

Abstract
The Nε-methyl lysine status of histones is important in the regulation of eukaryotic transcription. The Fe(ii) and 2-oxoglutarate (2OG) -dependent JmjC domain enzymes are the largest family of histone Nε-methyl lysine demethylases (KDMs). The human KDM4 subfamily of JmjC KDMs is linked with multiple cancers and some of its members are medicinal chemistry targets. We describe the use of combined molecular dynamics (MD) and Quantum Mechanical/Molecular Mechanical (QM/MM) methods to study the mechanism of KDM4A, which catalyzes demethylation of both tri- and di-methylated forms of histone H3 at K9 and K36. The results show that the oxygen activation at the active site of KDM4A is optimized towards the generation of the reactive Fe(iv)-oxo intermediate. Factors including the substrate binding mode, correlated motions of the protein and histone substrates, and molecular orbital control synergistically contribute to the reactivity of the Fe(iv)-oxo intermediate. In silico substitutions were performed to investigate the roles of residues (Lys241, Tyr177, and Asn290) in substrate orientation. The Lys241Ala substitution abolishes activity due to altered substrate orientation consistent with reported experimental studies. Calculations with a macrocyclic peptide substrate analogue reveal that induced conformational changes/correlated motions in KDM4A are sequence-specific in a manner that influences substrate binding affinity. Second sphere residues, such as Ser288 and Thr289, may contribute to KDM4A catalysis by correlated motions with active site residues. Residues that stabilize key intermediates, and which are predicted to be involved in correlated motions with other residues in the second sphere and beyond, are shown to be different in KDM4A compared to those in another JmjC KDM (PHF8), which acts on H3K9 di- and mono-methylated forms, suggesting that allosteric type inhibition is of interest from the perspective of developing selective JmjC KDM inhibitors.
The second sphere residues and regions of the protein in histone demethylase enzymes that makes correlated motion with the active site contribute to efficient catalysis.
1. Introduction
Epigenetic processes regulate eukaryotic development and are involved in diseases, including cancer, metabolic syndromes, and brain disorders.1 Post-translational modifications of histone tails in nucleosomes, including via reversible lysine- and arginine N-methylation and demethylation, are crucial mechanisms of transcriptional regulation (Scheme 1).2,3
Scheme 1. N ε-Lysine methylation and demethylation of histone H3 play important roles in the regulation of transcription.

There are nine groups of identified human Nε-methyl lysine demethylases (KDMs) (KDM1-9) which act on different Nε-methylation states and histone substrate sequence selectivities.3,4 The KDM1 subfamily, or lysine-specific demethylases (LSDs), are flavin-dependent enzymes that catalyze demethylation of di- and mono-methylated forms of lysine-4 in histone 3 (H3K4me2/me1) substrates.3a KDM2-9 are non-heme Fe(ii) – and 2-oxoglutarate (2OG) – dependent Jumonji C (JmjC) domain-containing oxygenases that are capable of catalyzing demethylation of all three Nε-lysine methylation states.3b–d KDM2-9 employ an Fe(ii) cofactor, and 2OG and O2 as cosubstrates. The chemical basis of the underlying differences in selectivity between the JmjC KDM subfamilies is poorly understood.
The human KDM4 subfamily comprises five multi-domain enzymes (KDM4A–E),5 which catalyze demethylation of the tri- and di-methylated lysine residue K9 in histone H3 N-terminal tails (H3K9me3/me2).5,6 KDM4A–C (which have >50% sequence identity over their core JmjC domain, JmjN, two plant homeodomains (PHD) and two hybrid Tudor domains)5 also catalyze H3K36me3/me2 demethylation, but KDM4D/E do not.7 Demethylation of H3K9me3 by the JmjC KDM4s facilitates an open chromatin state, contributing to transcriptional activation.8 Multiple common cancers, including breast,9 prostate,10 and lung11 cancer are associated with KDM4A gene overexpression.10c,11c,12–14 Thus, KDM4 and other JmjC KDMs are being pursued as targets for anti-cancer therapeutics.15
The JmjC KDM catalytic cycle is proposed to follow the consensus for 2OG oxygenases, as established by spectroscopic,16 kinetic,17 and computational studies,18 (Fig. 1). In the resting enzyme, the Fe(ii) is octahedrally-coordinated by His-188, Glu-190 and His-276 (for KDM4A) and two to three water molecules. 2OG binding to A generate B (Fig. 1), is followed by that of the substrate, then dioxygen. Work with multiple non-heme Fe(ii) enzymes reveals that the presence of a substrate is necessary for productive oxygen binding and reaction to form a ferric-superoxo complex (RC1, Fig. 1). Oxidative decarboxylation of 2OG results in CO2 and an Fe-linked succinyl peroxide intermediate IM1 (Fig. 1). Subsequent O–O homolysis gives a ferryl intermediate (IM2, Fig. 1) (S = 2, M = 5), which abstracts a hydrogen from the substrate to give IM3; a subsequent rebound (IM3 to PD) provides a hemiaminal, which eliminates formaldehyde to provide the demethylated product.19 Water ligation regenerates the resting state. The final step likely proceeds non-enzymatically, at least for some Fe(ii)/2OG oxygenases.19
Fig. 1. Outline of catalytic cycle for the JmjC KDMs.

The JmjC KDMs have a characteristic distorted double stranded beta-helix (DSBH) core fold, as conserved in 2OG oxygenases.20 Elements surrounding the core DSBH fold are subfamily characteristic and are involved in substrate binding.21 One end of the DSBH supports the active site containing the single Fe ion. The KDM4 catalytic domains also contain a Zn binding site which is structurally important.21a,c Single substitutions of residues, not in the immediate vicinity of the Fe-center, can impact substantially on KDM4 activity, e.g. second sphere residue substitutions in KDM4A (e.g. K241A, ST288-289TV/NV/GG) can ablate activity.22b Similarly, the ST288-289AI KDM4A substitution alters binding of both di- and tri-methylated H3-K9me2/3 substrates.22
Computational studies on KDMs have been focused on the conformational dynamics and the reaction mechanism in the KDM7 (ref. 23) enzymes and dioxygen binding in KDM4A.24 There is little understanding of the key interactions involved in directing the reaction mechanism of KDM4A and how these differ between KDMs in different subfamilies. Knowledge of long-range correlated motions important in catalysis is not only of basic mechanistic interest, but by identifying enzyme specific processes it may help enable the design of selective inhibitors.
We report studies aimed at testing the proposal that, although the catalytic domains of KDMs appear to employ the same mechanisms and share substantial structural similarity, catalytically important interactions unique to specific sets of KDMs exist. We envisaged that in addition to enzyme-specific first-sphere catalytic residues, there would be a broader enzyme-specific network of correlated motions and interactions between residues directly involved in catalysis and second sphere and beyond residues. We carried out molecular dynamics (MD) and quantum mechanical/molecular mechanical (QM/MM) calculations on the roles of structural dynamics and long-range correlated motions in KDM4A with two types of substrate – histone H3 peptides and a tight binding unnatural macrocyclic peptide substrate analogue.21c These results are further compared to those for KDM7B (PHF8) from previous work, and interesting differences are analyzed and related back to the substrate specificity of these enzymes.
2. Methodology
Crystal structures of the catalytic domain of KDM4A bound to the H3(7–12)K9me3 natural substrate (PDB ID: 2OQ6)21a or to a cyclic peptide substrate analogue/inhibitor (PDB ID: 5LY1) were used.21c A crystal structure of PHF8 complexed with an H3(1–14)K4me3K9me2 substrate fragment (PDB ID: 3KV4)21b was treated similarly to the KDM4 structures. Amber16 was used for the MD simulations.25 ChemShell suite of programs was used for QM/MM calculations.26,27 The QM calculations were performed with Turbomole,28 and MM with DL_POLY software.29 All geometries were optimized using the B3LYP30 density functional theory with the def2-SVP (split valence polarization) basis set (B1) which can reproduce accurate barriers and electronic structure for Fe metal containing systems.23a,31,32 Details are provided in ESI† of the article.
3. Results and discussion
3.1. Conformational flexibility and correlated motions in the reactant complex: Fe(iii)-superoxo complex with H3(7–12)K9me3
To date the proposed reactive intermediates in KDM4 catalysis have not been characterized experimentally, hence we carried out studies on the KDM4A Fe(iii)–O2·2OG·H3(7–12)K9me3 complex (RC1 in Fig. 1), initially by performing an MD simulation of 1 μs (Fig. 3). The KDM4A active site orients both 2OG and O2 for apparently productive reaction during the entire 1 μs MD simulation. Distance plots reflecting the relative orientation of the O2 derived atoms and 2OG (Fig. 3) and between the tri-methylated Nε-lysine and the proximal oxygen from O2 (Op–N) show only small variations, implying a stable enzyme–substrate complex. The 2OG and the substrate orientations are stabilized by hydrogen bonds and other interactions (Fig. 2 and S2c in ESI†). The C5 oxygens of 2OG (O3 and O4 in Fig. 3) are stabilized by polar interactions with Tyr132 and Lys206. Asn198 forms a stable hydrogen bond with O1 oxygen of 2OG. The results thus imply that Tyr132, Lys206, and Asn198, stabilize productive binding of 2OG to Fe, as implied by the crystal structures.21a,c
Fig. 3. Distances between the dioxygen derived proximal oxygen (Op) to the substrate N-methyl N (Op–N) (in red) and of the distal oxygen (Od) to the 2OG co-substrate (C2–Od) (in black) during the 1 μs simulation. The x-axis denotes time in ns, and the y-axis denotes distance in Å. Atom labels are shown in the inset.

Fig. 2. Views of the KDM4A·Fe(iii)·O2-substrate (H3(7–12)K9me3) complex. (a) Geometry of the optimized ferric-superoxo [Fe(iii)–O2] complex for KDM4A. (b) The quantum mechanical region used in calculations. Wiggly lines define the boundary of the QM/MM partition. The views were derived from a modeled structure of Fe(iii)–O2 complexed with H3(7–12)K9me3 (based PDB: 2OQ6) as described in Methods.

Due to correlated motions, second sphere residues not directly coordinated to the metal ion can influence catalysis.33 To investigate roles of correlated motions in KDM4A catalysis, we carried out dynamic cross-correlation analyses (DCCA)34 for the KDM4A Fe(iii)-superoxo·H3(7–12)K9me3 complex (RC1 in Fig. 2). The results (Fig. 4) reveal strong correlated motions between residues 258–300 and 188–213. His-188 and Glu-190 are two of the three Fe-coordinating residues (see Fig. 1); Ser-288 and Thr-289 are second sphere residues located on β15, which is one of the 8 DSBH β-strands (secondary structure elements of KDM4A are given in Fig. S1 of ESI†). The experimentally observed ablation of KDM4A activity with the ST288-289TV, NV, or GG variants and altered binding specificity for both dimethylated (H3-K9me2) and trimethylated (H3-K9me3) substrates by ST288-289AI may thus, at least in part, be due to alterations in correlated motions involving residues 288–289 along with other possible alterations in the electrostatic field.22b Principal Component Analysis (PCA) (Fig. 4b) shows the directions of motion of the protein and the relative flexibility of specific protein regions during the simulation.34 Backbone residues located at α5, β6 (158 to 170) and α7 (227 to 234) show relatively larger movements compared to other regions of the protein.
Fig. 4. Motions in the Fe(iii)–O2·2OG H3(7–12)K9me3 complex. (a) Dynamic cross-correlation diagram with important correlations circled. Residues 158–170 belong to α5 and β6; 227–234 belong to α7. (b) Principal component analysis showing the direction of motions yellow to blue.

3.2. Interactions and correlated motions in the Fe(iv) O complex with the substrate
As a result of O2 activation, a ferryl-oxo [Fe(iv)-oxo] intermediate (IM2 in Fig. 1), is generated in a manner established for other 2OG oxygenases (see ESI† for details of calculations). The Fe(iv)-oxo intermediate, IM2, abstracts a hydrogen from the substrate methyl group to give the radical cation IM3, which undergoes rebound to give the hydroxylated intermediate PD. To investigate the role of protein flexibility in reactivity of Fe(iv)-oxo intermediate IM2, a 1 μs simulation was performed (Fig. 5). The ground state of Fe(iv)-oxo intermediate IM2 in the non-heme iron oxygenases is in a high spin quintet state (S = 2, M = 5), as shown by studies on model complexes and other non-heme enzymes.35 The Fe(iv)-oxo intermediate prefers to abstract a substrate methyl hydrogen through an (Fe–O) 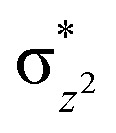 antibonding orbital due to exchange-enhanced reactivity, as shown by Shaik and coworkers.36 An electron shift diagram showing hydrogen atom transfer (HAT) through
antibonding orbital due to exchange-enhanced reactivity, as shown by Shaik and coworkers.36 An electron shift diagram showing hydrogen atom transfer (HAT) through 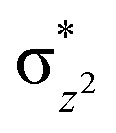 is given in Fig. 5a. The results imply that linear positioning of a substrate Nε-methyl lysine methyl group with respect to the Fe–O bond is important for efficient catalysis. Thus, the Fe–O–N bond angle (Fig. 5c) is a geometric determinant for HAT. The proximity of the methyl group hydrogens to the oxygen of Fe
O is another factor that will affect the barrier for reaction. Another possible electron shift is likely to the π* orbitals based on the angle of HAT trajectory that emanate from a nonlinear positioning of substrate with respect to Fe–O bond.18h,36,37 The exchange enhanced reactivity makes the σ pathway preferred over the π pathway.36,37
is given in Fig. 5a. The results imply that linear positioning of a substrate Nε-methyl lysine methyl group with respect to the Fe–O bond is important for efficient catalysis. Thus, the Fe–O–N bond angle (Fig. 5c) is a geometric determinant for HAT. The proximity of the methyl group hydrogens to the oxygen of Fe
O is another factor that will affect the barrier for reaction. Another possible electron shift is likely to the π* orbitals based on the angle of HAT trajectory that emanate from a nonlinear positioning of substrate with respect to Fe–O bond.18h,36,37 The exchange enhanced reactivity makes the σ pathway preferred over the π pathway.36,37
Fig. 5. Substrate dynamics for the Fe(iv)-oxo intermediate. (a) Fe(iv)-oxo intermediate with the substrate and MO diagram for a σ-trajectory HAT. (b) Distance of the Fe(iv)-oxo O to substrate N during the 1 μs simulation. The y-axis is distance (Å) and x-axis is time (ns). (c) Variation in the Fe–O–N angle for the 1 μs simulations. The y-axis is the angle (°).

MD simulations of the KDM4A Fe(iv)-oxo intermediate (IM2) show that Tyr132 and Lys241 are positioned to make hydrogen bonds with the C4 oxygens of succinate, and Asn198 is oriented to make a hydrogen bond with the non-coordinating C1 oxygen (O2) of the succinate (Fig. S6†). With KDM4A, Glu190 is positioned to make a hydrogen-bond with Asn290 in both ferric-superoxo and ferryl-oxo intermediates (Fig. S5 and S6 in ESI†).
Whilst analysis of the long-range correlated motions in the Fe(iv) O complex (IM2) show a similar pattern as those for the ferric-superoxo complex (Fig. S3†), there are differences. Overall, compared to the ferric-superoxo complex, the intensities of the correlated and anticorrelated motions in the Fe(iv) O complex (IM2) show lower intensities, e.g. in the N-terminal region (residues 7–200, including the Fe-ligands His188 and Glu190), and in the C-terminal region after residue 308 (Fig. S3†).
3.3. The rate of hydrogen atom transfer as a function of protein flexibility and geometric determinants
Hydroxylation begins with hydrogen atom transfer (HAT) followed by a rebound process (IM2 to IM3 to PD in Fig. 1). To evaluate the influence of protein dynamics on the reactivity of Fe(iv)-oxo intermediate, we carried out reaction modeling using five snapshots from the MD trajectory. The Fe–O–N angle during dynamics ranges around 150 ± 10° (Fig. 5). The potential energy profile for the reaction from a snapshot at 443 ns is given in Fig. 6b. The barrier at the QM(B2)/MM level of theory with zero-point energy correction (QM(B2+ZPE)/MM) is 21.6 kcal mol−1 and without ZPE correction is 25.9 kcal mol−1. HAT is thus rate-determining step (RDS) once substrate is bound (Fig. 1) and forms IM3 in a slightly endothermic process. In the optimized geometry of the transition state (TS) for the HAT step in the enzyme surroundings (Fig. 6a), the Fe–O bond elongates to 1.78 Å due to the transfer of an electron to the (Fe–O) 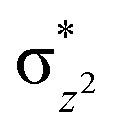 antibonding orbital. The spin natural orbitals (SNOs) of the TS are given in Fig. 6d. The Mulliken spin densities of Fe–O is 4.1, and the substrate is −0.5 at the TS_HAT transition state. The spin densities of Fe–O and substrate are 4.5 and −1.0, respectively, in IM3. Thus, the α electron from the substrate methyl C–H bond is transferred to the Fe–O, and the β-electron resides at the carbon radical. These processes are similar in the other HAT steps described below. The Mulliken spin densities and charges of intermediates and TSs are given in Tables S3–S6 (ESI†). The O–H and H–C distances at the TS_HAT transition state are 1.16 and 1.37 Å, respectively. The Fe–O–H angle remains 162.5°, which is reasonably close to the ideal σ trajectory of 180°. Since HAT is the RDS, any deviation of Fe–O–H angle from linearity or the O⋯H distances in the reactant complex (IM2) would affect catalytic efficiency. The roles of the Fe–O–H angle and O⋯H distances of IM2 on the barrier of HAT, are demonstrated in multiple snapshots representing the starting reactant complex (IM2). As seen from the inset in Fig. 6b, the barrier is directly related to the distance between the substrate methyl hydrogen and the Fe–O oxygen in IM2. Among the tested IM2 complexes, the tightly bound substrates with the O⋯H distances of 2.49 or 2.50 Å have relatively low barriers of 21.6 and 23.2 kcal mol−1. As the O⋯H distance in these complexes increases to 2.81 and 2.96 Å, respectively, the barriers increase to 28.3 and 26.1 kcal mol−1, respectively.
antibonding orbital. The spin natural orbitals (SNOs) of the TS are given in Fig. 6d. The Mulliken spin densities of Fe–O is 4.1, and the substrate is −0.5 at the TS_HAT transition state. The spin densities of Fe–O and substrate are 4.5 and −1.0, respectively, in IM3. Thus, the α electron from the substrate methyl C–H bond is transferred to the Fe–O, and the β-electron resides at the carbon radical. These processes are similar in the other HAT steps described below. The Mulliken spin densities and charges of intermediates and TSs are given in Tables S3–S6 (ESI†). The O–H and H–C distances at the TS_HAT transition state are 1.16 and 1.37 Å, respectively. The Fe–O–H angle remains 162.5°, which is reasonably close to the ideal σ trajectory of 180°. Since HAT is the RDS, any deviation of Fe–O–H angle from linearity or the O⋯H distances in the reactant complex (IM2) would affect catalytic efficiency. The roles of the Fe–O–H angle and O⋯H distances of IM2 on the barrier of HAT, are demonstrated in multiple snapshots representing the starting reactant complex (IM2). As seen from the inset in Fig. 6b, the barrier is directly related to the distance between the substrate methyl hydrogen and the Fe–O oxygen in IM2. Among the tested IM2 complexes, the tightly bound substrates with the O⋯H distances of 2.49 or 2.50 Å have relatively low barriers of 21.6 and 23.2 kcal mol−1. As the O⋯H distance in these complexes increases to 2.81 and 2.96 Å, respectively, the barriers increase to 28.3 and 26.1 kcal mol−1, respectively.
Fig. 6. Influence of protein dynamics on catalysis and overview of the whole catalytic events. (a) The QM(B1)/MM optimized TS geometry. (b) Potential energy profile (in kcal mol−1) for HAT and the rebound step at the QM(B2)/MM followed by QM(B2+ZPE)/MM level of theory. The inset shows the barrier for multiple HAT events at the QM(B2+ZPE)/MM level with starting geometry of the corresponding IM2. Distances are in Å; angles in degrees (°). (c) Superimposed geometries of the optimized reactants for the KDM4A·Fe(iv)-oxo·H3(7–12)K9me3 complex. (d) Spin Natural Orbitals (SNO) of HAT transition state with populations in parentheses. (e) Summary of important steps in the catalytic cycle with QM(B2+ZPE)/MM energies in kcal mol−1.

The Fe–O–H angle is a factor that further affects barrier for reaction of (IM2). Thus, the barriers for the two IM2 snapshots with comparable initial O⋯H distances of 2.49 and 2.50 Å are affected by the Fe–O–H angle, i.e. the former has an Fe–O–H angle of 150.0° and a barrier of 21.6 kcal mol−1, compared to the latter which has an Fe–O–H angle 140.4° and a barrier of 23.2 kcal mol−1. IM2 geometries with initial O⋯H distances of 2.81 and 2.96 Å manifest Fe–O–H angles of 170.7° and 138.5° and barriers of 26.1 kcal mol−1 and 28.3 kcal mol−1, respectively.
Comparison of the residues that interact with the IM2 or TS_HAT implies that the interactions are consistent in the reactant complex and the transition state (Fig. S7†). It is notable that in multiple snapshots used for HAT, the orientation of trimethylated lysine changes while the Fe-coordination sphere showed little change (Fig. 6c). The rebound step has a barrier of 16.4 kcal mol−1, which is lower than for the HAT step. Formation of the product (PD) from IM3 is exothermic by 41.8 kcal mol−1 at the QM(B2+ZPE)/MM level of theory. The geometry of the optimized TS of the rebound step is given in Fig. S8 of the ESI.† The final hydroxylated product in the catalytic cycle is a hemiaminal (carbinol amine) that can spontaneously dissociate to complete C–N bond cleavage.19,38
3.4. Catalytic roles of Lys241, Tyr177, and Asn290
Analysis of the reactant and optimized transition state at the active site (Fig. 6a) imply interactions with Lys241, Tyr177, and Asn290 directly affect substrate orientation (Fig. 6b). Thus, substitution of these residues may impact on activity. To investigate this, we performed MD simulations on the in silico mutated enzyme forms; all three substitutions with Ala affected reactivity. The Lys241Ala variant is of particular interest because experimental reports showed that it ablates activity.22a With Lys241Ala KDM4A, the reaction stalls at the succinate/ferryl intermediate stage, i.e. does not proceed to hydroxylation; the MD results show that the trimethylated lysine (M3L) adopts a new non-productive orientation with the each of the variants. Replacement of Lys241 with Ala apparently provides more space for M3L in which to move. Substitutions of Tyr177 and Asn290 with Ala also change the binding orientation of the substrate (Fig. 7). Hence any structural modification related to Tyr177 and Asn290 likely has a pronounced effect on hydroxylase activity, including complete loss of activity.
Fig. 7. KDM4A mutation causes changes in substrate orientation from productive to non-productive. (a) The first geometry of simulation for the (grey) variants is overlapped with the geometry at 1000 ns (blue for Tyr177Ala; green for Lys241Ala; red for Asn290Ala). (b) Distances for the Fe(iv) O to substrate Nε of wildtype and KDM4A variants. y-Axis: distance in Å; x-axis: time in ns. Colors: black for WT; blue for Tyr177Ala; green for Lys241Ala; red for Asn290Ala.

3.5. Correlated motions of the residues that stabilize the transition states
The QM/MM calculations reveal that Tyr177, Lys241 and Asn290 contribute to stabilization of the transition states for both O2-activation and the HAT abstraction steps (Fig. S5 and S6†). These three residues make correlated motions with residues belonging to the DSBH core fold, other β-strands, and α-helices in both the Fe(iii)-superoxo and Fe(iv) O complexes as detailed below. Tyr177 makes a correlated motion with the Ala286 from the DSBH (β15), and Asn290 makes correlated motion with residues from β9 (194–196) (Fig. S1† gives the KDM4A secondary structure elements). Similarly, Lys241 from β11 also makes strong correlations with the coordinating Zn metal binding region. Subtle differences are visible for the correlation of Tyr177 in the Fe(iii)-superoxo compared to the Fe(iv) O complex. Uniquely of these residues, Tyr177 correlates with substrate (363–364) residues in the Fe(iii)-superoxo complex (Fig. S3†). The results involving correlated motions of catalytically important residues reveal the roles of long-range interactions that might control formation of intermediates on the reaction coordinate.
3.6. The macrocyclic CP2(R6Kme3) peptide as an inhibitor and substrate
A synthetic macrocyclic peptide (CP2) efficiently and selectively inhibits KDM4A–C, over other KDMs including closely related KDM4D–E.21d We investigated the dynamics of demethylation employing a variant of CP2, i.e. the tight binding CP2(R6Kme3) substrate. CP2 has an Arg (R6), the side chain of which projects into the KDM4A active site in a manner resembling that of Kme3 substrates, but its overall binding mode differs substantially from that of natural substrates (Fig. 8a). With CP2R6Kme3, both demethylation and potent inhibition are observed, with the latter reflecting tight binding of both CP2R6Kme3 and its products.21d CP2R6Kme3 is thus an interesting model for studying dynamics during demethylation (independent of binding events). The experimentally determined binding constants for CP2 and CP2(R6Kme3) are ∼1000-fold higher than for the natural substrate.21c The tight binding of CP2(R6Kme3) and, probably, its hydroxylated/demethylated product(s) enable effective inhibition. Consistent with this, the binding energies calculated by MMGBSA39 (molecular mechanics, the generalized Born model and solvent accessibility method) imply CP2(R6Kme3) binds better than natural peptide at both the Fe(iii)-superoxo and Fe(iv)-oxo intermediate stages. Thus, the superior binding of CP2(R6Kme3) over natural substrate is shown for two key steps during catalysis. The binding energy of the Fe(iii)-superoxo complex with CP2(R6Kme3) is 5.4 kcal mol−1 higher than the natural substrate (H3(7–12)K9me3). Similarly, the binding energy for CP2(R6Kme3) is 16.4 kcal mol−1 higher than natural substrate (H3(7–12)K9me3) for Fe(iv)-oxo complex. Thus, the reported inhibition is governed by strong binding of CP2(R6Kme3). The potential energy profile for ferryl-oxo generation (O2 activation) in CP2(R6Kme3) bound KDM4A is very similar to that for the analogous natural substrate (Fig. S9 of the ESI†). Since HAT is the rate-determining step in the catalysis (Fig. 6), comparison of the dynamics of the conformationally constrained CP2(R6Kme3) at the ferryl-oxo intermediate stage with those of a natural substrate (H3(7–12)K9me3) are of interest (Fig. 8). The distance between Kme3 and the Fe–O oxygen in the CP2(R6Kme3) complex shows larger fluctuations compared to the H3(7–12)K9me3 substrate (red vs. black) (Fig. 8b). The Fe–O–N angle has a more pronounced difference where H3(7–12)K9me3 is more linear to Fe–O than the CP2(R6Kme3). The Fe–O–N angle in the H3(7–12)K9me3 is ∼150° and is ∼130° for CP2(R6Kme3). As discussed for H3(7–12)K9me3 above (Fig. 6b) the barrier for HAT relates to the proximity of the substrate N-methyl group to the Fe(iv)-oxygen and the Fe–O–H angle, with a more linear angle being favored.
Fig. 8. Comparisons of binding interactions in CP2(R6Kme3) with those for H3(7–12)K9me3. (a) Binding of the cyclic peptide (CP2(R6Kme3)) to KDM4A. (b) The distance of the O of the Fe O intermediate to the N of the substrate N-methyl group during 1 μs simulation for H3(7–12)K9me3 (black) and CP2(R6Kme3) (red). The y-axis is labeled with distance in Å and x-axis with time in ns. (c) Representation of Fe–O–N angle for H3(7–12)K9me3 (black) and CP2(R6Kme3) (red). y-Axis: angle in degrees (°).

The dependence of the barrier for HAT on conformational changes was calculated from multiple snapshots obtained from dynamics of CP2(R6Kme3)·Fe(iv)-oxo complex (Fig. 9). The barrier for the initial HAT step is 21.1 kcal mol−1 for the CP2(R6Kme3).KDM4A complex (for TS_HAT_inh in Fig. 9a). As with H3(7–12)K9me3, a correlation between the HAT barrier and the proximity and orientation of the substrate towards the Fe–O oxygen was observed with CP2(R6Kme3) (Fig. 9b). Thus, the barriers for HAT from CP2(R6Kme3) are lower for shorter O⋯H initial distance and are affected by the Fe–O–H angle, with more linear geometries lowering the barrier. Thus, even though CP2(R6Kme3) has a substantially different binding mode compared to H3(7–12)K9me3, the active site and second sphere residues synergize to enable effective demethylation.
Fig. 9. Influence of protein dynamics on the barrier in the CP2(R6Kme3) bound KDM4A. (a) Potential energy profile for HAT and rebound step for CP2(R6Kme3) substrate/inhibitor at the QM(B2)/MM followed by QM(B2+ZPE)/MM level of theory. (b) The barrier for multiple HAT at the QM(B2+ZPE)/MM level of theory with starting geometric details of the corresponding IM2. Barriers are in kcal mol−1, distances in Å and angles in degrees (°).

With KDM4A, the activation barriers for demethylation of H3(7–12)K9me3 and CP2(R6Kme3) manifest a dependence on the distance between the ferryl oxygen and the hydrogen of the methyl group of the substrate. This contrasts with the KDM PHF8 (ref. 23a) and two 2OG-dependent nucleic acid demethylases (AlkB/AkBH2 (ref. 40)) where such correlation was not found for the tested reactant complexes for O⋯H distances (below 3.2 Å) or for the Fe–O–H angle (range between 122–167°). The dependence of the barrier on the geometric determinants of the hydrogen abstraction trajectory may thus be oxygenase subfamily-specific and is affected by the protein environment.
A comparative MD study was performed for KDM4A complexed with CP2, which bears an arginine instead of the tri-methylated lysine of CP2(R6Kme3) in the position binding deepest at the active site as revealed by crystallographic analysis of KDM4A in complex with CP2 and Ni.21c The tri-methylated lysine (M3L) of CP2(R6Kme3) shows some changes in its active site interactions compared to CP2. The interactions of Ser288, Tyr177 and Lys241 with CP2 are weakened in the CP2(R6Kme3) complex. Interestingly, in our MD results, the guanidinium group of the CP2 arginine is positioned to make a strong electrostatic interaction with the non Fe-chelating carboxylate group of succinate when present (Fig. S10†). Note the interaction with succinate was not observed in the reported crystal structure of KDM4A complexed with CP2 which was obtained under non catalytic conditions in the absence of succinate,21c under inhibition conditions succinate will be produced by 2OG turnover. Overall, the calculations demonstrate that CP2R6Kme3 binds stronger than the natural substrate (H3(7–12)K9me3) to KDM4A not only in the initial complex in agreement with experimental studies21c but also in key intermediates during catalysis.
3.7. Comparison of correlated motions in KDM4A bound to CP2(R6Kme3) with those for H3(7–12)K9me3
Dynamic cross-correlation studies of the KDM4A–CP2(R6Kme3) complexes reveal that is cyclic/rigid nature likely causes changes in the binding orientation of the substrate N-methyl group, i.e. orienting it more perpendicular to the Fe(iv)-oxo, compared to the H3(7–12)K9me3 complex, (Fig. 8); this may intensify the extent of anticorrelated motions in the KDM4A–CP2(R6Kme3) complex. The overall patterns of correlated motions are similar, but not identical, in the CP2(R6Kme3) Fe(iii) and Fe(iv) intermediate complexes compared to those for H3(7–12)K9me3. In the Fe(iii)-superoxo complex with CP2(R6Kme3), there is larger area of anticorrelated motions between residues 60–120 (β3 and β4 of DSBH) and the first 50 N-terminal residues that belongs to the JmjN domain of KDM4A, compared to the analogous complex with H3(7–12)K9me3. The Fe(iv) O complex with CP2(R6Kme3) also shows some increased anticorrelations compared to the analogous complex with H3(7–12)K9me3, in particular between residues 180–200 (β7, β8 and β9 of DSBH, and mixed domain) with residues 20–150 (JmjN domain, β3 and β4 of DSBH).
The residues stabilizing the RCs and TSs in the O2-activation and the hydrogen atom abstraction reactions with CP2(R6Kme3) (Tyr177, Lys241 and Asn290) are the same as in H3(7–12)K9me3 (Fig. S3†). Comparison of the correlated motions for these residues with the H3(7–12)K9me3 and CP2(R6Kme3) complexes show similar patterns, with slightly more differences in the Fe(iii)–O2 complexes than in the Fe(iv)-oxo complexes. For example, the correlation between residues 31–35 (α2) helix and 356–358 (Zn, Fe and 2OG/succinate binding) are unique to H3(7–12)K9me3 compared to the Fe(iii)-superoxo complexes of CP2(R6Kme3). The differences in the correlated motions between the residues that stabilize the key catalytic species in KDM4A with different substrate types, raise the possibility of the identifying compounds that modulate KDM4A catalysis with a particular substrate.
3.8. Comparison of catalysis and correlated motions in KDM4A and PHF8
The KDM7 (PHF8) subfamily of JmjC KDMs act on H3K9me2/me1, in contrast to the H3K9me3/2 demethylase activity of the KDM4s. In KDM4A, the Nε-lysine trimethylated group (M3L) is bound by electrostatic interactions, including with Tyr177, Lys241 and Asn290. With PHF8, Ile191 (PHF8), Arg460 (H3R8), and Phe250 (PHF8) are involved in binding of the Nε-lysine di-methyl group.23a These and other active site differences between KDM4A and PHF8 mean the substrate's H3K9Me2 group is bound more snugly to PHF8 than the H3K9Me1–3 group of KDM4A, resulting in their different selectivities. PHF8 also has a smaller radius of gyration for the active site cavity than KDM4A (Fig. S11 of ESI†). Ile191 of PHF8 makes a strong correlation with non-DSBH beta strands through residues 157–161 that belong to β1III. In PHF8, the correlated motions in both the Fe(iii)–O2− substrate and Fe(iv)-oxo substrate complexes have similar intensity, whereas KDM4A shows slightly less intense correlations for the Fe(iv)-oxo compared to the Fe(iii)–O2− substrate complex. Overall, the general nature of correlated motions of crucial residues involved in catalysis with second sphere residues appear to be similar in general for KDM4A and PHF8. However, differences are apparent, where KDM4A shows more correlated motions with β15 and β9 of DSBH, while PHF8 with β1III at the active site entrance and a surface exposed loop region that connects α9 and α10.
4. Conclusions
The combined studies with MD and QM/MM calculations highlight the importance of correlated motions in both enabling substrate selectivity and efficient catalysis for 2OG oxygenases. Due to their ability to accept substrates with different sequence and N-methylation state selectivities, the JmjC KDMs, including the KDM4s, are of particular interest from the perspective of understanding structural dynamics during catalysis. The results imply correlated motions of protein domains are employed by KDM4A to enable strict molecular orbital control of reactive intermediates during the multiple steps in its mechanism.
KDM4A possesses pre-juxtaposed residues in its active site that hold its substrates in a manner that enforces a close to linear Fe–O⋯HC angle (with “CH” being the H-atom of the substrate to be activated) at the ferryl intermediate stage so enabling efficient hydrogen atom transfer (HAT). Lower HAT barriers were obtained for shorter O⋯H distances and more linear Fe–O–H angles. “Linear positioning” of the substrate thus facilitates electron transfer to the (Fe–O) 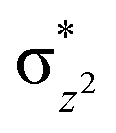 antibonding orbital, which is the favorable HAT trajectory in high spin non-heme ferryl-oxo intermediates.36
antibonding orbital, which is the favorable HAT trajectory in high spin non-heme ferryl-oxo intermediates.36
The results identify Lys241, Tyr177, and Asn290 as key second sphere residues that orient the substrate in the active site. In silico substitutions of these residues with Ala resulted in altered substrate Nε-methylated lysine side chain orientations with non-productive geometries for the HAT. The results of substitution of Lys241 correlate with experimental reports of loss of activity in corresponding variants, by implying the catalytic cycle will halt at the Fe(iv)-oxo stage without proceeding to substrate hydroxylation.22a Dynamic cross-correlation studies support the importance of second sphere residues in catalysis. Ser288 and Thr289 of KDM4A correlate with the Fe binding ligands. These correlated motions may reflect the experimental observations that ST 288–289 substitutions to TV, NV, or GG ablate KDM4A activity, whereas substitution to AI alters the binding specificity of dimethylated (H3-K9me2) and trimethylated (H3-K9me3) substrates.22b
Studies with a tight binding cyclic macrocyclic peptide (CP2(R6Kme3)) inhibitor/substrate21c complement the findings with the natural substrate. With CP2(R6Kme3), at the Fe(iv)-oxo stage, the Fe–O–H angle was found to deviate from linearity compared to the natural substrate, likely due to a non optimal orientation of Kme3 in CP2(R6Kme3) in the active site for demethylation, possibly relating to the tight binding and inhibitory nature of the cyclic peptide.21c The flexibility of the active site and second sphere residue contributions enable reaction of CP2(R6Kme3) to proceed at the active site with a similar rate compared to H3(7–12)K9me3. Given the different active site binding modes of CP2(R6Kme3) and the natural H3-K9me3/2 or H3-K36me3/2 substrates the results thus imply that KDM4A and likely other KDM4 enzymes have the potential to demethylate non-histone non Nε-methyl lysine substrates as proposed based on studies with multiple peptides.3e,41 This result also imply that the long-range motions related to catalysis will enable novel allosteric binding sites and that this knowledge can be further used in drug design.
The catalytically important residues in KDM4A differ from those in PHF8 and their correlated motions manifest differences. For example, the crucial active site residues in KDM4A shows pronounced correlated motions with β15 and β9 of DSBH, while in PHF8 with β1III and a surface-exposed loop that connects α9 and α10. The identification of differences in correlated motions for different JmjC KDMs raises the possibility of modulating their reactivity for particular substrate types, e.g. by mutation or of binding of ligands to specific regions of the protein that correlate with active site motions. Knowledge of how enzyme-specific residues and their correlated motions are involved in JmjC KDM catalysis could also help with the identification of improved inhibitors, including derivatives of substrate competitors such as CP2, to complement established inhibitors, which work by binding to the active site Fe(ii).6
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
CZC and TKC thank to Michigan Technological University for the start-up funding. C. J. S. thanks Cancer Research UK (C8717/A18245), the EPRC, and the Wellcome Trust (106244/Z/14/Z) for support. C. Z. C. acknowledges NSF grant 1904215 (testing the methodology).
Electronic supplementary information (ESI) available: Potential energy profiles, cross-correlation plots, orbital pictures, Mulliken spin and charges and Cartesian coordinates of the QM zone in QM/MM calculations are provided. See DOI: 10.1039/d0sc03713c
References
- (a) Ganesan A. Philos. Trans. R. Soc., B. 2018;373:1–7. [Google Scholar]; (b) Hauser A. T. Robaa D. Jung M. Curr. Opin. Chem. Biol. 2018;45:73–85. doi: 10.1016/j.cbpa.2018.03.003. [DOI] [PubMed] [Google Scholar]
- (a) Horton J. R., Gale M., Yan Q. and Cheng X., The molecular basis of histone demethylation, in DNA and Histone Methylation as Cancer Targets, ed. A. Kaneda and Y.-i. Tsukada, Cancer Drug Discovery and Development, 2017, pp. 151–219 [Google Scholar]; (b) Rotili D. Mai A. Genes Cancer. 2011;2:663–679. doi: 10.1177/1947601911417976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Shi Y. Lan F. Matson C. Mulligan P. Whetstine J. R. Cole P. A. Casero R. A. Shi Y. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]; (b) Tsukada Y. I. Fang J. Erdjument-Bromage H. Warren M. E. Borchers C. H. Tempst P. Zhang Y. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]; (c) Yamane K. Toumazou C. Tsukada Y. ichi. Erdjument-Bromage H. Tempst P. Wong J. Zhang Y. Cell. 2006;125:483–495. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]; (d) Whetstine J. R. Nottke A. Lan F. Huarte M. Smolikov S. Chen Z. Spooner E. Li E. Zhang G. Colaiacovo M. Shi Y. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]; (e) Walport L. J. Hopkinson R. J. Chowdhury R. Schiller R. Ge W. Kawamura A. Schofield C. J. Nat. Commun. 2016;7:11974. doi: 10.1038/ncomms11974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S. Eisenhaber F. O'Carroll D. Strahl B. D. Sun Z. W. Schmid M. Opravil S. Mechtier K. Ponting C. P. Allis C. D. Jenuwein T. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- Horton J. R., Gale M., Yan Q. and Cheng X., The Molecular Basis of Histone Demethylation, in DNA and Histone Methylation as Cancer Targets, Humana Press, Cham, 2017, pp. 151–219 [Google Scholar]
- Kaniskan H. Ü. Martini M. L. Jin J. Chem. Rev. 2018;118:989–1068. doi: 10.1021/acs.chemrev.6b00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillringhaus L. Yue W. W. Rose N. R. Ng S. S. Gileadi C. Loenarz C. Bello S. H. Bray J. E. Schofield C. J. Oppermann U. J. Biol. Chem. 2011;286:41616–41625. doi: 10.1074/jbc.M111.283689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra-Calderas L. González-Barrios R. Herrera L. A. Cantú de León D. Soto-Reyes E. Cancer Genet. 2015;208:215–224. doi: 10.1016/j.cancergen.2014.11.001. [DOI] [PubMed] [Google Scholar]
- (a) Ye Q. Holowatyj A. Wu J. Liu H. Zhang L. Suzuki T. Yang Z.-Q. Am. J. Cancer Res. 2015;5:1519–1530. [PMC free article] [PubMed] [Google Scholar]; (b) Li L. L. Xue A. M. Li B. X. Shen Y. W. Li Y. H. Luo C. L. Zhang M. C. Jiang J. Q. Xu Z. De. Xie J. H. Zhao Z. Q. Breast Cancer Res. 2014;16:R56. doi: 10.1186/bcr3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Cloos P. A. C. Christensen J. Agger K. Maiolica A. Rappsilber J. Antal T. Hansen K. H. Helin K. Nature. 2006;442:307–311. doi: 10.1038/nature04837. [DOI] [PubMed] [Google Scholar]; (b) Björkman M. Östling P. Härmä V. Virtanen J. Mpindi J. P. Rantala J. Mirtti T. Vesterinen T. Lundin M. Sankila A. Rannikko A. Kaivanto E. Kohonen P. Kallioniemi O. Nees M. Oncogene. 2012;31:3444–3456. doi: 10.1038/onc.2011.512. [DOI] [PubMed] [Google Scholar]; (c) Kim T. D. Jin F. Shin S. Oh S. Lightfoot S. A. Grande J. P. Johnson A. J. Van Deursen J. M. Wren J. D. Janknecht R. J. Clin. Invest. 2016;126:706–720. doi: 10.1172/JCI78132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Xu W. Jiang K. Shen M. Chen Y. Huang H. Y. Oncol. Rep. 2016;35:352–358. doi: 10.3892/or.2015.4349. [DOI] [PubMed] [Google Scholar]; (b) Mallette F. A. Richard S. Cell Rep. 2012;2:1233–1243. doi: 10.1016/j.celrep.2012.09.033. [DOI] [PubMed] [Google Scholar]; (c) Kogure M. Takawa M. Cho H. S. Toyokawa G. Hayashi K. Tsunoda T. Kobayashi T. Daigo Y. Sugiyama M. Atomi Y. Nakamura Y. Hamamoto R. Cancer Lett. 2013;336:76–84. doi: 10.1016/j.canlet.2013.04.009. [DOI] [PubMed] [Google Scholar]
- Hu C. E. Liu Y. C. Zhang H. D. Huang G. J. Biochem. Biophys. Res. Commun. 2014;449:1–7. doi: 10.1016/j.bbrc.2014.04.126. [DOI] [PubMed] [Google Scholar]
- Qiu M. T. Fan Q. Zhu Z. Kwan S. Y. Chen L. Chen J. H. Ying Z. L. Zhou Y. Gu W. Wang L. H. Cheng W. W. Zeng J. Wan X. P. Mok S. C. Wong K. K. Bao W. Oncotarget. 2015;6:31702–31720. doi: 10.18632/oncotarget.5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agger K. Miyagi S. Pedersen M. T. Kooistra S. M. Johansen J. V. Helin K. Genes Dev. 2016;30:1278–1288. doi: 10.1101/gad.280495.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Lin H. Li Q. Li Q. Zhu J. Gu K. Jiang X. Hu Q. Feng F. Qu W. Chen Y. Sun H. J. Enzyme Inhib. Med. Chem. 2018;33:777–793. doi: 10.1080/14756366.2018.1455676. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wozniak G. G. Strahl B. D. Biochim. Biophys. Acta. 2014;1839:1353–1361. doi: 10.1016/j.bbagrm.2014.03.002. [DOI] [PubMed] [Google Scholar]
- (a) Martinez S. Hausinger R. P. J. Biol. Chem. 2015;290:20702–20711. doi: 10.1074/jbc.R115.648691. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Solomon E. I. Light K. M. Liu L. V. Srnec M. Wong S. D. Acc. Chem. Res. 2013;46:2725–2739. doi: 10.1021/ar400149m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Solomon E. I. Goudarzi S. Sutherlin K. D. Biochemistry. 2016;55:6363–6374. doi: 10.1021/acs.biochem.6b00635. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mitchell A. J. Dunham N. P. Martinie R. J. Bergman J. A. Pollock C. J. Hu K. Allen B. D. Chang W. C. Silakov A. Bollinger J. M. Krebs C. Boal A. K. J. Am. Chem. Soc. 2017;139:13830–13836. doi: 10.1021/jacs.7b07374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Costas M. Mehn M. P. Jensen M. P. Que L. Chem. Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]; (b) Grzyska P. K. Ryle M. J. Monterosso G. R. Liu J. Ballou D. P. Hausinger R. P. Biochemistry. 2005;44:3845–3855. doi: 10.1021/bi048746n. [DOI] [PubMed] [Google Scholar]
- (a) Song X. Lu J. Lai W. Phys. Chem. Chem. Phys. 2017;19:20188–20197. doi: 10.1039/C7CP02687K. [DOI] [PubMed] [Google Scholar]; (b) Torabifard H. Cisneros G. A. Chem. Sci. 2017;8:6230–6238. doi: 10.1039/C7SC00997F. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Blomberg M. R. A. Borowski T. Himo F. Liao R. Z. Siegbahn P. E. M. Chem. Rev. 2014;114:3601–3658. doi: 10.1021/cr400388t. [DOI] [PubMed] [Google Scholar]; (d) Wang B. Usharani D. Li C. Shaik S. J. Am. Chem. Soc. 2014;136:13895–13901. doi: 10.1021/ja507934g. [DOI] [PubMed] [Google Scholar]; (e) Quesne M. G. Latifi R. Gonzalez-Ovalle L. E. Kumar D. de Visser S. P. Chem.–Eur. J. 2014;20:435–446. doi: 10.1002/chem.201303282. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Ye S. Riplinger C. Hansen A. Krebs C. Bollinger J. M. Neese F. Chem.–Eur. J. 2012;18:6555–6567. doi: 10.1002/chem.201102829. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Chen H. Cho K. B. Lai W. Nam W. Shaik S. J. Chem. Theory Comput. 2012;8:915–926. doi: 10.1021/ct300015y. [DOI] [PubMed] [Google Scholar]; (h) Neidig M. L. Decker A. Choroba O. W. Huang F. Kavana M. Moran G. R. Spencer J. B. Solomon E. I. Proc. Natl. Acad. Sci. U. S. A. 2006;103:12966–12973. doi: 10.1073/pnas.0605067103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B. Cao Z. Sharon D. A. Shaik S. ACS Catal. 2015;5:7077–7090. doi: 10.1021/acscatal.5b01867. [DOI] [Google Scholar]
- Markolovic S. Wilkins S. E. Schofield C. J. J. Biol. Chem. 2015;290:20712–20722. doi: 10.1074/jbc.R115.662627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Ng S. S. Kavanagh K. L. McDonough M. A. Butler D. Pilka E. S. Lienard B. M. R. Bray J. E. Savitsky P. Gileadi O. Von Delft F. Rose N. R. Offer J. Scheinost J. C. Borowski T. Sundstrom M. Schofield C. J. Oppermann U. Nature. 2007;448:87–91. doi: 10.1038/nature05971. [DOI] [PubMed] [Google Scholar]; (b) Horton J. R. Upadhyay A. K. Qi H. H. Zhang X. Shi Y. Cheng X. Nat. Struct. Mol. Biol. 2010;17:38–44. doi: 10.1038/nsmb.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kawamura A. Münzel M. Kojima T. Yapp C. Bhushan B. Goto Y. Tumber A. Katoh T. King O. N. F. Passioura T. Walport L. J. Hatch S. B. Madden S. Müller S. Brennan P. E. Chowdhury R. Hopkinson R. J. Suga H. Schofield C. J. Nat. Commun. 2017;8:14773. doi: 10.1038/ncomms14773. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Klose R. J. Kallin E. M. Zhang Y. Nat. Rev. Genet. 2006;7:715–727. doi: 10.1038/nrg1945. [DOI] [PubMed] [Google Scholar]; (e) Marmorstein R. Trievel R. C. Biochim. Biophys. Acta. 2009;1789:58–68. doi: 10.1016/j.bbagrm.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Hancock R. L. Abboud M. I. Smart T. J. Flashman E. Kawamura A. Schofield C. J. Hopkinson R. J. ChemBioChem. 2018;19:917–921. doi: 10.1002/cbic.201800002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen Z. Zang J. Whetstine J. Hong X. Davrazou F. Kutateladze T. G. Simpson M. Mao Q. Pan C. H. Dai S. Hagman J. Hansen K. Shi Y. Zhang G. Cell. 2006;125:691–702. doi: 10.1016/j.cell.2006.04.024. [DOI] [PubMed] [Google Scholar]
- (a) Chaturvedi S. S. Ramanan R. Lehnert N. Schofield C. J. Karabencheva-Christova T. G. Christov C. Z. ACS Catal. 2020;10:1195–1209. doi: 10.1021/acscatal.9b04907. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chaturvedi S. S. Ramanan R. Waheed S. O. Ainsley J. Evison M. P. Ames J. M. Schofield C. J. Karabencheva-Christova T. G. Christov C. Z. Chem.–Eur. J. 2019;20:5422–5426. doi: 10.1002/chem.201900492. [DOI] [PubMed] [Google Scholar]
- Cortopassi W. A. Simion R. Honsby C. E. França T. C. C. Paton R. S. Chem.–Eur. J. 2015;21:18983–18992. doi: 10.1002/chem.201502983. [DOI] [PubMed] [Google Scholar]
- Salomon-Ferrer R. Götz A. W. Poole D. Le Grand S. Walker R. C. J. Chem. Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- Sherwood P. De Vries A. H. Guest M. F. Schreckenbach G. Catlow C. R. A. French S. A. Sokol A. A. Bromley S. T. Thiel W. Turner A. J. Billeter S. Terstegen F. Thiel S. Kendrick J. Rogers S. C. Casci J. Watson M. King F. Karlsen E. Sjøvoll M. Fahmi A. Schäfer A. Lennartz C. J. Mol. Struct.: THEOCHEM. 2003;632:1–28. doi: 10.1016/S0166-1280(03)00285-9. [DOI] [Google Scholar]
- Metz S. Kästner J. Sokol A. A. Keal T. W. Sherwood P. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014;4:101–110. [Google Scholar]
- Ahlrichs R. Bär M. Häser M. Horn H. Kölmel C. Chem. Phys. Lett. 1989;162:165–169. doi: 10.1016/0009-2614(89)85118-8. [DOI] [Google Scholar]
- Smith W. Forester T. R. J. Mol. Graphics. 1996;14:136–141. doi: 10.1016/S0263-7855(96)00043-4. [DOI] [PubMed] [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- Schäfer A. Horn H. Ahlrichs R. J. Chem. Phys. 1992;97:2571–2577. doi: 10.1063/1.463096. [DOI] [Google Scholar]
- Ramanan R. Dubey K. D. Wang B. Mandal D. Shaik S. J. Am. Chem. Soc. 2016;138:6786–6797. doi: 10.1021/jacs.6b01716. [DOI] [PubMed] [Google Scholar]
- (a) McCammon J. A. and Harvey S. C., Dynamics of Proteins and Nucleic Acids, Cambridge University Press, 1987 [Google Scholar]; (b) Adcock S. A. McCammon J. A. Chem. Rev. 2006;106:1589–1615. doi: 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Karplus M. McCammon J. A. Nat. Struct. Biol. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- Grant B. J. Rodrigues A. P. C. ElSawy K. M. McCammon J. A. Caves L. S. D. Bioinformatics. 2006;22:2695–2696. doi: 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- (a) Borowski T. Bassan A. Siegbahn P. E. M. Chem.–Eur. J. 2004;10:1031–1041. doi: 10.1002/chem.200305306. [DOI] [PubMed] [Google Scholar]; (b) Wójcik A. Radoń M. Borowski T. J. Phys. Chem. A. 2016;120:1261–1274. doi: 10.1021/acs.jpca.5b12311. [DOI] [PubMed] [Google Scholar]
- (a) Usharani D. Janardanan D. Li C. Shaik S. Acc. Chem. Res. 2013;46:471–482. doi: 10.1021/ar300204y. [DOI] [PubMed] [Google Scholar]; (b) Shaik S. Chen H. Janardanan D. Nat. Chem. 2011;3:19–27. doi: 10.1038/nchem.943. [DOI] [PubMed] [Google Scholar]
- Geng C. Ye S. Neese F. Angew. Chem., Int. Ed. 2010;49:5717–5720. doi: 10.1002/anie.201001850. [DOI] [PubMed] [Google Scholar]
- Rose J. Castagnoli Jr N. Med. Res. Rev. 1983;3:73–88. doi: 10.1002/med.2610030105. [DOI] [PubMed] [Google Scholar]
- Miller B. R. McGee T. D. Swails J. M. Homeyer N. Gohlke H. Roitberg A. E. J. Chem. Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- Waheed S. O. Ramanan R. Chaturvedi S. S. Lehnert N. Schofield C. J. Christov C. Z. Karabencheva-Christova T. G. ACS Cent. Sci. 2020;6:795–814. doi: 10.1021/acscentsci.0c00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnaluri V. K. C. Vavilala D. T. Putty S. Gutheil W. G. Mukherji M. Biochem. Biophys. Res. Commun. 2009;390:280–284. doi: 10.1016/j.bbrc.2009.09.107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.