

ABSTRACT
NRBF2 is a component of the class III phosphatidylinositol 3-kinase (PtdIns3K) complex. Our previous study has revealed its role in regulating ATG14-associated PtdIns3K activity for autophagosome initiation. In this study, we revealed an unknown mechanism by which NRBF2 modulates autophagosome maturation and APP-C-terminal fragment (CTF) degradation. Our data showed that NRBF2 localized at autolysosomes, and loss of NRBF2 impaired autophagosome maturation. Mechanistically, NRBF2 colocalizes with RAB7 and is required for generation of GTP-bound RAB7 by interacting with RAB7 GEF CCZ1-MON1A and maintaining the GEF activity. Specifically, NRBF2 regulates CCZ1-MON1A interaction with PI3KC3/VPS34 and CCZ1-associated PI3KC3 kinase activity, which are required for CCZ1-MON1A GEF activity. Finally, we showed that NRBF2 is involved in APP-CTF degradation and amyloid beta peptide production by maintaining the interaction between APP and the CCZ1-MON1A-RAB7 module to facilitate the maturation of APP-containing vesicles. Overall, our study revealed a pivotal role of NRBF2 as a new RAB7 effector in modulating autophagosome maturation, providing insight into the molecular mechanism of NRBF2-PtdIns3K in regulating RAB7 activity for macroautophagy/autophagy maturation and Alzheimer disease-associated protein degradation..
Abbreviations: 3xTg AD, triple transgenic mouse for Alzheimer disease; Aβ, amyloid beta peptide; Aβ1-40, amyloid beta peptide 1–40; Aβ1-42, amyloid beta peptide 1–42; AD, Alzheimer disease; APP, amyloid beta precursor protein; APP-CTFs, APP C-terminal fragments; ATG, autophagy related; ATG5, autophagy related 5; ATG7, autophagy related 7; ATG14, autophagy related 14; CCD, coiled-coil domain; CCZ1, CCZ1 homolog, vacuolar protein trafficking and biogenesis associated; CHX, cycloheximide; CQ, chloroquine; DAPI, 4ʹ,6-diamidino-2-phenylindole; dCCD, delete CCD; dMIT, delete MIT; FYCO1, FYVE and coiled-coil domain autophagy adaptor 1; FYVE, Fab1, YGL023, Vps27, and EEA1; GAP, GTPase-activating protein; GDP, guanine diphosphate; GEF, guanine nucleotide exchange factor; GTP, guanine triphosphate; GTPase, guanosine triphosphatase; HOPS, homotypic fusion and vacuole protein sorting; ILVs, endosomal intralumenal vesicles; KD, knockdown; KO, knockout; LAMP1, lysosomal associated membrane protein 1; MAP1LC3/LC3, microtubule associated protein 1 light chain 3; MLVs, multilamellar vesicles; MON1A, MON1 homolog A, secretory trafficking associated; NRBF2, nuclear receptor binding factor 2; PtdIns3K, class III phosphatidylinositol 3-kinase; PtdIns3P, phosphatidylinositol-3-phosphate; RILP, Rab interacting lysosomal protein; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; SQSTM1/p62, sequestosome 1; UVRAG, UV radiation resistance associated; VPS, vacuolar protein sorting; WT, wild type.
KEYWORDS: Autophagy, CCZ1-MON1A, maturation, NRBF2, PI3KC3/VPS34, RAB7, trafficking
Introduction
Macroautophagy (henceforth to be referred to as autophagy) is a conserved pathway that is involved in the degradation of cytoplasmic materials including long-lived proteins, protein aggregates, cellular organelles (e.g., mitochondria), and infectious agents via lysosomes [1–5]. Autophagy involves the formation of cup-shaped double-membrane structures called phagophores, followed by extension and closure to form autophagosomes [1–5]. Finally, autophagosomes are delivered to the perinuclear region along microtubules in order to fuses with lysosome [6,7]. Upon the fusion of autophagosome with lysosome to form autolysosome, the sequestered contents inside the autophagosome are degraded by lysosome hydrolases, a process known as autophagosome maturation [1–5]. Though a variety of ATG (autophagy-related) proteins acting at different steps for autophagy regulation had been identified, the mechanisms of autophagosome maturation are not fully characterized.
PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3), the only class III phosphatidylinositol 3-kinase (PtdIns3K) in mammals, is a central node in autophagy process [8]. The main product of PtdIns3K is phosphatidylinositol-3-phosphate (PtdIns3P). The PtdIns3K complex core components include the catalytic subunit PIK3C3, regulatory subunit PIK3R4/VPS15, and BECN1 (beclin 1) [9]. The PtdIns3K complex is involved in both autophagosome formation and autophagosome maturation by binding with diverse partners to form distinct complexes [9]. We and others have previously identified NRBF2 as the fifth component of PtdIns3K complex 1 and showed that NRBF2 directly interacts with ATG14 [10–15]. Importantly, NRBF2 regulates autophagosome formation via modulation of ATG14-linked PIK3C3 kinase activity for autophagosome biogenesis [10]. Such findings are consistent with a report by Dr. Yoshinori Ohsumi’s group that Atg38, a yeast ortholog of NRBF2, is a component of PtdIns3K complex 1 and is required for the initiation of autophagy [16].
Fusion of autophagosomes with late endosomes/lysosomes is regulated by a variety of factors such as small GTPase RAB7, tethering proteins, and soluble N-ethylmaleimide sensitive factor attachment protein receptor (SNARE) complex [4,17–19]. RAB7 localizes on late endosomes/lysosomes and is important for both the fusion of autophagosomes with lysosomes and the subsequent degradation of cellular contents sequestered in the autophagosomes. RAB7 has been shown to regulate the proper bidirectional transport of autophagosomes through interaction with FYCO1 [7]. RAB7 is activated by specific guanine nucleotide exchange factors (GEFs) that drive GTP binding and is inhibited by GTPase activating proteins (GAPs) [20–22]. Thus, GEFs play a crucial role in regulating GTPase activity. CCZ1-MON1A [21,23–25], HOPS complex PLEKHM1 [26], EPG5 [27], RILP [28,29] and FYCO1 [7] have been identified as RAB7 effectors that are important for autophagosome-lysosome fusion. CCZ1-MON1A complex is a RAB7 GEF which promotes the exchange of GDP-RAB7 (inactive state) to GTP-RAB7 (active state). Gao et al. discovered that membrane-bound Atg8 can recruit Ccz1-Mon1 to membrane to promote Ypt7 activation in yeast [30]. Stephan Kiontke et al. identified the domains required for catalytic activity, complex assembly and localization of Ccz1-Mon1. The crystal structure of a catalytic Ccz1-Mon1 core complex bound to Ypt7 provides mechanistic insight into its function [25].
Though PtdIns3K complex 1 and its product PtdIns3P have been involved in regulating autophagosome formation, accumulating evidence indicates that PtdIns3K complex 1 and PtdIns3P also modulate autophagosome maturation [4,31]. For instance, PtdIns3K complex 2 component UVRAG directly binds to a component of the homotypic vacuole fusion and protein sorting (HOPS) complex VPS16 [32] to enhance RAB7 GTPase activity and subsequent autophagosome maturation. FYCO1 binds to LC3, PtdIns3P and RAB7, and is involved in autophagosome maturation [7]. Another PtdIns3P-binding protein TECPR1, is also implicated in autophagosome-lysosome fusion [33]. Although there is an increasing understanding of this progress, how PtdIns3K coordinates with RAB7 to regulate autophagosome maturation is largely unclear.
Autophagy dysfunction is known to be involved in a variety of diseases [34,35]. Impaired autophagy activity increases APP and CTF levels [36]. We previously showed that NRBF2-deficient mice demonstrate liver injury [10]. Additionally, we further identified that NRBF2 levels are reduced in the hippocampus region of an Alzheimer disease (AD) animal model and that NRBF2 interacts with APP (amyloid beta precursor protein) to regulate APP-CTFs degradation and Aβ homeostasis in an autophagy-dependent manner [37]. Interestingly, the reduction of NRBF2 levels is accompanied by autophagy flux inhibition in AD mice [37] indicating that NRBF2 may have a role in regulating autophagosome-lysosome fusion. Furthermore, we recently reported that NRBF2 expression was reduced in parahippocampal gyrus and hippocampus in late-onset AD postmortem brains and nrbf2-/- mice displayed spatial memory deficits with long-term potentiation impairment (LTP) [38]. Though these results indicate the potential involvement of NRBF2/Atg38 in autophagy regulation and AD, the molecular mechanisms of NRBF2 in regulating autophagic degradation of APP-CTF are not fully understood.
Here, we report a pivotal role of NRBF2, a key component of PtdIns3K-C1, in controlling autophagosome maturation apart from its known function of regulating autophagosome biogenesis. Our results show that this mechanism is related to the role of NRBF2 in regulating the GTP-form of RAB7 by mediating CCZ1-MON1A-PIK3C3 interaction for maintaining GEF activity. More interestingly, this process is required for APP-CTF degradation. Collectively, our study reveals the unexpected role of NRBF2 in modulating autophagosome maturation, providing insight into the molecular mechanism of NRBF2-PtdIns3K in autophagy regulation and AD association.
Results
NRBF2 is required for autophagosome maturation
Recently, we and others identified NRBF2 as a novel component of the PIK3C3 complex, and we found that it plays a critical role in modulating autophagosome biogenesis [10,13–15]. As the PIK3C3 complex is critical for regulating both autophagosome formation and autophagosome maturation, we wondered whether NRBF2 also plays a role in modulating autophagosome maturation. To answer this question, we generated nrbf2 knockout (KO) mouse neuroblastoma N2a cells by using the CRISPR-Cas9 system (Figure. S1A) and found that nrbf2 KO increased both LC3-II and SQSTM1 levels (Figure 1A-C). To further confirm the role of NRBF2 in other cell types, we detected the levels of SQSTM1 and LC3-II in SH-SY5Y and HEK293 cells treated with NRBF2 siRNA. As shown in Figure. S1B-I, transient transfection of Nrbf2 siRNA resulted in significant reduction in the protein levels of NRBF2 and accumulation of LC3-II and SQSTM1 levels in these cell lines. Additionally, in the brains of nrbf2-/- mice, both LC3-II and SQSTM1 levels were significantly increased (Figure 1D-F). SQSTM1 is a selective substrate for autophagy. Enhancing autophagy flux promotes the degradation of SQSTM1 [39].
Figure 1.
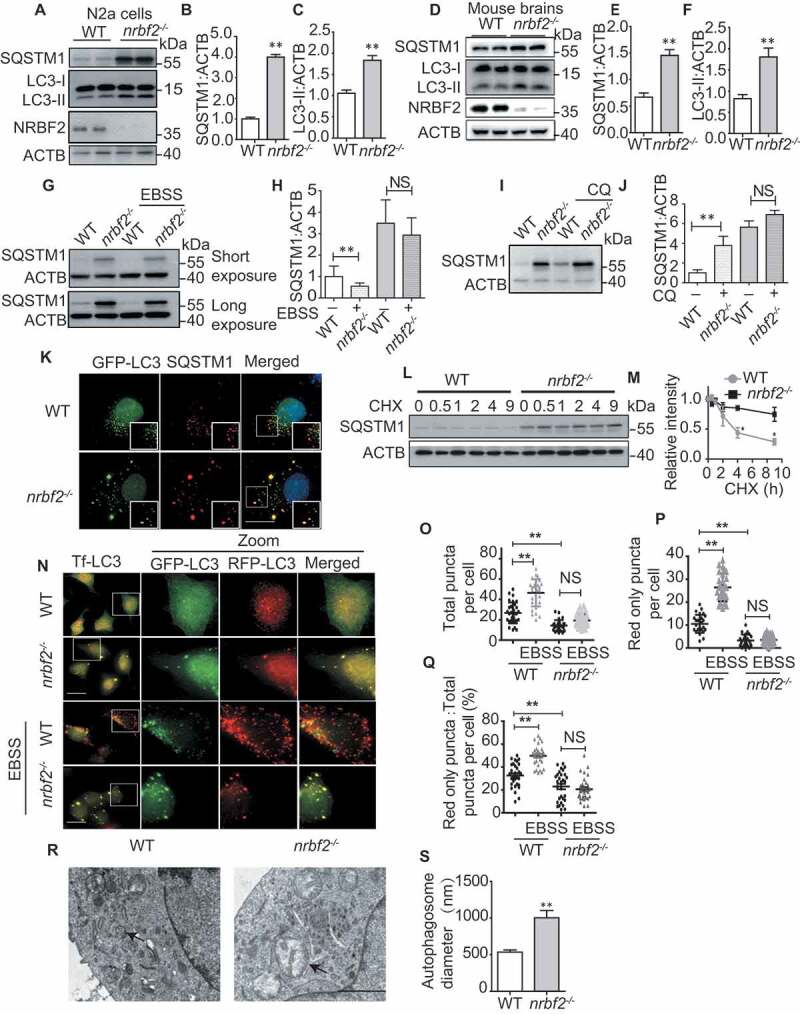
NRBF2 is required for autophagosome maturation. (A-C) Generation of nrbf2-/- N2a cells by CRISPR-Cas9 technique and WB analysis of LC3-II and SQSTM1 levels in WT and nrbf2-/- N2a cells. Data are quantified as mean ± SEM (n = 3). NS, not significant; *P < 0.05, **P < 0.01, vs. the relative control. (D-F) WB analysis of LC3-II and SQSTM1 levels in WT and nrbf2-/- mice brain. Data are quantified as mean ± SEM (n = 3). NS, not significant; *P < 0.05, **P < 0.01, vs. the relative control. (G and H) SQSTM1 levels were determined by WB under starvation (EBSS) condition both in WT and nrbf2-/- N2a cells. Data are quantified as mean ± SEM (n = 3). NS, not significant; *P < 0.05, **P < 0.01, vs. the relative control. (I and J) SQSTM1 levels were determined by WB under lysosome inhibition CQ condition both in WT and nrbf2-/- N2a cells. Data are quantified as mean ± SEM (n = 3). NS, not significant; *P < 0.05, **P < 0.01, vs. the relative control. (K) N2a cells were transiently transfected with GFP-Lc3, and the colocalization of GFP-LC3 with endogenous SQSTM1 was visualized under confocal microscopy. Quantification data were presented as the mean ± SEM, n = 20–25 cells from 3 independent experiments. Scale bar: 10 μm. (L and M) Degradation of SQSTM1 in the presence of protein synthesis inhibitor cycloheximide (CHX) was tracked in WT and nrbf2-/- N2a cells by WB. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, vs. the relative control. (N-Q) Autophagosome maturation in WT and nrbf2-/- N2a cells was determined by the tf-LC3 probe under both basal and starvation conditions (EBSS). Quantification data were presented as the mean ± SEM, n = 20–25 cells from 3 independent experiments. NS, not significant; *P < 0.05, **P < 0.01, vs. the relative control. Scale bar: 20 μm. (R and S) Abnormally enlarged autophagosomes were observed in nrbf2-/- N2a cells by EM. Quantification data were presented as the mean ± SEM, n = 20. *P < 0.05, **P < 0.01, vs. the relative control. Scale bar: 2 μm
To further confirm the function of NRBF2 in regulating autophagosome maturation, we investigated the role of NRBF2 in modulating SQSTM1 degradation. We enhanced autophagy flux by cell starvation (EBSS treatment). As showed in (Figure 1G and H), EBSS enhanced the degradation of SQSTM1 levels in wild type (WT) cells rather than that in nrbf2-/- N2a cells. Additionally, there was no dramatic increase of SQSTM1 levels after cells were treated with the lysosome inhibitor CQ in nrbf2-/- cells, though CQ significantly increased SQSTM1 levels in WT cell (Figure 1I and J), indicating the degradation of SQSTM1 by lysosome was impaired in NRBF2-depleted cells. Interestingly, almost all SQSTM1 colocalized with LC3 puncta after KO of nrbf2 (Figure 1K), indicating that nrbf2 KO may attenuate autophagy-dependent degradation of SQSTM1. As showed in (Figure 1L and M), the inhibition of protein synthesis with cycloheximide (CHX) causes the degradation of SQSTM1 in WT cells. In contrast, nrbf2 KO impaired the degradation of SQSTM1 (Figure 1L and M). The total ubiquitinated proteins were examined to confirm that the concentration of CHX is effective in chasing protein degradation (Figure. S1J).
Tf-Lc3 plasmid (GFP-RFP-Lc3 construct) is a valuable tool for monitoring autophagosome maturation based on the principle that GFP is more rapidly quenched than RFP at acidic environment in lysosome. To further confirm the role of NRBF2 in modulating autophagosome maturation, we transiently transfected N2a cells with tf-Lc3 plasmids and found that NRBF2 deficiency causes the accumulation of yellow autophagosomes (Figure 1N-Q). Upon induction of autophagy by starvation (EBSS) or torin 1 treatment, there were more red-only autolysosomes in WT cells than that in nrbf2-/- N2a cells (Figure 1N and S1K). Compared with WT N2a cells, the number of total puncta decreased (Figure 1O) and the number of red-only puncta (Figure 1P) also decreased, the ratio of red puncta to total puncta also decreased in nrbf2-/- cells (Figure 1Q), indicating an increased number of autophagosome not fused with the lysosome. Furthermore, transmission electron microscope images of WT N2a and nrbf2-/- N2a cells showed that autophagosome size was larger in nrbf2-/- N2a cells (Figure 1R and S). These data together demonstrate that nrbf2 KO represses autophagosome maturation.
NRBF2 localizes at autolysosomes and is-required for autolysosome maturation
Our and other’s previous reports showed that NRBF2 puncta partially colocalize with autophagic structures [10,12]. As NRBF2 also regulates autophagosome maturation, we asked whether NRBF2 localizes on late endosomes/lysosomes. As showed in (Figure 2A), endogenous NRBF2 puncta partially colocalized with LC3- and LAMP1-positive structures in N2a cells (Figure 2A). As shown in Figure. S2A, exogenous NRBF2 puncta partially colocalized with LC3- and LAMP1-positive structures in RPE19 cells (Figure. S2A). More interestingly, we found that colocalization between NRBF2 and LC3 or LAMP1 was increased after torin 1 (Figure 2A and S1L) and HBSS (Figure. S2A and S2B) treatment. The NRBF2 antibody has been verified by immunofluorescence staining of WT and nrbf2-/- cells (Figure. S2C). Furthermore, the colocalization of autophagosome marker LC3 with lysosome member protein LAMP1 was attenuated after nrbf2 KO (Figure 2B) which further confirmed that NRBF2 has a role in regulating autophagosome maturation. Autophagosome maturation includes the autophagosome trafficking to the lysosome and autophagosome fusion with lysosome/multivesicular body, forming autolysosomes. Trafficking of autophagosomes is particularly important in large, highly specialized cells, such as neurons, where the autophagosome needs to be transported to the soma for lysosomal degradation. We used an in vitro autophagosome-lysosome fusion experiment to determine whether NRBF2 affects the fusion [40]. From the results, we can see that nrbf2 KO does not affect direct autophagosome-lysosome fusion (Figure 2C). By a similar approach, we showed that nrbf2 KO does not affect direct MVB-lysosome fusion (Figure. S2D).
Figure 2.
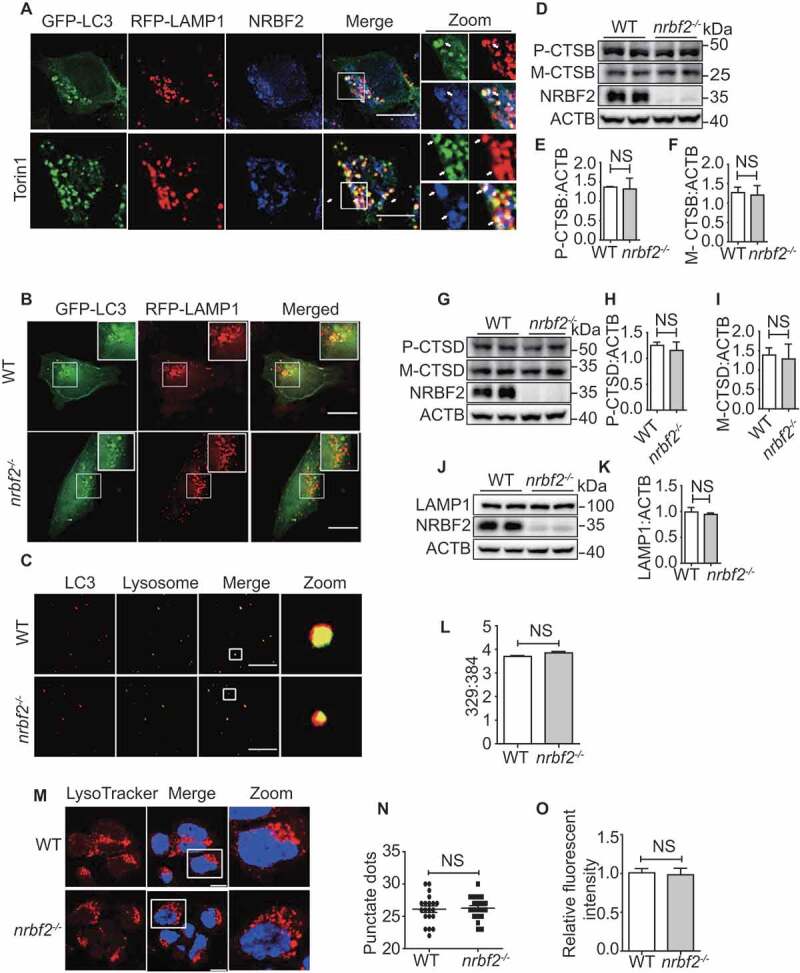
NRBF2 localizes at autolysosomes and is required for autolysosome maturation. (A) NRBF2 partially localizes with LC3- and LAMP1-positive autophagic structures under both basal and torin 1-treated conditions. N2a cells were transiently transfected with GFP-Lc3 and RFP-Lamp1, and then co-stained with NRBF2 antibody. Colocalization of NRBF2 and the autolysosome markers under normal or torin 1-treated conditions were visualized under confocal microscope. Scale bar: 10 μm. (B) WT and nrbf2-/- N2a cells were transiently transfected with GFP-Lc3 and RFP-Lamp1, the colocalization of autophagosomes and lysosomes was visualized under confocal microscope. Scale bar: 10 μm. (C) Purified autophagosome and lysosome isolated from starved WT and nrbf2-/- mice liver were incubated in an energy-regenerating buffer and stained with LC3 and LAMP1 antibody, respectively, to monitor the direct fusion between autophagosome and lysosome. Scale bar: 8.5 μm. (D-K) Lysosome proteins CTSB (cathepsin B), CTSD and LAMP1 were measured in WT and nrbf2-/- brain lysates. Data are quantified as mean ± SEM (n = 3). NS, not significant; vs. the relative control. (L) WT and nrbf2-/- N2a cells were stained with LysoSensor™ Yellow/Blue DND-160 and the Ex at 329 nm/384 nm were recorded with Em at 440 nm/540 nm to determine the acidification of lysosome. (M) WT and nrbf2-/- N2a cells were stained with LysoTracker (selective for acidic organelles, usually used to detect the number of lysosomes) to determine the quantity of lysosome. Scale bar: 200 μm. (N and O) Quantitative date from LysoTracker staining. Lysosomal numbers as reflected by LysoTracker staining dots and intensity. Quantification data were presented as the mean ±SEM, n = 20–25 cells from 3 independent experiments. NS, not significant
Alternatively, impairment of SQSTM1 degradation may result from lysosomal dysfunction in nrbf2-deficient cells. To test this possibility, we examined the effects of nrbf2 KO on the expression of lysosome enzymes, lysosomal numbers and lysosomal pH value. We found that there are no differences in the expression levels of lysosomal enzymes CTSB (cathepsin B) (Figure 2D-F) and CTSD (cathepsin D) (Figure 2G-I) in WT and nrbf2 KO conditions. NRBF2 deficiency does not affect the expression level of LAMP1 (Figure 2J and K), lysosome numbers (Figure 2M-O), and lysosome pH (Figure 2L). Overall, these results demonstrated that role of NRBF2 in regulating autophagosome maturation might be attributed to autophagosome trafficking rather than lysosome inhibition.
MIT domain of NRBF2 is required for maturation
The N-terminal MIT domain and the C-terminal CCD domain are two discrete motifs in NRBF2 [10,14]. We want to know which domain of NRBF2 is responsible for NRBF2-mediated modulation of autophagosome-lysosome fusion. After transfecting nrbf2-/- N2a with full-length NRBF2-CFP, NRBF2 mutants missing either the MIT domain (dMIT-CFP) or the CCD domain (dCCD-CFP), only MIT domain (MIT-CFP) or only CCD domain (CCD-CFP) (Figure 3A), we detected the SQSTM1 levels by immunoblotting. Our data showed that both full-length NRBF2-CFP and dCCD-CFP mutant are able to rescue nrbf2 KO-mediated increase of SQSTM1 levels in both basal and starvation conditions (Figure 3B and C). Additionally, tf-Lc3 staining showed that both NRBF2-CFP and dCCD-CFP could effectively promote autophagy flux in nrbf2-/- cells. Number of total puncta increases (Figure 3E) and the number of red-only puncta (Figure 3F) also increase. However, the MIT domain alone is not sufficient for rescuing the impairment of autophagosome maturation in nrbf2-/- cell (Figure 3D-F). We found that deletion of MIT domain changed the localization of NRBF2 on autophagosome. Immunofluorescence results show that dMIT-CFP cannot colocalize with RFP-LC3 in nrbf2-/- N2a cells (Figure. S2E). Collectively, these results strongly support the critical role of the MIT domain in regulating autophagosome-lysosome fusion.
Figure 3.
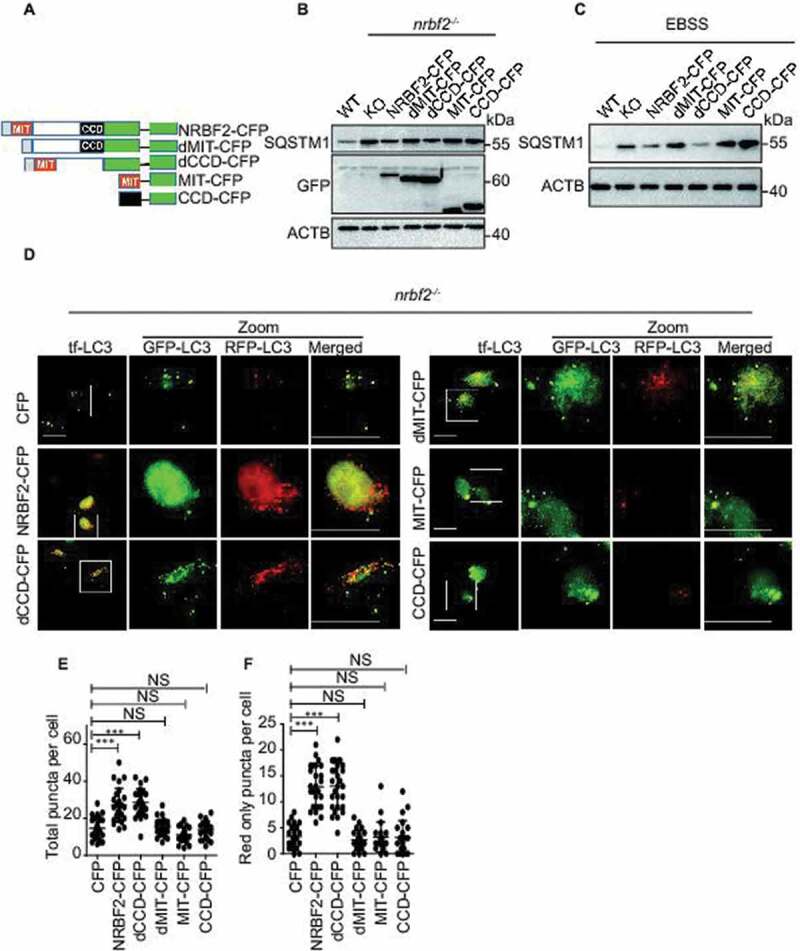
MIT domain of NRBF2 is required for maturation. (A) Schematic map of full-length and truncated NRBF2-CFP. (B and C) NRBF2 MIT domain is important for regulating SQSTM1 degradation. nrbf2-/- N2a cells were transiently transfected with CFP, Nrbf2-CFP, dMIT-CFP dCCD-CFP, CFP-MIT or CFP-CCD, the expression of SQSTM1 levels were detected in basal (B) and starvation conditions (EBSS) (C). (D-F) NRBF2 MIT domain is required for regulating autophagosome-lysosome fusion. nrbf2-/- N2a cells were transiently transfected with CFP, Nrbf2-CFP, dMIT-CFP, dCCD-CFP, CFP-MIT or CFP-CCD in the presence of RFP-GFP-Lc3 transfection. The autophagosome maturation was observed under confocal microscope. Quantification data were presented as the mean ±SEM, n = 20–25 cells from 3 independent experiments. *P < 0.05, **P < 0.01, vs. the relative control. Scale bar: 20 μm
NRBF2 is required for RAB7 activation to promote autophagosome maturation
To understand how NRBF2 regulates autophagosome-lysosome fusion, we investigated whether NRBF2 modulates small GTPase RAB7 activity, which is critical for autophagosome maturation [41–43]. By using GTP beads affinity-isolation assay as described previously [44], we found that depletion of NRBF2 significantly reduces GTP form RAB7 in N2a cells (Figure 4A and B), without affecting total RAB7 protein level (Figure 4A). This result was also confirmed in the brains of nrbf2-/- mice (Figure 4C and D). RILP (Rab interacting lysosomal protein) selectively binds RAB7-GTP [45,46]. We evaluated the activation state of RAB7 in the nrbf2-/- cells and mouse using a GST-tagged RAB7-binding domain (GST-R7BD) of RILP. Consistently, the GST-R7BD interaction with RAB7 (GTP-RAB7) decreased significantly in nrbf2-/- cell (Figure 4E and F) and nrbf2-/- mice tissues (Figure 4G and H). We examined the effects of nrbf2 KO on the expression of RILP and found that there are no differences in the expression level of RILP in WT and nrbf2 KO conditions (Figure. S2F-I). Finally, overexpression of NRBF2 could rescue decreased GTP-RAB7 (Figure 4I and J). The colocalization of RAB7 with LC3-positive autophagosomes is reduced in nrbf2-/- cells, and this phenotype can be rescued by re-introducing NRBF2 in nrbf2-/- cells (Figure 4M). Additionally, endogenous NRBF2 puncta partially colocalized with LC3- and RAB7-positive structures (Figure 4N and S2 J), indicating NRBF2 could localize at amphisome. Interestingly, we found that RAB7 and LC3 interaction with NRBF2 was increased under starvation-induced conditions (Figure. S2J). In order to confirm this result, we transfected mCherry-NRBF2 and RAB7 to RPE19 cell and observed that EBSS treatment increased the colocalization between NBRF2 and RAB7 (Figure. S2K). To confirm that nrbf2 KO impairs autophagy maturation through regulating RAB7 activity, we transiently transfected cells with RAB7Q67L (constitutively active form), RAB7T22N (constitutively inactive form) and RAB7 WT plasmid to the nrbf2-/- cells, and we found that only RAB7Q67L and RAB7 WT overexpression significantly decreased autophagy substrate SQSTM1 levels in nrbf2-/- cells, but RAB7T22N does not affect the SQSTM1 levels (Figure 4K and L). Moreover, we isolated autophagosomes from WT and nrbf2-/- mice liver (Figure. S3A and S3B) and brain (Figure. S3C and S3D) to perform the GST-R7BD affinity-isolation assay. The results indicate that active RAB7 is reduced on autophagosome in nrbf2-/- tissues. We isolated late endosomes, autophagosome and lysosome by density gradient centrifugation from C57 mouse liver and found that NRBF2 mainly distributed in autophagosomes fractions and also exist in late endosomes and lysosomes fractions (Figure. S3E).
Figure 4.
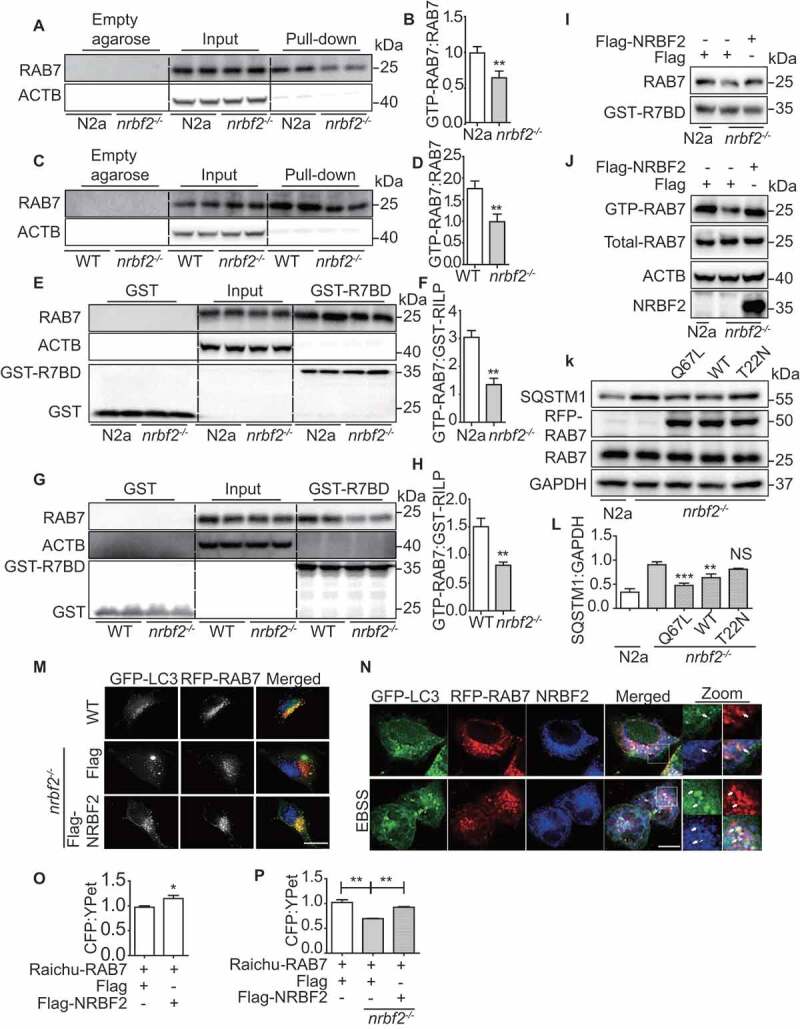
NRBF2 is required for RAB7 activation to promote autophagosome maturation. (A and B) GTP-RAB7 in WT and nrbf2-/- N2a cells was determined by GTP-beads affinity-isolation assay. The “empty agarose” indicated affinity isolation with empty agarose beads, the “pull-down” indicated affinity isolation with GTP-beads. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, vs. the relative control. (C and D) GTP-RAB7 in the WT and nrbf2-/- mice brain was determined by GTP-beads affinity-isolation assay. The “empty agarose” indicated affinity isolation with empty agarose beads, and the “pull-down” indicated affinity isolation with GTP-beads. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, vs. the relative control. (E and F) GST-R7BD affinity-isolation assay indicate that active RAB7 is reduced in nrbf2-/- cell. These findings are consistent with GTP-beads affinity-isolation assay. The “GST” indicated affinity isolation with GST only beads, and the “GST-R7BD” indicated affinity isolation with GST-R7BD beads. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, vs. the relative control. (G and H) GST-R7BD affinity-isolation assay indicates that active RAB7 is reduced in nrbf2-/-mouse. The GST means affinity isolation with GST-only beads, GST-R7BD means affinity isolation with GST-R7BD beads. These findings are consistent with GTP-beads affinity-isolation assay. (I and J) Rescue of decreased GTP-RAB7 in nrbf2-/- cells by overexpressing Flag-NRBF2. (K and L) Transiently overexpression of RAB7Q67L and RAB7 WT but not RAB7T22N promotes the degradation of SQSTM1 in nrbf2-/- cells. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, vs. the relative control. (M) Colocalization of GFP-LC3 and RFP-RAB7 in WT and nrbf2-/- N2a cells as seen under confocal microscope. Scale bar: 5 μm. (N) Colocalization of GFP-LC3, RFP-RAB7 and NRBF2 in N2a cells as seen under confocal microscope. Scale bar: 2.5 μm. (O) Transient overexpression of NRBF2 increases RAB7 activity as reflected by an assay using the RAB7 FRET sensor, Raichu-RAB7. The CFP/YFP ratio was obtained from microplate reader (Molecular devices FlexStation 3). (P) nrbf2 KO decreases RAB7 activity as reflected by an assay using the RAB7 FRET sensor, Raichu-RAB7. The CFP/YFP ratio was obtained from microplate reader (Molecular devices FlexStation 3). Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, vs. the relative control
Raichu-RAB7, a RAB7 FRET sensor, comprises YPet-GL, the RAB7-binding domain of Rabring7, super-enhanced CFP and RAB7. RAB7 GTPase activities can be detected by this sensor and are measured by the ratio between 525 nm and 475 nm emission [47]. In its GDP-bound inactive form, CFP and YPet within the probe are located remotely from each other. Upon GEF activation, GDP on the RAB7 is exchanged for GTP to induce the association of active GTP-bound RAB7 with the RAB7-binding domain of Rabring7. This intramolecular binding brings CFP in close proximity to YPet and, thereby causes FRET. Using confocal FRET imaging, we detected changes in fluorescence in live cell. From the results, we can see that in WT N2a cells the GDP-RAB7 can be converted to GTP-RAB7, while nrbf2 KO impaired the GDP-RAB7 conversion to GTP-RAB7 (Figure. S3F). We also used the microplate reader to measure the change of FRET signals. The result showed that NRBF2 overexpression increased RAB7 activity (Figure 4O), while nrbf2 KO decreased RAB7 activity (Figure 4P). The reliability of Raichu-RAB7 system was confirmed by adding the positive control (overexpression of CCZ1-MON1A, the GEF for RAB7) and negative control (CID1067700, inhibitor of RAB7 GTPase) (Figure. S3G and S3H). The Raichu-RAB7-NC (the RAB7-binding domain of Rabring7 was replaced with the RAB11-binding domain of RAB3 LIL1/GRAB) was used to confirm that NRBF2 do not affect the CFP/YPet ratio of Raichu-RAB7-NC (Figure. S3I and S3J). Collectively, these data revealed the important role of NRBF2 in modulating RAB7 activity for autophagosome maturation.
NRBF2 interacts and colocalizes with CCZ1-MON1A
To understand how NRBF2 regulates the RAB7 GTPase activity, we determined the interaction of NRBF2 with CCZ1-MON1A, a well-characterized guanine nucleotide exchange factor (GEF) for RAB7 [24]. As showed in (Figure 5A), transiently expressed Flag-NRBF2 was co-immunoprecipitated with GFP-CCZ1 and GFP-MON1A. Interestingly, we further demonstrated that endogenous NRBF2 interacts with endogenous CCZ1 and MON1A, and endogenous CCZ1 is also co-immunoprecipitated with endogenous NRBF2 (Figure 5B-D). Additionally, immunostaining results showed that transiently expressed HA-NRBF2 colocalizes with GFP-CCZ1 or GFP-MON1A in HeLa cells (Figure 5E and F). These results strongly support the conclusion that NRBF2 interacts with CCZ1-MON1A. To further confirm the direct interaction of NRBF2 with CCZ1-MON1A, we performed affinity-isolation assays using recombinant glutathione S-transferase (GST)-NRBF2 or GST immobilized on agarose beads, with recombinant MON1A and CCZ1 protein. Our results showed that GST-NRBF2 can pull down recombinant MON1A (Figure 5G) but not CCZ1 (data not shown), indicating that NRBF2 may directly interact with MON1A. We further found that endogenous CCZ1 is effectively co-immunoprecipitated with NRBF2-GFP or NRBF2 mutant without CCD domain (Figure 5H). Similarly, transiently expressed Flag-MON1A in nrbf2-/- cells was co-immunoprecipitated with NRBF2-GFP or NRBF2 mutant without CCD domain, but not NRBF2 mutant without MIT domain (Figure 5I), indicating that NRBF2 MIT domain is essential for its interaction with CCZ1-MON1A complex and for modulating RAB7 activity. More interestingly, we found that CCZ1-MON1A interaction with NRBF2 was increased in a starvation-induced autophagy condition (Figure 5J-L). This result showed that NRBF2 could promote the maturation of autophagy by regulating the activity of RAB7 during autophagy induction. Overall, these results indicate that NRBF2 interacts with CCZ1-MON1A, and this interaction is important for regulating RAB7 activation, especially in the autophagy-inducing condition.
Figure 5.
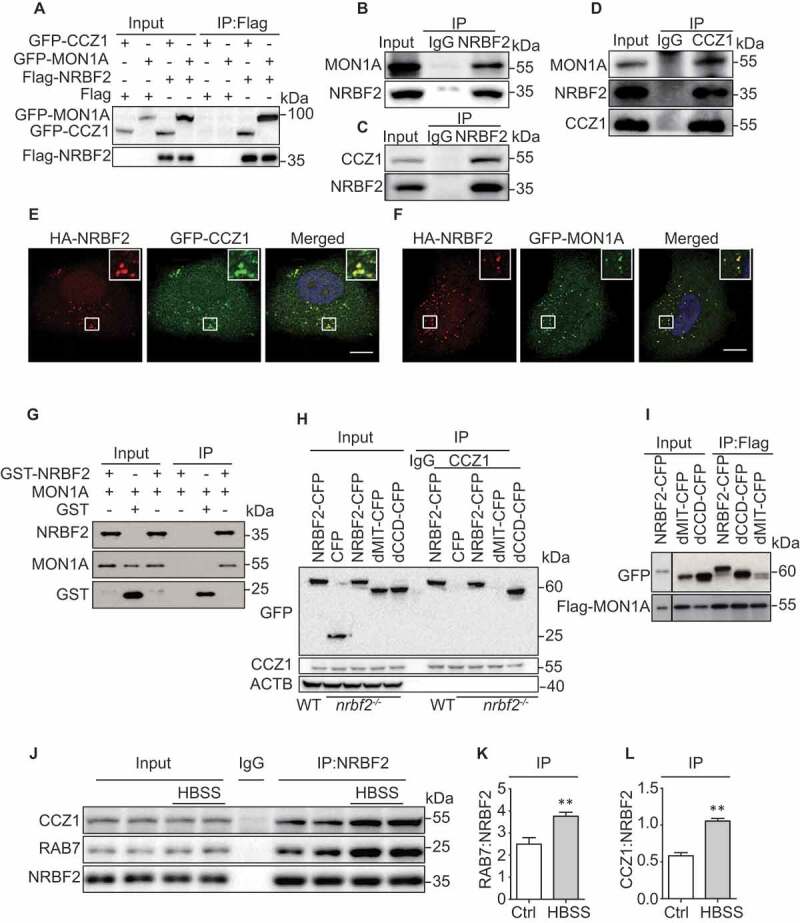
NRBF2 interacts and colocalizes with CCZ1-MON1A. (A) NRBF2 interacts with CCZ1 or MON1A. After N2a cells transiently transfected with Flag or Flag-Nrbf2 with GFP-Ccz1 or GFP-Mon1a, followed by immunoprecipitated (IP) with anti-Flag antibody; eluates were resolved by SDS-PAGE and analyzed by immunoblotting with the corresponding antibodies. (B and C) Endogenous NRBF2 interacts with endogenous CCZ1 and MON1A in N2a cells as reflected by immunoprecipitation. (D) Endogenous CCZ1 interacts with endogenous NRBF2 and MON1A in N2a cells as reflected by immunoprecipitation. (E and F) Colocalization of NRBF2 with CCZ1 or MON1A. After HeLa cells transiently co-expressed NRBF2-HA with GFP-MON1A or GFP-CCZ1, the colocalization was visualized and representative images are shown. Scale bar: 7.5 μm. (G) Recombinant GST-NRBF2 protein directly binding with recombinant MON1A as reflected by GST affinity-isolation assay. (H) Endogenous CCZ1 interacts with NRBF2-CFP and dCCD-CFP. After N2a cells transiently expressed CFP, NRBF2-CFP or NRNF2-CFP mutant either missing CCD or MIT domain plasmids in nrbf2-/- N2a cells, followed by immunoprecipitated (IP) with anti-CCZ1 antibody; eluates were resolved by SDS-PAGE and analyzed by immunoblotting with the corresponding antibodies. (I) Flag-MON1A interacts with NRBF2-CFP and dCCD-CFP in nrbf2-/- N2a cells as reflected by immunoprecipitation. Irrelevant blots were removed. (J-L) The interaction between NRBF2 and CCZ1/RAB7 under normal and starvation conditions is determined by IP with an anti-NRBF2 antibody. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, vs. the relative control
NRBF2 is required for CCZ1-MON1A GEF activity
We performed in vitro GEF activity assay using purified RAB7, CCZ1, MON1A and NRBF2 proteins. For this assay, RAB7 was preloaded with the fluorescent GDP analog mant-GDP. GEF-mediated displacement of mant-GDP from RAB7 results in a strong loss of fluorescence as a result of the changes in the microenvironment of the mant group, indicative of GEF activity [21]. It is somewhat surprising that adding NRBF2 did not increase the CCZ1-MON1A GEF activity (Figure 6A). Margarita Cabrera and her colleagues found that the GEF activity of the CCZ1-MON1A for RAB7 is strongly stimulated on the membranes and is promoted by phosphatidylinositol-3-phosphate (PtdIns3P), which also supports membrane association of GEF complex [24]. We confirmed this result by GEF assay on multilamellar vesicles (MLVs) mimicked by PtdIns3P or PI (Figure 6B). To investigate whether NRBF2 promotes GEF activity via other participators, we purified CCZ1-MON1A complex from WT and nrbf2-/- mouse brain by immunoprecipitation under non-denaturing conditions and performed the in vitro GEF activity assay. Notably, the GEF activity of CCZ1-MON1A purified from nrbf2-/- mice was decreased obviously compared with that purified from the WT mice (Figure 6C). To quantify the protein amount, we used different amounts of purified recombinant CCZ1 protein as quantitative standard and estimated the amount of CCZ1-MON1A complex (around 4 ng in each reaction) in the assay (Figure. S3K and S3L). Similarly, we purified CCZ1-MON1A complex from N2a cells, nrbf2-/- N2a cells and N2a cells treated with SAR405, and the result showed that nrbf2 KO or PIK3C3 inhibitor could decrease CCZ1-MON1A GEF activity significantly (Figure 6D). After nrbf2-/- N2a cells were transfected with full-length NRBF2-CFP, NRBF2 mutants missing either the MIT domain (dMIT-CFP) or the CCD domain (dCCD-CFP), we detected CCZ1-MON1A GEF activity. Our data showed that both full-length NRBF2-CFP and dCCD-CFP mutant, but not dMIT-CFP mutant, are able to rescue nrbf2 KO-mediated CCZ1-MON1A GEF activity decrease (Figure 6E), indicating that MIT domain is essential for promoting CCZ1-MON1A GEF activity. More interestingly, we tested NRBF2-associated GEF activity during autophagy induction and found that NRBF2-associated GEF activity was increased in a starvation-induced autophagy condition (Figure 6F). Overall, these results indicate that NRBF2 is required for CCZ1-MON1A GEF activity to promote RAB7 activation, especially under autophagy inducing condition.
Figure 6.
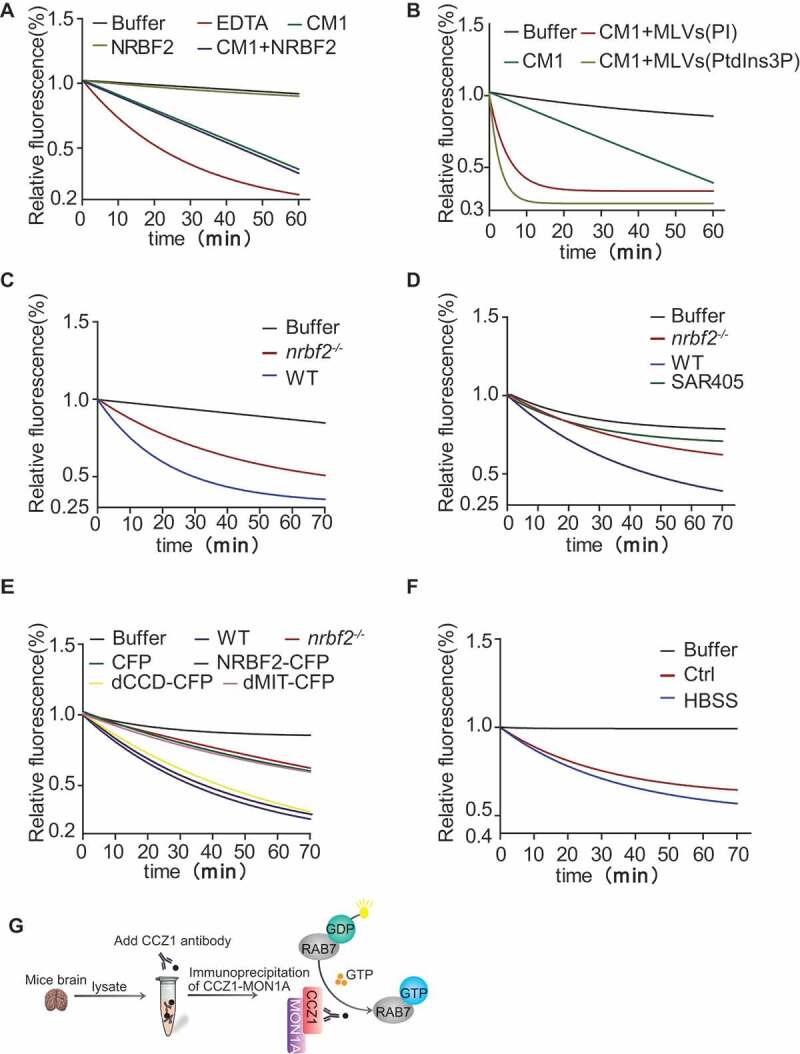
NRBF2 is required for CCZ1-MON1A GEF activity. (A) GEF activity assay was used to determine whether NRBF2 enhances GEF activity of CCZ1-MON1A (CM1). Mant-GDP bound RAB7 were diluted to a concentration of 50 nM in exchange buffer (20 mM HEPES [pH 7.5], 150 mM NaCl, and 0.5 mM MgCl2). The dissociation of mant-GDP from RAB7 was determined by measuring the decrease in fluorescence signal that accompanies the release of mant-GDP in the presence of a large excess of GTP. Nucleotide release reactions were initiated by the addition of GTP (1 mM final concentration) in the reaction buffer containing CCZ1 (500 pmol) and MON1A (500 pmol), with or without adding NRBF2 (500 pmol). Samples are excited at 360 nm, and the emission was monitored at 440 nm. (B) GEF activity of CCZ1-MON1A toward RAB7 is increased in the presence of PtdIns3P (PtdIns3P concentration is 20 pmol). (C) GEF activity of the CCZ1-MON1A purified from nrbf2-/- mice was decreased obviously compared with that from WT group. Immunopurified CCZ1-MON1A protein by CCZ1 antibody from WT or nrbf2-/- mice were used to perform the GEF assay. (D) GEF activity of the CCZ1-MON1A purified from nrbf2-/- and SAR405-treated cells were decreased compared with that purified from WT or untreated group. Immunopurified CCZ1-MON1A protein by CCZ1 antibody from WT or nrbf2-/- cells were used to perform the GEF assay. (E) NRBF2 dCCD domain is important for regulating CCZ1-MON1A GEF activity. nrbf2-/- N2a cells were transiently transfected with CFP, Nrbf2-CFP, dMIT-CFP or dCCD-CFP, Immunopurified CCZ1-MON1A protein by CCZ1 antibody were used to perform the GEF assay. (F) NRBF2-associated GEF activity was increased in a starvation-induced autophagy condition. (G) Schematic model for NRBF2 regulating CCZ1-MON1A GEF activity
NRBF2 is required for CCZ1-MON1A-associated PIK3C3 kinase activity and complex formation
As the PtdIns3K complex has been reported to regulate RAB7 activation and produce PtdIns3P, we firstly examined the physical interaction of PIK3C3, a core component of the PtdIns3K complex, with CCZ1-MON1A [21,23–25]. By using the immunoprecipitation assay, we found that endogenous CCZ1 is co-immunoprecipitated with endogenous PIK3C3 in mouse brains, as well as in N2a cells (Figure 7A and B). As expected, NRBF2 also interacted with RAB7 and MON1A (Figure 7C). Interestingly, the interaction of CCZ1 with PIK3C3 and RAB7 was impaired in nrbf2-/- mouse brains (Figure 7C-E). The impaired CCZ1-MON1A interaction with PIK3C3 can be further supported by the reduced colocalization of CCZ1-MON1A with PtdIns3P probe GFP-FYVE by immunostaining (Figure 7F and G). The GEF activity of the CCZ1-MON1A is strongly stimulated on the membranes, especially in the presence of PtdIns3P. It is likely that PtdIns3P facilitates the RAB7-GEF interaction by optimally positioning the GEF complex relative to the RAB7. We showed that nrbf2 KO reduced colocalization of RAB7 with GFP-2XFYVE (PtdIns3P probe) (Figure. S4A), indicating that NRBF2 may serve as a “bridge” to connect the PtdIns3K complex and CCZ1-MON1A for subsequent RAB7 activation.
Figure 7.
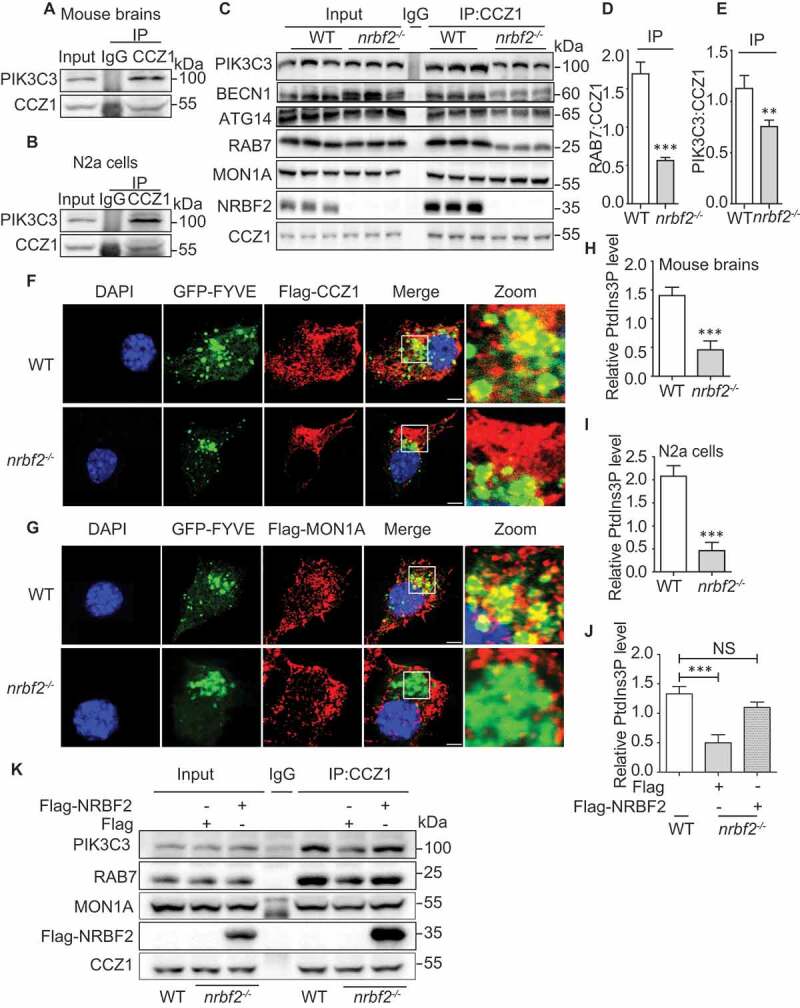
NRBF2 is required for CCZ1-MON1A-associated PIK3C3 kinase activity and complex formation. (A) Endogenous CCZ1 interacts with endogenous PIK3C3 in the mouse brain as reflected by immunoprecipitation. (B) Endogenous CCZ1 interacts with endogenous PIK3C3 in N2a cells as reflected by immunoprecipitation. (C-E) CCZ1 interacts with PIK3C3, and nrbf2 KO attenuates the interaction of CCZ1 with PIK3C3 in mouse brains. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. the relative control. (F and G) nrbf2 KO attenuates the colocalization of CCZ1-MON1A with GFP-2xFYVE, a chimeric protein that specifically binds PtdIns3P. After N2a cells were transiently transfected with GFP-2xFYVE plus FLAG-CCZ1 or FLAG-MON1A plasmids, the colocalization was visualized and representative images were shown. Scale bar: 2.5 μm. (H) Quantification of CCZ1-IPed PIK3C3 kinase activity in WT and nrbf2-/- mice brain. (I and J) Quantification of CCZ1-IPed PIK3C3 kinase activity in WT and nrbf2-/- N2a cells, and the rescue of impaired CCZ1-associated PIK3C3 kinase activity by overexpressing Flag-NRBF. (K) Rescue of impaired interaction between CCZ1 and PIK3C3, RAB7 in nrbf2-/- cells by overexpressing Flag-NRBF2
Because PIK3C3 is mainly responsible for producing PtdIns3P, and nrbf2 KO affects the interaction of CCZ1 with PIK3C3, we wondered whether CCZ1-associated PIK3C3 kinase activity is reduced in nrbf2-/- cells. We used PtdIns3P ELISA kit to test CCZ1-linked PIK3C3 kinase activity. From the result, we can see CCZ1-linked PIK3C3 kinase activity was markedly decreased in nrbf2-/- mice and nrbf2-/- cells (Figure 7H and I). Overexpression of NRBF2 could rescue decreased CCZ1-linked PIK3C3 kinase activity in nrbf2-/-cells (Figure 7J). Finally, overexpression of NRBF2 could rescue impaired interaction of CCZ1 with PIK3C3 and RAB7 in nrbf2-/- cells (Figure 7K).
NRBF2 regulates APP-CTF degradation by recruiting CCZ1-MON1A-RAB7 module to APP-containing vesicles
Our previous study confirmed that NRBF2 expression levels are reduced in the hippocampus of 5XFAD mice and that NRBF2 interacts with APP in vivo [37]. NRBF2 overexpression promotes degradation of APP-CTF and Aβ in N2S cells via lysosomal pathway [37]. To further confirm the finding, we overexpressed or depleted nrbf2 in primary cortical neurons isolated from 3xTg AD transgenic mice. We found that overexpression of GFP-NRBF2 but not GFP decreases APP-CTFs and Aβ levels (Figure 8A-F), while depletion of NRBF2 increases APP-CTFs and Aβ levels (Figure 8G-M). We also showed that NRBF2 colocalizes with the BACE (beta-secretase) cleaved APP-CTF (CTFβ) in autophagosomes, and CQ treatment led to CTFβ accumulation in autophagosomes (Figure 8N).
Figure 8.
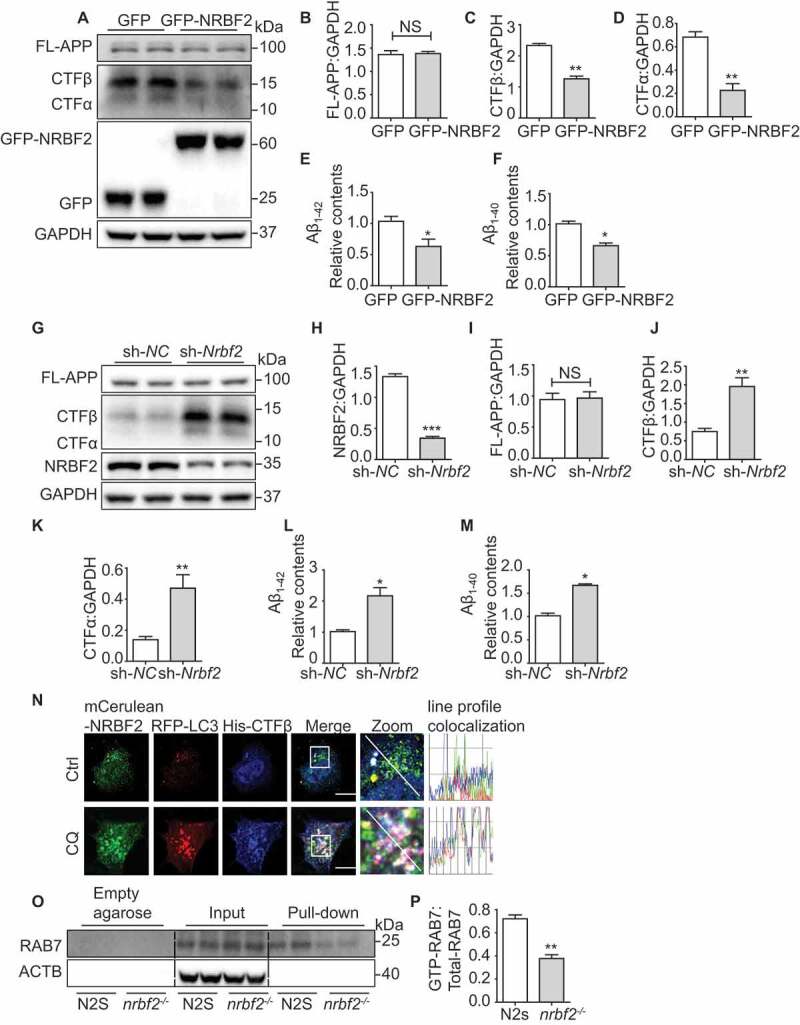
NRBF2 regulates APP-CTF degradation via CCZ1-MON1A-mediated RAB7 activation. (A-D) NRBF2 overexpression reduces APP-CTFs and intracellular Aβ1-40, Aβ1-42 levels in primary neuron culture. Primary cortical neurons isolated from 3xTg AD mouse were transfected with Recombinant lentivirus packed GFP-NRBF2 or GFP, the expression of NRBF2, FL-APP, APP-CTFs were examined by immunoblotting. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. the relative control. (E and F) ELISA results demonstrated that NRBF2 overexpression significantly reduced intracellular Aβ. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. the relative control. (G-K) Silencing Nrbf2 increased APP-CTFs and intracellular Aβ. Primary cortical neurons were transfected with lentiviral Nrbf2 shRNA or nontargeting shRNA. After knocking down (KD) Nrbf2, the level of FL-APP, APP-CTFs were detected by immunoblotting. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. the relative control. (L and M) ELISA results demonstrated that Nrbf2 KD significantly increased intracellular Aβ level. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. the relative control. (N) N2a cells were transfected with mCerulean-Nrbf2, RFP-Lc3 and CTFβ-His under basal or CQ-treated conditions. CTFβ-His was stained with anti-His antibody and the colocalization of mCerulean-NRBF2, RFP-LC3 and CTFβ-His were visualized under confocal microscope. Scale bar: 7.5 μm. (O and P) GTP-RAB7 in N2 S and nrbf2-/- N2 S cells were determined by GTP-beads affinity-isolation assay, we used empty agarose beads as equivalent control. The empty agarose beads cannot pull down RAB7. The “empty agarose” indicated affinity isolation with empty agarose beads, and the “pull-down” indicated affinity isolation with GTP-beads. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. the relative control
Similar to WT N2a cells, depletion of nrbf2 significantly reduced GTP-form RAB7 in a Swedish mutant APP overexpression N2a cells (N2S) (Figure 8O and P). To demonstrate whether RAB7 inactivation is involved in APP-CTFs and Aβ accumulation in nrbf2-/- N2S cells, we overexpressed WT, as well as the constantly inactive (T22N) and active (Q67L) forms of RAB7 in nrbf2-/- N2S cells and observed dramatic reduction of APP-CTFs and Aβ in WT and RAB7Q67L overexpression group, but not in the RAB7T22N overexpression group (Figure 9A-F). Next, we treated RAB7Q67L overexpression cells with bafilomycin A1 to inhibit lysosomal degradation and found that the RAB7Q76L overexpression-mediated reduction of APP-CTFs was attenuated by bafilomycin A1 (Figure. S4B-E)
Figure 9.
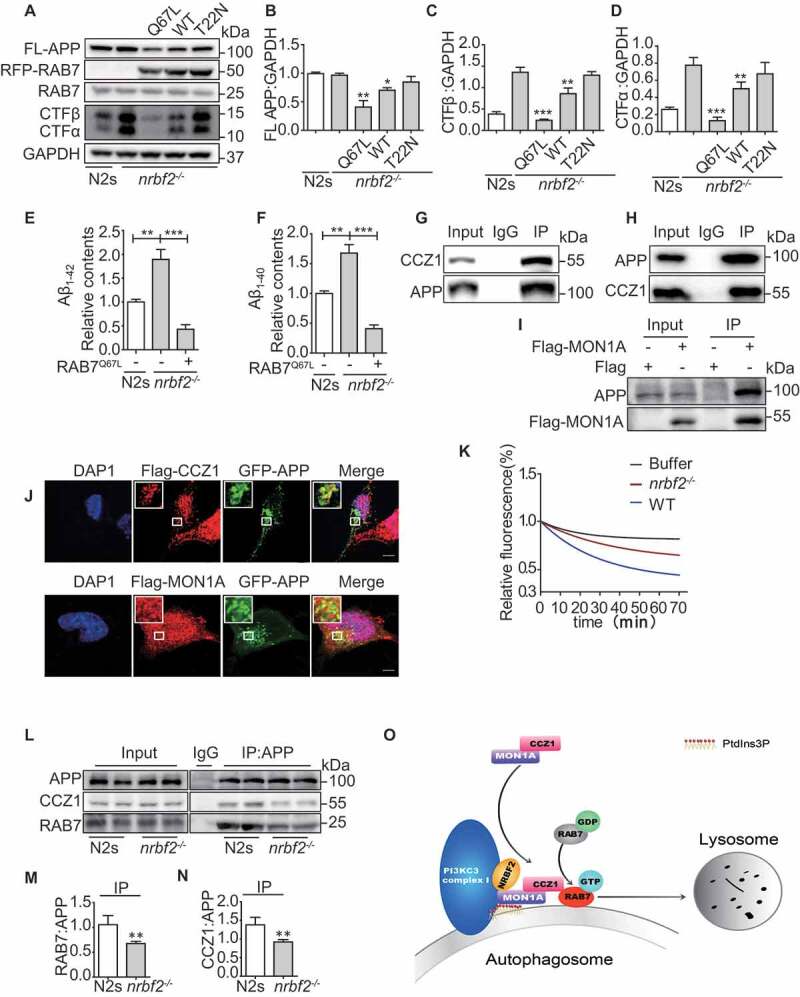
NRBF2 regulates APP-CTF degradation via CCZ1-MON1A-mediated RAB7 activation. (A-D) Overexpression of WT, RAB7Q67L and RAB7T22N in nrbf2-/- N2S cells and measurement of FL-APP and APP-CTFs by WB. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. the relative control. (E and F) ELISA results demonstrated that active RAB7 overexpression significantly reduced intracellular Aβ. Data are quantified as mean ± SEM (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 vs. the relative control. (G) Endogenous APP interacts with CCZ1-MON1A. N2S cells were lysed and immunoprecipitated (IP) with APP antibody; eluates were resolved by SDS-PAGE and analyzed by immunoblotting with the CCZ1 antibodies. (H) CCZ1 interacts with APP in N2S cells. N2S cells were lysed and immunoprecipitated (IP) with CCZ1 antibody; eluates were resolved by SDS-PAGE and analyzed by immunoblotting with the APP antibodies. (I) Flag-MON1A interacts with APP in N2S cells. FLAG-MON1A were transfected into N2S, then N2S cells were lysed and immunoprecipitated (IP) with Flag antibody; eluates were resolved by SDS-PAGE and analyzed by immunoblotting with the APP antibodies. (J) FLAG-CCZ1 and FLAG-MON1A were transfected into N2a cells together with GFP-APP, the colocalization of Flag-CCZ1 or GFP-APP and Flag-MON1A or GFP-APP were visualized under confocal microscope. Scale bar: 5 μm. (K) APP-associated GEF activity was significantly reduced in nrbf2-/- N2S cells. (L-N) Interaction between APP and CCZ1 or RAB7 was determined by IP in WT and nrbf2-/- N2S cells. nrbf2 KO impaired interaction between CCZ1 and APP, RAB7. (O) Schematic model for the NRBF2 function in regulating autophagosome maturation. NRBF2 may serve as an enhancer for PtdIns3K complex I and MON1A-CCZ1 complex association to activate the GEF activity and subsequent RAB7 activation on the autophagosome, so as to promote the autophagosome maturation
Our previous research found that nrbf2 KO resulted in the accumulation of APP and APP-CTFs in RAB5-positive early endosomes and impaired the distribution of APP and APP-CTFs to the LC3- and LAMP1-positive autophagosome/lysosomes. Therefore, the accumulation of APP, APP-CTFs in nrbf2-/- N2s cells deposited in the form of vesicles, we called APP-containing vesicles [37]. Interestingly, we found that APP interacts and colocalizes with CCZ1 and MON1A in N2S cells (Figure 9G-J), and that APP-associated GEF activity was significantly reduced in nrbf2-/- N2S cells (Figure 9K). Finally, we showed that the interaction between APP and CCZ1 or RAB7 was significantly reduced in nrbf2-/- N2s cells (Figure 9L-N), indicating that NRBF2 plays a specific role in regulating APP degradation by recruiting CCZ1-MON1A-RAB7 to APP-containing vesicles. The data reveal a previously unknown role for NRBF2 in regulating CCZ1-MON1A GEF activity on APP containing vesicles to activate RAB7 for lysosome degradation.
Discussion
The data presented in this study reveals a previously uncharacterized role of NRBF2, a component of the PtdIns3K complex, in regulating autophagy-lysosome fusion. Mechanistic studies indicate that NRBF2 is required for autophagosome trafficking toward the lysosome via regulating RAB7 activity by interacting with GEF CCZ1-MON1A and facilitating GEF activity. Interestingly, this mechanism is associated with APP-CTFs degradation and Aβ production. Loss of NRBF2 impaired the interaction between APP and CCZ1-MON1A to reduce APP-associated GFE activity required for APP-containing vesicle fusion with lysosome. Collectively, our results indicate that NRBF2 is important for regulating autophagosome-lysosome fusion and APP-CTF degradation, providing insight into the function of NRBF2-PtdIns3K complex in autophagy regulation as well as in the pathogenesis of AD.
We and others have recently identified NRBF2 as a novel component of PtdIns3K complex [10–12,16]. Though we and others found that NRBF2 is a positive regulator of autophagosome biogenesis, which is consistent with the function of Atg38, the yeast homolog of NRBF2 [16], there was also a controversial report [12]. Nevertheless, previous results have indicated that NRBF2 plays a major role in regulating autophagosome biogenesis. Here, our results showed that Nrbf2 depletion increases both LC3-II and SQSTM1 levels in neuronal cells and mouse brains. Tf-LC3 staining, and LC3, LAMP1 colocalization, all support the conclusion that nrbf2 KO impairs autophagosome maturation. Initially, ATG14-containing PtdIns3K complex is believed to specifically regulate autophagosome biogenesis, while UVRAG-associated PtdIns3K complex 2 controls autophagosome-lysosome fusion. Our findings about NRBF2, together with recent findings that ATG14 regulates autophagosome-lysosome fusion [10,48], reveal the diverse and complex involvement of the PtdIns3K complex in autophagy regulation, expanding our knowledge of PtdIns3K in autophagy regulation. Future studies for determining how the PtdIns3 K complex, such as ATG14, UVRAG and NRBF2, coordinates with each other for regulating autophagosome biogenesis and autophagosome-lysosome fusion are required.
NRBF2 is a component of PtdIns3K complex and directly interacts with ATG14. Our previous results indicated that NRBF2 controls autophagosome biogenesis via regulating ATG14-containing PIK3C3 lipid kinase activity [10]. Interestingly, we showed that the NRBF2 MIT domain is important for regulating both autophagosome biogenesis and the autophagosome maturation. However, how the NRBF2 MIT domain regulates both autophagosome biogenesis and autophagosome maturation is not well understood currently.
Small GTPase RAB7 plays a key role in regulating autophagosome-lysosome fusion [41–43]. Several proteins, such as UVRAG, EPG5, HOPS and FYCO1, have been reported to regulate autophagosome-lysosome fusion via modulating RAB7 activity [49]. Here, we found that NRBF2 regulates autophagosome-lysosome fusion via modulating RAB7 activity. Mechanistically, NRBF2 interacts with RAB7 GEF CCZ1-MON1A and is required for GEF activity to activate RAB7. Interestingly, reconstitution experiment showed that NRBF2 does not directly affect GEF activity. It is well-known that both PtdIns3K and RAB7 play important roles in regulating autophagosome maturation [1–3,8]. However, how PtdIns3K and RAB7 coordinate with each other to regulate autophagosome-lysosome fusion is still largely unclear. The fact that PtdIns3K is mainly responsible for the production of PtdIns3P and previous findings suggesting that CCZ1-MON1A binds with PtdIns3P for RAB7 activation, indicate that there may be a connection between PtdIns3K and RAB7 activation. Here, we showed that CCZ1 interacts with PIK3C3, and nrbf2 KO attenuates the interaction of CCZ1 with PIK3C3, implying that NRBF2 serves as a “bridge” for linking PtdIns3K with the CCZ1-MON1A-RAB7 module. In accordance with this notion, we found that loss of NRBF2 inhibited CCZ1-associated PIK3C3 kinase activity and reduced the colocalization of CCZ1-MON1A with PtdIns3P. In contrast, the in vitro reconstitution experiment showed that addition of PtdIns3P greatly enhanced CCZ1-MON1A GEF activity. Thus, we propose that NRBF2 is required for CCZ1-MON1A interaction with PtdIns3K so that CCZ1-MON1A can efficiently bind to and be activated by the PtdIns3P generated by PtdIns3K for RAB7 activation and subsequent autophagosome maturation. Overall, our results provide important information for understanding how PtdIns3K coordinates with RAB7 for autophagy regulation. However, this point does not exclude other mechanisms by which CCZ1-MON1A are recruited to the autophagosome, for example, via interacting with LC3 [30].
PIK3C3 component UVRAG also regulates autophagosome-lysosome fusion via competing with RUBCN to interact with the HOPS complex for RAB7 activation [32]. Though both NRBF2 and UVRAG modulate RAB7 activity, these two molecules use different mechanisms. Interestingly, another PtdIns3K component ATG14 promotes autophagosome-lysosome fusion via another unique mechanism involving SNARE proteins [48]. These results suggest that PtdIns3K controls autophagosome-lysosome fusion via different and multiple mechanisms. However, how cells dynamically regulate this process by using different components of PtdIns3K complex needs to be further explored.
The PtdIns3K complex has been implicated in the pathogenesis of a variety of diseases, including AD. For instance, brain tissues from AD patients and AD mouse models are selectively deficient in PtdIns3P, the product of PtdIns3K [50]. BECN1, the major component of the PtdIns3K complex, is decreased in brain tissues from AD patients, and its deficiency leads to amyloid plaque formation in AD mice models [51–53]. We recently showed that NRBF2 expression is decreased in the brains of AD mice, and NRBF2 is required for the degradation of APP-C terminal fragments (APP-CTFs) for Aβ homeostasis in an autophagy-dependent manner [37]. However, the detailed mechanism is not fully understood. In this study, we showed that APP interacts with CCZ1-MON1A, and that NRBF2 is required for this interaction as well as for APP-associated CCZ1-MON1A GEF activity. The accumulation of APP-CTFs in nrbf2-/- N2S cells can be alleviated by over-expressing active and WT RAB7 but not inactive RAB7. The data reveal a previously unknown role of NRBF2 in regulating CCZ1-MON1A GEF activity on APP-containing vesicles to activate RAB7 for lysosome fusion. Disruption of this process may lead to the accumulation of APP-CTFs and Aβ.
Overall, our finding of NRBF2 in regulating autophagosome maturation and APP processing significantly expands current knowledge of PtdIns3K in regulating autophagy and AD development. Given the importance of autophagy and the PtdIns3K complex in a variety of human disease, this study will provide critical information for the evaluation of targeting NRBF2-PtdIns3K as a therapeutic strategy for the treatment of autophagy-related diseases such as AD.
Materials and methods
Reagents
Antibody information: for western blot and IP, anti-ACTB/β-actin (sc-47,778), anti-CCZ1 antibody (sc-514,290), anti-CTSB/cathepsin B (sc-365,558) and normal mouse IgG (sc-2025) were purchased from Santa Cruz Biotechnology; anti-Flag antibody (14,794), anti-GFP (2956), anti-GST antibody (2622), anti-NRBF2 (8633), anti-His (12,698), anti-ubiquitin (P4D1) antibody (3936), anti-RAB7 antibody (9367), anti-rabbit IgG (7074), anti-Mouse IgG (7076) and normal rabbit IgG (2729) were purchased from Cell Signaling Technology. Anti-LC3B (NB100-2220) antibody, anti-LAMP1 antibody (6E2; NBP2-25,155), anti-MON1A (NBP1-52,007) were purchased from Novus Biologicals; anti-NRBF2 antibody (A301-851A) was purchased from Bethyl Laboratories; anti-hPIK3C3/VPS34 antibody (Z-R016 and Z-R015) was purchased from Echelon Bioscience; anti-SQSTM1/p62 (P0067) was purchased from Sigma-Aldrich. For immunofluorescence, Alexa Fluor® 488 goat anti-rabbit/mouse IgG (A-11,008, A-11,001), Alexa Fluor® 555 goat anti-rabbit/mouse IgG (A-21,429, A-21,424) and Alexa Fluor® 647 goat anti-rabbit/mouse IgG (A-21,244, A-21,235) were purchased from Thermo Fisher Scientific. For ELISA, anti-Aβ1-16 monoclonal antibody (6E10, 803,001) and anti-beta amyloid polyclonal antibody (CT695, 51–2700) were purchased from Thermo Fisher; anti-beta-amyloid 1–42 antibody (AB5078P) was purchased from Millipore; anti-beta-amyloid 1–40 antibody (0060-100/bA4(40)-5C3) was purchased from Nanotools. DMEM (11,965–126), LysoTracker red DND-99 (L7528), dynabeads® protein G for immunoprecipitation (10003D), fetal bovine serum (10,270–106), opti-MEM (31,985–070), G418 (10,131–035), lipofectamine™ 3000 Transfection Reagent (L3000008), mant-GDP (M12414) and GTP (18,332,015) were purchased from Thermo Fisher Scientific. GST-NRBF2 protein (BC001345), 6XHis-MON1A (BC047022), and 6XHis-CCZ1 (BC132749) were obtained from Proteintech. Recombinant RAB7 protein was purchased from Abcam (ab103507). Chloroquine (C6628) was purchased from Sigma-Aldrich
Cell culture
Mouse neuroblastoma N2a cells (ATCC, CCL-131) and HeLa (ATCC, CCL-2) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM). All the mediums were supplemented with 10% FBS, penicillin and streptomycin (Thermo Fisher Scientific, 15,140,163). N2S maintained in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher Scientific, 11,965–126) 10% FBS, penicillin and streptomycin and 200 μg/mL G418. For starvation, after cells washed with phosphate-buffered saline (PBS; Thermo Fisher Scientific, 70,011,044) twice; EBSS/HBSS medium (Earle’s Balanced Salt Solution/Hanks balanced salt solution; Thermo Fisher SCIENTIFIC, 24,010,043, 14,025,092) was added, and incubated for 4 h. Cells were incubated at 37°C in a humid 5% CO2:95% air environment.
Stable cell lines construction
nrbf2-/- N2a cells were established by using the lentivirus-mediated CRISPR/Cas9 system. Briefly, the nrbf2 sgRNA/Cas9 plasmid, psPAX2 (Addgene, 12,260; deposited by Didier Trono) and pCMV-VSV-G (Addgene, 8454; deposited by Bob Weinberg) were co-transfected into HEK293T cells for 72 h to generate virus particles. Virus-containing medium was collected and filtered through a 0.45 μm filter (Pall, KA2DBLP6 G), followed by infected to N2a cell. After selection by puromycin and isolation of single-cell clones, KO of nrbf2 in the expanded colonies was confirmed by immunoblotting. nrbf2-/- N2S cells were used as described previously [37].
Cell transfection
Cells were transfected with plasmids using Lipofectamine® 3000 Transfection Reagent (L300008) from ThermoFisher according to the manufacturer’s instructions. 24–72 h after transfection, cell was used for analyzes.
Mice feeding
nrbf2-/- mice and 3xTG AD mice were housed in individually ventilated cages on standardized rodent bedding. The room temperature was kept at approximately 22°C and the relative humidity between 40 and 70%. Mice were housed under constant light-cycle (12 h light/dark). All experiments were carried out in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Committee on the Ethics of Animal Experiments of the University of Macau.
Primary cortical neuronal culture
Cortical neurons were obtained from 3xTg AD mice. On embryonic day 15, cerebral cortices were dissected and digested with trypsin. Single neuron suspension was generated by pipetting and filtering through 40-µm nylon strainer (Sigma, CLS431750). The neurons were cultured in neurobasal medium (Gibco, Carlsbad, CA, USA) supplemented with B27, 1% L-glutamine (Thermo Fisher Scientific, 25,030,081), and 1% penicillin/streptomycin. The medium was changed every other day.
Cell imaging
Cells were cultured on coverslips using 24-well plates followed by transient transfection with indicated plasmids for 24–72 h. Cells were fixed in 4% formaldehyde for 10 min at room temperature and permeabilized in 0.2% Triton X-100 (used APP and CCZ1/MON1A colocalization staining permeabilized) (Sigma, T8787) or and 50 µg/ml digitonin (used in permeabilization for all immunostaining experiments except APP and CCZ1/MON1A colocalization staining; Sigma, D141). Cells were blocked with 3% BSA (Sigma, B2046) to prevent antibodies from adsorbing to nonspecific sites, followed by staining with appropriate primary antibodies overnight at 4°C. Staining with fluorescent secondary antibodies (Alexa Fluor® 488, Alexa Fluor® 555 or Alexa Fluor® 647 secondary antibodies) for 2 h at room temperature. Cell images were acquired using a DeltaVision Deconvolution microscope (GE Healthcare) or confocal microscope.
Immunoblotting and immunoprecipitation
Cell proteins were extracted using ice-cold RIPA buffer (Cell Signaling Technology, 9806) with complete protease inhibitor mixture (Roche Applied Science, 04693124001). Proteins were resolved by gel electrophoresis in 10–15% SDS-polyacrylamide gels and subsequently transferred onto polyvinylidene difluoride (PVDF) membranes (Bio-Rad, 1,704,156). Following blocking with TBS-T (Tris-buffered saline with 0.1% Tween-20 [Sigma, P1379]) buffer containing 5% (w:v) nonfat milk powder (Bio-Rad, 1,706,404) the blots were probed with the corresponding primary antibodies and secondary antibody. Blots were visualized using the Pierce ECL kit (Pierce, 32,106). We used ImageJ to get quantitative results from our western blot bands. It is essential to ensure that the bands are not in the saturation range and the background correction method selected actually makes sense. The specific method is executed according to the description of ImageJ (National Institutes of Health).
For immunoprecipitation, cells lysates or animal brains were lysed in Nonidet P-40 (NP-40) cell lysis buffer (25 mM Tris, pH 7.6, 100 mM NaCl, 0.5% NP40 [Sigma, 74,385], 1 mM EDTA (Sigma, E9884), 10% glycerol [Affymetrix/USB – J16374] with protease inhibitors). Then immunoprecipitation was performed using the indicated antibodies. Generally, 5 µg of antibody was added to 500–1000 µg of cell lysate and incubated at 4°C overnight. After addition of protein A/G-agarose beads (Thermo Fisher Scientific, 10003D), incubation was continued for 2 h, and then immunocomplexes were washed five times using lysis buffer, resolved by SDS-PAGE, and analyzed by immune blot. The clean-blot IP detection kit (Thermofisher, 21,232) was used to avoid the interference of IP primary antibody. The Clean-Blot IP Reagent and Kit eliminate detection-interference from both heavy-chain (approx. 50 kDa) and light-chain (25 kDa) IgG-fragments of antibodies used for the initial immunoprecipitation assay.
Subcellular fractionation and in vitro fusion assay
Subcellular fractionation was undertaken as described [40]. Briefly, livers of mice were dissected and finely minced. Liver tissue was rinsed with cold PBS and resuspended in 7–8 ml homogenization solution (10 mM HEPES (Thermofisher, 15,630,080), 250 mM sucrose (Sigma, S8501), 1 mM EDTA, pH 7.4, protease inhibitor cocktail). Lysates were centrifuged at 1,000 g for 10 min at 4°C then removed the floating fat, and the supernatant was saved. Resuspended the pellet, gently homogenized for another 3–4 strokes followed by centrifugation. Supernatants were combined and centrifuged at 20,000 g at 4°C for 20 min. Supernatants were saved as the cytosolic fraction. The pellet was resuspended in 2.25 mL homogenization solution and mixed with 3.75 mL 80% (w:v) Histodenz TM (Sigma Aldrich, D2158) stock solution (80% Histodenz TM, 1 mM EDTA, 10 mM HEPES, pH 7.4). A discontinuous Histodenz TM gradient (26%, 24%, 20%, 15%) with protease inhibitor was carefully layered on top of the lysates, followed by centrifuging at 25,000 g at 4°C for 4 h. Autophagosome fraction and lysosome fraction at the interfaces of gradients were collected by syringe. Diluted the fraction to 0.01 μg/μl protein in fusion buffer (10 mM HEPES pH 7.0, 10 mM KCl, 1.5 mM MgCl2, 1 mM DTT, 250 mM sucrose, and a protease inhibitor cocktail). Autophagosome were labeled by an anti-LC3 antibody and CY3-conjugated secondary antibody, while lysosome was labeled by an anti-LAMP1 antibody and Alexa488-conjugated secondary antibody. Autophagosome fusion with lysosome was performed in an energy-regenerating system (3 mM ATP, 2 mM GTP, 2 mM CaCl2, 8 mM phosphocreatine [Sigma, P1937], 0.16 mg/ml creatine phosphokinase [Sigma, C7886], and a protease inhibitor cocktail) at 37°C for 40 min. The reaction was stopped by 8% paraformaldehyde on ice for 15 min. Images were acquired using a DeltaVision Deconvolution microscope (GE Healthcare) or confocal microscope.
GTP-agarose affinity isolation for RAB7 activity assays
For RAB7 activity assay, we measured the amount of GTP form of RAB7 present using GTP agarose beads (Sigma, G9768) following a published protocol described previously, with minor modification [44]. Briefly, cells or animal brains were collected and lysed in a buffer containing 50 mM Tris-HCl pH7.5, 250 mM NaCl, 5 mM MgCl2, 0.5% Triton X-100, and protease inhibitors. Equal amounts of protein lysates were incubated with GTP-agarose beads overnight at 4°C. The GTP-agarose beads were then washed with cell lysis buffer and boiled in SDS-loading buffer before SDS-PAGE for immunoblotting. The amount of RAB7 bound to GTP was measured by blotting with a RAB7-specific antibody and appropriate secondary antibody. Total RAB7 was measured by immunoblotting and detected by a RAB7 antibody.
GST-R7BD affinity isolation
Purification of GST-R7BD (GST-RILP) from bacterial [54]. GST-R7BD was transformed into Escherichia coli strain BL21(Takara, 9126). The 300 ml 2XYT culture was incubated for additional 12 h at 30°C, Spin down the bacterial at 5000 g at 4°C for 10 min, washed with cold PBS, resuspended in 8 ml of cold lysis buffer (50 mM Tris HCl PH7.5, 1 mm EGTA, 1% TritonX 100, 0.27 M sucrose, 0.1% beta-ME (Sigma, M6250) 1:4000 DNAase I (Thermofisher, EN0521) with protease inhibitors). Protein was purified by 500 μl of glutathione-sepharose 4B beads (GE Healthcare GE17-0756-01). Mammalian cells lysed in the lysis (pull down) buffer (20 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 1% Triton X-100, protease inhibitors). Each affinity isolation was performed in 1 ml with 500 µg of cell lysate and 100 µl of beads pre-equilibrated in affinity-isolation buffer. Beads were rocked overnight at 4°C, washed twice with cold affinity-isolation buffer, and bound proteins were eluted by adding 1× sample buffer incubating at 95°C for 8 min. The amount of RAB7 bound to GTP was measured by blotting with a RAB7 specific antibody and appropriate secondary antibody.
In vitro affinity-isolation assay
Purified proteins were used to detect interactions of NRBF2 with CCZ1-MON1A proteins. GST or GST-tagged NRBF2 protein (2 μg; Proteintech, Ag21403) was incubated with GST beads in 500 μl PBS buffer containing 0.5% NP-40 for 2 h min at 4°C. Next, His-CCZ1 or His-MON1A recombinant proteins were added; then, the supernatants were removed, and the bound proteins were boiled in 1× SDS sample loading buffer, then separated by SDS-PAGE and finally visualized by immunoblotting.
ELISA assay of Aβ1-40 and Aβ1-42
The cells, culture medium and brain tissue homogenates, which were lysed in lysis buffer. One day before the experiment, coat the ELISA plates (Nunc) with the 6E10 antibody and incubated overnight at 4°C. Wash plates with PBST (0.05% Tween 20) and block with Block Ace (4 times diluted from the stock in PBS) for 2 h at room temperature. Wash plates with PBST for two times. Dispense conditioned media (sample) or standard samples and incubate at 37°C for 2 h. Discard the samples and wash plates with PBST three times. Added secondary antibody incubated for 2 h at 37°C. Wash the plates thoroughly for 4 times with PBST. Dilute streptavidin-conjugated horseradish peroxidase to 1:4000 in assay diluant and dispense 100 μl/well. Incubate for 1 h at 37°C. Wash plates for 4 times with PBST and removed all traces of liquid. Dilute buffer A and B in 1:1 ratio and add 100 μl/well and incubate for 30 min in the dark. Stop the reaction by addition of 2 M H2 SO4 (Millipore, 100,731) (100 μl/well) for 15–30 min or after seeing the desired signal. Read the absorbance values at 450 nm.
Statistical analysis
Each experiment was performed at least 3 times, and the results are presented as mean ± SD. One-way analysis of variance (ANOVA) was followed by the Student-Newman-Keuls test using the Sigma Plot 11.0 software package. A probability value of P < 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Mitsunori Fukuda at Tohoku University for the MON1A-GFP and CCZ1-GFP plasmids, Dr. Takeshi Nakamura at Tokyo University of Science for the Raichu-RAB7 plasmid and Dr. Qing Jun Wang at University of Kentucky for the mCerulean-NRBF2 and mCherry-NRBF2 plasmid. The authors would like to thank Dr. Carol Chu for her kind technique support and Dr. Martha Dahlen for her English editing of this manuscript.
Funding Statement
This work was supported by the Ministry of Science and Technology of the People’s Republic of China [MoST-2017YFE0120100]; National Natural Science Foundation of China [31871024, 8103487, 81773926]; Science and Technology Development Fund, Macau SAR [024/2017/AMJ, 022/2015/A1]; University of Macau [MYRG2019-00129-ICMS]; Health and Medical Research Fund [17182541, 17182551]; Shenzhen Science and Technology Innovation Commission [JCYJ20180302174028790, JCYJ20180507184656626]; GRF [HKBU12100618]; Hong Kong Baptist University [RC-IRCs/17-18/03].
Disclosure statement
The authors declare no competing interests.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Bento CF, Renna M, Ghislat G, et al. Mammalian autophagy: how does it work? Annu Rev Biochem. 2016;85(1):685–713. [DOI] [PubMed] [Google Scholar]
- [2].Hurley JH, Young LN.. Mechanisms of autophagy initiation. Annu Rev Biochem. 2017;225–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ktistakis NT, Tooze SA. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016;26:624–635. [DOI] [PubMed] [Google Scholar]
- [4].Nakamura S, Yoshimori T. New insights into autophagosome–lysosome fusion. J Cell Sci. 2017;130:1209–1216. [DOI] [PubMed] [Google Scholar]
- [5].Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759–774. [DOI] [PubMed] [Google Scholar]
- [6].Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33:109–122. [DOI] [PubMed] [Google Scholar]
- [7].Pankiv S, Alemu EA, Brech A, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188:253–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wirth M, Joachim J, Tooze SA. Autophagosome formation—the role of ULK1 and Beclin1–PI3KC3 complexes in setting the stage. Sem Cancer Biol. 2013. [DOI] [PubMed] [Google Scholar]
- [9].Levine B, Liu R, Dong X, et al. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol. 2015;25:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lu J, He L, Behrends C, et al. NRBF2 regulates autophagy and prevents liver injury by modulating Atg14L-linked phosphatidylinositol-3 kinase III activity. Nat Commun. 2014;5(1):3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cao Y, Wang Y, Saab WFA, et al. NRBF2 regulates macroautophagy as a component of Vps34 Complex I. Biochem J. 2014;461(2):315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zhong Y, Morris DH, Jin L, et al. Nrbf2 protein suppresses autophagy by modulating Atg14L protein-containing Beclin 1-Vps34 complex architecture and reducing intracellular phosphatidylinositol-3 phosphate levels. J Biol Chem. 2014;289(38):26021–26037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ohashi Y, Soler N, García Ortegón M, et al. Characterization of Atg38 and NRBF2, a fifth subunit of the autophagic Vps34/PIK3C3 complex. Autophagy. 2016;12(11):2129–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Young LN, Cho K, Lawrence R, et al. (2016) Dynamics and architecture of the NRBF2-containing phosphatidylinositol 3-kinase complex I of autophagy, Proceedings of the National Academy of Sciences. 113, 8224–8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ma X, Zhang S, He L, et al. MTORC1-mediated NRBF2 phosphorylation functions as a switch for the class III PtdIns3K and autophagy. Autophagy. 2017;13(3):592–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Araki Y, Ku W-C, Akioka M, et al. Atg38 is required for autophagy-specific phosphatidylinositol 3-kinase complex integrity. J Cell Biol. 2013;203(2):299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256–1269. [DOI] [PubMed] [Google Scholar]
- [18].Wang C-W, Klionsky DJ. The molecular mechanism of autophagy. Mol Med. 2003;9:65–76. [PMC free article] [PubMed] [Google Scholar]
- [19].Shi Y, Tan S-H, Ng S, et al. Critical role of CAV1/caveolin-1 in cell stress responses in human breast cancer cells via modulation of lysosomal function and autophagy. Autophagy. 2015;11:769–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hyttinen JM, Niittykoski M, Salminen A, et al. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim Biophys Acta-Mol Cell Res. 2013;1833:503–510. [DOI] [PubMed] [Google Scholar]
- [21].Nordmann M, Cabrera M, Perz A, et al. The Mon1-Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr Biol. 2010;20:1654–1659. [DOI] [PubMed] [Google Scholar]
- [22].Jaber N, Mohd-Naim N, Wang ZQ, et al. Vps34 regulates Rab7 and late endocytic trafficking through recruitment of the GTPase-activating protein armus. J Cell Sci. 2016;129:4424–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hegedűs K, Takáts S, Boda A, et al. The Ccz1-Mon1-Rab7 module and Rab5 control distinct steps of autophagy. Mol Biol Cell. 2016;27:3132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cabrera M, Nordmann M, Perz A, et al. The Mon1–Ccz1 GEF activates the Rab7 GTPase Ypt7 via a longin-fold–Rab interface and association with PI3P-positive membranes. J Cell Sci. 2014;127:1043–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kiontke S, Langemeyer L, Kuhlee A, et al. Architecture and mechanism of the late endosomal Rab7-like Ypt7 guanine nucleotide exchange factor complex Mon1–Ccz1. Nat Commun. 2017;8:14034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].McEwan DG, Popovic D, Gubas A, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. 2015;57:39–54. [DOI] [PubMed] [Google Scholar]
- [27].Wang Z, Miao G, Xue X, et al. The Vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol Cell. 2016;63:781–795. [DOI] [PubMed] [Google Scholar]
- [28].Jordens I, Fernandez-Borja M, Marsman M, et al. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr Biol. 2001;11:1680–1685. [DOI] [PubMed] [Google Scholar]
- [29].Cantalupo G, Alifano P, Roberti V, et al. Rab‐interacting lysosomal protein (RILP): the Rab7 effector required for transport to lysosomes. Embo J. 2001;20:683–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Gao J, Langemeyer L, Kummel D, et al. Molecular mechanism to target the endosomal Mon1-Ccz1 GEF complex to the pre-autophagosomal structure. eLife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yu X, Long YC, Shen H-M. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy. 2015;11:1711–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liang C, Lee J-S, Inn K-S, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol. 2008;10:776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Chen D, Fan W, Lu Y, et al. A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate. Mol Cell. 2012;45:629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci. 2015;16:345–357. [DOI] [PubMed] [Google Scholar]
- [35].Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gonzalez AE, Munoz VC, Cavieres VA, et al. Autophagosomes cooperate in the degradation of intracellular C-terminal fragments of the amyloid precursor protein via the MVB/lysosomal pathway. Faseb J. 2017;31:2446–2459. [DOI] [PubMed] [Google Scholar]
- [37].Yang CB, Cai CZ, Song JX, et al. NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy. 2017;13:2028–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lachance V, Wang Q, Sweet E, et al. Autophagy protein NRBF2 has reduced expression in Alzheimer’s brains and modulates memory and amyloid-beta homeostasis in mice. Mol Neurodegener. 2019;14:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ichimura Y, Komatsu M. Selective degradation of p62 by autophagy. Sem Immunopathol. 2010. [DOI] [PubMed] [Google Scholar]
- [40].Wang RX, Tan JQ, Chen TT, et al. ATP13A2 facilitates HDAC6 recruitment to lysosome to promote autophagosome-lysosome fusion. J Cell Biol. 2019;218:267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bucci C, Thomsen P, Nicoziani P, et al. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000;11:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gutierrez MG, Munafó DB, Berón W, et al. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–2697. [DOI] [PubMed] [Google Scholar]
- [43].Jäger S, Bucci C, Tanida I, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117:4837–4848. [DOI] [PubMed] [Google Scholar]
- [44].Zhang K, Kenan RFB, Osakada Y, et al. Defective axonal transport of Rab7 GTPase results in dysregulated trophic signaling. J Neurosci. 2013;33:7451–7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rosales KR, Peralta ER, Guenther GG, et al. Rab7 activation by growth factor withdrawal contributes to the induction of apoptosis. Mol Biol Cell. 2009;20:2831–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sun J, Deghmane AE, Bucci C, et al. Detection of activated Rab7 GTPase with an immobilized RILP probe. Methods Mol Biol. 2009;531:57–69. [DOI] [PubMed] [Google Scholar]
- [47].Yasuda S, Morishita S, Fujita A, et al. Mon1-Ccz1 activates Rab7 only on late endosomes and dissociates from the lysosome in mammalian cells. J Cell Sci. 2016;129:329–340. [DOI] [PubMed] [Google Scholar]
- [48].Diao J, Liu R, Rong Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015;520:563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhao YG, Zhang H. Autophagosome maturation: an epic journey from the ER to lysosomes. J Cell Biol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Morel E, Chamoun Z, Lasiecka ZM, et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat Commun. 2013;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lucin KM, O’Brien CE, Bieri G, et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer’s disease. Neuron. 2013;79:873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest. 2008;118:2190–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ejlerskov P, Ashkenazi A, Rubinsztein DC. Genetic enhancement of macroautophagy in vertebrate models of neurodegenerative diseases. Neurobiol Dis. 2018. [DOI] [PubMed] [Google Scholar]
- [54].Peralta ER, Martin BC, Edinger AL. Differential effects of TBC1D15 and mammalian Vps39 on Rab7 activation state, lysosomal morphology, and growth factor dependence. J Biol Chem. 2010;285:16814–16821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.