

Abstract
Fluorogenic protein tagging systems have been less developed for prokaryotes than for eukaryotic cell systems. Here, we extend the concept of noncovalent fluorogenic protein tags in bacteria by introducing transcription factor-based tags, namely, LmrR and RamR, for probe binding and fluorescence readout under aerobic and anaerobic conditions. We developed two chemogenetic protein tags that impart fluorogenicity and a longer fluorescence lifetime to reversibly bound organic fluorophores, hence the name Chemogenetic Tags with Probe Exchange (CTPEs). We present an extensive characterization of 30 fluorophores reversibly interacting with the two different CTPEs and conclude that aromatic planar structures bind with high specificity to the hydrophobic pockets of these tags. The reversible binding of organic fluorophores to the CTPEs and the superior photophysical properties of organic fluorophores enable long-term fluorescence microscopy of living bacterial cells. Our protein tags provide a general tool for investigating (sub)cellular protein localization and dynamics, protein–protein interactions, and prolonged live-cell microscopy, even under oxygen-free conditions.
Introduction
Biochemistry is evolving from mostly in vitro studies of macromolecules to analyses of complex processes in living cells, wherein macromolecules and multiprotein complexes are mapped three-dimensionally with high spatial and temporal resolution and full functionality. To attain this, fluorescence live-cell imaging techniques have traditionally relied on tagging specific proteins with genetically encoded fluorescent proteins (FPs), such as green fluorescent protein (GFP) and analogous proteins.1,2 FPs are target-specific but often fall short in photophysical characteristics when benchmarked against organic fluorophores. Not surprisingly, significant efforts are being made to develop strategies to make smaller FPs with improved photophysical characteristics.3−5 Organic fluorophores are alternatives to FPs as they typically have better photophysical characteristics such as greater photostability, longer fluorescent lifetimes, higher quantum yields, and a wider spectral range.1,6−10 Additionally, organic fluorophores do not require oxygen, whereas FPs do so to fold correctly, limiting their applicability in anaerobic environments. One drawback of using organic fluorophores compared to FPs is that they can interact nonspecifically with cellular components. To minimize the background from nonspecific interactions, strategies employing organic fluorophores with enhanced specificity and fluorogenicity (enhanced fluorescence upon binding target) have been developed.11,12 However, such strategies require specific chemistry for ligand binding and ligands that irreversibly or covalently bind to the modified fluorophore as exemplified in the case of peptide tags like SNAP-tag and HaloTag.6,7,13,14 The covalent linkage of the fluorophores in SNAP-tag and HaloTag does not allow replacement of the bound dye by a non-photobleached one. Improvements in long-term imaging have been achieved with the introduction of fluorogen-activating proteins (FAPs),15−19 fluorescence-activating and absorption-shifting tags (FASTs),20−23 flavin mononucleotide (FMN)-based fluorescent proteins (FbFPs),3,24 and a bilirubin-binding green fluorescent protein (UnaG).25 To date, the potential offered by the aforementioned fluorogenic systems has been exploited predominantly in eukaryotes15−19 and much less in prokaryotes20,25,26 (see Supplementary Table S1). In this report, we extend the concept of noncovalent, oxygen-independent fluorogenic protein tags to a new class of protein–dye reporters that are based on bacterial transcription factors.
We introduce a self-labeling protein tagging system that combines the best of genetic tags and organic fluorophores developed through a chemogenetic approach. The straightforward labeling strategy ensures fluorogenicity, longer fluorescence lifetime, and target recognition of noncovalently and reversibly bound dyes in various organisms and alleviates the need to synthesize or modify commercially available fluorophores. We have exploited the biochemical properties of two small bacterial transcription factors, namely, resistance antibiotic multiple regulator27,28 (RamR) and lactococcal multidrug resistance repressor29−31 (LmrR), that differ in sequence, molecular weight, and structure (Supplementary Figure S1). Both RamR (from the Gram-negative bacterium Salmonella typhimurium(28)) and LmrR (from the Gram-positive bacterium Lactococcus lactis(32)) are homodimeric proteins that contain hydrophobic pocket(s) where planar organic compounds bind noncovalently with high affinity. Under native conditions, RamR and LmrR act by repressing the synthesis of multidrug efflux pumps, and this effect is removed upon binding to organic compounds such as antibiotics. The hydrophobic pocket(s) are attractive scaffolds for developing chemogenetic tags for probe exchange (CTPEs) since they bind a variety of aromatic molecules.
Results and Discussion
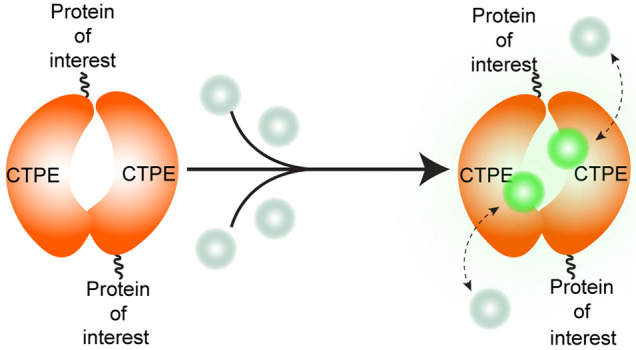
We envisioned that binding of planar organic fluorophores into the hydrophobic binding pockets of LmrR33 and RamR (hereafter named CTPEs) would improve their photophysical properties. Furthermore, engineered variants of LmrR were used to minimize interactions of the tags with DNA (see Methods section). Indeed, spectroscopic characterization of 30 organic fluorophores, several of which have applications in super-resolution microscopy and single-particle tracking, demonstrated that Bodipy, rhodamine-based fluorophores, and SNAP- and Halo-tag conjugated dyes show a significant increase in fluorescence (up to 35-fold) and absorbance (up to 10-fold) when exposed to the chemical environment of the CTPEs (Figure 1a–c, Table 1, and Supplementary Figures S2–18). Thus, the dye-binding pockets are promiscuous for a range of organic dyes, which attain fluorogenicity in hydrophobic environments.
Figure 1.

In vitro characterization of CTPEs. (a) Working principle of CTPEs: RamR (green) and LmrR (magenta). (b) Fluorescence emission spectra depicting fold change in fluorescence emission intensity of Bodipy495 upon addition of CTPEs. (c) Absorption spectra depicting fold change in the absorption of Bodipy495 after the addition of CTPEs. (d–f) Fold change in Bodipy495 fluorescence emission intensity across pH (d), NaCl concentration (e), and temperature (f) using 5 μM protein and 0.5 μM Bodipy495 in 20 mM Na-MOPS, 150 mM NaCl buffered at pH 7.0. The dotted line in panel f indicates the onset of precipitation of RamR. The solid lines in panels d–f are spline fits, and the color-shaded regions represent SD over three independent measurements. (g) Correlation plots of fold-change in absorbance and fluorescence for 7 fluorogenic dyes. Gray dotted lines connect the values for the respective dye. (h, i) Effect of oxygen on the fluorogenicity of dyes in the presence of CTPEs; panel h, RamR; panel i, LmrR. The fold change in fluorescence in the absence of oxygen (faded points) is comparable to that in the presence of oxygen (solid points). Fold-change in fluorescence of Bodipy625 in the absence of oxygen (indicated with asterisks) could not be measured due to the lack of the appropriate excitation source.
Table 1. Spectral Properties of Fluorophores in the Presence of CTPEsa.
| dye | MW (Da) | ε (M–1 cm–1) | λex/λem | F/F0 (RamR) | F/F0 (LmrR) | A/A0 (RamR) | A/A0 (LmrR) | Kd (RamR) | Kd (LmrR) | τdye (ns) | τRamR (ns) | τLmrR (ns) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DFHBI | 252.2 | 24271 | 420/495 | 1.9 | 35.1 | 2.5 | 1.7 | >10 | >10 | 0.3 | 0.4 | 1.8 |
| Bodipy488 | 262.0 | 79000 | 488/503 | 5.8 | 2.4 | 3.4 | 2.6 | 1.2 | 3.5 | 5.6 | 6.8 | 7.1 |
| Bodipy495 | 324.0 | 45000 | 495/508 | 17.4 | 6.2 | 8.9 | 6.0 | 0.2 | 2.2 | 3.8 | 6.8 | 5.1 |
| Rhodamine 6G | 479.0 | 116000 | 530/555 | 4.5 | 4.1 | 1.9 | 1.7 | 0.4 | 0.3 | 3.8 | 4.2 | 3.8 |
| Rose Bengal | 1017.6 | 90400 | 559/568 | 10.0 | 5.0 | 2.4 | 3.1 | 0.7 | 1.7 | 0.3 | 1.5 | 1.7 |
| Bodipy589 | 424.2 | 69000 | 589/622 | 2.3 | 0.6 | 3.7 | 2.3 | 0.1 | b | 4.9 | 5.9 | 5.8 |
| Bodipy625 | 450.3 | 97000 | 628/642 | 16.1 | 8.7 | 5.7 | 4.3 | 5.7 | 0.6 | 3.2 | 4.7 | 4.6 |
| Riboflavin | 376.4 | 12544 | 450/540 | 0.8 | 0.4 | b | b | b | b | 4.1 | 5.2 | 5.2 |
| AlexaFluor488 | 643.4 | 73000 | 494/519 | 0.9 | 1.0 | b | b | b | b | 3.9 | 3.9 | 4.0 |
| Bodipy FL COOH | 292.1 | 80000 | 503/511 | 0.6 | 0.7 | b | b | b | b | 5.5 | 6.3 | 5.6 |
| Eosin Y | 647.9 | 112000 | 524/543 | 1.5 | 0.8 | b | b | b | b | 1.1 | 2.4 | 2.7 |
| Bodipy R6G COOH | 340.1 | 70000 | 530/548 | 0.5 | 0.8 | b | b | b | b | 5.2 | 6.2 | 5.1 |
| 6-TAMRA | 430.5 | 92000 | 543/575 | 1.0 | 0.9 | b | b | b | b | 2.5 | 2.5 | 2.5 |
| Bodipy558 | 346.2 | 84400 | 561/569 | 0.6 | 0.6 | b | b | b | b | 5.2 | 5.6 | 5.0 |
| AlexaFluor647 | 1025.2 | 270000 | 651/672 | 11 | 0.8 | b | b | b | b | 1.1 | 1.9 | 2.6 |
| probe 6 | 682.3 | 22000 | 555/578 | 2.5 | 1.9 | b | b | b | b | 3.5 | 3.7 | 3.8 |
| probe 10 | 788.3 | 3500 | 556/576 | 4.2 | 3.8 | b | b | b | b | 2.5 | 2.7 | 2.7 |
| probe 11 | 635.3 | 58000 | 555/578 | 3.1 | 2.2 | b | b | b | b | 2.9 | 4.1 | 3.8 |
| probe 15 | 742.3 | 5200 | 555/578 | 1.8 | 1.3 | b | b | b | b | 2.5 | 2.8 | 2.8 |
| probe 22 | 579.2 | 40000 | 505/527 | 2.7 | 2.5 | b | b | b | b | 4.1 | 4.1 | 4.1 |
| probe 23 | 685.2 | 4500 | 510/531 | 4.2 | 5.1 | b | b | b | b | 3.8 | 4.2 | 4.2 |
| probe 29 | 661.3 | 109000 | 615/635 | 2.2 | 2.0 | b | b | b | b | 3.2 | 3.6 | 3.3 |
| probe 33 | 767.4 | 260 | 618/635 | 1.1 | 1.0 | b | b | b | b | 3.1 | 3.2 | 3.3 |
| EtBr | 394.3 | 5450 | 360/618 | 3.6 | 1.5 | b | b | b | b | b | b | b |
| Hoechst 33342 | 616.0 | 47000 | 361/460 | 33.5 | 65.4 | b | b | b | b | b | b | b |
| DPH | 461.6 | 88000 | 350/395 | 17.9 | 13.0 | b | b | b | b | b | b | b |
| NPN | 219.3 | 26000 | 350/420 | 36.5 | 7.0 | b | b | b | b | b | b | b |
| DAPI | 277.3 | 27000 | 358/461 | 2.7 | 1.4 | b | b | b | b | b | b | b |
| ANS | 299.3 | 8000 | 380/470 | 15.1 | 6.2 | b | b | b | b | b | b | b |
| Thioflavin T | 318.9 | 26600 | 450/480 | 27.5 | 20.6 | b | b | b | b | b | b | b |
DFHBI, 3,5-difluoro-4-hydroxybenzylidene imidazolinone; probes 6, 10, 11, 15, 22, 23, 29, and 33 are MaP probes developed by Prof. Kai Johnsson’s laboratory13 for protein with SNAP- or Halo-Tags but show fluorogenic behavior with CTPEs. EtBr, ethidium bromide; DPH, 1,6-diphenyl-1,3,5-hexatriene; NPN, 1-N-phenylnaphthylamine; DAPI, 4′,6-diamidino-2-phenylindole; ANS, 8-anilinonaphthalene-1-sulfonic acid (ANS). The dyes in the last group, although fluorogenic, are nonspecific intercalators in living cells and therefore not useful in our technology. The ε values are given by the manufacturer and correspond to values in organic solvents (methanol, ethanol, etc.). F/F0 (RamR/LmrR), fold change in fluorescence of dye in the presence of purified CTPE compared to dye alone; A/A0 (RamR/LmrR), fold change in absorbance of dye in the presence of purified CTPE compared to dye alone; Kd (RamR/LmrR), dissociation constants in μM; τdye, fluorescence lifetime of dye alone; τRamR/LmrR, fluorescence lifetime of dye in the presence of purified CTPE. Measurements were done in 20 mM Na-MOPS, 150 mM NaCl buffered at pH 7.0 with 50 μM CTPEs, and a dye concentration of 1 μM. Data shown is an average of three independent measurements.
Not determined.
First, we characterized the physicochemical robustness of the purified CTPEs in detail in vitro. Using Bodipy495, the dye with the highest fluorogenicity, we show that both CTPEs are insensitive to pH in the range from 4 to 8, salt concentrations up to 880 mM, temperatures up to 55 °C (Figure 1d–f), and crowding agent Ficoll70 (Supplementary Figure S19), allowing applications in diverse cellular and noncellular environments. Next, we evaluated in-depth the spectral properties of 30 commonly available organic dyes with our CTPEs (Table 1). A representative data set of CTPEs with Bodipy625 is shown in Figure 2; the same characterization was done for 29 other dyes and is shown in Supplementary Figures S2–18. We observe marked effects of CTPE tags on the fluorogenic behavior of seven dyes (Table 1, first seven dyes), which are accompanied by corresponding increases in absorbance and fluorescence lifetime (Figure 1g, and Supplementary Figure S26), and the dyes remain fluorescent in the absence and presence of oxygen (Figure 1h,i). With most dyes (except DFHBI), the fluorescence enhancement, increased absorbance, and longer lifetimes are higher with RamR than LmrR.
Figure 2.

Representative characterization of CTPEs (RamR, green, and LmrR, magenta) with Bodipy625. (a) Structure of Bodipy625. (b) Excitation (dotted line) and emission (solid line) spectra of Bodipy625. (c) Fluorescence fold change of Bodipy625 on titration with CTPEs. (d) Fluorescence emission spectra of Bodipy625 with CTPEs at a protein/dye molar ratio of 50:1. (e) Fluorescence lifetime spectra of Bodipy625 with CTPEs at a protein/dye molar ratio of 25:1 fit with a monoexponential decay function (solid lines). (f) Bound fraction of Bodipy625 with CTPEs was obtained from a Hill fit. (g) Absorbance fold-change of Bodipy625 on titration with CTPEs. Solid lines represent spline fits, and shaded regions represent SD over three independent measurements. (h) Absorption spectra of Bodipy625 with CTPEs at a protein/dye molar ratio of 25:1. All experiments were performed at 30 °C in 20 mM K-MOPS, 150 mM NaCl buffered at pH 7.0 with 50 μM CTPEs and a dye concentration of 1 μM, unless indicated differently (panels c, f, and g).
The characterization of 30 organic fluorophores suggests the applicability of our tagging strategy in living cells for at least seven of the tested dyes based on fluorogenicity and longer fluorescence lifetime. To illustrate this, we tagged and labeled proteins in the cytoplasm, inner membrane (penicillin-binding protein 5, PBP5), and periplasm (osmotically inducible protein Y, OsmY) of Escherichia coli (Figure 3a). We chose Bodipy495 and Bodipy625 for these measurements since they show the highest fluorogenic behavior (Table 1), negligible background staining, and high cell permeability (Supplementary Figures S20). Although we observe some enhanced peripheral staining with Bodipy495 in E. coli expressing CTPEs (Figure 3a, Cytoplasm), this is not seen in cells without CTPEs (Supplementary Figure S20, Control), ruling out interference from nonspecific binding of Bodipy495 and other fluorogenic dyes to endogenous components of E. coli (Supplementary Figure S20). Furthermore, flow cytometry experiments do not show nonspecific interactions with the seven best fluorogenic dyes used in this study (Table 2 and Supplementary Figure S22). Hence, we conclude that the in vivo fluorogenic behavior of Bodipy495 and other organic dyes in E. coli results from specific binding to CTPEs. We observed some nonspecific binding of Bodipy488 and Bodipy495 in testing the application of CTPEs in the Gram-positive bacterium Lactococcus lactis but not with the five other fluorogenic dyes (Supplementary Figures S21 and S23). A summary of the pros and cons of the seven best-performing dyes for E. coli and L. lactis is given in Table 2.
Figure 3.

Compartmental labeling and oxygen-free imaging of CTPEs in E. coli. (a) Confocal and accompanying differential interference contrast (DIC) micrographs of live E. coli cells expressing CTPEs (LmrR or RamR) in the cytoplasm; CTPE fused to OsmY, which is freely diffusing in periplasm; and CTPE fused to the inner membrane-bound protein PBP5. The cells were labeled with 15 μM Bodipy495. (b) Diffusion coefficients obtained by FRAP measurements of the aforementioned cytoplasmic, periplasmic, and inner membrane proteins benchmarked against fluorescent SuperFolder mTurquiose2ox (SfTq2) and mNeongreen (mNG). Whiskers represent SD, and median values are indicated within. (c) Photobleaching of fluorescence in E. coli cells expressing cytoplasmic LmrR (magenta; supplemented with Bodipy495) or the fluorescent protein mNeongreen (mNG). (d, e) Confocal images and corresponding differential interference contrast (DIC) micrographs of E. coli cells expressing cytoplasmic CTPEs labeled with 15 μM Bodipy625 under strictly anaerobic conditions (d) and expressing cytoplasmic mNeongreen (mNG) (e); the integrated fluorescence histograms of panel e are shown in panel f. (g) Western blots of CTPEs (15 and 23 kDa for LmrR and RamR, respectively) and mNG protein (26.6 kDa) expressed in the E. coli cytoplasm under aerobic and anaerobic conditions. Scale bars are 3 μm.
Table 2. Applicability of Seven Fluorogenic Dyes with CTPEs in Living Cellsa.
| CTPE tag | DFHBI | Bodipy488 | Bodipy495 | Rhod6G | Rose bengal | Bodipy589 | Bodipy625 | |
|---|---|---|---|---|---|---|---|---|
| E. coli | LmriR | yes (12%) | yes (40%) | yes (54%) | yes (21%) | limited (6%) | yes (45%) | yes (76%) |
| RamR | limited (1%) | yes (38%) | yes (75%) | yes (23%) | limited (5%) | yes (44%) | yes (82%) | |
| L. lactis | LmriR | no | b (13%) | b (15%) | limited (3%) | yes (56%) | yes (54%) | yes (95%) |
| RamR | no | b (9%) | b (13%) | limited (2%) | yes (77%) | yes (31%) | yes (91%) | |
| S. cerevisiae | LmrR | no | no | no | no | no | no | yes (53%) |
| RamR | no | no | no | no | no | no | yes (32%) |
E. coli and L. lactis were labeled using 15 μM of the corresponding dye. We denote the applicability for dyes that label <10% of live cells as limited. The numbers in parentheses indicate the percentages of labeled cells as determined by flow cytometry (Supplementary Figures S22–24).
Nonspecific binding.
It is important to emphasize that the fluorescence enhancements of the subcompartments of E. coli are between 4- and 190-fold of the background signal in all cases (Supplementary Figure S20, S21). While higher fluorescence enhancements improve contrast and minimize the need to remove the unbound probe, existing literature data indicate that 1 order of magnitude enhancement is enough to discern specific labeling in vivo.34,35 We also confirmed specific targeting and labeling of the test proteins by performing fluorescence recovery after photobleaching (FRAP) measurements to compare the diffusion coefficients of the test proteins fused to CTPE–dye conjugates with the same test proteins fused to mTurquiose2ox (SfTq2) and mNeongreen (mNG) (Figure 3b). Indeed, we find that the diffusion coefficients of the fusion proteins with the CTPEs and SfTq2 are comparable, with no indications of higher oligomer formation or protein aggregation. Membrane proteins are expected to diffuse slower than periplasmic and cytoplasmic proteins because of the relatively high membrane viscosity. The average mobility of CTPEs in the cytoplasm is a little slower than that of mNG, presumably due to weak interactions with cell components, as reported previously for engineered GFPs.36−38
We demonstrate the superior photostability of the dyes (Bodipy495) in live E. coli cells by benchmarking the fluorescence against that of mNeongreen (mNG) (Figure 3c), one of the brightest and most photostable fluorescent proteins currently in use.39,40 The oxygen-independent fluorescence enhancement in E. coli cells expressing CTPEs under strictly anaerobic growth conditions (Figure 3d) and purified CTPEs (Figure 1h,i) is comparable to that under aerobic conditions. In contrast, under our experimental conditions, mNG fluorescence was completely absent under anaerobic conditions, and the fluorescence developed upon exposure of the cells to oxygen (Figure 3e,f). Both mNG and CTPEs expressed well under aerobic or anaerobic conditions (Figure 3g). The cell-to-cell fluorescence intensity variation in Figure 3a,d is most likely due to differences in protein expression as we observe it with both CTPE–dye conjugates and SfTq2. The expression of CTPEs and their subsequent labeling with organic dyes do not affect the cell morphology of exponentially growing Gram-negative (E. coli) and Gram-positive bacteria (L. lactis) and Saccharomyces cerevisiae (Supplementary Figure S25).
We further illustrate the usefulness of our method by targeting proteins with Bodipy625 in the cytoplasm of live E. coli, L. lactis, and S. cerevisiae cells (Figure 4). For S. cerevisiae, only Bodipy625 entered the cytoplasm in adequate amounts, sufficient for labeling. We observed 32% labeled cells with LmrR and 53% with RamR when using Bodipy625 (Supplementary Figure S24). We also tested the application of our CTPE-labeling technology in mammalian cells but observed significant background fluorescence with Bodipy625 in HEK cells; Bodipy625 accumulated in mitochondria even in nontransfected cells (Supplementary Figure S24b,c). The CTPEs natively form high-affinity dimers, but we did not observe any fluorescent punctate spots in live bacterial and yeast cells (irrespective of growth phase), and thus we have no indication that our tags cause protein aggregation (Supplementary Figures S20 and S21). Dynamic nonspecific interactions with other proteins and RNA/DNA cannot be ruled out completely, but we have no indications that they have posed a problem in our measurements.
Figure 4.

Live-cell imaging of CTPEs in prokaryotic and eukaryotic cells. (a) Fluorescence confocal images and the corresponding differential interference contrast (DIC) micrographs of E. coli, L. lactis, and S. cerevisiae cytoplasm with Bodipy625 (yellow) from at least two independent biological replicates. Live cells were labeled using 15 μM Bodipy625, and the unbound dye was washed away before fluorescence imaging (see Methods). White bars across arbitrarily picked cells indicate the CTPE-induced fluorescence enhancement, which is given as a ratio relative to the background fluorescence. Control panels are corresponding live cells lacking CTPEs. The scale bars are 3 μm.
The fluorogenic nature of the dyes upon binding to CTPEs circumvents the need to remove external fluorophores, a highly desirable feature for long-term imaging demonstrated in very few studies.13,41 The presence of an excess of dye in the cell and or medium allows the exchange of photobleached for fresh fluorophore (Figure 5a–d). To test the exchange of noncovalently bound dyes, E. coli cells expressing cytoplasmic or periplasmic CTPEs were incubated with Bodipy625, Bodipy495, or DFHBI and imaged directly, without washing of the cells. Complete binding was observed within 3 min irrespective of the tag used. We then photobleached the entire cell, using a higher laser output but observed complete recovery within 3 min. In addition, we demonstrate in E. coli that one dye can be exchanged for another one (Figure 5d), utilizing the differences in relative binding affinities of DFHBI and Bodipy625 (Table 1 and Supplementary Figures S6 and S8). Overall, these experiments show that CTPEs can exchange organic dyes irrespective of the subcompartment, cytoplasm, or periplasm of E. coli, wherein the proteins are expressed, which enables prolonged live-cell imaging relative to methods that use covalently bound organic fluorophores.
Figure 5.

Wash-free live-cell imaging and organic fluorophore exchange. (a) Fluorescence confocal images of E. coli cells expressing cytoplasmic RamR in the presence of 0.5 μM Bodipy625 (yellow) or 0.5 μM Bodipy495 (magenta) or cytoplasmic LmrR in the presence of 2.0 μM DFHBI (red). Scale bar: 3 μm. White bars across arbitrarily picked cells depict signal to background fluorescence ratios. (b, c) Repetitive photobleaching of E. coli expressing periplasmic CPTEs in the presence of 0.5 μM Bodipy625 and Bodipy495 dyes was followed by complete fluorescence recovery within 3 min for RamR (b) and LmrR (c). The scale bar is 1 μm. The first trace (I*) in panels b and c shows the uptake and binding of the indicated dyes. Red notches on the x-axis in panels b and c represent bleaching times of 60 s needed to bleach the majority of Bodipy dyes in the cell. (d) Dye-swapping experiment using DFHBI (relatively low-affinity binding) and Bodipy625 (relatively high-affinity binding) in E. coli cells expressing cytoplasmic LmrR. Shaded regions indicate the standard deviation based on 10 cells.
Finally, we show the applicability of our tagging system for screening of expression differences or mutant analyses on agar plates, using E. coli expressing CTPEs and grown on LB agar plates that were incubated with organic dyes postgrowth (Figure 6). Depending on the expression of the CPTEs, the bacterial colonies become fluorescent upon the binding of the dyes to the CPTE tags. This method can hence be used for screening of expression of proteins tagged with the CPTEs. Compared to the well-known binary Xgal blue-white screening, the fluorescence will be proportional to the expression levels of CPTE and thus makes it possible to better distinguish bacteria with high and low expression. The CTPE system thus provides a robust and straightforward method for visualizing proteins and, for example, expression screening in living cells.42,43
Figure 6.

In situ colony labeling of E. coli cells expressing CTPEs and grown on LB-agar plates with 0.1% arabinose to induce expression of CTPEs. Following overnight growth, the cells expressing CTPEs were stained with Bodipy495 (green), rhodamine 6G (yellow), or Bodipy625 (red). Enhanced fluorescence from E. coli colonies is seen in the top and middle panels. The middle panel shows E. coli growing in the shape of the text “Membrane Enzymology” labeled with the same dyes as shown in the top panel. The bottom panel shows E. coli with a control plasmid, that is, without expression of CTPEs.
The promiscuity of CTPEs in binding planar organic dyes stems from their biological function as transcriptional factors, proteins that bind drug-like molecules promoting the transcription of drug-exporting proteins. We have analyzed 30 planar organic dyes and observed an array of spectral effects upon the binding of the dyes to the CTPEs LmrR and RamR. For some of the dyes, an increase in fluorescence emission intensity is observed in the presence of both CTPEs, which correlates with an accompanying increase in dye absorbance and fluorescence lifetime. These fluorophores bind with moderate to high affinity to the CTPEs, accompanied by a significant increase in fluorescence lifetime (Table 1 and Supplementary Figure S26a,b), allowing high contrast of labeled proteins in fluorescence lifetime imaging microscopy44 without washing away the unbound dye. In general, the enhanced fluorescence of the probes is more pronounced upon binding to RamR than LmrR, except for DFHBI, likely due to (i) a single dye-binding pocket in LmrR (formed at the dimer interface) vs two apparent dye-binding pockets in RamR (Figure 1a), (ii) different microenvironments in the binding pockets of RamR and LmrR, and (iii) possible excited-state cross-talk between the two bound dye molecules in RamR. In dyes wherein the emission fluorescence is either quenched or remains unaffected, the correlation between absorption, fluorescence, and fluorescent lifetimes is not apparent. A peculiar case is eosin Y, wherein fluorescence lifetimes are equally increased in RamR and LmrR but the fluorescence emission intensity is quenched for LmrR and enhanced for RamR. The structural promiscuity of the binding sites in both proteins allows the capturing of organic molecules not limited to this study but opens up possibilities of testing other fluorophores with desired photochemical properties. For instance, most of the MaP dyes13 exhibit fluorogenicity with our CTPEs (2–5-fold) and a longer fluorescence lifetime, circumventing the need for specific chemistry between the tag and the ligand (Table 1 and Supplementary Figure S18).
Although the promiscuity of the binding sites of the CTPEs enables the use of a wide range of dyes, it also precludes us from delineating precisely the underlying processes and interactions that contribute to the fluorogenicity and longer fluorescence lifetime. The selective interaction of CTPEs with the organic dyes arises likely from the geometry and hydrophobicity of the binding pockets that are more accessible or hydrophobic than other cellular structures. We observe fluorescence enhancements of well-known intercalators (EtBr, Hoechst 33342, DPH, NPN, DAPI, ANS, and thioflavin T) with CTPEs consistent with those typically observed in nonpolar environments. The stability of the interactions between CTPEs and organic dyes at salt concentrations up to 800 mM NaCl further suggests that CTPEs interact with the dyes predominantly through hydrophobic contacts. Compared to each other, the two CTPE proteins impose a somewhat different (hydrophobic) environment to the dyes, which could be exploited further by protein engineering. In this respect, future studies aim to design monomeric variants of RamR by mutating residues at the dimer interface and decrease the size of the nonessential regions unimportant for dye binding.
In addition to hydrophobic interactions, π–π stacking involving aromatic residues (Phe155 in RamR and Trp96 in LmrR) is critical for dye binding.27−29 Indeed, the crystal structure of RamR with rhodamine 6G (PDB ID 3VVZ) confirms the nonpolar nature of the binding pocket (Supplementary Figure S26c,d). We show that the transition of the fluorogenic dyes from an aqueous medium to a relatively nonpolar medium cannot solely explain the spectral enhancements observed with CTPEs (Supplementary Figure S27). The presence of tryptophan, phenylalanine, and other hydrophobic amino acids in the binding pockets of CTPEs results in multiple hydrophobic contacts and π–π stacking interactions with the electron-donating or electron-withdrawing groups of the organic dye. These interactions likely perturb the existing electron densities and concomitantly the excitation and emission transition dipoles. The resulting properties are additionally influenced by several factors, including changes in radiative and nonradiative decay rates (affecting quantum yields), viscosity, dye conformational changes in the binding pocket not limited to tautomeric changes, reduced rotational freedom due to steric hindrance, or forced planarization of out-of-plane twisted moieties influencing the HOMO–LUMO gap in the organic dye. The overall net outcome of these interactions results in a specific spectral signature for each dye–CTPE pair. Notably, the CTPEs are stable across a wide range of physicochemical conditions, including pH, temperature, ionic strength, and oxygen and environments mimicking in vivo crowding (excluded volume effects); the latter has been mimicked by using Ficoll70 as a synthetic macromolecular crowding agent. While the affinity of binding of dyes to CTPEs is not affected by Ficoll70, the fluorogenicity is decreased (Supplementary Figure S19).
In conclusion, we report the development and extensive characterization of two chemogenetic protein tags that enable the use of organic fluorophores for live-cell imaging and dynamic studies in bacterial and lower eukaryotic cells and facilitate in vitro applications under a wide range of conditions. Our method allows the use of cheap and widely available organic fluorophores spanning the ultraviolet–visible–infrared spectrum for fast and noncovalent labeling and direct application in fluorescence lifetime imaging studies. We also demonstrate the exchange of dyes, which allows replacing photobleached dyes for fresh fluorophores and thus prolonged live-cell imaging. The stability of our CTPEs across a wide physiological range provides an imaging tool for single-molecule studies in anaerobic gut microbes and other cells and organelles in environments low in oxygen and visualization and physiological studies of bacteria (and their subcompartments) living in extreme environments. The current inapplicability of our CTPEs to mammalian cells calls for explorative studies with fluorogenic dyes not yet tested with our system, for example, malachite green and thiazole orange derivatives, or modification of existing dyes to reduce nonspecific background interactions inside the cells. We envision CTPEs to be applicable in the burgeoning field of cellular cartography, in particular for (a) real-time fluorescence monitoring for high-throughput screening of protein production and dynamics in anaerobic gut microbes45 and extremophiles,46 (b) real-time monitoring of protein stability and turnover,47 (c) acquisition of long single-molecule trajectories to characterize the diffusion of proteins in the membranes, and (d) super-resolution imaging48 and FLIM studies of cells in a wide range of environments.49
Methods
Construction of CTPE Plasmids
Escherichia coli
The pET-17b plasmids encoding lmrR and ramR under an IPTG inducible T7 promoter were used for protein expression and purification experiments. The DNA-binding capability of the LmrR was removed by substituting two lysine residues (K55 and K59) in the DNA-binding region of the protein with the negatively charged aspartic acid and neutral glutamine, respectively.50 The resulting LmrR (K55D/K59Q) allowed easier purification of the protein and has reduced interactions with DNA. For facilitating studies in E. coli BW25113, the corresponding plasmids for lmrR and ramR expression in the cytoplasm were created under the control of an arabinose-inducible promoter on a pBAD24 vector. Fusion constructs of lmrR and ramR with the periplasmic protein OsmY at the N-terminus of the resulting fusion were created on a pBAD24 vector harboring an arabinose-inducible promoter. Fusion constructs of lmrR and ramR with the inner membrane protein PBP5 (penicillin-binding protein 5) were created on the pNM077 vector harboring an IPTG-inducible trc promoter provided by Prof. Tanneke den Blaauwen (University of Amsterdam). Amplification of lmrR and ramR genes and corresponding plasmid backbones for USER based cloning51 was performed using forward and reverse primers that added a uracil residue instead of a thymine residue at flanking regions (see Appendix 2 for primer sequences). The USER reaction for ligating fragments was performed as per the manufacturer’s instructions,51 followed by the heat-shock transformation of chemically competent E. coli MC1061. Positive colonies were selected on LB–ampicillin (100 μg·mL–1) plates, and isolated plasmids were confirmed by DNA sequencing (Eurofins Genomics Germany GmbH). Thereafter the sequence-verified plasmids were transformed into E. coli BW25113 and stored as glycerol stocks (30% glycerol) until further use. The DNA sequences for constructs are provided in Appendix 3.
Lactococcus lactis
All experiments were performed on the L. lactis strain NZ9000 ΔlmrR.32 NZ9000 is a L. lactis MG1363 strain derivative containing the pepN::nisR/K(52) substitution, which in the presence of the inducer, nisin A, switches on the expression of genes from the nisA promoter. By using the pNZ8048 vector housing the nisA promoter, pNZ8048-lmrR was constructed.32 The strain NZ9000 ΔlmrR(32) lacks a chromosomal copy of the lmrR gene and served as a control. Both NZ9000 ΔlmrR(32) strain and the plasmid pNZ8048-lmrR were provided by Prof. Arnold Driessen (University of Groningen). For the construction of nisin A-inducible plasmids for ramR, we cloned the native ramR gene into a pNZC3GH vector harboring a nisin A-inducible nisA promoter. However, we failed to observe any protein expression with this construct, which possibly arose because of codon-mismatch from the GC-rich native ramR sequence (Appendix 3). Next, we codon-optimized the ramR gene for L. lactis using the graphical codon usage analyzer tool53 and synthesized the ramR gene fragment (Geneart, Regensburg, Germany). The amplification of the ramR gene and corresponding plasmid pNZC3GH backbone for USER based cloning51 was performed using forward and reverse primers that added a uracil residue instead of a thymine residue at flanking regions. The resulting plasmid pNZC3GH-ramR was confirmed by DNA sequencing (Eurofins Genomics Germany GmbH) and transformed into the L. lactis strain NZ9000 ΔlmrR(32) by electroporation. The native and codon-optimized DNA sequences for ramR are provided in Appendix 3.
Saccharomyces cerevisiae
We chose the cytoplasmic protein adenylosuccinate synthase (Ade12) for studies with our CTPEs because of uniform cytoplasmic fluorescence.54 For plasmid cloning, E. coli MC1061 was used for cloning and plasmid storage. The ade12 gene was amplified using PCR from S. cerevisiae BY4742 chromosomal DNA using forward and reverse primers that added a uracil residue instead of a thymine residue at flanking regions. For plasmid backbone amplification, the multicopy plasmid pRSII426 (housing the selectable ura3 gene and allowing expression of a target protein from a constitutive ADH1 promoter) was used with forward and reverse primers that added a uracil residue instead of a thymine residue at flanking regions. The USER reaction was performed as per the manufacturer’s instructions,51 followed by the heat-shock transformation of chemically competent E. coli MC1061. Positive colonies were selected on LB chloramphenicol (32 μg·mL–1) plates, and isolated plasmids were confirmed by DNA sequencing (Eurofins Genomics Germany GmbH).
The correct plasmids were then transformed to S. cerevisiae strain BY4709 lacking the ura3 gene enabling uracil based selection. Transformation of plasmids into S. cerevisiae was performed as described elsewhere55 with some minor modifications. In short, single colonies were inoculated into 5 mL of yeast extract peptone dextrose (YPD) media and incubated at 30 °C, 200 rpm overnight. The following day cells were diluted to OD600 ≈ 0.1 in 50 mL of media and grown at 30 °C, 200 rpm until a target OD600 ≈ 0.4–0.6 was reached. Once the target OD600 was reached, cells were pelleted, the supernatant was removed, and pellets were resuspended in sterile H2O. Cells were kept on ice throughout the transformation procedure. The wash step was repeated, and then cells were resuspended in 1 mL of 0.1 M lithium acetate. Cells were pelleted and resuspended in the required volume of 0.1 M lithium acetate before 50 μL was added to 50% (w/v) PEG4000, and samples were vortexed until homogeneous. Twenty-five microliters of 2 mg·mL–1 single-stranded salmon sperm DNA was added to the cell suspension, and samples were vortexed again. Finally, 50 μL of corresponding plasmid DNA (250 ng to 1 μg) was added to the cell suspension before vortexing and incubation at 30 °C with shaking for 30 min. Heat-shock was carried out for 25 min at 42 °C with shaking before cells were pelleted, resuspended in 200 μL of sterile H2O, and plated onto uracil lacking agar plates. After plates were incubated at 30 °C for 48–72 h, single colonies were selected for restreaking on selective agar plates to attain a monoclonal population. Single colonies from the monoclonal population were then selected to confirm positive clones by plasmid isolation and subsequent sequencing of the coding region (Eurofins Genomics Germany GmbH).
HEK-293T Cells
pmTurquoise2-Mito was a gift from Dorus Gadella56 (Addgene plasmid no. 36208). Cox8A-RmrR-FLAG is a fusion construct comprising of 29 amino acids of COX8A (a mitochondrial targeting signal), RamR codon-optimized for mammalian expression, and a FLAG-tag. Expression was in pcDNA3.1 from the constitutive CMV (human cytomegalovirus) promoter. The complete sequence of the RamR construct used is given in Appendix 3.
Fluorescence Confocal Microscopy and Phase Contrast Microscopy
Preparation of Glass Slides
To ensure the immobility of E. coli cells, we used (3-aminopropyl)triethoxysilane (APTES)-treated glass cover slides. The glass slides were first cleaned by sonicating them for 1 h in 5 M KOH, followed by rinsing at least 10 times with Milli-Q treated water and blowing off the remaining Milli-Q water with pressurized nitrogen. Next, the glass slides were immersed in acetone containing 2% v/v APTES for 30 min at RT. Thereafter we removed the acetone and APTES and rinsed the slides 10 times with Milli-Q water. Again, the remaining Milli-Q water was blown off with pressurized nitrogen. The glass slides were used within 2 days of preparation. The cells were concentrated to OD600 ≈ 1, and after addition of 20 μL of cell suspension on the APTES slide, a clean object slide was put on the top, and the whole assembly was placed inverted onto the microscope stage.
For L. lactis cells, the glass slides were first cleaned by sonicating them for 1 h in 5 M KOH, followed by rinsing at least 10 times with Milli-Q water and blowing off the remaining Milli-Q water with pressurized nitrogen. The glass slides were used within 2 days of preparation. The cells were concentrated to OD600 ≈ 1, and after addition of 20 μL of cell suspension on the cleaned slide, a clean object slide was put on the top, and the whole assembly was placed inverted onto the microscope stage.
For S. cerevisiae cells, a similar glass slide cleaning protocol as described for L. lactis cells was followed, and the cleaned slides were used in a stick-Slide 8 well chamber (Ibidi GmbH, cat. 80828). The cells were concentrated to OD600 ≈ 0.5, and 100 μL of cell suspension was added in each chamber.
Preparation of E. coli for Fluorescence Confocal Microscopy and Phase Contrast Microscopy
For each experiment, a glycerol stock of E. coli BW25113 with desired LmrR/RamR variant on a pBAD plasmid was stabbed with a sterile pipet tip and deposited in 3 mL of lysogeny broth (LB Lennox: 10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) containing 0.2% v/v glycerol and 100 μg·mL–1 ampicillin. The LB medium was then incubated at 37 °C with 200 rpm shaking. The next day, the saturated LB culture was diluted 100-fold in a 3 mL of fresh LB medium containing 0.2% v/v glycerol and 100 μg·mL–1 ampicillin. The LB medium was then incubated at 37 °C with 200 rpm shaking until the culture reached an OD600 of 0.7–0.8. At this stage, protein expression was induced using 0.1% w/v arabinose, and the cultures were then incubated at 30 °C with 200 rpm shaking overnight. The following day, the saturated LB culture was diluted 100-fold in a 3 mL of fresh LB medium containing 0.2% v/v glycerol, 0.1% arabinose, 100 μg·mL–1 ampicillin and allowed to grow until the culture reached OD600 of 0.4–0.6. These cells were now directly used for labeling and consecutive imaging. For dye labeling, 0.5 mL of OD600 = 0.6 cultures were centrifuged at 11000g for 1 min, and a final concentration of 15 μM dye was added. The pellet was gently resuspended, and the cell suspension was kept at 30 °C for 30 min. Thereafter, the suspension was centrifuged at 11000g for 1 min, and the cell pellet was washed 3 times with 1 mL of LB medium. The washing step was repeated 3 times to ensure the removal of free dye. The resulting suspension was now used for confocal fluorescence microscopy and phase-contrast microscopy.
Preparation of L. lactis for Fluorescence Confocal Microscopy and Phase Contrast Microscopy
For experiments with LmrR, a glycerol stock of Lactococcus lactis NZ9000 ΔlmrR cells with desired LmrR/RamR variant on a nisin-inducible plasmid was stabbed with a sterile pipet tip to obtain a small number of cells. Lactococcus lactis NZ9000 was grown in M17 medium (Difco, Franklin Lakes, NJ, USA) supplemented with 1% (w/v) glucose (GM17) and 5 μg·mL–1 chloramphenicol at 30 °C without shaking. We incubated the cultures at 30 °C, without shaking since L. lactis is facultatively anaerobic. The next day, the saturated culture was diluted 100× in 3 mL if fresh GM17 medium containing 5 μg·mL–1 chloramphenicol and incubated at 30 °C without shaking until OD600 reached 0.3. At this stage, we added 2 μL of nisin A solution (filtered supernatant from L. lactis NZ9700 culture), and the culture was allowed to grow overnight at 30 °C without shaking. On the morning of the next day, about 100 μL of culture was added to 4 mL of fresh GM17 medium containing 5 μg·mL–1 chloramphenicol and 2 μL of nisin A solution, to yield OD600 ≈ 0.1. The cultures were then incubated at 30 °C until OD600 reached 0.4. These cells were now directly used for labeling and consecutive imaging. For dye labeling, 0.5 mL of OD600 = 0.6 cultures was centrifuged at 11000g for 1 min, and a final concentration of 15 μM dye was added. The pellet was gently resuspended, and the cell suspension was kept at 30 °C for 30 min. Thereafter, the suspension was centrifuged at 11000g for 1 min, and the cell pellet was washed 3 times with 1 mL of LB medium. The washing step was repeated 3 times to ensure the removal of free dye. The resulting suspension was now used for confocal fluorescence microscopy and phase-contrast microscopy.
Preparation of S. cerevisiae for Fluorescence Confocal Microscopy, Phase Contrast Microscopy, and Flow Cytometry Experiments
S. cerevisiae was grown in a minimal synthetic defined (SD) media, including a yeast nitrogen base lacking riboflavin and folic acid, ammonium sulfate, and 2% glucose as a carbon source. Riboflavin was not added to prevent interactions with CTPEs. For each experiment, single colonies from uracil lacking synthetic defined (SD URA–) plates were inoculated into 5 mL of SD URA– media and incubated at 30 °C, 200 rpm overnight. The following day cells were diluted to OD600 ≈ 0.02 in 5 mL of media, grown at 30 °C, 200 rpm, and maintained in exponential phase for three consecutive days. On the day of the experiment, once the OD600 reached 0.5, cells were pelleted at 8000g for 1 min in 1.5 mL sterile Eppendorf tubes, the supernatant was removed, and pellets were resuspended in sterile SD URA– media to a final OD600 = 1. These cells were now directly used for labeling and consecutive imaging. For dye labeling, 0.5 mL of OD600 = 0.6 cultures was centrifuged at 11000g for 1 min, and a final concentration of 15 μM dye was added. The pellet was gently resuspended, and the cell suspension was kept at 30 °C for 30 min. Thereafter, the suspension was centrifuged at 11000g for 1 min, and the cell pellet was washed 3 times with 1 mL of LB medium. The washing step was repeated 3 times to ensure the removal of free dye. The resulting suspension was now used for confocal fluorescence microscopy and phase-contrast microscopy.
Preparation of Mammalian Cells (HEK293T) for Transfection and Fluorescence Confocal Microscopy
HEK293T cells (100000 cells) were cultured in a 35 mm imaging dish with a glass bottom and an imprinted 50 μm cell location grid (Ibidi; cat. no. 81148) in DMEM medium supplemented with 1% (v/v) sodium pyruvate, 1% (v/v) antibiotics, 1% (v/v) glutamine, and 10% (v/v) fetal calf serum (FCS). JetPEI (PolyPlus; cat. no. 101-10N) was used to cotransfect cells with pmTurquoise2-Mito and Cox8-RamR-FLAG constructs. Sixteen hours post-transfection, the cells were washed with phenol-red free, serum-free, antibiotic-free RPMI imaging medium (ThermoFisher; cat. no. 11835030) containing 1% glutamine. Bodipy625 was diluted in the imaging medium to a final concentration of 450 nM. After 15 min of incubation at 37 °C, the free dye was washed away with the imaging medium. Cells were imaged live at 37 °C by confocal imaging, and their positions on the grid were marked. Subsequently, the cells were washed with PBS, fixed with 4% paraformaldehyde (PAF) for 15 min, and permeabilized with 0.1% Triton-X100 in PBS for 5 min. Cells were immunolabeled with mouse IgG1 Anti-Flag antibody (Sigma; cat. no. F1804) overnight at dilution 1:200 in PBS. Next, cells were washed with PBS and labeled with secondary donkey anti-mouse antibody conjugated to Alexa Fluor 568 (ThermoFisher; cat. no. A10037) at dilution 1:400 in PBS for 30 min. Finally cells located at the stored positions on the grid were imaged by confocal microscopy.
Imaging
Confocal laser scanning microscopy (LSM 710, Carl Zeiss AG Jena, Germany) equipped with a C-Apochromat 40×/1.2 NA objective was used for in vivo fluorescence imaging of live E. coli, L. lactis, and S. cerevisiae cells. Lasers (405, 488, 543, and 632 nm) were employed for fluorescence excitation. For all measurements, data were acquired within 20 min, and thereafter a fresh slide was used. The stage temperature was maintained at 30 °C. We recorded 16-bit images at randomized positions on the glass slide with 512 × 512 pixels (34.19 μm × 34.19 μm) and analyzed at least 100 cells for each dye with a corresponding CTPE. All images were collected under identical conditions of power and gain for a given dye. For anaerobic fluorescence imaging, experiments were performed in a sterile glovebox always maintained under a 5% CO2 environment. Control experiments to verify oxygen unavailability were performed by assessing mNG fluorescence in E. coli BW25113 housing a pBAD-mNG plasmid in a TECAN multiwell plate reader inside the glovebox. To avoid any recovery of the fluorescent protein mNeonGreen (mNG) fluorescence during cell harvesting, all steps until slide preparation were done in the sterile glovebox. The same samples, when exposed to air, gained fluorescence, which saturated over time. For anaerobic imaging, cytoplasmic LmrR and RamR proteins were expressed from a pBAD24 plasmid under conditions identical to those performed in the presence of oxygen.
For wash-free live-cell confocal imaging, E. coli cells expressing cytoplasmic CTPEs were loaded onto APTES coated slides, and 0.5 μM Bodipy495, 0.5 μM Bodipy625, or 2.0 μM DFHBI dye was added. The cells were imaged a few frames before adding the dyes and thereafter every 5 s. For repetitive bleaching measurements, E. coli cells expressing periplasmic CTPEs were imaged in the presence of 0.5 μM Bodipy495, 0.5 μM Bodipy625, or 2.0 μM DFHBI. A binding trace was first obtained (I* in Figure 5b,c), and thereafter cells were photobleached for 1 min (15 mW), and recovery of fluorescence was measured for about 3 min. For dye-swapping measurements, E. coli cells expressing cytoplasmic LmrR were first labeled with 2.0 μM DFHBI, and 0.5 μM of Bodipy625 was added. For photobleaching measurements comparing mNeongreen and Bodipy495, continuous time-lapse images were acquired at a laser power of 2 mW (512 × 512 pixels, 1 μs pixel dwell time, 26.57 μm × 26.57 μm frame size) for 2 min.
For labeling colonies on LB agar plates (grown in the presence of 100 μg·mL–1 ampicillin plus 0.1% (w/v) arabinose), 0.5 μM Bodipy495, Bodipy625, or rhodamine 6G was added postgrowth. The cells expressing TfCP tags and negative controls (E. coli cells housing an empty pBAD plasmid) were incubated for 15 min prior to imaging on a Typhoon fluorescence scanner with laser excitation at 488 nm (520 nm filter with a bandpass of 40 nm) for Bodipy495, at 532 nm (555 nm filter with a bandpass of 20 nm) for rhodamine 6G, and at 633 nm (670 nm filter with a bandpass of 30 nm) for Bodipy625. Images were acquired at a scanning rate of 100 μm min–1.
Phase-contrast images were acquired using an Axio Observer Z1 microscope (Carl Zeiss, Jena, Germany) equipped with a C-Apochromat 100×/1.49 NA objective for imaging of live E. coli, L. lactis, and S. cerevisiae cells. For all measurements, data were acquired within 20 min, and thereafter a fresh slide was used. The stage temperature was maintained at 30 °C. We recorded 16-bit images at randomized positions on the glass slide with 1024 × 1024 pixels (66.05 μm × 66.05 μm) and analyzed at least 100 cells for each dye with a corresponding CTPE. Cell aspect ratios (length/width) were obtained using the MicrobeJ plugin in Fiji. Fiji57 was used for all image analysis of confocal and phase-contrast microscopy images.
For imaging HEK cells, a confocal laser-scanning microscope (LSM800, Carl Zeiss AG Jena, Germany) equipped with a 63× oil immersion objective was used. A 640 nm laser was employed for Bodipy625 excitation. The stage temperature was maintained at 30 °C. All images were collected under identical conditions of power and gain for a given dye.
Protein Expression and Purification
Chemically competent BL21(DE3) E. coli cells were transformed with a pET17b expression vectors carrying cyto-LmrR and cyto-RamR constructs under the control of a T7 RNA polymerase promoter (p(T7)). Single colonies were picked and inoculated into a starter culture of 10 mL of fresh lysogeny broth (LB) medium (10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl) containing 100 μg·mL–1 ampicillin and grown at 37 °C with 180 rpm shaking overnight. The following day, the saturated LB culture was diluted 100-fold in 500 mL of fresh LB medium in a 2 L Erlenmeyer flask containing 100 μg·mL–1 ampicillin and allowed to grow at 37 °C with 180 rpm shaking until the culture reached OD600 = 0.84–0.90. At this stage, isopropyl β-d-1-thiogalactopyranoside (IPTG) at a final concentration of 1 mM was added to induce the expression of the target protein, and expressions were carried out at 30 °C with 180 rpm shaking overnight. The next day, cells were harvested by centrifugation (6000 rpm, JA10, 20 min, 4 °C, Beckman). The pellet was resuspended in 15–20 mL of 50 mM NaH2PO4, pH 8.0, 150 mM NaCl with half a tablet of mini complete EDTA-free protease inhibitor cocktail (Roche) and 1 mM of the serine protease inhibitor phenylmethanesulfonyl fluoride (PMSF). Next, DnaseI (final concentration, 0.1 mg·mL–1) and MgCl2 (final concentration, 10 mM) were added. Sonication was carried out (tip diameter 6 mm, 75% (200 W)) for 8 min (10 s on, 15 s off). Additional sheer forcing with a syringe and a long needle was applied at least 2 times. Henceforth, all steps were carried out at 4 °C.
The cell lysates obtained after sonication were centrifuged (16000 rpm, JA-17, 45 min, 4 °C, Beckman) to remove unlysed cells and other high molecular weight debris. The cell-free extract was then filtered and equilibrated with 5 mL of pre-equilibrated Strep-Tactin column material for 1 h (mixed at 200 rpm on a rotary shaker). The column was washed with 3 × 2 CV (column volume) of resuspension buffer (same as buffer used before) and eluted multiple times with 0.5 CV (6–7 times) of elution buffer (resuspension buffer containing 5 mM desthiobiotin). Fractions were analyzed on a 12% polyacrylamide SDS-Tris Tricine gel followed by Coomassie Blue staining. Fractions containing protein (excluding the first elution) were concentrated in centrifugal filters and rebuffered to 20 mM K-MOPS, pH 7.0, 150 mM NaCl using dialysis (reduces DNA contamination in elution fractions). The concentration of purified LmrR and RamR was determined using the calculated extinction coefficient obtained from Protparam on the Expasy server (ε280 for LmrR monomer = 25440 M–1 cm–1 and that of RamR monomer = 29450 M–1 cm–1). Expression yields typically were 30–40 mg/L for LmrR and 40–50 mg/L for RamR. Aliquots of 500 μL were flash-freezed using liquid nitrogen until use. Thawed protein samples for analysis were not frozen a second time and were used within 24 h.
For purification of HisTag containing proteins, a Ni2+-Sepharose resin was used. The resin was pre-equilibrated in 50 mM KPi, 150 mM NaCl, pH 7.0, with 10 mM imidazole. The cell-free extract was added to the Ni2+-Sepharose resin (0.5 mL of bed volume per 10 mg of total protein) and nutated for 3 h, after which the resin was washed with 20 column volumes of 50 mM KPi, 150 mM NaCl, pH 7.0, with 50 mM imidazole. Proteins were then eluted with 50 mM KPi, 150 mM NaCl, pH 7.0, with 500 mM imidazole in 500 μL aliquots. The most concentrated fractions were run on a Superdex15 Increase 10/300 GL size-exclusion column (GE Healthcare) in 20 mM K-MOPS, pH 7.0, with 150 mM NaCl. Protein containing fractions were pooled and concentrated to 5 mg·mL–1 in a Vivaspin 500 (3 kDa) centrifugal concentrator (Sartorius AG), after which they were aliquoted, flash-frozen in 500 μL aliquots, and stored at −80 °C.
Absorption and Fluorescence Spectroscopy
Organic dyes were prepared as stock solutions (2.5–5 mM) in DMSO and diluted for spectroscopy to a final concentration of 1 μM in 20 mM K-MOPS, 150 mM NaCl, pH 7.0, such that the DMSO concentration did not exceed 0.5% (v/v). Purified RamR and LmrR were then added and incubated for less than 1 min before measuring absorption and fluorescence spectra. All measurements (3 independent replicates) were taken at 30 °C. Samples were incubated for 1–2 min after mixing gently with a pipet and thereafter measured in black polystyrene, μClear bottom, 96-well plates (Greiner Bio-One, cat. 655096) and for samples having excitation wavelength <400 nm on 96-well plates with a UV-compatible optical bottom (Greiner Bio-One, cat. 655801) on a TECAN Spark 10M microplate reader. Reported values are averages of 3 independent experiments. The obtained results are concisely summarized in Table 1 in the main text. Experiments to determine the bound fraction were performed for each dye by keeping the dye concentration constant at 1 μM to avoid any inner filter effects at high dye concentrations. The protein concentrations were increased until the fluorescence emission intensity saturated, resulting in an apparent binding curve. The dissociation constant, Kd, was then estimated by fitting the data points to a Hill model. The same samples were also used for absorption spectroscopy to validate if the spectral changes observed in fluorescence emission emanated from absorption changes. Fluorescence emission and absorption spectra of the organic dyes in the presence of CTPEs were normalized against that of the organic dyes alone, giving a fold-change (from peak values) in absorption and fluorescence emission (panels c and e in Supplementary Figures 2–17). At saturation values of CTPEs with the organic dyes, normalized fluorescence emission spectra and absorption spectra are depicted in panels d and f in Supplementary Figures 2–17. The excitation and emission bandwidths were set to 5 nm for all measurements.
For pH, NaCl, and temperature scans, purified RamR and LmrR were used at a final concentrations of 4 μM and 50 μM, respectively, in 20 mM MOPS, 150 mM NaCl, pH 7.0, with Bodipy495. The buffer pH was adjusted using KOH (K-MOPS). Buffer exchange for pH scan measurements was performed using Thermo Scientific Zeba Spin desalting columns, and the pH was verified subsequently using a pH meter. For pH values in the range 4–7, a citric acid-Na2HPO4 buffer was used supplemented with 150 mM NaCl. For pH values between pH values 6.5 and 8, 50 mM sodium phosphate buffers supplemented with 150 mM NaCl were used at 30 °C, keeping the dye and CTPE concentration constant. To probe the stability of our CTPEs with increasing ionic strengths, we modulated the concentration of NaCl in 20 mM K-MOPS, 150 mM NaCl, pH 7.0, buffer keeping the dye and CTPE concentration constant. The color-shaded regions represent the standard deviation (SD) over three independent measurements.
Temperature scan measurements were performed in Teflon sealed quartz cuvettes in an FP-8300 spectrofluorimeter (Jasco, Inc.) equipped with a temperature modulation system (Julabo GmbH). The temperature rise gradient was 1 °C min–1, and an additional minute was allowed to equilibrate samples at a given temperature. Samples were excited at 480 nm, and the emission spectra were acquired from 495 to 600 nm with a 1 nm data interval and 5 °C temperature intervals from 20 to 60 °C. Excitation and emission bandwidths were kept constant at 5 nm. Reported values are averages of independent experiments. For temperature scan, beyond 55 °C, visible white precipitates could be observed for CTPEs indicated with a dotted line in Figure 1f.
For measurements under strictly anaerobic conditions, 20 mM K-MOPS, 150 mM NaCl buffered at pH 7.0 was prepared and equilibrated in the CO2 hood at least a week before measurements.
Fluorescence Lifetime
The fluorescence lifetime measurements were acquired at a 10 MHz repetition rate for 30 s on a MicroTime 200 confocal microscope (PicoQuant, Berlin, Germany) on glass-bottom dishes (Willco Wells, cat. HBST-3522). Purified RamR and LmrR were used at a final concentrations of 4 μM and 50 μM, respectively, in 20 mM K-MOPS, 150 mM NaCl, pH 7.0, with 1 μM organic dye. The exponential-tail fitting of the lifetime decay was done with the SymphoTime software. Reported values are averages of independent experiments, and the accompanying error bars represent SD at ambient temperature (∼25 °C). The laser excitation modules employed in the MicroTime 200 confocal microscope were 440, 485, 532, 595, and 640 nm.
Fluorescence Recovery after Photobleaching (FRAP)
We performed fluorescence recovery after photobleaching (FRAP; see Figure 3b) on an LSM710 Zeiss confocal laser scanning microscope (Zeiss, Oberkochen, Germany) as reported previously by our group,38,58 based on a previously described method.59 We programmed the microscope to take three images (prebleach), then photobleached the cell at one of the poles, and finally recorded the recovery of the fluorescence over time. We ensured that we picked cells lying flat on one position without exhibiting any rotational or translational motion during measurement, not undergoing cell division, and having no neighbors that would obscure the analysis.
Flow Cytometry
Live cells were prepared and labeled identically for confocal fluorescence microscopy. For E. coli, L. lactis, and S. cerevisiae cells, first using the FSC/SSC gating, cell debris was removed from the main cell population. A positivity threshold gate for each sample was defined based on unlabeled (0%) and labeled control cells expressing no protein (<3%). An identical positivity threshold gate was applied to all samples for a given organic dye. Samples were measured on an LSR-II flow cytometer (BD Bioscience) with 10000 events for each sample and analyzed with Kaluza Analysis 2.1 software (Beckman Coulter, CA, USA). The data was acquired (18 bits digitalization in 5 decades) using DIVA 8.0 software and saved as FCS 3.0 or 3.1 files. The following laser and corresponding filter sets (dye, laser, peak/bandwidth) were employed: (a) DHFBI, 405, 525/15; (b) Bodipy488 and Bodipy495, 488, 530/30; (c) rhodamine 6G and Rose Bengal, 561, 585/15, (d) Bodipy589, 561, 615/20; (e) Bodipy625, 635, 660/30. At least two independent biological replicate measurements were performed for each sample. For DFHBI, the excitation laser and the fluorescence filter sets were not ideal, resulting in underestimating the fraction of labeled cells. The gating strategy is given in Appendix 4. For HEK293T cells, a starting cell population per sample was collected with the stopping rule of 30000 events per preliminary gate drawn in FSC/SSC in a CytoFlex LX (Beckman Coulter) flow cytometer. Next, the cells were analyzed, placing gates on PE (phycoerythrin) channel indicating FLAG expression labeled with Alexa Fluor 568. APC (allophycocyanin) filter was used to detect Bodipy625 fluorescence. Negative/high background populations were defined by unstained cells visible in the PE channel and untransfected but Bodipy625 pulsed cells in the APC channel. All experimental data were analyzed in Kaluza Analysis 2.1 software.
Size-Exclusion Chromatography with Multiangle Laser Light Scattering (SEC-MALLS) Detection
The column was equilibrated with 20 mM K-MOPS, 150 mM NaCl (pH 7.0). The Superdex 200 column used for the SEC-MALLS analysis was equilibrated with 20 mM K-MOPS, 150 mM NaCl buffered at pH 7.0, filtered through 0.1 μm pore size VVLP filters (Millipore). Subsequently, the buffer was recirculated through the system for 16 h at 0.5 mL–1. This allowed the buffer to pass several times through the degasser and the preinjection filter, thereby thoroughly removing air and particles and allowing a judgment of the stability of detector baselines. Protein solution (400 μL of 0.4 mg·mL–1) was injected, and the data from the three detectors were imported by the ASTRA software package, version 5.3.2.10 (Wyatt Technologies). For instrument calibration and analysis, aldolase protein was used as an internal standard.
Gel Electrophoresis and Western Blot
E. coli cells expressing cytoplasmic LmrR, RamR, and mNG tagged with a C-terminal 6-HisTag were initially centrifuged at 11000g to remove spent media, and the cell pellet was resuspended in 50 mM KPi, 100 mM NaCl (PBS) buffered at pH 7.0 to a final OD600 = 6. Samples were then mixed with 5× SDS loading buffer, heated at 90 °C for 5 min, and separated using a 15% SDS-polyacrylamide gel. Proteins were blotted onto poly(vinylidene difluoride) (PVDF) membranes for 35 min at a constant current of 0.08 A. Blots were blocked for 2 h in freshly prepared 0.2% (w/v) I-block in 0.2% (w/v) Tween20 containing 50 mM KPi, 100 mM NaCl (PBST) buffered at pH 7.0. HRP-conjugated anti-HisTag antibodies were incubated for 1 h at 1:6000 dilution in PBST buffer and washed 3 times for 5 min in 0.2% (w/v) I-block in PBST buffer, followed by washing 3 times for 5 min in PBS buffer. For the chemiluminescent readout, the Super Signal West Pico (Thermo Scientific) substrate was used according to the manufacturer’s instructions. Chemiluminescent detection was performed on a LAS-4000 mini (Fujifilm, Düsseldorf, Germany).
Acknowledgments
We thank our colleagues in the Membrane Enzymology group at the University of Groningen for their valuable suggestions, colleagues in the Molecular Immunology group for help with the cultivation and analysis of cells under strictly anaerobic conditions, den Blaauwen (University of Amsterdam) who kindly provided the fluorescent SuperFolder mTurquoise (sfTq2) and PBP5-sfTq2 constructs, G. Mesander of the University Medical Center Groningen for assistance in fluorescence-activated cell sorting measurements, V. Krasnikov for assistance in fluorescence lifetime measurements, K. Johnsson (Max Planck Institute for Medical Research, Germany) for providing the MaP dyes, A. Driessen (University of Groningen) for kindly providing the pNSC8048 plasmid (carrying the lmrR gene) and L. lactis lmrR knockout strain, and M. Partipilo for providing the pNZC3GH plasmid. This research was funded by two ERC Advanced grants (ABCVolume, no. 670578, and DENZUAC, no. 885396).
Glossary
Abbreviations
- Bodipy
4,4-difluoro-4-bora-3a,4a-diaza-s-indacene
- SD
standard deviation
- HOMO
highest occupied molecular orbital
- DFHBI
3,5-difluoro-4-hydroxybenzylidene imidazolinone
- LUMO
lowest unoccupied molecular orbital
- APC
allophycocyanin
- EtBr
ethidium bromide
- DPH
1,6-diphenyl-1,3,5-hexatriene
- NPN
1-N-phenylnaphthylamine
- DAPI
4′,6-diamidino-2-phenylindole
- ANS
8-anilinonaphthalene-1-sulfonic acid
- Xgal
5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.1c00100.
Complete in vitro and in vivo characterization of 30 organic dyes with CTPEs, representative images of labeled E. coli, L. lactis, S. cerevisiae, and HEK cells with 7 fluorogenic dyes, the effect of Ficoll70 on fluorogenicity, the gating strategy for flow cytometry, and accompanying data with the fraction of labeled cells, the effect of dye labeling on cell morphology, and the strains and plasmids used in the study (PDF)
Author Contributions
A.I., S.C., G.R., R.V., G.v.d.B., and B.P. conceived the project and designed the experiments. A.I. and S.C. designed and performed cloning experiments in E. coli, L. lactis, and S. cerevisiae and purified CTPEs. A.I. performed most of the bacterial cloning and microscopy experiments and analyzed the data. A.I. and A.J.F. designed and performed cloning and microscopy experiments in S. cerevisiae. M.B. designed and performed experiments in mammalian cells. A.I. and B.P. wrote the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Shaner N. C.; Steinbach P. A.; Tsien R. Y. (2005) A Guide to Choosing Fluorescent Proteins. Nat. Methods 2 (12), 905–909. 10.1038/nmeth819. [DOI] [PubMed] [Google Scholar]
- Rodriguez E. A.; Campbell R. E.; Lin J. Y.; Lin M. Z.; Miyawaki A.; Palmer A. E.; Shu X.; Zhang J.; Tsien R. Y. (2017) The Growing and Glowing Toolbox of Fluorescent and Photoactive Proteins. Trends Biochem. Sci. 42, 111. 10.1016/j.tibs.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drepper T.; Eggert T.; Circolone F.; Heck A.; Krauß U.; Guterl J. K.; Wendorff M.; Losi A.; Gärtner W.; Jaeger K. E. (2007) Reporter Proteins for in Vivo Fluorescence without Oxygen. Nat. Biotechnol. 25 (4), 443–445. 10.1038/nbt1293. [DOI] [PubMed] [Google Scholar]
- Nagai T.; Ibata K.; Park E. S.; Kubota M.; Mikoshiba K.; Miyawaki A. (2002) A Variant of Yellow Fluorescent Protein with Fast and Efficient Maturation for Cell-Biological Applications. Nat. Biotechnol. 20 (1), 87–90. 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- Dippel A. B.; Anderson W. A.; Park J. H.; Yildiz F. H.; Hammond M. C. (2020) Development of Ratiometric Bioluminescent Sensors for in Vivo Detection of Bacterial Signaling. ACS Chem. Biol. 15 (4), 904–914. 10.1021/acschembio.9b00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing C.; Cornish V. W. (2011) Chemical Tags for Labeling Proteins inside Living Cells. Acc. Chem. Res. 44 (9), 784–792. 10.1021/ar200099f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocaoglu O.; Carlson E. E. (2016) Progress and Prospects for Small-Molecule Probes of Bacterial Imaging. Nat. Chem. Biol. 12 (7), 472–478. 10.1038/nchembio.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cranfill P. J.; Sell B. R.; Baird M. A.; Allen J. R.; Lavagnino Z.; De Gruiter H. M.; Kremers G. J.; Davidson M. W.; Ustione A.; Piston D. W. (2016) Quantitative Assessment of Fluorescent Proteins. Nat. Methods 13 (7), 557–562. 10.1038/nmeth.3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleza E.; Kim J. M.; Cluzel P. (2018) Systematic Characterization of Maturation Time of Fluorescent Proteins in Living Cells. Nat. Methods 15 (1), 47–51. 10.1038/nmeth.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm J. B.; English B. P.; Choi H.; Muthusamy A. K.; Mehl B. P.; Dong P.; Brown T. A.; Lippincott-Schwartz J.; Liu Z.; Lionnet T.; Lavis L. D. (2016) Bright Photoactivatable Fluorophores for Single-Molecule Imaging. Nat. Methods 13 (12), 985–988. 10.1038/nmeth.4034. [DOI] [PubMed] [Google Scholar]
- Kowada T.; Maeda H.; Kikuchi K. (2015) BODIPY-Based Probes for the Fluorescence Imaging of Biomolecules in Living Cells. Chem. Soc. Rev. 44 (14), 4953–4972. 10.1039/C5CS00030K. [DOI] [PubMed] [Google Scholar]
- Song W.; Strack R. L.; Svensen N.; Jaffrey S. R. (2014) Plug-and-Play Fluorophores Extend the Spectral Properties of Spinach. J. Am. Chem. Soc. 136 (4), 1198–1201. 10.1021/ja410819x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Tran M.; D’Este E.; Roberti J.; Koch B.; Xue L.; Johnsson K. (2020) A General Strategy to Develop Cell Permeable and Fluorogenic Probes for Multicolour Nanoscopy. Nat. Chem. 12 (2), 165–172. 10.1038/s41557-019-0371-1. [DOI] [PubMed] [Google Scholar]
- Grimm J. B.; Muthusamy A. K.; Liang Y.; Brown T. A.; Lemon W. C.; Patel R.; Lu R.; Macklin J. J.; Keller P. J.; Ji N.; Lavis L. D. (2017) A General Method to Fine-Tune Fluorophores for Live-Cell and in Vivo Imaging. Nat. Methods 14 (10), 987–994. 10.1038/nmeth.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szent-Gyorgyi C.; Schmidt B. A.; Creeger Y.; Fisher G. W.; Zakel K. L.; Adler S.; Fitzpatrick J. A. J.; Woolford C. A.; Yan Q.; Vasilev K. V.; Berget P. B.; Bruchez M. P.; Jarvik J. W.; Waggoner A. (2008) Fluorogen-Activating Single-Chain Antibodies for Imaging Cell Surface Proteins. Nat. Biotechnol. 26, 235. 10.1038/nbt1368. [DOI] [PubMed] [Google Scholar]
- Yan Q.; Schwartz S. L.; Maji S.; Huang F.; Szent-Gyorgyi C.; Lidke D. S.; Lidke K. A.; Bruchez M. P. (2014) Localization Microscopy Using Noncovalent Fluorogen Activation by Genetically Encoded Fluorogen-Activating Proteins. ChemPhysChem 15, 687. 10.1002/cphc.201300757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick J. A. J.; Yan Q.; Sieber J. J.; Dyba M.; Schwarz U.; Szent-Gyorgyi C.; Woolford C. A.; Berget P. B.; Waggoner A. S.; Bruchez M. P. (2009) STED Nanoscopy in Living Cells Using Fluorogen Activating Proteins. Bioconjugate Chem. 20, 1843. 10.1021/bc900249e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telmer C. A.; Verma R.; Teng H.; Andreko S.; Law L.; Bruchez M. P. (2015) Rapid, Specific, No-Wash, Far-Red Fluorogen Activation in Subcellular Compartments by Targeted Fluorogen Activating Proteins. ACS Chem. Biol. 10, 1239. 10.1021/cb500957k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özhalici-Ünal H.; Pow C. L.; Marks S. A.; Jesper L. D.; Silva G. L.; Shank N. I.; Jones E. W.; Burnette J. M.; Berget P. B.; Armitage B. A. (2008) A Rainbow of Fluoromodules: A Promiscuous ScFv Protein Binds to and Activates a Diverse Set of Fluorogenic Cyanine Dyes. J. Am. Chem. Soc. 130, 12620. 10.1021/ja805042p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plamont M. A.; Billon-Denis E.; Maurin S.; Gauron C.; Pimenta F. M.; Specht C. G.; Shi J.; Quérard J.; Pan B.; Rossignol J.; Morellet N.; Volovitch M.; Lescop E.; Chen Y.; Triller A.; Vriz S.; Le Saux T.; Jullien L.; Gautier A.; Moncoq K. (2016) Small Fluorescence-Activating and Absorption-Shifting Tag for Tunable Protein Imaging in Vivo. Proc. Natl. Acad. Sci. U. S. A. 113, 497. 10.1073/pnas.1513094113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tebo A. G.; Moeyaert B.; Thauvin M.; Carlon-Andres I.; Böken D.; Volovitch M.; Padilla-Parra S.; Dedecker P.; Vriz S.; Gautier A. (2021) Orthogonal Fluorescent Chemogenetic Reporters for Multicolor Imaging. Nat. Chem. Biol. 17, 30. 10.1038/s41589-020-0611-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineev K. S.; Goncharuk S. A.; Goncharuk M. V.; Povarova N. V. (2020) NanoFAST: Structure-Based Design of a Small Fluorogen-Activating Protein with Only 98 Amino Acids. bioRxiv 10.1101/2020.12.29.424647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Plamont M. A.; Sladitschek H. L.; Rodrigues V.; Aujard I.; Neveu P.; Le Saux T.; Jullien L.; Gautier A. (2017) Dynamic Multicolor Protein Labeling in Living Cells. Chem. Sci. 8, 5598. 10.1039/C7SC01364G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.; Gustafson W. C.; Han C.; Lafaye C.; Noirclerc-Savoye M.; Ge W. P.; Thayer D. A.; Huang H.; Kornberg T. B.; Royant A.; Jan L. Y.; Jan Y. N.; Weiss W. A.; Shu X. (2014) An Improved Monomeric Infrared Fluorescent Protein for Neuronal and Tumour Brain Imaging. Nat. Commun. 5, 3626. 10.1038/ncomms4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A.; Ando R.; Miyatake H.; Greimel P.; Kobayashi T.; Hirabayashi Y.; Shimogori T.; Miyawaki A. (2013) A Bilirubin-Inducible Fluorescent Protein from Eel Muscle. Cell 153 (7), 1602–1611. 10.1016/j.cell.2013.05.038. [DOI] [PubMed] [Google Scholar]
- Saurabh S.; Perez A. M.; Comerci C. J.; Shapiro L.; Moerner W. E. (2016) Super-Resolution Imaging of Live Bacteria Cells Using a Genetically Directed, Highly Photostable Fluoromodule. J. Am. Chem. Soc. 138, 10398. 10.1021/jacs.6b05943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki S.; Nikaido E.; Nakashima R.; Sakurai K.; Fujiwara D.; Fujii I.; Nishino K. (2013) The Crystal Structure of Multidrug-Resistance Regulator RamR with Multiple Drugs. Nat. Commun. 4 (1), 2078. 10.1038/ncomms3078. [DOI] [PubMed] [Google Scholar]
- Yamasaki S.; Nakashima R.; Sakurai K.; Baucheron S.; Giraud E.; Doublet B.; Cloeckaert A.; Nishino K. (2019) Crystal Structure of the Multidrug Resistance Regulator RamR Complexed with Bile Acids. Sci. Rep. 9, 177. 10.1038/s41598-018-36025-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelfes G. (2019) LmrR: A Privileged Scaffold for Artificial Metalloenzymes. Acc. Chem. Res. 52 (3), 545–556. 10.1021/acs.accounts.9b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K.; Tokunaga Y.; Imai M.; Takahashi H.; Shimada I. (2015) Dynamic Multidrug Recognition by Multidrug Transcriptional Repressor LmrR. Sci. Rep. 4, 1–12. 10.1038/srep06922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madoori P. K.; Agustiandari H.; Driessen A. J. M.; Thunnissen A. M. W. H. (2009) Structure of the Transcriptional Regulator LmrR and Its Mechanism of Multidrug Recognition. EMBO J. 28, 156. 10.1038/emboj.2008.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agustiandari H.; Lubelski J.; Van Den Berg Van Saparoea H. B.; Kuipers O. P.; Driessen A. J. M. (2008) LmrR Is a Transcriptional Repressor of Expression of the Multidrug ABC Transporter LmrCD in Lactococcus Lactis. J. Bacteriol. 190 (2), 759–763. 10.1128/JB.01151-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejías S. H.; Roelfes G.; Browne W. R. (2020) Impact of Binding to the Multidrug Resistance Regulator Protein LmrR on the Photo-Physics and -Chemistry of Photosensitizers. Phys. Chem. Chem. Phys. 22 (21), 12228–12238. 10.1039/D0CP01755H. [DOI] [PubMed] [Google Scholar]
- Wieczorek A.; Werther P.; Euchner J.; Wombacher R. (2017) Green- to Far-Red-Emitting Fluorogenic Tetrazine Probes-Synthetic Access and No-Wash Protein Imaging inside Living Cells. Chem. Sci. 8, 1506. 10.1039/C6SC03879D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozma E.; Estrada Girona G.; Paci G.; Lemke E. A.; Kele P. (2017) Bioorthogonal Double-Fluorogenic Siliconrhodamine Probes for Intracellular Super-Resolution Microscopy. Chem. Commun. 53, 6696. 10.1039/C7CC02212C. [DOI] [PubMed] [Google Scholar]
- Schavemaker P. E.; Boersma A. J.; Poolman B. (2018) How Important Is Protein Diffusion in Prokaryotes?. Frontiers in Molecular Biosciences 93. 10.3389/fmolb.2018.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M.; Mommer M. S.; Sourjik V. (2010) Mobility of Cytoplasmic, Membrane, and DNA-Binding Proteins in Escherichia Coli. Biophys. J. 98, 552. 10.1016/j.bpj.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schavemaker P. E.; Śmigiel W. M.; Poolman B. (2017) Ribosome Surface Properties May Impose Limits on the Nature of the Cytoplasmic Proteome. eLife 6, e30084. 10.7554/eLife.30084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botman D.; de Groot D. H.; Schmidt P.; Goedhart J.; Teusink B. (2019) In Vivo Characterisation of Fluorescent Proteins in Budding Yeast. Sci. Rep. 9, 2234. 10.1038/s41598-019-38913-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner N. C.; Lambert G. G.; Chammas A.; Ni Y.; Cranfill P. J.; Baird M. A.; Sell B. R.; Allen J. R.; Day R. N.; Israelsson M.; Davidson M. W.; Wang J. (2013) A Bright Monomeric Green Fluorescent Protein Derived from Branchiostoma Lanceolatum. Nat. Methods 10 (5), 407–409. 10.1038/nmeth.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukinavičius G.; Reymond L.; Umezawa K.; Sallin O.; D’Este E.; Göttfert F.; Ta H.; Hell S. W.; Urano Y.; Johnsson K. (2016) Fluorogenic Probes for Multicolor Imaging in Living Cells. J. Am. Chem. Soc. 138, 9365. 10.1021/jacs.6b04782. [DOI] [PubMed] [Google Scholar]
- Geertsma E. R.; Groeneveld M.; Slotboom D. J.; Poolman B. (2008) Quality Control of Overexpressed Membrane Proteins. Proc. Natl. Acad. Sci. U. S. A. 105, 5722. 10.1073/pnas.0802190105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew D.; Lerch M.; Kunji E.; Slotboom D. J.; de Gier J. W. (2006) Optimization of Membrane Protein Overexpression and Purification Using GFP Fusions. Nat. Methods 3, 303. 10.1038/nmeth0406-303. [DOI] [PubMed] [Google Scholar]
- Berezin M. Y.; Achilefu S. (2010) Fluorescence Lifetime Measurements and Biological Imaging. Chem. Rev. 110 (5), 2641–2684. 10.1021/cr900343z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geva-Zatorsky N.; Alvarez D.; Hudak J. E.; Reading N. C.; Erturk-Hasdemir D.; Dasgupta S.; von Andrian U. H.; Kasper D. L. (2015) In Vivo Imaging and Tracking of Host-Microbiota Interactions via Metabolic Labeling of Gut Anaerobic Bacteria. Nat. Med. 21 (9), 1091–1100. 10.1038/nm.3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maslov I.; Bogorodskiy A.; Mishin A.; Okhrimenko I.; Gushchin I.; Kalenov S.; Dencher N. A.; Fahlke C.; Büldt G.; Gordeliy V.; Gensch T.; Borshchevskiy V. (2018) Efficient Non-Cytotoxic Fluorescent Staining of Halophiles. Sci. Rep. 8, 2549. 10.1038/s41598-018-20839-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatova Z.; Gierasch L. M. (2004) Monitoring Protein Stability and Aggregation in Vivo by Real-Time Fluorescent Labeling. Proc. Natl. Acad. Sci. U. S. A. 101 (2), 523–528. 10.1073/pnas.0304533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Frei M. S.; Salim A.; Johnsson K. (2019) Small-Molecule Fluorescent Probes for Live-Cell Super-Resolution Microscopy. J. Am. Chem. Soc. 141 (7), 2770–2781. 10.1021/jacs.8b11134. [DOI] [PubMed] [Google Scholar]
- Adhikari S.; Moscatelli J.; Smith E. M.; Banerjee C.; Puchner E. M. (2019) Single-Molecule Localization Microscopy and Tracking with Red-Shifted States of Conventional BODIPY Conjugates in Living Cells. Nat. Commun. 10, 3400. 10.1038/s41467-019-11384-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos J.; García-Herraiz A.; Roelfes G. (2013) An Enantioselective Artificial Metallo-Hydratase. Chem. Sci. 4 (9), 3578–3582. 10.1039/c3sc51449h. [DOI] [Google Scholar]
- Bitinaite J.; Rubino M.; Varma K. H.; Schildkraut I.; Vaisvila R.; Vaiskunaite R. (2007) USERTM Friendly DNA Engineering and Cloning Method by Uracil Excision. Nucleic Acids Res. 35 (6), 1992–2002. 10.1093/nar/gkm041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ruyter P. G. G. A.; Kuipers O. P.; De Vos W. M. (1996) Controlled Gene Expression Systems for Lactococcus Lactis with the Food- Grade Inducer Nisin. Appl. Environ. Microbiol. 62 (10), 3662–3667. 10.1128/AEM.62.10.3662-3667.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann M.; Hausherr A.; Ferbitz L.; Schödl T.; Heitzer M.; Hegemann P. (2004) Monitoring Dynamic Expression of Nuclear Genes in Chlamydomonas Reinhardtii by Using a Synthetic Luciferase Reporter Gene. Plant Mol. Biol. 55 (6), 869–881. 10.1007/s11103-005-2150-1. [DOI] [PubMed] [Google Scholar]
- Munder M. C.; Midtvedt D.; Franzmann T.; Nüske E.; Otto O.; Herbig M.; Ulbricht E.; Müller P.; Taubenberger A.; Maharana S.; Malinovska L.; Richter D.; Guck J.; Zaburdaev V.; Alberti S. (2016) A PH-Driven Transition of the Cytoplasm from a Fluid- to a Solid-like State Promotes Entry into Dormancy. eLife 5, e09347. 10.7554/eLife.09347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew D.; Newstead S.; Sonoda Y.; Kim H.; von Heijne G.; Iwata S. (2008) GFP-Based Optimization Scheme for the Overexpression and Purification of Eukaryotic Membrane Proteins in Saccharomyces Cerevisiae. Nat. Protoc. 3 (5), 784–798. 10.1038/nprot.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J.; Von Stetten D.; Noirclerc-Savoye M.; Lelimousin M.; Joosen L.; Hink M. A.; Van Weeren L.; Gadella T. W. J.; Royant A. (2012) Structure-Guided Evolution of Cyan Fluorescent Proteins towards a Quantum Yield of 93%. Nat. Commun. 3, 3. 10.1038/ncomms1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J.; Arganda-Carreras I.; Frise E.; Kaynig V.; Longair M.; Pietzsch T.; Preibisch S.; Rueden C.; Saalfeld S.; Schmid B.; Tinevez J. Y.; White D. J.; Hartenstein V.; Eliceiri K.; Tomancak P.; Cardona A. (2012) Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 9 (7), 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika J. T.; Schavemaker P. E.; Krasnikov V.; Poolman B. (2014) Impact of Osmotic Stress on Protein Diffusion in Lactococcus Lactis. Mol. Microbiol. 94 (4), 857–870. 10.1111/mmi.12800. [DOI] [PubMed] [Google Scholar]
- Elowitz M. B.; Surette M. G.; Wolf P. E.; Stock J. B.; Leibler S. (1999) Protein Mobility in the Cytoplasm of Escherichia Coli. J. Bacteriol. 181 (1), 197–203. 10.1128/JB.181.1.197-203.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.