

Abstract
The high selectivity and affinity of antibody binding have made antibodies all-pervasive tools in therapy, diagnosis, and basic science. A plethora of chemogenetic approaches has been devised to make antibodies responsive to stimuli ranging from light to enzymatic activity, temperature, pH, ions, and effector molecules. Within a single decade, the field of activatable antibodies has yielded marketed therapeutics capable of engaging antigens that could not be targeted with traditional antibodies, as well as new tools to control intracellular protein location and investigate biological processes. Many opportunities remain untapped, waiting for more efficient and generally applicable masking strategies to be developed at the interface between chemistry and biotechnology.
Short abstract
The combination of chemistry and genetic modification has yielded within the past decade a plethora of antibodies responsive to environmental cues. Activatable antibodies enable engaging therapeutic antigens that could not be targeted with traditional antibodies and provide new tools to investigate biological systems.
1. Activatable Antibodies for Every Stimulus
Antibodies are extensively exploited in therapy, diagnosis, and basic science due to their high affinity and selectivity.1 The field of antibody engineering appeared to reach its maturity with the discovery of directed evolution to optimize binding, with humanization to decrease immunogenicity, and, more recently, with the possibility to target cytotoxic molecules to cancer cells and to engineer bispecific molecules that engage immune cells.2 However, in the past decade, several technologies that render antibodies activatable under particular stimuli have unravelled new avenues for antibodies as therapeutics and as probes to investigate biological systems.
A plethora of approaches, many of them combining chemistry and genetic modification, has been devised to make antibodies responsive to endogenous or exogenous cues ranging from light to enzymatic activity, temperature, pH, ions, effector molecules, and antigen combinations (Figure 1). Remarkably, many antibodies sensitive to pH and proteases are in clinical development, and two of them have recently been approved for clinical use. Other antibodies responsive to light, effector molecules, and the presence of antigen combinations show great promise in the control of molecular and cellular processes. Alternative approaches to modulate binding of antibodies without modifying them, such as engineering of activatable antigens3 and encapsulation4,5 are beyond the scope of this review.
Figure 1.
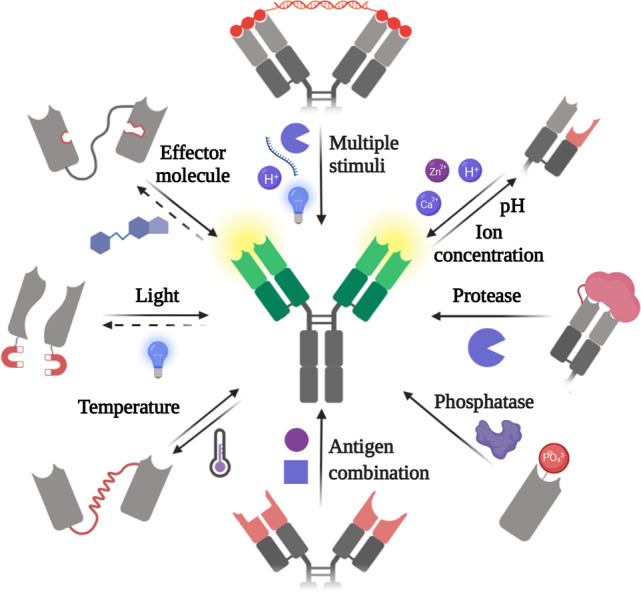
Very diverse molecular engineering strategies have been applied to generate antibodies responsive to a variety of stimuli.
Herein, we will provide a critical assessment of the chemogenetic approaches that have been used to generate activatable antibodies, discussing their applicability in therapy and basic research. Therapeutic applications have mainly focused on full-length immunoglobulins G (IgGs), which are the most widely used format in pharmacology due to their long circulation times and the additional immune cytotoxicity they may trigger against target cells, especially relevant in the treatment of cancer.2 However, full-length antibodies require sophisticated engineering and expression systems, are complex to characterize, and have slow diffusion in tissues. Hence, for therapeutic applications involving transport across biological barriers and tissue penetration, and for basic research applications, a wide variety of smaller antibody derivatives and mimetics has been developed.7 Although many complex combinations of antibody domains have been engineered, the three basic classes of antibody derivatives are antigen-binding fragments (Fab), single-chain variable fragments (scFvs), and single-domain antibodies (sdAbs, such as nanobodies). Beyond structures directly derived from antibodies, antibody mimetics include any protein that can be easily evolved to bind different antigens, such as monobodies, affibodies, and designed ankyrin repeat proteins (DARPins). Selection of the antibody or antibody mimetic class may have a profound effect on the type of chemical and genetic modifications required to enable stimulus sensitivity, as discussed throughout the text.
In this Outlook, we analyse strategies and applications for every one of the main cues used for antibody activation. We start focusing on the stimuli that have been utilized to engineer antibodies both for the study of biological systems and pharmacology, i.e., light and effector molecules, and then we transition to the endogenous cues that have enabled the design of antibodies mostly intended for therapy, i.e., pH, proteases, phosphatases, and antigen combination. Finally, we provide a horizontal perspective comparing all strategies and highlighting the trends in the field.
2. Light Activation
Light enables high temporal and spatial resolution in the study of molecules and living systems, and in photopharmacology. Hence, several approaches have been devised to confer photosensitive binding to antibodies (Figure 2), which can be classified into (i) incorporation of sterically demanding photocaging groups8−10 and (ii) insertion of photosensitive domains for antibody structure modulation.11−13 Divalent peptide–DNA masks, which have been engineered to be sensitive to several stimuli including light,14 will be described in section 3.
Figure 2.

Light-induced activation can be engineered through various mechanisms for therapeutic or cell biology applications. (A) Random nucleophile photocaging enables generating selective T-cell engagers.23 (B) Genetic encoding of photocaged tyrosine (ONBY) provides selective targeting and cell biology tools such as protein dimerizers. A bispecific nanobody–photobody construct is used as a model dimerizer to recruit GFP from the extracellular space and bring it close to mCherry-EGFR upon UV-light irradiation.10 (C) A photosensitive variant of the peptide–DNA lock described in section 3.1 could be applied in photopharmacology.14 (D) Split nanobody assembly via optical Magnets controls protein location to investigate cellular processes. In the confocal images shown, the two split parts of the antibody colocalize with Mito-GFP upon blue light irradiation. (E) Encoding of the LOV domain enables intracellular control of protein location and immunopurification.11,13 Adapted with permission from refs (10, 12, 13, and 22). Copyright 2020 Wiley-VCH, and 2019 and 2020 Springer Nature.
2.1. Photocaging Groups Hindering Binding
2.1.1. Randomly Photocaged Nucleophilic Side Chains
Photolabile protecting groups, i.e., photocaging groups, have been used for selective light activation of biologically active compounds since the late 1970s.15 Not surprisingly, this was the first activation strategy to be applied to antibodies.8,16 The field of activatable antibodies was pioneered by Self and Thompson, who developed a method to protect random nucleophilic side chains on the antibody surface with 1-(2-nitrophenyl) ethanol (NPE, Figure 2A). Although good nucleophiles such as lysine residues have low abundance on antibody complementary determining regions (CDRs),17 photocaging these residues can affect multiple interactions in their vicinity via steric hindrance. NPE coupling conditions require optimization for every antibody, resulting in 20–50 nitrophenyl molecules per antibody.8,18 Deprotection of NPE is achieved using UV-A light.
The applicability of this approach has been demonstrated by generating a bispecific masked T-cell engager for the treatment of aggressive ovarian carcinoma.18−23 Anti-CD3 bispecifics are highly promising therapeutics but may cause peripheral activation of T-cells, which results in T-cell depletion and off-target effects such as cytokine storms. By photocaging the anti-CD3 T-cell engaging domain, a significant decrease of primary tumor growth and of liver metastases in a mouse model was achieved.22
2.1.2. Genetically Encoded Photocaged Amino Acid Residues
Although random photocaging of lysine residues with NPE is simple and generally applicable, the inactivation (from background levels to 30% native binding) and activation (25–75% native binding) efficiencies are highly variable.8,18,20 Moreover, in antibody–drug conjugates, random functionalization of surface lysines has been shown to result into lower therapeutic indexes compared to site-specific modification.24 Aiming to produce homogeneous antibody conjugates activated by light, two research groups have recently reported the genetic encoding of a photocaged tyrosine derivative (Figure 2B).9,10
Tyrosine was selected because it is the amino acid residue most commonly involved in antigen binding.17O-Nitrobenzyl tyrosine (ONBY) was encoded into nanobody genes via amber suppression using an evolved orthogonal tyrosyl tRNA synthetase/tRNA pair.9,10,25 Binding was reduced 10 000-fold in the presence of ONBY and was restored almost completely in all cases (76–95%) upon decaging with UV-A light.10 Computational tools may help rationalizing the selection of residues to be photocaged.9
The utility of encoding ONBY has been shown with nanobodies targeting therapeutically relevant receptors such as EGFR and HER2. These photocaged ligands can be applied for targeting small molecules to cancer cells as proven by Sachdeva and collaborators.9 Mootz and co-workers have shown that light-activated ONBY antibodies can also be applied for the generation of molecular biology tools, such as light-induced protein dimerizers that can be deprotected within seconds on cells (Figure 2B).10
Although studies with ONBY have focused on nanobodies, the same technology should be applicable to any other antibody format or mimetic. Full-length antibodies or Fabs, which have larger paratopes, may require screening a larger number of positions or encoding more than one photocaged residue. Red-shifted or two-photon caging groups, which enable deeper tissue penetration, would facilitate translation of this technology into animals. Recent advances in genetic code expansion may enable the efficient incorporation of noncanonical amino acids bearing these protecting groups and others into antibodies.26,27
2.2. Photoresponsive Protein Domains Modulating Antibody Conformation
2.2.1. Optogenetic Dimerization of Split Antibody Fragments
Photosensitive antibodies may have broad applicability in cell biology because, despite the numerous tools developed in the field of optogenetics, controlling untagged target proteins remains elusive. Although genetic encoding of photocaged residues is an effective solution to this problem, approaches that offer reversible activation and do not require nonproteinogenic amino acids might facilitate a more extended usage. Very recently, two strategies exploiting photoresponsive protein domains have been engineered to meet these challenges, opening new horizons for the precise perturbation of cellular processes to investigate timing, location, and identity of active proteins.
Do Heo, Park, and co-workers have applied optical dimerization for the first time to produce a photosensitive antibody (Figure 2D).12 Their approach consists of splitting a nanobody or an scFv into two parts, each fused to complementary Magnet optical dimerization proteins, which only bind upon blue light (488 nm) irradiation. Blue light enables longer exposure times to monitor living cells compared to the UV light required by current photocaging groups. The Magnet system is based on engineered variants of the fungal photoreceptor VVD that selectively heterodimerize through electrostatic interactions upon light irradiation.28 Such dimerization is mediated by the formation of a photoadduct between Cys108 and the flavin adenine dinucleotide.
Although Magnet-mediated dimerization is theoretically reversible, the high affinity of the nanobody for its antigen (KD = 7.27 × 10–8 M) prevents dissociation. The applicability of the anti-GFP “optobody” was first shown by rendering the protein degradation method deGraFP light-inducible. deGraFP is a system that ubiquitinates GFP-tagged proteins in cells, targeting them for proteasomal degradation. This approach relies on the fusion of an anti-GFP nanobody with an F-box protein, which defines substrate specificity in E3 ubiquitin ligase complexes. By exchanging the nanobody for an optobody in this system, degradation of any GFP-tagged protein can be controlled with light. The authors further demonstrated the broad scope of their method by developing optobodies that control protein intracellular location, thereby manipulating cell processes such as gelosin-induced cell migration and beta-2 adrenergic receptor signaling.
2.2.2. Conformational Switching via a Photosensitive Domain
Despite the number of methods to control antibody–antigen binding that have recently emerged, few enable toggling between on and off states. Harnessing the power of a natural protein switch, Avalos and Toettcher groups have jointly created antibody mimetics dubbed “optobinders” (Figure 2E).13 The basis for their design is the insertion of a light-oxygen-voltage domain from Avena sativa phototropin 1 (AsLOV2) into the structure of nanobodies (OptoNBs)11 and monobodies (OptoMBs).13 The AsLOV2 domain exerts switching under blue light (447 nm) via a large conformational change triggered by the covalent conjugation of photoexcited flavin chromophore FMN to a conserved cysteine. This reaction causes the disruption of the C-terminal Jα helix in AsLOV2, which results in a looser conformation of the protein generally inducing inactivation. Surprisingly, in OptoNBs, irradiation with light results in a binding affinity increase or decrease depending on the loop in which AsLOV2 is inserted.
To prove the applicability of this approach in controlling downstream cellular functions, an anti-mCherry OptoNB was fused to the catalytic domain of the SOS protein, and membrane localization of this domain was used to regulate Ras activity and thereby control the Erk mitogen-protein kinase (MAPK) cascade. Erk activity could be reversibly toggled on and off with both light- and dark-inducible OptoNBs. Despite the successful application of OptoNBs in cells, only up to a 5.5-fold change in binding affinity between light and dark states was achieved. By contrast, OptoMBs reached a 330-fold affinity increase, which proved highly advantageous in light-controlled affinity chromatography. Since the positions selected to insert LOV domains are conserved across the respective antibody mimetic families, this method may be transferable to other antigen specificities.
3. Effector Molecule Activation
Light needs an external source, and spatial and temporal control often require complex setups. By contrast, control using soluble compounds may be easier to implement in different experimental settings and may be modulated with concentration. Moreover, activation triggered by particular endogenous molecules could enable smart systems with intrinsic selectivity. Although most systems have been tested with molecules added exogenously, many of the engineered antibodies may be adapted to interact with endogenous biomolecules or metabolites. Effector molecules (Figure 3), including oligonucleotides,29 peptides,30 and small molecules,31−33 have been used both in the investigation of biological systems and in therapeutics. These molecules can mediate activation via displacement of a moiety masking the paratope or via recovery of the antibody binding conformation.
Figure 3.

Small molecules, peptides, and oligonucleotides can be used to switch antibody binding in diverse applications. (A) A peptide–DNA lock may be used in the construction of molecular logic gates.29 (B) Chemogenetic antibody activation enables intracellular control of protein location and manipulation of biological systems.33 (C) scFvs activated with calmodulin-binding peptides could provide useful immunoaffinity purification tools.30 (D) Rapamycin-activated antibodies may be used to investigate biological systems.12 (E) Chemical rescue of binding conformation could enable prodrug-activated antibodies.32 Adapted with permission from refs (30, 32, and 33). Copyright 2020 American Chemical Society and 2020 Springer Nature.
3.1. Paratope Masking Moieties
3.1.1. Displacement of a Bivalent Peptide–dsDNA Lock
A family of effector molecules that is particularly interesting because of its predictable binding properties and biological significance is oligonucleotides. These molecules may enable the construction of molecular logical circuits for the production of autonomous signal processing systems in synthetic biology and molecular diagnostics. By using a peptide–double-strand DNA (peptide–dsDNA) lock, Merkx and co-workers have expanded the scope of activatable antibodies toward this direction (Figure 3A).29 Antibody inactivation relies on the bivalent binding of a dsDNA with a peptide at the 5′ end of each strand. The peptide sequence acts as a mimotope, i.e., an epitope-mimetic, and binds to the antibody with such an affinity as to enable dissociation when monomeric and binding when dimerized with the dsDNA. The 3′ end of one or both DNA strands is elongated with an overhanging DNA sequence called the “toehold”; in this way, a strand that binds this toehold and part of the dsDNA sequence can dissociate the dsDNA linker. The installation of a toehold on each DNA strand enables the generation of AND and OR gates, opening the doors to programmed activation of antibody-based systems. This approach is highly versatile and may be rendered sensitive to a second stimulus such as proteases or light by adding different sensing modules in the peptide–dsDNA linker (Figures 2C and 5B). However, the efficacy of the lock is limited by the delicate affinity equilibrium required between the monomeric and dimeric state to enable mimotope release upon dsDNA dissociation or cleavage. Amenability to in vivo applications may be further limited by dsDNA stability. While this approach is only applicable to full-length IgGs, other effector molecule strategies are mostly limited to smaller antibody derivatives.
Figure 5.

Protease overexpression enables selective antibody activation in diseased tissues. (A) In the probody approach, extending the N-terminus with a mimotope that can be removed by proteolytic cleavage shows great versatility and decreases off-target binding, thereby enhancing antibody circulation time.58−61 The graph schematically represents results from ref (61). (B) Epitope-mimetics and idiotypic masks enable good inactivation efficiency at the expense of transferability to to different antigen specificities.62−65 (C) Approaches relying on the interaction with more conserved regions in the Fv could enable transferability to other antibody specificities and formats.66,67 (D) Several strategies generally applicable to certain antibody formats have been developed relying on steric hindrance with variable degrees of inactivation efficiency.68−72 (E) Masking via N-terminal coiled-coil domains is efficient and suggests high transferability to other antigen specificities.73 Adapted with permission from refs (58 and 73). Copyright 2013 American Association for the Advancement of Science and 2019 Springer Nature.
3.1.2. Chemogenetic Unmasking
Aiming to provide a new tool that enables control over cellular functions with both endogenous and exogenous molecules via allosteric regulation, Johnsson and collaborators have recently engineered ligand-modulated antibody fragments (LAMAs) (Figure 3B).33 Their approach consists in inserting a circularly permutated bacterial dihydrofolate reductase (cpDHFR) in the CDR3 of a nanobody. In the “on” state, apo-cpDHFR is partly unfolded and enables binding to the antigen with single-digit nanomolar affinity. In the “off” state, triggered by the presence of its cofactor nicotinamide adenine dinucleotide phosphate (NADPH) and small-molecule inhibitors such as trimethoprim (TMP), the cpDHFR becomes fully folded, sterically blocking antibody binding. Remarkably, binding is reversible, and the affinity difference with and without both small-molecule effectors is over 1000-fold. Since NADPH is present in the cytosol and TMP is cell-permeable and has low toxicity, this system is readily implemented in living cells. The utility of this approach to study biological systems was shown by controlling the intracellular location and thereby the function of Mad2L1, a key component of the mitotic checkpoint complex. Transferability of this strategy was demonstrated by creating LAMAs against other targets, although their response was variable and required screening alternative cpDHFR insertion sites.
3.2. Conformational Inactivation
3.2.1. Peptide-Sensitive scFv Linker
Rescue of the antibody function can be achieved not only via unmasking as shown in LAMAs or the peptide–dsDNA lock but also through allosteric regulation of the conformation of the antibody itself. Thie, Dübel, and co-workers devised a way to modulate scFv binding exploiting the large conformational change of calmodulin upon interaction with calmodulin-binding peptides (Figure 3C).30 The characteristic (Gly4Ser)3 linker present in scFvs was replaced by circularly permutated calmodulins. Upon binding the peptide ligand, the alpha helical central domain of calmodulin folds wrapping around the peptide, which introduces significant strain in the scFv linker. The system was responsive to a variety of peptides; curiously, some of them had opposite switching behavior. This strategy could find applicability in high specificity affinity chromatography requiring mild elution conditions. Despite the potential of this approach, further improvement of the modulation strength should be sought since the affinity decrease measured for the most promising candidate is limited to 4.1-fold.
3.2.2. Chemical Rescue of Antibody Structure
In addition to peptides, small molecules have also been used to allosterically control antibody conformation. This has been achieved via split nanobodies fused to rapamycin dimerization domains12 (Figure 3D) in a similar manner as described for the light-inducible Magnet fusions (see section 2.2 and Figure 2D). Although rapamycin dimerizers have been applied in vivo,34 antibodies intended for therapy may benefit from activation strategies independent of large switching domains, which may affect pharmacokinetics and induce immunogenicity. Along these lines, Karanicolas and collaborators have envisioned a strategy that relies on engineering antibodies with a cavity that precludes them from adopting the active conformation. Activity can then be rescued by fitting a small molecule in the cavity (Figure 3E).31,32 A computationally guided design for allosteric antibody activation, which alters 2–3 residues to form the cavity, enabled creating an antibody with a 10-fold affinity increase in the presence of micromolar concentrations of 6-(benzyloxy)indazole.32 This approach should be transferable to scaffolds that contain the three mutated residues, which account for 78% of entries in the structure database SAbDab.
Chemical rescue via cavity engineering might be applicable to Fabs despite their larger interaction surface taking into account that the knobs-in-holes strategy enables the generation of full-length IgG bispecifics with a single cavity.35 Regarding translation of this approach into therapeutical applications, two important improvements suggested by the authors are required: (i) enhancing the molecule affinity for the cavity so that lower concentrations of the effector molecule are needed and (ii) turning the effector molecule into a prodrug that is only activated at the target site.
4. pH Activation
Although light and effector molecule activation provides promising opportunities in pharmacology, pH-activatable antibodies are the ones that have progressed the most as therapeutics to date. pH is an attractive endogenous cue for strategies aiming to enhance antibody bioavailability and therapeutic window. In the past decade, the difference in pH between late endocytic vesicles and the extracellular space has been applied to enable antibody “recycling”,37 to enhance antibody–drug conjugate (ADC) cytotoxicity,38 and to boost antibody transcytosis across the blood–brain barrier.39 Two pH-sensitive recycling antibodies are currently used in clinical practice, and many others are under clinical and preclinical development. In addition, the pH difference between healthy and solid tumors has been exploited to increase the selectivity of antibody therapeutics.40,41 pH-sensitive antibodies have also been used in immunoaffinity chromatography.42
In all cases, antibodies are rendered pH sensitive by introducing histidine residues in the CDRs and framework (Figure 4A, top). Capitalizing on a pKa near 6, the imidazole ring on histidine can be protonated in intra- and extracellular microenvironments where the pH is significantly lower than physiological values (7.2–7.4), especially in the endosomes and in the tumor tissue. When histidine is protonated, it is assumed to cause an electrostatic repulsion between the histidine-rich paratope and the positively charged basic residues in the epitope. Histidines have been engineered into pH-dependent antibodies via structure-based design and via directed evolution techniques such as phage and yeast display.43−45 Full-length IgG has been the preferred format for pH-sensitive antibodies given its advantageous pharmacokinetic properties for therapeutic applications.
Figure 4.
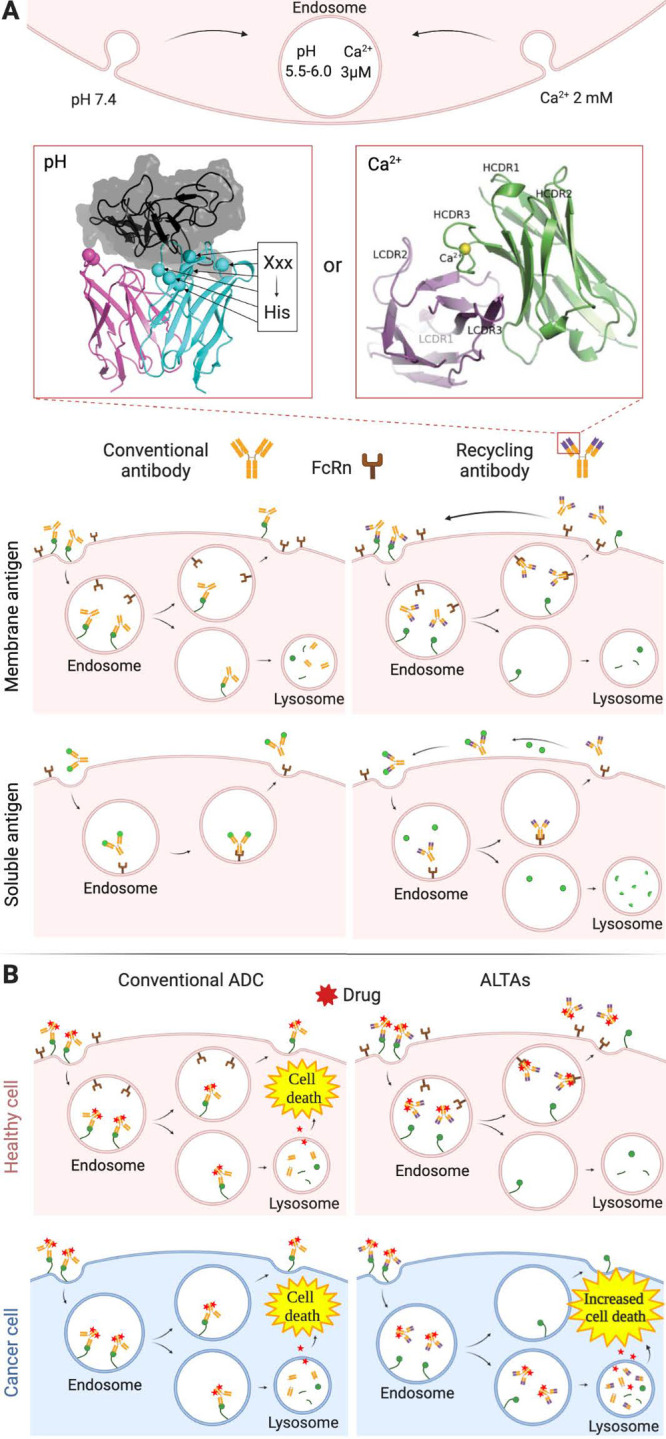
pH sensitivity enhances the therapeutic potential of antibodies. (A) Recycling antibodies have an enhanced half-life and degradation of soluble antigens.46 Such antibodies may be engineered by introducing histidine residues to induce pH-sensitivity40 or by evolving Ca2+-sensing loops in the CDRs.57 (B) ADCs with decreased affinity at the lower endosomal pH have enhanced efficacy and selectivity for the tumor tissue.38 Adapted with permission from refs (40 and 57). Copyright 2020 MAbs.
4.1. Activation in the Endosome
4.1.1. Recycling Antibodies
Although antibodies have high affinity and selectivity, they can engage their targets only once during the antibody’s circulation lifetime. Therefore, antigens with high abundance and turnover rates cannot be efficiently engaged using acceptable antibody administration frequency and doses. In order to generate antibodies that could bind more than one target in their lifetime, Igawa and collaborators proposed the concept of “recycling antibodies” (Figure 4A, bottom).46,47 These antibodies are engineered with reduced affinity at endosomal pH (5.5–6.0) so that they dissociate from their antigens only in the endosome. Consequently, antigens may be sorted to lysosomal degradation, while free antibodies bind the neonatal Fc receptor (FcRn) and are recycled back to the surface. Since the FcRn-antibody affinity at near-neutral pH is very low, antibodies are subsequently released into the extracellular space. This phenomenon reduces antibody lysosomal degradation and enables an antibody molecule to repeatedly neutralize target antigen molecules, thereby displaying high efficacy even in substoichiometric amounts relative to the antigen. Increasing the affinity of antibodies for the FcRn at acidic or neutral pH, such as in “sweeping antibodies”, may further increase their efficacy as recently reviewed elsewhere.48
A seminal study in 2010 revealed that about 75% of endocytosed acid-switched antibodies against interleukin 6 receptor (IL-6R) were recycled. By contrast, a non-pH-sensitive version of the same antibody was almost fully degraded in the lysosome.46 The pH-switched antibody had a 6.3-fold change in affinity between pH 7.4 and 6.0. This feature enabled the release from membrane-bound IL-6R in the endosome, eventually leading to a 20-fold increase in the antibody concentration in blood plasma after 78 h. Moreover, the soluble form of IL-6R was reduced 40-fold with the acid-switched antibody compared to the non-pH-sensitive antibody at day 4. This anti-IL-6R recycling antibody, under the name of satralizumab,49 and ravulizumab,50 an acid-sensitive antibody against the complement component C5, have recently been approved for their use in clinics. The first is used to treat neuromyelitis optica and the latter for paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. Other antibodies using this strategy are in clinical and preclinical development, including antibodies targeting PCSK9 to treat cardiovascular diseases; TFPI for hemophilia; TNF for inflammatory diseases; and CEA, HER2, and CTLA-4 for cancer therapies.45,51−56
pH is a well-known feature that differentiates late endosomes and the extracellular environment. Moreover, evolving pH-sensitive antibodies is well established. However, calcium sensitivity is a promising alternative to pH-sensitivity in recycling antibodies (Figure 4A, top) because (i) the calcium concentration difference between endosomes and blood plasma is roughly 650-fold, while the proton concentration difference is on the order of 50-fold, (ii) Ca2+ has greater capacity to destabilize binding because of the much larger electrostatic potential compared to protonated histidines, and (iii) divalent ions bring into proximity negatively charged residues, which repel each other in the absence of the ion. Allosteric antibodies, in which the CDR3 loop on the heavy chain is structured by coordinating Ca2+, have been evolved via phage display. Although antibodies with sensitivity to calcium have shown substantial enhancement of IL-6R clearance in vivo,57 this approach has so far been applied in very few instances compared to pH switching.
4.1.2. ADCs with Increased Lysosomal Degradation
A recent study by Ward, Ober, and co-workers has shown that pH sensitivity can increase ADC lysosomal degradation instead of recycling in cancer cells. This phenomenon increases toxin release in target cells, thereby enhancing ADC efficacy (Figure 4B).38 The inability of these ADCs to be recycled is rationalized by the presence of low levels of FcRn in tumors such as breast, prostate, and lung cancers. In the absence of FcRn, the soluble contents of late endosomes, including ADCs dissociated from their membrane-bound antigens, are fully degraded. In this study, pH-sensitive antibodies against HER2 derived from pertuzumab were conjugated to the microtubule polymerization inhibitor monomethyl auristatin E (MMAE). The higher efficacy of these ADCs with increased lysosomal-trafficking activity (ALTAs) was confirmed in two mouse models of HER2+ breast cancer, inhibiting growth of xenografts more effectively than pertuzumab-MMAE and the FDA-approved trastuzumab-MMAE.
4.2. Activation in the Tumor Microenvironment
ALTAs have enhanced efficiency against cancer cells by capitalizing on the pH difference between the endosome and the extracellular space and the low abundance of the FcRn. However, there is another pH difference that can be exploited to enhance tumor selectivity: the one existing between the tumor microenvironment (pH 6.0–6.8) and healthy tissues (7.2–7.4). The lower pH in the tumor is due to several factors such as poor vascular perfusion, regional hypoxia, and fermentative glycolysis.74 By contrast to the affinity decrease at lower pH required in ALTAs and recycling antibodies to achieve endosomal release, tumor targeting requires an affinity increase. Antibodies with low affinity at pH 7.2–7.4 and high affinity at pH 6.0–6.8 will display minimal target engagement in healthy tissue and maximal binding in the tumor.
Sulea, Zwaagstra, and collaborators have engineered an acid-switched antibody that can selectively engage its HER2 antigen in the tumor.40 Via a de novo structure-based approach, the authors obtained antibodies with a 10-fold weaker binding at pH 7.3 compared to pH 6.8, while preserving an affinity below 30 nM in the pH range of 6.0–6.8. Variants formatted as full-length IgG1/k antibodies inhibited the growth of tumor spheroids at a level comparable to the gold standard anti-HER2 antibody, trastuzumab, at acidic pH and had a significantly reduced effect at physiological pH. The substantial improvement in efficacy of these therapies still needs to be confirmed in vivo.
5. Protease Activation
Alongside the pH, the main internal cue exploited for antibody activation is the activity of specific enzymes. In this field, research groups and companies have mainly focused on the activity of proteases.
Altered protease expression and activity are a hallmark of cancer and many other pathologies such as autoimmune, cardiovascular, and neurodegenerative diseases.75 Conversely, in healthy tissues protease activity is tightly regulated at different levels. Most proteases are expressed as zymogens and require post-translational modifications to be activated. Moreover, once activated, their functioning can be further controlled by endogenous protease inhibitors. Such efficient and redundant control mechanisms ensure that protease-dependent prodrugs are mainly activated in diseased tissues, enhancing their specificity and reducing side effects.76 Applicability to a wide variety of diseases, paired with the promising results shown by small-molecule prodrugs, have drawn the interest of several companies, which have contributed to the development and advancement toward clinical application of this subclass of switchable antibodies. Protease-activatable antibodies, mostly full-length IgGs, have been engineered utilizing a number of strategies based on the introduction of different types of masking moieties, including (i) mimotopes and idiotypic masks; (ii) proteins binding the variable domain and extending over the paratope or disrupting the binding conformation; and (iii) nonbinding bulky moieties inserted near the paratope and sterically hindering antigen binding (Figure 5).
5.1. Mimotopes and Idiotypic Masks
The most widely used approach consists of a mimotope or epitope-mimetic peptide tethered via a protease-labile linker to the N-terminus of the antibody or antibody derivative (Figure 5A). In this form, interaction with the antigen is impaired, and, only in the presence of proteases, the linker is cleaved and the masking peptide is released restoring antigen-binding ability. This strategy was first presented by Williams and Rodeck (2009) in the form of cross-masked scFvs (Figure 5B)64 and then became the core technology of Probody Therapeutics (CytomX), which has mainly focused on the development of masked full-length IgGs.75,77,78 The probody approach has been applied to target a variety of receptors, including vascular cell-adhesion molecule 1 (VCAM-1), EGFR, PD-L1, as well as receptors considered undruggable because of their presence in many tissues, such as CD166 and CD71.58,60,75,79−81 Affinity changes provided by the mimotope mask range from 40- to 300 000-fold,61,75 and several of these products are currently in clinical trials.60,82
One of the main advantages of the probody strategy is its great versatility across antibody formats, including antibody–drug conjugates, chimeric antigen receptors (CARs), and bispecifics. Probody drug conjugates (PDCs) against CD166 and CD71 have shown tumor regression and are well tolerated in patients with advanced solid tumors.60 More recently, the probody technology has been applied to CARs, synthetic receptors expressed on the surface of T-cells that allow activation of these cells upon tumor antigen engagement.59 Regarding bispecifics, a T-cell engager probody targeting EGFR and CD3 has shown an increased maximum tolerated dose in clinical trials.61
Probody epitope-mimetic N-terminal extensions are not the only mimotope-based strategies. In the field of multispecific antibodies for T-cell activation, Harpoon Therapeutics has developed an activatable form of its trispecific T-cell activating construct (proTriTAC). In proTriTAC (Figure 5B), the CD3-targeting moiety is masked by an anti-albumin single-domain antibody fragment modified to specifically interact with the anti-CD3 CDRs.63 The masked construct has a 250-fold reduced affinity in addition to an enhanced half-life due to albumin binding. Another alternative to using mimotopes is the use of anti-idiotypic binders. Löfblom and collaborators proved that an inactivating anti-idiotypic affibody can be fused to an anti-HER2 affibody, reducing apparent affinity over 1000-fold.65
Although using epitope-mimetics or anti-idiotypic binders provides outstanding inactivation efficiency, each mask can only be applied for one antigen specificity. Furthermore, generation of the masking domain requires a challenging and time-consuming affinity fine-tuning involving bacterial or phage display selections.
5.2. Masking Moieties Binding the Variable Domain
To improve transferability to other antigen specificities, moieties that bind more conserved regions in the variable domain (Fv) and extend over the paratope or disrupt the binding conformation could be exploited (Figure 5C).
Conditional T-cell engagers named COBRAs (Maverick Therapeutics) rely on the cleavage of an inactivating domain and subsequent dimerization of the active domains of an scFv to allow binding to CD3.67 To the best of our knowledge, this is the only protease-sensitive strategy that involves allosteric disruption of the binding conformation rather than masking of the paratope. Despite its complexity, this system is functional in vivo as proven by its capacity to target T-cell-dependent cytotoxicity to HT-29 xenograft murine models, inducing complete tumor remission at modest doses.
Easier transferability across some antigen specificities can be achieved with protein M from Mycoplasma genitalium. Chen and co-workers engineered a protease-cleavable version of this protein that recognizes a conserved region in the Fv and extends over the paratope, thus blocking binding in a full-length IgG.66 Interestingly, this is one of the few approaches that does not require modification of the antibody, which may enable easier transferability to other IgGs and antibody derivatives.
5.3. Nonbinding Masking Moieties
A more universal approach would require inactivation exclusively via steric hindrance. Several strategies have been developed aiming toward this ambitious goal by extending the N-termini of full-length antibodies or derivatives with different moieties displaying varying degrees of efficiency and transferability (Figure 5D).
A requirement for the steric mask is low immunogenicity. Taking into account this criteria, several protein moieties have been N-terminally fused to antibodies, including endogenously derived inhibitory domains (i.e., LAP domain from pro-TGF-beta),68 peptides able to recruit albumin,69 and intrinsically disordered XTEN polypeptides.70 The first two approaches enable 2- and 10-fold affinity decrease, respectively, and have not been reported in animals studies. Conversely, XTENylated protease-activated T-cell engagers (XPATs) show a significant increase in the maximal tolerated dose in cynomolgus monkeys and minimal effect on antibody pharmacokinetics.
Aiming to provide a more stringent lock by elongating both N-termini with interacting polypeptide chains, antibody fusions have been engineered so that one masks the other decreasing antigen-binding affinity up to 3000-fold. In one instance, a disulfide-stabilized Fv was fused to the heavy chain of a full-length IgG, which acted as a masking moiety.71 In another instance, the intrinsically lower binding affinity in the “inner domain” of a dual variable domain antibody was exploited.72
More recently, two strategies have been proposed that substantially reduce the length of dimerizing N-terminal extensions while still blocking antigen binding efficiently. While Cheng and co-workers used the disulfide-stabilized hinge region of an IgG1 as a lock,84 Levengood and collaborators applied high-affinity noncovalent interactions between leucin-zipper coiled-coil domains.73 The latter strategy enables a 750-fold reduced affinity in an anti-CD19 antibody, which results in a lower engagement of systemic antigen and prolonged half-life (Figure 5E). Remarkably, this antibody displays enhanced efficacy in lymphoma xenograft models. This general protease-sensitive approach could be easily transferred to virtually any Fab or full-length antibody.
6. Phosphatase Activation
Protease expression and activity in several pathologies is well characterized. However, the expression of other enzymes is also altered in diseased tissues, and interest in the role of phosphatases in diseases has recently increased. Intra- and extracellular phosphatases are involved in a wide variety of cellular processes, and their activity can be altered in numerous diseases, including cancer,85 Alzheimer’s disease,86 and hypophosphatasia.87 Although the expression and activity of phosphatases in many pathologies need to be further characterized, dephosphorylation could be an interesting stimulus to activate antibodies.
Davis and collaborators developed an anti-lysozyme sdAb reversibly inactivated through a phosphate group in the paratope.88 This phosphate group was located at a site that mediates a hydrophobic interaction with the antigen, taking advantage of a suggested evolved switching mechanism of phosphorylation. Post-translational inactivation was achieved via a “tag-and-modify” approach. First, the sulfhydryl group of a Cys residue in the CDR3 loop was site-specifically eliminated to yield dehydroalanine. Next, the orthogonally reactive tag was chemically phosphorylated into a phosphocysteine (Figure 6A). Several alkaline phosphatases were able to remove the phosphate group and restore binding from background to almost prephosphorylation levels. Moreover, ex vivo, such a construct was able to selectively stain lysozyme-positive cells transplanted into mice brains only after expression of secreted embryonic alkaline phosphatase (SEAP) was induced by a lipopolysaccharide (LPS)-triggered TNF-α response (Figure 6B). Further investigation is needed to evaluate the therapeutic potential of this novel strategy. In particular, taking into account the broad substrate specificity of many phosphatases,89 stability of the inactivating group in circulation and diffusion in healthy tissues should be evaluated. Interestingly, the post-translational installation of dehydroalanine allows for site-selective modification of proteins and antibodies with a wide array of groups that could be sensitive to other stimuli.
Figure 6.
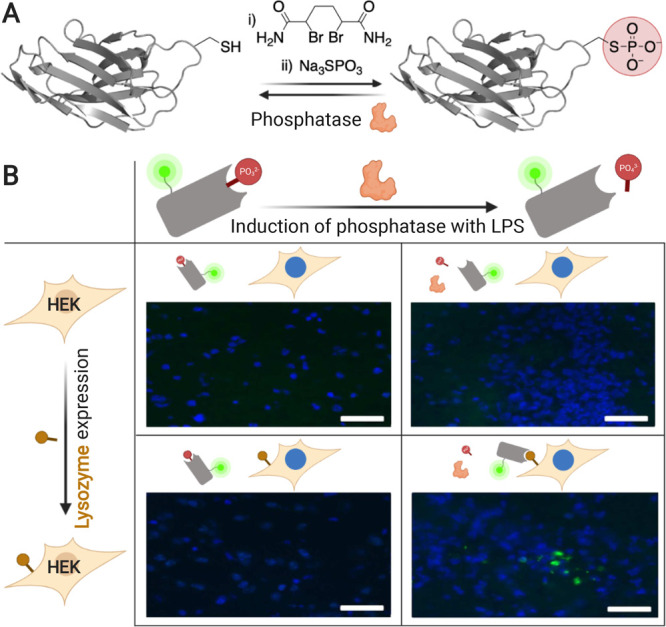
Antibody phosphorylation provides selective activation on mice tissues ex vivo. (A) Chemical phosphorylation of a cysteine on an anti-lysozyme nanobody via a dehydroalanine intermediate. (B) Lysozyme-expressing cells implanted in mice brain are selectively stained ex vivo only in response to LPS-induced secretion of SEAP. Adapted with permission from ref (88). Copyright 2014 Nature Communication.
7. Activation via Antigen Combination
The advantage of rendering therapeutic antibodies activatable arises from the scarcity of antigens exclusively expressed on diseased tissues. All strategies introduced up to this point rely on a reversibly inactive antibody that is only able to engage its antigen when activated at the target site by an environmental cue. An alternative way to enhance selectivity is to design antibodies that only bind target cells expressing certain membrane antigen combinations. This concept was first materialized in bispecific antibodies that only bind cells expressing two particular antigens.90 Although such antibodies do not have clearly defined active/binding and inactive/nonbinding states, they are activated (able to engage target cells) only in the presence of their two cognate antigens in close proximity. Bispecific antibodies targeting oncogenic receptor tyrosine kinases have been used to enhance selectivity and counteract the onset of the most common resistance mechanisms.91 Affinity of individual arms, as well as overall avidity, needs to be fine-tuned to reduce binding to single-positive nontarget tissues, which is a complex and delicate process.92,93
Baker and collaborators have recently developed a system that overcomes the challenge of fine-tuning bispecific antibody affinity and enables recognition of more complex antigen combinations, bringing selectivity to the next level.94 This system, named LOCKR, includes three main components (Figure 7): first, a “cage” protein sequestering a functional peptide in an inactive conformation through a “latch” domain; second, a “key” protein that can bind the cage when in close proximity but not in high dilution. Upon binding to the cage, the key displaces the functional peptide, thus allowing it to recruit the third component, an “effector” protein. Target specificity is conferred to the cage and key proteins by fusing them to DARPINs or scFvs with native affinity. Adding to this basic system either a second key protein or a decoy protein, which can sequester the key, enables implementation of AND–OR and AND–NOT Boolean logic, respectively. The authors show that the LOCKR system can retarget CAR-T cells recognizing the functional peptide in its active form to selectively kill Raji cells presenting the right combination of antigens in vitro.
Figure 7.
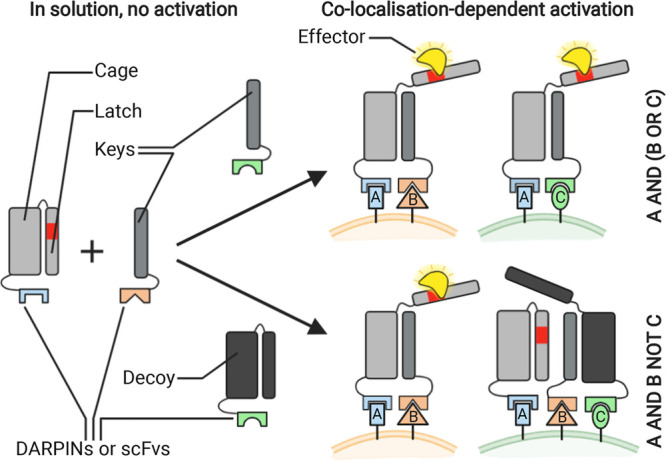
LOCKR system enables recruitment of an effector protein mediated by antigen colocalization. Increasing the number of keys and/or decoys present enables complex Boolean logic operation and enhances target specificity.
The LOCKR system enables integration of binding inputs based on complex Boolean logic operations without relying on fine-tuned antibody affinity or the cellular machinery. Moreover, it provides enhanced specificity dependent on membrane-protein expression patterns. Nonetheless, many challenges lie ahead for this complex system to be applied in therapy, including potential immunogenicity of the cage and key components, as well as selectivity and stability in physiologic media of the numerous protein–protein interactions involved.
8. Conclusions and Outlook
A plethora of strategies have been applied to generate activatable antibodies responsive to a variety of environmental cues for diverse applications (Table 1). Activation stimuli are closely related to applications, which can be broadly classified into therapy and basic research.
Table 1. Selected Approaches for Antibody Activation.
| activation cue | inactivation strategy (name) | format used (potential) | applicability | selected advantages (+) and disadvantages (−) | refs |
|---|---|---|---|---|---|
| UV-A light | genetically encoded photocaged Tyr | nanobody (all) | cell biology, targeted delivery | + small modification, format transferability, 10 000-fold affinity change | (9, 10) |
| – known binding residues, UV light | |||||
| blue light | optical magnet split antibodies (optobodies) | nanobody, scFv | cell biology | + blue light, potentially reversible | (12) |
| – transferability may be challenging | |||||
| blue light | conformational modulation via light-oxygen-voltage domain (optobinders) | nanobody, monobody | cell biology, affinity purification | + reversible, activation or inactivation, blue light, 330-fold affinity increase monobodies | (11, 13) |
| – <6-fold affinity increase nanobodies | |||||
| oligonucleotide | divalent peptide–dsDNA lock | IgG | logic gating | + no genetic modification required, possible combination with other stimuli | (29) |
| – small affinity change, relative instability | |||||
| small molecules | folded dihydrofolate reductase blocking CDRs (LAMAs) | nanobody | cell biology | + reversible, 1000-fold affinity change | (33) |
| – transferability may be challenging | |||||
| small molecule | conformational disruption via cavity | scFv (Fab) | therapeutic | + no potentially immunogenic appendage | (32) |
| – 10-fold affinity change | |||||
| pH decrease | electrostatic repulsion upon protonation | IgG | therapeutic | + reversible, increased antibody half-life, two antibodies in clinical use | (46) |
| – only applicable to IgGs (FcRn binding) | |||||
| pH decrease | electrostatic repulsion upon protonation (ALTAs) | antibody–drug conjugates | therapeutic | + reversible, increased efficacy | (38) |
| – only in tumors with low levels of FcRn | |||||
| protease activity | N-terminal epitope-mimetic masking peptide | IgG, bispecific IgG, CAR-T (all) | therapeutic, diagnostic | + up to 300000-fold affinity change, advanced clinical trials, format transferability | (58−61) |
| – complex fine-tuning of masking peptide affinity | |||||
| protease activity | masking N-terminal coiled-coil domains | IgG (Fab) | therapeutic | + 750-fold affinity change, highly transferable across antigen specificities | (73) |
| – only applicable to IgGs and Fabs | |||||
| antigen combination | functional peptide recruited by cage domain | DARPINs, scFvs (all) | therapeutic, diagnostic | + complex Boolean logic operations possible, modularity | (94) |
| – complex multicomponent system, possible immunogenicity |
Antibody derivatives and mimetics activatable with light and effector molecules such as optobodies, optobinders, LAMAs, and genetically encoded photocaged tyrosines provide powerful opto- and chemogenetic tools to investigate biological processes. The reversibility displayed by optobinders and LAMAs may be particularly interesting for cell biology applications. In addition, many stimuli, including light and effector ions and molecules, have been harnessed for the generation of effective immunoaffinity purification systems and may also find applicability in sensing devices. Temperature could also be applied in molecular transducers; however, the only strategy reported to date relies on an scFv with an elastin-based interdomain linker, which has a high critical temperature (49 °C) and small affinity change (<2-fold).95,96 Although the activation strategies reported to study biological systems have so far been tested in vitro, some of them might be amenable to experiments in animals. For instance, large protein modulatory domains depending on biologically orthogonal stimuli generally display a powerful and selective affinity switching. Other methods would need substantial refinement for in vivo applications, including the peptide–DNA lock strategy and the protein M steric blocking. Although these two strategies are highly appealing because they require neither chemical nor genetic modification of the antibody, they display limited masking capacity and stability.
Activatable antibodies that have advanced the most in therapy, i.e., “recycling antibodies” and “probodies”, are based on endogenous stimuli and relatively simple and robust strategies. Moreover, they are applied on full-length antibodies and preserve or boost IgG outstanding pharmacokinetic properties. Although other activation approaches based on Ca2+ concentration, light, effector molecules, and membrane antigen combinations provide interesting therapeutic options, they generally require more elaborate engineering, have mostly been applied on small antibody derivatives, and may need further refinement for an extended usage.
Apart from the special case of antigen-combination-mediated activation, reversible inactivation mechanisms described here can be broadly classified in three categories: (i) structural disruption of small antibody derivatives or mimetics, (ii) alteration of the charge on the CDR loops, and (iii) steric blockage of antibody paratopes. Disruption of the structure of antibody derivatives has been achieved with very slight changes, such as the suppression of a few residues forming a chemically rescuable cavity, to major structural modifications. Such major rearrangements include the introduction of photoreceptor domains, splitting nanobodies fused to photo- or chemodimerizing domains, producing scFvs with a cleavable inactivating domain and reengineering the interdomain linker of scFvs. The latter strategy is very versatile and has been used to enable sensitivity to temperature, Ca2+, and calmodulin-binding peptides. By contrast to small antibody derivatives, conformational modulation of IgGs is difficult to achieve. Therefore, control of these antibodies has mostly relied either on CDR charge alteration, as in pH- and phosphatase-sensitive antibodies, or via insertion of moieties sterically hindering antigen binding. Blockage of paratopes has been materialized in very diverse formats, including mimotopes and masking moieties solely relying on steric hindrance, anchored in the CDRs or close to them. Mimotopes have mainly been used as N-terminal fusions in probodies and in the peptide–dsDNA lock. Remarkably, the latter strategy (Figures 2C, 3A, and 4B) utilizes a chemically synthesized linker and can thus be engineered to be sensitive to virtually any stimulus, including the presence of specific oligonucleotides,29 proteases,62 light,14 and pH.41 Masks that work only via steric hindrance range from a single small light-sensitive o-nitrophenyl group, to the extension of the N-termini with protease-labile coiled-coil peptides or the insertion of chemosensitive domains such as cpDHFR in LAMAs.
Some of the inactivation strategies, such as blockage via nonbinding N-terminal extensions or replacement of the flexible interdomain linker in scFvs, are easily transferable to antibodies with different antigen specificities. However, most approaches require a higher degree of engineering and some knowledge of the structure. For instance, using modulatory domains inserted in the CDRs and N-terminal extensions with epitope-mimetics requires screening of many mutants or directed evolution methods, respectively. Transferability across antigen specificities has almost an inverse correlation with applicability to other antibody derivatives and mimetics. In this regard, addition of mimotopes and introduction of histidines, photocaged amino acids, or modulatory domains in the CDRs could be applicable to any binder ranging from full-length IgGs to affibodies. By contrast, IgG N-terminal extension with coiled-coil peptides and the scFv linker engineering by modulatory domains have a very limited applicability in other antibodies or mimetics. The LOCKR system overcomes this limitation to some extent by uncoupling activation from the ability of its individual components to engage their cognate antigens. Therefore, LOCKR may utilize virtually any type of antibody derivative or mimetic to engage the target antigen combination.
Overall, within a single decade, research into activatable antibodies has yielded marketed therapeutics capable of targeting antigens that could not be engaged with traditional antibodies, as well as new tools to control untagged proteins and investigate cellular processes. Yet, many opportunities in this expanding field remain untapped. New activation mechanisms at the interface between chemistry and biology, together with recently reported tools for directed evolution,98,99 may render more efficient and generally applicable masking strategies.
Acknowledgments
B.O.-S. acknowledges funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 844441. R.L. is grateful to IQS for a Ph.D. fellowship (2019−2020) and to the Spanish Ministry of Science, Innovation and Universities for an FPU fellowship (FPU19/03216). We acknowledge support from the Agència de Gestió d’Ajuts Universitaris i de Recerca (Generalitat de Catalunya) under the grant agreement SGR 2017 1559. All the figures in this article have been created with BioRender.
The authors declare no competing financial interest.
References
- Meulenberg E. P.Antibodies Applications and New Development; Meulenberg E. P., Ed.; Bentham Science Publishers Ltd., 2012. [Google Scholar]
- Carter P. J.; Lazar G. A. Next Generation Antibody Drugs : Pursuit of the ‘ High-Hanging Fruit. Nat. Rev. Drug Discovery 2018, 17 (3), 197–223. 10.1038/nrd.2017.227. [DOI] [PubMed] [Google Scholar]
- Tang S.; Wan Z.; Gao Y.; Zheng J. S.; Wang J.; Si Y. Y.; Chen X.; Qi H.; Liu L.; Liu W. Total Chemical Synthesis of Photoactivatable Proteins for Light-Controlled Manipulation of Antigen-Antibody Interactions. Chem. Sci. 2016, 7 (3), 1891–1895. 10.1039/C5SC03404C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas S. M.; Bachelet I.; Church G. M. A Logic-Gated Nanorobot for Targeted Transport of Molecular Payloads. Science (Washington, DC, U. S.) 2012, 335, 831–834. 10.1126/science.1214081. [DOI] [PubMed] [Google Scholar]
- Wen J.; Wu D.; Qin M.; Liu C.; Wang L.; Xu D.; Vinters H. V.; Liu Y.; Kranz E.; Guan X.; Sun G.; Sun X.; Lee Y.; Martinez-Maza O.; Widney D.; Lu Y.; Chen I. S.; Kamata M. Sustained Delivery and Molecular Targeting of a Therapeutic Monoclonal Antibody to Metastases in the Central Nervous System of Mice. Nat. Biomed Eng. 2019, 3 (9), 706–716. 10.1038/s41551-019-0434-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards D. A. Exploring Alternative Antibody Scaffolds: Antibody Fragments and Antibody Mimics for Targeted Drug Delivery. Drug Discovery Today: Technol. 2018, 30, 35–46. 10.1016/j.ddtec.2018.10.005. [DOI] [PubMed] [Google Scholar]
- Self C. H.; Thompson S. Light Activatable Antibodies: Models for Remotely Activatable Proteins. Nat. Med. 1996, 2 (7), 817–820. 10.1038/nm0796-817. [DOI] [PubMed] [Google Scholar]
- Bridge T.; Shaikh S. A.; Thomas P.; Botta J.; McCormick P. J.; Sachdeva A. Site-Specific Encoding of Photoactivity in Antibodies Enables Light-Mediated Antibody–Antigen Binding on Live Cells. Angew. Chem. Int. Ed. 2019, 58 (50), 17986–17993. 10.1002/ange.201908655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedlitzke B.; Yilmaz Z.; Dörner W.; Mootz H. D. Photobodies: Light-Activatable Single-Domain Antibody Fragments. Angew. Chem., Int. Ed. 2020, 59 (4), 1506–1510. 10.1002/anie.201912286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil A. A.; Carrasco-López C.; Zhu L.; Zhao E. M.; Ravindran P. T.; Wilson M. Z.; Goglia A. G.; Avalos J. L.; Toettcher J. E. Optogenetic Control of Protein Binding Using Light-Switchable Nanobodies. Nat. Commun. 2020, 11 (1), 4044. 10.1038/s41467-020-17836-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D.; Lee H.; Hong J.; Jung H.; Jo Y. J.; Oh B. H.; Park B. O.; Do Heo W. Optogenetic Activation of Intracellular Antibodies for Direct Modulation of Endogenous Proteins. Nat. Methods 2019, 16 (11), 1095–1100. 10.1038/s41592-019-0592-7. [DOI] [PubMed] [Google Scholar]
- Carrasco-López C.; Zhao E. M.; Gil A. A.; Alam N.; Toettcher J. E.; Avalos J. L. Development of Light-Responsive Protein Binding in the Monobody Non-Immunoglobulin Scaffold. Nat. Commun. 2020, 11 (1), 4045. 10.1038/s41467-020-17837-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wouters S. F. A.; Wijker E.; Merkx M. Optical Control of Antibody Activity by Using Photocleavable Bivalent Peptide–DNA Locks. ChemBioChem 2019, 20 (19), 2463–2466. 10.1002/cbic.201900241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieke C.; Rohrbach F.; Gottschalk A.; Mayer G.; Heckel A. Light-Controlled Tools. Angew. Chem., Int. Ed. 2012, 51, 8446–8476. 10.1002/anie.201202134. [DOI] [PubMed] [Google Scholar]
- Thompson S.; Fawcett M. C.; Spoors J. A.; Self C. H. The Modulation of Protein A-IgG(Fc) Binding by the Reversible Addition of 2-Nitrobenzyl Groups. Biochem. Soc. Trans. 1995, 23 (2), 155S. 10.1042/bst023155s. [DOI] [PubMed] [Google Scholar]
- Ramaraj T.; Angel T.; Dratz E. A.; Jesaitis A. J.; Mumey B. Antigen–Antibody Interface Properties: Composition, Residue Interactions, and Features of 53 Non-Redundant Structures. Biochim. Biophys. Acta, Proteins Proteomics 2012, 1824 (3), 520–532. 10.1016/j.bbapap.2011.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S.; Dessi J.; Self C. H. The Construction and in Vitro Testing of Photo-Activatable Cancer Targeting Folated Anti-CD3 Conjugates. Biochem. Biophys. Res. Commun. 2008, 366 (2), 526–531. 10.1016/j.bbrc.2007.11.158. [DOI] [PubMed] [Google Scholar]
- Self C. H.; Self A. C.; Smith J. A.; Self D. J.; Thompson S. Light-Directed Activation of Human T-Cells. ChemMedChem 2007, 2 (11), 1587–1590. 10.1002/cmdc.200700200. [DOI] [PubMed] [Google Scholar]
- Thompson S.; Fawcett M. C.; Self C. H. The Construction of a Functional Photoactivatable Cancer Targeting Bispecific Antibody Conjugate. ChemMedChem 2007, 2 (8), 1162–1164. 10.1002/cmdc.200700078. [DOI] [PubMed] [Google Scholar]
- Thompson S.; Stewart R.; Smith J. A.; Self C. H. Light Activation of Anti-CD3 in Vivo Reduces the Growth of an Aggressive Ovarian Carcinoma. ChemMedChem 2007, 2 (11), 1591–1593. 10.1002/cmdc.200700116. [DOI] [PubMed] [Google Scholar]
- Thompson S.; Dessi J.; Self C. H. Preclinical Evaluation of Light-Activatable, Bispecific Anti-Human CD3 Antibody Conjugates as Anti-Ovarian Cancer Therapeutics. MAbs 2009, 1 (4), 348–356. 10.4161/mabs.1.4.9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S.; Self A. C.; Self C. H. Light-Activated Antibodies in the Fight against Primary and Metastatic Cancer. Drug Discovery Today 2010, 15 (11–12), 468–473. 10.1016/j.drudis.2010.04.006. [DOI] [PubMed] [Google Scholar]
- Beck A.; Goetsch L.; Dumontet C.; Corvaïa N. Strategies and Challenges for the next Generation of Antibody–Drug Conjugates. Nat. Rev. Drug Discovery 2017, 16 (5), 315–337. 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- Deiters A.; Groff D.; Ryu Y.; Xie J.; Schultz P. G. A Genetically Encoded Photocaged Tyrosine. Angew. Chem. Int. Ed. 2006, 45 (17), 2728–2731. 10.1002/anie.200600264. [DOI] [PubMed] [Google Scholar]
- Xiao H.; Schultz P. G. At the Interface of Chemical and Biological Synthesis: An Expanded Genetic Code. Cold Spring Harbor Perspect. Biol. 2016, 8 (9), a023945. 10.1101/cshperspect.a023945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oller-Salvia B.; Kym G.; Chin J. W. Rapid and Efficient Generation of Stable Antibody–Drug Conjugates via an Encoded Cyclopropene and an Inverse-Electron-Demand Diels–Alder Reaction. Angew. Chem., Int. Ed. 2018, 57 (11), 2831–2834. 10.1002/anie.201712370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano F.; Suzuki H.; Furuya A.; Sato M. Engineered Pairs of Distinct Photoswitches for Optogenetic Control of Cellular Proteins. Nat. Commun. 2015, 6, 6256. 10.1038/ncomms7256. [DOI] [PubMed] [Google Scholar]
- Janssen B. M. G.; van Rosmalen M.; van Beek L.; Merkx M. Antibody Activation Using DNA-Based Logic Gates. Angew. Chem. Int. Ed. 2015, 54 (8), 2530–2533. 10.1002/ange.201410779. [DOI] [PubMed] [Google Scholar]
- Kellmann S. J.; Dübel S.; Thie H. A Strategy to Identify Linker-Based Modules for the Allosteric Regulation of Antibody-Antigen Binding Affinities of Different ScFvs. MAbs 2017, 9 (3), 404–418. 10.1080/19420862.2016.1277302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser C. E.; Rincon Pabon J. P.; Khowsathit J.; Castaldi M. P.; Kazmirski S. L.; Weis D. D.; Zhang A. X.; Karanicolas J. Modulating Antibody Structure and Function through Directed Mutations and Chemical Rescue. ACS Synth. Biol. 2018, 7 (4), 1152–1162. 10.1021/acssynbio.8b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khowsathit J.; Bazzoli A.; Cheng H.; Karanicolas J. Computational Design of an Allosteric Antibody Switch by Deletion and Rescue of a Complex Structural Constellation. ACS Cent. Sci. 2020, 6 (3), 390–403. 10.1021/acscentsci.9b01065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrants H.; Tarnawski M.; Müller T. G.; Otsuka S.; Hiblot J.; Koch B.; Kueblbeck M.; Kräusslich H. G.; Ellenberg J.; Johnsson K. Chemogenetic Control of Nanobodies. Nat. Methods 2020, 17 (3), 279–282. 10.1038/s41592-020-0746-7. [DOI] [PubMed] [Google Scholar]
- Stavrou M.; Philip B.; Traynor-White C.; Davis C. G.; Onuoha S.; Cordoba S.; Thomas S.; Pule M. A Rapamycin-Activated Caspase 9-Based Suicide Gene. Mol. Ther. 2018, 26 (5), 1266–1276. 10.1016/j.ymthe.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merchant A. M.; Zhu Z.; Yuan J. Q.; Goddard A.; Adams C. W.; Presta L. G.; Carter P. An Efficient Route to Human Bispecific IgG. Nat Biotechnol. 1998, 16, 677–681. 10.1038/nbt0798-677. [DOI] [PubMed] [Google Scholar]
- Igawa T.; Maeda A.; Haraya K.; Tachibana T.; Iwayanagi Y.; Mimoto F.; Higuchi Y.; Ishii S.; Tamba S.; Hironiwa N.; Nagano K.; Wakabayashi T.; Tsunoda H.; Hattori K. Engineered Monoclonal Antibody with Novel Antigen-Sweeping Activity In Vivo. PLoS One 2013, 8 (5), e63236. 10.1371/journal.pone.0063236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J. C.; Sun W.; Khare P.; Karimi M.; Wang X.; Shen Y.; Ober R. J.; Ward E. S. Engineering a HER2-Specific Antibody–Drug Conjugate to Increase Lysosomal Delivery and Therapeutic Efficacy. Nat. Biotechnol. 2019, 37 (5), 523–526. 10.1038/s41587-019-0073-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade H.; Baumgartner C.; Hugenmatter A.; Moessner E.; Freskgård P. O.; Niewoehner J. A Human Blood-Brain Barrier Transcytosis Assay Reveals Antibody Transcytosis Influenced by pH-Dependent Receptor Binding. PLoS One 2014, 9 (4), e96340. 10.1371/journal.pone.0096340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulea T.; Rohani N.; Baardsnes J.; Corbeil C. R.; Deprez C.; Cepero-Donates Y.; Robert A.; Schrag J. D.; Parat M.; Duchesne M.; Jaramillo M. L.; Purisima E. O.; Zwaagstra J. C. Structure-Based Engineering of PH-Dependent Antibody Binding for Selective Targeting of Solid-Tumor Microenvironment. mAbs 2020, 12 (1), e1682866–e1682880. 10.1080/19420862.2019.1682866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelen W.; Zhu K.; Subedi N.; Idili A.; Ricci F.; Tel J.; Merkx M. Programmable Bivalent Peptide-DNA Locks for PH-Based Control of Antibody Activity. ACS Cent. Sci. 2020, 6 (1), 22–31. 10.1021/acscentsci.9b00964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelek W. M.; Stott M.; Walters D.; Harris C. L.; Morgan B. P. Characterizing a PH-Switch Anti-C5 Antibody as a Tool for Human and Mouse Complement C5 Purification and Cross-Species Inhibition of Classical and Reactive Lysis. Immunology 2018, 155 (3), 396–403. 10.1111/imm.12982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murtaugh M. L.; Fanning S. W.; Sharma T. M.; Terry A. M.; Horn J. R. A Combinatorial Histidine Scanning Library Approach to Engineer Highly PH-Dependent Protein Switches. Protein Sci. 2011, 20 (9), 1619–1631. 10.1002/pro.696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonvin P.; Venet S.; Fontaine G.; Ravn U.; Gueneau F.; Kosco-Vilbois M.; Proudfoot A. E. I.; Fischer N. De Novo Isolation of Antibodies with PH-Dependent Binding Properties. MAbs 2015, 7 (2), 294–302. 10.1080/19420862.2015.1006993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröter C.; Günther R.; Rhiel L.; Becker S.; Toleikis L.; Doerner A.; Becker J.; Schönemann A.; Nasu D.; Neuteboom B.; Kolmar H.; Hock B. A Generic Approach to Engineer Antibody PH-Switches Using Combinatorial Histidine Scanning Libraries and Yeast Display. MAbs 2015, 7 (1), 138–151. 10.4161/19420862.2014.985993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igawa T.; Ishii S.; Tachibana T.; Maeda A.; Higuchi Y.; Shimaoka S.; Moriyama C.; Watanabe T.; Takubo R.; Doi Y.; Wakabayashi T.; Hayasaka A.; Kadono S.; Miyazaki T.; Haraya K.; Sekimori Y.; Kojima T.; Nabuchi Y.; Aso Y.; Kawabe Y.; Hattori K. Antibody Recycling by Engineered PH-Dependent Antigen Binding Improves the Duration of Antigen Neutralization. Nat. Biotechnol. 2010, 28 (11), 1203–1207. 10.1038/nbt.1691. [DOI] [PubMed] [Google Scholar]
- Igawa T.; Haraya K.; Hattori K. Sweeping Antibody as a Novel Therapeutic Antibody Modality Capable of Eliminating Soluble Antigens from Circulation. Immunol. Rev. 2016, 270 (1), 132–151. 10.1111/imr.12392. [DOI] [PubMed] [Google Scholar]
- Ward E. S.; Ober R. J. Targeting FcRn to Generate Antibody-Based Therapeutics. Trends Pharmacol. Sci. 2018, 39 (10), 892–904. 10.1016/j.tips.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo Y. A. Satralizumab: First Approval. Drugs 2020, 80 (14), 1477–1482. 10.1007/s40265-020-01380-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeage K. Ravulizumab: First Global Approval. Drugs 2019, 79 (3), 347–352. 10.1007/s40265-019-01068-2. [DOI] [PubMed] [Google Scholar]
- Chaparro-Riggers J.; Liang H.; DeVay R. M.; Bai L.; Sutton J. E.; Chen W.; Geng T.; Lindquist K.; Casas M. G.; Boustany L. M.; Brown C. L.; Chabot J.; Gomes B.; Garzone P.; Rossi A.; Strop P.; Shelton D.; Pons J.; Rajpal A. Increasing Serum Half-Life and Extending Cholesterol Lowering in Vivo by Engineering Antibody with PH-Sensitive Binding to PCSK9. J. Biol. Chem. 2012, 287 (14), 11090–11097. 10.1074/jbc.M111.319764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levisetti M.; Joh T.; Wan H.; Liang H.; Forgues P.; Gumbiner B.; Garzone P. D. A Phase I Randomized Study of a Specifically Engineered, PH-Sensitive PCSK9 Inhibitor RN317 (PF-05335810) in Hypercholesterolemic Subjects on Statin Therapy. Clin. Transl. Sci. 2017, 10 (1), 3–11. 10.1111/cts.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan D.; Rode F.; Cao Y. A Systems Pharmacokinetic/Pharmacodynamic Model for Concizumab to Explore the Potential of Anti-TFPI Recycling Antibodies. Eur. J. Pharm. Sci. 2019, 138, 105032. 10.1016/j.ejps.2019.105032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxlmayr M. W.; Lobner E.; Hasenhindl C.; Stadlmayr G.; Oostenbrink C.; Rüker F.; Obinger C. Construction of PH-Sensitive Her2-Binding IgG1-Fc by Directed Evolution. Biotechnol. J. 2014, 9 (8), 1013–1022. 10.1002/biot.201300483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Du X.; Liu M.; Tang F.; Zhang P.; Ai C.; Fields J. K.; Sundberg E. J.; Latinovic O. S.; Devenport M.; Zheng P.; Liu Y. Hijacking Antibody-Induced CTLA-4 Lysosomal Degradation for Safer and More Effective Cancer Immunotherapy. Cell Res. 2019, 29 (8), 609–627. 10.1038/s41422-019-0184-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler F. A.; Polli J. R.; Li T.; An B.; Otteneder M.; Qu J.; Balthasar J. P. “Catch-and-Release” Anti-Carcinoembryonic Antigen Monoclonal Antibody Leads to Greater Plasma and Tumor Exposure in a Mouse Model of Colorectal Cancer. J. Pharmacol. Exp. Ther. 2018, 366 (1), 205–219. 10.1124/jpet.117.246900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hironiwa N.; Ishii S.; Kadono S.; Iwayanagi Y.; Mimoto F.; Habu K.; Igawa T.; Hattori K. Calcium-Dependent Antigen Binding as a Novel Modality for Antibody Recycling by Endosomal Antigen Dissociation. MAbs 2016, 8 (1), 65–73. 10.1080/19420862.2015.1110660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desnoyers L. R.; Vasiljeva O.; Richardson J. H.; Yang A.; Menendez E. E. M.; Liang T. W.; Wong C.; Bessette P. H.; Kamath K.; Moore S. J.; Sagert J. G.; Hostetter D. R.; Han F.; Gee J.; Flandez J.; Markham K.; Nguyen M.; Krimm M.; Wong K. R.; Liu S.; Daugherty P. S.; West J. W.; Lowman H. B. Tumor-Specific Activation of an EGFR-Targeting Probody Enhances Therapeutic Index. Sci. Transl. Med. 2013, 5 (207), 207ra144. 10.1126/scitranslmed.3006682. [DOI] [PubMed] [Google Scholar]
- Han X.; Bryson P. D.; Zhao Y.; Cinay G. E.; Li S.; Guo Y.; Siriwon N.; Wang P. Masked Chimeric Antigen Receptor for Tumor-Specific Activation. Mol. Ther. 2017, 25 (1), 274–284. 10.1016/j.ymthe.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boni V.; Burris H. A. III; Liu J. F.; Spira A. I.; Arkenau H.-T.; Fidler M. J.; Rosen L. S.; Sweis R. F.; Uboha N. V.; Sanborn R. E.; O’Neil B.; Harding J. J.; LoRusso P.; Weise A. M.; Garcia-Corbacho J.; Victoria I.; Frye J. W.; Li R.; Stroh M.; Meric-Bernstam F. CX-2009, a CD166-Directed Probody Drug Conjugate (PDC): Results from the First-in-Human Study in Patients (Pts) with Advanced Cancer Including Breast Cancer (BC). J. Clin. Oncol. 2020, 38 (15_suppl), 526–526. 10.1200/JCO.2020.38.15_suppl.526. [DOI] [Google Scholar]
- Boustany L. M.; Wong L.; White C. W.; Diep L.; Huang Y.; Liu S.; Richardson J. H.; Kavanaugh W. M.; Irving B. A. EGFR-CD3 Bispecific Probody Therapeutic Induces Tumor Regressions and Increases Maximum Tolerated Dose >60-Fold in Preclinical Studies. Molecular Cancer Therapeutics 2018, 17, Abstract A164. 10.1158/1535-7163.targ-17-a164. [DOI] [Google Scholar]
- Janssen B. M. G.; Lempens E. H. M.; Olijve L. L. C.; Voets I. K.; van Dongen J. L. J.; de Greef T. F. A.; Merkx M. Reversible Blocking of Antibodies Using Bivalent Peptide–DNA Conjugates Allows Protease-Activatable Targeting. Chem. Sci. 2013, 4 (4), 1442–1450. 10.1039/c3sc22033h. [DOI] [Google Scholar]
- Lin S. J.; Dayao M. R.; Kim K. J.; Rocha S. S.; Kwant K.; Yu T.; Evans T.; Yu S.; Cremin M.; Aaron W.; Gamez-Guerrero M.; Callihan E.; Hemmati G.; Wright K. J.; Xiao Y.; Barath M.; Law C.-L.; Lemon B.; Austin R.; Wesche H. ProTriTAC: A Protease-Activatable T Cell Engager Platform That Links Half-Life Extension to Functional Masking and expands therapeutic window to enable targeting of broadly expressed tumor antigen. Journal for ImmunoTherapy of Cancer 2018, 6 (Supplement 1), 115–Abstract P608. 10.1186/s40425-018-0423-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson J. M.; Kari C.; Fragoso R. C.; Rodeck U.; Williams J. C. Design and Development of Masked Therapeutic Antibodies to Limit Off-Target Effects: Application to Anti-EGFR Antibodies. Cancer Biol. Ther. 2009, 8 (22), 2147–2152. 10.4161/cbt.8.22.9765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandersjöö L.; Jonsson A.; Löfblom J. A New Prodrug Form of Affibody Molecules (pro-Affibody) Is Selectively Activated by Cancer-Associated Proteases. Cell. Mol. Life Sci. 2015, 72 (7), 1405–1415. 10.1007/s00018-014-1751-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Gaynor A.; Chen W. Tunable Modulation of Antibody-Antigen Interaction by Protease Cleavage of Protein M. Biotechnol. Bioeng. 2019, 116 (11), 2834–2842. 10.1002/bit.27111. [DOI] [PubMed] [Google Scholar]
- Panchal A.; Seto P.; Wall R.; Hillier B. J.; Zhu Y.; Krakow J.; Datt A.; Pongo E.; Bagheri A.; Chen T. H. T.; Degenhardt J. D.; Culp P. A.; Dettling D. E.; Vinogradova M. V.; May C.; DuBridge R. B. COBRA: A Highly Potent Conditionally Active T Cell Engager Engineered for the Treatment of Solid Tumors. MAbs 2020, 12 (1), 1792130. 10.1080/19420862.2020.1792130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I. J.; Chuang C. H.; Hsieh Y. C.; Lu Y. C.; Lin W. W.; Huang C. C.; Cheng T. C.; Cheng Y. A.; Cheng K. W.; Wang Y. T.; Chen F. M.; Cheng T. L.; Tzou S. C. Selective Antibody Activation through Protease-Activated pro-Antibodies That Mask Binding Sites with Inhibitory Domains. Sci. Rep. 2017, 7, 11587. 10.1038/s41598-017-11886-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowman H. B.; Liu S.. Activatable Antibodies Having Non-Binding Steric Moieties and Methods of Using the Same. US9856314B2, 2018.
- Cattaruzza F.; Nazeer A.; Lange Z.; Hammond M.; Koski C.; Dao-Pick T.; Henkensiefken A.; Derynck M. K.; Irving B. A.; Schellenberger V.. HER2-XPAT and EGFR-XPAT: Pro-Drug T Cell Engagers (TCEs) Engineered to Address On-Target, Off-Tumor Toxicity With Potent Efficacy in Vitro and in Vivo and Large Safety Margins in NHP. In AACR Virtual Annual Meeting II; American Association for Cancer Research, 2020; p 69.
- Metz S.; Panke C.; Haas A. K.; Schanzer J.; Lau W.; Croasdale R.; Hoffmann E.; Schneider B.; Auer J.; Gassner C.; Bossenmaier B.; Umana P.; Sustmann C.; Brinkmann U. Bispecific Antibody Derivatives with Restricted Binding Functionalities That Are Activated by Proteolytic Processing. Protein Eng., Des. Sel. 2012, 25 (10), 571–580. 10.1093/protein/gzs064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuoha S. C.; Ferrari M.; Sblattero D.; Pitzalis C. Rational Design of Antirheumatic Prodrugs Specific for Sites of Inflammation. Arthritis Rheumatol. 2015, 67 (10), 2661–2672. 10.1002/art.39232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trang V. H.; Zhang X.; Yumul R. C.; Zeng W.; Stone I. J.; Wo S. W.; Dominguez M. M.; Cochran J. H.; Simmons J. K.; Ryan M. C.; Lyon R. P.; Senter P. D.; Levengood M. R. A Coiled-Coil Masking Domain for Selective Activation of Therapeutic Antibodies. Nat. Biotechnol. 2019, 37 (7), 761–765. 10.1038/s41587-019-0135-x. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Lin Y.; Gillies R. J. Tumor PH and Its Measurement. J. Nucl. Med. 2010, 51 (8), 1167–1170. 10.2967/jnumed.109.068981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erster O.; Thomas J. M.; Hamzah J.; Jabaiah A. M.; Getz J. A.; Schoep T. D.; Hall S. S.; Ruoslahti E.; Daugherty P. S. Site-Specific Targeting of Antibody Activity in Vivo Mediated by Disease-Associated Proteases. J. Controlled Release 2012, 161 (3), 804–812. 10.1016/j.jconrel.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poreba M. Protease-Activated Prodrugs: Strategies, Challenges, and Future Directions. FEBS J. 2020, 287 (10), 1936–1969. 10.1111/febs.15227. [DOI] [PubMed] [Google Scholar]
- Autio K. A.; Boni V.; Humphrey R. W.; Naing A. Probody Therapeutics: An Emerging Class of Therapies Designed to Enhance On-Target Effects with Reduced Off-Tumor Toxicity for Use in Immuno-Oncology. Clin. Cancer Res. 2020, 26 (5), 984–989. 10.1158/1078-0432.CCR-19-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh W. M. Antibody Prodrugs for Cancer. Expert Opin. Biol. Ther. 2020, 20 (2), 163–171. 10.1080/14712598.2020.1699053. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Guo Q.; Xia M.; Li Y.; Peng X.; Liu T.; Tong X.; Xu J.; Guo H.; Qian W.; Hou S.; Dai J.; Wang H.; Liu R.; Guo Y. Generation and Characterization of a Target-Selectively Activated Antibody against Epidermal Growth Factor Receptor with Enhanced Anti-Tumor Potency. MAbs 2015, 7 (2), 440–450. 10.1080/19420862.2015.1008352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesen D.; Broer L. N.; Lub-de Hooge M. N.; Popova I.; Howng B.; Nguyen M.; Vasiljeva O.; de Vries E. G. E.; Pool M. Probody Therapeutic Design of 89 Zr-CX-072 Promotes Accumulation in PD-L1–Expressing Tumors Compared to Normal Murine Lymphoid Tissue. Clin. Cancer Res. 2020, 26 (15), 3999–4009. 10.1158/1078-0432.CCR-19-3137. [DOI] [PubMed] [Google Scholar]
- Johnson M. L.; El-Khoueiry A. B.; Hafez N.; Lakhani N. J.; Mamdani H.; Rodon Ahnert J.; Sanborn R. E.; Ho T.; Li R.; Waldes J.; Spira A. I. CX-2029, a PROBODY Drug Conjugate Targeting CD71 (Transferrin Receptor): Results from a First-in-Human Study (PROCLAIM-CX-2029) in Patients (Pts) with Advanced Cancer. J. Clin. Oncol. 2020, 38, 3502–3502. 10.1200/JCO.2020.38.15_suppl.3502. [DOI] [Google Scholar]
- Naing A.; Thistlethwaite F. C.; Spira A. I.; Garcia-Corbacho J.; Randhawa M.; Eskens F.; O’Neil B.; Lavernia J.; Uboha N. V.; Hamid O.; El-Khoueiry A. B.; Benson B. A.; Garner W.; Huels V. J.; Arkenau H.-T.; LoRusso P. CX-072, a PD-L1 Probody Therapeutic, as Monotherapy in Patients with Advanced Solid Tumors: Preliminary Results of PROCLAIM-CX-072. J. Clin. Oncol. 2019, 37 (15_suppl), 2513–2513. 10.1200/JCO.2019.37.15_suppl.2513. [DOI] [Google Scholar]
- Lu Y.; Chuang C. H.; Chuang K. H.; Chen I. J.; Huang B. C.; Lee W. H.; Wang H. E.; Li J. J.; Cheng Y. A.; Cheng K. W.; Wang J. Y.; Hsieh Y. C.; Lin W. W.; Cheng T. L. Specific Activation of Pro-In Iximab Enhances Selectivity and Safety of Rheumatoid Arthritis Therapy. PLoS Biol. 2019, 17 (6), e3000286. 10.1371/journal.pbio.3000286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan G.; Kalaitzidis D.; Neel B. G. The Tyrosine Phosphatase Shp2 (PTPN11) in Cancer. Cancer Metastasis Rev. 2008, 27, 179–192. 10.1007/s10555-008-9126-y. [DOI] [PubMed] [Google Scholar]
- Tian Q.; Wang J. Role of Serine/Threonine Protein Phosphatase in Alzheimer’s Disease. Neurosignals 2002, 11 (5), 262–269. 10.1159/000067425. [DOI] [PubMed] [Google Scholar]
- Whyte M. P. Hypophosphatasia-Aetiology, Nosology, Pathogenesis, Diagnosis and Treatment. Nat. Rev. Endocrinol. 2016, 12, 233–246. 10.1038/nrendo.2016.14. [DOI] [PubMed] [Google Scholar]
- Gunnoo S. B.; Finney H. M.; Baker T. S.; Lawson A. D.; Anthony D. C.; Davis B. G. Creation of a Gated Antibody as a Conditionally Functional Synthetic Protein. Nat. Commun. 2014, 5, 4388. 10.1038/ncomms5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks J. W.; Brautigan D. L. Molecular Basis for Substrate Specificity of Protein Kinases and Phosphatases. Int. J. Biochem. 1986, 18 (6), 497–504. 10.1016/0020-711X(86)90159-X. [DOI] [PubMed] [Google Scholar]
- Labrijn A. F.; Janmaat M. L.; Reichert J. M.; Parren P. W. H. I. Bispecific Antibodies: A Mechanistic Review of the Pipeline. Nat. Rev. Drug Discovery 2019, 18 (8), 585–608. 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- Mazor Y.; Oganesyan V.; Yang C.; Hansen A.; Wang J.; Liu H.; Sachsenmeier K.; Carlson M.; Gadre D. V.; Borrok M. J.; Yu X. Q.; Dall’Acqua W.; Wu H.; Chowdhury P. S. Improving Target Cell Specificity Using a Novel Monovalent Bispecific IgG Design. MAbs 2015, 7 (2), 377–389. 10.1080/19420862.2015.1007816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazor Y.; Sachsenmeier K. F.; Yang C.; Hansen A.; Filderman J.; Mulgrew K.; Wu H.; Dall’Acqua W. F. Enhanced Tumor-Targeting Selectivity by Modulating Bispecific Antibody Binding Affinity and Format Valence. Sci. Rep. 2017, 7, 40098. 10.1038/srep40098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellmann C.; Doerner A.; Knuehl C.; Rasche N.; Sood V.; Krah S.; Rhiel L.; Messemer A.; Wesolowski J.; Schuette M.; Becker S.; Toleikis L.; Kolmar H.; Hock B. Balancing Selectivity and Efficacy of Bispecific Epidermal Growth Factor Receptor (EGFR) x c-MET Antibodies and Antibody-Drug Conjugates. J. Biol. Chem. 2016, 291 (48), 25106–25119. 10.1074/jbc.M116.753491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie M. J.; Boyken S. E.; Salter A. I.; Bruffey J.; Rajan A.; Langan R. A.; Olshefsky A.; Muhunthan V.; Bick M. J.; Gewe M.; Quijano-Rubio A.; Johnson J. L.; Lenz G.; Nguyen A.; Pun S.; Correnti C. E.; Riddell S. R.; Baker D. Designed Protein Logic to Target Cells with Precise Combinations of Surface Antigens. Science (Washington, DC, U. S.) 2020, 369 (6511), 1637–1643. 10.1126/science.aba6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megeed Z.; Winters R. M.; Yarmush M. L. Modulation of Single-Chain Antibody Affinity with Temperature-Responsive Elastin-like Polypeptide Linkers. Biomacromolecules 2006, 7 (4), 999–1004. 10.1021/bm0507002. [DOI] [PubMed] [Google Scholar]
- Blenner M. A.; Banta S. Characterization of the 4D5Flu Single-Chain Antibody with a Stimulus-Responsive Elastin-like Peptide Linker: A Potential Reporter of Peptide Linker Conformation. Protein Sci. 2008, 17 (3), 527–536. 10.1110/ps.073257308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oller-Salvia B.; Chin J. W. Efficient Phage Display with Multiple Distinct Non-Canonical Amino Acids Using Orthogonal Ribosome-Mediated Genetic Code Expansion. Angew. Chem., Int. Ed. 2019, 58 (32), 10844–10848. 10.1002/anie.201902658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Badran A. H.; Huang T. P.; Liu D. R. Continuous Directed Evolution of Proteins with Improved Soluble Expression. Nat. Chem. Biol. 2018, 14 (10), 972–980. 10.1038/s41589-018-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]