

Abstract
In the global health emergency caused by coronavirus disease 2019 (COVID-19), efficient and specific therapies are urgently needed. Compared with traditional small-molecular drugs, antibody therapies are relatively easy to develop; they are as specific as vaccines in targeting severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2); and they have thus attracted much attention in the past few months. This article reviews seven existing antibodies for neutralizing SARS-CoV-2 with 3D structures deposited in the Protein Data Bank (PDB). Five 3D antibody structures associated with the SARS-CoV spike (S) protein are also evaluated for their potential in neutralizing SARS-CoV-2. The interactions of these antibodies with the S protein receptor-binding domain (RBD) are compared with those between angiotensin-converting enzyme 2 and RBD complexes. Due to the orders of magnitude in the discrepancies of experimental binding affinities, we introduce topological data analysis, a variety of network models, and deep learning to analyze the binding strength and therapeutic potential of the 14 antibody–antigen complexes. The current COVID-19 antibody clinical trials, which are not limited to the S protein target, are also reviewed.
Keywords: COVID-19, SARS-CoV-2, antibody therapy, binding affinity, persistent homology, deep learning, network analysis
1. INTRODUCTION
The coronavirus disease 2019 (COVID-19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has rapidly spread around the world. By June 17, 2020, more than 8.2 million individuals were infected, and 443,000 fatalities had been reported. Currently, there are neither specific drugs nor effective vaccines available (8). Traditional drug discovery involves a long and costly process, requiring more than 10 years on average to put a drug on the market. Vaccine development typically takes more than one year. In contrast, developing potent SARS-CoV-2-specified antibodies that are produced from blood B cells in response to and counteracting SARS-CoV-2 antigens is a relatively less time-consuming and more efficient strategy for combating the ongoing pandemic (8, 31, 36, 49, 56, 65, 73, 74, 81, 85, 88).
Antibody, also called immunoglobulin (Ig),is a large, Y-shaped protein that typically consists of two identical heavy chains and two identical light chains. A heavy chain can be separated into two regions, a constant region and a variable region. Moreover, each light chain has two successive domains, a constant domain and a variable domain. The two heavy and two light chains of an antibody are connected through disulfide bonds within the constant region (52). An antibody binds to the antigenic determinant (also called the epitope) through the variable regions in the tips of the heavy and light chains. Each of these chains contains three complementarity-determining regions (CDRs), which are located in the tips of each variable domain. Most of the differences among antibodies are generated within the CDRs, which determine the specificity of individual antibodies.
Benefitting from the broad specificity of antibodies, antibody therapies have been proven to be a promising way to fight cancer; autoimmune disease; neurological disorders; and infectious viruses such as human immunodeficiency virus (HIV), Ebola, and Middle East respiratory syndrome (MERS) (12, 58, 76). Recently, several studies have shown that the convalescent plasma of SARS-CoV-2 patients, which contains neutralizing antibodies created by adaptive immune responses, can effectively improve patient survival rates (7, 9, 59). However, plasma-based therapeutics cannot be produced on a large scale. Therefore, seeking potent industrial-scale antibody therapies has become one of the most feasible strategies to fight against SARS-CoV-2. The spike (S) protein, a multifunctional molecular machine that binds to the human cell receptor angiotensin-converting enzyme 2 (ACE2), is one important target of neutralizing antibodies and the focus of therapeutic and vaccine design efforts (67). Many researchers have reported the binding affinities of SARS-CoV-2 S protein in complex with antibody candidates and ACE2. However, these reported values may vary by two orders of magnitude for a given antibody due to different experimental methods, conditions, calibrations, and human errors, which hinders the development of antibody therapies for SARS-CoV-2. Therefore, the development of a unified paradigm for ranking the potency of SARS-CoV-2 antibodies is a pressing need.
In this article, we review seven existing SARS-CoV-2 antibody therapeutic candidates from the literature. As molecular structures are able to reveal the molecular mechanism of antibody–antigen interactions, we only focus on the SARS-CoV-2 S protein antibodies that have 3D structures released in the Protein Data Bank (PDB). Since antibodies may directly compete with ACE2 for binding to the S protein receptor-binding domain (RBD), the structure and binding affinity of ACE2 and S protein complexes have been studied to understand the efficiency of antibodies. Moreover, since the S proteins of SARS-CoV and SARS-CoV-2 share 80% amino acid sequence identity (70), SARS-CoV S protein antibodies are potential candidates for treatment of COVID-19. Therefore, we review five existing SARS-CoV S protein antibodies and analyze their binding affinities with the SARS-CoV-2 S protein. Furthermore, we employ topological data analysis (TDA), artificial intelligence, and a variety of network models to address literature controversy and provide a unified paradigm for ranking the potency of all antibodies. Finally, we review the current clinical trials of COVID-19 antibody candidates.
2. AN OVERVIEW OF ANTIBODY STRUCTURES, FUNCTIONS, AND THERAPIES
An antibody can be divided into different parts according to its functions. Specifically, the arms of the Y-shaped protein contain sites that can recognize and bind to specific antigens. This region of the antibody is called a fragment, antigen-binding (Fab) region and is composed of one constant domain and one variable domain from each heavy and light chain of the antibody (52). Figure 1 illustrates the structure of the antibody. The variable domain (Fv) region is the most important region for binding to antigens. On light and heavy chains, CDRs composed of three variable loops of β-strands are responsible for binding to a specific antigen. The CDRs are incredibly variable, allowing a large number of antibodies with slightly different tip structures, or antigen-binding sites, to exist. Each of these variants can bind to a different antigen; thus, the enormous diversity of antibody paratopes on the antigen-binding fragments allows the immune system to recognize an equally wide variety of antigens (43). This antibody paratope diversity is generated by random recombination events of a set of gene segments that encode different antigen-binding sites (or paratopes), followed by random mutations in this area of the antibody gene to create further diversity (16, 42). It has been estimated that humans generate approximately 10 billion different antibodies (24). The base of the Y plays a role in modulating immune cell activity. This region is named the fragment, crystallizable (Fc) region and is composed of two heavy chains. The Fc region ensures that each antibody generates an appropriate immune response for a given antigen by binding to a specific class of Fc receptors or other immune molecules. This process activates different physiological effects, including recognition of opsonized particles; lysis of cells; and degranulation of mast cells, basophils, and eosinophils (33, 80).
Figure 1.
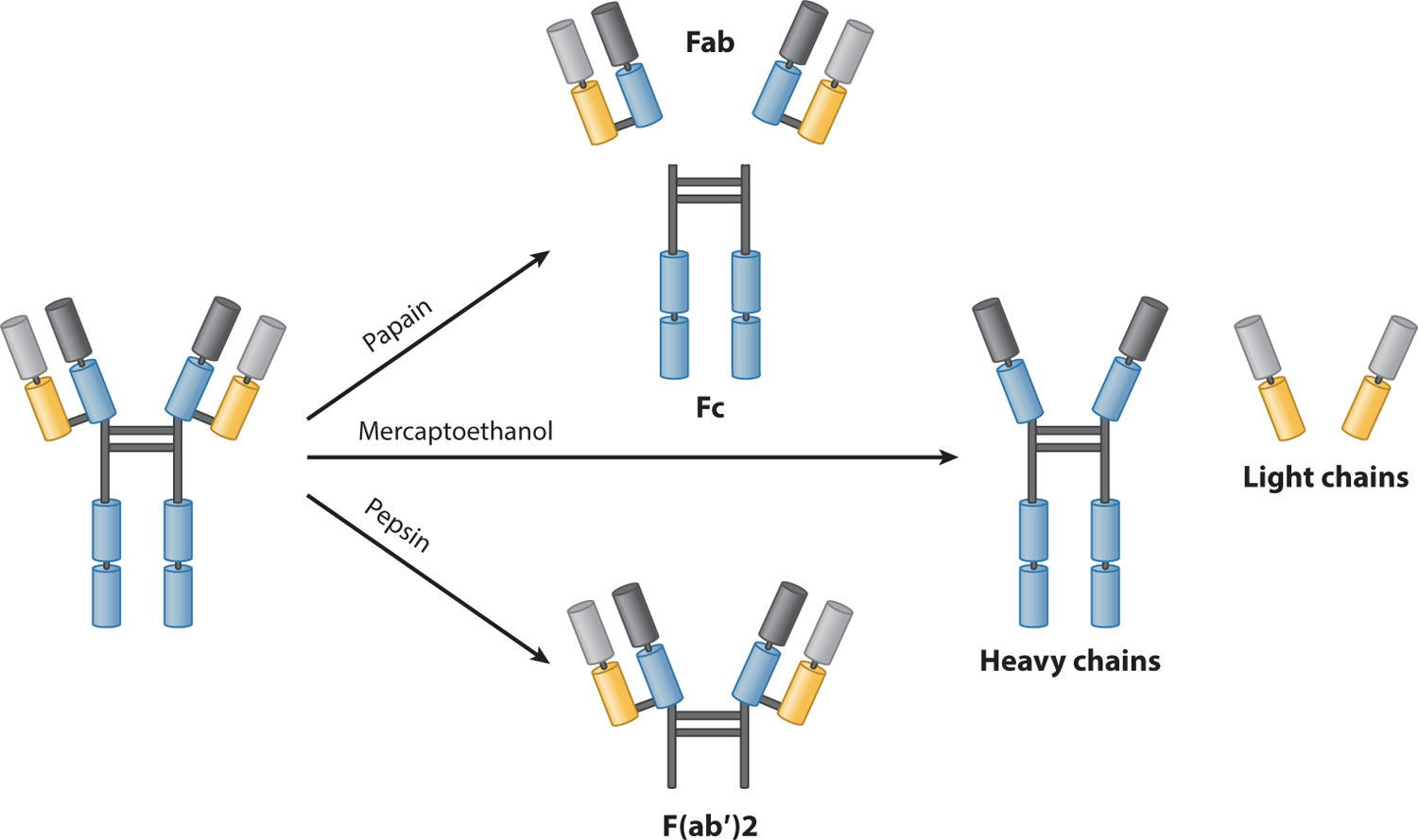
A schematic illustration of an antibody. Abbreviations: Fab, fragment, antigen-binding; Fc, fragment, crystallizable.
In addition to conventional antibodies, camelids also produce heavy-chain-only antibodies (HCAbs). HCAbs, also referred to as nanobodies, contain a single variable domain (VHH) that makes up the equivalent Fab of conventional immunoglobulin G (IgG) antibodies (29). This single variable domain typically can acquire affinity and specificity for antigens comparable to those of conventional antibodies. VHHs can easily be constructed into multivalent formats and have higher thermal stability and chemostability than do most antibodies (14, 17, 27, 40, 57, 68). Another advantage of VHHs is that they are less susceptible to steric hindrances than are large conventional antibodies (25).
In immune systems, antibodies are generated and secreted by B cells, usually differentiated B cells, including plasma cells or memory B cells (Figure 2). Antibodies have two physical forms, a membrane-bound form called the B-cell receptor (BCR), which is found to attach to the surface of B cells, and a soluble form that moves freely in the blood plasma. The BCR facilitates the activation and subsequent differentiation of B cells into either plasma or memory B cells. The activation of B cells has two mechanisms: T cell–dependent (TD) activation and T cell–independent (TI) activation (44). In TD activation, once a BCR binds a TD antigen, the antigen is taken up into the B cell through receptor-mediated endocytosis, degraded, and presented to T helper (TH) cells as peptide pieces in complex with major histocompatibility complex-II (MHC-II) molecules on the cell membrane (2). TH cells recognize and bind these MHC-II–peptide complexes through their T cell receptor (TCR). Following TCR–MHC-II–peptide binding, T cells express the surface protein CD40L, as well as cytokines such as IL-4 and IL-21. These signals promote B cell proliferation, immunoglobulin class switching, somatic hypermutation, and guide differentiation. Upon receipt of these signals, B cells are activated (13). In TI activation, T cells are absent, and B cells receive signals from recognition and binding of a common microbial constituent to toll-like receptors (TLRs) or extensive cross-linking of BCRs to repeated epitopes on a bacterial cell (44). TI activation is rapid, but antibodies generated from it tend to have a lower affinity and are also less functionally versatile than those from TD activation (44). After being activated, B cells can be differentiated into plasma cells or memory B cells to generate and secrete antibodies. Memory B cells can even survive in a human body for years to remember the same antigen and trigger a fast response upon future exposure (4).
Figure 2.
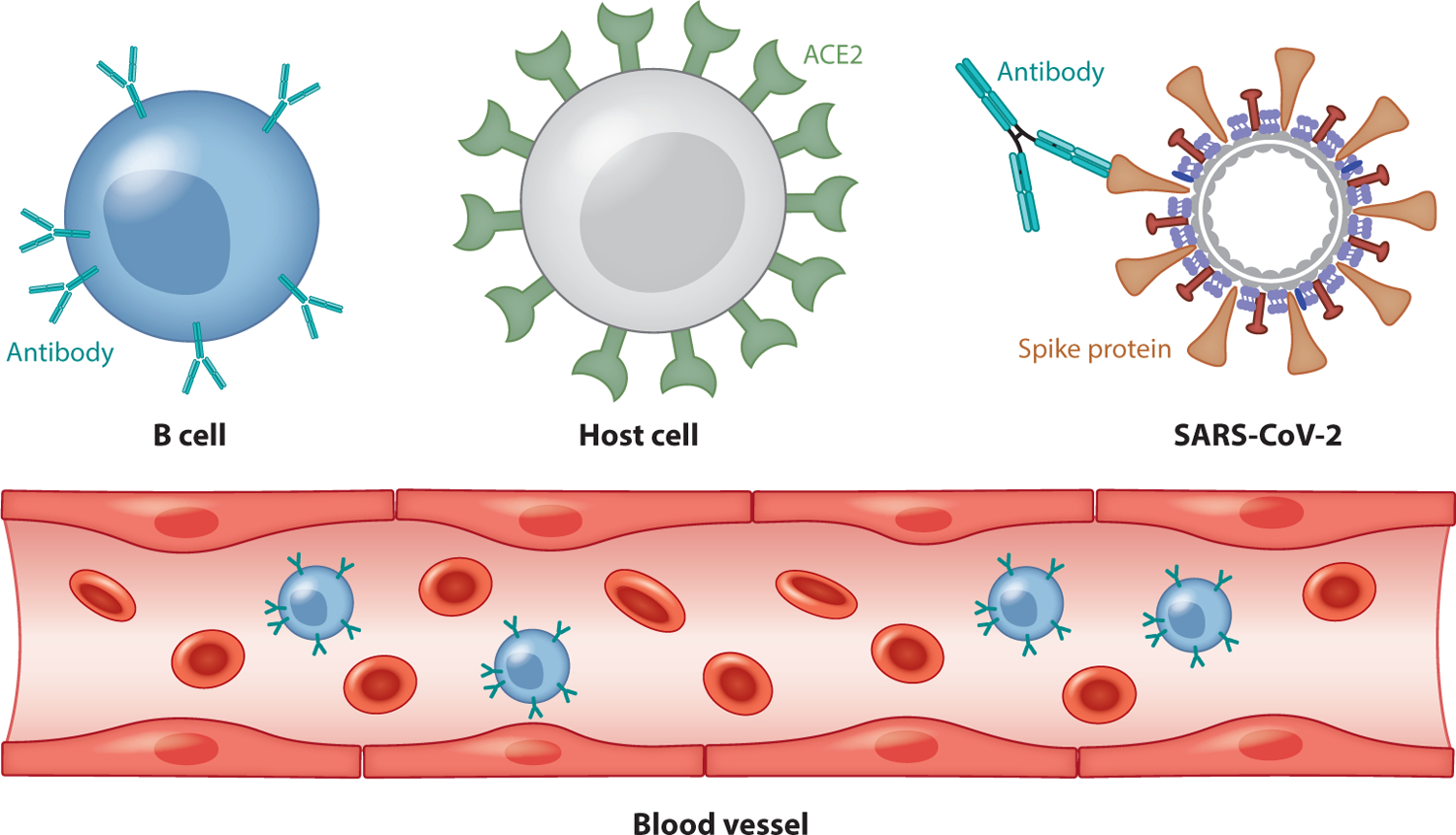
A schematic illustration of antibody therapy. Abbreviations: ACE2, angiotensin-converting enzyme 2; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Antibodies protect our health in four ways: First, their Fab regions can bind to pathogens and thus prevent pathogens from entering or damaging cells; second, they trigger the removal of pathogens by macrophages and other cells via coating of the pathogen; third, they cause the destruction of pathogens by stimulating other immune responses such as a complement pathway (53); and last, antibodies can also lead to vasoactive amine degranulation against certain types of antigens such as helminths and allergens (33).
The antibody mechanism enlightens the development of vaccines and antibody therapies. A vaccine is typically made of weakened or killed forms of a microbe, its toxins, or one of its surface proteins that resemble a disease-causing microorganism. These forms cannot cause an infection, but the immune system still regards them as foreign objects and produces antibodies in response. After the threat has passed, most of the antibodies will break down, but memory B cells remain and remember the antigens in the vaccine.
Antibodytherapiesweredevelopedinthe1970s, following the discovery of the structures of antibodies and the development of the hybridoma technology that provided the first reliable source of monoclonal antibodies (mAbs) (5, 38). Rather than being extracted from convalescent patient plasma, mAbs are made from identical immune cells that are all clones of a unique parent cell; thus, they can have a monovalent affinity to the same epitope. As a result, the most significant advantage of mAbs over conventional small-molecular drugs is their high specificity, which facilitates precise action (32). A second advantage is their long half-life, which allows infrequent dosing (41). Third, molecular engineering technologies have enabled the structure of mAbs to be fine-tuned for specific therapeutic actions and to minimize immunogenicity (28, 46, 51, 69), thus enhancing their safety. This is reflected in mAbs’ approval rate of approximately 20%, compared to 5% for new small-molecular entities (54, 55). Finally, mAbs can be developed in a short time period, e.g., 5–6 months (37). Currently, mAbs have already established their therapeutic and prophylactic efficacy against cancer; autoimmune disease; neurological disorders; and infectious viruses such as HIV, Ebola, and MERS (12, 58, 76). However, there are adverse effects, mostly related to immunomodulation, and therapeutic mAbs (32), such as antibody-dependent enhancement (66) and cytokine storm (11), can be associated with infection.
3. SARS-CoV-2 ANTIBODY THERAPEUTIC CANDIDATES
Both SARS-CoV and SARS-CoV-2 belong to lineage B of the betacoronavirus genus and have four structural proteins, known as the S, envelope, membrane, and nucleocapsid proteins (84, 89). The nucleocapsid protein holds the single-stranded RNA genome. Together with the membrane, S, envelope, and membrane proteins create the viral envelope (83). The S protein, which forms homotrimers protruding from the viral surface, mediates the entry of coronaviruses into host cells when it binds with ACE2 (67). More specifically, the S protein comprises two functional subunits: the S1, which is responsible for binding to the host cell receptor, and the S2, which promotes the fusion of the virus and cellular membranes (71, 72).
ACE2 is a single-pass transmembrane protein with its active domain exposed on the cell surface and is expressed in lungs and many other tissues (30). ACE2 serves as the main cell entry point for SARS-CoV, SARS-CoV-2, and some other coronaviruses (73). Notably, the equilibrium dissociation constant (Kd) of the binding between ACE2 and the S protein is significantly increased in SARS-CoV-2 compared to SARS-CoV (65, 70). Moreover, SARS-CoV-2 may also use basigin to assist in cell entry (75). Therefore, SARS-CoV-2 is more infectious than SARS-CoV.
Antibody therapy is a promising means of fighting COVID-19. Figure 2 provides a schematic illustration of antibody therapy for COVID-19. Notably, neutralizing monoclonal antibodies (mAbs) isolated from convalescent patient memory B cells provides an effective intervention for SARS-CoV-2 due to the safety, scalability, and therapeutic effectiveness of these antibodies (7, 9, 59). As the S protein mediates host cell entry, it is the target of neutralizing antibodies and the main focus of therapeutic and vaccine design efforts (67).
Antibodies can target different SARS-CoV-2 S protein positions. Although a potent N-terminal domain (NTD) antibody has been reported (10), most known antibodies target the SARS-CoV-2 S-protein RBD. Table 1 provides a summary of SARS-CoV-2 and SARS-CoV S protein RBDs in complexes with existing antibodies and ACE2 structures. The structures, functions, and properties of these complexes are analyzed below.
Table 1.
A summary of monoclonal antibodies targeting the SARS-CoV-2 or SARS-CoV S protein with the 3D experimental structures of their complexes available in PDB
| Protein or antibody | Target | Kd (nM)/Method | PDB ID | Resolution (A)˚ |
|---|---|---|---|---|
| ACE2 | SARS-CoV-2 RBD | 1.2 / BLI (70) 34.6 / BLI (82) 15.2 / BLI (65) |
6M0J (39) | 2.45 |
| S309 | SARS-CoV-2 RBD | IgG = 0.104 Fab = 1.98 / BLI (49) | 6WPS, 6WPT (49) | 3.10, 3.70 |
| CR3022 | SARS-CoV-2 RBD | IgG = 6.28 / BLI (65) IgG < 0.1 Fab = 115 / BLI (88) |
6W41 (88) | 3.08 |
| CB6 | SARS-CoV-2 RBD | IgG = 2.49 / SPR (60) | 7C01 | 2.85 |
| P2B-2F6 | SARS-CoV-2 RBD | IgG = 5.14 /SPR (36) | 7BWJ (36) | 2.85 |
| B38 | SARS-CoV-2 RBD | IgG = 70.1 /SPR(85) | 7BZ5 (85) | 1.84 |
| H11-D4 | SARS-CoV-2 RBD | NA | 6Z43 | 3.30 |
| BD23 | SARS-CoV-2 RBD | NA | 7BYR (8) | 3.84 |
| ACE2 | SARS-CoV RBD | 5.0 / BLI (70) 325.8 / BLI (82) 1.70 / BLI (61) |
3D0G | 2.80 |
| CR3022 | SARS-CoV RBD | IgG < 0.1 Fab = 1 / BLI (88) | NA | NA |
| S309 | SARS-CoV RBD | IgG = 0.12 Fab = 1.81 / BLI (49) | NA | NA |
| m396 | SARS-CoV RBD | IgG = 0.005 Fab = 20 / BLI (50) | 2DD8 (50) | 2.30 |
| S230 | SARS-CoV RBD | IgG = 0.06 / BLI (73) | 6NB6 (73) | 4.30 |
| VHH-72a | SARS-CoV RBD | IgG = 1.15 / SPR (81) | 6WAQ (81) | 2.20 |
| 80R | SARS-CoV RBD | IgG = 1.59 / BLI (61) | 2GHW (34) | 2.30 |
| F26G19 | SARS-CoV RBD | Fab = 26 / SPR (47) | 3BGF (47) | 3.00 |
The binding affinity of VHH-72 with the SARS-CoV-2 RBD is Fab = 54 nM.
Abbreviations: ACE2, angiotensin-converting enzyme 2; BLI, biolayer interferometry; Fab, fragment, antigen-binding; NA, not applicable; PDB, Protein Data Bank; RBD, receptor-binding domain; S, spike; SARS-CoV, severe acute respiratory syndrome coronavirus; SPR, surface plasmon resonance.
As summarized in Table 1, 12 mAbs targeting the SARS-CoV-2 or SARS-CoV S-protein RBD have been reported that have their 3D experimental complex structures available in the PDB. According to Pinto et al. (49), the most promising is S309, which shows almost equal neutralization potency against SARS-CoV and SARS-CoV-2. Pinto et al. state that 19 out of 24 residues of the S309’s epitope are strictly unchanged from SARS-CoV and SARS-CoV-2, and the other five residues are conservatively or semiconservatively substituted (49). However, some other researchers are still concerned about the claimed cross-effectiveness against both SARS-CoV and SARS-CoV-2 (74). Notably, two experimental structures of the S309 and SARS-CoV-2 S-protein complex have been released, one having a closed conformation of the S protein and the other having an open conformation. The binding affinity of S309 and S-protein RBD complex is not sensitive to closed or open conformations (49) of the S protein.
CR3022 is another potentially potent antibody that may bind to both SARS-CoV and SARS-CoV-2 (65,88). It has also been shown that, compared with m396,a SARS-CoV-specific antibody, CR3022 has a significantly stronger binding signal to SARS-CoV-2. However,itsaffinity to SARS-CoV-2 is much weaker than its affinity to SARS-CoV (88). It has also been suggested that CR3022 can only access the open conformation of the S-protein RBD (88).
Other promising SARS-CoV-2 antibodies include CB6 and P2B-2F6, which are specific human mAbs extracted from convalescent COVID-19 patients (36,60). VHH-72 cross-reacts with SARS-CoV-2 and SARS-CoV S proteins, but its binding affinity to SARS-CoV-2 is much lower than that to SARS-CoV (81).B38 also shows direct competition with ACE2 in binding to the SARS-CoV-2 S protein (85).
3.1. Three-Dimensional Structure Alignment
All of the available 3D structures of the SARS-CoV-2 S-protein RBD in complex with antibodies are aligned to ACE2. Figures 3 and 4 show the alignment of SARS-CoV-2 and SARS-CoV antibodies, respectively. The PDB ID of these complexes can be found in Table 1.
Figure 3.
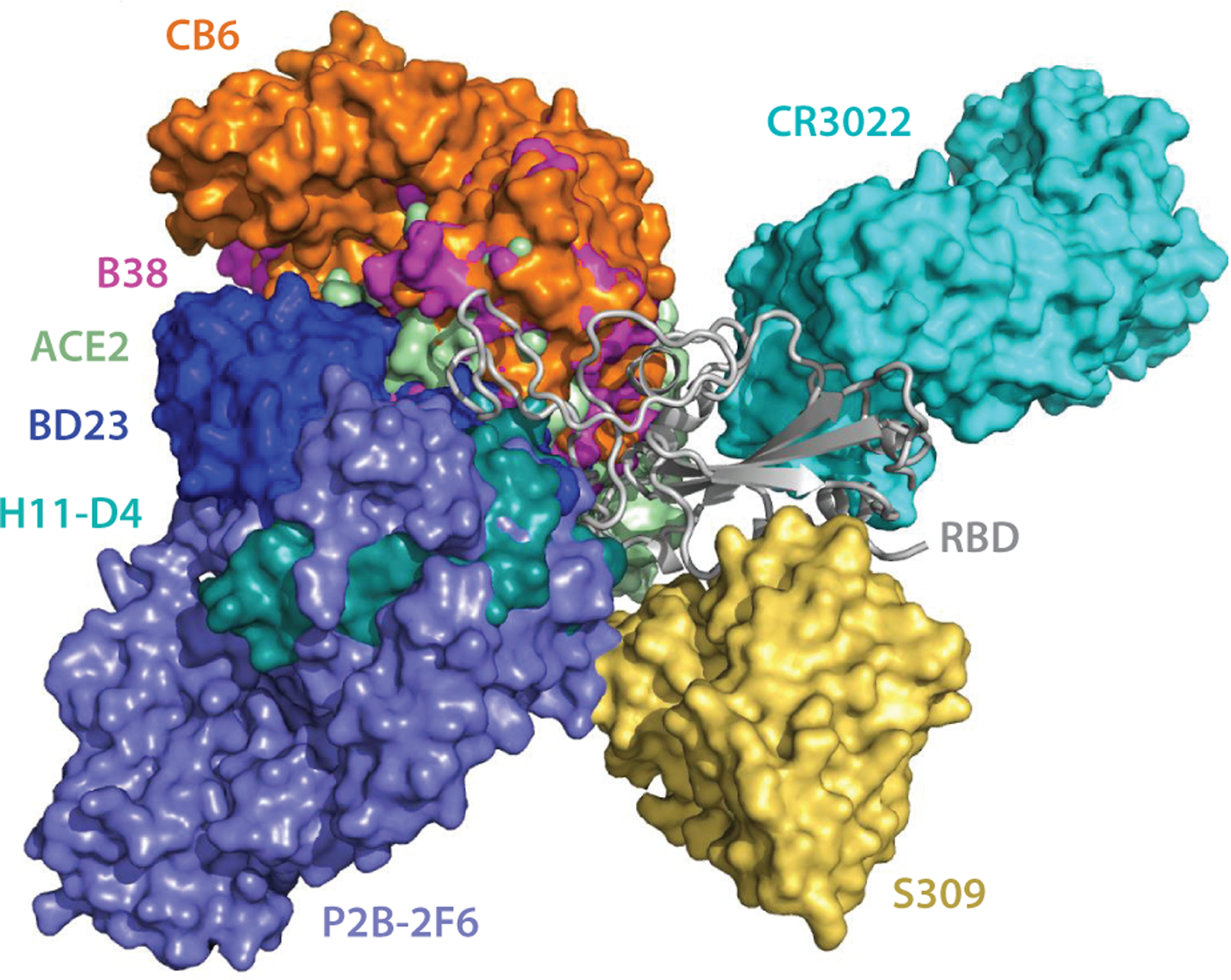
The alignment of the available 3D structures of SARS-CoV-2 S-protein RBD in binding complexes with antibodies, as well as with ACE2. Abbreviations: ACE2, angiotensin-converting enzyme 2; RBD, receptor-binding domain; S, spike; SARS-CoV-2, severe acute respiratory syndrome coronavirus-2.
Figure 4.
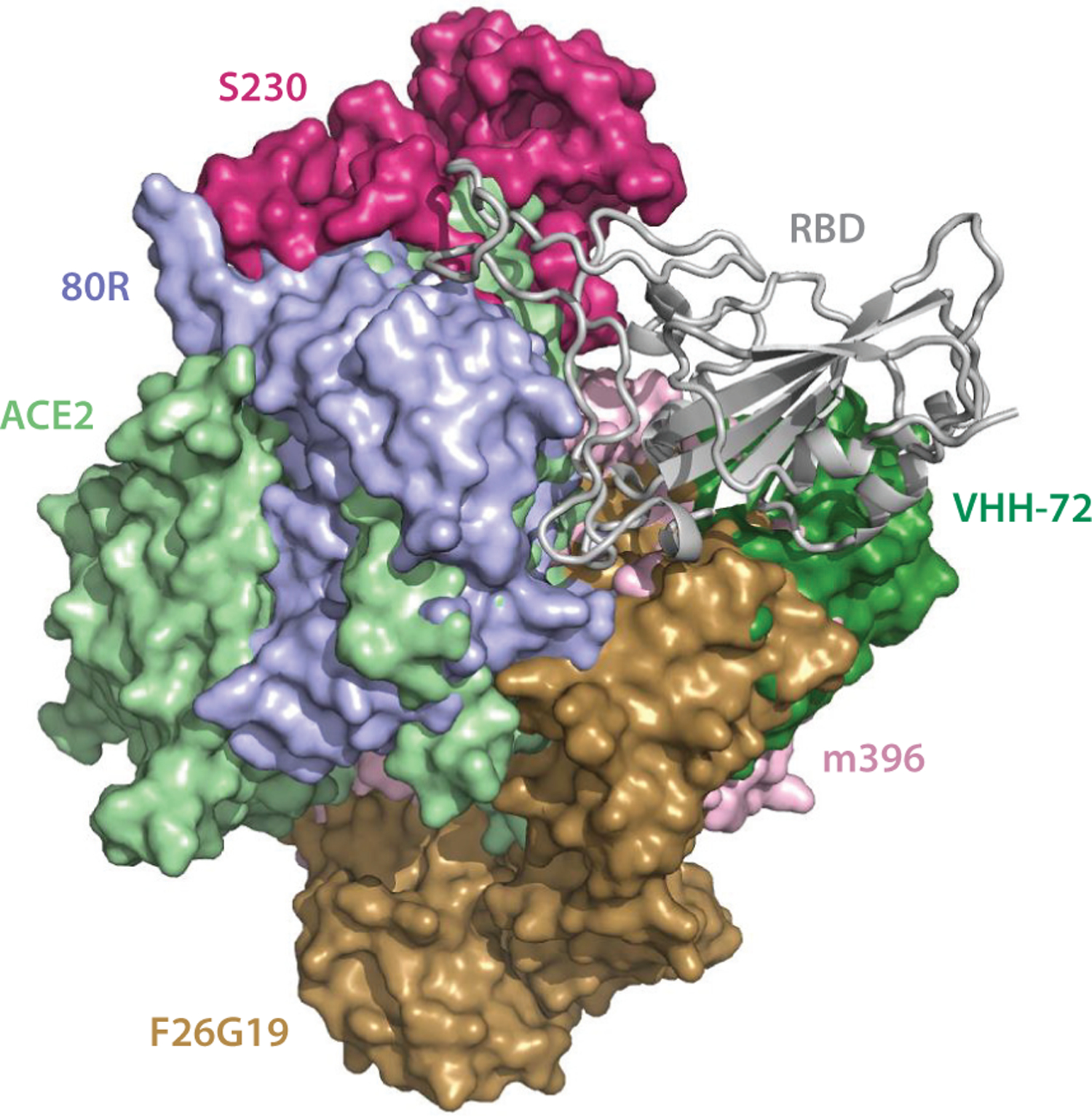
The alignment of the available 3D structures of the SARS-CoV S-protein RBD in binding complexes with antibodies, as well as with ACE2. Abbreviations: ACE2, angiotensin-converting enzyme 2; RBD, receptor-binding domain; S, spike; SARS-CoV, severe acute respiratory syndrome coronavirus.
As shown in Figure 3, the antibodies CB6, B38, H11-D4, and P2B-2F6 have their epitopes overlapping with the ACE2 binding site, suggesting that their bindings are in direct competition with that of ACE2. Theoretically, this direct competition reduces the viral infection rate. For an antibody with a strong binding ability, it will directly neutralize SARS-CoV-2 without the need of antibody-dependent cell cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) mechanisms. However, the binding sites of epitopes of S309 and CR3022 are far from that of ACE2, leading to the absence of binding competition (49, 65). One study shows that the ADCC and ADCP mechanisms contribute to the viral control conducted by S309 in infected individuals (49). Research indicates that CR3022 neutralizes the virus in a synergistic fashion (63).
Figure 4 shows that, on the SARS-CoV RBD, the epitopes of antibodies S230, 80R, F26G19, and m396 overlap with that of ACE2. VHH-72 locates slightly away from the ACE2 binding site but still sterically clashes with the ACE2 binding. They all lead to binding competition that neutralizes the virus.
3.2. Alignment of Antibody and ACE2 Epitopes on Spike Protein 2D Sequences
Figure 5 highlights the contact regions of antibodies and ACE2 epitopes on SARS-CoV-2 RBD or SARS-CoV RBD 2D sequences. Consistent with the 3D alignment, with the exception of S309, CR3022, and VHH-72, all of the antibodies have epitopes overlapping with the ACE2 binding site, especially the residues from 486 to 505 of the SARS-CoV-2 RBD (corresponding to the residues 472 to 491 of the SARS-CoV RBD). Although the VHH-72 epitope residues do not overlap with the ACE2 binding site, VHH72 occupies parts of the space of the ACE2 binding site, which disrupts ACE2 binding with RBD. Therefore, VHH-72 also has a competitive binding ability against ACE2. Figure 5 also shows that these epitope residues have many mutations from the SARS-CoV-2 RBD to the SARS-CoV RBD, which could explain why most of the antibodies lack cross-reaction to both SARS-CoV-2 and SARS-CoV. We return to this below.
Figure 5.
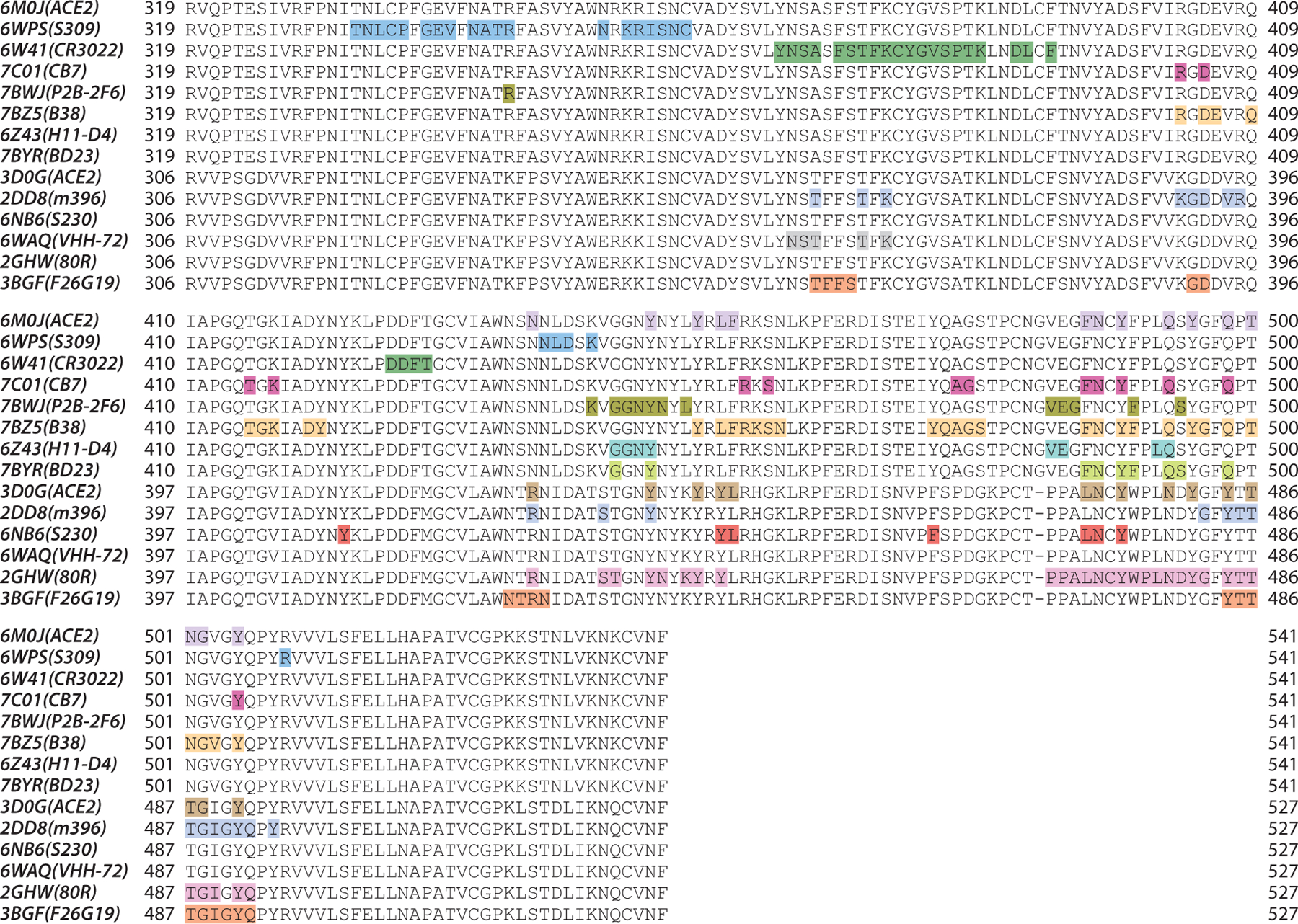
Illustration of the contact positions of antibody and ACE2 epitopes with SARS-CoV-2 and SARS-CoV S-protein RBDs on RBD 2D sequences. The proteins in the structures of 6M0J, 6WPS, 6W41, 7C01, 7BWJ, 7BZ5, 6Z43, and 7BYR are in complexes with the SARS-CoV-2 S protein, while the proteins in the structures of 3D0G, 2DD8, 6NB6, 6WAQ, 2GHW, and 3BGF are in complexes with the SARS-CoV S protein. Abbreviations: ACE2, angiotensin-converting enzyme 2; RBD, receptor-binding domain; S, spike; SARS-CoV, severe acute respiratory syndrome coronavirus.
4. EXPERIMENTAL PITFALLS IN THE AFFINITY MEASUREMENTS OF ANTIBODY BINDING WITH S PROTEIN RECEPTOR BINDING DOMAIN
Table 1 clearly shows the discrepancies in reported experimental Kd values for ACE2 in complexes with the SARS-CoV-2 S protein [i.e., 1.2 nM (70), 15.2 nM (65), and 34.6 nM (82)]. Moreover, a 191-fold difference in magnitude has been reported in experimental Kd values for the ACE2 and SARS-CoV S protein complex [i.e., 5.0 nM (70), 325.8 nM (82), and 1.70 nM (61)].
The inconsistency in experimental values is not isolated. The experimental Kd values for CR3022 binding with the SARS-CoV-2 S-protein RBD have been reported as 6.28 nM (65) and <0.1 nM (88). This level of discrepancy in reported experimental values makes it impossible to select antibody candidates appropriately.
As shown in Table 1, two binding assay techniques are used to measure Kd values of antibody–antigen interactions. The discrepancies in Kd values for CR3022 are based on biolayer interferometry (BLI) measurements. BLI detects the surface changes on biosensor tips induced by protein–protein association and dissociation by analyzing the interference pattern of white light reflected from the surface. BLI results are sensitive to biosensor preparation, stability of the light source, temperature control, calibration, and human errors (62). Surface plasmon resonance (SPR) has also been employed for determining the Kd values of antibody and RBD complexes, as shown in Table 1. This method detects the reflectivity change induced by molecular adsorption, such as that of polymers, DNA, or proteins, using changes in reflection angles. Similarly, SPR is also sensitive to the preparation of conjugated antigens, stability of the light source, temperature control, calibration, and human errors (1). The widespread inconsistency in reported antibody and S protein binding affinities motivated us to carry out the computational analysis of existing antibody–S protein complexes described below.
5. COMPUTATIONAL ANALYSIS OF ANTIBODY–SARS-CoV-2 INTERACTIONS
To create a unified assessment and ranking of S-protein RBD binding complexes with antibodies and ACE2, we utilize TDA, graph theory, network models, and machine learning to analyze the 3D complexes presented in Table 1. We also evaluate the potential of repurposing SARS-CoV antibodies for SARS-CoV-2 using the topology-based network tree (TopNetTree) model (77).
5.1. Ranking of ACE2 and Antibodies
The prediction results and network descriptors are presented in Tables 2 and 3 for SARS-CoV-2 complexes and SARS-CoV complexes, respectively. In Table 2, the complexes are ranked according to their predicted binding affinities, G, followed by flexibility–rigidity indexes (FRIs) (45, 86), which have the highest covariance. They are computed based on all of the Cα atoms on the RBD and all of the Cα atoms in antibodies or ACE2. The FRI Rη indicates the measurement of geometric compactness and topological connectivity of protein–protein interactions (PPIs) at each residue, such that the larger η is, the longer the range of pairwise influence will be. In comparison with the predicted energy, a strong binding affinity corresponds to a large rigidity index. The summation of binding affinity changes computed with the TopNetTree model (77) by modifying the RBD residues to glycine (G) are presented; S10 and S8 stand for those residues within 10 Å and 8 Å, respectively, of any Cα of antibodies. The binding affinity change following each mutation to glycine (G) is calculated by the TopNetTree model for PPIs (77), where a positive binding affinity change ΔΔG means a stronger binding affinity for the mutant and vice versa. Therefore, a summation of considered residues in the RBD with smaller values indicates a strong binding affinity of the wild type.
Table 2.
Graph network descriptor consisting of SARS-CoV-2 S-protein RBD, ACE2, and antibodies
| Molecule | CR3022 | B38 | CB6 | ACE2 | BD23 | H11-D4 | S309 | P2B-2F6 |
|---|---|---|---|---|---|---|---|---|
| PDB ID | 6W41 | 7BZ5 | 7C01 | 6M0J | 7BYR | 6Z43 | 6WPS | 7BWJ |
| ΔG | −15.4 | −14.7 | −13.5 | −11.9 | −10.8 | −10.3 | −9.9 | −9.6 |
| R10 | 335 | 349 | 338 | 279 | 227 | 201 | 256 | 211 |
| R8 | 134 | 138 | 132 | 105 | 106 | 82 | 97 | 82 |
| S10 | 19.15 | 30.17 | 36.56 | 20.83 | 10.39 | 8.91 | 18.28 | 17.74 |
| S8 | 11.69 | 12.39 | 13.36 | 16.60 | 5.49 | 4.41 | 7.04 | 5.97 |
| d | 0.070 | 0.069 | 0.072 | 0.072 | 0.069 | 0.077 | 0.071 | 0.074 |
| ρ | 0.0192 | 0.0190 | 0.0196 | 0.0185 | 0.0171 | 0.0190 | 0.0206 | 0.0196 |
| 〈L〉 | 13.69 | 14.26 | 13.75 | 13.85 | 14.80 | 13.59 | 13.86 | 13.98 |
| 〈Cb〉 | 0.0109 | 0.0111 | 0.0110 | 0.0113 | 0.0130 | 0.0113 | 0.0112 | 0.0117 |
| 〈Ce〉 | 0.052 | 0.050 | 0.052 | 0.051 | 0.053 | 0.054 | 0.054 | 0.053 |
| 〈Cs〉 | 1590955 | 2397825 | 2010826 | 2105421 | 813061 | 2248985 | 1387110 | 1562243 |
| 〈M〉 | 847509 | 1237464 | 1096771 | 1132331 | 413572 | 1239452 | 753625 | 855641 |
| 〈Θ〉 | 0.0192 | 0.0190 | 0.0196 | 0.0185 | 0.0171 | 0.0190 | 0.0206 | 0.0196 |
ΔG indicates the predicted binding affinity (kcal/mol) [the predictions are made using the Prodigy web server (87)]; R10 and R8 indicate the FRI with η of 10 and 8, respectively; S10 and S8 indicate the summation of binding affinity changes (ΔΔG kcal/mol) by mutating RBD residues within 10 Å and 8 Å to any Cα of antibodies to glycine; d indicates edge density; ρ indicates degree heterogeneity; 〈L〉 indicates average path length; 〈Cb〉 indicates average betweenness centrality; 〈Ce〉 indicates average eigencentrality; 〈Cs〉 indicates average subgraph centrality; 〈M〉 indicates average communicability; and 〈Θ〉 indicates average communicability angle.
Abbreviations: FRI, flexibility–rigidity index; PDB, Protein Data Bank; RBD, receptor-binding domain; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
Table 3.
Graph network descriptor consisting of SARS-CoV S-protein RBD, ACE2, and antibodies
| Molecule | 80R | ACE2 | VHH-72 | m396 | S230 | F26G19 |
|---|---|---|---|---|---|---|
| PDB ID | 2GHW | 3D0G | 6WAQ | 2DD8 | 6NB6 | 3BGF |
| ΔG | −17.3 | −11.5 | −9.7 | −9.4 | −7.7 | −6.7 |
| R10 | 378 | 270 | 255 | 304 | 195 | 254 |
| R8 | 157 | 101 | 103 | 119 | 72 | 101 |
| S10 | 20.35 | 13.05 | 13.37 | 10.79 | 11.48 | 14.01 |
| S8 | 8.92 | 1.56 | 7.49 | 6.47 | 8.09 | 7.84 |
| d | 0.070 | 0.070 | 0.074 | 0.073 | 0.078 | 0.072 |
| ρ | 0.0206 | 0.0187 | 0.0193 | 0.0186 | 0.0197 | 0.0190 |
| 〈L〉 | 13.35 | 12.91 | 13.46 | 12.90 | 12.96 | 12.63 |
| 〈Cb〉 | 0.0120 | 0.0108 | 0.0109 | 0.0113 | 0.0119 | 0.0113 |
| 〈Ce〉 | 0.053 | 0.053 | 0.053 | 0.054 | 0.054 | 0.056 |
| 〈Cs〉 | 2693776 | 1418662 | 1607217 | 2383597 | 3167175 | 1897873 |
| 〈M〉 | 1446506 | 730809 | 915061 | 1311299 | 1714397 | 1039376 |
| 〈Θ〉 | 0.0206 | 0.0187 | 0.0192 | 0.0186 | 0.0196 | 0.0190 |
ΔG indicates the predicted binding affinity (kcal/mol) [the predictions are made using the Prodigy web server (87)]; R10 and R8 indicate FRI with η of 10 and 8, respectively; S10 and S8 indicate the summation of binding affinity changes (ΔΔG kcal/mol) by mutating RBD residues within 10 Å and 8 Å to any Cα of antibodies to glycine; d indicates edge density; ρ indicates degree heterogeneity; 〈L〉 indicates average path length; 〈Cb〉 indicates average betweenness centrality; 〈Ce〉 indicates average eigencentrality; 〈Cs〉 indicates average subgraph centrality; 〈M〉 indicates average communicability; and 〈Θ〉 indicates average communicability angle.
Abbreviations: FRI, flexibility–rigidity index; PDB, Protein Data Bank; RBD, receptor-binding domain; SARS-CoV-2, severe acute respiratory syndrome coronavirus 2.
The rest of the table gives the Cα-based complex analysis from multiple network descriptors, including edge density (d), degree heterogeneity (ρ) (19), average path length (〈L〉) (79), average betweenness centrality (〈Cb〉) (26), average eigencentrality (〈Ce〉) (3), average subgraph centrality (〈Cs〉) (23), average network communicability (〈M〉) (21), and average network communicability angle (〈Θ〉) (22). With the exception of the degree heterogeneity, which is calculated based only on the RBD Cα atoms, other descriptors are calculated from all Cα atoms on the RBD and antibody (or ACE2) Cα atoms within 10 Å from any Cα atom on the RBD. The degree heterogeneity demonstrates antibody or ACE2 influence to the RBD such that close degree heterogeneity numbers would have similar impacts. For example, molecules B38 and H11-D4 have degree heterogeneity values that are close to ACE2, as well as sharing the same receipt domain. The average betweenness centrality (26) and average eigencentrality (3) values are correlated quite well to the predicted binding affinities.
Table 3 shows the results of predictions and network descriptors for the SARS-CoV S-protein complex. Again, the predicted binding affinities have high correlations to FRIs. For degree heterogeneity, m396 has a similar impact to ACE2. Molecule 80R (PDB 2GHW) has the highest rigidity index both for η = 10 and η = 8, which indicates a more rigid complex structure between 80R and the RBD. In the comparison of SARS-CoV S-protein RBD and SARS-CoV-2 S-protein RBD in Table 2, descriptors are close to each other except for the summation of binding affinity changes, which includes more biological and chemical information than do the other rows. Thus, the network structures for the SARS-CoV RBD and SARS-CoV-2 RBD complexes are similar.
5.2. Repositioning of SARS-CoV Antibody Candidates for SARS-CoV-2
In this section, we predict the binding affinities of SARS-CoV antibodies when they are applied to SARS-CoV-2 neutralization using the TopNetTree model (77). Specifically, we compute the binding affinity changes following the mutations from the SARS-CoV RBD to the SARS-CoV-2 RBD. These changes can be very significant. One study showed that a single mutation (V367F) can lead to a 10-fold increase in IC50 for a particular antibody (56).
Figure 6 shows both predicted binding affinities of each SARS-CoV complex and predicted binding affinities of each molecule with the SARS-CoV-2 RBD, which are calculated by accumulating binding affinities of single mutations from SARS-CoV RBD to SARS-CoV-2 RBD and adding the sum to the binding affinities of SARS-CoV complexes. Obviously, antibody 80R (PDB 2GHW) has the largest binding energy change in SARS-CoV ranking, as well as in SARS-CoV-2 ranking, among these SARS-CoV antibodies. Molecule VHH-72 (6WAQ) has a smaller binding affinity than does ACE2 (3D0G) for the SARS-CoV RBD but an equivalent binding affinity for the SARS-CoV-2 RBD. Molecules m396, S230, and F26G19 have weaker binding affinities after modifying from the SARS-CoV RBD to the SARS-CoV-2 RBD. Finally, the binding affinity of the SARS-CoV RBD with ACE2 following mutations to SARS-CoV-2 is slightly higher than the binding affinity of the SARS-CoV RBD with ACE2, indicating that SARS-CoV-2 is more infectious than SARS-CoV. This is consistent with experimental reports (70, 82).
Figure 6.
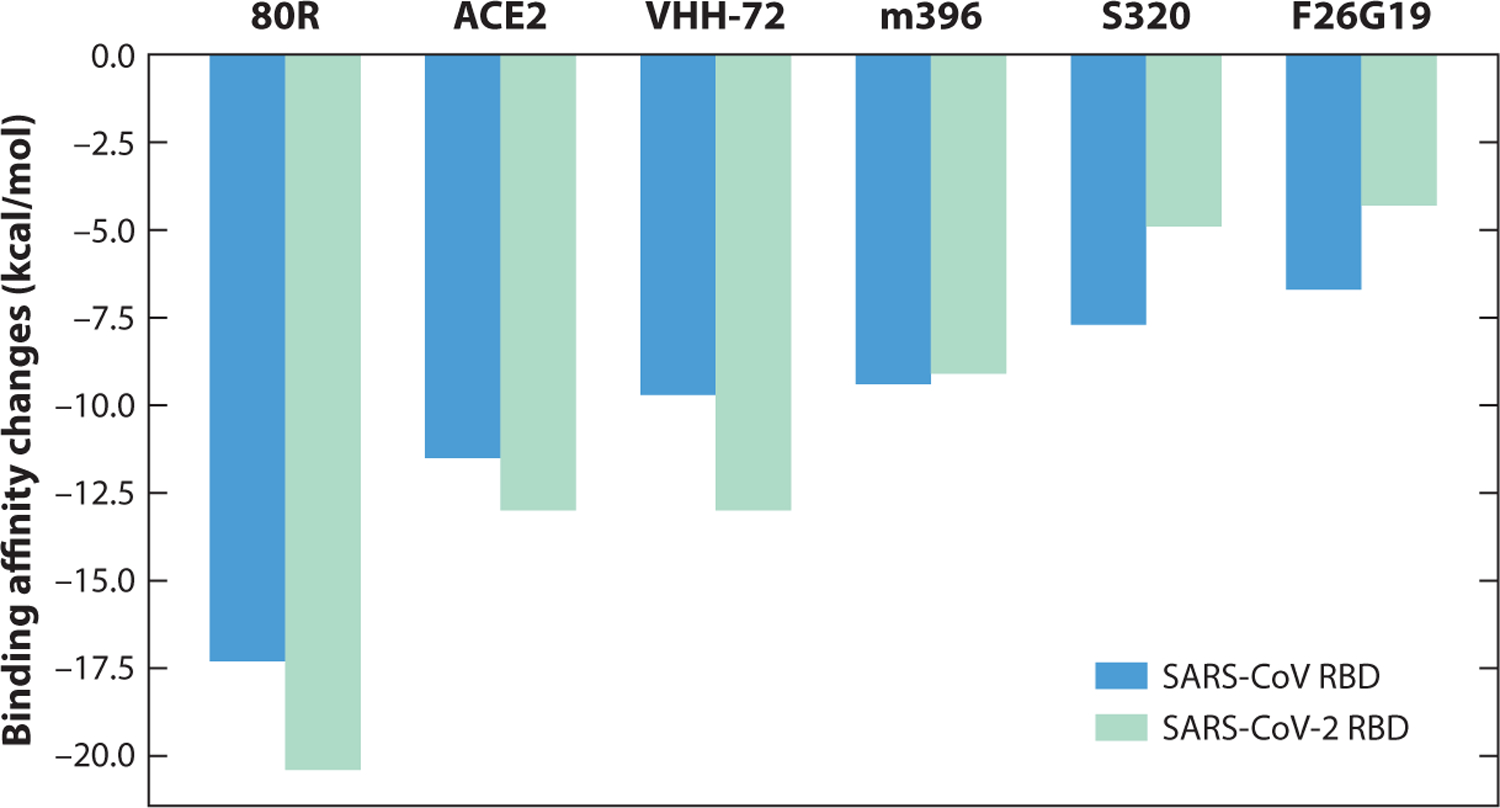
An illustration of the binding affinities of antibodies with SARS-CoV and SARS-CoV-2 RBDs. The molecular names of these antibodies are 80R (PDB 2GHW), ACE2 (3D0G), VHH-72 (6WAQ), m396 (2DD8), S230 (6NB6), and F26G19 (3BGF). Abbreviations: ACE2, angiotensin-converting enzyme 2; RBD, receptor-binding domain; SARS-CoV, severe acute respiratory syndrome coronavirus.
Figures 7 and 8 show the binding affinity changes on individual mutations of the SARS-CoV S-protein RBD, where more precise trends can be observed. In Figure 7, the molecule 80R has a similar trend to ACE2; both of them share the most receptive binding domain of the SARS-CoV-2 S protein. In Figure 8, most of the binding affinity changes following mutations in the receptor binding motif (RBM) of ACE2 are negative, which indicates stronger binding affinities with the SARS-CoV RBD.
Figure 7.
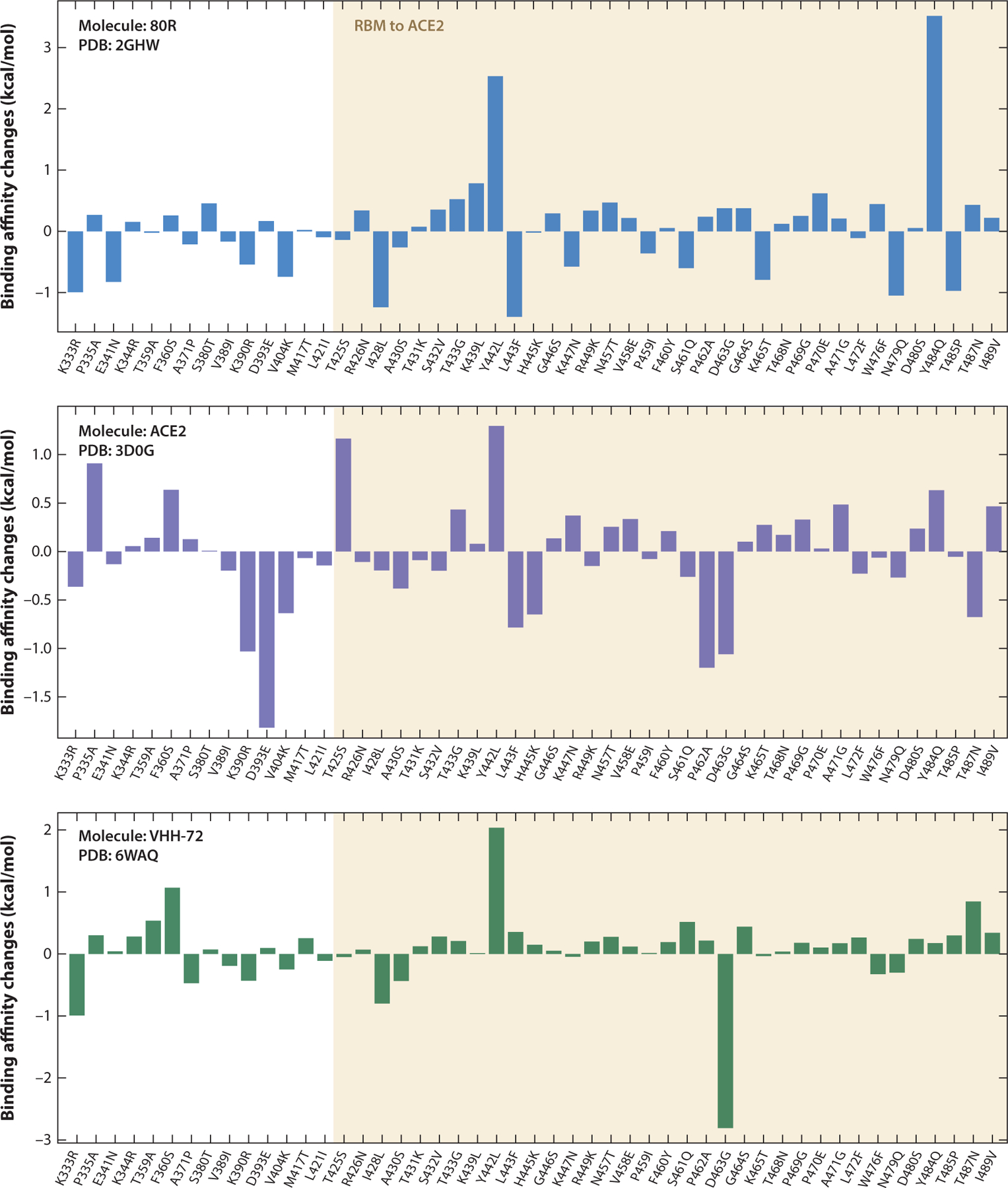
Overall binding affinity changes following the mutations of S protein RBD from SARS-CoV to SARS-CoV-2 for molecules 80R, ACE2, and VHH-72. The x axis records the wild type to the mutant type at the specific residue position. The tan region marks the RBM corresponding to ACE2. The height of each bar indicates the predicted binding affinity changes. A positive change indicates a strengthening of binding. Abbreviations: ACE2, angiotensin-converting enzyme 2; PDB, Protein Data Bank; RBD, receptor-binding domain; RBM, receptor-binding motif; S, spike; SARS-CoV, severe acute respiratory syndrome coronavirus.
Figure 8.
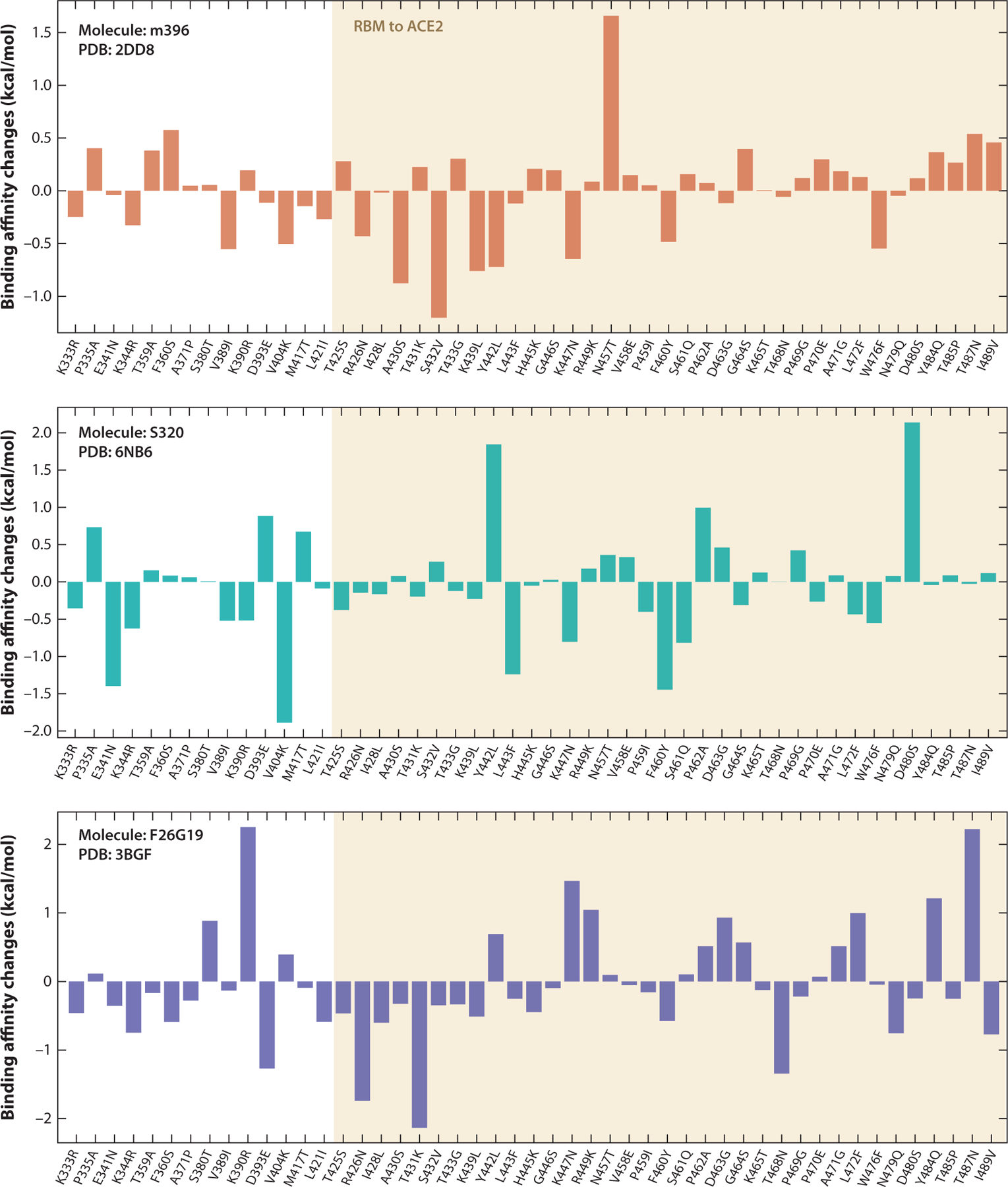
Overall binding affinity changes following the mutations of S protein RBD from SARS-CoV to SARS-CoV-2 for molecules m396, S320, and F26G19. The x axis records the wild type to the mutant type at the specific residue position. The tan region marks the RBM corresponding to ACE2. The height of each bar indicates the predicted binding affinity changes. A positive change indicates a strengthening of binding. Abbreviations: ACE2, angiotensin-converting enzyme 2; PDB, Protein Data Bank; RBD, receptor-binding domain; RBM, receptor-binding motif; S, spike; SARS-CoV, severe acute respiratory syndrome coronavirus.
5.3. Network Analysis of Antibody–Antigen Complexes
Various network models have been employed to analyze the structure and function of the main SARS-CoV and SARS-CoV-2 proteases (20). We utilize network models to illustrate interactions between the binding complexes of the RBD of SARS-CoV or SARS-CoV-2 and antibodies or ACE2.
In the microscopy of each single residue, the performances on the network models reveal the similarities and differences between complexes. In Figure 9, the SARS-CoV RBD with ACE2 (PDB 3D0G), SARS-CoV-2 RBD with ACE2 (PDB 6M0J),and SARS-CoV-2 RBD with CR3022 (PDB: 6W41) are listed and aligned; 6W41 has the strongest predicted binding affinity and the largest deviation to 6M0J, as shown in Tables 2 and 3. It is interesting to observe that the domains that have high rigidity index values are similar in all three complexes. In the comparison of betweenness centralities of the three structures, although 3D0G has many large values, it has the lowest average betweenness centrality among the structures, as shown in Tables 2 and 3. Analogously to the rigidity index, all three complexes have the same regions of high individual eigencentrality values and subgraph centrality values. Overall, the differences between 3D0G and 6M0J are quite small in their network analysis. However, the betweenness centrality reflects their difference such that a higher average value would indicate a stronger binding affinity. Moreover, all three complexes coincidentally have similar regions of high values for network descriptors, which suggests that this region would play a key role in PPIs.
Figure 9.
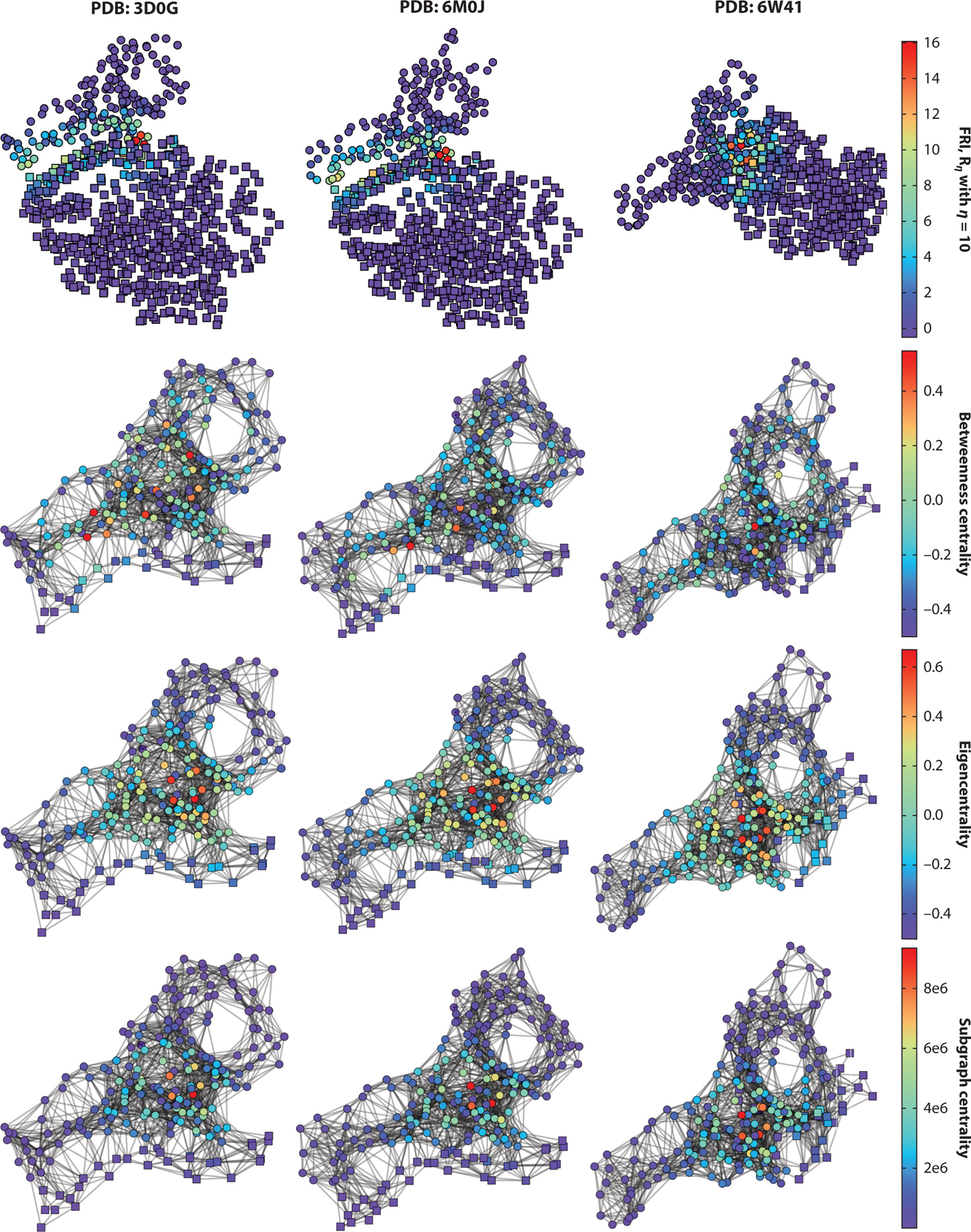
Cα network analysis of three antibody–antigen complexes. Circles indicate antigen (spike-protein receptor-binding domain), and cubes represent antibody or angiotensin-converting enzyme 2. Columns list complexes 3D0G, 6M0J, and 6W41. Rows represent FRI, betweenness centrality, eigencentrality, and subgraph centrality. Abbreviations: FRI, flexibility–rigidity index; PDB, Protein Data Bank.
6. CLINICAL TRIALS OF COVID-19 ANTIBODY THERAPEUTIC CANDIDATES
Table 4 summarizes the currently ongoing clinical trials of COVID-19 antibody therapeutic candidates in the United States, China, and Europe. These data are collected from the National Institutes of Health (NIH) (https://www.nih.gov/coronavirus), the European Medicines Agency (EMA) (https://www.clinicaltrialsregister.eu/ctr-search/search?query=covid-19), and media reports.
Table 4.
Summary of ongoing clinical trials of COVID-19 antibody therapeutic candidates
| Antibody ID | Manufacturer | Target | Trial location | Trial phase | Start date |
|---|---|---|---|---|---|
| Lanadelumaba | Shire | pKal | Radboud University Medical Center, Nijmegen, Netherlands | 4 | / |
| Octagama | Pfizer | Antibody mixture | Sharp Memorial Hospital, San Diego, California, United States | 4 | 4/28/2020 |
| Sarilumaba,b | Regeneron Pharmaceuticals and Sanofi | IL-6 | Assistance Publique—Hôpitaux de Paris, Paris, France | 3 | 3/25/2020 |
| VA Boston Healthcare System, Boston, Massachusetts, United States | 2 | 4/10/2020 | |||
| Sirukumaba,b | Janssen Biotech | IL-6 | Sanofi-Aventis Recherche et Développement, Chilly-Mazarin, France | 3 | 3/26/2020 |
| Loyola University Medical Center, Maywood, Illinois, United States | 2 | 4/24/2020 | |||
| Canakinumaba,b | Novartis | Interleukin-1β | Novartis Investigative Site, Glendale, California, United States | 3 | 4/30/2020 |
| Novartis Pharma GmbH, Nürnberg, Germany | 3 | 4/29/2020 | |||
| IFX-1b | InflaRx | C5a | InflaRx GmbH, Jena, Germany | 3 | 3/29/2020 |
| Lenzilumaba | Humanigen | GM-CSF | Mayo Clinic, Phoenix, Arizona, United States | 3 | 4/30/2020 |
| Mylotargb | Celltech and Wyeth | CD33 | UK Research and Innovation, United Kingdom | 3 | 5/5/2020 |
| Ravulizumabb | Alexion Pharmaceuticals | C5 | Alexion Europe SAS, Levallois-Perret, France | 3 | 5/7/2020 |
| Tocilizumaba,b | Roche | IL-6 | Queen’s Medical Center, Honolulu, Hawaii, United States | 3 | 6/1/2020 |
| F. Hoffmann-La Roche Ltd., Basel, Switzerland | 3 | 4/6/2020 | |||
| Avdoralimabb | Innate Pharma | C5a | Assistance Publique Hôpitaux De Marseille, Marseille, France | 2 | 4/23/2020 |
| Bevacizumabb | Genentech | VEGF-A | Fundación para la Investigación Biomédica de Córdoba, Córdoba, Spain | 2 | 4/24/2020 |
| CERC 002a | Cerecor | LIGHT | Cape Fear Valley Medical Center, Fayetteville, North Carolina, United States | 2 | 6/9/2020 |
| Clazakizumaba | Bristol Myers Squibb and Alder Biopharmaceuticals | IL-6 | Cedars-Sinai Medical Center, Los Angeles, California, United States | 2 | 4/24/2020 |
| Gimsilumaba | Eisai Inc | GM-CSF | UCLA Ronald Reagan Medical Center, Los Angeles, California, United States | 2 | 4/12/2020 |
| IC14a | Scripps Research | CD14 | University of Washington, Seattle, Washington, United States | 2 | 7/2020 |
| Infliximaba | Janssen Biotech | TNFα | Tufts Medical Center, Boston, Massachusetts, United States | 2 | 6/1/2020 |
| Leronlimaba | CytoDyn | CCR5 | University of California, Los Angeles, California, United States | 2 | 4/1/2020 |
| LY3127804a | Eli Lilly and Company | Ang2 | NorthShore University HealthSystem, Evanston, Illinois, United States | 2 | 4/20/2020 |
| LY3819253a | Eli Lilly and Company | S protein | Cedars-Sinai Medical Center, Los Angeles, California, United States | 2 | 6/13/2020 |
| Mavrilimumaba | MedImmune | GM-CSF | Cleveland Clinic Health System, Cleveland, Ohio, United States | 2 | 5/20/2020 |
| MSTT1041Aa | Genentech | ST2 | eStudySite-Chula Vista-PPDS, Chula Vista, California, United States | 2 | 6/2/2020 |
| Nivolumabb | Bristol-Myers Squibb | PD-1 | Centre Léon Bérard, Léon, France | 2 | 4/1/2020 |
| Otilimaba,b | MorphoSys | GM-CSF | GSK Investigational Site, Saint Louis Park, Minnesota, United States | 2 | 5/28/2020 |
| GlaxoSmithKline Research Development Ltd., Brentford, United Kingdom | 2 | 5/20/2020 | |||
| Siltuximabb | Eusapharma | IL-6 | Fundacié Clínic per a la Recerca Biomédica, Barcelona, Spain | 2 | 4/7/2020 |
| SNDX-6352a | Syndax Pharmaceuticals | CSF-1R | HonorHealth, Scottsdale, Arizona, United States | 2 | 5/30/2020 |
| ARGX-117b | Argenx | C2 | Argenx BV, Zwijnaarde, Belgium | 1 | 4/21/2020 |
| TJ003234a | / | GM-CSF | GW Medical Faculty Associates, Washington, District of Columbia, United States | 1 | 4/11/2020 |
| JS016c | Junshi Biosciences | S protein | Huashan Hospital Affiliated to Fudan University, Shanghai, China | 1 | 6/7/2020 |
Notably, most of the current COVID-19 antibody therapeutic clinical trial candidates are aimed at targets other than the S protein. These antibodies were initially developed for other diseases and have been repurposed for treating COVID-19, and they could alleviate some COVID-19 symptoms such as cytokine storm and inflammation instead of killing the virus directly.
Nonetheless, two antibody candidates under clinical trials are targeting the S protein and block the SARS-CoV-2 entry into human cells. One of them is LY3819253, developed by Eli Lilly and Company in the United States, which is in phase II clinical trials and already highlighted in The Scientist (90). The other is JS016,developed by Junshi Biosciences in China (60),which is currently in phase I clinical trials.
7. MATERIAL AND METHODS
7.1. Sequences and Structures
All of the sequences and 3D structures that we used were downloaded from the PDB (https://www.rcsb.org). The sequences were extracted from FASTA files, while 3D structures were obtained from PDB files.
3D alignments and graphs were created using PyMOL (15).2D sequence alignments were calculatedbyclustalw(https://www.genome.jp/tools-bin/clustalw)(64),and2Dalignmentgraphs were generated by Jalview (78).
7.2. Topology-Based Network Tree Model for Protein–Protein Interaction Binding Affinity Changes Upon Mutation
In this section, we describe the TopNetTree model, which predicts binding affinity changes following mutation ΔΔG for PPIs (77). This method is based on structures regarded as topological features and a supervised machine learning model, gradient boosting tree (GBT), and convolution neural network (CNN). In Figure 10, the train and predicting processes are elucidated, involving two major modules: topology-based feature generation and CNN-assisted GBT model. In feature generation, the element-specific and site-specific persistent homology is the key mathematical technique that can simplify the structural complexity of protein–protein complexes and translate the biological information into topological invariants. The first step of the TopNetTree process uses a CNN as a preprocessing model to extract topological features. Assembling CNN-pretrained features and other features, the GBT model predicts PPI binding affinity changes. Other features such as chemical and physical information that have not been absorbed into topological features can improve the proposed model’s predicting ability (for more details, see 77).
Figure 10.
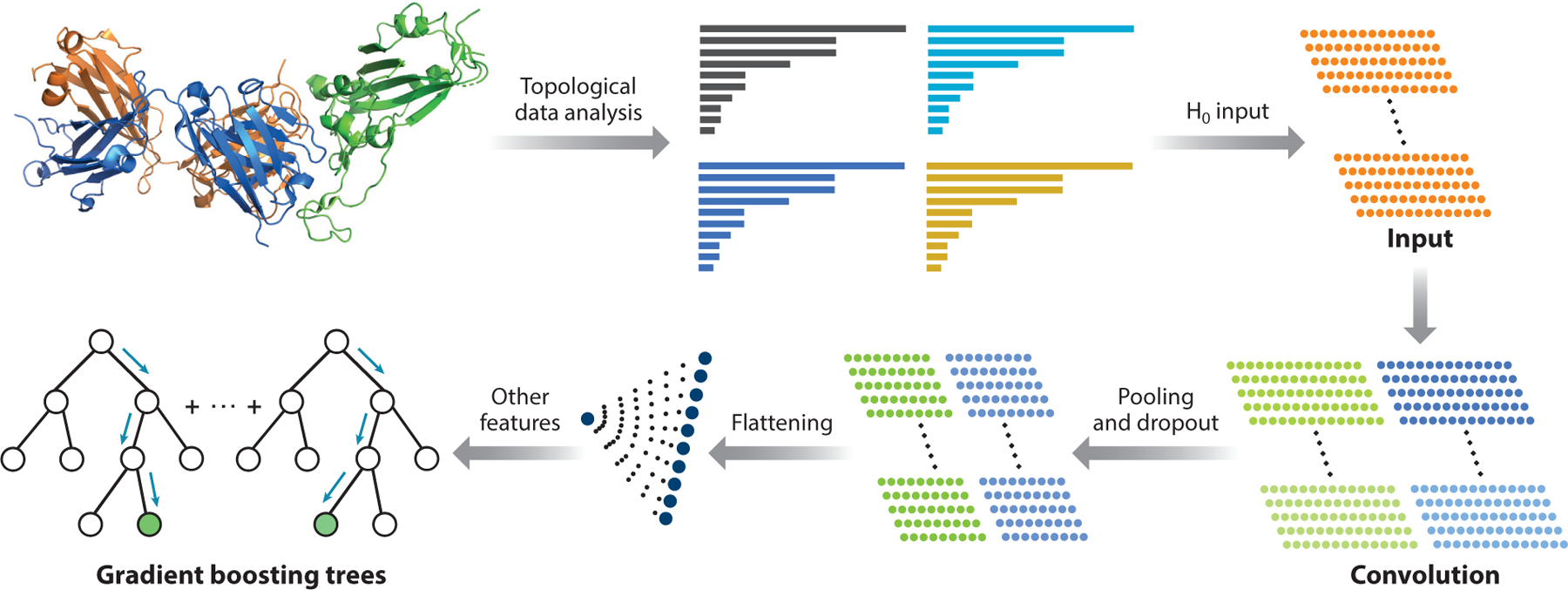
An illustration of the TopNetTree model (77). Protein structure shown in the plot is an antibody (PDB 7BZ5) (blue indicates heavy chain, orange indicates light chain) and SARS-CoV-2 S protein RBD (green) complex. H0 are the 0-dimensional topological input features for the machine learning model. Abbreviations: PDB, Protein Data Bank; RBD, receptor-binding domain; S, spike; SARS-CoV, severe acute respiratory syndrome coronavirus.
7.2.1. Topology-based feature generation of protein–protein interactions.
Persistence homology is the key mathematical theory behind topology-based feature generation. As a subtopic of algebraic topology, persistence homology is built on the simplicial complex and filtration on discrete data sets under various settings. For example, the set of atoms in PPIs forms the discrete data set. When building the constructions, a variety of simplicial complexes are built on point clouds such that the Vietoris-Rips (VR) complex and alpha complex are widely used (18); this applied to our approach. After a simplicial complex is set up, the topological invariants of the point-cloud data set can be identified and are enumerated by counting the numbers referred to as Betti-0 (H0), Betti-1 (H1), and Betti-2 (H2) for components, rings, and cavities of the data set, respectively. Obviously, the complex protein–protein structure is simplified to its geometric and topological characteristics for data features, while redundant and uninformative features or calculations are fully abandoned. Moreover, making filtration dependent on the simplicial complex turns the 3D point-cloud data set of PPIs into topological bar codes, which record the birth and death of each topological invariant. The topological features simplify the PPI complex in many directions. However, it is also essential to have better construction to reflect different biological or chemical properties. Various subsets for PPI complex constructions are defined as follows:
: atoms of the mutation site;
: atoms in the neighborhood of the mutation site within a cut-off distance rÅ;
: antibody atoms within rÅ of any atoms of antigen;
: antigen atoms within rÅ of any atoms of antibody; and
: atoms in the system that has atoms of element type E
Therefore, the distance matrix is defined based on atom sets such that it excludes the interactions in the same set. For interactions between atoms ai and aj in set and/or set , the modified distance is defined as
| 1. |
where De(ai, aj) is the Euclidian distance between ai and aj. Then, the persistence homology can be constructed to be element and site specific.
If one has atomic coordinates, then one can carry out their topological analysis and analysis of their properties via simplices and simplicial complexes. A set of k + 1 affinely independent points, v0, v1, v2, …, vk in , is a k-simplex denoted σi, such that a 0-, 1-, 2-, or 3-simplex in geometry representation is a vertex, an edge, a triangle, or a tetrahedron, respectively. The finite collection of the simplex is a simplicial complex K = {σi}, which is true if a subset (also called a face) τ of a k-simplex σi of K is also in K, τ ⊆ σi and σi ∊ K imply τ ∊ K, and the nonempty intersection of any two simplices in K is a face of both. Furthermore, a k-chain is a finite formal sum of all simplices in K, , where αi is a coefficient in , and p is a chosen prime number. The set of all k-chains of the simplicial complex K equipped with an algebraic field forms an Abelian group .
A boundary operator ∂k : Ck → Ck − 1 for a k-simplex σk is homomorphism defined as
where is a (k − 1)-simplex excluding vi from the vertex set. An important property of the boundary operator, ∂k ‒ 1 ∂k = Ø, follows from the fact that boundaries are boundaryless. Moreover, the kernel of the boundary operator is Zk = ker∂k = {c ∊ Ck|∂kc = ∅}, whose elements are called k-cycles; the kth boundary group is the image of ∂k + 1 denoted as . The algebraic construction to connect a sequence of complexes by boundary maps is called a chain complex:
and the kth homology group is the quotient group defined by Hk = Zk/Bk. Obviously, boundary operators imply Bk ⊆ Zk ⊆ Ck. The Betti numbers are defined by the number of basis in kth homology group Hk, which counts k-dimensional holes. For example, Betti-0, β0 = rank(H0) reflects the number of connected components; Betti-1, β1 = rank(H1) reflects the number of loops; and Betti-2, β2 = rank(H2) reveals the number of voids or cavities. Together, the set of Betti numbers {β0, β1, β2, …} indicates the intrinsic topological property of a system. Computational boundary operators directly work on the distance matrices generated on different atom groups, and the Betti number can be calculated by counting the number of zero eigenvalues of corresponding boundary operators.
The model is interested in the evolution of a simplicial complex and in tracking topological characteristics that vary as the simplicial complex changes such that each object (the atomic set of a PPI complex) can be classified and represented as a machine learning feature. In persistent homology, a filtration of a topology space Kisa nested sequence ofsubspaces {Kt}t = 0, …, m of K such that . Considering the complex group in this sequence, we can have a sequence of chain complexes by homomorphisms, , and a corresponding homology sequence, . The p-persistent kth homology group of Kt is defined as , where . Thus, the homology group reveals that the homology classes of Kt persist until Kt+p. In the filtration process, the persistent homology bar codes recording the birth and death of topological invariants can be generated along the spacial changing of radius on the point-cloud data set. The machine learning feature vectors, as a consequence, can be constructed from these sets of filtration bar code intervals.
The filtration parameter interval is discretized into bins, which can model the behavior of bar codes in each bin (6). Thus, these bins are packaged as features for advanced machine learning algorithms directly. Then, the number of persistence intervals is counted for each bin to record birth events and death events. Three feature vectors (H0, H1, and H2) are generated for each topological bar code for the machine learning method. The Betti-0 (H0) bar code is obtained from the VR filtration,and the Betti-1 (H1) and Betti-2 (H2) bar codes are obtained from alpha complex filtration, where Betti-1 and Betti-2 are sparser and more stable than the Betti-0 bar codes. Thus, the Betti-0 bar code is incorporated into CNN models, and the Betti-1 and Betti-2 bar codes are for GBT training. Intuitively, features generated by binned bar code vectorization can reflect the structure of the protein–protein complex and its biological and chemical properties, such as the strength of atom bonds or van der Waals interactions. Meanwhile, the statistics of bar lengths, birth values, and death values, such as maximum, minimum, mean, etc., can be set as features for the machine learning process.
7.2.2. Machine learning models.
Predicting the binding affinity changes following mutations for PPIs is very challenging due to the complex data set and different 3D structures. To overcome this challenge, one can use a hybrid machine learning algorithm that integrates a CNN and GBT to predict the binding affinity changes. The vectorized H0 bar code feature is converted into concise features by the CNN method. Then, CNN-trained features are combined with the rest features as the full feature set to train a GBT module for a robust predictor with effective control of overfitting.
CNN is considered to be the most successful architecture as a class of deep neural networks. CNN is a regularized case of a multilayer connected neural network. Each neuron is connected locally to the next convolution layer neurons, and the weights are shared in different locations. In TopNetTree, CNN is an intermediate model that applies vectorized H0 features into a higher-level abstract feature for the GBT method. GBT is an ensemble method that builds a powerful module for regression and classification problems as weak learners. The method sums the weak learners to eliminate the overall error based on the assumption that each learner is likely to make different mistakes. According to the current prediction error on the training data set, the ensemble method is built on a decision tree structure. GBT with topological features (TopGBT) is relatively robust against hyperparameter tuning and overfitting and is suitable for a moderate number of features. Our work uses the GBT package provided by scikit-learn (v. 0.23.0) (48).
Finally, TopNetTree follows a process (Figure 10) in which a supervised CNN model is trained for extracting high-level features from H0 barcodes, where the PPI ΔΔG is a label. Then, the flattened layer neural outputs of CNN are ranked according to their importance in a GBT model. Based on the importance, the whole set of features consists of an ordered subset of CNN-trained features, high-dimensional topological bar codes, H1 and H2, and auxiliary features for the final GBT model. As for the parameters, an optimal parameter setting with the best result of the 10-fold evaluation is selected from the experiments with different parameter settings.
7.2.3. Cross-validation of TopNetTree.
The TopNetTree method is trained on the SKEMPI 2.0 data set (35), which has 4,169 variants in 319 different complexes. A set S8338 with 8,338 variants was derived from the SKEMPI 2.0 data set by setting the reverse mutation energy changes to the negative values of the original energy changes. To address the reliability of the TopNetTree method, we performed a 10-fold cross-validation on the SKEMPI 2.0 data set with the Pearson correlation coefficients Rp, Kendall’s τ, and the root mean square error (RMSE) being 0.98, 0.89, and 0.37 kcal/mol, respectively. As shown in Table 5, these metrics are based on the average of 10 10-fold cross-validations, which indicates that TopNetTree is well trained. The performance test of 10-fold cross-validation on a data set gives Rp = 0.84, τ = 0.60, and RMSE =1.06 kcal/mol, which is the same level of accuracy as the best results in the literature (77).
Table 5.
Ten-fold cross-validation of the TopNetTree on the SKEMPI 2.0 data set
| Rp | τ | RMSE (kcal/mol) | Rp | τ | RMSE (kcal/mol) | ||
|---|---|---|---|---|---|---|---|
| Fold 1 (Train) | 0.981 | 0.884 | 0.366 | Fold 6 (Train) | 0.983 | 0.904 | 0.353 |
| Fold 1 (Test) | 0.835 | 0.595 | 1.065 | Fold 6 (Test) | 0.836 | 0.594 | 1.064 |
| Fold 2 (Train) | 0.982 | 0.902 | 0.360 | Fold 7 (Train) | 0.983 | 0.904 | 0.356 |
| Fold 2 (Test) | 0.839 | 0.600 | 1.061 | Fold 7 (Test) | 0.838 | 0.594 | 1.060 |
| Fold 3 (Train) | 0.982 | 0.887 | 0.366 | Fold 8 (Train) | 0.979 | 0.878 | 0.392 |
| Fold 3 (Test) | 0.837 | 0.595 | 1.068 | Fold 8 (Test) | 0.840 | 0.596 | 1.061 |
| Fold 4 (Train) | 0.981 | 0.880 | 0.369 | Fold 9 (Train) | 0.982 | 0.902 | 0.362 |
| Fold 4 (Test) | 0.841 | 0.596 | 1.059 | Fold 9 (Test) | 0.838 | 0.596 | 1.069 |
| Fold 5 (Train) | 0.982 | 0.906 | 0.365 | Fold 10 (Train) | 0.982 | 0.886 | 0.367 |
| Fold 5 (Test) | 0.839 | 0.594 | 1.062 | Fold 10 (Test) | 0.835 | 0.596 | 1.064 |
| Average (Train) | 0.982 | 0.893 | 0.366 | ||||
| Average (Test) | 0.838 | 0.596 | 1.063 |
Abbreviation: RMSE, root mean square error.
7.3. Graph Network Analysis
Graph networks represent interactions between pairs of units in biomolecular systems. The quantity of unique characteristics of the networks can be measured for descriptions and comparisons of different networks. When the PPIs are considered as networks, each descriptor evaluates the network properties and measures how proteins connect. For instance, a fixed domain of S-protein RBD and antibodies forms a network, where residues from 320 to 518 on SARS-CoV and residues from 329 to 530 on SARS-CoV-2 are considered in terms of Cα atoms. As discussed above, interaction subsets or similar subsets for Cα of each amino acid are defined as follows:
: antibody Cα atoms within r Å of any Cα of the antigen, where r = ∞ is for all Cα atoms on the antibody, and
: antigen Cα atoms within r Å of any Cα of the antibody, where r = ∞ is for all Cα atoms on the antigen.
With these definitions, network descriptors are defined below.
7.3.1. Flexibility–rigidity index.
The FRI is a great tool to illustrate the elasticity between atoms for molecular interaction prediction (45, 86). The molecular rigidity index is defined as a summation of all the atomic rigidity index μη,i as
| 2. |
where ri are atom positions; NAB and NAG are the numbers of atoms of antibody and antigen, respectively; and r =∞ for all Cα atoms such that and . The molecular rigidity index is used to describe the behavior of the dynamics and elastostatics of the biomolecular elasticity, where η controls the influence range between atoms. In PPIs, the elasticity between antibody and antigen, especially long-range impacts, is studied by calculating the FRI of the network consisting of Cα atoms.
7.3.2. Degree heterogeneity.
The degree heterogeneity is an index that evaluates the heterogeneity of a network on different distribution (19). The degree distribution ki is the number of ith nodes that have ki connections to other nodes. Therefore, the degree heterogeneity reflects the distributions of a network on different impacts, which is defined as
| 3. |
In this case, Ne represents the number of edges. In our case, we study two networks consisting of all Cα atoms in , where one network consists of Cα atoms from the SARS-CoV RBD, and the other consists of Cα atoms from the SARS-CoV-2 RBD. The degree heterogeneity illustrates the impacts of ACE2 or antibodies on these networks.
The rest descriptors are built on the network consisting of Cα atoms from and .
7.3.3. Edge density.
The edge density is defined as
| 4. |
where Ne is the number of edges, and Nv is the number of vertices for Cα atoms from and . The edge density is also called the average degree centrality. For a complete network in which each every pair of network vertices is connected, the edge density is equal to one. A noncomplete network has an edge density smaller than one. With the same number of residues in the RBD for each PPI, a higher edge density stands for a firm connection between RBD and ACE2 or antibodies.
7.3.4. Average path length.
The characteristic path length indicates the typical separation between two vertices in the network. It was used to study infectious disease spread in the so-called small-world networks (79). The shortest path distance d(i,j) is defined as the shortest path between the corresponding pairs of vertices i and j. In PPIs, the path length between two atoms reflects how ACE2 or antibodies connect to the RBD. The average path length is defined as
| 5. |
for Cα atoms from and (10). In this case, Nv represents the number of vertices.
7.3.5. Average betweenness centrality.
The concept of betweenness centrality illustrates communications in a network (26). The betweenness centrality of a vertex vk is given as
| 6. |
and the average betweenness centrality is given as
| 7. |
where gij(vk) is defined as the number of geodesics linking vertex vi and vj that pass vk, and gij considers all the paths between vi and vj. Nv indicates the number of vertices.
7.3.6. Average eigencentrality.
The eigenvector centrality represents the elements of the eigenvector Vmax with respect to the largest eigenvalue of the adjacency matrix A(3). It describes the probability of starting at and returning to the same point for infinite-length walks. Thus, the average eigenvector centrality is
| 8. |
where ei are elements of Vmax. The average eigenvalue centrality stands for the average impact spread of vertices beyond its neighborhood for an infinite walk.
7.3.7. Average subgraph centrality.
The following descriptors are built on the exponential of the adjacency matrix, E = eA. The average subgraph centrality is defined as
| 9. |
which indicates that the vertex participates in all subgraphs of the graphs (20, 23). In this case, E(k, k) indicates the element located at the kth row and kth column. Subgraph centrality is the summation of weighted closed walks of all lengths starting and ending at same node. The long path length has a small contribution.
7.3.8. Average communicability and average communicability angle.
The final two descriptors are average communicability, given as
| 10. |
and average communicability angle, given as
| 11. |
where , and E is the exponential of the adjacency matrix. The average communicability measures how much two vertices can communicate by using all of the possible paths in the network, where the shorter path has more weight (20, 21). The average communicability angle evaluates the efficiency of two vertices passing impacts to each other in the network with all possible paths (20, 22).
8. CONCLUSION
Developing effective therapies for combating COVID-19 caused by SARS-CoV-2 has become a vital task for human health and the world economy. Although designing new anti-SARS-CoV-2 drugs is of paramount importance, traditional drug discovery might take many years. Effective vaccines typically require more than a year to develop. Therefore, a more efficient strategy in fighting COVID-19 is to look for antibody therapies, which is a relatively easier technique compared to the development of small-molecular drugs or vaccines. The search for possible antibody drugs has attracted increasing attention in recent months. Moreover, CDRs, which are located in the tip of the antibody, determine the specificity of antibodies and make antibody therapies a promising way to fight COVID-19. Above, we analyze the structure, function, and therapeutic potential of seven SARS-CoV-2-neutralizing antibody candidates that have 3D structures available in the PDB. In a comparative study, we also review five antibody 3D structures associated with SARS-CoV, as well as two ACE2 3D structures, one associated with SARS-CoV-2 and the other with SARS-CoV. All antibody and ACE2 structures form complexes with viral S proteins. The multiple-order-of-magnitude discrepancies in reported experimental binding affinities for these complexes motivates us to carry out a systematic computational analysis of these 14 complexes. Using computational topology, machine learning, and wide class network models, we put all of the complexes on an equal footing to evaluate binding and interactions. Additionally, we evaluate the repositioning potentials of five SARS-CoV antibodies for treating COVID-19 by predicting their binding affinity changes following the mutations from SARS-CoV to SARS-CoV-2 at the S-protein RBD. Finally, we summarize all of the currently ongoing clinical antibody trails for COVID-19, which have many targets, including the S protein. In a nutshell, we provide a review of existing antibody therapies for COVID-19 and introduce many theoretical models to rank the potency and analyze the properties of antibodies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health grant GM126189; National Science Foundation grants DMS-1721024, DMS-1761320, and IIS1900473; the Michigan Economic Development Corporation; George Mason University award PD45722; Bristol-Myers Squibb; and Pfizer. The authors thank the IBM TJ Watson Research Center, the COVID-19 High Performance Computing Consortium, NVIDIA, and the Michigan State University High Performance Computing Center for computational assistance.
Footnotes
SUPPORTING MATERIAL
Supporting materials are available in the Supplemental Appendix. Notably, the Supplemental Appendix includes an analysis of antibodies that became available after the acceptance of this review.
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Berggård T, Linse S, James P. 2007. Methods for the detection and analysis of protein–protein interactions. Proteomics 7(16):2833–42 [DOI] [PubMed] [Google Scholar]
- 2.Blum JS, Wearsch PA, Cresswell P.2013.Pathways of antigen processing. Annu. Rev. Immunol 31:443–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonacich P 1987. Power and centrality: a family of measures. Am. J. Sociol 92(5):1170–82 [Google Scholar]
- 4.Borghesi L, Milcarek C. 2006. From B cell to plasma cell. Immunol. Res 36(1–3):27–32 [DOI] [PubMed] [Google Scholar]
- 5.Breedveld F 2000. Therapeutic monoclonal antibodies. Lancet 355(9205):735–40 [DOI] [PubMed] [Google Scholar]
- 6.Cang Z, Mu L, Wei G-W. 2018. Representability of algebraic topology for biomolecules in machine learning based scoring and virtual screening. PLOS Comput. Biol 14(1):e1005929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao X 2020. COVID-19: immunopathology and its implications for therapy. Nat. Rev. Immunol 20(5):269–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao Y, Su B, Guo X, Sun W, Deng Y, et al. 2020. Potent neutralizing antibodies against SARS-CoV-2 identified by high-throughput single-cell sequencing of convalescent patients’ B cells. Cell 182. In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Xiong J, Bao L, Shi Y. 2020. Convalescent plasma as a potential therapy for COVID-19. Lancet Infect. Dis 20(4):398–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chi X, Yan R, Zhang J, Zhang G, Zhang Y, et al. 2020. A neutralizing human antibody binds to the N-terminal domain of the spike protein of SARS-CoV-2. Science In press [DOI] [PMC free article] [PubMed]
- 11.Clark IA. 2007. The advent of the cytokine storm. Immunol. Cell Biol 85(4):271–73 [DOI] [PubMed] [Google Scholar]
- 12.Corti D, Misasi J, Mulangu S, Stanley DA, Kanekiyo M, et al.2016.Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science 351(6279):1339–42 [DOI] [PubMed] [Google Scholar]
- 13.Crotty S 2015. A brief history of T cell help to B cells. Nat. Rev. Immunol 15(3):185–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Vlieger D, Ballegeer M, Rossey I, Schepens B, Saelens X. 2019. Single-domain antibodies and their formatting to combat viral infections. Antibodies 8(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeLano WL. 2002. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr 40(1):82–92 [Google Scholar]
- 16.Diaz M, Casali P. 2002. Somatic immunoglobulin hypermutation. Curr. Opin. Immunol 14(2):235–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dumoulin M, Conrath K, Van Meirhaeghe A, Meersman F, Heremans K, et al. 2002. Single-domain antibody fragments with high conformational stability. Protein Sci 11(3):500–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Edelsbrunner H, Letscher D, Zomorodian A. 2000. Topological persistence and simplification. In Proceedings of the 41st Annual Symposium on Foundations of Computer Science, pp. 454–63. Piscataway, NJ: IEEE [Google Scholar]
- 19.Estrada E 2010. Quantifying network heterogeneity. Phys. Rev. E 82(6):066102. [DOI] [PubMed] [Google Scholar]
- 20.Estrada E 2020. Topological analysis of SARS-CoV-2 main protease. Chaos Interdiscip. J. Nonlinear Sci 30(6):061102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Estrada E, Hatano N. 2008. Communicability in complex networks. Phys. Rev. E 77(3):036111. [DOI] [PubMed] [Google Scholar]
- 22.Estrada E, Hatano N. 2016. Communicability angle and the spatial efficiency of networks. SIAM Rev 58(4):692–715 [Google Scholar]
- 23.Estrada E, Rodriguez-Velazquez JA. 2005. Subgraph centrality in complex networks. Phys. Rev. E 71(5):056103. [DOI] [PubMed] [Google Scholar]
- 24.Fanning LJ, Connor AM, Wu GE. 1996. Development of the immunoglobulin repertoire. Clin. Immunol. Immunopathol 79(1):1–14 [DOI] [PubMed] [Google Scholar]
- 25.Forsman A, Beirnaert E, Aasa-Chapman MM, Hoorelbeke B, Hijazi K, et al. 2008. Llama antibody fragments with cross-subtype human immunodeficiency virus type 1 (HIV-1)-neutralizing properties and high affinity for HIV-1 gp120. J. Virol 82(24):12069–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Freeman LC. 1978. Centrality in social networks conceptual clarification. Soc. Netw 1(3):215–39 [Google Scholar]
- 27.Govaert J, Pellis M, Deschacht N, Vincke C, Conrath K, et al. 2012. Dual beneficial effect of interloop disulfide bond for single domain antibody fragments. J. Biol. Chem 287(3):1970–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hale G 2006. Therapeutic antibodies: delivering the promise? Adv. Drug Deliv. Rev 58(5–6):633–39 [DOI] [PubMed] [Google Scholar]
- 29.Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hammers C, et al. 1993. Naturally occurring antibodies devoid of light chains. Nature 363(6428):446–48 [DOI] [PubMed] [Google Scholar]
- 30.Hamming I, Timens W, Bulthuis M, Lely A, Navis G, van Goor H. 2004. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus: a first step in understanding SARS pathogenesis. J. Pathol 203(2):631–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanke L, Vidakovics MLP, Sheward D, Das H, Schulte T, et al. 2020. An alpaca nanobody neutralizesSARS-CoV-2 by blocking receptor interaction. bioRxiv 130161. 10.1101/2020.06.02.130161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. 2010. The safety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov 9(4):325–38 [DOI] [PubMed] [Google Scholar]
- 33.Heyman B 1996. Complement and Fc-receptors in regulation of the antibody response. Immunol. Lett 54(2–3):195–99 [DOI] [PubMed] [Google Scholar]
- 34.Hwang WC, Lin Y, Santelli E, Sui J, Jaroszewski L, et al. 2006. Structural basis of neutralization by a human anti-severe acute respiratory syndrome spike protein antibody, 80R. J. Biol. Chem 281(45):34610–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jankauskaitė J, Jiménez-García B, Dapkūnas J, Fernández-Recio J, Moal IH.2019.Skempi 2.0: an updated benchmark of changes in protein–protein binding energy, kinetics and thermodynamics upon mutation. Bioinformatics 35(3):462–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ju B, Zhang Q, Ge J, Wang R, Sun J, et al. 2020. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature In press [DOI] [PubMed] [Google Scholar]
- 37.Kelley B 2020. Developing therapeutic monoclonal antibodies at pandemic pace. Nat. Biotechnol 38(5):540–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Köhler G, Milstein C. 1975. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256(5517):495–97 [DOI] [PubMed] [Google Scholar]
- 39.Lan J, Ge J, Yu J, Shan S, Zhou H, et al. 2020. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581:215–20 [DOI] [PubMed] [Google Scholar]
- 40.Laursen NS, Friesen RH, Zhu X, Jongeneelen M, Blokland S, et al. 2018. Universal protection against influenza infection by a multidomain antibody to influenza hemagglutinin. Science 362(6414):598–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leader B, Baca QJ, Golan DE. 2008.Protein therapeutics: a summary and pharmacological classification. Nat. Rev. Drug Discov 7(1):21–39 [DOI] [PubMed] [Google Scholar]
- 42.Market E, Papavasiliou FN.2003.V(D)J recombination and the evolution of the adaptive immune system. PLOS Biol 1(1):e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mian IS, Bradwell AR, Olson AJ. 1991. Structure, function and properties of antibody binding sites. J. Mol. Biol 217(1):133–51 [DOI] [PubMed] [Google Scholar]
- 44.Murphy K, Weaver C. 2016. Janeway’s Immunobiology New York: Garland Sci. [Google Scholar]
- 45.Nguyen DD, Xia K, Wei G-W.2016.Generalized flexibility-rigidity index. J. Chem. Phys 144(23):234106. [DOI] [PubMed] [Google Scholar]
- 46.Nissim A, Chernajovsky Y. 2008. Historical development of monoclonal antibody therapeutics. In Therapeutic Antibodies, ed. Chernajovsky Y, Nissim A, pp. 3–18. Berlin: Springer; [DOI] [PubMed] [Google Scholar]
- 47.Pak JE, Sharon C, Satkunarajah M, Auperin TC, Cameron CM, et al. 2009. Structural insights into immune recognition of the severe acute respiratory syndrome coronavirus S protein receptor binding domain. J. Mol. Biol 388(4):815–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, et al. 2011. Scikit-learn: machine learning in python. J. Mach. Learn. Res 12:2825–30 [Google Scholar]
- 49.Pinto D, Park Y-J, Beltramello M, Walls AC, Tortorici MA, et al. 2020. Structural and functional analysis of a potent sarbecovirus neutralizing antibody. bioRxiv 023903 10.1101/2020.04.07.023903 [DOI] [Google Scholar]
- 50.Prabakaran P, Gan J, Feng Y, Zhu Z, Choudhry V, et al. 2006. Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J. Biol. Chem 281(23):15829–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Presta LG. 2008. Molecular engineering and design of therapeutic antibodies. Curr. Opin. Immunol 20(4):460–70 [DOI] [PubMed] [Google Scholar]
- 52.Putnam FW, Liu Y, Low T.1979.Primary structure of a human IgA1 immunoglobulin. IV. Streptococcal IgA1 protease, digestion, Fab and Fc fragments, and the complete amino acid sequence of the alpha 1 heavy chain. J. Biol. Chem 254(8):2865–74 [PubMed] [Google Scholar]
- 53.Ravetch JV, Bolland S. 2001. Igg Fc receptors. Annu. Rev. Immunol 19:275–90 [DOI] [PubMed] [Google Scholar]
- 54.Reichert JM, Dewitz MC. 2006. Anti-infective monoclonal antibodies: perils and promise of development. Nat. Rev. Drug Discov 5(3):191–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. 2005. Monoclonal antibody successes in the clinic. Nat. Biotechnol 23(9):1073–78 [DOI] [PubMed] [Google Scholar]
- 56.Rogers TF, Zhao F, Huang D, Beutler N, Burns A, et al. 2020. Rapid isolation of potent SARS-CoV2 neutralizing antibodies and protection in a small animal model. bioRxiv 088674 10.1101/2020.05.11.088674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rotman M, Welling MM, van den Boogaard ML, Moursel LG, van der Graaf LM, et al. 2015. Fusion ofhIgG1-Fc to 111In-anti-amyloid single domain antibody fragment VHH-pa2H prolongs blood residential time in APP/PS1 mice but does not increase brain uptake. Nuclear Med. Biol 42(8):695–702 [DOI] [PubMed] [Google Scholar]
- 58.Scheid JF, Mouquet H, Feldhahn N, Seaman MS, Velinzon K, et al. 2009. Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 458(7238):636–40 [DOI] [PubMed] [Google Scholar]
- 59.Shen C, Wang Z, Zhao F, Yang Y, Li J, et al. 2020. Treatment of 5 critically ill patients with COVID-19 with convalescent plasma. JAMA 323(16):1582–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shi R, Shan C, Duan X, Chen Z, Liu P, et al. 2020. A human neutralizing antibody targets the receptor binding site of SARS-CoV-2. Nature In press [DOI] [PubMed]
- 61.Sui J, Li W, Murakami A, Tamin A, Matthews LJ, et al. 2004. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. PNAS 101(8):2536–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sultana A, Lee JE. 2015. Measuring protein-protein and protein-nucleic acid interactions by biolayer interferometry. Curr. Protoc. Protein Sci 79(1):19–25 [DOI] [PubMed] [Google Scholar]
- 63.Ter Meulen J, Van Den Brink EN, Poon LL, Marissen WE, Leung CS, et al. 2006. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLOS Med 3(7):e237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thompson JD, Gibson TJ, Higgins DG. 2003. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform (1):2–3 [DOI] [PubMed] [Google Scholar]
- 65.Tian X, Li C, Huang A, Xia S, Lu S, et al. 2020. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerging Microbes Infect 9(1):382–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tirado SMC, Yoon K-J. 2003. Antibody-dependent enhancement of virus infection and disease. Viral Immunol 16(1):69–86 [DOI] [PubMed] [Google Scholar]
- 67.Tortorici MA, Veesler D. 2019. Structural insights into coronavirus entry. Adv. Virus Res 105:93–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van der Linden R, Frenken L, De Geus B, Harmsen M, Ruuls R, et al. 1999. Comparison of physical chemical properties of llama VHH antibody fragments and mouse monoclonal antibodies. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol 1431(1):37–46 [DOI] [PubMed] [Google Scholar]
- 69.Waldmann H, Hale G. 2005. Campath: from concept to clinic. Philos. Trans. R. Soc. B 360(1461):1707–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. 2020. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181:281–92.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Walls AC, Tortorici MA, Bosch B-J, Frenz B, Rottier PJ, et al. 2016. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 531(7592):114–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Walls AC, Tortorici MA, Snijder J, Xiong X, Bosch B-J, et al. 2017. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. PNAS 114(42):11157–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Walls AC, Xiong X, Park Y-J, Tortorici MA, Snijder J, et al. 2019.Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell 176(5):1026–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wan J, Xing S, Ding L, Wang Y, Zhu D, et al. 2020. Human IgG neutralizing monoclonal antibodies block SARS-CoV-2 infection. bioRxiv 104117. 10.1101/2020.05.19/104117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang K, Chen W, Zhou Y-S, Lian J-Q, Zhang Z, et al. 2020. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv 988345 10.1101/2020.03.14.988345 [DOI] [Google Scholar]
- 76.Wang L, Shi W, Chappell JD, Joyce MG, Zhang Y, et al. 2018. Importance of neutralizing monoclonal antibodies targeting multiple antigenic sites on the Middle East respiratory syndrome coronavirus spike glycoprotein to avoid neutralization escape. J. Virol 92(10):e02002–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang M, Cang Z, Wei G-W. 2020. A topology-based network tree for the prediction of protein–protein binding affinity changes following mutation. Nat. Mach. Intel 2(2):116–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2: a multiple sequence alignment editor and analysis workbench. Bioinformatics 25(9):1189–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Watts DJ, Strogatz SH. 1998. Collective dynamics of “small-world” networks. Nature 393(6684):440–42 [DOI] [PubMed] [Google Scholar]
- 80.Woof JM, Burton DR. 2004. Human antibody–Fc receptor interactions illuminated by crystal structures. Nat. Rev. Immunol 4(2):89–99 [DOI] [PubMed] [Google Scholar]
- 81.Wrapp D, De Vlieger D, Corbett KS, Torres GM, Wang N, et al. 2020. Structural basis for potent neutralization of betacoronaviruses by single-domain camelid antibodies. Cell 181:1004–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh C-L, et al. 2020. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367(6483):1260–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu C, Liu Y, Yang Y, Zhang P, Zhong W, et al. 2020. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 10:766–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu F, Zhao S, Yu B, Chen Y-M, Wang W, et al. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579(7798):265–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wu Y, Wang F, Shen C, Peng W, Li D, et al. 2020. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 368:1274–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xia K, Opron K, Wei G-W. 2013. Multiscale multiphysics and multidomain models: flexibility and rigidity. J. Chem. Phys 139(19):194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone A. 2016. Prodigy: a web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 32(23):3676–78 [DOI] [PubMed] [Google Scholar]
- 88.Yuan M, Wu NC, Zhu X, Lee C-CD, So RT, et al. 2020. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 368(6491):630–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, et al. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zimmer K 2020.First antibody trial launched in COVID-19 patients. The Scientist, June 2. https://www.the-scientist.com/news-opinion/first-antibody-trial-launched-in-covid-19-patients-67604 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.