

Abstract
How far can we push the limits in removing stereoelectronic protection from an unstable intermediate? We address this question by exploring the interplay between the primary and secondary stereoelectronic effects in the Baeyer–Villiger (BV) rearrangement by experimental and computational studies of γ-OR-substituted γ-peroxylactones, the previously elusive non-strained Criegee intermediates (CI). These new cyclic peroxides were synthesized by the peroxidation of γ-ketoesters followed by in situ cyclization using a BF3·Et2O/H2O2 system. Although the primary effect (alignment of the migrating C–Rm bond with the breaking O–O bond) is active in the 6-membered ring, weakening of the secondary effect (donation from the OR lone pair to the breaking C–Rm bond) provides sufficient kinetic stabilization to allow the formation and isolation of stable γ-hydroperoxy-γ-peroxylactones with a methyl-substituent in the C6-position. Furthermore, supplementary protection is also provided by reactant stabilization originating from two new stereoelectronic factors, both identified and quantified for the first time in the present work. First, an unexpected boat preference in the γ-hydroperoxy-γ-peroxylactones weakens the primary stereoelectronic effects and introduces a ∼2 kcal mol−1 Curtin–Hammett penalty for reacquiring the more reactive chair conformation. Second, activation of the secondary stereoelectronic effect in the TS comes with a ∼2–3 kcal mol−1 penalty for giving up the exo-anomeric stabilization in the 6-membered Criegee intermediate. Together, the three new stereoelectronic factors (inverse α-effect, misalignment of reacting bonds in the boat conformation, and the exo-anomeric effect) illustrate the richness of stereoelectronic patterns in peroxide chemistry and provide experimentally significant kinetic stabilization to this new class of bisperoxides. Furthermore, mild reduction of γ-hydroperoxy-γ-peroxylactone with Ph3P produced an isolable γ-hydroxy-γ-peroxylactone, the first example of a structurally unencumbered CI where neither the primary nor the secondary stereoelectronic effect are impeded. Although this compound is relatively unstable, it does not undergo the BV reaction and instead follows a new mode of reactivity for the CI – a ring-opening process.
Protecting stereoelectronic effects prevent Baeyer–Villiger rearrangement and stabilize γ-OX-γ-peroxylactones (X = H, OH), the previously elusive non-strained Criegee intermediates.
Introduction
The Baeyer–Villiger (BV) reaction was discovered by Adolf von Baeyer and Victor Villiger in 1899, more than one hundred and twenty years ago.1,2 This oxidative transformation opens synthetic access to an ester from a ketone or to a lactone from a cyclic ketone, using peroxyacids as an oxidant.3 Thousands of studies are devoted to this reaction4–8 and its regio- and stereoselective versions were developed.9–12 An important monomer for polyesters and polyamides – caprolactone is produced via BV reaction in industry.13,14 Throughout its history, several mechanisms were suggested for the BV oxidation.15,16 Presently, a mechanism passing through a tetrahedral Criegee intermediate (CI) is generally accepted (Scheme 1).17–20 Surprisingly, this intermediate has not been structurally characterized until our initial report.21 Only three partially characterized examples of “protected” CIs have been known at that point.22–24
Scheme 1. Postulated mechanism of Baeyer–Villiger oxidation.

The hydroxyl peroxyesters, Criegee intermediates of the BV rearrangement, remained elusive due to their high reactivity (Scheme 1).25 However, the key to understanding the BV rearrangement mechanism and to the design a stereo- and regioselective BV processes lies in the intimate details of the CI structure, a notion that provides motivation for this research.
An additional broader incentive behind this work is to expand conceptual understanding of chemistry of organic peroxides. In comparison to the textbook organic oxygen-containing functionalities, for which the vast body of chemical knowledge has been accumulated, it is often impossible to predict whether a suggested organic peroxide would have more than fleeting stability. Because the experimental data for this neglected O-containing functionality is still relatively scarce, computational analysis becomes essential for evaluating synthetic routes to the new classes of organic peroxides and for advancing conceptual understanding of their structure and reactivity. Our work continues to underscore the utility of stereoelectronic thinking as the bridge between experimental data and understanding and predicting organic peroxide chemistry. Herein, we will show that the classic repertoire of stereoelectronic effects in the BV reaction is incomplete and that, in addition to two classic stereoelectronic effects that facilitate BV by activating CIs, there are also effects that prevent BV by protecting and/or stabilizing CIs. Two of such effects are quantified for the first time in the present work.
The assistance of two stereoelectronic effects is well-documented in the transformation of the Criegee intermediates into the final BV product.26–29 The key participants of these effects are the p-type lone pair of O1, the breaking C2–Rm bond and the O3–O4 acceptor (Scheme 2). The “primary stereoelectronic effect” requires that the breaking O–O bond and the migrating C2–Rm bond are antiperiplanar.30 The “secondary effect” is operative when the lone pair of the O1H group aligns with the breaking C2–Rm bond.31,32 When both effects take place, an uninterrupted electron flow from the donor (O1) to the acceptor assures that donation from the O1 lone pair assists in breaking the C2–Rm bond by stabilizing the incipient cationic center as the Rm group moves to O3 and the O3O4 bond breaks. As the result, the O1 C2 and Rm–O3 bonds are formed.
Scheme 2. The toolbox of stereoelectronic effects related to the BV of Criegee intermediate.

In our initial report of a stable Criegee intermediates, we have built two “stereoelectronic traps” by selective deactivation of these classic stereoelectronic effects (Scheme 2).21 The “first stereoelectronic trap” increased the stability of CI by constraining it in a five-membered cycle with the goal of preventing antiperiplanarity of the breaking O3–O4 bond and the migrating C2–Rm bond.26,33 The “second stereoelectronic trap” weakens the donation of electron density from a lone pair of the O1H group to the breaking C2–Rm bond. We disclosed that this is achieved via the replacement of the O1H group by the O1OH group. The change activates the “inverse α-effect”,34 a new stereoelectronic effect introduced previously for the control of CI stability. This recently discovered effect accounts for the lower donor ability of peroxides in intramolecular hyperconjugative interactions with the adjacent acceptors.35
The essence of inverse α-effect is shown in the Scheme 3, which illustrates that the two oxygen atoms of a peroxide moiety do not stabilize an adjacent cationic center or a stereoelectronically aligned σ-acceptor as much as a single oxygen atom of an ether. This effect has the potential to unlock many unusual aspects of peroxide chemistry.34 For example, it can impose ∼10 kcal mol−1 penalty for the generation of simple peroxycarbenium ions relative to their oxacarbenium analogues. The power and broad utility of inverse α-effect in peroxide chemistry was illustrated by using it to discover previously invisible chemistry of peroxycarbenium cations,36 and to understand the paradoxical situations where a peroxide is more stable than its mono-oxygen counterpart.21
Scheme 3. The magnitude of inverse α-effect in carbenium ions (a) and anomeric systems (b). Comparison of NBO 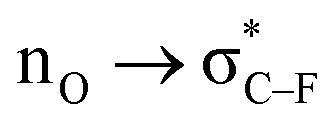 interactions (c) (energies are in kcal mol−1).
interactions (c) (energies are in kcal mol−1).

Five-membered peroxy-Criegee intermediates (β-hydroperoxy-β-peroxylactones) were protected from Baeyer–Villiger rearrangement by disrupting both stereoelectronic effects in BV by (a) breaking antiperiplanarity of O3–O4 and C2–Rm bond by cycle formation, and by (b) lowering donor ability of O1 atom in O1OH group by inverse α-effect (Scheme 4).21 However, the protection in β-hydroxy-β-peroxylactones (five-membered Criegee intermediates) was provided mostly by deactivation of the primary stereoelectronic effect (Scheme 4).21
Scheme 4. Fine-tuning the stability of Criegee intermediates via construction and partial deactivation of stereoelectronic traps. Arrows – the effect is activated. Crossed out arrows – the effect is deactivated.

In the present work, we remove another level of protective armor by restoring the unstrained 6-membered cycle for reactivating the primary stereoelectronic effect as an assistant to BV rearrangement. Our goal herein is to determine if attenuation of the secondary stereoelectronic effect alone can also save the Criegee intermediate from following the BV reaction (Scheme 4). We have introduced this protection by weakening the donor ability of the O1 atom in the six-membered CIs via the inverse α-effect. Furthermore, we will illustrate that even the combination of the three aforementioned effects was incomplete and that exo-anomeric effect is the fourth kinetically significant component of this system of intertwined stereoelectronic effects. Finally, we will also identify a new way to control the “primary stereoelectronic effect” with the boat/chair conformational transition in the six-membered cyclic peroxides.
In addition to providing a deeper insight into the key intermediate of the BV reaction, the goal of this work is to develop an approach to a previously unavailable class of organic peroxides. The recent renaissance in chemistry of organic peroxides has catalyzed the discovery of antimalarial (e.g., arterolane and artemisinin),37–45 anticancer,46–48 anthelmintic,49–51 antiviral52–54 and antimicrobial55–59 peroxides (in addition to the traditional applications as precursors,3 oxidizers,60–63 polymerization initiators, vulcanizing agents,64–66 and explosives).67 Despite the long history of peroxide chemistry, their selective synthesis and the inaccessibility of certain classes of peroxides remains a fundamental problem.68–70
Based on our earlier findings, we designed new methods for synthesis of stable 5-membered cyclic Criegee intermediates (β-hydroxy-β-peroxylactones),21 hydroperoxy-analogs of Criegee intermediates (β-hydroperoxy-β-peroxylactones),71 and alkoxy-analogs of Criegee intermediates (β-alkoxy-β-peroxylactones)36 (Scheme 4). The new insights in the nature of factors controlling the stability of the γ-hydroxy-γ-peroxylactone core in the new Criegee intermediates allowed us to design a synthetic approach to γ-hydroperoxy-γ-peroxylactones, a novel class of organic peroxides (Scheme 4). It should be noted that the 6-membered γ-hydroxy-γ-peroxylactones were postulated earlier to be unstable and highly reactive.33,72
Results and discussion
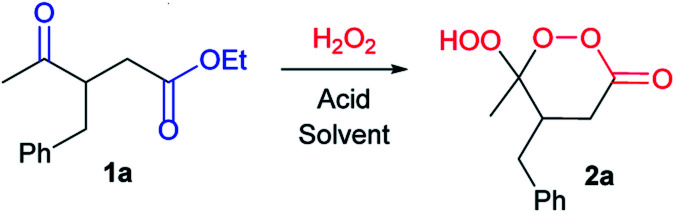
Even in the absence of a γ-hydroxy or γ-hydroperoxy groups, synthetic access to γ-peroxylactones is relatively difficult. The known approaches to these compounds, such as a peroxidation of γ-hydroxy amides,73,74 lactones,75,76 and γ-hydroxy esters,77 ozonolysis78 or singlet oxygen treatment79 of allylic esters, and few scattered examples,80–83 have limited utility. In contrast, the combination of dicarbonyl compounds and hydrogen peroxide as starting materials benefits from simplicity and affordability. In this paper, we disclose how these simple conditions can be used for peroxidation of γ-ketoesters with in situ cyclization into γ-hydroperoxy-γ-peroxylactones.
In the initial screening step, we studied the reaction of ethyl-3-benzyl-4-oxopentanoate (1a) with H2O2 in the presence of various acids and solvents (Table 1).
Screening conditions for γ-hydroperoxy-γ-peroxylactone 2a synthesisa.
| |||
|---|---|---|---|
| Entry | Eq. H2O2 | Acid, eq. | Yield 2a, % |
| 1 | 10 | BF3·Et2O, 10 | 54 |
| 2 | 10 | SnCl4, 5 | 46 |
| 3 | 10 | HClO4, 10 | 13 |
| 4 | 10 | HBF4, 10 | 17 |
| 5 | 10 | TsOH·H2O, 10 | 24 |
| 6 | 10 | PMA, 1 | 15 |
| 7 | 10 | BF3·Et2O, 5 | 38 |
| 8 | 10 | BF3·Et2O, 2 | 19 |
| 9b | 10 | BF3·Et2O, 10 | 35 |
| 10 | 5 | BF3·Et2O, 10 | 53 |
| 11 | 3 | BF3·Et2O, 10 | 27 |
General procedure: an ethereal solution of H2O2 (4.30 M, 0.698–2.326 mL, 3.0–10.0 mmol, 3.0–10.0 eq.) was added with stirring to a solution of 1a (234.3 mg, 1.00 mmol, 1.0 eq.) in Et2O (3.5 mL). The mixture was cooled to 0 °C and acid was added dropwise with stirring. The reaction mixture was then stirred at 20–25 °C for 24 h.
CH3CN as solvent.
We began our study with the use of 10 eq. of an ethereal solution of H2O2 and 10 eq. of BF3·Et2O (Table 1, entry 1) according to previously optimized conditions for β-hydroperoxy-β-peroxylactone synthesis.21,71 Yield of γ-hydroperoxy-γ-peroxylactone 2a was 54% (Table 1, entry 1). Attempts to use other Lewis and Brønsted acids were counterproductive, the yield of 2a decreased to 13–46% (Table 1, entries 2–6). Decreasing the amount of BF3·Et2O led to decreasing the yield of 2a to 19% (Table 1, entries 7, 8). Substitution of diethyl ether to acetonitrile yields target peroxide 2a in moderate yield (Table 1, entry 9). Using less H2O2 (5 eq. and 3 eq. in entries 10, 11) results in the formation of peroxide 2a in 53% and 27% yield, respectively.
With optimized conditions in hand (Table 1, entry 10) we investigated the influence of substituents in the C2 and C3 positions of starting γ-ketoesters 1 on the outcome of the peroxidation reaction (Scheme 5).
Scheme 5. Scope of γ-hydroperoxy-γ-peroxylactones 2 synthesized from γ-ketoesters 1.

As shown in Scheme 5, a range of 1,4-ketoesters 1a–l, with various R1 substituent – electron-donating and electron-withdrawing groups in aryl ring of benzyl substitutes 1a–i and alkyl groups 1j–l, were viable in the peroxidation reaction, γ-hydroperoxy-γ-peroxylactones 2a–l were obtained with from moderate (44%, 2d) to good (72%, 2j) yields. The exception is a hexyl substituted γ-ketoester 1k where the yield of γ-hydroperoxy-γ-peroxylactone 2k is 26%. Unsubstituted and C4-methyl-substituted γ-hydroperoxy-γ-peroxylactones 2m and 2n were synthesized in good yields, 60%, and 83%, respectively.
The γ-hydroperoxy-γ-peroxylactones 2a–l are produced as a mixture of two diastereomers with the predominance of the trans isomer. For example, the peroxylactone 2a was formed in the 22 : 78 cis : trans ratio (see ESI†). This finding disagreed with the greater calculated thermodynamic stability of the cis isomer of 2a in the chair conformation (1.4 kcal mol−1, Scheme 6). This discrepancy led us to explore conformational behavior of this system deeper. Surprisingly, calculations revealed that both diastereomers prefer the boat conformation where the trans-conformer is 0.7 kcal mol−1 more stable in a full agreement with the experimental data. Furthermore, the boat conformation was found in the X-ray structure of related peroxylactone 2m (Scheme 6). This finding will have significant stereoelectronic consequences for our subsequent mechanistic discussions.
Scheme 6. Relative stability of the four forms of γ-hydroperoxy-γ-peroxylactone 2.

When the substituent at the C4 position of γ-ketoesters is either a primary alkyl or the phenyl group (1o, 1p), the corresponding γ-peroxylactones were not obtained. For these substrates, the observed reaction products 6 and 7 result from the Baeyer–Villiger rearrangement and subsequent hydrolysis (Scheme 7).
Scheme 7. Transformation of γ-ketoesters with primarily alkyl or phenyl substituent in C4 position 1o, 1p in optimal conditions.

Interesting results were obtained by treatment of γ-hydroperoxy-γ-peroxylactone 2a with Ph3P (Scheme 8). The reaction of 2a with 1.1 eq. Ph3P for 5 min followed by chromatographic purification resulted in quite unstable γ-hydroxy-γ-peroxylactone 3a in 25% yield, the remaining reaction mass was a mixture of γ-ketoacid 5a and a new compound that we tentatively assign the structure of γ-ketoperacid 4a (see the ESI†). Previously, the γ-hydroxy-γ-peroxylactone was considered to be non-isolable due to the preferred Baeyer–Villiger rearrangement.33,72 The reaction of 2a with 1.1 eq. Ph3P for 1 day gave only a mixture of the ring-opened products 5a and 4a, according to NMR analysis (see ESI†). The treatment of γ-hydroperoxy-γ-peroxylactone 2a by 2.0 eq. Ph3P leads to the formation of γ-ketoacid 5a as the only product by NMR (68% isolated yield, Scheme 8). Thus, γ-hydroperoxy-γ-peroxylactone under the reductive conditions prefers C–O scission that leads to the peroxide ring opening rather than to Baeyer–Villiger rearrangement. The products of Baeyer–Villiger rearrangement were not observed in all cases.
Scheme 8. Reduction of γ-hydroperoxy-γ-peroxylactone 2a.

Computational analysis
As the starting point, we have calculated the activation barriers for the BV rearrangement of the six-membered Criegee intermediate and its hydroperoxy version (Scheme 9).84
Scheme 9. Computational analysis of the BV 1,2-shift in cyclic CIs and their hydroperoxyl analogs for the chair conformations.

Both of the barriers were found to be considerably lower than the barriers for five-membered analogues illustrating that the C–Rm/O–O bond alignment (the primary BV stereoelectronic effect) is a powerful protecting force for the Criegee intermediate.21 Increasing the cycle size removes this protecting force and makes these compounds much more vulnerable to the BV rearrangement. This vulnerability explains why preparation of the six-membered CIs has been so challenging.
Comparison of computed barriers for the chair conformations of the six-membered CI and its hydroperoxy version also illustrates that, in this case, the protective power of inverse α-effect is smaller than the effect of a five-membered ring. In particular, the free energy barrier for Me group migration is increased by only 3 kcal mol−1 for the chair conformations of the OOH–CI relative to its OH-counterpart (Scheme 9). For the Et group migration, the difference is even smaller, ∼1 kcal mol−1. Interestingly, even with this extra protection, the BV barrier for the Et/OOH derivative is still lower than it is for the Me/OH derivative.
Additional complexity – the chair/boat equilibrium
However, the conformational profile of the six-membered peroxylactones has an additional peculiarity that our computations have, for the first time, identified as a supplementary source of kinetic protection for the six-membered CIs. As mentioned earlier, the preferred conformation of these species is the boat, but the BV reaction proceeds through a chair TS. Although the chair TS is lower in the absolute free energy than the boat TS (∼1.6 and 0.1 kcal mol−1 for the OH and OOH derivatives, respectively), the need to adopt the reactive chair conformation is an additional Curtin–Hammett penalty that can be evaluated as ∼1.8 and 2.4 kcal mol−1 for the OH and OOH derivatives, respectively (Scheme 10). Such effect is not negligible since it should lead to >10-fold deceleration of the 1,2-methyl shift for the OH derivative. Furthermore, the presence of two energetically close but geometrically different transition states for the 1,2-shift should have implications for the design of stereoselective Baeyer–Villiger rearrangements of the six-membered peroxides (Scheme 11).
Scheme 10. Computational analysis of the BV 1,2-shift in cyclic CIs and their hydroperoxyl analogs for the boat conformations.

Scheme 11. Computational analysis of the BV 1,2-shift in cyclic CIs and their hydroperoxyl analogs for the boat and chair conformations.

Origin of the boat/chair effect on the BV barrier – interfering with the “primary stereoelectronic effect”
The RmCOO dihedral in hydroperoxides with R = Me chair is −174.1 and boat is 171.4 (similar to peroxylactones in Scheme 12). Although, this is not a large change but it suggests one more way to weaken the primary stereoelectronic effect in BV reaction – to start from a boat, rather than a chair conformation.
Scheme 12. The activation barriers for the BV rearrangement of the six- and five-membered Criegee intermediates.

However, this is not the end of this stereoelectronic story because a more detailed computational analysis of the reaction path reveals one more “hidden” stereoelectronic factor related to the second BV stereoelectronic effect. We will discuss this effect in the following section.
Competition with the C–O scission – the two roles of the anomeric effect in the Curtin–Hammett scenario
In the BV reaction, the lone pair of oxygen has to give up (sacrifice) its stabilizing anomeric interaction with the 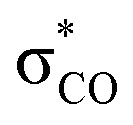 orbital in order to start assisting with the migration of the Rm (i.e., participate in the “secondary stereoelectronic effect of BV reaction”). This is an example of “orthogonal” stereoelectronic effect on reactivity, or “unproductive ground state stabilization”.85 Because loss of the anomeric effect is an additional penalty that every BV reaction should go through, anomeric effect is the additional stereoelectronic force that protects the Criegee intermediates from following the BV path.
orbital in order to start assisting with the migration of the Rm (i.e., participate in the “secondary stereoelectronic effect of BV reaction”). This is an example of “orthogonal” stereoelectronic effect on reactivity, or “unproductive ground state stabilization”.85 Because loss of the anomeric effect is an additional penalty that every BV reaction should go through, anomeric effect is the additional stereoelectronic force that protects the Criegee intermediates from following the BV path.
In order to evaluate the extent of anomeric stabilization that our systems have to lose when the lone pair of the exocyclic oxygen atoms is aligned with the C–R bond instead of the C–O(OR) bond, we have optimized these molecules with the OOCR dihedral constrained to their value in the respective BV transition states. The energy difference between this conformation and the most stable conformation of the reactant evaluates the degree of anomeric stabilization that the molecule sacrifices in order to satisfy the “secondary stereoelectronic effect in the BV”. Scheme 13 summarizes the energetic consequences of anomeric effect for the competition between BV rearrangement and simple C–O scission that leads to the opening of the peroxide ring. C–O scission, the observed reaction path, wouldn't need to sacrifice the AE effect (the 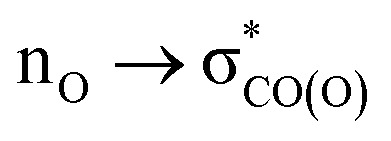 interaction can continue to get stronger in the process of C⋯O(O) bond scission).86–88 On the other hand, the OH (or OOH) group has to give up the AE stabilization in order to have an appropriate conformation for participation in the “secondary stereoelectronic effect” in BV rearrangement. In the ground state, the lone pair is aligned with the endocyclic C–O bond. However, the lone pair of the adjacent oxygen realigns with the breaking C–C bond to the migrating group in the BV TS when there a significant positive charge develops at the migrating group. Development of positive charge at the methyl group is difficult, as illustrated by the instability of the methyl cation. Hence, the lone pair does not offer its assistance to the C–C scission and the C–O scission is observed instead.
interaction can continue to get stronger in the process of C⋯O(O) bond scission).86–88 On the other hand, the OH (or OOH) group has to give up the AE stabilization in order to have an appropriate conformation for participation in the “secondary stereoelectronic effect” in BV rearrangement. In the ground state, the lone pair is aligned with the endocyclic C–O bond. However, the lone pair of the adjacent oxygen realigns with the breaking C–C bond to the migrating group in the BV TS when there a significant positive charge develops at the migrating group. Development of positive charge at the methyl group is difficult, as illustrated by the instability of the methyl cation. Hence, the lone pair does not offer its assistance to the C–C scission and the C–O scission is observed instead.
Scheme 13. Anomeric effect as “the fourth stereoelectronic effect” in the BV rearrangement. R is the migrating substituent. The scheme illustrates the cost of giving up the anomerically stabilized conformation in order to adopt a reactive conformation that satisfies the BV “primary stereoelectronic effect”. The energies of the OH and OOH CIs are taken the same for illustration purposes.

Conclusions
The protective power of inverse α-effect weakens upon transition from a five-membered to a six-membered CI (from 4.4 to 2.2 kcal mol−1 for the parent compounds) (Scheme 14). However, this power is still sufficient for preventing the Baeyer–Villiger rearrangement of γ-hydroperoxy-γ-peroxylactones with methyl-substituent in the C6-position. Such compounds were prepared selectively via in situ peroxidation/cyclization of γ-ketoesters with a BF3·Et2O/H2O2 system.
Scheme 14. The free energy activation barriers for the non-catalyzed BV rearrangement of the six- and five-membered Criegee intermediates depending on the layers of protection.

A variety of γ-hydroperoxy-γ-peroxylactones with a methyl-group at the C6-position and a wide scope of substituents at the C5-position were isolated in moderate to good yields. Attempts to prepare γ-hydroperoxy-γ-peroxylactones with a primary alkyl or a aryl group at C6 led to Baeyer–Villiger products. Treatment of γ-hydroperoxy-γ-peroxylactone 2a by Ph3P led to selective reduction of hydroperoxyl-group with the formation of γ-hydroxy-γ-peroxylactone 3a. Although the latter is quite unstable and transformed into a mixture of γ-ketoacid and, probably, γ-ketoperacid, we were able to isolate and characterize 3a. This cyclic peroxide provides the first example of the Criegee intermediate constrained in a six-membered ring, where it is not protected by pronounced misalignment of the two breaking bonds (C–Rm and O–O).
These surprising observations motivated us to explore these systems deeper and led to the discovery of two new stereoelectronic effects important for the BV process – the chair/boat transition and a hidden penalty for the loss of anomeric effect along the reaction path. These effects can amplify the protective power of inverse α-effect in the peroxy-derivatives and provide marginal stability to the OH–CIs to the extent where the C–O bond scission starts to be observed. Interestingly, this is a new reactivity direction for CIs under the neutral conditions.
We have also identified a hidden role that the anomeric effect plays in the fate of Criegee intermediate – the loss of the reactant anomeric 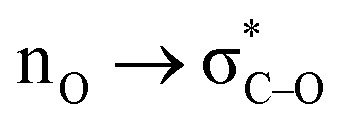 stabilization has to happen in order to activate the “second stereoelectronic effect” (i.e., to realign the lone pair of the exocyclic oxygen with the breaking C–R bond). This sacrifice adds an additional level of kinetic protection for the peroxy CI.
stabilization has to happen in order to activate the “second stereoelectronic effect” (i.e., to realign the lone pair of the exocyclic oxygen with the breaking C–R bond). This sacrifice adds an additional level of kinetic protection for the peroxy CI.
In summary, this work further expanded the growing list of stereoelectronic effects in peroxide chemistry.89–91 We have identified and compared contributions of four effects of potential importance for the BV rearrangement: (a) “primary and secondary stereoelectronic effects”, – two effects that stabilize the TS of the final 1,2-alkyl shift (or withhold this stabilization either in the presence of structural constraints or due to the inverse α-effect) and (b) boat/chair conversion and exo-anomeric effect – the two effects that stabilize the reactant but have to be sacrificed in order to reach the TS (Scheme 15). By understanding the interplay of these stereoelectronic factors, a much deeper understanding of the possible CI transformations is possible including the newly found switch from 1,2-alkyl shift to C–O bond scission. The discovery of a simple method for synthesis of previously elusive γ-hydroperoxy-γ-peroxylactones expands the list of possible synthetic routes to cyclic peroxides with promising spectrum of potential biological activity.
Scheme 15. The three stereoelectronic factors in the BV rearrangement working together to protect the six-membered Criegee intermediate (AE = anomeric effect).

Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
Synthesis and experimental studies of peroxides were supported by the Russian Science Foundation (Grant No. 18-73-00315). X-ray study of 2m was carried out with financial support from Ministry of Science and Higher Education of the Russian Federation using the equipment of Center for molecular composition studies of INEOS RAS. Computational analysis at FSU was supported by the U.S. National Science Foundation (CHE-1800329, I. V. A.).
Electronic supplementary information (ESI) available. CCDC 1980721. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc01025a
Notes and references
- Baeyer A. Villiger V. Ber. Dtsch. Chem. Ges. 1899;32:3625–3633. doi: 10.1002/cber.189903203151. [DOI] [Google Scholar]
- Baeyer A. Villiger V. Ber. Dtsch. Chem. Ges. 1900;33:124–126. doi: 10.1002/cber.19000330115. [DOI] [Google Scholar]
- Yaremenko I. A. Vil' V. A. Demchuk D. V. Terent'ev A. O. Beilstein J. Org. Chem. 2016;12:1647–1748. doi: 10.3762/bjoc.12.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Brink G. J. Arends I. W. C. E. Sheldon R. A. Chem. Rev. 2004;104:4105–4124. doi: 10.1021/cr030011l. [DOI] [PubMed] [Google Scholar]
- Renz M. Meunier B. Eur. J. Org. Chem. 1999;1999:737–750. doi: 10.1002/(SICI)1099-0690(199904)1999:4<737::AID-EJOC737>3.0.CO;2-B. [DOI] [Google Scholar]
- Hassall C. H., in Organic Reactions, John Wiley & Sons, Inc., 2004, 10.1002/0471264180.or009.03 [DOI] [Google Scholar]
- Strukul G. Angew. Chem., Int. Ed. 1998;37:1198–1209. doi: 10.1002/(SICI)1521-3773(19980518)37:9<1198::AID-ANIE1198>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Krow G. R., in Organic Reactions, John Wiley & Sons, Inc., 2004, 10.1002/0471264180.or043.03 [DOI] [Google Scholar]
- Bolm C. Schlingloff G. Weickhardt K. Angew. Chem., Int. Ed. Engl. 1994;33:1848–1849. doi: 10.1002/anie.199418481. [DOI] [Google Scholar]
- Xu S. Wang Z. Zhang X. Zhang X. Ding K. Angew. Chem., Int. Ed. 2008;47:2840–2843. doi: 10.1002/anie.200705932. [DOI] [PubMed] [Google Scholar]
- Walsh C. T. Chen Y.-C. J. Angew. Chem., Int. Ed. Engl. 1988;27:333–343. doi: 10.1002/anie.198803331. [DOI] [Google Scholar]
- Wu W. Cao W. Hu L. Su Z. Liu X. Feng X. Chem. Sci. 2019;10:7003–7008. doi: 10.1039/C9SC01563A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig S. D. Avenell P. J. Bird P. A. Gallati P. Lander K. S. Lye G. J. Wohlgemuth R. Woodley J. M. Biotechnol. Prog. 2002;18:1039–1046. doi: 10.1021/bp0200954. [DOI] [PubMed] [Google Scholar]
- Chen C. Peng J. Li B. Wang L. Catal. Lett. 2009;131:618. doi: 10.1007/s10562-009-9967-1. [DOI] [Google Scholar]
- Doering W. v. E. Speers L. J. Am. Chem. Soc. 1950;72:5515–5518. doi: 10.1021/ja01168a041. [DOI] [Google Scholar]
- Doering W. v. E. Dorfman E. J. Am. Chem. Soc. 1953;75:5595–5598. doi: 10.1021/ja01118a035. [DOI] [Google Scholar]
- Itoh Y. Yamanaka M. Mikami K. Org. Lett. 2003;5:4803–4806. doi: 10.1021/ol0358298. [DOI] [PubMed] [Google Scholar]
- Itoh Y. Yamanaka M. Mikami K. J. Org. Chem. 2013;78:146–153. doi: 10.1021/jo302151r. [DOI] [PubMed] [Google Scholar]
- Bach R. D. J. Org. Chem. 2012;77:6801–6815. doi: 10.1021/jo300727w. [DOI] [PubMed] [Google Scholar]
- Schweitzer-Chaput B. Kurtén T. Klussmann M. Angew. Chem., Int. Ed. 2015;54:11848–11851. doi: 10.1002/anie.201505648. [DOI] [PubMed] [Google Scholar]
- Vil' V. A. Gomes G. d. P. Bityukov O. V. Lyssenko K. A. Nikishin G. I. Alabugin I. V. Terent'ev A. O. Angew. Chem., Int. Ed. 2018;57:3372–3376. doi: 10.1002/anie.201712651. [DOI] [PubMed] [Google Scholar]
- Griesbaum K. Krieger-Beck P. Beck J. Chem. Ber. 1991;124:391–396. doi: 10.1002/cber.19911240224. [DOI] [Google Scholar]
- Anderson L. R. Young D. E. Young W. B. J. Fluorine Chem. 1976;7:491–500. doi: 10.1016/S0022-1139(00)82565-8. [DOI] [Google Scholar]
- Saito I. Nagata R. Yuba K. Matsuura T. Tetrahedron Lett. 1983;24:1737–1740. doi: 10.1016/S0040-4039(00)81758-X. [DOI] [Google Scholar]
- Criegee R. Justus Liebigs Ann. Chem. 1948;560:127–135. doi: 10.1002/jlac.19485600106. [DOI] [Google Scholar]
- Crudden C. M. Chen A. C. Calhoun L. A. Angew. Chem., Int. Ed. 2000;39:2851–2855. doi: 10.1002/1521-3773(20000818)39:16<2851::AID-ANIE2851>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Hannachi H. Anoune N. Arnaud C. Lantéri P. Longeray R. Chermette H. J. Mol. Struct.: THEOCHEM. 1998;434:183–191. doi: 10.1016/S0166-1280(98)00096-7. [DOI] [Google Scholar]
- Li G. Garcia-Borràs M. Fürst M. J. L. J. Ilie A. Fraaije M. W. Houk K. N. Reetz M. T. J. Am. Chem. Soc. 2018;140:10464–10472. doi: 10.1021/jacs.8b04742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paryzek Z. Koenig H. Tetrahedron. 2012;68:9061–9067. doi: 10.1016/j.tet.2012.08.060. [DOI] [Google Scholar]
- Goodman R. M. Kishi Y. J. Am. Chem. Soc. 1998;120:9392–9393. doi: 10.1021/ja982188g. [DOI] [Google Scholar]
- Noyori R. Sato T. Kobayashi H. Tetrahedron Lett. 1980;21:2569–2572. doi: 10.1016/0040-4039(80)80132-8. [DOI] [Google Scholar]
- Noyori R. Kobayashi H. Sato T. Tetrahedron Lett. 1980;21:2573–2576. doi: 10.1016/0040-4039(80)80133-X. [DOI] [Google Scholar]
- Chandrasekhar S. Roy C. D. J. Chem. Soc., Perkin Trans. 2. 1994:2141–2143. doi: 10.1039/P29940002141. [DOI] [Google Scholar]
- Juaristi E. Gomes G. d. P. Terent'ev A. O. Notario R. Alabugin I. V. J. Am. Chem. Soc. 2017;139:10799–10813. doi: 10.1021/jacs.7b05367. [DOI] [PubMed] [Google Scholar]
- Comment: Similar to the earlier work,21,34 we use the term “inverse α-effect” to describe intramolecular stereoelectronic interactions in systems with two adjacent heteroatoms. It is conceptually related to the well-known “α-effect”, i.e., the enhancement of nucleophilicity due to the presence of an adjacent (alpha) atom in intermolecular reactions. The fundamental question at the heart of both α-effects is whether the lone pairs of two directly connected heteroatoms can combine into a more powerful donor than each of the lone pairs taken separately. In a systematic study of the intramolecular α-effects,34 we found that contrary to the expectations based on the simple orbital mixing model, the lone pairs in a pair of directly connected heteroatoms are not raised in energy to become stronger donors towards adjacent acceptors
- Vil' V. A. Barsegyan Y. A. Barsukov D. V. Korlyukov A. A. Alabugin I. V. Terent'ev A. O. Chem.–Eur. J. 2019;25:14460–14468. doi: 10.1002/chem.201903752. [DOI] [PubMed] [Google Scholar]
- Zhou W.-S. Xu X.-X. Acc. Chem. Res. 1994;27:211–216. doi: 10.1021/ar00043a005. [DOI] [Google Scholar]
- White N. J. Science. 2008;320:330–334. doi: 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- Haynes R. K. Vonwiller S. C. Acc. Chem. Res. 1997;30:73–79. doi: 10.1021/ar950058w. [DOI] [Google Scholar]
- Kumar V. Mahajan A. Chibale K. Biorg. Med. Chem. 2009;17:2236–2275. doi: 10.1016/j.bmc.2008.10.072. [DOI] [PubMed] [Google Scholar]
- Meshnick S. R. Jefford C. W. Posner G. H. Avery M. A. Peters W. Parasitol. Today. 1996;12:79–82. doi: 10.1016/0169-4758(96)80660-0. [DOI] [PubMed] [Google Scholar]
- Vil' V. A. Yaremenko I. A. Ilovaisky A. I. Terent'ev A. O. Molecules. 2017;22:1881. doi: 10.3390/molecules22111881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y. Dong Y. Vennerstrom J. L. Med. Res. Rev. 2004;24:425–448. doi: 10.1002/med.10066. [DOI] [PubMed] [Google Scholar]
- Jefford C. W. Drug Discovery Today. 2007;12:487–495. doi: 10.1016/j.drudis.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Opsenica D. M. Šolaja B. A. J. Serb. Chem. Soc. 2009;74:1155–1193. doi: 10.2298/JSC0911155O. [DOI] [Google Scholar]
- Dembitsky V. M. Eur. J. Med. Chem. 2008;43:223–251. doi: 10.1016/j.ejmech.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Chaturvedi D. Goswami A. Pratim Saikia P. Barua N. C. Rao P. G. Chem. Soc. Rev. 2010;39:435–454. doi: 10.1039/B816679J. [DOI] [PubMed] [Google Scholar]
- Liu D.-Z. Liu J.-K. Nat. Prod. Bioprospect. 2013;3:161–206. doi: 10.1007/s13659-013-0042-7. [DOI] [Google Scholar]
- Keiser J. Utzinger J. Trends Parasitol. 2007;23:555–562. doi: 10.1016/j.pt.2007.07.012. [DOI] [PubMed] [Google Scholar]
- Muraleedharan K. M. Avery M. A. Drug Discovery Today. 2009;14:793–803. doi: 10.1016/j.drudis.2009.05.008. [DOI] [PubMed] [Google Scholar]
- Panic G. Duthaler U. Speich B. Keiser J. Int. J. Parasitol. 2014;4:185–200. doi: 10.1016/j.ijpddr.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efferth T. Marschall M. Wang X. Huong S.-M. Hauber I. Olbrich A. Kronschnabl M. Stamminger T. Huang E.-S. J. Mol. Med. 2002;80:233–242. doi: 10.1007/s00109-001-0300-8. [DOI] [PubMed] [Google Scholar]
- Efferth T. Romero M. R. Wolf D. G. Stamminger T. Marin J. J. G. Marschall M. Clin. Infect. Dis. 2008;47:804–811. doi: 10.1086/591195. [DOI] [PubMed] [Google Scholar]
- Jia M. Zhao R. Xu B. Yan W. Chu F. Gu H. Xie T. Xiang H. Ren J. Chen D. Wang P. Lei H. MedChemComm. 2017;8:148–151. doi: 10.1039/C6MD00344C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitis M. Environ. Int. 2004;30:47–55. doi: 10.1016/S0160-4120(03)00147-8. [DOI] [PubMed] [Google Scholar]
- Chassot A. L. C. Poisl M. I. P. Samuel S. M. W. Braz. Dent. J. 2006;17:117–121. doi: 10.1590/S0103-64402006000200006. [DOI] [PubMed] [Google Scholar]
- Baldry M. G. C. French M. S. Water Sci. Technol. 1989;21:203–206. doi: 10.2166/wst.1989.0100. [DOI] [Google Scholar]
- Alvaro J. E. Moreno S. Dianez F. Santos M. Carrasco G. Urrestarazu M. J. Food Eng. 2009;95:11–15. doi: 10.1016/j.jfoodeng.2009.05.003. [DOI] [Google Scholar]
- Luukkonen T. Pehkonen S. O. Crit. Rev. Environ. Sci. Technol. 2017;47:1–39. doi: 10.1080/10643389.2016.1272343. [DOI] [Google Scholar]
- Wu X.-F. Gong J.-L. Qi X. Org. Biomol. Chem. 2014;12:5807–5817. doi: 10.1039/C4OB00276H. [DOI] [PubMed] [Google Scholar]
- Schmidt R. J. Appl. Catal., A. 2005;280:89–103. doi: 10.1016/j.apcata.2004.08.030. [DOI] [Google Scholar]
- Zhu Y. Wang Q. Cornwall R. G. Shi Y. Chem. Rev. 2014;114:8199–8256. doi: 10.1021/cr500064w. [DOI] [PubMed] [Google Scholar]
- Fisher T. J. Dussault P. H. Tetrahedron. 2017;73:4233–4258. doi: 10.1016/j.tet.2017.03.039. [DOI] [Google Scholar]
- Gaylord N. G. Mandal B. M. Martan M. J. Polym. Sci., Polym. Lett. Ed. 1976;14:555–559. doi: 10.1002/pol.1976.130140908. [DOI] [Google Scholar]
- Emami S. H. Salovey R. Hogen-Esch T. E. J. Polym. Sci., Part A: Polym. Chem. 2002;40:3021–3026. doi: 10.1002/pola.10367. [DOI] [Google Scholar]
- Russell K. E. Prog. Polym. Sci. 2002;27:1007–1038. doi: 10.1016/S0079-6700(02)00007-2. [DOI] [Google Scholar]
- Klapötke T. M. and Wloka T., in PATAI'S Chemistry of Functional Groups, John Wiley & Sons, Ltd, 2009, 10.1002/9780470682531.pat0879 [DOI] [Google Scholar]
- Swern D., Organic peroxides, Wiley-Interscience, 1970 [Google Scholar]
- The Chemistry of Peroxides, ed. J. F. Liebman and A. Greer, John Wiley & Sons, 2006 [Google Scholar]
- Schulz M., in Peroxide Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2005, pp. 1–38, 10.1002/3527600396.ch1 [DOI] [Google Scholar]
- Vil' V. A. Gomes G. d. P. Ekimova M. V. Lyssenko K. A. Syroeshkin M. A. Nikishin G. I. Alabugin I. V. Terent'ev A. O. J. Org. Chem. 2018;83:13427–13445. doi: 10.1021/acs.joc.8b02218. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar S. Deo Roy C. Tetrahedron Lett. 1987;28:6371–6372. doi: 10.1016/S0040-4039(01)91376-0. [DOI] [Google Scholar]
- Adam W. Szendrey L. M. J. Am. Chem. Soc. 1974;96:7135–7137. doi: 10.1021/ja00829a070. [DOI] [Google Scholar]
- Adam W. Szendrey L. J. Chem. Soc. D. 1971:1299–1300. doi: 10.1039/C29710001299. [DOI] [Google Scholar]
- Gibson D. H. Joseph J. T. Tetrahedron Lett. 1972;13:3483–3486. doi: 10.1016/S0040-4039(01)94078-X. [DOI] [Google Scholar]
- Adam W. Kliem U. Peters E.-M. Peters K. von Schnering H.-G. J. Prakt. Chem. 1988;330:391–405. doi: 10.1002/prac.19883300309. [DOI] [Google Scholar]
- Xu Z.-J. Tan D.-X. Wu Y. Org. Lett. 2015;17:5092–5095. doi: 10.1021/acs.orglett.5b02599. [DOI] [PubMed] [Google Scholar]
- Kukovinets O. S. Kasradze V. G. Galin F. Z. Spirikhin L. V. Zainullin R. A. Kislitsyn M. I. Abdullin M. I. Kunakova R. V. Tolstikov G. A. Russ. J. Org. Chem. 2002;38:511–518. doi: 10.1023/A:1016543021664. [DOI] [Google Scholar]
- Wilson S. L. Schuster G. B. J. Org. Chem. 1986;51:2056–2060. doi: 10.1021/jo00361a023. [DOI] [Google Scholar]
- Sun X. Li X. Song S. Zhu Y. Liang Y.-F. Jiao N. J. Am. Chem. Soc. 2015;137:6059–6066. doi: 10.1021/jacs.5b02347. [DOI] [PubMed] [Google Scholar]
- Takayoshi I. Jiro T. Hideo T. Bull. Chem. Soc. Jpn. 2009;82:737–742. doi: 10.1246/bcsj.82.737. [DOI] [Google Scholar]
- Wu A.-B. Cheng H.-W. Hu C.-M. Chen F.-A. Chou T.-C. Chen C.-Y. Tetrahedron Lett. 1997;38:621–622. doi: 10.1016/S0040-4039(96)02379-9. [DOI] [Google Scholar]
- Najjar F. Gorrichon L. Baltas M. André-Barrès C. Vial H. Org. Biomol. Chem. 2005;3:1612–1614. doi: 10.1039/B503402G. [DOI] [PubMed] [Google Scholar]
- One should keep in mind that the BV rearrangement is greatly accelerated by acid catalysis. Hence, the intrinsic trends in reactivity discussed in this section can be further amplified in the presence of Brønsted or Lewis acids
- Alabugin I. V., Stereoelectronic Effects: A Bridge Between Structure and Reactivity, John Wiley & Sons, Ltd., 2016 [Google Scholar]
- Mora-Diez N. Keller S. Alvarez-Idaboy J. R. Org. Biomol. Chem. 2009;7:3682–3690. doi: 10.1039/B906058H. [DOI] [PubMed] [Google Scholar]
- Carlqvist P. Eklund R. Brinck T. J. Org. Chem. 2001;66:1193–1199. doi: 10.1021/jo001278c. [DOI] [PubMed] [Google Scholar]
- Snowden M. Bermudez A. Kelly D. R. Radkiewicz-Poutsma J. L. J. Org. Chem. 2004;69:7148–7156. doi: 10.1021/jo0491307. [DOI] [PubMed] [Google Scholar]
- Gomes G. d. P. Vil' V. A. Terent'ev A. O. Alabugin I. V. Chem. Sci. 2015;6:6783–6791. doi: 10.1039/C5SC02402A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes G. d. P. Yaremenko I. A. Radulov P. S. Novikov R. A. Chernyshev V. V. Korlyukov A. A. Nikishin G. I. Alabugin I. V. Terent'ev A. O. Angew. Chem., Int. Ed. 2017;56:4955–4959. doi: 10.1002/anie.201610699. [DOI] [PubMed] [Google Scholar]
- Yaremenko I. A. Gomes G. d. P. Radulov P. S. Belyakova Y. Y. Vilikotskiy A. E. Vil' V. A. Korlyukov A. A. Nikishin G. I. Alabugin I. V. Terent'ev A. O. J. Org. Chem. 2018;83:4402–4426. doi: 10.1021/acs.joc.8b00130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.