

Abstract
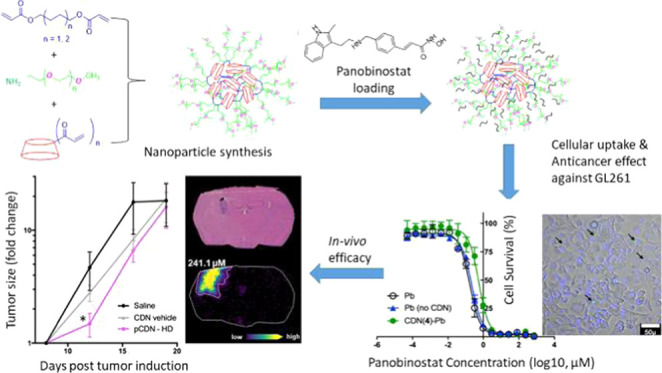
Therapeutic development of histone deacetylase inhibitors (HDACi) has been hampered by a number of barriers to drug delivery, including poor solubility and inadequate tissue penetration. Nanoparticle encapsulation could be one approach to improve the delivery of HDACi to target tissues; however, effective and generalizable loading of HDACi within nanoparticle systems remains a long-term challenge. We hypothesized that the common terminally ionizable moiety on many HDACi molecules could be capitalized upon for loading in polymeric nanoparticles. Here, we describe the simple, efficient formulation of a novel library of β-cyclodextrin-poly (β-amino ester) networks (CDN) to achieve this goal. We observed that network architecture was a critical determinant of CDN encapsulation of candidate molecules, with a more hydrophobic core enabling effective self-assembly and a PEGylated surface enabling high loading (up to ∼30% w/w), effective self-assembly of the nanoparticle, and slow release of drug into aqueous media (up to 24 days) for the model HDACi panobinostat. We next constructed a library of CDNs to encapsulate various small, hydrophobic, terminally ionizable molecules (panobinostat, quisinostat, dacinostat, givinostat, bortezomib, camptothecin, nile red, and cytarabine), which yielded important insights into the structural requirements for effective drug loading and CDN self-assembly. Optimized CDN nanoparticles were taken up by GL261 cells in culture and a released panobinostat was confirmed to be bioactive. Panobinostat-loaded CDNs were next administered by convection-enhanced delivery (CED) to mice bearing intracranial GL261 tumors. These studies confirm that CDN encapsulation enables a higher deliverable dose of drug to effectively slow tumor growth. Matrix-assisted laser desorption/ionization (MALDI) analysis on tissue sections confirms higher exposure of tumor to drug, which likely accounts for the therapeutic effects. Taken in sum, these studies present a novel nanocarrier platform for encapsulation of HDACi via both ionic and hydrophobic interactions, which is an important step toward better treatment of disease via HDACi therapy.
Keywords: drug delivery, nanomedicine, nanoparticle, HDACi, convection enhanced delivery
1. Introduction
Histone deacetylase inhibitors (HDACi) are a class of small molecules that promote hyperacetylation of core histones, leading to relaxation of chromatin and therapeutic effects in a multitude of disease models.1−10 Unfortunately, the utility of HDACi is plagued by delivery problems, including rapid clearance and poor tissue distribution when molecules are administered in free form.11−13 We and others have developed nanoparticle encapsulation strategies to improve drug tolerability, pharmacokinetics, and site-specific delivery of HDAC inhibitors.14−17 However, each of these reports has developed approaches for encapsulation of individual HDACi, requiring development of unique loading strategies for individual drugs. These intensive efforts have thus far yielded only modest loading. HDACi loading in and controlled release from nanocarrier systems remains unoptimized, and a generalizable strategy for drug delivery for this class of molecules is lacking. Hence, addressing this problem would not only provide us a generic high-capacity loading strategy for HDAC inhibitors but also enable us to overcome the challenges of low delivery efficiency of polymeric nanoparticles.18
The majority of HDAC inhibitors bear a characteristic hydroxamic acid (zinc ion chelating domain), which is linked to the capping moiety by a spacer of appropriate chain length.19 This hydroxamic acid enables manipulation of charge and thus drug solubility through changes in solution pH. We previously loaded the HDAC inhibitor quisinostat (JNJ-26481585) onto polymeric nanoparticles composed of poly(lactic acid)–poly(ethylene glycol) (PLA–PEG) through a pH manipulation and ionization strategy.14 Although these quisinostat-loaded PLA–PEG nanoparticles were highly loaded and useful for treating intracranial glioblastoma by intravenous injection, in vitro studies demonstrated that drug was released rapidly once nanoparticles were exposed to an aqueous environment. Presumably, this rapid release occurred because quisinostat was only associated with the surface of the nanoparticle, instead of being embedded within a particle core.
Here, we sought to develop β-cyclodextrin-poly (β-amino ester) (cyclodextrin networks, or CDNs) for the delivery of HDAC inhibitors. We hypothesized that the common terminally ionizable moiety in HDACi molecules could be leveraged for loading this class of agents into nanoparticles to achieve sustained release. To test this hypothesis, we describe the development of a small library of CDN materials that self-assemble into drug-loaded nanoparticles. We predicted that dual loading via both ionic and hydrophobic interactions would confer favorable characteristics for drug loading. Two uniquely surface-functionalized (aminated and PEGylated) versions of CDNs were generated via simple three-component Michael addition reactions. Three subtypes of amine-CDN were developed, each with a successively increasing and flexible hydrophobic core, to obtain a total of four unique CDN structures. HDAC inhibitors (panobinostat, quisinostat, dacinostat, givinostat) and other small, hydrophobic molecules (bortezomib, camptothecin, nile red) were passively doped into the resultant particles. Some of these particles possess biophysical properties favorable for drug delivery applications, such as small size, close to neutral surface charge, and high loading (up to ∼30%). Detailed structure–property investigation revealed that the key structural aspects of CDNs enabling self-assembly and effective drug incorporation include cross-linker length, surface charge density, and the accessibility of the hydrophobic cyclodextrin core. Importantly, we observed that the encapsulated molecule needed to be terminally ionizable with a flexible core to enable effective loading in CDNs, and this was generalizable across molecules. Evaluation of spatial distribution of drug and tumor growth in mice demonstrates that CDNs enable a higher quantity of drug to be delivered to treat orthotopic glioblastoma. Thus, the nanoparticle platform described here offers new opportunities for nanomedicine development to deliver HDACi and other terminally ionizable agents (Figure 1).
Figure 1.

Schematic representation of the synthesis of (i) acrylated β-cyclodextrin; (ii) blank; and (iii) panobinostat-loaded CDN nanoparticles.
2. Materials and Methods
2.1. Materials
The following reagents were obtained from Alfa Aesar: β-cyclodextrin, acrolyl chloride, chloroform (99%, ACS reagent), ethyl acetate (99%, ACS reagent), and dimethyl sulfoxide (99%, ACS reagent). 1-Methyl-2-pyrrolidone (NMP) and diethylether (99+%) were purchased from Acros Organics. 1,4-Butanediol diacrylate and 1,6-hexanediol diacrylate were obtained from Sigma-Aldrich. Methoxy-terminated poly(ethylene glycol) (MW 550, mPEG550)-amine was obtained from Creative Peg Works (Chapel Hill, NC). N,N-Dimethylethylenediamine was obtained from Oakwood chemicals (Estill, SC). Phosphate-buffered saline (PBS, pH 7.4) and Polyester Transwell inserts (96-well plate, 0.4 μm pore size) were obtained from Costar (Cambridge, MA). Dialysis cassettes (MWCO 3500 Dalton) were obtained from Thermo Fisher Scientific (Waltham, MA). CellTiter-Glo was purchased from Promega (Madison, WI). Costar 96-well plates were purchased from VWR International (Radnor, PA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), trypan blue, and 0.25% trypsin-EDTA were purchased from Gibco Invitrogen (Carlsbad, CA). Quisinostat (JNJ-26481585) and panobinostat (LBH589) were purchased from ApexBio (Houston, TX). Camptothecin and dacinostat (LAQ824) were purchased from Selleckchem (Houston, TX). Nile red was purchased from TCI America. All of these chemicals were used as received without further purification unless otherwise noted.
2.2. Synthesis of Acrylated-CD
β-Cyclodextrin was oven dried for 12 h at 110 °C. N-Methyl-2-pyrrolidone (NMP) was stored with oven-parched molecular sieves (4A) for at least 24 h. β-Cyclodextrin (2.3 g, 2.0 mmol) was measured and stirred in NMP in a 25 mL round-bottom flask. The solution was cooled with ice-cold water, and acrolyl chloride (2.0 mL, 24.7 mmol) was added via a syringe. The resultant solution was stirred under the ice-cold condition for 1 h, after which the solution was heated up to 21 °C. Stirring was continued for 48 h, after which the reaction mixture was poured into approximately 200 mL of distilled water. The precipitated solid was homogenously bath-sonicated, followed by filtration. The collected residue was washed twice with distilled water and then allowed to dry over 48 h. The dry white powder recovered was weighed out (3.59 g) and subsequently analyzed by 1H NMR and matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) spectrometry (see the Supporting Information).
2.3. Synthesis of Blank CDN Nanoparticles
2.3.1. Amine-CDNs (CDNs 1–3)
β-Cyclodextrin-poly(β-amino ester) CDNs with various structures were synthesized employing Michael addition of a three-component mixture. Acrylated β-cyclodextrin (cyclodextrin precursor; 89.8 mg; 0.06 mmol), 1,4-butanediol or 1,6-hexanediol diacrylate (cross-linking polyesters; 1.3 and 2.7 mmol), and N,N-dimethylethylenediamine (207 μL; 1.9 mmol) were dissolved in a solvent mixture of EtOAc/CHCl3 (3:7) and heated at 65 °C under stirring at 600 rpm for 12–14 h. The reaction mixture was subsequently dried under reduced pressure, and the resultant crude was redissolved in chloroform (approx. 2 mL). This solution was dispersed in 15 mL (repeated twice with 5 mL) of diethylether to precipitate the insoluble network. The supernatant was discarded, and the obtained precipitate was filtered, dried, and redispersed into fresh DI water (40 mL). The aqueous dispersion of the network was subsequently washed via ultrafiltration through two Amicon Ultra-15 centrifugal filters (10 kDa MWCO, 15 mL capacity) for 20 min (×2) spins at 5000 RCF. Aliquots were frozen and lyophilized to determine recovery and biophysical characteristics (size and ζ-potential).
2.3.2. mPEG550-CDN (CDN-4)
Acrylated β-cyclodextrin (cyclodextrin precursor; 22.5 mg; 0.015 mmol), 1,4-butanediol diacrylate (cross-linking polyesters; 126 μL; 0.675 mmol), and mPEG550-amine (260 mg; 0.475 mmol) were dissolved in a solvent mixture of EtOAc/CHCl3 (3:7) and heated at 65 °C under stirring at 600 rpm for 12 h. The reaction mixture was subsequently dried under reduced pressure, and the resultant crude was redispersed into fresh DI water (40 mL). The aqueous dispersion was subsequently washed via ultrafiltration through Amicon Ultra-15 centrifugal filters (10 kDa MWCO, 15 mL capacity) for 20 min (×2) spins at 5000 RCF. Aliquots were frozen and lyophilized to determine recovery and biophysical characteristics (size and ζ-potential).
Average recovery for each CDN was calculated for at least three independent experimental repeats: CDN-1 = 69.3 (±7.8) mg; CDN-2 = 84.2 (±8.8) mg; CDN-3 = 88.5 (±8.1) mg; and CDN-4 = 82.9 (±13.8) mg.
2.4. Synthesis of BODIPY-Labeled CDN Nanoparticles
Acrylated β-cyclodextrin (cyclodextrin precursor; 22.5 mg; 0.015 mmol), 1,4-butanediol diacrylate (cross-linking polyesters; 126 μL; 0.675 mmol), mPEG550-amine (0.354 mmol), and amino-PEG12-propionic acid (0.121 mmol) were dissolved in a solvent mixture of EtOAc/CHCl3 (3:7) and heated at 65 °C under stirring at 600 rpm for 12 h. The reaction mixture was subsequently dried under reduced pressure, and the crude obtained was redispersed into fresh DI water (40 mL). The aqueous dispersion was subsequently washed via ultrafiltration through Amicon Ultra-15 mL centrifugal filters (10 kDa MWCO, 15 mL capacity) for 20 min (×2) spins at 5000 RCF. Aliquots were frozen and lyophilized overnight. For labeling, 5.0 mg of the lyophilized sample was further treated with N-hydroxysuccinimide (6.2 mg), EDC (9.5 μL), and BDP-FL-amine (0.44 μmol) in dimethyl sulfoxide (DMSO) (500 μL) at room temperature for 24 h. The reaction mixture was subsequently washed with DI water via ultrafiltration through Amicon Ultra-2 mL centrifugal filters (3 kDa MWCO, 2 mL capacity) for three, 20 min spins at 5000 RCF. Aliquots were frozen and lyophilized overnight. The recovery of BODIPY-CDN-4 was calculated to be 4.6 mg.
2.5. MALDI-TOF Mass Spectrometry
Typically, 20 mg/mL solutions were separately made for analyte (acrylated-CD) and matrix (2,5-dihydroxybenzoic acid) in acetonitirile/water (1/9) mixture. A mixture of 4 μL of analyte, 14 μL of matrix, and 2 μL of sodium acetate as cationization agent (1 mg/mL aqueous solution) was homogenized by vortexing. Two microliters of the mixture was transferred onto a MALDI target plate, followed by air-drying to prepare a thin matrix/analyte film. Mass spectra were obtained using a MS Bruker Autoflex MALDI-TOF mass spectrometer equipped with a nitrogen laser delivering 2 ns laser pulses at 337 nm with positive ion ToF detection performed using an accelerating voltage of 25 kV.
2.6. NMR Spectroscopy
Lyophilized CDN samples were dispersed in dimethyl sulfoxide-d6 (DMSO-d6). 1H NMR spectra were obtained using a 300 MHz Bruker Avance NMR spectrometer. The spectra were compared to those of the individual components employed in the synthesis of CDNs.
2.7. Scanning Electron Microscopy (SEM)
Lyophilized nanoparticles were suspended in an aqueous solution to a final concentration of 20 mg/mL. Five microliter droplets of the above solution were placed onto aluminum stubs with carbon adhesive. Samples were allowed to air-dry prior to coating. Samples were sputter-coated using a Denton Desk-V Sputter system with gold at 20 mA for 20 s and imaged using a FEI Quanta 400 environmental scanning electron microscope with an ETD detector at 20 kV and a 4 mm working distance.
2.8. Dynamic Light Scattering (DLS)
One hundred microliters of CDN1-3 or 50 μL of CDN-4 aqueous solutions (20 mg/mL) were added to approximately 3 mL of distilled water in a clean dry cuvette. The dispersion was homogenized by repeated agitation and then allowed to stabilize for 2 min. Mean hydrodynamic sizes of the particles were measured using NanoBrook 90 Plus Zeta particle size analyzer (Brookhaven Instruments, Holtsville, NY). Results are reported as the average of at least three separate readings.
2.9. ζ-Potential
Sixty microliters of CDN (CDNs 1–4) aqueous solutions (20 mg/mL) were added to approximately 2 mL of KCl (1 mM aq. solution) in a clean dry cuvette. The dispersion was homogenized by repeated agitation and then allowed to stabilize for 5 min. ζ-Potential measurements were carried out using NanoBrook 90 Plus Zeta particle size analyzer (Brookhaven Instruments, Holtsville, NY). Results are reported as the average of at least three separate readings.
2.10. Drug-Loaded CDN Nanoparticles
Aqueous dispersions (2.0 mL) of blank CDN nanoparticles (10 mg/mL) were doped with various drugs dissolved in DMSO (50 μL). The aqueous dispersion was left to agitate for 18 h and then subjected to concentration via ultrafiltration through Amicon Ultra-15 centrifugal filters (3 kDa MWCO, 0.5 mL capacity) for 4 and 20 min spins at 5000 RCF. Aliquots were frozen and lyophilized prior to characterization. Drug concentration in loaded CDNs was quantified by absorbance (310 nm for quisinostat, panobinostat, and dacinostat; 285 nm for givinostat; 365 nm for camptothecin; 550 nm for Nile Red) on a Tecan plate reader. Lyophilized nanoparticles were dissolved at 5 mg/mL in DMSO. Samples were plated in triplicate (40 μL of nanoparticles and 10 μL of DMSO per well) in a clear, flat bottom 96-well assay plate. A control curve was constructed in technical triplicate by adding 40 μL of blank nanoparticles per well and spiking with 10 μL of known drug concentration in DMSO. Forty microliters of bortezomib-loaded nanoparticles (5 mg/mL aqueous solution) was extracted with 100 μL of ethyl acetate. The ethyl acetate layer was collected and dried under nitrogen. The contents were dissolved in 40 μL of DMSO and plated in triplicate. A control curve was constructed in technical triplicate by spiking with 10 μL of known drug concentration in DMSO. Absorbance was measured at 285 nm. For all formulations, theoretical drug loading was calculated as the mass of drug added divided by mass of blank CDN nanoparticles (w/w%), and experimental drug loading was calculated as mass of drug measured divided by the mass of nanoparticles (w/w%). Subsequent characterizations are reported primarily as a function of theoretical drug loading, which is a convenient way to label CDNs formulated under different conditions since there is variability in experimental drug loading.
2.11. Controlled Release
Lyophilized nanoparticles were dispersed in an aqueous solution to a final drug concentration of 1 mg/mL (i.e., 20 mg/mL panobinostat for a sample with 5% experimental loading) and 400 μL was transferred to a 3.5 k MWCO Slide-A-Lyzer Dialysis cassette (Thermo Fisher Scientific, Waltham, MA). The cassette was immersed in 4 L of PBS (pH 7, replaced at every 24 h time point) at 37 °C with gentle stirring (75 rpm). At each time point, 30 μL of nanoparticles was removed from the cassette and dissolved in 120 μL of DMSO. Fifty microliters of dissolved nanoparticles was added in triplicate to a clear, flat bottom, 96-well plate, and the amount of drug remaining was quantified by absorbance.
2.12. Stability Studies for Drug-Loaded CDN Nanoparticles
One hundred microliters of panobinostat-loaded (20 mg/mL, 20% theoretical) CDN-3 or 50 μL of panobinostat-loaded (20 mg/mL, 20% theoretical) CDN-4 were prepared in three separate aqueous media—acidic (pH = 4.0), PBS (pH = 7.4), and basic (pH = 10.2). A set of five DLS measurements were recorded for each sample over several days to track the stability of the colloidal system.
2.13. In Vitro Experiments
For IC50 experiments, GL261 cells were seeded at 3000 cells/100 μL into 96-well plates containing Dulbecco’s modified Eagle’s medium (DMEM) and 10% fetal bovine serum (FBS). Cells were given 4 h to adhere prior to drug treatments. Initially, stock solutions of free panobinostat, drug-loaded, and blank CDN-4 (21% drug-loaded) were made in 2.5% DMSO in cell culture media at 10 mg/mL drug concentration. Equivalent doses (10 μL of the stock) were added to the 96-well plates at 16 serial dilutions with concentrations from 0 to 70 μM, followed by incubation at 37 °C with 5% CO2 for 72 h. The final DMSO concentration in the well plates was approximately 0.25 vol %. This DMSO concentration was determined in control experiments to be well below the concentration that exerts toxic effects on GL261 cell lines (data not shown). Cell viability was determined with an IC50 value obtained from a CellTiter-Glo Luminescence assay and calculated using GraphPad Prism (San Diego, CA). For uptake experiments, GL261 cells were seeded at 90 000 cells/1 mL into 48-well plates containing DMEM and 10% FBS. BODIPY-labeled CDN-4 nanoparticles (250 μg) were incubated with cells for 48 h, after which cells were thoroughly washed, fixed with 4% paraformaldehyde, and counterstained with DAPI. Images were collected on an upright Nikon Ti2 microscope.
2.14. In Vivo Experiments
All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science at Houston in accordance with all relevant guidelines. A total of 57 male, Bl6/C57 mice between the ages of 12 and 14 weeks were obtained from the Jackson Laboratory. Mice were anesthetized with 2% isoflurane and mounted on a Kopf stereotactic frame. A burr hole was drilled to target the dorsal striatum (+2.0 mm lateral and −0.1 mm posterior to bregma). A Hamilton syringe was loaded with GL261 cells, lowered to −3.2 mm, and raised to −2.8 mm. Cells (50 000 cells per 2 μL) were infused over 2 min, the surgical incisions were closed with staples, and mice were returned to recovery cages.
Treatments were administered 8 days following induction to target the same stereotactic location as the original tumor, returning through the burr hole to infuse a total volume of 5 μL at a rate of 0.67 μL/min. The needle was allowed to remain in place for 5 min before removal to prevent backflow. Mice were imaged the day prior to treatment for allocation to treatment groups, which included saline (n = 9), cyclodextrin vehicle (CD vehicle, n = 9), CDN vehicle (CDN vehicle, n = 9), panobinostat solubilized within cyclodextrin at a 1 μg dose (pCD, n = 10), panobinostat encapsulated in CDN-4 at a 1 μg of dose (pCDN, n = 9), and panobinostat encapsulated in CDN-4 at a 30 μg dose (pCDN-HD, n = 10). The CD vehicle was matched to the polymer concentration in the pCD group, and the CDN vehicle was matched to the polymer concentration in the pCDN-HD group. All treatments were provided in the same volume of saline, i.e., 5 μL. Mice were weighed and evaluated daily, receiving bioluminescent imaging every 3–4 days. Imaging was conducted on an IVIS Xenogen system under isoflurane anesthesia following a subcutaneous injection of luciferin (15 mg/kg). One mouse was excluded from the study after it died due to reasons that were unrelated to treatment or tumor burden. Mice were euthanized upon loss of 20% or greater body weight or on the appearance of neurological symptoms. To quantify the IVIS data, regions of interest (ROI) were drawn to encompass the entire tumor signal for each mouse. Flux values obtained from the ROI analysis were normalized to the pre-dose average for each group, which enables the IVIS data to be expressed as a fold change in tumor size following treatment.
2.15. MALDI Mass Spectrometry Imaging (MALDI MSI) Analysis
2.15.1. MALDI Tissue Preparation
For MALDI MSI analysis of drug distribution, mice bearing tumors were treated as described above and euthanized by rapid decapitation 1 h after infusion of either pCD or pCDN-HD. The brains were quickly extracted and snap-frozen, mounted onto a specimen chuck, and sectioned coronally at 10 μm thickness (Microm HM550, Thermo Scientific, Waltham, MA). The tissue sections were thaw-mounted onto indium tin oxide (ITO) slides. A tissue microarray (TMA) mold was used to prepare a tissue mimetic model for panobinostat quantitation by MALDI MSI. Panobinostat concentrations ranging from 1.0 to 50 μM were spiked into normal mouse brain tissue homogenate, and the spiked tissue mixtures were dispensed into 1.5 mm core diameter channels of the 40% gelatin TMS mold and frozen at −80 °C. The tissue mimetic model was sectioned and thaw-mounted adjacent to the mouse brain tissue sections for analysis. Serial sections of the mimetic and mouse brains were mounted on regular microscopy slides for hematoxylin and eosin (H&E) staining. The ITO slides mounted with the tissue and mimetic model sections were placed in a vacuum desiccator before matrix deposition. 2,5-Dihydroxybenzoic acid (160 mg/mL) matrix was dissolved in 70:30 methanol: 0.1% TFA with 1% DMSO and applied using a TM sprayer (HTX Technologies, Chapel Hill, NC) at a flow rate (0.18 mL/min), spray nozzle velocity (1200 mm/min), nitrogen gas pressure (10 psi), spray nozzle temperature (75 °C), and track spacing (2 mm) for two passes. Optical microscopy images of the H&E-stained serial tissue sections were acquired using a 10× objective (Zeiss Observer Z.1, Oberkochen, Germany).
2.15.2. MALDI Multiple Reaction Monitoring (MALDI MRM) Mass Spectrometry Imaging
A multiple reaction monitoring (MRM) approach was used for quantitative imaging of panobinostat by monitoring the transition of the precursor ion to product ion (334.155 → 317.152) using a dual source timsTOF fleX mass spectrometer (Bruker Daltonics, Billerica, MA) in positive ion mode. Data was acquired between m/z 100 and 650. The fragment ion from the precursor corresponds to the loss of a methyl group. The method was optimized using ESI with an infusion of panobinostat to adjust the ion-transfer funnels, quadrupole, collision cell, and focus pre-TOF parameters, and an Agilent tune mix solution (Agilent Technologies, Santa Clara, CA) was used to calibrate the mass range. Tandem MS parameters were set for a collision energy of 23 eV with an isolation width of 3 m/z. MALDI MS images were acquired with a laser repetition set to 5000 Hz with 2000 laser shots per 100 μm pixel. SCiLS Lab software (version 2020a premium, Bruker Daltonics, Billerica, MA) was used for data visualization without data normalization. The average ion intensity for each spiked TMA sectioned core area was plotted against corresponding panobinostat concentration from 1.0 to 50 μM for calibration of the MALDI MS signal, resulting in a limit of detection (LOD) of 8.41 μM (S/N ratio of >3), and a limit of quantification (LOQ) of 28.0 μM (S/N ratio of >10).
3. Results and Discussion
3.1. Cyclodextrin Network (CDN) Architectures
Cyclodextrins are a class of cyclic oligosaccharides that have been used widely for solubilization of hydrophobic molecules to enable delivery to aqueous environments, including by us and others for the purpose of delivering HDAC inhibitors.20,21 Cyclodextrins are useful for enabling the administration of lipophilic molecules from aqueous media but do not, in unmodified form, provide sustained release. Synthetically modified amphiphilic cyclodextrins have been shown to not only form drug-loaded nanoparticles22 but also to outperform traditional polyester particles for certain drugs in terms of drug loading and controlled release.23 In addition, advanced cyclodextrin-based cross-linked24 and linear polymeric architectures25 have been reported in the literature for their potential application as therapeutics.
β-Cyclodextrin-poly (β-amino ester) nanoparticles (composition of CDN-1) were previously reported by Lowe and colleagues as a blood–brain barrier (BBB) permeable platform to support the sustained release of doxorubicin.26 Here, we engineered the constituent polymer to develop a library of new materials possessing different cross-linker concentration, length, and amine functionalities (CDN-2, CDN-3, and CDN-4, respectively). This library was designed to study the complex structure–function relationships between loading of small molecules and network compositions and to identify conditions under which CDNs could be highly loaded with therapeutic molecules. The 1,4-butanediol diacrylate cross-linker was employed at two different concentrations [1.3 mmol (1.0 equiv) and 2.7 mmol (2.0 equiv) for CDN-1 and CDN-2, respectively], whereas CDN-3 was synthesized from 1,6-hexanediol diacrylate [2.7 mmol, (2.0 equiv)]. CDN-4 was synthesized from mPEG550-amine with 1,4-butanediol diacrylate cross-linker (2.7 mmol, [2.0 equiv]). The synthesis of materials incorporating expected chemical moieties was confirmed by NMR (see the Supporting Information), while the nanoparticle formation was determined by DLS and SEM images (see the Supporting Information). The overall biophysical characterization of the CDNs without any encapsulated payload are elucidated in Figure 2A–C. Comparing CDN-1 against CDN-2, the latter has more surface-amine functionalities than the former. CDN-2 also has an expanded and flexible hydrophobic core. CDN-3 possesses a greater cross-linked length but self-assembles into smaller-sized nanoparticles than CDNs 1 and 2. Finally, CDN-4 possesses a PEG-modified surface and close to neutral surface charge, which could confer favorable properties for in vivo applications.
Figure 2.

Drug-empty CDNs self-assemble into nanoparticles. (A) Table displaying the mean size, PDI, and ζ-potential for drug-empty nanoparticles formed by CDNs 1–4. (B) SEM image of representative Amine-CDN-3. (C) SEM image of representative CDN-4, which bears mPEG550 surface functionality. Scale bars = 1 μm. Errors are reported as standard deviations of at least three independently formulated batches.
3.2. Panobinostat-Loaded CDNs
To assess the drug loading capacity in CDNs, we first focused on the HDAC inhibitor panobinostat (LBH589). Panobinostat is a pan-HDACi that is of interest for the treatment of a multitude of diseases, including solid and hematological cancers, neuroinflammation, and traumatic brain injury.27−30 Similar to other HDACi, panobinostat has several delivery challenges that are expected to reduce its therapeutic potential. First, panobinostat is poorly water-soluble (0.064 mg/mL at a pH 7.6) and difficult to administer in free form. Second, both peripheral and central pharmacokinetic analyses show that it is cleared rapidly from fluid compartments.31 Therefore, we focused on panobinostat as a candidate molecule for nanoparticle delivery based on the expectation that solubilization within a carrier system capable of sustained release could be significant for its use to treat human disease. Nanoparticles were formulated by doping a given concentration of the drug with a polymer in 2.5 vol % DMSO-aqueous medium. Panobinostat was added at different theoretical loadings, and the amount of panobinostat incorporated into the network after thorough washing was quantified by absorbance (Figure 3). Panobinostat was found to incorporate effectively into all four CDNs. Drug loading varied starkly for the different structural variations of the networks, with both the diacrylate cross-linkers and the functionality of the amine affecting drug loading capacity (Figure 3). Drug loading increased across CDNs 1–3, which was associated with a larger and more flexible hydrophobic core of the nanoparticles imparted by the hexyl cross-linkers (CDN-3).
Figure 3.
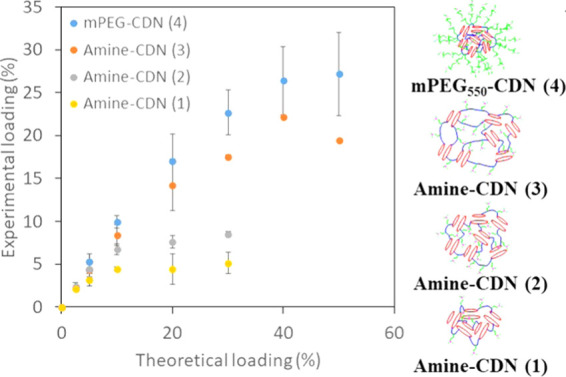
Plot of measured panobinostat loading for CDNs 1–4 as a function of different theoretical loadings. Error bars show standard deviations of at least three independently formulated batches.
Loading was significantly higher in CDN-4 compared to other nanoparticles. High loading of panobinostat in CDN-4 is consistent with our prior report of favorable interaction of ionized quisinostat, another HDACi, with PEG present at the surface of polyester nanoparticles.14 To better understand the mechanisms by which a drug incorporates into the CDN network, we engaged in detailed biophysical characterization of each formulation.
The maximum panobinostat loading achieved for CDN-1 was approximately 5% (∼22% encapsulation efficiency), and the experimental drug loading did not increase with the addition of more drugs (Figure 4A). Nanoparticles produced from panobinostat-loaded CDN-1 were micron-sized, as evident from the DLS size measurements. The ζ-potential of the particles was found to increase linearly with increasing drug loading (Figure 4B). The larger size is likely due to the formation of particle or drug aggregates and eventual network collapse at increasing drug concentration (Figure 4C). The linear increase in ζ-potential can be explained based on an acid–base interaction between the drug (hydroxamic acid) and the surface-amine functionalities (Lewis base), thereby aiding drug solubility. Thus, CDN-1 could incorporate panobinostat in the micron range but did not self-assemble into stably condensed nanoparticles (Figure 4D).
Figure 4.

Plot of average (A) experimental loading, (B) average ζ-potential, and (C) mean hydrodynamic size of CDN-1 nanoparticles against theoretical loading of panobinostat. (D) Aggregate formation as a mechanism of drug stabilization by the CDN-1 nanoparticles. Error bars show the standard deviation of three separate batches.
Upon increasing the hydrophobic core of the network in CDN-2, experimental loading for panobinostat was found to increase up to approximately 8% (∼35% encapsulation efficiency) (Figure 5A). This increase in drug loading was associated with a rise in ζ-potential for up to 5% theoretical loading and a steady increase in particle size with increasing drug incorporation (Figure 5B). While the same acid–base interaction of the drug and surface-amine functionalities holds true in this case, we speculate that a larger hydrophobic core assists in stabilizing the increasing amount of drug in the network compared to CDN-1 as drug incorporation exceeds 5% (Figure 5C). Thus, CDN-2 was capable of self-assembling into relatively stable nanoparticles with modest, saturable loading capacity (Figure 5D).
Figure 5.

Plot of average (A) experimental loading, (B) average ζ-potential, and (C) mean hydrodynamic size of CDN-2 nanoparticles against theoretical loading of panobinostat. (D) Stable drug–nanoparticle interactions. Error bars show the standard deviation of three separate formulations.
CDN-3 employed a longer length cross-linker (1,6-hexanediol diacrylate) that enabled the self-assembly of polymer and drug into smaller and more consistently sized nanoparticles than CDN-1 or CDN-2, with the highest experimental loading of ∼20% (∼71% encapsulation efficiency) (Figure 6A). Presumably, this increased stability is due to the more flexible network with a more hydrophobic core that can accept even greater quantities of drug. The ζ-potential was observed to increase as loading increased (Figure 6B). Interestingly, the aqueous diameter was observed to increase for nanoparticles formed from CDN-3 with a 5% theoretical loading compared to other loadings (Figure 6C). This observation of an outlying high diameter at 5% theoretical loading was reproducible in at least three independent experimental replicates.
Figure 6.

Plot of (A) average experimental loading, (B) average ζ-potential, and (C) mean hydrodynamic size of CDN-3 nanoparticles against theoretical loading of panobinostat. (D) Expanded hydrophobic core stabilizing drug complexation with cyclodextrin cavity. Error bars show the standard deviation of three separate formulations.
This could likely be ascribed to cyclodextrin–drug complexation at lower theoretical loadings owing to an expanded hydrophobic core, which facilitates easier access to cyclodextrin units (Figure 6D).
CDN-4, synthesized with mPEG550-amine, exhibited the highest extent of panobinostat incorporation, with measured loading of up to ∼30% (Figure 7A). This high level of experimental loading was achieved at only a moderate cost to encapsulation efficiency (∼82% encapsulation efficiency). The particle size was found to decrease steadily with increased drug loading, while the near-neutral ζ-potential decreased slightly with increasing drug loading (Figure 7B,C). This is likely due to the association of ionized panobinostat molecules with the mPEG550 shell of the particles (Figure 7D). We speculate that CDN-4 condenses into a more tightly assembled nanoparticle upon incorporation of additional panobinostat.
Figure 7.

Plot of average (A) experimental loading, (B) average ζ-potential, and (C) mean hydrodynamic size of CDN-4 nanoparticles against theoretical loading of panobinostat. (D) mPEG550–drug interaction stabilizing higher-loaded CDN-4 nanoparticles. Error bars show the standard deviation of three separate formulations.
Comparison of the loading, size, and surface charge of particles formed by these different CDNs for a single encapsulated drug yields important insight into the mechanisms of loading and self-assembly that enables such a high degree of drug incorporation. For CDNs 1–3 (amine-functionalized CDNs), the ζ-potential was observed to increase with increased drug loading. This observation supports an ionic interaction of panobinostat with surface-exposed amine groups, thereby facilitating drug solubilization. Comparing CDN-1, CDN-2, and CDN-3, the experimental drug loading was found to increase with an expansion of the predicted size of the hydrophobic core of the nanoparticles, supporting additional hydrophobic interactions as being significant for loading drug and promoting self-assembly. These hydrophobic interactions likely explain the aggregate formation in CDN-1, which can be contrasted with the stability of nanoparticles formed by CDNs 2 and 3 that possess a greater capacity to incorporate drug. The high loading of panobinostat in nanoparticles formed by CDN-4 (mPEG550 functionalized) suggests an interaction of panobinostat with the mPEG550 outer shell, which could be a similar loading mechanism to what we have previously reported for the HDAC inhibitor quisinostat and PEG-coated polyester nanoparticles.14 The near-neutral ζ-potential of these particles precludes the possibility of an acid–base interaction between panobinostat and the particle surface. We therefore attribute the drug-loading induced negative ζ-potential to the alignment of ionized drug molecules aligning along the mPEG550 exterior shell of the particle. This drug–polymer interaction would account for the substantially higher drug loading (and minimal saturation) observed for CDN-4 compared to other structural variations.
3.3. Controlled Release
Controlled release of panobinostat was studied in vitro for two different theoretical loadings of CDN-2, 3, and 4 in PBS at 37 °C. We describe these data as a function of theoretical rather than measured loading for simplicity of data description, with measured loading for each formulation described in Figures 5–7. Controlled release from CDN-1 was not studied due to the large and highly variable size of resultant particles, which makes it unsuitable for any further development. All CDNs studied provided effective controlled release, with release times ranging between 5 and 24 days for different CDN structures. CDN-4 was found to have the slowest release kinetics in comparison to CDNs 2 and 3, presumably due to slowed diffusion of panobinostat through or enhanced partitioning within the mPEG550 layer. Experimental loading was observed to affect controlled release. For instance, highly loaded CDN-2 was observed to completely release panobinostat within 4 days (Figure 8; purple triangle) as compared to 8 days (blue circle) for the less highly loaded formulation. Similarly, highly loaded CDN-4 was found to completely release panobinostat within 13 days as compared to 24 days for the less highly loaded formulation (see the Supporting Information). The apparently slower release kinetics for less highly loaded particles for CDN-2 and 4 suggests that drug may exhibit a greater surface association when particles are highly loaded. In contrast, CDN-3 exhibits similar release kinetics for both higher and lower drug-loaded samples (red square vs orange diamond), signifying the role of an expanded hydrophobic core in enabling stable incorporation of larger quantities of drug.
Figure 8.

Controlled release of 5% (w/w) and 20% (w/w) theoretically drug-loaded CDNs 2–4 samples. While CDN-2 completely released the drug within 4–8 days, CDN-3 and CDN-4 were found to completely release the drug within 7–8 and 13–24 days (inset), respectively.
3.4. Library of Drug-Loaded CDNs
Initial studies with panobinostat suggested that the loading mechanism includes both hydrophobic (within the cyclodextrin/polymer core) and ionic (possibly within the amine or PEG layer) interactions. We predicted that these interactions would be useful for the solubility, as well as encapsulation of other terminally ionizable molecules. To test this prediction, we attempted to load other HDACi’s containing terminally ionizable hydroxamic acids (quisinostat, dacinostat, and givinostat), as well as nonpolar molecules that are either nonionizable (nile red), possess reduced ionization capacity (camptothecin), or contain an alternative terminally ionizable boronic acid group (bortezomib). All drugs were formulated at a concentration (1 mg/mL), which is expected to be well above their aqueous solubility limits. As a control hydrophilic drug, we also attempted to encapsulate cytarabine with all of the CDNs, but this yielded poor experimental loadings (data not shown).
Drugs with terminally ionizable moieties in their molecular structure (quisinostat, panobinostat, dacinostat, givinostat, and bortezomib) loaded particularly well in CDNs 1–3, supporting the role of surface-amine functionality enabling drug interaction and solubilization via H-bonding (Figure 9). Drug loading was found to increase from CDN-1 to CDN-3 for all of the HDACi’s investigated except for givinostat (ITF2357). Given that givinostat possesses the bulkiest, inflexible hydrophobic end, these data support the role of a flexible hydrophobic core of the CDNs for effective encapsulation of drugs. CDN-4 was found to load all of the HDACis and bortezomib except dacinostat. The molecular structures of quisinostat, panobinostat, givinostat, and bortezomib are relatively slender, flexible, and polar. However, the structure of dacinostat (LAQ824) bears an ionizable (hydroxyl) offshoot midway throughout the molecule, which perturbs the hydrophobic end. We speculate that the flexible, slender structures of these other HDAC inhibitors facilitate efficient association with the exterior mPEG550 layer of the particle, which is not possible in the case of dacinostat owing to its steric incompatibility. These results highlight ionizability as being the most significant factor for solubilization and drug incorporation in particles formed from CDNs 1 to 3, while steric compatibility as being an essential requirement for drug loading into particles formed from CDN-4. Although some of these molecules possess amine groups that are capable of being ionized (e.g., for panobinostat, secondary amine (-benzylethyl-) and heterocyclic amine (2-methyl indole) with pKa ≈ 10 and pKa > 10 respectively), they are not expected to play a significant role in enabling drug encapsulation at the neutral pHs used for nanoparticle self-assembly. Camptothecin and nile red were found to be predominantly insoluble in the CDNs, which posited because they lack the terminally ionizable moieties necessary for effective drug–nanoparticle interactions. Given that the mechanism of drug loading involves ionization, it is possible that drug release could be influenced by microenvironmental pH, which will be an interesting avenue for future work.
Figure 9.

Library of drug-loaded CDN nanoparticles. The average experimental drug loading values of various small molecules for CDNs 1–4 are shown for a theoretical loading of 10% (w/w), including an assortment of HDACi and non-HDACi molecules: (A) quisinostat, (B) panobinostat, (C) dacinostat, (D) givinostat, (E) bortezomib, (F) camptothecin, and (G) nile red. Molecular structures of compounds illustrate the hydrophobic (red) and terminally ionizable (green) moieties. Error bars are reported as the standard deviation of three separate experimental repeats.
3.5. Stability, Tolerability, and Potency of Panobinostat-Loaded CDNs
The ζ-potential of colloidal materials plays an essential role in how particles interact with tissues and cells, governing biodistribution, cellular uptake, and drug lifespan in vivo.32 Tumor cell membranes tend to be more negative in charge than healthy cells, which some investigators have utilized for the purposes of targeting nanoparticle delivery. However, less negative surface charges can promote nonspecific cellular uptake or be plagued by aggregation due to the absorption of plasma proteins.33 As an alternative strategy, PEGylation of nanoparticles can improve their plasma half-life and shield the encapsulated drug during circulation, which can increase the drug’s lifespan.34 Thus, to move toward the assessment of nanoparticle CDNs for drug delivery, we focused on a comparison of CDN-3, bearing free amines and slightly positive surface charge, and CDN-4, for which PEGylation has brought the surface charge close to neutral.
We first focused on colloidal stability, assessing changes in the hydrodynamic diameter of panobinostat-loaded CDN-3 and CDN-4 under three different aqueous media conditions—acidic (pH = 4.0), PBS (pH = 7.4), and basic (pH = 10.2) (see the Supporting Information). CDN-3 was found to be particularly stable in acidic conditions over a longer period (up to 10 days). Under basic conditions, the particles were found to swell over time resulting, eventually yielding aggregate formations. This apparent behavior is likely due to the protonated amine functionalities on the surface, which renders colloidal stability under normal acidic conditions. We also observed that drug-empty CDN-3 exhibited toxicity against cells and was poorly tolerated in vivo, yielding neurological effects and death immediately after administration at the doses that would be needed to achieve drug effects. In contrast, drug-loaded CDN-4 nanoparticles were observed to maintain a small size when suspended in pure water but formed aggregates over time in PBS and both pHs.
Large aggregates (>1000 nm) were observed in acidic media (pH = 4.0) within 24 h, which could be considered either a detriment or a benefit to the system (for example, it is possible that CDNs could aggregate or collapse to deliver drug within the acidic microenvironment of a tumor, which could enhance tumor exposure to drug).35 Following administration to healthy mice, CDN-4 was observed to be highly biocompatible at therapeutically relevant concentrations for intravenous (we tested up to 80 mg/kg CDN-4 via lateral tail vein injection), intrathecal (we tested up to 3 μg of CDN-4 via cisterna magna injection), and intratumoral (we tested up to 100 μg of CDN-4 via convection-enhanced delivery (CED)) routes. In fact, CED of CDN-4 enabled a much higher deliverable dose of panobinostat (we tested up to 30 μg drug) compared to panobinostat solubilized within free cyclodextrins (maximum injectable dose of 2 μg of drug due to the limitations of pCD solubility). Thus, given the small size, near-neutral surface charge, and good tolerability, we were motivated to move forward with CDN-4.
Bioactivity of panobinostat-loaded CDN-4 was evaluated in GL261 cells, a murine glioblastoma line. Following 72 h of incubation, free panobinostat exhibited an IC50 value of 0.17 μM, while panobinostat-loaded CDN-4 resulted in IC50 values of 0.56 μM (Figure 10A). To test whether panobinostat loses activity as a consequence of formulation, we subjected panobinostat without CDN materials to the complete formulation process and assessed IC50; non-CDN formulated panobinostat possessed an IC50 value of 0.23 μM, suggesting that the apparent increase in IC50 for panobinostat-loaded CDN-4 is not due to degradation of the drug. When BODIPY-labeled CDN-4 nanoparticles were incubated with cells for 48 h, a perinuclear fluorescence signal was observed, confirming that nanoparticles are effectively internalized by cells (Figure 10B). Thus, the apparent increase in IC50 observed for panobinostat loaded into CDN-4 likely represents retention of panobinostat within the nanoparticle core. Increased IC50 following drug encapsulation due to reduced cellular availability of the encapsulated drug has been reported for other nanoparticle preparations but does not necessarily represent a problem for in vivo development of these systems, since nanoparticle encapsulation will confer advantages in vivo that are not captured in cellular assays.36−39
Figure 10.
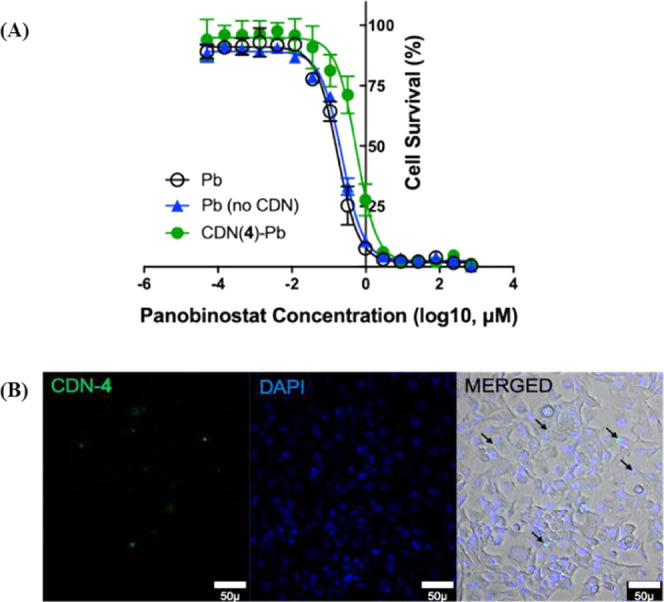
(A) In vitro GL261 cell viability assay for panobinostat-loaded CDN-4 formulation. IC50 values after 72 h of incubation: free Pb = 0.17 μM, CDN-4-Pb = 0.56 μM, and non-CDN-formulated Pb = 0.23 μM. Error bars are reported as the standard deviation of three separate experiments. Freely solubilized panobinostat is abbreviated as Pb. (B) Association of BODIPY dye-labeled CDN-4 (λex = 503 nm; λem = 509 nm) to GL261 cells confirmed via fluorescence microscopy experiments.
3.6. Drug Delivery and Efficacy of Panobinostat-Loaded CDNs
For therapeutic evaluation, we focused on the treatment of intracranial tumors via intratumoral injection or CED. We based our dose selection on prior work demonstrating the efficacy of cyclodextrin-solubilized panobinostat (referred to here as pCD) in the rat models of diffuse intrinsic pontine glioma (DIPG).40 In our study, mice received either a normal dose formulated in free cyclodextrin or CDN nanoparticles (pCD or pCDN, 1 μg of panobinostat) or a high dose that was only possible for the nanoparticle drug (pCDN-HD, 30 μg of panobinostat). All treatments were well tolerated, and no significant weight loss was observed following administration of any treatment, confirming tolerability. Neither pCD nor pCDN exerted any effect on tumor growth when delivered at the normal dose (see the Supporting Information). In contrast, the treatment of tumors with pCDN-HD yielded a significant slowing of tumor growth compared to CDN vehicle or saline-injected controls following treatment administration (Figure 11A). MALDI MSI analysis was conducted on coronal brain tissue sections, which confirmed higher exposure of tumor to drug: 1 h after treatment administration, pCDN-HD achieved a concentration of 241 μM of panobinostat in the tumor site compared to only 33 μM for pCD administered at the normal dose (Figure 11B). Panobinostat concentration was below the LOD and LOQ in nontumor brain regions for either treatment. Ultimately, median survival was 20 days for saline and CDN vehicle-treated mice compared to 22 days for pCDN-HD-treated mice (see the Supporting Information). Thus, treatments did not significantly prolong survival in this model, although it is worth noting that higher or more frequent dosing is possible and would be expected to enhance therapy. Recent work by Tosi and colleagues showed that CED of panobinostat in mice was ineffective at prolonging survival when provided as a single dose, but the same concentration of panobinostat was highly effective when dosed repeatedly.41 It will be the subject of future studies in our group to determine dose level, dose frequency, and ideal infusion parameters to completely optimize therapy. The delivery and efficacy studies described here highlight the impact of high drug loading within nanoparticles for the effective dosing of therapeutic compounds to the central nervous system, thus opening new avenues for future therapeutic development.
Figure 11.

(A) Tumor growth determined by the change in the tumor size (mean ± standard error of the mean (SEM)) for saline (n = 9), CDN vehicle (n = 9), and pCDN-HD (n = 10)-treated mice. The fold change in tumor size was significantly decreased 4 days following treatment for pCDN-HD mice compared to CDN vehicle control (p = 0.03, two-tailed Student’s t-test). (B) Mass spectrometry imaging of pCD and pCDN-HD distribution in the GL261 mouse model following 1 h after intratumoral convection-enhanced delivery (8 days post tumor induction). Brightfield microscopy imaging of hematoxylin and eosin (H&E) stained tissue sections shows the tumor in striatum. The molecular ion images of panobinostat from an animal treated with pCD (1 μg dose) show drug level below the lower limit of quantification (LOQ) for the nontumor brain region (3.6 μM) with a localized 33.0 μM concentration in the tumor. The pCDN-HD (30 μg dose)-dosed animal also showed drug levels below the LOQ for the nontumor brain region (6.0 μM) and a localized 241.1 μM concentration in the tumor region.
These studies present a novel approach for generating nanoparticles that are highly loaded with HDACi drugs or other molecules bearing terminally ionizable moieties. To our knowledge, the maximum reported HDACi loading in the polymeric system is ∼9%,14 which in turn is higher than most other lipid-derived systems (∼2.5 to 5.0%).42−44 The particle system described herein exhibits a maximum observed loading of ∼30% and also sustains drug release for prolonged periods of time in vitro (13–24 days). Importantly, all of the tested HDACi could be loaded at a relatively high capacity in the CDN systems, which confirms the utility of our dual loading mechanism. Electrolyte/cosolvent induced ionic drug loading45 and synthetic polymer constructs with ionizable backbone46 have been widely reported in the literature as reliable ionic drug loading strategies. Nevertheless, the general utility of these strategies is questionable, as the choice of the inorganic salt/electrolyte is highly drug specific and requires exhaustive optimization to achieve good drug loading. Our work directly addresses the problem of finding a generalizable strategy for loading drugs bearing terminally ionizable moieties into polymeric nanoparticles. The modular nature of the CDN platform described here also provides the advantage of enabling easy modification to both the internal and external structures, thereby tuning the material properties for drug loading and sustained release. Further, focusing specifically on CDN-4, we demonstrate that the system is biocompatible and well-tolerated for administration by various routes, and we show a proof-of-concept that the high loading of CDN-4 enables panobinostat efficacy in a murine model of glioblastoma. Ultimately, these studies focused on the therapeutic evaluation of the CDN platform in glioblastoma, which bears significant translational relevance. Panobinostat solubilized in cyclodextrin is being evaluated in phase I clinical trial by us for the treatment of recurrent pediatric medulloblastoma via intrathecal administration (NCT04315064) as well as by others for the treatment of DIPG via CED (NCT03566199, NCT04264143). Better formulation approaches thus may have the opportunity to positively impact patient care in the future.
4. Conclusions
We have demonstrated the general utility of the β-cyclodextrin-poly (β-amino ester) network as an efficient platform for loading and delivery of hydrophobic drugs bearing terminally ionizable moieties. The architectures of these networks are key to their drug loading capacities and biophysical characteristics, as revealed by structure–property relationships. While both the linker and the surface functionality determine the experimental loadings of the drug, the amine-CDNs (1, 2, and 3) displayed a positive surface charge and the mPEG550-CDN (CDN-4) exhibited a near-neutral surface charge. We discovered that a slender, terminally ionizable drug molecular structure (e.g., panobinostat and quisinostat) maximizes drug encapsulation in these networks. While CDN-3 completely releases the drug over 7 days, CDN-4 reduces the release kinetics to affect the complete release within 13–24 days, depending on the drug loading. CDN-4 nanoparticles are internalized by cells and deliver bioactive panobinostat to GL261 cells. Panobinostat incorporated in CDN-4 enabled treatment of orthotopic glioblastoma via improved drug dosing that enhanced exposure of tumor to drug. In sum, these studies describe the development of a generalizable strategy for nanoparticle encapsulation of terminally ionizable, hydrophobic molecules that will be an important step forward in developing HDACi nanomedicine for the treatment of disease.
Acknowledgments
This project was supported by funding from the National Institutes of Health (R21NS107985, R01NS116657, R01NS111292, and R01HD099543) and the Ian’s Friends Foundation. We are thankful to the Nuclear Magnetic Resonance and Mass Spectrometry facility at M.D. Anderson Cancer Center and BioScience Research Collaborative (BRC) at Rice University, respectively. We also thank Dr. Shwetal Mehta of Barrow Neurological Institute for providing us with the GL261-LucNeo cells. MALDI MSI studies were funded in part by NIH U54 CA210180 MIT/Mayo Physical Science Oncology Center for Drug Distribution and Drug Efficacy in Brain Tumors, the Ferenc Jolesz National Center for Image Guided Therapy NIH P41-EB-015898, and by the Pediatric Low-Grade Astrocytoma Program at the Pediatric Brain Tumor Foundation. During this study, SAS was in receipt of an NIH Fellowship (T32EB025823).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.0c22587.
MALDI-TOF spectra of acrylated β-cyclodextrin; 1H NMR spectra; SEM images of CDN nanoparticles; controlled release study of CDN-4; stability studies of panobinostat-loaded CDNs; MALDI MSI calibration; and survivals, weight changes, and imaging data for therapeutic studies (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Campos B.; Bermejo J. L.; Han L.; Felsberg J.; Ahmadi R.; Grabe N.; Reifenberger G.; Unterberg A.; Herold-Mende C. Expression of Nuclear Receptor Corepressors and Class I Histone Deacetylases in Astrocytic Gliomas. Cancer Sci. 2011, 102, 387–392. 10.1111/j.1349-7006.2010.01792.x. [DOI] [PubMed] [Google Scholar]
- Ceccacci E.; Minucci S. Inhibition of Histone Deacetylases in Cancer Therapy: Lessons from Leukaemia. Br. J. Cancer 2016, 114, 605–611. 10.1038/bjc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorakova M.; Vanek T. Histone Deacetylase Inhibitors for the Treatment of Cancer Stem Cells. Med. Chem. Commun. 2016, 7, 2217–2231. 10.1039/C6MD00297H. [DOI] [Google Scholar]
- Giudice F. S.; Pinto D. S.; Nör J. E.; Squarize C. H.; Castilho R. M. Inhibition of Histone Deacetylase Impacts Cancer Stem Cells and Induces Epithelial-Mesenchyme Transition of Head and Neck Cancer. PLoS One 2013, 8, e58672 10.1371/journal.pone.0058672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornago M.; Garcia-Alberich C.; Blasco-Angulo N.; Vall-llaura N.; Nager M.; Herreros J.; Comella J. X.; Sanchis D.; Llovera M. Histone Deacetylase Inhibitors Promote Glioma Cell Death by G2 Checkpoint Abrogation Leading to Mitotic Catastrophe. Cell Death Dis. 2014, 5, e1435 10.1038/cddis.2014.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone R. W. Histone-Deacetylase Inhibitors: Novel Drugs for the Treatment of Cancer. Nat. Rev. Drug Discovery 2002, 1, 287–299. 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- Cote S.; Rosenauer A.; Bianchini A.; Seiter K.; Vandewiele J.; Nervi C.; Miller W. H. Jr. Response to Histone Deacetylase Inhibition of Novel PML/RARalpha Mutants Detected in Retinoic Acid-Resistant APL Cells. Blood 2002, 100, 2586–2596. 10.1182/blood-2002-02-0614. [DOI] [PubMed] [Google Scholar]
- Pandolfi P. P. Transcription Therapy for Cancer. Oncogene 2001, 20, 3116–3127. 10.1038/sj.onc.1204299. [DOI] [PubMed] [Google Scholar]
- Fischer A.; Sananbenesi F.; Wang X.; Dobbin M.; Tsai L. H. Recovery of Learning and Memory is Associated with Chromatin Remodelling. Nature 2007, 447, 178–182. 10.1038/nature05772. [DOI] [PubMed] [Google Scholar]
- Dash P. K.; Orsi S. A.; Moore A. N. HDAC Inhibition Combined with Behavioral Therapy Enhances Learning and Memory Following Traumatic Brain Injury. Neuroscience 2009, 163, 1–8. 10.1016/j.neuroscience.2009.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arts J.; King P.; Mariën A.; Floren W.; Beliën A.; Janssen L.; Pilatte I.; Roux B.; Decrane L.; Gilissen R.; Hickson I.; Vreys V.; Cox E.; Bol K.; Talloen W.; Goris I.; Andries L.; Jardin M. D.; Janicot M.; Page M.; van Emelen K.; Angibaud P. JNJ-26481585, a Novel “Second-Generation” Oral Histone Deacetylase Inhibitor Shows Broad-Spectrum Preclinical Antitumoral Activity. Clin. Cancer Res. 2009, 15, 6841–6851. 10.1158/1078-0432.CCR-09-0547. [DOI] [PubMed] [Google Scholar]
- Carol H.; Gorlick R.; Kolb E. A.; Morton C. L.; Manesh D. M.; Keir S. T.; Reynolds C. P.; Kang M. H.; Maris J. M.; Wozniak A.; Hickson I.; Lyalin D.; Kurmasheva R. T.; Houghton P. J.; Smith M. A.; Lock R. Initial testing (Stage 1) of the Histone Deacetylase Inhibitor, Quisinostat (JNJ-26481585), by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2014, 61, 245–252. 10.1002/pbc.24724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal B.; Baird R.; Kristeleit R. S.; Plummer R.; Cowan R.; Stewart A.; Fourneau N.; Hellemans P.; Elsayed Y.; Mcclue S.; Smit J. W.; Forslund A.; Phelps C.; Camm J.; Evans T. R. J.; de Bono J. S.; Banerji U. A phase I Study of Quisinostat (JNJ-26481585), an Oral Hydroxamate Histone Deacetylase Inhibitor with Evidence of Target Modulation and Antitumor Activity, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2013, 19, 4262–4272. 10.1158/1078-0432.CCR-13-0312. [DOI] [PubMed] [Google Scholar]
- Householder K. T.; DiPerna D. M.; Chung E. P.; Luning A. R.; Nguyen D. T.; Stabenfeldt S. E.; Mehta S.; Sirianni R. W. pH driven Precipitation of Quisinostat onto PLA-PEG Nanoparticles Enables Treatment of Intracranial Glioblastoma. Colloids Surf., B 2018, 166, 37–44. 10.1016/j.colsurfb.2018.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E. C.; Min Y.; Palm R. C.; Fiordalisi J. J.; Wagner K. T.; Hyder N.; Cox A. D.; Caster J.; Tian X.; Wang A. Z. Nanoparticle Formulations of Histone Deacetylase Inhibitors for Effective Chemoradiotherapy in Solid Tumors. Biomaterials 2015, 51, 208–215. 10.1016/j.biomaterials.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed E. A.; Zhao Y.; Meshali M. M.; Remsberg C. M.; Borg T. M.; Foda A. M. M.; Takemoto J. K.; Sayre C. L.; Martinez S. E.; Davies N. M.; Forrest M. L. Vorinostat with Sustained Exposure with High Solubility in Poly(ethylene glycol)-b-poly(d, l-lactic acid) Micelle Nanocarriers: Characterization and Effects on Pharmacokinetics in Rat Serum and Urine. J. Pharm. Sci. 2012, 101, 3787–3798. 10.1002/jps.23265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Liang Y.; Liu X.; Zhou S.; Liu L.; Zhang F.; Xie C.; Cai S.; Wei J.; Zhu Y.; Hou W. PLGA-PEG Nanoparticles Coated with Anti-CD45RO and Loaded with HDAC Plus Protease Inhibitors Activate Latent HIV and Inhibit Viral Spread. Nanoscale Res. Lett. 2015, 10, 413 10.1186/s11671-015-1112-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm S.; Tavares A. J.; Dai Q.; Ohta S.; Audet J.; Dvorak H. F.; Chan W. C. W. Analysis of Nanoparticle Delivery to Tumors. Nat. Rev. Mater. 2016, 1, 16014 10.1038/natrevmats.2016.14. [DOI] [Google Scholar]
- Somoza J. R.; Skene R. J.; Katz B. A.; Mol C.; Ho J. D.; Jennings A. J.; Luong C.; Arvai A.; Buggy J. J.; Chi E.; Tang J.; Sang B. C.; Verner E.; Wynands R.; Leahy E. M.; Dougan D. R.; Snell G.; Navre M.; Knuth M. W.; Swanson R. V.; McRee D. E.; Tari L. W. Structural Snapshots of Human HDAC8 Provide Insights into the Class I Histone Deacetylases. Structure 2004, 12, 1325. 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Davis M. E.; Brewster M. E. Cyclodextrin-based Pharmaceutics: Past, Present and Future. Nat. Rev. Drug Discovery 2004, 3, 1023–1035. 10.1038/nrd1576. [DOI] [PubMed] [Google Scholar]
- Coulter T.; Damment S.; Pace A.; Palmer D.. Cyclodextrin-Panbinostat Adduct, WO Patent WO2017167837A12017.
- Bonnet V.; Gervaise C.; Djedaïni-Pilard F.; Furlan A.; Sarazin C. Cyclodextrin Nanoassemblies: A Promising Tool for Drug Delivery. Drug Discovery Today 2015, 20, 1120–1126. 10.1016/j.drudis.2015.05.008. [DOI] [PubMed] [Google Scholar]
- Çirpanli Y.; Bilensoy E.; Doğan A. L.; Calis S. Comparative Evaluation of Polymeric and Amphiphilic Cyclodextrin Nanoparticles for Effective Camptothecin Delivery. Eur. J. Pharm. Biopharm. 2009, 73, 82–89. 10.1016/j.ejpb.2009.04.013. [DOI] [PubMed] [Google Scholar]
- Caldera F.; Tannous M.; Cavalli R.; Zanetti M.; Trotta F. Evolution of Cyclodextrin Nanosponges. Int. J. Pharm. 2017, 531, 470–479. 10.1016/j.ijpharm.2017.06.072. [DOI] [PubMed] [Google Scholar]
- Clark A. J.; Wiley D. T.; Zuckerman J. E.; Webster P.; Chao J.; Lin J.; Yen Y.; Davis M. E. CRLX101 Nanoparticles Localize in Human Tumors and Not in Adjacent, Nonneoplastic Tissue after Intravenous Dosing. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 3850–3854. 10.1073/pnas.1603018113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil E. S.; Wu L.; Xu L.; Lowe T. L. β-Cyclodextrin-poly (β-Amino Ester) Nanoparticles for Sustained Drug Delivery across the Blood–Brain Barrier. Biomacromolecules 2012, 13, 3533–3541. 10.1021/bm3008633. [DOI] [PubMed] [Google Scholar]
- Fortunati N.; Marano F.; Bandino A.; Frairia R.; Catalano M. G.; Boccuzzi G. The Pan- Histone Deacetylase Inhibitor LBH589 (Panobinostat) Alters the Invasive Breast Cancer Cell Phenotype. Int J. Oncol. 2014, 44, 700–708. 10.3892/ijo.2013.2218. [DOI] [PubMed] [Google Scholar]
- Terpos E. The Synergistic Effect of Panobinostat (LBH589) with Melphalan or Doxorubicin on Multiple Myeloma Cells; Rationale for the Use of Combination Regimens in Myeloma Patients. Leuk. Res. 2011, 35, 295–296. 10.1016/j.leukres.2010.10.019. [DOI] [PubMed] [Google Scholar]
- Shao W.; Growney J. D.; Feng Y.; O’Connor G.; Pu M.; Zhu W.; Yao Y.-M.; Kwon P.; Fawell S.; Atadja P. Activity of Deacetylase Inhibitor Panobinostat (LBH589) in Cutaneous T-cell Lymphoma Models: Defining Molecular Mechanisms of Resistance. Int. J. Cancer 2010, 127, 2199–2208. 10.1002/ijc.25218. [DOI] [PubMed] [Google Scholar]
- Pei Y.; Liu K.-W.; Wang J.; Garancher A.; Tao R.; Esparza L. A.; Maier D. L.; Udaka Y. T.; Murad N.; Morrissy S.; Seker-Cin H.; Brabetz S.; Qi L.; Kogiso M.; Schubert S.; Olson J. M.; Cho Y.-J.; Li X.-N.; Crawford J. R.; Levy M. L.; Kool M.; Pfister S. M.; Taylor M. D.; Wechsler-Reya R. J. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. 10.1016/j.ccell.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra V.; Quinti L.; Khanna P.; Paganetti P.; Kuhn R.; Young A. B.; Kazantsev A. G.; Hersch S. LBH589, A Hydroxamic Acid-Derived HDAC Inhibitor, is Neuroprotective in Mouse Models of Huntington’s Disease. J. Huntington’s Dis. 2016, 5, 347–355. 10.3233/JHD-160226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C.; Hu Y.; Yin L.; Tang C.; Yin C. Effects of Particle Size and Surface Charge on Cellular Uptake and Biodistribution of Polymeric Nanoparticles. Biomaterials 2010, 31, 3657–3666. 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- Honary S.; Zahir F. Effect of Zeta Potential on the Properties of Nano-Drug Delivery Systems - A Review (Part 1). Trop. J. Pharm. Res. 2012, 12, 255–264. 10.4314/tjpr.v12i2.19. [DOI] [Google Scholar]
- Hamidi M.; Azadi A.; Rafiei P. Pharmacokinetic Consequences of Pegylation. Drug Delivery 2006, 13, 399–409. 10.1080/10717540600814402. [DOI] [PubMed] [Google Scholar]
- He X.; Li J.; An S.; Jiang C. pH-sensitive Drug-delivery Systems for Tumor Targeting. Ther. Delivery 2013, 4, 1499–1510. 10.4155/tde.13.120. [DOI] [PubMed] [Google Scholar]
- Lundberg B. B.; Griffiths G.; Hansen H. J. Cellular Association and Cytotoxicity of Doxorubicin-Loaded Immunoliposomes Targeted via Fab’ Fragments of an Anti-CD74 Antibody. Drug Delivery 2007, 14, 171–175. 10.1080/10717540601036831. [DOI] [PubMed] [Google Scholar]
- Duan J.; Zhang Y.; Han S.; Chen Y.; Li B.; Liao M.; Chen W.; Deng X.; Zhao J.; Huang B. Synthesis and In vitro/In vivo Anti-cancer Evaluation of Curcumin-loaded Chitosan/Poly (butyl cyanoacrylate) Nanoparticles. Int. J. Pharm. 2010, 400, 211–220. 10.1016/j.ijpharm.2010.08.033. [DOI] [PubMed] [Google Scholar]
- Li Q.; Sun Y.; Sun Y.-L.; Wen J.; Zhou Y.; Bing Q.; Isaacs L. D.; Jin Y.; Gao H.; Yang Y. Mesoporous Silica Nanoparticles Coated by Layer-by-Layer Self-assembly Using Cucurbit[7]uril for in Vitro and in Vivo Anticancer Drug Release. Chem. Mater. 2014, 26, 6418–6431. 10.1021/cm503304p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin H.; Jiang X.; Gu J.; Sha X.; Chen L.; Law K.; Chen Y.; Wang X.; Jiang Y.; Fang X. Angiopep-conjugated Poly(ethylene glycol)-co-poly(e-caprolactone) Nanoparticles as Dual- Targeting Drug Delivery System for Brain Glioma. Biomaterials 2011, 32, 4293–4305. 10.1016/j.biomaterials.2011.02.044. [DOI] [PubMed] [Google Scholar]
- Singleton W. G. B.; Bienemann A. S.; Woolley M.; Johnson D.; Lewis O.; Wyatt M. J.; Damment S. J. P.; Boulter L. J.; Killick-Cole C. L.; Asby D. J.; Gill S. S. The Distribution, Clearance, and Brainstem Toxicity of Panobinostat Administered by Convection-Enhanced Delivery. J. Neurosurg. Pediatr. 2018, 22, 288–296. 10.3171/2018.2.PEDS17663. [DOI] [PubMed] [Google Scholar]
- Tosi U.; Kommidi H.; Adeuyan O.; Guo H.; Maachani U. B.; Chen N.; Su T.; Zhang G.; Pisapia D. J.; Dahmane N.; Ting R.; Souweidane M. M. PET, Image-guided HDAC Inhibition of Pediatric Diffuse Midline Glioma Improves Survival in Murine Models. Sci. Adv. 2020, 6, eabb4105 10.1126/sciadv.abb4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbinati G.; Marsaud V.; Plassat V.; Fattal E.; Lesieur S.; Renoir J.-M. Liposomes Loaded with Histone Deacetylase Inhibitors for Breast Cancer Therapy. Int. J. Pharm. 2010, 397, 184–193. 10.1016/j.ijpharm.2010.06.046. [DOI] [PubMed] [Google Scholar]
- Tran T. H.; Chu D. T.; Truong D. H.; Tak J. W.; Jeong J.; Hoang V. L.; Yong C. S.; Kim J. O. Development of Lipid Nanoparticles for a Histone Deacetylases Inhibitor as a Promising Anticancer Therapeutic. Drug Delivery 2016, 23, 1335–1343. 10.3109/10717544.2014.991432. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Tu S.; Steffen D.; Xiong M. P. Iron Complexation to Histone Deacetylase Inhibitors SAHA and LAQ824 in PEGylated Liposomes Can Considerably Improve Pharmacokinetics in Rats. J. Pharm. Pharm. Sci. 2014, 17, 583–602. 10.18433/J3TS4V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Maaieh A.; Flanagan D. R. Salt and Cosolvent Effects on Ionic Drug Loading into Microspheres Using an O/W Method. J. Controlled Release 2001, 70, 169–181. 10.1016/S0168-3659(00)00347-3. [DOI] [PubMed] [Google Scholar]
- Neugebauer D.; Mielanczyk A.; Bielas R.; Odrobinska J.; Kupczak M.; Niesyto K. Ionic Polymethacrylate Based Delivery Systems: Effect of Carrier Topology and Drug Loading. Pharmaceutics 2019, 11, 337 10.3390/pharmaceutics11070337. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.