

Abstract
Psoriasis and type 2 diabetes (T2D) are complex conditions with significant impact on health. Psoriasis patients have higher risk of type 2 diabetes (~1.5 Odds Ratio) and vice versa, controlling for body mass index (BMI), yet there has been limited study comparing their genetic architecture. We hypothesized there are shared genetic components between psoriasis and T2D. Trans-disease meta-analysis (TDMA) was applied to 8,016,731 well-imputed genetic markers from large-scale meta-analyses of psoriasis (11,024 cases and 16,336 controls) and T2D adjusted for BMI (74,124 cases and 824,006 controls). We confirmed our findings in a hospital-based study (42,112 patients) and tested for causal relationships with multi-variable Mendelian randomization. Mendelian randomization identified a causal relationship between psoriasis and T2D (p=1.6x10−4, OR=1.01), and highlighted the impact of BMI. TDMA further revealed 4 genome-wide significant loci (p<5x10−8) with evidence of colocalization, and shared directions of effect between psoriasis and T2D not present in BMI. Proteins coded by genes in these loci (ACTR2, ERLIN1, TRMT112, and BECN1) are connected through NF-κB signaling. Our results provide insight into the immunological components which connect immune-mediated skin conditions and metabolic diseases, independent of confounding factors.
Keywords: Psoriasis, Genome-Wide Association Studies (GWAS), Epidemiology, Inflammation, Bioinformatics
Introduction
Psoriasis is a complex skin disease, which affects over 7 million adults in the USA (Rachakonda et al.,2014), causing painful lesions and itching. Furthermore, psoriasis comorbidities account for more than half the direct healthcare costs (Brezinski et al., 2015, Feldman et al., 2017, Pilon et al., 2019, Vanderpuye-Orgle et al., 2015). Diabetes, the seventh leading cause of death in the USA (posing an economic burden of over $300 billion per year (American Diabetes Association, 2018)), was to our knowledge among the first psoriasis comorbidities identified (Strauss, 1897, Takeshita et al., 2017). Epidemiological studies show type 2 diabetes (T2D) and psoriasis are significantly associated, with reported Odds Ratios (ORs) of approximately 1.5 (Dubreuil et al., 2014, Hemminki et al., 2015), after controlling for Body Mass Index (BMI) and other covariates. Psoriasis patients exhibit reduced incretin effect (Gyldenlove et al., 2015), insulin secretion in response to oral glucose, which is considered to be indicative of prediabetes. Notably, patients with higher severity of psoriasis (i.e. more extensive skin involvement) were found to be at greater risk of developing T2D (Wan et al., 2018).
The high incidence of specific comorbidities for patients with chronic diseases (Ellinghaus et al., 2016, Hu et al., 2016, Meghani et al., 2013, Pefoyo et al., 2015) suggests potential genetic relationships among these conditions. While large-scale genome-wide association studies have been conducted to reveal disease susceptibility loci for psoriasis (Patrick et al., 2018, Tsoi et al., 2017) and T2D (Mahajan et al., 2018) separately, so far there has been very limited study into the shared genetic signals between these conditions. Wang et al. (2017) genotyped the lead markers for 51 T2D loci in psoriasis cases and controls, and found two (in proximity to ST6GAL1 and JAZF1, respectively) to be significantly associated with psoriasis among the Chinese population. However, no other Chinese studies of psoriasis have replicated these loci and we were unable to replicate the associations (even at nominal significance) in our own Caucasian cohorts (Patrick et al., 2018, Tsoi et al., 2017). Another study (Quaranta et al., 2009) explored T2D and psoriasis signals in proximity to CDKAL1 for Caucasian patients, but concluded that, despite their close location, the association signals were completely independent (r2=0.04). However, these previous studies may be limited by their “candidate gene” approach and by not considering potential confounding factors such as BMI. More broadly, psoriasis and T2D are similarly mediated by Th1 signaling, as well as cytokines TNF and IL-6, with further possible links including leptin, adiponectin, VEGF and IGF-II (Davidovici et al., 2010). These shared molecular pathways merit further attention as potential contributors to genetic similarities between the diseases.
By conducting a trans-disease meta-analysis (TDMA) of nearly 1 million individuals from genome-wide association studies (GWAS) of previously established consortia for the two diseases, our aim is to assess the genetic similarities between T2D and psoriasis to help us understand the molecular mechanisms these conditions have in common. In addition to revealing four shared loci between psoriasis and T2D, we used Mendelian randomization to unravel causal relationships and identified putative mechanisms involving TNF receptor associated factor TRAF6 that may explain these connections. Ultimately, our findings enhance our understanding of genetic risk factors and their associated pathways, thus improving precision health care for individuals suffering from psoriasis and/or T2D.
Results
We performed TDMA (Figure 1a) between psoriasis (11,024 cases and 16,336 controls from our recent meta-analysis (Patrick et al., 2018)) and T2D adjusted for BMI (74,124 cases and 824,006 controls from the DIAGRAM consortium (Mahajan et al., 2018)), using 8,016,731 well-imputed markers common to both cohorts. We then performed multi-variable Mendelian randomization, with BMI summary statistics from 806,834 samples in the GIANT consortium as a covariate (Pulit et al., 2019).
Figure 1: Trans-disease Meta-analysis.
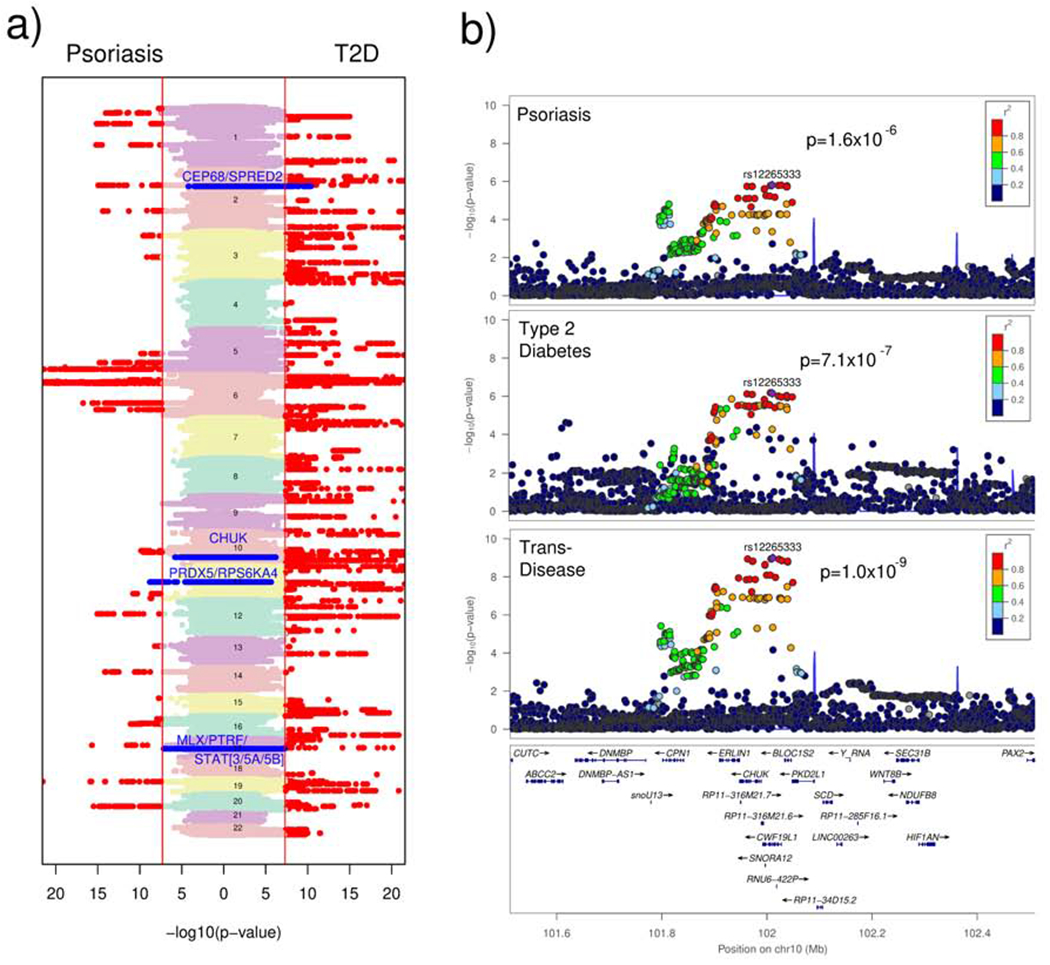
a) Vertical Manhattan plots of the meta-analysis association results for psoriasis and T2D, showing genome-wide significant (p≤5x10−8) markers in red and shared loci identified by our trans-disease meta-analysis in blue. b) Regional association plots for the chromosome 10 locus in psoriasis and T2D (with the lead marker in purple). The locus is suggestive significant for each disease and genome-wide significant in the trans-disease meta-analysis.
Shared Loci Identified
43 “shared loci” (Supplementary Table 1) and 32 “opposing loci” (Supplementary Table 2) were genome-wide significant (p<5x10−8) in TDMA, when combining effect in the same or opposing direction across the two diseases, respectively. Among these associations, 11 shared and 4 opposing loci had stronger evidence of association (more significant p-value) in TDMA than the individual meta-analysis for each disease.
We investigated the loci with suggestive evidence of association (p<1x10−4) in each of the two diseases. Four shared disease loci (Table 1; Supplementary Figure 1) met this criterion: 2p14 (p=9.6x10−9); 10q24.31 (p=1.0x10−9); 11q13.1 (p=1.0x10−); and 17q21.2 (p=1.5x10−9). We further note these loci have significantly higher colocalization (between psoriasis and T2D) posterior probability (Mann-Whitney p=5.7x10−4) than the 59 other previously reported loci for psoriasis (Supplemental Figure 2), for example the signal in chromosome 10 has 0.97 posterior probability of being a shared locus. The chromosome 10 locus (Figure 1b) is suggestive significant in each disease meta-analysis (p=1.6x10−6 for psoriasis and p=7.1x10−7 for T2D) but genome-wide significant (p=1.0x10−9) in TDMA. This locus is of particular interest as the lead trans-disease marker (rs12265333) is the top signal for psoriasis and the third most significant signal for T2D in the individual disease association studies. Crucially, only one of the loci identified by our approach (chromosome 11, TRMT112) was significantly associated with body mass index (BMI) in summary statistics from the GIANT consortium (Pulit et al., 2019) and the effect of the BMI signal is in the opposite direction to that of the psoriasis and T2D signal, suggesting they are not on the same haplotype.
Table 1:
Trans-disease meta-analysis results for loci meeting criteria
| Marker ID | Chr | Position (hg19) | Alleles (risk/non-risk) | Meta-analysis P-values | Meta-analysis Odds Ratios | Minor Allele Frequenciesa | Nearby Gene | BMI Locus?b | Newc GWAS Locus?b | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P(Psoriasis) | P(T2D) | P(Both) | OR(Psoriasis) | OR(T2D) | OR(Both) | Psoriasis | T2D | |||||||
| rs840967 | 2 | 65701757 | C/A | 6.6x10−5 | 4.4x10−8 | 9.6x10−9 | 1.08 | 1.04 | 1.06 | 0.40 | 0.41 | SPRED2 | No | Psoriasis |
| rs12265333 | 10 | 102011211 | A/G | 1.6x10−6 | 7.1x10−7 | 1.0x10−9 | 1.12 | 1.04 | 1.08 | 0.49 | 0.49 | CHUK | No | No |
| rs685870 | 11 | 64111928 | T/C | 8.5x10−9 | 7.8x10−5 | 1.0x10−11 | 1.13 | 1.03 | 1.08 | 0.31 | 0.30 | PRDX5 | Yesd | T2D |
| rs2292749 | 17 | 40818584 | T/C | 4.3x10−6 | 1.5x10−6 | 1.5x10−9 | 1.10 | 1.04 | 1.07 | 0.29 | 0.30 | STAT3 | No | No |
Abbreviations are as follows: Chr, chromosome; OR, odds ratio; p, p-value; T2D, type 2 diabetes; BMI, body mass index; GWS, genome-wide significant (i.e. p<5x10−8).
Minor alleles are provided for each entire meta-analysis (cases and controls), rather than specigically for each disease.
We determine whether a locus is previously known to be associated with BMI, psoriasis, or T2D according to data from the GIANT and DIAGRAM consortium, as well as a search of the available literature.
To our knowledge, previously unreported.
The lead marker of the chromosome 11 locus (rs685870) is significantly associated with BMI in the GIANT consortium meta-analysis (2.3x10−9), but its effect (OR=1.01) is in the opposite direction to the signal we identified in our trans-disease meta-analysis (i.e. the risk allele is C rather than T), suggesting it is not in the same haplotype.
In the MGI data, the risk allele frequencies (RAF) for the lead marker at each of the four loci were higher (8% on average) for patients with both diseases than controls (Table 2). Interestingly, for all but the 2p14 loci, the RAF is higher for controls than patients with only one of the diseases, suggesting our TDMA approach performs as intended by selecting association signals driven by a shared mechanism, rather than being dominated by one or the other disease. The four shared loci are all in proximity to known signals for psoriasis and/or T2D (Mahajan et al., 2018, Tsoi et al., 2017), however, the chromosome 2 locus (2p14) is, to our knowledge, a previously unreported genome-wide significant signal for psoriasis (p=6.6x10−5 in psoriasis meta-analysis), as is the chromosome 11 locus (11q13.1) for T2D (p=7.8x10−5 in T2D meta-analysis).
Table 2:
Frequencies of the Shared Loci in the Michigan Genomics Initiative
| Marker ID | Chr | Position (hg19) | Alleles (risk/non-risk) | Risk Allele Frequency | |||
|---|---|---|---|---|---|---|---|
| Control | Psoriasis (only) | T2D (only) | Both | ||||
| rs840967 | 2 | 65701757 | C/A | 0.398 | 0.416 | 0.404 | 0.434 |
| rs12265333 | 10 | 102011211 | A/G | 0.510 | 0.511 | 0.502 | 0.515 |
| rs685870 | 11 | 64111928 | T/C | 0.306 | 0.301 | 0.307 | 0.331 |
| rs2292749 | 17 | 40818584 | T/C | 0.284 | 0.282 | 0.293 | 0.325 |
Abbreviations are as follows: Chr, chromosome; T2D, type 2 diabetes.
Functional Interpretation of Shared Loci
To investigate which cell types are affected by the shared psoriasis/T2D genetic signals, we applied GARFIELD (Iotchkova et al., 2019) to evaluate the overlap between different chromatin marks with the LD-independent significant markers in the TDMA (analysis restricted to markers more significant in TDMA than both individual diseases). We first studied the enrichment across 1,005 different features representing cell types and annotation marks. Strong enrichment was observed in blood tissue (Supplementary Figure 3), with 48 out of 60 (80%) DNase-seq experiments having their hypersensitive sites significantly overlapping with disease-shared loci after Bonferroni correction (i.e. p<9.7x10−5), indicating potential immune-cell involvement.
We repeated the enrichment analysis after excluding the MHC region and found blood was still the most enriched tissue (Figure 2a), with four of the top five most significantly enriched features pertaining to GM12878 lymphoblastoid cells (Supplementary Table 3). We acknowledge that GM12878 possesses more epigenomic data than other immune cells, however it allows us to highlight the involvement of the regulatory mechanisms in immune cell in general. We then applied GARFIELD to active regulatory elements measured by H3K27ac marks in different immune cells (Farh et al., 2015). Interestingly, Th17 cells were now the most significant (p=4.8x10−8), overlapping in particular our chromosome 10 and 17 loci, and other immune-cell types were also more enriched (Figure 2b) than lymphoblastoids in GARFIELD's default annotation set. These results illustrate specific immune-cell involvement shared between the diseases, with Th17 cells having a key role in the development of psoriasis (Di Cesare et al.,2009) and suggested involvement in type 2 diabetes (Ip et al., 2016).
Figure 2: Cell Type Enrichment for Trans-Disease Meta-Analysis Markers Outside the MHC.
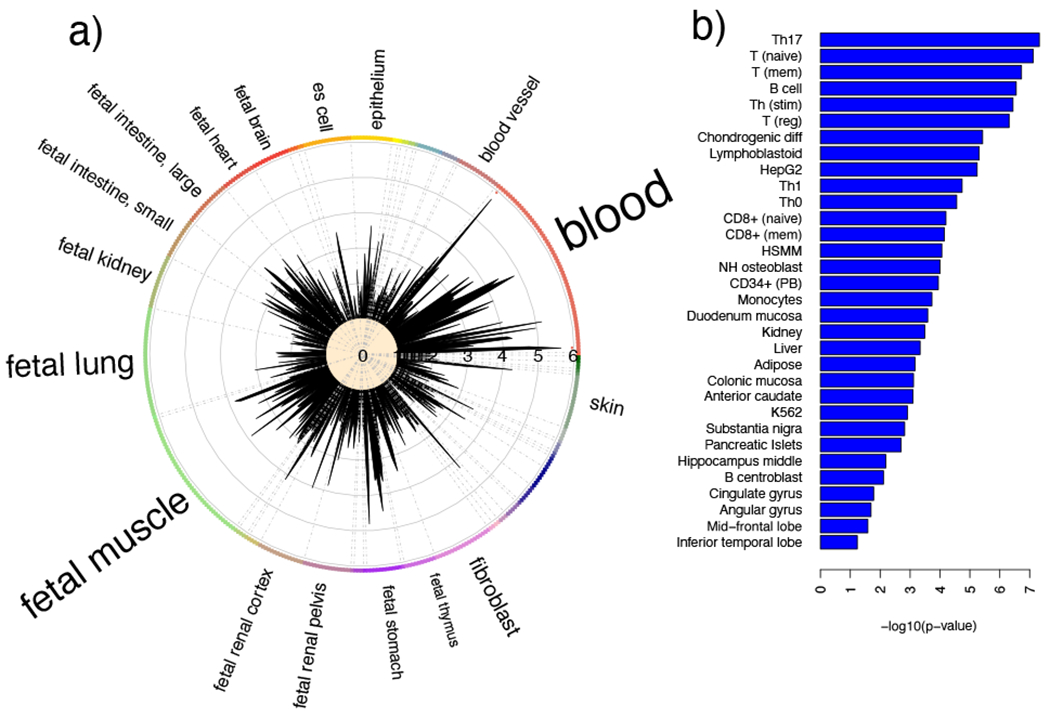
a) Calculated using DNAse hotspots in GARFIELD. b) Using H3K27ac marks from Farh et al.(Farh et al., 2015) The enrichment results illustrate immune-cell involvement.
Three of the four genetic signals identified by our approach carry missense mutations: the chromosome 17 lead marker is a missense variant for TUBG2, the chromosome 11 lead marker is a missense variant for CCDC88B and the chromosome 10 lead marker is in high linkage disequilibrium (r2=0.94) with a missense variant for CHUK. However, SIFT (Kumar et al., 2009) and PolyPhen-2 (Adzhubei et al., 2013) suggest these mutations are unlikely to have strong deleterious effect on protein function.
Therefore, instead of focusing on the best signal in the trans-disease meta-analysis, we broadened our investigation to the Bayesian credible sets for each locus for their potential biological effect. Interestingly, the 95% Bayesian credible sets contained fewer markers for trans-disease meta-analysis (TDMA) than either of the individual traits at 3 of the 4 loci (Supplementary Table 4). We then recorded the CADD (Combined Annotation Dependent Depletion) score for each marker in the intersection of the credible sets (Rentzsch et al., 2019) (TDMA/psoriasis/T2D) for each locus (Supplementary Table 5), and identified all their significant cis-eQTL gene targets (FDR<0.05) in three different eQTL datasets for blood (Jansen et al., 2017, Vosa et al., 2018, Westra et al., 2013). The PHRED-scaled CADD score for 6 of the markers (in chromosome 2 and 10) was higher than 10.0, indicating they are within the top 10% most deleterious variants in the genome, and for one marker (the missense variant rs2230804 in chromosome 10) the score was 21.4, indicating it is in the top 1%. According to the three blood eQTL databases, the expression of 34 genes in blood is associated with at least one marker in the intersection of the credible sets, with capture Hi-C results showing contacts between the promoters for many of these genes and the credible set markers in lymphoblastoid cells, pancreas, adipose, liver, skeletal muscle, thymus and keratinocytes (Supplementary Table 6). Significantly, the PHRED-scaled CADD score for the most significant eQTL of each gene (mean: 7.1; SD: 2.6) is significantly higher (Wilcoxon rank sum test p=2.76x10−3) than that of the full set of intersected markers (mean: 5.5; SD: 4.5). Furthermore, all but two of the genes (GPR137 and TRPT1) we identified using the eQTLGen database (94%) have higher mean PHRED-scaled CADD score for eQTLs in the intersection of the credible sets than the full set of eQTLs for each gene from the database (Supplementary Table 7), suggesting the markers identified by our approach are more likely to have larger biological effect.
Interestingly, four proteins encoded by genes from each of the four loci (ACTR2, ERLIN1, TRMT112, and BECN1) are presented in a protein-protein interaction (PPI) dataset (Chen et al., 2012) to interact with the hub protein TRAF6 (enrichment p=1.3x10−6). TRAF6 mediates NF-κB expression and has previously been implicated in the development of both psoriasis (Huffmeier et al., 2010) and type 2 diabetes (Balasubramanyam et al., 2011). ACTR2 (p=5.0x10−18, fold change=1.85) and TRMT112 (p=5.4x10−13, fold change=1.57) were found to be up-regulated in microarray (Gudjonsson et al., 2009) and RNA-seq (Tsoi et al., 2019) differential expression analysis, respectively (Supplementary Table 8) for lesional psoriatic compared to healthy skin. ACTR2 was also up-regulated in skeletal muscle (Wu et al., 2007) (p=8.7x10−12, fold change=1.76) and the subcutaneous adipose tissue (Soronen et al., 2012) (p=2.4x10−2, fold change=1.58) from type 2 diabetes patients compared to healthy controls, but down-regulated in the pancreas (Dominguez et al., 2011)(p=1.6x10−2, fold change=0.31). Similarly, BECN1 was up-regulated in skeletal muscle (p=5.4x10−7, fold change=1.70) and down-regulated in pancreas (p=3.3x10−2, fold change=0.48), and both TRMT112 (p=4.8x10−3, fold change=0.63) and ERLIN1 (p=2.7x10−3, fold change=0.57) were down-regulated in the pancreas of T2D patients compared to healthy controls.
Mendelian randomization (MR) to infer causal relationship
We next evaluated whether there is a causal relationship between psoriasis and T2D, after taking into account the effect of BMI. Rather than disease incidence (which can be heavily affected by confounders), Mendelian randomization uses genetic markers associated with functions or traits of interest to model the effect of one or more exposures on the outcome. We used genetic associations for BMI from 806,834 samples in the GIANT consortium (Pulit et al., 2019) as a covariate, in addition to the T2D and psoriasis summary statistics, selecting genetic markers (instruments) by linkage disequilibrium (LD) clumping, which considers both the association and the LD pattern for each locus. We performed MR in a multivariable design (estimating the effect of psoriasis and BMI on T2D, as well as T2D and BMI on psoriasis), to simultaneously estimate the causal effect of genetic variants for each trait (Table 3), taking the union of genetic markers (instruments) from the exposures (for completeness, we also provide the single-variable results in Supplementary Table 9). When applying MR using established regions (p<5x10−), BMI is observed to have a significant causal effect on both T2D (p=4.8x10−73, OR=2.73) and psoriasis (p=1.4x10−4, OR=1.73), but we were unable to identify a significant causal relationship between the diseases. By contrast, when using genome-wide information (all loci), we have power to identify a significant but modest causal relationship for psoriasis on T2D (p=1.6x10−4, OR=1.01), and a nominally significant causal relationship for T2D on psoriasis (p=0.014, OR=1.05). The difference in effect sizes between established loci and genome-wide information is negligible (Table 3), suggesting the results are not biased from using all loci.
Table 3:
Multi-Variable Mendelian Randomization
| Approach | Psoriasis + BMI on T2D | T2D + BMI on Psoriasis | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Num. Markers | Psoriasis | BMI | Num. Markers | T2D | BMI | ||||||
| OR | P | OR | P | OR | P | OR | P | ||||
| Established (p<5x10−8) loci | IVW (standard) | 548 | 1.01 | 0.528 | 2.73 | 4.84x10−73 | 650 | 1.06 | 0.095 | 1.37 | 1.44x10−4 |
| MR-Egger | 1.01 | 0.506 | 2.73 | 7.70x10−73 | 1.06 | 0.096 | 1.37 | 1.59x10−4 | |||
| MR-RAPS | 1.02 | 0.363 | 2.60 | 2.07x10−70 | 1.04 | 0.215 | 1.35 | 4.49x10−4 | |||
| Genome-wide information | IVW (standard) | 3,749 | 1.01 | 1.59x10−4 | 2.59 | 3.07x10−304 | 3,703 | 1.05 | 0.014 | 1.35 | 1.40x10−7 |
| MR-Egger | 1.01 | 1.36x10−4 | 2.59 | 1.90x10−303 | 1.05 | 0.016 | 1.35 | 1.64x10−7 | |||
| MR-RAPS | 1.01 | 0.0128 | 2.26 | 1.37x10−174 | 1.04 | 0.116 | 1.29 | 2.70x10−5 | |||
Abbreviations are as follows: IVW, inverse variance weighted; Num., number of; T2D, type 2 diabetes; BMI, body mass index; OR, odds ratio; p, p-value.
To assess the potential impact of any further pleiotropy (besides that due to BMI), we applied two variations of MR beyond the standard (inverse-variance weighted) approach. MR-Egger (Bowden et al., 2015) differs from standard MR in that the intercept is included in the model to test and control for pleiotropy, since when the effect of the exposures is zero, the outcome should be zero as well. MR-RAPS (Zhao et al., 2018) uses a random effect model to control for pleiotropy and takes into account the variance in the effect sizes used for the exposures. We observed the effect sizes to be consistent when including the intercept in the model and using MR-RAPS. The p-values were similar when including or not including the intercept, but their significance was reduced when using MR-RAPS (p=0.0128 for psoriasis on T2D and p=0.116 for T2D on psoriasis). This random effect-based model also supports a modest but significant causal effect of psoriasis on the risk of T2D independent of BMI. There may also be a causal effect of T2D on the risk of psoriasis, but despite the larger effect size, its significance was not as high as for psoriasis on T2D.
Discussion
The relationship between immune-mediated skin diseases and metabolic disorders is highly complex. Metabolic pathways modulate immune responses and influence immune cell differentiation/activation (Buck et al., 2017, Jung et al., 2019) through competition for resources (such as glucose and oxygen). Drugs which target metabolism can also reduce inflammation (Stathopoulou et al., 2019): for example, rapamycin is an immunosuppressant used for preventing transplant rejection (Thomson et al., 2009), but operates by inhibiting mTOR, a kinase coordinator of metabolic pathways; similarly metformin is a T2D drug (targeting AMP-activated protein kinase) but recent studies have suggested it can help treat skin disorders (Badr et al., 2013).
Previous studies have used Mendelian randomization to identify a causal relationship between BMI and psoriasis (Budu-Aggrey et al., 2019, Ogawa et al., 2019) as well as between BMI and T2D (Corbin et al., 2016, Holmes et al., 2014). Indeed, we found BMI to have a stronger impact on psoriasis and T2D than either disease has on each other. Our work represents, to our knowledge, the first genome-wide based genetic study and reveals four shared loci and a potential causal relationship between psoriasis and T2D independent of BMI. Our Mendelian randomization results suggest psoriasis may have a causal effect on T2D, but are less clear about the effect of T2D on psoriasis. Indeed, psoriasis is believed to have an underlying systemic component (Reich, 2012), and this can increase the risk of T2D (Duncan et al., 2003). While the causal effect of psoriasis on T2D independent of BMI is small (OR=1.01), the high impact and prevalence of T2D makes even a small effect important to consider. Including genome-wide information (as opposed to only established loci) allowed us to increase the power of our analysis; we confirmed pleiotropy had been taken into account using MR-Egger and addressed the weak instrument bias using MR-RAPS. Nevertheless, selection bias (Haycock et al., 2016) could mean we underestimate the significance of the causal relationship and we need to consider the potential impact of disparity in number of loci for each trait on the weak instrument bias (BMI has 516 genome-wide significant loci, while T2D has 176 and psoriasis has 32). Future studies may wish to use a separate selection dataset, and/or try different strategies to equalize the number of loci used for each trait, to be confident in achieving accurate measurements of effect size.
As an additional means to investigate the genetic correlation between psoriasis and T2D, we applied LD score regression, excluding the MHC due to the high LD in this region. Using the T2D summary statistics that have not been adjusted for BMI, the genetic correlation with psoriasis was rg=0.157 (p=1.0x10−5), while using the T2D summary statistics adjusted for BMI, the genetic correlation with psoriasis was rg=0.077 (p=0.064). We believe this confirms our conclusion from Mendelian Randomization that BMI is the dominant factor in the relationship between psoriasis and T2D, and it supports our decision to use the summary statistics adjusted for BMI in our trans-disease meta-analysis. It is also interesting that the genetic correlation is close to being nominally significant when adjusting for BMI. While LD score regression and Mendelian randomization are ‘broad brush’ approaches, our trans-disease meta-analysis approach and colocalization operate at the level of each locus.
For the 4 shared genetic loci identified, the gene targets are largely involved in immune processes, suggesting there may be a link between psoriasis and T2D independent of obesity (Supplementary Note 1). Interestingly, while the locus in chromosome 11 is negatively associated with BMI, it is positively associated with BMI-adjusted waist-to-hip ratio (p=1.6x10−14, OR=1.01) (Pulit et al., 2019). Waist-to-hip ratio has been associated with various health conditions including type 2 diabetes (Emdin et al., 2017), and it is believed the distribution of fat can have a significant impact on its role in cardiometabolic disease. We should also consider the potential for patients with type 1 diabetes (T1D) being misdiagnosed as T2D, however only the chromosome 17 locus has a previously reported for T1D signal within 500kb (according to the EBI GWAS Catalog) and it is not in LD with the locus we identified (R2=0.003 in Europeans).
We applied equally weighted TDMA (see Methods), rather than the inverse-variance weighted (IVW) approach typical of meta-analyses for a single trait, to avoid biasing results towards T2D, which has a larger sample size compared with the psoriasis cohorts. Compared to other meta-analysis approaches, TDMA revealed the most loci (Supplementary Table 10). Nevertheless, the results are largely consistent, with all techniques revealing the chromosome 10 and chromosome 17 loci, and all but ASSET revealing the chromosome 11 locus. ASSET also identifies a locus in the MHC (rs9273366), which fits with our hypothesis on the immune basis for shared psoriasis/T2D genetics, but the lead marker in our approach was not significant for T2D.
Requiring the p-value to be more significant in TDMA than each disease was designed to avoid false positives. For example, the region around CDKAL1 contains genome-wide significant signals for both psoriasis and T2D but previous research has shown these signals are independent (Quaranta et al., 2009). Our approach does not identify this locus, as it is not as significant in TDMA as it is in the individual disease meta-analysis. Weakening the suggestive significance threshold for each disease to p<1x10−3 would allow us to report three more shared loci (Supplementary Table 11), one of which has a gene target (TRAFD1) in the PPI set for TRAF6. However, psoriasis-only patients in MGI (Supplementary Table 12) have higher risk allele frequency than those with both diseases for the other two loci, suggesting they may be driven by psoriasis rather than representing a shared mechanism, thus supporting our decision to use the current, more conservative p-value threshold (i.e. p<1x10−4).
The MGI risk allele frequency for patients with only one of the diseases was lower than that of controls for some loci and this could be due to interaction with other partially correlated signals from the same region (rather than demonstrating a shared mechanism as we suggested). For example, in chromosome 2, there is another T2D signal ~400kb away which does not occur in psoriasis (Supplementary Figure 1). However, LD between this signal and the one identified by TDMA is low (R2=0.02 in Europeans) and the RAF for this locus is higher in both T2D and psoriasis patients than controls.
Although we focused on the differential expression of genes in the TRAF6 PPI set, other eQTL targets are differentially expressed. Notably, STAT3 is upregulated in psoriatic skin compared to controls (fold change=2.13 in the microarray data and 3.34 in RNA-seq), as well as in liver (fold change=2.58) and adipose (fold change=1.74). STAT3 binds to NF-κB in competition with I-κB (Yang et al., 2007); CHUK (IKKa) was downregulated in T2D pancreas (fold change=0.44) and liver (fold change=0.59) compared to controls and activates NF-κB through phosphorylation of I-κB (Hacker and Karin, 2006). Development of insulin resistance has been linked to IKKP/NF-κB (Shoelson et al., 2007), however, CHUK was not differentially expressed in psoriasis. CEP68 and DNMBP are both downregulated in psoriasis and T2D liver, with CEP68 being upregulated skeletal muscle (fold change=1.8) and DNMBP in adipose (fold change=2.17). These genes are involved in centrosome cohesion (Thompson et al., 2004) and centrosome amplification is increased in T2D (Wang et al., 2018). Interestingly, the lead marker from our chromosome 17 locus is a genome-wide significant eQTL (p=3.64x10−8) in glomerulus (Qiu et al., 2018) for TUBG2 (in addition to being a missense variant for this gene) and a suggestive significant eQTL (p=0.048) in glomerulus (Gillies et al., 2018) for BRCA1, another gene involved in centrosome regulation. However, we are not able to reveal this as an eQTL signal in normal skin tissue.
By combining summary statistics from large T2D and psoriasis GWAS, we have identified four genome-wide significant trans-disease loci (two of which are, to our knowledge, previously unreported findings for one of the diseases). Enrichment analysis suggests these loci are involved in immune regulation, and this is supported by Mendelian randomization’s detection of a small but significant causal effect of psoriasis on T2D independent of BMI (although the impact of BMI is much larger). We have suggested some potential mechanisms by which the loci may impact psoriasis and T2D, such as the regulation of NF-kB expression through TRAF6. Our work provides a starting point through which efforts can be made at precision medicine to improve the treatment of patients with one or both of these diseases. Overall, while our results suggest the observed relationship between T2D and psoriasis is largely driven by BMI, the BMI-independent T2D/psoriasis shared loci revealed by our approach hint at a potential direct causal link between the two conditions.
Materials and Methods
Data processing
Data were collected and processed with quality control procedures described in the paper for the GWAS meta-analysis of each trait (Mahajan et al., 2018, Patrick et al., 2018, Yengo et al., 2018). Since T2D and psoriasis have previously been found to be associated with BMI (Takeshita et al., 2017), we used the BMI-adjusted version of the T2D meta-analysis from the DIAGRAM consortium (Mahajan et al., 2018) for TDMA. To measure the causal effect of BMI, we used the unadjusted version of the T2D meta-analysis results in Mendelian randomization. All samples are of Caucasian descent and samples were excluded if they had substantial non-European admixture. Relatedness testing was performed within each meta-analysis, to ensure only independent samples were used, but not between studies, due to access limitations for the individual-level data used by the DIAGRAM and GIANT consortia.
For the identified signals, we calculated the risk allele frequencies (RAF) of the lead marker at each locus in 42,112 Caucasian individuals from the Michigan Genomics Initiative (MGI) (Fritsche et al., 2018), which contains genotypes linked to electronic health records, allowing us to evaluate the loci in a hospital-based study. By using ICD9/10 codes to define disease status (Supplementary Note 2), we identified 8,622 patients with T2D only, 783 patients with psoriasis only, 344 patients with both psoriasis and T2D, and 32,363 controls with neither disease. To the best of our knowledge, written informed consent was obtained in the MGI project (Fritsche et al., 2018) and in each of the cohorts included in the summary statistics (Mahajan et al., 2018, Patrick et al., 2018, Yengo et al., 2018).
Trans-disease meta-analysis (TDMA)
We avoided biasing results towards the disease with largest sample size (T2D) by conducting TDMA using an equally weighted combination of effect sizes and variances from the meta-analysis for each disease (Supplementary Note 3). To our knowledge, our study represents the first time this approach has been applied. We then filtered these results to only select loci for which the TDMA lead marker is: 1) genome-wide significant (p<5x10−8) in TDMA; 2) suggestive significant (p<1x10−4) in the individual meta-analyses; and 3) more significant in TDMA than both the individual meta-analyses. We also compared our approach with existing meta-analysis methods. Colocalization was performed between the psoriasis and T2D summary statistics for each locus using COLOC (Giambartolomei et al., 2014). The identified loci were interpreted through chromatin marks, eQTLs, differential expression, promoter capture Hi-C and PPI enrichment (Supplementary Note 4).
Mendelian randomization
We applied Mendelian randomization to test for causal relationship between psoriasis and T2D. MR was performed using MR-Base (Hemani et al., 2016) (an R package which envelops a wide range of MR techniques) and MR-RAPS (Zhao et al., 2018) (a recent technique shown to be effective and robust when used with genome-wide information). Mendelian randomization was performed using both uni- and multi-variable analysis (i.e. including BMI as a covariate), on markers identified through linkage disequilibrium (LD) clumping using the 1000 Genomes European samples (LD≥0.001, window size=10Mbp) and the p-values in the summary statistics for each trait on the intersection of genetic markers across the traits. For the MR-Base approach, all the exposures were fitted in a multi-variable regression to model the outcome (e.g. βPsV ~ βT2D + βBMI).
Data Availability
Data from the DIAGRAM (https://www.diagram-consortium.org) and GIANT (https://portals.broadinstitute.org/collaboration/giant) consortium may be found on their respective websites. The psoriasis summary statistics are available upon request.
Supplementary Material
Acknowledgments
This work was supported by the Arthritis National Research Foundation and the National Psoriasis Foundation (L.C.T., and M.T.P.), and awards from the National Institutes of Health (R01AR042742, R01AR050511, R01AR054966, R01AR063611, R01AR065183 to J.T.E.; K01AR072129 to L.C.T.; and DK062370 to M.B.). L.C.T. was also supported by the Dermatology Foundation and M.T.P was supported by a Precision Health Scholars Award from the University of Michigan. L.C.T., P.E.S., J.E.G., J.J.V., R.P.N. and J.T.E. are supported by the Dawn and Dudley Holmes Foundation and the Babcock Memorial Trust. J.E.G. was supported by Doris Duke Foundation (Grant #:2013106) and the National Institute of Health (K08AR060802 and R01AR06907) and the Taubman Medical Research Institute as the Frances and Kenneth Eisenberg Emerging Scholar. L.C.T. and J.E.G. are also supported by the NIAMS grant UMSBDRC 1P30AR075043 and the Taubman Institute Innovation Project. J.T.E. is supported by the Ann Arbor Veterans Affairs Hospital. X.J.Z was supported by Beijing Natural Science Foundation (Z190023) and the University of Michigan Health System–Peking University Health Science Center Joint Institute for Translational and Clinical Research (BMU2017JI007).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
JEG received research grants from AbbVie, AnaptysBio, Pfizer, Novartis, Celgene, and Eli Lilly, and serves as advisory board in Novartis, AbbVie, Eli Lilly, MiRagen, and Almirall. NNM is a full-time US government employee and has served as a consultant for Amgen, Eli Lilly, and Leo Pharma receiving grants/other payments; as a principal investigator and/or investigator for AbbVie, Celgene, Janssen Pharmaceuticals, Inc, and Novartis receiving grants and/or research funding; and as a principal investigator for the National Institute of Health receiving grants and/or research funding.
References
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013;7:Unit7.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes Association. Economic Costs of Diabetes in the U.S. in 2017. Diabetes Care 2018;41(5):917–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badr D, Kurban M, Abbas O. Metformin in dermatology: an overview. Journal of the European Academy of Dermatology and Venereology : JEADV 2013;27(11):1329–35. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam M, Aravind S, Gokulakrishnan K, Prabu P, Sathishkumar C, Ranjani H, et al. Impaired miR-146a expression links subclinical inflammation and insulin resistance in Type 2 diabetes. Molecular and cellular biochemistry 2011;351(1–2):197–205. [DOI] [PubMed] [Google Scholar]
- Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44(2):512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brezinski EA, Dhillon JS, Armstrong AW. Economic Burden of Psoriasis in the United States: A Systematic Review. JAMA Dermatol 2015;151(6):651–8. [DOI] [PubMed] [Google Scholar]
- Buck MD, Sowell RT, Kaech SM, Pearce EL. Metabolic Instruction of Immunity. Cell 2017;169(4):570–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budu-Aggrey A, Brumpton B, Tyrrell J, Watkins S, Modalsli EH, Celis-Morales C, et al. Evidence of a causal relationship between body mass index and psoriasis: A mendelian randomization study. PLoS Med 2019;16(1):e1002739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Xu H, Gordonov S, Lim MP, Perkins MH, Ma’ayan A. Expression2Kinases: mRNA profiling linked to multiple upstream regulatory layers. Bioinformatics 2012;28(1):105–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbin LJ, Richmond RC, Wade KH, Burgess S, Bowden J, Smith GD, et al. BMI as a Modifiable Risk Factor for Type 2 Diabetes: Refining and Understanding Causal Estimates Using Mendelian Randomization. Diabetes 2016;65(10):3002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidovici BB, Sattar N, Prinz J, Puig L, Emery P, Barker JN, et al. Psoriasis and systemic inflammatory diseases: potential mechanistic links between skin disease and co-morbid conditions. The Journal of investigative dermatology 2010;130(7):1785–96. [DOI] [PubMed] [Google Scholar]
- Di Cesare A, Di Meglio P, Nestle FO. The IL-23/Th17 axis in the immunopathogenesis of psoriasis. The Journal of investigative dermatology 2009;129(6):1339–50. [DOI] [PubMed] [Google Scholar]
- Dominguez V, Raimondi C, Somanath S, Bugliani M, Loder MK, Edling CE, et al. Class II phosphoinositide 3-kinase regulates exocytosis of insulin granules in pancreatic beta cells. J Biol Chem 2011;286(6):4216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil M, Rho YH, Man A, Zhu Y, Zhang Y, Love TJ, et al. Diabetes incidence in psoriatic arthritis, psoriasis and rheumatoid arthritis: a UK population-based cohort study. Rheumatology (Oxford, England) 2014;53(2):346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan BB, Schmidt MI, Pankow JS, Ballantyne CM, Couper D, Vigo A, et al. Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes 2003;52(7):1799–805. [DOI] [PubMed] [Google Scholar]
- Ellinghaus D, Jostins L, Spain SL, Cortes A, Bethune J, Han B, et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat Genet 2016;48(5):510–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emdin CA, Khera AV, Natarajan P, Klarin D, Zekavat SM, Hsiao AJ, et al. Genetic Association of Waist-to- Hip Ratio With Cardiometabolic Traits, Type 2 Diabetes, and Coronary Heart Disease. Jama 2017;317(6):626–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015;518(7539):337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman SR, Tian H, Gilloteau I, Mollon P, Shu M. Economic burden of comorbidities in psoriasis patients in the United States: results from a retrospective U.S. database. BMC Health Serv Res 2017;17(1):337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche LG, Gruber SB, Wu Z, Schmidt EM, Zawistowski M, Moser SE, et al. Association of Polygenic Risk Scores for Multiple Cancers in a Phenome-wide Study: Results from The Michigan Genomics Initiative. American journal of human genetics 2018;102(6):1048–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 2014;10(5):e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies CE, Putler R, Menon R, Otto E, Yasutake K, Nair V, et al. An eQTL Landscape of Kidney Tissue in Human Nephrotic Syndrome. American journal of human genetics 2018;103(2):232–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudjonsson JE, Ding J, Li X, Nair RP, Tejasvi T, Qin ZS, et al. Global gene expression analysis reveals evidence for decreased lipid biosynthesis and increased innate immunity in uninvolved psoriatic skin. J Invest Dermatol 2009;129(12):2795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyldenlove M, Vilsboll T, Zachariae C, Holst JJ, Knop FK, Skov L. Impaired incretin effect is an early sign of glucose dysmetabolism in nondiabetic patients with psoriasis. Journal of internal medicine 2015;278(6):660–70. [DOI] [PubMed] [Google Scholar]
- Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Science’s STKE : signal transduction knowledge environment 2006;2006(357):re13. [DOI] [PubMed] [Google Scholar]
- Haycock PC, Burgess S, Wade KH, Bowden J, Relton C, Davey Smith G. Best (but oft-forgotten) practices: the design, analysis, and interpretation of Mendelian randomization studies. The American journal of clinical nutrition 2016;103(4):965–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemani G, Zheng J, Wade KH, Laurin C, Elsworth B, Burgess S, et al. MR-Base: a platform for systematic causal inference across the phenome using billions of genetic associations. bioRxiv 2016:078972. [Google Scholar]
- Hemminki K, Liu X, Forsti A, Sundquist J, Sundquist K, Ji J. Subsequent Type 2 Diabetes in Patients with Autoimmune Disease. Scientific reports 2015;5:13871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MV, Lange LA, Palmer T, Lanktree MB, North KE, Almoguera B, et al. Causal effects of body mass index on cardiometabolic traits and events: a Mendelian randomization analysis. Am J Hum Genet 2014;94(2):198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JX, Thomas CE, Brunak S. Network biology concepts in complex disease comorbidities. Nat Rev Genet 2016;17(10):615–29. [DOI] [PubMed] [Google Scholar]
- Huffmeier U, Uebe S, Ekici AB, Bowes J, Giardina E, Korendowych E, et al. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nature genetics 2010;42(11):996–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iotchkova V, Ritchie GRS, Geihs M, Morganella S, Min JL, Walter K, et al. GARFIELD classifies diseaserelevant genomic features through integration of functional annotations with association signals. Nat Genet 2019;51(2):343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip B, Cilfone NA, Belkina AC, DeFuria J, Jagannathan-Bogdan M, Zhu M, et al. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFalpha production. Obesity (Silver Spring, Md) 2016;24(1):102–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R, Hottenga JJ, Nivard MG, Abdellaoui A, Laport B, de Geus EJ, et al. Conditional eQTL analysis reveals allelic heterogeneity of gene expression. Hum Mol Genet 2017;26(8):1444–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Zeng H, Horng T. Metabolism as a guiding force for immunity. Nat Cell Biol 2019;21(1):85–93. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4(7):1073–81. [DOI] [PubMed] [Google Scholar]
- Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nature genetics 2018;50(11):1505–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meghani SH, Buck HG, Dickson VV, Hammer MJ, Rabelo-Silva ER, Clark R, et al. The conceptualization and measurement of comorbidity: a review of the interprofessional discourse. Nurs Res Pract 2013;2013:192782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K, Stuart PE, Tsoi LC, Suzuki K, Nair RP, Mochizuki H, et al. A Transethnic Mendelian Randomization Study Identifies Causality of Obesity on Risk of Psoriasis. J Invest Dermatol 2019;139(6):1397–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick MT, Stuart PE, Raja K, Gudjonsson JE, Tejasvi T, Yang J, et al. Genetic signature to provide robust risk assessment of psoriatic arthritis development in psoriasis patients. Nature communications 2018;9(1):4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pefoyo AJ, Bronskill SE, Gruneir A, Calzavara A, Thavorn K, Petrosyan Y, et al. The increasing burden and complexity of multimorbidity. BMC Public Health 2015;15:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilon D, Teeple A, Zhdanava M, Ladouceur M, Ching Cheung H, Muser E, et al. The economic burden of psoriasis with high comorbidity among privately insured patients in the United States. J Med Econ 2019;22(2):196–203. [DOI] [PubMed] [Google Scholar]
- Pulit SL, Stoneman C, Morris AP, Wood AR, Glastonbury CA, Tyrrell J, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Human molecular genetics 2019;28(1):166–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu C, Huang S, Park J, Park Y, Ko YA, Seasock MJ, et al. Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nature medicine 2018;24(11):1721– 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaranta M, Burden AD, Griffiths CE, Worthington J, Barker JN, Trembath RC, et al. Differential contribution of CDKAL1 variants to psoriasis, Crohn’s disease and type II diabetes. Genes and immunity 2009;10(7):654–8. [DOI] [PubMed] [Google Scholar]
- Rachakonda TD, Schupp CW, Armstrong AW. Psoriasis prevalence among adults in the United States. Journal of the American Academy of Dermatology 2014;70(3):512–6. [DOI] [PubMed] [Google Scholar]
- Reich K The concept of psoriasis as a systemic inflammation: implications for disease management. J Eur Acad Dermatol Vener 2012;26:3–11. [DOI] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47(D1):D886–d94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007;132(6):2169–80. [DOI] [PubMed] [Google Scholar]
- Soronen J, Laurila PP, Naukkarinen J, Surakka I, Ripatti S, Jauhiainen M, et al. Adipose tissue gene expression analysis reveals changes in inflammatory, mitochondrial respiratory and lipid metabolic pathways in obese insulin-resistant subjects. BMC Med Genomics 2012;5:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopoulou C, Nikoleri D, Bertsias G. Immunometabolism: an overview and therapeutic prospects in autoimmune diseases. Immunotherapy 2019;11(9):813–29. [DOI] [PubMed] [Google Scholar]
- Strauss H Zur Lehre von der neurogenen und der thyreogenen Glykosurie. Dtsch Med Wochenschr 1897;20:309–12. [Google Scholar]
- Takeshita J, Grewal S, Langan SM, Mehta NN, Ogdie A, Van Voorhees AS, et al. Psoriasis and comorbid diseases: Epidemiology. J Am Acad Dermatol 2017;76(3):377–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson HM, Cao H, Chen J, Euteneuer U, McNiven MA. Dynamin 2 binds gamma-tubulin and participates in centrosome cohesion. Nature cell biology 2004;6(4):335–42. [DOI] [PubMed] [Google Scholar]
- Thomson AW, Turnquist HR, Raimondi G. Immunoregulatory functions of mTOR inhibition. Nature reviews Immunology 2009;9(5):324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi LC, Rodriguez E, Degenhardt F, Baurecht H, Wehkamp U, Volks N, et al. Atopic Dermatitis Is an IL-13-Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J Invest Dermatol 2019;139(7):1480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi LC, Stuart PE, Tian C, Gudjonsson JE, Das S, Zawistowski M, et al. Large scale meta-analysis characterizes genetic architecture for common psoriasis associated variants. Nature communications 2017;8:15382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderpuye-Orgle J, Zhao Y, Lu J, Shrestha A, Sexton A, Seabury S, et al. Evaluating the economic burden of psoriasis in the United States. J Am Acad Dermatol 2015;72(6):961–7. e5. [DOI] [PubMed] [Google Scholar]
- Võsa U, Claringbould A, Westra H-J, Bonder MJ, Deelen P, Zeng B, et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv 2018:447367. [Google Scholar]
- Wan MT, Shin DB, Hubbard RA, Noe MH, Mehta NN, Gelfand JM. Psoriasis and the risk of diabetes: A prospective population-based cohort study. Journal of the American Academy of Dermatology 2018;78(2):315–22. e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang Z, Rani PL, Fu X, Yu W, Bao F, et al. Identification of PTPN22, ST6GAL1 and JAZF1 as psoriasis risk genes demonstrates shared pathogenesis between psoriasis and diabetes. Experimental dermatology 2017;26(11):1112–7. [DOI] [PubMed] [Google Scholar]
- Wang P, Lu YC, Wang J, Wang L, Yu H, Li YF, et al. Type 2 Diabetes Promotes Cell Centrosome Amplification via AKT-ROS-Dependent Signalling of ROCK1 and 14-3-3sigma. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology 2018;47(1):356–67. [DOI] [PubMed] [Google Scholar]
- Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet 2013;45(10):1238–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Wang J, Cui X, Maianu L, Rhees B, Rosinski J, et al. The effect of insulin on expression of genes and biochemical pathways in human skeletal muscle. Endocrine 2007;31(1):5–17. [DOI] [PubMed] [Google Scholar]
- Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes & development 2007;21(11):1396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Human molecular genetics 2018;27(20):3641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Chen Y, Wang J, Small DS. Powerful genome-wide design and robust statistical inference in two-sample summary-data Mendelian randomization. arXiv e-prints 2018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from the DIAGRAM (https://www.diagram-consortium.org) and GIANT (https://portals.broadinstitute.org/collaboration/giant) consortium may be found on their respective websites. The psoriasis summary statistics are available upon request.