

Abstract
Cutaneous squamous cell carcinoma (cSCC) ranks second in the frequency of all skin cancers. The balance between keratinocyte proliferation and differentiation is disrupted in the pathological development of cSCC. DLX3 is a homeobox transcription factor which plays pivotal roles in embryonic development and epidermal homeostasis. To investigate the impact of DLX3 expression on cSCC prognosis, we carried out clinicopathologic analysis of DLX3 expression which showed statistical correlation between tumors of higher pathologic grade and levels of DLX3 protein expression. Further, Kaplan-Meier survival curve analysis demonstrated that low DLX3 expression correlated with poor patient survival. To model the function of Dlx3 in skin tumorigenesis, a two-stage dimethylbenzanthracene (DMBA)/12-O-tetradecanoylphorbol 13-acetate (TPA) study was performed on mice genetically depleted of Dlx3 in skin epithelium (Dlx3cKO). Dlx3cKO mice developed significantly more tumors, with more rapid tumorigenesis compared to control mice. In Dlx3cKO mice treated only with DMBA, tumors developed after ~16 weeks suggesting that loss of Dlx3 has a tumor promoting effect. Whole transcriptome analysis of tumor and skin tissue from our mouse model revealed spontaneous activation of the EGFR-ERBB2 pathway in the absence of Dlx3. Together, our findings from human and mouse model system support a tumor suppressive function for DLX3 in skin and underscore the efficacy of therapeutic approaches that target EGFR-ERBB2 pathway.
Introduction
Cutaneous squamous cell carcinoma (cSCC) is the second most common form of skin cancer [1]. Although cSCCs have a generally favorable prognosis, approximately 1.5%–2% of patients die from the disease [2, 3]. cSCC constitutes a spectrum of progressively advanced malignancies, ranging from precursor actinic keratosis (AK), to squamous cell carcinoma (SCC) in situ, invasive SCC, and metastatic SCC. Development and progression of cSCC is known to be regulated by a group of traditional oncogenic and tumor suppressive proteins (e.g., RAS, p53) [4]; however, emerging evidence suggests that homeoproteins can act as modulators of tumor initiation and progression through regulation of proliferation, migration and survival pathways [5][6][7, 8, 9] where they act as tumor suppressors or proto-oncogenes depending on cancer type. Additionly, dysregulation of homeobox genes has been associated with increased cancer risk and poor survival rate in cancer patients [7, 9].
DLX3 is a homeobox transcription factor known to regulate keratinocyte differentiation [10, 11]. Epidermal-specific deletion of Dlx3 in murine skin leads to epidermal hyperplasia, accompanied by barrier disruption [12] and an associated development of an IL17 dependent inflammatory response via activation of STAT3 signaling [13]. Dlx3 promotes p53-dependent p21 transcription to modulate cell cycle exit in the skin, functioning as a proliferative brake [14]. The role of DLX3 in cell cycle regulation and absence of expression in human SCCs supports that this transcription factor plays a crucial role in tumorigenesis [14].
Dysregulated growth factors and their receptors have impact on tumor initiation and progression [15]. The epidermal growth factor receptor family of tyrosine kinases (EGFR, also known as ERBB1/HER1, ERBB2/HER2/neu, ERBB3/HER3, and ERBB4/HER4) becomes activated by receptor overexpression as well as ligand-dependent and ligand-independent mechanisms. Skin‐specific Erbb2 [16] and Erbb3 [17] knockout mice have shown that both receptors play a major role in non-melanoma skin cancer promotion, and EGFR plays a crucial role in skin carcinogenesis [18, 19]. ERBB2 signaling is mostly transduced by the phosphatidylinositol 3-kinase/Rac-alpha serine/threonine-protein kinase (PI3K/AKT) and the mitogen-activated protein kinase (MAPK) pathways [16]. ERBB2 is frequently mutated or overexpressed in a range of solid tumors [20], and two-stage skin carcinogenesis indicated that ERBB2 inhibition may be suitable for suppressing tumor promotion and is a highly effective target in medical oncology [21].
In this report, we show the expression profile of DLX3 in human SCCs and its prognostic significance in the patients survival. Furthermore, we demonstrate DLX3’s tumor suppressive role in epithelial tumor development and tumor progression, and identify specific mechanisms of Dlx3-mediated skin carcinogenesis via upregulation of EGFR ligands and activation of EGFR/ERBB2/MAPK pathway.
Materials and Methods
Patients
Investigation has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and national and international guidelines. 121 patients with SCC and 6 benign or intermediate skin tumors (verruca vulgaris, actinic keratosis and Bowen’s disease) underwent surgical resection and/or chemotherapy with/without radiation therapy at Gunma University Hospital. Healthy skin tissue collected for skin graft was used as control. Clinical stages were defined according to American Joint Committee on Cancer (AJCC) 8th edition. This study was approved by the Research Ethics Committee of Gunma University (HS2019–071).
Immunohistochemical and immunofluorescence staining
For histological and immunohistochemical analysis, human tissues were fixed with 4% PFA and embedded in paraffin. 4 μm sections were deparaffinized and antigens retrieved with 10mM sodium citrate. Sections were treated with endogenous peroxidase-blocking reagent (Dako) for 5 minutes and protein block (Dako) for 10 minutes at room temperature. Sections were incubated with DLX3 antibody (Abcam ab178428, 1:500 dilution) overnight at 4°C, followed by incubation with horseradish peroxidase-labeled secondary Abs (ENVISION: Dako) and visualized with 3, 3′-diaminobenzidine tetra-hydrochloride, counterstained with Mayer’s hematoxylin.
For immunofluorescence staining, sections were blocked with 3% dry milk-PBS supplemented with 5% normal goat serum for 1 hour at room temperature. Antibody details are in Supplementary Table 1. Sections were counterstained with 4,6-diamidino-2-phenylindole (DAPI) to visualize nuclei.
Proliferative activity in tumors was assessed by calculating the percentage of PCNA-positive nuclei examined using laser-scanning confocal microscope Zeiss LSM780. The number of positive cells was expressed as a percentage of the total number counted to give a PCNA score. Two independent sections per animal were stained and analyzed. A minimum of five microscopic fields per section were counted manually by two independent investigators and the mean was used for analysis.
Histopathological analysis
Pathological grade of SCC was performed by a clinical histopathologist at the University of Gunma and classified into Grade; (1), Well differentiated, (2), Moderately differentiated and (3), Poorly differentiated. The DLX3 expression scores were assessed as follows: 0, ≤ 10% of tumor area stained; 1, 11–25% stained; 2, 26–50% stained; 3, ≥51 % stained. Tumors scoring 2 and 3 were defined as high-expression tumors.
Survival analysis according to DLX3 expression
Overall survival analyses according to the expression level of Dlx3 transcripts was done using the Kaplan-Meier plotter (n = 500 patients), and the OncoLnc (n = 497 patients) hnSCC datasets. The median of the expression distribution was used to establish the low and high expression groups and, subsequently, the Mantel-Cox test was applied to statistically validate the differences between the survival distributions.
Dlx3cKO mice and genotyping
Male and female Dlx3cKO mice were generated and genotyped by PCR at the age of 3 weeks as previously described [10]. Mice of mixed CD-1 and C57BL/6 background, 8 to 10 weeks of age, were used according to the animal procedures approved by the Institutional Animal Care and Use Committee of the NIAMS.
Chemical Skin Carcinogenesis
Groups of 8–10 weeks old mice were subjected to the DMBA/TPA tumor induction regimen as previously detailed [22]. The Dlx3cKO experimental group consisted of eleven mice and the control group had twenty-three mice. Two days after shaving, a single initiating dose of topical DMBA (100ug/200ul acetone, Sigma-Aldrich) was applied to the dorsal skin. One week later, tumor promoter TPA (5 ug in 200 uL acetone/mouse) was applied two times per week for 5 weeks. The onset of tumor formation, number and size of tumors were recorded weekly. Following termination of the TPA treatments, conversion of benign tumors to SCCs were monitored. Tumor tissues were harvested for histological verification at week 40 post-initiation with DMBA.
DMBA only treatments were done in two protocols. For the first protocol, the dorsal skin of seven Dlx3cKO and nine control mice was topically treated with a single dose of DMBA (100ug/200ul acetone/mouse). Animals were terminated 34 weeks after the initiation of the protocol and tumors were collected. In the second protocol, three doses of DMBA (100ug/200ul) were topically administered on the dorsal side of twelve Dlx3cKO and ten control mice. The time interval between treatments was one week. The number of tumors exceeding 1mm in diameter was monitored weekly. Animals were euthanized 31 weeks after the initiation of the protocol, and tumors collected. For tumor size (papilloma burden), we first took average of all tumors on one mouse and then plot it along tumor size (two categories: small papilloma, 2–4mm and large papilloma, 4–6mm). Tissue samples were either snap-frozen in liquid nitrogen or fixed in fresh phosphate-buffered 4% paraformaldehyde for 24 hours at 4°C and embedded in paraffin.
Genome-wide transcriptome analysis by RNA sequencing (RNA-Seq)
Total RNA from mouse skin was used to create ribosomal RNA-depleted cDNA libraries (NEBNext® Ultra™ RNA Library Prep Kit). Each sample was sequenced to 40 million raw reads in a single end 75bp sequencing run on the Illumina NextSeq500, demultiplexed and converted to FastQ data. The NIAMS Biodata Mining and Discovery Section mapped FastQ data to the mouse genome build mm10 using TopHat 2.1.1. Gene expression values (RPKM, reads per kilobase exon per million mapped reads) were calculated and log2 transformed (with a 0.1 offset). Differentially expressed genes (DEG) were determined by ANOVA, and a DEG heat map was created with Partek Genomic Suite. DEGs were defined as having FDR (q-value) ≤ 0.05 and with raw FC ≥ 1.8. Ingenuity Pathway Analysis (Qiagen: www.ingenuity.com) used Fishers exact test to detect significantly enriched (P-value < 0.05) upstream regulators and disease pathways from expression data.
Accession numbers
Raw and analyzed RNA-Seq data have been deposited in the Gene Expression Omnibus (GEO) site GSE161372.
RNA extraction and qRT-PCR
Total RNA was isolated by RNeasy Mini Kits (Qiagen) and was subjected to reverse transcription using a SuperScript III First-Strand Synthesis System (Invitrogen). Quantitative real-time PCR was performed with Taqman system (Applied Biosystems, Foster City, CA) using StepOnePlus real-time PCR system (Applied Biosystems). TaqMan Gene Expression Assays and Master Mix were obtained from Thermo Fisher; Dlx3-Mm00438428_m1, Elf3-Mm001295975_m1, Elf5-Mm00468732_m1, Epgn-Mm00504344_m1,Hbegf-Mm00439306_m1, Ereg-Mm00514794_m1, Areg-Mm01354339_m1 Erbb3-Mm01159999_m1, Egfr-Mm01187858_m1, Erbb2-Mm00658541_m1. Relative expression was normalized against the housekeeping gene Gapdh or β-actin. Fold changes were calculated by the Ct method as described previously [23]. Statistical comparisons were performed using Two-way ANOVA.
Western blot analysis
Dorsal mouse skins were treated with trypsin overnight at 4°C for epidermis and dermis separation [24]. Epidermis was sonicated for lysis in denaturing buffer composed of 300 mM NaCl, 2 mM EDTA, 20 mM Hepes, 1% SDS, 0.1 mM hemin chloride, 5 mM NEM, 20 mM NaF, 100mM PMSF. 30 ug of protein lysate were loaded on 4–12% SDS-PAGE gels (Invitrogen, Carlsbad, CA, USA). After electrophoresis, transfer to PVDF membrane (Invitrogen, Carlsbad, CA, USA), and incubation in 5% non fat dry milk, membranes were probed with primary antibody. Blots were rinsed in TBST and incubated in peroxidase-conjugated secondary antibodies, accordingly. Proteins were visualized using Bio-Rad Chemi Doc XRS with ECL (Pierce Biotech, Waltham, MA, USA). Antibodies used are detailed in Supplementary Table 2.
Cell culture and treatments
Primary keratinocytes were isolated from FVB/N mice as previously described [24], and were plated in low calcium medium (8% chelated fetal calf serum, 0.05 mM Ca2+). siGENOME SMARTpool (Dharmacon, Lafayette, CO) for mouse Egfr small interfering RNA (M-040411-01-0010), non-targeting control small interfering RNA (D-001206-13-05) and ON-TARGETplus Mouse Dlx3 (L-041957-01-0010), ON-Target plus Mouse Erbb2 (L-064147-00-0010) were obtained and transfected using RNAiMAX (Life Technologies) at final concentration of 20nM. Small interfering RNA experiments were performed during 24–48 hours after transfection.
Proliferation assay
Mouse primary keratinocytes were seeded onto 6-well plates. Images cell confluency were automatically acquired with nine images per well every 6 hours for 48 hours within the CO2 incubator by the IncuCyteS3 instrument software (Essen BioScience, Ann Arbor, MI).
Statistical analysis
Pathway enrichment statistics were calculated within the Ingenuity software package using Fisher’s exact test with Benjamini-Hochberg correction for multiple testing. Upstream regulators and gene ontology enrichment P-values were similarly calculated within IPA using Fisher’s exact test. Data analysis was performed with GraphPad Prism, version 8.4.1. P-values were calculated using the Student t test (two-sided) or by analysis of two-way analysis of variance. P-value < 0.05 was considered statistically significant. Error bars represent standard errors of the mean (s.e.m) and numbers of experiments (n) are as indicated.
Results
Loss of DLX3 expression in tumors is associated with poor survival in SCC patients
To examine how DLX3 might contribute to skin cancer pathogenesis and progression, we analyzed the expression levels of DLX3 in skin lesions from 121 patients with SCC, 6 benign or intermediate skin tumors and matched healthy skin tissue collected for skin graft. Nuclear DLX3 detection is reduced as skin tumors progressed from precancer stage to advanced SCCs (Fig. 1a). The clinicopathological features of the 121 patients included in this study are shown in Table 1. The group was composed of 75 males and 46 females. The median age was 77 years (range 50–97) for men and 84 years (range 54–96) for women. The most frequent anatomical site affected by SCC was the face and lower limb, involved in 80% of patients, while the remaining 20% were divided as follows: 5% to the trunk and 14% the upper limb. Of the 121 primary tumors, 63% were Grade 1 (well-differentiated), 29% were Grade 2 (moderately differentiated), and 8% were poorly differentiated Grade 3 tumors. Statistical correlation revealed that pathologic tumor grade correlated with low DLX3 expression. Age, sex and pathologic differentiation stage were not correlated with DLX3 expression (Table 1).
Figure 1. Decreased expression of DLX3 correlates with skin cancer progression and poor survival in patients.
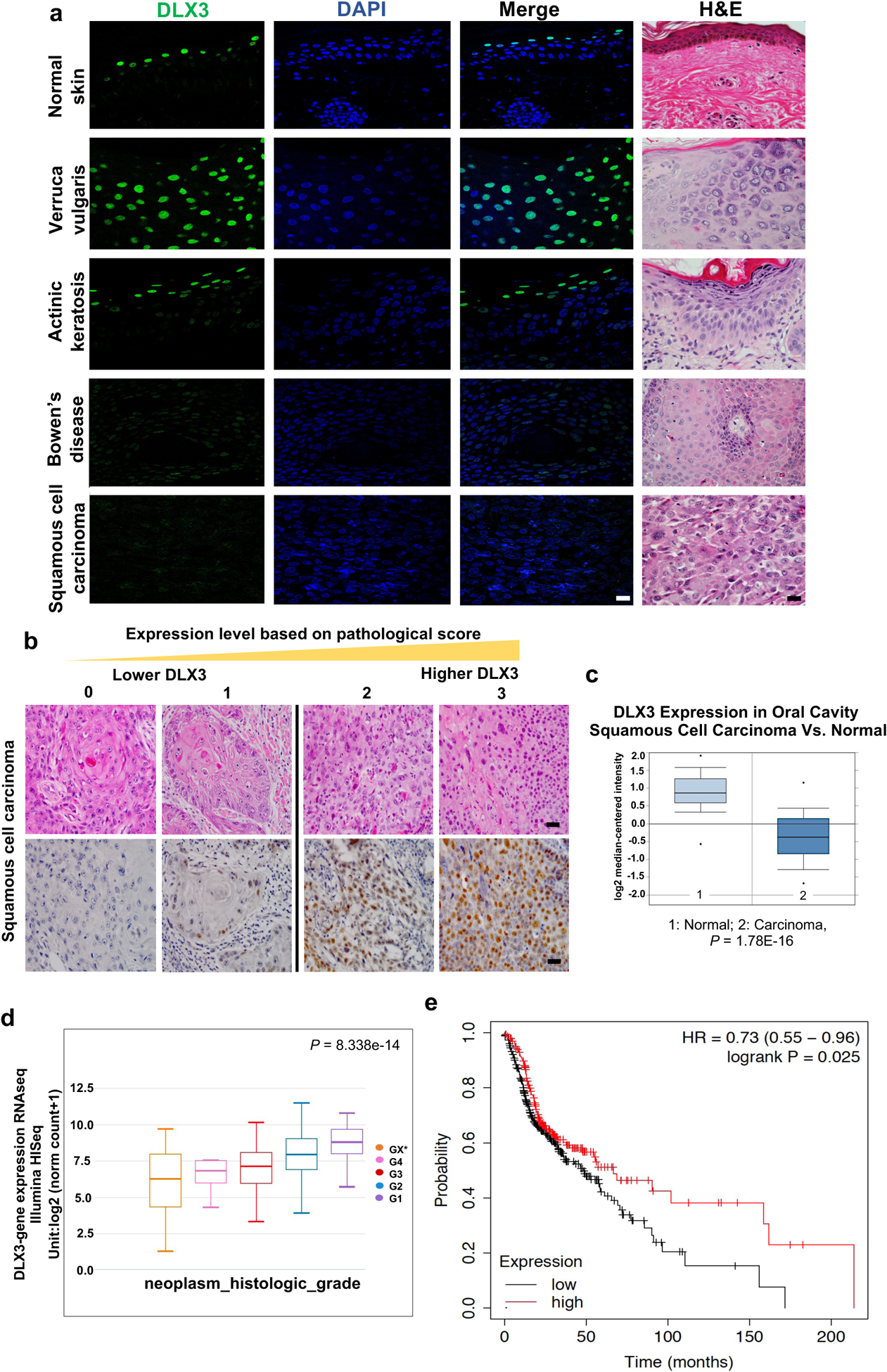
(a) Immunofluorescence and Hematoxylin/eosin (H&E) staining using antibodies specific for DLX3. (Scale bar: 25μm). A total of 121 SCC patient samples and six benign skin tumors were stained and analyzed. (b) Representative H&E and immunohistochemical staining of DLX3 in SCC with a reactivity score of 0 to 3. (Scale bar: 50μm). Pathological score was defined as: 0, ≤ 10% of tumor area stained; 1, 11–25% stained; 2, 26–50% stained; 3, ≥51 % stained. Tumors scoring 2 and 3 were defined as high-expression tumors. (c) Box-plots showing DLX3 expression level is significantly decreased in Oral Cavity Squamous Cell Carcinoma [1=Normal; 2=Squamous Cell Carcinoma] as determined by TCGA Database. (d) Box-plots showing Dlx3 expression in HNSCC samples determined by TCGA Database. G1 to G4 indicate increase in tumor stages. *Grading could not be defined. [P-value=8.338e–14]. (e) Survival of hnSCC patients according to the expression profile of Dlx3 mRNA in 500 patients with available clinical data (Source data: Kaplan-Meier Plotter, Pan-cancer RNA-seq, https://kmplot.com/analysis/index.php?p=service&cancer=pancancer_rnaseq). The logrank P-value is indicated on the graph.
Table 1.
Clinical features of SCC patients with low or high DLX3 expression
| DLX3 | ||||||
|---|---|---|---|---|---|---|
| Low | High | |||||
| (n=33) | (n=88) | |||||
| Variables | P | |||||
| Age (years; mean ± SE) | 78.3 ± 1.9 | 77.7 ± 1 | > 0.99 | |||
| Sex | Male (%) | 57.60 | (19/33) | 63.60 | (56/88) | 0.4686 |
| Female (%) | 42.40 | (14/33) | 36.40 | (32/88) | ||
| Part | Face (%) | 69.70 | (23/33) | 63.60 | (56/88) | |
| Trunk (%) | 9.10 | (3/33) | 3.40 | (3/88) | ||
| Upper (%) | 6.10 | (2/33) | 17.10 | (15/88) | 0.0249 | |
| Lower (%) | 15.10 | (5/33) | 15.90 | (14/88) | ||
| Grade | G1 (%) | 54.50 | (18/33) | 65.90 | (58/88) | |
| G2 (%) | 27.30 | (9/33) | 30.70 | (27/88) | ||
| G3 (%) | 18.20 | (6/33) | 3.40 | (3/88) | 0.0008 | |
| Stage | I (%) | 36.30 | (12/33) | 38.60 | (34/88) | > 0.99 |
| II (%) | 39.40 | (13/33) | 48.90 | (43/88) | ||
| III (%) | 15.10 | (5/33) | 8.00 | (7/88) | ||
| IV (%) | 9.10 | (3/33) | 4.50 | (4/88) | ||
P values were obtained by the Fisher exact test.
We next found using standard immunohistochemical technique that of the 121 patients with SCC evaluated, 33 showed score-0 or −1 (25%), 50 showed score-2 (38.1%), and 38 showed score-3 (29%) (Fig. 1b). Based on this scoring, tumors were grouped into low (n=33) and high (n=88) DLX3 expression cohorts. We had previously evaluated DLX3 mRNA levels using Oncomine and Cancer Genome Atlas (TCGA) databases [14]. We re-evaluated these results with more recent databases accessible in TCGA, and show that DLX3 mRNA levels were reduced in 75 out of 116 (63.7%) in oral cavity squamous cell carcinoma (Fold-Change: −5.895; P-value = 1.78E-16) (Fig. 1c). DLX3 expression also exhibited a negative correlation with the increased tumor grade malignancy in hnSCC tumors from TCGA studies (P-value = 8.338e-14) (Fig. 1d). We next used Kaplan-Meier plotter and OncoLnc in silico analysis tool to show that levels of the DLX3 mRNA directly correlated with poor hnSCC patient prognosis in independent datasets (Fig. 1e and Supplementary Fig. S1).
Dlx3 deficiency enhances skin carcinogenesis in a two-stage skin cancer model
To analyze DLX3 function in skin carcinogenesis in vivo, we used Dlx3cKO mice (Supplementary Fig. S2a–c) [10]. Western blots verified absence of DLX3 protein in Dlx3cKO mice (Supplementary Fig. S2d).
We next examined the effect of Dlx3 deletion using a mouse two-stage skin carcinogenesis protocol [22]. Mice were treated once with DMBA (100ug/200ul) and subsequently with TPA (5ug/200ul) twice a week for 5 weeks (Fig. 2a). In control mice, tumors began to appear at 7 weeks after TPA treatment. However, tumors appeared at 3 weeks in Dlx3cKO mice (Fig. 2b). Histopathologically, tumors were defined as papillomas, carcinoma in situ and squamous cell carcinoma (Fig. 2c). It is noteworthy that Dlx3 deficiency also correlated with higher incidence of tumor numbers (Fig. 2d). Furthermore, the average number of tumor-bearing mice was higher in the Dlx3cKO mice compared to control mice. By 7 weeks, ~43% of the null mice had developed first tumors versus ~5% of control mice. By 35 weeks, 100% of null mice versus ~24% of control mice had developed tumors and this percent remained consistent for 40 weeks after which the animals were euthanized and tumors collected (Fig. 2e). The average number of tumors that appeared in Dlx3-deficient mice at week 26 was increased (3.9 tumors/mouse) compared to control (0.9 tumors/mouse) (Fig. 2f). This difference remained significant from week 21 until the end (Fig. 2d–f). Moreover, 16 out of 23 control-treated mice did not develop any tumors during the entire protocol (Fig. 2d). To evaluate whether the higher number of tumors observed in the Dlx3-deficient mice presented increased proliferation in epidermal cells, we performed immunofluorescence analysis with PCNA (Fig. 2g) and showed a significantly higher level of PCNA-positive tumor cells (P-value <0.001) (Fig. 2g, h). All Dlx3cKO tumors presented strong de novo expression of KRT13 (Fig. 2g), a cytokeratin whose expression correlates with malignant conversion in mouse chemical carcinogenesis [25]. Histological analysis of DMBA-TPA treated Dlx3cKO skin showed extensive epidermal thickness compared to control (Supplementary Fig. S3a). Moreover, treated skin and tumors of Dlx3-deficient mice presented significantly more proliferating cells than the treated skin and tumors of control animals (Supplementary Fig. S3b). We observed an increased number of inflammatory (CD45-positive) cells infiltrating the dermis of Dlx3cKO mice skin and tumors (Supplementary Fig. S3c–e), corroborating what had been previously observed after TPA topical application [26].
Figure 2. Dlx3 deficiency enhances the development of DMBA/TPA-induced skin tumors.
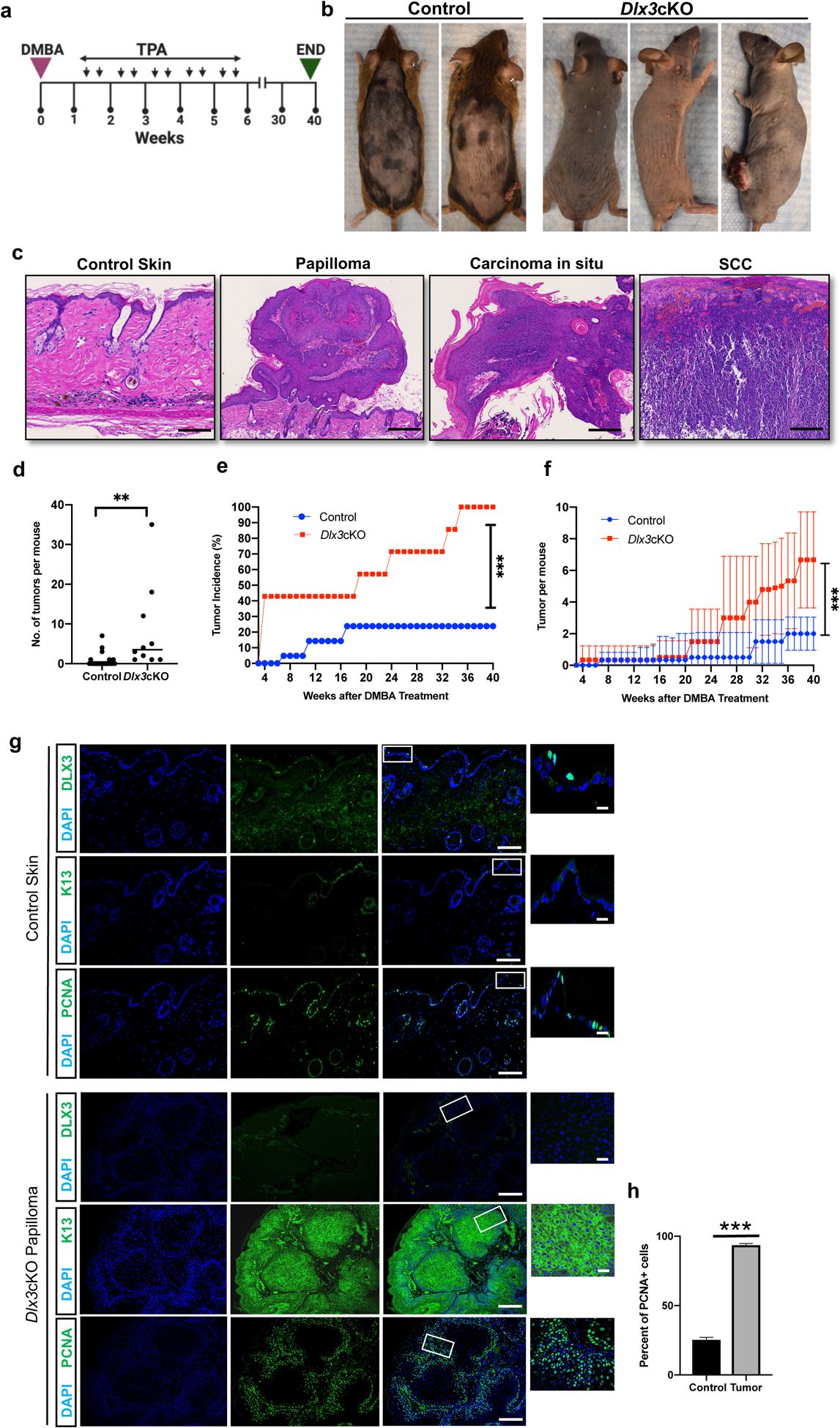
(a) Experimental design for a chemically induced skin tumorigenesis study. After DMBA treatment, mice were applied with TPA twice a week for 5 weeks. Tumors larger than 1 mm in diameter were counted weekly. At week 40, tumors were collected for analysis. Dlx3cKO; n=11: Control; n=23 (b) Dorsal view of control and Dlx3cKO mice at week 40. (c) H&E staining of wild type skin, papilloma, carcinoma in situ and SCC (Scale bar, 100μm). (d) Number of tumors per mouse. (e) Tumor incidence; percentage (%) of mice that developed any tumors. (f) Average number of tumors per mouse in the total number of mice tested. (g) Representative immunohistochemistry of DLX3, K13 and PCNA (Scale bars, 100μm and 10μm). As shown DLX3 expression is absent in the tumors. Higher PCNA expression and elevated K13 expression in cytoplasm was detected in tumors from Dlx3cKO mice. (h) Detection of proliferating cell nuclear antigen (PCNA)-positive cells in tumor tissue with the control skin. Data are presented as mean ± standard error of the mean. (*** P-value <0.001). n=number of mice.
Skin lacking Dlx3 promotes tumor development for DMBA-initiated carcinogenesis
We next examined the effect of DMBA only on Dlx3-deficient skin tissue in the absence of chemical tumor promoter. Initiating doses of single DMBA treatment, while mutagenic to mouse skin, does not cause tumorigenesis without a promotion agent or repeated DMBA applications [22]. In protocol 1 of this study, Dlx3cKO or control mice were applied a single initiating dose of DMBA and monitored for tumor formation over 34 weeks (Fig. 3a, b). Tumors were examined and histologically classified (Fig. 3c). In Dlx3cKO mice, tumors appeared at ~19 weeks, and by week 33, 100% Dlx3cKO mice had developed tumors (Fig. 3d–f). Control mice showed no tumors within the 34 week experimental timeline (Fig. 3d–f). Assessment of KRT13 and PCNA expression was performed on Dlx3cKO papillomas and control-treated skin, and showed elevated detection of KRT13 and significant increased proliferation in absence of Dlx3 (Fig. 3g).
Figure 3. Dlx3 deficiency drives tumor promotion in single dose DMBA-only model.
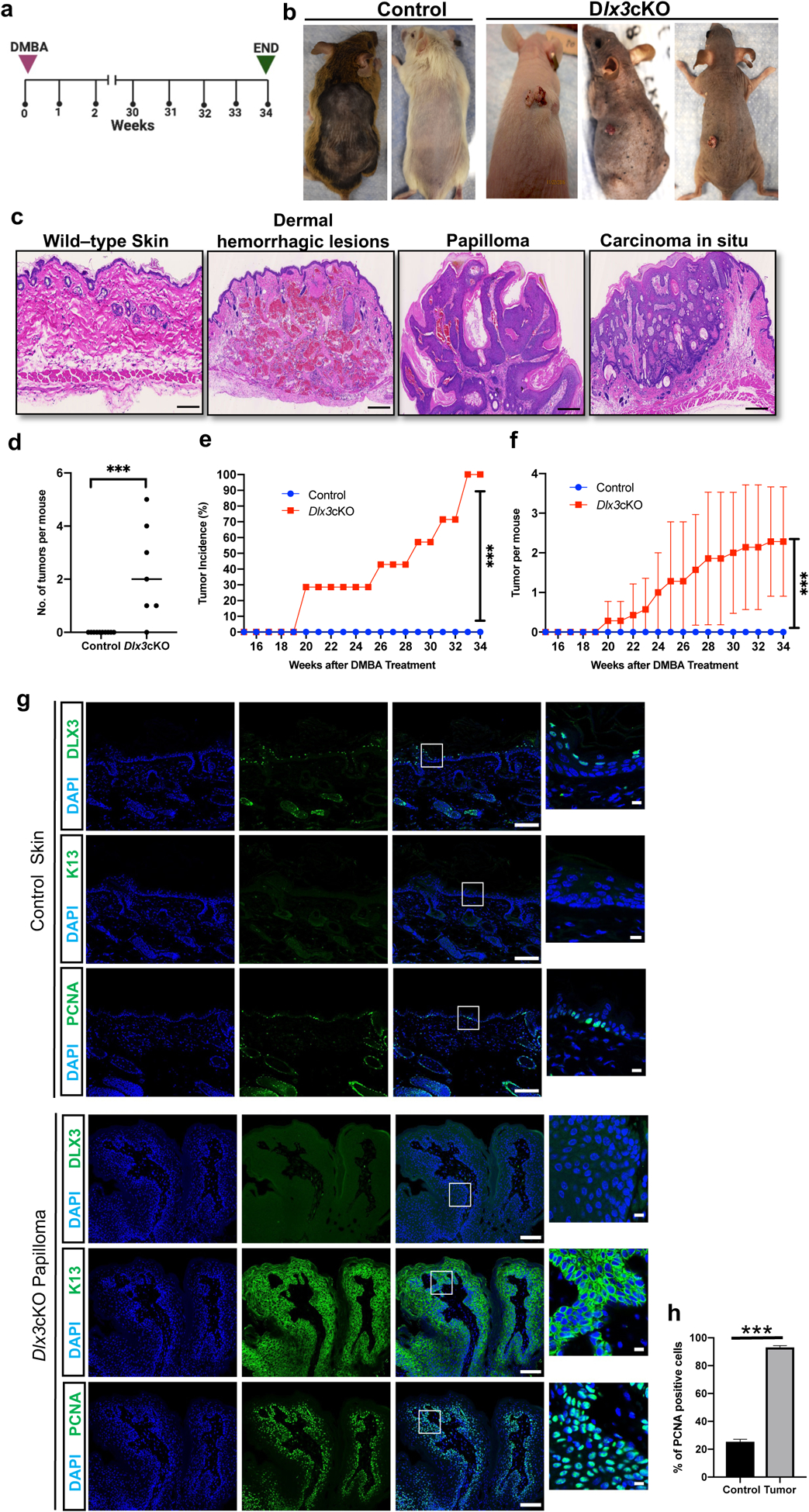
(a) Experimental design for a chemically induced skin tumorigenesis study. Mice were treated with a single DMBA dose. Tumors larger than 1 mm in diameter were counted weekly. At week 34, tumors were collected for analysis. Dlx3cKO; n=7: Control; n=9 (b) Dorsal view of control and Dlx3cKO mice after 34 weeks of DMBA treatment. (c) H&E staining of sections of skin biopsies 34 weeks after DMBA treatment shows that single dose of DMBA treatment significantly increases dermal hemorrhagic lesions, papilloma and carcinoma in situ of Dlx3cKO but not that of control animals (Scale bars, 100μm). (d) Number of tumors per mouse (e) Tumor incidence: Comparison of control and Dlx3cKO incidence curves. The difference in tumor incidence between control and Dlx3cKO mice becomes significant from week 20 onwards; ***P-value <0.001, Student’s t-test. (f) Frequencies of skin tumors: DMBA-only treatment increased numbers of skin tumors only in Dlx3cKO mice. (g) Representative immunohistochemistry of DLX3, K13 and PCNA (Scale bars, 100μm and 10μm). As shown DLX3 expression is absent in the skin papilloma. Higher PCNA expression in nucleus and elevated K13 expression in cytoplasm were detected in papillomas from Dlx3cKO mice. (h) Detection of proliferating cell nuclear antigen (PCNA)-positive cells in tumor tissue with the control skin. Data are presented as mean ± standard error of the mean. (*** P-value <0.001).
n=number of mice.
In protocol 2 of the DMBA-only study, three DMBA doses (instead of one DMBA dose) were applied and the mice were monitored for tumor formation over 31 weeks (Fig. 4a). Only Dlx3cKO mice developed tumors, with no tumors developing in the control-treated mice (Fig. 4b). Histopathologically, tumors were defined as papillomas and carcinoma in situ (Fig. 4c). Significantly, Dlx3cKO mice exhibited increased susceptibility to skin carcinogenesis with markedly accelerated papilloma development (Fig. 4d–f).
Figure 4. Epidermal specific deletion of Dlx3 accelerates tumor formation and tumor numbers in triple dose DMBA only model.
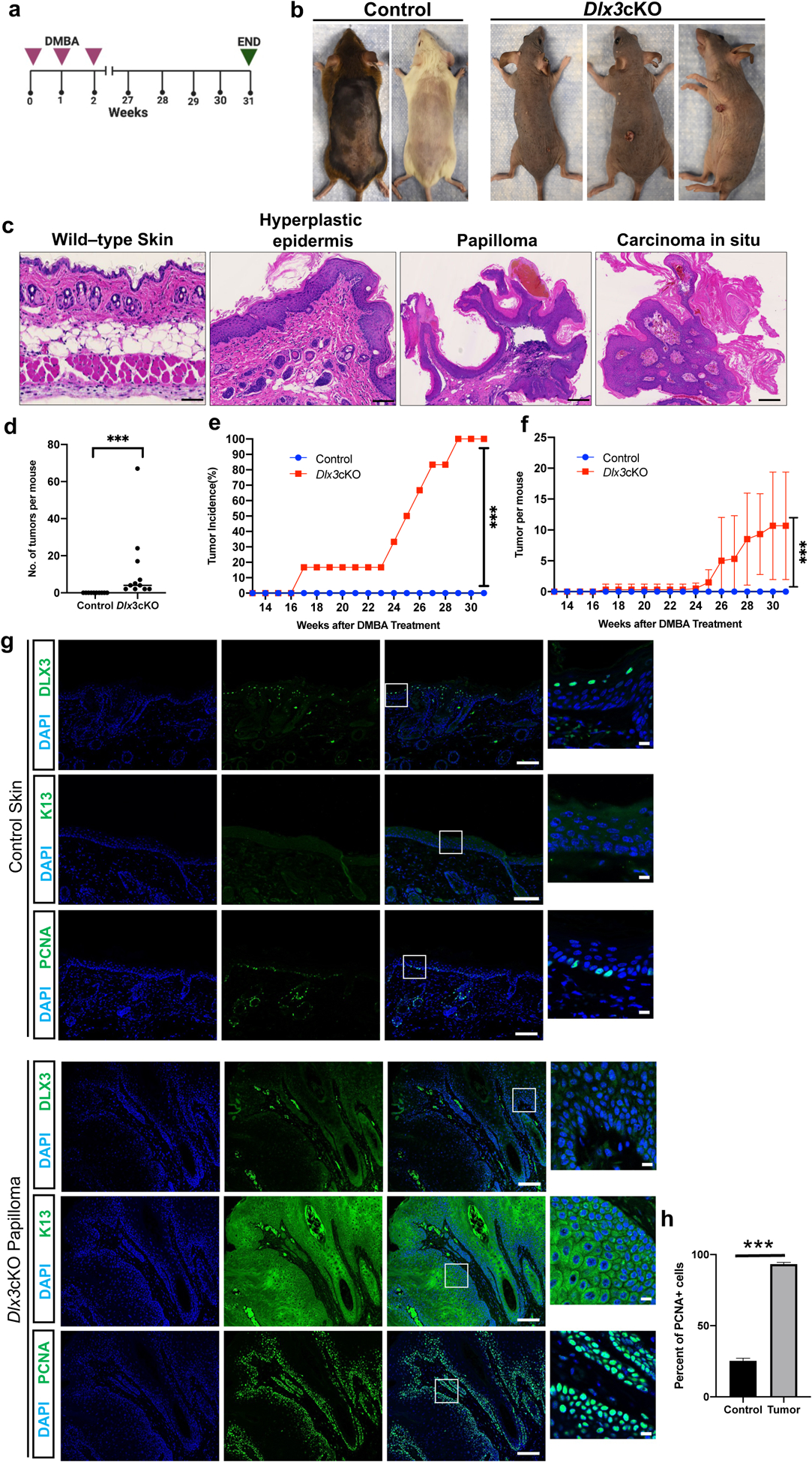
(a) Experimental design for a chemically induced skin tumorigenesis study. Mice were treated with three DMBA doses for three consecutive weeks. Tumors larger than 1 mm in diameter were counted weekly. At week 31, tumors were collected for analysis. Dlx3cKO; n=12: Control; n=10 (b) Dorsal view of control and Dlx3cKO mice after 31 weeks of DMBA treatment. (c) H&E staining of sections of skin biopsies 31 weeks after DMBA treatment shows that three doses of DMBA treatment significantly increases hyperplastic epidermis, papilloma and carcinoma in situ of Dlx3cKO, but not that of control animals (Scale bars, 1mm). (d) Number of tumors per mouse. (e) Tumor incidence: Comparison of control and Dlx3cKO incidence curves. The difference in tumor incidence between wild-type and knock-out mice becomes significant from week 17 onwards; ***P-value <0.001, Student’s t-test. (f) Frequencies of skin tumors: 3x DMBA-only treatment increased numbers of skin tumors only in Dlx3cKO mice. (g) Representative immunohistochemistry of DLX3, K13 and PCNA (Scale bars, 100μm and 10μm). As shown DLX3 expression is absent in the skin papilloma. Higher PCNA expression in nucleus and elevated K13 expression in cytoplasm were detected in papillomas from Dlx3cKO mice. (h) Detection of proliferating cell nuclear antigen (PCNA)-positive cells in tumor tissue with the control skin. Data are presented as mean ± standard error of the mean. (*** P-value <0.001). n=number of mice.
104 tumors from Dlx3-deficient mice were classified by two independent pathologists as: hyperplastic epidermis (n = 10), small papilloma (n = 47), papilloma (n = 30), large papilloma (n = 10) and carcinoma in situ (n = 7). Representative histological images are shown in Fig. 4c. While the tumors did not present detectable expression of DLX3, control skin showed the previously reported suprabasal staining for DLX3 (Fig. 4g). As for tumors generated from one-dose DMBA, the percentage of PCNA-positive cells in Dlx3cKO tumors was significantly higher than that for the control group (Fig. 4g). Expression of KRT13 was only detectable in the Dlx3cKO papillomas (Fig. 4g).
These findings support that lack of DLX3 function is associated with elevated skin tumorigenesis and enhanced cell proliferation. Thus, in the absence of a chemical promoter, DLX3 deficiency provides a promotive environment to incipient tumor cells suggesting that Dlx3 functions as a tumor suppressor in skin.
Epidermal tumors generated in Dlx3cKO mice are in the early stages of malignant conversion
Loss of keratin KRT10 and the concomitant detection of KRT13 (normally not expressed in epidermis), are indicators of early stages of malignant conversion [25]. To determine if the papillomas generated in the DMBA-only protocol in Dlx3cKO mice were at the cusp of malignant conversion, we separated the papillomas based on their size, small (2–4 mm), and large (4–6 mm) (Fig. 5a, b). Immunofuorescence was performed using keratins KRT5, KRT10, and KRT13 antibodies (Fig. 5c). KRT5 was detected in the basal cell layer in healthy skin and in papillomas of all sizes. We observed increased expression of KRT13 in Dlx3cKO tumors, suggesting that DLX3-ablated tumors have a more aggressive phenotype (Figs.3g and 4g). Tumors expressing KRT13 were more frequent in the large papilloma category, with 70% of the tumor area presenting KRT13 expression (Fig. 5c). KRT10 expression was concordantly diminished in large tumors. Additionally, control skin and tumors were stained with other epidermal differentiation-specifc markers Loricrin and Filaggrin. Expression of these differentiation markers was also decreased in Dlx3cKO tumors compared to the control skin (Supplementary Fig. S4).
Figure 5. Dlx3 deficiency facilitates conversion of low risk papillomas to high risk papillomas.
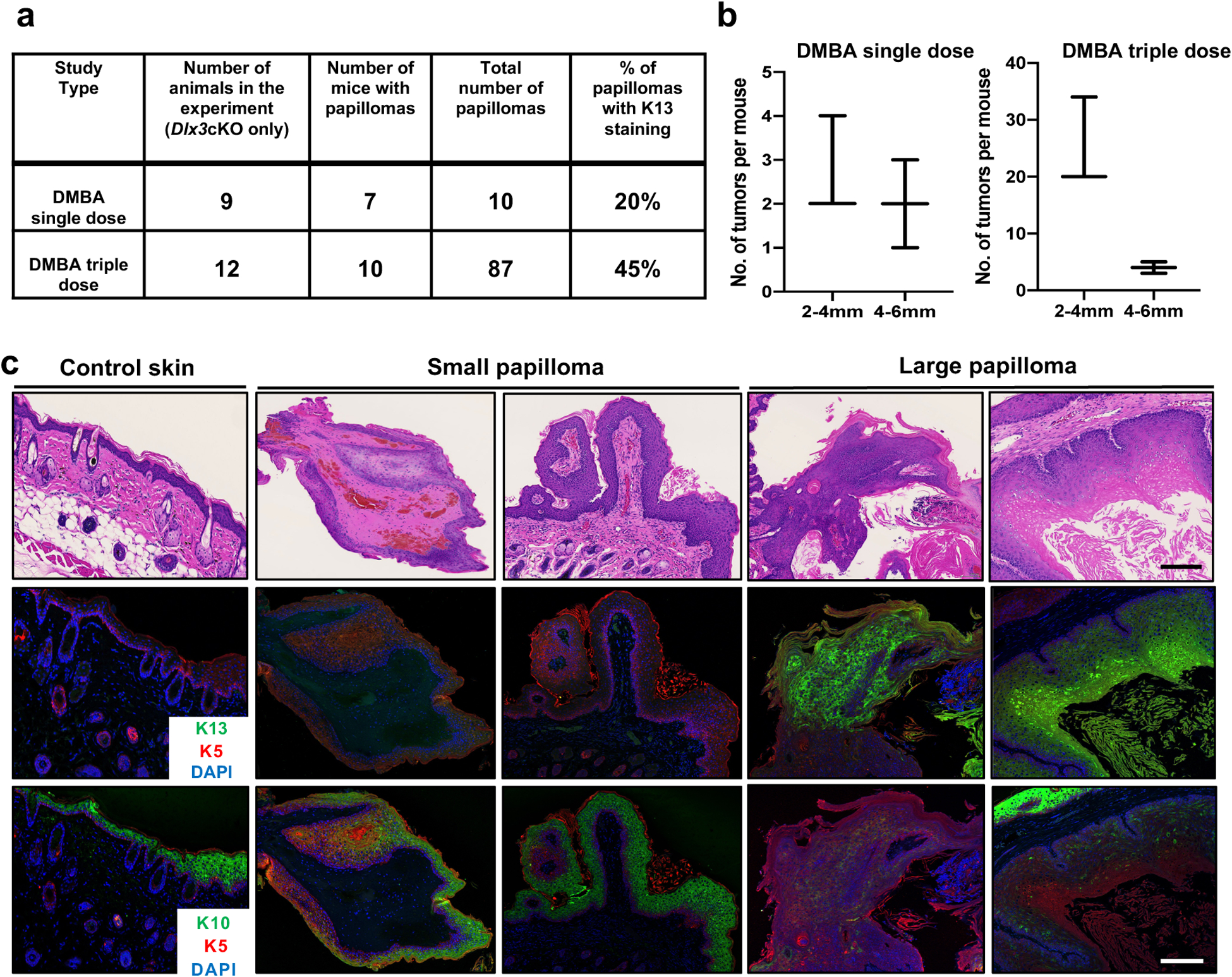
(a) Summary of the results. The table shows the total number of papillomas developed and percentage of them with K13 staining. (b) Size distribution of papillomas. The length of a papilloma was used to represent its size. Small papilloma (2–4mm) and Large papilloma (4–6 mm) (c) Representative images of control skin, small papilloma, and large papilloma from Dlx3cKO mice. Dual immunofluorescence staining was performed for K5 (red) and K13 (green). The nuclei were counterstained with DAPI (blue). The same sections were used for hematoxylin and eosin (H&E) staining. K10 staining was performed with other sections from the same biopsies. Scale bars: 100μm.
Transcriptional profiling identifies downstream mediators of DLX3 function.
Molecular evaluation of the Dlx3cKO tumor-permissive properties was done by performing RNAseq on tumors, Dlx3cKO unaffected whole skin, and control skin from the triple dose DMBA-only protocol (Fig. 6a). We compared differentially expressed genes (DEG) from Dlx3cKO/control treated skins against Tumor/Dlx3cKO skin (Fig. 6b). Ingenuity Pathway Analysis (IPA) identified significantly regulated biological functions or disease phenotypes in both DEG datasets (Fig. 6c). Dlx3cKO/control exhibited 58 significantly activated biological functions, 44 of which were related to the proliferation, activation, and migration of immunocytes. 28 of the 61 Tumor/Dlx3cKO DEG activated disease states were associated with cancer, predominantly carcinomas and epithelial neoplasms (Fig. 6c). Upstream regulator predictive analysis identified Egfr and Ebbb2 as active in Dlx3cKO skin compared to control, with Ebbb2 activation conserved and increased in tumors as compared to Dlx3cKO skin (Fig. 6d). Ebbb2 was the only regulator predicted to be activated in Dlx3cKO skin and further activated upon neoplasm formation.
Figure 6. Gene expression profiling of mouse skin and tumor samples from DMBA-only skin carcinogenesis.
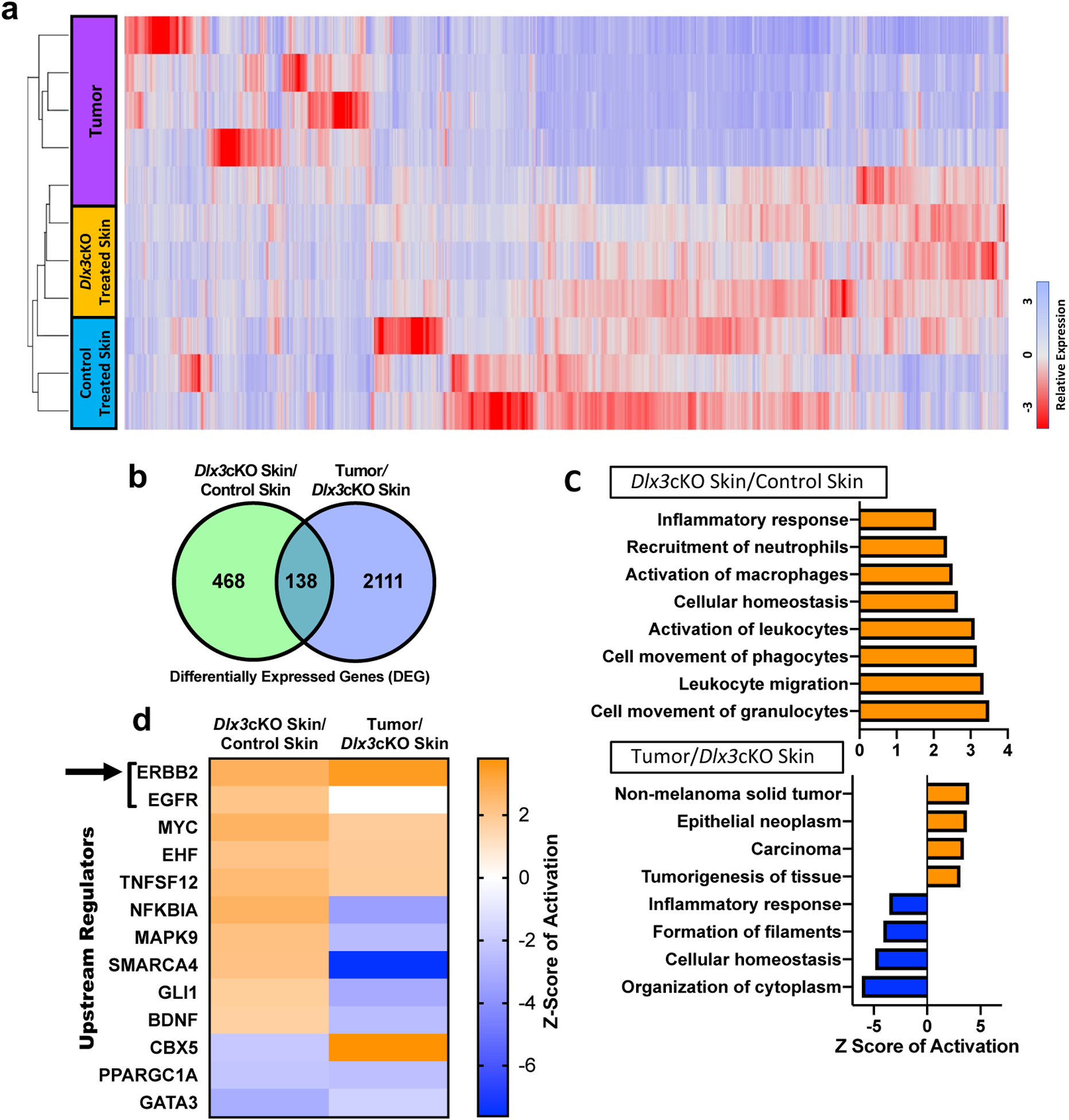
(a) A cluster diagram of RNA-seq data from five tumors and, skin tissues from three treated control and Dlx3cKO mice, respectively. The color bars represent relative expression levels: blue indicates higher than average expression and red indicates lower than average expression. (b) Venn diagram of RNA-seq analysis of two comparisons: Dlx3cKO treated skin vs control treated skin and Tumor vs Dlx3cKO treated skin. (c) Top enriched GO processes from commonly regulated genes in Dlx3cKO treated skin vs control treated skin and Tumor vs Dlx3cKO treated skin demonstrates processes involved in inflammation and carcinogenesis to be deregulated in DLX3cKO skin and tumors, respectively. (d) Ingenuity pathway software analysis identifies statistically significant upstream regulators for the cluster of genes showed in panel. ERBB2 and EGFR are the two topmost significantly upregulated upstream regulators in both the Dlx3cKO skin and tumor (indicated by black arrow).
DEG groups displayed distinct downstream profiles for ERBB2 activation as classified by IPA, with Ereg, Elf3, Mybl2, Il1α, Pgm1, Krt81, and Slc2a1 (Glut1) as the seven conserved DEG between the two groups (Fig. 7a). Within the skin-specific profile, upregulation of other EGFR ligands such as Hbegf, Areg and Epgn, as well as an Erbb2-Ehf-Elf3 axis, were predicted by IPA and validated by qPCR (Fig. 7b). EGFR and ERBB2 were phosphorylated in Dlx3cKO adult epidermis (Figs. 7c and Supplementary Fig. S5).
Figure 7. DLX3 downregulates the activation of EGFR and ERBB2 by moderating the expression of EGFR ligands in healthy epidermis.
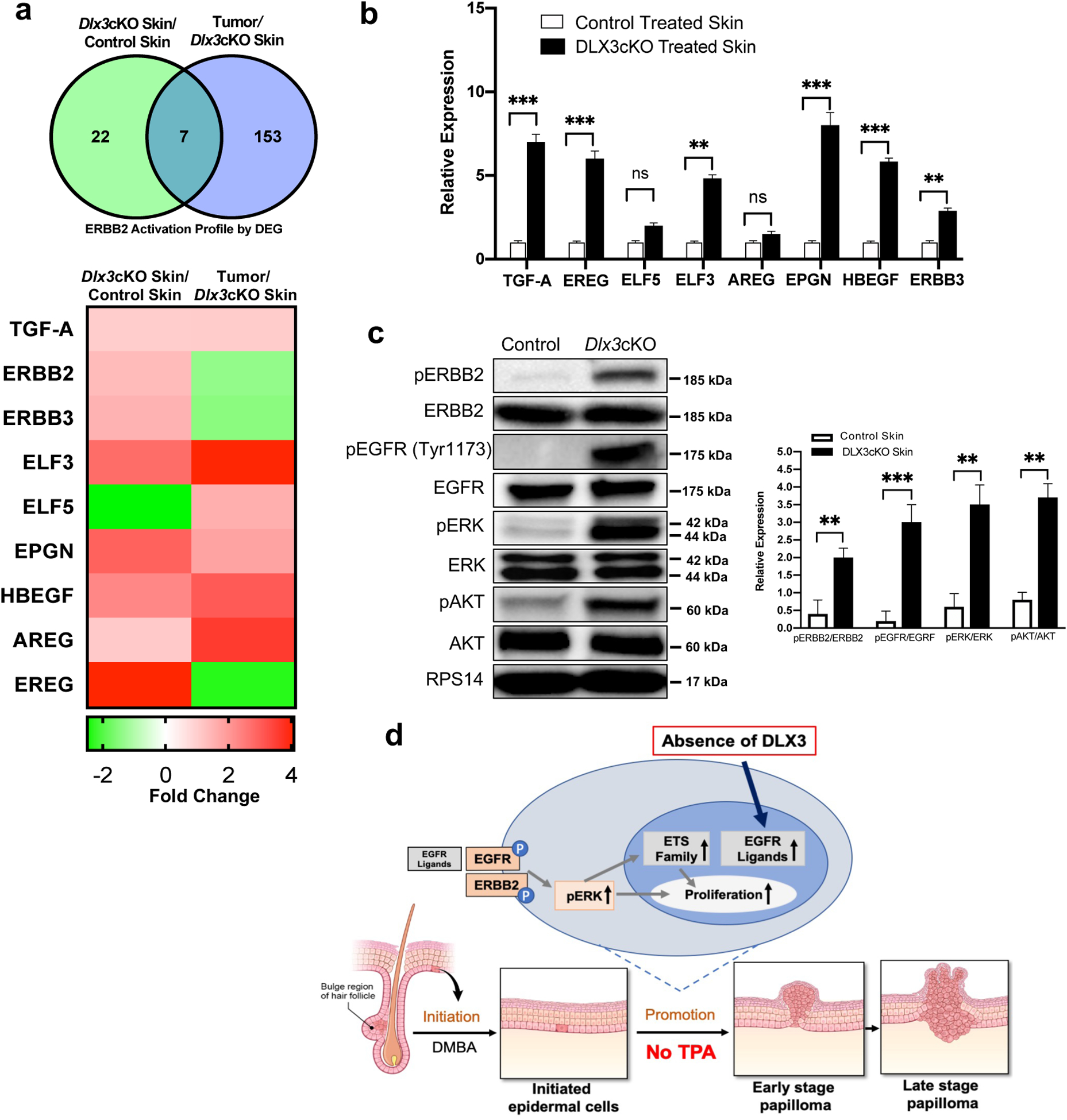
(a) Top: Venn diagram summarizing IPA-classified DEGs (Differentially expressed genes) downstream of the activated ERBB2 receptor in comparisons of treated mouse skin and tumor samples. Bottom: Fold changes for specific DEGs including EGFR ligands and the ETS family. (b) qPCR validation of previously highlighted DEG expression changes from treated Dlx3cKO and control skin. Data were measured in triplicate and represent mean ± SEM. (c) Western blot analysis of isolated epidermis from 6–8 weeks old Dlx3cKO and control mice. The total and the phosphorylated levels of the receptors ERBB2, EGFR (Tyr1173), and of the downstream signaling proteins ERK1/2 and AKT are shown. The densitometric quantification and normalization are shown at the bottom. n = 4, **P-value < 0.01, ***P-value < 0.001. (d) The diagram summarizes our findings and shows a model displaying that, Dlx3 deficiency in the skin acts as a promoting event in mouse chemically induced carcinogenesis. Initiation is achieved by topical application of the carcinogen DMBA, and no promoting agent is used. It demonstrates loss of Dlx3 activates ERBB2 and EGFR signaling and also upregulates the expression level of EGFR ligands and ETS factors. The activated ligands in a positive feedback loop can also further enhance EGFR and its downstream signaling pathways, such as ERK1/2 and AKT. The result of the activation of this signaling profile is leading to clonal expansion of initiated cells, an effect similar to application of TPA.
To further validate the mechanism that loss of Dlx3 tumor suppressive function promotes progression of tumor through EGFR-ERBB2 pathway, we knocked down Dlx3, Egfr and Erbb2 using small interfering RNAs (siRNAs) (Supplementary Fig. S6a, b). We examined the effect of siRNA-treatment on the proliferation capacity of skin keratinocytes in vitro and show that knocking down Egfr and Erbb2 in si-Dlx3 treated cells significantly diminished the enhancement of cell proliferation (Supplementary Fig. S6c, d). These results indicate that loss of Dlx3 promotes the proliferation of keratinocytes via the EGFR/ERBB2 pathway. We also showed that knockdown of EGFR/ERBB2 pathway affects the downstream signaling pathway AKT and ERK, as treatment significantly diminished the levels of pAKT and pERK in Dlx3 knocked down cells co-treated with si-Erbb2 and si-Egfr (Supplementary Fig. S6e–g).
Altogether, our results support a model in which DLX3 downregulates the activation of EGFR and ERBB2 by moderating the expression of EGFR ligands in healthy epidermis. Upon loss of Dlx3, EGFR ligand production activates the EGFR pathway, leading to enhanced proliferation and self-promotion in initiated keratinocytes. The innate activation of this pathway in Dlx3cKO skin is conserved through tumorigenesis (Fig. 7d).
Discussion
Cancer and normal development have a great deal in common, as both processes involve shifts between cell proliferation and differentiation [27]. The deregulation of many HOX genes has been found in a variety of cancers, and often reported in association with an increased cancer risk and poor survival rate of cancer patients [28, 29, 30][31]. In clinical settings, lower expression of homeobox genes is greatly associated with elevated tumor invasion, metastasis, and patient mortality [29]. We demonstrated gradual loss of nuclear DLX3 expression in clinical specimens as the skin cancer progressed from in situ stage to advanced SCC stage. Further analysis indicated that Dlx3 expression was downregulated in other SCC types, and that this downregulation correlates with poor prognosis in the case of hnSCC patients. We have previously reported that low levels of Dlx3 mRNA correlates with reduced expression of p53 and modulation of other tumor-associated markers [14].
The pathology of the two-stage chemically induced skin carcinogenesis is similar to the development of human skin cancers and thus offers an ideal model to study skin cancer initiation and growth [32, 33]. Single DMBA treatment alone, without any chemical promoter induced papillomas in Dlx3-deficient mice. These findings suggest that tumor promotion activity associated with DLX3 deficiency predisposes skin to carcinogenesis initiated by DMBA.
Chronic inflammation and sustained epidermal hyperplasia reportedly promotes tumor progression in mice and humans [34, 35]. Consistent with this, Dlx3-deficient murine skin presents epidermal hyperproliferation, impaired terminal differentiation and development of IL17 dependent skin inflammation [12] and STAT3 activation that results in rapid immune cell infiltration and development of skin inflammation [13].
The alteration of the keratin pattern in the skin carcinogenesis model appears to be a sensitive marker to determine the different stages in the progression of a papilloma to a malignancy [36] [37] [38]. We observed increased expression of KRT13, suggesting that DLX3-ablated tumors have a more aggressive phenotype. We showed a switch from a KRT13Lo/KRT10Hi to a KRT13Hi/KRT10Lo phenotype in our DMBA-only skin carcinogenesis model that correlated with conversion of low risk to high risk papillomas.
Genome-wide transcriptome analysis identified ErbB receptors as mediators of DLX3’s tumor promotional activity. EGFR, a prototypical member of the ErbB network, is activated by autophosphorylation and/or transphosphorylation and plays essential roles in regulating downstream targets, such as MAPK, PI3-Akt and JAK-Stat 3/5 pathways [39, 40]. There are several critical tyrosine phosphorylation sites in the intracellular domain of EGFR [39]. Y992, 1068, 1086 and 1173 are generally linked to MAPK and AKT signaling pathways [41], while Y1045 phosphorylation regulates EGFR ubiquitination and degradation [42]. In our study we found that Dlx3cKO skin showed a robust increase in phosphorylated EGFR (P-EGFR, Tyr1173) and also significant difference in the phosphorylated EGFR at Tyr1068 (Supplementary Fig. S4) which resulted in ERK1/2 and AKT phosphorylation in Dlx3cKO skin. These findings suggest that loss of DLX3 function acts as a promotional event in cutaneous carcinogenesis by activating EGFR and its downstream targets.
A number of human tumors express high levels of EGFR and its ligands, and correlate with poor prognosis for cancer patients [43]. Diverse tumor promoters lead to elevated levels of EGFR ligands including transforming growth factor α (TGFα), amphiregulin, and heparin-binding EGF-like growth factor (HBEGF) [44]. Dlx3cKO skin presents upregulation of EGFR ligands, indicating that DLX3 absence can substitute for the promotion stage of multistage skin carcinogenesis.
Contrary to EGFR, not much is known about the role of ERBB2 in skin carcinogenesis. Herein, we demonstrated activation of ERBB2 in Dlx3cKO skin. Activation of both EGFR and ERBB2 and the presence of EGFR:ERBB2 heterodimers has been reported in TPA-treated mouse epidermis [45]. These results support that in absence of Dlx3, ERBB2 may facilitate signaling through the EGFR during the tumor promotion stage. Several drugs have been approved for targeting ERBB2 in the treatment of many cancer types [46] and our data provide evidence to potentially repurpose them for cutaneous cancer treatments.
Altogether, we show loss of Dlx3 expression in human skin cancer and our in silico analyses of gene expression data support a correlation to poor patient survival. Loss of Dlx3 tumor suppressive function acts as a tumor promoter through regulation of ERBB2 and EGFR signaling and impairing the ERBB2/EGFR signaling attenuates the cellular proliferation and downstream signalling pathway in vitro. Our findings also support an important inhibitory role of Dlx3 in skin cancer promotion and progression, that may potentially be used as a promising prognostic marker.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH (ZIA-AR041124 to M.I.M.). We thank Gutierrez-Cruz and S. Dell’Orso of the NIAMS Genome Analysis Core Facility and members of the NIAMS Light Imaging Core Facility. This work used the computational resources of the NIH High-Performance Computing Biowulf Cluster. We also thank all members of our laboratories for their continuous support. BioRender was used to create experimental design schematic.
Footnotes
Competing interests statement
The authors state no conflict of interest.
References
- 1.Que SK, Zwald F, Schmults C. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J Am Acad Dermatol. 2018; 78: 237–47. [DOI] [PubMed] [Google Scholar]
- 2.Karia PS, Han J, Schmults CD. Cutaneous squamous cell carcinoma: estimated incidence of disease, nodal metastastasis, and deaths from disease in the United States, 2012. J Am Acad Dermatol. 2013; 68: 957–966. [DOI] [PubMed] [Google Scholar]
- 3.Lallas A, Pyne J, Kyrgidis A, Andreani S, Argenziano G, Cavaller A, et al. The clinical and dermoscopic features of invasive cutaneous squamous cell carcinoma depend on the histopathological grade of differentiation. Br J Dermatol. 2015; 172: 1308–1315. [DOI] [PubMed] [Google Scholar]
- 4.Cillo C, Faiella A, Cantile M, Boncinelli E. Homeobox genes and cancer. Experimental Cell Research. 1999; 248: 1–9. [DOI] [PubMed] [Google Scholar]
- 5.Abate-Shen C Deregulated homeobox gene expression in cancer: cause or consequence? Nature reviews Cancer. 2002; 2: 777–785. [DOI] [PubMed] [Google Scholar]
- 6.Sun Y, Zhou B, Mao F, Xu J, Miao H, Zou Z, et al. HOXA9 Reprograms the Enhancer Landscape to Promote Leukemogenesis. Cancer Cell. 2018; 34: 643–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah N, Sukumar S The hox genes and their roles in oncogenesis. Nat. Rev. Cancer 2010; 10: 361–371. [DOI] [PubMed] [Google Scholar]
- 8.Samuel S, Naora H. Homeobox gene expression in cancer: insights from developmental regulation and deregulation. Eur J Cancer. 2005; 41: 2428–37. [DOI] [PubMed] [Google Scholar]
- 9.Luo Z, Rhie SK, Farnham PJ. The enigmatic hox genes: Can we crack their code? Cancers. 2019; 11: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hwang J, Mehrani T, Millar SE, Morasso MI. Dlx3 is a crucial regulator of hair follicle differentiation and cycling. Development. 2008; 135: 3149–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morasso MI, Radoja N. Dlx genes, p63, and ectodermal dysplasias. Birth defects research Part C, Embryo today: reviews. 2005; 75: 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hwang J, Kita R, Kwon HS, Choi EH, Lee SH, Udey MC, et al. Epidermal ablation of Dlx3 is linked to IL-17-associated skin inflammation. PNAS. 2011; 108: 11566–11571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhattacharya S, Kim JC, Ogawa Y, Nakato G, Nagle V, Brooks SR, et al. DLX3-Dependent STAT3 Signaling in Keratinocytes Regulates Skin Immune Homeostasis. J Invest Dermatol. 2018; 138: 1052–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palazzo E, Kellett M, Cataisson C, Gormley A, Bible PW, Pietroni V, et al. The homeoprotein DLX3 and tumor suppressor p53 co-regulate cell cycle progression and squamous tumor growth. Oncogene. 2016; 35: 3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Witsch E, Sela M, Yarden Y. Roles for growth factors in cancer progression. Physiology (Bethesda) 2010; 25: 85–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dahlhoff M, Muzumdar S, Schafer M and Schneider MR. ERBB2 is essential for the growth of chemically induced skin tumors in mice. J Invest Dermatol. 2017; 137: 921–930. [DOI] [PubMed] [Google Scholar]
- 17.Dahlhoff M, Schafer M, Muzumdar S, Rose C and Schneider MR. ERBB3 is required for tumor promotion in a mouse model of skin carcinogenesis. Mol Oncol. 2015; 9: 1825–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan KS, Carbajal S, Kiguchi K, Clifford J, Sano S and DiGiovanni J. Epidermal growth factor receptor‐mediated activation of Stat3 during multistage skin carcinogenesis. Cancer Res. 2004; 64: 2382–2389. [DOI] [PubMed] [Google Scholar]
- 19.Dahlhoff M, Rose C, Wolf E and Schneider MR. Decreased incidence of papillomas in mice with impaired EGFR function during multi‐stage skin carcinogenesis. Exp Dermatol. 2011; 20: 290–293. [DOI] [PubMed] [Google Scholar]
- 20.Subramanian J, Katta A, Masood A, Vudem DR, Kancha RK. Emergence of ERBB2 Mutation as a Biomarker and an Actionable Target in Solid Cancers. Oncologist. 2019; 24: e1303–e1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiguchi K, Kitamura T, Moore T, Rumi M, Chang HC, Treece D, et al. Dual inhibition of the epidermal growth factor receptor and erbB2 effectively inhibits the promotion of skin tumors during two-stage carcinogenesis. Cancer Prev Res. 2010; 3: 940–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abel EL, Angel JM, Kiguchi K, DiGiovanni J. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc. 2009; 4: 1350–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 24.Lichti U, Anders J, Yuspa SH. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc. 2008; 3: 799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darwiche N, Ryscavage A, Perez-Lorenzo R, Wright L, Bae D-S, Hennings H, et al. Expression profile of skin papillomas with high cancer risk displays a unique genetic signature that clusters with squamous cell carcinomas and predicts risk for malignant conversion. Oncogene 2007; 26: 6885–6895. [DOI] [PubMed] [Google Scholar]
- 26.Palazzo E, Kellett MD, Cataisson C, Bible PW, Bhattacharya S, Sun HW, et al. A novel DLX3-PKC integrated signaling network drives keratinocyte differentiation. Cell Death Differ. 2017; 4: 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010; 10: 361–371. [DOI] [PubMed] [Google Scholar]
- 28.Plowright L, Harrington K, Pandha H, Morgan R. HOX transcription factors are potential therapeutic targets in non-small-cell lung cancer (targeting HOX genes in lung cancer). Br J Cancer. 2009; 100: 470–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilbert PM, Mouw JK, Unger MA, Lakins JN, Gbegnon MK, Clemmer VB, et al. Hoxa9 regulates brca1 expression to modulate human breast tumor phenotype. J. Clin. Investig 2010; 120: 1535–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo Z, Rhie SK, Farnham PJ. The enigmatic hox genes: Can we crack their code? Cancers. 2019; 11: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, Jaffee E, et al. Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature. 2000; 405: 974–978. [DOI] [PubMed] [Google Scholar]
- 32.Wheeler DL, Verma AK, Denning MF. Mouse models of the skin: models to define mechanisms of skin carcinogenesis. J. Skin Cancer 2013; 971495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilker E, Lu J, Rho O, Carbajal S, Beltrán L, DiGiovanni J. Role of PI3K/Akt signaling in insulin-like growth factor-1 (IGF-1) skin tumor promotion. Mol. Carcinog 2005; 44: 137–145. [DOI] [PubMed] [Google Scholar]
- 34.Cataisson C, Ohman R, Patel G, Pearson A, Tsien M, Jay S, et al. Inducible cutaneous inflammation reveals a protumorigenic role for keratinocyte CXCR2 in skin carcinogenesis. Cancer Res. 2009; 69: 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mueller MM. Inflammation in epithelial skin tumours: old stories and new ideas. Eur J Cancer. 2006; 42: 735–744. [DOI] [PubMed] [Google Scholar]
- 36.Gimenez-Conti I, Aldaz CM, Bianchi AB, Roop DR, Slaga TJ, Conti CJ. Early expression of type I K13 keratin in the progression of mouse skin papillomas. Carcinogenesis. 1990; 11: 1995–1999. [DOI] [PubMed] [Google Scholar]
- 37.Toftgard R, Yuspa SH and Roop DR. Keratin gene expression in mouse skin tumors and in mouse skin treated with 12-Otetradecanoylphorbol-13-acetate. Cancer Res, 1985; 45: 5845–5850. [PubMed] [Google Scholar]
- 38.Nischt R, Roop DR, Mehrel T, Yuspa SH, Rentrop M, Winter H, et al. Aberrant expression during two-stage mouse skin carcinogenesis of a type I 47-kDa keratin, K13, normally associated with terminal differentiation of internal stratified epithelia. Mol. Carcinog 1988; 1: 96–108. [DOI] [PubMed] [Google Scholar]
- 39.Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004; 305: 1163–1167. [DOI] [PubMed] [Google Scholar]
- 40.Nyati MK, Morgan MA, Feng FY, Lawrence TS. Integration of EGFR inhibitors with radiochemotherapy [published correction appears in Nat Rev Cancer. 2006 Dec;6(12):974]. Nat Rev Cancer. 2006; 6: 876–885. [DOI] [PubMed] [Google Scholar]
- 41.Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009; 315: 683–696. [DOI] [PubMed] [Google Scholar]
- 42.Herbst RS, Shin DM. Monoclonal antibodies to target epidermal growth factor receptor-positive tumors: a new paradigm for cancer therapy. Cancer. 2002; 94: 1593–1611. [DOI] [PubMed] [Google Scholar]
- 43.Kiguchi K, Beltrán L, Rupp T, DiGiovanni J. Altered expression of epidermal growth factor receptor ligands in tumor promoter-treated mouse epidermis and in primary mouse skin tumors induced by an initiation-promotion protocol. Mol Carcinog. 1998; 22: 73–83. [PubMed] [Google Scholar]
- 44.El-Abaseri TB, Fuhrman J, Trempus C, Shendrik I, Tennant RW, Hansen LA. Chemoprevention of UV light-induced skin tumorigenesis by inhibition of the epidermal growth factor receptor. Cancer Res. 2005; 65: 3958–3965. [DOI] [PubMed] [Google Scholar]
- 45.Xian W, Rosenberg MP, DiGiovanni J. Activation of erbb2 and c-src in phorbol ester-treated mouse epidermis: possible role in mouse skin tumor promotion. Oncogene. 1997; 14: 1435–1444. [DOI] [PubMed] [Google Scholar]
- 46.Schneider M, Yarden Y. The EGFR-HER2 module: a stem cell approach to understanding a prime target and driver of solid tumors. Oncogene. 2016; 35: 2949–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.