

Abstract
Alzheimer’s disease (AD) is the most prevalent form of dementia in the elderly population and has worldwide impact. The etiology of the disease is complex and results from the confluence of multiple mechanisms ultimately leading to neuronal loss and cognitive decline. Among risk factors, aging is the most relevant and accounts for several pathogenic events that contribute to disease-specific toxic mechanisms. Accumulating evidence linked the alterations of the mammalian target of rapamycin (mTOR), a serine/threonine protein kinase playing a key role in the regulation of protein synthesis and degradation, to age-dependent cognitive decline and pathogenesis of AD.
To date, growing studies demonstrated that aberrant mTOR signaling in the brain affects several pathways involved in energy metabolism, cell growth, mitochondrial function and proteostasis. Recent advances associated alterations of the mTOR pathway with the increased oxidative stress. Disruption of all these events strongly contribute to age-related cognitive decline including AD. The current review discusses the main regulatory roles of mTOR signaling network in the brain, focusing on its role in autophagy, oxidative stress and energy metabolism. Collectively, experimental data suggest that targeting mTOR in the CNS can be a valuable strategy to prevent/slow the progression of AD.
Keywords: mTOR, Alzheimer’s disease, proteostasis, oxidative stress, protein aggregation
1. Introduction
Alzheimer disease (AD), the most wide-spread and prevalent form of dementia, is a neurodegenerative disorder affecting the elderly population. The number of people globally living with AD and other dementias has duplicated from 1990 up to date, mainly due to increased lifespan of the population. Neuropathologically, AD is characterized by the presence of two initial hallmarks: formation of senile plaques by extracellular deposition of amyloid β-peptide (Aβ) and neurofibrillary degeneration caused by aggregation of hyper-phosphorylated Tau protein. Further, progression is marked by selective neuronal death at selective vulnerable regions of the brain.
Amyloid β-peptide is a 39–43 amino acid peptide that undergoes misfolding and self-assembly in the form of oligomers, protofibrils, and fibrillar deposits in brains of AD patients and an earlier stage of this disorder, mild cognitive impairment (MCI). Aβ40 is the major Aβ species, whereas Aβ42, though being a minor species, readily aggregates into the above-listed aggregation states, and oligomeric Aβ42 is the more toxic of the two Aβ species. Both Aβ40 and Aβ42 are found in senile plaques [1]. Aβ is generated from the proteolytic cleavage of β-amyloid precursor protein (APP) through two divergent pathways [2]: the major (around 90% of APP) non amyloidogenic pathway normally involves the initial cleavage of APP by two membrane-bound proteases, α-secretase followed by γ-secretase; the minor - which is amyloidogenic - involves the action of β- and γ-secretases. Following its formation, Aβ can be degraded by proteolysis or cleared from the brain into the peripheral blood circulation.
The currently revised theory on Aβ-driven AD onset [3] has evolved from the consideration of amyloid fibrils as the predominant toxic form of Aβ towards smaller and soluble oligomers (AβO) or protofibrils [4]. This updated revision has gained increasing consensus based on the finding that the correlation between the numbers of senile plaques and the severity of dementia is poor, while correlation between small soluble AβO and degree of dementia is high.
The second protein associated with AD pathology is Tau, a structural protein that regulates the stability of tubulin assemblies that play a critical role in axonal transport [5]. Six tau isoforms are expressed in the adult human brain as a result of mRNA alternative splicing; 3R- and 4R-tau isoforms are located mainly in axons of adult neurons under normal physiological conditions. In AD brains, 3R and 4R tau is accumulated in a hyperphosphorylated state in pathological inclusions, referred to as neurofibrillary tangles (NFTs) [6]. Furthermore, Tau inclusions are thought to contribute to AD pathogenesis due to their occurrence in brain regions with altered function, and NFTs formation correlates with the duration and progression of the disease.
The clinical classification of AD is based on the severity of cognitive decline and the histopathological alterations [7]. Currently, five stages are described: i) preclinical: PCAD is characterized by by the absence of clinical signs and symptoms of AD (both typical or atypical phenotypes) and the presence of at least one biomarker of Alzheimer’s pathology [8] ii) amnestic mild cognitive impairment (MCI): characterized by lower levels of AD pathology, marked loss of memory, but normal activities of daily living [9]. The earliest pathological changes begin and are present in the entorhinal cortex and hippocampus iii) mild AD: cognitive symptoms are first manifested. The pathological alterations reach the cerebral cortex. Along with memory loss there is an inability to remember new information and disturbance in executive functioning. Subjects show personality changes, confusion and disorientation. iv) moderate AD: the symptom severity further increases. The pathological damage spreads to the areas responsible for language, reasoning and sensory processing (cerebral cortex). Behavioral problems, language disorder and impairment of visuo-spatial skills start to appear. v) severe AD: subjects completely lose their ability to execute daily activities. The pathological damage cover most of the cortical areas. Cognitive abilities significantly decrease, and systemic symptoms also appear (olfactory dysfunction, sleep disturbances, dystonia, and akathisia symptoms). AD pathology occurs approximately two decades before symptoms appear.
Other staging systems were developed for AD, including the Braak system [10], that is based on topographical staging of neurofibrillary tangles. This Braak classification categorizes AD progression in 6 stages and is an integral part of the National Institute on Aging and Reagan Institute neuropathological criteria for the diagnosis of AD [11].
In addition to the well-characterized plaque and tangle lesions, early deficits in synaptic function with later loss of neurons are considered to be crucial events in the onset and progression of AD. Both synapse and neuronal loss account for macroscopic cortical atrophy [12]. Increasing evidence shows that the loss of functional synapses represents an early defect in AD pathogenesis and that this synaptic loss correlates with memory impairment, even before evident neuronal loss [13]. Further, in AD NMDA and AMPA receptor-mediated excitotoxicity and defective compensatory GABAergic brain circuitry seem to occur concomitantly with synaptic dysfunction [14]. During early phases of neurodegeneration, AβO disrupt synaptic membranes resulting into a deficit in the long-term potentiation (LTP) accompanied by loss of memory and learning [15].
Synapse loss correlates with increased oxidative stress (OS) levels thus accounting for pathological changes observed during the clinically silent prodromal phases of AD [16]. Indeed, OS has been widely recognized as a prodromal factor associated with AD neurodegeneration [17]. Multiple factors mainly related to mitochondrial dysfunction and energy metabolism deficit contribute to elevate cellular OS. Brain structure, significant unsaturated lipid composition, high oxygen consumption, and rapid metabolic rate all contribute to brain susceptibility to toxic effects of OS. Moreover, the natural process of aging is associated with a physiological increase of OS over time [18, 19]. As an individual ages, ROS are integrated, to some extent, in the aging brain, disrupting redox-related communication and leading to cellular alterations, such as senescence and cell death, due to the inability to maintain redox homeostasis. This equilibrium in the cell is particularly important to keep the balanced microenvironment needed for multiple biological processes, from bioenergetics to vesicle transport and intracellular signaling. It is well-known that oxidative damage is greater in aged brains than it is in younger brains [20–23]. Notably, this “physiological accumulation” of oxidative damage with aging is exacerbated during neurodegeneration and is associated with loss of cognitive functions [24]. At the neuronal level, oxidative damage related to aging, strongly impairs the synaptic components involved in neuronal plasticity, cytoskeletal dynamics and cellular communication, among other alterations [25, 26].
In the context of AD neuropathology, several studies showed that OS can interfere with Aβ and Tau protein processing and functioning [27, 28]. Moreover, Aβ peptides themselves are involved in the production of ROS, which in turn causes mitochondrial dysfunction. The biomarkers of OS are well documented in the brain from AD patients. Among OS markers, enhanced levels of carbonylated proteins, mainly in parietal cortex and hippocampal region in brain, have been demonstrated by several authors [29–32]. Membrane proteins are more susceptible to undergo oxidation as compared to cytosolic proteins because radical reactions initiated at the membrane level produce highly reactive and neurotoxic lipid peroxidation products. Indeed, OS induces lipid bilayer modification, promotes lipid peroxidation thus increasing the levels 4-hydroxy-2-nonenal (4-HNE). In turn, protein modification can take place as a result of indirect oxidation by formation of protein adducts with 4-HNE. Increased levels of 4-HNE were reported in brain regions showing histopathological alterations of AD in this disorder and its earlier stage, MCI [31–36]. Oxidative modification of lipoic acid by 4-HNE was altered in AD hippocampus, with implications for pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase enzymatic activities [37]. In addition, the elevated protein carbonyl content in specific brain regions rich in senile plaques indicates a close association between oxidative damage and AD lesions [38]. Indeed, the brains of patients with MCI and AD also have increased oxidative alterations, such as protein nitration and nucleic acid modifications [39]. Fluorodeoxyglucose-positive emission tomography (FDG-PET) analysis revealed reduced brain glucose metabolism in MCI and AD, suggesting a role of mitochondria, the powerhouse of the cell [40].
In particular, redox proteomics studies from Butterfield’s laboratory allowed to identify the most susceptible proteins target of irreversible oxidative modifications, including carbonylation, nitration and HNE-adducts, reviewed in [41–43]. The group performed a comprehensive redox proteome analysis of different brain regions from late-stage AD (LAD), MCI subjects and also from subjects with EAD and PCAD [44, 45]. These results showed a map of specific oxidized proteins in the different stages of the disease. Collectively, these studies provided valuable insights into the molecular mechanisms involved both in the pathogenesis and progression of AD. The impairment of numerous cellular processes including energy production, cellular structure, signal transduction, synaptic function, mitochondrial function, cell cycle progression, and degradative systems are closely linked to oxidative damage to selected components of these pathways [46]. Considering that these cellular functions normally contribute to keep healthy neurons, loss of one or more of these functions could contribute to the pathology and clinical presentation of AD.
Further, the oxidation of nuclear and mitochondrial DNA has been reported in AD with amplified levels of oxidized bases like 8-hydroxyadenine, 8-oxo-2-dehydroguaninie and 5-hydroxyuracil inside frontal, parietal and temporal lobes [47]. Higher levels of 8-hydroxyguanine are detected inside hippocampus of patients with AD [48].
2. Protein oxidation, aggregation and degradation: a dangerous cocktail in the aging brain
Among toxic reactions initiated by ROS, oxidative modifications of proteins likely result in the loss of protein function, as discussed above. Further, protein carbonylation of hydrophobic amino acids buried in the structure of globular proteins lead to a significant dipole moment associated with the carbonyl moiety. This drives the carbonylated hydrophobic amino acids to the surface of the proteins, where the hydrophobic surfaces of different protein molecules aggregate in an attempt to exclude water, accounting for their aggregation [49, 50]. Overall, it is likely that free-radical mediated protein modification takes place in misfolding/aggregation process therefore accelerating dangerous protein deposition [51]. Proteins that are structured efficiently in the form of amyloid fibers or disorganized as amorphous aggregates and plaques are a common toxic aspect of these diseases. Though with diverse clinical manifestations Aβ, α-synuclein and Tau protein are key examples of chemical fingerprint underlying protein aggregation that has been demonstrated to strongly contribute to the pathogenesis and progression of neurodegenerative diseases [52].
However, in addition to protein misfolding and aggregation process, failure of protein quality control (PQC) mechanisms, such as chaperone or proteasome complex and autophagy, is a key pathological feature of age-associated neurodegenerative disorders [53]. When severely damaged, proteins cannot be refolded into their native shapes and the cell utilizes multiple security measure systems to maintain their functions. Chaperones will first act to rescue unfolded protein, and if unsuccessful, they can activate different cellular programs, including the unfolded protein response (UPR), heat shock response, ubiquitin-proteasome system (UPS), and endoplasmic reticulum-associated degradation to either solve the problem or eliminate the unfolded protein [54–56].
This latter drastic proteolytic removal of abnormal proteins is usually performed by the proteasome, a large macromolecular complex that degrades polyubiquitinylated, damaged proteins following removal of the poly(ubiquitin) chain, one residue at a time from the carboxy-terminal end employing the enzyme, ubiquitin carboxy-terminal hydrolase L-1 (UCHL-1). In AD, UCHL-1 is oxidatively dysfunctional [57, 58]. Alternatively, misfolded proteins with specific motif can be guided by HSP into the lysosome via translocation through the lysosomal-associated membrane transporter (LAMP2A), a process known as chaperone-mediated autophagy (CMA) [59, 60]. However, during aging, these systems may become less efficient and may be overwhelmed by the proteotoxic stress [61]. In particular, it is well-known that reduced proteasome activity may occur during aging and neurodegeneration, thus contributing to elevate the intracellular levels of oxidized/aggregated proteins [62, 63]. In AD, the UPS system is dysfunctional [64]. It is worth to be mentioned that at the same time oxidized/aggregated proteins may themselves inhibit the proteasome thus evading their removal [58, 65].
In parallel to proteasomal pathway, the lysosomal system is the most important “digestive mechanism” of the cell acting on different extracellular and intracellular substrates [60]. To do that, lysosomes, which are membrane-enclosed organelles, contain a variety of enzymes, including hydrolases, proteases, lipases, peptidases, glycosidases, nucleases, phosphatases, and sulfatases, able to digest all types of biological materials. Soluble, long-lived proteins, extracellular material, large protein aggregates, and even organelles can be targeted to the lysosome in a process called autophagy. The term autophagy derives from the Greek word “self-eating” and was coined in 1960s based on the observation of double-membrane vesicles entrapping organelles and proteins [66]. As a whole, autophagy is a self-degradative process that is crucial for balancing different sources of energy during starvation, both in development and in response to nutrient deprivation. Autophagy also plays a housekeeping role in removing misfolded/aggregated proteins, removing damaged organelles, such as mitochondria, endoplasmic reticulum and peroxisomes, as well as eliminating intracellular pathogens [67]. Thus, autophagy is generally considered a survival mechanism, though its aberrant regulation has been linked to non-apoptotic cell death.
The concerted function of UPS and autophagy may benefit cells by eliminating misfolded, dangerous proteins that accumulate and form intracellular aggregate structures in many major neurodegenerative diseases. Thus, understanding the molecular signaling signature that regulates both processes is essential to understand which event may contribute to disturbance of their correct equilibrium.
Among putative candidates, the mammalian target of rapamycin (mTOR) signaling pathway seems to be a key determinant in nutrient sensing and in regulating cell growth and autophagy [68]. The complexity of mTOR signaling, at the crossroad of multiple intracellular pathways, is discussed in the following sections, in particular focusing on the redox-mediated mechanisms in Alzheimer’s neurodegeneration.
3. Role of mTOR in physiology and in AD neuropathology
mTOR is a serine-threonine kinase that controls several important aspects of mammalian cell function. mTOR activity is modulated by various intra- and extra-cellular factors and its main function is to monitor whether cell resources and cell health are sufficient to respond to extracellular stimuli [69, 70]. Increasing evidence indicates that mTOR acts as a ‘master switch’ of cellular anabolic and catabolic processes, being involved in the regulation of a wide repertoire of cellular functions including cell growth and proliferation, cytoskeleton organization, and response to hypoxia. In the central nervous system (CNS), mTOR is significantly involved in synaptic plasticity, memory preservation, and neuronal recovery, while the dysregulation of mTOR is emerging as a common theme in a large number of human diseases, including cancer, metabolic syndromes and neurological disorders [69–71]. Recently, great attention has been placed on the role of mTOR in the development of AD since the alteration of mTOR activity was observed in AD brain and in AD mouse models thereof, supporting the notion that aberrant mTOR activity may be one of the leading events contributing to the onset and progression of AD hallmarks [72–77].
3.1. mTOR signaling
mTOR is a ubiquitously expressed protein complex, mainly localized in the cytoplasm whose catalytic activity is regulated by the phosphorylation at Thr-2446, Ser-2448 and Ser-2481 within the kinase catalytic (KIN) domains [69]. mTOR is known to be part of two different protein complexes: mTORC1 and mTORC2, which differ in some components, in upstream and downstream signaling, and in responsiveness to rapamycin treatment. Adjacent to the KIN domain is the FKBP12 rapamycin-binding domain, the site of inhibitory interaction between rapamycin and mTOR. Rapamycin disturbs mTOR–protein complex formation, thereby impairing mTOR activity. The mTORC1 core complex contains mTOR, regulatory associated protein of TOR (raptor), DEP-domain containing mTOR interacting protein (DEPTOR), proline–rich Akt substrate 40kDa (PRAS40), and mammalian lethal with sec-13 protein 8 (mLST8) [78]. In contrast, mTORC2 contains mTOR, mLST8, DEPTOR, rapamycin insensitive companion of mTOR (rictor), protein observed with rictor (Protor), and stress-activated protein kinase-interacting protein 1 (mSin1). The association of mTOR with either Raptor or Rictor determines the formation of mTORC1 or mTORC2. The heterodimer consisting of tuberous sclerosis 1 and 2 (TSC1 and TSC2) is a key upstream regulator of mTOR. TSC1/2 activation constitutively inhibit mTORC1/2 via Rheb (Ras-homolog expressed in brain) by stimulating the conversion of active Rheb-GTP to inactive Rheb-GDP. TSC1/2 activities rely on heterodimer formation and are controlled through by phosphorylation by a number of different kinases [70, 77, 79]. Among the upstream pathway that can activate mTORC1 by phosphorylation-dependent inhibition of TSC1/TSC2 complex is worth to mention PI3K/Akt and Ras/Erk, which are activated by growth factors like insulin or Akt which also inhibit PRAS40 by phosphorylation [70, 80–83]. A primary inhibitory-feedback loop exists among these pathways whereby sustained activation of mTOR/p70S6K suppresses AKT activity.
The above-described mechanism acts through mTORC1-mediated phosphorylation of IRS-1 serine, to induce IRS-1 inactivation and degradation, thus eliminating the coupling of PI3-K/Akt to insulin and IGF-1 receptors. This process results in and is a major cause of insulin resistance [84–87]. mTORC2 phosphorylates Akt to activate the pathway, but also promotes Akt degradation. Similarly, JNK, TNFα and other inflammatory molecules antagonize and turn off PI3-K/Akt axis, deregulating IRS-1 activity. In addition, Parkin7 (DJ-1), under acute oxidative stress, can activate the ERK pathway, which increase mTORC1 activity by directly inhibiting TSC1/2, or by indirectly interacting with p70S6K [77].
mTORC1 is involved in the response to cellular energy demand through AMPK. During low cellular energy, AMPK inhibits mTORC1 through the direct phosphorylation of raptor, which disrupts the mTORC1 complex, and through the phosphorylation of TSC1/2 [81, 88]. Moreover, increasing cellular amino acid or glucose concentration activates mTORC1 through the Rag family of GTPases. Growth factors can activate mTORC2 via the PI3K signaling pathway, increasing the association between mTORC2 and ribosomes. mTORC2 is involved in cellular metabolism and cell shape interacting with actin through PKC-α and Rho GTPase, and in cellular proliferation regulating AKT. mTORC2 is also modulated by increased glucose though rictor acetylation [69, 70, 89].
p70S6K and 4EBP, which regulate protein translation, and Ulk1 and Atg13, which suppress autophagy, are the best-characterized substrates for mTORC1. During availability of high cellular energy resources, mTORC1 promotes ribosome biogenesis and mRNA translation through phosphorylation and activation of p70S6K and 4EBPs. mTORC1- activated p70S6K phosphorylates substrates to promote translation and elongation (i.e., 40S ribosomal subunit proteins, eiF4B and eEF2K), while mTORC1-phosphorylated 4EBPs promote cap-dependent translation through their participation in the eIF4F complex. mTORC1 also regulates lipid biosynthesis through activation of SREBP1, a major lipogenic transcription factor that controls genes involved in fatty acid and cholesterol synthesis [90–92]. In physiological conditions mTORC1 directly interacts with the Ulk1 complex (Ulk1-Atg13-FIP200-Atg101), thus controlling autophagy at the stage of phagophore formation. Autophagy allows the clearance of obsolete proteins, misfolded or overexpressed protein aggregates, and whole organelles [69]. The mTORC1 complex phosphorylates and inhibits Ulk1 and its interacting partner Atg13 as well as AMBRA1, the key link between Ulk1 and Beclin-1 complexes. Under starvation conditions or rapamycin treatment, mTORC1-mediated phosphorylation of Atg13 and Ulk1 is inhibited, leading to dephosphorylation-dependent activation of Ulk1 and Ulk1-mediated phosphorylation of Atg13, FIP200, and Ulk1 itself that triggers autophagy initiation [82, 93, 94]. Furthermore, nutrient deprivation or mTORC1pharmacological inhibition promote the induction of the energy-sensing kinase AMPK, which interact and activate Ulk1, but also directly inhibit mTOR through phosphorylation of TSC2 [95]. Recent studies demonstrated that mTORC1 regulates lysosomes through the transcription factor EB (TFEB), which controls many genes whose associated proteins play key roles in lysosomal function. mTORC1 inhibition favor the nuclear accumulation of TFEB and thus its activity, promoting autophagy induction when nutrient levels are low [96].
3.2. mTOR in brain development and surveillance
Although the precise mechanisms are still not fully understood, regulated and coordinated activities of mTORC1 and of mTORC2 are necessary for the development of CNS [69]. During embryonic development, mTOR signaling functions as a powerful neuronal survival and division signal in response to growth factors and guidance cues. In brain maturation, the mTOR pathway promotes extension of neurites (dendrites and axons) and, through the induction of protein and lipid synthesis, increases cellular mass with expansion of plasma membrane, thus favoring axon guidance [97, 98]. mTORC2, by affecting actin dynamics, may be a putative facilitator of growth cone motility, including neurite path finding and elongation. The same mTOR-mediated regulatory mechanisms that control division/migration programs during brain development evolve in adult individuals to control synaptic plasticity, neuronal polarity, neurotransmission, metabolic control, proteostasis and stress responses. Therefore, in post-mitotic neurons the mTOR pathway has significant functional impact on synaptic processes underlying memory and learning. mTOR is crucial in almost all kinds of neuronal plasticity (e.g., LTP, LTD) and different learning and memory processes [70, 77, 99]. mTOR activity also contributes to nervous system regeneration. The activity of mTOR is essential in terms of normal cognition, whereas hyperactivity of mTOR can be harmful to the brain functions [79]. Furthermore, mTOR is also important for feeding and body weight control, circadian rhythm, and nociception [70].
In aged brain mTOR/autophagy axis plays a dual role in the cellular response to stress and in the regulation of the proteostasis network [90–92]. Increasing evidence suggests that reduced autophagy could lead to accumulation of toxic aggregates and oxidized proteins [56, 100–102]. In addition, the age-dependent reduction in autophagy is responsible of the build-up of severely damaged mitochondria, thus exacerbating oxidative stress and tissue damage with age. In agreement with its role in promoting cell survival and increasing life span during physiological conditions, a deregulated mTOR/autophagy axis under stress conditions (e.g., oxidative stress) may result in cell death [103–105].
3.3. mTOR in the development of AD
In the last two decades mTOR signaling has been extensively analyzed in AD brains and in brains from AD mouse models, demonstrating its aberrant up-regulation during neurodegeneration [74, 106–108]. Data from AD brains indicate that mTOR phosphorylation (both Ser-2448 and at Ser-2481) and the regulation of its downstream targets, p70S6K and eIF4E, are increased in the hippocampus and in other brain areas [109–113]. In addition, mTOR hyperactivity correlated with Braak stages and/or cognitive severity of AD patients. Increased mTOR activity was also observed in the brain from MCI individuals but not in preclinical AD (PCAD) subjects [74]. Further, the dysregulation of the mTOR signaling was observed in peripheral lymphocytes of patients with AD, which correlated with the progression of the disease [114].
Consistent with mTOR data, several studies demonstrated the impairment of the PI3K/Akt axis in AD brain as well as ERK1/2 signaling. The sustained activation of neuronal PI3K/Akt/mTOR pathway in AD and MCI brain and in AD models was associated with insulin receptor substrate 1 (IRS1) inhibition, disabling normal activation of PI3K/Akt by insulin and inducing insulin resistance [74, 87, 103, 115–117]. The hyperactivation of the PI3K/Akt/mTOR axis is also associated with reduced autophagy in AD brain leading to the accumulation of protein aggregates. Several markers of autophagy initiation and execution or autophagosome formation (e.g., LC3 II/I) and of autophagic flux are reduced in AD brain and in AD early stages [92, 97, 113].
Consonant with the above discussion, pharmacological reduction of mTOR hyperactivity in the brains of AD and of AD-like mouse models was shown to restore autophagy [118–122]. The reduction of autophagy in AD also was shown to be influenced by a number of different events, such as increased OS, but not directly related to mTOR hyperactivation [60, 73, 102, 123]. The deterioration of neuronal autophagy, observed in AD, most likely allows increased Aβ production and aggregation, and contributes to the malfunction of Aβ elimination from the brain. Indeed, the autophagy-lysosome pathway is an important regulator of APP processing and clearence and also one of the major cellular pathways for the removal of Aβ aggregates [56, 91, 92, 103, 107, 123, 124]. In addition, the analysis of the brain from AD subjects and mouse models of the disease indicated a strong link between increased mTOR signalling and tau neuropathology comprising the formation of tau oligomers and insoluble aggregates [74, 75, 112, 125]. In a small number of studies concerning the analysis of mTOR signaling in AD animal models contrasting results were shown. Indeed, the analysis of APP/PS1 double transgenic mice and of Tg2576 mice demonstrated the inhibition of mTOR signaling, thus hypothesizing that mTOR alterations results might depend on the strain of the models analyzed and/or on the age of the mice [126, 127].
4. The crosstalk between mTOR and mitochondria in AD
Mitochondria play an essential role in energy generation, cell signaling, differentiation, death, and senescence in eukaryotic cells. In particular, the role of mitochondria in cellular senescence and neurodegeneration is widely associated with their significant function as generators of ROS- mediated random molecular damage. Indeed, ROS have been shown to induce genomic damage [128], accelerate telomere shortening [129], impair actin dynamics and synaptic plasticity mechanisms [130, 131], and to act as drivers of signa ling networks important for the maintenance of the senescent phenotype [132]. Accumulation of ROS such as hydrogen peroxide (H2O2) is an oxidative stress response, which induces various defense mechanisms or programmed cell death [103, 125, 133, 134]. As one of the major types of programmed cell death, autophagy has been observed in response to several anticancer drugs and demonstrated to be responsible for cell death. However, the exact mechanism by which ROS regulates autophagy is still poorly understood. The role of mTOR appears crucial in regulating mitochondrial functions and ROS production. Baker et al. [135] proposed that mitochondria and their capacity to generate free radicals near mitochondrial DNA (mtDNA) are one of the most studied possible mechanisms of aging. In addition to that, Correira-Melo et al. proposed a mechanism by which the DNA damage response, a major driver of senescence, interacts with mitochondria to develop a senescent phenotype. In particular, Akt and mTORC1 phosphorylation cascade would play a key role as an effector of the DNA damage response. Indeed, mTOR activity was decreased in senescent mitochondria- depleted cells [136].
4.1. mTOR, mitochondria and oxidative stress
Many investigations have consistently shown that all the interventions that increase longevity in mammals, i.e., dietary, protein or methionine restriction are associated with reduced mTOR activation, decreased mitochondrial ROS production, and decreased oxidative-derived damage possibly contributing to longevity extension and reduced neurodegeneration [135]. Moreover, as reported by our group both in AD and DS brain the accumulation of protein oxidative damage results from the increased free radical production, mainly related to metabolic alterations, mitochondrial degeneration, and amyloid-β oligomers, and aberrant activity of protein degradative systems, including mTOR-dependent autophagy [72, 74, 78, 80, 84, 137]. In this context, the early aberrant hyper-phosphorylation of mTOR coupled with the reduction of autophagosome formation was found to be associated with increased levels of 3-NT and 4-HNE Ts65Dn mice (a well-known model to study DS), suggesting the potential involvement of altered autophagy in the buildup of protein oxidative damage [138, 139]. Data obtained in vitro strengthened the protective role of autophagy in reducing protein oxidation [138].
In addition, concurrent phosphorylation of AMPK and mTOR was observed in AD brains with high colocalization with hyperphosphorylated Tau [140]. Mitochondrial antioxidant enzymes e.g., SOD2, p1, and p4 were substantially decreased in p-AMPK, p-mTOR, and p-tau positive cells along with higher levels of DNA and protein oxidation [140]. Similarly, a novel molecular mechanism favoring the disruption of the AMPK/mTOR axis and the impairment of autophagy along with the accumulation of oxidatively-damaged proteins and lipids within the brain was recently reported [81]. Together, these lines of evidence suggest that AMPK and mTOR metabolic axis has a crucial role in AD brains and that their dysfunction leads to increased oxidative stress levels in AD. As originally proposed by our group [32, 74] and expanded by Kim et al. [141], Aβ peptides may be crucial determinants linking ROS, mitochondria, and mTOR in the AD brain. These authors showed that Aβ treatment leads to increased ROS generation and mitochondrial fission, accompanied by dysfunction of mitochondria such as loss of membrane potential and ATP production in neuronal cells [141]. These events were coupled with the sustained Akt activation, that induced not only the fragmentation of mitochondria but also the activation of mTOR, eventually suppressing autophagy. Indeed, inhibition of autophagic clearance of Aβ led to increased ROS levels and aggravating mitochondrial defects, which were blocked by rapamycin (noted above to be a mTOR inhibitor). Hence, sustained phosphorylation of Akt by Aβ directly inhibits autophagy through the mTOR pathway and these changes elicit abundant mitochondrial fragmentation resulting in ROS-mediated neuronal apoptosis [141].
Among the strategies to clarify the role of mTOR hyperactivation in mediating mitochondrial defects and increased oxidative damage leading to brain dysfunctions and AD, rapamycin (an mTOR inhibitor) administration appears to be of great interest. While the number of studies addressing the beneficial effects of rapamycin in neurodegenerative disorders is consistently increasing in the last years, only a limited number of papers, including those from our group, addressed a potential role for rapamycin in ameliorating mitochondrial defects in AD and are discussed in the current review. Rapamycin administration promoted autophagy and markedly reversed Aβ1–42-induced impaired redox homeostasis by decreasing the levels of prooxidants-ROS generation, intracellular Ca2+ flux, and lipoperoxides, and increasing the levels of antioxidants, i.e., SOD, catalase, and GSH in adult rats [82]. Autophagy activation following rapamycin treatment also provided significant neuroprotection against Aβ1–42-induced synaptic dysfunction by increasing the expression of synapsin-I, synaptophysin, and PSD95; and neurotransmission dysfunction by increasing the levels of CHRM2, DAD2 receptor, NMDA receptor, and AMPA receptor; and ultimately improved cognitive ability in rats [82]. Moreover, rapamycin abolished the age-associated increase of mitochondrial ROS production and percent free radical leak at complex I, accumulation of mtDNA fragments inside nuclear DNA, mitochondrial protein lipoxidation, and lipofuscin accumulation in mice. Rapamycin also decreased RAPTOR and increased PGC1-α and ATG13 proteins. These findings support the possibility that a decrease in the rate of free radical generation at mitochondria and increased autophagy coordinately contribute to the increase in longevity and to reduced neurodegenerative processes induced by rapamycin in aged CB6F1 mice [94]. Our group also reported that intranasal administration of rapamycin to target the brain ameliorate Alzheimer-like cognitive decline and reduce the oxidative damage in the brain of a mouse model of DS, by reducing mTOR hyperactivation [118, 137].
An interesting observation comes from the work performed by Till et al. [142], highlighting a role for GABA in mediating mTOR activation, autophagy impairment, and mitochondrial defects in the brain. Indeed, autophagy operates specifically to degrade particular proteins or organelles, such as peroxisomes (pexophagy) [142], mitochondria (mitophagy) [143], or ribosomes (ribophagy) [144]. Much of the same core machinery used for general autophagy also overlaps in the selective autophagy pathways. In this picture, GABA was proposed as a regulator of mitophagy and pexophagy via mTOR activation. When GABA levels are increased, cells are unable to specifically degrade mitochondria and peroxisomes via these selective autophagy pathways during starvation conditions, leading to oxidative stress due to the accumulation of these organelles. Indeed, GABA administration leads to an increase of ROS, which can be prevented by co-treatment with glutathione, but even more by rapamycin, thus suggesting that GABA increases cellular oxidative stress, probably due to the presence of longer- lived or damaged peroxisomes and mitochondria because impaired autophagy [145].
Also, lysosomes are essential for autophagy, and autophagic clearance of dysfunctional mitochondria represents an important element of mitochondrial quality control. Indeed, alterations in lysosomal function can affect upstream autophagic functions. Loss of ATP13A2, which encodes a lysosomal P-type ATPase, was shown to affect mitochondrial function through mTOR [146]. Knockdown of ATP13A2 led to an increase in mitochondrial mass in primary mouse cortical neurons and in SH-SY5Y cells forced into mitochondrial dependence. ATP13A2-deficient cells exhibited increased oxygen consumption without a significant change in steady-state levels of ATP. Mitochondria in knockdown cells exhibited increased fragmentation and increased production of ROS. ATP13A2 knockdown cells exhibited decreased autophagic flux, associated with increased levels of phospho-mTOR, and resistance to autophagy induction by rapamycin. The effects of reduced ATP13A2 levels on oxygen consumption, mitochondrial mass and ROS production are mimicked by inhibiting autophagy induction. Hence, hyper-active mTOR-dependent decreased autophagy associated with ATP13A2 deficiency impairs mitochondrial quality control, resulting in increased ROS production [146]. These data, therefore, suggest that lysosomal impairment affects mitochondrial homeostasis further supporting the evidence for mitochondrial dysregulation in AD neuropathology [103]. In vitro, rapamycin inhibited S6 phosphorylation events, mitochondrial ROS production in hippocampal neuronal cell line HT22, thus suggesting a role for mTOR in regulating mitochondrial bioenergetics in the brain [83].
4.2. mTOR, mitochondria and energy metabolism
Human aging is characterized by a gradual reduction in the ability to coordinate cellular energy expenditure and storage (crucial to maintain energy homeostasis), and by a gradual decrease in the ability to mount a successful stress response [147, 148]. These physiological changes are typically associated with changes in body composition (i.e., increase in fat mass and the decline in fat-free mass), and with a chronic state of oxidative stress with important consequences on health status [149–151]. In particular, mitochondria have a crucial role in the supply of energy to the brain. Mitochondrial alterations other than an increased ROS production can lead to detrimental consequences to the function of brain cells and for that reason are thought to have a pivotal role in the pathogenesis of several neurologic disorders. Within the brain, mitochondrial functions deputed to energy production are needed to regulate synaptic transmission, neurotransmitter recycling, dendritic and axonal transport, ion channels, and ion pump activity, which are processes with high energetic requirements [152]. In this sense, mitochondrial bioenergetic failure has been pinpointed as a mechanistic event underlying brain malfunction, senescence and/or neurodegeneration [103, 153].
Redox proteomics techniques in AD brain allowed the identification of specific mitochondrial proteins, including some in the mitochondrial oxidative phosphorylation machinery, that are targets of oxidative modifications during neurodegenerative processes and thereby become dysfunctional thus impairing mitochondrial bioenergetics [134]. ATP synthase alpha, manganese superoxide dismutase (MnSOD), malate dehydrogenase and VDAC were identified modified by both protein carbonyls, 3-NT and HNE-bound, in the early stages and in the late stage of AD, in MCI and EAD [134, 154]. Indeed, Eno1 and ATPase activities are reduced in AD brain [155]. ATP synthase, the last complex of the electron transport chain (aka complex V), is an essential enzyme in the inner mitochondrial membrane and plays a key role in energy metabolism. Its oxidative modifications modify its conformation leading to the inactivation of the complex. The defective function of ATP synthase activity secondary to oxidative modification significantly contributes to lower ATP levels, possibly resulting in electron leakage and increased ROS production [45]. Dysfunction of single complexes of the respiratory system is frequently accompanied by deleterious side effects like loss of mitochondrial membrane potential (MMP) and consequently decreased ATP levels, but also the production of ROS [156]. Dysfunction of single enzyme complexes, ROS production, mitochondrial permeability transition pore (mPTP) opening, elevated apoptosis, in addition to structural alterations, and a diminished mitochondrial content are believed to critically contribute to the onset and progression of neurodegenerative pathology in AD [157–160].
Consonant with the above, deficiency in several mitochondrial key enzymes is well documented in AD. These include enzymes involved in the TCA cycle, such as aconitase, ketoglutarate dehydrogenase complex and pyruvate dehydrogenase complex as well as those involved in the electron transport chain of oxidative phosphorylation such as cytochrome oxidase [41]. Targeting brain metabolism and mitochondrial function are relevant to hypometabolism and impaired mitochondrial bioenergetics that are among the earliest pathogenic events in AD pathology [134]. While the involvement of mitochondrial failure in AD has been extensively reviewed, also by our group [161], in the current review we focus on the involvement of mTOR in regulating mitochondrial bioenergetics in AD. In particular, in light of the fact (as described in the section above) that pharmacological agents that inhibit mTOR can protect neurons against dysfunction and degeneration in animal models of acute brain injury and neurodegenerative disorders. Therefore, a better understanding of such adaptive responses of neurons to bioenergetic challenges may lead to the development of novel approaches for promoting optimal brain function and for preventing and treating neurodegenerative disorders.
Among the processes identified to impair mitochondrial bioenergetics favoring increased oxidative stress during aging and AD the dysregulation of uncoupling proteins (UCPs) emerge as a pivotal event. In humans, UCPs are a group of five mitochondrial inner membrane transporters with variable tissue expression, which seem to function as regulators of energy homeostasis and antioxidants [162, 163]. In particular, these proteins uncouple respiration from ATP production, allowing stored energy to be released as heat. Data from experimental models have previously suggested that UCPs may play an important role in the rate of aging and consequent lifespan. Mitochondrial uncoupling imposes an energetic stress on cells by causing a proton leak across the inner membrane which reduces the membrane potential and uncouples substrate oxidation from ADP phosphorylation, thereby increasing energy expenditure [162, 163]. Mitochondrial uncoupling is a physiologically regulated process that plays important roles in the adaptive responses of organisms to changing environmental conditions. Mitochondrial uncoupling proteins regulate multiple physiological processes including thermogenesis, mitochondrial redox balance and free radical production, cellular calcium homeostasis, and autophagy/mitophagy. Thus, by promoting fatty acid oxidation, and by reducing ATP and ROS production, the induction of mitochondrial uncoupling through UCPs may also be a critical pathway in the modulation of the rate of aging and lifespan [164–166].
A potential role for mTOR in regulating UCPs was proposed by Mattson and co-workers [167]. These authors showed that mild mitochondrial uncoupling enhances cellular energy expenditure in mitochondria and can be induced with 2,4-dinitrophenol (DNP), a proton ionophore previously used for weight loss. DNP treatment reduces mitochondrial membrane potential, increases intracellular Ca2+ levels and reduces oxidative stress in cerebral cortical neurons. Gene expression profiling of the cerebral cortex of DNP-treated mice revealed reprogramming of signaling cascades that included the suppression of mTOR and insulin-PI3K-MAPK pathways, and up-regulation of tuberous sclerosis complex 2, a negative regulator of mTOR [88]. Genes encoding proteins involved in autophagy processes were up-regulated in response to DNP. CREB signaling, Arc and brain-derived neurotrophic factor, which play important roles in synaptic plasticity and adaptive cellular stress responses, were up-regulated in response to DNP, and DNP-treated mice exhibited improved performance in a test of learning and memory [167]. Hence, these findings suggest that mild mitochondrial uncoupling triggers an integrated signaling response in brain cells characterized by reprogramming of mTOR and insulin signaling, and up-regulation of pathways involved in adaptive stress responses, molecular waste disposal, and synaptic plasticity.
Physiological bioenergetic challenges such as exercise and fasting also can enhance neuroplasticity and protect neurons against injury and neurodegeneration. Conversely, in AD brains the UCPs gene expression decreased significantly relative to control brains, and the mean levels tended to be lower in AD brains with higher grades of neurodegeneration. Considering that mTOR hyperactivation has been reported in AD and its earlier stage, MCI [74], we speculate about a possible involvement for mTOR in repressing UCPs expression. Overall, the failure to maintain normal levels or to increase the expression of UCPs may potentiate oxidative stress leading to progressive mitochondrial DNA damage and energy depletion [168]. As recently reported by Montesanto et al. [169], rs9472817-C/G, an intronic variant of neuronal mitochondrial uncoupling protein-4 (UCP4/SLC25A27) gene affects the risk of late-onset Alzheimer’s disease (LOAD), and that the variant’s effect is strongly dependent on APOE-ε4 status [169].
Another pathway including mTOR as a key player and involved in the regulation of mitochondrial bioenergetics that was found to be impaired in AD is the insulin signaling cascade [85]. Insulin signaling activation triggers multiple effects, including synthesis of proteins involved in neuronal glucose metabolism, anti-apoptotic mechanisms, and antioxidant defense [85, 170]. It is worthy of mention that insulin (a) prevents the disruption of mitochondrial membrane potential and cytochrome c release, thus promoting neuronal survival; and (b) enhances mitochondrial membrane potential, ATP levels, nicotinamide adenine dinucleotide phosphate (NADPH) redox state, and hexokinase activity, thus favoring mitochondrial bioenergetics [85, 170]. In light of these roles, the development of brain insulin resistance was proposed among the mechanisms responsible for increased oxidative damage in the brain [43, 139, 171] as well as for the development of AD [85, 172]. Brain insulin resistance mediates synaptotoxic effects in several ways leading to (1) synapses loss, impaired autophagy and increased neuronal apoptosis [85, 115, 173]; (2) increased production and secretion of beta-amyloid peptides (Aβ) [85, 174]; as well as (3) an increased Tau phosphorylation [85, 125, 175]. These observations collected in vitro and in AD mouse models have been also strengthened by clinical studies reporting that the failure in brain energy metabolism responsible for cognitive decline during aging or AD could be driven by the development of brain insulin resistance particularly at early stages [85, 176]. On the other hand, strategies to overcome brain insulin resistance, such as intranasal insulin administration, were shown to ameliorate memory and cognitive functions [170]. Similar results were obtained with rapamycin [118, 137] treatment, or by genetically reducing mTOR hyperactivation [86, 177], suggesting a role for mTOR in the metabolic effects mediated by insulin in the brain. Taken together, these findings support a crucial role for mTOR in mediating brain insulin effects on several processes that are essential for healthy aging.
Aberrant mTOR phosphorylation was proposed to be a key driver for IRS1 inhibition and brain insulin resistance [85, 103, 178, 179]. Indeed, increased mTOR activation and IRS1 inhibition were reported in both MCI and AD brain [74] as well as in mouse models of AD [103, 171, 174, 180]. Among the pathways responsible for mTOR hyperactivation in AD, our group identified the impairment of biliverdin reductase-A as a crucial event responsible for the disruption of the AMPK/mTOR axis resulting in mTOR hyper-activation in the brain [81, 84, 171]. Within the insulin signaling pathway, mTOR hyper-activation would be responsible for mitochondrial damage and reduced energy production. As reported by Swerdlow and co-workers [159], hybrid cells, modeled by transferring mitochondria from MCI, AD and control subject platelets to mtDNA-depleted SH-SY5Y cells, were characterized by altered mTOR along with an altered mitochondrial dynamic and bioenergetics, as well as, reduced glycolytic flux [181]. Moreover, Norambuena et al. [182] showed that Aβ oligomers (AβOs) activate mTORC1 at the plasma membrane in neurons, which leads to cell cycle re- entry, a frequent prelude to the death of neurons [87]. In doing so, AβOs-mediated mTOR hyperactivation also interferes and suppresses the nutrient- induced mitochondrial activity, thus generating a sort of prolonged starvation phenotype in AD neurons. As noted above, AβOs in the membrane bilayer led to lipid peroxidation with production of neurotoxic HNE [183]. Therefore, AβOs initiate two parallel, interconnected pathways that together lead to neuronal dysfunction in AD [182]. Rather, this effect of AβOs reportedly can be blocked by insulin and nutrients, like amino acids [87].
Finally, a significant decrease in the protein levels of LC3-II and a significant increase in protein levels of mTOR phosphorylated at serine residue 2448 was observed in GK rats (a model for type 2 diabetes mellitus) suggesting suppression of autophagy in the diabetic brain cortex. No significant alterations were observed in the parameters related to mitochondrial biogenesis. Altogether, these results demonstrate that during the early stages of T2D, brain mitochondrial function is maintained in part due to a delicate balance between mitochondrial fusion-fission and biogenesis and autophagy [184]. Moreover, these events are precedent to an age-related impairment of the respiratory chain and uncoupling of oxidative phosphorylation documented in brain mitochondria isolated from 12- to 24-month GK rats [185].
4.3. mTOR, mitochondria and lifespan
Aging is the major risk factor for developing AD and is the most complex phenomenon in nature, where an inevitable time-dependent, progressive functional decline of diverse physiological functions finally results in death [103]. Aberrant mitochondrial function is a major characteristic of aging cells and is thought to contribute to neurodegeneration observed in AD [186]. Remarkably, genetic models of mitochondrial dysfunction (such as mutation of mitochondrial DNA polymerase in mice) correlate with reduced life span [187]. Surprisingly, many interventions that extend life span, e.g., caloric restriction and rapamycin, by modulating mTOR activity lead to reduced energy intake and decreased mitochondrial functions [135]. These findings revealed complex and antagonistic functions of mitochondria in aging and have been at least partially reconciled by biphasic modeling of mitochondrial dysregulation [135]. This model proposes that alterations of mitochondrial functions are not linear with aging of the organism, but rather that they increase and peak in middle age, followed by a decline in older age [135]. Mitochondrial dysfunction in aging cells is characterized by several factors, including elevated ROS production and mitochondrial DNA mutations; decreased electron transport function, membrane potential, and ATP production; altered mitochondrial dynamics; or dysregulated mitophagy [186, 188]. Functionally, altered mitochondrial activity participates in inducing cellular senescence [189], chronic inflammation, and a decline in stem cell activity associated with aging [187]. Nevertheless, while excessive ROS production is detrimental for neurons, low ROS levels could promote the activation of AMPK through S-glutathionylation of reactive cysteines located at the α- and β-subunits of AMPK [102]. This modification would further lead to phosphorylation and activation of the tuberous sclerosis complex1 (TSC1)/TSC2 and consequent inhibition mTOR activity [88].
This mechanism seems to be at the base of the interventions that slow aging and prevent chronic disease, including caloric restriction (CR). CR impacts on the major nutrient-sensing pathways that are mTOR, AMPK, sirtuins and insulin/IGF-1, which sense the availability of macronutrients (glucose, amino acids and lipids) or the energy status of the cell (AMP/ATP or NAD+/NADH ratio). These pathways regulate several cellular processes such as autophagy, metabolism, oxidative stress and gene expression that, in the end, are beneficial also for the improvement of cognitive and learning functions [190]. Considerable lines of evidence suggest that CR-associated benefits are mediated at the molecular level by the modulation of mTOR, which normally responds to variation of amino acid availability, oxygen, and growth signaling [79, 191]. Lower levels of mTOR activity, as would be expected to be encountered in CR, resulting in reduced protein and lipid synthesis, increased autophagy and stress- defense activity, enhanced regenerative capacity, and longer life in various organisms [192]. Several studies also indicate that mTOR influences feeding behavior and formation of memory in the hippocampus [193]. In Caenorhabditis elegans CR lifespan extension depended upon a group of regulators that are involved in stress responses and mTOR signaling, e.g., FOXO, Nrf1/2/3, FOXA, AMPK. CR reduced total oxygen consumption but increased the proportion of respiration devoted to ATP production. Apparently, CR drives the activity of key NAD+- associated mechanisms through a shift toward oxidative metabolism but does not necessarily increase overall respiration rates. In particular, CR-mediated extension in longevity was promoted by the up-regulation of FOXO, Nrf1/2/3, FOXA, AMPK, which are normally inhibited by mTOR signaling. The proposed importance of mTOR in CR also fits with the importance of the low- energy sensor AMPK [194], which inhibits mTOR in many species [79]. Hence, these findings support the view that CR extends lifespan in part through a reduction in mTOR signaling, and possibly insulin/IGF1 pathways [195].
Recent studies in mice also demonstrated that the reduction of insulin signaling by the action of CR shifts the energy metabolism in the brain from glucose to ketone bodies. CR additionally alleviates insulin resistance and through the inhibition of mTOR pathway, it enhances the clearance of misfolded protein including Aβ [196]. Therefore, CR was proposed to retard the progression of AD and is beneficial for healthy brain aging. Similar results were collected in ApoE-deficient mice (ApoE−/−), serving as a model of neurodegeneration [95]. ApoE−/− mice showed upon CR vs ad libitum feeding increased phosphorylation of AMPK and reduced activity of mTOR in the brain, which were associated with decreased Tau-phosphorylation [95]. Moreover, CR-mediated neuroprotective effects in ApoE−/− were characterized by increased numbers of PSD95-positive neurons and better cognitive performance [95].
Other than the above-cited effects mediated by CR, the role for mTOR in regulating healthy-aging and lifespan was highlighted by profiling genes differentially expressed in a successful model of aging. A genomic and transcriptomic data from 17 rodent species revealed a set of about 250 positively selected genes (PSGs) that are differentially regulated between long-lived naked mole-rats and short-lived rats [197]. In particular, long-lived rats show higher levels of these PSGs enriched for genes known to be related to aging. Among these enrichments were “cellular respiration” and “metal ion homeostasis”, as well as functional terms associated with processes regulated by the mTOR pathway: translation, autophagy and inflammation. Remarkably, among PSGs are RHEB, a regulator of mTOR, and IGF1, both central components of aging-relevant pathways [197]. Indeed, mTOR can be activated by RHEB either on the surface of the peroxisome [198]–in response to reactive oxygen species (ROS)–or on the surface of the lysosome [89]–in response to amino acids. Furthermore, among mTOR-regulated processes that are relevant for both growth and aging are translation and cellular respiration [79]. Consistent with the observed antagonistic expression patterns of PSGs in the long-lived naked mole-rat and short-lived rat, lower expression of genes related to these processes as well as pharmacological inhibition of the respective gene products were shown to be associated with longer lifespan [197].
Regarding human data, little research evaluated the potential role of diet on AD progression. There is evidence for certain dietary practices, such as Mediterranean diet and specific vitamin supplementation, offering protection against neurodegenerative disease development, but the effects of dietary intervention on the management of AD are relatively unknown [199, 200]. A few small clinical studies reportedly have shown a relationship between the ketogenic diet and improved cognition in AD patients [201]. These findings seem to be supported by studies showing AD patients consistently exhibit reductions in cerebral glucose utilization without alteration in brain ketone metabolism [202]. The application of CR to human studies has begun. To an extent, chronic CR has shown to be beneficial to human health such that moderate CR without malnutrition causes a protective effect against obesity, type-II diabetes, inflammation, hypertension and cardiovascular disease, all of which are major causes of morbidity, disability and mortality [203]. However, there remain valid concerns against CR since the duration and severity required for optimal health and anti-aging benefits is not feasible for most people over long periods of time and could lead to undesirable side effects [204].
5. Redox regulation of protein quality control networks in AD
mTOR signaling is strongly associated with neurodegeneration through its controlling protein homeostasis, which holds an important role in maintenance of brain structure and functions [101, 137, 205–207]. Protein homeostasis is preserved by a network of cellular apparati that control expression, localization, folding and interactions of proteins from their synthesis until their degradation [103, 208, 209]. The UPR operates to ensure the proper folding of proteins during the life of neurons [104, 210, 211], but if refolding strategies fail, misfolded/unfolded proteins are shifted to the UPS and autophagic degradation pathways [104, 105, 212–214]. AD is characterized by abnormal accumulation of aggregated proteins in postmitotic neurons, and the alteration of protein synthesis, folding, surveillance and degradation are implicated in this phenomenon [60, 105, 210, 215, 216]. The induction and functionality of protein quality control systems, and the preservation of proteostasis is closely associated with the redox balance of the brain [217, 218]. Low amounts of ROS can activate the adaptive cellular machinery to increase the organism’s stress resistance. This response involves the enhancement of antioxidant strategies and the induction of UPR, UPS and autophagy. Conversely, a chronic increase of ROS can on one side overwhelm the responsive capacity of these pathways and on the other side induce the oxidation of specific component of UPR, UPS or autophagy, thus exacerbating the accumulation of unfolded/misfolded proteins [60, 154] with deleterious consequences for cells, including neurons (Figure 1).
Figure 1.
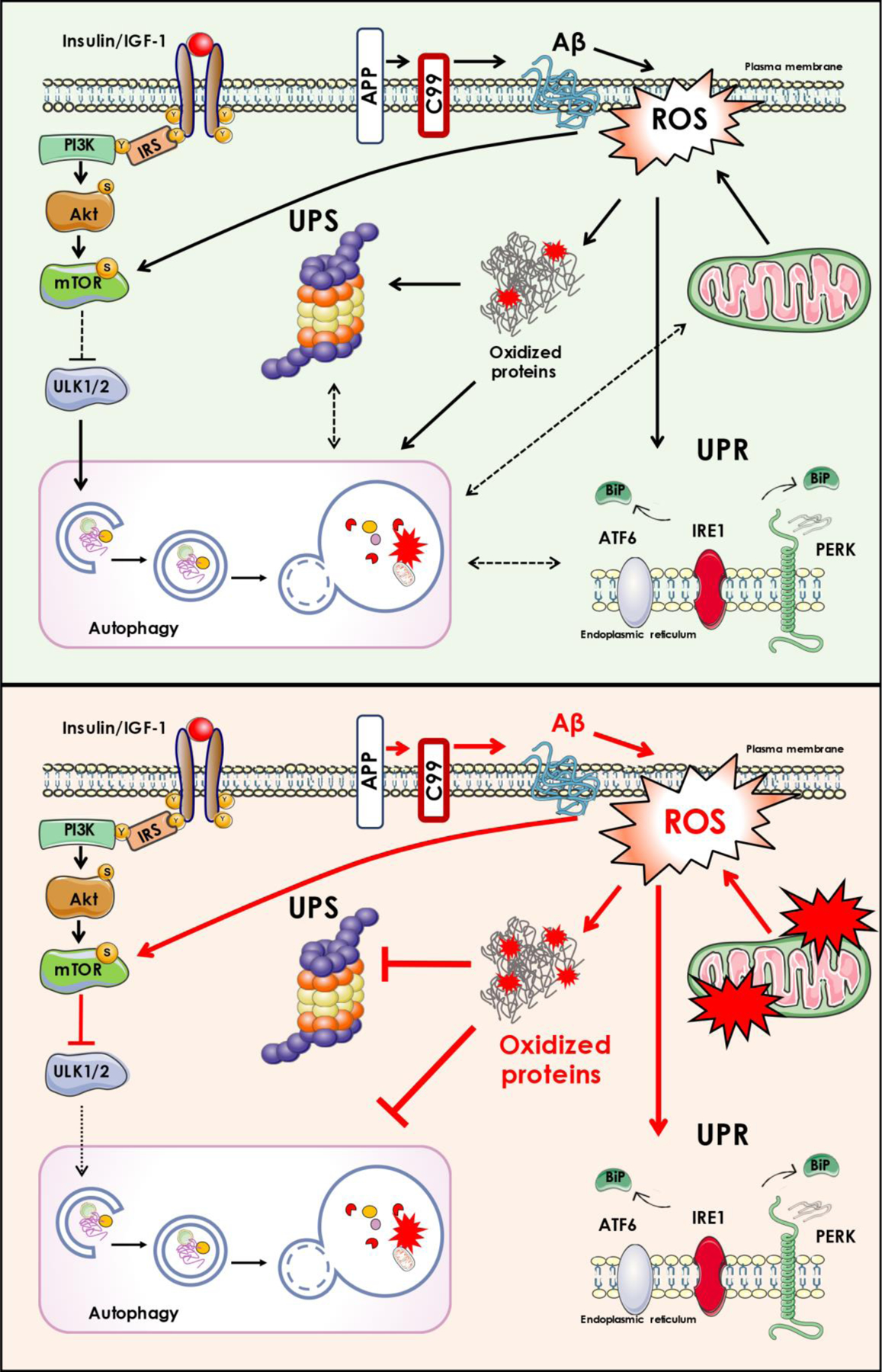
In physiological conditions (depicted in the upper part of the figure), several oxidant stimuli, such as low levels of free radicals (ROS) and low amount of amyloid beta peptide (Aß), lead to coordinated stress responses (UPR, UPS and autophagy) for the removal of damaged molecules/organelles. In pathological conditions (depicted in the lower part of the figure), the overproduction of oxidants overwhelms the protein quality control, in part due to the oxidation/inactivation of some of its members, resulting in the accumulation of oxidized/dysfunctional proteins that are harmful for neuronal survival. In this scenario, mTOR hyperactivation, as it occurs in AD brain, not only contributes to impair autophagy machinery, but also dysregulates insulin signaling (by feedback mechanisms).
5.1. mTOR/autophagy
The crosstalk among OS and the mTOR/autophagy axis in AD remains understated, even though several studies highlighted the relevance of their association either in physiological or pathological conditions [60, 73, 102, 219]. It is well recognized that ROS induce autophagy and that autophagy, in turn, operates to reduce oxidative damage [217, 218, 220]. In particular, SH-SY5Y neuroblastoma cells exposed to OS accumulated more Aβ-enriched lysosomes than the control group, indicating the regulation of autophagy through OS [221]. Other authors have suggested an OS-induced improvement of APP processing and a decrease in Aβ elimination because of either reduced lysosomal degradation or Aβ structural modification [222]. Acute OS increase stimulates the initiation of autophagy, in association with stress signal pathways, through the posttranslational modifications of regulatory proteins [217]. Studies in transgenic mouse models of AD and AD-like dementia demonstrated that, in turn, the rescue of autophagic deficits led to a reduction of OS and brain deposition of Aβ while learning and memory deficits were prevented [118, 119, 223]. The redox proteomic analysis of AD brain samples demonstrates that the oxidative modification of key components of the protein quality control (PQC) pathways, as result of Aβ-induced increase of OS, might contribute to aberrant protein homeostasis and consequently to the deposition of senile plaques and NFTs [45, 103, 134, 154, 155, 224]. With respect to mTOR/autophagy, redox proteomics data suggest that increased OS, through the oxidative modification of autophagy components, might represent the link between the accumulation of Aβ, the alteration of mTOR signaling pathways and vice versa [224–226]. Indeed, the increase of oxidative stress appears to affect the final step of autophagic flux, which is involved in the degradation of the autophagosome cargo by lysosomal hydrolases. This final degradative process relies on the activity of two proteins: the vacuolar [H+] ATPase (V0-ATPase) and cathepsin D, which were found oxidized in the brain of DS individuals at risk of developing AD, by redox proteomics approach [224–227]. In addition, V0-ATPase is necessary for amino acids to activate mTOR, thus supporting that V0-ATPase is an indirect, but integral, component of the mTOR pathway [228].
5.2. UPR
Accumulating evidence supports the concept of dysregulated UPR as key mechanism of neurodegeneration [213, 229]. Indeed, under persistent activation the UPR could foster the long-term memory and synaptic plasticity defects in AD [208, 210]. UPR is initiated upon disturbances of ER homeostasis and involves the activation of three transmembrane proteins in the ER membrane that serve as sensor proteins: the protein kinase-like endoplasmic reticulum kinase (PERK), the activating transcription factor 6 (ATF6), and the inositol-requiring kinase 1 (IRE1) [43, 230]. IRE1, PERK, and ATF6 pathways coordinate a complex network of stress signals to the cytoplasm and the nucleus that result in the inhibition of protein translation and control the expression of specific transcription factors to regulates folding and degradation of proteins. In this fashion, the UPR is finely linked to the proteolytic pathways, including the UPS and autophagy [231–235]. All three arms of the UPR are activated after BiP/Grp78 dissociation to allow the folding of accumulated unfolded proteins in the ER lumen. Despite its protective role, prolonged UPR induction is considered to promote the progression of neuropathological mechanisms [236]. Specific markers of UPR activation are increased in AD brain tissue. BiP/Grp78 expression levels are increased in the hippocampus and temporal cortex of AD and several studies have shown the increased presence of phosphorylated PERK, IRE1, and eIF2α in AD neurons [208, 210, 211, 237–239]. These observations imply that the UPR play a pathological role in the development of AD, since early stages. The adaptive response induced by UPR can modulate ROS production within the ER by reducing the folding demand and upregulating the expression of antioxidant factors [236, 240]. The control of the redox balance by UPR is essentially linked to IRE1 and PERK pathways through the induction of Nrf2-related antioxidant response and ATF4, which plays a key role in glutathione (GSH) synthesis [208, 230, 239]. However, studies on AD brain highlighted that under a condition of chronic UPR activation, an uncoupling between PERK and Nrf2 signals occurs, while ATF4 switches from pro-survival to pro-apoptotic pathways [211]. These data support the inability of UPR to induce stress responses, favoring the build-up of pro-oxidant species and the damage of biological components. Consistent with these findings, redox proteomic studies reported increased HNE modification of BiP/Grp78 during AD pathology [224], which may cause the failure to bind to misfolded proteins thus contributing to the dysfunction of the UPR. In addition, the oxidation of eIF2α, observed in MCI IPL, might lead to its impaired activity leading to UPR defects and exacerbating the accumulation of unfolded/misfolded proteins [241]. Emerging studies also demonstrated a mechanistic link between the ER stress and autophagy in which the activation of PERK and its downstream targets induce autophagy [242] by regulating the gene transcription of autophagy components such as BECN1, MAP1LC3B, ATG5, ATG7 and ATG12. Therefore, autophagy is induced under conditions of ER stress and reports on neurons from human brain demonstrated that high autophagic flux is associated with activated UPR. Furthermore, the UPS overload observed during ER stress conditions in AD may trigger the induction of autophagy to degrade ubiquitinylated proteins [208, 210, 234]. Within this context, AD is characterized by the establishment of a vicious cycle that arises by the increased activation of the UPR combined with decreased efficiency of autophagy, as well as other parts of the proteostasis network, due to the aging process and the increased OS [60, 209, 230].
5.3. UPS
UPS represents one of the main pathways of protein clearance in eukaryotic cells, which not only digests misfolded, oxidized, or damaged proteins, but also eliminates proteins involved in a plethora of intracellular processes [104]. Experimental evidence indicates that disturbance of UPS plays a major role in AD [57, 58, 212, 216, 231, 232]. The activity of proteasome is significantly decreased in the brain of AD [243], and it was proven that proteasome inhibition leads to the accumulation of Aβ and tau. Increased OS/NS levels were suggested as one of the major factors contributing to the impairment of UPS in AD [244]. Indeed, if from one side moderately oxidized proteins were preferentially recognized and degraded by the proteasome, severely oxidized proteins cannot be easily degraded and, instead, inhibit the proteasome [245, 246]. Therefore, the overload of unfolded/oxidized substrates coupled with the oxidative damage of UPS constituents may lead to accumulation of abnormal proteins and to selective degeneration of neurons. The proficiency of the UPS also depends on the activity of deubiquitinating enzymes that play an essential role in determining the rate of protein clearance in cells [58]. Among deubiquitinating enzymes, ubiquitin carboxy-terminal hydrolase L1 was found oxidatively modified in AD brain, and as a consequence its activity was found to be reduced [57, 155, 224, 225, 247]. In addition, data obtained by Saito et al. showed that the E3 ubiquitin ligase—HRD1—was precipitated from solution by OS, [248]. Cecarini et al. [249] demonstrated a significant reduction in proteasome-mediated degradation of oxidized proteins in MCI and AD subjects, and such decreased proteolytic activities were associated with the increase of oxidative modifications such as carbonylation, HNE-modification, and neuroprostane conjugation, thereby confirming a role for OS as a causative factor.
Despite that UPS and autophagy were often considered as two independent proteolytic pathways, recent advances strongly suggest that their activities are carefully orchestrated by multiple crosstalk mechanisms [214]. Indeed, both the pathways share common substrates such as ubiquitinylated proteins and, under specific conditions, they both can selectively degrade short-lived proteins and/or long-lived proteins [104]. In neurodegenerative conditions, where the accumulation of toxic species is profound, cells can reorganize proteolysis regulating the communication between the two protein degrading pathways [90, 159, 210, 213, 214, 219]. In principle, when one proteolytic system is damaged and shows a reduced functionality, the enhanced activity of the other pathway may become a compensatory mechanism necessary to protect neuronal cells against the accumulation of toxic species [231, 250]. An example of the inter-regulation between the UPS and autophagy is the observation that impairment in UPS-mediated degradation leads to an increased autophagic function. The activation of this compensatory mechanism involving autophagy activation allows cells to reduce the number of aggregates formed in response to proteasomal inhibition [104, 124, 208]. The analysis of SH-SY5Y cells overexpressing either the wild-type AβPP gene or the 717 valine-to-glycine AβPP-mutated gene demonstrated that, in addition to the increase of oxidative stress, neuronal cells demonstrated a reorganization of the cellular proteolytic machineries with marked inhibition of proteasome activities, impairment in the autophagic flux and increased expression of compensatory mechanisms as an attempt to rescue proteolysis [251]. The analysis of AD post-mortem brain suggests that the accumulation of amyloid-β plaques arises by the concomitant impairment of both the UPS system and autophagy and a prominent role in this cascade of events might be played by the increased oxidative damage of protein degradation machineries components [57, 74, 91, 92, 103, 161, 216, 232, 252].
6. Conclusions
Several studies have demonstrated the complex etiology of Alzheimer disease as the result of multifaceted toxic mechanisms. This likely explains the failure of several therapies that if correctly target a particular pathological pathway, still leaves ongoing pathological events that act independently from the “selected target”. In this scenario, it becomes a future challenge to design therapeutic strategies that have the potential to intersect multiple pathways if we are able to identify the correct “hub”. Collecting studies suggest that a major cellular hub is represented by OS and mTOR signaling. This signaling network is complex, with several downstream physiological outputs, and the mechanisms underlying its age-related effects still remain enigmatic. These pathways are intimately linked within a complex network of signals that are essential for a healthy longevity. It is likely that alteration of one or more of the components of the mTOR pathway is able to modulate the entire system.
Growing evidence suggest that hyperactivation mTOR signaling pathway play a key role in major pathological mechanisms underlying AD. This aberrant activation correlates with dysfunction of energy metabolism, reduced ATP production and increased oxidative stress levels in a sort of self-sustaining cycle. Understanding the precise molecular mechanisms that foster this vicious cycle is essential to develop mTOR-targeted therapies not only to treat Alzheimer disease but also other neurodegenerative disorders.
HIGHLIGHTS:
Oxidative stress in brain is critically involved in Alzheimer disease progression.
mTORC1 activation in AD & MCI brains leads to inhibition of protein quality control
mTORC1 and mitochondrial functions have significant crosstalk, including metabolism
mTORC1 activation is related to brain insulin resistance and neuronal death in AD.
Selective mTORC1 inhibition activation may be a promising AD therapeutic strategy.
Acknowledgement
This work was supported in part by a NIH grant [AG060056]
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, DeLucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T, Abeta42 is essential for parenchymal and vascular amyloid deposition in mice, Neuron 47(2) (2005) 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].O’Brien RJ, Wong PC, Amyloid precursor protein processing and Alzheimer’s disease, Annu Rev Neurosci 34 (2011) 185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hayden EY, Teplow DB, Amyloid beta-protein oligomers and Alzheimer’s disease, Alzheimers Res Ther 5(6) (2013) 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Selkoe DJ, Hardy J, The amyloid hypothesis of Alzheimer’s disease at 25 years, EMBO Mol Med 8(6) (2016) 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Iqbal K, Liu F, Gong CX, Grundke-Iqbal I, Tau in Alzheimer disease and related tauopathies, Curr Alzheimer Res 7(8) (2010) 656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Medeiros R, Baglietto-Vargas D, LaFerla FM, The role of tau in Alzheimer’s disease and related disorders, CNS Neurosci Ther 17(5) (2011) 514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kelley BJ, Petersen RC, Alzheimer’s disease and mild cognitive impairment, Neurol Clin 25(3) (2007) 577–609, v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Morris JC, Mild cognitive impairment and preclinical Alzheimer’s disease, Geriatrics Suppl (2005) 9–14. [PubMed] [Google Scholar]
- [9].Langa KM, Levine DA, The diagnosis and management of mild cognitive impairment: a clinical review, JAMA 312(23) (2014) 2551–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K, Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry, Acta Neuropathol 112(4) (2006) 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Murayama S, Saito Y, Neuropathological diagnostic criteria for Alzheimer’s disease, Neuropathology 24(3) (2004) 254–60. [DOI] [PubMed] [Google Scholar]
- [12].Chen XQ, Mobley WC, Alzheimer Disease Pathogenesis: Insights From Molecular and Cellular Biology Studies of Oligomeric Abeta and Tau Species, Front Neurosci 13 (2019) 659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Henstridge CM, Hyman BT, Spires-Jones TL, Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis, Nat Rev Neurosci 20(2) (2019) 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu J, Chang L, Song Y, Li H, Wu Y, The Role of NMDA Receptors in Alzheimer’s Disease, Front Neurosci 13 (2019) 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Skaper SD, Facci L, Zusso M, Giusti P, Synaptic Plasticity, Dementia and Alzheimer Disease, CNS Neurol Disord Drug Targets 16(3) (2017) 220–233. [DOI] [PubMed] [Google Scholar]
- [16].Kamat PK, Kalani A, Rai S, Swarnkar S, Tota S, Nath C, Tyagi N, Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies, Mol Neurobiol 53(1) (2016) 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA, Oxidative damage is the earliest event in Alzheimer disease, J Neuropathol Exp Neurol 60(8) (2001) 759–67. [DOI] [PubMed] [Google Scholar]
- [18].Halliwell B, Oxidative stress and neurodegeneration: where are we now?, J Neurochem 97(6) (2006) 1634–58. [DOI] [PubMed] [Google Scholar]
- [19].Di Domenico F, Pupo G, Giraldo E, Badia MC, Monllor P, Lloret A, Schinina ME, Giorgi A, Cini C, Tramutola A, Butterfield DA, Vina J, Perluigi M, Oxidative signature of cerebrospinal fluid from mild cognitive impairment and Alzheimer disease patients, Free Radic Biol Med 91 (2016) 1–9. [DOI] [PubMed] [Google Scholar]
- [20].Head E, Oxidative damage and cognitive dysfunction: antioxidant treatments to promote healthy brain aging, Neurochem Res 34(4) (2009) 670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sohal RS, Ku HH, Agarwal S, Forster MJ, Lal H, Oxidative damage, mitochondrial oxidant generation and antioxidant defenses during aging and in response to food restriction in the mouse, Mech Ageing Dev 74(1–2) (1994) 121–33. [DOI] [PubMed] [Google Scholar]
- [22].Liu R, Liu IY, Bi X, Thompson RF, Doctrow SR, Malfroy B, Baudry M, Reversal of age-related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics, Proc Natl Acad Sci U S A 100(14) (2003) 8526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stadtman ER, Berlett BS, Reactive oxygen-mediated protein oxidation in aging and disease, Chem Res Toxicol 10(5) (1997) 485–94. [DOI] [PubMed] [Google Scholar]
- [24].Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, Bohr VA, Ageing as a risk factor for neurodegenerative disease, Nat Rev Neurol 15(10) (2019) 565–581. [DOI] [PubMed] [Google Scholar]
- [25].Mattson MP, Magnus T, Ageing and neuronal vulnerability, Nat Rev Neurosci 7(4) (2006) 278–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Perluigi M, Barone E, Di Domenico F, Butterfield DA, Aberrant protein phosphorylation in Alzheimer disease brain disturbs pro-survival and cell death pathways, Biochim Biophys Acta 1862(10) (2016) 1871–82. [DOI] [PubMed] [Google Scholar]
- [27].Zhao Y, Zhao B, Oxidative stress and the pathogenesis of Alzheimer’s disease, Oxid Med Cell Longev 2013 (2013) 316523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mondragon-Rodriguez S, Perry G, Zhu X, Moreira PI, Acevedo-Aquino MC, Williams S, Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: implications for Alzheimer’s disease, Oxid Med Cell Longev 2013 (2013) 940603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sultana R, Perluigi M, Newman SF, Pierce WM, Cini C, Coccia R, Butterfield DA, Redox proteomic analysis of carbonylated brain proteins in mild cognitive impairment and early Alzheimer’s disease, Antioxid Redox Signal 12(3) (2010) 327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mangialasche F, Polidori MC, Monastero R, Ercolani S, Camarda C, Cecchetti R, Mecocci P, Biomarkers of oxidative and nitrosative damage in Alzheimer’s disease and mild cognitive impairment, Ageing Res Rev 8(4) (2009) 285–305. [DOI] [PubMed] [Google Scholar]
- [31].Butterfield DA, Lauderback CM, Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress, Free Radic Biol Med 32(11) (2002) 1050–60. [DOI] [PubMed] [Google Scholar]
- [32].Butterfield DA, Drake J, Pocernich C, Castegna A, Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide, Trends Mol Med 7(12) (2001) 548–54. [DOI] [PubMed] [Google Scholar]
- [33].Sultana R, Perluigi M, Butterfield DA, Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain, Free Radic Biol Med 62 (2013) 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR, Evidence of increased oxidative damage in subjects with mild cognitive impairment, Neurology 64(7) (2005) 1152–6. [DOI] [PubMed] [Google Scholar]
- [35].Butterfield DA, Reed T, Perluigi M, De Marco C, Coccia R, Cini C, Sultana R, Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment, Neurosci Lett 397(3) (2006) 170–3. [DOI] [PubMed] [Google Scholar]
- [36].Montine TJ, Neely MD, Quinn JF, Beal MF, Markesbery WR, Roberts LJ, Morrow JD, Lipid peroxidation in aging brain and Alzheimer’s disease, Free Radic Biol Med 33(5) (2002) 620–6. [DOI] [PubMed] [Google Scholar]
- [37].Hardas SS, Sultana R, Clark AM, Beckett TL, Szweda LI, Murphy MP, Butterfield DA, Oxidative modification of lipoic acid by HNE in Alzheimer disease brain, Redox Biol 1 (2013) 80–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hensley K, Hall N, Subramaniam R, Cole P, Harris M, Aksenov M, Aksenova M, Gabbita SP, Wu JF, Carney JM, Lovell M, Markesbery WR, Butterfield DA, Brain Regional Correspondence between Alzheimers-Disease Histopathology and Biomarkers of Protein Oxidation, Journal of Neurochemistry 65(5) (1995) 2146–2156. [DOI] [PubMed] [Google Scholar]
- [39].Bhatt S, Puli L, Patil CR, Role of reactive oxygen species in the progression of Alzheimer’s disease, Drug Discov Today (2020). [DOI] [PubMed] [Google Scholar]
- [40].Marcus C, Mena E, Subramaniam RM, Brain PET in the diagnosis of Alzheimer’s disease, Clin Nucl Med 39(10) (2014) e413–22; quiz e423–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Butterfield DA, Boyd-Kimball D, Redox proteomics and amyloid beta-peptide: insights into Alzheimer disease, J Neurochem 151(4) (2019) 459–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Butterfield DA, Brain lipid peroxidation and alzheimer disease: Synergy between the Butterfield and Mattson laboratories, Ageing Res Rev 64 (2020) 101049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Butterfield DA, Halliwell B, Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease, Nat Rev Neurosci 20(3) (2019) 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Aluise CD, Robinson RA, Beckett TL, Murphy MP, Cai J, Pierce WM, Markesbery WR, Butterfield DA, Preclinical Alzheimer disease: brain oxidative stress, Abeta peptide and proteomics, Neurobiol Dis 39(2) (2010) 221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Aluise CD, Robinson RA, Cai J, Pierce WM, Markesbery WR, Butterfield DA, Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical Alzheimer’s disease: insights into memory loss in MCI, J Alzheimers Dis 23(2) (2011) 257–69. [DOI] [PubMed] [Google Scholar]
- [46].Sayre LM, Perry G, Smith MA, Oxidative stress and neurotoxicity, Chem Res Toxicol 21(1) (2008) 172–88. [DOI] [PubMed] [Google Scholar]
- [47].Santos RX, Correia SC, Zhu X, Lee HG, Petersen RB, Nunomura A, Smith MA, Perry G, Moreira PI, Nuclear and mitochondrial DNA oxidation in Alzheimer’s disease, Free Radic Res 46(4) (2012) 565–76. [DOI] [PubMed] [Google Scholar]
- [48].Lovell MA, Markesbery WR, Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease, Nucleic Acids Res 35(22) (2007) 7497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Levy E, El Banna N, Baille D, Heneman-Masurel A, Truchet S, Rezaei H, Huang ME, Beringue V, Martin D, Vernis L, Causative Links between Protein Aggregation and Oxidative Stress: A Review, Int J Mol Sci 20(16) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chao CC, Ma YS, Stadtman ER, Modification of protein surface hydrophobicity and methionine oxidation by oxidative systems, Proc Natl Acad Sci U S A 94(7) (1997) 2969–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Tanase M, Urbanska AM, Zolla V, Clement CC, Huang L, Morozova K, Follo C, Goldberg M, Roda B, Reschiglian P, Santambrogio L, Role of Carbonyl Modifications on Aging-Associated Protein Aggregation, Sci Rep 6 (2016) 19311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, Vendruscolo M, Hayer-Hartl M, Hartl FU, Vabulas RM, Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions, Cell 144(1) (2011) 67–78. [DOI] [PubMed] [Google Scholar]
- [53].Chhangani D, Mishra A, Protein quality control system in neurodegeneration: a healing company hard to beat but failure is fatal, Mol Neurobiol 48(1) (2013) 141–56. [DOI] [PubMed] [Google Scholar]
- [54].Higuchi-Sanabria R, Frankino PA, Paul JW 3rd, Tronnes SU, Dillin A, A Futile Battle? Protein Quality Control and the Stress of Aging, Dev Cell 44(2) (2018) 139–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pickering AM, Davies KJ, Degradation of damaged proteins: the main function of the 20S proteasome, Prog Mol Biol Transl Sci 109 (2012) 227–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nixon RA, Yang DS, Autophagy failure in Alzheimer’s disease--locating the primary defect, Neurobiol Dis 43(1) (2011) 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, Klein JB, Pierce WM, Booze R, Markesbery WR, Butterfield DA, Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1, Free Radic Biol Med 33(4) (2002) 562–71. [DOI] [PubMed] [Google Scholar]
- [58].Tramutola A, Di Domenico F, Barone E, Perluigi M, Butterfield DA, It Is All about (U)biquitin: Role of Altered Ubiquitin-Proteasome System and UCHL1 in Alzheimer Disease, Oxid Med Cell Longev 2016 (2016) 2756068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kaushik S, Cuervo AM, Chaperone-mediated autophagy: a unique way to enter the lysosome world, Trends Cell Biol 22(8) (2012) 407–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hohn A, Tramutola A, Cascella R, Proteostasis Failure in Neurodegenerative Diseases: Focus on Oxidative Stress, Oxid Med Cell Longev 2020 (2020) 5497046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Morimoto RI, Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging, Genes Dev 22(11) (2008) 1427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Thibaudeau TA, Anderson RT, Smith DM, A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers, Nat Commun 9(1) (2018) 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Vilchez D, Saez I, Dillin A, The role of protein clearance mechanisms in organismal ageing and age-related diseases, Nat Commun 5 (2014) 5659. [DOI] [PubMed] [Google Scholar]
- [64].Dasuri K, Ebenezer P, Zhang L, Fernandez-Kim SO, Bruce-Keller AJ, Markesbery WR, Keller JN, Increased protein hydrophobicity in response to aging and Alzheimer disease, Free Radic Biol Med 48(10) (2010) 1330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bonet-Costa V, Pomatto LC, Davies KJ, The Proteasome and Oxidative Stress in Alzheimer’s Disease, Antioxid Redox Signal 25(16) (2016) 886–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Glick D, Barth S, Macleod KF, Autophagy: cellular and molecular mechanisms, J Pathol 221(1) (2010) 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Anding AL, Baehrecke EH, Cleaning House: Selective Autophagy of Organelles, Dev Cell 41(1) (2017) 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Yuan HX, Xiong Y, Guan KL, Nutrient sensing, metabolism, and cell growth control, Mol Cell 49(3) (2013) 379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Perluigi M, Di Domenico F, Butterfield DA, mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy, Neurobiology of Disease 84 (2015) 39–49. [DOI] [PubMed] [Google Scholar]
- [70].Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, Jaworski J, Molecular neurobiology of mTOR, Neuroscience 341 (2017) 112–153. [DOI] [PubMed] [Google Scholar]
- [71].Maiese K, Targeting the core of neurodegeneration: FoxO, mTOR, and SIRT1, Neural Regen Res 16(3) (2021) 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Di Domenico F, Tramutola A, Foppoli C, Head E, Perluigi M, Butterfield DA, mTOR in Down syndrome: Role in A beta and tau neuropathology and transition to Alzheimer disease-like dementia, Free Radical Bio Med 114 (2018) 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Butterfield DA, Di Domenico F, Tramutola A, Head E, Perluigi M, Oxidative Stress and mTOR Activation in Down Syndrome Brain: Roles in A beta 42 and Tau Neuropathology and Transition to Alzheimer Disease-Like Dementia, Free Radical Bio Med 112 (2017) 70–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, Perluigi M, Butterfield DA, Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD, Journal of Neurochemistry 133(5) (2015) 739–749. [DOI] [PubMed] [Google Scholar]
- [75].Mueed Z, Tandon P, Maurya SK, Deval R, Kamal MA, Poddar NK, Tau and mTOR: The Hotspots for Multifarious Diseases in Alzheimer’s Development, Front Neurosci 12 (2018) 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Van Skike CE, Galvan V, A Perfect sTORm: The Role of the Mammalian Target of Rapamycin (mTOR) in Cerebrovascular Dysfunction of Alzheimer’s Disease: A Mini-Review, Gerontology 64(3) (2018) 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Cai ZY, Chen GH, He WB, Xiao M, Yan LJ, Activation of mTOR: a culprit of Alzheimer’s disease?, Neuropsych Dis Treat 11 (2015) 1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Tramutola A, Lanzillotta C, Di Domenico F, Targeting mTOR to reduce Alzheimer-related cognitive decline: from current hits to future therapies, Expert Rev Neurother 17(1) (2017) 33–45. [DOI] [PubMed] [Google Scholar]
- [79].Johnson SC, Rabinovitch PS, Kaeberlein M, mTOR is a key modulator of ageing and age-related disease, Nature 493(7432) (2013) 338–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Perluigi M, Pupo G, Tramutola A, Cini C, Coccia R, Barone E, Head E, Butterfield DA, Di Domenico F, Neuropathological role of PI3K/Akt/mTOR axis in Down syndrome brain, Bba-Mol Basis Dis 1842(7) (2014) 1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Lanzillotta C, Zuliani I, Vasavda C, Snyder SH, Paul BD, Perluigi M, Di Domenico F, Barone E, BVR-A Deficiency Leads to Autophagy Impairment through the Dysregulation of AMPK/mTOR Axis in the Brain-Implications for Neurodegeneration, Antioxidants (Basel) 9(8) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Singh AK, Kashyap MP, Tripathi VK, Singh S, Garg G, Rizvi SI, Neuroprotection Through Rapamycin-Induced Activation of Autophagy and PI3K/Akt1/mTOR/CREB Signaling Against Amyloid-beta-Induced Oxidative Stress, Synaptic/Neurotransmission Dysfunction, and Neurodegeneration in Adult Rats, Mol Neurobiol 54(8) (2017) 5815–5828. [DOI] [PubMed] [Google Scholar]
- [83].Liu Q, Qiu J, Liang M, Golinski J, van Leyen K, Jung JE, You Z, Lo EH, Degterev A, Whalen MJ, Akt and mTOR mediate programmed necrosis in neurons, Cell Death Dis 5 (2014) e1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Barone E, Di Domenico F, Cassano T, Arena A, Tramutola A, Lavecchia MA, Coccia R, Butterfield DA, Perluigi M, Impairment of biliverdin reductase-A promotes brain insulin resistance in Alzheimer disease: A new paradigm, Free Radic Biol Med 91 (2016) 127–42. [DOI] [PubMed] [Google Scholar]
- [85].Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang HY, Ahima RS, Craft S, Gandy S, Buettner C, Stoeckel LE, Holtzman DM, Nathan DM, Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums, Nat Rev Neurol 14(3) (2018) 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Caccamo A, Belfiore R, Oddo S, Genetically reducing mTOR signaling rescues central insulin dysregulation in a mouse model of Alzheimer’s disease, Neurobiol Aging 68 (2018) 1. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [87].Norambuena A, Wallrabe H, McMahon L, Silva A, Swanson E, Khan SS, Baerthlein D, Kodis E, Oddo S, Mandell JW, Bloom GS, mTOR and neuronal cell cycle reentry: How impaired brain insulin signaling promotes Alzheimer’s disease, Alzheimers Dement 13(2) (2017) 152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Liu H, Qiu H, Xiao Q, Le W, Chronic Hypoxia-Induced Autophagy Aggravates the Neuropathology of Alzheimer’s Disease through AMPK-mTOR Signaling in the APPSwe/PS1dE9 Mouse Model, J Alzheimers Dis 48(4) (2015) 1019–32. [DOI] [PubMed] [Google Scholar]
- [89].Betz C, Hall MN, Where is mTOR and what is it doing there?, J Cell Biol 203(4) (2013) 563–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Di Domenico F, Zuliani I, Tramutola A, Shining a light on defective autophagy by proteomics approaches: implications for neurodegenerative illnesses, Expert Rev Proteomic 16(11–12) (2019) 951–964. [DOI] [PubMed] [Google Scholar]
- [91].Uddin MS, Stachowiak A, Mamun AA, Tzvetkov NT, Takeda S, Atanasov AG, Bergantin LB, Abdel-Daim MM, Stankiewicz AM, Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications, Front Aging Neurosci 10 (2018) 04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Li Q, Liu Y, Sun M, Autophagy and Alzheimer’s Disease, Cell Mol Neurobiol 37(3) (2017) 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Meng T, Lin S, Zhuang H, Huang H, He Z, Hu Y, Gong Q, Feng D, Recent progress in the role of autophagy in neurological diseases, Cell Stress 3(5) (2019) 141–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Martinez-Cisuelo V, Gomez J, Garcia-Junceda I, Naudi A, Cabre R, Mota-Martorell N, Lopez-Torres M, Gonzalez-Sanchez M, Pamplona R, Barja G, Rapamycin reverses age-related increases in mitochondrial ROS production at complex I, oxidative stress, accumulation of mtDNA fragments inside nuclear DNA, and lipofuscin level, and increases autophagy, in the liver of middle-aged mice, Exp Gerontol 83 (2016) 130–8. [DOI] [PubMed] [Google Scholar]
- [95].Ruhlmann C, Wolk T, Blumel T, Stahn L, Vollmar B, Kuhla A, Long-term caloric restriction in ApoE-deficient mice results in neuroprotection via Fgf21-induced AMPK/mTOR pathway, Aging (Albany NY) 8(11) (2016) 2777–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Napolitano G, Esposito A, Choi H, Matarese M, Benedetti V, Di Malta C, Monfregola J, Medina DL, Lippincott-Schwartz J, Ballabio A, mTOR-dependent phosphorylation controls TFEB nuclear export, Nat Commun 9(1) (2018) 3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Heras-Sandoval D, Perez-Rojas JM, Hernandez-Damian J, Pedraza-Chaverri J, The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration, Cell Signal 26(12) (2014) 2694–701. [DOI] [PubMed] [Google Scholar]
- [98].Meijer AJ, Lorin S, Blommaart EF, Codogno P, Regulation of autophagy by amino acids and MTOR-dependent signal transduction, Amino Acids 47(10) (2015) 2037–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Tyagi A, Kamal MA, Poddar NK, Integrated Pathways of COX-2 and mTOR: Roles in Cell Sensing and Alzheimer’s Disease, Front Neurosci 14 (2020) 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Rahman MA, Rahman MS, Rahman MDH, Rasheduzzaman M, Mamun-Or-Rashid A, Uddin MJ, Rahman MR, Hwang H, Pang MG, Rhim H, Modulatory Effects of Autophagy on APP Processing as a Potential Treatment Target for Alzheimer’s Disease, Biomedicines 9(1) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Park H, Kang JH, Lee S, Autophagy in Neurodegenerative Diseases: A Hunter for Aggregates, International Journal of Molecular Sciences 21(9) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Filomeni G, De Zio D, Cecconi F, Oxidative stress and autophagy: the clash between damage and metabolic needs, Cell Death Differ 22(3) (2015) 377–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Di Domenico F, Barone E, Perluigi M, Butterfield DA, The Triangle of Death in Alzheimer’s Disease Brain: The Aberrant Cross-Talk Among Energy Metabolism, Mammalian Target of Rapamycin Signaling, and Protein Homeostasis Revealed by Redox Proteomics, Antioxid Redox Sign 26(8) (2017) 364–387. [DOI] [PubMed] [Google Scholar]
- [104].Chondrogianni N, Petropoulos I, Grimm S, Georgila K, Catalgol B, Friguet B, Grune T, Gonos ES, Protein damage, repair and proteolysis, Mol Aspects Med 35 (2014) 1–71. [DOI] [PubMed] [Google Scholar]
- [105].Chung KM, Hernandez N, Sproul AA, Yu WH, Alzheimer’s disease and the autophagic-lysosomal system, Neurosci Lett 697 (2019) 49–58. [DOI] [PubMed] [Google Scholar]
- [106].Kaeberlein M, Galvan V, Rapamycin and Alzheimer’s disease: Time for a clinical trial?, Sci Transl Med 11(476) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Uddin MS, Mamun AA, Labu ZK, Hidalgo-Lanussa O, Barreto GE, Ashraf GM, Autophagic dysfunction in Alzheimer’s disease: Cellular and molecular mechanistic approaches to halt Alzheimer’s pathogenesis, J Cell Physiol 234(6) (2019) 8094–8112. [DOI] [PubMed] [Google Scholar]
- [108].Talboom JS, Velazquez R, Oddo S, The mammalian target of rapamycin at the crossroad between cognitive aging and Alzheimer’s disease, Npj Aging Mech Dis 1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O’Connor R, O’Neill C, Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology, J Neurochem 93(1) (2005) 105–17. [DOI] [PubMed] [Google Scholar]
- [110].Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ, Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain, FEBS J 272(16) (2005) 4211–20. [DOI] [PubMed] [Google Scholar]
- [111].Pei JJ, Hugon J, mTOR-dependent signalling in Alzheimer’s disease, J Cell Mol Med 12(6B) (2008) 2525–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Pei JJ, Bjorkdahl C, Zhang H, Zhou X, Winblad B, p70 S6 kinase and tau in Alzheimer’s disease, J Alzheimers Dis 14(4) (2008) 385–92. [DOI] [PubMed] [Google Scholar]
- [113].Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, Perluigi M, Butterfield DA, Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD, J Neurochem 133(5) (2015) 739–49. [DOI] [PubMed] [Google Scholar]
- [114].Paccalin M, Pain-Barc S, Pluchon C, Paul C, Besson MN, Carret-Rebillat AS, Rioux-Bilan A, Gil R, Hugon J, Activated mTOR and PKR kinases in lymphocytes correlate with memory and cognitive decline in Alzheimer’s disease, Dement Geriatr Cogn Disord 22(4) (2006) 320–6. [DOI] [PubMed] [Google Scholar]
- [115].Spinelli M, Fusco S, Grassi C, Brain Insulin Resistance and Hippocampal Plasticity: Mechanisms and Biomarkers of Cognitive Decline, Front Neurosci 13 (2019) 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].O’Neill C, Kiely AP, Coakley MF, Manning S, Long-Smith CM, Insulin and IGF-1 signalling: longevity, protein homoeostasis and Alzheimer’s disease, Biochem Soc Trans 40(4) (2012) 721–7. [DOI] [PubMed] [Google Scholar]
- [117].O’ Neill C, PI3-kinase/Akt/mTOR signaling: impaired on/off switches in aging, cognitive decline and Alzheimer’s disease, Exp Gerontol 48(7) (2013) 647–53. [DOI] [PubMed] [Google Scholar]
- [118].Tramutola A, Lanzillotta C, Barone E, Arena A, Zuliani I, Mosca L, Blarzino C, Butterfield DA, Perluigi M, Di Domenico F, Intranasal rapamycin ameliorates Alzheimer-like cognitive decline in a mouse model of Down syndrome, Transl Neurodegener 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Caccamo A, Majumder S, Richardson A, Strong R, Oddo S, Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments, J Biol Chem 285(17) (2010) 13107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Majumder S, Richardson A, Strong R, Oddo S, Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits, PLoS One 6(9) (2011) e25416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V, Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease, PLoS One 5(4) (2010) e9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Cassano T, Magini A, Giovagnoli S, Polchi A, Calcagnini S, Pace L, Lavecchia MA, Scuderi C, Bronzuoli MR, Ruggeri L, Gentileschi MP, Romano A, Gaetani S, De Marco F, Emiliani C, Dolcetta D, Early intrathecal infusion of everolimus restores cognitive function and mood in a murine model of Alzheimer’s disease, Exp Neurol 311 (2019) 88–105. [DOI] [PubMed] [Google Scholar]
- [123].Butterfield DA, Boyd-Kimball D, Oxidative Stress, Amyloid-beta Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease, J Alzheimers Dis 62(3) (2018) 1345–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Cheng J, North BJ, Zhang T, Dai X, Tao K, Guo J, Wei W, The emerging roles of protein homeostasis-governing pathways in Alzheimer’s disease, Aging Cell 17(5) (2018) e12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Sharma N, Tramutola A, Lanzillotta C, Arena A, Blarzino C, Cassano T, Butterfield DA, Di Domenico F, Perluigi M, Barone E, Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease, Neurobiol Dis 125 (2019) 176–189. [DOI] [PubMed] [Google Scholar]
- [126].Lafay-Chebassier C, Perault-Pochat MC, Page G, Rioux Bilan A, Damjanac M, Pain S, Houeto JL, Gil R, Hugon J, The immunosuppressant rapamycin exacerbates neurotoxicity of Abeta peptide, J Neurosci Res 84(6) (2006) 1323–34. [DOI] [PubMed] [Google Scholar]
- [127].Lafay-Chebassier C, Paccalin M, Page G, Barc-Pain S, Perault-Pochat MC, Gil R, Pradier L, Hugon J, mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease, J Neurochem 94(1) (2005) 215–25. [DOI] [PubMed] [Google Scholar]
- [128].Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J, Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts, Nat Cell Biol 5(8) (2003) 741–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].von Zglinicki T, Oxidative stress shortens telomeres, Trends Biochem Sci 27(7) (2002) 339–44. [DOI] [PubMed] [Google Scholar]
- [130].Barone E, Mosser S, Fraering PC, Inactivation of brain Cofilin-1 by age, Alzheimer’s disease and gamma-secretase, Biochim Biophys Acta 1842(12 Pt A) (2014) 2500–9. [DOI] [PubMed] [Google Scholar]
- [131].Hoffmann L, Rust MB, Culmsee C, Actin(g) on mitochondria - a role for cofilin1 in neuronal cell death pathways, Biol Chem 400(9) (2019) 1089–1097. [DOI] [PubMed] [Google Scholar]
- [132].Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, Saretzki G, Rudolph KL, Kirkwood TB, von Zglinicki T, Feedback between p21 and reactive oxygen production is necessary for cell senescence, Mol Syst Biol 6 (2010) 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Mancuso C, Siciliano R, Barone E, Curcumin and Alzheimer disease: this marriage is not to be performed, J Biol Chem 286(3) (2011) le3; author reply le4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Butterfield DA, Di Domenico F, Swomley AM, Head E, Perluigi M, Redox proteomics analysis to decipher the neurobiology of Alzheimer-like neurodegeneration: overlaps in Down’s syndrome and Alzheimer’s disease brain, Biochem J 463(2) (2014) 177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Baker DJ, Peleg S, Biphasic Modeling of Mitochondrial Metabolism Dysregulation during Aging, Trends Biochem Sci 42(9) (2017) 702–711. [DOI] [PubMed] [Google Scholar]
- [136].Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, Rushton MD, Charles M, Jurk D, Tait SW, Czapiewski R, Greaves L, Nelson G, Bohlooly YM, Rodriguez-Cuenca S, Vidal-Puig A, Mann D, Saretzki G, Quarato G, Green DR, Adams PD, von Zglinicki T, Korolchuk VI, Passos JF, Mitochondria are required for pro-ageing features of the senescent phenotype, EMBO J 35(7) (2016) 724–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Di Domenico F, Tramutola A, Barone E, Lanzillotta C, Defever O, Arena A, Zuliani I, Foppoli C, Iavarone F, Vincenzoni F, Castagnola M, Butterfield DA, Perluigi M, Restoration of aberrant mTOR signaling by intranasal rapamycin reduces oxidative damage: Focus on HNE-modified proteins in a mouse model of down syndrome, Redox Biol 23 (2019) 101162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Tramutola A, Lanzillotta C, Arena A, Barone E, Perluigi M, Di Domenico F, Increased Mammalian Target of Rapamycin Signaling Contributes to the Accumulation of Protein Oxidative Damage in a Mouse Model of Down’s Syndrome, Neurodegener Dis 16(1–2) (2016) 62–8. [DOI] [PubMed] [Google Scholar]
- [139].Lanzillotta C, Tramutola A, Di Giacomo G, Marini F, Butterfield DA, Di Domenico F, Perluigi M, Barone E, Insulin resistance, oxidative stress and mitochondrial defects in Ts65dn mice brain: A harmful synergistic path in down syndrome, Free Radic Biol Med 165 (2021) 152–170. [DOI] [PubMed] [Google Scholar]
- [140].Majd S, Power JHT, Oxidative Stress and Decreased Mitochondrial Superoxide Dismutase 2 and Peroxiredoxins 1 and 4 Based Mechanism of Concurrent Activation of AMPK and mTOR in Alzheimer’s Disease, Curr Alzheimer Res 15(8) (2018) 764–776. [DOI] [PubMed] [Google Scholar]
- [141].Kim DI, Lee KH, Gabr AA, Choi GE, Kim JS, Ko SH, Han HJ, Abeta-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis, Biochim Biophys Acta 1863(11) (2016) 2820–2834. [DOI] [PubMed] [Google Scholar]
- [142].Till A, Lakhani R, Burnett SF, Subramani S, Pexophagy: the selective degradation of peroxisomes, Int J Cell Biol 2012 (2012) 512721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kanki T, Klionsky DJ, Mitophagy in yeast occurs through a selective mechanism, J Biol Chem 283(47) (2008) 32386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Kraft C, Deplazes A, Sohrmann M, Peter M, Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease, Nat Cell Biol 10(5) (2008) 602–10. [DOI] [PubMed] [Google Scholar]
- [145].Lakhani R, Vogel KR, Till A, Liu J, Burnett SF, Gibson KM, Subramani S, Defects in GABA metabolism affect selective autophagy pathways and are alleviated by mTOR inhibition, EMBO Mol Med 6(4) (2014) 551–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Gusdon AM, Zhu J, Van Houten B, Chu CT, ATP13A2 regulates mitochondrial bioenergetics through macroautophagy, Neurobiol Dis 45(3) (2012) 962–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Wilson MM, Morley JE, Invited review: Aging and energy balance, J Appl Physiol (1985) 95(4) (2003) 1728–36. [DOI] [PubMed] [Google Scholar]
- [148].Frisard M, Ravussin E, Energy metabolism and oxidative stress: impact on the metabolic syndrome and the aging process, Endocrine 29(1) (2006) 27–32. [DOI] [PubMed] [Google Scholar]
- [149].Fried LP, Tangen CM, Walston J, Newman AB, Hirsch C, Gottdiener J, Seeman T, Tracy R, Kop WJ, Burke G, McBurnie MA, Cardiovascular G Health Study Collaborative Research, Frailty in older adults: evidence for a phenotype, J Gerontol A Biol Sci Med Sci 56(3) (2001) M146–56. [DOI] [PubMed] [Google Scholar]
- [150].Roberts SB, Rosenberg I, Nutrition and aging: changes in the regulation of energy metabolism with aging, Physiol Rev 86(2) (2006) 651–67. [DOI] [PubMed] [Google Scholar]
- [151].Frisard MI, Broussard A, Davies SS, Roberts LJ 2nd, Rood J, de Jonge L, Fang X, Jazwinski SM, Deutsch WA, Ravussin E, Louisiana S Healthy Aging, Aging, resting metabolic rate, and oxidative damage: results from the Louisiana Healthy Aging Study, J Gerontol A Biol Sci Med Sci 62(7) (2007) 752–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Kann O, Kovacs R, Mitochondria and neuronal activity, Am J Physiol Cell Physiol 292(2) (2007) C641–57. [DOI] [PubMed] [Google Scholar]
- [153].Correia SC, Santos RX, Carvalho C, Cardoso S, Candeias E, Santos MS, Oliveira CR, Moreira PI, Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer’s disease and diabetes interrelation, Brain Res 1441 (2012) 64–78. [DOI] [PubMed] [Google Scholar]
- [154].Butterfield DA, Gu LQ, Di Domenico F, Robinson RAS, Mass Spectrometry and Redox Proteomics: Applications in Disease, Mass Spectrom Rev 33(4) (2014) 277–301. [DOI] [PubMed] [Google Scholar]
- [155].Sultana R, Boyd-Kimball D, Poon HF, Cai J, Pierce WM, Klein JB, Merchant M, Markesbery WR, Butterfield DA, Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: an approach to understand pathological and biochemical alterations in AD, Neurobiol Aging 27(11) (2006) 1564–76. [DOI] [PubMed] [Google Scholar]
- [156].Murphy MP, How mitochondria produce reactive oxygen species, Biochem J 417(1) (2009) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Bilsland LG, Nirmalananthan N, Yip J, Greensmith L, Duchen MR, Expression of mutant SOD1 in astrocytes induces functional deficits in motoneuron mitochondria, J Neurochem 107(5) (2008) 1271–83. [DOI] [PubMed] [Google Scholar]
- [158].Casley CS, Land JM, Sharpe MA, Clark JB, Duchen MR, Canevari L, Beta-amyloid fragment 25–35 causes mitochondrial dysfunction in primary cortical neurons, Neurobiol Dis 10(3) (2002) 258–67. [DOI] [PubMed] [Google Scholar]
- [159].Weidling IW, Swerdlow RH, Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis, Experimental Neurology 330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Tran M, Reddy PH, Defective Autophagy and Mitophagy in Aging and Alzheimer’s Disease, Front Neurosci 14 (2020) 612757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Lanzillotta C, Di Domenico F, Perluigi M, Butterfield DA, Targeting Mitochondria in Alzheimer Disease: Rationale and Perspectives, Cns Drugs 33(10) (2019) 957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Starkov AA, Protein-mediated energy-dissipating pathways in mitochondria, Chem Biol Interact 163(1–2) (2006) 133–44. [DOI] [PubMed] [Google Scholar]
- [163].Echtay KS, Mitochondrial uncoupling proteins--what is their physiological role?, Free Radic Biol Med 43(10) (2007) 1351–71. [DOI] [PubMed] [Google Scholar]
- [164].Brand MD, Uncoupling to survive? The role of mitochondrial inefficiency in ageing, Exp Gerontol 35(6–7) (2000) 811–20. [DOI] [PubMed] [Google Scholar]
- [165].Wolkow CA, Iser WB, Uncoupling protein homologs may provide a link between mitochondria, metabolism and lifespan, Ageing Res Rev 5(2) (2006) 196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Mookerjee SA, Divakaruni AS, Jastroch M, Brand MD, Mitochondrial uncoupling and lifespan, Mech Ageing Dev 131(7–8) (2010) 463–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Liu D, Zhang Y, Gharavi R, Park HR, Lee J, Siddiqui S, Telljohann R, Nassar MR, Cutler RG, Becker KG, Mattson MP, The mitochondrial uncoupler DNP triggers brain cell mTOR signaling network reprogramming and CREB pathway up-regulation, J Neurochem 134(4) (2015) 677–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].de la Monte SM, Wands JR, Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease, J Alzheimers Dis 9(2) (2006) 167–81. [DOI] [PubMed] [Google Scholar]
- [169].Montesanto A, Crocco P, Dato S, Geracitano S, Frangipane F, Colao R, Maletta R, Passarino G, Bruni AC, Rose G, Uncoupling protein 4 (UCP4) gene variability in neurodegenerative disorders: further evidence of association in Frontotemporal dementia, Aging (Albany NY) 10(11) (2018) 3283–3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Kellar D, Craft S, Brain insulin resistance in Alzheimer’s disease and related disorders: mechanisms and therapeutic approaches, Lancet Neurol 19(9) (2020) 758–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Barone E, Tramutola A, Triani F, Calcagnini S, Di Domenico F, Ripoli C, Gaetani S, Grassi C, Butterfield DA, Cassano T, Perluigi M, Biliverdin Reductase-A Mediates the Beneficial Effects of Intranasal Insulin in Alzheimer Disease, Mol Neurobiol 56(4) (2019) 2922–2943. [DOI] [PubMed] [Google Scholar]
- [172].Kapogiannis D, Mustapic M, Shardell MD, Berkowitz ST, Diehl TC, Spangler RD, Tran J, Lazaropoulos MP, Chawla S, Gulyani S, Eitan E, An Y, Huang CW, Oh ES, Lyketsos CG, Resnick SM, Goetzl EJ, Ferrucci L, Association of Extracellular Vesicle Biomarkers With Alzheimer Disease in the Baltimore Longitudinal Study of Aging, JAMA Neurol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Butterfield DA, Di Domenico F, Barone E, Elevated risk of type 2 diabetes for development of Alzheimer disease: a key role for oxidative stress in brain, Biochim Biophys Acta 1842(9) (2014) 1693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Triani F, Tramutola A, Di Domenico F, Sharma N, Butterfield DA, Head E, Perluigi M, Barone E, Biliverdin reductase-A impairment links brain insulin resistance with increased Abeta production in an animal model of aging: Implications for Alzheimer disease, Biochim Biophys Acta Mol Basis Dis 1864(10) (2018) 3181–3194. [DOI] [PubMed] [Google Scholar]
- [175].Imamura T, Yanagihara YT, Ohyagi Y, Nakamura N, Iinuma KM, Yamasaki R, Asai H, Maeda M, Murakami K, Irie K, Kira JI, Insulin deficiency promotes formation of toxic amyloid-beta42 conformer co-aggregating with hyper-phosphorylated tau oligomer in an Alzheimer’s disease model, Neurobiol Dis 137 (2020) 104739. [DOI] [PubMed] [Google Scholar]
- [176].Eren E, Hunt JFV, Shardell M, Chawla S, Tran J, Gu J, Vogt NM, Johnson SC, Bendlin BB, Kapogiannis D, Extracellular vesicle biomarkers of Alzheimer’s disease associated with sub-clinical cognitive decline in late middle age, Alzheimers Dement 16(9) (2020) 1293–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Caccamo A, De Pinto V, Messina A, Branca C, Oddo S, Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature, J Neurosci 34(23) (2014) 7988–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Copps KD, White MF, Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2, Diabetologia 55(10) (2012) 2565–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].White MF, Copps KD, The Mechanisms of Insulin Action, Endocrinology: Adult and Pediatric (Seventh Edition), 20162016, pp. 556–585.e13. [Google Scholar]
- [180].Caccamo A, Branca C, Talboom JS, Shaw DM, Turner D, Ma L, Messina A, Huang Z, Wu J, Oddo S, Reducing Ribosomal Protein S6 Kinase 1 Expression Improves Spatial Memory and Synaptic Plasticity in a Mouse Model of Alzheimer’s Disease, J Neurosci 35(41) (2015) 14042–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Silva DF, Selfridge JE, Lu J, Roy LE,N, Hutfles L, Burns JM, Michaelis EK, Yan S, Cardoso SM, Swerdlow RH, Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines, Hum Mol Genet 22(19) (2013) 3931–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Norambuena A, Wallrabe H, Cao R, Wang DB, Silva A, Svindrych Z, Periasamy A, Hu S, Tanzi RE, Kim DY, Bloom GS, A novel lysosome-to-mitochondria signaling pathway disrupted by amyloid-beta oligomers, EMBO J 37(22) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Lauderback CM, Hackett JM, Huang FF, Keller JN, Szweda LI, Markesbery WR, Butterfield DA, The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Abeta1–42, J Neurochem 78(2) (2001) 413–6. [DOI] [PubMed] [Google Scholar]
- [184].Santos RX, Correia SC, Alves MG, Oliveira PF, Cardoso S, Carvalho C, Seica R, Santos MS, Moreira PI, Mitochondrial quality control systems sustain brain mitochondrial bioenergetics in early stages of type 2 diabetes, Mol Cell Biochem 394(1–2) (2014) 13–22. [DOI] [PubMed] [Google Scholar]
- [185].Moreira PI, Santos MS, Moreno AM, Seica R, Oliveira CR, Increased vulnerability of brain mitochondria in diabetic (Goto-Kakizaki) rats with aging and amyloid-beta exposure, Diabetes 52(6) (2003) 1449–56. [DOI] [PubMed] [Google Scholar]
- [186].Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G, The hallmarks of aging, Cell 153(6) (2013) 1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG, Premature ageing in mice expressing defective mitochondrial DNA polymerase, Nature 429(6990) (2004) 417–23. [DOI] [PubMed] [Google Scholar]
- [188].Dhillon RS, Denu JM, Using comparative biology to understand how aging affects mitochondrial metabolism, Mol Cell Endocrinol 455 (2017) 54–61. [DOI] [PubMed] [Google Scholar]
- [189].Moiseeva O, Bourdeau V, Roux A, Deschenes-Simard X, Ferbeyre G, Mitochondrial dysfunction contributes to oncogene-induced senescence, Mol Cell Biol 29(16) (2009) 4495–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Koubova J, Guarente L, How does calorie restriction work?, Genes Dev 17(3) (2003) 313–21. [DOI] [PubMed] [Google Scholar]
- [191].Fontana L, Partridge L, Longo VD, Extending healthy life span--from yeast to humans, Science 328(5976) (2010) 321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Hwangbo DS, Lee HY, Abozaid LS, Min KJ, Mechanisms of Lifespan Regulation by Calorie Restriction and Intermittent Fasting in Model Organisms, Nutrients 12(4) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Garelick MG, Kennedy BK, TOR on the brain, Exp Gerontol 46(2–3) (2011) 155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Greer EL, Brunet A, Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans, Aging Cell 8(2) (2009) 113–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Moroz N, Carmona JJ, Anderson E, Hart AC, Sinclair DA, Blackwell TK, Dietary restriction involves NAD(+) -dependent mechanisms and a shift toward oxidative metabolism, Aging Cell 13(6) (2014) 1075–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Guo J, Bakshi V, Lin AL, Early Shifts of Brain Metabolism by Caloric Restriction Preserve White Matter Integrity and Long-Term Memory in Aging Mice, Front Aging Neurosci 7 (2015) 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Sahm A, Bens M, Szafranski K, Holtze S, Groth M, Gorlach M, Calkhoven C, Muller C, Schwab M, Kraus J, Kestler HA, Cellerino A, Burda H, Hildebrandt T, Dammann P, Platzer M, Long-lived rodents reveal signatures of positive selection in genes associated with lifespan, PLoS Genet 14(3) (2018) e1007272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Zhang J, Kim J, Alexander A, Cai S, Tripathi DN, Dere R, Tee AR, Tait-Mulder J, Di Nardo A, Han JM, Kwiatkowski E, Dunlop EA, Dodd KM, Folkerth RD, Faust PL, Kastan MB, Sahin M, Walker CL, A tuberous sclerosis complex signalling node at the peroxisome regulates mTORC1 and autophagy in response to ROS, Nat Cell Biol 15(10) (2013) 1186–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Mendiola-Precoma J, Berumen LC, Padilla K, Garcia-Alcocer G, Therapies for Prevention and Treatment of Alzheimer’s Disease, Biomed Res Int 2016 (2016) 2589276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].McGrattan AM, McGuinness B, McKinley MC, Kee F, Passmore P, Woodside JV, McEvoy CT, Diet and Inflammation in Cognitive Ageing and Alzheimer’s Disease, Curr Nutr Rep 8(2) (2019) 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Rusek M, Pluta R, Ulamek-Koziol M, Czuczwar SJ, Ketogenic Diet in Alzheimer’s Disease, Int J Mol Sci 20(16) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Taylor MK, Swerdlow RH, Sullivan DK, Dietary Neuroketotherapeutics for Alzheimer’s Disease: An Evidence Update and the Potential Role for Diet Quality, Nutrients 11(8) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Fontana L, Meyer TE, Klein S, Holloszy JO, Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans, Proc Natl Acad Sci U S A 101(17) (2004) 6659–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Longo VD, Antebi A, Bartke A, Barzilai N, Brown-Borg HM, Caruso C, Curiel TJ, de Cabo R, Franceschi C, Gems D, Ingram DK, Johnson TE, Kennedy BK, Kenyon C, Klein S, Kopchick JJ, Lepperdinger G, Madeo F, Mirisola MG, Mitchell JR, Passarino G, Rudolph KL, Sedivy JM, Shadel GS, Sinclair DA, Spindler SR, Suh Y, Vijg J, Vinciguerra M, Fontana L, Interventions to Slow Aging in Humans: Are We Ready?, Aging Cell 14(4) (2015) 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Nixon RA, Autophagy, amyloidogenesis and Alzheimer disease, J Cell Sci 120(Pt 23) (2007) 4081–91. [DOI] [PubMed] [Google Scholar]
- [206].Hands SL, Proud CG, Wyttenbach A, mTOR’s role in ageing: protein synthesis or autophagy?, Aging (Albany NY) 1(7) (2009) 586–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Garcia-Arencibia M, Hochfeld WE, Toh PP, Rubinsztein DC, Autophagy, a guardian against neurodegeneration, Semin Cell Dev Biol 21(7) (2010) 691–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Matus S, Lisbona F, Torres M, Leon C, Thielen P, Hetz C, The stress rheostat: An interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration, Curr Mol Med 8(3) (2008) 157–172. [DOI] [PubMed] [Google Scholar]
- [209].Lanzillotta C, Di Domenico F, Stress Responses in Down Syndrome Neurodegeneration: State of the Art and Therapeutic Molecules, Biomolecules 11(2) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Scheper W, Nijholt DAT, Hoozemans JJM, The unfolded protein response and proteostasis in Alzheimer disease Preferential activation of autophagy by endoplasmic reticulum stress, Autophagy 7(8) (2011) 910–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Lanzillotta C, Zuliani I, Tramutola A, Barone E, Blarzino C, Folgiero V, Caforio M, Valentini D, Villani A, Locatelli F, Butterfield DA, Head E, Perluigi M, Abisambra JF, Di Domenico F, Chronic PERK induction promotes Alzheimer-like neuropathology in Down syndrome: Insights for therapeutic intervention, Prog Neurobiol 196 (2021). [DOI] [PubMed] [Google Scholar]
- [212].Weng FL, He L, Disrupted ubiquitin proteasome system underlying tau accumulation in Alzheimer’s disease, Neurobiol Aging 99 (2021) 79–85. [DOI] [PubMed] [Google Scholar]
- [213].Cuanalo-Contreras K, Mukherjee A, Soto C, Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging, Int J Cell Biol 2013 (2013) 638083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Cecarini V, Bonfili L, Cuccioloni M, Mozzicafreddo M, Angeletti M, Keller JN, Eleuteri AM, The fine-tuning of proteolytic pathways in Alzheimer’s disease, Cell Mol Life Sci 73(18) (2016) 3433–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Giorgi C, Bouhamida E, Danese A, Previati M, Pinton P, Patergnani S, Relevance of Autophagy and Mitophagy Dynamics and Markers in Neurodegenerative Diseases, Biomedicines 9(2) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Tramutola A, Triani F, Di Domenico F, Barone E, Cai J, Klein JB, Perluigi M, Butterfield DA, Poly-ubiquitin profile in Alzheimer disease brain, Neurobiology of Disease 118 (2018) 129–141. [DOI] [PubMed] [Google Scholar]
- [217].Pajares M, Jimenez-Moreno N, Dias IHK, Debelec B, Vucetic M, Fladmark KE, Basaga H, Ribaric S, Milisav I, Cuadrado A, Redox control of protein degradation, Redox Biol 6 (2015) 409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Hagiwara M, Nagata K, Redox-dependent protein quality control in the endoplasmic reticulum: folding to degradation, Antioxid Redox Signal 16(10) (2012) 1119–28. [DOI] [PubMed] [Google Scholar]
- [219].Llanos-Gonzalez E, Henares-Chavarino AA, Pedrero-Prieto CM, Garcia-Carpintero S, Frontinan-Rubio J, Sancho-Bielsa FJ, Alcain FJ, Peinado JR, Rabanal-Ruiz Y, Duran-Prado M, Interplay Between Mitochondrial Oxidative Disorders and Proteostasis in Alzheimer’s Disease, Front Neurosci-Switz 13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Di Domenico F, Sultana R, Ferree A, Smith K, Barone E, Perluigi M, Coccia R, Pierce W, Cai J, Mancuso C, Squillace R, Wiengele M, Dalle-Donne I, Wolozin B, Butterfield DA, Redox proteomics analyses of the influence of co-expression of wild-type or mutated LRRK2 and Tau on C. elegans protein expression and oxidative modification: relevance to Parkinson disease, Antioxid Redox Signal 17(11) (2012) 1490–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Zheng L, Terman A, Hallbeck M, Dehvari N, Cowburn RF, Benedikz E, Kagedal K, Cedazo-Minguez A, Marcusson J, Macroautophagy-generated increase of lysosomal amyloid beta-protein mediates oxidant-induced apoptosis of cultured neuroblastoma cells, Autophagy 7(12) (2011) 1528–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [222].Muche A, Arendt T, Schliebs R, Oxidative stress affects processing of amyloid precursor protein in vascular endothelial cells, PLoS One 12(6) (2017) e0178127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Di Domenico F, Tramutola A, Barone E, Lanzillotta C, Defever O, Arena A, Zuliani I, Foppoli C, Iavarone F, Vincenzoni F, Castagnola M, Butterfieldg DA, Perluigi M, Restoration of aberrant mTOR signaling by intranasal rapamycin reduces oxidative damage: Focus on HNE-modified proteins in a mouse model of down syndrome, Redox Biology 23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Di Domenico F, Pupo G, Tramutola A, Giorgi A, Schinina ME, Coccia R, Head E, Butterfield DA, Perluigi M, Redox proteomics analysis of HNE-modified proteins in Down syndrome brain: clues for understanding the development of Alzheimer disease, Free Radic Biol Med 71 (2014) 270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Di Domenico F, Coccia R, Cocciolo A, Murphy MP, Cenini G, Head E, Butterfield DA, Giorgi A, Schinina ME, Mancuso C, Cini C, Perluigi M, Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: redox proteomics analysis of human brain, Biochim Biophys Acta 1832(8) (2013) 1249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Di Domenico F, Tramutola A, Perluigi M, Cathepsin D as a therapeutic target in Alzheimer’s disease, Expert Opin Ther Tar 20(12) (2016) 1393–1395. [DOI] [PubMed] [Google Scholar]
- [227].Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA, Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease, J Neurosci 28(27) (2008) 6926–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM, mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase, Science 334(6056) (2011) 678–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Wang S, Kaufman RJ, The impact of the unfolded protein response on human disease, J Cell Biol 197(7) (2012) 857–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Walter P, Ron D, The unfolded protein response: from stress pathway to homeostatic regulation, Science 334(6059) (2011) 1081–6. [DOI] [PubMed] [Google Scholar]
- [231].Zhang Y, Chen X, Zhao Y, Ponnusamy M, Liu Y, The role of ubiquitin proteasomal system and autophagy-lysosome pathway in Alzheimer’s disease, Rev Neurosci 28(8) (2017) 861–868. [DOI] [PubMed] [Google Scholar]
- [232].Cao J, Zhong MB, Toro CA, Zhang L, Cai D, Endo-lysosomal pathway and ubiquitin-proteasome system dysfunction in Alzheimer’s disease pathogenesis, Neurosci Lett 703 (2019) 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Ruggiano A, Foresti O, Carvalho P, Quality control: ER-associated degradation: protein quality control and beyond, J Cell Biol 204(6) (2014) 869–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K, Autophagy is activated for cell survival after endoplasmic reticulum stress, Mol Cell Biol 26(24) (2006) 9220–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Bernales S, McDonald KL, Walter P, Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response, PLoS biology 4(12) (2006) e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ, Endoplasmic reticulum dysfunction in neurological disease, Lancet Neurol 12(1) (2013) 105–18. [DOI] [PubMed] [Google Scholar]
- [237].Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Eikelenboom P, Scheper W, The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus, Am J Pathol 174(4) (2009) 1241–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W, The unfolded protein response is activated in Alzheimer’s disease, Acta Neuropathol 110(2) (2005) 165–72. [DOI] [PubMed] [Google Scholar]
- [239].Hoozemans JJM, Scheper W, Endoplasmic reticulum: The unfolded protein response is tangled in neurodegeneration, Int J Biochem Cell B 44(8) (2012) 1295–1298. [DOI] [PubMed] [Google Scholar]
- [240].Chakrabarti A, Chen AW, Varner JD, A review of the mammalian unfolded protein response, Biotechnology and bioengineering 108(12) (2011) 2777–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, Butterfield DA, Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease, Neurobiol Dis 30(1) (2008) 107–20. [DOI] [PubMed] [Google Scholar]
- [242].Cai Y, Arikkath J, Yang L, Guo ML, Periyasamy P, Buch S, Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders, Autophagy 12(2) (2016) 225–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Keller JN, Dimayuga E, Chen Q, Thorpe J, Gee J, Ding Q, Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain, Int J Biochem Cell Biol 36(12) (2004) 2376–91. [DOI] [PubMed] [Google Scholar]
- [244].Butterfield DA, Perluigi M, Sultana R, Oxidative stress in Alzheimer’s disease brain: new insights from redox proteomics, European journal of pharmacology 545(1) (2006) 39–50. [DOI] [PubMed] [Google Scholar]
- [245].Grune T, Reinheckel T, Davies KJ, Degradation of oxidized proteins in mammalian cells, FASEB journal : official publication of the Federation of American Societies for Experimental Biology 11(7) (1997) 526–34. [PubMed] [Google Scholar]
- [246].Reinheckel T, Sitte N, Ullrich O, Kuckelkorn U, Davies KJ, Grune T, Comparative resistance of the 20S and 26S proteasome to oxidative stress, Biochem J 335 (Pt 3) (1998) 637–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Choi J, Levey AI, Weintraub ST, Rees HD, Gearing M, Chin LS, Li L, Oxidative modifications and down-regulation of ubiquitin carboxyl-terminal hydrolase L1 associated with idiopathic Parkinson’s and Alzheimer’s diseases, J Biol Chem 279(13) (2004) 13256–64. [DOI] [PubMed] [Google Scholar]
- [248].Saito R, Kaneko M, Kitamura Y, Takata K, Kawada K, Okuma Y, Nomura Y, Effects of oxidative stress on the solubility of HRD1, a ubiquitin ligase implicated in Alzheimer’s disease, PLoS One 9(5) (2014) e94576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [249].Cecarini V, Ding Q, Keller JN, Oxidative inactivation of the proteasome in Alzheimer’s disease, Free Radic Res 41(6) (2007) 673–80. [DOI] [PubMed] [Google Scholar]
- [250].Desai S, Juncker M, Kim C, Regulation of mitophagy by the ubiquitin pathway in neurodegenerative diseases, Exp Biol Med 243(6) (2018) 554–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Cecarini V, Bonfili L, Cuccioloni M, Mozzicafreddo M, Rossi G, Buizza L, Uberti D, Angeletti M, Eleuteri AM, Crosstalk between the ubiquitin-proteasome system and autophagy in a human cellular model of Alzheimer’s disease, Biochim Biophys Acta 1822(11) (2012) 1741–51. [DOI] [PubMed] [Google Scholar]
- [252].Tramutola A, Sharma N, Barone E, Lanzillotta C, Castellani A, Iavarone F, Vincenzoni F, Castagnola M, Butterfield DA, Gaetani S, Cassano T, Perluigi M, Di Domenico F, Proteomic identification of altered protein O-GlcNAcylation in a triple transgenic mouse model of Alzheimer’s disease, Bba-Mol Basis Dis 1864(10) (2018) 3309–3321. [DOI] [PubMed] [Google Scholar]