

Abstract
Myocardial fibrosis, the expansion of the cardiac interstitium through deposition of extracellular matrix proteins, is a common pathophysiologic companion of many different myocardial conditions. Fibrosis may reflect activation of reparative or maladaptive processes. Activated fibroblasts and myofibroblasts are the central cellular effectors in cardiac fibrosis, serving as the main source of matrix proteins. Immune cells, vascular cells and cardiomyocytes may also acquire a fibrogenic phenotype under conditions of stress, activating fibroblast populations. Fibrogenic growth factors (such as transforming growth factor-β and platelet-derived growth factors), cytokines [including tumour necrosis factor-α, interleukin (IL)-1, IL-6, IL-10, and IL-4], and neurohumoral pathways trigger fibrogenic signalling cascades through binding to surface receptors, and activation of downstream signalling cascades. In addition, matricellular macromolecules are deposited in the remodelling myocardium and regulate matrix assembly, while modulating signal transduction cascades and protease or growth factor activity. Cardiac fibroblasts can also sense mechanical stress through mechanosensitive receptors, ion channels and integrins, activating intracellular fibrogenic cascades that contribute to fibrosis in response to pressure overload. Although subpopulations of fibroblast-like cells may exert important protective actions in both reparative and interstitial/perivascular fibrosis, ultimately fibrotic changes perturb systolic and diastolic function, and may play an important role in the pathogenesis of arrhythmias. This review article discusses the molecular mechanisms involved in the pathogenesis of cardiac fibrosis in various myocardial diseases, including myocardial infarction, heart failure with reduced or preserved ejection fraction, genetic cardiomyopathies, and diabetic heart disease. Development of fibrosis-targeting therapies for patients with myocardial diseases will require not only understanding of the functional pluralism of cardiac fibroblasts and dissection of the molecular basis for fibrotic remodelling, but also appreciation of the pathophysiologic heterogeneity of fibrosis-associated myocardial disease.
Keywords: Fibrosis, Myofibroblast, Growth factor, Heart failure, Extracellular matrix
1. Introduction
The term fibrosis is used to describe the excessive deposition of extracellular matrix (ECM) proteins in parenchymal tissues, and typically reflects inappropriate or unrestrained activation of a reparative programme. Fibrosis is an important cause of organ dysfunction in many different diseases, including interstitial lung diseases, liver cirrhosis, progressive systemic sclerosis, and diabetic nephropathy.1
Myocardial fibrosis, the expansion of the cardiac interstitium due to net accumulation of ECM proteins, accompanies most cardiac pathologic conditions.2,3 Although in most myocardial diseases, the extent of cardiac fibrosis predicts adverse outcome, fibrosis is not necessarily the primary cause of dysfunction. The adult mammalian heart has negligible regenerative capacity and heals through formation of a scar. Thus, in many cases, cardiac fibrosis is reparative, reflecting replacement of dead cardiomyocytes with a collagen-based scar. Myocardial infarction (MI) is a typical example of reparative fibrosis, as sudden death of a large number of cardiomyocytes stimulates inflammation and subsequent activation of reparative myofibroblasts, leading to formation of a scar. The scar lacks contractile capacity, but serves a critical protective role, maintaining the structural integrity of the chamber, and in the case of a transmural infarction, preventing catastrophic mechanical complications, such as cardiac rupture.4 In other cardiac diseases, fibrosis predominantly involves the interstitium, and develops insidiously in the absence of significant cardiomyocyte loss. For example, systemic hypertension is associated with progressive interstitial and perivascular deposition of ECM proteins that increase myocardial stiffness and cause diastolic dysfunction. Ageing, obesity, and diabetes can also promote myocardial interstitial fibrosis and reduce ventricular compliance, potentially contributing to the pathogenesis of heart failure with preserved ejection fraction (HFpEF).
Thus, cardiac fibrosis is not a single disease entity that would predictably benefit from a standardized therapeutic intervention, but rather a pathologic response that may be appropriate or inappropriate, depending on the pathophysiologic context. This review article discusses the pathophysiologic heterogeneity of myocardial fibrotic responses, focusing on the functional consequences of cardiac fibrosis, the key cellular effectors, and the main molecular mechanisms. The article also highlights the unique characteristics of fibrotic remodelling in various myocardial diseases and discusses the benefits and perils of possible therapeutic interventions.
2. The normal cardiac interstitium
In adult mammalian hearts, cardiomyocytes occupy approximately 75% of the myocardial volume and are organized into laminae, 2–5 cells thick. These layers of cardiomyocytes are surrounded by an interstitial ECM network, comprised predominantly of fibrillar collagens.5,6 The cardiac ECM not only serves as a mechanical scaffold but is also important for transmission of contractile force. In addition to type I collagen (the most abundant protein in the cardiac ECM) and type III collagen,7,8 the cardiac ECM also contains a wide range of glycoproteins, glycosaminoglycans, and proteoglycans, and a reservoir of stored latent growth factors and proteases, that can be rapidly activated following injury to stimulate repair. Several different cell types are enmeshed within the interstitial ECM. Vascular endothelial cells are the most abundant non-cardiomyocytes in adult mouse hearts, reflecting the rich microvasculature of the myocardium.9 Smaller populations of immune cells (macrophages, mast cells, and dendritic cells), and vascular mural cells (smooth muscle cells and pericytes) are also found in the normal cardiac interstitium.10–13 Cardiac fibroblasts, the main matrix-producing cells, form one of the largest cell populations in normal mammalian hearts and are typically enmeshed in the endomysium and perimysium.14,15 The abundance of fibroblasts in uninjured hearts may suggest an important homeostatic role; however, the functions of cardiac fibroblasts in the absence of injury remain poorly defined. Resident cardiac fibroblasts may serve to maintain the structural integrity of the matrix network,16 regulating collagen turnover, a process that requires ongoing synthesis and degradation of ECM proteins.17,18 Fibroblast-specific baseline activation of Smad3 signalling has been reported to maintain normal ECM content in adult mouse hearts,19 whereas platelet-derived growth factor receptor (PDGFR)-α signalling was found to be important for fibroblast survival and optimal support of the normal microvascular network.20 However, these actions had no significant effects on baseline organ function. It has been suggested that cardiac fibroblast subpopulations may also support cardiomyocyte survival and function; however, robust evidence documenting such actions in normal adult hearts is lacking. In uninjured mouse hearts, fibroblasts exhibit heterogeneity and can be subclassified into several subsets on the basis of their transcriptomic profile.21 Whether these subsets exhibit distinct roles in cardiac homeostasis remains unknown.
3. Classification of myocardial fibrotic lesions
Basic histopathological analysis allows classification of cardiac fibrotic lesions into three distinct forms (Figure 1). In MI, necrotic cardiomyocytes are replaced by collagen-based scar, causing ‘replacement fibrosis’. ‘Interstitial fibrosis’ describes the expansion of the endomysial and perimysial space, caused by net accumulation of ECM proteins in the absence of significant cardiomyocyte loss. The term ‘perivascular fibrosis’ is used to describe the expansion of the microvascular adventitia. This simple and crude classification has major implications regarding the pathophysiologic basis of the fibrotic lesion and its effects on cardiac function. Replacement fibrosis is typically reparative, reflecting the absence of regenerative capacity of the adult mammalian heart in response to primary necrotic injury. Interstitial and perivascular fibrosis on the other hand may result from prolonged activation of fibrogenic stimuli and may represent primary injurious processes. Perivascular fibrotic changes are prominent in hypertensive heart disease and may reflect fibroblast conversion or fibrogenic activation of vascular endothelial or mural cells,22,23 or stimulation of adventitial fibroblasts.24 Whether perivascular and interstitial fibrotic changes have distinct cell biological origins remains unknown. Replacement fibrosis is predominantly associated with systolic dysfunction. In contrast, interstitial and perivascular fibrosis typically reduce left ventricular compliance perturbing diastolic function with less pronounced or late effects on systolic function. Moreover, perivascular fibrosis may perturb coronary follow and induce microvascular dysfunction25,26 precipitating diastolic dysfunction and worsening the severity of ischaemia.
Figure 1.

Histological types of cardiac fibrosis. Images show sections of adult mouse hearts stained with picrosirius red in order to label collagen fibres. (A) The normal adult mammalian heart contains an intricate network of collagenous matrix. Every cardiomyocyte is surrounded by a thin network of endomysial collagen. Thicker perimysial collagen fibres (arrows) enwrap bundles of cardiomyocytes. (B) Following myocardial infarction, dead cardiomyocytes are replaced by collagen-based scar. Replacement fibrosis is shown in a mouse model of reperfused myocardial infarction (arrows, 1 h ischaemia–7 days reperfusion). Infarction is midmyocardial; interstitial fibrosis is noted in remodelling subendocardial and subepicardial areas (arrowheads). (C) Interstitial fibrosis (arrows) is noted in a model of left ventricular pressure overload, induced through transverse aortic constriction (7 days). (D) Perivascular fibrosis (arrows) is also noted in the pressure-overloaded myocardium, with large amounts of collagen surrounding an intracoronary vessel (*). Scalebar = 80 µm.
More sophisticated analysis of the cellular composition, molecular and biochemical profile of fibrotic lesions could be used to gain additional information with mechanistic, diagnostic and potentially also prognostic significance. The relative abundance of immune cells, the phenotype and level of activation of fibroblasts and myofibroblasts, the expression of matricellular proteins and the levels of ECM cross-linking could provide key insights into the pathogenesis, activity and reversibility of myocardial fibrosis. In human patients with ischaemic cardiomyopathy undergoing aortocoronary bypass surgery, highly cellular interstitial fibrosis, characterized by increased inflammatory activity, and higher levels of the matricellular protein tenascin-C was associated with more dynamic lesions and with an increased likelihood of recovery after aortocoronary bypass surgery.27,28 On the other hand, higher levels of ECM cross-linking and a hypocellular interstitium may mark fibrotic lesions with low potential reversibility.29
4. The cell biology of cardiac fibrosis
Activated fibroblasts are the central cellular effectors of myocardial fibrosis, serving as the main ECM-producing cells. Conversion of fibroblasts into secretory, matrix-producing and contractile cells, called myofibroblasts, is a crucial cellular event in many fibrotic conditions (Figure 2). Activated myofibroblasts are the main source of structural ECM proteins in fibrotic hearts,31 produce large amounts of matricellular proteins,32,33 and can also contribute to the regulation of matrix remodelling by producing proteases, such as matrix metalloproteinases (MMPs), and their inhibitors.34 The expansion and activation of fibroblasts in remodelling hearts may involve direct effects of fibrogenic mediators, or upstream stimulation of other cell types, including immune cells, vascular cells and cardiomyocytes. Immune cells (macrophages, lymphocytes, mast cells, and eosinophils) may promote fibroblast activation by secreting cytokines, growth factors, and matricellular proteins. Vascular endothelial cells and pericytes may also secrete fibroblast-activating mediators, and have been reported to undergo myofibroblast conversion. Injured cardiomyocytes are also capable of producing fibrogenic mediators in response to stress, contributing to fibroblast activation. Moreover, in MI, death of cardiomyocytes and other myocardial cells results in release of damage-associated molecular patterns that activate inflammation, ultimately leading to fibrosis. The relative role of various cell types in the fibrotic response is dependent on the type of myocardial injury.
Figure 2.
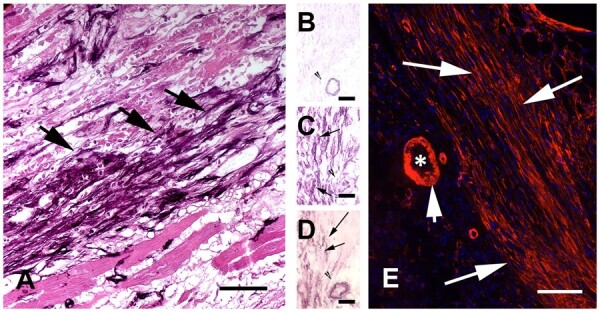
The myofibroblasts are the main cellular effectors of fibrosis in injured and remodelling hearts. Myofibroblasts are activated fibroblasts that express contractile proteins, such as α-SMA and secrete large amounts of ECM proteins. Images show abundant myofibroblasts in infarcted hearts in both large animal and rodent models. (A) Immunohistochemical staining identifies non-vascular α-SMA+ myofibroblasts in the border zone of a reperfused canine infarct (arrows, 1 h ischaemia/7 days reperfusion). (B–D) Serial staining of frozen sections from the infarcted canine heart show that border zone infarct myofibroblasts express α-SMA (D) and the embryonal isoform of smooth muscle myosin (SMemb) (C), but not the SM2 isoform, a marker of mature smooth muscle cells (B). Thus, these cells (arrows) are myofibroblasts and not smooth muscle cells. Arrowhead shows an arteriole, exhibiting expression of α-SMA (D) and SM2 (B). (E) Abundant α-SMA+ myofibroblasts are found in the border zone of a mouse infarct (7 days ischaemia). The short arrow points to the vascular smooth muscle cells on the arteriolar (*) media, which are also strongly positive for α-SMA. Scalebar = 80 µm. Original data published in ref.30
4.1 Fibroblasts and myofibroblasts: the central effector cells in cardiac fibrosis
The term fibroblast is used to describe cells of mesenchymal origin that populate connective tissues, lack a basement membrane and are capable of producing significant amounts of ECM proteins. Under this broad definition, a wide range of cardiac interstitial cells can be identified as fibroblasts. Fibroblasts can be distinguished from other myocardial interstitial cells on the basis of the expression of discoidin domain-containing receptor 2, PDGFR-α, and the transcription factor Tcf21.15 Identification and characterization of fibroblast populations is further complicated by their dynamic phenotypic alterations in injured tissues.35 In infarcted and stressed hearts, fibroblasts undergo conversion to myofibroblasts, cells that exhibit a prominent endoplasmic reticulum (indicating a synthetically active phenotype), while expressing contractile proteins, such as α-smooth muscle actin (α-SMA).36,37 Infiltration of the heart with activated myofibroblasts has been reported in a wide range of pathologic conditions, including MI,30,38 non-infarctive ischaemic cardiomyopathy,27,39 myocarditis,40 diseases associated with cardiac pressure or volume overload,41,42 and alcoholic cardiomyopathy.43 However, myofibroblast conversion is not required for activation of a pro-fibrotic programme. In a model of diabetic fibrotic cardiomyopathy, deposition of interstitial collagen and acquisition of a matrix-synthetic phenotype by cardiac fibroblasts was found to be independent of myofibroblast transdifferentiation.44 Moreover, single-cell transcriptomic analysis in a model of angiotensin II-induced fibrosis identified fibroblast subsets expressing Cartilage Intermediate layer protein 1 (Cilp) or Thrombospondin-4 (Thbs4) as the predominant fibrogenic cell types, in the absence of α-SMA-expressing myofibroblasts.45
Moreover, a growing body of evidence suggests that myofibroblasts exhibit a range of phenotypic profiles. Although α-SMA expression is often used to identify ‘differentiated myofibroblasts’ in injured tissues, early stage myofibroblasts may not express α-SMA, but may have a robust stress fibre network and form focal adhesions. These cells were termed ‘proto-myofibroblasts’,46,47 and may be prominent in early interstitial fibrotic lesions triggered by mechanical stress. Transcriptomic analysis suggests that in myocardial injury sites, infiltration with activated fibroblasts expressing the matricellular protein periostin precedes their conversion to myofibroblasts.21 Whether these activated periostin+/α-SMA− cells represent proto-myofibroblasts remains unknown.
In the era of single-cell transcriptomics, it is increasingly appreciated that both fibroblasts and myofibroblasts are heterogeneous populations, comprised of several subsets with distinct transcriptomic profiles. In healing infarcts subpopulations of myofibroblasts with high expression of fibrogenic mediators, such as transforming growth factor (TGF)-β, have been identified, along with other myofibroblasts that expressed transcripts encoding proteins with anti-fibrotic properties, such as the matricellular gene Wisp/CCN5 and the Wnt antagonist Sfrp.21 Whether these transcriptomic profiles are associated with pro- vs. anti-fibrotic properties remains unknown.
4.2 Where do activated cardiac myofibroblasts come from?
The abundant fibroblasts that reside in the normal cardiac interstitium are capable of activation following injury and can account for the injury-associated expansion of matrix-secreting myofibroblasts. Despite this fact, several studies have suggested that other cell types, including haematopoietic fibroblast progenitors,48–51 macrophages,52 and endothelial cells22,53 may also contribute to myofibroblast accumulation in injured hearts by transdifferentiating to fibroblasts or myofibroblasts. The relative contribution of these cells remains controversial. Several robust studies combining lineage tracing strategies, bone marrow transplantation experiments, and parabiosis models have demonstrated that the vast majority of activated myofibroblasts in both infarctive and in pressure overload-induced cardiac fibrosis are derived from resident fibroblast populations, and that the contributions of vascular endothelial cells and haematopoietic cells are very limited.54–57 Several factors may explain the conflicting observations. First, some of the studies use associative data based on co-expression of various markers to imply haematopoietic or endothelial cell origin of fibroblasts. Second, in some studies the use of non-specific markers for identification of fibroblasts and myofibroblasts, such as fibroblast-specific protein-1 which is also highly expressed by macrophages and vascular cells,58 may explain erroneous conclusions regarding cellular identity. Third, in some cases, the use of non-specific Cre drivers to trace endothelial cells or leucocytes may limit the value of the observations. Fourth, conflicting findings may reflect disease-specific and context-dependent alterations in the cellular composition of the cardiac interstitium. Generalizing cell biological paradigms to all types of myocardial injury are inherently problematic, and do not take into account the distinct profile of cellular responses caused by various injurious stimuli. For example, in inflammatory myocardial diseases, such as myocarditis, the intense chemokine-dependent infiltration of the heart with immune cells may also recruit abundant circulating progenitors that may contribute to fibroblast expansion, independently of the resident fibroblast population. Experiments using bone marrow chimeric mice in a model of autoimmune myocarditis suggested that a large fraction of activated fibroblasts originate from a population of prominin+ haematopoietic cells that infiltrate the myocardium.59 On the other hand, in MI, contributions of various cell types may be context-dependent. In a reperfused infarct, most fibroblasts in the area at risk likely survive the ischaemic event and may account for myofibroblast expansion. In contrast, when the coronary artery is permanently occluded, the majority of cardiac interstitial cells may die along with the neighbouring cardiomyocytes; thus, limiting their contribution to the reparative myofibroblast population, which may be derived predominantly from fibroblasts recruited from non-infarcted segments, or from other remote cell types (including circulating haematopoietic cells). Unfortunately, studies on myocardial cell death following infarction have focused almost exclusively on cardiomyocytes; information on the fate of fibroblasts and other interstitial cells is extremely limited. The unique cell biological characteristics of disease-specific fibrotic myocardial responses remain underappreciated and understudied.
4.3 Mechanisms involved in myofibroblast activation in fibrotic hearts
Activated myofibroblasts contribute to matrix remodelling, not only by secreting large amounts of structural ECM proteins but also by releasing proteases and their inhibitors and by producing enzymes involved in matrix processing. Although the mechanisms of myofibroblast activation are dependent on the specific type of injury causing the myocardial fibrotic response, some common patterns have emerged. Neurohumoral mediators, cytokines and growth factors are released and activated following myocardial injury and bind to cell surface receptors, transducing fibrogenic intracellular signalling cascades. Moreover, specialized matrix proteins and matricellular macromolecules enrich the matrix network and serve to locally activate fibrogenic growth factors at the site of injury providing spatial regulation of the fibroblast-activating signals.
In addition to the fibroblasts, several other cell types contribute to the fibrogenic environment. Immune cells, including macrophages, mast cells, and lymphocytes are recruited and activated in remodelling hearts and may play an important role in fibroblast activation by secreting a wide range of fibrogenic mediators, including cytokines, growth factors, and matricellular proteins.60–65 Endothelial cells, pericytes, and vascular smooth muscle cells may also secrete molecular signals that regulate fibroblast behaviour.66 Cardiomyocytes are also highly sensitive to microenvironmental changes and typically respond to injurious stimuli by activating inflammatory and fibrogenic programmes.67 It should be emphasized that the actions of leucocytes, vascular cells and cardiomyocytes on fibroblast phenotype are not necessarily activating. Several studies have demonstrated injury-associated induction and release of anti-fibrotic mediators in leucocytes,68 endothelial cells,69,70 and cardiomyocytes71 that may serve to restrain fibrogenic responses.
5. Fibroblast-activating neurohumoral pathways
5.1 The renin–angiotensin–aldosterone system
5.1.1 The angiotensin II/AT1 axis
The renin–angiotensin–aldosterone system is activated in fibrotic and remodelling hearts regardless of the underlying aetiology and plays an important role in myofibroblast activation. Stressed cardiomyocytes, immune cells and fibroblasts produce renin and angiotensin-converting enzyme (ACE), contributing to angiotensin II generation72–74 (Figure 3). When released in the cardiac interstitium, angiotensin II is a potent activator of cardiac fibroblasts. In vitro studies have demonstrated that angiotensin II stimulates cardiac fibroblast proliferation,75 inhibits fibroblast apoptosis,76 stimulates fibroblast migration77,78 inducing integrin expression,79 promotes myofibroblast conversion,80 and markedly increases synthesis of structural ECM proteins.81,82 All these activating effects of angiotensin II involve signalling through the type 1 angiotensin receptor (AT1). In contrast, the type 2 receptor AT2 acts as a negative regulator of angiotensin II-mediated fibrogenic responses, inhibiting AT1 actions and suppressing fibroblast proliferation and matrix synthesis.83,84
Figure 3.

Fibrogenic effects of angiotensin II. Angiotensin II generated in injured or remodelling myocardium exerts potent fibrogenic actions on cardiac fibroblasts through interactions with the AT1 receptor, promoting myofibroblast conversion, fibroblast proliferation and survival and stimulating ECM synthesis. In contrast, signalling through the AT2 receptor inhibits the pro-fibrotic effects of AT1. Pro-inflammatory cytokines, such as TNF-α, induce AT1 in cardiac fibroblasts and may accentuate the activating effects of angiotensin II. Fibrogenic effects of angiotensin II are mediated through activation of PKC and Erk signalling. Some of the effects of angiotensin II may involve downstream induction and activation of TGF-β.
The activating effects of the angiotensin II/AT1 axis in cardiac fibroblasts are mediated at least in part through activation of p38 mitogen-activated protein kinase (MAPK), protein kinase C (PKC) and extracellular signal-regulated kinase (Erk) cascades.75,85–87 Some fibrogenic angiotensin II actions require induction,88,89 or activation of TGF-β.90 Moreover, responsiveness of fibroblasts to angiotensin II is modulated by a wide range of mediators that regulate expression of the angiotensin receptors. For example, pro-inflammatory cytokines may make fibroblasts more responsive to the effects of angiotensin II by inducing AT1 synthesis.91 In contrast, angiotensin II has been reported to downmodulate AT1 expression,92 providing a negative feedback mechanism that may restrain uncontrolled fibrogenic actions.
There is abundant in vivo evidence, predominantly derived from pharmacologic interventions, supporting the role of AT1 signalling in the pathogenesis of cardiac fibrosis. AT1 inhibition attenuated interstitial fibrosis in models of MI93 and left ventricular pressure overload.94 Despite the lack of evidence documenting the role of fibroblast-specific AT1 signalling in vivo using cell-specific genetic approaches, the potent in vitro effects of the angiotensin II/AT1 axis suggest that cardiac fibroblasts may be major targets of angiotensin II. Thus, the beneficial effects of ACE inhibition and AT1 blockade in heart failure patients may reflect, at least in part, attenuation of angiotensin-mediated fibrosis. Evidence in isolated cardiomyocytes has suggested that AT1 signalling may not always require angiotensin II stimulation but may be directly activated by mechanical stress, stimulating a hypertrophic response.95 Whether angiotensin-independent AT1 signalling is involved in myofibroblast activation and in the pathogenesis of cardiac fibrosis remains unknown.
5.1.2 The fibrogenic actions of aldosterone
Extensive evidence suggests that aldosterone may play an important role in fibroblast activation following myocardial injury.96,97 In isolated cardiac fibroblasts, aldosterone triggers a proliferative response,98 stimulates migration,99 and promotes a matrix-synthetic phenotype,100 at least in part through activation of MAPK cascades.101 Whether these actions are responsible for the fibrogenic actions of aldosterone in vivo is unclear. In addition to its effects on fibroblasts, aldosterone has been suggested to modulate cardiomyocyte, macrophage, lymphocyte, and vascular cell phenotype, leading to activation of a fibrogenic programme. Experiments using cell-specific mineralocorticoid receptor knockout mice suggested that aldosterone actions on myeloid cells,102 T lymphocytes,103 and cardiomyocytes104 may contribute to the pathogenesis of cardiac fibrosis.
5.2 The β-adrenergic response in fibroblast activation
Adrenergic stimulation activates fibroblasts and induces fibrotic cardiac remodelling. In vivo, infusion of the non-specific β adrenergic receptor (AR) agonist isoproterenol,105 or the α1 AR agonist phenylephrine,106 and transgenic overexpression of β2-ARs107 cause marked myocardial fibrosis. The pro-fibrotic effects of adrenergic stimulation may be indirect, reflecting reparative fibrosis in response to cardiomyocyte necrosis, release of fibrogenic mediators by stimulated cardiomyocytes, or immune cell activation.105,108–110 Although direct effects of adrenergic agonists on cardiac fibroblasts are well-documented, their in vivo significance is unclear. β-AR activation stimulates proliferation of cardiac fibroblasts,111,112 activating phosphatidylinositol 3-kinase (PI-3K)113 and MAPK114 signalling Moreover, proliferative effects of β-AR stimulation in fibroblasts have been attributed to epidermal growth factor receptor (EGFR) transactivation and downstream activation of ERK signalling.115
The relative role of β1- and β2-AR responses in regulating the fibrotic response is unknown. Although in some experimental animal models of heart failure, non-selective β-AR inhibition and administration of β1-AR blockers attenuated cardiac fibrosis,116,117 such effects may represent an epiphenomenon reflecting reduced blood pressure or attenuated cardiomyocyte death. Moreover, β2-AR signalling in myofibroblasts has been reported to exert pro-hypertrophic actions through paracrine mechanisms.118 Although the role of β3-AR signalling in fibroblasts is unknown, recent evidence suggests that cardiomyocyte-specific β3-AR signalling may exert anti-fibrotic actions by downmodulating expression of cardiomyocyte-derived fibrogenic proteins, such as the matricellular protein CCN2.119
AR signalling induces conformational changes in G protein βγ subunits, ultimately resulting in activation of G protein-coupled receptor kinase 2 (GRK2). Activation of GRK2 in fibroblasts has been implicated in cardiac fibrosis in experimental models of MI.120,121 However, the molecular targets responsible for the fibrogenic actions of GRK2 remain poorly understood.
5.3 Inflammatory mediators as regulators of fibroblast function
Inflammatory cytokines, chemokines, and growth factors have been implicated in the pathogenesis of cardiac fibrosis through direct actions on fibroblasts, by stimulating recruitment and activation of fibrogenic macrophages and lymphocytes, and by triggering a fibrogenic programme in vascular cells and cardiomyocytes. Moreover, prolonged chronic inflammation may cause cardiomyocyte necrosis, triggering a reparative form of fibrosis.
5.3.1 Chemokines as regulators of cardiac fibrosis
Chemokines are chemotactic cytokines with a prominent role in leucocyte trafficking.122 From a structural perspective, chemokines are classified into four subfamilies (CC, CXC, CX3C, and XC), on the basis of the number of aminoacids between their first two cysteine residues. This classification has important functional implications: CC chemokines are predominantly mononuclear cell chemoattractants, whereas CXC chemokines, and in particular the subgroup containing the conserved ELR sequence motif (glutamic acid–leucine–arginine) near the aminoternimal end, are involved in recruitment of neutrophils. Actions of chemokines in regulation of fibrosis are predominantly mediated through chemotactic recruitment of leucocytes. Although several members of the family may directly modulate fibroblast phenotype and function, the in vivo significance of these effects in cardiac fibrotic remodelling remains unclear.123
5.3.2 CC chemokines
Extensive experimental evidence suggests that the CC chemokine CCL2/monocyte chemoattractant protein-1 is involved in the pathogenesis of cardiac fibrosis in a wide range of models, including ischaemic cardiomyopathy, pressure overload-induced myocardiac remodelling and inflammatory heart diseases (Table 1).125,126,129 The fibrogenic actions of CCL2 has been predominantly attributed to recruitment and activation of monocytes and macrophages expressing the main CCL2 receptor, CCR2, resulting in up-regulation of fibrogenic mediators, such as TGF-β and osteopontin.128,129,137 Although CCL2 has been suggested to promote direct fibrogenic actions in hepatic, cutaneous and pulmonary fibroblasts,138–140 in isolated cardiac fibroblasts, no significant effects of CCL2 were noted on profibrotic gene expression profile and proliferative activity.129 Some studies have suggested that CCL2 may promote fibrosis by recruiting circulating fibroblast progenitors.49,50 However, the evidence supporting this notion is associative and based on co-expression of haematopoietic cell and fibroblast markers by the same cells. Considering the robust evidence from several studies using lineage tracing strategies suggesting that most activated fibroblasts in the remodelling heart are derived from resident populations and not from haematopoietic cells55–57 the role of circulating fibroblast progenitors in cardiac fibrosis remains controversial.141
Table 1.
The CCL2/CCR2 axis in cardiac fibrosis
| Experimental model of cardiac fibrosis | Findings | Cellular target and molecular mechanism | References |
|---|---|---|---|
| Transgenic CCL2 overexpression | Cardiomyocyte-specific CCL2 overexpression in mice induced myocardial inflammation and fibrotic changes. | Fibrogenic actions presumed due to inflammatory cell recruitment and activation. | 124 |
| Rat model of cardiovascular remodelling through chronic inhibition of NO synthesis | Treatment with anti-CCL2 antibody attenuated inflammation and reduced coronary vascular remodelling but did NOT affect fibrosis. | NA | 125 |
| Rat model of pressure overload through suprarenal aortic constriction | Anti-CCL2 antibody attenuated myocardial fibrosis. | Anti-fibrotic effects were presumed due to decreased macrophage recruitment and attenuated TGF-β release | 126 |
| Mouse model of non-reperfused myocardial infarction | Anti-CCL2 gene therapy attenuated interstitial fibrosis without affecting infarct size. | Anti-fibrotic effects were attributed to decreased macrophage infiltration and attenuated TNF-α and TGF-β levels. | 127 |
| Mouse model of reperfused myocardial infarction | CCL2 KO mice had attenuated myofibroblast proliferation and delayed granulation tissue formation after MI, associated with reduced dilative remodelling. | Anti-fibrotic effects were attributed to reduced macrophage recruitment and impaired macrophage activation, associated with markedly decreased expression of TGF-β and osteopontin. | 128 |
| Mouse model of ischaemic cardiomyopathy induced through brief repetitive ischaemia/reperfusion | CCL2 KO mice had attenuated interstitial fibrosis, associated with reduced macrophage recruitment and decreased fibroblast proliferation. | CCL2 did not have direct effects on proliferation or expression of fibrosis-associated genes by cardiac fibroblasts. Thus, the profibrotic effects of CCL2 were attributed to actions on macrophages. | 129 |
| Mouse model of angiotensin II-induced fibrosis | CCL2 KO mice had attenuated fibrosis. | Fibrogenic effects of CCL2 were attributed to recruitment of circulating fibroblast precursors. This notion was based on the presence of CD34+/CD45+ collagen-expressing fibroblast-like cells in the myocardium. | 50 |
| Mouse model of angiotensin II-induced fibrosis | CCR2 KO mice had attenuated angiotensin II-mediated cardiac fibrosis. | Fibrogenic effects of CCR2 were attributed to recruitment of bone marrow-derived fibroblast precursors. This notion was based on the presence of CD34+/CD45+ collagen-expressing fibroblast-like cells in the myocardium. | 130 |
| Mouse model of angiotensin II-induced fibrosis | CCR2 KO mice had attenuated angiotensin II-mediated cardiac fibrosis. | Fibrogenic effects of CCR2 were attributed to attenuated recruitment of fibroblast progenitors. This notion was based on detection of CD45/α-SMA+ cells. CCR2-mediated fibrosis was attributed to actions of CCL12 (and not CCL2), based on associative data. | 131 |
| Mouse model of mineralocorticoid receptor-mediated cardiac fibrosis | In a deoxycorticosterone (DOC)/salt model, CCL2 KO mice had 35% less myocardial fibrosis. | Fibrogenic effects of CCL2 were attributed to actions on macrophage recruitment and activation. However, CCL2 KO mice also had lower blood pressure, suggesting that the fibrogenic actions of CCL2 may be indirect, reflecting effects on blood pressure regulation. | 132 |
| Mouse model of ageing | Aged CCL2 KO mice had reduced fibrosis and attenuated diastolic dysfunction. | Attenuated fibrosis in CCL2 KO mice was associated with reduced leucocyte infiltration. | 133 |
| Mouse model of angiotensin-induced fibrosis | Administration of a CCR2 antagonist attenuated myocardial inflammation and fibrosis. | CCL2 up-regulation was stimulated by angiotensin-induced CaMKIIδ activation. The mechanisms for the fibrogenic effects of the CCL2/CCR2 axis were not investigated. | 134 |
| Mouse model of left ventricular pressure overload | Depletion of CCR2+ monocytes ameliorated cardiac fibrosis. | The findings highlighted the role of the monocytes recruited through CC chemokines in the pathogenesis of myocardial fibrosis. | 135 |
| Mouse model of streptozotocin-induced diabetes | Diabetic CCR2 KO mice had attenuated myocardial fibrosis. | The fibrogenic actions were attributed to macrophage-driven inflammation and oxidative stress. | 136 |
5.3.3 CXC chemokines
Several members of the CXC chemokine family have been implicated in the pathogenesis of cardiac fibrosis. CXC chemokines that bind to the CXCR2 receptor may exert fibrogenic actions through recruitment of neutrophils or monocytes with fibrogenic properties. In spontaneously hypertensive rats, CXCR2 inhibition was found to attenuate fibrosis142; however, CXCR2-mediated fibrosis may involve effects on blood pressure regulation, rather than pro-fibrotic actions. CXCL1, one of the CXCR2 ligands has been reported to contribute to the development of angiotensin-induced cardiac fibrosis through recruitment of a fibrogenic monocyte subpopulation.143
CXCL12 has a broad range of actions in injured and remodelling hearts and has been involved in angiogenesis, recruitment of inflammatory and progenitor cells, and in regulation of cardiomyocyte survival following injury.144,145 Several studies have suggested that CXCL12 may exert fibrogenic actions through CXCR4 activation. CXCR4 antagonism reduced cardiac fibrosis in a genetic model of murine cardiomyopathy,146 and in models of diabetic fibrosis147 and cardiorenal syndrome.148 The fibrogenic actions of CXCL12 were attributed to direct pro-migratory effects on fibroblasts, to recruitment of fibroblast progenitors, or to activation of macrophages.149
The ELR-negative CXC chemokines do not stimulate neutrophil chemotaxis, but recruit lymphocyte subsets and have been suggested to exert actions on fibroblasts and endothelial cells. The ELR-CXC chemokine CXCL10/interferon-γ-inducible protein (IP)-10 is consistently up-regulated following cardiac injury and has been suggested to exert anti-fibrotic actions. Inhibition of fibrosis by CXCL10 may involve recruitment of yet unidentified leucocyte subsets that suppress fibroblast function, or direct de-activating effects on cardiac fibroblasts.69,70In vitro, CXCL10 was found to inhibit growth-factor-mediated fibroblast migration,69 through interactions with proteoglycans that were independent of the main CXCL10 receptor, CXCR3.150
5.3.4 The pro-inflammatory cytokines in cardiac fibrosis
Levels of the pro-inflammatory cytokines tumour necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6 are markedly increased in many myocardial pathologic conditions associated with fibrosis. Several clinical studies have demonstrated correlations between pro-inflammatory cytokine levels and fibrosis-related endpoints. For example, in patients with dilated cardiomyopathy, elevated myocardial IL-6 and TNF-α mRNA levels were associated with collagen deposition and gelatinase expression,151 and in transplanted human hearts myocardial IL-6 levels were associated with fibrotic remodelling.152 These associations suggest a link between pro-inflammatory cytokines and fibroblast activation. The fibrogenic effects of TNF-α, IL-1, and IL-6 may involve direct actions on fibroblasts, or indirect effects, related to recruitment of macrophages, induction of matricellular proteins and up-regulation of growth factors with prominent fibroblast-activating properties, such as TGF-β.
5.3.5 TNF-α
Abundant in vivo evidence suggests an important role for TNF-α in cardiac fibrosis (Table 2). Transgenic mice with myocardial TNF-α overexpression develop heart failure associated with fibrotic changes.153,154 The time course of ECM remodelling in TNF-α overexpressing hearts suggests that early protease-mediated matrix degradation is followed by late up-regulation of TGF-βs and activation of a matrix-preserving programme.155 Experiments using loss-of-function models have implicated endogenous TNF-α in the pathogenesis of fibrosis induced through pressure overload157,163 through actions involving the type 1 TNF receptor (TNFR1).161
Table 2.
The role of TNF-α in cardiac fibrosis
| Model | Findings | Cellular mechanism of fibrosis | References |
|---|---|---|---|
| Cardiac-specific TNF-α overexpression | Terminally ill mice with myocardial overexpression of TNF-α had biventricular fibrosis. This was accompanied by chamber dilation, cardiomyocyte apoptosis, and transmural myocarditis. | Not studied. TNF-induced fibrosis may reflect activation of immune cells that release fibrogenic cytokines, cardiomyocyte death activating a reparative programme, or direct effects of TNF-α on fibroblasts. | 153 |
| Cardiac-specific TNF-α overexpression | Cardiac overexpression of TNF-α caused heart failure associated with cardiac fibrosis. | Fibrosis was associated with marked induction of collagen synthesis and activationg of matrix-degrading proteases (MMP2 and MMP9). The primary cellular targets of TNF-α are unclear. | 154 |
| Cardiac-specific TNF-α overexpression | Cardiomyocyte-specific TNF-α overexpression caused progressive ventricular dilation and fibrosis. | Early activation of matrix-degrading MMPs was followed by late increases in TGF-β levels and fibrillar collagen content. Thus, TNF-α may promote an early matrix-degrading response, followed by a late TGF-β-driven matrix-preserving process. | 155 |
| Global TNFR1 and TNFR2 KO mice undergoing non-reperfused MI protocols. | Interstitial fibrosis was increased in TNFR2, but not in TNFR1 KO mice. Infarct size was comparable between groups. | Unclear cellular targets. TNFR2 loss was associated with increased post-infarction IL-1β and IL-6 levels that may account for fibrogenic actions. | 156 |
| TNF-α KO mice undergoing transverse aortic constriction (TAC) protocols | TNF-α loss attenuated cardiac fibrosis following TAC, associated with improved cardiac function. | TNF-α mediates fibrosis and protease activation in the pressure-overloaded heart. Cellular targets unclear. | 157 |
| Anti-TNF monoclonal antibody treatment in streptozotocin-induced diabetic cardiomyopathy | TNF-α inhibition reduced myocardial fibrosis and improved LV function. | TNF-α mediates fibrosis activating a matrix-synthetic programme. Cellular targets unclear. | 158 |
| TNF-α KO mice undergoing TAC protocols | TNF loss attenuated cardiac fibrosis following TAC. | Profibrotic effects of TNF-α were attributed to activation of a superoxide/MMP axis more prominently in cardiomyocytes and at a lower level in fibroblasts. | 159 |
| Cardiac-specific TNF-α overexpression | TNF overexpression was associated with mast cell expansion and subsequent fibroblast activation. Mast cell deficiency abrogated TNF-induced fibrosis. | Profibrotic effects of TNF-α may involve mast cell growth and activation, and subsequent release of mast-cell-derived fibrogenic mediators. | 160 |
| TNFR1 and TNFR2 KO mice undergoing angiotensin II infusion protocols | TNFR1 loss, but not TNFR2 absence, attenuated cardiac fibrosis. | Fibrogenic actions of TNF-α/TNFR1 signalling were attributed to recruitment of circulating fibroblast progenitors. This notion was based on identification of CD34+/CD45+ fibroblast-like cells. | 161 |
| Mdx dystrophic mice receiving anti-TNF treatment (Remicade) | Remicade attenuated cardiac fibrosis but worsened systolic function. | The basis for the effects of TNF blockade was not studied. | 162 |
| TNF-α KO mice undergoing angiotensin II infusion protocols | TNF-α KO mice had reduced perivascular and interstitial fibrosis following angiotensin II infusion. | Fibrogenic actions were attributed to effects on oxidative stress-dependent MAPK, TGF-β, and nuclear factor (NF)-κB signalling. Fibrogenic effects may be in part related to hypertensive actions of TNF-α. | 163 |
TNF-α may promote fibrosis through direct actions on cardiac fibroblasts, or through effects on other myocardial cell types. It should be noted that TNF-α does not directly induce a matrix-synthetic programme in cardiac fibroblasts, but rather decreases collagen synthesis and stimulates MMP expression.164 Thus, TNF-α-mediated fibrosis may reflect a response to ECM degradation. In vitro experiments have suggested several additional mechanisms that may be responsible for TNF-α-induced fibroblast activation. TNF-α stimulated synthesis of the matricellular protein CCN4 and subsequent fibroblast proliferation,165 induced TGF-β expression,166 and may potentiate fibrogenic effects of angiotensin II, by stimulating AT1 receptor synthesis.91 TNF-α may also promote fibrogenic activation of immune cell populations in the injured myocardium. In TNF-α− overexpressing mice, the fibrogenic actions of TNF-α in the myocardium have been attributed, at least in part, to expansion of cardiac mast cells.160
5.3.6 The fibrogenic actions of the IL-1 family of cytokines
Studies in experimental models of MI have implicated IL-1 in the pathogenesis of cardiac fibrosis. Global loss of IL-1 signalling in mice lacking the type 1 IL-1 receptor (IL-1R1) attenuated adverse fibrotic remodelling in a model of reperfused MI.167 Moreover, in a model of viral myocarditis local inhibition of IL-1 through overexpression of IL-1 receptor antagonist (IL-1Ra) significantly reduced fibrosis.168 Much like for TNF-α, the fibrogenic actions of IL-1 likely involve effects in both fibroblasts and immune cells. Experiments in animals with fibroblast-specific loss of the IL1R1 receptor documented the significance of direct actions of IL-1 on cardiac fibroblasts. Fibroblast-specific IL-1R1 KO mice had lower myocardial collagen levels in the infarcted heart, and exhibited improved function and attenuated remodelling following MI.169In vitro studies suggest that the fibrogenic effects of IL-1 are not mediated through direct activation of myofibroblast-driven ECM synthesis. In fact, both IL-1α and IL-1β inhibit conversion of fibroblasts into myofibroblasts,170–172 inhibit fibroblast proliferation,173 and decrease collagen synthesis by cardiac fibroblasts.164 IL-1-mediated fibrosis may be due to primary activation of pro-inflammatory and matrix-degrading pathways. IL-1 potently stimulates MMP expression and activity,167,170 while reducing synthesis of MMP inhibitors.174 Generation of matrix fragments and induction of TGF-βs by IL-1 may be responsible for indirect activation of a fibrogenic programme.
IL-33, another member of the IL-1 family of cytokines has been implicated in the regulation of fibrosis in several different organs.175 In human patients with end-stage heart failure, IL-33 and ST2 up-regulation were associated with fibrotic changes.176 In the pressure-overloaded myocardium, IL-33 was found to exert cardioprotective and anti-fibrotic actions, attributed to its binding to the ST2 receptor in cardiomyocytes and fibroblasts.177 However, considering the potent effects of the IL-33/ST2 axis on cardiomyocytes, macrophages and lymphocytes, whether the protective effects of IL-33 in the remodelling heart are mediated through direct actions on fibroblasts remains unknown. In vitro, IL-33 stimulation did not affect collagen synthesis by cardiac fibroblasts, but inhibited their migratory capacity and enhanced expression of inflammatory genes.178
5.3.7 The IL-6 family of cytokines
The IL-6 family of cytokines (also referred as the gp130 family, indicating the common signal transducer of these molecules) includes IL-6, IL-11, oncostatin-M, leukaemia-inhibitory factor and cardiotrophin-1 and has been implicated in the pathogenesis of cardiac fibrosis. The bulk of experimental evidence suggests that IL-6 exerts fibrogenic actions. Genetic absence of IL-6 attenuated cardiac fibrosis and dysfunction in models of left ventricular pressure overload,179,180 MI,181 and diabetic cardiomyopathy.182 IL-6 neutralization has also been reported to exert anti-fibrotic actions. In models of aldosterone-mediated cardiac remodelling, left ventricular pressure overload, and genetic cardiomyopathy induced through Hsp20 overexpression, IL-6 neutralization reduced fibrotic changes and attenuated fibroblast activation.183–185 The fibrogenic effects of IL-6 have been attributed to STAT3-dependent stimulation of collagen synthesis by cardiac fibroblasts,186 or to induction of TGF-β.182
Recent studies have suggested that IL-11 may play a critical role in the pathogenesis of fibrosis not only in the myocardium187 but also in the lung and kidney,188,189 serving as a downstream fibrogenic signal, induced by TGF-β. IL-11 inhibition attenuated TGF-β-mediated fibroblast activation in vitro, and global loss of IL-11Ra in vivo reduced interstitial fibrotic remodelling following pressure overload. The effects of IL-11 on fibroblast activation were attributed to post-transcriptional mechanisms involving ERK signalling. To what extent IL-11 exerts pro-fibrotic actions in human patients remains unclear. Recombinant human IL-11 has been extensively used for the treatment of thrombocytopenias with a good safety record and without any reports of fibrotic disease.190
It should be emphasized that studies examining the role of other gp130 cytokines do not support a consistent fibrogenic role of gp130 signalling. Oncostatin M has been suggested to exert anti-fibrotic actions in the myocardium by inhibiting fibrogenic TGF-β signalling cascades.191 Experimental studies in fibrotic responses of other organs have produced conflicting findings suggesting both pro-fibrotic192 and anti-fibrotic effects193 of oncostatin M. The conflicting data likely reflect the context-dependent actions of gp130 cytokines in vivo, which are driven by the wide range of their actions in many different cell types.
5.3.8 The fibrogenic actions of the anti-inflammatory cytokines IL-4, IL-10, and IL-13
The anti-inflammatory cytokines IL-4, IL-10, and IL-13 have been implicated in the pathogenesis of fibrosis in several different organs.194,195 Extensive in vivo and in vitro evidence supports the role of endogenous IL-4 in cardiac fibrosis. IL-4 up-regulation has been reported in experimental models of cardiac injury, predominantly localized in mast cells,196 lymphocytes, and macrophages. In myocarditis, eosinophils may be an additional major source of IL-4.197 In a model of angiotensin II infusion, genetic loss of IL-4 reduced collagen deposition in the myocardium by 60%.198 Moreover, in a model of pressure overload induced through TAC, IL-4 neutralization attenuated fibrotic changes.199 IL-4 may exert direct fibrogenic actions by stimulating collagen synthesis in cardiac fibroblasts through activation of STAT6.198
In contrast, the evidence on the role of IL-13 in cardiac fibrosis is more limited. Induction of IL-13 has been documented in infarcted200 and in fibrotic senescent mouse hearts201 and may be localized in Th2 lymphocytes. IL-13 can activate a matrix-synthetic programme in fibroblasts,202 stimulating TGF-β expression and activation,203 and may promote a fibrogenic phenotype in monocytes and macrophages.201 However, the in vivo contribution of these fibrogenic actions of IL-13 in the cardiac fibrotic response has not been documented.
IL-10 up-regulation has been documented in both rodent and large animal models of reparative and pressure overload-induced fibrosis and is localized in T lymphocytes and subsets of macrophages infiltrating the injured and remodelling heart.204–206 In addition to its well-documented anti-inflammatory actions, IL-10 may also regulate the cardiac fibrotic response. Unfortunately, data from in vivo studies are conflicting, suggesting both pro-fibrotic and anti-fibrotic actions of IL-10. In a model of cardiac remodelling induced through unilateral nephrectomy, salt loading and aldosterone infusion, IL-10 was found to critically contribute to fibroblast activation and to the pathogenesis of diastolic dysfunction.206 The fibrogenic effects of IL-10 were indirect, and were attributed to acquisition of a fibrogenic programme by macrophages. These findings are consistent with in vitro observations suggesting that IL-10 does not significantly affect fibroblast gene expression profile,207 but promotes a matrix-preserving phenotype in macrophages through up-regulation of TIMP-1 synthesis.204 In contrast, other studies suggested that endogenous IL-10 may not significantly contribute to fibrosis, or may even exert anti-fibrotic actions. In a model of reperfused infarction, genetic loss of IL-10 had no significant effects on scar formation, myofibroblast infiltration and adverse remodelling.207 In a model of non-reperfused MI, IL-10 was found to inhibit fibrosis.208 Moreover, in a model of pressure overload, endogenous IL-10 derived from haematopoietic cells inhibited fibrosis, through actions attributed to attenuated maturation of fibroblast progenitors.209 What is the basis for the conflicting evidence? The in vivo effects of IL-10 in the myocardial fibrotic response may be dependent on the balance between anti-inflammatory and pro-fibrotic actions. IL-10-mediated suppression of pro-inflammatory cytokine expression may inhibit downstream activation of fibrogenic pathways and, depending on the context, may outweigh any pro-fibrotic actions.
5.4 The TGF-β superfamily
5.4.1 TGF-βs in cardiac fibrosis
TGF-β is the best characterized fibrogenic growth factor.210,211 In mammals, the TGF-β subfamily is comprised of three isoforms (TGF-β1, 2, and 3)212 that signal through the same receptors and share common cellular targets, but exhibit distinct patterns of regulation and different affinities for their receptors and co-receptors. Thus, the three isoforms likely play different roles in the pathophysiology of fibrotic conditions. Unfortunately, isoform-specific actions have not been systematically studied in vivo. Thus, our knowledge on the role of TGF-β in fibrotic conditions is primarily derived from studies investigating the common downstream signalling pathways of the three isoforms. TGF-β induction and activation has been consistently demonstrated in several different models of cardiac fibrosis,205,213,214 and in human hearts with fibrotic cardiomyopathic changes.215,216
Although many different cell types (including macrophages, fibroblasts, cardiomyocytes, and platelets) can produce de novo large amounts of TGF-β in the injured heart, TGF-β signalling in injured tissues is predominantly regulated through activation. The mammalian heart contains latent stores of TGF-β that cannot associate with TGF-β receptors. Injury results in rapid activation of TGF-β, providing a mechanism for appropriate spatially-restricted stimulation of the reparative TGF-β response. Pericellular activation of TGF-β in sites of injury involves several different molecular signals, including proteases,217–219 matricellular proteins,214,220 and integrins.221 The relative contribution of specific TGF-β activating signals in the fibrotic heart has not been investigated.
The role of TGF-β in the pathogenesis of cardiac fibrosis is supported by several lines of evidence. First, gain-of-function studies showed that myocardial overexpression of TGF-β1, and constitutive expression of an activated mutant TGF-β protein trigger ventricular fibrosis, associated with de novo deposition of structural ECM proteins and activation of a matrix-preserving programme.222–224 Surprisingly, transgenic mice with a large proportion of constitutively active TGF-β1 due to a mutation that blocks covalent tethering of the TGF-β1 latent complex to the ECM exhibited only atrial fibrosis,225 possibly reflecting exaggerated responses of atrial fibroblasts to the fibrogenic effects of TGF-β. Second, genetic loss-of-function studies suggest that endogenous TGF-β signalling plays an important role in cardiac fibrosis. Heterozygous TGF-β1+/− deficient mice had delayed interstitial fibrotic changes as they aged.226 Moreover, mice with fibroblast-specific loss of the type I or type II TGF-β receptors (TβRI or TβRII) had reduced cardiac fibrosis in a model of left ventricular pressure overload227 suggesting that fibrotic remodelling involves the activation of the TGF-β signalling cascade. Third, pharmacologic TGF-β inhibition strategies attenuated fibrosis in a model of pressure overload,228 and in the fibrotic cardiomyopathy due to Chagas’ disease.229
TGF-β exerts a broad range of direct effects on fibroblasts that may contribute to the pathogenesis of fibrosis. TGF-β stimulation potently induces myofibroblast conversion,230 and increases ECM protein synthesis by activated fibroblasts.231 Moreover, TGF-β increases expression of integrins,232 and shifts the protease/anti-protease balance towards a matrix-preserving phenotype by inducing protease inhibitors, such as Plasminogen Activator Inhibitor (PAI)-1 and tissue inhibitor of metalloproteinase (TIMP)1212,233 and by suppressing MMP synthesis.234 It should be noted that the effects of TGF-β on cardiac fibroblast proliferation are dependent on the context. Some studies have reported that TGF-β stimulates proliferation of cardiac fibroblasts,235 whereas other investigations demonstrated anti-proliferative effects.231,236 The contrasting findings likely reflect differences in the state of differentiation of fibroblast populations,237 the presence or absence of other growth factors, and the matrix environment.
Considering the broad effects of TGF-β in many cellular responses, and the need to develop strategies to specifically inhibit its fibrogenic actions, dissection of pro-fibrotic TGF-β-driven molecular signals has attracted significant interest. TGF-β signals through a series of intracellular effector proteins, the Smads, and through Smad-independent pathways (Figure 4). In vitro and in vivo evidence suggests that both canonical Smad-dependent cascades and non-canonical Smad-independent signalling contribute to fibroblast activation in fibrotic hearts. In vivo studies have demonstrated that Smad3 signalling is critically involved in activation of reparative fibroblasts following MI, inducing ECM protein synthesis, integrin transcription, and α-SMA expression,231,233 and contributing to the formation of an organized scar.232 Smad3 signalling in infarct myofibroblasts restrained cell proliferation,231–233 generating well-aligned arrays of activated myofibroblasts that preserve the structural integrity of the infarcted heart through activation of a reparative integrin-reactive oxygen species (ROS) axis.232 These findings highlight the reparative function of fibroblasts in the presence of significant cardiomyocyte loss. In contrast, Smad2 does not seem to play a major role in mediating fibroblast activation in fibrotic hearts.238 The contrasting effects of Smad2 and Smad3 may reflect differences in their patterns of activation and nuclear translocation following TGF-β stimulation, or distinct interactions with transcriptional regulators in the nucleus. On the other hand, some actions of TGF-β on cardiac fibroblasts may involve activation of Smad-independent p38 MAPK, ERK, or Transforming Growth Factor-β-activated kinase (TAK)1 signalling.114,239,240 However, considering the wide range of mediators that signal through these common kinase pathways, the relative contribution of TGF-β in their activation is unclear.
Figure 4.

Regulation of TGF-β signalling in cardiac fibrosis. Active TGF-β binds to type II and type I receptors, activating downstream Smad-dependent signalling cascades and Smad-independent pathways. TGF-β binding to the ALK5 type 1 receptor and downstream activation of Smad3 signalling, induces a matrix-preserving programme in cardiac fibroblasts and plays an important role in their activation following cardiac injury. In contrast, the role of ALK1/Smad1/5 signalling in regulation of fibroblast phenotype is poorly understood. Activation of Smad-independent pathways, including RhoA and MAPK signalling, mediates some of the effects of TGF-β in cardiac fibroblasts. Endogenous pathways for negative regulation of TGF-β cascades may protect from excessive or unrestrained fibrotic responses. The inhibitory Smads (Smad6/7), pseudoreceptors such as BAMBI, and soluble endoglin may serve as endogenous inhibitors of TGF-β signalling, limiting pro-fibrotic responses.
In addition to the direct actions of TGF-β on transcription of fibrosis-associated genes, some TGF-β-mediated fibrogenic effects may be mediated through induction of other secreted mediators. It has been suggested that TGF-β-mediated fibrosis may require downstream release of the matricellular protein CCN2/connective tissue growth factor.241 However, experiments using both loss- and gain-of-function approaches have challenged the in vivo significance of CCN2 in mediating the pro-fibrotic actions of TGF-β.224,242 The gp130 cytokine IL-11 has also been suggested to act as a critical downstream signal mediating TGF-β-driven fibrosis not only in the heart187 but also in other organs.188,189
Considering its broad cellular targets, some of the in vivo fibrogenic actions of TGF-β may involve indirect activation of other cell types. Direct in vivo evidence supporting this notion is lacking. Although myeloid cell-specific loss of Smad3 markedly attenuated TGF-β synthesis by macrophages, collagen deposition in the infarct and interstitial fibrosis in non-infarcted areas was not affected in a model of non-reperfused MI.243
5.4.2 Other members of the TGF-β superfamily
Information on the role of other members of the TGF-β superfamily in cardiac fibrosis is limited. Several members of the bone morphogenetic protein (BMP) subfamily, including BMP2, BMP4, and BMP6, are up-regulated in infarcted hearts and may exert pro-inflammatory actions244; however, their potential involvement in the fibrotic response has not been investigated. Two members of the BMP subfamily, BMP7 and BMP10, have been reported to exert anti-fibrotic effects. Several independent investigations suggested that BMP7 administration attenuates collagen deposition in models of pressure overload and diabetic cardiomyopathy.22,245,246 The antifibrotic effects were attributed to blockade of TGF-β signalling and were associated with attenuated endothelial to mesenchymal transition.22,247 Moreover, gain-of-function studies have suggested that BMP10 may exert anti-fibrotic actions in the isoproterenol-treated heart.248 Whether these effects involve direct modulation of fibroblast phenotype, or indirect actions involving other cell types is unclear.
The follistatins bind with high affinity with TGF-β superfamily proteins (such as activins, BMPs and growth differentiation factors) disrupting their function. In addition to these effects, several members of the follistatin family have been implicated in the pathogenesis of cardiac fibrosis through direct activation of cardiac fibroblasts. Follistatin-like 1 activated a reparative programme in infarct fibroblasts through ERK signalling and protected the infarcted ventricle from cardiac rupture.249In vitro studies have suggested that follistatin-like 3 may also have fibroblast-activating effects.250
5.5 The PDGFs
The PDGFs (PDGF-AA, -BB, AB, CC, and DD) are homo- or hetero-dimeric growth factors that signal through two different receptors: PDGFR-α and PDGFR-β. Abundant evidence suggests that PDGF isoforms play a role in activation of the fibrotic response following myocardial injury. PDGF-A, PDGF-C, and PDGF-D have potent fibrogenic effects on the myocardium, mediated through direct actions and, at least in part, through up-regulation of TGF-β.251In vitro, PDGF-A and PDGF-D potently stimulate cardiac fibroblast proliferation and ECM protein synthesis.252,253In vivo, overexpression of PDGF-A or PDGF-D cause marked fibrotic cardiomyopathy associated with fibroblast activation.254,255 PDGF isoforms and PDGFRs are up-regulated in fibrotic hearts256 and have been implicated in the pathogenesis of fibrotic responses. Consistent with its high expression in fibroblasts, PDGFR-α activation has been consistently involved in myocardial fibrosis. In a model of reperfused MI, treatment with a neutralizing anti-PDGFR-α antibody attenuated collagen deposition.256 Moreover, in pressure-overloaded hearts, PDGFR-α blockade reduced atrial fibrosis and protected from atrial fibrillation.257 Although such effects likely reflect direct inhibition of PDGFR − α-driven fibroblast activation, actions on other cell types cannot be excluded. Moreover, broad PDGFR blockade through administration of the non-specific small molecule kinase inhibitor imatinib was found to reduce interstitial fibrosis in models of MI258 and viral myocarditis.259
The PDGF-BB/PDGFR-β axis on the other hand is predominantly activated in vascular mural cells; thus, its fibrogenic actions may be more limited. PDGFR-β activation through an integrin β1-mediated mechanism was suggested to augment fibroblast proliferation and matrix synthesis in a model of cardiac pressure overload.260 However, mice overexpressing PDGF-B had only focal fibrotic changes, much less severe than PDGF-A overexpressing animals.255 Neutralization of PDGFR-β signalilng in infarcted mice reduced collagen deposition, but had much more impressive effects in perturbation of vascular maturation, highlighting the significance of PDGFRβ actions on vascular mural cells.256,261
5.6 The Wnt/β-catenin axis
In mammals, the Wnt signalling pathway is involved in embryonic development, but becomes quiescent in adult low turnover tissues, such as the heart. Members of the Wnt family of secreted glycoproteins are secreted following injury,53,262 bind to Frizzled family transmembrane receptors and initiate signalling through canonical pathways involving β-catenin, or through non-canonical pathways. Activation of the canonical β-catenin pathway requires Wnt-mediated dissociation of the β-catenin degradation complex, stabilization and accumulation of β-catenin in the cytoplasm, followed by nuclear translocation and interactions with transcription factors and transcriptional co-activators that regulate gene expression (Figure 5). Wnt/β-catenin activation has broad effects on a wide range of cellular responses,263 involved in repair, remodelling and regeneration.
Figure 5.

The Wnt/β-catenin pathway plays an important role in cardiac fibrosis. In the absence of Wnt, cytoplasmic β-catenin is continuously degraded by the Axin complex, which involves casein kinase 1 (CK1), adenomatous polyposis coli (APC) and glycogen synthase kinase 3 (GSK3). Wnts are released in injured and remodelling myocardium and bind to Frizzled transmembrane receptors and their co-receptors LRP5/6. This complex recruits the scaffolding protein Dishevelled (Dvl) and inhibits axin-mediated β-catenin phosphorylation, which is required for ubiquitination and proteasomal degradation. Thus, through the effects of Wnts, β-catenin is stabilized and translocates to the nucleus, where it interacts with T cell Factor (TCF), regulating gene expression. Activation of β-catenin in cardiac fibroblasts has been implicated in fibrogenic signalling and in paracrine hypertrophic effects on cardiomyocytes. Secreted frizzled-related proteins (sFRP) bind to Wnts and inhibit their actions.
In the injured heart, Wnt signalling is activated in many different cell types and has been suggested to contribute to reparative, regenerative and fibrotic responses. Secretion of Wnt proteins may be mediated through growth factors, such as TGF-β,40 and requires post-translational modification by the acyltransferase Porcupine (Porcn), a process that supplies a single fatty acid adduct necessary for Wnt protein secretion. In vitro, Wnt family members promote conversion of cardiac fibroblasts to myofibroblasts and stimulate cytokine synthesis.264,265In vivo, administration of a small molecule inhibitor that disables Porcn, thus preventing secretion of Wnt proteins, reduced fibrosis in a model of MI.266 Wnt-mediated fibroblast activation in the injured heart likely involves signalling through the β-catenin pathway.267,268 Cell-specific genetic loss-of-function studies demonstrated that fibroblast-specific activation of β-catenin mediates both fibrosis and cardiomyocyte hypertrophy in the pressure-overloaded heart.268 The downstream molecular targets of the Wnt/β-catenin axis remain unknown. It should be emphasized that Wnt-induced fibrogenic actions may not be due exclusively to direct fibroblast activation, but may also involve effects on other cell types, including cardiomyocytes, vascular cells, and immune cells.269,270
5.7 Endothelin-1
Endothelin (ET)-1 exerts potent fibrogenic effects, acting downstream of cytokines and neurohumoral mediators,271 thus linking inflammation and fibrosis.272 Angiotensin II and TGF-β induce ET-1 in vitro,273 and in experimental models of cardiac fibrosis.274 Up-regulation of ET-1 is consistently noted in many fibrosis-associated cardiac pathologies, including MI, heart failure, hypertensive heart disease, diabetes, and ageing.275–277 Fibrogenic effects of ET-1 in myocardial disease are suggested by both genetic models and by pharmacologic inhibition studies. Cardiac ET-1 overexpression in mice induced myocardial fibrosis associated with both systolic and diastolic dysfunction.278 Moreover, endothelial-specific loss of ET-1 attenuated fibrosis in a model of angiotensin II infusion.279 In experimental animal models of hypertensive heart disease and of reparative myocardial fibrosis, ET-1 inhibition reduced fibrotic remodelling.280,281 The anti-fibrotic effects of ET-1 blockade may have therapeutic implications. Endothelin receptor antagonists reduced myocardial stiffness, suppressing collagen I deposition and attenuating interstitial and perivascular fibrosis in rat and mouse models of aldosterone infusion that recapitulate features of human HFpEF.282,283
To what extent the fibrogenic effects of ET-1 are due to direct activation of fibroblasts vs. indirect actions on other cellular targets, such as endothelial cells and cardiomyocytes, remains unknown. In vitro, cardiac fibroblasts are highly responsive to ET-1 stimulation, exhibiting increased proliferative activity,284 accentuated ECM protein synthesis,285 and resistance to apoptosis.286 In a model of type 1 diabetes, ET-1-mediated EndMT has been suggested to contribute to the myocardial fibrotic response.287 Unfortunately, the evidence supporting this specific cellular mechanism was limited to dual staining with non-specific markers for fibroblasts and endothelial cells.
5.8 The ECM as a modulator of the fibrosis-associated cellular response
The profound changes in the amount and biochemical profile of the ECM proteins in fibrotic hearts, not only has consequences on cardiac function and conduction of the electrical impulse, but may also play a major role in regulation of cellular responses and in transduction of fibrogenic signalling pathways. Generation of matrix fragments (termed matricryptins and matrikines) by activated proteases ,288 deposition of provisional matrix proteins (such as fibronectin and fibrin), and induction of matricellular macromolecules that enrich the interstitial matrix are the best-documented mechanisms for matrix-mediated regulation of fibrogenic cellular responses in injured hearts. Cardiac remodelling is intricately regulated by this crosstalk between the cells and the surrounding ECM: the cells secrete ECM molecules with regulatory properties, but also respond to changes in the matrix environment by interacting with specific functional domains of ECM macromolecules.65,289
5.8.1 Matrikines and matricryptins
Many forms of cardiac injury, including myocardial ischaemia and pressure overload, induce rapid release and activation of proteases that degrade the native ECM, generating bioactive peptides termed matrikines and matricryptins.290,291 Matrikines are fragments of ECM macromolecules with biological properties distinct from those of the full-length form of the molecule. The term matricryptin is used to describe a matrikine that requires proteolytic processing to expose a bioactive functional domain.292 These matrix fragments bind to cell surface receptors on fibroblasts, vascular cells or immune cells, and can modulate the fibrotic response. In a protease-rich environment, all constituents of the cardiac ECM, (including collagens I, IV, and XVIII, elastin, fibronectin, hyaluronan, and tenascin-C) can generate fragments.288,293 Most of the evidence on the role of ECM fragments in tissue injury suggests actions that regulate angiogenesis, and effects on immune cells that activate inflammatory signalling cascades,291,294 and may indirectly promote fibrosis. In addition to these actions, in vitro studies have demonstrated that matrix fragments may also modulate fibroblast phenotype, potentially exerting direct actions in cardiac fibrosis.295–298 However, the in vivo role of specific matrix fragments in fibrotic myocardial conditions has not been investigated.
5.8.2 The provisional matrix components: fibrin and fibronectin
Myocardial injury increases microvascular permeability, causing extravasation of plasma fibrinogen and fibronectin and generating a provisional matrix network that is particularly prominent during the inflammatory and proliferative phase of MI.299 The provisional matrix provides a dynamic substrate for interactions between integrins expressed by fibroblasts and immune cells, and functional domains of fibrin and fibronectin, thus facilitating cell migration and transducing activating signals.300,301 The ED-A variant of cellular fibronectin plays an important role in activation of TGF-β and in subsequent myofibroblast conversion.47,302–304 Moreover, the plasma-derived provisional matrix is later enriched with cell-derived matrix components, including non-fibrillar collagens, cellular fibronectin, proteoglycans and a wide range of matricellular macromolecules.
5.8.3 Deposition and cross-linking of fibrillar collagens
Secretion of the major fibrillar collagens, collagen I and collagen III is the hallmark of cardiac fibrosis.8,31 The relative amounts of type I vs type III collagen fibres may be important in regulation of the mechanical properties of the myocardium. Collagen I fibres are thicker and stiffer; in contrast the finer reticular collagen III fibres are more compliant. The accentuated and prolonged up-regulation of type I collagen in hypertensive hearts has been suggested to increase myocardial stiffness.305,306 Moreover, in patients with dilated cardiomyopathy enrichment of the cardiac ECM with collagen I fibres was associated with reduced ventricular compliance.307 Other members of the fibrillar collagen subfamily may play important regulatory roles in cardiac fibrosis. Collagen V is known to regulate collagen fibril diameter and assembly. Recent studies in a model of MI suggested that collagen V induction in healing scars restrains fibroblast activation by regulating expression of mechanosensitive integrins.308
Following their secretion by activated fibroblasts and myofibroblasts, procollagen chains are processed, assembled into fibrils and cross-linked. Cross-linking enzymes, such as the members of the lysyl-oxidase (LOX) and transglutaminase (TG) families play a crucial role in formation of intermolecular covalent links between collagen chains. The LOX family is the main enzymatic system responsible for cross-linking of extracellular collagen in fibrotic hearts309–313; its members act on specific lysine or hydroxylysine residues, catalysing allysine aldehyde formation, leading to subsequent spontaneous formation of the cross-links. TG2, (also known as tissue transglutaminase) is the highest-expressed member of the transglutaminase family in remodelling hearts314–316 and catalyzes formation of an isopeptide bond between the ε-amino group of a lysine and the γ-carboxamide group of a glutamine. In addition to its enzymatic effects, TG2 may also regulate fibroblast phenotype through non-enzymatic mechanisms, modulating integrin-dependent responses.317 Cross-linking is a major determinant of collagen fibre stiffness and resistance to degradation and has major implications in regulation of cardiac function.318 Although some matrix cross-linking activity is required to provide mechanical support and to prevent chamber dilation under conditions of stress319 excessive collagen cross-linking increases myocardial stiffness and can contribute to the pathogenesis of diastolic dysfunction.320
5.8.4 Non-fibrillar collagens
In addition to the increased synthesis and deposition of structural fibrillar collagens, cardiac fibrosis is also associated with overexpression of several non-fibrillar collagens, including collagen IV, VI, and VIII.321,322 Fibrogenic growth factors, such as TGF-β, are responsible for induction of non-fibrillar collagens in activated fibroblasts and in fibrotic hearts. Collagen VI and collagen VIII have been implicated in fibroblast activation in experimental models of cardiac remodelling. Collagen VI promotes myofibroblast conversion,323 and enhances fibrosis in experimental MI.324 Collagen VIII stimulates fibroblast migration and enhances TGF-β synthesis, accentuating fibrosis in the pressure-overloaded heart.325
5.8.5 Matricellular proteins, galectins and proteoglycans
Matricellular proteins are a group of structurally unrelated proteins that do not play a major role in tissue homeostasis, but are up-regulated following injury and modulate cellular responses.65 Matricellular proteins bind to structural ECM proteins and to cell surface receptors, thus transducing or modulating signalling cascades, and may also regulate activity of proteases, cytokines and growth factors. Several matricellular macromolecules, including the thrombospondins (TSPs), SPARC (secreted protein acidic and rich in cysteine), osteopontin, periostin, the tenascins-C and -X, and the members of the CCN family have been implicated in the pathogenesis of cardiac fibrosis, acting as key regulators of fibroblast activity and of immune cell function. Table 3 summarizes the role of specific matricellular proteins in fibrotic hearts. Localized expression of matricellular proteins in areas of injury controls the spatial localization of the reparative response in infarcted hearts. On the other hand, inappropriate, excessive, or unrestrained induction of matricellular proteins may promote persistent fibrosis.
Table 3.
Functions of the prototypical matricellular proteins in cardiac fibrosis
| Matricellularprotein | Biochemical properties | Proposed functions in models of cardiac remodelling and fibrosis | References |
|---|---|---|---|
| Thrombospondin (TSP)-1 | Homotrimeric glycoprotein with multiple functional domains. Characterized by the presence of three properdin-like repeats, called type 1 domains. | TGF-β activation and subsequent myofibroblast conversion. Inhibition of MMPs and stabilization of the matrix network. Inhibition of angiogenesis. Direct effects on collagen fibril formation. | 65 , 214 , 326–328 |
| Tenascin C | Hexameric multimodular glycoportein (hexabrachion). Not expressed in normal hearts, but induced and deposited in remodelling myocardium. | Pro-inflammatory actions. Recruitment of fibrogenic macrophages. Activation of fibrogenic integrin signalling inmacrophages. Fibrogenic actions though accentuation of growth factor signalling. Regenerative actions in fish and amphibians. |
329–334 |
| SPARC (secreted protein acidic and rich in cysteine) | Multimeric glycoprotein with a high-capacity calcium-binding domain, a follistatin-like domain and high affinity to collagen. | Regulates post-synthetic collagen processing. May exert fibrogenic actions through ADAMTS1 induction. Accentuates growth factor signalling, promoting fibroblast activation and angiogenesis. |
335–338 |
| Osteopontin | Acidic glycoprotein expressed predominantly in macrophages, but also in other cell types, with a central RGD sequence that binds integrins. Also interacts with CD44 through RGD-independent mechanisms. | Regulates matrix assembly. Activates growth factor signalling, promoting fibroblast activation and myofibroblast conversion. May stimulate integrin-dependent fibrogenic actions. May induce MMPs. May induce expression and activity of the crosslinking enzyme lysyl-oxidase. May induce galectin-3 expression in macrophages. Has been suggested to exert fibrogenic actions through effects on mitochondrial function. |
339–348 |
| Periostin | Fasciclin domain-containing adhesive glycoprotein, highly expressed by activated fibroblasts and secreted into the ECM following injury. | Regulates matrix assembly. Activates a fibrogenic programme in fibroblasts. May have regenerative functions in neonatal mouse hearts. |
349–351 |
The galectins are β-galactoside-binding lectins involved in cell proliferation, differentiation, adhesion and migration that play important roles in regulation of inflammatory and immune responses.352 Galectin-3, the best-studied member of the family, has been implicated in the pathogenesis of cardiac fibrosis. Galectin-3 is markedly and consistently up-regulated in fibrotic hearts and is predominantly expressed by activated macrophages.353,354 Macrophage-derived galectin-3 may bind to the ECM acting as a matricellular protein, or may have cytokine-like actions. In vitro, galectin-3 activates fibroblasts, stimulating their proliferation, and collagen synthesis,353 but may also promote a fibrogenic phenotype in macrophages.355–357 Experiments on the in vivo role of galectin-3 in cardiac fibrosis have produced conflicting data. Some studies using galectin-3 knockout mice have suggested a critical involvement of galectin-3 in fibrosis and dysfunction of the pressure-overloaded heart,358,359 while other investigations using very similar models reported no significant effects of galectin-3 in cardiac fibrosis and remodelling.354,360 Due to its marked up-regulation and secretion in fibrotic cardiac conditions, galectin-3 may be a promising biomarker, and a useful predictor of adverse outcome in patients with heart failure.361,362
The fibrotic heart is also rich in proteoglycans, with a profile that is dependent on topography. Heparan sulfate proteoglycans (HSPGs) are typically found close to the cell surface and accumulate in the pericellular matrix. Further away from the cells, chondroitin- and dermatan sulfate-containing proteoglycans (CSPGs and DSPGs) predominate.363 Growth factor stimulation induces expression and deposition of CSPGs in the fibrotic myocardium.364 The cardiac ECM also contains small leucine rich proteoglycans (SLRPs), such as decorin, biglycan, fibromodulin, lumican, and osteoglycin. SLRPs have been implicated in the organization of the structural collagen-based ECM, in regulation of growth factor activation, and in signalling responses involving receptor tyrosine kinases. In vivo studies, has implicated several members of the SLRP family in regulation of the fibrotic response. Biglycan and osteoglycin have fibrogenic actions and contribute to the organization of the structural matrix in reparative fibrosis.365–367 Lumican and decorin on the other hand may be anti-fibrotic, inhibiting neurohumoral and TGF-β-driven fibrogenic responses.368,369 Fibromodulin also had anti-fibrotic effects in vitro; however, did not significantly affect fibrosis in an in vivo model of pressure overload.370 Whether the effects of SLRPs in regulation of fibrosis are mediated through direct actions on fibroblasts, or represent effects on other cell types remains unknown.
5.9 The syndecans
The syndecans are transmembrane proteoglycans that contribute to signal transduction in growth factor-mediated fibrogenic processes. Syndecans can also undergo proteolysis, shedding their ectodomain and acting as soluble HSPGs. The bulk of the evidence from loss-of-function mouse models suggests important fibrogenic roles for syndecan-1 and -4 in myocardial diseases. Syndecan-1 is implicated in activation of reparative fibroblasts following MI, protecting the ventricle from rupture,371 and plays a similar fibroblast-activating role in angiotensin II-induced fibrosis, promoting dysfunction.372 Syndecan-4 has also been implicated in myofibroblast activation in infarction373 and pressure overload models.374 The fibrogenic actions of syndecans may be mediated through activation of TGF-β signalling cascades,372,375 or through activation of integrin-dependent signalling.376 Shedding of syndecans and release of soluble ectodomains with distinct properties further complicates the biological actions of syndecans.377,378 Syndecans may mediate fibrotic conditions not only through direct actions on cardiac fibroblasts but also through effects on immune cell recruitment and function,379,380 and on cardiomyocyte survival.381
5.10 CD44
The ubiquitously expressed glycoprotein CD44 is involved in regulation of a wide range of cellular functions, including cell adhesion, migration, proliferation and differentiation.382 In fibrotic conditions, ECM proteins such as hyaluronan, and osteopontin can bind to CD44 and transduce fibrogenic intracellular cascades. In a model of reparative fibrosis,383 and in a model of angiotensin II-induced fibrotic cardiomyopathy,384 CD44 transduced signals that activated cardiac fibroblasts, triggering cell proliferation and promoting activation of TGF-β cascades. Fibrogenic actions of CD44 may also be mediated through effects on immune cells.383,385 Alternatively spliced CD44 isoforms with distinct pro- or anti-fibrotic properties contribute additional layers of complexity to the effects of this multifunctional glycoprotein.386
5.11 Mechanosensitive signalling pathways in cardiac fibrosis
In addition to their reparative response to injury and death of surrounding myocardial cells, which is mediated predominantly through secreted mediators, or through changes in the composition and stiffness of the ECM, fibroblasts can also become activated by mechanical stress. Fibroblasts sense mechanical forces through mechanosensitive receptors, such as integrins, ion channels, G-protein coupled receptors and growth factor receptors,387 and activate downstream pathways that promote matrix transcription (Figure 6). Mechanosensitive activation of fibroblasts may have evolved to protect the integrity of the tissue, preventing catastrophic effects of mechanical forces on tissue structure. However, in the heart prolonged mechanical tension triggers persistent fibroblast activation and excessive deposition of collagens, leading to maladaptive changes in myocardial structure and functional impairment.
Figure 6.

Mechanosensitive pathways play a critical role in fibroblast activation in many cardiac pathologic conditions. Integrins, mechanosenstive ion channels, growth factor receptor activation, and activation of G-protein coupled receptors stimulate FAK, MAPK, RhoA/ROCK, and PI-3K signalling, mediating responses of fibroblasts to mechanical stress. Cytoskeletal changes are also sensed through the MRTF/SRF axis or the YAP/TAZ system and regulate transcription of fibrogenic genes. TF, transcription factors.
5.11.1 Integrins
Integrins are transmembrane receptors involved in transduction of mechanonensitive, matricellular and growth factor-mediated signals. In injured and remodelling hearts, integrins are induced and activated in fibroblasts, cardiomyocytes and immune cells, and transduce signalling cascades with crucial roles in cardiac fibrosis.388,389 Growth factors, such as TGF-β and neurohumoral mediators, such as angiotensin II induce and activate integrins in cardiac fibroblasts.79,232 Activated integrins interact with a wide range of binding partners and transduce downstream signalling cascades, promoting a migratory, matrix-synthetic and proliferative fibroblast phenotype. Integrins β1 and αV are the best-studied members of the family in cardiac fibrosis, and have been implicated in fibroblast activation in several different pathologic conditions. Integrin β1 is induced and activated in fibroblasts and immune cells following cardiac injury,390 and mediates fibroblast activation in pressure-overloaded hearts.391 In a model of angiotensin-mediated cardiac remodelling, αv integrin expressed in perivascular PDGFRβ+ cells was implicated in the development of fibrosis.392 Whether this finding reflects conversion of PDGFRβ+ mural cells to fibroblasts is unclear. αV integrins may also exert fibrogenic actions by activating latent TGF-β.393 Mechanosensitive integrin-mediated fibrogenic actions may involve activation of focal adhesion kinase (FAK), Ras homologue gene family/Rho-associated coiled-coil containing kinases (Rho/ROCK) and MAPK signalling.
5.11.2 FAK
FAK, a tyrosine kinase without a receptor protein, is activated in response to mechanical stress and represents a critical molecular link between mechanosensitive or growth factor-mediated integrin activation, and acquisition of a matrix-synthetic myofibroblast phenotype.394–396In vivo, pharmacologic inhibition of FAK attenuated cardiac fibrosis in a model of MI,397,398 and FAK siRNA knockdown reduced collagen deposition in a model of pressure overload.399 Whether these effects reflect critical FAK actions on cardiac fibroblasts remains unknown. FAK has broad effects on cardiomyocytes, vascular and interstitial cells400,401; thus, FAK-mediated fibrosis may be due to actions on several different cell types. Cell-specific loss-of-function approaches are needed to document the in vivo role of FAK in mechanosensitive or growth factor-mediated activation of fibroblasts.
5.11.3 MAPKs
In the injured and remodelling myocardium, MAPKs are activated in many different cell types by a wide range of stimuli, including mechanical stress, secreted mediators (cytokines, growth factors, and neurohumoral mediators) and matricellular proteins. Although extensive in vitro evidence suggests important roles for MAPK pathways in fibroblast activation, the in vivo role of MAPKs in fibroblast responses is less convincingly documented. p38α MAPK is the major isoform expressed in cardiac fibroblasts402 and was found to promote myofibroblast conversion upon ischaemic injury or neurohumoral stimulation. The fibrogenic actions of p38 MAPK were attributed to actions involving the transcription factor serum response factor (SRF) and calcineurin.114,239 In contrast, in vivo studies investigating fibroblast-specific actions of ERK and JNK MAPKs are lacking.
5.11.4 The RhoA/ROCK pathway
Activation of tyrosine kinases, G-protein couple receptors and integrins recruits Rho guanine nucleotide-exchange factors (RhoGEFs), leading to activation of the small GTP-binding protein RhoA. Subsequently RhoA signals through the Rho-associated coiled-coil containing kinases (ROCKs), ROCK1 and ROCK2 and plays an important role in generation of actin-myosin contractility and in regulation of cytoskeletal dynamics. The effects of ROCK on the actin cytoskeleton have profound impact on many cellular functions, including cell migration, proliferation, differentiation and survival, proliferation, migration and differentiation through actions on the actin cytoskeleton.403 The RhoA/ROCK system may play an important role in fibrosis through effects on several different cell types, including fibroblasts, cardiomyocytes, immune cells, and vascular cells.404 In fibrotic hearts RhoA activation may be mediated through mechanosensitive cascades or angiotensin II stimulation and has been suggested to involve p63RhoGEF.405 The in vivo evidence suggesting a role for RhoA/ROCK in fibrotic remodelling of the heart is based on both pharmacologic inhibition and genetic studies. Inhibition of the RhoA-ROCK pathway reduced fibrosis of the pressure-overloaded heart.406 Whether ROCK-mediated fibrosis reflects direct actions on fibroblasts, or indirect effects in macrophages or cardiomyocytes remains unknown. In the pressure overload model, cardiomyocyte-specific RhoA signalling triggers fibrogenic signalling through yet-unidentified paracrine effects.407 The relative role of the ROCK isoforms in cardiac fibrosis remains poorly understood; evidence suggests a role for both ROCK1 and ROCK2 in fibroblast activation. Global hemizygous ROCK1 +/− mice and homozygous ROCK1 null animals had attenuated cardiac fibrosis in models of left ventricular pressure overload and ischaemic cardiomyopathy.408,409 Moreover, fibroblast-specific ROCK2 was found to contribute to angiotensin II-induced fibrosis through CCN2 and Fibroblast Growth Factor (FGF)2 induction.410
5.11.5 Mechanosensitive ion channels
Emerging evidence suggests an important role for mechanosensitive ion channels in activation of fibroblasts and in the pathogenesis of myocardial fibrosis. Fibrogenic mediators, such as angiotensin II and TGF-β activate members of the transient receptor potential (TRP) family of cationic channels, such as TRPC6, TRPM7, and TRPV4.411–414 All three TRP channels have been implicated in myofibroblast conversion411–414; however, their in vivo role is not uniformly fibrogenic. TRPM7 is a channel fused to a kinase and has been reported to exert both pro- and anti-fibrotic actions. Although the bulk of the in vitro evidence suggests a role for TRPM7 in fibroblast activation,413in vivo the kinase domain of TRPM7 was found to protect from fibrosis, presumably through effects on macrophage inflammatory activity.415 The potassium channel TREK1 has also been involved in fibroblast activation in pressure-overloaded hearts through effects that involve JNK activation.416 Activation of stretch-sensitive ion channels in cardiac fibroblasts results in influx of Ca2+ and other ions. In vitro, angiotensin II Ca2+ oscillations have been broadly implicated in both the contractile function of myofibroblasts and in regulation of ECM synthesis417–420 through activation of multiple signalling pathways, including MAPKs and CAMKII. However, in vivo evidence on the dynamics and role of Ca2+ in cardiac fibrosis is scarce.
5.11.6 YAP/TAZ
The homologous transcriptional coactivators yes-associated protein (YAP) and TAZ (transcriptional co-activator with PDZ-binding motif) are key regulators of cellular responses to mechanical stress.421 The dynamics of the actin cytoskeleton, cadherin-mediated junctions and focal adhesions play an important role in YAP/TAZ activation. YAP/TAZ are regulated both by the Hippo pathway and through Hippo-independent mechanisms. The Hippo pathway negatively regulates YAP/TAZ through phosphorylation and activation of the large tumor suppressor (LATS)1/2 kinases. Upon activation, LATS1/2 phosphorylate YAP and TAZ to promote their nuclear export and degradation, thus abrogating their transcriptional effects. YAP/TAZ actions have profound effects on cellular phenotype, modulating cell differentiation, proliferation and survival. YAP/TAZ has been implicated in fibrotic responses in many different tissues, by accentuating TGF-β-driven activation of Smad2/3422 and by stimulating fibroblast proliferation and synthesis of soluble fibrogenic factors.423 In the myocardium, emerging evidence suggests that the YAP/TAZ may exert fibrogenic actions. Fibroblast-specific deletion of LATS1/2 caused fibrosis in normal hearts and accentuated matrix deposition following infarction, suggesting an important role for YAP/TAZ in activation of a secretory myofibroblast phenotype.424 Moreover, experiments in zebrafish and in rat fibroblasts suggested that YAP/TAZ may promote activation of myofibroblasts and scar formation following MI.425 It should be emphasized that YAP/TAZ-may regulate cardiac fibrosis, not only through direct activating effects on fibroblasts but also by promoting fibrogenic effects in cardiomyocytes,426 or by inhibiting inflammatory activation of a fibrogenic response in epicardial cells.427
5.11.7 The myocardin-related transcription factor (MRTF)/SRF axis
Much like YAP/TAZ, the MRTF pathway is also regulated by cytoskeletal dynamics. Changes in the mechanical environment or growth factor signalling activate the RhoA/ROCK system and trigger F-actin polymerization, leading to MRTF translocation to the nucleus. MRTF interacts with the ubiquitously expressed transcription factor SRF, activating promoters harbouring the DNA element CArG box.428 Interactions between SRF and members of the myocardin family of transcriptional co-activators have been implicated in stimulation of transcription of genes encoding smooth muscle cell contractile proteins. In a similar manner, in fibroblasts, the SRF/MRTF axis plays a dominant role in regulation of α-SMA transcription and subsequent myofibroblast conversion, through interactions that may involve TGF-β/Smad and RhoA/ROCK.429–431In vivo studies showed that mice with global loss of MRTF-A had attenuated fibrosis following MI and in response to angiotensin II infusion.432 Whether these observations reflect abrogation of MRTF-dependent effects on fibroblasts remains unclear, considering that MRTF-A may also modulate cardiomyocyte and vascular cell phenotype and function.433,434
5.12 Oxidative stress
Mitochondrial ROS production is increased in activated fibroblasts and may critically regulate their phenotype and function, contributing to the pathogenesis of fibrotic conditions.435 ROS are generated in ischaemic and remodelling hearts and may represent a common link between many different forms of cardiac stress and fibrosis. Several studies have suggested that the pro-fibrotic effects of fibrogenic growth factors, such as TGF-β, angiotensin II and aldosterone may be mediated through ROS generation.436–439 TGF-β stimulation generates mitochondrial ROS440 and also induces nicotinamide adenine dinucleotide phosphate (NADPH) oxidases that further increase ROS levels in the cells.232,441 Increased ROS levels in activated fibroblasts stimulate ECM gene transcription, but also regulate post-translational modifications of ECM components. Both matrix-preserving and matrix-degrading effects of ROS have been reported. In addition to their effects on collagen synthesis, ROS also critically regulate synthesis and activity of MMPs involved in ECM degradation and remodelling.442,443
5.13 Epigenetic regulation of cardiac fibrosis
The term epigenetic regulation refers to effects of gene expression mediated through chemical modifications of the nucleosomal DNA and histone tails. In fibroblasts, post-translational modifications of histones, including acetylation and methylation, and DNA methylation have well-documented effects on expression of fibrosis-associated genes.444–447 Histone tail lysine residues can be acetylated by histone acetyltransferases (HATs). Although non-specific pharmacologic inhibitors of HAT were reported to attenuate cardiac fibrosis in a mouse model of pressure overload,448 robust evidence on the role of HATs in cardiac fibrotic remodelling is lacking. Acetyl groups are removed from histone tails are by a family of enzymes, the histone deacetylases (HDACs).449 Extensive evidence derived through the use of small molecule inhibitors suggests an important fibrogenic role for HDAC activity in experimental models.450–452 HDAC inhibition has been demonstrated to protect from diastolic dysfunction in both mouse and large animal models of cardiac ageing, hypertension, or slow progressive pressure overload.453–455 The protective actions may be due to inhibition of fibroblast activation and proliferation, but may also involve improved myofibril relaxation in cardiomyocytes.455,456 In addition to placement (writing) and removal (erasing) of histone modifications, a third epigenetic step involves the ‘reading’ of the specific histone marks. Histone acetylation creates docking sites for the ‘acetyl-readers’, the members of the bromodomain and extraterminal (BET) family, which recognize acetylated lysine residues through two tandem aminoterminal bromodomains.457 BRD4 is the best studied member of the BET family in fibrotic conditions and regulates gene expression of fibrosis-associated genes in many organs.458 In cardiac fibroblasts, BRD4 is a central regulator of the activated fibroblast phenotype.459In vivo, BET bromodomain inhibitors inhibited fibrosis in mouse models of pressure overload, MI,460 and laminopathy.461 To what extent these actions reflect direct inhibition of fibroblast activity remains unclear.
5.14 Non-coding RNAs in cardiac fibrosis
A large and growing body of evidence suggests that noncoding RNA transcripts, including microRNAs (miRNAs) and long non coding RNAs (lncRNAs), are involved in the pathogenesis of cardiac fibrosis by regulating gene expression at the post-transcriptional level.462,463 miRNAs have been suggested to target several crucial fibrogenic cascades, including TGF-β/Smad, angiotensin II/MAPK, RhoA/ROCK, MRTF/SRF, and the cationic ion channels.464 It has been suggested that in some cases fibroblast-derived miRNAs may also mediate paracrine effects through their secretion in the cardiac interstitium as exosome cargo, and subsequent uptake by other cell types, such as cardiomyocytes and immune cells.465 Understanding of the role of miRNAs in fibrotic conditions is hampered by the complexity of their actions in many different cell types, and by the broad range of their molecular targets. In a study examining the in vitro effects of 194 different miRNAs, differentially expressed in inflamed hearts, on cellular function, a large percentage of the corresponding mimics (25.8%) modulated fibroblast function.466 Effects of the mimics on other cell types (including cardiomyocytes and macrophages) highlighted the need for in vivo studies in order to determine the role of miRNAs in the fibrotic response. In vivo data have suggested that several miRNAs, including miR-15, miR-29, miR-101, and miR-1954, may be involved in negative regulation of cardiac fibrosis through inhibitory effects on pathways activated by fibrogenic growth factors or matricellular proteins.467–470 MiR-21, miR671-5p, and miR-144-3p on the other hand, have been suggested to exert fibrogenic actions.471–473 The basis for this remarkable complexity in miRNA-mediated regulation of fibrogenic processes remains unclear.
An emerging body of evidence suggests than lncRNAs represent an additional layer of complexity in the regulation of cardiac fibrosis, exerting broad effects on fibroblast phenotype and function.474 Maternally expressed gene 3 (Meg3),475 Wisp2 super-enhancer associated RNA (Wisper),476 pro-cardiac fibrotic lncRNA,477 and plasmacytoma variant translocation 1 (PVT1) have been suggested to exert fibrogenic actions on the myocardium. Much like miRNAs, lncRNAs may also be implicated in paracrine actions that regulate fibrosis. Hypoxic and stressed cardiomyocytes were found to secrete vesicles containing fibrogenic lncRNAs, such as Neat1, that may promote fibroblast activation.478 Whether lncRNAs are involved in the pathogenesis of human conditions associated with cardiac fibrosis remains unknown; however, associative studies have shown a correlation between expression of lncRNAs and ECM genes in ischaemic cardiomyopathy patients.479
5.15 Endogenous mechanisms involved in negative regulation of cardiac fibrosis
Endogenous signals restraining fibroblast expansion and inhibiting fibroblast function have evolved to prevent prolonged, excessive or expanding fibrogenic responses in healing tissues. In the infarcted heart, scar maturation is associated with apoptosis of significant numbers of myofibroblasts.480 Many other fibroblast-like cells survive481 but become quiescent and may reduce their levels of expression of ECM proteins. Deactivation of infarct myofibroblasts may involve in part passive removal of fibrogenic mediators, such as clearance of matricellular proteins and growth factors, but may also require active processes and induction of anti-fibrotic signals. Perturbations of these anti-fibrotic pathways may be involved in the pathogenesis of fibrosis following injury. Unfortunately, our understanding of the specific molecular signals involved in negative regulation of fibrosis remains limited, and their involvement in human myocardial diseases is uncertain. TGF-β is a major target of anti-fibrotic signals and TGF-β signalling cascades are tightly regulated by endogenous inhibitory mediators that act at the receptor level, or by inhibiting downstream intracellular cascades.482 In the failing heart, release of a cleaved soluble form of the accessory TGF-β receptor endoglin was found to act as a negative regulator of TGF-β signalling.483 In the pressure-overloaded heart, expression of the TGF-β pseudo-receptor BAMBI (Bone Morphogenetic Protein/BMP and activin membrane-bound inhibitor) has been suggested to downmodulate TGF-β signalling, attenuating its profibrotic actions,484 and serving as a molecular sink for TGF-β. Induction of the inhibitory Smads, such as Smad7 and Smad6, may restrain pro-fibrotic TGF-β responses through interactions with the TGF-β receptors, or the receptor-activated Smads.485 Other anti-fibrotic signals may inhibit responses to neurohumoral activators of fibrosis. Apelin signalling has been proposed as a negative regulator of angiotensin II-mediated fibrotic remodelling.486 Moreover, fibroblasts may modulate their response to the fibrogenic actions of angiotensin II by altering their relative expression of the pro-fibrotic AT1 receptor vs. the anti-fibrotic AT2.83,84 Activation of a p53- and p16-mediated senescence programme in cardiac fibroblasts may also be implicated in negative regulation of cardiac fibrosis.487 As previously discussed, up-regulation of anti-fibrotic miRNAs may serve an important inhibitory mechanism, preventing excessive fibroblast activation in remodelling hearts.467–469 Whether such inhibitory signals are selectively induced in subsets of suppressive interstitial cells with an anti-fibrotic phenotype remains unknown.
6 Fibrosis in human myocardial diseases
In most clinical studies and in a wide range of myocardial pathologies, the extent of cardiac fibrosis predicts adverse outcome.488 However, this does not necessarily imply causation. Depending on the underlying disease and the pathophysiologic context, the presence of cardiac fibrosis may contribute to dysfunction and adverse outcome, or simply reflect activation of a reparative process.
6.1 Fibrosis in myocardial infarction
MI is typically caused by rapid cessation of coronary blood flow, due to rupture of an atherosclerotic plaque. Occlusion of the vessel causes prolonged ischaemia and activates a ‘wavefront’ of cardiomyocyte death extending from the more susceptible subendocardium to the subepicardium.489 Because the adult mammalian heart has negligible regenerative capacity, massive cardiomyocyte necrosis triggers an inflammation-driven reparative fibrotic response that leads to formation of a scar.490 Thus, infarctive fibrosis is reparative; formation of a scar following infarction is crucial for protection of the ventricle from catastrophic mechanical complications, such as cardiac rupture. Clearly, patients with larger scars have worse prognosis.491,492 However, this association likely reflects the relation between more extensive loss of functional cardiomyocytes and poor outcome, rather than the adverse consequences of a primary fibrotic process.
Although a collagen-based scar cannot replace the rhythmic contractile function of healthy cardiomyocytes, the biochemical composition of the infarct is a major determinant of cardiac mechanics and affects outcome. Decades of experimental work have suggested that inflammation and fibrosis following infarction need to be tightly regulated in order to protect the ventricle from catastrophic structural failure, while limiting the development of heart failure. Formation of myofibroblast arrays that form an organized network of collagen fibres is crucial to prevent cardiac rupture.232 Timely activation and suppression of inflammatory and pro-fibrotic signals, and spatial containment of the reparative process within the area of injury are critical to protect from adverse remodelling and dysfunction. Prolonged, unrestrained or excessive activation of fibroblast-driven matrix deposition can contribute to the development of post-infarction heart failure.493 Overactive inflammatory signalling may promote protease activation, causing ECM degradation, reduction of the tensile strength of the scar and ventricular dilation.494 On the other hand, overactive matrix-synthetic pathways may increase stiffness promoting diastolic dysfunction.
Identification of associations between scar composition and prognosis in patients with MI poses major challenges, considering the limitations of current imaging technologies and biomarker-based approaches. Some studies have suggested associations between cardiac magnetic resonance (CMR)-derived scar composition parameters and clinical outcome in patients surviving MI. Magnetic resonance measurements reflecting the texture of the scar were associated with an increased incidence of arrhythmias following infarction.495 Moreover, high ‘entropy’ within the scar (an indicator of inhomogeneous scar composition) was associated with mortality and increased arrhythmogenicity.496 The potential relation between these crude imaging-derived parameters and specific cellular or biochemical changes remains unknown.
As the scar matures, infarct fibroblasts lose their myofibroblast characteristics30,481 and may undergo apoptosis.480 However, in large infarcts, pressure and volume overload due to the massive loss of contractile cells may result in progressive interstitial fibrosis in the border zone and in the remote remodelling myocardium.497 Associative evidence in experimental models suggests that these interstitial fibrotic changes may contribute to systolic and diastolic dysfunction leading to post-infarction heart failure. In animal models, the beneficial effects of ACE inhibition and AT1 receptor blockade following infarction were associated with attenuated interstitial collagen accumulation in non-infarcted myocardial segments.498 However, data on the extent and significance of fibrotic remodelling of non-infarcted segments in patients with MI are conflicting. In a biochemical study using explanted hearts and autopsy material, no significant increases in collagen content were noted in non-infarcted myocardium remote from the scar.499 In contrast, in patients with reperfused ST-elevation MI, CMR showed expansion of the extracellular volume in non-infarcted segments that was associated with progression of contractile dysfunction.500
6.2 Fibrosis in chronic heart failure
Myocardial fibrotic remodelling is found in many pathologic conditions associated with heart failure. In many cases, it is unclear whether fibrosis represents the main causative cellular process responsible for dysfunction, or simply reflects a reparative response to a primary injurious insult. However, in most conditions, the presence and severity of myocardial fibrosis carries prognostic information and predicts adverse outcome. Prominent fibrosis is found in patients with both heart failure with reduced ejection fraction (HFrEF) and HFpEF and is associated with all major pathophysiologic conditions that cause heart failure, including chronic ischaemia, genetic cardiomyopathies, pressure and volume overload, and metabolic diseases.
6.3 Cardiac fibrosis and HFpEF
Nearly half of heart failure patients have a preserved ejection fraction. HFpEF patients tend to be older, have a high prevalence of hypertension, obesity and diabetes, and typically exhibit reduced ventricular and vascular compliance. Considering the close association between fibrosis and tissue stiffness (both myocardial and vascular), it is tempting to hypothesize that deposition of cross-linked collagen in the cardiac interstitium or in the vascular wall may promote diastolic dysfunction and play a prominent role in the pathogenesis of HFpEF.501 A large body of evidence from human patients suggests that at least a subset of HFpEF patients exhibits cardiac fibrosis (Table 4). In some studies, the association between HFpEF and fibrosis was documented through histological analysis of biopsied or autopsied myocardial samples503,510; however, in most studies CMR-based measurements of extracellular volume was used to assess fibrotic changes. HFpEF patients with evidence of fibrosis tend to have a higher stiffness constant in comparison to those without significant fibrosis505 and exhibit acute increases in myocardial stiffness with transient elevations of blood pressure.506 Moreover, some investigations have suggested that fibrosis may precede the development of HFpEF in high-risk patients, thus supporting a possible causative relation.509
Table 4.
Clinical evidence on the link between HFpEF and cardiac fibrosis
| Patient population | Fibrosis marker | Finding | References |
|---|---|---|---|
| Hypertensives | Serum markers of collagen turnover (MMP2, CITP, PIIINP) | In a population of hypertensive subjects, markers of collagen turnover (circulating levels of MMP2, CITP, PIIINP) identified patients with diastolic dysfunction and HFpEF. | 502 |
| Heart failure (HFpEF and HFrEF) | Histology | In a heart failure population, histologically assessed fibrosis was comparable in HFpEF and HFrEF groups. | 503 |
| Heart failure (HFpEF and HFrEF) | Circulating biomarkers of fibrosis, CMR | HFpEF patients had elevated plasma biomarkers of inflammation and fibrosis (including TN-C, MMP3, 7 8 and GDF15) in comparison to controls. Biomarker levels were associated with CMR-assessed fibrosis (extracellular volume, ECV). However, indicators of inflammation and fibrosis were comparable between HFrEF and HFpEF patients. | 504 |
| HFpEF | CMR-assessed ECV | On the basis of ECV assessment, two subpopulations of HFpEF patients could be identified. Patients with higher ECV had a higher stiffness constant, whereas those with lower ECV had evidence of prolonged active LV relaxation. | 505 |
| HFpEF | Histology | HFpEF patients exhibited varying levels of fibrosis. Myocardial stiffness increased acutely following manoeuvres associated with transient elevations in systolic blood pressure (such as handgrip). This increase correlated with the levels of myocardial collagen deposition. | 506 |
| HFpEF with and without diabetes | Circulating biomarkers of ECM remodelling. | HFpEF patients with diabetes had increased plasma biomarkers of inflammation (IL-8 and TNFR1) and fibrosis (MMP7, TIMP1), in comparison to non-diabetic HFpEF patients. | 507 |
| HFpEF | CMR-assessed ECV | In HFpEF patients, CMR-assessed LV fibrosis was associated with impaired diastolic function. | 508 |
| HFpEF and patients at risk for HFpEF | CMR-assessed ECV | Similar levels of CMR-assessed myocardial fibrosis were present in patients with HFpEF and those ‘at risk’ for HFpEF (based on elevated B-type natriuretic peptide (BNP) levels). Thus, fibrosis may precede development of HFpEF. | 509 |
| Patients dying with HFpEF or conditions unrelated to heart failure | Histology | In an autopsy study, HFpEF subjects had more severe left ventricular fibrosis and capillary rarefaction than age-matched controls with non-cardiac death and no heart failure. The HFpEF group also had more severe coronary artery disease (CAD), and a higher prevalence of hypertension and diabetes that may account for the increased fibrosis. However, myocardial fibrosis was comparable in HFpEF patients with and without CAD. | 510 |
Extensive evidence suggests that fibrosis in HFpEF has prognostic implications (Table 5), indicating that HFpEF patients without fibrosis have a more favourable outcome.509 It should be emphasized that in practically all studies suggesting prognostic implications, fibrosis was measured using CMR or circulating serum biomarkers. In contrast, a study using histological analysis failed to reveal a relation between fibrosis and prognosis in HFpEF patients.503 The lack of histological evidence to support the prognostic role of fibrosis in HFpEF may not necessarily reflect the absence of a relation, but may reflect the challenges in performing adequately powered biopsy studies in HFpEF patients.
Table 5.
The prognostic implications of fibrosis in HFpEF patients
| Fibrosis-associated marker | Findings | References |
|---|---|---|
| Histology | Fibrosis predicted adverse outcome in HFrEF patients, but NOT in HFpEF patients. | 503 |
| Serum galectin-3 | In a HFpEF population, galectin-3 levels were modestly elevated, were inversely associated with functional performance, and were independently associated with adverse outcome. | 511 |
| Molli-ECV | In HFpEF patients MOLLI-ECV (CMR-assessed) correlated with histological evidence of fibrosis (assessed through trichrome staining in a smaller number of patients). However, ECV-associated fibrosis was not independently associated with adverse outcome. | 512 |
| Serum biomarkers of collagen turnover | In a HFpEF patient population, circulating levels of collagen turnover biomarkers and osteopontin levels were not independently associated with outcome (mortality and hospitalizations), despite association with single-variable analysis. | 513 |
| Serum TSP-2 | Serum TSP-2 levels predicted adverse outcome in HFpEF patients. | 514 |
| Multiple serum biomarkers, including markers of ECM remodelling. | A multimarker approach combining biomarkers reflecting calcification, inflammation, metabolism, angiogenesis and matrix turnover predicted outcome in a HFpEF population. | 515 |
| LGE (late gadolinium enhanced)-MRI | In a HFpEF population, myocardial fibrosis assessed through LGE-MRI was associated with adverse outcome. | 516 |
| CMR | In HFpEF patients, CMR-assessed fibrosis was associated with worse outcome. | 517 |
| CMR | Myocardial fibrosis was associated with worse outcome in both HFpEF patients and in subjects ‘at risk’ for HFpEF (based on high BNP levels). HFpEF patients without fibrosis had better outcome. | 509 |
| CMR | Myocardial fibrosis assessed through CMR-based extracellular volume assessment was increased in HFpEF patients and was an independent predictor of outcome. | 518 |
6.4 Cardiac fibrosis and HFrEF
Fibrotic changes are typically observed in patients with HFrEF regardless of the underlying aetiology. Extensive evidence suggests that the severity of fibrosis in HFrEF patients is associated with worse outcome. In heart failure patients, myocardial fibrosis quantified by CMR was associated with subsequent hospitalizations and mortality across the spectrum of ejection fraction and heart failure stage.519 Moreover, in new onset HFrEF, the absence of fibrosis was associated with favourable prognosis.520 Although the effects of fibrosis on myocardial stiffness and diastolic function are intuitive, causative links between fibrotic interstitial changes and systolic dysfunction are less convincingly documented. Perturbations of the interstitial matrix network may depress systolic function through several distinct mechanisms. First, expansion of the interstitial matrix may disrupt excitation-contraction coupling. Second, certain forms of fibrosis may be associated with prominent activation of proteases, leading to disruption of key molecular links between the matrix and the sarcomeric contractile apparatus, and leading to systolic dysfunction.521 Generation of matrix fragments in protease-rich fibrotic lesions may stimulate pro-inflammatory signalling, further exacerbating systolic dysfunction.234 Third, perturbations in the native matrix network may deprive cardiomyocytes from crucial matrix-dependent molecular signals that promote cardiomyocyte survival, resulting in a form of cell death termed ‘anoikis’.522 Fourth, development of perivascular fibrosis may result in microvascular dysfunction and reduce perfusion of cardiomyocytes under conditions of stress, leading to depression of systolic function.
6.5 Cardiac fibrosis in chronic ischaemic cardiomyopathy
Patients with ischaemic cardiomyopathy often have evidence of replacement fibrosis, even in the absence of a history of MI, reflecting in many cases silent coronary events. However, both experimental and clinical studies suggest that episodic demand ischaemia can cause fibrosis, in the absence of infarction. In a subset of patients dying with severe coronary artery disease, severe interstitial and perivascular fibrosis was found in the absence of grossly visible acute or healed MI.523 Moreover, in patients with ischaemic cardiomyopathy undergoing aortocoronary bypass surgery, segments without evidence of prior MI, but perfused through a partially occluded coronary, exhibited interstitial fibrosis, associated with contractile dysfunction27,28 (Figure 7). The characteristics of the fibrotic lesions predicted recovery of function following revascularization. Segments with a highly cellular interstitium, and more abundant myofibroblasts and inflammatory cells were more likely to recover after revascularization; in contrast segments with hypocellular interstitial fibrosis and increased collagen deposition had persistent dysfunction. In a mouse model, brief repetitive myocardial ischaemia and reperfusion-induced fibrosis in the absence of infarction, by activating an ROS-dependent chemokine-mediated fibrogenic programme.39,129 Thus, brief ischaemic insults, or low perfusion states associated with episodic demand ischaemia may be sufficient to stimulate interstitial myocardial fibrosis, contributing to the pathogenesis of ischaemic cardiomyopathy.
Figure 7.

Fibrotic changes in patients with ischaemic cardiomyopathy in the absence of myocardial infarction. Patients with ischaemic cardiomyopathy undergoing aortocoronary bypass surgery underwent transesophageal echocardiography (TEE)-guided biopsy to sample non-infarcted segments perfused by partially occluded coronaries. (A) Prominent interstitial fibrosis (arrows), and occasional small foci of replacement fibrosis (arrowheads) were typically noted in these segments. (B) Immunohistochemical staining shows interstitial deposition of the matricellular protein tenascin-C (arrows), an indicator of active remodelling. C. α-SMA staining identifies myofibroblasts in the cardiac interstitium (arrows). The findings of the study (originally published in ref.28 and ref.27) suggested that segments with an active interstitum, characterized by more inflammatory cells, more myofibroblasts, but less mature collagen had a higher chance of recovery following revascularization. These observations may support a role for interstitial cells in resolution and regression of fibrosis. Scalebar = 50 µm.
6.6 Fibrosis in genetic cardiomyopathies
Although extensive associative data suggest that fibrosis often accompanies cardiomyocyte changes in patients with genetic cardiomyopathies, the role of fibrotic remodelling in mediating dysfunction and disease progression is unclear. Limited data from animal model studies using cell-specific loss-of-function approaches support the notion that fibroblasts in certain genetic cardiomyopathies do not simply respond to injury in a reparative manner, but may undergo activation, secreting large amounts of ECM proteins and accentuating dysfunction. In sarcomere mutation carriers, elevations of circulating biomarkers of collagen synthesis were noted prior to development of overt hypertrophic cardiomyopathy (HCM),524 suggesting that fibrogenic activation may precede the clinical manifestations. In transgenic mice with cardiomyocyte-specific overexpression of a truncated cardiac myosin binding protein C (a mouse model that recapitulates features of a relatively common human form of HCM), abrogation of TGF-β signalling specifically in myofibroblasts attenuated fibrosis, reduced cardiomyocyte hypertrophy, and prolonged lifespan.525 Some genetic forms of dilated cardiomyopathy (DCM) are also associated with severe and diffuse fibrosis,526 and in mouse models of genetic DCM, fibrogenic activation occurs early and has been suggested to contribute to disease progression.527,528 Moreover, in patients with arrhythmogenic right ventricular cardiomyopathy (ARVC), mutations in genes encoding desmosome proteins trigger expansion and activation of fibro-adipocytes, leading to collagen deposition and fatty infiltration529 and may also activate a fibrogenic programme in cardiomyocytes.530,531
Whether certain genetic conditions can cause cardiomyopathy in human patients through primary activation of fibroblasts is unknown. Patients with mutations in desmoplakin, the primary force transducer between cardiac desmosomes and intermediate filaments, exhibited a cardiomyopathy characterized by early onset of left ventricular fibrosis that preceded systolic dysfunction and was associated with arrhythmias.532 In autopsies of young patients with sudden cardiac death, cardiac fibrosis is often found in the absence of any identifiable cardiomyocyte pathology.533 In many of these patients mutations in cardiomyocyte-specific genes are identified,534 supporting the notion that fibrotic remodelling involves paracrine cardiomyocyte-derived signals, and not primary activation of interstitial cells.
6.7 Fibrosis in heart failure associated with pressure overload
Left ventricular pressure overload induces interstitial and perivascular fibrosis in several common pathologic conditions, including hypertensive heart disease and in the cardiomyopathy associated with aortic stenosis. Long-standing hypertension triggers collagen deposition in the interstitium and in perivascular areas, increasing myocardial stiffness and contributing to the pathogenesis of HFpEF.535,536 Moreover, in patients with aortic stenosis, fibrosis assessed through CMR was more extensive in women,537 and was independently associated with mortality.538–540 Although concomitant ischaemic heart disease may be responsible for fibrosis in aortic stenosis patients, in many cases, significant fibrotic lesions are noted in the absence of coronary atherosclerosis,541 reflecting the consequences of chronic pressure overload.
In animal models, left ventricular pressure overload induces early hypertrophy, fibrosis and diastolic dysfunction, followed by decompensation, chamber dilation, and development of systolic dysfunction.542,543 Although the severity of fibrotic changes and the time course of cellular events are dependent on the specific characteristics of the pressure overload model, the alterations recapitulate many of the geometric and functional changes observed in patients with untreated malignant hypertension, or uncorrected critical aortic stenosis. Several mechanisms co-operate to activate fibroblasts in the pressure overloaded heart. First neurohumoral activation plays an essential role in promoting myofibroblast conversion and acquisition of a matrix-synthetic programme.544 Second, induction of matricellular proteins locally activates growth factor-mediated signalling in fibroblasts, stimulating ECM protein synthesis.65,545 Third, high intracardiac pressures directly activate mechanosensitive cascades, triggering fibrogenic signalling in cardiac fibroblasts.546 Fourth, inflammatory cytokines and growth factors released by immune cells, or by stressed cardiomyocytes may significantly contribute to fibroblast activation. The critical contribution of lymphocytes and macrophages in the pathogenesis of fibrosis and dysfunction following pressure overload has been documented by several experimental studies.157,547,548 Activation of immune cells in the pressure-overloaded myocardium occurs in the absence of significant cardiomyocyte necrosis and may be mediated through stimulation of neurohumoral pathways, generation of matrix fragments, or ROS-dependent pro-inflammatory signalling.
6.8 Fibrosis in conditions associated with volume overload
Left ventricular volume overload is the dominant pathophysiologic perturbation in patients with severe valvular regurgitant lesions. In patients with severe aortic regurgitation fibrotic remodelling has been extensively documented.549–551 Animal models and in vitro experiments have provided interesting insights into the unique effects of volume overload on fibroblast function and ECM remodelling. In contrast to the net collagen deposition typically observed in pressure-overloaded hearts, volume overload was found to cause a marked loss of interstitial collagen,552 associated with MMP induction.552,553 Moreover, it has been suggested that volume overload induces fibronectin synthesis in cardiac fibroblasts without significantly affecting collagen production.554 Activation of a matrix-degrading environment may explain the pronounced chamber dilation observed in volume-overloaded hearts. The molecular basis for predominant activation of MMPs in volume overloaded hearts remains unknown. Lesions causing volume overload likely generate a unique mechanical environment with distinct effects on cardiomyocytes and interstitial cells. However, the network of molecular cascades activated by volume overload-induced mechanical stress has not been characterized.
6.9 Fibrosis in obesity and diabetes
Patients with diabetes and severe obesity develop fibrosis that is not limited to the myocardium, but may also affect other organs, such as the kidney. The basis for accentuated fibroblast activation in diabetic patients remains unclear. Animal model studies have suggested that expansion of the cardiac interstitium and collagen deposition in obese diabetic mice does not require myofibroblast conversion, but is associated with activation of a matrix-synthetic programme in cardiac fibroblasts.44 Hyperglycaemia has direct activating effects on cardiac fibroblasts, stimulating fibrogenic TGF-β cascades,555 leading to accumulation of advanced glycation end-products (AGEs) that crosslink the cardiac ECM, transducing ROS-dependent fibrogenic signals, and triggering receptor for AGE-mediated fibroblast activation.213,556–558 Glucose-independent pathways may also stimulate fibroblast activation through the effects of adipokines, neurohumoral pathways, or ET-1 signalling.557 Microvascular inflammation is also induced and may play an important role in fibroblast activation in diabetics.559,560 Moreover, metabolic dysregulation in diabetic subjects has been found to induce matricellular proteins with a prominent role in activation of fibrogenic cascades, such as TSP-1.326,561
In human patients, obesity and diabetes are associated with fibrotic cardiomyopathic changes562 and with a marked increase in the risk of HFpEF, that is independent of the presence of ischaemic heart disease.563,564 Changes in the interstitial ECM may contribute to the distinct obesity-related phenotype of human HFpEF,565 which is also associated with increased plasma volume, increased hypertrophy and accentuated right ventricular dilatation, despite lower plasma levels of natriuretic peptides.565,566
6.10 Fibrosis in the ageing heart
Ageing is typically associated with diastolic dysfunction and preserved systolic contractility, thus contributing to the pathogenesis of HFpEF. In both human patients and in experimental models, ageing is associated with interstitial deposition of ECM proteins in both ventricles and atria.567 The cell biological basis of ageing-related fibrosis involves expansion of resident fibroblasts of epicardial origin that infiltrate the cardiac interstitium.568 Several activating signals may promote fibroblast proliferation and activation in the ageing heart, including generation of ROS, neurohumoral activation, TGF-β, and inflammatory chemokines and cytokines.569,570 Single-cell transcriptomics in mouse models demonstrated that ageing is associated with alterations in cardiac fibroblast phenotype, characterized by increased expression of osteogenesis-associated genes, impaired angiogenic activation of endothelial cells, and a pro-inflammatory phenotype.571 To what extent, ageing-associated fibrotic changes contribute to the functional perurbations in senescent hearts remains unclear. In senescent mice, exercise training improved diastolic function without affecting cardiac fibrosis,572 highlighting the importance of fibrosis-independent processes in the cardiomyopathy associated with ageing. It should be emphasized that, although senescent mice have higher baseline myocardial collagen levels, they also exhibit an impaired fibroblast response following injury. In a model of reperfused infarction, senescent mouse hearts had impaired healing, reduced deposition of collagen in the scar and accentuated cardiac dilation.573 Perturbations in reparative fibroblast functions in senescent mice may involve impaired responses to fibrogenic growth factors.573
6.11 Fibrosis in dysrhythmias
In addition to its effects on cardiac function, fibrosis may perturb propagation of the electrical impulse, and may generate re-entry circuits, thus contributing to the pathogenesis of dysrhythmias.574,575 Links between fibrosis and increased risk for ventricular arrhythmias have been documented in practically every cardiac pathologic condition using CMR for assessment of fibrotic remodelling576 Fibrosis was found to predict ventricular arrhythmias and/or sudden cardiac death in patients with ischaemic and dilated cardiomyopathy,577 HCM,578 mitral valve prolapse,579 and myocarditis.580 The relation between specific cellular and biochemical characteristics of the fibrotic lesions and the susceptibility to arrhythmias remains poorly understood. It is likely that the biochemical profile of the interstitial ECM, as well as the composition of the interstitial cellular infiltrate may be important determinants of arrhythmogenic potential in fibrotic hearts. Animal model investigations suggest that infarct myofibroblasts may be electrically coupled with border zone cardiomyocytes; albeit with slower voltage responses, reflecting less effective conduction across heterocellular junctions.581,582 α-SMA expression and TGF-β stimulation have been suggested to increase fibroblast arrhythmogenic actions in the injured heart.583,584 Because even small changes in conduction and impulse propagation may greatly impact susceptibility to ventricular arrhythmias, it has been suggested that manipulation of non-excitable fibroblasts in the infarct may have profound effects on the arrhythmogenic process.585 On the other hand, deposition of ECM in fibrotic regions with a low cellular content may be associated with marked disruption of impulse propagation.
The arrhythmogenic effects of cardiac fibrosis are not limited to the left ventricular myocardium. In patients with pulmonary hypertension, right ventricular fibrosis may not only perturb function, but also contribute to the pathogenesis of arrhythmias.586,587 In patients with ARVC, prominent fibrosis is typically associated with a high susceptibility to ventricular arrhythmias.588,589
On the other hand, fibrotic atrial remodelling is often associated with elevated filling pressures in patients with heart failure and predisposes to initiation and maintenance of atrial fibrillation.590–594 The severity of atrial fibrosis has also been linked to the effectiveness of treatment interventions for atrial fibrillation: more severe fibrotic changes reduce the chances of success.595 A large body of evidence suggests that atrial fibrosis can be useful for risk stratification in patients with atrial fibrillation, serving as a predictor of recurrence, persistence, heart failure and stroke.596 The relation between the biochemical characteristics of the fibrotic response and the susceptibility to atrial fibrillation has not been systematically studied. It has been suggested that, although early and evolving fibrotic lesions may contribute to the pathogenesis of atrial fibrillation, very extensive fibrosis may decrease atrial excitability, reducing atrial fibrillation burden.597 Data supporting this interesting hypothesis are lacking.
7. Targeting the fibrotic response in myocardial disease
Anti-fibrotic strategies have been proposed as promising therapeutic approaches for patients with myocardial diseases. Unfortunately, the rationale for implementation of such approaches is often based on oversimplified concepts and a misguided view of ‘cardiac fibrosis’ as a single disease entity that resembles primary fibrotic conditions in other systems, such as idiopathic pulmonary fibrosis or scleroderma. It should be emphasized that cardiac fibrosis is not a specific disease, but rather a common pathologic abnormality that accompanies most myocardial diseases, and often represents an appropriate secondary reparative response to a primary insult. Because the adult mammalian heart has negligible regenerative potential, myocardial insults causing cardiomyocyte necrosis are repaired through generation of fibrous tissue. Even in conditions associated with predominant interstitial and perivascular fibrosis, in the absence of significant cardiomyocyte death (such as hypertensive heart disease, or the cardiomyopathy associated with diabetes and obesity), the relative contribution of fibrosis in dysfunction and adverse outcome is unclear. Although in many cases, fibrotic changes are likely contributors to functional impairment and arrhythmogenesis, there is no human myocardial disease with a well-documented primary fibrotic cause. Thus, recommendations regarding anti-fibrotic interventions should avoid generalizations and simplistic views, while taking into account the pathophysiologic heterogeneity of myocardial disease and the distinct profile of fibrotic changes found in each type of myocardial injury.
7.1 Can we reverse myocardial fibrosis?
A key question that needs to be answered when designing anti-fibrotic strategies for patients with myocardial disease is whether established cardiac fibrosis can be reversed, in the absence of an effective regenerative intervention. The answer is currently based more on intuition and clinical experience, than on experimental evidence. Reversibility is likely dependent on the aetiology of fibrosis, the extent of fibrotic remodelling, and the biochemical properties of the ECM, which are typically dependent on the age of the lesions. Predicting reversal of fibrosis seems straight-forward for lesions in the extremes of the pathophysiologic spectrum. For example, established replacement fibrosis due to a large MI typically would require major advances in myocardial regeneration, a visionary goal of modern cardiovascular research. On the other hand, early evolving interstitial or perivascular fibrotic changes are more likely to be reversible. In an experimental mouse model of ischaemic interstitial fibrosis due to brief repetitive ischaemia and reperfusion in the absence of MI, discontinuation of the ischaemic insults after 7 or 28 days resulted in reversal of fibrosis.39 Moreover, pharmacologic treatment with ACE inhibitors in experimental models reversed hypertensive fibrosis.598 Clinical studies have generated inconclusive and, in some cases, conflicting data, reflecting the heterogeneity of fibrotic lesions. In a small population of hypertensive patients with left ventricular hypertrophy, a 6-month course of lisinopril reduced fibrosis and attenuated diastolic dysfunction.599 Moreover, in patients with aortic stenosis, imaging studies showed a significant reduction in matrix volume, accompanied by a proportionally greater reduction of cell volume, but consistent with the possibility of regression of diffuse fibrotic lesions.600,601 In contrast, in patients with advanced end-stage heart failure, aggressive unloading through a left ventricular assist device did not affect the extent of cardiac fibrosis602 and in patients with severe aortic stenosis, valve replacement did not reverse established fibrotic lesions.603 The conflicting findings likely reflect distinct responses of various types of fibrotic lesions, but also the methodology used to assess reversal and the timing of assessment after the unloading procedure.
The cellular basis for reversal of cardiac fibrotic lesions remains enigmatic. Clearance of collagen and other matrix proteins from the fibrotic heart would be expected to require release and activation of proteases from specialized populations of anti-fibrotic interstitial cells. However, whether specific subsets of ‘fibrosis-resolving’ immune cells or fibroblasts are implicated in regression of fibrotic lesions remains unknown. In a group of patients with ischaemic cardiomyopathy undergoing aortocoronary bypass surgery, recovery of function after revascularization was more likely in myocardial segments with a higher cellular content, increased numbers of macrophages, higher levels of matricellular proteins, more myofibroblasts and lower collagen content.27,28 Although improved dysfunction does not necessarily imply reduced fibrosis, such observations may suggest that subpopulations of macrophages and fibroblasts may be required for resorption and resolution of fibrotic lesions.
7.2 Strategies for inhibition of cardiac fibrosis in chronic heart failure
Independently of our capability to reverse established fibrotic lesions, inhibition of active fibrotic responses may benefit subsets of patients with chronic heart failure. Based on the abundant experimental evidence implicating specific cytokines, growth factors, matricellular proteins, integrins and mechanosensitive signalling pathways in activation of cardiac fibroblasts, it is not surprising that a vast range of ‘anti-fibrotic’ pharmacologic approaches has been proposed as promising therapy for heart failure patients. Some established heart failure therapies targeting neurohumoral pathways (such as ACE inhibition, AT1 receptor blockade, mineralocorticoid antagonism, and β-adrenergic blockade) may reduce mortality and event rates in heart failure patients, at least in part through inhibition of fibrosis. Experimental studies have suggested that several additional anti-fibrotic interventions may hold therapeutic promise, including inhibition of fibrogenic pro-inflammatory cytokines (such as IL-1 and CCL2),129,604,605 anti-TGF-β approaches, αV or β1integrin inhibition, strategies inhibiting ECM cross-linking enzymes,316 and blockade of common kinase pathways. Recently, a fibroblast ablation strategy using adoptive transfer of CD8+ T cells engineered to expressed a chimeric antigen receptor against fibroblast activating protein reduced fibrosis and improved dysfunction in a mouse model of cardiac remodelling induced through neurohumoral activation.606 Unfortunately, the path towards clinical translation for anti-fibrotic approaches in myocardial disease is long and uncertain, as therapeutic implementation is hampered by several major challenges.
First, most of these approaches would require very early therapeutic intervention at a pre-clinical stage. In animal models, these interventions are typically initiated at an early stage of a dynamic model of rapidly progressive fibrotic disease. This is impractical in heart failure patients who are likely to present with more advanced fibrotic lesions.
Second, many of these pathways have important protective or reparative functions in response to a variety of injurious stimuli. Thus, chronic targeting of these pathways over the multi-year period typically required for progression of myocardial fibrosis may not be safe.
Third, the reparative nature of cardiac fibroblast activation and the pathophysiologic heterogeneity of human myocardial conditions associated with fibrosis greatly complicate implementation of anti-fibrotic strategies. We currently do not know to what extent myocardial dysfunction is caused by fibrosis. In many patients, fibrotic myocardial responses reflect appropriate activation of reparative or protective mechanisms. Abrogation of these responses may carry significant risks.
7.3 The need for pathophysiologic stratification of patients with fibrotic changes
In contrast to the well-defined injurious role of fibrogenic signalling in primary fibrotic conditions, such as progressive systemic sclerosis or idiopathic pulmonary fibrosis, the contribution of fibrotic remodelling to dysfunction, arrhythmogenesis and adverse outcome in patients with myocardial disease is uncertain and context-dependent. Thus, there is a need for pathophysiologic stratification of heart failure patients, in order to identify subpopulations with inappropriate, progressive or unrestrained fibrogenic responses that may be candidates for anti-fibrotic interventions.
Considering the potential risks of chronic anti-fibrotic therapies, targeting fibrogenic activation may be most attractive for patients with dynamic, rapidly evolving disease processes, such as MI, that could benefit from a brief course of therapy. The combined use of biomarkers reflecting inflammatory activation and ECM metabolism,607,608 or imaging studies609 may identify patient subpopulations with excessive and progressive ECM deposition or degradation that may benefit from modulatory therapy. Patients with prominent and inappropriate ECM deposition (that is typically associated with increased ventricular stiffness and diastolic dysfunction) may benefit from brief inhibition of fibrogenic growth factors. On the other hand, patients with accentuated and prolonged inflammatory activation typically exhibit enhanced protease activation leading to dilative remodelling and systolic dysfunction. These patients may be candidates for targeted anti-inflammatory therapy, such as IL-1 inhibition, or CCL2 antagonism.493,610
8. Conclusions
The pathophysiologic heterogeneity of fibrotic myocardial conditions and the complexity of fibroblast responses to injury greatly complicate development of anti-fibrotic strategies in myocardial disease. Despite the clear association between the severity of cardiac fibrosis and adverse outcome in patients with heart disease, documentation of causative relations remains challenging. Fibroblast activation is a critical reparative response following myocardial injury and may preserve structure and function not only in MI but also in chronic heart failure. Progress in the field requires new knowledge in several different directions. First, considering the heterogeneity of interstitial cells, characterization of fibroblast subsets in health and disease and identification of relations between distinct transcriptomic or proteomic profiles and functional properties is of critical significance. Second, systematic study of cell-specific actions in vivo will provide us with insights into the cell biological role of these multifunctional cells. Third, considering the co-existence of cardiomyocyte and interstitial pathology in most human myocardial conditions, we need to understand to what extent primary fibroblast activation contributes to dysfunction, arrhythmias and adverse outcome. Because the relative contribution of fibrosis is likely dependent on contextual factors, there is a need to develop strategies for identification of patient subsets with maladaptive or excessive fibrotic responses that may benefit from interventions targeting fibrogenic signalling.
Conflict of interest: none declared.
Funding
N.G.F. laboratory is supported by NIH grants R01 HL76246, R01 HL85440, and R01 HL149407 and by U.S. Department of Defense grants PR151134, PR151029, and PR181464.
References
- 1. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 2008;214:199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Frangogiannis NG. Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med 2019;65:70–99. [DOI] [PubMed] [Google Scholar]
- 3. Berk BC, Fujiwara K, Lehoux S.. ECM remodeling in hypertensive heart disease. J Clin Invest 2007;117:568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gao XM, White DA, Dart AM, Du XJ.. Post-infarct cardiac rupture: recent insights on pathogenesis and therapeutic interventions. Pharmacol Ther 2012;134:156–179. [DOI] [PubMed] [Google Scholar]
- 5. Borg TK, Caulfield JB.. The collagen matrix of the heart. Fed Proc 1981;40:2037–2041. [PubMed] [Google Scholar]
- 6. Bowers SL, Banerjee I, Baudino TA.. The extracellular matrix: at the center of it all. J Mol Cell Cardiol 2010;48:474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation 2003;108:1395–1403. [DOI] [PubMed] [Google Scholar]
- 8. Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol 1989;13:1637–1652. [DOI] [PubMed] [Google Scholar]
- 9. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD.. Revisiting cardiac cellular composition. Circ Res 2016;118:400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gersch C, Dewald O, Zoerlein M, Michael LH, Entman ML, Frangogiannis NG.. Mast cells and macrophages in normal C57/BL/6 mice. Histochem Cell Biol 2002;118:41–49. [DOI] [PubMed] [Google Scholar]
- 11. Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, Epelman S, Kreisel D, Liu Y, Itoh A, Shankar TS, Selzman CH, Drakos SG, Lavine KJ.. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med 2018;24:1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL.. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014;40:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee JS, Jeong SJ, Kim S, Chalifour L, Yun TJ, Miah MA, Li B, Majdoubi A, Sabourin A, Keler T, Guimond JV, Haddad E, Choi EY, Epelman S, Choi JH, Thibodeau J, Oh GT, Cheong C.. Conventional dendritic cells impair recovery after myocardial infarction. J Immunol 2018;201:1784–1798. [DOI] [PubMed] [Google Scholar]
- 14. Camelliti P, Borg TK, Kohl P.. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc Res 2005;65:40–51. [DOI] [PubMed] [Google Scholar]
- 15. Tallquist MD. Cardiac fibroblast diversity. Annu Rev Physiol 2020;82:63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eghbali M, Blumenfeld OO, Seifter S, Buttrick PM, Leinwand LA, Robinson TF, Zern MA, Giambrone MA.. Localization of types I, III and IV collagen mRNAs in rat heart cells by in situ hybridization. J Mol Cell Cardiol 1989;21:103–113. [DOI] [PubMed] [Google Scholar]
- 17. Brown RD, Ambler SK, Mitchell MD, Long CS.. The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol 2005;45:657–687. [DOI] [PubMed] [Google Scholar]
- 18. Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev 2007;87:1285–1342. [DOI] [PubMed] [Google Scholar]
- 19. Huang S, Chen B, Humeres C, Alex L, Hanna A, Frangogiannis NG.. The role of Smad2 and Smad3 in regulating homeostatic functions of fibroblasts in vitro and in adult mice. Biochim Biophys Acta Mol Cell Res 2020;1867:118703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ivey MJ, Kuwabara JT, Riggsbee KL, Tallquist MD.. Platelet-derived growth factor receptor-alpha is essential for cardiac fibroblast survival. Am J Physiol Heart Circ Physiol 2019;317:H330–H344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JW, Nordon RE, Harvey RP.. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 2019;8:e43882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R.. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 2007;13:952–961. [DOI] [PubMed] [Google Scholar]
- 23. Kramann R, Schneider RK, DiRocco DP, Machado F, Fleig S, Bondzie PA, Henderson JM, Ebert BL, Humphreys BD.. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015;16:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ieronimakis N, Hays AL, Janebodin K, Mahoney WM Jr, Duffield JS, Majesky MW, Reyes M.. Coronary adventitial cells are linked to perivascular cardiac fibrosis via TGFbeta1 signaling in the mdx mouse model of Duchenne muscular dystrophy. J Mol Cell Cardiol 2013;63:122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dai Z, Aoki T, Fukumoto Y, Shimokawa H.. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J Cardiol 2012;60:416–421. [DOI] [PubMed] [Google Scholar]
- 26. Kalkman E, Bilgin Y, Haren P, Suylen R, Saxena P, Schoemaker R.. Determinants of coronary reserve in rats subjected to coronary artery ligation or aortic banding. Cardiovasc Res 1996;32:1088–1095. [DOI] [PubMed] [Google Scholar]
- 27. Frangogiannis NG, Shimoni S, Chang SM, Ren G, Dewald O, Gersch C, Shan K, Aggeli C, Reardon M, Letsou GV, Espada R, Ramchandani M, Entman ML, Zoghbi WA.. Active interstitial remodeling: an important process in the hibernating human myocardium. J Am Coll Cardiol 2002;39:1468–1474. [DOI] [PubMed] [Google Scholar]
- 28. Frangogiannis NG, Shimoni S, Chang SM, Ren G, Shan K, Aggeli C, Reardon MJ, Letsou GV, Espada R, Ramchandani M, Entman ML, Zoghbi WA.. Evidence for an active inflammatory process in the hibernating human myocardium. Am J Pathol 2002;160:1425–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Frangogiannis NG. Can myocardial fibrosis be reversed? J Am Coll Cardiol 2019;73:2283–2285. [DOI] [PubMed] [Google Scholar]
- 30. Frangogiannis NG, Michael LH, Entman ML.. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb). Cardiovasc Res 2000;48:89–100. [DOI] [PubMed] [Google Scholar]
- 31. Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ.. Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol 1995;147:325–338. [PMC free article] [PubMed] [Google Scholar]
- 32. Ashizawa N, Graf K, Do YS, Nunohiro T, Giachelli CM, Meehan WP, Tuan TL, Hsueh WA.. Osteopontin is produced by rat cardiac fibroblasts and mediates A(II)-induced DNA synthesis and collagen gel contraction. J Clin Invest 1996;98:2218–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Komatsubara I, Murakami T, Kusachi S, Nakamura K, Hirohata S, Hayashi J, Takemoto S, Suezawa C, Ninomiya Y, Shiratori Y.. Spatially and temporally different expression of osteonectin and osteopontin in the infarct zone of experimentally induced myocardial infarction in rats. Cardiovasc Pathol 2003;12:186–194. [DOI] [PubMed] [Google Scholar]
- 34. Cleutjens JP, Kandala JC, Guarda E, Guntaka RV, Weber KT.. Regulation of collagen degradation in the rat myocardium after infarction. J Mol Cell Cardiol 1995;27:1281–1292. [DOI] [PubMed] [Google Scholar]
- 35. Shinde AV, Humeres C, Frangogiannis NG.. The role of alpha-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim Biophys Acta 2017;1863:298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol 2007;127:526–537. [DOI] [PubMed] [Google Scholar]
- 37. Hinz B. The myofibroblast: paradigm for a mechanically active cell. J Biomech 2010;43:146–155. [DOI] [PubMed] [Google Scholar]
- 38. Willems IE, Havenith MG, De Mey JG, Daemen MJ.. The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol 1994;145:868–875. [PMC free article] [PubMed] [Google Scholar]
- 39. Dewald O, Frangogiannis NG, Zoerlein M, Duerr GD, Klemm C, Knuefermann P, Taffet G, Michael LH, Crapo JD, Welz A, Entman ML.. Development of murine ischemic cardiomyopathy is associated with a transient inflammatory reaction and depends on reactive oxygen species. Proc Natl Acad Sci USA 2003;100:2700–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Blyszczuk P, Muller-Edenborn B, Valenta T, Osto E, Stellato M, Behnke S, Glatz K, Basler K, Luscher TF, Distler O, Eriksson U, Kania G.. Transforming growth factor-beta-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur Heart J 2017;38:1413–1425. [DOI] [PubMed] [Google Scholar]
- 41. Leslie KO, Taatjes DJ, Schwarz J, vonTurkovich M, Low RB.. Cardiac myofibroblasts express alpha smooth muscle actin during right ventricular pressure overload in the rabbit. Am J Pathol 1991;139:207–216. [PMC free article] [PubMed] [Google Scholar]
- 42. Wang J, Chen H, Seth A, McCulloch CA.. Mechanical force regulation of myofibroblast differentiation in cardiac fibroblasts. Am J Physiol Heart Circ Physiol 2003;285:H1871–H1881. [DOI] [PubMed] [Google Scholar]
- 43. Law BA, Levick SP, Carver WE.. Alterations in cardiac structure and function in a murine model of chronic alcohol consumption. Microsc Microanal 2012;18:453–461. [DOI] [PubMed] [Google Scholar]
- 44. Alex L, Russo I, Holoborodko V, Frangogiannis NG.. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol 2018;315:H934–H949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McLellan MA, Skelly DA, Dona MSI, Squiers GT, Farrugia GE, Gaynor TL, Cohen CD, Pandey R, Diep H, Vinh A, Rosenthal NA, Pinto AR.. High-resolution transcriptomic profiling of the heart during chronic stress reveals cellular drivers of cardiac fibrosis and hypertrophy. Circulation 2020;142:1448–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA.. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002;3:349–363. [DOI] [PubMed] [Google Scholar]
- 47. Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G.. The myofibroblast: one function, multiple origins. Am J Pathol 2007;170:1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mollmann H, Nef HM, Kostin S, von Kalle C, Pilz I, Weber M, Schaper J, Hamm CW, Elsasser A.. Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc Res 2006;71:661–671. [DOI] [PubMed] [Google Scholar]
- 49. Haudek SB, Xia Y, Huebener P, Lee JM, Carlson S, Crawford JR, Pilling D, Gomer RH, Trial J, Frangogiannis NG, Entman ML.. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc Natl Acad Sci USA 2006;103:18284–18289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial J, Taffet GE, Entman ML.. Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol 2010;49:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Trial J, Entman ML, Cieslik KA.. Mesenchymal stem cell-derived inflammatory fibroblasts mediate interstitial fibrosis in the aging heart. J Mol Cell Cardiol 2016;91:28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Haider N, Boscá L, Zandbergen HR, Kovacic JC, Narula N, González-Ramos S, Fernandez-Velasco M, Agrawal S, Paz-García M, Gupta S, DeLeon-Pennell K, Fuster V, Ibañez B, Narula J.. Transition of macrophages to fibroblast-like cells in healing myocardial infarction. J Am Coll Cardiol 2019;74:3124–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aisagbonhi O, Rai M, Ryzhov S, Atria N, Feoktistov I, Hatzopoulos AK.. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis Model Mech 2011;4:469–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, Kamran P, Müller AMS, Volz KS, Tang Z, Red-Horse K, Ardehali R.. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res 2014;115:625–635. [DOI] [PubMed] [Google Scholar]
- 55. Moore-Morris T, Guimaraes-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM.. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest 2014;124:2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, J. Lin S-C, Aronow BJ, Tallquist MD, Molkentin JD.. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun 2016;7:12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Moore-Morris T, Cattaneo P, Guimarães-Camboa N, Bogomolovas J, Cedenilla M, Banerjee I, Ricote M, Kisseleva T, Zhang L, Gu Y, Dalton ND, Peterson KL, Chen J, Pucéat M, Evans SM.. Infarct fibroblasts do not derive from bone marrow lineages. Circ Res 2018;122:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kong P, Christia P, Saxena A, Su Y, Frangogiannis NG.. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am J Physiol Heart Circ Physiol 2013;305:H1363–H1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kania G, Blyszczuk P, Stein S, Valaperti A, Germano D, Dirnhofer S, Hunziker L, Matter CM, Eriksson U.. Heart-infiltrating prominin-1+/CD133+ progenitor cells represent the cellular source of transforming growth factor beta-mediated cardiac fibrosis in experimental autoimmune myocarditis. Circ Res 2009;105:462–470. [DOI] [PubMed] [Google Scholar]
- 60. Okyere AD, Tilley DG.. Leukocyte-dependent regulation of cardiac fibrosis. Front Physiol 2020;11:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Carlson S, Helterline D, Asbe L, Dupras S, Minami E, Farris S, Stempien-Otero A, Cardiac macrophages adopt profibrotic/M2 phenotype in infarcted hearts: Role of urokinase plasminogen activator. J Mol Cell Cardiol 2017;108:42–49. [DOI] [PubMed] [Google Scholar]
- 62. Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velázquez F, Aronovitz M, Kapur NK, Karas RH, Blanton RM, Alcaide P.. Left ventricular T-cell recruitment contributes to the pathogenesis of heart failure. Circ Heart Fail 2015;8:776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Saxena A, Dobaczewski M, Rai V, Haque Z, Chen W, Li N, Frangogiannis NG.. Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am J Physiol Heart Circ Physiol 2014;307:H1233–H1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, Adachi H, Yashiro K, Suzuki K.. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest 2016;126:2151–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev 2012;92:635–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sun X, Nkennor B, Mastikhina O, Soon K, Nunes SS.. Endothelium-mediated contributions to fibrosis. Semin Cell Dev Biol 2020;101:78–86. [DOI] [PubMed] [Google Scholar]
- 67. Turner NA. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). J Mol Cell Cardiol 2016;94:189–200. [DOI] [PubMed] [Google Scholar]
- 68. Deniset JF, Belke D, Lee W-Y, Jorch SK, Deppermann C, Hassanabad AF, Turnbull JD, Teng G, Rozich I, Hudspeth K, Kanno Y, Brooks SR, Hadjantonakis A-K, O’Shea JJ, Weber GF, Fedak PWM, Kubes P.. Gata6(+) pericardial cavity macrophages relocate to the injured heart and prevent cardiac fibrosis. Immunity 2019;51:131–140.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bujak M, Dobaczewski M, Gonzalez-Quesada C, Xia Y, Leucker T, Zymek P, Veeranna V, Tager AM, Luster AD, Frangogiannis NG.. Induction of the CXC chemokine interferon-gamma-inducible protein 10 regulates the reparative response following myocardial infarction. Circ Res 2009;105:973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Frangogiannis NG, Mendoza LH, Lewallen M, Michael LH, Smith CW, Entman ML.. Induction and suppression of interferon-inducible protein 10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB J 2001;15:1428–1430. [DOI] [PubMed] [Google Scholar]
- 71. Sassi Y, Ahles A, Truong DJ, Baqi Y, Lee SY, Husse B, Hulot JS, Foinquinos A, Thum T, Muller CE, Dendorfer A, Laggerbauer B, Engelhardt S.. Cardiac myocyte-secreted cAMP exerts paracrine action via adenosine receptor activation. J Clin Invest 2014;124:5385–5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC.. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol 2013;10:15–26. [DOI] [PubMed] [Google Scholar]
- 73. Hokimoto S, Yasue H, Fujimoto K, Yamamoto H, Nakao K, Kaikita K, Sakata R, Miyamoto E.. Expression of angiotensin-converting enzyme in remaining viable myocytes of human ventricles after myocardial infarction. Circulation 1996;94:1513–1518. [DOI] [PubMed] [Google Scholar]
- 74. Sadoshima J, Xu Y, Slayter HS, Izumo S.. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell 1993;75:977–984. [DOI] [PubMed] [Google Scholar]
- 75. Schorb W, Booz GW, Dostal DE, Conrad KM, Chang KC, Baker KM.. Angiotensin II is mitogenic in neonatal rat cardiac fibroblasts. Circ Res 1993;72:1245–1254. [DOI] [PubMed] [Google Scholar]
- 76. Tian B, Liu J, Bitterman P, Bache RJ.. Angiotensin II modulates nitric oxide-induced cardiac fibroblast apoptosis by activation of AKT/PKB. Am J Physiol Heart Circ Physiol 2003;285:H1105–1112. [DOI] [PubMed] [Google Scholar]
- 77. Ehanire T, Ren L, Bond J, Medina M, Li G, Bashirov L, Chen L, Kokosis G, Ibrahim M, Selim A, Blobe GC, Levinson H.. Angiotensin II stimulates canonical TGF-beta signaling pathway through angiotensin type 1 receptor to induce granulation tissue contraction. J Mol Med 2015;93:289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Siddesha JM, Valente AJ, Sakamuri SS, Yoshida T, Gardner JD, Somanna N, Takahashi C, Noda M, Chandrasekar B.. Angiotensin II stimulates cardiac fibroblast migration via the differential regulation of matrixins and RECK. J Mol Cell Cardiol 2013;65:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Stawowy P, Margeta C, Blaschke F, Lindschau C, Spencer-Hansch C, Leitges M, Biagini G, Fleck E, Graf K.. Protein kinase C epsilon mediates angiotensin II-induced activation of beta1-integrins in cardiac fibroblasts. Cardiovasc Res 2005;67:50–59. [DOI] [PubMed] [Google Scholar]
- 80. Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA.. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc Natl Acad Sci USA 2005;102:437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sadoshima J, Izumo S.. Molecular characterization of angiotensin II-induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res 1993;73:413–423. [DOI] [PubMed] [Google Scholar]
- 82. Crabos M, Roth M, Hahn AW, Erne P.. Characterization of angiotensin II receptors in cultured adult rat cardiac fibroblasts. Coupling to signaling systems and gene expression. J Clin Invest 1994;93:2372–2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ohkubo N, Matsubara H, Nozawa Y, Mori Y, Murasawa S, Kijima K, Maruyama K, Masaki H, Tsutumi Y, Shibazaki Y, Iwasaka T, Inada M.. Angiotensin type 2 receptors are reexpressed by cardiac fibroblasts from failing myopathic hamster hearts and inhibit cell growth and fibrillar collagen metabolism. Circulation 1997;96:3954–3962. [DOI] [PubMed] [Google Scholar]
- 84. Kurisu S, Ozono R, Oshima T, Kambe M, Ishida T, Sugino H, Matsuura H, Chayama K, Teranishi Y, Iba O, Amano K, Matsubara H.. Cardiac angiotensin II type 2 receptor activates the kinin/NO system and inhibits fibrosis. Hypertension 2003;41:99–107. [DOI] [PubMed] [Google Scholar]
- 85. Sano M, Fukuda K, Sato T, Kawaguchi H, Suematsu M, Matsuda S, Koyasu S, Matsui H, Yamauchi-Takihara K, Harada M, Saito Y, Ogawa S.. ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res 2001;89:661–669. [DOI] [PubMed] [Google Scholar]
- 86. Booz GW, Baker KM.. Molecular signalling mechanisms controlling growth and function of cardiac fibroblasts. Cardiovasc Res 1995;30:537–543. [PubMed] [Google Scholar]
- 87. Booz GW, Baker KM.. Protein kinase C in angiotensin II signalling in neonatal rat cardiac fibroblasts. Role in the mitogenic response. Ann NY Acad Sci 1995;752:158–167. [DOI] [PubMed] [Google Scholar]
- 88. Kagami S, Border WA, Miller DE, Noble NA.. Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-beta expression in rat glomerular mesangial cells. J Clin Invest 1994;93:2431–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Campbell SE, Katwa LC.. Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol 1997;29:1947–1958. [DOI] [PubMed] [Google Scholar]
- 90. Zhou Y, Poczatek MH, Berecek KH, Murphy-Ullrich JE.. Thrombospondin 1 mediates angiotensin II induction of TGF-beta activation by cardiac and renal cells under both high and low glucose conditions. Biochem Biophys Res Commun 2006;339:633–641. [DOI] [PubMed] [Google Scholar]
- 91. Gurantz D, Cowling RT, Varki N, Frikovsky E, Moore CD, Greenberg BH.. IL-1beta and TNF-alpha upregulate angiotensin II type 1 (AT1) receptors on cardiac fibroblasts and are associated with increased AT1 density in the post-MI heart. J Mol Cell Cardiol 2005;38:505–515. [DOI] [PubMed] [Google Scholar]
- 92. Everett AD, Heller F, Fisher A.. AT1 receptor gene regulation in cardiac myocytes and fibroblasts. J Mol Cell Cardiol 1996;28:1727–1736. [DOI] [PubMed] [Google Scholar]
- 93. Schieffer B, Wirger A, Meybrunn M, Seitz S, Holtz J, Riede UN, Drexler H.. Comparative effects of chronic angiotensin-converting enzyme inhibition and angiotensin II type 1 receptor blockade on cardiac remodeling after myocardial infarction in the rat. Circulation 1994;89:2273–2282. [DOI] [PubMed] [Google Scholar]
- 94. Regan CP, Anderson PG, Bishop SP, Berecek KH.. Pressure-independent effects of AT1-receptor antagonism on cardiovascular remodeling in aortic-banded rats. Am J Physiol 1997;272:H2131–2138. [DOI] [PubMed] [Google Scholar]
- 95. Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I.. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol 2004;6:499–506. [DOI] [PubMed] [Google Scholar]
- 96. Lijnen P, Petrov V.. Induction of cardiac fibrosis by aldosterone. J Mol Cell Cardiol 2000;32:865–879. [DOI] [PubMed] [Google Scholar]
- 97. Campbell SE, Janicki JS, Weber KT.. Temporal differences in fibroblast proliferation and phenotype expression in response to chronic administration of angiotensin II or aldosterone. J Mol Cell Cardiol 1995;27:1545–1560. [DOI] [PubMed] [Google Scholar]
- 98. Neumann S, Huse K, Semrau R, Diegeler A, Gebhardt R, Buniatian GH, Scholz GH.. Aldosterone and D-glucose stimulate the proliferation of human cardiac myofibroblasts in vitro. Hypertension 2002;39:756–760. [DOI] [PubMed] [Google Scholar]
- 99. Somanna NK, Yariswamy M, Garagliano JM, Siebenlist U, Mummidi S, Valente AJ, Chandrasekar B.. Aldosterone-induced cardiomyocyte growth, and fibroblast migration and proliferation are mediated by TRAF3IP2. Cell Signal 2015;27:1928–1938. [DOI] [PubMed] [Google Scholar]
- 100. Brilla CG, Zhou G, Matsubara L, Weber KT.. Collagen metabolism in cultured adult rat cardiac fibroblasts: response to angiotensin II and aldosterone. J Mol Cell Cardiol 1994;26:809–820. [DOI] [PubMed] [Google Scholar]
- 101. Stockand JD, Meszaros JG.. Aldosterone stimulates proliferation of cardiac fibroblasts by activating Ki-RasA and MAPK1/2 signaling. Am J Physiol Heart Circ Physiol 2003;284:H176–184. [DOI] [PubMed] [Google Scholar]
- 102. Rickard AJ, Morgan J, Tesch G, Funder JW, Fuller PJ, Young MJ.. Deletion of mineralocorticoid receptors from macrophages protects against deoxycorticosterone/salt-induced cardiac fibrosis and increased blood pressure. Hypertension 2009;54:537–543. [DOI] [PubMed] [Google Scholar]
- 103. Li C, Sun X-N, Zeng M-R, Zheng X-J, Zhang Y-Y, Wan Q, Zhang W-C, Shi C, Du L-J, Ai T-J, Liu Y, Liu Y, Du L-L, Yi Y, Yu Y, Duan S-Z.. Mineralocorticoid receptor deficiency in T cells attenuates pressure overload-induced cardiac hypertrophy and dysfunction through modulating T-cell activation. Hypertension 2017;70:137–147. [DOI] [PubMed] [Google Scholar]
- 104. Rickard AJ, Morgan J, Bienvenu LA, Fletcher EK, Cranston GA, Shen JZ, Reichelt ME, Delbridge LM, Young MJ.. Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt-mediated inflammation and cardiac fibrosis. Hypertension 2012;60:1443–1450. [DOI] [PubMed] [Google Scholar]
- 105. Benjamin IJ, Jalil JE, Tan LB, Cho K, Weber KT, Clark WA.. Isoproterenol-induced myocardial fibrosis in relation to myocyte necrosis. Circ Res 1989;65:657–670. [DOI] [PubMed] [Google Scholar]
- 106. Farivar RS, Crawford DC, Chobanian AV, Brecher P.. Effect of angiotensin II blockade on the fibroproliferative response to phenylephrine in the rat heart. Hypertension 1995;25:809–813. [DOI] [PubMed] [Google Scholar]
- 107. Nguyen MN, Kiriazis H, Ruggiero D, Gao XM, Su Y, Jian A, Han LP, McMullen JR, Du XJ.. Spontaneous ventricular tachyarrhythmias in beta2-adrenoceptor transgenic mice in relation to cardiac interstitial fibrosis. Am J Physiol Heart Circ Physiol 2015;309:H946–H957. [DOI] [PubMed] [Google Scholar]
- 108. Xiao H, Li H, Wang JJ, Zhang JS, Shen J, An XB, Zhang CC, Wu JM, Song Y, Wang XY, Yu HY, Deng XN, Li ZJ, Xu M, Lu ZZ, Du J, Gao W, Zhang AH, Feng Y, Zhang YY.. IL-18 cleavage triggers cardiac inflammation and fibrosis upon beta-adrenergic insult. Eur Heart J 2018;39:60–69. [DOI] [PubMed] [Google Scholar]
- 109. Noh H, Yu MR, Kim HJ, Lee JH, Park BW, Wu IH, Matsumoto M, King GL.. Beta 2-adrenergic receptor agonists are novel regulators of macrophage activation in diabetic renal and cardiovascular complications. Kidney Int 2017;92:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Nuamnaichati N, Sato VH, Moongkarndi P, Parichatikanond W, Mangmool S.. Sustained beta-AR stimulation induces synthesis and secretion of growth factors in cardiac myocytes that affect on cardiac fibroblast activation. Life Sci 2018;193:257–269. [DOI] [PubMed] [Google Scholar]
- 111. Calderone A, Thaik CM, Takahashi N, Chang DL, Colucci WS.. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J Clin Invest 1998;101:812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Turner NA, Porter KE, Smith WH, White HL, Ball SG, Balmforth AJ.. Chronic beta2-adrenergic receptor stimulation increases proliferation of human cardiac fibroblasts via an autocrine mechanism. Cardiovasc Res 2003;57:784–792. [DOI] [PubMed] [Google Scholar]
- 113. Colombo F, Noel J, Mayers P, Mercier I, Calderone A.. beta-Adrenergic stimulation of rat cardiac fibroblasts promotes protein synthesis via the activation of phosphatidylinositol 3-kinase. J Mol Cell Cardiol 2001;33:1091–1106. [DOI] [PubMed] [Google Scholar]
- 114. Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, Gunaje J, Otsu K, Davis J.. Fibroblast-specific genetic manipulation of p38 mitogen-activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation 2017;136:549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kim J, Eckhart AD, Eguchi S, Koch WJ.. Beta-adrenergic receptor-mediated DNA synthesis in cardiac fibroblasts is dependent on transactivation of the epidermal growth factor receptor and subsequent activation of extracellular signal-regulated kinases. J Biol Chem 2002;277:32116–32123. [DOI] [PubMed] [Google Scholar]
- 116. Le DE, Pascotto M, Leong-Poi H, Sari I, Micari A, Kaul S.. Anti-inflammatory and pro-angiogenic effects of beta blockers in a canine model of chronic ischemic cardiomyopathy: comparison between carvedilol and metoprolol. Basic Res Cardiol 2013;108:384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Ye H, Ling S, Castillo AC, Thomas B, Long B, Qian J, Perez-Polo JR, Ye Y, Chen X, Birnbaum Y.. Nebivolol induces distinct changes in profibrosis microRNA expression compared with atenolol, in salt-sensitive hypertensive rats. Hypertension 2013;61:1008–1013. [DOI] [PubMed] [Google Scholar]
- 118. Imaeda A, Tanaka S, Tonegawa K, Fuchigami S, Obana M, Maeda M, Kihara M, Kiyonari H, Conway SJ, Fujio Y, Nakayama H.. Myofibroblast beta2 adrenergic signaling amplifies cardiac hypertrophy in mice. Biochem Biophys Res Commun 2019;510:149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Hermida N, Michel L, Esfahani H, Dubois-Deruy E, Hammond J, Bouzin C, Markl A, Colin H, Steenbergen AV, De Meester C, Beauloye C, Horman S, Yin X, Mayr M, Balligand JL.. Cardiac myocyte beta3-adrenergic receptors prevent myocardial fibrosis by modulating oxidant stress-dependent paracrine signaling. Eur Heart J 2018;39:888–898. [DOI] [PubMed] [Google Scholar]
- 120. Woodall MC, Woodall BP, Gao E, Yuan A, Koch WJ.. Cardiac fibroblast GRK2 deletion enhances contractility and remodeling following ischemia/reperfusion injury. Circ Res 2016;119:1116–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Travers JG, Kamal FA, Valiente-Alandi I, Nieman ML, Sargent MA, Lorenz JN, Molkentin JD, Blaxall BC.. Pharmacological and activated fibroblast targeting of gbetagamma-GRK2 after myocardial ischemia attenuates heart failure progression. J Am Coll Cardiol 2017;70:958–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Rollins BJ. Chemokines. Blood 1997;90:909–928. [PubMed] [Google Scholar]
- 123. Dobaczewski M, Frangogiannis NG.. Chemokines and cardiac fibrosis. Front Biosci 2009;S1:391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kolattukudy PE, Quach T, Bergese S, Breckenridge S, Hensley J, Altschuld R, Gordillo G, Klenotic S, Orosz C, Parker-Thornburg J.. Myocarditis induced by targeted expression of the MCP-1 gene in murine cardiac muscle. Am J Pathol 1998;152:101–111. [PMC free article] [PubMed] [Google Scholar]
- 125. Koyanagi M, Egashira K, Kitamoto S, Ni W, Shimokawa H, Takeya M, Yoshimura T, Takeshita A.. Role of monocyte chemoattractant protein-1 in cardiovascular remodeling induced by chronic blockade of nitric oxide synthesis. Circulation 2000;102:2243–2248. [DOI] [PubMed] [Google Scholar]
- 126. Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, Egashira K, Imaizumi T.. Hypertensive myocardial fibrosis and diastolic dysfunction: another model of inflammation? Hypertension 2004;43:739–745. [DOI] [PubMed] [Google Scholar]
- 127. Hayashidani S, Tsutsui H, Shiomi T, Ikeuchi M, Matsusaka H, Suematsu N, Wen J, Egashira K, Takeshita A.. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation 2003;108:2134–2140. [DOI] [PubMed] [Google Scholar]
- 128. Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG.. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res 2005;96:881–889. [DOI] [PubMed] [Google Scholar]
- 129. Frangogiannis NG, Dewald O, Xia Y, Ren G, Haudek S, Leucker T, Kraemer D, Taffet G, Rollins BJ, Entman ML.. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation 2007;115:584–592. [DOI] [PubMed] [Google Scholar]
- 130. Xu J, Lin SC, Chen J, Miao Y, Taffet GE, Entman ML, Wang Y.. CCR2 mediates the uptake of bone marrow-derived fibroblast precursors in angiotensin II-induced cardiac fibrosis. Am J Physiol Heart Circ Physiol 2011;301:H538–H547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Falkenham A, Sopel M, Rosin N, Lee TD, Issekutz T, Légaré J-F.. Early fibroblast progenitor cell migration to the AngII-exposed myocardium is not CXCL12 or CCL2 dependent as previously thought. Am J Pathol 2013;183:459–469. [DOI] [PubMed] [Google Scholar]
- 132. Shen JZ, Morgan J, Tesch GH, Fuller PJ, Young MJ.. CCL2-dependent macrophage recruitment is critical for mineralocorticoid receptor-mediated cardiac fibrosis, inflammation, and blood pressure responses in male mice. Endocrinology 2014;155:1057–1066. [DOI] [PubMed] [Google Scholar]
- 133. Trial J, Heredia CP, Taffet GE, Entman ML, Cieslik KA.. Dissecting the role of myeloid and mesenchymal fibroblasts in age-dependent cardiac fibrosis. Basic Res Cardiol 2017;112:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Willeford A, Suetomi T, Nickle A, Hoffman HM, Miyamoto S, Heller Brown J.. CaMKIIδ-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight 2018;3:e97054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, Prabhu SD.. CCR2(+) monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. JACC Basic Transl Sci 2018;3:230–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Tan X, Hu L, Shu Z, Chen L, Li X, Du M, Sun D, Mao X, Deng S, Huang K, Zhang F.. Role of CCR2 in the development of streptozotocin-treated diabetic cardiomyopathy. Diabetes 2019;68:2063–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Sakai N, Wada T, Furuichi K, Shimizu K, Kokubo S, Hara A, Yamahana J, Okumura T, Matsushima K, Yokoyama H, Kaneko S.. MCP-1/CCR2-dependent loop for fibrogenesis in human peripheral CD14-positive monocytes. J Leukoc Biol 2006;79:555–563. [DOI] [PubMed] [Google Scholar]
- 138. Kruglov EA, Nathanson RA, Nguyen T, Dranoff JA.. Secretion of MCP-1/CCL2 by bile duct epithelia induces myofibroblastic transdifferentiation of portal fibroblasts. Am J Physiol Gastrointest Liver Physiol 2006;290:G765–G771. [DOI] [PubMed] [Google Scholar]
- 139. Yamamoto T, Eckes B, Mauch C, Hartmann K, Krieg T.. Monocyte chemoattractant protein-1 enhances gene expression and synthesis of matrix metalloproteinase-1 in human fibroblasts by an autocrine IL-1 alpha loop. J Immunol 2000;164:6174–6179. [DOI] [PubMed] [Google Scholar]
- 140. Gharaee-Kermani M, Denholm EM, Phan SH.. Costimulation of fibroblast collagen and transforming growth factor beta1 gene expression by monocyte chemoattractant protein-1 via specific receptors. J Biol Chem 1996;271:17779–17784. [DOI] [PubMed] [Google Scholar]
- 141. Alex L, Frangogiannis NG.. The cellular origin of activated fibroblasts in the infarcted and remodeling myocardium. Circ Res 2018;122:540–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Zhang YL, Geng C, Yang J, Fang J, Yan X, Li PB, Zou LX, Chen C, Guo SB, Li HH, Liu Y.. Chronic inhibition of chemokine receptor CXCR2 attenuates cardiac remodeling and dysfunction in spontaneously hypertensive rats. Biochim Biophys Acta Mol Basis Dis 2019;1865:165551. [DOI] [PubMed] [Google Scholar]
- 143. Wang L, Zhang YL, Lin QY, Liu Y, Guan XM, Ma XL, Cao HJ, Liu Y, Bai J, Xia YL, Du J, Li HH.. CXCL1-CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur Heart J 2018;39:1818–1831. [DOI] [PubMed] [Google Scholar]
- 144. Doring Y, Pawig L, Weber C, Noels H.. The CXCL12/CXCR4 chemokine ligand/receptor axis in cardiovascular disease. Front Physiol 2014;5:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Penn MS, Pastore J, Miller T, Aras R.. SDF-1 in myocardial repair. Gene Ther 2012;19:583–587. [DOI] [PubMed] [Google Scholar]
- 146. Chu PY, Joshi MS, Horlock D, Kiriazis H, Kaye DM.. CXCR4 antagonism reduces cardiac fibrosis and improves cardiac performance in dilated cardiomyopathy. Front Pharmacol 2019;10:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Chu PY, Walder K, Horlock D, Williams D, Nelson E, Byrne M, Jandeleit-Dahm K, Zimmet P, Kaye DM.. CXCR4 antagonism attenuates the development of diabetic cardiac fibrosis. PLoS One 2015;10:e0133616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Chu PY, Zatta A, Kiriazis H, Chin-Dusting J, Du XJ, Marshall T, Kaye DM.. CXCR4 antagonism attenuates the cardiorenal consequences of mineralocorticoid excess. Circ Heart Fail 2011;4:651–658. [DOI] [PubMed] [Google Scholar]
- 149. Chu PY, Mariani J, Finch S, McMullen JR, Sadoshima J, Marshall T, Kaye DM.. Bone marrow-derived cells contribute to fibrosis in the chronically failing heart. Am J Pathol 2010;176:1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Saxena A, Bujak M, Frunza O, Dobaczewski M, Gonzalez-Quesada C, Lu B, Gerard C, Frangogiannis NG.. CXCR3-independent actions of the CXC chemokine CXCL10 in the infarcted myocardium and in isolated cardiac fibroblasts are mediated through proteoglycans. Cardiovasc Res 2014;103:217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Sivakumar P, Gupta S, Sarkar S, Sen S.. Upregulation of lysyl oxidase and MMPs during cardiac remodeling in human dilated cardiomyopathy. Mol Cell Biochem 2007;307:159–167. [DOI] [PubMed] [Google Scholar]
- 152. Vanhoutte D, van Almen GC, Van Aelst LN, Van Cleemput J, Droogne W, Jin Y, Van de Werf F, Carmeliet P, Vanhaecke J, Papageorgiou AP, Heymans S.. Matricellular proteins and matrix metalloproteinases mark the inflammatory and fibrotic response in human cardiac allograft rejection. Eur Heart J 2013;34:1930–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Bryant D, Becker L, Richardson J, Shelton J, Franco F, Peshock R, Thompson M, Giroir B.. Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation 1998;97:1375–1381. [DOI] [PubMed] [Google Scholar]
- 154. Li YY, Feng YQ, Kadokami T, McTiernan CF, Draviam R, Watkins SC, Feldman AM.. Myocardial extracellular matrix remodeling in transgenic mice overexpressing tumor necrosis factor alpha can be modulated by anti-tumor necrosis factor alpha therapy. Proc Natl Acad Sci USA 2000;97:12746–12751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Sivasubramanian N, Coker ML, Kurrelmeyer KM, MacLellan WR, DeMayo FJ, Spinale FG, Mann DL.. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation 2001;104:826–831. [DOI] [PubMed] [Google Scholar]
- 156. Monden Y, Kubota T, Inoue T, Tsutsumi T, Kawano S, Ide T, Tsutsui H, Sunagawa K.. Tumor necrosis factor-alpha is toxic via receptor 1 and protective via receptor 2 in a murine model of myocardial infarction. Am J Physiol Heart Circ Physiol 2007;293:H743–H753. [DOI] [PubMed] [Google Scholar]
- 157. Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, Kassiri Z, Kirshenbaum LA, Arnold M, Khokha R, Liu PP.. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation 2007;115:1398–1407. [DOI] [PubMed] [Google Scholar]
- 158. Westermann D, Linthout S, Dhayat S, Dhayat N, Schmidt A, Noutsias M, Song X-Y, Spillmann F, Riad A, Schultheiss H-P, Tschöpe C.. Tumor necrosis factor-alpha antagonism protects from myocardial inflammation and fibrosis in experimental diabetic cardiomyopathy. Basic Res Cardiol 2007;102:500–507. [DOI] [PubMed] [Google Scholar]
- 159. Awad AE, Kandalam V, Chakrabarti S, Wang X, Penninger JM, Davidge ST, Oudit GY, Kassiri Z.. Tumor necrosis factor induces matrix metalloproteinases in cardiomyocytes and cardiofibroblasts differentially via superoxide production in a PI3Kgamma-dependent manner. Am J Physiol Cell Physiol 2010;298:C679–C692. [DOI] [PubMed] [Google Scholar]
- 160. Zhang W, Chancey AL, Tzeng HP, Zhou Z, Lavine KJ, Gao F, Sivasubramanian N, Barger PM, Mann DL.. The development of myocardial fibrosis in transgenic mice with targeted overexpression of tumor necrosis factor requires mast cell-fibroblast interactions. Circulation 2011;124:2106–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Duerrschmid C, Crawford JR, Reineke E, Taffet GE, Trial J, Entman ML, Haudek SB, TNF receptor 1 signaling is critically involved in mediating angiotensin-II-induced cardiac fibrosis. J Mol Cell Cardiol 2013;57:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Ermolova NV, Martinez L, Vetrone SA, Jordan MC, Roos KP, Sweeney HL, Spencer MJ.. Long-term administration of the TNF blocking drug Remicade (cV1q) to mdx mice reduces skeletal and cardiac muscle fibrosis, but negatively impacts cardiac function. Neuromuscul Disord 2014;24:583–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Sriramula S, Francis J.. Tumor necrosis factor-alpha is essential for angiotensin II-induced ventricular remodeling: role for oxidative stress. PLoS One 2015;10:e0138372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Siwik DA, Chang DL, Colucci WS.. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res 2000;86:1259–1265. [DOI] [PubMed] [Google Scholar]
- 165. Venkatachalam K, Venkatesan B, Valente AJ, Melby PC, Nandish S, Reusch JE, Clark RA, Chandrasekar B.. WISP1, a pro-mitogenic, pro-survival factor, mediates tumor necrosis factor-alpha (TNF-alpha)-stimulated cardiac fibroblast proliferation but inhibits TNF-alpha-induced cardiomyocyte death. J Biol Chem 2009;284:14414–14427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Voloshenyuk TG, Hart AD, Khoutorova E, Gardner JD.. TNF-alpha increases cardiac fibroblast lysyl oxidase expression through TGF-beta and PI3Kinase signaling pathways. Biochem Biophys Res Commun 2011;413:370–375. [DOI] [PubMed] [Google Scholar]
- 167. Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, Frangogiannis NG.. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol 2008;173:57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Lim B-K, Choe S-C, Shin J-O, Ho S-H, Kim J-M, Yu S-S, Kim S, Jeon E-S.. Local expression of interleukin-1 receptor antagonist by plasmid DNA improves mortality and decreases myocardial inflammation in experimental coxsackieviral myocarditis. Circulation 2002;105:1278–1281. [PubMed] [Google Scholar]
- 169. Bageghni SA, Hemmings KE, Yuldasheva NY, Maqbool A, Gamboa-Esteves FO, Humphreys NE, Jackson MS, Denton CP, Francis S, Porter KE, Ainscough JF, Pinteaux E, Drinkhill MJ, Turner NA.. Fibroblast-specific deletion of interleukin-1 receptor-1 reduces adverse cardiac remodeling following myocardial infarction. JCI Insight 2019;5:e125074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N, Frangogiannis NG.. IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol 2013;191:4838–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Bronnum H, Eskildsen T, Andersen DC, Schneider M, Sheikh SP.. IL-1beta suppresses TGF-beta-mediated myofibroblast differentiation in cardiac fibroblasts. Growth Factors 2013;31:81–89. [DOI] [PubMed] [Google Scholar]
- 172. van Nieuwenhoven FA, Hemmings KE, Porter KE, Turner NA.. Combined effects of interleukin-1alpha and transforming growth factor-beta1 on modulation of human cardiac fibroblast function. Matrix Biol 2013;32:399–406. [DOI] [PubMed] [Google Scholar]
- 173. Palmer JN, Hartogensis WE, Patten M, Fortuin FD, Long CS.. Interleukin-1 beta induces cardiac myocyte growth but inhibits cardiac fibroblast proliferation in culture. J Clin Invest 1995;95:2555–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Li J, Schwimmbeck PL, Tschope C, Leschka S, Husmann L, Rutschow S, Reichenbach F, Noutsias M, Kobalz U, Poller W, Spillmann F, Zeichhardt H, Schultheiss HP, Pauschinger M.. Collagen degradation in a murine myocarditis model: relevance of matrix metalloproteinase in association with inflammatory induction. Cardiovasc Res 2002;56:235–247. [DOI] [PubMed] [Google Scholar]
- 175. Kotsiou OS, Gourgoulianis KI, Zarogiannis SG.. IL-33/ST2 axis in organ fibrosis. Front Immunol 2018;9:2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Tseng CCS, Huibers MMH, van Kuik J, de Weger RA, Vink A, de Jonge N.. The interleukin-33/ST2 pathway is expressed in the failing human heart and associated with pro-fibrotic remodeling of the myocardium. J Cardiovasc Trans Res 2018;11:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Sanada S, Hakuno D, Higgins LJ, Schreiter ER, McKenzie AN, Lee RT.. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J Clin Invest 2007;117:1538–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Zhu J, Carver W.. Effects of interleukin-33 on cardiac fibroblast gene expression and activity. Cytokine 2012;58:368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Zhao L, Cheng G, Jin R, Afzal MR, Samanta A, Xuan YT, Girgis M, Elias HK, Zhu Y, Davani A, Yang Y, Chen X, Ye S, Wang OL, Chen L, Hauptman J, Vincent RJ, Dawn B.. Deletion of interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ Res 2016;118:1918–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Gonzalez GE, Rhaleb NE, D'Ambrosio MA, Nakagawa P, Liu Y, Leung P, Dai X, Yang XP, Peterson EL, Carretero OA.. Deletion of interleukin-6 prevents cardiac inflammation, fibrosis and dysfunction without affecting blood pressure in angiotensin II-high salt-induced hypertension. J Hypertens 2015;33:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Jing R, Long TY, Pan W, Li F, Xie QY.. IL-6 knockout ameliorates myocardial remodeling after myocardial infarction by regulating activation of M2 macrophages and fibroblast cells. Eur Rev Med Pharmacol Sci 2019;23:6283–6291. [DOI] [PubMed] [Google Scholar]
- 182. Zhang Y, Wang JH, Zhang YY, Wang YZ, Wang J, Zhao Y, Jin XX, Xue GL, Li PH, Sun YL, Huang QH, Song XT, Zhang ZR, Gao X, Yang BF, Du ZM, Pan ZW.. Deletion of interleukin-6 alleviated interstitial fibrosis in streptozotocin-induced diabetic cardiomyopathy of mice through affecting TGFbeta1 and miR-29 pathways. Sci Rep 2016;6:23010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Chou CH, Hung CS, Liao CW, Wei LH, Chen CW, Shun CT, Wen WF, Wan CH, Wu XM, Chang YY, Wu VC, Wu KD, Lin YH, Group TS; The TAIPAI Study Group. IL-6 trans-signalling contributes to aldosterone-induced cardiac fibrosis. Cardiovasc Res 2018;114:690–702. [DOI] [PubMed] [Google Scholar]
- 184. Kumar S, Wang G, Zheng N, Cheng W, Ouyang K, Lin H, Liao Y, Liu J.. HIMF (hypoxia-induced mitogenic factor)-IL (interleukin)-6 signaling mediates cardiomyocyte-fibroblast crosstalk to promote cardiac hypertrophy and fibrosis. Hypertension 2019;73:1058–1070. [DOI] [PubMed] [Google Scholar]
- 185. Gardner GT, Travers JG, Qian J, Liu GS, Haghighi K, Robbins N, Jiang M, Li Y, Fan GC, Rubinstein J, Blaxall BC, Kranias EG.. Phosphorylation of Hsp20 promotes fibrotic remodeling and heart failure. JACC Basic Transl Sci 2019;4:188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Datta R, Bansal T, Rana S, Datta K, Datta Chaudhuri R, Chawla-Sarkar M, Sarkar S.. Myocyte-derived Hsp90 modulates collagen upregulation via biphasic activation of STAT-3 in fibroblasts during cardiac hypertrophy. Mol Cell Biol 2017;37:e00611–e00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Schafer S, Viswanathan S, Widjaja AA, Lim WW, Moreno-Moral A, DeLaughter DM, Ng B, Patone G, Chow K, Khin E, Tan J, Chothani SP, Ye L, Rackham OJL, Ko NSJ, Sahib NE, Pua CJ, Zhen NTG, Xie C, Wang M, Maatz H, Lim S, Saar K, Blachut S, Petretto E, Schmidt S, Putoczki T, Guimaraes-Camboa N, Wakimoto H, van Heesch S, Sigmundsson K, Lim SL, Soon JL, Chao VTT, Chua YL, Tan TE, Evans SM, Loh YJ, Jamal MH, Ong KK, Chua KC, Ong BH, Chakaramakkil MJ, Seidman JG, Seidman CE, Hubner N, Sin KYK, Cook SA.. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017;552:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Corden B, Adami E, Sweeney M, Schafer S, Cook SA.. IL-11 in cardiac and renal fibrosis: late to the party but a central player. Br J Pharmacol 2020;177:1695–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Ng B, Dong J, D'Agostino G, Viswanathan S, Widjaja AA, Lim WW, Ko NSJ, Tan J, Chothani SP, Huang B, Xie C, Pua CJ, Chacko AM, Guimaraes-Camboa N, Evans SM, Byrne AJ, Maher TM, Liang J, Jiang D, Noble PW, Schafer S, Cook SA.. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci Transl Med 2019;11:eaaw1237. [DOI] [PubMed] [Google Scholar]
- 190. Liu Z, Wang Y, Yan J, Liu J, Chen B, Zhang L, Cheng L.. Efficacy and safety of recombinant human interleukin-11 in the treatment of acute leukaemia patients with chemotherapy-induced thrombocytopenia: a systematic review and meta-analysis. J Eval Clin Pract 2020;26:262–271. [DOI] [PubMed] [Google Scholar]
- 191. Abe H, Takeda N, Isagawa T, Semba H, Nishimura S, Morioka MS, Nakagama Y, Sato T, Soma K, Koyama K, Wake M, Katoh M, Asagiri M, Neugent ML, Kim J-W, Stockmann C, Yonezawa T, Inuzuka R, Hirota Y, Maemura K, Yamashita T, Otsu K, Manabe I, Nagai R, Komuro I.. Macrophage hypoxia signaling regulates cardiac fibrosis via Oncostatin M. Nat Commun 2019;10:2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Matsuda M, Tsurusaki S, Miyata N, Saijou E, Okochi H, Miyajima A, Tanaka M.. Oncostatin M causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatology 2018;67:296–312. [DOI] [PubMed] [Google Scholar]
- 193. Huguier V, Giot JP, Simonneau M, Levillain P, Charreau S, Garcia M, Jegou JF, Bodet C, Morel F, Lecron JC, Favot L.. Oncostatin M exerts a protective effect against excessive scarring by counteracting the inductive effect of TGFbeta1 on fibrosis markers. Sci Rep 2019;9:2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Gieseck RL 3rd, Wilson MS, Wynn TA.. Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol 2018;18:62–76. [DOI] [PubMed] [Google Scholar]
- 195. Steen EH, Wang X, Balaji S, Butte MJ, Bollyky PL, Keswani SG.. The role of the anti-inflammatory cytokine interleukin-10 in tissue fibrosis. Adv Wound Care (New Rochelle) 2020;9:184–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Levick SP, McLarty JL, Murray DB, Freeman RM, Carver WE, Brower GL.. Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension 2009;53:1041–1047. [DOI] [PubMed] [Google Scholar]
- 197. Diny NL, Baldeviano GC, Talor MV, Barin JG, Ong SFey, Bedja D, Hays AG, Gilotra NA, Coppens I, Rose NR, Čiháková D.. Eosinophil-derived IL-4 drives progression of myocarditis to inflammatory dilated cardiomyopathy. J Exp Med 2017;214:943–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Peng H, Sarwar Z, Yang X-P, Peterson EL, Xu J, Janic B, Rhaleb N, Carretero OA, Rhaleb N-E.. Profibrotic role for interleukin-4 in cardiac remodeling and dysfunction. Hypertension 2015;66:582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Kanellakis P, Ditiatkovski M, Kostolias G, Bobik A.. A pro-fibrotic role for interleukin-4 in cardiac pressure overload. Cardiovasc Res 2012;95:77–85. [DOI] [PubMed] [Google Scholar]
- 200. Hofmann U, Knorr S, Vogel B, Weirather J, Frey A, Ertl G, Frantz S.. Interleukin-13 deficiency aggravates healing and remodeling in male mice after experimental myocardial infarction. Circ Heart Fail 2014;7:822–830. [DOI] [PubMed] [Google Scholar]
- 201. Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial JAnn, Entman ML.. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J Mol Cell Cardiol 2011;50:248–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Kolodsick JE, Toews GB, Jakubzick C, Hogaboam C, Moore TA, McKenzie A, Wilke CA, Chrisman CJ, Moore BB.. Protection from fluorescein isothiocyanate-induced fibrosis in IL-13-deficient, but not IL-4-deficient, mice results from impaired collagen synthesis by fibroblasts. J Immunol 2004;172:4068–4076. [DOI] [PubMed] [Google Scholar]
- 203. Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, Shipley JM, Gotwals P, Noble P, Chen Q, Senior RM, Elias JA.. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med 2001;194:809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Frangogiannis NG, Mendoza LH, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, Entman ML.. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol 2000;165:2798–2808. [DOI] [PubMed] [Google Scholar]
- 205. Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, Tincey S, Michael LH, Entman ML, Frangogiannis NG.. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol 2004;164:665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Hulsmans M, Sager HB, Roh JD, Valero-Muñoz M, Houstis NE, Iwamoto Y, Sun Y, Wilson RM, Wojtkiewicz G, Tricot B, Osborne MT, Hung J, Vinegoni C, Naxerova K, Sosnovik DE, Zile MR, Bradshaw AD, Liao R, Tawakol A, Weissleder R, Rosenzweig A, Swirski FK, Sam F, Nahrendorf M.. Cardiac macrophages promote diastolic dysfunction. J Exp Med 2018;215:423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Zymek P, Nah DY, Bujak M, Ren G, Koerting A, Leucker T, Huebener P, Taffet G, Entman M, Frangogiannis NG.. Interleukin-10 is not a critical regulator of infarct healing and left ventricular remodeling. Cardiovasc Res 2007;74:313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Krishnamurthy P, Lambers E, Verma S, Thorne T, Qin G, Losordo DW, Kishore R.. Myocardial knockdown of mRNA-stabilizing protein HuR attenuates post-MI inflammatory response and left ventricular dysfunction in IL-10-null mice. FASEB J 2010;24:2484–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Verma SK, Garikipati VNS, Krishnamurthy P, Schumacher SM, Grisanti LA, Cimini M, Cheng Z, Khan M, Yue Y, Benedict C, Truongcao MM, Rabinowitz JE, Goukassian DA, Tilley D, Koch WJ, Kishore R.. Interleukin-10 inhibits bone marrow fibroblast progenitor cell-mediated cardiac fibrosis in pressure-overloaded myocardium. Circulation 2017;136:940–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Frangogiannis NG. Transforming growth factor (TGF)-beta in tissue fibrosis. J Exp Med 2020;217:e20190103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Lodyga M, Hinz B.. TGF-beta1—a truly transforming growth factor in fibrosis and immunity. Semin Cell Dev Biol 2020;101:123–139. [DOI] [PubMed] [Google Scholar]
- 212. Schiller M, Javelaud D, Mauviel A.. TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci 2004;35:83–92. [DOI] [PubMed] [Google Scholar]
- 213. Biernacka A, Cavalera M, Wang J, Russo I, Shinde A, Kong P, Gonzalez-Quesada C, Rai V, Dobaczewski M, Lee D-W, Wang X-F, Frangogiannis NG.. Smad3 signaling promotes fibrosis while preserving cardiac and aortic geometry in obese diabetic mice. Circ Heart Fail 2015;8:788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Xia Y, Dobaczewski M, Gonzalez-Quesada C, Chen W, Biernacka A, Li N, Lee DW, Frangogiannis NG.. Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension 2011;58:902–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Li RK, Li G, Mickle DA, Weisel RD, Merante F, Luss H, Rao V, Christakis GT, Williams WG.. Overexpression of transforming growth factor-beta1 and insulin-like growth factor-I in patients with idiopathic hypertrophic cardiomyopathy. Circulation 1997;96:874–881. [DOI] [PubMed] [Google Scholar]
- 216. Nagaraju CK, Robinson EL, Abdesselem M, Trenson S, Dries E, Gilbert G, Janssens S, Van Cleemput J, Rega F, Meyns B, Roderick HL, Driesen RB, Sipido KR.. Myofibroblast phenotype and reversibility of fibrosis in patients with end-stage heart failure. J Am Coll Cardiol 2019;73:2267–2282. [DOI] [PubMed] [Google Scholar]
- 217. Annes JP, Munger JS, Rifkin DB.. Making sense of latent TGFbeta activation. J Cell Sci 2003;116:217–224. [DOI] [PubMed] [Google Scholar]
- 218. Ignotz RA, Massagué J.. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem 1986;261:4337–4345. [PubMed] [Google Scholar]
- 219. Rifkin DB, Mazzieri R, Munger JS, Noguera I, Sung J.. Proteolytic control of growth factor availability. APMIS 1999;107:80–85. [DOI] [PubMed] [Google Scholar]
- 220. Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML.. The critical role of endogenous Thrombospondin (TSP)-1 in preventing expansion of healing myocardial infarcts. Circulation 2005;111:2935–2942. [DOI] [PubMed] [Google Scholar]
- 221. Asano Y, Ihn H, Yamane K, Jinnin M, Tamaki K.. Increased expression of integrin alphavbeta5 induces the myofibroblastic differentiation of dermal fibroblasts. Am J Pathol 2006;168:499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Seeland U, Haeuseler C, Hinrichs R, Rosenkranz S, Pfitzner T, Scharffetter-Kochanek K, Böhm M.. Myocardial fibrosis in transforming growth factor-beta(1) (TGF-beta(1)) transgenic mice is associated with inhibition of interstitial collagenase. Eur J Clin Invest 2002;32:295–303. [DOI] [PubMed] [Google Scholar]
- 223. Rosenkranz S, Flesch M, Amann K, Haeuseler C, Kilter H, Seeland U, Schluter KD, Bohm M.. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1). Am J Physiol Heart Circ Physiol 2002;283:H1253–H1262. [DOI] [PubMed] [Google Scholar]
- 224. Accornero F, van Berlo JH, Correll RN, Elrod JW, Sargent MA, York A, Rabinowitz JE, Leask A, Molkentin JD.. Genetic analysis of connective tissue growth factor as an effector of transforming growth factor beta signaling and cardiac remodeling. Mol Cell Biol 2015;35:2154–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Nakajima H, Nakajima HO, Salcher O, Dittie AS, Dembowsky K, Jing S, Field LJ.. Atrial but not ventricular fibrosis in mice expressing a mutant transforming growth factor-beta(1) transgene in the heart. Circ Res 2000;86:571–579. [DOI] [PubMed] [Google Scholar]
- 226. Brooks WW, Conrad CH.. Myocardial fibrosis in transforming growth factor beta(1)heterozygous mice. J Mol Cell Cardiol 2000;32:187–195. [DOI] [PubMed] [Google Scholar]
- 227. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, Karch J, Molkentin JD.. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest 2017;127:3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T.. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 2002;106:130–135. [DOI] [PubMed] [Google Scholar]
- 229. Ferreira RR, Abreu RDS, Vilar-Pereira G, Degrave W, Meuser-Batista M, Ferreira NVC, da Cruz Moreira O, da Silva Gomes NL, Mello de Souza E, Ramos IP, Bailly S, Feige JJ, Lannes-Vieira J, de A-JT, Waghabi MC.. TGF-beta inhibitor therapy decreases fibrosis and stimulates cardiac improvement in a pre-clinical study of chronic Chagas' heart disease. PLoS Negl Trop Dis 2019;13:e0007602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G.. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 1993;122:103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang X-F, Frangogiannis NG.. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res 2010;107:418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, Conway SJ, Graff JM, Frangogiannis NG.. Opposing actions of fibroblast and cardiomyocyte Smad3 signaling in the infarcted myocardium. Circulation 2018;137:707–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, Wang X-F, Frangogiannis NG.. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 2007;116:2127–2138. [DOI] [PubMed] [Google Scholar]
- 234. Russo I, Cavalera M, Huang S, Su Y, Hanna A, Chen B, Shinde AV, Conway SJ, Graff J, Frangogiannis NG.. Protective effects of activated myofibroblasts in the pressure-overloaded myocardium are mediated through smad-dependent activation of a matrix-preserving program. Circ Res 2019;124:1214–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Kapoun AM, Liang F, O’Young G, Damm DL, Quon D, White RT, Munson K, Lam A, Schreiner GF, Protter AA.. B-type natriuretic peptide exerts broad functional opposition to transforming growth factor-beta in primary human cardiac fibroblasts: fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ Res 2004;94:453–461. [DOI] [PubMed] [Google Scholar]
- 236. Petrov VV, Fagard RH, Lijnen PJ.. Transforming growth factor-beta(1) induces angiotensin-converting enzyme synthesis in rat cardiac fibroblasts during their differentiation to myofibroblasts. J Renin Angiotensin Aldosterone Syst 2000;1:342–352. [DOI] [PubMed] [Google Scholar]
- 237. Driesen RB, Nagaraju CK, Abi-Char J, Coenen T, Lijnen PJ, Fagard RH, Sipido KR, Petrov VV.. Reversible and irreversible differentiation of cardiac fibroblasts. Cardiovasc Res 2014;101:411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Huang S, Chen B, Su Y, Alex L, Humeres C, Shinde AV, Conway SJ, Frangogiannis NG.. Distinct roles of myofibroblast-specific Smad2 and Smad3 signaling in repair and remodeling of the infarcted heart. J Mol Cell Cardiol 2019;132:84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Bageghni SA, Hemmings KE, Zava N, Denton CP, Porter KE, Ainscough JFX, Drinkhill MJ, Turner NA.. Cardiac fibroblast-specific p38alpha MAP kinase promotes cardiac hypertrophy via a putative paracrine interleukin-6 signaling mechanism. FASEB J 2018;32:4941–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Wang H, Leinwand LA, Anseth KS.. Roles of transforming growth factor-beta1 and OB-cadherin in porcine cardiac valve myofibroblast differentiation. FASEB J 2014;28:4551–4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Mori T, Kawara S, Shinozaki M, Hayashi N, Kakinuma T, Igarashi A, Takigawa M, Nakanishi T, Takehara K.. Role and interaction of connective tissue growth factor with transforming growth factor-beta in persistent fibrosis: a mouse fibrosis model. J Cell Physiol 1999;181:153–159. [DOI] [PubMed] [Google Scholar]
- 242. Zhang Y, Yan H, Guang GC, Deng ZR.. Overexpressed connective tissue growth factor in cardiomyocytes attenuates left ventricular remodeling induced by angiotensin II perfusion. Clin Exp Hypertens 2017;39:168–174. [DOI] [PubMed] [Google Scholar]
- 243. Chen B, Huang S, Su Y, Wu YJ, Hanna A, Brickshawana A, Graff J, Frangogiannis NG.. Macrophage Smad3 protects the infarcted heart, stimulating phagocytosis and regulating inflammation. Circ Res 2019;125:55–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Sanders LN, Schoenhard JA, Saleh MA, Mukherjee A, Ryzhov S, McMaster WG Jr, Nolan K, Gumina RJ, Thompson TB, Magnuson MA, Harrison DG, Hatzopoulos AK.. BMP antagonist Gremlin 2 limits inflammation after myocardial infarction. Circ Res 2016;119:434–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Merino D, Villar AV, Garcia R, Tramullas M, Ruiz L, Ribas C, Cabezudo S, Nistal JF, Hurle MA.. BMP-7 attenuates left ventricular remodelling under pressure overload and facilitates reverse remodelling and functional recovery. Cardiovasc Res 2016;110:331–345. [DOI] [PubMed] [Google Scholar]
- 246. Urbina P, Singla DK.. BMP-7 attenuates adverse cardiac remodeling mediated through M2 macrophages in prediabetic cardiomyopathy. Am J Physiol Heart Circ Physiol 2014;307:H762–H772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R.. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med 2003;9:964–968. [DOI] [PubMed] [Google Scholar]
- 248. Qu X, Liu Y, Cao D, Chen J, Liu Z, Ji H, Chen Y, Zhang W, Zhu P, Xiao D, Li X, Shou W, Chen H.. BMP10 preserves cardiac function through its dual activation of SMAD-mediated and STAT3-mediated pathways. J Biol Chem 2019;294:19877–19888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Maruyama S, Nakamura K, Papanicolaou KN, Sano S, Shimizu I, Asaumi Y, Hoff MJ, Ouchi N, Recchia FA, Walsh K.. Follistatin-like 1 promotes cardiac fibroblast activation and protects the heart from rupture. EMBO Mol Med 2016;8:949–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Panse KD, Felkin LE, Lopez-Olaneta MM, Gomez-Salinero J, Villalba M, Munoz L, Nakamura K, Shimano M, Walsh K, Barton PJ, Rosenthal N, Lara-Pezzi E.. Follistatin-like 3 mediates paracrine fibroblast activation by cardiomyocytes. J Cardiovasc Trans Res 2012;5:814–826. [DOI] [PubMed] [Google Scholar]
- 251. Tuuminen R, NykäNen AI, Krebs R, Soronen J, Pajusola K, KeräNen MAI, Koskinen PK, Alitalo K, LemströM KB, PDGF-A, -C, and -D but not PDGF-B increase TGF-beta1 and chronic rejection in rat cardiac allografts. ATVB 2009;29:691–698. [DOI] [PubMed] [Google Scholar]
- 252. Simm A, Nestler M, Hoppe V.. Mitogenic effect of PDGF-AA on cardiac fibroblasts. Basic Res Cardiol 1998;93:40–43. [DOI] [PubMed] [Google Scholar]
- 253. Zhao T, Zhao W, Chen Y, Li VS, Meng W, Sun Y.. Platelet-derived growth factor-D promotes fibrogenesis of cardiac fibroblasts. Am J Physiol Heart Circ Physiol 2013;304:H1719–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Pontén A, Bergsten Folestad E, Pietras K, Eriksson U.. Platelet-derived growth factor D induces cardiac fibrosis and proliferation of vascular smooth muscle cells in heart-specific transgenic mice. Circ Res 2005;97:1036–1045. [DOI] [PubMed] [Google Scholar]
- 255. Gallini R, Lindblom P, Bondjers C, Betsholtz C, Andrae J.. PDGF-A and PDGF-B induces cardiac fibrosis in transgenic mice. Exp Cell Res 2016;349:282–290. [DOI] [PubMed] [Google Scholar]
- 256. Zymek P, Bujak M, Chatila K, Cieslak A, Thakker G, Entman ML, Frangogiannis NG.. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol 2006;48:2315–2323. [DOI] [PubMed] [Google Scholar]
- 257. Liao CH, Akazawa H, Tamagawa M, Ito K, Yasuda N, Kudo Y, Yamamoto R, Ozasa Y, Fujimoto M, Wang P, Nakauchi H, Nakaya H, Komuro I.. Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J Clin Invest 2010;120:242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Liu C, Zhao W, Meng W, Zhao T, Chen Y, Ahokas RA, Liu H, Sun Y.. Platelet-derived growth factor blockade on cardiac remodeling following infarction. Mol Cell Biochem 2014;397:295–304. [DOI] [PubMed] [Google Scholar]
- 259. Leipner C, Grun K, Muller A, Buchdunger E, Borsi L, Kosmehl H, Berndt A, Janik T, Uecker A, Kiehntopf M, Bohmer FD.. Imatinib mesylate attenuates fibrosis in coxsackievirus b3-induced chronic myocarditis. Cardiovasc Res 2008;79:118–126. [DOI] [PubMed] [Google Scholar]
- 260. Saraswati S, Lietman CD, Li B, Mathew S, Zent R, Young PP.. Small proline-rich repeat 3 is a novel coordinator of PDGFRβ and integrin β1 crosstalk to augment proliferation and matrix synthesis by cardiac fibroblasts. FASEB J 2020;34:7885–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Alex L, Frangogiannis NG.. Pericytes in the infarcted heart. Vasc Biol 2019;1:H23–H31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Bastakoty D, Young PP.. Wnt/beta-catenin pathway in tissue injury: roles in pathology and therapeutic opportunities for regeneration. FASEB J 2016;30:3271–3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Wang Y, Li YP, Paulson C, Shao JZ, Zhang X, Wu M Chen W.. Wnt and the Wnt signaling pathway in bone development and disease. Front Biosci 2014;19:379–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Abraityte A, Vinge LE, Askevold ET, Lekva T, Michelsen AE, Ranheim T, Alfsnes K, Fiane A, Aakhus S, Lunde IG, Dahl CP, Aukrust P, Christensen G, Gullestad L, Yndestad A, Ueland T.. Wnt5a is elevated in heart failure and affects cardiac fibroblast function. J Mol Med 2017;95:767–777. [DOI] [PubMed] [Google Scholar]
- 265. Carthy JM, Garmaroudi FS, Luo Z, McManus BM.. Wnt3a induces myofibroblast differentiation by upregulating TGF-beta signaling through SMAD2 in a beta-catenin-dependent manner. PLoS One 2011;6:e19809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Moon J, Zhou H, Zhang L-S, Tan W, Liu Y, Zhang S, Morlock LK, Bao X, Palecek SP, Feng JQ, Williams NS, Amatruda JF, Olson EN, Bassel-Duby R, Lum L.. Blockade to pathological remodeling of infarcted heart tissue using a porcupine antagonist. Proc Natl Acad Sci USA 2017;114:1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Duan J, Gherghe C, Liu D, Hamlett E, Srikantha L, Rodgers L, Regan JN, Rojas M, Willis M, Leask A, Majesky M, Deb A.. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J 2012;31:429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Xiang FL, Fang M, Yutzey KE.. Loss of beta-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat Commun 2017;8:712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Meyer IS, Jungmann A, Dieterich C, Zhang M, Lasitschka F, Werkmeister S, Haas J, Muller OJ, Boutros M, Nahrendorf M, Katus HA, Hardt SE, Leuschner F.. The cardiac microenvironment uses non-canonical WNT signaling to activate monocytes after myocardial infarction. EMBO Mol Med 2017;9:1279–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Paik DT, Rai M, Ryzhov S, Sanders LN, Aisagbonhi O, Funke MJ, Feoktistov I, Hatzopoulos AK.. Wnt10b gain-of-function improves cardiac repair by arteriole formation and attenuation of fibrosis. Circ Res 2015;117:804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Leask A. Potential therapeutic targets for cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF, partners in fibroblast activation. Circ Res 2010;106:1675–1680. [DOI] [PubMed] [Google Scholar]
- 272. Alvarez D, Briassouli P, Clancy RM, Zavadil J, Reed JH, Abellar RG, Halushka M, Fox-Talbot K, Barrat FJ, Buyon JP.. A novel role of endothelin-1 in linking Toll-like receptor 7-mediated inflammation to fibrosis in congenital heart block. J Biol Chem 2011;286:30444–30454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Shi-Wen X, Kennedy L, Renzoni EA, Bou-Gharios G, Du Bois RM, Black CM, Denton CP, Abraham DJ, Leask A.. Endothelin is a downstream mediator of profibrotic responses to transforming growth factor beta in human lung fibroblasts. Arthritis Rheum 2007;56:4189–4194. [DOI] [PubMed] [Google Scholar]
- 274. Liu J, Zhuang T, Pi J, Chen X, Zhang Q, Li Y, Wang H, Shen Y, Tomlinson B, Chan P, Yu Z, Cheng Y, Zheng X, Reilly M, Morrisey E, Zhang L, Liu Z, Zhang Y.. Endothelial forkhead box transcription factor P1 regulates pathological cardiac remodeling through transforming growth factor-beta1-endothelin-1 signal pathway. Circulation 2019;140:665–680. [DOI] [PubMed] [Google Scholar]
- 275. Tsutamoto T, Wada A, Maeda K, Mabuchi N, Hayashi M, Tsutsui T, Ohnishi M, Sawaki M, Fujii M, Matsumoto T, Horie H, Sugimoto Y, Kinoshita M.. Transcardiac extraction of circulating endothelin-1 across the failing heart. Am J Cardiol 2000;86:524–528. [DOI] [PubMed] [Google Scholar]
- 276. Yamamoto K, Masuyama T, Sakata Y, Mano T, Nishikawa N, Kondo H, Akehi N, Kuzuya T, Miwa T, Hori M.. Roles of renin-angiotensin and endothelin systems in development of diastolic heart failure in hypertensive hearts. Cardiovasc Res 2000;47:274–283. [DOI] [PubMed] [Google Scholar]
- 277. Wang X, Guo Z, Ding Z, Khaidakov M, Lin J, Xu Z, Sharma SG, Jiwani S, Mehta JL.. Endothelin-1 upregulation mediates aging-related cardiac fibrosis. J Mol Cell Cardiol 2015;80:101–109. [DOI] [PubMed] [Google Scholar]
- 278. Mueller EE, Momen A, Masse S, Zhou YQ, Liu J, Backx PH, Henkelman RM, Nanthakumar K, Stewart DJ, Husain M.. Electrical remodelling precedes heart failure in an endothelin-1-induced model of cardiomyopathy. Cardiovasc Res 2011;89:623–633. [DOI] [PubMed] [Google Scholar]
- 279. Adiarto S, Heiden S, Vignon-Zellweger N, Nakayama K, Yagi K, Yanagisawa M, Emoto N.. ET-1 from endothelial cells is required for complete angiotensin II-induced cardiac fibrosis and hypertrophy. Life Sci 2012;91:651–657. [DOI] [PubMed] [Google Scholar]
- 280. Ammarguellat F, Larouche I, Schiffrin EL.. Myocardial fibrosis in DOCA-salt hypertensive rats: effect of endothelin ET(A) receptor antagonism. Circulation 2001;103:319–324. [DOI] [PubMed] [Google Scholar]
- 281. Mulder P, Richard V, Derumeaux G, Hogie M, Henry JP, Lallemand F, Compagnon P, Mace B, Comoy E, Letac B, Thuillez C.. Role of endogenous endothelin in chronic heart failure: effect of long-term treatment with an endothelin antagonist on survival, hemodynamics, and cardiac remodeling. Circulation 1997;96:1976–1982. [DOI] [PubMed] [Google Scholar]
- 282. Park JB, Schiffrin EL.. Cardiac and vascular fibrosis and hypertrophy in aldosterone-infused rats: role of endothelin-1. Am J Hypertens 2002;15:164–169. [DOI] [PubMed] [Google Scholar]
- 283. Valero-Munoz M, Li S, Wilson RM, Boldbaatar B, Iglarz M, Sam F.. Dual endothelin-A/endothelin-B receptor blockade and cardiac remodeling in heart failure with preserved ejection fraction. Circ Heart Fail 2016;9:e003381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Piacentini L, Gray M, Honbo NY, Chentoufi J, Bergman M, Karliner JS.. Endothelin-1 stimulates cardiac fibroblast proliferation through activation of protein kinase C. J Mol Cell Cardiol 2000;32:565–576. [DOI] [PubMed] [Google Scholar]
- 285. Guarda E, Katwa LC, Myers PR, Tyagi SC, Weber KT.. Effects of endothelins on collagen turnover in cardiac fibroblasts. Cardiovasc Res 1993;27:2130–2134. [DOI] [PubMed] [Google Scholar]
- 286. Kulasekaran P, Scavone CA, Rogers DS, Arenberg DA, Thannickal VJ, Horowitz JC.. Endothelin-1 and transforming growth factor-beta1 independently induce fibroblast resistance to apoptosis via AKT activation. Am J Respir Cell Mol Biol 2009;41:484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N, Yagi K, Miyagawa K, Rikitake Y, Suzuki T, Kisanuki YY, Yanagisawa M, Hirata K.. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 2010;121:2407–2418. [DOI] [PubMed] [Google Scholar]
- 288. Frangogiannis NG, Kovacic JC.. Extracellular matrix in ischemic heart disease, part 4/4: JACC Focus Seminar. J Am Coll Cardiol 2020;75:2219–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG.. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol 2010;48:504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Gaggar A, Weathington N.. Bioactive extracellular matrix fragments in lung health and disease. J Clin Invest 2016;126:3176–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Wells JM, Gaggar A, Blalock JE.. MMP generated matrikines. Matrix Biol 2015;44-46:122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Ricard-Blum S, Salza R.. Matricryptins and matrikines: biologically active fragments of the extracellular matrix. Exp Dermatol 2014;23:457–463. [DOI] [PubMed] [Google Scholar]
- 293. Bras Lec Frangogiannis NG. Extracellular matrix-derived peptides in tissue remodeling and fibrosis. Matrix Biol 2020;91–92:176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Senior RM, Griffin GL, Mecham RP.. Chemotactic activity of elastin-derived peptides. J Clin Invest 1980;66:859–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Lindsey ML, Iyer RP, Zamilpa R, Yabluchanskiy A, DeLeon-Pennell KY, Hall ME, Kaplan A, Zouein FA, Bratton D, Flynn ER, Cannon PL, Tian Y, Jin Y-F, Lange RA, Tokmina-Roszyk D, Fields GB, de Castro Brás LE.. A novel collagen matricryptin reduces left ventricular dilation post-myocardial infarction by promoting scar formation and angiogenesis. J Am Coll Cardiol 2015;66:1364–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Okada M, Oba Y, Yamawaki H.. Endostatin stimulates proliferation and migration of adult rat cardiac fibroblasts through PI3K/Akt pathway. Eur J Pharmacol 2015;750:20–26. [DOI] [PubMed] [Google Scholar]
- 297. Okada M, Murata N, Yamawaki H.. Canstatin stimulates migration of rat cardiac fibroblasts via secretion of matrix metalloproteinase-2. Am J Physiol Cell Physiol 2017;312:C199–C208. [DOI] [PubMed] [Google Scholar]
- 298. Sugiyama A, Okada M, Yamawaki H.. Pathophysiological roles of canstatin on myofibroblasts after myocardial infarction in rats. Eur J Pharmacol 2017;807:32–43. [DOI] [PubMed] [Google Scholar]
- 299. Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG.. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res 2006;324:475–488. [DOI] [PubMed] [Google Scholar]
- 300. Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL.. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest 2004;113:1596–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. Rybarczyk BJ, Lawrence SO, Simpson-Haidaris PJ.. Matrix-fibrinogen enhances wound closure by increasing both cell proliferation and migration. Blood 2003;102:4035–4043. [DOI] [PubMed] [Google Scholar]
- 302. Arslan F, Smeets MB, Riem Vis PW, Karper JC, Quax PH, Bongartz LG, Peters JH, Hoefer IE, Doevendans PA, Pasterkamp G, de Kleijn DP.. Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circ Res 2011;108:582–592. [DOI] [PubMed] [Google Scholar]
- 303. Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G.. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol 1998;142:873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Klingberg F, Chau G, Walraven M, Boo S, Koehler A, Chow ML, Olsen AL, Im M, Lodyga M, Wells RG, White ES, Hinz B.. The fibronectin ED-A domain enhances recruitment of latent TGF-beta-binding protein-1 to the fibroblast matrix. J Cell Sci 2018;131:jcs201293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Mukherjee D, Sen S.. Alteration of cardiac collagen phenotypes in hypertensive hypertrophy: role of blood pressure. J Mol Cell Cardiol 1993;25:185–196. [DOI] [PubMed] [Google Scholar]
- 306. Whittaker P, Boughner DR, Kloner RA.. Analysis of healing after myocardial infarction using polarized light microscopy. Am J Pathol 1989;134:879–893. [PMC free article] [PubMed] [Google Scholar]
- 307. Marijianowski MM, Teeling P, Mann J, Becker AE.. Dilated cardiomyopathy is associated with an increase in the type I/type III collagen ratio: a quantitative assessment. J Am Coll Cardiol 1995;25:1263–1272. [DOI] [PubMed] [Google Scholar]
- 308. Yokota T, McCourt J, Ma F, Ren S, Li S, Kim TH, Kurmangaliyev YZ, Nasiri R, Ahadian S, Nguyen T, Tan XHM, Zhou Y, Wu R, Rodriguez A, Cohn W, Wang Y, Whitelegge J, Ryazantsev S, Khademhosseini A, Teitell MA, Chiou PY, Birk DE, Rowat AC, Crosbie RH, Pellegrini M, Seldin M, Lusis AJ, Deb A.. Type V collagen in scar tissue regulates the size of scar after heart injury. Cell 2020;182:545–562.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Zibadi S, Vazquez R, Moore D, Larson DF, Watson RR.. Myocardial lysyl oxidase regulation of cardiac remodeling in a murine model of diet-induced metabolic syndrome. Am J Physiol Heart Circ Physiol 2009;297:H976–H982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310. Adam O, Theobald K, Lavall D, Grube M, Kroemer HK, Ameling S, Schafers HJ, Bohm M, Laufs U.. Increased lysyl oxidase expression and collagen cross-linking during atrial fibrillation. J Mol Cell Cardiol 2011;50:678–685. [DOI] [PubMed] [Google Scholar]
- 311. El Hajj EC, El Hajj MC, Ninh VK, Gardner JD.. Cardioprotective effects of lysyl oxidase inhibition against volume overload-induced extracellular matrix remodeling. Exp Biol Med (Maywood) 2016;241:539–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. López B, González A, Hermida N, Valencia F, de Teresa E, Díez J.. Role of lysyl oxidase in myocardial fibrosis: from basic science to clinical aspects. Am J Physiol Heart Circ Physiol 2010;299:H1–H9. [DOI] [PubMed] [Google Scholar]
- 313. Yang J, Savvatis K, Kang JS, Fan P, Zhong H, Schwartz K, Barry V, Mikels-Vigdal A, Karpinski S, Kornyeyev D, Adamkewicz J, Feng X, Zhou Q, Shang C, Kumar P, Phan D, Kasner M, López B, Diez J, Wright KC, Kovacs RL, Chen P-S, Quertermous T, Smith V, Yao L, Tschöpe C, Chang C-P.. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat Commun 2016;7:13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314. Shinde AV, Dobaczewski M, de Haan JJ, Saxena A, Lee KK, Xia Y, Chen W, Su Y, Hanif W, Kaur Madahar I, Paulino VM, Melino G, Frangogiannis NG.. Tissue transglutaminase induction in the pressure-overloaded myocardium regulates matrix remodelling. Cardiovasc Res 2017;113:892–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Shinde AV, Frangogiannis NG.. Tissue transglutaminase in the pathogenesis of heart failure. Cell Death Differ 2018;25:453–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Shinde AV, Su Y, Palanski BA, Fujikura K, Garcia MJ, Frangogiannis NG.. Pharmacologic inhibition of the enzymatic effects of tissue transglutaminase reduces cardiac fibrosis and attenuates cardiomyocyte hypertrophy following pressure overload. J Mol Cell Cardiol 2018;117:36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. Akimov SS, Krylov D, Fleischman LF, Belkin AM.. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol 2000;148:825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Gonzalez A, Lopez B, Ravassa S, San Jose G, Diez J.. The complex dynamics of myocardial interstitial fibrosis in heart failure. Focus on collagen cross-linking. Biochim Biophys Acta Mol Cell Res 2019;1866:1421–1432. [DOI] [PubMed] [Google Scholar]
- 319. Woodiwiss AJ, Tsotetsi OJ, Sprott S, Lancaster EJ, Mela T, Chung ES, Meyer TE, Norton GR.. Reduction in myocardial collagen cross-linking parallels left ventricular dilatation in rat models of systolic chamber dysfunction. Circulation 2001;103:155–160. [DOI] [PubMed] [Google Scholar]
- 320. Lopez B, Querejeta R, Gonzalez A, Larman M, Diez J.. Collagen cross-linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure: potential role of lysyl oxidase. Hypertension 2012;60:677–683. [DOI] [PubMed] [Google Scholar]
- 321. Gil-Cayuela C, Roselló-LLetí E, Ortega A, Tarazón E, Triviño JC, Martínez-Dolz L, González-Juanatey JR, Lago F, Portolés M, Rivera M.. New altered non-fibrillar collagens in human dilated cardiomyopathy: role in the remodeling process. PLoS One 2016;11:e0168130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Chute M, Aujla P, Jana S, Kassiri Z.. The non-fibrillar side of fibrosis: contribution of the basement membrane, proteoglycans, and glycoproteins to myocardial fibrosis. J Cardiovasc Dev Dis 2019;6:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Naugle JE, Olson ER, Zhang X, Mase SE, Pilati CF, Maron MB, Folkesson HG, Horne WI, Doane KJ, Meszaros JG.. Type VI collagen induces cardiac myofibroblast differentiation: implications for postinfarction remodeling. Am J Physiol Heart Circ Physiol 2006;290:H323–H330. [DOI] [PubMed] [Google Scholar]
- 324. Luther DJ, Thodeti CK, Shamhart PE, Adapala RK, Hodnichak C, Weihrauch D, Bonaldo P, Chilian WM, Meszaros JG.. Absence of type VI collagen paradoxically improves cardiac function, structure, and remodeling after myocardial infarction. Circ Res 2012;110:851–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Skrbic B, Engebretsen KV, Strand ME, Lunde IG, Herum KM, Marstein HS, Sjaastad I, Lunde PK, Carlson CR, Christensen G, Bjornstad JL, Tonnessen T.. Lack of collagen VIII reduces fibrosis and promotes early mortality and cardiac dilatation in pressure overload in mice. Cardiovasc Res 2015;106:32–42. [DOI] [PubMed] [Google Scholar]
- 326. Gonzalez-Quesada C, Cavalera M, Biernacka A, Kong P, Lee DW, Saxena A, Frunza O, Dobaczewski M, Shinde A, Frangogiannis NG.. Thrombospondin-1 induction in the diabetic myocardium stabilizes the cardiac matrix in addition to promoting vascular rarefaction through angiopoietin-2 upregulation. Circ Res 2013;113:1331–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Rosini S, Pugh N, Bonna AM, Hulmes DJS, Farndale RW, Adams JC.. Thrombospondin-1 promotes matrix homeostasis by interacting with collagen and lysyl oxidase precursors and collagen cross-linking sites. Sci Signal 2018;11:eaar2566. [DOI] [PubMed] [Google Scholar]
- 328. Belmadani S, Bernal J, Wei CC, Pallero MA, Dell'italia L, Murphy-Ullrich JE, Berecek KH.. A thrombospondin-1 antagonist of transforming growth factor-beta activation blocks cardiomyopathy in rats with diabetes and elevated angiotensin II. Am J Pathol 2007;171:777–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Chablais F, Jazwinska A.. The regenerative capacity of the zebrafish heart is dependent on TGFbeta signaling. Development 2012;139:1921–1930. [DOI] [PubMed] [Google Scholar]
- 330. Mercer SE, Odelberg SJ, Simon HG.. A dynamic spatiotemporal extracellular matrix facilitates epicardial-mediated vertebrate heart regeneration. Dev Biol 2013;382:457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Tamaoki M, Imanaka-Yoshida K, Yokoyama K, Nishioka T, Inada H, Hiroe M, Sakakura T, Yoshida T.. Tenascin-C regulates recruitment of myofibroblasts during tissue repair after myocardial injury. Am J Pathol 2005;167:71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Podesser BK, Kreibich M, Dzilic E, Santer D, Forster L, Trojanek S, Abraham D, Krssak M, Klein KU, Tretter EV, Kaun C, Wojta J, Kapeller B, Goncalves IF.. Trescher K Kiss A. Tenascin-C promotes chronic pressure overload-induced cardiac dysfunction, hypertrophy and myocardial fibrosis. J Hypertens 2018;36:847–856. [DOI] [PubMed] [Google Scholar]
- 333. Abbadi D, Laroumanie F, Bizou M, Pozzo J, Daviaud D, Delage C, Calise D, Gaits-Iacovoni F, Dutaur M, Tortosa F, Renaud-Gabardos E, Douin-Echinard V, Prats AC, Roncalli J, Parini A, Pizzinat N.. Local production of tenascin-C acts as a trigger for monocyte/macrophage recruitment that provokes cardiac dysfunction. Cardiovasc Res 2018;114:123–137. [DOI] [PubMed] [Google Scholar]
- 334. Shimojo N, Hashizume R, Kanayama K, Hara M, Suzuki Y, Nishioka T, Hiroe M, Yoshida T, Imanaka-Yoshida K.. Tenascin-C may accelerate cardiac fibrosis by activating macrophages via the integrin alphaVbeta3/nuclear factor-kappaB/interleukin-6 axis. Hypertension 2015;66:757–766. [DOI] [PubMed] [Google Scholar]
- 335. Schellings MW, Vanhoutte D, Swinnen M, Cleutjens JP, Debets J, van Leeuwen RE, d'Hooge J, Van de Werf F, Carmeliet P, Pinto YM, Sage EH, Heymans S.. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J Exp Med 2009;206:113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. McCurdy SM, Dai Q, Zhang J, Zamilpa R, Ramirez TA, Dayah T, Nguyen N, Jin YF, Bradshaw AD, Lindsey ML.. SPARC mediates early extracellular matrix remodeling following myocardial infarction. Am J Physiol Heart Circ Physiol 2011;301:H497–H505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Toba H, de Castro Bras LE, Baicu CF, Zile MR, Lindsey ML, Bradshaw AD.. Increased ADAMTS1 mediates SPARC-dependent collagen deposition in the aging myocardium. Am J Physiol Endocrinol Metab 2016;310:E1027–E1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338. Bradshaw AD, Baicu CF, Rentz TJ, Van Laer AO, Boggs J, Lacy JM, Zile MR.. Pressure overload-induced alterations in fibrillar collagen content and myocardial diastolic function: role of secreted protein acidic and rich in cysteine (SPARC) in post-synthetic procollagen processing. Circulation 2009;119:269–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Trueblood NA, Xie Z, Communal C, Sam F, Ngoy S, Liaw L, Jenkins AW, Wang J, Sawyer DB, Bing OH, Apstein CS, Colucci WS, Singh K.. Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ Res 2001;88:1080–1087. [DOI] [PubMed] [Google Scholar]
- 340. Yousefi K, Irion CI, Takeuchi LM, Ding W, Lambert G, Eisenberg T, Sukkar S, Granzier HL, Methawasin M, Lee DI, Hahn VS, Kass DA, Hatzistergos KE, Hare JM, Webster KA, Shehadeh LA.. Osteopontin promotes left ventricular diastolic dysfunction through a mitochondrial pathway. J Am Coll Cardiol 2019;73:2705–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Caballero EP, Santamaria MH, Corral RS.. Endogenous osteopontin induces myocardial CCL5 and MMP-2 activation that contributes to inflammation and cardiac remodeling in a mouse model of chronic Chagas heart disease. Biochim Biophys Acta Mol Basis Dis 2018;1864:11–23. [DOI] [PubMed] [Google Scholar]
- 342. Lopez B, Gonzalez A, Lindner D, Westermann D, Ravassa S, Beaumont J, Gallego I, Zudaire A, Brugnolaro C, Querejeta R, Larman M, Tschope C, Diez J.. Osteopontin-mediated myocardial fibrosis in heart failure: a role for lysyl oxidase? Cardiovasc Res 2013;99:111–120. [DOI] [PubMed] [Google Scholar]
- 343. Dahiya S, Givvimani S, Bhatnagar S, Qipshidze N, Tyagi SC, Kumar A.. Osteopontin-stimulated expression of matrix metalloproteinase-9 causes cardiomyopathy in the mdx model of Duchenne muscular dystrophy. J Immunol 2011;187:2723–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Psarras S, Mavroidis M, Sanoudou D, Davos CH, Xanthou G, Varela AE, Panoutsakopoulou V, Capetanaki Y.. Regulation of adverse remodelling by osteopontin in a genetic heart failure model. Eur Heart J 2012;33:1954–1963. [DOI] [PubMed] [Google Scholar]
- 345. Sam F, Xie Z, Ooi H, Kerstetter DL, Colucci WS, Singh M, Singh K.. Mice lacking osteopontin exhibit increased left ventricular dilation and reduced fibrosis after aldosterone infusion. Am J Hypertens 2004;17:188–193. [DOI] [PubMed] [Google Scholar]
- 346. Collins AR, Schnee J, Wang W, Kim S, Fishbein MC, Bruemmer D, Law RE, Nicholas S, Ross RS, Hsueh WA.. Osteopontin modulates angiotensin II-induced fibrosis in the intact murine heart. J Am Coll Cardiol 2004;43:1698–1705. [DOI] [PubMed] [Google Scholar]
- 347. Matsui Y, Jia N, Okamoto H, Kon S, Onozuka H, Akino M, Liu L, Morimoto J, Rittling SR, Denhardt D, Kitabatake A, Uede T.. Role of osteopontin in cardiac fibrosis and remodeling in angiotensin II-induced cardiac hypertrophy. Hypertension 2004;43:1195–1201. [DOI] [PubMed] [Google Scholar]
- 348. Lenga Y, Koh A, Perera AS, McCulloch CA, Sodek J, Zohar R.. Osteopontin expression is required for myofibroblast differentiation. Circ Res 2008;102:319–327. [DOI] [PubMed] [Google Scholar]
- 349. Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, Lorts A, Brunskill EW, Dorn GW, Conway SJ, Aronow BJ, Robbins J, Molkentin JD.. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res 2007;101:313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, Saito M, Fukuda K, Nishiyama T, Kitajima S, Saga Y, Fukayama M, Sata M, Kudo A.. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med 2008;205:295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Chen Z, Xie J, Hao H, Lin H, Wang L, Zhang Y, Chen L, Cao S, Huang X, Liao W, Bin J, Liao Y.. Ablation of periostin inhibits post-infarction myocardial regeneration in neonatal mice mediated by the phosphatidylinositol 3 kinase/glycogen synthase kinase 3beta/cyclin D1 signalling pathway. Cardiovasc Res 2017;113:620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Johannes L, Jacob R, Leffler H.. Galectins at a glance. J Cell Sci 2018;131:jcs208884. [DOI] [PubMed] [Google Scholar]
- 353. Sharma UC, Pokharel S, van Brakel TJ, van Berlo JH, Cleutjens JP, Schroen B, Andre S, Crijns HJ, Gabius HJ, Maessen J, Pinto YM.. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004;110:3121–3128. [DOI] [PubMed] [Google Scholar]
- 354. Frunza O, Russo I, Saxena A, Shinde AV, Humeres C, Hanif W, Rai V, Su Y, Frangogiannis NG.. Myocardial galectin-3 expression is associated with remodeling of the pressure-overloaded heart and may delay the hypertrophic response without affecting survival, dysfunction, and cardiac fibrosis. Am J Pathol 2016;186:1114–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Mackinnon AC, Gibbons MA, Farnworth SL, Leffler H, Nilsson UJ, Delaine T, Simpson AJ, Forbes SJ, Hirani N, Gauldie J, Sethi T.. Regulation of transforming growth factor-beta1-driven lung fibrosis by galectin-3. Am J Respir Crit Care Med 2012;185:537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. MacKinnon AC, Farnworth SL, Hodkinson PS, Henderson NC, Atkinson KM, Leffler H, Nilsson UJ, Haslett C, Forbes SJ, Sethi T.. Regulation of alternative macrophage activation by galectin-3. J Immunol 2008;180:2650–2658. [DOI] [PubMed] [Google Scholar]
- 357. Frangogiannis NG. Galectin-3 in the fibrotic response: cellular targets and molecular mechanisms. Int J Cardiol 2018;258:226–227. [DOI] [PubMed] [Google Scholar]
- 358. Yu L, Ruifrok WPT, Meissner M, Bos EM, van Goor H, Sanjabi B, van der Harst P, Pitt B, Goldstein IJ, Koerts JA, van Veldhuisen DJ, Bank RA, van Gilst WH, Silljé HHW, de Boer RA.. Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ Heart Fail 2013;6:107–117. [DOI] [PubMed] [Google Scholar]
- 359. Martínez-Martínez E, Calvier L, Fernández-Celis A, Rousseau E, Jurado-López R, Rossoni LV, Jaisser F, Zannad F, Rossignol P, Cachofeiro V, López-Andrés N.. Galectin-3 blockade inhibits cardiac inflammation and fibrosis in experimental hyperaldosteronism and hypertension. Hypertension 2015;66:767–775. [DOI] [PubMed] [Google Scholar]
- 360. Nguyen MN, Su Y, Kiriazis H, Yang Y, Gao XM, McMullen JR, Dart AM, Du XJ.. Upregulated galectin-3 is not a critical disease mediator of cardiomyopathy induced by beta2-adrenoceptor overexpression. Am J Physiol Heart Circ Physiol 2018;314:H1169–H1178. [DOI] [PubMed] [Google Scholar]
- 361. de Boer RA, Lok DJ, Jaarsma T, van der Meer P, Voors AA, Hillege HL, van Veldhuisen DJ.. Predictive value of plasma galectin-3 levels in heart failure with reduced and preserved ejection fraction. Ann Med 2011;43:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362. Filipe MD, Meijers WC, Rogier van der Velde A, de Boer RA.. Galectin-3 and heart failure: prognosis, prediction & clinical utility. Clin Chim Acta 2015;443:48–56. [DOI] [PubMed] [Google Scholar]
- 363. Iozzo RV, Schaefer L.. Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol 2015;42:11–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364. Zhao RR, Ackers-Johnson M, Stenzig J, Chen C, Ding T, Zhou Y, Wang P, Ng SL, Li PY, Teo G, Rudd PM, Fawcett JW, Foo RSY.. Targeting chondroitin sulfate glycosaminoglycans to treat cardiac fibrosis in pathological remodeling. Circulation 2018;137:2497–2513. [DOI] [PubMed] [Google Scholar]
- 365. Westermann D, Mersmann J, Melchior A, Freudenberger T, Petrik C, Schaefer L, LüLlmann-Rauch R, Lettau O, Jacoby C, Schrader J, Brand-Herrmann S-M, Young MF, Schultheiss HP, Levkau B, Baba HA, Unger T, Zacharowski K, TschöPe C, Fischer JW, Biglycan is required for adaptive remodeling after myocardial infarction. Circulation 2008;117:1269–1276. [DOI] [PubMed] [Google Scholar]
- 366. Beetz N, Rommel C, Schnick T, Neumann E, Lother A, Monroy-Ordonez EB, Zeeb M, Preissl S, Gilsbach R, Melchior-Becker A, Rylski B, Stoll M, Schaefer L, Beyersdorf F, Stiller B, Hein L.. Ablation of biglycan attenuates cardiac hypertrophy and fibrosis after left ventricular pressure overload. J Mol Cell Cardiol 2016;101:145–155. [DOI] [PubMed] [Google Scholar]
- 367. Van Aelst LNL, Voss S, Carai P, Van Leeuwen R, Vanhoutte D, Sanders-van Wijk S, Eurlings L, Swinnen M, Verheyen FK, Verbeken E, Nef H, Troidl C, Cook SA, Brunner-La Rocca H-P, Möllmann H, Papageorgiou A-P, Heymans S.. Osteoglycin prevents cardiac dilatation and dysfunction after myocardial infarction through infarct collagen strengthening. Circ Res 2015;116:425–436. [DOI] [PubMed] [Google Scholar]
- 368. Chen SW, Tung YC, Jung SM, Chu Y, Lin PJ, Kao WW, Chu PH.. Lumican-null mice are susceptible to aging and isoproterenol-induced myocardial fibrosis. Biochem Biophys Res Commun 2017;482:1304–1311. [DOI] [PubMed] [Google Scholar]
- 369. Yan W, Wang P, Zhao CX, Tang J, Xiao X, Wang DW.. Decorin gene delivery inhibits cardiac fibrosis in spontaneously hypertensive rats by modulation of transforming growth factor-beta/Smad and p38 mitogen-activated protein kinase signaling pathways. Hum Gene Ther 2009;20:1190–1200. [DOI] [PubMed] [Google Scholar]
- 370. Andenæs K, Lunde IG, Mohammadzadeh N, Dahl CP, Aronsen JM, Strand ME, Palmero S, Sjaastad I, Christensen G, Engebretsen KVT, Tønnessen T.. The extracellular matrix proteoglycan fibromodulin is upregulated in clinical and experimental heart failure and affects cardiac remodeling. PLoS One 2018;13:e0201422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371. Vanhoutte D, Schellings MWM, GöTte M, Swinnen M, Herias V, Wild MK, Vestweber D, Chorianopoulos E, Cortés V, Rigotti A, Stepp M-A, Van de Werf F, Carmeliet P, Pinto YM, Heymans S, Increased expression of syndecan-1 protects against cardiac dilatation and dysfunction after myocardial infarction. Circulation 2007;115:475–482. [DOI] [PubMed] [Google Scholar]
- 372. Schellings MW, Vanhoutte D, van Almen GC, Swinnen M, Leenders JJ, Kubben N, van Leeuwen RE, Hofstra L, Heymans S, Pinto YM.. Syndecan-1 amplifies angiotensin II-induced cardiac fibrosis. Hypertension 2010;55:249–256. [DOI] [PubMed] [Google Scholar]
- 373. Matsui Y, Ikesue M, Danzaki K, Morimoto J, Sato M, Tanaka S, Kojima T, Tsutsui H, Uede T.. Syndecan-4 prevents cardiac rupture and dysfunction after myocardial infarction. Circ Res 2011;108:1328–1339. [DOI] [PubMed] [Google Scholar]
- 374. Herum KM, Lunde IG, Skrbic B, Louch WE, Hasic A, Boye S, Unger A, Brorson SH, Sjaastad I, Tonnessen T, Linke WA, Gomez MF, Christensen G.. Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc Res 2015;106:217–226. [DOI] [PubMed] [Google Scholar]
- 375. Chen L, Klass C, Woods A.. Syndecan-2 regulates transforming growth factor-beta signaling. J Biol Chem 2004;279:15715–15718. [DOI] [PubMed] [Google Scholar]
- 376. McQuade KJ, Beauvais DM, Burbach BJ, Rapraeger AC.. Syndecan-1 regulates alphavbeta5 integrin activity in B82L fibroblasts. J Cell Sci 2006;119:2445–2456. [DOI] [PubMed] [Google Scholar]
- 377. Kato M, Wang H, Kainulainen V, Fitzgerald ML, Ledbetter S, Ornitz DM, Bernfield M.. Physiological degradation converts the soluble syndecan-1 ectodomain from an inhibitor to a potent activator of FGF-2. Nat Med 1998;4:691–697. [DOI] [PubMed] [Google Scholar]
- 378. Herum KM, Romaine A, Wang A, Melleby AO, Strand ME, Pacheco J, Braathen B, Duner P, Tonnessen T, Lunde IG, Sjaastad I, Brakebusch C, McCulloch AD, Gomez MF, Carlson CR, Christensen G.. Syndecan-4 protects the heart from the profibrotic effects of thrombin-cleaved osteopontin. J Am Heart Assoc 2020;9:e013518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Lipphardt M, Song JW, Ratliff BB, Dihazi H, Muller GA, Goligorsky MS.. Endothelial dysfunction is a superinducer of syndecan-4: fibrogenic role of its ectodomain. Am J Physiol Heart Circ Physiol 2018;314:H484–H496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Strand ME, Aronsen JM, Braathen B, Sjaastad I, Kvaloy H, Tonnessen T, Christensen G, Lunde IG.. Shedding of syndecan-4 promotes immune cell recruitment and mitigates cardiac dysfunction after lipopolysaccharide challenge in mice. J Mol Cell Cardiol 2015;88:133–144. [DOI] [PubMed] [Google Scholar]
- 381. Echtermeyer F, Harendza T, Hubrich S, Lorenz A, Herzog C, Mueller M, Schmitz M, Grund A, Larmann J, Stypmann J, Schieffer B, Lichtinghagen R, Hilfiker-Kleiner D, Wollert KC, Heineke J, Theilmeier G.. Syndecan-4 signalling inhibits apoptosis and controls NFAT activity during myocardial damage and remodelling. Cardiovasc Res 2011;92:123–131. [DOI] [PubMed] [Google Scholar]
- 382. Senbanjo LT, Chellaiah MA.. CD44: a multifunctional cell surface adhesion receptor is a regulator of progression and metastasis of cancer cells. Front Cell Dev Biol 2017;5:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383. Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, Haudek S, Thakker G, Frangogiannis NG.. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J Immunol 2008;180:2625–2633. [DOI] [PubMed] [Google Scholar]
- 384. Yang LW, Qin DZ, James E, McKallip RJ, Wang NP, Zhang WW, Zheng RH, Han QH, Zhao ZQ.. CD44 deficiency in mice protects the heart against angiotensin II-induced cardiac fibrosis. Shock 2018;51:372–380. [DOI] [PubMed] [Google Scholar]
- 385. Govindaraju P, Todd L, Shetye S, Monslow J, Pure E.. CD44-dependent inflammation, fibrogenesis, and collagenolysis regulates extracellular matrix remodeling and tensile strength during cutaneous wound healing. Matrix Biol 2019;75–76:314–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Midgley AC, Oltean S, Hascall V, Woods EL, Steadman R, Phillips AO, Meran S.. Nuclear hyaluronidase 2 drives alternative splicing of CD44 pre-mRNA to determine profibrotic or antifibrotic cell phenotype. Sci Signal 2017;10:eaao1822. [DOI] [PubMed] [Google Scholar]
- 387. Barnes LA, Marshall CD, Leavitt T, Hu MS, Moore AL, Gonzalez JG, Longaker MT, Gurtner GC.. Mechanical forces in cutaneous wound healing: emerging therapies to minimize scar formation. Adv Wound Care (New Rochelle) 2018;7:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Chen C, Li R, Ross RS, Manso AM.. Integrins and integrin-related proteins in cardiac fibrosis. J Mol Cell Cardiol 2016;93:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389. Leask A. Integrin 1: a mechanosignaling sensor essential for connective tissue deposition by fibroblasts. Adv Wound Care (New Rochelle) 2013;2:160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Sun M, Opavsky MA, Stewart DJ, Rabinovitch M, Dawood F, Wen WH, Liu PP.. Temporal response and localization of integrins beta1 and beta3 in the heart after myocardial infarction: regulation by cytokines. Circulation 2003;107:1046–1052. [DOI] [PubMed] [Google Scholar]
- 391. Takawale A, Zhang P, Patel VB, Wang X, Oudit G, Kassiri Z.. Tissue inhibitor of matrix metalloproteinase-1 promotes myocardial fibrosis by mediating CD63-integrin beta1 interaction. Hypertension 2017;69:1092–1103. [DOI] [PubMed] [Google Scholar]
- 392. Murray IR, Gonzalez ZN, Baily J, Dobie R, Wallace RJ, Mackinnon AC, Smith JR, Greenhalgh SN, Thompson AI, Conroy KP, Griggs DW, Ruminski PG, Gray GA, Singh M, Campbell MA, Kendall TJ, Dai J, Li Y, Iredale JP, Simpson H, Huard J, Peault B, Henderson NC.. alphav integrins on mesenchymal cells regulate skeletal and cardiac muscle fibrosis. Nat Commun 2017;8:1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA.. Latent TGF-beta structure and activation. Nature 2011;474:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Leask A. Focal adhesion kinase: a key mediator of transforming growth factor beta signaling in fibroblasts. Adv Wound Care (New Rochelle) 2013;2:247–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395. Zhang P, Wang W, Wang X, Wang X, Song Y, Zhang J, Zhao H.. Focal adhesion kinase mediates atrial fibrosis via the AKT/S6K signaling pathway in chronic atrial fibrillation patients with rheumatic mitral valve disease. Int J Cardiol 2013;168:3200–3207. [DOI] [PubMed] [Google Scholar]
- 396. Chan MW, Arora PD, Bozavikov P, McCulloch CA.. FAK, PIP5KIgamma and gelsolin cooperatively mediate force-induced expression of alpha-smooth muscle actin. J Cell Sci 2009;122:2769–2781. [DOI] [PubMed] [Google Scholar]
- 397. Fan GP, Wang W, Zhao H, Cai L, Zhang PD, Yang ZH, Zhang J, Wang X.. Pharmacological inhibition of focal adhesion kinase attenuates cardiac fibrosis in mice cardiac fibroblast and post-myocardial-infarction models. Cell Physiol Biochem 2015;37:515–526. [DOI] [PubMed] [Google Scholar]
- 398. Zhang J, Fan G, Zhao H, Wang Z, Li F, Zhang P, Zhang J, Wang X, Wang W.. Targeted inhibition of focal adhesion kinase attenuates cardiac fibrosis and preserves heart function in adverse cardiac remodeling. Sci Rep 2017;7:43146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399. Clemente CF, Tornatore TF, Theizen TH, Deckmann AC, Pereira TC, Lopes-Cendes I, Souza JR, Franchini KG.. Targeting focal adhesion kinase with small interfering RNA prevents and reverses load-induced cardiac hypertrophy in mice. Circ Res 2007;101:1339–1348. [DOI] [PubMed] [Google Scholar]
- 400. Cheng Z, Sundberg-Smith LJ, Mangiante LE, Sayers RL, Hakim ZS, Musunuri S, Maguire CT, Majesky MW, Zhou Z, Mack CP, Taylor JM.. Focal adhesion kinase regulates smooth muscle cell recruitment to the developing vasculature. Arterioscler Thromb Vasc Biol 2011;31:2193–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401. Hakim ZS, DiMichele LA, Rojas M, Meredith D, Mack CP, Taylor JM.. FAK regulates cardiomyocyte survival following ischemia/reperfusion. J Mol Cell Cardiol 2009;46:241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. Sinfield JK, Das A, O’Regan DJ, Ball SG, Porter KE, Turner NA.. p38 MAPK alpha mediates cytokine-induced IL-6 and MMP-3 expression in human cardiac fibroblasts. Biochem Biophys Res Commun 2013;430:419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 403. Loirand G, Sauzeau V, Pacaud P.. Small G proteins in the cardiovascular system: physiological and pathological aspects. Physiol Rev 2013;93:1659–1720. [DOI] [PubMed] [Google Scholar]
- 404. Shimizu T, Liao JK.. Rho kinases and cardiac remodeling. Circ J 2016;80:1491–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405. Ongherth A, Pasch S, Wuertz CM, Nowak K, Kittana N, Weis CA, Jatho A, Vettel C, Tiburcy M, Toischer K, Hasenfuss G, Zimmermann WH, Wieland T, Lutz S.. p63RhoGEF regulates auto- and paracrine signaling in cardiac fibroblasts. J Mol Cell Cardiol 2015;88:39–54. [DOI] [PubMed] [Google Scholar]
- 406. Phrommintikul A, Tran L, Kompa A, Wang B, Adrahtas A, Cantwell D, Kelly DJ, Krum H.. Effects of a Rho kinase inhibitor on pressure overload induced cardiac hypertrophy and associated diastolic dysfunction. Am J Physiol Heart Circ Physiol 2008;294:H1804–H1814. [DOI] [PubMed] [Google Scholar]
- 407. Lauriol J, Keith K, Jaffre F, Couvillon A, Saci A, Goonasekera SA, McCarthy JR, Kessinger CW, Wang J, Ke Q, Kang PM, Molkentin JD, Carpenter C, Kontaridis MI.. RhoA signaling in cardiomyocytes protects against stress-induced heart failure but facilitates cardiac fibrosis. Sci Signal 2014;7:ra100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Rikitake Y, Oyama N, Wang CY, Noma K, Satoh M, Kim HH, Liao JK.. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/- haploinsufficient mice. Circulation 2005;112:2959–2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Haudek SB, Gupta D, Dewald O, Schwartz RJ, Wei L, Trial J, Entman ML.. Rho kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovasc Res 2009;83:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Shimizu T, Narang N, Chen P, Yu B, Knapp M, Janardanan J, Blair J, Liao JK.. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight 2017;2:e93187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411. Nishida M, Onohara N, Sato Y, Suda R, Ogushi M, Tanabe S, Inoue R, Mori Y, Kurose H.. Galpha12/13-mediated up-regulation of TRPC6 negatively regulates endothelin-1-induced cardiac myofibroblast formation and collagen synthesis through nuclear factor of activated T cells activation. J Biol Chem 2007;282:23117–23128. [DOI] [PubMed] [Google Scholar]
- 412. Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD.. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell 2012;23:705–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B, Yue L.. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ Res 2010;106:992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414. Adapala RK, Thoppil RJ, Luther DJ, Paruchuri S, Meszaros JG, Chilian WM, Thodeti CK.. TRPV4 channels mediate cardiac fibroblast differentiation by integrating mechanical and soluble signals. J Mol Cell Cardiol 2013;54:45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415. Rios FJ, Zou ZG, Harvey AP, Harvey KY, Nosalski R, Anyfanti P, Camargo LL, Lacchini S, Ryazanov AG, Ryazanova L, McGrath S, Guzik TJ, Goodyear CS, Montezano AC, Touyz RM.. Chanzyme TRPM7 protects against cardiovascular inflammation and fibrosis. Cardiovasc Res 2020;116:721–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Abraham DM, Lee TE, Watson LJ, Mao L, Chandok G, Wang H-G, Frangakis S, Pitt GS, Shah SH, Wolf MJ, Rockman HA.. The two-pore domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction. J Clin Invest 2018;128:4843–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Mukherjee S, Ayaub EA, Murphy J, Lu C, Kolb M, Ask K, Janssen LJ.. Disruption of calcium signaling in fibroblasts and attenuation of bleomycin-induced fibrosis by nifedipine. Am J Respir Cell Mol Biol 2015;53:450–458. [DOI] [PubMed] [Google Scholar]
- 418. Janssen LJ, Mukherjee S, Ask K.. Calcium homeostasis and ionic mechanisms in pulmonary fibroblasts. Am J Respir Cell Mol Biol 2015;53:135–148. [DOI] [PubMed] [Google Scholar]
- 419. Follonier Castella L, Gabbiani G, McCulloch CA, Hinz B.. Regulation of myofibroblast activities: calcium pulls some strings behind the scene. Exp Cell Res 2010;316:2390–2401. [DOI] [PubMed] [Google Scholar]
- 420. Godbout C, Follonier Castella L, Smith EA, Talele N, Chow ML, Garonna A, Hinz B.. The mechanical environment modulates intracellular calcium oscillation activities of myofibroblasts. PLoS One 2013;8:e64560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Dasgupta I, McCollum D.. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J Biol Chem 2019;294:17693–17706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422. Szeto SG, Narimatsu M, Lu M, He X, Sidiqi AM, Tolosa MF, Chan L, De Freitas K, Bialik JF, Majumder S, Boo S, Hinz B, Dan Q, Advani A, John R, Wrana JL, Kapus A, Yuen DA.. YAP/TAZ are mechanoregulators of TGF-beta-Smad signaling and renal fibrogenesis. JASN 2016;27:3117–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423. Jorgenson AJ, Choi KM, Sicard D, Smith KM, Hiemer SE, Varelas X, Tschumperlin DJ.. TAZ activation drives fibroblast spheroid growth, expression of profibrotic paracrine signals, and context-dependent ECM gene expression. Am J Physiol Cell Physiol 2017;312:C277–C285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424. Xiao Y, Hill MC, Li L, Deshmukh V, Martin TJ, Wang J, Martin JF.. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes Dev 2019;33:1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Flinn MA, Jeffery BE, O’Meara CC, Link BA.. Yap is required for scar formation but not myocyte proliferation during heart regeneration in zebrafish. Cardiovasc Res 2019;115:570–577. [DOI] [PubMed] [Google Scholar]
- 426. Byun J, Del Re DP, Zhai P, Ikeda S, Shirakabe A, Mizushima W, Miyamoto S, Brown JH, Sadoshima J.. Yes-associated protein (YAP) mediates adaptive cardiac hypertrophy in response to pressure overload. J Biol Chem 2019;294:3603–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Ramjee V, Li D, Manderfield LJ, Liu F, Engleka KA, Aghajanian H, Rodell CB, Lu W, Ho V, Wang T, Li L, Singh A, Cibi DM, Burdick JA, Singh MK, Jain R, Epstein JA.. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J Clin Invest 2017;127:899–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428. Norman C, Runswick M, Pollock R, Treisman R.. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell 1988;55:989–1003. [DOI] [PubMed] [Google Scholar]
- 429. Tomasek JJ, McRae J, Owens GK, Haaksma CJ.. Regulation of alpha-smooth muscle actin expression in granulation tissue myofibroblasts is dependent on the intronic CArG element and the transforming growth factor-beta1 control element. Am J Pathol 2005;166:1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430. Lighthouse JK, Small EM.. Transcriptional control of cardiac fibroblast plasticity. J Mol Cell Cardiol 2016;91:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431. Small EM. The actin-MRTF-SRF gene regulatory axis and myofibroblast differentiation. J Cardiovasc Trans Res 2012;5:794–804. [DOI] [PubMed] [Google Scholar]
- 432. Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K, Olson EN.. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ Res 2010;107:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Weng X, Yu L, Liang P, Chen D, Cheng X, Yang Y, Li L, Zhang T, Zhou B, Wu X, Xu H, Fang M, Gao Y, Chen Q, Xu Y.. Endothelial MRTF-A mediates angiotensin II induced cardiac hypertrophy. J Mol Cell Cardiol 2015;80:23–33. [DOI] [PubMed] [Google Scholar]
- 434. Trembley MA, Quijada P, Agullo-Pascual E, Tylock KM, Colpan M, Dirkx RA Jr, Myers JR, Mickelsen DM, de Mesy Bentley K, Rothenberg E, Moravec CS, Alexis JD, Gregorio CC, Dirksen RT, Delmar M, Small EM.. Mechanosensitive gene regulation by myocardin-related transcription factors is required for cardiomyocyte integrity in load-induced ventricular hypertrophy. Circulation 2018;138:1864–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435. Grosche J, Meissner J, Eble JA.. More than a syllable in fib-ROS-is: the role of ROS on the fibrotic extracellular matrix and on cellular contacts. Mol Aspects Med 2018;63:30–46. [DOI] [PubMed] [Google Scholar]
- 436. Cheng TH, Cheng PY, Shih NL, Chen IB, Wang DL, Chen JJ.. Involvement of reactive oxygen species in angiotensin II-induced endothelin-1 gene expression in rat cardiac fibroblasts. J Am Coll Cardiol 2003;42:1845–1854. [DOI] [PubMed] [Google Scholar]
- 437. Ohtsu H, Frank GD, Utsunomiya H, Eguchi S.. Redox-dependent protein kinase regulation by angiotensin II: mechanistic insights and its pathophysiology. Antioxid Redox Signal 2005;7:1315–1326. [DOI] [PubMed] [Google Scholar]
- 438. Lijnen P, Papparella I, Petrov V, Semplicini A, Fagard R.. Angiotensin II-stimulated collagen production in cardiac fibroblasts is mediated by reactive oxygen species. J Hypertens 2006;24:757–766. [DOI] [PubMed] [Google Scholar]
- 439. Iglarz M, Touyz RM, Viel EC, Amiri F, Schiffrin EL.. Involvement of oxidative stress in the profibrotic action of aldosterone. Interaction wtih the renin-angiotension system. Am J Hypertens 2004;17:597–603. [PubMed] [Google Scholar]
- 440. Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, Feghali-Bostwick C, Mutlu GM, Budinger GR, Chandel NS.. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J Biol Chem 2013;288:770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441. Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D.. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res 2005;97:900–907. [DOI] [PubMed] [Google Scholar]
- 442. Siwik DA, Pagano PJ, Colucci WS.. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol 2001;280:C53–C60. [DOI] [PubMed] [Google Scholar]
- 443. Siwik DA, Colucci WS.. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail Rev 2004;9:43–51. [DOI] [PubMed] [Google Scholar]
- 444. Zhang X, Hu M, Lyu X, Li C, Thannickal VJ, Sanders YY.. DNA methylation regulated gene expression in organ fibrosis. Biochim Biophys Acta 2017;1863:2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Schuetze KB, McKinsey TA, Long CS.. Targeting cardiac fibroblasts to treat fibrosis of the heart: focus on HDACs. J Mol Cell Cardiol 2014;70:100–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446. Tao H, Song ZY, Ding XS, Yang JJ, Shi KH, Li J.. Epigenetic signatures in cardiac fibrosis, special emphasis on DNA methylation and histone modification. Heart Fail Rev 2018;23:789–799. [DOI] [PubMed] [Google Scholar]
- 447. Felisbino MB, McKinsey TA.. Epigenetics in cardiac fibrosis: emphasis on inflammation and fibroblast activation. JACC Basic Transl Sci 2018;3:704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448. Shimizu K, Sunagawa Y, Funamoto M, Wakabayashi H, Genpei M, Miyazaki Y, Katanasaka Y, Sari N, Shimizu S, Katayama A, Shibata H, Iwabuchi Y, Kakeya H, Wada H, Hasegawa K, Morimoto T.. The synthetic curcumin analogue GO-Y030 effectively suppresses the development of pressure overload-induced heart failure in mice. Sci Rep 2020;10:7172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Seto E, Yoshida M.. Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb Perspect Biol 2014;6:a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450. Stratton MS, McKinsey TA.. Epigenetic regulation of cardiac fibrosis. J Mol Cell Cardiol 2016;92:206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, Demos-Davies K, Yeager ME, Stenmark KR, McKinsey TA.. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J Mol Cell Cardiol 2014;67:112–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452. Nural-Guvener HF, Zakharova L, Nimlos J, Popovic S, Mastroeni D, Gaballa MA.. HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenesis Tissue Repair 2014;7:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453. Jeong MY, Lin YH, Wennersten SA, Demos-Davies KM, Cavasin MA, Mahaffey JH, Monzani V, Saripalli C, Mascagni P, Reece TB, Ambardekar AV, Granzier HL, Dinarello CA, McKinsey TA.. Histone deacetylase activity governs diastolic dysfunction through a nongenomic mechanism. Sci Transl Med 2018;10:eaao0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454. Blakeslee WW, Lin Y-H, Stratton MS, Tatman PD, Hu T, Ferguson BS, McKinsey TA.. Class I HDACs control a JIP1-dependent pathway for kinesin-microtubule binding in cardiomyocytes. J Mol Cell Cardiol 2017;112:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455. Wallner M, Eaton DM, Berretta RM, Liesinger L, Schittmayer M, Gindlhuber J, Wu J, Jeong MY, Lin YH, Borghetti G, Baker ST, Zhao H, Pfleger J, Blass S, Rainer PP, von Lewinski D, Bugger H, Mohsin S, Graier WF, Zirlik A, McKinsey TA, Birner-Gruenberger R, Wolfson MR, Houser SR.. HDAC inhibition improves cardiopulmonary function in a feline model of diastolic dysfunction. Sci Transl Med 2020;12:eaay7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456. Schuetze KB, Stratton MS, Blakeslee WW, Wempe MF, Wagner FF, Holson EB, Kuo YM, Andrews AJ, Gilbert TM, Hooker JM, McKinsey TA.. Overlapping and divergent actions of structurally distinct histone deacetylase inhibitors in cardiac fibroblasts. J Pharmacol Exp Ther 2017;361:140–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Borck PC, Guo LW, Plutzky J.. BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ Res 2020;126:1190–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458. Stratton MS, Haldar SM, McKinsey TA.. BRD4 inhibition for the treatment of pathological organ fibrosis. F1000Res 2017;6:1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Stratton MS, Bagchi RA, Felisbino MB, Hirsch RA, Smith HE, Riching AS, Enyart BY, Koch KA, Cavasin MA, Alexanian M, Song K, Qi J, Lemieux ME, Srivastava D, Lam MPY, Haldar SM, Lin CY, McKinsey TA.. Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation. Circ Res 2019;125:662–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460. Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, Huang Y, Zhang R, Sahadevan A, Lemieux ME, Brown JD, Srivastava D, Bradner JE, McKinsey TA, Haldar SM.. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med 2017;9:eaah5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461. Auguste G, Rouhi L, Matkovich SJ, Coarfa C, Robertson MJ, Czernuszewicz G, Gurha P, Marian AJ.. BET bromodomain inhibition attenuates cardiac phenotype in myocyte-specific Lamin A/C-deficient mice. J Clin Invest 2020;130:4740–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462. Creemers EE, van Rooij E.. Function and therapeutic potential of noncoding RNAs in cardiac fibrosis. Circ Res 2016;118:108–118. [DOI] [PubMed] [Google Scholar]
- 463. Thum T. Noncoding RNAs and myocardial fibrosis. Nat Rev Cardiol 2014;11:655–663. [DOI] [PubMed] [Google Scholar]
- 464. Piccoli MT, Bar C, Thum T.. Non-coding RNAs as modulators of the cardiac fibroblast phenotype. J Mol Cell Cardiol 2016;92:75–81. [DOI] [PubMed] [Google Scholar]
- 465. Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A, Just A, Remke J, Zimmer K, Zeug A, Ponimaskin E, Schmiedl A, Yin X, Mayr M, Halder R, Fischer A, Engelhardt S, Wei Y, Schober A, Fiedler J, Thum T.. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest 2014;124:2136–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466. Verjans R, Derks WJA, Korn K, Sonnichsen B, van Leeuwen REW, Schroen B, van Bilsen M, Heymans S.. Functional screening identifies microRNAs as multi-cellular regulators of heart failure. Sci Rep 2019;9:6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN.. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci USA 2008;105:13027–13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468. Tijsen AJ, van der Made I, van den Hoogenhof MM, Wijnen WJ, van Deel ED, de Groot NE, Alekseev S, Fluiter K, Schroen B, Goumans MJ, van der Velden J, Duncker DJ, Pinto YM, Creemers EE.. The microRNA-15 family inhibits the TGFbeta-pathway in the heart. Cardiovasc Res 2014;104:61–71. [DOI] [PubMed] [Google Scholar]
- 469. Pan Z, Sun X, Shan H, Wang N, Wang J, Ren J, Feng S, Xie L, Lu C, Yuan Y, Zhang Y, Wang Y, Lu Y, Yang B.. MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-beta1 pathway. Circulation 2012;126:840–850. [DOI] [PubMed] [Google Scholar]
- 470. Chiasson V, Takano APC, Guleria RS, Gupta S.. Deficiency of microRNA miR-1954 promotes cardiac remodeling and fibrosis. J Am Heart Assoc 2019;8:e012880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S.. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008;456:980–984. [DOI] [PubMed] [Google Scholar]
- 472. Schimmel K, Jung M, Foinquinos A, José GS, Beaumont J, Bock K, Grote-Levi L, Xiao K, Bär C, Pfanne A, Just A, Zimmer K, Ngoy S, López B, Ravassa S, Samolovac S, Janssen-Peters H, Remke J, Scherf K, Dangwal S, Piccoli M-T, Kleemiss F, Kreutzer FP, Kenneweg F, Leonardy J, Hobuß L, Santer L, Do Q-T, Geffers R, Braesen JH, Schmitz J, Brandenberger C, Müller DN, Wilck N, Kaever V, Bähre H, Batkai S, Fiedler J, Alexander KM, Wertheim BM, Fisch S, Liao R, Diez J, González A, Thum T.. Natural compound library screening identifies new molecules for the treatment of cardiac fibrosis and diastolic dysfunction. Circulation 2020;141:751–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473. Yuan X, Pan J, Wen L, Gong B, Li J, Gao H, Tan W, Liang S, Zhang H, Wang X.. MiR-144-3p enhances cardiac fibrosis after myocardial infarction by targeting PTEN. Front Cell Dev Biol 2019;7:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474. Jiang X, Zhang F.. Long noncoding RNA: a new contributor and potential therapeutic target in fibrosis. Epigenomics 2017;9:1233–1241. [DOI] [PubMed] [Google Scholar]
- 475. Piccoli MT, Gupta SK, Viereck J, Foinquinos A, Samolovac S, Kramer FL, Garg A, Remke J, Zimmer K, Batkai S, Thum T.. Inhibition of the cardiac fibroblast-enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ Res 2017;121:575–583. [DOI] [PubMed] [Google Scholar]
- 476. Micheletti R, Plaisance I, Abraham BJ, Sarre A, Ting CC, Alexanian M, Maric D, Maison D, Nemir M, Young RA, Schroen B, Gonzalez A, Ounzain S, Pedrazzini T.. The long noncoding RNA Wisper controls cardiac fibrosis and remodeling. Sci Transl Med 2017;9:eaai9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477. Sun F, Zhuang Y, Zhu H, Wu H, Li D, Zhan L, Yang W, Yuan Y, Xie Y, Yang S, Luo S, Jiang W, Zhang J, Pan Z, Lu Y.. LncRNA PCFL promotes cardiac fibrosis via miR-378/GRB2 pathway following myocardial infarction. J Mol Cell Cardiol 2019;133:188–198. [DOI] [PubMed] [Google Scholar]
- 478. Kenneweg F, Bang C, Xiao K, Boulanger CM, Loyer X, Mazlan S, Schroen B, Hermans-Beijnsberger S, Foinquinos A, Hirt MN, Eschenhagen T, Funcke S, Stojanovic S, Genschel C, Schimmel K, Just A, Pfanne A, Scherf K, Dehmel S, Raemon-Buettner SM, Fiedler J, Thum T.. Long noncoding RNA-enriched vesicles secreted by hypoxic cardiomyocytes drive cardiac fibrosis. Mol Ther Nucleic Acids 2019;18:363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479. Huang ZP, Ding Y, Chen J, Wu G, Kataoka M, Hu Y, Yang JH, Liu J, Drakos SG, Selzman CH, Kyselovic J, Qu LH, Dos Remedios CG, Pu WT, Wang DZ.. Long non-coding RNAs link extracellular matrix gene expression to ischemic cardiomyopathy. Cardiovasc Res 2016;112:543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480. Takemura G, Ohno M, Hayakawa Y, Misao J, Kanoh M, Ohno A, Uno Y, Minatoguchi S, Fujiwara T, Fujiwara H.. Role of apoptosis in the disappearance of infiltrated and proliferated interstitial cells after myocardial infarction. Circ Res 1998;82:1130–1138. [DOI] [PubMed] [Google Scholar]
- 481. Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, Molkentin JD.. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest 2018;128:2127–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482. Itoh S, ten Dijke P.. Negative regulation of TGF-beta receptor/Smad signal transduction. Curr Opin Cell Biol 2007;19:176–184. [DOI] [PubMed] [Google Scholar]
- 483. Kapur NK, Wilson S, Yunis AA, Qiao X, Mackey E, Paruchuri V, Baker C, Aronovitz MJ, Karumanchi SA, Letarte M, Kass DA, Mendelsohn ME, Karas RH.. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation 2012;125:2728–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484. Villar AV, Garcia R, Llano M, Cobo M, Merino D, Lantero A, Tramullas M, Hurle JM, Hurle MA, Nistal JF.. BAMBI (BMP and activin membrane-bound inhibitor) protects the murine heart from pressure-overload biomechanical stress by restraining TGF-beta signaling. Biochim Biophys Acta 2013;1832:323–335. [DOI] [PubMed] [Google Scholar]
- 485. Miyazawa K, Miyazono K.. Regulation of TGF-beta family signaling by inhibitory Smads. Cold Spring Harb Perspect Biol 2017;9:a022095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486. Zhang ZZ, Wang W, Jin HY, Chen X, Cheng YW, Xu YL, Song B, Penninger JM, Oudit GY, Zhong JC.. Apelin is a negative regulator of angiotensin II-mediated adverse myocardial remodeling and dysfunction. Hypertension 2017;70:1165–1175. [DOI] [PubMed] [Google Scholar]
- 487. Meyer K, Hodwin B, Ramanujam D, Engelhardt S, Sarikas A.. Essential role for premature senescence of myofibroblasts in myocardial fibrosis. J Am Coll Cardiol 2016;67:2018–2028. [DOI] [PubMed] [Google Scholar]
- 488. Wong TC, Piehler K, Meier CG, Testa SM, Klock AM, Aneizi AA, Shakesprere J, Kellman P, Shroff SG, Schwartzman DS, Mulukutla SR, Simon MA, Schelbert EB.. Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short-term mortality. Circulation 2012;126:1206–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489. Reimer KA, Lowe JE, Rasmussen MM, Jennings RB.. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation 1977;56:786–794. [DOI] [PubMed] [Google Scholar]
- 490. Prabhu SD, Frangogiannis NG.. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res 2016;119:91–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491. Di Bella G, Siciliano V, Aquaro GD, Molinaro S, Lombardi M, Carerj S, Landi P, Rovai D, Pingitore A.. Scar extent, left ventricular end-diastolic volume, and wall motion abnormalities identify high-risk patients with previous myocardial infarction: a multiparametric approach for prognostic stratification. Eur Heart J 2013;34:104–111. [DOI] [PubMed] [Google Scholar]
- 492. Kelle S, Roes SD, Klein C, Kokocinski T, de Roos A, Fleck E, Bax JJ, Nagel E.. Prognostic value of myocardial infarct size and contractile reserve using magnetic resonance imaging. J Am Coll Cardiol 2009;54:1770–1777. [DOI] [PubMed] [Google Scholar]
- 493. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 2014;11:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494. Richardson WJ, Clarke SA, Quinn TA, Holmes JW.. Physiological implications of myocardial scar structure. Compr Physiol 2015;5:1877–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495. Gibbs T, Villa ADM, Sammut E, Jeyabraba S, Carr-White G, Ismail TF, Mullen G, Ganeshan B, Chiribiri A.. Quantitative assessment of myocardial scar heterogeneity using cardiovascular magnetic resonance texture analysis to risk stratify patients post-myocardial infarction. Clin Radiol 2018;73:1059.e17–1059.e26. [DOI] [PubMed] [Google Scholar]
- 496. Androulakis AFA, Zeppenfeld K, Paiman EHM, Piers SRD, Wijnmaalen AP, Siebelink HJ, Sramko M, Lamb HJ, van der Geest RJ, de Riva M, Tao Q.. Entropy as a novel measure of myocardial tissue heterogeneity for prediction of ventricular arrhythmias and mortality in post-infarct patients. JACC Clin Electrophysiol 2019;5:480–489. [DOI] [PubMed] [Google Scholar]
- 497. van den Borne SW, Isobe S, Zandbergen HR, Li P, Petrov A, Wong ND, Fujimoto S, Fujimoto A, Lovhaug D, Smits JF, Daemen MJ, Blankesteijn WM, Reutelingsperger C, Zannad F, Narula N, Vannan MA, Pitt B, Hofstra L, Narula J.. Molecular imaging for efficacy of pharmacologic intervention in myocardial remodeling. JACC Cardiovasc Imaging 2009;2:187–198. [DOI] [PubMed] [Google Scholar]
- 498. Wollert KC, Studer R, Doerfer K, Schieffer E, Holubarsch C, Just H, Drexler H.. Differential effects of kinins on cardiomyocyte hypertrophy and interstitial collagen matrix in the surviving myocardium after myocardial infarction in the rat. Circulation 1997;95:1910–1917. [DOI] [PubMed] [Google Scholar]
- 499. Marijianowski MM, Teeling P, Becker AE.. Remodeling after myocardial infarction in humans is not associated with interstitial fibrosis of noninfarcted myocardium. J Am Coll Cardiol 1997;30:76–82. [DOI] [PubMed] [Google Scholar]
- 500. Garg P, Broadbent DA, Swoboda PP, Foley JRJ, Fent GJ, Musa TA, Ripley DP, Erhayiem B, Dobson LE, McDiarmid AK, Haaf P, Kidambi A, Crandon S, Chew PG, van der Geest RJ, Greenwood JP, Plein S.. Extra-cellular expansion in the normal, non-infarcted myocardium is associated with worsening of regional myocardial function after acute myocardial infarction. J Cardiovasc Magn Reson 2017;19:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501. Nagueh SF. Heart failure with preserved ejection fraction: insights into diagnosis and pathophysiology. Cardiovasc Res 2020. [DOI] [PubMed] [Google Scholar]
- 502. Martos R, Baugh J, Ledwidge M, O'Loughlin C, Murphy NF, Conlon C, Patle A, Donnelly SC, McDonald K.. Diagnosis of heart failure with preserved ejection fraction: improved accuracy with the use of markers of collagen turnover. Eur J Heart Fail 2009;11:191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503. Aoki T, Fukumoto Y, Sugimura K, Oikawa M, Satoh K, Nakano M, Nakayama M, Shimokawa H.. Prognostic impact of myocardial interstitial fibrosis in non-ischemic heart failure. -Comparison between preserved and reduced ejection fraction heart failure. Circ J 2011;75:2605–2613. [DOI] [PubMed] [Google Scholar]
- 504. Kanagala P, Arnold JR, Singh A, Chan DCS, Cheng ASH, Khan JN, Gulsin GS, Yang J, Zhao L, Gupta P, Squire IB, Ng LL, McCann GP.. Characterizing heart failure with preserved and reduced ejection fraction: An imaging and plasma biomarker approach. PLoS One 2020;15:e0232280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505. Rommel KP, von Roeder M, Latuscynski K, Oberueck C, Blazek S, Fengler K, Besler C, Sandri M, Lucke C, Gutberlet M, Linke A, Schuler G, Lurz P.. Extracellular volume fraction for characterization of patients with heart failure and preserved ejection fraction. J Am Coll Cardiol 2016;67:1815–1825. [DOI] [PubMed] [Google Scholar]
- 506. Pérez del Villar C, Savvatis K, López B, Kasner M, Martinez-Legazpi P, Yotti R, González A, Díez J, Fernández-Avilés F, Tschöpe C, Bermejo J.. Impact of acute hypertension transients on diastolic function in patients with heart failure with preserved ejection fraction. Cardiovasc Res 2017;113:906–914. [DOI] [PubMed] [Google Scholar]
- 507. Gulsin GS, Kanagala P, Chan DCS, Cheng ASH, Athithan L, Graham-Brown MPM, Singh A, Yang J, Li Z, Khunti K, Davies MJ, Arnold JR, Squire IB, Ng LL, McCann GP.. Differential left ventricular and left atrial remodelling in heart failure with preserved ejection fraction patients with and without diabetes. Ther Adv Endocrinol 2019;10:204201881986159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508. Su MY, Lin LY, Tseng YH, Chang CC, Wu CK, Lin JL, Tseng WY.. CMR-verified diffuse myocardial fibrosis is associated with diastolic dysfunction in HFpEF. JACC Cardiovasc Imaging 2014;7:991–997. [DOI] [PubMed] [Google Scholar]
- 509. Schelbert EB, Fridman Y, Wong TC, Abu Daya H, Piehler KM, Kadakkal A, Miller CA, Ugander M, Maanja M, Kellman P, Shah DJ, Abebe KZ, Simon MA, Quarta G, Senni M, Butler J, Diez J, Redfield MM, Gheorghiade M.. Temporal relation between myocardial fibrosis and heart failure with preserved ejection fraction: association with baseline disease severity and subsequent outcome. JAMA Cardiol 2017;2:995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510. Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM.. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015;131:550–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511. Edelmann F, Holzendorf V, Wachter R, Nolte K, Schmidt AG, Kraigher-Krainer E, Duvinage A, Unkelbach I, Dungen HD, Tschope C, Herrmann-Lingen C, Halle M, Hasenfuss G, Gelbrich G, Stough WG, Pieske BM.. Galectin-3 in patients with heart failure with preserved ejection fraction: results from the Aldo-DHF trial. Eur J Heart Fail 2015;17:214–223. [DOI] [PubMed] [Google Scholar]
- 512. Duca F, Kammerlander AA, Zotter-Tufaro C, Aschauer S, Schwaiger ML, Marzluf BA, Bonderman D, Mascherbauer J.. Interstitial fibrosis, functional status, and outcomes in heart failure with preserved ejection fraction: insights from a prospective cardiac magnetic resonance imaging study. Circ Cardiovasc Imaging 2016;9:e005277. [DOI] [PubMed] [Google Scholar]
- 513. Krum H, Elsik M, Schneider HG, Ptaszynska A, Black M, Carson PE, Komajda M, Massie BM, McKelvie RS, McMurray JJ, Zile MR, Anand IS.. Relation of peripheral collagen markers to death and hospitalization in patients with heart failure and preserved ejection fraction: results of the I-PRESERVE collagen substudy. Circ Heart Fail 2011;4:561–568. [DOI] [PubMed] [Google Scholar]
- 514. Kimura Y, Izumiya Y, Hanatani S, Yamamoto E, Kusaka H, Tokitsu T, Takashio S, Sakamoto K, Tsujita K, Tanaka T, Yamamuro M, Kojima S, Tayama S, Kaikita K, Hokimoto S, Ogawa H.. High serum levels of thrombospondin-2 correlate with poor prognosis of patients with heart failure with preserved ejection fraction. Heart Vessels 2016;31:52–59. [DOI] [PubMed] [Google Scholar]
- 515. Chirinos JA, Orlenko A, Zhao L, Basso MD, Cvijic ME, Li Z, Spires TE, Yarde M, Wang Z, Seiffert DA, Prenner S, Zamani P, Bhattacharya P, Kumar A, Margulies KB, Car BD, Gordon DA, Moore JH, Cappola TP.. Multiple plasma biomarkers for risk stratification in patients with heart failure and preserved ejection fraction. J Am Coll Cardiol 2020;75:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516. Kato S, Saito N, Kirigaya H, Gyotoku D, Iinuma N, Kusakawa Y, Iguchi K, Nakachi T, Fukui K, Futaki M, Iwasawa T, Taguri M, Kimura K, Umemura S.. Prognostic significance of quantitative assessment of focal myocardial fibrosis in patients with heart failure with preserved ejection fraction. Int J Cardiol 2015;191:314–319. [DOI] [PubMed] [Google Scholar]
- 517. Roy C, Slimani A, de Meester C, Amzulescu M, Pasquet A, Vancraeynest D, Beauloye C, Vanoverschelde JL, Gerber BL, Pouleur AC.. Associations and prognostic significance of diffuse myocardial fibrosis by cardiovascular magnetic resonance in heart failure with preserved ejection fraction. J Cardiovasc Magn Reson 2018;20:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518. Kanagala P, Cheng ASH, Singh A, Khan JN, Gulsin GS, Patel P, Gupta P, Arnold JR, Squire IB, Ng LL, McCann GP.. Relationship between focal and diffuse fibrosis assessed by CMR and clinical outcomes in heart failure with preserved ejection fraction. JACC Cardiovasc Imaging 2019;12:2291–2301. [DOI] [PubMed] [Google Scholar]
- 519. Schelbert EB, Piehler KM, Zareba KM, Moon JC, Ugander M, Messroghli DR, Valeti US, Chang CC, Shroff SG, Diez J, Miller CA, Schmitt M, Kellman P, Butler J, Gheorghiade M, Wong TC.. Myocardial fibrosis quantified by extracellular volume is associated with subsequent hospitalization for heart failure, death, or both across the spectrum of ejection fraction and heart failure stage. J Am Heart Assoc 2015;4:e002613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520. Gulati A, Japp AG, Raza S, Halliday BP, Jones DA, Newsome S, Ismail NA, Morarji K, Khwaja J, Spath N, Shakespeare C, Kalra PR, Lloyd G, Mathur A, Cleland JGF, Cowie MR, Assomull RG, Pennell DJ, Ismail TF, Prasad SK.. Absence of myocardial fibrosis predicts favorable long-term survival in new-onset heart failure. Circ Cardiovasc Imaging 2018;11:e007722. [DOI] [PubMed] [Google Scholar]
- 521. Iwanaga Y, Aoyama T, Kihara Y, Onozawa Y, Yoneda T, Sasayama S.. Excessive activation of matrix metalloproteinases coincides with left ventricular remodeling during transition from hypertrophy to heart failure in hypertensive rats. J Am Coll Cardiol 2002;39:1384–1391. [DOI] [PubMed] [Google Scholar]
- 522. Michel JB. Anoikis in the cardiovascular system: known and unknown extracellular mediators. ATVB 2003;23:2146–2154. [DOI] [PubMed] [Google Scholar]
- 523. Atkinson JB, Virmani R.. Congestive heart failure due to coronary artery disease without myocardial infarction: clinicopathologic description of an unusual cardiomyopathy. Hum Pathol 1989;20:1155–1162. [DOI] [PubMed] [Google Scholar]
- 524. Ho CY, López B, Coelho-Filho OR, Lakdawala NK, Cirino AL, Jarolim P, Kwong R, González A, Colan SD, Seidman JG, Díez J, Seidman CE.. Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med 2010;363:552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 525. Meng Q, Bhandary B, Bhuiyan MS, James J, Osinska H, Valiente-Alandi I, Shay-Winkler K, Gulick J, Molkentin JD, Blaxall BC, Robbins J.. Myofibroblast-specific TGFbeta receptor II signaling in the fibrotic response to cardiac myosin binding protein C-induced cardiomyopathy. Circ Res 2018;123:1285–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526. van Tintelen JP, Tio RA, Kerstjens-Frederikse WS, van Berlo JH, Boven LG, Suurmeijer AJ, White SJ, den Dunnen JT, Te Meerman GJ, Vos YJ, van der Hout AH, Osinga J, van den Berg MP, van Veldhuisen DJ, Buys CH, Hofstra RM, Pinto YM.. Severe myocardial fibrosis caused by a deletion of the 5' end of the lamin A/C gene. J Am Coll Cardiol 2007;49:2430–2439. [DOI] [PubMed] [Google Scholar]
- 527. Burke MA, Chang S, Wakimoto H, Gorham JM, Conner DA, Christodoulou DC, Parfenov MG, DePalma SR, Eminaga S, Konno T, Seidman JG, Seidman CE.. Molecular profiling of dilated cardiomyopathy that progresses to heart failure. JCI Insight 2016;1:e86898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528. Chen SN, Lombardi R, Karmouch J, Tsai J-Y, Czernuszewicz G, Taylor MRG, Mestroni L, Coarfa C, Gurha P, Marian AJ.. DNA damage response/TP53 pathway is activated and contributes to the pathogenesis of dilated cardiomyopathy associated with LMNA (Lamin A/C) mutations. Circ Res 2019;124:856–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529. Tandri H, Saranathan M, Rodriguez ER, Martinez C, Bomma C, Nasir K, Rosen B, Lima JA, Calkins H, Bluemke DA.. Noninvasive detection of myocardial fibrosis in arrhythmogenic right ventricular cardiomyopathy using delayed-enhancement magnetic resonance imaging. J Am Coll Cardiol 2005;45:98–103. [DOI] [PubMed] [Google Scholar]
- 530. Lombardi R, Chen SN, Ruggiero A, Gurha P, Czernuszewicz GZ, Willerson JT, Marian AJ.. Cardiac fibro-adipocyte progenitors express desmosome proteins and preferentially differentiate to adipocytes upon deletion of the desmoplakin gene. Circ Res 2016;119:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531. Dubash AD, Kam CY, Aguado BA, Patel DM, Delmar M, Shea LD, Green KJ.. Plakophilin-2 loss promotes TGF-beta1/p38 MAPK-dependent fibrotic gene expression in cardiomyocytes. J Cell Biol 2016;212:425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532. Smith ED, Lakdawala NK, Papoutsidakis N, Aubert G, Mazzanti A, McCanta AC, Agarwal PP, Arscott P, Dellefave-Castillo LM, Vorovich EE, Nutakki K, Wilsbacher LD, Priori SG, Jacoby DL, McNally EM, Helms AS.. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation 2020;141:1872–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533. Hookana E, Junttila MJ, Puurunen VP, Tikkanen JT, Kaikkonen KS, Kortelainen ML, Myerburg RJ, Huikuri HV.. Causes of nonischemic sudden cardiac death in the current era. Heart Rhythm 2011;8:1570–1575. [DOI] [PubMed] [Google Scholar]
- 534. Junttila MJ, Holmstrom L, Pylkas K, Mantere T, Kaikkonen K, Porvari K, Kortelainen ML, Pakanen L, Kerkela R, Myerburg RJ, Huikuri HV.. Primary myocardial fibrosis as an alternative phenotype pathway of inherited cardiac structural disorders. Circulation 2018;137:2716–2726. [DOI] [PubMed] [Google Scholar]
- 535. Schwartzkopff B, Frenzel H, Diekerhoff J, Betz P, Flasshove M, Schulte HD, Mundhenke M, Motz W, Strauer BE.. Morphometric investigation of human myocardium in arterial hypertension and valvular aortic stenosis. Eur Heart J 1992;13 Suppl D:17–23. [DOI] [PubMed] [Google Scholar]
- 536. van Hoeven KH, Factor SM.. A comparison of the pathological spectrum of hypertensive, diabetic, and hypertensive-diabetic heart disease. Circulation 1990;82:848–855. [DOI] [PubMed] [Google Scholar]
- 537. Tastet L, Kwiecinski J, Pibarot P, Capoulade R, Everett RJ, Newby DE, Shen M, Guzzetti E, Arsenault M, Bedard E, Larose E, Beaudoin J, Dweck M, Clavel MA.. Sex-related differences in the extent of myocardial fibrosis in patients with aortic valve stenosis. JACC Cardiovasc Imaging 2020;13:699–711. [DOI] [PubMed] [Google Scholar]
- 538. Musa TA, Treibel TA, Vassiliou VS, Captur G, Singh A, Chin C, Dobson LE, Pica S, Loudon M, Malley T, Rigolli M, Foley JRJ, Bijsterveld P, Law GR, Dweck MR, Myerson SG, McCann GP, Prasad SK, Moon JC, Greenwood JP.. Myocardial scar and mortality in severe aortic stenosis. Circulation 2018;138:1935–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539. Singh A, Musa TA, Treibel TA, Vassiliou VS, Captur G, Chin C, Dobson LE, Pica S, Loudon M, Malley T, Rigolli M, Foley JRJ, Bijsterveld P, Law GR, Dweck MR, Myerson SG, Prasad SK, Moon JC, Greenwood JP, McCann GP.. Sex differences in left ventricular remodelling, myocardial fibrosis and mortality after aortic valve replacement. Heart 2019;105:1818–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540. Herrmann S, Fries B, Salinger T, Liu D, Hu K, Gensler D, Strotmann J, Christa M, Beer M, Gattenlohner S, Stork S, Voelker W, Bening C, Lorenz K, Leyh R, Frantz S, Ertl G, Weidemann F, Nordbeck P.. Myocardial fibrosis predicts 10-year survival in patients undergoing aortic valve replacement. Circ Cardiovasc Imaging 2018;11:e007131. [DOI] [PubMed] [Google Scholar]
- 541. Hutchins GM, Kuhajda FP, Moore GW.. Myocardial injury in patients with aortic stenosis. Am J Cardiovasc Pathol 1987;1:31–37. [PubMed] [Google Scholar]
- 542. Xia Y, Lee K, Li N, Corbett D, Mendoza L, Frangogiannis NG.. Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem Cell Biol 2009;131:471–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543. Conrad CH, Brooks WW, Hayes JA, Sen S, Robinson KG, Bing OH.. Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation 1995;91:161–170. [DOI] [PubMed] [Google Scholar]
- 544. Brilla CG, Maisch B.. Regulation of the structural remodelling of the myocardium: from hypertrophy to heart failure. Eur Heart J 1994;15 Suppl D:45–52. [DOI] [PubMed] [Google Scholar]
- 545. Schellings MW, Pinto YM, Heymans S.. Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc Res 2004;64:24–31. [DOI] [PubMed] [Google Scholar]
- 546. Saucerman JJ, Tan PM, Buchholz KS, McCulloch AD, Omens JH.. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat Rev Cardiol 2019;16:361–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547. Salvador AM, Nevers T, Velazquez F, Aronovitz M, Wang B, Abadia Molina A, Jaffe IZ, Karas RH, Blanton RM, Alcaide P.. Intercellular adhesion molecule 1 regulates left ventricular leukocyte infiltration, cardiac remodeling, and function in pressure overload-induced heart failure. J Am Heart Assoc 2016;5:e003126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548. Tokuda K, Kai H, Kuwahara F, Yasukawa H, Tahara N, Kudo H, Takemiya K, Koga M, Yamamoto T, Imaizumi T.. Pressure-independent effects of angiotensin II on hypertensive myocardial fibrosis. Hypertension 2004;43:499–503. [DOI] [PubMed] [Google Scholar]
- 549. Lee JKT, Franzone A, Lanz J, Siontis GCM, Stortecky S, Grani C, Roost E, Windecker S, Pilgrim T.. Early detection of subclinical myocardial damage in chronic aortic regurgitation and strategies for timely treatment of asymptomatic patients. Circulation 2018;137:184–196. [DOI] [PubMed] [Google Scholar]
- 550. Mannacio V, Guadagno E, Mannacio L, Cervasio M, Antignano A, Mottola M, Gagliardi C, Vosa C.. Comparison of left ventricular myocardial structure and function in patients with aortic stenosis and those with pure aortic regurgitation. Cardiology 2015;132:111–118. [DOI] [PubMed] [Google Scholar]
- 551. Villari B, Sossalla S, Ciampi Q, Petruzziello B, Turina J, Schneider J, Turina M, Hess OM.. Persistent diastolic dysfunction late after valve replacement in severe aortic regurgitation. Circulation 2009;120:2386–2392. [DOI] [PubMed] [Google Scholar]
- 552. Zheng J, Chen Y, Pat B, Dell'italia LA, Tillson M, Dillon AR, Powell PC, Shi K, Shah N, Denney T, Husain A, Dell'Italia LJ.. Microarray identifies extensive downregulation of noncollagen extracellular matrix and profibrotic growth factor genes in chronic isolated mitral regurgitation in the dog. Circulation 2009;119:2086–2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553. Nagatomo Y, Carabello BA, Coker ML, McDermott PJ, Nemoto S, Hamawaki M, Spinale FG.. Differential effects of pressure or volume overload on myocardial MMP levels and inhibitory control. Am J Physiol Heart Circ Physiol 2000;278:H151–H161. [DOI] [PubMed] [Google Scholar]
- 554. Borer JS, Truter S, Herrold EM, Falcone DJ, Pena M, Carter JN, Dumlao TF, Lee JA, Supino PG.. Myocardial fibrosis in chronic aortic regurgitation: molecular and cellular responses to volume overload. Circulation 2002;105:1837–1842. [DOI] [PubMed] [Google Scholar]
- 555. Li JH, Huang XR, Zhu HJ, Johnson R, Lan HY.. Role of TGF-beta signaling in extracellular matrix production under high glucose conditions. Kidney Int 2003;63:2010–2019. [DOI] [PubMed] [Google Scholar]
- 556. Zhao J, Randive R, Stewart JA.. Molecular mechanisms of AGE/RAGE-mediated fibrosis in the diabetic heart. World J Diabetes 2014;5:860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557. Russo I, Frangogiannis NG.. Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol 2016;90:84–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558. Cavalera M, Wang J, Frangogiannis NG.. Obesity, metabolic dysfunction, and cardiac fibrosis: pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Transl Res 2014;164:323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559. Paulus WJ, Tschöpe C.. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 2013;62:263–271. [DOI] [PubMed] [Google Scholar]
- 560. Packer M. Derangements in adrenergic-adipokine signalling establish a neurohormonal basis for obesity-related heart failure with a preserved ejection fraction. Eur J Heart Fail 2018;20:873–878. [DOI] [PubMed] [Google Scholar]
- 561. Kong P, Gonzalez-Quesada C, Li N, Cavalera M, Lee DW, Frangogiannis NG.. Thrombospondin-1 regulates adiposity and metabolic dysfunction in diet-induced obesity enhancing adipose inflammation and stimulating adipocyte proliferation. Am J Physiol Endocrinol Metab 2013;305:E439–E450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562. Regan TJ, Lyons MM, Ahmed SS, Levinson GE, Oldewurtel HA, Ahmad MR, Haider B.. Evidence for cardiomyopathy in familial diabetes mellitus. J Clin Invest 1977;60:884–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563. Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan RS.. Obesity and the risk of heart failure. N Engl J Med 2002;347:305–313. [DOI] [PubMed] [Google Scholar]
- 564. Packer M, Kitzman DW.. Obesity-related heart failure with a preserved ejection fraction: the mechanistic rationale for combining inhibitors of aldosterone, neprilysin, and sodium-glucose cotransporter-2. JACC Heart Fail 2018;6:633–639. [DOI] [PubMed] [Google Scholar]
- 565. Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA.. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation 2017;136:6–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566. Buckley LF, Canada JM, Del Buono MG, Carbone S, Trankle CR, Billingsley H, Kadariya D, Arena R, Van Tassell BW, Abbate A.. Low NT-proBNP levels in overweight and obese patients do not rule out a diagnosis of heart failure with preserved ejection fraction. ESC Heart Fail 2018;5:372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567. Biernacka A, Frangogiannis NG.. Aging and cardiac fibrosis. Aging Dis 2011;2:158–173. [PMC free article] [PubMed] [Google Scholar]
- 568. Quijada P, Misra A, Velasquez LS, Burke RM, Lighthouse JK, Mickelsen DM, Dirkx RA Jr, Small EM.. Pre-existing fibroblasts of epicardial origin are the primary source of pathological fibrosis in cardiac ischemia and aging. J Mol Cell Cardiol 2019;129:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 569. Chen W, Frangogiannis NG.. The role of inflammatory and fibrogenic pathways in heart failure associated with aging. Heart Fail Rev 2010;15:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570. Trial J, Cieslik KA.. Changes in cardiac resident fibroblast physiology and phenotype in aging. Am J Physiol Heart Circ Physiol 2018;315:H745–H755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571. Vidal R, Wagner JUG, Braeuning C, Fischer C, Patrick R, Tombor L, Muhly-Reinholz M, John D, Kliem M, Conrad T, Guimaraes-Camboa N, Harvey R, Dimmeler S, Sauer S.. Transcriptional heterogeneity of fibroblasts is a hallmark of the aging heart. JCI Insight 2019;4: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 572. Roh JD, Houstis N, Yu A, Chang B, Yeri A, Li H, Hobson R, Lerchenmuller C, Vujic A, Chaudhari V, Damilano F, Platt C, Zlotoff D, Lee RT, Shah R, Jerosch-Herold M, Rosenzweig A.. Exercise training reverses cardiac aging phenotypes associated with heart failure with preserved ejection fraction in male mice. Aging Cell 2020;19:e13159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 573. Bujak M, Kweon HJ, Chatila K, Li N, Taffet G, Frangogiannis NG.. Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol 2008;51:1384–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574. Khan R, Sheppard R.. Fibrosis in heart disease: understanding the role of transforming growth factor-beta in cardiomyopathy, valvular disease and arrhythmia. Immunology 2006;118:10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575. Nguyen MN, Kiriazis H, Gao XM, Du XJ.. Cardiac fibrosis and arrhythmogenesis. Compr Physiol 2017;7:1009–1049. [DOI] [PubMed] [Google Scholar]
- 576. Disertori M, Mase M, Ravelli F.. Myocardial fibrosis predicts ventricular tachyarrhythmias. Trends Cardiovasc Med 2017;27:363–372. [DOI] [PubMed] [Google Scholar]
- 577. Disertori M, Rigoni M, Pace N, Casolo G, Mase M, Gonzini L, Lucci D, Nollo G, Ravelli F.. Myocardial fibrosis assessment by LGE is a powerful predictor of ventricular tachyarrhythmias in ischemic and nonischemic LV dysfunction: a meta-analysis. JACC Cardiovasc Imaging 2016;9:1046–1055. [DOI] [PubMed] [Google Scholar]
- 578. Mc LA, Ellims AH, Prabhu S, Voskoboinik A, Iles LM, Hare JL, Kaye DM, Macciocca I, Mariani JA, Kalman JM, Taylor AJ, Kistler PM.. Diffuse ventricular fibrosis on cardiac magnetic resonance imaging associates with ventricular tachycardia in patients with hypertrophic cardiomyopathy. J Cardiovasc Electrophysiol 2016;27:571–580. [DOI] [PubMed] [Google Scholar]
- 579. Kitkungvan D, Nabi F, Kim RJ, Bonow RO, Khan MA, Xu J, Little SH, Quinones MA, Lawrie GM, Zoghbi WA, Shah DJ.. Myocardial fibrosis in patients with primary mitral regurgitation with and without prolapse. J Am Coll Cardiol 2018;72:823–834. [DOI] [PubMed] [Google Scholar]
- 580. Ekstrom K, Lehtonen J, Kandolin R, Raisanen-Sokolowski A, Salmenkivi K, Kupari M.. Incidence, risk factors, and outcome of life-threatening ventricular arrhythmias in giant cell myocarditis. Circ Arrhythm Electrophysiol 2016;9:e004559. [DOI] [PubMed] [Google Scholar]
- 581. Rubart M, Tao W, Lu XL, Conway SJ, Reuter SP, Lin SF, Soonpaa MH.. Electrical coupling between ventricular myocytes and myofibroblasts in the infarcted mouse heart. Cardiovasc Res 2018;114:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582. Nguyen TP, Xie Y, Garfinkel A, Qu Z, Weiss JN.. Arrhythmogenic consequences of myofibroblast-myocyte coupling. Cardiovasc Res 2012;93:242–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583. Salvarani N, Maguy A, De Simone SA, Miragoli M, Jousset F, Rohr S.. TGF-beta1 (transforming growth factor-beta1) plays a pivotal role in cardiac myofibroblast arrhythmogenicity. Circ Arrhythm Electrophysiol 2017;10:e004567. [DOI] [PubMed] [Google Scholar]
- 584. Rosker C, Salvarani N, Schmutz S, Grand T, Rohr S.. S. Abolishing myofibroblast arrhythmogeneicity by pharmacological ablation of alpha-smooth muscle actin containing stress fibers. Circ Res 2011;109:1120–1131. [DOI] [PubMed] [Google Scholar]
- 585. Roell W, Klein AM, Breitbach M, Becker TS, Parikh A, Lee J, Zimmermann K, Reining S, Gabris B, Ottersbach A, Doran R, Engelbrecht B, Schiffer M, Kimura K, Freitag P, Carls E, Geisen C, Duerr GD, Sasse P, Welz A, Pfeifer A, Salama G, Kotlikoff M, Fleischmann BK.. Overexpression of Cx43 in cells of the myocardial scar: correction of post-infarct arrhythmias through heterotypic cell-cell coupling. Sci Rep 2018;8:7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586. Andersen S, Nielsen-Kudsk JE, Vonk Noordegraaf A, de Man FS.. Right ventricular fibrosis. Circulation 2019;139:269–285. [DOI] [PubMed] [Google Scholar]
- 587. Frangogiannis NG. Fibroblasts and the extracellular matrix in right ventricular disease. Cardiovasc Res 2017;113:1453–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588. Hamilton RM. Arrhythmogenic right ventricular cardiomyopathy. Pacing Clin Electrophysiol 2009;32 Suppl 2:S44–S51. [DOI] [PubMed] [Google Scholar]
- 589. Turrini P, Angelini A, Thiene G, Buja G, Daliento L, Rizzoli G, Nava A.. Late potentials and ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol 1999;83:1214–1219. [DOI] [PubMed] [Google Scholar]
- 590. Thomas L, Abhayaratna WP.. Left atrial reverse remodeling: mechanisms, evaluation, and clinical significance. JACC Cardiovasc Imaging 2017;10:65–77. [DOI] [PubMed] [Google Scholar]
- 591. Tanaka K, Zlochiver S, Vikstrom KL, Yamazaki M, Moreno J, Klos M, Zaitsev AV, Vaidyanathan R, Auerbach DS, Landas S, Guiraudon G, Jalife J, Berenfeld O, Kalifa J.. Spatial distribution of fibrosis governs fibrillation wave dynamics in the posterior left atrium during heart failure. Circ Res 2007;101:839–847. [DOI] [PubMed] [Google Scholar]
- 592. Nattel S. How does fibrosis promote atrial fibrillation persistence: in silico findings, clinical observations, and experimental data. Cardiovasc Res 2016;110:295–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593. Hanif W, Alex L, Su Y, Shinde AV, Russo I, Li N, Frangogiannis NG.. Left atrial remodeling, hypertrophy, and fibrosis in mouse models of heart failure. Cardiovasc Pathol 2017;30:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 594. Dzeshka MS, Lip GY, Snezhitskiy V, Shantsila E.. Cardiac fibrosis in patients with atrial fibrillation: mechanisms and clinical implications. J Am Coll Cardiol 2015;66:943–959. [DOI] [PubMed] [Google Scholar]
- 595. Seitz J, Horvilleur J, Lacotte J, O H-Ici D, Mouhoub Y, Maltret A, Salerno F, Mylotte D, Monchi M, Garot J.. Correlation between AF substrate ablation difficulty and left atrial fibrosis quantified by delayed-enhancement cardiac magnetic resonance. Pacing Clin Electrophysiol 2011;34:1267–1277. [DOI] [PubMed] [Google Scholar]
- 596. King JB, Azadani PN, Suksaranjit P, Bress AP, Witt DM, Han FT, Chelu MG, Silver MA, Biskupiak J, Wilson BD, Morris AK, Kholmovski EG, Marrouche N.. Left atrial fibrosis and risk of cerebrovascular and cardiovascular events in patients with atrial fibrillation. J Am Coll Cardiol 2017;70:1311–1321. [DOI] [PubMed] [Google Scholar]
- 597. Burashnikov A, Antzelevitch C.. Is extensive atrial fibrosis in the setting of heart failure associated with a reduced atrial fibrillation burden? Pacing Clin Electrophysiol 2018;41:1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598. Brilla CG, Matsubara L, Weber KT.. Advanced hypertensive heart disease in spontaneously hypertensive rats. Lisinopril-mediated regression of myocardial fibrosis. Hypertension 1996;28:269–275. [DOI] [PubMed] [Google Scholar]
- 599. Brilla CG, Funck RC, Rupp H.. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 2000;102:1388–1393. [DOI] [PubMed] [Google Scholar]
- 600. Everett RJ, Tastet L, Clavel MA, Chin CWL, Capoulade R, Vassiliou VS, Kwiecinski J, Gomez M, van Beek EJR, White AC, Prasad SK, Larose E, Tuck C, Semple S, Newby DE, Pibarot P, Dweck MR.. Progression of hypertrophy and myocardial fibrosis in aortic stenosis: a multicenter cardiac magnetic resonance study. Circ Cardiovasc Imaging 2018;11:e007451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601. Treibel TA, Kozor R, Schofield R, Benedetti G, Fontana M, Bhuva AN, Sheikh A, Lopez B, Gonzalez A, Manisty C, Lloyd G, Kellman P, Diez J, Moon JC.. Reverse myocardial remodeling following valve replacement in patients with aortic stenosis. J Am Coll Cardiol 2018;71:860–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602. Farris SD, Don C, Helterline D, Costa C, Plummer T, Steffes S, Mahr C, Mokadam NA, Stempien-Otero A.. Cell-specific pathways supporting persistent fibrosis in heart failure. J Am Coll Cardiol 2017;70:344–354. [DOI] [PubMed] [Google Scholar]
- 603. Weidemann F, Herrmann S, StöRk S, Niemann M, Frantz S, Lange V, Beer M, GattenlöHner S, Voelker W, Ertl G, Strotmann JrgM, Impact of myocardial fibrosis in patients with symptomatic severe aortic stenosis. Circulation 2009;120:577–584. [DOI] [PubMed] [Google Scholar]
- 604. Kraft L, Erdenesukh T, Sauter M, Tschope C, Klingel K.. Blocking the IL-1beta signalling pathway prevents chronic viral myocarditis and cardiac remodeling. Basic Res Cardiol 2019;114:11. [DOI] [PubMed] [Google Scholar]
- 605. Bujak M, Frangogiannis NG.. The role of IL-1 in the pathogenesis of heart disease. Arch Immunol Ther Exp 2009;57:165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606. Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, Wang T, Bedi K, Morley MP, Linares Saldana RA, Bolar NA, McDaid K, Assenmacher CA, Smith CL, Wirth D, June CH, Margulies KB, Jain R, Pure E, Albelda SM, Epstein JA.. Targeting cardiac fibrosis with engineered T cells. Nature 2019;573:430–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607. Lopez B, Gonzalez A., Diez J.. Circulating biomarkers of collagen metabolism in cardiac diseases. Circulation 2010;121:1645–1654. [DOI] [PubMed] [Google Scholar]
- 608. Perea RJ, Morales-Ruiz M, Ortiz-Perez JT, Bosch X, Andreu D, Borras R, Acosta J, Penela D, Prat-González S, de Caralt TM, Martínez M, Morales-Romero B, Lasalvia L, Donnelly J, Jiménez W, Mira A, Mont L, Berruezo A.. Utility of galectin-3 in predicting post-infarct remodeling after acute myocardial infarction based on extracellular volume fraction mapping. Int J Cardiol 2016;223:458–464. [DOI] [PubMed] [Google Scholar]
- 609. Hervas A, Ruiz-Sauri A, Gavara J, Monmeneu JV, de Dios E, Rios-Navarro C, Perez-Sole N, Perez I, Monleon D, Morales JM, Minana G, Nunez J, Bonanad C, Diaz A, Vila JM, Chorro FJ, Bodi V.. A multidisciplinary assessment of remote myocardial fibrosis after reperfused myocardial infarction in swine and patients. J Cardiovasc Trans Res 2016;9:321–333. [DOI] [PubMed] [Google Scholar]
- 610. Huang S, Frangogiannis NG.. Anti-inflammatory therapies in myocardial infarction: failures, hopes and challenges. Br J Pharmacol 2018;175:1377–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]