., Armonk, NY, USA) software. The Kruskal-Wallis test was applied for comparison among groups. P < 0.05 denoted statistically significant difference.
Results
Treatment With E212 Induced SOD Activity and Reduced ROS Production
In Figure 2A, ROS produced in the ganglion cell layer were stained with CellROX in red, whereas the RGCs were labeled with FluoroGold in green. ROS were produced in the RGC layer after the occurrence of AION. The percentage of RGC-produced ROS (FluoroGold/CellROX double-positive cell) increased significantly in the PBS-treated group compared with the sham group (89% vs. 25%, respectively, P < 0.05). In the E212-treated group, the percentage of CellROX-positive RGCs significantly decreased to 39% compared with that recorded in the PBS-treated group (P < 0.05). As shown in Figure 2B, the relative SOD activity of the retina at day 1 after infarct was 481.5, 455.2, and 557.9 in the sham, PBS-treated, and E212-treated groups, respectively. The relative SOD activity in the E212-treated group was 1.23-fold higher than that noted in the PBS-treated group (P = 0.0219).
Figure 2.

Oxidative stress levels in retinal tissues at day 1 after rAION. (A) Representative fluorescence images of frozen sections showing ROS-producing cells (shown in red) and FG-positive cells (shown in green) in the GCL. Scale bar: 50 µm. (B) The relative SOD activity in the retinal tissues at day 1 after infarct. The relative SOD activity in the E212-treated group was 1.23-fold higher than that in the PBS-treated group (P < 0.01). Data are expressed as the mean ± SD. FG, FluoroGold; GCL, ganglion cell layer.
Treatment With E212 Improved Visual Function by the FVEP
At week 4 after AION, the amplitude of the P1-N2 wave of the FVEP was 54.3 ± 10.1 µV, 23.7 ± 8.4 µV, and 41.6 ± 9.4 µV in the sham, PBS-treated, and E212-treated groups, respectively (Fig. 3A). The amplitude of the P1-N2 wave in the E212-treated group was 1.75-fold higher than that observed in the PBS-treated group (P < 0.05) (Fig. 3B). These results indicated that treatment with E212 preserved the visual function after AION.
Figure 3.

Effect of E212 on FVEP recording at week 4 after infarct. (A) Representative FVEP wavelet in each group. (B) Quantitative data showed that the amplitude of the P1-N2 wave in the E212-treated group was 1.75-fold higher than that in the PBS-treated group (*P < 0.05, n = 6 per group). Data are expressed as the mean ± SD.
Treatment With E212 Preserved the RGC Density and Reduced the Apoptosis of RGCs
The density of RGCs in the central retina of the sham, PBS-treated, and E212-treated groups at week 4 after infarct was 1923.3 ± 143.3/mm2, 645.1 ± 220.0/mm2, and 1319.3 ± 160.7/mm2, respectively (Fig. 4A). The RGC density in the E212-treated group was significantly higher than that measured in the PBS-treated group after rAION (P < 0.05) (Fig. 4B).
Figure 4.

Effect of E212 on morphometry of RGCs at week 4 after infarct. (A) Representative RGC density of the central retinas in each group. (B) Bar chart showing that the RGC density in the E212-treated group was significantly higher than that in the PBS-treated group after rAION (*P < 0.05, n = 6 per group; scale bar: 200 µm).
The TUNEL assay detected 0.6 ± 0.4, 12.3 ± 4.5, and 4.2 ± 2.8 positive cells/HPF on the RGC layers in the sham-operated, PBS-treated, and E212-treated groups, respectively (Fig. 5A). The number of TUNEL-positive cells/HPF in the E212-treated group was 2.93-fold lower than that detected in the PBS-treated group (P < 0.05) (Fig. 5B).
Figure 5.
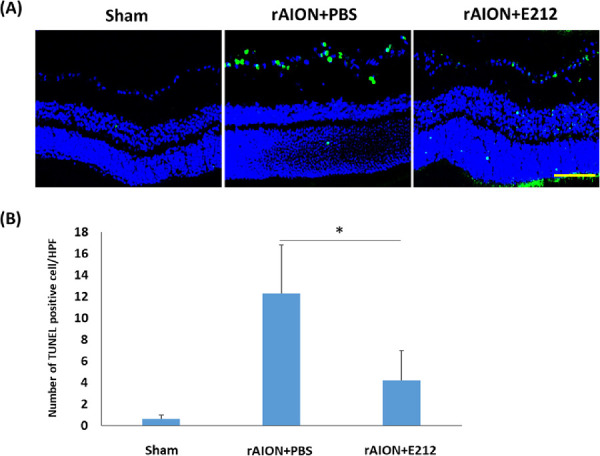
Analysis of RGC apoptosis in the RGC layer through the TUNEL assay at week 4 after rAION induction. (A) Representative images of apoptotic cells in the RGC layers in the sham, PBS-treated, and E-212-treated group at week 4 after AION induction. The TUNEL-positive cells (green) and the nuclei of RGCs (blue) were stained with DAPI staining. (B) Quantification of TUNEL-positive cells per HPF. The number of apoptotic cells in the E-212-treated group were significantly lower (2.93-fold, *P < 0.05, n = 6 per group, scale bar: 100 µm) than that in the PBS-treated group. Data are expressed as the mean ± SD. HPF, high-power field; rAION, rodent model of anterior ischemic optic neuropathy; RGC, retinal ganglion cell; TUNEL, TdT-dUTP nick end-labeling.
Treatment With E212 Maintained the Blood-Retinal Barrier (BRB) Integrity After ON Infarct
The signal of ZO-1 protein in the RPE layer was barely detectable in the PBS-treated group 3 days after rAION induction in contrast to that observed in the sham-operated group. Treatment with E212 maintained the ZO-1 expression and RPE polarity at three days after AION (Fig. 6). This result indicated that treatment with E212 may prevent the disruption of BRB after the occurrence of an ischemic insult on the ON.
Figure 6.
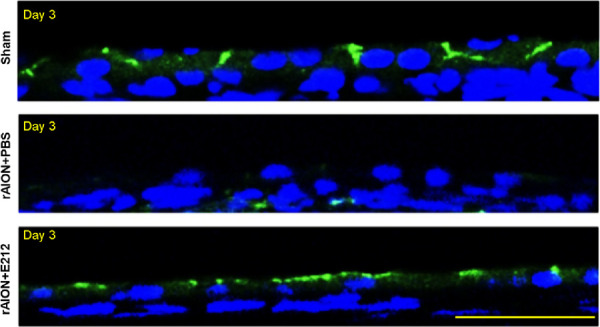
ZO-1 staining for BRB integrity. The representative images display ZO-1 staining in the RPE layer at day 3 after rAION induction. The ZO-1 was labeled with FITC (green), and the nuclei were stained with DAPI (blue). Scale bar: 50 µm. BRB, blood-retinal barrier; DAPI, 4, 6-diamidino-2-phenylindole; FITC, fluorescein isothiocyanate; rAION, rodent model of anterior ischemic optic neuropathy; RPE, retinal pigment epithelium; ZO-1, zona occludens 1.
Treatment With E212 Reduced Extrinsic Macrophage Infiltration and Induced Macrophage/Microglia Polarization
At week 4 after AION, the number of ED1-positive cells per HPF was 1.6 ± 0.5, 58.7 ± 12.4, and 23.1 ± 7.6 in the sham, PBS-treated, and E212-treated groups, respectively (Fig. 7A). A significant difference was observed between the PBS-treated group and E212-treated group (P < 0.05) (Fig. 7B). These results indicated that treatment with E212 inhibited macrophage infiltration into the ON after rAION.
Figure 7.

Anti-inflammatory effect of E212 on macrophage infiltration and polarization. (A) Representative images display ED1 staining in the longitudinal sections of the ON at week 4 after rAION induction. The ED1-positive cells were labeled with FITC (green), and the nuclei were stained with DAPI (blue). Scale bar: 200 µm. (B) Quantification of ED1-positive cells per HPF. The extrinsic macrophage recruitment was significantly lower in the E-212-treated group compared with the PBS-treated group (*P < 0.05, n = 6 per group). Data are expressed as the mean ± SD. DAPI, 4, 6-diamidino-2-phenylindole; ED1, ectodermal dysplasia 1; FITC, fluorescein isothiocyanate; ON, optic nerve.
The protein levels of p-STAT3, CD206, and TNF-α in the ON samples were analyzed by conducting Western blotting analysis (Fig. 7C). The protein levels of p-STAT3 and CD206 was increased by 2.13- and 4.08-fold, respectively, after treatment with E212 versus PBS (P < 0.05) (Fig. 7D). The protein level of TNF-α was significantly decreased by 2.13-fold in the E212-treated group versus the PBS-treated group (P < 0.05) (Fig. 7D). Therefore an intravitreal injection of E212 can activate STAT3 and induce M2 macrophage/microglia polarization in the rAION model.
ROCK Inhibitor E212 Regulated the Phosphatase and Tensin Homolog (PTEN)/AKT1/CRMP2 Axis
The protein levels of phosphorylated-phosphatase and tensin homolog (p-PTEN), p-AKT1, and p-CRMP2 in the retinal samples were analyzed using western blotting (Fig. 8A). The levels of p-PTEN and p-CRMP2 were significantly decreased by 2.97- and 2.87-fold, respectively, in the E212-treated group compared with the PBS-treated group (P < 0.05) (Fig. 8B). The level of p-AKT1 was significantly increased by 3.44-fold in the E212-treated group versus the PBS-treated group (P < 0.05) (Fig. 8B).
Figure 8.

Western blotting analysis of p-PTEN, p-AKT1, and p-CRMP2 expression. (A) Representative photograph of the western blotting analysis of the retinal sample at day 3 after rAION induction. (B) Each value was normalized to the level of GAPDH. The levels of p-PTEN and p-CRMP2 were significantly decreased by 2.97- and 2.87-fold, respectively, in the E212-treated group compared with the PBS-treated group (P < 0.05). The level of p-AKT1 was significantly increased by 3.44-fold in the E212-treated group compared with the PBS-treated group (P < 0.05). Data are expressed as the mean ± SD. GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Discussion
Intravitreal injection of E212 immediately after the occurrence of rAION significantly preserved the visual function and prevented RGC death. In addition, it maintained the BRB and decreased the degree of oxidative stress in retinas and ON inflammation. Treatment with E212 induced M2 macrophage/microglia polarization as evidenced by the upregulation of CD206 expression, and suppressed ON inflammation by activating STAT3. Moreover, treatment with E212 manipulated the PTEN/AKT1/CRMP2 axis to prevent cell death.
Oxidative stress is a critical factor that contributes to neuronal damage during cerebral ischemia. The cellular redox homeostasis is broken by increasing the production of ROS through the activation of cytoplasmic oxidases and decreasing ROS elimination through inhibition of antioxidant mechanisms. As a result, excessive levels of ROS lead to necrosis and apoptosis via several pathways.49 Previous studies have reported that oxidative stress is involved in RGC death after an ON injury.18,36 ROCK inhibitors suppress oxidative stress through the NOX pathway to prevent RGC death in an optic nerve crush model.36 In a mouse model of rAION, SOD-1 (oxidative-stress-related) decreased to 82% of the control levels (uninjured eye) at day 1 after infarct.50 In the present study, we first demonstrated that ON infarct can induce ROS production in the RGCs at 1 day after induction of AION. Treatment with E212 markedly reduced the production of ROS in the RGCs and increased the SOD activity in the retinas. Taken together, this ROCK inhibitor possesses an antioxidative ability to reduce the oxidative stress-related damage in rAION.
ROCK activation has been associated with blood-brain barrier disruption. In an animal ischemic stroke model, a ROCK inhibitor enhanced the integrity of blood-brain barrier by regulating endothelial cell oxidative homeostasis and reduced brain edema.51 In the acute phase of NAION, both cytotoxic and vasogenic edema caused damage in the ON and the surrounding retina.52 In the present study, the BRB was examined by performing ZO-1 staining at day 3 after AION, showing that E212 maintained the RPE polarity and tight junction after an ischemic insult. This result indicated that E212 can potentially protect the integrity of the vessel barrier after an ON infarct. Our previous reports have demonstrated that breakdown of the blood-optic nerve barrier causes extrinsic macrophage invasion in the experimental optic nerve ischemia model.48,53–55 Infiltration of extrinsic macrophages (ED1+) in the ON occurred three days after the occurrence of AION, and the extrinsic macrophages continued to accumulate (>30 days), suggesting that AION causes chronic inflammation of the ON.15,56 Treatment with E212 decreased the infiltration of extrinsic macrophages, which may be associated with the effect of ROCK inhibitors on the preservation of the vessel barrier. This is one of the possible mechanisms underlying the suppression of neuroinflammation after an ON infarct.
Microglia or macrophages can be induced by TNF-α to an M1 phenotype to produce proinflammatory cytokines, such as TNF-α, IL1β, IL6, and IL23, resulting in neurotoxicity.57,58 Interestingly, treatment with E212 induced the activation of STAT3 and expression of CD206 in rAION. The STAT3 pathway is crucial in regulating the polarization of macrophages in numerous experimental models.59–62 Moreover, the ROCK inhibitor could shift the macrophage phenotype from M1 to M2 for neuroprotection.63,64 These findings indicate that ROCK inhibitors have an anti-inflammatory effect, reducing the number of proinflammatory cytokines by polarizing M2 macrophage/microglia in the rAION model.
The present findings showed that the ROCK inhibitor suppressed PTEN activation, whereas it induced that of AKT1. The expression of phosphorylated CRMP2 in retinas was reduced after treatment with E212. The downregulation of ROCK activates AKT, increasing the survival of RGCs.39 A recent study also demonstrated that the PTEN/AKT/glycogen synthase kinase 3 beta (GSK-3β)/CRMP-2 axis is involved in neuronal apoptosis and axonal injury in early brain injury after the occurrence of subarachnoid hemorrhage in rats.65 Inhibition of PTEN expression can induce AKT activation to enhance axon regeneration and neural repair; however, p-CRMP2 inhibits axonal growth in neurons, leading to a neurological deficit in middle cerebral artery occlusion models.66–69 Moreover, another study demonstrated that Y-27632 (a ROCK inhibitor) could decrease the level of p-CRMP2 and reverse the loss of neurological function in a rat middle cerebral artery occlusion model.69 An in vitro study also demonstrated that CRMP2 phosphorylation by ROCK caused growth cone collapse.70 Taken together, the present results indicate the new ROCK inhibitor E212 is a potential drug that may regulate the PTEN/AKT/CRMP2 axis to improve axonal growth and neuronal survival in individuals with ischemic optic neuropathy.
Conclusions
The present study indicated that intravitreal injection of E212 immediately after the induction of rAION prevents RGC death and improves visual function. The administration of E212 maintained the integrity of the BRB, decreased the level of oxidative stress in the retina, and reduced inflammation in ON. E212 also activated STAT3 to modulate macrophage/microglia polarization on the ON and inhibited PTEN and PRMP2 activation to enhance RGC survival.
Supplementary Material
{kind=link}
Acknowledgments
Disclosure: Y.-T. Wen, None; C.-W. Huang, None; C.-P. Liu, None; C.-H. Chen, None; C.-M. Tu, None; C.-S. Hwang, None; Y.-H. Chen, None; W.-R. Chen, None; K.-L. Lin, None; Y.-C. Ho, None; T.-C. Chen, None; R.-K. Tsai, None
References
- 1. Johnson LN, Arnold AC.. Incidence of nonarteritic and arteritic anterior ischemic optic neuropathy. Population-based study in the state of Missouri and Los Angeles County, California. J Neuroophthalmol. 1994; 14: 38–44. [PubMed] [Google Scholar]
- 2. Hattenhauer MG, Leavitt JA, Hodge DO, Grill R, Gray DT.. Incidence of nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol. 1997; 123: 103–107. [DOI] [PubMed] [Google Scholar]
- 3. Kapupara K, Wen YT, Tsai RK, Huang SP.. Soluble P-selectin promotes retinal ganglion cell survival through activation of Nrf2 signaling after ischemia injury. Cell Death and Disease. 2017; 8: e3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee YC, Wang JH, Huang TL, Tsai RK.. Increased risk of stroke in patients with nonarteritic anterior ischemic optic neuropathy: a nationwide retrospective cohort study. Am J Ophthalmol. 2016; 170: 183–189. [DOI] [PubMed] [Google Scholar]
- 5. Biousse V, Newman NJ.. Ischemic optic neuropathies. N Engl J Med. 2015; 372: 2428–2436. [DOI] [PubMed] [Google Scholar]
- 6. Arnold AC. Pathogenesis of nonarteritic anterior ischemic optic neuropathy. J Neuroophthalmol. 2003; 23: 157–163. [DOI] [PubMed] [Google Scholar]
- 7. Hayreh SS, Zimmerman MB, Podhajsky P, Alward WL.. Nocturnal arterial hypotension and its role in optic nerve head and ocular ischemic disorders. Am J Ophthalmol. 1994; 117: 603–624. [DOI] [PubMed] [Google Scholar]
- 8. Hayreh SS. Ischemic optic neuropathy. Prog Retin Eye Res. 2009; 28: 34–62. [DOI] [PubMed] [Google Scholar]
- 9. Kerr NM, Chew SS, Danesh-Meyer HV. Non-arteritic anterior ischaemic optic neuropathy: a review and update. J Clin Neurosci. 2009; 16: 994–1000. [DOI] [PubMed] [Google Scholar]
- 10. Hayreh SS. Management of ischemic optic neuropathies. Indian J Ophthalmol. 2011; 59: 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burde RM. Optic disk risk factors for nonarteritic anterior ischemic optic neuropathy. Am J Ophthalmol. 1993; 116: 759–764. [DOI] [PubMed] [Google Scholar]
- 12. Levin LA, Louhab A.. Apoptosis of retinal ganglion cells in anterior ischemic optic neuropathy. Arch Ophthalmol. 1996; 114: 488–491. [DOI] [PubMed] [Google Scholar]
- 13. Zhang C, Guo Y, Miller NR, Bernstein SL.. Optic nerve infarction and post-ischemic inflammation in the rodent model of anterior ischemic optic neuropathy (rAION). Brain Res. 2009; 1264: 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang C, Guo Y, Slater BJ, Miller NR, Bernstein SL.. Axonal degeneration, regeneration and ganglion cell death in a rodent model of anterior ischemic optic neuropathy (rAION). Exp Eye Res. 2010; 91: 286–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Salgado C, Vilson F, Miller NR, Bernstein SL.. Cellular inflammation in nonarteritic anterior ischemic optic neuropathy and its primate model. Arch Ophthalmol. 2011; 129: 1583–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nicholson JD, Leiba H, Goldenberg-Cohen N.. Translational preclinical research may lead to improved medical management of non-arteritic anterior ischemic optic neuropathy. Front Neurol. 2014; 5: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wei Y, Gong J, Yoshida T, et al.. Nrf2 has a protective role against neuronal and capillary degeneration in retinal ischemia–reperfusion injury. Free Radic Biol Med. 2011; 51: 216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Himori N, Yamamoto K, Maruyama K, et al.. Critical role of Nrf2 in oxidative stress-induced retinal ganglion cell death. J Neurochem. 2013; 127: 669–680. [DOI] [PubMed] [Google Scholar]
- 19. Xu Z, Cho H, Hartsock MJ, et al.. Neuroprotective role of Nrf2 for retinal ganglion cells in ischemia-reperfusion. J Neurochem. 2015; 133: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hall A. Rho GTPases and the control of cell behaviour. Biochem Soc Trans. 2005; 33: 891–895. [DOI] [PubMed] [Google Scholar]
- 21. Bach LA. Rho kinase inhibition: a new approach for treating diabetic nephropathy? Diabetes. 2008; 57: 532–533. [DOI] [PubMed] [Google Scholar]
- 22. Budzyn K, Marley PD, Sobey CG.. Targeting Rho and Rho-kinase in the treatment of cardiovascular disease. Trends Pharmacol Sci. 2006; 27: 97–104. [DOI] [PubMed] [Google Scholar]
- 23. Ramamoorthy M, Sykora P, Scheibye-Knudsen M, et al.. Sporadic Alzheimer disease fibroblasts display an oxidative stress phenotype. Free Radic Biol Med. 2012; 53: 1371–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chiba Y, Kuroda S, Shichinohe H, et al.. Synergistic effects of bone marrow stromal cells and a Rho kinase (ROCK) inhibitor, fasudil on axon regeneration in rat spinal cord injury. Neuropathology. 2010; 30: 241–250. [DOI] [PubMed] [Google Scholar]
- 25. Cheng CI, Lin YC, Tsai TH, et al.. The prognostic values of leukocyte Rho kinase activity in acute ischemic stroke. Biomed Res Int. 2014; 2014: 214587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. LoGrasso PV, Feng Y.. Rho kinase (ROCK) inhibitors and their application to Inflammatory Disorders. Curr Top Med Chem. 2009; 9: 704–723. [DOI] [PubMed] [Google Scholar]
- 27. Challa P, Arnold JJ.. Rho-kinase inhibitors offer a new approach in the treatment of glaucoma. Expert Opin Investig Drugs. 2014; 23: 81–95. [DOI] [PubMed] [Google Scholar]
- 28. Fard MA, Ebrahimi KB, Miller NR.. RhoA activity and post-ischemic inflammation in an experimental model of adult rodent anterior ischemic optic neuropathy. Brain Res. 2013; 1534: 76–86. [DOI] [PubMed] [Google Scholar]
- 29. Feng Y, LoGrasso PV, Defert O, Li R.. Rho kinase (ROCK) inhibitors and their therapeutic potential. J Med Chem. 2016; 59: 2269–2300. [DOI] [PubMed] [Google Scholar]
- 30. Schaafsma D, Gosens R, Zaagsma J, Halayko AJ, Meurs H.. Rho kinase inhibitors: a novel therapeutical intervention in asthma? Eur J Pharmacol. 2008; 585: 398–406. [DOI] [PubMed] [Google Scholar]
- 31. Rath N, Olson MF.. Rho-associated kinases in tumorigenesis: re-considering ROCK inhibition for cancer therapy. EMBO Rep. 2012; 13: 900–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Albersen M, Shindel AW, Mwamukonda KB, Lue TF.. The future is today: emerging drugs for the treatment of erectile dysfunction. Expert Opin Emerg Drugs. 2010; 15: 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lee DH, Shi J, Jeoung NH, et al.. Targeted disruption of ROCK1 causes insulin resistance in vivo. J Biol Chem. 2009; 284: 11776–11780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Komers R, Oyama TT, Beard DR, et al.. Rho kinase inhibition protects kidneys from diabetic nephropathy without reducing blood pressure. Kidney Int . 2011; 79: 432–442. [DOI] [PubMed] [Google Scholar]
- 35. Mueller BK, Mack H, Teusch N.. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov. 2005; 4: 387–398. [DOI] [PubMed] [Google Scholar]
- 36. Yamamoto K, Maruyama K, Himori N, et al.. The novel Rho kinase (ROCK) inhibitor K-115: a new candidate drug for neuroprotective treatment in glaucoma. Invest Ophthalmol Vis Sci. 2014; 55: 7126–7136. [DOI] [PubMed] [Google Scholar]
- 37. Van de Velde S, De Groef L, Stalmans I, Moons L, Van Hove I. Towards axonal regeneration and neuroprotection in glaucoma: Rho kinase inhibitors as promising therapeutics. Prog Neurobiol. 2015; 131: 105–119. [DOI] [PubMed] [Google Scholar]
- 38. Rodriguez-Perez AI, Dominguez-Meijide A, Lanciego JL, Guerra MJ, Labandeira-Garcia JL.. Inhibition of Rho kinase mediates the neuroprotective effects of estrogen in the MPTP model of Parkinson's disease. Neurobiol Dis. 2013; 58: 209–219. [DOI] [PubMed] [Google Scholar]
- 39. Koch JC, Tönges L, Barski E, Michel U, Bähr M, Lingor P.. ROCK2 is a major regulator of axonal degeneration, neuronal death and axonal regeneration in the CNS. Cell Death Dis. 2014; 5: e1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sanjari N, Pakravan M, Nourinia R, et al.. Intravitreal injection of a Rho-kinase inhibitor (fasudil) for recent-onset nonarteritic anterior ischemic optic neuropathy. J Clin Pharmacol. 2016; 56: 749–753. [DOI] [PubMed] [Google Scholar]
- 41. Georgiou T, Wen YT, Chang CH, et al.. Neuroprotective effects of Omega-3 polyunsaturated fatty acids in a rat model of anterior ischemic optic neuropathy. Invest Ophthalmol Vis Sci. 2017; 58: 1603–1611. [DOI] [PubMed] [Google Scholar]
- 42. Huang TL, Chang CH, Chang SW, Lin KH, Tsai RK.. Efficacy of intravitreal injections of antivascular endothelial growth factor agents in a rat model of anterior ischemic optic neuropathy. Invest Ophthalmol Vis Sci. 2015; 56: 2290–2296. [DOI] [PubMed] [Google Scholar]
- 43. Huang TL, Wen YT, Chang CH, Chang SW, Lin KH, Tsai RK.. Efficacy of intravitreal injections of triamcinolone acetonide in a rodent model of nonarteritic anterior ischemic optic neuropathy. Invest Ophthalmol Vis Sci. 2016; 57: 1878–1884. [DOI] [PubMed] [Google Scholar]
- 44. Bernstein SL, Johnson MA, Miller NR.. Nonarteritic anterior ischemic optic neuropathy (NAION) and its experimental models. Prog Retin Eye Res. 2011; 30: 167–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsai RK, Chang CH, Wang HZ.. Neuroprotective effects of recombinant human granulocyte colony-stimulating factor (G-CSF) in neurodegeneration after optic nerve crush in rats. Exp Eye Res. 2008; 87: 242–250. [DOI] [PubMed] [Google Scholar]
- 46. Tsai RK, Chang CH, Sheu MM, Huang ZL.. Anti-apoptotic effects of human granulocyte colony-stimulating factor (G-CSF) on retinal ganglion cells after optic nerve crush are PI3K/AKT-dependent. Exp Eye Res. 2010; 90: 537–545. [DOI] [PubMed] [Google Scholar]
- 47. Huang TL, Chang CH, Lin KH, Sheu MM, Tsai RK.. Lack of protective effect of local administration of triamcinolone or systemic treatment with methylprednisolone against damages caused by optic nerve crush in rats. Exp Eye Res. 2011; 92: 112–119. [DOI] [PubMed] [Google Scholar]
- 48. Huang TL, Huang SP, Chang CH, Lin KH, Chang SW, Tsai RK.. Protective effects of systemic treatment with methylprednisolone in a rodent model of non-arteritic anterior ischemic optic neuropathy (rAION). Exp Eye Res. 2015; 131: 69–76. [DOI] [PubMed] [Google Scholar]
- 49. Manzanero S, Santro T, Arumugam TV.. Neuronal oxidative stress in acute ischemic stroke: sources and contribution to cell injury. Neurochem Int. 2013; 62: 712–718. [DOI] [PubMed] [Google Scholar]
- 50. Avraham-Lubin BC, Dratviman-Storobinsky O, El SD, Hasanreisoglu M, Goldenberg-Cohen N. Neuroprotective effect of hyperbaric oxygen therapy on anterior ischemic optic neuropathy. Front Neurol. 2011; 2: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gibson CL, Srivastava K, Sprigg N, Bath PM, Bayraktutan U.. Inhibition of Rho-kinase protects cerebral barrier from ischaemia-evoked injury through modulations of endothelial cell oxidative stress and tight junctions. J Neurochem. 2014; 129: 816–826. [DOI] [PubMed] [Google Scholar]
- 52. Levin M, Danesh-Meyer HV. Hypothesis: A venous etiology for nonarteritic anterior ischemic optic neuropathy. Arch Ophthalmol. 2008; 126: 1582–1585. [DOI] [PubMed] [Google Scholar]
- 53. Zhang C, Lam TT, Tso MO.. Heterogeneous populations of microglia/macrophages in the retina and their activation after retinal ischemia and reperfusion injury. Exp Eye Res. 2005; 81: 700–709. [DOI] [PubMed] [Google Scholar]
- 54. Wen YT, Huang TL, Huang SP, Chang CH, Tsai RK.. Early applications of granulocyte colony-stimulating factor (G-CSF) can stabilize the blood-optic-nerve barrier and ameliorate inflammation in a rat model of anterior ischemic optic neuropathy (rAION). Dis Model Mech. 2016; 9: 1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Huang TL, Wen YT, Chang CH, Chang SW, Lin KH, Tsai RK.. Early methylprednisolone treatment can stabilize the blood-optic nerve barrier in a rat model of anterior ischemic optic neuropathy (rAION). Invest Ophthalmol Vis Sci. 2017; 58: 1628–1636. [DOI] [PubMed] [Google Scholar]
- 56. Chen CS, Johnson MA, Flower RA, Slater BJ, Miller NR, Bernstein SL.. A primate model of nonarteritic anterior ischemic optic neuropathy. Invest Ophthalmol Vis Sci. 2008; 49: 2985–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Boche D, Perry VH, Nicoll JAR.. Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol. 2013; 39: 3–18. [DOI] [PubMed] [Google Scholar]
- 58. Orihuela R, McPherson CA, Harry GJ.. Microglial M1/M2 polarization and metabolic states. Br J Pharmacol. 2016; 173: 649–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yuan F, Fu X, Shi H, Chen G, Dong P, Zhang W.. Induction of murine macrophage M2 polarization by cigarette smoke extract via the JAK2/STAT3 pathway. PLoS One. 2014; 9: e107063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cui J, Zhang F, Cao W, et al.. Erythropoietin alleviates hyperglycaemia-associated inflammation by regulating macrophage polarization via the JAK2/STAT3 signalling pathway. Mol Immunol. 2018; 101: 221–228. [DOI] [PubMed] [Google Scholar]
- 61. Yang X, He G, Hao Y, et al.. The role of the JAK2-STAT3 pathway in pro-inflammatory responses of EMF-stimulated N9 microglial cells. J Neuroinflammation. 2010; 7: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang X, Xu F, Liu L, et al.. (+)-Borneol improves the efficacy of edaravone against DSS-induced colitis by promoting M2 macrophages polarization via JAK2-STAT3 signaling pathway. Int Immunopharmacol. 2017; 53: 1–10. [DOI] [PubMed] [Google Scholar]
- 63. Liu C, Li Y, Yu J, et al.. Targeting the shift from M1 to M2 macrophages in experimental autoimmune encephalomyelitis mice treated with fasudil. PLoS One. 2013; 8: e54841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen J, Yin W, Tu Y, et al.. L-F001, a novel multifunctional ROCK inhibitor, suppresses neuroinflammation in vitro and in vivo: involvement of NF-kappaB inhibition and Nrf2 pathway activation. Eur J Pharmacol. 2017; 806: 1–9. [DOI] [PubMed] [Google Scholar]
- 65. Chen H, Zhou C, Zheng J, et al.. PTEN and AKT/GSK-3β/CRMP-2 signaling pathway are involved in neuronal apoptosis and axonal injury in early brain injury after SAH in rats [published online ahead of print June 18, 2020]. Genes Dis, 10.1016/j.gendis.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K.. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005; 120: 137–149. [DOI] [PubMed] [Google Scholar]
- 67. Gim SA, Sung JH, Shah FA, Kim MO, Koh PO.. Ferulic acid regulates the AKT/GSK-3β/CRMP-2 signaling pathway in a middle cerebral artery occlusion animal model. Lab Anim Res. 2013; 29: 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang T, Wu X, Yin C, Klebe D, Zhang JH, Qin X.. CRMP-2 is involved in axon growth inhibition induced by RGMa in vitro and in vivo. Mol Neurobiol. 2013; 47: 903–913. [DOI] [PubMed] [Google Scholar]
- 69. Li XB, Ding MX, Ding CL, Li LL, Feng J, Yu XJ.. TollLike receptor 4 promotes the phosphorylation of CRMP2 via the activation of Rhokinase in MCAO rats. Mol Med Rep. 2018; 18: 342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Arimura N, Ménager C, Kawano Y, et al.. Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol Cell Biol. 2005; 25: 9973–9984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.