

Abstract
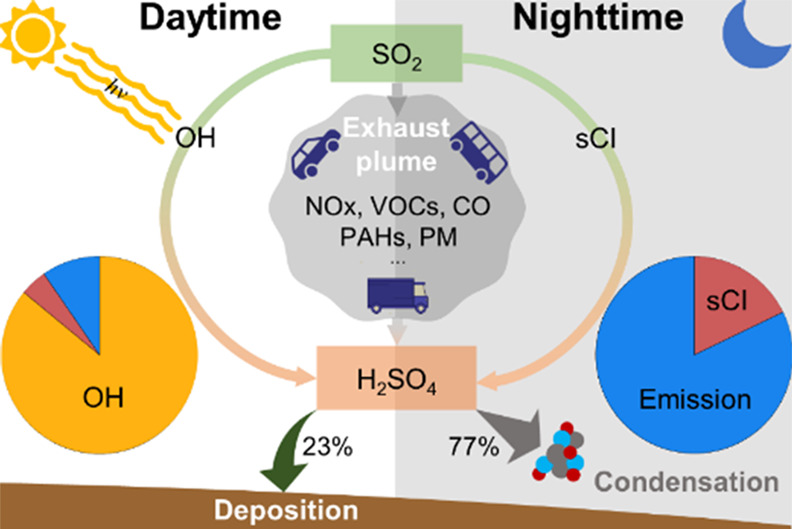
Gaseous sulfuric acid (H2SO4) is a crucial precursor for secondary aerosol formation, particularly for new particle formation (NPF) that plays an essential role in the global number budget of aerosol particles and cloud condensation nuclei. Due to technology challenges, global-wide and long-term measurements of gaseous H2SO4 are currently very challenging. Empirical proxies for H2SO4 have been derived mainly based on short-term intensive campaigns. In this work, we performed comprehensive measurements of H2SO4 and related parameters in the polluted Yangtze River Delta in East China during four seasons and developed a physical proxy based on the budget analysis of gaseous H2SO4. Besides the photo-oxidation of SO2, we found that primary emissions can contribute considerably, particularly at night. Dry deposition has the potential to be a non-negligible sink, in addition to condensation onto particle surfaces. Compared with the empirical proxies, the newly developed physical proxy demonstrates extraordinary stability in all the seasons and has the potential to be widely used to improve the understanding of global NPF fundamentally.
Keywords: sulfuric acid, proxy, budget analysis, primary emission, dry deposition
Short abstract
A budget analysis based physical proxy of H2SO4 is established in polluted environments, that can help improve the global understanding of new particle and aerosol sulfate formation.
Introduction
Atmospheric aerosols play an essential role in air quality,1 human health,2 and climate change.3 New particle formation (NPF), which can be observed worldwide,4 determines the global budget of aerosol particles in terms of their number concentrations.5 Gaseous sulfuric acid (H2SO4) is the key precursor for particle nucleation via a series of processes, including H2SO4–H2O binary nucleation, H2SO4–ammonia (NH3)–H2O ternary nucleation,6,7 H2SO4–amines–H2O nucleation,8,9 H2SO4–organic–H2O nucleation,10,11 and H2SO4–NH3–organic nucleation.12 However, measuring gaseous H2SO4 has been challenging due to its extremely low atmospheric concentrations. Long-term measurements of ambient H2SO4 have only been reported in very few studies,13−18 which limits the understanding of global NPF.
Gaseous H2SO4 was long believed to be formed only from the reaction between SO2 and OH. More recently, stabilized Criegee intermediates (sCI), formed in the ozonolysis of alkenes, were demonstrated to be able to oxidize SO2 and contribute to gaseous H2SO4.19,20 This was found to be particularly important for the nighttime H2SO4 formation.21 In the coastal marine atmosphere, SO2 oxidation by small Criegee intermediates (CI), produced possibly in photochemical reactions, and SO3 formed from dimethyl sulfide (DMS) oxidation reaction with water might be non-negligible sources of H2SO4.17,22,23 Recently, Olin et al.24 indicated a potential source of H2SO4 from traffic emissions in urban Helsinki based on the observed positive correlation between H2SO4 and NOx concentrations. Because of its weak chemical reactivity, the main sinks for gaseous H2SO4 are expected to be condensation onto aerosol particles or dry deposition to the ground or other surfaces.
Several proxies for gaseous H2SO4 were developed to enhance our global understanding of H2SO4 and NPF. Petäjä et al.25 and Mikkonen et al.16 built such proxies by considering the SO2–OH reaction as the only source, and condensation onto pre-existing aerosol particles as the only sink, for H2SO4. Lu et al.26 took into account O3 and HONO concentrations to differentiate between OH originating from the O3 and HONO photolysis. Dada et al.18 added the reaction of SO2 with sCI as an additional H2SO4 source to the proxy. These efforts largely improved the understanding of H2SO4 in different environments, however, with obvious limitations. First, they all are empirical proxies based on mainly short-term and site-specific measurements, casting doubts on their general applicability. Second, nighttime H2SO4 is rarely considered in these proxies. Here, we carried out comprehensive measurements of H2SO4 and related parameters during four seasons at the SORPES station, East China, and conducted a budget analysis for gaseous H2SO4 by taking all possible sources and sinks into consideration. We then developed a new physical proxy accordingly and evaluated its stability during different seasons.
Materials and Methods
Field Observations
The observations were carried out at the Station for Observing Regional Processes of the Earth System (SORPES) in the northeastern part of Nanjing, China (118°57′E, 32°07′N), which is the regional background station upwind from downtown Nanjing, during winter (from December 11, 2017, to January 17, 2018), spring (from April 13, 2018, to April 23, 2018), summer (from July 11, 2018, to August 9, 2018), and autumn (all of November 2018). The surrounding environment of the site is given in Figure S1 and detailed descriptions can be found elsewhere.27−30
H2SO4 was measured with a chemical ionization mass spectrometer (CIMS) equipped with a nitrate ionization source.31 Ambient H2SO4 molecules were charged by reacting with NO3– reagent ions in the reaction chamber of the inlet. Then, the concentration is calculated from the measured ion signals according to:
| 1 |
where C is the calibration coefficient, which was obtained seasonally by the known concentration of H2SO4 calculated from the SO2–OH reaction here with 33% systematic uncertainty.32 Also, this instrument provides a high signal-to-noise ratio and the limit of detection for H2SO4 was reported to be close to 2 × 104 mol/cm3.31
During the whole four campaigns, we measured benzene and isoprene by proton-transfer-reaction mass spectrometry33 (PTR-ToF-MS). Trace gases (SO2, O3, NOx, and CO) were measured continuously using a Thermo TEI 43i, TEI 49i, TEI 42i, and TEI 48i, respectively. Typical anthropogenic alkenes were obtained by gas chromatography and mass spectrometry34 (TT24-2 GCMS) from November 26, 2019, to January 4, 2020. Ultraviolate B (UVB) radiation intensity was measured using a radiometer (Kipp & Zonen UVS-B-T). Wind speeds were measured at six levels using GILL Wind Sonic, and here, we used data from the height of 4 m. The particle number size distribution between 6 and 800 nm was measured using a differential mobility particle sizer (DMPS), and for the sub-6 nm particle and particles larger than 800 nm, a scanning mobility particle sizer (SMPS, 4–495.8 nm) and aerodynamic particle sizer (APS, 0.54–19.8 μm) were used respectively from January 1, 2019, to June 30, 2019. In this work, the daytime window was defined from 08:00 to 16:00 and the nighttime window from 20:00 to 04:00 on the following day. In addition to the correlation coefficient (R), the relative error (RE) is used to evaluate the performance of proxies in the statistical analysis and can be written as follows:
| 2 |
Here, X denotes the selected species and the subscript of 0.5 denotes the median number. More details are described in the Supporting Information.
Calculation Methods
The Proxy of Alkenes
Long-term measurement of complex alkenes is challenging, especially for those from anthropogenic sources. In this study, volatile organic compounds (VOCs) were measured simultaneously with H2SO4 during four seasons using PTR-ToF-MS, which is not sensitive to short-chain alkenes. Therefore, we derived a proxy based on a relatively short-term GC–MS measurement, during which ethylene, propene, and 1-butene were the dominating anthropogenic species and correlated linearly to benzene (Figure S3) with the slope of 1.922 and relative error of 28.2%. Since the estimated isoprene emissions are much larger than those of monoterpenes over the Yangtze River Delta,35 we used isoprene concentration to represent the total biogenic alkenes. Therefore, the total alkene concentration can be obtained by the following equation:
| 3 |
Calculation of Dry Deposition
Dry deposition, as the ultimate path by which trace gases and particles are removed from the atmosphere in the absence of precipitation, is governed by three factors: atmospheric turbulence, the physical and chemical properties of the depositing species, and the nature of the surface. Since simulating such a variety of complex processes is generally impractical, dry deposition is usually simplified as a single parameter, the deposition velocity (Vd).
Here, a dry deposition resistance model36 was used to estimate the deposition losses of H2SO4 indirectly based on measurements by an eddy-covariance system (EC3000, Campbell Scientific) at the height of 3 m. For gases, Vd in this dry deposition model is computed from a formula analogous to Ohm’s law in electrical circuits:37
| 4 |
The term Ra, governed by turbulent transport, represents aerodynamic resistance and Rbi represents the quasi-laminar resistance dependent on molecular properties of the substance and surface characteristics. Rc is the surface resistance, but it is negligible for H2SO4 due to its extremely high Henry’s law coefficient. The detailed calculation can be found in the Supporting Information.
Calculation of Condensation Sink (CS)
CS was calculated using the method proposed by Kulmala et al.38 based on measurements of the particle number size distribution between 6 and 800 nm using DMPS. Also, the contribution of sub-6 nm particles and particles larger than 800 nm to CS was estimated to be less than 10% based on measurements of SMPS and APS from January 1, 2019, to June 30, 2019, as described in the Supporting Information.
Simulation of Clustering Processes
Due to the lack of most H2SO4 clusters measurements, a kinetic model was used to estimate clustering processes. The distribution of molecular clusters in acid–base nucleation can be simulated using the explicit solution of the general dynamic equations. Detailed descriptions for the model can be found elsewhere.39
Estimation of Hydroxyl Radical (•OH)
For the difficulty to measure the OH radical continuously, we calculated the daytime OH concentration by applying the empirical formula proposed by Rohrer and Berresheim,40 which demonstrated that the OH radical is linearly correlated to the photolysis frequency J(O1D). We calculated J(O1D) using the tropospheric ultraviolet and visible (TUV) radiation model and corrected it by the observed UVB.41 Detailed information can be found in the Supporting Information.
Results and Discussion
Overall Observations and Empirical Formula
Seasonal patterns of the daytime H2SO4 concentration and related parameters are illustrated in Figure 1. The median daytime concentrations of H2SO4 were significantly higher in autumn (9.02 × 106 cm–3) and spring (7.35 × 106 cm–3) than in summer (4.47 × 106 cm–3) and winter (1.85 × 106 cm–3). A typical diurnal cycle shows the noontime peaks of H2SO4 to follow UVB in spring, autumn, and winter. In summer, SO2 concentrations, owing to the long-term emission reduction in China,30 were low enough to be the limiting factor for H2SO4 production, and the resulting H2SO4 concentration peaked at about 10:00 am when SO2 had its maximum value. We then used a traditional approach analogous to most previous works, developed empirical proxies of H2SO4 separately for the four seasons, and verified their suitability. Note that the traditional proxy excludes the formation of H2SO4 in the oxidation by sCI and, therefore, we only derived the daytime H2SO4 proxy. The proxy formula is as follows:
| 5 |
Figure 1.

Daytime variation of UVB, SO2, measured H2SO4, and calculated H2SO4 by empirical formulas in (a) winter, (b) spring, (c) summer, and (d) autumn. The daytime window is defined from 08:00 to 16:00. The levels of UVB and H2SO4 are displayed as their median concentrations. The blue horizontal lines show the median SO2, blue boxes show 25th and 75th percentile values, and whiskers show outlier cutoffs. The red points show the mean concentration of SO2. The bottom panel shows measured H2SO4 and calculated H2SO4 from four proxies based on different seasons. Blue lines, orange dotted lines, yellow dotted lines, purple dotted lines, and green dotted lines denote measured H2SO4 and calculated H2SO4 based on Proxywinter, Proxyspring, Proxysummer, and Proxyautumn, respectively. We provide a time series of related parameters in Figure S2, including UVB, PM2.5, NOx, O3, SO2, and H2SO4.
In Table S1, we summarize the proxy parameters of this work and previous studies. We can see that the fitting parameters (k0, a, b, and c) vary remarkably from site to site, as well as between the different seasons, challenging their general applicability. For example, the pre-exponential coefficient k0 varied from 0.01 to 2.33 × 103. To verify this, we used the proxy obtained from the measurement in one season to the other three seasons, showing the results and relative errors in Figure 1 and Table S2. Comparisons between the four proxies are shown in Figure S5. It is obvious that the empirical proxy derived from one season cannot capture the variation of H2SO4 in the other three seasons. Especially for Proxyautumn and Proxywinter, they either greatly overestimate or underestimate the H2SO4 concentration for the other campaigns, causing significant relative errors (even up to 240%). Furthermore, since these parameters are obtained from a mathematical fitting and are not independent of each other, the empirical proxy would not help understand the budget of H2SO4. Our results suggest that the empirical proxy has strong limitations and is incapable of being widely used.
Budget Analysis
Based on current knowledge, gaseous H2SO4 can only be formed from the reactions of SO2 with OH or sCI, and it can be lost to aerosol surfaces (condensation sink), ground surface (dry deposition), and newly formed particles by forming H2SO4 clusters. Given a very short lifetime, the budget of H2SO4 can be described by the following equation
| 6 |
During the daytime, the H2SO4 formation is dominated by the oxidation of SO2 by OH, which is extremely difficult to be measured but has been demonstrated to be well reproduced by J(O1D).40 During the nighttime, SO2 oxidation by sCI and OH, produced from the ozonolysis of alkenes, is believed to control the formation of H2SO4.21 In polluted urban areas, alkenes have complex species, and it is generally impractical to make long-term, full-spectrum measurements. Therefore, the alkene ozonolysis source is simplified to be a “bulk” item, being expressed as k1[O3][alkenes][SO2]. Here, k1 is an apparent reaction rate constant that takes into account the rate constants between O3 and alkenes, the yield of OH radical and sCI, and their reaction with SO2.
Most previous studies considered CS as the only important sink for gaseous H2SO4. However, the measurement site of H2SO4 is usually close to a ground surface (as well as other surfaces) so that dry deposition has the potential to be an important sink. Here, we estimated the loss of H2SO4 via dry deposition with a resistance model. Due to the extremely high Henry’s law coefficient of H2SO4, its dry deposition is limited by atmospheric turbulence. Accurate micrometeorological parameters are needed, especially the friction velocity, which was measured in real time based on the eddy-covariance system in our experiments. We found that the calculated H2SO4 loss onto the ground surface could be up to 30% of that onto aerosol particles with the sampling inlet height of 1.5 m, suggesting a considerable sink for the near-surface H2SO4, particularly during the noontime when the boundary layer mixing tends to decrease CS while, at the same time, enhancing dry deposition (Figure 2).
Figure 2.
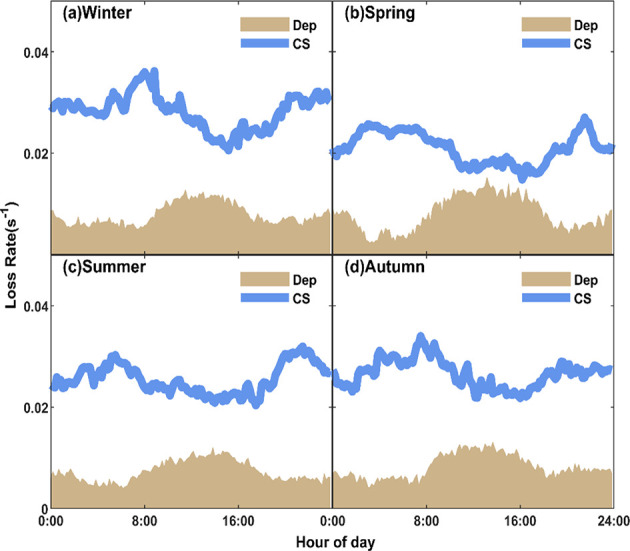
Diurnal variation of dry deposition rate and condensation sink in (a) winter, (b) spring, (c) summer, and (d) autumn.
In addition, clustering processes could be a considerable loss for the H2SO4 monomer but were difficult to be evaluated due to the limitation on measuring most clusters, i.e., (H2SO4)>2.7,11,42 Here, we deployed a kinetic model to simulate and estimate the clustering processes.39 Given that dimethylamine (DMA) is the crucial stabilizer to form H2SO4 clusters, especially in East China,8 we simulated the daytime clustering processes in four seasons with different DMA concentrations, as well as varied temperatures. The results showed that H2SO4 clustering processes at the SORPES station in summer and winter tended to the 10 ppt DMA scenario, while spring and autumn matched the 5 ppt DMA scenario (Figure S6).
Clustering processes were simplified as β[H2SO4]2. As shown in Figure 3, higher H2SO4 and DMA concentrations and lower CS and temperature tend to elevate the value of β. In our study, the majority of data points fall in the left side of the lines in Figure 3, where clustering losses contributed less than 10% of CS on removing the H2SO4 monomer. However, it was non-negligible in some time in spring, autumn, and winter. Therefore, we take this term into consideration in budget analysis, with β of 5.69 × 10–10 cm3 s–1 in winter, 1.37 × 10–10 cm3 s–1 in spring, 2.55 × 10–11 cm3 s–1 in summer, and 1.15 × 10–10 cm3 s–1 in autumn.
Figure 3.

Dependences of β on H2SO4 and CS with 10 ppt of DMA at different temperatures (a–c) and 5 ppt of DMA at different temperatures (d–f). Gray dots denote daytime measurements during winter, summer, and autumn. Black dots denote daytime measurements during spring.
By assuming the H2SO4 concentration to be at a pseudo-steady state (the left-hand side of eq 6 can be approximated to be zero), we fitted the data for the four seasons. The coefficients k1 and k2 were in the ranges of 2.21 × 10–30 to 5.91 × 10–30 cm6 s–1 and 5.82 × 10–2 to 18.00 × 10–2, respectively (Table S3), i.e., they were relatively stable between the different seasons. The SORPES station is located on the southern edge of East China, and it can be influenced by both anthropogenic and biogenic emissions.29,100 Since anthropogenic alkenes consist mainly of short-chain alkenes, we could take these three above-mentioned dominating alkenes as representative examples of anthropogenic alkenes, and isoprene as a representative for biogenic alkenes, to estimate the range of k1. The rate constants of the alkene–O3 reaction are 1.59 × 10–18 cm3 s–1 for ethene, 1.01 × 10–17 cm3 s–1 for propene, 9.64 × 10–18 cm3 s–1 for 1-butene, and 1.28 × 10–17 cm3 s–1 for isoprene.43 The rate constant of reaction between their derived sCI and SO2 varies from 2.4 × 10–11 to 6.7 × 10–11 cm3 s–1,44−47 and the yield of sCI and OH radical varies from 0.17 to 0.56.46,48−52 The unimolecular decomposition of sCI and reactions between sCI and atmospheric trace gas species, due to its strong chemical reactivity with them, result in a high loss rate of sCI, and we used the value of 32 s–1 for this loss rate based on previous studies.50,53 Therefore, k1 is considered to vary from 2.03 × 10–31 to 2.2 × 10–28 cm6 s–1 in theory, which well covered coefficients obtained in our experiments and reported in Beijing.21 It should be pointed out that in the real atmosphere, k1 can vary over a larger range due to the complexity of sCI chemistry.
k2 is also an apparent coefficient by considering the rate constant of the OH–SO2 reaction (kOH–SO2) and the pre-exponential coefficient (a, see Table S5) of the J(O1D)–OH nearly linear relationship, which can be described as [OH] = a×J(O1D)/10–5 s–1. kOH–SO2 was reported to be in the range from 3.39 × 10–13 to 1.24 × 10–12 cm3 s–1,54−57 and the value of coefficient a varied from 2 × 106 to 4.8 × 106 cm–3 in different observations (see Table S5). Therefore, the theoretical value of k2 should range from 0.068 to 0.595. Here, the fitted k2 varied from 0.058 to 0.18 for the different seasons, mostly within the theoretical value range.
Although the fitted k1 is within the theoretical range, calculations accordingly underestimated the nighttime H2SO4 significantly (Figure 4 and Figure S7) when the reaction of SO2 with sCI or OH from ozonolysis of alkenes was assumed to be the major source. This is particularly the truth during winter when simulated H2SO4 was one order of magnitude lower than the measured value.
Figure 4.
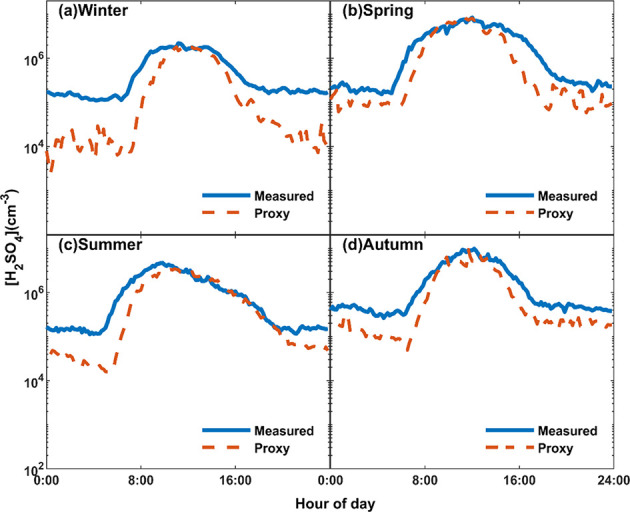
(a–d) Diurnal variation of measured SA and simulated SA based on eq 6. Coefficients in four seasons are listed in Table S3.
As shown in Figure 5a, the correlation between the loss of nighttime H2SO4 and ([SO2][O3][alkenes]) is poor, indicating additional sources other than alkene ozonolysis contributing to the nighttime H2SO4. We then investigated nighttime H2SO4 events, defined by two criteria: (a) H2SO4 rises, reaching a distinct peak, instead of a continuous decrease from 20:00 to 04:00 in the following day; (b) the maximum concentration exceeds 1 × 106 cm–3. In total, 14 events were selected (Figure S8), only 3 of which can be explained by the source of alkene ozonolysis. However, in 6 events, the H2SO4 concentration correlated strongly with the benzene concentration (Figure S9), indicating a direct emission source related to benzene. Here, we used the toluene-to-benzene (T/B) ratio to indicate the sources of VOCs. The T/B in nighttime benzene-related H2SO4 events has a range of 1–2, very close to road-side and tunnel research studies.58,59 SO2 is a byproduct of the combustion of most fossil fuels and has been reported to be emitted from on-road transportation.60 Since OH is also largely produced via burning processes,61 H2SO4 is then able to be emitted from vehicles or produced in the freshly emitted plumes by reacting with ambient SO2.
Figure 5.

(a) Scatter plot of loss term ([SA]CS + [SA]Dep + β[SA]2) and the source term ([SO2][O3][alkenes]). The data is colored with O3 concentration. (b) Scatter plot of benzene and unexplainable nighttime H2SO4. Gray dots denote all unexplainable nighttime H2SO4. Red triangles denote points with WS ≥ 1.5 m/s and CS ≤ 0.02 s–1 and the correlation coefficient (Spearman type) is 0.563. (c) Relationship between the nonlinear proxy of emissions and nighttime H2SO4 unexplained by the alkene ozonolysis source. The correlation coefficient (Pearson type) is 0.457 and the relative error is 68%.
To estimate the quantitative contribution from direct emissions, we selected the data points that cannot be explained by the alkene ozonolysis source, locating in the left of the line with k1 = 5 × 10–29 cm6 s–1 in Figure 5a. O3 concentrations for these data points were very low, probably due to a strong titration by freshly emitted NO. Because of the very short lifetime of gaseous H2SO4 in polluted air, plumes with freshly emitted H2SO4 need to transport quickly enough and have low pre-existing particle loading to overcome the loss of H2SO4 before arriving at the sampling site. Here, we separated the plumes using the wind speed threshold of 1.5 m/s and 0.02 s–1 for CS. Under high wind speed and low condensation sink, a positive correlation between the H2SO4 and benzene concentrations was observed (Figure 5b), suggesting that direct emissions related to benzene play an important role in nighttime H2SO4 formation. However, we cannot observe the signal of direct emission at other times. As a matter of fact, the observed H2SO4 concentration from emission is governed by both the intensity of emissions and losses along the transmission path. Since this term was found to be related to benzene, we could assume the intensity of primary emissions to be proportional to the concentration of benzene. Also, with wind speed and condensation sink into consideration, the emission term of H2SO4 can be expressed as
| 7 |
Here, the pre-exponential coefficient, k0, reflects the dependence on the intensity of emissions. The exponents a and b reflect the effects of wind speed and condensation sink, respectively. We further fitted these coefficients based on the nighttime data that cannot be explained by the alkene ozonolysis source. The values of k0, a, and b were 2.591 × 10–5, 1.398, and −1.404, respectively. Figure 5c shows the good performance of the proxy; the simulated emission term was well correlated with the unexplainable nighttime H2SO4. It needs to be noted that the contribution of direct emission to the observed H2SO4 concentration should be site/location-dependent. Primary H2SO4 has the potential to be co-emitted with SO2 from power plants, industry, international ships, residential emissions, and transportation on a global scale.62 Parameters, including the distance of emission sources and meteorological conditions, would have significant impacts on this term.
Proxy Development and Its Stability
Based on the above budget analysis of H2SO4, we developed a physical proxy for H2SO4 as follows, by considering all the above-mentioned sources and sinks:
| 8 |
Here, k1 is an apparent reaction rate constant, including the rate constant of the O3–alkenes reaction, the yield of OH radical and sCI, and the fraction and rate constants of their reaction with SO2; k2 is also an apparent coefficient that takes into account the rate constant of the OH–SO2 reaction and the pre-exponential coefficient of the nearly linear relationship between J(O1D) and OH. We first selected nighttime data points with O3 concentrations higher than 10 ppb in different seasons to obtain k1. As shown in Figure S10, the loss term of nighttime H2SO4 was strongly and positively correlated with the source term ([SO2][O3][alkenes]) at high concentrations of O3, suggesting that the alkene ozonolysis contributed significantly to the nighttime H2SO4 concentration. We then estimated k1 by fitting the 10th percentile data points to eliminate the impact of emissions to the extent possible. The estimated value of k1 ranged from 6.67 × 10–31 to 5.27 × 10–30 cm6 s–1 in different seasons, within the range of the theoretical value. The fitted k2 varied from 0.08 to 0.18 in different seasons (see Table S4), also within the range of the theoretical value.
The developed proxies captured well the measured values in different seasons (Figure S12a–d) and surprisingly correlated with each other very well (Figure S13), with correlation coefficients larger than 0.94, suggesting the strong stability of this method and the possibility to develop a more widely used proxy based on the data from all the seasons. The fitted values of k1 and k2 were 2.49 × 10–30 cm6 s–1 and 0.15, respectively, so the proxy can be written as
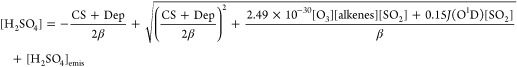 |
9 |
This final proxy showed a good performance in such a complex environment (Figure 6a), the simulated values being very close to the measured ones (Figure 6a,b), with a relative error of 58% and the correlation coefficient of 0.71. Given the systematic uncertainty in H2SO4 concentration discussed in the method part, our results suggest a nearly close relationship between measurements and proxy. In particular, the simulation of H2SO4 in the nighttime and early morning was significantly improved with the consideration of direct emission. Meanwhile, due to negligible clustering processes at most times, the proxy involving the dominant influencing factor can be simplified as eq 10.
| 10 |
Figure 6.

H2SO4 proxy based on data points during the total period (eq 9). (a) Diurnal variation of H2SO4 proxy and measured concentrations; (b) relationship between proxy H2SO4 and measured H2SO4; (c) fraction contribution of each source term to H2SO4 concentration during the nighttime and daytime.
We finally investigated the contribution from different sources quantitatively in different seasons based on the newly developed proxy (Figure 6c). As expected, the oxidation of SO2 by OH dominated the daytime H2SO4 formation with a contribution larger than 75% in all the seasons. Therefore, there is a possibility to derive a proxy based on the light-dependent source in the absence of more data. Ozonolysis of alkenes gave a small, about 6% contribution in both summer and autumn and smaller than 4% contribution in winter and spring. In winter daytime, primary emissions contributed considerably by more than 10%. During the nighttime, direct emission contributed much more than alkene ozonolysis, up to more than 90% during winter and about 80% during summer.
Uncertainty Analysis
Here, we discussed
the uncertainty
of the final proxy. For the light-dependent source, the uncertainty
of OH calculation consists of two main components: calculation of J(O1D) using the TUV radiation model and the
precision of OH calculation based on J(O1D). The relative error between the modeled and measured J(O1D) was estimated to be within 10%, except for the dust
event days.63 Here, we corrected the calculated J(O1D) by the observed UVB to further lower the
uncertainty, assuming 5% (the uncertainty of UVB measurement). Also,
the precision of the J(O1D) calculation
of OH was reported to be 7.8%.40 Therefore,
the total uncertainty of OH estimation should be around 9% (calculated
from 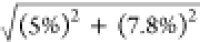 ). For the alkenes
ozonolysis source, the
uncertainty mainly comes from the estimation of total alkenes. First,
relative precisions of measurements are 12% for isoprene and 10% for
benzene.64 Second, the proxy of anthropogenic
alkene causes a relative error of 28%. The relative error of the emission
term was estimated as 68% (Figure 5c).
). For the alkenes
ozonolysis source, the
uncertainty mainly comes from the estimation of total alkenes. First,
relative precisions of measurements are 12% for isoprene and 10% for
benzene.64 Second, the proxy of anthropogenic
alkene causes a relative error of 28%. The relative error of the emission
term was estimated as 68% (Figure 5c).
The CS was underestimated by about 8% because we used the particle size distribution from 6 to 800 nm instead of a full range (Figure S3). In the dry deposition model, the extremely high Henry’s law coefficient of H2SO4 causes surface resistance, relying heavily on empirical data, which is negligible compared with aerodynamic and quasi-laminar resistance. However, the performance of the large-scale deposition model applied to such local measurements is not clear,37 and the uncertainty of dry deposition was assumed to be 100%. Direct measurement of dry deposition is encouraged in the future to compare with the model-based estimation.
Environmental Implication
Gaseous sulfuric acid (H2SO4) is essential for new particle formation (NPF) and therefore for the global budget of aerosol particles and cloud condensation nuclei. In situ measurements of H2SO4 are very rare, largely limiting our understanding of global NPF mechanisms. Previous efforts on building empirical proxies for H2SO4 have been demonstrated to be not broadly applicable. A physical proxy was believed to have a wide application, e.g., rebuild the long-term variation of H2SO4 in various environments. The detailed budget analysis of H2SO4 can shed some insights into improving the simulation of H2SO4, and nanoparticles, in the current regional and global air quality models that can help improve the global understanding of new particle and aerosol sulfate formation. In addition, a considerable contribution of primary emissions to H2SO4 indicates a new connection between human activities and climate change through anthropogenic H2SO4-induced secondary aerosol formation.
Acknowledgments
This work was mainly funded by the National Key R&D Program of China (2016YFC0202000 and 2016YFC0200500) and the National Natural Science Foundation of China (NSFC) project (41675145 and 91644218). Data analysis was also supported by other NSFC projects (41875175, 41605098, 42075101, 41875004, and 92044301), Jiangsu Province Key R&D Program Major Technology Demonstration (BE 2019704), Academy of Finland via Center of Excellence in Atmospheric Sciences (project no. 307331), and European Research Council via ATM-GTP (742206).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.1c00738.
Detailed descriptions of measurements of H2SO4, estimation of the hydroxyl radical, calculation of condensation sink, and dry deposition; map of the measurement of site (Figure S1); time series of H2SO4 and related parameters in four seasons (Figure S2); relationship between dominating alkenes and benzene (Figure S3); correlation between CS contributed by 6–800 nm particles and CS contributed by 4–19,800 nm particles (Figure S4); relationship between H2SO4 estimated by different proxies based on the empirical formulas in different seasons (Figure S5); comparison between daytime data in four seasons and simulations under different temperatures and DMA concentrations (Figure S6); performance of physical proxy without an emission term (Figure S7); time series of measured H2SO4, simulated H2SO4, and benzene during selected nighttime H2SO4 events (Figure S8); relationship between H2SO4 and benzene in emission cases (Figure S9); development of proxy with an emission term for four seasons (Figures S10 and S11); diurnal variations of measured H2SO4 and H2SO4 obtained from the respective steady-state equilibrium method with emissions into consideration (Figure S12); correlation between H2SO4 estimated by physical proxies in different seasons (Figure S13); development of the final proxy (Figure S14); results of the nonlinear proxy in daytime and comparison with previous works (Table S1); relative errors of traditional proxies (Table S2); coefficients of the physical proxy without an emission term (Table S3); coefficients of proxy based on seasonal data with O3 ≥ 10 ppb (Table S4); summary of the coefficients of the OH–J(O1D) nearly linear relationship in observations (Table S5) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Huang R.-J.; Zhang Y.; Bozzetti C.; Ho K.-F.; Cao J.-J.; Han Y.; Daellenbach K. R.; Slowik J. G.; Platt S. M.; Canonaco F.; Zotter P.; Wolf R.; Pieber S. M.; Bruns E. A.; Crippa M.; Ciarelli G.; Piazzalunga A.; Schwikowski M.; Abbaszade G.; Schnelle-Kreis J.; Zimmermann R.; An Z.; Szidat S.; Baltensperger U.; El Haddad I.; Prévôt A. S. H. High secondary aerosol contribution to particulate pollution during haze events in China. Nature 2014, 514, 218–222. 10.1038/nature13774. [DOI] [PubMed] [Google Scholar]
- Chow J. C.; Watson J. G.; Mauderly J. L.; Costa D. L.; Wyzga R. E.; Vedal S.; Hidy G. M.; Altshuler S. L.; Marrack D.; Heuss J. M.; Wolff G. T.; Arden Pope C. III; Dockery D. W. Health effects of fine particulate air pollution: Lines that connect. J. Air Waste Manage. Assoc. 2006, 56, 1368–1380. 10.1080/10473289.2006.10464545. [DOI] [PubMed] [Google Scholar]
- Charlson R. J.; Schwartz S. E.; Hales J. M.; Cess R. D.; Coakley J. A.; Hansen J. E.; Hofmann D. J. Climate Forcing by Anthropogenic Aerosols. Science 1992, 255, 423–430. 10.1126/science.255.5043.423. [DOI] [PubMed] [Google Scholar]
- Kerminen V.-M.; Chen X.; Vakkari V.; Petäjä T.; Kulmala M.; Bianchi F. Atmospheric new particle formation and growth: review of field observations. Environ. Res. Lett. 2018, 13, 103003. 10.1088/1748-9326/aadf3c. [DOI] [Google Scholar]
- Yue D.; Hu M.; Wu Z.; Wang Z.; Guo S.; Wehner B.; Nowak A.; Achtert P.; Wiedensohler A.; Jung J.; Kim Y. J.; Liu S. Characteristics of aerosol size distributions and new particle formation in the summer in Beijing. J. Geophys. Res.: Atmos. 2009, 114, 13. 10.1029/2008JD010894. [DOI] [Google Scholar]
- Ball S. M.; Hanson D. R.; Eisele F. L.; McMurry P. H. Laboratory studies of particle nucleation: Initial results for H2SO4, H2O, and NH3 vapors. J. Geophys. Res.: Atmos. 1999, 104, 23709–23718. 10.1029/1999JD900411. [DOI] [Google Scholar]
- Kirkby J.; Curtius J.; Almeida J.; Dunne E.; Duplissy J.; Ehrhart S.; Franchin A.; Gagné S.; Ickes L.; Kürten A.; Kupc A.; Metzger A.; Riccobono F.; Rondo L.; Schobesberger S.; Tsagkogeorgas G.; Wimmer D.; Amorim A.; Bianchi F.; Breitenlechner M.; David A.; Dommen J.; Downard A.; Ehn M.; Flagan R. C.; Haider S.; Hansel A.; Hauser D.; Jud W.; Junninen H.; Kreissl F.; Kvashin A.; Laaksonen A.; Lehtipalo K.; Lima J.; Lovejoy E. R.; Makhmutov V.; Mathot S.; Mikkilä J.; Minginette P.; Mogo S.; Nieminen T.; Onnela A.; Pereira P.; Petäjä T.; Schnitzhofer R.; Seinfeld J. H.; Sipilä M.; Stozhkov Y.; Stratmann F.; Tomé A.; Vanhanen J.; Viisanen Y.; Vrtala A.; Wagner P. E.; Walther H.; Weingartner E.; Wex H.; Winkler P. M.; Carslaw K. S.; Worsnop D. R.; Baltensperger U.; Kulmala M. Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation. Nature 2011, 476, 429–433. 10.1038/nature10343. [DOI] [PubMed] [Google Scholar]
- Yao L.; Garmash O.; Bianchi F.; Zheng J.; Yan C.; Kontkanen J.; Junninen H.; Mazon S. B.; Ehn M.; Paasonen P.; Sipilä M.; Wang M.; Wang X.; Xiao S.; Chen H.; Lu Y.; Zhang B.; Wang D.; Fu Q.; Geng F.; Li L.; Wang H.; Qiao L.; Yang X.; Chen J.; Kerminen V. M.; Petäjä T.; Worsnop D. R.; Kulmala M.; Wang L. Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity. Science 2018, 361, 278–281. 10.1126/science.aao4839. [DOI] [PubMed] [Google Scholar]
- Almeida J.; Schobesberger S.; Kürten A.; Ortega I. K.; Kupiainen-Määttä O.; Praplan A. P.; Adamov A.; Amorim A.; Bianchi F.; Breitenlechner M.; David A.; Dommen J.; Donahue N. M.; Downard A.; Dunne E.; Duplissy J.; Ehrhart S.; Flagan R. C.; Franchin A.; Guida R.; Hakala J.; Hansel A.; Heinritzi M.; Henschel H.; Jokinen T.; Junninen H.; Kajos M.; Kangasluoma J.; Keskinen H.; Kupc A.; Kurtén T.; Kvashin A. N.; Laaksonen A.; Lehtipalo K.; Leiminger M.; Leppä J.; Loukonen V.; Makhmutov V.; Mathot S.; McGrath M. J.; Nieminen T.; Olenius T.; Onnela A.; Petäjä T.; Riccobono F.; Riipinen I.; Rissanen M.; Rondo L.; Ruuskanen T.; Santos F. D.; Sarnela N.; Schallhart S.; Schnitzhofer R.; Seinfeld J. H.; Simon M.; Sipilä M.; Stozhkov Y.; Stratmann F.; Tomé A.; Tröstl J.; Tsagkogeorgas G.; Vaattovaara P.; Viisanen Y.; Virtanen A.; Vrtala A.; Wagner P. E.; Weingartner E.; Wex H.; Williamson C.; Wimmer D.; Ye P.; Yli-Juuti T.; Carslaw K. S.; Kulmala M.; Curtius J.; Baltensperger U.; Worsnop D. R.; Vehkamäki H.; Kirkby J. Molecular understanding of sulphuric acid-amine particle nucleation in the atmosphere. Nature 2013, 502, 359–363. 10.1038/nature12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R.; Suh I.; Zhao J.; Zhang D.; Fortner E. C.; Tie X.; Molina L. T.; Molina M. J. Atmospheric new particle formation enhanced by organic acids. Science 2004, 304, 1487–1490. 10.1126/science.1095139. [DOI] [PubMed] [Google Scholar]
- Riccobono F.; Schobesberger S.; Scott C. E.; Dommen J.; Ortega I. K.; Rondo L.; Almeida J.; Amorim A.; Bianchi F.; Breitenlechner M.; David A.; Downard A.; Dunne E. M.; Duplissy J.; Ehrhart S.; Flagan R. C.; Franchin A.; Hansel A.; Junninen H.; Kajos M.; Keskinen H.; Kupc A.; Kurten A.; Kvashin A. N.; Laaksonen A.; Lehtipalo K.; Makhmutov V.; Mathot S.; Nieminen T.; Onnela A.; Petaja T.; Praplan A. P.; Santos F. D.; Schallhart S.; Seinfeld J. H.; Sipila M.; Spracklen D. V.; Stozhkov Y.; Stratmann F.; Tome A.; Tsagkogeorgas G.; Vaattovaara P.; Viisanen Y.; Vrtala A.; Wagner P. E.; Weingartner E.; Wex H.; Wimmer D.; Carslaw K. S.; Curtius J.; Donahue N. M.; Kirkby J.; Kulmala M.; Worsnop D. R.; Baltensperger U. Oxidation Products of Biogenic Emissions Contribute to Nucleation of Atmospheric Particles. Science 2014, 344, 717–721. 10.1126/science.1243527. [DOI] [PubMed] [Google Scholar]
- Lehtipalo K.; Yan C.; Dada L.; Bianchi F.; Xiao M.; Wagner R.; Stolzenburg D.; Ahonen L. R.; Amorim A.; Baccarini A.; Bauer P. S.; Baumgartner B.; Bergen A.; Bernhammer A. K.; Breitenlechner M.; Brilke S.; Buchholz A.; Mazon S. B.; Chen D.; Chen X.; Dias A.; Dommen J.; Draper D. C.; Duplissy J.; Ehn M.; Finkenzeller H.; Fischer L.; Frege C.; Fuchs C.; Garmash O.; Gordon H.; Hakala J.; He X.; Heikkinen L.; Heinritzi M.; Helm J. C.; Hofbauer V.; Hoyle C. R.; Jokinen T.; Kangasluoma J.; Kerminen V. M.; Kim C.; Kirkby J.; Kontkanen J.; Kürten A.; Lawler M. J.; Mai H.; Mathot S.; Mauldin R. L. III; Molteni U.; Nichman L.; Nie W.; Nieminen T.; Ojdanic A.; Onnela A.; Passananti M.; Petäjä T.; Piel F.; Pospisilova V.; Quéléver L. L. J.; Rissanen M. P.; Rose C.; Sarnela N.; Schallhart S.; Schuchmann S.; Sengupta K.; Simon M.; Sipilä M.; Tauber C.; Tomé A.; Tröstl J.; Väisänen O.; Vogel A. L.; Volkamer R.; Wagner A. C.; Wang M.; Weitz L.; Wimmer D.; Ye P.; Ylisirniö A.; Zha Q.; Carslaw K. S.; Curtius J.; Donahue N. M.; Flagan R. C.; Hansel A.; Riipinen I.; Virtanen A.; Winkler P. M.; Baltensperger U.; Kulmala M.; Worsnop D. R. Multicomponent new particle formation from sulfuric acid, ammonia, and biogenic vapors. Sci. Adv. 2018, 4, eaau5363. 10.1126/sciadv.aau5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmili W.; Berresheim H.; Plass-Dülmer C.; Elste T.; Gilge S.; Wiedensohler A.; Uhrner U. The Hohenpeissenberg aerosol formation experiment (HAFEX): a long-term study including size-resolved aerosol, H2SO4, OH, and monoterpenes measurements. Atmos. Chem. Phys. 2003, 3, 361–376. 10.5194/acp-3-361-2003. [DOI] [Google Scholar]
- Erupe M. E.; Benson D. R.; Li J.; Young L.-H.; Verheggen B.; al-Refai M.; Tahboub O.; Cunningham V.; Frimpong F.; Viggiano A. A.; Lee S.-H. Correlation of aerosol nucleation rate with sulfuric acid and ammonia in Kent, Ohio: An atmospheric observation. J. Geophys. Res.: Atmos. 2010, 115, 18. 10.1029/2010JD013942. [DOI] [Google Scholar]
- Riipinen I.; Sihto S.-L.; Kulmala M.; Arnold F.; Dal Maso M.; Birmili W.; Saarnio K.; Teinilä K.; Kerminen V.-M.; Laaksonen A.; Lehtinen K. E. J. Connections between atmospheric sulphuric acid and new particle formation during QUEST III–IV campaigns in Heidelberg and Hyytiälä. Atmos. Chem. Phys. 2007, 7, 1899–1914. 10.5194/acp-7-1899-2007. [DOI] [Google Scholar]
- Mikkonen S.; Romakkaniemi S.; Smith J. N.; Korhonen H.; Petäjä T.; Plass-Duelmer C.; Boy M.; McMurry P. H.; Lehtinen K. E. J.; Joutsensaari J.; Hamed A.; Mauldin Iii R. L.; Birmili W.; Spindler G.; Arnold F.; Kulmala M.; Laaksonen A. A statistical proxy for sulphuric acid concentration. Atmos. Chem. Phys. 2011, 11, 11319–11334. 10.5194/acp-11-11319-2011. [DOI] [Google Scholar]
- Berresheim H.; Adam M.; Monahan C.; O’Dowd C.; Plane J. M. C.; Bohn B.; Rohrer F. Missing SO2 oxidant in the coastal atmosphere? - observations from high-resolution measurements of OH and atmospheric sulfur compounds. Atmos. Chem. Phys. 2014, 14, 12209–12223. 10.5194/acp-14-12209-2014. [DOI] [Google Scholar]
- Dada L.; Ylivinkka I.; Baalbaki R.; Li C.; Guo Y.; Yan C.; Yao L.; Sarnela N.; Jokinen T.; Daellenbach K. R.; Yin R.; Deng C.; Chu B.; Nieminen T.; Wang Y.; Lin Z.; Thakur R. C.; Kontkanen J.; Stolzenburg D.; Sipilä M.; Hussein T.; Paasonen P.; Bianchi F.; Salma I.; Weidinger T.; Pikridas M.; Sciare J.; Jiang J.; Liu Y.; Petäjä T.; Kerminen V. M.; Kulmala M. Sources and sinks driving sulfuric acid concentrations in contrasting environments: implications on proxy calculations. Atmos. Chem. Phys. 2020, 20, 11747–11766. 10.5194/acp-20-11747-2020. [DOI] [Google Scholar]
- Mauldin Iii R. L.; Berndt T.; Sipilä M.; Paasonen P.; Petäjä T.; Kim S.; Kurtén T.; Stratmann F.; Kerminen V. M.; Kulmala M. A new atmospherically relevant oxidant of sulphur dioxide. Nature 2012, 488, 193–196. 10.1038/nature11278. [DOI] [PubMed] [Google Scholar]
- Kim S.; Guenther A.; Lefer B.; Flynn J.; Griffin R.; Rutter A. P.; Gong L.; Cevik B. K. Potential Role of Stabilized Criegee Radicals in Sulfuric Acid Production in a High Biogenic VOC Environment. Environ. Sci. Technol. 2015, 49, 3383–3391. 10.1021/es505793t. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Yan C.; Li C.; Ma W.; Feng Z.; Zhou Y.; Lin Z.; Dada L.; Stolzenburg D.; Yin R.; Kontkanen J.; Daellenbach K. R.; Kangasluoma J.; Yao L.; Chu B.; Wang Y.; Cai R.; Bianchi F.; Liu Y.; Kulmala M. Formation of nighttime sulfuric acid from the ozonolysis of alkenes in Beijing. Atmos. Chem. Phys. 2021, 21, 5499–5511. 10.5194/acp-21-5499-2021. [DOI] [Google Scholar]
- Berresheim H.; Elste T.; Tremmel H. G.; Allen A. G.; Hansson H.-C.; Rosman K.; Dal Maso M.; Mäkelä J. M.; Kulmala M.; O’Dowd C. D. Gas-aerosol relationships of H2SO4, MSA, and OH: Observations in the coastal marine boundary layer at Mace Head, Ireland. J. Geophys. Res.: Atmos. 2002, 107, PAR 5-1–PAR 5-12. 10.1029/2000JD000229. [DOI] [Google Scholar]
- Davis D.; Chen G.; Kasibhatla P.; Jefferson A.; Tanner D.; Eisele F.; Lenschow D.; Neff W.; Berresheim H. DMS oxidation in the Antarctic marine boundary layer: Comparison of model simulations and field observations of DMS, DMSO, DMSO2, H2SO4(g), MSA(g), and MSA(p). J. Geophys. Res.: Atmos. 1998, 103, 1657–1678. 10.1029/97JD03452. [DOI] [Google Scholar]
- Olin M.; Kuuluvainen H.; Aurela M.; Kalliokoski J.; Kuittinen N.; Isotalo M.; Timonen H. J.; Niemi J. V.; Rönkkö T.; Dal Maso M. Traffic-originated nanocluster emission exceeds H2SO4-driven photochemical new particle formation in an urban area. Atmos. Chem. Phys. 2020, 20, 1–13. 10.5194/acp-20-1-2020. [DOI] [Google Scholar]
- Petäjä T.; Mauldin R. L. III; Kosciuch E.; McGrath J.; Nieminen T.; Paasonen P.; Boy M.; Adamov A.; Kotiaho T.; Kulmala M. Sulfuric acid and OH concentrations in a boreal forest site. Atmos. Chem. Phys. 2009, 9, 7435–7448. 10.5194/acp-9-7435-2009. [DOI] [Google Scholar]
- Lu Y.; Yan C.; Fu Y.; Chen Y.; Liu Y.; Yang G.; Wang Y.; Bianchi F.; Chu B.; Zhou Y.; Yin R.; Baalbaki R.; Garmash O.; Deng C.; Wang W.; Liu Y.; Petäjä T.; Kerminen V.-M.; Jiang J.; Kulmala M.; Wang L. A proxy for atmospheric daytime gaseous sulfuric acid concentration in urban Beijing. Atmos. Chem. Phys. 2019, 19, 1971–1983. 10.5194/acp-19-1971-2019. [DOI] [Google Scholar]
- Ding A. J.; Fu C. B.; Yang X. Q.; Sun J. N.; Zheng L. F.; Xie Y. N.; Herrmann E.; Nie W.; Petäjä T.; Kerminen V. M.; Kulmala M. Ozone and fine particle in the western Yangtze River Delta: an overview of 1 yr data at the SORPES station. Atmos. Chem. Phys. 2013, 13, 5813–5830. 10.5194/acp-13-5813-2013. [DOI] [Google Scholar]
- Nie W.; Ding A. J.; Xie Y. N.; Xu Z.; Mao H.; Kerminen V. M.; Zheng L. F.; Qi X. M.; Huang X.; Yang X. Q.; Sun J. N.; Herrmann E.; Petäjä T.; Kulmala M.; Fu C. B. Influence of biomass burning plumes on HONO chemistry in eastern China. Atmos. Chem. Phys. 2015, 15, 1147–1159. 10.5194/acp-15-1147-2015. [DOI] [Google Scholar]
- Ding A.; Nie W.; Huang X.; Chi X.; Sun J.; Kerminen V.-M.; Xu Z.; Guo W.; Petäjä T.; Yang X.; Kulmala M.; Fu C. Long-term observation of air pollution-weather/climate interactions at the SORPES station: a review and outlook. Front. Environ. Sci. Eng. 2016, 10, 15. 10.1007/s11783-016-0877-3. [DOI] [Google Scholar]
- Xu Z. N.; Nie W.; Liu Y. L.; Sun P.; Huang D. D.; Yan C.; Krechmer J.; Ye P. L.; Xu Z.; Qi X. M.; Zhu C. J.; Li Y. Y.; Wang T. Y.; Wang L.; Huang X.; Tang R. Z.; Guo S.; Xiu G. L.; Fu Q. Y.; Worsnop D.; Chi X. G.; Ding A. J. Multifunctional Products of Isoprene Oxidation in Polluted Atmosphere and Their Contribution to SOA. Geophysical Research Letters 2021, 48, e2020GL089276. 10.1029/2020GL089276. [DOI] [Google Scholar]
- Ding A.; Huang X.; Nie W.; Chi X.; Xu Z.; Zheng L.; Xu Z.; Xie Y.; Qi X.; Shen Y.; Sun P.; Wang J.; Wang L.; Sun J.; Yang X.-Q.; Qin W.; Zhang X.; Cheng W.; Liu W.; Pan L.; Fu C. Significant reduction of PM2.5 in eastern China due to regional-scale emission control: evidence from SORPES in 2011-2018. Atmos. Chem. Phys. 2019, 19, 11791–11801. 10.5194/acp-19-11791-2019. [DOI] [Google Scholar]
- Jokinen T.; Sipilä M.; Junninen H.; Ehn M.; Lönn G.; Hakala J.; Petäjä T.; Mauldin R. L. III; Kulmala M.; Worsnop D. R. Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF. Atmos. Chem. Phys. 2012, 12, 4117–4125. 10.5194/acp-12-4117-2012. [DOI] [Google Scholar]
- Kürten A.; Rondo L.; Ehrhart S.; Curtius J. Calibration of a Chemical Ionization Mass Spectrometer for the Measurement of Gaseous Sulfuric Acid. J. Phys. Chem. A 2012, 116, 6375–6386. 10.1021/jp212123n. [DOI] [PubMed] [Google Scholar]
- Yuan B.; Koss A. R.; Warneke C.; Coggon M.; Sekimoto K.; de Gouw J. A. Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences. Chem. Rev. 2017, 117, 13187–13229. 10.1021/acs.chemrev.7b00325. [DOI] [PubMed] [Google Scholar]
- Wang J. L.; Chang C. C.; Lee K. Z. In-line sampling with gas chromatography-mass spectrometry to monitor ambient volatile organic compounds. J. Chromatogr. A 2012, 1248, 161–168. 10.1016/j.chroma.2012.05.091. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Li L.; An J.; Huang L.; Yan R.; Huang C.; Wang H.; Wang Q.; Wang M.; Zhang W. Estimation of biogenic VOC emissions and its impact on ozone formation over the Yangtze River Delta region, China. Atmos. Environ. 2018, 186, 113–128. 10.1016/j.atmosenv.2018.05.027. [DOI] [Google Scholar]
- Wesely M. L. Parameterization of surface resistances to gaseous dry deposition in regional-scale numerical models. Atmos. Environ. 1989, 23, 1293–1304. 10.1016/0004-6981(89)90153-4. [DOI] [Google Scholar]
- Wesely M. L.; Hicks B. B. A review of the current status of knowledge on dry deposition. Atmos. Environ. 2000, 34, 2261–2282. 10.1016/S1352-2310(99)00467-7. [DOI] [Google Scholar]
- Kulmala M.; Petäjä T.; Nieminen T.; Sipilä M.; Manninen H. E.; Lehtipalo K.; Dal Maso M.; Aalto P. P.; Junninen H.; Paasonen P.; Riipinen I.; Lehtinen K. E. J.; Laaksonen A.; Kerminen V.-M. Measurement of the nucleation of atmospheric aerosol particles. Nat. Protoc. 2012, 7, 1651–1667. 10.1038/nprot.2012.091. [DOI] [PubMed] [Google Scholar]
- Xiao M.; Hoyle C. R.; Dada L.; Stolzenburg D.; Kürten A.; Wang M.; Lamkaddam H.; Garmash O.; Mentler B.; Molteni U.; Baccarini A.; Simon M.; He X.-C.; Lehtipalo K.; Ahonen L. R.; Baalbaki R.; Bauer P. S.; Beck L.; Bell D.; Bianchi F.; Brilke S.; Chen D.; Chiu R.; Dias A.; Duplissy J.; Finkenzeller H.; Gordon H.; Hofbauer V.; Kim C.; Koenig T. K.; Lampilahti J.; Lee C. P.; Li Z.; Mai H.; Makhmutov V.; Manninen H. E.; Marten R.; Mathot S.; Mauldin R. L.; Nie W.; Onnela A.; Partoll E.; Petäjä T.; Pfeifer J.; Pospisilova V.; Quéléver L. L. J.; Rissanen M.; Schobesberger S.; Schuchmann S.; Stozhkov Y.; Tauber C.; Tham Y. J.; Tomé A.; Vazquez-Pufleau M.; Wagner A. C.; Wanger R.; Wang Y.; Weitz L.; Wimmer D.; Wu Y.; Yan C.; Ye P.; Ye Q.; Zha Q.; Zhou X.; Amorim A.; Carslaw K.; Curtius J.; Hansel A.; Volkamer R.; Winkler P. M.; Flagan R. C.; Kulmala M.; Worsnop D. R.; Kirkby J.; Donahue N. M.; Baltensperger U.; El Haddad I.; Dommen J. The driving factors of new particle formation and growth in the polluted boundary layer. Atmos. Chem. Phys. Discuss 2021, 1–28. 10.5194/acp-2020-1323. [DOI] [Google Scholar]
- Rohrer F.; Berresheim H. Strong correlation between levels of tropospheric hydroxyl radicals and solar ultraviolet radiation. Nature 2006, 442, 184–187. 10.1038/nature04924. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Nie W.; Xu Z.; Wang T.; Wang R.; Li Y.; Wang L.; Chi X.; Ding A. Semi-quantitative understanding of source contribution to nitrous acid (HONO) based on 1 year of continuous observation at the SORPES station in eastern China. Atmos. Chem. Phys. 2019, 19, 13289–13308. 10.5194/acp-19-13289-2019. [DOI] [Google Scholar]
- Hirsikko A.; Nieminen T.; Gagné S.; Lehtipalo K.; Manninen H. E.; Ehn M.; Hõrrak U.; Kerminen V.-M.; Laakso L.; McMurry P. H.; Mirme A.; Mirme S.; Petäjä T.; Tammet H.; Vakkari V.; Vana M.; Kulmala M. Atmospheric ions and nucleation: a review of observations. Atmos. Chem. Phys. 2011, 11, 767–798. 10.5194/acp-11-767-2011. [DOI] [Google Scholar]
- Atkinson R. Gas-phase tropospheric chemistry of volatile organic compounds .1. Alkanes and alkenes. J. Phys. Chem. Ref. Data 1997, 26, 215–290. 10.1063/1.556012. [DOI] [Google Scholar]
- Welz O.; Savee J. D.; Osborn D. L.; Vasu S. S.; Percival C. J.; Shallcross D. E.; Taatjes C. A. Direct Kinetic Measurements of Criegee Intermediate (CH2OO) Formed by Reaction of CH2I with O-2. Science 2012, 335, 204–207. 10.1126/science.1213229. [DOI] [PubMed] [Google Scholar]
- Taatjes C. A.; Welz O.; Eskola A. J.; Savee J. D.; Scheer A. M.; Shallcross D. E.; Rotavera B.; Lee E. P. F.; Dyke J. M.; Mok D. K. W.; Osborn D. L.; Percival C. J. Direct Measurements of Conformer-Dependent Reactivity of the Criegee Intermediate CH3CHOO. Science 2013, 340, 177–180. 10.1126/science.1234689. [DOI] [PubMed] [Google Scholar]
- Newland M. J.; Rickard A. R.; Vereecken L.; Muñoz A.; Ródenas M.; Bloss W. J. Atmospheric isoprene ozonolysis: impacts of stabilised Criegee intermediate reactions with SO2, H2O and dimethyl sulfide. Atmos. Chem. Phys. 2015, 15, 9521–9536. 10.5194/acp-15-9521-2015. [DOI] [PubMed] [Google Scholar]
- Stone D.; Blitz M.; Daubney L.; Howes N. U. M.; Seakins P. Kinetics of CH2OO reactions with SO2, NO2, NO, H2O and CH3CHO as a function of pressure. Phys. Chem. Chem. Phys. 2014, 16, 1139–1149. 10.1039/C3CP54391A. [DOI] [PubMed] [Google Scholar]
- Alam M. S.; Camredon M.; Rickard A. R.; Carr T.; Wyche K. P.; Hornsby K. E.; Monks P. S.; Bloss W. J. Total radical yields from tropospheric ethene ozonolysis. Phys. Chem. Chem. Phys. 2011, 13, 11002–11015. 10.1039/c0cp02342f. [DOI] [PubMed] [Google Scholar]
- Malkin T. L.; Goddard A.; Heard D. E.; Seakins P. W. Measurements of OH and HO2 yields from the gas phase ozonolysis of isoprene. Atmos. Chem. Phys. 2010, 10, 1441–1459. 10.5194/acp-10-1441-2010. [DOI] [Google Scholar]
- Novelli A.; Vereecken L.; Lelieveld J.; Harder H. Direct observation of OH formation from stabilised Criegee intermediates. Phys. Chem. Chem. Phys. 2014, 16, 19941–19951. 10.1039/C4CP02719A. [DOI] [PubMed] [Google Scholar]
- Hasson A. S.; Ho A. W.; Kuwata K. T.; Paulson S. E. Production of stabilized Criegee intermediates and peroxides in the gas phase ozonolysis of alkenes: 2. Asymmetric and biogenic alkenes. J. Geophys. Res.-Atmos. 2001, 106, 34143–34153. 10.1029/2001JD000598. [DOI] [Google Scholar]
- Campos-Pineda M.; Zhang J. Product yields of stabilized Criegee intermediates in the ozonolysis reactions of cis-2-butene, 2-methyl-2-butene, cyclopentene, and cyclohexene. Sci. China-Chem. 2018, 61, 850–856. 10.1007/s11426-017-9229-0. [DOI] [Google Scholar]
- Novelli A.; Hens K.; Ernest C. T.; Martinez M.; Nölscher A. C.; Sinha V.; Paasonen P.; Petäjä T.; Sipilä M.; Elste T.; Plass-Dülmer C.; Phillips G. J.; Kubistin D.; Williams J.; Vereecken L.; Lelieveld J.; Harder H. Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument. Atmos. Chem. Phys. 2017, 17, 7807–7826. 10.5194/acp-17-7807-2017. [DOI] [Google Scholar]
- Wine P. H.; Thompson R. J.; Ravishankara A. R.; Semmes D. H.; Gump C. A.; Torabi A.; Nicovich J. M. Kinetics of the Reaction OH + SO2 + M .fwdarw. HOSO2 + M. Temperature and Pressure Dependence in the Fall-off Region. J. Phys. Chem. 1984, 88, 2095–2104. 10.1021/j150654a031. [DOI] [Google Scholar]
- Long B.; Bao J. L.; Truhlar D. G. Reaction of SO2 with OH in the atmosphere. Phys. Chem. Chem. Phys. 2017, 19, 8091–8100. 10.1039/C7CP00497D. [DOI] [PubMed] [Google Scholar]
- Blitz M. A.; Salter R. J.; Heard D. E.; Seakins P. W. An Experimental and Master Equation Study of the Kinetics of OH/OD + SO2: The Limiting High-Pressure Rate Coefficients. J. Phys. Chem. A 2017, 121, 3184–3191. 10.1021/acs.jpca.7b01295. [DOI] [PubMed] [Google Scholar]
- Blitz M. A.; Salter R. J.; Heard D. E.; Seakins P. W. An Experimental Study of the Kinetics of OH/OD(v=1,2,3) + SO2: The Limiting High-Pressure Rate Coefficients as a Function of Temperature. J. Phys. Chem. A 2017, 121, 3175–3183. 10.1021/acs.jpca.7b01294. [DOI] [PubMed] [Google Scholar]
- Gentner D. R.; Worton D. R.; Isaacman G.; Davis L. C.; Dallmann T. R.; Wood E. C.; Herndon S. C.; Goldstein A. H.; Harley R. A. Chemical Composition of Gas-Phase Organic Carbon Emissions from Motor Vehicles and Implications for Ozone Production. Environ. Sci. Technol. 2013, 47, 11837–11848. 10.1021/es401470e. [DOI] [PubMed] [Google Scholar]
- Gelencsér A.; Siszler K.; Hlavay J. Toluene-benzene concentration ratio as a tool for characterizing the distance from vehicular emission sources. Environ. Sci. Technol. 1997, 31, 2869–2872. 10.1021/es970004c. [DOI] [Google Scholar]
- Fu X.; Wang S.; Zhao B.; Xing J.; Cheng Z.; Liu H.; Hao J. Emission inventory of primary pollutants and chemical speciation in 2010 for the Yangtze River Delta region, China. Atmos. Environ. 2013, 70, 39–50. 10.1016/j.atmosenv.2012.12.034. [DOI] [Google Scholar]
- Blocquet M.; Schoemaecker C.; Amedro D.; Herbinet O.; Battin-Leclerc F.; Fittschen C. Quantification of OH and HO2 radicals during the low-temperature oxidation of hydrocarbons by Fluorescence Assay by Gas Expansion technique. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20014–20017. 10.1073/pnas.1314968110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Q.; Shen H.; Yun X.; Chen Y.; Ren Y.’a.; Xu H.; Shen G.; Du W.; Meng J.; Li W.; Ma J.; Tao S. Global Sulfur Dioxide Emissions and the Driving Forces. Environ. Sci. Technol. 2020, 54, 6508–6517. 10.1021/acs.est.9b07696. [DOI] [PubMed] [Google Scholar]
- Balis D. S.; Zerefos C. S.; Kourtidis K.; Bais A. F.; Hofzumahaus A.; Kraus A.; Schmitt R.; Blumthaler M.; Gobbi G. P. Measurements and modeling of photolysis rates during the Photochemical Activity and Ultraviolet Radiation (PAUR) II campaign. J. Geophys. Res.: Atmos. 2002, 107, PAU 5-1. 10.1029/2000JD000136. [DOI] [Google Scholar]
- Müller M.; Mikoviny T.; Feil S.; Haidacher S.; Hanel G.; Hartungen E.; Jordan A.; Märk L.; Mutschlechner P.; Schottkowsky R.; Sulzer P.; Crawford J. H.; Wisthaler A. A compact PTR-ToF-MS instrument for airborne measurements of volatile organic compounds at high spatiotemporal resolution. Atmos. Meas. Tech. 2014, 7, 3763–3772. 10.5194/amt-7-3763-2014. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.