

ABSTRACT
Antibodies against coronavirus spike protein potently protect against infection and disease, but whether such protection can be extended to variant coronaviruses is unclear. This is exemplified by a set of iconic and well-characterized monoclonal antibodies developed after the 2003 SARS outbreak, including mAbs m396, CR3022, CR3014 and 80R, which potently neutralize SARS-CoV-1, but not SARS-CoV-2. Here, we explore antibody engineering strategies to change and broaden their specificity, enabling nanomolar binding and potent neutralization of SARS-CoV-2. Intriguingly, while many of the matured clones maintained specificity of the parental antibody, new specificities were also observed, which was further confirmed by X-ray crystallography and cryo-electron microscopy, indicating that a limited set of VH antibody domains can give rise to variants targeting diverse epitopes, when paired with a diverse VL repertoire. Our findings open up over 15 years of antibody development efforts against SARS-CoV-1 to the SARS-CoV-2 field and outline general principles for the maturation of antibody specificity against emerging viruses.
KEYWORDS: Monoclonal antibodies, antibody maturation, antibody engineering, phage display, SARS-CoV-2, structural studies
Introduction
The emergence of at least three coronaviruses (SARS-CoV-1, SARS-CoV-2 and MERS) in the human population in the past two decades has highlighted the need for rapid and sustained development of prophylactic and therapeutic modalities. Among such modalities, antibody reagents blocking the interaction of the viral spike protein with human receptors (angiotensin converting enzyme 2 (ACE2) in the case of SARS-CoV-1 and CoV-2, and dipeptidyl peptidase 4 (DPP4) in the case of MERS) are the most promising.1–3 Various approaches have been used to identify neutralizing antibodies, including the identification of B cells from convalescent patients,4,5 the immunization of humanized transgenic mice,6 or through the use of in vitro library display approaches against viral spike protein (or more commonly its receptor-binding domain (RBD)).7,8
Here we used a different approach based on the re-engineering and maturation of previously reported antibodies against SARS-CoV-1. Although such antibodies generally do not bind and neutralize SARS-CoV-2, we speculated that the relatively high level of sequence identity of the RBDs of the two viruses (76% amino acid identity9,10) would allow us to shift antibody specificity through limited changes in antibody variable regions.
We focused our attention on four well-characterized monoclonal antibodies (m396,11 CR3022,12 CR301413 and 80 R14) that bind and neutralize SARS-CoV-1 with equilibrium binding (KD) and half maximum inhibitory (IC50) constants in the nanomolar range. Crystal structures have been reported for m396,15 CR302216 and 80R17 in complex with RBD; these reveal binding to a diverse set of epitopes, with m396 and 80 R binding to distinct, but adjacent, epitopes overlapping with the ACE2 binding site (Figure 1(a)). Although no structural information has been reported for CR3014, the antibody has been shown to block ACE2 binding.12 In marked contrast, CR3022 binds to an epitope distant from the ACE2 binding site that is largely conserved between SARS-CoV-1 and SARS-CoV-2.16 Unlike m396, CR3014 and 80R, CR3022 displays residual binding to SARS-CoV-2 RBD; however, it does not detectably neutralize live SARS-CoV-2 virus.16,18
Figure 1.
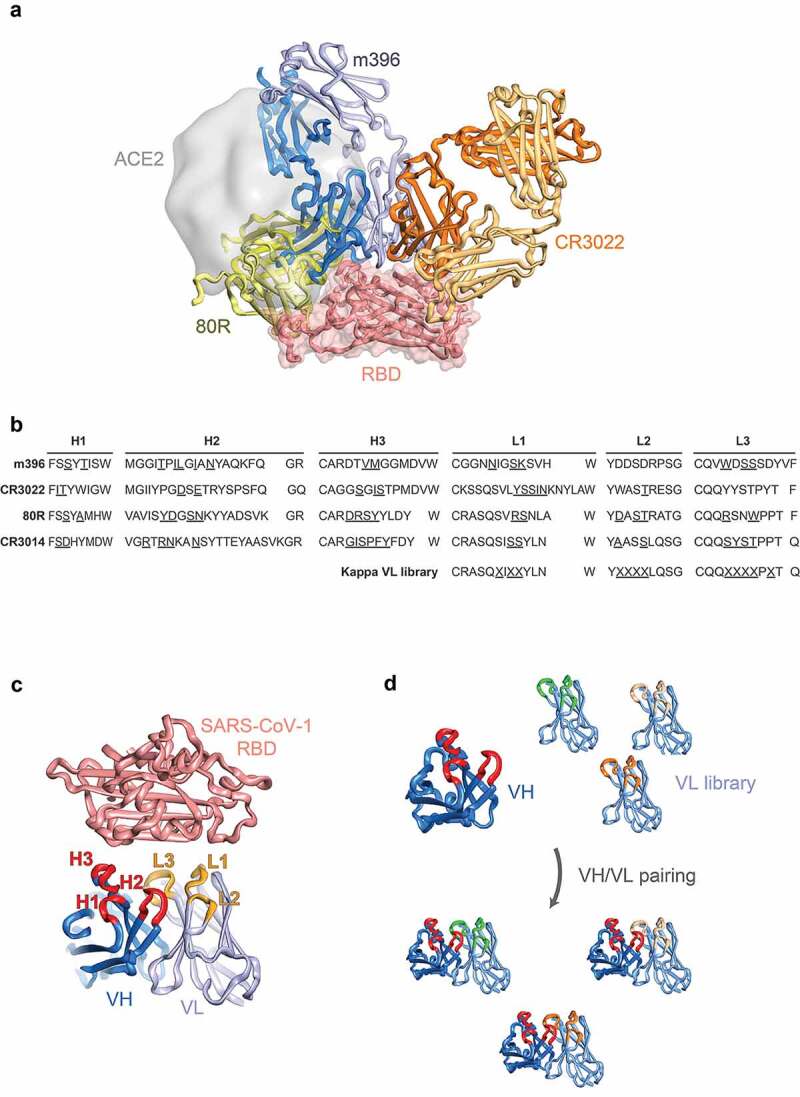
Design of antibody libraries. (a) Structures of SARS-CoV-1 antibodies m396 (Fab format, blue, PDB entry 2dd8), CR3022 (Fab format, orange, PDB entry 6w41) and 80R (scFv format, green, PDB entry 2ghw) superposed on the surface of SARS-CoV-2 RBD (salmon surface and cartoon) with ACE2 highlighted (gray surface, PDB entry 6m0j). (b) CDRs of SARS-CoV-1 antibodies with randomized position underlined. (c) Site-directed mutagenesis strategy with targeted antibody CDRs highlighted (VH in red, VL in orange). (d) Light chain shuffling strategy with variant kappa VL domains highlighted
For the re-engineering strategy, we focused on two well-established in vitro methods for antibody affinity maturation: 1) site-directed mutagenesis of complementarity-determining regions (CDRs) of human variable domains;19 and 2) light chain shuffling20 (Figure 1(b–d)). Library design based on the reported structures of m396, CR3022 and 80R in complex with RBD was used for the construction of site-directed mutagenesis repertoires, with antibody contact residues with antigen targeted for diversification (residues underlined in Figure 1(b)). For the alternative light chain shuffling approach, a previously described highly diverse synthetic antibody library based on a single Vκ1 framework was used.21,22 Both library classes were then selected for binding to SARS-CoV-2 RBD using iterative selections on biotinylated antigen (100 nM to 500 pM range). Using these approaches, we rapidly identified human antibody variants with potent affinity and neutralization potential for SARS-CoV-2.
Results
Generation and selection of SARS-CoV-2 binding antibodies by site-directed mutagenesis
For the design of site-directed mutagenesis libraries, we used previously reported crystal structures of antibodies developed against SARS-CoV-1 in complex with either cognate RBD (80 R – PDB entry 2ghw;17 m396 – PDB entry 2dd815) or SARS-CoV-2 RBD in the case of the cross-specific CR3022 antibody (PDB entry 6w4116). Based on the structures, we selected contact and proximal residues in the CDRs of the variable heavy (VH) and variable light (VL) domains and introduced targeted diversification by Kunkel mutagenesis19 (Figure 1(a–c) and Supplementary Table 2; all six CDRs were targeted for CR3014 for which no structural information has been reported). Library construction was carried out in an antibody single-chain variable fragment (scFv) format, resulting in 6.1 x 108, 2.3 x 107, 3.4 × 107 and 5.7 × 107 clones for m396, CR3022, CR3014 and 80R, respectively. We performed four rounds of phage display selection, using decreasing amounts of SARS-CoV-2 RBD for selection (see Methods); this resulted in enrichment of SARS-CoV-2 specific binders for the libraries (with the exception of CR3014), as indicated by polyclonal phage enzyme-linked immunosorbent assay (ELISA) (Figure 2(a)). Screening of individual clones by monoclonal soluble ELISA was performed after round 4, followed by sequencing and cloning of non-redundant variants into an IgG expression vector. After production in Chinese hamster ovary (CHO) cells, monoclonal antibodies were characterized for binding to recombinant RBD by biolayer-interferometry (BLI) and for neutralization of live SARS-CoV-2 virus in Vero E6 cells.
Figure 2.
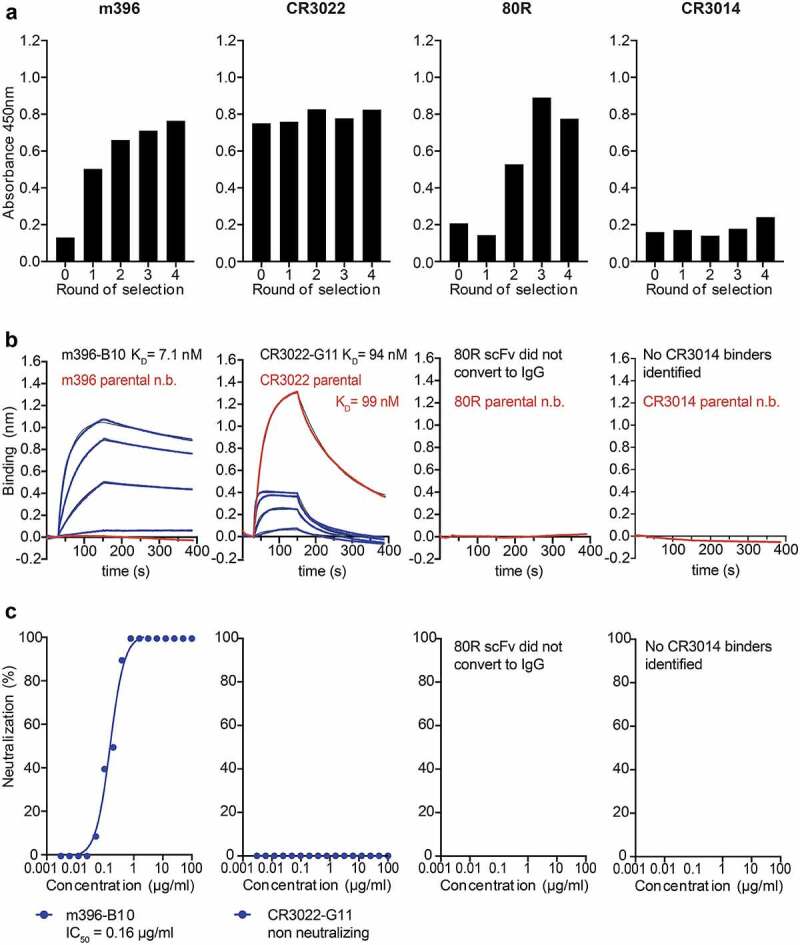
Selection of SARS-CoV-2 specific antibodies from site-directed mutagenesis libraries of SARS-CoV-1 binders. (a) Enrichment of scFv antibody binders by phage display (polyclonal phage ELISA). (b) Biolayer interferometry affinity measurements of soluble SARS-CoV-2 RBD (at 400 nM, 200 nM, 100 nM, 50 nM; highest concentration only shown for parental antibodies) binding to immobilized antibody (see Methods). (c) Neutralization of live SARS-CoV-2 virus in Vero E6 cells (IgG format)
In the case of m396, two variants (designated B10 and C4) were chosen for further characterization, with both antibody variants encoding several mutations in VH and VL (Supplementary Sequences). Both variants displayed high monovalent binding affinity to soluble SARS-CoV-2 RBD with KDs in the low nanomolar range (7 nM in the case of m396-B10 and 13 nM in the case of m396-C4) (Figure 2(b), Supplementary Fig. 1a and Table 1). Both variants also potently neutralized live SARS-CoV-2 virus with IC50s of 160 ng/ml and 340 ng/ml, respectively (Figure 2(c) and Supplementary Fig. 1e). In addition to live virus, m396-B10 also potently neutralized both SARS-CoV-1 and SARS-CoV-2 pseudoparticles (with IC50s of 2.2 and 0.3 μg/ml, respectively) (Supplementary Fig. 1l).
Table 1.
Affinity of monoclonal antibodies (biolayer interferometry)
| Antibody variant | KD (nM) | ka (M−1.s−1) | kd (s−1) | Neutralization IC50 (µg/ml) |
|---|---|---|---|---|
| m396 parental | nd | nd | nd | nd |
| m396-C4 | 13.0 | 8.13 x 104 | 1.06 x 10−3 | 0.34 |
| m396-B10 | 7.1 | 1.01 x 105 | 7.18 x 10−4 | 0.16 |
| CR3022 parental | 99.2 | 7.19 x 104 | 7.13 x 10−3 | nd |
| CR3022-B11 | 131.0 | 1.02 x 105 | 1.33 x 10−2 | nd |
| CR3022-G11 | 93.6 | 2.41 x 105 | 2.26 x 10−2 | nd |
| CR3022-B6 | 261.0 | 7.62 x 104 | 1.99 x 10−2 | 4.43 |
| CR3022-M | 188.0 | 7.94 x 104 | 1.49 x 10−2 | 0.35 |
| 80R parental | nd | nd | nd | nd |
| 80R-A2 | 61.0 | 6.58 x 104 | 4.01 x 10−3 | 17.83 |
| CR3014 parental | nd | nd | nd | nd |
| CR3014-D1 | 50.8 | 7.95 x 104 | 4.04 x 10−3 | >100 |
| CR3014-C8 | 61.1 | 5.84 x 104 | 3.57 x 10−3 | nd |
nd: no binding/neutralization detected
The B10 variant of m396 was further selected for structural characterization by crystallography in an antigen-binding fragment (Fab) format, both in isolation and in complex with SARS-CoV-2 RBD. Crystals were obtained for the unliganded m396-B10 Fab, which diffracted to 2.3 Å (Supplementary Table 3). Although no crystals were obtained for m396-B10 in complex with SARS-CoV-2 RBD, analysis of the structure of the m396 parent bound to SARS-CoV-1 RBD (PDB entry 2dd8 – Supplementary Fig. 2a) 15 reveals that the bulk of the contact surface is contributed by heavy chain interactions (517 Å2 buried surface vs 370 Å2 for the light chain) in which CDR H1 and H2 line one side of a cleft, whilst H3 lines the other side, into which a loop of SARS-CoV-1 RBD projects (residues 484–492 SARS-CoV-1 numbering, residues 498–506 SARS-CoV-2 numbering). The m396-B10 clone contains several heavy chain CDR mutations relative to the parental m396 antibody: two in H1, four in H2, and two in H3 (Supplementary Sequences). Although the overall RBD fold is conserved between SARS-CoV-1 and SARS-CoV-2 (Supplementary Fig. 2, colored in yellow and salmon, respectively), the loop bound by the m396 heavy chain cleft comprises a local divergence hotspot containing multiple substitutions: Y498Q, T499P, T501N, I503V (all SARS-CoV-2 numbering), considerably more divergent than the overall RBD. The crystal structure of the m396-B10 Fab described here lacks electron density at most of these CDR positions, indicating conformational plasticity in the unliganded state. In contrast to the heavy chain, light chain residues form more limited contacts in the m396 parental complex, with CDR L1 and L3 contacting a surface with considerably greater conservation between SARS-CoV-1 and SARS-CoV-2. A total of two mutations were selected in the m396-B10 L3 region (Supplementary Sequences), which contact the RBD in the parental m396 complex, with no mutations observed in the L1 and L2 regions.
To further define the epitope of m396-B10, we carried out epitope binning by BLI, which indicated competition with recombinant ACE2 (Supplementary Fig. 3a). In addition, we generated a triple mutant within the ACE2 binding site of SARS-CoV-2 RBD (comprising T500A, N501A and Y505A, SARS-CoV-2 numbering; Figure surface a), predicted to interfere with parental m396 binding. Mutation of this region in the RBD resulted in the complete loss of binding of m396-B10 (as well as for m396-C4) (Supplementary Fig. 3b), suggesting that these variants bind to an epitope within the ACE2 binding site, as previously demonstrated for the parental m396 SARS-CoV-1 RBD interaction.11
In addition to the m396 variants, we selected two variants of CR3022 for further characterization (clones G11 and B11). When expressed in an IgG format both antibodies displayed similar KDs for SARS-CoV-2 RBD as the parental CR3022 antibody: 94 nM for B11 and 131 nM for G11, respectively, compared to 99 nM for CR3022 (Figure 2(b), Table 1 and Supplementary Fig. 1b). However, both the parental CR3022 IgG, as well as the G11 and B11 variants, did not detectably neutralize SARS-CoV-2 virus (Figure 2(c) and Supplementary Fig. 1f-g).
In the case of the 80 R selection, polyclonal ELISA indicated the selection of binders, which could also be detected by soluble ELISA in scFv format (Supplementary Fig. 4). Three of the selected clones, designated C9, D1 and D10 were converted into an IgG format, but, as observed for prior scFv to IgG conversions,23 binding was lost upon format conversion and the clones did not display detectable neutralization activity (Figure 2(b, c) and Supplementary Fig. 1h-j).
In contrast to the m396, CR3022 and 80 R phage display selections, no enrichment was observed for the selection of the CR3014 site-directed mutagenesis library (Figure 2(a)).
Generation and selection of SARS-CoV-2 binding antibodies by light chain shuffling
In addition to the site-directed mutagenesis approach described above, we investigated light chain shuffling as a strategy for shifting the specificity of antibodies from SARS-CoV-1 toward SARS-CoV-2.20 We used splice overlap extension PCR24 to pair DNA encoding variable heavy domains of each of the four antibodies analyzed here (m396, CR3022, CR3014 and 80 R) with a kappa light chain library on phage in an scFv format (Figure 1(d)). The synthetic light chain library used is based on the human Vκ1 framework, with human natural diversity introduced at the CDR L1, L2 and L3 positions.21 After ligation and electroporation into E. coli TG1, light chain shuffled libraries of 5 x 107, 1 x 108, 4 × 107 and 1 × 108 clones were obtained for m396, CR3022, CR3014 and 80 R, respectively. Three rounds of phage display selection were performed using decreasing amounts of SARS-CoV-2 RBD antigen for selection (see Methods); this resulted in enrichment of binders for all of the libraries except m396, as indicated by polyclonal phage ELISA (Figure 3(a)).
Figure 3.
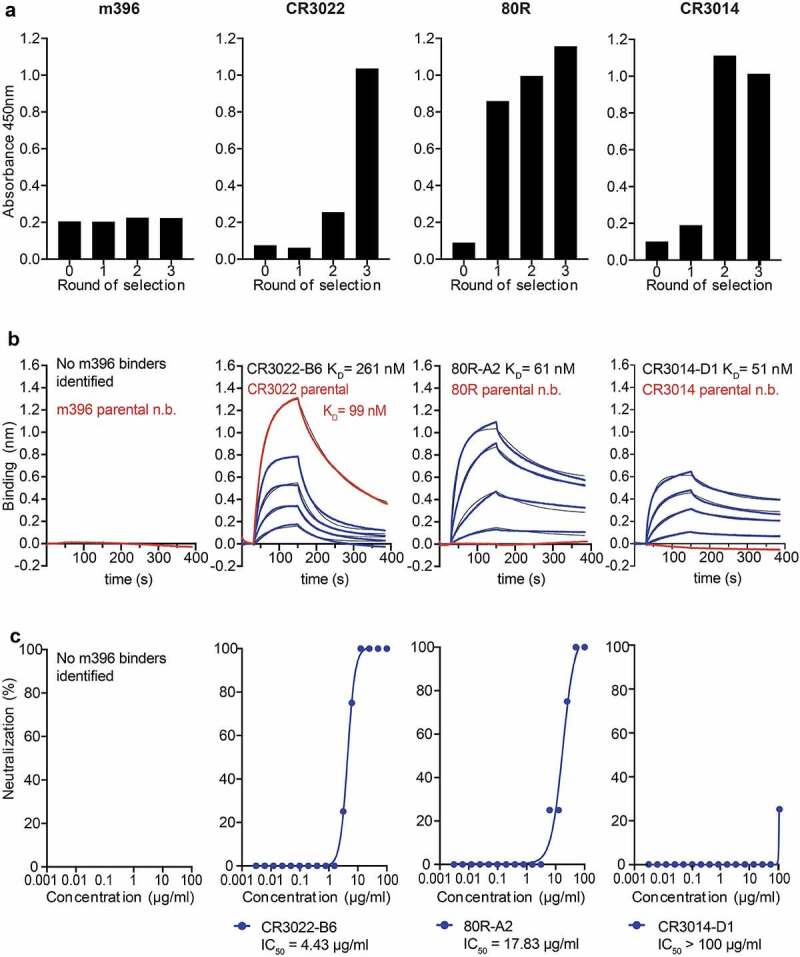
Selection of SARS-CoV-2 specific antibodies from light chain shuffled libraries of SARS-CoV-1 binders. (a) Enrichment of scFv antibody binders by phage display (polyclonal phage ELISA). (b) Biolayer interferometry affinity measurements of soluble SARS-CoV-2 RBD (at 400 nM, 200 nM, 100 nM, 50 nM; highest concentration only shown for parental antibodies) binding to immobilized antibody (see Methods). (c) Neutralization of live SARS-CoV-2 virus in Vero E6 cells (IgG format)
In the case of the CR3022 selection, after three rounds, binders were dominated by a single clone designated B6. No additional binders were identified when screening earlier selection rounds. The CR3022-B6 variant was expressed in an IgG format in CHO cells, and further characterized for binding by BLI and for neutralization in Vero E6 cells. These analyses revealed that CR3022-B6 IgG bound to soluble recombinant SARS-CoV-2 RBD with a KD of 290 nM, higher than observed for the parental CR3022 antibody (99 nM) (Figure 3(b) and Table 1). Intriguingly, and unlike parental CR3022, B6 was capable of neutralizing live SARS-CoV-2 virus with an IC50 of 4.4 µg/ml (Figure 3(c)). RBD mutagenesis was used to further characterize the CR3022-B6 epitope by targeting the parental CR3022-RBD interface through a K378S mutation designed to disrupt the interaction (Figure 4(d), surface c). While parental CR3022 binding was abolished through the mutation, CR3022-B6 fully maintained binding affinity (Supplementary Fig. 3b), indicating that the binding mode of the variant may have changed compared to the parent antibody. CR3022-B6 was further improved through affinity maturation by targeting all six CDRs for diversification using Kunkel mutagenesis and off-rate selections on phage using biotinylated SARS-CoV-2 RBD (see Methods). This resulted in an affinity-matured clone, designated CR3022-M, with moderately increased affinity (188 nM vs 290 nM for CR3022-B6) and considerably increased neutralization potential (0.35 µg/ml vs 4.4 µg/ml for CR3022-B6) (Supplementary Fig. 5).
Figure 4.
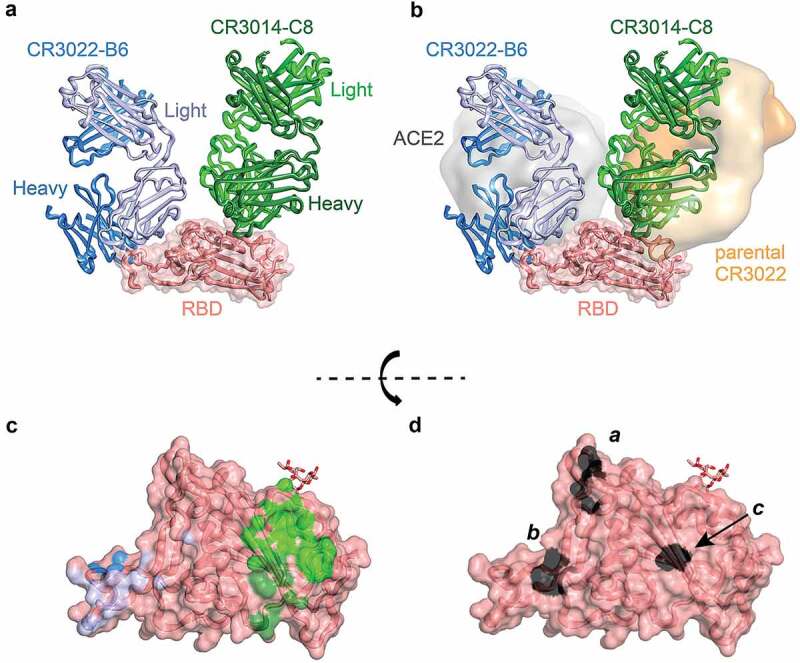
Crystal structure of CR3022-B6 and CR3014-C8 in complex with SARS-CoV-2 RBD. (a) CR3022-B6 (blue) and CR3014-C8 (green) Fabs bound to SARS-CoV-2 RBD (salmon); (b) CR3022-B6 binding to an RBD epitope overlapping with the ACE2 (gray surface, PDB entry 6m0j) interface and different to the parental CR3022 (orange surface, PDB entry 6w41) epitope; CR3014-C8 binding to an epitope distant from the ACE2 interface; (c) Antibody contact surfaces on RBD for CR3022-B6 (blue) and CR3014-C8 (green). The majority of the RBD surface is buried by antibody VL domains (light blue and light green), with more limited VH interactions (dark blue and dark green); (d) RBD surface with residues targeted for epitope mapping in black; surface a (T500, N501 and Y505) ACE2 binding interface; surface b (L455 and F456) CR3022-B6 and CR3014-D1 interface (adjacent to the ACE2 binding site); surface c (K378) parental CR3022 binding interface
In the case of the 80 R selections, later rounds were dominated by a set of variants with closely related CDR sequences. A representative clone (80 R-A2) was expressed in an IgG format and analyzed by BLI and neutralization, which revealed that the 80 R-A2 variant had acquired the capacity to bind RBD and neutralize SARS-CoV-2 with a KD of 61 nM and an IC50 of 17.8 µg/ml (Figure 3(b, c) and Table 1). Parental 80 R has been shown to block the interaction of SARS-CoV-1 RBD with ACE2.14 Similarly, 80 R-A2 binding to SARS-CoV-2 RBD was disrupted through T500A/N501A/Y505A triple substitutions targeting the ACE2 binding site as described above (Figure 4(d) surface A and Supplementary Fig. 3B).
In the case of the CR3014 selections, libraries became dominated by two clones (D1 and C8) after three rounds of selection. When converted into IgG format (and unlike the parental CR3014 IgG, which did not detectably bind to SARS-CoV-2 RBD), both clones bound with mid-nanomolar affinity (KDs of 51 nM and 61 nM, respectively) (Figure 3(b) and Table 1). Similar to the observation for the CR3022-B6/CR3022-parental pair, only CR3014-D1 (but not CR3014-C8) could detectably neutralize live virus (although weakly, with an IC50 > 100 µg/ml) (Figure 3(c)).
Structural characterization of light chain shuffled antibodies
In parallel to the affinity maturation of CR3022-B6, we crystallized this variant in a Fab format, both alone (1.7 Å) and as part of a ternary complex. The complex containing CR3022-B6 Fab, CR3014-C8 Fab and SARS-CoV-2 RBD diffracted to 2.8 Å (Figure 4(a)). Although electron density for the CR3022-B6 Fab component in the ternary complex was weak (reflected in high average B factors – Supplementary Table 3 and Supplementary Fig. 6A for interface and representative electron density), it was evident that the interaction was distant from the previously described binding site of parental CR3022 (Figure 4(b) and Supplementary Fig. 7). This observation was further confirmed by targeting the CR3022-B6 RBD interface observed here with a double mutation in the RBD (L455A/F456A), which completely abolished binding to the variant (but not parental CR3022) (Figure 4(d) surface b and Supplementary Fig. 3b). We also used cryo-electron microscopy (cryo-EM) to position the CR3022-B6 Fab onto the surface of SARS-CoV-2 spike trimer (Figure 5(a) and Supplementary Table 4). In the absence of antibody, classification of the single particles indicated that the majority of spike trimers harbored two RBD domains in the down conformation, with the remaining RBD in the up position (Supplementary Fig. 8).23 When incubated with an equimolar equivalent of CR3022-B6 Fab, ~77% of spike trimer particles were visualized with a Fab attached to RBD in an up conformation (Figure 5(a) and Supplementary Fig. 8) and the remaining ~23% were in the single RBD up conformation. Comparison to the RBD-CR3022-B6 crystallographic complex resulted in good agreement with the cryo-EM map, highlighting binding of the CR3022-B6 Fab in proximity to the RBD ACE2 binding site, but distant from the parental CR3022 binding site (Figures 4(b) and 5(a)).
Figure 5.
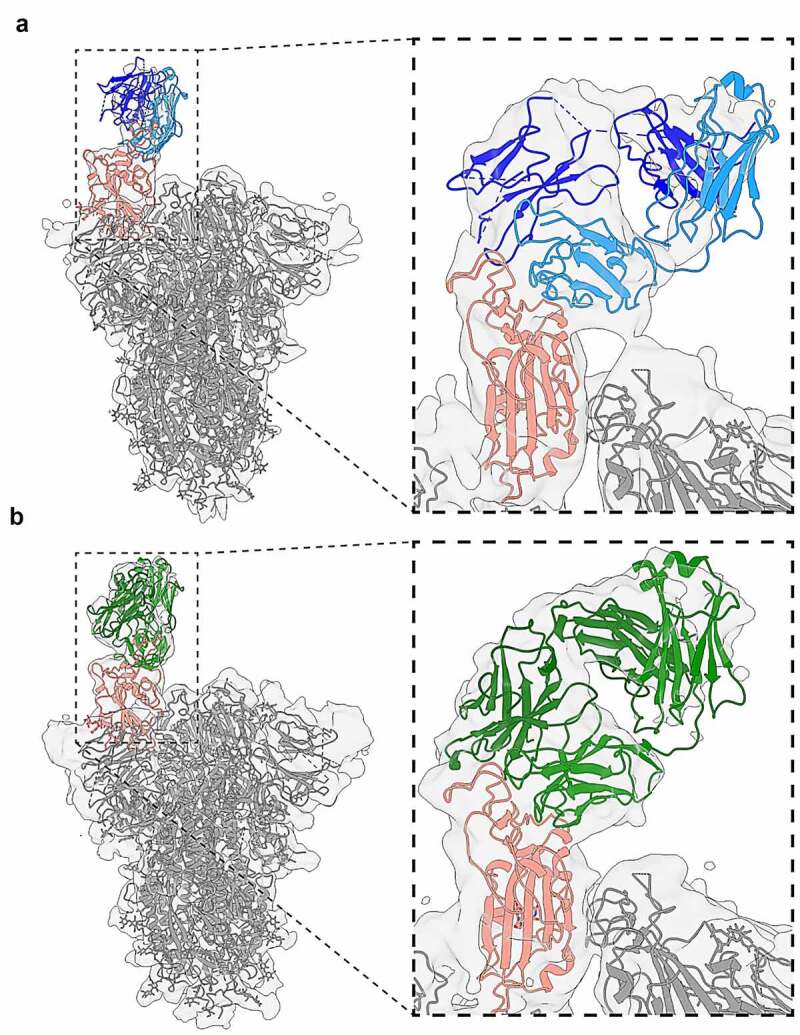
Cryo-electron microscopy of Fab-spike trimer complexes. (a) Binding of CR3022-B6 Fab (blue) to SARS-CoV-2 trimer (gray with RBD highlighted in salmon). A single Fab molecule bound to the RBD domain of a spike protomer in the ‘up’ conformation was resolved. (b) Binding of CR3014-D1 Fab (green) to SARS-CoV-2 trimer (gray with RBD highlighted in salmon). Both antibody Fab bound with similar stoichiometry (with two protomers in the ‘down’ and one in the ‘up’ conformation), and epitope specificity (overlapping with the ACE2 binding interface)
In contrast to CR3022-B6, electron density of the CR3014-C8 Fab component of the ternary complex crystal structure was well defined, highlighting an epitope distant from the ACE2 binding site (Figure 4(a) and Supplementary Fig. 6B for interface and representative electron density). Indeed, the epitope of CR3014-C8 closely resembled that of the CR3022 antibody (Figure 4(b) and Supplementary Fig. 7). Further structural information for a second, but neutralizing, CR3014 variant (CR3014-D1) was obtained by cryo-EM. When incubated with an equimolar equivalent of CR3014-D1 Fab, ~45% of spike particles were visualized with a single Fab attached to an up-conformation RBD domain (Figure 5(b) and Supplementary Fig. 8), with the remaining particles in the single RBD up conformation. Superposition of the CR3022-B6 and CR3014-D1 cryo-EM spike-Fab structures revealed that both Fabs bound to highly similar and overlapping RBD epitopes (Supplementary Fig. 9). These observations were in agreement with mutagenesis experiments, with both CR3022-B6 and CR3014-D1 binding abolished by mutations (L455A/F456A) adjacent to the RBD ACE2 binding site (Figure 4(d), surface b), while CR3014-C8 binding was abolished by a mutation centered on the RBD CR3022 binding site (K378S) (Figure 4(d), surface c, and Supplementary Fig. 3b).
Discussion
Here we describe a rapid and straightforward in vitro strategy for the generation of antibodies that potently bind and neutralize SARS-CoV-2. Using two complementary strategies, site-directed mutagenesis and light chain shuffling, we introduced diversity into the variable domain regions of four well-characterized monoclonal antibodies that had been developed after the 2003 SARS outbreak (Figure 1(a, b)).
From the site-directed mutagenesis libraries, we identified variants of antibodies m396, CR3022 and 80 R that bound to SARS-CoV-2 RBD by soluble ELISA (no binders were obtained for CR3104 selections). A variant of m396, m396-B10 was further characterized by BLI. These experiments revealed that while the parental m396 antibody displayed no detectable binding, m396-B10 bound to SARS-CoV-2 RBD with 7 nM monovalent affinity (Figure 2; compared to 20 nM for the m396-parent/SARS-CoV-1 RBD interaction15). Neutralization of live SARS-CoV-2 virus in Vero E6 cells confirmed potent neutralization, with an IC50 of 160 ng/ml (a neutralization potency highly similar to antibodies with m396-like CDR H3 identified in SARS-CoV-2 patients through next generation sequencing).25 Neutralization was also observed in SARS-CoV-2 and SARS-CoV-1 pseudoparticle assays, indicating potent cross-specificity of m396-B10 (Supplementary Fig. 1l).
In contrast, no detectable increase of equilibrium-binding affinity was observed for variants of CR3022, although two of the analyzed variants displayed increased kinetic association constants for SARS-CoV-2 binding compared to the parental CR3022 antibody. However, none of the CR3022 variants generated by site-directed mutagenesis displayed detectable viral neutralization (Figure 2(c)).
From the kappa light chain shuffled libraries, we identified variants of antibodies CR3022, 80 R and CR3014 that bound to SARS-CoV-2 RBD (no binders were obtained for m396 selections, presumably due to the presence of lambda light chain in the parental antibody). The CR3022 selections were dominated by a single clone, CR3022-B6, which bound SARS-CoV-2 with reduced affinity compared to the parental CR3022 antibody (290 nM compared to 99 nM) (Figure 3(b) and Table 1). Intriguingly, and unlike wild-type CR3022, CR3022-B6 effectively neutralized SARS-CoV-2 live virus with an IC50 of 4.4 µg/ml. This apparent discrepancy between affinity and neutralization potential in otherwise closely related variants was further confirmed through epitope mapping. Mutation of the SARS-CoV-2 RBD CR3022 binding site abolished binding of parent CR3022, but not of CR3022-B6 (Supplementary Fig. 3b). In contrast, CR3022-B6 binding was abolished by mutation of the RBD adjacent to the ACE2 binding site (L455A/F456A, Figure 4(d) surface B), which did not affect binding of CR3022 wild type (Supplementary Fig. 3b). The suggestion that CR3022-B6 binds a different epitope to its parent was confirmed by a crystal structure of CR3022-B6 in complex with SARS-CoV-2 RBD (along with CR3014-C8 Fab in a ternary complex), and through cryo-EM analysis of CR3022-B6 in complex with SARS-CoV-2 trimeric spike. Both structural analyses confirmed binding of CR3022-B6 in proximity to the RBD ACE2 interaction surface, and distant to the original CR3022 binding site, providing a rationale for its observed neutralization activity (Figures 4 and 5(a)).
The observation that a human antibody, in its wild-type and light chain shuffled form, can bind completely different epitopes is intriguing, and in marked contrast to previous observations.26 This observation was not limited to CR3022, with similarly distinct epitopes observed among two variants of the CR3014 antibody (CR3014-C8 and CR3014-D1). Despite the absence of structural information for the parental CR3014 antibody, mutagenesis, crystallography and cryo-EM clearly highlight the considerable difference in epitope specificity for these two light chain shuffled clones (Figures 4 and 5(b)). The difference in specificity also correlates well with neutralization potential: clones binding to a surface proximal to the ACE2 binding site (CR3022-B6 and CR3014-D1) display neutralization activity against live SARS-CoV-2, while clones binding distant to the ACE2 binding site are not detectably neutralizing (parental CR3022 and CR3014-C8).
How are antibodies with identical heavy chain, but different light chains, capable of binding to completely different epitopes? Further inspection of the CR3022-B6 and CR3014-C8 interactions with SARS-CoV-2 RBD, and the relative contributions of their heavy and light chains to the interaction, provide an intriguing insight into this question: while the majority of the interface (and binding energy) in most antibody–antigen interactions is dominated by VH domains, this is not the case in the variant structures reported here (Figure 4(c)). In the case of CR3022-B6, a total of 480 Å2 of buried surface is observed for VL, with only 230 Å2 observed for VH (relative contact surfaces in shades of blue in Figure 4(c)), calculated using PBDePisa.27 This is in marked contrast to the parental CR3022 antibody, where the interaction is dominated by heavy chain contacts (VH: 592 Å2 and VL: 415 Å2). Similar ratios were observed for the CR3014-C8 interaction with SARS-CoV-2 (VH: 240 Å2 and VL: 460 Å2; Figure 4(c), relative contact surfaces in shades of green). In the case of CR3022-B6, the increase of VL contact surface is accompanied by a considerable reduction in the length of CDR L1, which is elongated in the parental CR3022 structure, resulting in a more shallow and extended interface and allowing for additional interactions (Supplementary Fig. 2b).
Taken together, our results demonstrate that high-affinity antibodies against a variant coronavirus can be generated through maturation of previously reported antibodies. Several of the selected antibodies potently neutralized live SARS-CoV-2 virus with IC50s in the therapeutic range. Indeed, the monoclonal antibodies developed here (with KDs and IC50s as low as 7 nM and 0.16 ug/ml) fall broadly within the affinity and neutralization range of the US Food and Drug Administration (FDA)-approved anti-respiratory syncytial virus monoclonal antibody palivizumab (Synagis®) (at 1 nM and 0.5 ug/ml),28 as well as the SARS-CoV-2 monoclonal etesevimab (at 6 nM and 0.046 μg/mL)29 (Eli Lilly), which has received emergency use authorization by the FDA. Further increases of affinity and neutralization potential could conceivably be obtained by affinity maturation on phage (as demonstrated for CR3022-B6 as below). Importantly, the binding of several of the antibodies developed here was unaffected by mutations observed in emerging variants of concern, including B.1.1.7 (N501Y) and B.1.351 (N501Y, K417N, E484K)25 (Supplementary Table 1). In the case of m396-derived antibodies, this was observed despite the fact that the N501Y mutation is in proximity of CDR H2 in the parental SARS-CoV-1 RBD m396 complex, and may be related to the fact that the side chain at position 501 is oriented away from the antibody interface.
While the use of site-directed mutagenesis libraries resulted in the selection of potent antibody variants with conserved epitope specificity, the use of light chain shuffling also resulted in the generation of antibodies with completely new specificities. The discovery of such dual specificity antibody pairs with identical heavy chains, but different light chains, is intriguing and may enable the generation of bispecific reagents with improved resistance against mutational escape.30
The observation that a limited number of CDR mutations can endow nanomolar affinity binding and potent neutralization onto antibodies originally raised against a different variant coronavirus (SARS-CoV-1), also raises important implications for natural immunity and vaccine design. While the potential of antibody maturation against variant antigens has been demonstrated using haptens31 and viral model antigens,32 insights into the mutational plasticity of coronavirus antibodies had remained unclear. We conclude that protein engineering and in vitro maturation provides a rapid pathway for the identification of potent antibody reagents against emerging viruses.
Materials and methods
Generation of site-directed mutagenesis antibody libraries
m396, CR3022, CR3014, and 80 R scFv were gene synthesized (Genscript) and cloned into the pHEN1 phagemid vector. Site-directed mutagenesis was carried out by Kunkel mutagenesis.19 In brief, phagemid vectors were transformed into E. coli CJ236, and a single colony grown in 2xYT media supplemented with 100 µg/mL ampicillin, 10 µg/mL chloramphenicol and 2% glucose until reaching an OD600nm of 0.4. Bacteria were then infected with KM13 helper phage and grown overnight at 30°C in 2xYT media supplemented with 100 µg/mL ampicillin, 10 µg/mL chloramphenicol, 50 µg/mL kanamycin and 0.25 µg/mL uridine. Phage particles were precipitated from the culture media using polyethylene glycol (PEG)/NaCl, and uridine-containing single-stranded DNA (dU-ssDNA) extracted using a QIAprep spin M13 kit (Qiagen). Mutagenesis was carried out by annealing degenerated oligonucleotides to the dU-ssDNA, followed by synthesis of the covalently closed circular DNA (cccDNA) with T7 DNA polymerase and T4 ligase (NEB). Finally, the cccDNA was transformed into electro-competent E. coli TG1 and bacteria titrated onto 2xYT agar plates containing 2% glucose and 100 µg/ml ampicillin to determine library sizes. Bacteria were harvested from the agar plates, grown in 2xYT media supplemented with 100 µg/mL ampicillin, and 2% glucose until reaching an OD600nm of 0.4. At this point, bacteria were infected with KM13 helper phage and grown overnight at 30°C in 2xYT media supplemented with 100 µg/mL ampicillin, and 50 µg/mL kanamycin. Phage antibody libraries were precipitated from culture media using PEG/NaCl and stored at 4°C.
Generation of light chain shuffled antibody libraries
DNA encoding SARS-CoV-1 VH regions was amplified by PCR (using Q5 polymerase – NEB), where J segments were modified as required to match the following protein sequence (GTLVTVSS). DNA encoding V kappa library regions were amplified from a pHEN1 scFv library, comprising the end of the VH J segment, glycine-serine linker and VL regions. The resulting light chain shuffled library was generated by splice-overlapping extension PCR and cloned into pHEN1 in an scFv format using NcoI and NotI restriction sites. DNA was transformed into electro-competent TG1, and phage produced and precipitated as above.
Phage display selections
For phage display selection, we biotinylated SARS-CoV-2 RBD using a terminal AviTag and BirA biotin ligase (Avidity) according to the manufacturer’s protocol. Phage display selections were carried out at room temperature (unless stated otherwise), alternating between capture of the antigen on neutravidin coated wells on Maxisorp plates (Nunc) and streptavidin magnetic beads (Invitrogen).33 For Maxisorp plate selection, neutravidin was coated overnight at 50 µg/mL in carbonate coating buffer at 4°C, then biotinylated RBD captured and blocked in phosphate-buffered saline (PBS) supplemented with 4% skim milk and 0.1% Tween-20 (MPBST) at room temperature. 1 × 1012 phage were blocked in MPBST, added to the wells containing antigen and incubated for 1 h. The wells were washed once with PBST, and once with PBS. Phage were eluted with 100 µg/mL trypsin for 1 h, then used to infect TG1 bacteria at an OD600nm of 0.4. Infected TG1 were plated onto 2xYT agar plates supplemented with 100 µg/mL ampicillin and 2% glucose. For streptavidin beads selection, phages were blocked as described above and incubated with biotinylated RBD at room temperature. 30 µl of streptavidin magnetic beads (Invitrogen) were blocked in PBST supplemented with 4% BSA (Sigma), then incubated for 15 min with the phage/antigen mix. Magnetic beads were washed with PBST and PBS and phage eluted as described above. For site-directed mutagenesis libraries, we used 100 nM, 50 nM, 5 nM and 0.5 nM of biotinylated RBD for selection rounds 1 to 4, and 100 nM, 50 nM, 25 nM and 10 nM for light chain shuffled libraries. Phage titers used for selection were reduced to 1 × 1011 for rounds 2 and 3 and 1 × 1010 for round 4.
Affinity maturation was carried out using off-rate selections and streptavidin magnetic beads. Selections were performed essentially as previously described,34 with the following adjustments: phage were incubated with the biotinylated RBD for 1 h, excess unbiotinylated RBD was added (100x and 350x for rounds 2 and 3) and further incubated for 2/8 h for rounds 2/3 before capture on magnetic streptavidin beads.
Polyclonal phage and monoclonal soluble ELISA
For polyclonal ELISA, all steps were carried out at room temperature, unless stated otherwise. Maxisorp plates were coated with neutravidin overnight at 4°C, then 100 nM of biotinylated RBD was subsequently captured at room temperature. 1 × 109 purified phage were blocked in MPBST and incubated in each well for 1 h. Plates were washed with PBST, incubated with horseradish peroxidase (HRP)-conjugated anti-M13 antibody (GE Healthcare, CMYC-45P-Z) for 1 h and washed again. The plate was finally incubated with TMB substrate (Perkin Elmer), the reaction quenched with HCl and the plate read at Abs450nm (ClarioStar – BMG Labtech). For monoclonal soluble ELISA, individual colonies from the selection titration plates were inoculated in 96-well plates and incubated at 37°C overnight. The bacteria were re-inoculated the following day at 1:50 and incubated at 37°C for 4 h. The plates were then spun down, the culture media discarded, bacteria resuspended in 2xYT supplemented with 100 µg/ml ampicillin and 1 mM isopropyl β-d-1-thiogalactopyranoside and incubated overnight at 30°C. For ELISA, Maxisorp plates were coated with neutravidin overnight at 4°C, and 100 nM of biotinylated RBD subsequently captured at room temperature. The plates were then incubated with 50 µl of culture media, clarified by centrifugation, for 1 h and then washed with PBST. The plates were subsequently incubated with HRP-conjugated chicken anti c-myc antibody (ICL Lab 27942101) for 1 h and washed again. The plate was finally incubated with TMB substrate (Perkin Elmer), the reaction quenched HCl and the plate read at Abs450nm (ClarioStar – BMG Labtech).
Monoclonal antibody production and purification
DNA encoding antibody variable domains was amplified by PCR from the pHEN1 phage display vector and cloned into a human IgG1 expression vector based on pCEP4 (Invitrogen). After validation of the cloning by Sanger sequencing, the plasmids were transfected into ExpiCHO cells (Thermo Scientific) according to the manufacturer’s protocol (1 µg DNA/ml of cells; 2:1 ratio of heavy chain to light chain) and following the max titer protocol. After 14 days, cell culture media were clarified by centrifugation and the IgG captured using Protein G resin (Genscript). IgG were eluted from the resin using 100 mM glycine pH 3.0, eluate was dialyzed against PBS and the purity assessed by SDS-PAGE. For Fab production, DNA encoding VH and VL regions was cloned into a pCEP4 based vector encoding a C-terminal His tag on the heavy chain. Production was carried out in ExpiCHO cell as above. After 14 days, cell culture media were clarified by centrifugation, dialyzed against PBS and Fab protein captured using Talon resin (Thermo Scientific). Fab protein was eluted with 150 mM imidazole in PBS, dialyzed with PBS and the purity assessed by visualization on SDS-PAGE. In the case of m396-B10, Fab was generated through proteolytic cleavage of IgG using papain and purified using protein A affinity chromatography.
Affinity measurements using biolayer interferometry
Purified monoclonal antibodies (Fab/IgG) were buffer exchanged into PBS using equilibrated ZebaSpin columns (Thermo Fisher Scientific). The protein concentration was determined and the antibodies biotinylated by incubating for 30 min at room temperature with EZ-Link NHS-PEG4-Biotinylation reagent (Thermo Fisher Scientific) at a 10:1 biotin-to-protein ratio. Free biotin was removed from the samples by repeating the buffer exchange step in a second ZebaSpin column equilibrated with PBS. Affinity of interactions between biotinylated antibodies and purified soluble RBD proteins were measured by BLI (BLItz, ForteBio), at room temperature. Streptavidin biosensors were rehydrated in PBS containing 0.1% w/v BSA for 1 h at room temperature. Biotinylated antibody was loaded onto the sensors “on-line” using an advanced kinetics protocol, and global fits were obtained for the binding kinetics by running associations and dissociations of RBD proteins at a suitable range of molar concentrations (2-fold serial dilution ranging from 800 nM to 50 nM). The global dissociation constant (KD) for each 1:1 binding interaction was determined using the BlitzPro 1.2.1.3 software. Human IgG1 was used for all measurements, except for 80 R-A2, CR3014-D1, CR3014-C8 for which Fab was used. For ACE2 competition assays, biotinylated ACE2-Fc was loaded onto the streptavidin sensors on-line, and the binding kinetics determined using either 500 nM of soluble RBD, or 500 nM of soluble RBD pre-incubated with 1 µM of IgG for 5 min, using the advanced kinetics protocol.
Antigen production and purification
DNA encoding SARS-Cov-2 RBD (residues 319–541) was gene synthesized (Genscript) and cloned into the pCEP4 mammalian expression vector with a N-terminal IgG leader sequence and C-terminal Avitag and His tag. For the RBD variants, the mutations were introduced by splice-overlapping PCR, and cloned back into pCEP4 vector. The plasmids were transfected into Expi293 cells (Thermo Scientific) according to the manufacturer’s protocol and the protein expressed for 7 days at 37°C, 5% CO2. The cell culture was clarified by centrifugation, dialyzed with PBS and the protein captured with Talon resin. The RBD was eluted with 150 mM imidazole in PBS, dialyzed with PBS and the purity assessed by visualization on SDS-PAGE. The plasmid encoding the spike protein with C-terminal trimerization domain and His tag was a gift from the Krammer lab (BEI Resources). The plasmid was transfected into Expi293 cells and protein expressed for 3 days at 37°C, 5% CO2. The protein was purified using the His tag as for the RBD purification. The protein was further purified on a Superose 6 gel filtration column (GE Healthcare) using an AKTA Pure FPLC instrument (GE Healthcare) to isolate the trimeric protein and remove S2 pre-fusion protein.
SARS-CoV-2 neutralization assays
Serial 2-fold dilutions of test monoclonal antibody were prepared in 96-well plates in octuplicate (n = 8). The serial dilutions were incubated for 1 h at 37°C with an equal volume of SARS-CoV-2 isolate containing 200 TCID50 (infectious dose). A Vero E6 suspension containing 2 × 104 cells was added to each well, and plates were incubated at 37°C, 5% CO2. After 3 days, the plates were observed for cytopathic effect (CPE) and IC50 values were calculated from four parameter dose-response curves (GraphPad Prism). The CPE was determined by bright field microscopy (IN Cell Analyzer – Cytiva) in a binary mode; percentage neutralization was determined by the number of positive wells for each replicate. All dilution steps of antibody, virus, and cells were performed in culture media: MEM supplemented with 2% fetal bovine serum, L-glutamine and penicillin-streptomycin.
SARS-CoV-1 pseudovirus neutralization assays
Cells were cultured at 37°C, 5% CO2 in DMEM high glucose (ThermoFisher Scientific) supplemented with 10% fetal bovine serum (ThermoFisher Scientific). Retroviral SARS-CoV-1 and SARS-CoV-2 pseudoparticles (SARS-2pp) were generated by co-transfecting expression plasmids containing SARS-CoV-1 or SARS-CoV-1 spike (kindly provided by Prof Gary Whitaker (Cornell) and Dr Markus Hoffmann (German Primate Center), respectively) and the MLV gag/pol and luciferase vectors (kindly provided by Prof. Francois-Loic Cosset (CNRS), in CD81KO 293 T cells (kindly provided by Dr Joe Grove (UCL)30), using a mammalian Calphos transfection kit (Takara Bio). Culture supernatants containing SARS-2pp were harvested 48 h post transfection and clarified of cellular debris by centrifugation at 500 g for 10 min. SARS-2pp were concentrated 10-fold using 100,000 MWCO Vivaspin centrifugal concentrators (Sartorius) by centrifugation at 2000 g and stored at −80°C. For neutralization assays, the infectivity of SARSpp were diluted in media to 1000–5000-fold more infectious than negative background (based on pseudoparticles lacking SARS-CoV-1 Spike). Diluted pseudoparticles were incubated for 1 h with monoclonal antibodies, followed by the addition of polybrene at a final concentration of 4 µg/mL (Sigma-Aldrich), prior to addition to 293 T-ACE2 over-expressed cells (kindly provided by A/Prof Jesse Bloom). 293 T-ACE2 cells were seeded 24 h earlier at 1.5 × 104 cells per well in 96-well white flat-bottom plates (Sigma-Aldrich). Cells were spinoculated at 800 g for 2 h and incubated for 2 h at 37°C, prior to media change. After 72 h, the cells were lysed with lysis buffer (Promega) and Bright Glo reagent (Promega) was added at a 1:1 ratio. Luminescence (RLU) was measured using CLARIOstar microplate reader (BMG Labtech). Neutralization assays were performed in triplicate and outliers were excluded using the modified z-score method. Percentage neutralization of SARSpp was calculated as (1 – RLUtreatment/RLUno treatment) × 100. The 50% inhibitory concentration (IC50) titer was calculated using non-linear regression model (GraphPad Prism).
X-ray crystallography
Gel-filtration chromatography purified SARS-CoV-2 RBD (residues 333–528), and light chain shuffled Fabs CR3022-B6 and CR3014-C8 (in 25 mM Tris pH 8.0, 200 mM NaCl) were combined in a 1:1:1 molar ratio at a concentration of ~4 mg/mL, from which equal volumes (2 µL) were combined with well solution (100 mM Tris pH 8.0, 200 mM NaCl and 20% PEG3350) in a hanging drop format. After several weeks, crystals of sword-like morphology appeared growing out of precipitate. Due to their small size crystals were harvested without a cryoprotection regime and plunge-frozen in liquid nitrogen. Large crystals of CR3022-B6 Fab alone were grown by combining equal volumes (2 µL) of protein (9.6 mg/mL) with well solution comprising 200 mM sodium citrate (pH 6.65) and 24% PEG3350. Crystals of m396-B10 were grown by combining equal volumes of protein (5.95 mg/ml) with well solution comprising 200 mM NaCl, 100 mM BisTris (pH 5.95) and 25% (v/v) PEG 3350. Cryoprotection for CR3022-B6 crystals was achieved by briefly (5–10 s) swimming crystals in well solution supplemented with glycerol (to ~25% v/v) prior to looping and snap freezing. Diffraction data were collected at the Australian Synchrotron on beamline MX2 using a Dectris Eiger X16M detector. In all cases, a 360° sweep of data were deconvoluted into 3600 × 0.1° oscillation images which were indexed and integrated by XDS.35 Space groups were determined with Pointless36 and scaling and merging performed with Aimless,37 both components of CCP4.38
Structures were determined by molecular replacement using Phaser,39 and structure refinement was performed using Refmac5,40 with torsion libration screw parameterization. The search model for the CR3022-B6 structure was the CR3022 Fab component of PDB entry 6w41,16 split into variable domain (VH + VL) and constant domain (CH1 + CL) pairings. For the double-Fab 1:1:1 complex (RBD + CR3014-C8 + CR3022-B6), cell content analysis suggested that, should all components be present in the expected stoichiometric ratio, the solvent content would be 50%. The search model for the RBD was also derived from PDB entry 6w41. The search model for the Fab components were the same VH + VL and CH1 + CL pairings as used for the CR3022-B6 structure alone. Molecular replacement could place two Fabs without clashing on the surface of a single RBD, although the second Fab returned a much smaller log likelihood gain than the first. This was reflected in electron density for one Fab being unambiguous and well resolved (clearly CR3014-C8), whilst the other was weak, suggesting either incomplete occupancy or conformational/positional motion of this Fab, which otherwise lacked significant crystal contacts within the lattice. CR3022-B6 was modeled into this second Fab position.
Interestingly, the CR3014-C8 Fab bound RBD where wild-type CR3022 would have been expected to bind, whilst CR3022-B6 bound to a surface of RBD consistent with neutralization assay data suggesting it was in fact neutralizing (unlike parental CR3022). Additionally, the bulk of buried surface for both interactions was dominated by the light chains, which for these Fabs are very similar, raising the issue of whether the CR3022-B6 Fab was in fact correctly modeled (the VH region, in particular, was very poorly resolved). That CR3022-B6 does bind this second position was confirmed by a combination of: 1) fo-fc difference maps, suggesting CR3022-B6 was a better fit, 2) molecular replacement yielding stronger solutions with CR3022-B6 placement relative to an initial CR3104-C8 placement, 3) mutagenesis of the RBD epitope eliminating CR3022-B6 binding, and 4) cryo-EM returning a spike + CR3022-B6 model consistent with CR3022-B6 binding this epitope. A short, branched-chain carbohydrate was clearly present attached to Asn343 of the RBD, and has been modeled as two N-acetyl glucosamine (NAG) sugars connected by an beta(1–4) glycosidic bond, with a fucose (FUC) residue attached to the N-linked NAG via an alpha(1–6) glycosidic linkage. For the m396-B10 structure, the search model was the Fab component of PDB entry 2g75. Diffraction data and model refinement statistics are shown in Supplementary Table 3.
Cryo-electron microscopy
For sample preparation, either spike trimer alone or 1:1 Fab:monomer (molar ratio – for the CR3022-B6 or CR3014-D1 datasets) was incubated at room temperature for 1 h before applying to holey gold grids and freezing. 3.5 μl of each sample was applied to 1.2/1.3 Ultrfoil Au grids (Quantifoil), which had been glow-discharged for 1 min at 19 mA. Plunge freezing was performed using a Vitrobot Mark IV (ThermoFisher) with 0 blot force, 4 s blot time and 100% humidity at 22 °C. For data collection, grids were transferred to a Talos Arctica Electron Microscope (ThermoFisher) operating at 200 kV equipped with a FalconIII direct detector. Movies were recorded using EPU software with a calibrated pixel size of 0.986 Å, a total dose of 40 electrons spread over 29 frames and a total exposure time of 60 s. For data processing, motion correction, contrast transfer function (CTF) estimation41 and blob particle picking were performed in cryoSPARC.42 Extracted particles were subjected to multiple rounds of 2-dimentional (2D) classification and ab initio reconstruction in cryoSPARC before their locations were exported to Relion 3.0.43 Motion correction and CTF estimation was then implemented in Relion 3.0 and particles were re-extracted and again subjected to 2D classification before 3D auto-refinement and Bayesian polishing.44 3D classification was then used to sort the particles based on whether density (attributed to Fab) was present above on of the RBDs. The final Fab bound trimer particles were then imported back to cryoSPARC for NU-3D refinement. Supplementary Fig. 8 provides a flowchart to describe this workflow along with FSC curves. Supplementary Table 4 provides a summary of the data collection and refinement statistics.
Supplementary Material
Acknowledgments
We thank the UTS Biologics Innovation Facility for help on SARS-CoV-2 spike protein production, the Victor Chang Cardiac Research Institute Innovation Centre (funded by the NSW Government) and the Electron Microscope Unit at UNSW Sydney (funded in part by the NSW Government), the MX2 beamline at the Australian Synchrotron, as well as Pamela J. Bjorkman and Christopher Barnes (Caltech) for advice and help with early structural studies. This work was supported by the Medical Research Future Fund 2020 Antiviral Development Call (2001739); by Program Grants 1016953 and 1113904; Project Grant 1108800; Fellowships 585490, 1157744, 190774, 1176351, 1159347 and 1081858 from the National Health and Medical Research Council; Discovery Grant 160104915; DECRA Fellowship DE190100985 from the Australian Research Council.
Author contributions
R.R., O.M., P.S., and D.C. designed the project; R.R. performed light chain shuffling, selection and maturation; O.M. performed site-directed mutagenesis and selection; D.B.L. performed X-ray crystallography; M.S. and A.G.S. performed cryo-EM; G.W., A.A., R.A.B. and W.D.R. performed neutralization experiments; O.M., R.R., J.J., S.U. and J.Y.H. performed protein production and purification; H.L. and P.S. performed affinity measurements; R.R., D.B.L. and D.C. wrote the manuscript with input from all other authors.
Disclosure of Potential Conflicts of Interest
The authors declare no competing financial interest.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Abbreviations:
ACE2: angiotensin converting enzyme 2; VH: antibody variable heavy ; VL: antibody variable light; scFv: antibody single-chain variable fragment; BLI: biolayer-interferometry; CHO: chinese hamster ovary cells; CDRs: complementarity-determining regions; cryo-EM: cryo-electron microscopy; DPP4: dipeptidyl peptidase 4; ELISA: enzyme-linked immunosorbent assay; RBD: receptor binding domain.
References
- 1.Fung TS, Liu DX.. Human coronavirus: host-pathogen interaction. Annu Rev Microbiol. 2019. September 8;73(1):529–14. PubMed PMID: 31226023. doi: 10.1146/annurev-micro-020518-115759. [DOI] [PubMed] [Google Scholar]
- 2.Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J, Huhn G, Cardona J, Mocherla B, Stosor V, et al. SARS-CoV-2 neutralizing antibody LY-CoV555 in outpatients with Covid-19. N Engl J Med. 2020. October 28. PubMed PMID: 33113295. doi: 10.1056/NEJMoa2029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Du L, Zhao G, Yang Y, Qiu H, Wang L, Kou Z, Tao X, Yu H, Sun S, Tseng CT, et al. A conformation-dependent neutralizing monoclonal antibody specifically targeting receptor-binding domain in Middle East respiratory syndrome coronavirus spike protein. J Virol. 2014. June;88(12):7045–53. PubMed PMID: 24719424; PubMed Central PMCID: PMCPMC4054355. doi: 10.1128/JVI.00433-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rogers TF, Zhao F, Huang D, Beutler N, Burns A, He WT, Limbo O, Smith C, Song G, Woehl J, et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science. 2020. August 21;369(6506):956–63. PubMed PMID: 32540903; PubMed Central PMCID: PMCPMC7299280. doi: 10.1126/science.abc7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan M, Liu H, Wu NC, Lee CD, Zhu X, Zhao F, Huang D, Yu W, Hua Y, Tien H, et al. Structural basis of a shared antibody response to SARS-CoV-2. Science. 2020. August 28;369(6507):1119–23. PubMed PMID: 32661058; PubMed Central PMCID: PMCPMC7402627. doi: 10.1126/science.abd2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Murphy AJ, Macdonald LE, Stevens S, Karow M, Dore AT, Pobursky K, Huang TT, Poueymirou WT, Esau L, Meola M, et al. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc Natl Acad Sci U S A. 2014. April 8;111(14):5153–58. PubMed PMID: 24706856; PubMed Central PMCID: PMCPMC3986188. doi: 10.1073/pnas.1324022111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noy-Porat T, Makdasi E, Alcalay R, Mechaly A, Levy Y, Bercovich-Kinori A, Zauberman A, Tamir H, Yahalom-Ronen Y, Israeli M, et al. A panel of human neutralizing mAbs targeting SARS-CoV-2 spike at multiple epitopes. Nat Commun. 2020. August 27;11(1):4303. PubMed PMID: 32855401; PubMed Central PMCID: PMCPMC7452893. doi: 10.1038/s41467-020-18159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeng X, Li L, Lin J, Li X, Liu B, Kong Y, Zeng S, Du J, Xiao H, Zhang T, et al. Isolation of a human monoclonal antibody specific for the receptor binding domain of SARS-CoV-2 using a competitive phage biopanning strategy. Antib Ther. 2020;3(2):95–100. doi: 10.1093/abt/tbaa008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dutta NK, Mazumdar K, Gordy JT. The nucleocapsid protein of SARS-CoV-2: a target for vaccine development. J Virol. 2020. June 16;94(13). PubMed PMID: 32546606; PubMed Central PMCID: PMCPMC7307180. doi: 10.1128/JVI.00647-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020. March;579(7798):270–73. PubMed PMID: 32015507; PubMed Central PMCID: PMCPMC7095418. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu Z, Chakraborti S, He Y, Roberts A, Sheahan T, Xiao X, Hensley LE, Prabakaran P, Rockx B, Sidorov IA, et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc Natl Acad Sci U S A. 2007. July 17;104(29):12123–28. PubMed PMID: 17620608; PubMed Central PMCID: PMCPMC1924550. doi: 10.1073/pnas.0701000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ter Meulen J, Van Den Brink EN, Poon LL, Marissen WE, Leung CS, Cox F, Cheung CY, Bakker AQ, Bogaards JA, Van Deventer E, et al. Human monoclonal antibody combination against SARS coronavirus: synergy and coverage of escape mutants. PLoS Med. 2006. July;3(7):e237. PubMed PMID: 16796401; PubMed Central PMCID: PMCPMC1483912. doi: 10.1371/journal.pmed.0030237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ter Meulen J, Bakker AB, Van Den Brink EN, Weverling GJ, Martina BE, Haagmans BL, Kuiken T, De Kruif J, Preiser W, Spaan W, et al. Human monoclonal antibody as prophylaxis for SARS coronavirus infection in ferrets. Lancet. 2004. June 26;363(9427):2139–41. PubMed PMID: 15220038. doi: 10.1016/S0140-6736(04)16506-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sui J, Li W, Murakami A, Tamin A, Matthews LJ, Wong SK, Moore MJ, Tallarico AS, Olurinde M, Choe H, et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc Natl Acad Sci U S A. 2004. February 24;101(8):2536–41. PubMed PMID: 14983044; PubMed Central PMCID: PMCPMC356985. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prabakaran P, Gan J, Feng Y, Zhu Z, Choudhry V, Xiao X, Ji X, Dimitrov DS. Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J Biol Chem. 2006. June 9;281(23):15829–36. PubMed PMID: 16597622. doi: 10.1074/jbc.M600697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuan M, Wu NC, Zhu X, Lee CD, So RTY, Lv H, Mok CKP, Wilson IA. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science. 2020. May 8;368(6491):630–33. PubMed PMID: 32245784; PubMed Central PMCID: PMCPMC7164391. doi: 10.1126/science.abb7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hwang WC, Lin Y, Santelli E, Sui J, Jaroszewski L, Stec B, Farzan M, Marasco WA, Liddington RC. Structural basis of neutralization by a human anti-severe acute respiratory syndrome spike protein antibody, 80R. J Biol Chem. 2006. November 10;281(45):34610–16. PubMed PMID: 16954221. doi: 10.1074/jbc.M603275200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian X, Li C, Huang A, Xia S, Lu S, Shi Z, Lu L, Jiang S, Yang Z, Wu Y, et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg Microbes Infect. 2020. December 9;1:382–85. PubMed PMID: 32065055. doi: 10.1080/22221751.2020.1729069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rouet R, Dudgeon K, Christ D. Generation of human single domain antibody repertoires by Kunkel mutagenesis. Methods Mol Biol. 2012;907:195–209. PubMed PMID: 22907352; eng. doi: 10.1007/978-1-61779-974-7_10. [DOI] [PubMed] [Google Scholar]
- 20.Ohlin M, Owman H, Mach M, Borrebaeck CA. Light chain shuffling of a high affinity antibody results in a drift in epitope recognition. Mol Immunol. 1996. January;33(1):47–56. PubMed PMID: 8604223. doi: 10.1016/0161-5890(95)00123-9. [DOI] [PubMed] [Google Scholar]
- 21.Rouet R, Langley DB, Schofield P, Christie M, Roome B, Porebski BT, Buckle AM, Clifton BE, Jackson CJ, Stock D, et al. Structural reconstruction of protein ancestry. Proc Natl Acad Sci U S A. 2017. April 11;114(15):3897–902. PubMed PMID: 28356519; PubMed Central PMCID: PMCPMC5393204. doi: 10.1073/pnas.1613477114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dudgeon K, Rouet R, Kokmeijer I, Schofield P, Stolp J, Langley D, Stock D, Christ D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc Natl Acad Sci U S A. 2012. July 3;109(27):10879–84. PubMed PMID: 22745168; eng. doi: 10.1073/pnas.1202866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steinwand M, Droste P, Frenzel A, Hust M, Dubel S, Schirrmann T. The influence of antibody fragment format on phage display based affinity maturation of IgG. MAbs. 2014. Jan-Feb;6(1):204–18. PubMed PMID: 24262918; PubMed Central PMCID: PMCPMC3929444. doi: 10.4161/mabs.27227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16(15):7351–67. PubMed PMID: 3045756; PubMed Central PMCID: PMC338413. eng. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walensky RP, Walke HT, Fauci AS. SARS-CoV-2 variants of concern in the United States-challenges and opportunities. JAMA. 2021. March 16;325(11):1037–38. PubMed PMID: 33595644. doi: 10.1001/jama.2021.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jespers LS, Roberts A, Mahler SM, Winter G, Hoogenboom HR. Guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen. Biotechnology. 1994. September;12(9):899–903. PubMed PMID: 7521646. doi: 10.1038/nbt0994-899. [DOI] [PubMed] [Google Scholar]
- 27.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007. September 21;372(3):774–97. PubMed PMID: 17681537. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 28.Van Mechelen L, Luytjes W, De Haan CA, Wicht O. RSV neutralization by palivizumab, but not by monoclonal antibodies targeting other epitopes, is augmented by Fc gamma receptors. Antiviral Res. 2016. August;132:1–5. PubMed PMID: 27185625. doi: 10.1016/j.antiviral.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 29. https://www.fda.gov/media/145802/download [cited 2001 Jan 21].
- 30.Fischer N, Elson G, Magistrelli G, Dheilly E, Fouque N, Laurendon A, Gueneau F, Ravn U, Depoisier JF, Moine V, et al. Exploiting light chains for the scalable generation and platform purification of native human bispecific IgG. Nat Commun. 2015. February 12;6:6113. PubMed PMID: 25672245; PubMed Central PMCID: PMCPMC4339886. doi: 10.1038/ncomms7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith KG, Light A, Nossal GJ, Tarlinton DM. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. Embo J. 1997. June 2;16(11):2996–3006. PubMed PMID: 9214617; PubMed Central PMCID: PMCPMC1169918. doi: 10.1093/emboj/16.11.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Purtha WE, Tedder TF, Johnson S, Bhattacharya D, Diamond MS. Memory B cells, but not long-lived plasma cells, possess antigen specificities for viral escape mutants. J Exp Med. 2011. December 19;208(13):2599–606. PubMed PMID: 22162833; PubMed Central PMCID: PMCPMC3244041. doi: 10.1084/jem.20110740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee CM, Iorno N, Sierro F, Christ D. Selection of human antibody fragments by phage display. Nat Protoc. 2007;2(11):3001–08. PubMed PMID: 18007636; eng. doi: 10.1038/nprot.2007.448. [DOI] [PubMed] [Google Scholar]
- 34.Zahnd C, Sarkar CA, Pluckthun A. Computational analysis of off-rate selection experiments to optimize affinity maturation by directed evolution. Protein Eng Des Sel. 2010. April;23(4):175–84. PubMed PMID: 20130104. doi: 10.1093/protein/gzp087. [DOI] [PubMed] [Google Scholar]
- 35.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010. February;66(Pt 2):125–32. PubMed PMID: 20124692; PubMed Central PMCID: PMCPMC2815665. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr. 2011. April;67(Pt 4):282–92. PubMed PMID: 21460446; PubMed Central PMCID: PMCPMC3069743. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr Sect D. 2013;69(7):1204–14. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011. April;67(Pt 4):235–42. PubMed PMID: 21460441; PubMed Central PMCID: PMC3069738. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCoy AJ. Solving structures of protein complexes by molecular replacement with phaser. Acta Crystallogr D Biol Crystallogr. 2007. January;63(Pt 1):32–41. PubMed PMID: 17164524; PubMed Central PMCID: PMCPMC2483468. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011. April;67(Pt 4):355–67. PubMed PMID: 21460454; PubMed Central PMCID: PMCPMC3069751. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang K. Gctf: real-time CTF determination and correction. J Struct Biol. 2016. January;193(1):1–12. PubMed PMID: 26592709; PubMed Central PMCID: PMCPMC4711343. doi: 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods. 2017. March;14(3):290–96. PubMed PMID: 28165473. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- 43.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012. December;180(3):519–30. PubMed PMID: 23000701; PubMed Central PMCID: PMCPMC3690530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol. 2019. October 1;75(Pt10):861–77. PubMed PMID: 31588918; PubMed Central PMCID: PMCPMC6778852. doi: 10.1107/S2059798319011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.