

Summary:
A mechanistically guided approach is developed to predict electrophile compatibility in the palladium/norbornene (Pd/NBE) cooperative catalysis for the ipso/ortho difunctionalization of aryl halides. A key challenge in these reactions is to identify orthogonal electrophile and aryl hali de starting materials that react selectively with different transition metal complexes in separate oxidative addition events in the catalytic cycle. We performed detailed experimental and computational mechanistic studies to identify the catalytically active Pd(II) intermediate and the substrate-dependent mechanisms in reactions with various types of carbon and nitrogen electrophiles. We introduced the concept of electrophile compatibility score (ECS) to rationally select electrophiles based on the orthogonal reactivity of electrophile and aryl halide towards the Pd(0) and Pd(II) complexes. This approach was applied to predict electrophile compatibility in the Pd/NBE cooperative catalysis with a variety of electrophilic coupling partners used in alkylation, arylation, amination, and acylation reactions.
The Bigger Picture:
Developing catalytic methods for novel types of bond formation using readily available starting materials has been a long-term goal in organic synthesis. While traditional cross-coupling reactions take place between a nucleophile and an electrophile, various coupling reactions involving two different electrophiles have recently emerged. These reactions provide promising platforms for synthesizing functionalized molecules, considering the large variety of commercially available carbon and heteroatom electrophiles. However, it is not trivial to identify electrophiles that are compatible with each other, because they must have orthogonal reactivity that allows them to react selectively with different organometallic species in different elementary steps of the catalytic cycle. Here, we describe a mechanistically guided approach to rationally predict electrophiles that meet these criteria and can be potentially used in multi-electrophile coupling reactions.
Graphical Abstract
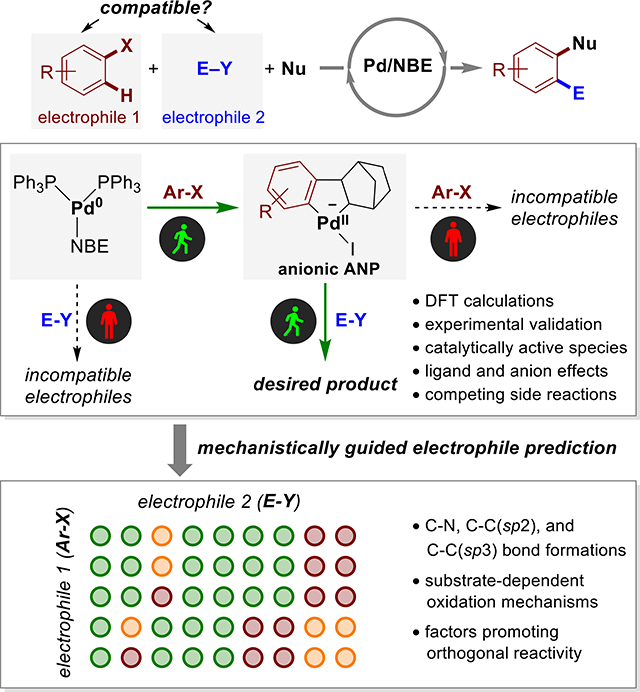
A mechanistically guided approach is developed for predicting effective electrophiles in Pd/NBE cooperative catalysis. Computational and experimental mechanistic studies revealed an anionic aryl-norbornyl-palladacycle (ANP) as the catalytically active species. These insights are used to establish a computational procedure to rapidly predict compatibility between aryl halides and electrophiles by evaluating their orthogonal reactivity in the Pd(0)/(II) and Pd(II)/(IV) oxidative addition steps.
INTRODUCTION
A variety of multicomponent cross-coupling reactions and cross electrophile-electrophile coupling reactions have emerged over the past decade (Scheme 1b–d).1–7 These catalytic transformations often involve more than one electrophilic starting materials, and thus offer unique ways for bond formation that are complementary to traditional cross-coupling reactions between an electrophilic and a nucleophilic coupling partner. The central challenge in developing such reactions is the effective control of chemoselectivity. The electrophiles must react selectively with different transition metal complexes at separate oxidative addition events in the catalytic cycle while not interfering with elementary steps involving other electrophiles (Scheme 1a). To achieve such orthogonal reactivity, it is imperative to utilize new paradigms of reactivity control that go beyond the classical understanding of steric and electronic effects on electrophile reactivity.
Scheme 1.

Multi-electrophile coupling reactions and the orthogonal reactivity of electrophiles
The complexity of rational electrophile selection is exemplified in the palladium/norbornene (Pd/NBE) cooperative catalysis, also known as Catellani-type reactions.6–12 These reactions offer an effective approach for the synthesis of poly-substituted aromatics through selective functionalization of both ortho and ipso positions of aryl halides (Ar‒X) with an electrophile and a nucleophile, respectively (Scheme 1d).13–16 Because the catalytic cycle involves two different oxidation events between Ar‒X and Pd(0) and between the electrophile (E‒Y) and a putative aryl-norbornyl-palladacycle (ANP) species17–22 (Scheme 1e), the choice of compatible electrophiles becomes an intricate process. The electrophile and the aryl halide must have orthogonal reactivities with Pd(0) and Pd(II) species (Scheme 1f): E‒Y should be less reactive than Ar‒X in the oxidative addition with Pd0; while E‒Y must be more reactive than Ar‒X when reacting with ANP. In addition, the reaction of electrophile with ANP to form intermediate III must outcompete several side reactions (e.g. formations of I and II, Scheme 1f). Despite the emergence of Pd/NBE cooperative catalysis as a powerful synthetic strategy, effective electrophiles that are known to meet these criteria are still rather limited. The lack of knowledge in discovering compatible electrophiles has hampered the development of new catalytic reactions with other electrophilic coupling partners and limited the application of Pd/NBE cooperative catalysis in natural product functionalization23 and synthesis of pharmaceutical compounds.24
The rational selection of compatible electrophiles must overcome several practical challenges. First, the crucial step involving the reaction of ANP with the electrophile is still mechanistically ambiguous. Although several ANP analogs have been synthesized using stoichiometric reactions and were found viable of generating the coupling product,17–19 the actual form of the active ANP species under catalytic conditions may be different from the isolated complexes. In particular, phosphine and anionic ligands (e.g. I−)25 may affect the reactivity and lead to completely different chemoselectivity of the ANP species. Second, because a number of structurally distinct electrophiles have been employed in arene ortho alkylation,9,26–30 arylation,31–33 amination,14,34,35 acylation,36–39 and thiolation40–42 reactions (Scheme 1d), it is not clear whether the identity of the electrophile will affect the mechanisms of these reactions, in particular, the key step of reacting with the ANP intermediate. Last but not least, there lacks a practical approach to tune the reactivity of the electrophile according to a given aryl halide starting material to achieve the orthogonal reactivity. Because most known Pd/NBE cooperative catalysis use aryl iodides as starting materials, electrophiles that are compatible with other aryl halides, such aryl bromides14 and triflates, are especially desirable.
We surmised that the rational selection of compatible electrophiles in the Pd/NBE cooperative catalysis and other multi-electrophile coupling reactions can be achieved via a two-step, mechanistically-guided approach. This process would initiate via detailed computational and experimental mechanistic studies to elucidate the exact transition metal complexes involved in reactions with different electrophiles. Then, the electrophile compatibility score between two electrophilic starting materials is calculated based on the activation free energies of the possible reactions between the electrophiles and the catalytically active transition metal complexes (Scheme 1a). In this manuscript, we demonstrate the application of this approach to predict the compatibilities between various electrophiles and aryl halide starting materials in Pd/NBE cooperative catalysis. We expect that this predictive approach can be extended to guide the electrophile design in other multi-electrophile coupling reactions.
RESULTS AND DISCUSSION
Experimental and computational mechanistic studies to identify the catalytically active ANP species
Before exploring the compatibility between aryl halides and various electrophiles in Pd/NBE cooperative catalysis, it is necessary to identify the catalytically active ANP species and to investigate the mechanisms of the reaction between ANP and electrophiles. We initiated our mechanistic investigations by a combined computational and experimental study on the Pd/NBE-catalyzed ortho-amination of aryl iodides using N-benzoyloxyamines as electrophilic coupling partners (Fig. 1a).14,34 The DFT-computed energy profile revealed that a bisphosphine-supported ANP complex (ANP-1) is generated through oxidative addition of aryl iodide (ArX-1) to bisphosphine-ligated Pd(0) species (TS-1),43–46 exo-insertion of NBE (TS-2),47 and concerted metallation-deprotonation (CMD) of the ortho C–H bond (TS-3) (Fig. 1b, see Fig. S3 for an alternative CMD pathway and Fig. S14 for alternative oxidative addition pathways). The relatively low barriers of these three steps indicate the ANP complex formation is facile under the experimental conditions (~100 °C). A stoichiometric reaction of ANP-1 and morpholino benzoate EY-1 gave aryl amination product 5 in 66% yield (Fig. 1c). These computational and experimental results suggest that ANP-1 is a potential intermediate of the catalytic amination reaction, consistent with the previous experimental mechanistic studies from Catellani and Lautens that indicated ANPs can react with other electrophiles, including alkyl halides and aryl halides.17–19 However, the stoichiometric reaction with ANP-1 yielded the C(sp3)–N reductive elimination side product 5b in 28% yield, which was not formed under catalytic conditions. This indicates that the subtle difference of reaction conditions (e.g. the relative concentrations of Pd, PPh3, and anionic ligands) significantly impacts the product selectivity. Because several three- and four-coordinated ANP species supported by phosphine or anionic ligands may exist in equilibrium under the catalytic conditions (Fig. 2a), we surmised that the active ANP complex that reacts with the electrophile under catalytic conditions may not be the bisphosphine-bound ANP-1.25 Therefore, we performed careful computational analysis on the possible ANP complexes formed under the experimental conditions and investigated the reactivity and chemoselectivity of these ANP complexes.
Fig. 1.

Pd/NBE-catalyzed arene C–H amination with N-benzoyloxyamine.
Fig. 2.

Possible mechanisms for the reaction of ANP with morpholino benzoate (EY-1).
Our DFT calculations indicated that at least six ANP complexes could exist in equilibrium because their relative energies are within several kcal/mol (Fig. 2a). We calculated the reaction pathways of these ANP intermediates with electrophile EY-1 via several possible mechanisms,48–50 including the concerted oxidative addition via either a three- or five-membered cyclic transition state,49 the SN2-type oxidative addition,22 and the aromatic electrophilic substitution (SEAr) (Fig. 2b). The SEAr transition state has an extremely high energy barrier (ΔG‡ = 49.9 kcal/mol for the reaction between ANP-6 and EY-1) and thus can be ruled out. Furthermore, the transmetalation mechanism, in which two independent Pd(0)/Pd(II) cycles activate the aryl halide and the electrophile, respectively, followed by transemtalation between the two Pd(II) complexes,51 can also be ruled out because of the high barrier to oxidative addition of EY-1 to Pd(0).52 The different oxidative addition pathways of EY-1 with the neutral ANP complexes (ANP-1 and ANP-2) and the iodide-bound anionic ANP complexes (ANP-3 and ANP-4) are summarized in Fig. 3a and 3b, respectively.53 The reaction pathways with the benzoate anion coordinated anionic ANP species (ANP-5 and ANP-6) are discussed in the Supporting Information because their reactivity trend is similar to those of ANP-3 and ANP-4.
Fig. 3.

Possible pathways of the reaction of morpholino benzoate (EY-1) with (a) neutral ANP species and (b) anionic ANP species. All energies are with respect to ANP-1.
Due to the lack of available coordination sites in the four-coordinated ANP complex ANP-1, either one or both of the PPh3 ligands on ANP-1 have to be replaced before reacting with the electrophile. Starting from the three-coordinate neutral ANP complex ANP-2 (Fig. 3a), the five-centered oxidative addition (OA) transition state (TS-4) and the SN2-type OA transition state (TS-6) require similar activation free energies.54 An alternative, slightly more favorable OA pathway involves the ligand exchange to replace the PPh3 ligand in ANP-2 with EY-1 to form a square-planar complex 9 with both the N and the carbonyl O atoms of EY-1 binding to the Pd. From 9, oxidative addition occurs through a five-membered cyclic transition state TS-7. These oxidative addition pathways lead to Pd(IV) intermediates 10 and 6, which then undergo facile C(sp2)−N reductive elimination via TS-8 and TS-5, respectively, to form alkyl Pd(II) complexes 11 and 7. The final steps in the catalytic cycle, β-C elimination, and hydride transfer, both require lower barriers than the oxidative addition (ΔG‡ = 19.5 and 22.3 kcal/mol, respectively. See Fig. S12).
The reaction of the four-coordinated anionic complex ANP-3 with EY-1 occurs via an SN2-type OA transition state (TS-9) with a relatively high barrier of 31.1 kcal/mol (Fig. 3b). On the other hand, the three-coordinated anionic ANP complex ANP-4 is much more reactive. The SN2-type transition state TS-11 has an activation free energy of 21.9 kcal/mol with respect to ANP-1, which is significantly lower than all other competing OA processes.55 Similar to the reaction pathways with neutral ANP complexes, the subsequent steps, including C(sp2)–N reductive elimination (TS-12), β-carbon elimination, and hydride transfer are all relatively facile (see Fig. S12).
Taken together, the DFT calculations revealed the catalytically active species in the amination is an anionic ANP complex ANP-4. Its reactivity is fundamentally different from that of the neutral ANP complex ANP-1, which was isolated from stoichiometric reactions. The anionic ANP not only reacts faster with the N-benzoyloxyamine electrophile but also favors a different, SN2-type OA mechanism. Natural population analysis (NPA) charge calculations indicated that this unique reactivity is due to the enhanced charge transfer from the anionic ANP complex to the electrophile in the SN2-type OA transition state (0.70e in TS-11, see Fig. 3b). The charge transfer promotes the reaction by facilitating the benzoate anion dissociation via the heterolytic cleavage of the N–O bond.56 On the other hand, the SN2-type OA with the neutral ANP complex has a much smaller amount of charge transfer (0.33e in TS-6, see Fig. S19) and thus requires a much higher barrier.
Experimental and computational validation of anionic ANP as the catalytically active species
Identifying the catalytically active species involved in reactions with electrophiles is a critical step in predicting the electrophile compatibility score. To confirm the computational prediction that the anionic ANP complex is catalytically active, we performed a series of experimental and computational validations. Because it is known experimentally that the reaction between the active ANP species and the N-benzoyloxyamine electrophile will suppress two undesired reaction pathways (Scheme 1f), the C–C reductive elimination from ANP8,26 and the C(sp3)–E reductive elimination from the Pd(IV) oxidative addition intermediate, we then investigated the effects of anionic ligands (e.g. I−, OBz−) on these competing pathways (Fig. 4). Our DFT calculations indicated the C–C reductive elimination from the neutral ANP-1 requires a relatively low activation free energy of 20.2 kcal/mol, which is 7.6 kcal/mol lower than the reaction of the same ANP complex with EY-1 (via TS-7). The low barrier to the C–C reductive elimination suggests that ANP-1 cannot be the dominant form of ANP in an effective Pd/NBE catalysis system. On the other hand, the anionic ANP complex ANP-3 is predicted to be much less susceptible to C–C reductive elimination.57 Because the C–C reductive elimination from ANP-3 requires a higher barrier than the reaction with EY-1 (via TS-11, Fig. 3b), the undesired formation of benzocyclobutene derivative would be disfavored if the anionic ANP species, rather than the neutral ANP, dominates. These computational findings are corroborated by control experiments (Fig. 4b) that suggest ANP-1 is converted to the C–C reductive elimination product 18 in 98% yield in 50 minutes at 60 °C, which is at a lower temperature than those of typical Pd/NBE-catalyzed reactions. This result confirmed that the neutral ANP-1 cannot be the catalytically active species. When 110 mol% tetrabutylammonium benzoate (TBAOBz) or tetrabutylammonium iodide (TBAI) was added to the same reaction, the yield of 18 decreased dramatically to 16% and 25%, respectively, and a large amount of ANP-1 was recovered, indicating the presence of benzoate or iodide anions under catalytic conditions may prevent the formation of 18 by converting ANP-1 to anionic ANP species. On the other hand, the use of cesium carbonate had a negligible effect on inhibiting the C–C reductive elimination, potentially due to its low solubility.58
Fig. 4.

Anion effects on the C–C bond reductive elimination and the pathway selectivity of C–N reductive elimination. All activation free energies are in kcal/mol with respect to the corresponding intermediate before the transition state.
Next, we investigated the effects of anionic ligands on the regioselectivity of the C–N reductive elimination from the Pd(IV) oxidative addition intermediate. In the stoichiometric reaction of ANP-1 with EY-1, products 5 and 5b were formed in a 2:1 ratio (Fig. 1c) and the structure of the aryl Pd(II) intermediate leading to the undesired product 5b was confirmed by X-ray crystallography (Fig. 4c). However, the C(sp3)–N reductive elimination product 5b was not observed under catalytic conditions (Fig. 1). We surmised that under the catalytic conditions with higher concentrations of anions (e.g. I−, OBz−), anionic ANP complexes are the active species reacting with the N-benzoyloxyamine electrophile to form an anionic Pd(IV) intermediate (IV, L = I−, Fig. 4c). By contrast, under the stoichiometric conditions, neutral Pd(IV) species (IV, L = PPh3) is formed. Our computational results revealed that the C(sp2)–N and C(sp3)–N reductive elimination transition states from the neutral IV (L = PPh3) have comparable activation free energies, while the C(sp2)–N reductive elimination is favored by 7.5 kcal/mol from the anionic IV (L = I−). These experimental and computational studies provided additional support to the hypothesis that the in-situ formed anionic ANP complex is the catalytically active species, because it can react efficiently with the N-benzoyloxyamine electrophile and suppress both the C–C reductive elimination from ANP and the C(sp3)–N reductive elimination from the Pd(IV) intermediate.
Compatibility score for evaluating the orthogonal reactivity of electrophiles and aryl halides
After identifying the catalytically active Pd(0) and Pd(II) species (Pd0 and ANP-4, respectively), we are now able to evaluate the compatibility between an aryl halide starting material and an electrophile using the electrophile compatibility score. The electrophile compatibility score is calculated based on the activation barriers of four competing oxidative addition events in the catalytic system: ΔG‡(Pd0+ArX), ΔG‡(Pd0+EY), ΔG‡(ANP+ArX), and ΔG‡(ANP+EY) (Scheme 2). A compatible system requires that Pd0 reacts faster with ArX than with EY (ΔG‡(Pd0+EY) − ΔG‡(Pd0+ArX) > 0) and ANP reacts faster with EY than with ArX (ΔG‡(ANP+ArX) − ΔG‡(ANP+EY) > 0). Therefore, a positive compatibility score indicates the electrophile EY meets both criteria and thus is compatible with ArX in Pd/NBE catalysis, and a negative score indicates the electrophile is incompatible because side reactions would outcompete the desired reaction pathways. For example, the computed electrophile compatibility score between ArX-1 and EY-1 is 3.3 kcal/mol, indicating these two starting materials are highly compatible, which is in agreement with the effective reactions involving these two compounds in the ortho-amination reactions.
Scheme 2.

Electrophile compatibility score to predict the orthogonal reactivity of aryl halide and electrophile
We used this procedure to calculate the compatibility of nine representative electrophiles (EY-1~EY-9) when reacting with ArX-1 (Fig. 5). The computed electrophile compatibility scores (ECS) are in good agreement with previous experimental results: EY-1~EY-6 have positive compatibility scores with ArX-1 and have been successfully employed in Catellani-type amination,34 alkylation,26 arylation,31 and acylation36,38 reactions with ortho-alkyliodobenzenes;59 on the other hand, the low yield in the reaction of EY-8 with 2-iodotoluene31 can be attributed to their low compatibility (ECS = −4.1 kcal/mol). In these reactions, the electrophile compatibility is determined in the second OA step, because all electrophiles investigated are less reactive than ArX-1 when reacting with Pd0, indicating that they will not interfere with the first OA step between ArX-1 and Pd0. In the second OA step, EY-1~EY-7 have comparable or lower barriers than ArX-1, indicating they can react effectively with ANP in the presence of the aryl iodide, while EY-8 and EY-9 are both incompatible because of the high barriers (ΔG‡(ANP+EY) = 25.0 and 27.8 kcal/mol, respectively) when reacting with ANP-4.
Fig. 5.

Computational predictions of compatibility of electrophiles (EY) in the Pd/NBE-catalyzed reactions with ortho-ethyl iodobenzene (ArX-1). a Ar1 = 2,6-dimethylphenyl. b All energies are in kcal/mol with respect to Pd0 or ANP-4. c Electrophile compatibility score between each electrophile and ArX-1. d Experimental yields from refs. 26, 38, 34, 31, 39 were from reactions with 2-iodotoluene unless otherwise noted. e Experimental yield was from the reaction with 2-bromoanisole.
To validate whether the compatibility scores can be used to identify effective electrophiles in reactions with other aryl halides, we computed the compatibility of EYs with 2-bromoanisole (ArX-4) and compared with previous experimental results (Fig. 6). Because of the lower reactivity of the aryl bromide when reacting with ANP (ΔG‡(ANP+ArX) = 27.9 kcal/mol), the four electrophiles all react faster with ANP than ArX-4. Therefore, the electrophile compatibility in these reactions are determined in the first OA step, where the electrophiles need to be less reactive with Pd(0) than ArX-4. The computed ECS values are in good agreement with experiment (Fig. 6)14: n-butyliodide (EY-2) is incompatible with ArX-4 because it reacts faster with Pd(0) than ArX-4 and electrophiles EY-7, EY-1, and EY-10 are all compatible because they react slower with Pd(0) than ArX-4. The above computational predictions highlighted the importance of considering both criteria, ΔΔG‡(Pd0) and ΔΔG‡(ANP), when evaluating the electrophile compatibility.
Fig. 6.

Computationally predicted electrophile compatibility scores (ECS) of electrophiles with 2-bromoanisole (ArX-4). a All energies are in kcal/mol with respect to Pd0 or ANP-4. b Electrophile compatibility score between each electrophile and ArX-4. c Experimental yields from ref. 14.
Next, we further expanded the computational predictions to all nine electrophiles in reactions with five different aryl halides (Fig. 7). The computed electrophile compatibility scores not only provided a straightforward approach to rapidly predict compatible reactions but also revealed mechanistic insights that may be used in future electrophile design. Several factors leading to compatible and incompatible systems (indicated as C-1~C-3 and I-1~I-2 in Fig. 7) can be generalized.60 First, electrophiles with an sp3-hybridized electrophilic atom (e.g. N-benzyloxyamine EY-1, alkyl iodide EY-2, and alkyl bromide EY-7) are privileged coupling partners because of their high reactivity with the negatively charged ANP via the SN2-type OA transition states61–64 (see “C-1”). As discussed above, these reactions are promoted by the charge transfer from the anionic ANP to the electrophile which facilitates leaving group dissociation. Second, anhydrides (e.g. EY-3 and EY-6. See “C-2”) are another type of effective electrophiles in Pd/NBE catalyzed arene acylation and alkoxycarbonylation reactions36–39 because they react more effectively with ANP via a five-centered OA transition state.20 The five-centered OA is more favorable in the Pd(II) to Pd(IV) oxidation than in the Pd(0) to Pd(II) oxidation because of the chelation of the anhydride carbonyl group with the higher valent Pd.65,66 On the other hand, carbonyl coordination with Pd(PPh3)2 is disfavored because the Pd(0) is more electron-rich and the large P–Pd–P angle prevents the more sterically encumbered five-centered transition state.67 A similar chelation effect promotes the oxidative addition of EY-4 to ANP, in which the ortho-ester group coordinates to the Pd(II) center (see “C-2”, Fig. 7),68 and thus makes EY-4 an effective coupling partner in arylation (Fig. 5).31 Third, electron-deficient aryl bromides (e.g. EY-5) are effective electrophiles in arylation reactions (see “C-3”) because they have relatively low activation energies in the OA with ANP (see Fig. S22 for details).69 These reactions are promoted by the d(Pd)→π*(Ar-Br) orbital interactions between the negatively charged ANP and the electron-deficient aryl bromide.70,71 On the other hand, aryl bromides EY-8 and EY-9 are not effective electrophiles (see “I-2”)31 because of their low reactivity towards ANP resulting from steric effects72 of the o-CF3 group (ΔG‡ = 25.0 kcal/mol) and electron-donating effects of the p-OMe (ΔG‡ = 27.8 kcal/mol), respectively. The above examples indicated that the substrate-dependent mechanisms of the OA to ANP (factors “C-1” and “C-2”) and steric and electronic effects of the electrophile (factors “C-3” and “I-2”) can both affect the compatibility of electrophiles in the Pd/NBE cooperative catalysis.
Fig. 7.

Predicted compatibility scores of electrophiles in reactions with different aryl halides and factors determining electrophile compatibility.
Comparisons of reactions with aryl iodide (ArX-1 ~ ArX-3) and aryl bromide (ArX-4 and ArX-5) starting materials revealed the identity of halides as another key factor determining electrophile compatibility. Based on the computed ECS values, the amination and acylation reactions with EY-1, EY-3, and EY-6 are expected to proceed with either aryl iodides or aryl bromides.14,34 However, the alkylation using alkyl iodide (EY-2) and arylation using aryl bromides EY-4 and EY-5 as electrophiles require the use of more reactive aryl iodides as the Ar-X substrate. Due to the lower reactivity of aryl bromides with Pd(0), the Pd(0) complex would react preferentially with the electrophile if aryl bromides (ArX-4 or ArX-5) were used in these reactions (labeled as “I-1” in Fig. 7). This prediction is consistent with the experimental observations that the reaction of ArX-4 with EY-2 did not form the arene alkylation product (Fig. 6).14 It is noteworthy that the computational results predicted that n-butylbromide EY-7 is not an effective electrophile in the alkylation of aryl iodides (ArX-1 ~ ArX-3) because of the low reactivity of EY-7 with ANP, while these reactions gave high yields experimentally.73 These results suggest that the alkyl bromide electrophiles were converted to the more reactive alkyl iodides in-situ via halogen substitution74 prior to the oxidative addition to ANP.
CONCLUSIONS
We describe a mechanistically driven approach to predict compatible electrophiles in the Pd/NBE cooperative catalysis. This approach uses the electrophile compatibility score (ECS) between two electrophilic starting materials to predict whether a reaction may proceed effectively. In the Pd/NBE cooperative catalysis, the Pd(0) catalyst needs to react selectively with the aryl halide starting material in the presence of the electrophile, while the Pd(II) ANP intermediate needs to react faster with the electrophile than with the aryl halide. The electrophile compatibility score evaluates whether such orthogonal reactivity can be achieved in the two oxidative addition events with the Pd(0) and Pd(II) species, respectively. We have demonstrated the application of this approach to predict whether an electrophile would be effective in a variety of Pd/NBE-catalyzed aryl halide functionalizations, including the amination, arylation, alkylation, and acylation of different aryl halide starting materials. We expect that this method can be extended to facilitate the rational design of electrophiles in other multi-electrophile coupling reactions.
The key to this mechanistically driven approach is to identify the organometallic species that react with electrophiles at different stages of the catalytic cycle. Through a combined computational and experimental mechanistic study, we revealed that, in Pd/NBE cooperative catalysis, the oxidative addition with electrophiles takes place with an anionic ANP intermediate with an iodide or a benzoate ligand bound to the Pd, rather than the previously proposed neutral ANP complexes. The anionic ANP is not only more reactive in the oxidative addition of electrophile but also suppresses two undesired side reaction pathways, the C–C reductive elimination from ANP and the C(sp3)–N reductive elimination from the Pd(IV) intermediate.
Based on these mechanistic insights, the compatibility scores of 45 pairs of aryl halide and electrophile starting materials were computed using their activation energies with Pd(0) and the anionic ANP species. The predicted electrophile compatibility scores are in good agreement with experiments for those have been previously investigated. This mechanistically driven approach also uncovered several general factors affecting the electrophile compatibility. Coupling partners with an sp3-hybridized electrophilic center and anhydrides are privileged electrophiles because they react with ANP via substrate-dependent mechanisms (SN2-type OA and five-centered OA, respectively) that are often more favorable than the three-centered OA of aryl halide to ANP. In addition, the compatibility of the electrophiles may be altered electronically. For example, electron-deficient aryl bromides are good electrophiles in the arylation of aryl iodides because the anionic ANP reacts efficiently with electron-deficient aryl bromides, which are promoted by d→π* interactions. Moreover, electrophiles that may chelate with the Pd(II) during ANP oxidation, such as aryl bromides with an ortho-ester group, are also good coupling partners.
Computational methods
Geometry optimizations and single-point energy calculations were carried out using Gaussian 09.75 All DFT calculations were performed on Pitt CRC, XSEDE,76 and TACC Frontera supercomputers. The geometries of intermediates and transition states were optimized using the B3LYP functional77,78 with a mixed basis set of LANL2DZ for Pd, Cs, I and 6–31G(d) for other atoms in the gas phase. Vibrational frequency calculations were performed for all stationary points to confirm if each optimized structure is a local minimum or a transition state structure. Solvation energy corrections were calculated in toluene solvent with the SMD continuum solvation model.79 The M06 functional80,81 with a mixed basis set of SDD for Pd, Cs, and I and 6–311+G(d,p) for other atoms were used for the single-point energy calculations in toluene. The stability of the DFT wavefunction was tested for all computed structures. Most of the intermediates and transition states are closed-shell singlet except for several oxidative addition transition states of EY-1 to ANP (TS-4, TS-6, TS-9, TS-11, and TS-13), which are open-shell singlet. The comparison between open-shell and closed-shell singlet structures of these transition states is included in the Supporting Information.
ACCESSION NUMBERS
The X-ray crystallographic data of VIII (L = PPh3) reported in this article has been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC: 2005718.
Supplementary Material
Highlights:
Mechanistically guided prediction of effective electrophiles in multicomponent coupling
Identification of active intermediate by computational/experimental mechanistic studies
Orthogonal reactivity of electrophiles achieved via substrate-dependent mechanisms
ACKNOWLEDGMENTS
We thank the NSF (CHE-1654122) (P.L.) and NIGMS (1R01GM124414-01A1) (G.D.) for financial support for this work. DFT calculations were performed at the Center for Research Computing at the University of Pittsburgh, the TACC Frontera supercomputer, and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the National Science Foundation grant number ACI-1548562. Dr. Ki-Young Yoon is acknowledged for X-ray crystallography.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Additional computational results (Figs. S1–S30 and Table S1) and experimental procedures (Fig. S31).
Table S2: Cartesian coordinates and energies of all computed structures.
REFERENCES AND NOTES
- 1.Everson DA, and Weix DJ (2014). Cross-electrophile coupling: principles of reactivity and selectivity. J. Org. Chem 79, 4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ackerman LK, Lovell MM, and Weix DJ (2015). Multimetallic catalysed cross-coupling of aryl bromides with aryl triflates. Nature 524, 454–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weix DJ (2015). Methods and Mechanisms for Cross-Electrophile Coupling of Csp(2) Halides with Alkyl Electrophiles. Acc. Chem. Res 48, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia-Dominguez A, Li Z, and Nevado C (2017). Nickel-Catalyzed Reductive Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc 139, 6835–6838. [DOI] [PubMed] [Google Scholar]
- 5.Yang T, Chen X, Rao W, and Koh MJ (2020). Broadly Applicable Directed Catalytic Reductive Difunctionalization of Alkenyl Carbonyl Compounds. Chem 6, 738–751. [Google Scholar]
- 6.Wang J, and Dong G (2019). Palladium/Norbornene Cooperative Catalysis. Chem. Rev 119, 7478–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J, Qin C, Lumb J-P, and Luan X (2020). Regioselective Synthesis of Polyfunctional Arenes by a 4-Component Catellani Reaction. Chem [Google Scholar]
- 8.Catellani M (2005). Novel Methods of Aromatic Functionalization Using Palladium and Norbornene as a Unique Catalytic System. Top. Organomet. Chem 14, 21–53. [Google Scholar]
- 9.Catellani M, Frignani F, and Rangoni A (1997). A Complex Catalytic Cycle Leading to a Regioselective Synthesis ofo,o′-Disubstituted Vinylarenes. Angew. Chem. Int. Ed 36, 119–122. [Google Scholar]
- 10.Della Ca N, Fontana M, Motti E, and Catellani M (2016). Pd/Norbornene: A Winning Combination for Selective Aromatic Functionalization via C-H Bond Activation. Acc. Chem. Res 49, 1389–1400. [DOI] [PubMed] [Google Scholar]
- 11.Martins A, Mariampillai B, and Lautens M (2010). Synthesis in the Key of Catellani: Norbornene-Mediated ortho C–H Functionalization. Top. Curr. Chem 292, 1–33. [DOI] [PubMed] [Google Scholar]
- 12.Cheng H-G, Chen S, Chen R, and Zhou Q (2019). Palladium(II)-Initiated Catellani-Type Reactions. Angew. Chem. Int. Ed 58, 5832–5844. [DOI] [PubMed] [Google Scholar]
- 13.Catellani M, Motti E, and Della Ca N (2008). Catalytic sequential reactions involving palladacycle-directed aryl coupling steps. Acc. Chem. Res 41, 1512–1522. [DOI] [PubMed] [Google Scholar]
- 14.Dong Z, Lu G, Wang J, Liu P, and Dong G (2018). Modular ipso/ortho Difunctionalization of Aryl Bromides via Palladium/Norbornene Cooperative Catalysis. J. Am. Chem. Soc 140, 8551–8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferraccioli R (2013). Palladium-Catalyzed Synthesis of Carbo- and Heterocycles through Norbornene-Mediated ortho C-H Functionalization. Synthesis 45, 581–591. [Google Scholar]
- 16.Zhao YB, Mariampillai B, Candito DA, Laleu B, Li M, and Lautens M (2009). Exploiting the divergent reactivity of aryl-palladium intermediates for the rapid assembly of fluorene and phenanthrene derivatives. Angew. Chem. Int. Ed 48, 1849–1852. [DOI] [PubMed] [Google Scholar]
- 17.Catellani M, and Chiusoli GP (1988). Palladium-(II) and -(IV) complexes as intermediates in catalytic C−C bond-forming reactions. J. Organomet. Chem 346, C27–C30. [Google Scholar]
- 18.Bocelli G, Catellani M, and Ghelli S (1993). Regioselective ring opening of a palladium(IV) alkylaromatic metallacycle by benzyl group migration from palladium to the aromatic carbon and X-ray structure of the resulting palladium(II) complex. J. Organomet. Chem 458, C12–C15. [Google Scholar]
- 19.Chai DI, Thansandote P, and Lautens M (2011). Mechanistic studies of Pd-catalyzed regioselective aryl C-H bond functionalization with strained alkenes: origin of regioselectivity. Chem.-Eur. J 17, 8175–8188. [DOI] [PubMed] [Google Scholar]
- 20.Liang Y, Jiang YY, Liu Y, and Bi S (2017). Mechanism of Pd-catalyzed acylation/alkenylation of aryl iodide: a DFT study. Org. Biomol. Chem 15, 6147–6156. [DOI] [PubMed] [Google Scholar]
- 21.Yang T, Kong C, Yang S, Yang Z, Yang S, and Ehara M (2019). Reaction mechanism, norbornene and ligand effects, and origins of meta-selectivity of Pd/norbornene-catalyzed C–H activation. Chem. Sci [Google Scholar]
- 22.Zhang H, Wang H-Y, Luo Y, Chen C, Cao Y, Chen P, Guo Y-L, Lan Y, and Liu G (2018). Regioselective Palladium-Catalyzed C-H Bond Trifluoroethylation of Indoles: Exploration and Mechanistic Insight. ACS Catal 8, 2173–2180. [Google Scholar]
- 23.Norseeda K, Gasser V, and Sarpong R (2019). A Late-Stage Functionalization Approach to Derivatives of the Pyrano[3,2- a]carbazole Natural Products. J. Org. Chem 84, 5965–5973. [DOI] [PubMed] [Google Scholar]
- 24.Littleson MM, Campbell AD, Clarke A, Dow M, Ensor G, Evans MC, Herring A, Jackson BA, Jackson LV, Karlsson S, et al. (2019). Synthetic Route Design of AZD4635, an A2AR Antagonist. Org. Process Res. Dev 23, 1407–1419. [Google Scholar]
- 25.A cesium-iodide salt (I-Cs2HCO3) was used as the ligand on ANP in a previous computational study of the mechanism of Pd/NBE catalyzed acylation/alkenylation reactions. See ref. 20.
- 26.Catellani M, and Cugini F (1999). A catalytic process based on sequential ortho-alkylation and vinylation of ortho-alkylaryl iodides via palladacycles. Tetrahedron 55, 6595–6602. [Google Scholar]
- 27.Gericke KM, Chai DI, Bieler N, and Lautens M (2009). The norbornene shuttle: multicomponent domino synthesis of tetrasubstituted helical alkenes through multiple C--H functionalization. Angew. Chem. Int. Ed 48, 1447–1451. [DOI] [PubMed] [Google Scholar]
- 28.Jiao L, Herdtweck E, and Bach T (2012). Pd(II)-catalyzed regioselective 2-alkylation of indoles via a norbornene-mediated C-H activation: mechanism and applications. J. Am. Chem. Soc 134, 14563–14572. [DOI] [PubMed] [Google Scholar]
- 29.Jiao L, and Bach T (2013). Palladium-catalyzed direct C-H alkylation of electron-deficient pyrrole derivatives. Angew. Chem. Int. Ed 52, 6080–6083. [DOI] [PubMed] [Google Scholar]
- 30.Gao Q, Shang Y, Song F, Ye J, Liu ZS, Li L, Cheng HG, and Zhou Q (2019). Modular Dual-Tasked C-H Methylation via the Catellani Strategy. J. Am. Chem. Soc 141, 15986–15993. [DOI] [PubMed] [Google Scholar]
- 31.Faccini F, Motti E, and Catellani M (2004). A new reaction sequence involving palladium-catalyzed unsymmetrical aryl coupling. J. Am. Chem. Soc 126, 78–79. [DOI] [PubMed] [Google Scholar]
- 32.Candito DA, and Lautens M (2009). Palladium-catalyzed domino direct arylation/N-arylation: convenient synthesis of phenanthridines. Angew. Chem. Int. Ed 48, 6713–6716. [DOI] [PubMed] [Google Scholar]
- 33.Sui X, Zhu R, Li G, Ma X, and Gu Z (2013). Pd-catalyzed chemoselective Catellani ortho-arylation of iodopyrroles: rapid total synthesis of rhazinal. J. Am. Chem. Soc 135, 9318–9321. [DOI] [PubMed] [Google Scholar]
- 34.Dong Z, and Dong G (2013). Ortho vs ipso: site-selective Pd and norbornene-catalyzed arene C-H amination using aryl halides. J. Am. Chem. Soc 135, 18350–18353. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Li R, Dong Z, Liu P, and Dong G (2018). Complementary site-selectivity in arene functionalization enabled by overcoming the ortho constraint in palladium/norbornene catalysis. Nature Chem 10, 866–872. [DOI] [PubMed] [Google Scholar]
- 36.Dong Z, Wang J, Ren Z, and Dong G (2015). Ortho C-H Acylation of Aryl Iodides by Palladium/Norbornene Catalysis. Angew. Chem. Int. Ed 54, 12664–12668. [DOI] [PubMed] [Google Scholar]
- 37.Huang Y, Zhu R, Zhao K, and Gu Z (2015). Palladium-Catalyzed Catellani ortho-Acylation Reaction: An Efficient and Regiospecific Synthesis of Diaryl Ketones. Angew. Chem. Int. Ed 54, 12669–12672. [DOI] [PubMed] [Google Scholar]
- 38.Zhou P-X, Ye Y-Y, Liu C, Zhao L-B, Hou J-Y, Chen D-Q, Tang Q, Wang A-Q, Zhang J-Y, Huang Q-X, et al. (2015). Palladium-Catalyzed Acylation/Alkenylation of Aryl Iodide: A Domino Approach Based on the Catellani–Lautens Reaction. ACS Catal 5, 4927–4931. [Google Scholar]
- 39.Wang J, Zhang L, Dong Z, and Dong G (2016). Reagent-Enabled ortho-Alkoxycarbonylation of Aryl Iodides via Palladium/Norbornene Catalysis. Chem 1, 581–591. [Google Scholar]
- 40.Cai W, and Gu Z (2019). Selective Ortho Thiolation Enabled by Tuning the Ancillary Ligand in Palladium/Norbornene Catalysis. Org. Lett 21, 3204–3209. [DOI] [PubMed] [Google Scholar]
- 41.Li R, Zhou Y, Yoon KY, Dong Z, and Dong G (2019). Sulfenamide-enabled ortho thiolation of aryl iodides via palladium/norbornene cooperative catalysis. Nat. Commun 10, 3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun F, Li M, He C, Wang B, Li B, Sui X, and Gu Z (2016). Cleavage of the C(O)-S Bond of Thioesters by Palladium/Norbornene/Copper Cooperative Catalysis: An Efficient Synthesis of 2-(Arylthio)aryl Ketones. J. Am. Chem. Soc 138, 7456–7459. [DOI] [PubMed] [Google Scholar]
- 43.Amatore C, Jutand A, Khalil F, M’Barki MA, and Mottier L (1993). Rates and mechanisms of oxidative addition to zerovalent palladium complexes generated in situ from mixtures of Pd0(dba)2 and triphenylphosphine. Organometallics 12, 3168–3178. [Google Scholar]
- 44.Vikse K, Naka T, McIndoe JS, Besora M, and Maseras F (2013). Oxidative Additions of Aryl Halides to Palladium Proceed through the Monoligated Complex. ChemCatChem 5, 3604–3609. [Google Scholar]
- 45.McMullin CL, Fey N, and Harvey JN (2014). Computed ligand effects on the oxidative addition of phenyl halides to phosphine supported palladium(0) catalysts. Dalton Trans 43, 13545–13556. [DOI] [PubMed] [Google Scholar]
- 46.Niemeyer ZL, Milo A, Hickey DP, and Sigman MS (2016). Parameterization of phosphine ligands reveals mechanistic pathways and predicts reaction outcomes. Nature Chem. 8, 610–617. [DOI] [PubMed] [Google Scholar]
- 47.The computed barrier of NBE migratory insertion is in good agreement with previous kinetic studies. See ref. 19.
- 48.Yoo EJ, Ma S, Mei T-S, Chan KS, and Yu J-Q (2011). Pd-catalyzed intermolecular C-H amination with alkylamines. J. Am. Chem. Soc 133, 7652–7655. [DOI] [PubMed] [Google Scholar]
- 49.Anand M, Sunoj RB, and Schaefer HF (2015). Palladium–Silver Cooperativity in an Aryl Amination Reaction through C–H Functionalization. ACS Catal 6, 696–708. [Google Scholar]
- 50.Zhou Y, and Bao X (2016). DFT Study of Pd(0)-Promoted Intermolecular C-H Amination with O-Benzoyl Hydroxylamines. Org. Lett 18, 4506–4509. [DOI] [PubMed] [Google Scholar]
- 51.Maestri G, Motti E, Della Ca N, Malacria M, Derat E, and Catellani M (2011). Of the ortho effect in palladium/norbornene-catalyzed reactions: a theoretical investigation. J. Am. Chem. Soc. 133, 8574–8585. [DOI] [PubMed] [Google Scholar]
- 52.Our calculations (Fig. 5) revealed that the oxidative addition of EY-1 to Pd(0) has a 10.1 kcal/mol higher activation free energy than that with ArX-1, which means the oxidative addition of EY-1 to Pd(0) to generate the Pd(II) intermediate necessary for transmetalation would be very difficult in the presence of ArX-1, which reacts much faster with Pd(0). In addition, the transmetalation mechanism can be similarly ruled out for other types of functionalization reactions investigated in this study because of the much higher barriers of oxidative addition of the electrophiles to Pd(0) than aryl iodides (see Fig. 5). These results are consistent with previous experimental and computational mechanistic studies that provided evidencecs against the transmetalation mechanism. See refs. 18 and 51.
- 53.Only the most stable cis/trans isomers of each transition state are shown in these figures. The less stable isomers are included in the SI.
- 54.Three-centered OA transition states cannot be located from these ANP complexes. The five-centered OA transition states are expected to be more favorable than three-centered OA, based on our results with ANP-4 (see later) and previous computational studies of reactions between Pd(II) complexes and morpholino benzoate. See refs. 49 and 50.
- 55.The three-centered OA transition state with ANP-4 requires a high activation energy (41.4 kcal/mol). See Figure S6 for details.
- 56.Diefenbach A, de Jong GT, and Bickelhaupt FM (2005). Activation of H-H, C-H, C-C and C-Cl Bonds by Pd and PdCl(−). Understanding Anion Assistance in C-X Bond Activation. J. Chem. Theory Comput 1, 286–298. [DOI] [PubMed] [Google Scholar]
- 57.Our DFT calculations indicate that the coordination of a benzoate anion to ANP has a similar inhibiting effect on the C–C reductive elimination (see Figure S13 for details). This effect of anionic ligands is consistent with Hartwig’s mechanistic studies that anionic pyrrolyl Pd(II) complexes disfavor the C–N reductive elimination. See; (a)Mann G, Hartwig JF, Driver MS, and Fernández-Rivas C (1998). Palladium-catalyzed C−N(sp2) bond formation: N-arylation of aromatic and unsaturated nitrogen and the reductive elimination chemistry of palladium azolyl and methyleneamido complexes. J. Am. Chem. Soc 120, 827–828.;Hartwig JF (1998). Carbon−heteroatom bond-forming reductive eliminations of amines, ethers, and sulfides. Acc. Chem. Res 31, 852–860.
- 58.Campeau LC, Parisien M, Jean A, and Fagnou K (2006). Catalytic direct arylation with aryl chlorides, bromides, and iodides: intramolecular studies leading to new intermolecular reactions. J. Am. Chem. Soc 128, 581–590. [DOI] [PubMed] [Google Scholar]
- 59.2-Iodotoluene (ArX-2) was a more commonly used substrate in the experimental studies. We expect its reactivity to be similar to that of ortho-ethyl iodobenzene (ArX-1) used in the calculations.
- 60.Several other effects on electrophile compatibility were also considered, including electronic effects of aryl halides and anhydride electrophiles, and the effects of phosphine ligands. These factors have relatively minor impact on the compatibility. See SI (Figs. S25–S27) for detailed discussions.
- 61.Gourlaouen C, Ujaque G, Lledos A, Medio-Simon M, Asensio G, and Maseras F (2009). Why is the Suzuki-Miyaura cross-coupling of sp3 carbons in alpha-bromo sulfoxide systems fast and stereoselective? A DFT study on the mechanism. J. Org. Chem 74, 4049–4054. [DOI] [PubMed] [Google Scholar]
- 62.Mollar C, Besora M, Maseras F, Asensio G, and Medio-Simon M (2010). Competitive and selective Csp3-Br versus Csp2-Br bond activation in palladium-catalysed Suzuki cross-coupling: an experimental and theoretical study of the role of phosphine ligands. Chem.-Eur. J 16, 13390–13397. [DOI] [PubMed] [Google Scholar]
- 63.Noverges Pedro B, Medio-Simón M, and Jutand A (2017). Influence of the Ligand of Palladium(0) Complexes on the Rate of the Oxidative Addition of Aryl and Activated Alkyl Bromides: Csp2−Br versus Csp3−Br Reactivity and Selectivity. ChemCatChem 9, 2136–2144. [Google Scholar]
- 64.Besora M, and Maseras F (2019). The diverse mechanisms for the oxidative addition of C-Br bonds to Pd(PR3) and Pd(PR3)2 complexes. Dalton Trans 48, 16242–16248. [DOI] [PubMed] [Google Scholar]
- 65.Ji C-L, and Hong X (2017). Factors Controlling the Reactivity and Chemoselectivity of Resonance Destabilized Amides in Ni-Catalyzed Decarbonylative and Nondecarbonylative Suzuki-Miyaura Coupling. J. Am. Chem. Soc 139, 15522–15529. [DOI] [PubMed] [Google Scholar]
- 66.Quasdorf KW, Antoft-Finch A, Liu P, Silberstein AL, Komaromi A, Blackburn T, Ramgren SD, Houk KN, Snieckus V, and Garg NK (2011). Suzuki-Miyaura cross-coupling of aryl carbamates and sulfamates: experimental and computational studies. J. Am. Chem. Soc 133, 6352–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.The oxidative addition of anhydride to Pd(0) favors three-centered TS. See Figure S14 for details.
- 68. The chelation of ortho-ester group with high valent Pd is observed in the ANP-4 oxidative addition transition state. Chelation with the ortho-ester is not observed in the oxidative addition to Pd(0) due to the lack of available coordination site in the four-coordinated transition state. [Google Scholar]
- 69.Ariafard A, and Lin Z (2006). Understanding the Relative Easiness of Oxidative Addition of Aryl and Alkyl Halides to Palladium(0). Organometallics 25, 4030–4033. [Google Scholar]
- 70.Legault CY, Garcia Y, Merlic CA, and Houk KN (2007). Origin of regioselectivity in palladium-catalyzed cross-coupling reactions of polyhalogenated heterocycles. J. Am. Chem. Soc 129, 12664–12665. [DOI] [PubMed] [Google Scholar]
- 71.Ahlquist M, and Norrby P-O (2007). Oxidative Addition of Aryl Chlorides to Monoligated Palladium(0): A DFT-SCRF Study. Organometallics 26, 550–553. [Google Scholar]
- 72.Another sterically hindered electrophile, iPr2N-OBz, also suffers from low compatibility with ArX-1 (ECS = –9.2), which is due to the higher barrier to the oxidative addition of the electrophile with ANP (ΔG‡ = 30.1 kcal/mol). Experimentally, this electrophile was ineffective in amination of ArX-1. See Fig. S28 in the Supporting Information for detailed computational and experimental results.
- 73.Catellani M, Motti E, and Minari M (2000). Symmetrical and unsymmetrical 2,6-dialkyl-1,1’-biaryls by combined catalysis of aromatic alkylation via palladacycles and Suzuki-type coupling. Chem. Commun, 157–158. [Google Scholar]
- 74.Kim S, Goldfogel MJ, Gilbert MM, and Weix DJ (2020). Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Chlorides with Primary Alkyl Chlorides. J. Am. Chem. Soc 142, 9902–9907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. , Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2013. [Google Scholar]
- 76.Towns J, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A, Hazlewood V, Lathrop S, Lifka D, Peterson GD, et al. (2014). XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng 16, 62–74. [Google Scholar]
- 77.Lee C, Yang W, and Parr RG (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 78.Becke AD (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 98, 5648–5652. [Google Scholar]
- 79.Marenich AV, Cramer CJ, and Truhlar DG (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- 80.Valero R, Costa R, de PRMI, Truhlar DG, and Illas F (2008). Performance of the M06 family of exchange-correlation functionals for predicting magnetic coupling in organic and inorganic molecules. J. Chem. Phys 128, 114103. [DOI] [PubMed] [Google Scholar]
- 81.Zhao Y, and Truhlar DG (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc 120, 215–241. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.