

SUMMARY
Although an imbalance between neuronal excitation and inhibition underlies seizures, clinical approaches that target these mechanisms are insufficient in containing seizures in patients with epilepsy, raising the need for alternative approaches. Brain-resident microglia contribute to the development and stability of neuronal structure and functional networks that are perturbed during seizures. However, the extent of microglial contributions in response to seizures in vivo remain to be elucidated. Using two-photon in vivo imaging to visualize microglial dynamics, we show that severe seizures induce formation of microglial process pouches that target but rarely engulf beaded neuronal dendrites. Microglial process pouches are stable for hours, although they often shrink in size. We further find that microglial process pouches are associated with a better structural resolution of beaded dendrites. These findings provide evidence for the structural resolution of injured dendrites by microglia as a form of neuroprotection.
In brief
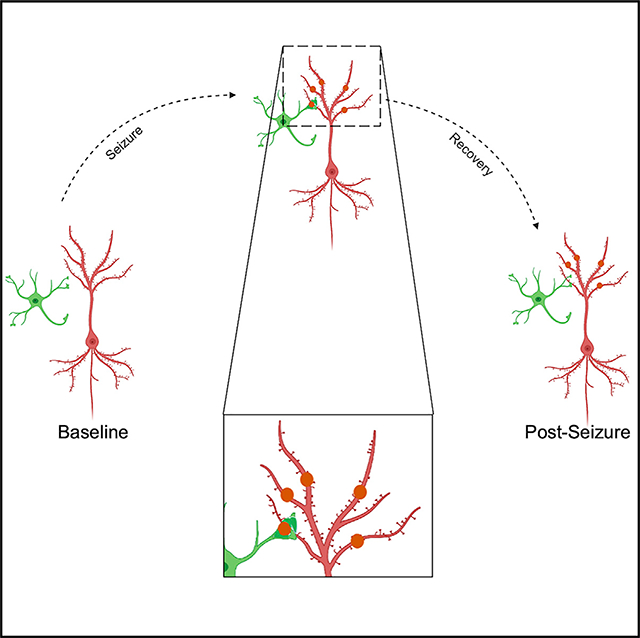
Eyo et al. report that microglia form unique spherical structures in an experimental seizure model. Using live imaging, the authors characterize these structure that they call “microglial process pouches” and show that they colocalize with injured neuronal dendrites and are strongly correlated with the structural resolution of these injured dendrites.
Graphical Abstract
INTRODUCTION
One-third of patients with epilepsy remain resistant to existing drugs, which is a number that has not changed in 30 years (Fisher et al., 2014; Hauser and Hesdorffer, 1990). Antiepileptic drug development has been focused on targeting known neuronal mechanisms. Seizures are the most obvious manifestation of epilepsy. In mouse preclinical models of seizures, prolonged seizures can cause dendritic injury (Jiang et al., 1998; Multani et al., 1994; Swann et al., 2000) and seriously compromise recovery because dendritic structures facilitate synaptic interactions and neuronal function (Rochefort and Konnerth, 2012; Sala and Segal, 2014). Therefore, processes facilitating the resolution of seizure-induced dendritic structural injury would be therapeutically beneficial.
Microglia are the primary brain-resident immune cells whose importance in brain development, homeostasis, and synaptic plasticity have now been well established (Eyo and Wu, 2013; Sierra et al., 2019). Contrary to traditional views that associated microglia with detrimental roles in pathology, recent findings indicate that they can be neuroprotective in chronic demyelinating diseases such multiple sclerosis (Gao et al., 2017; Tanabe et al., 2019) and acute injury conditions like ischemia (Szalay et al., 2016), as well as seizure disorders (Araki et al., 2019; Eyo et al., 2014, 2017; Waltl et al., 2018; Wu et al., 2020). Furthermore, microglial dysfunction has been implicated in Alzheimer’s disease in which microglia lose their homeostatic functions and facilitate disease progression (Sarlus and Heneka, 2017). Moreover, microglia-specific dysfunction was recently implicated in spontaneous seizure generation (Zhao et al., 2018). Therefore, microglia are an attractive target for modulating disease pathology in general and seizure disorders in specific.
Microglia are highly dynamic brain cells (Davalos et al., 2005; Eyo et al., 2018; Nimmerjahn et al., 2005) that physically interact with neurons in hyperactive or hypoactive contexts (Eyo et al., 2015; Kato et al., 2016; Liu et al., 2019; Umpierre et al., 2020). Indeed, microglial activity limits neuronal hyperactivity (Liu et al., 2020; Merlini et al., 2021). However, real-time microglial dynamics following seizures in vivo have not been determined. To do so, we performed two-photon imaging of microglia and neurons in transgenic mice following kainic acid (KA)-induced seizures. Here, we report a microglial phenotype that is directed toward beaded dendrites following severe seizures. Real-time and chronic imaging of these structures suggest against a primary phagocytic function for them. Hence, we call them “microglial process pouches” (MPPs) Interestingly, these pouches are maintained for extensive periods of time and strongly correlated with better structural resolutions of beaded dendrites. Our results suggest a restorative function for microglia through physical interactions with neuronal dendrites following severe seizures.
RESULTS
Severe seizures induce microglial process pouch formation
Using in vivo two-photon microscopy, we first confirmed with previous studies (Guo et al., 2012, 2016; Zeng et al., 2007) that prolonged stage 5 (but not mild stage 3) seizures induced dendritic beads in Thy1YFP mice (Figures 1A and 1B). Interestingly, swollen bead density decreased with increasing cortical depth (Figure 1C). In CX3CR1GFP/+ mice, KA treatment that resulted in severe seizures elicited a phenomenon with microglia processes that we called microglial process pouches (MPPs) following severe (but not mild) seizures at 6 h and 24 h of treatment (Figures 1D and 1E). No differences were observed between male and female mice (Figure 1F). Spatially, MPPs were more abundant in the superficial cortex than in the deeper cortex (Figures 1G and 1H; Video S1). Chronic imaging showed that the majority of pouches formed by 6 h of KA were gone by 24 h, indicating that pouches are not permanent (Figures 1I and 1J). As with results at 6 h, pouches were more abundant in the superficial cortex than in the deeper cortex at 24 h (Figure 1K; Figure S1A). To rule out the possibility that MPPs were induced by the craniotomy procedure, we visualized microglia in both freshly prepared live and fixed cortical brain slices generated without a craniotomy and got similar results (Figures S1B and S1C). These results indicate that severe KA-induced seizures trigger the formation of MPPs predominantly in the superficial cortex.
Figure 1. Severe seizures induce microglial process pouch formation.
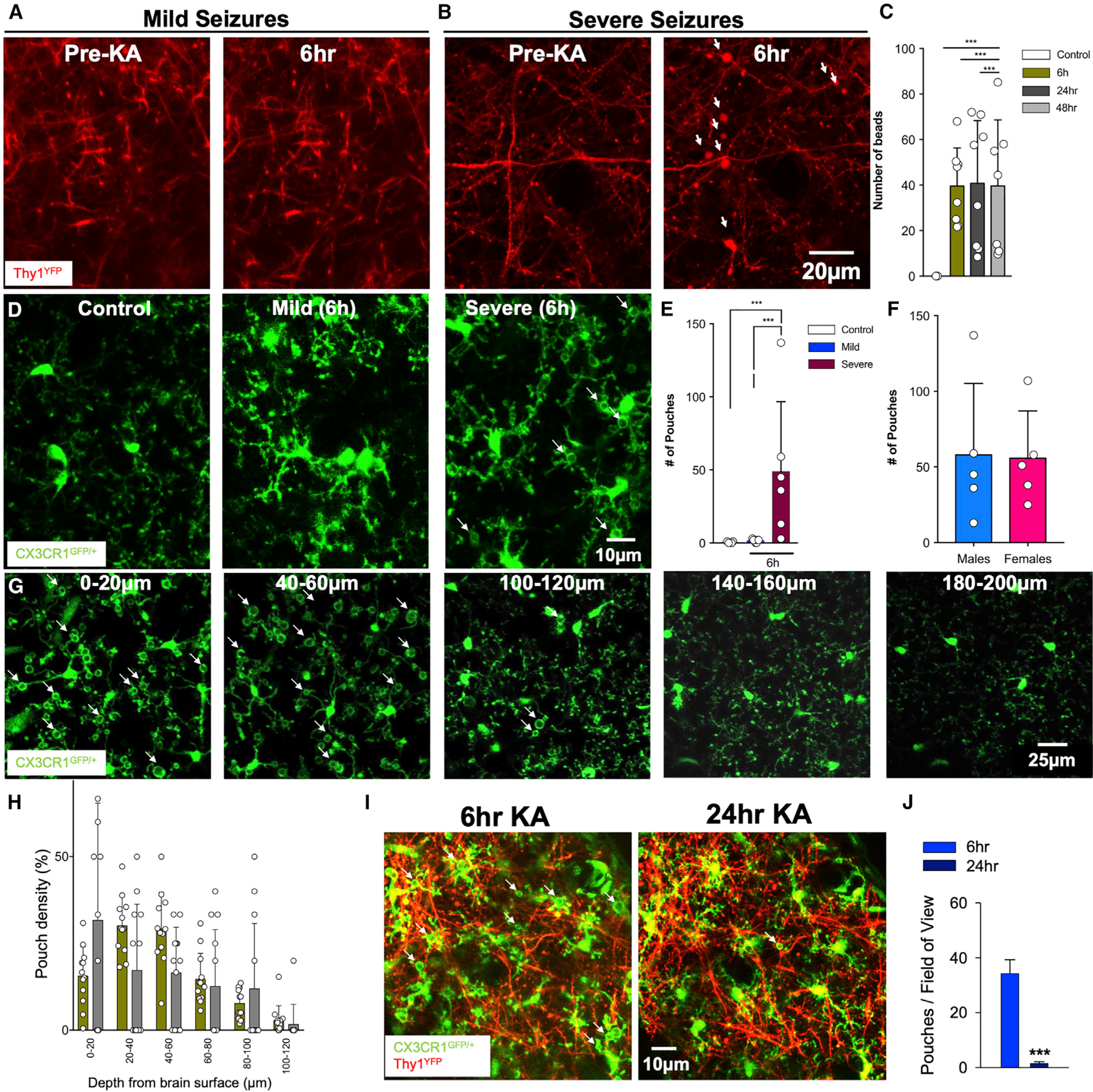
(A) Representative two-photon in vivo images through a chronically implanted cranial window from a Thy1YFP mouse before and following mild (stage 3) seizures with unperturbed dendrites.
(B) Representative two-photon in vivo images through a chronically implanted cranial window from a Thy1YFP mouse before and following severe (stage 5) seizures with beaded dendrites (white arrows). Scale bar: 20 μm.
(C) Quantification of dendritic beads at different times of KA-induced seizures.
(D) Representative two-photon in vivo images from CX3CR1GFP/+ mice following an acute craniotomy showing microglia in the superficial somatosensory cortex of control, mild, and severe seizures following kainic acid (KA) treatment. Microglial process pouches are identified (white arrows). Scale bar: 10 μm.
(E) Quantification of microglial pouches under different conditions.
(F) Quantification of microglial pouches in male and female mice.
(G) Representative two-photon in vivo images at 6 h of KA treatment at different cortical depths showing microglial process pouches (white arrows). Scale bar: 25 μm.
(H) Quantification of the percent of pouches at different depths at 6 h and 24 h of KA treatment.
(I) Representative two-photon in vivo images through a chronically implanted window showing microglia in a specific field of view before and after severe KA-induced seizures. Pouches (arrows) formed at 6 h are mostly resolved by 24 h. Scale bar: 10 μm.
(J) Quantification of pouches in the same field of view between 6 h and 24 h (n = 3 mice each). Data are presented as mean ± SEM.
Real-time dynamics of seizure-induced MPPs
Structures similar to MPPs have be previously described and are suggested to be phagocytic with rapid engulfment from tens of minutes to up to 2 h (Abiega et al., 2016; Petersen and Dailey, 2004; Sierra et al., 2010; Zabel et al., 2016; Zhao et al., 2015), although they have rarely been investigated in vivo. Thus, we performed time-lapse imaging in vivo. Microglia exhibited various pouch formation and resolution dynamics. Formation occurred rapidly following initial “frisking” activity that eventually resulted in a stable pouch within 10–30 min (Figure S2A). Sometimes, pouches failed to be stable after initial frisking activity and therefore collapsed rapidly (Figure S2B). Extant pouches sometimes underwent resolution either by a gradual dissociation for which processes retracted from the contact material (Figure S2C) or by a “crunch” for which the pouch space collapsed, and the pouch edges became condensed (Figure S2D). This usually occurred rapidly, taking less than 20 min.
Interestingly, 4-h in vivo imaging revealed that the majority of MPPs are maintained (~70%) rather than showing engulfing activity. Furthermore, only a few pouches translocated toward the microglial somata from which the process bearing the pouch was located (~20%). The remaining ~10% of pouches were newly formed during the course of imaging (Figures 2A–2D; Video S2).
Figure 2. Real-time dynamics of seizure-induced microglial process pouches.
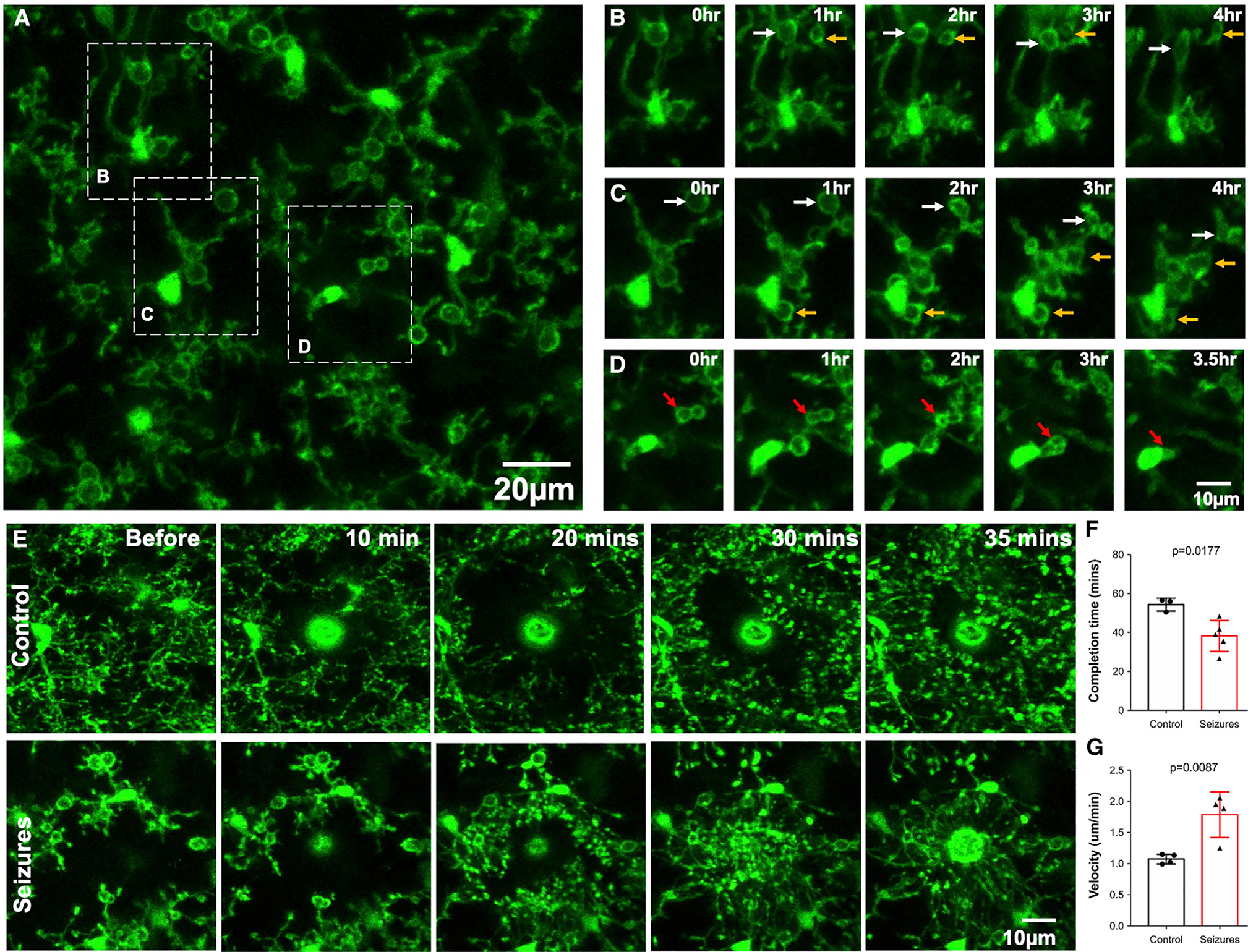
(A) Representative two-photon in vivo image from a CX3CR1GFP/+ mouse following an acute craniotomy at 6 h of seizures with pouches in the superficial somatosensory cortex. Scale bar: 20 μm.
(B) Representative time-lapse images from a field of view in (A) showing pouches that are present and maintained from the beginning of imaging (white arrows) and new pouches that formed during the course of imaging and were maintained (yellow arrows).
(C) Representative time-lapse images from a field of view in (A) showing pouches that are present and maintained from the beginning of imaging (white arrows) and new pouches that formed during the course of imaging and were maintained (yellow arrows). Note: pouches identified with white and yellow arrows also shrink during the course of imaging.
(D) Representative time-lapse images from a field of view in (A) showing pouches that are engulfed during the course of imaging (red arrows). Scale bar: 10 μm.
(E) Representative two-photon in vivo time-lapse images in brains from a control mouse (top) and a mouse at 6 h of seizures (bottom) following a laser-induced injury. Scale bar: 10 μm.
(F) Quantification of the completion time to chemotaxis around the laser injury in control and seizure mice (n = 3–5 mice each).
(G) Quantification of chemotaxis velocity in response to a laser injury in control and seizure mice (n = 3–5 mice each). Data are presented as mean ± SEM.
The paucity of microglial engulfment of pouch contents in vivo may have been a result of our imaging under isoflurane anesthesia or following a craniotomy. Therefore, we performed two sets of experiments, as follows: (1) intermittent imaging through an implanted window in vivo and (2) time-lapse imaging in brain slices ex vivo. When mice were imaged at 6 h, allowed to wake and resume normal activities without anesthesia, and re-imaged at 12 h, 63.5% ± 3.2% (n = 6 fields of view from 3 mice) of pouches were maintained over this 6-h period (Figure S3B). Similarly, MPPs in excised slices from mice that previously underwent severe seizures were maintained for long periods of time without engulfment (Figures S3C–S3F). Together, these results indicate that seizure-induced MPPs exhibit prolonged maintenance independent of anesthesia or craniotomy surgery.
Seizure-induced MPPs retain motile and chemotactic abilities
Pouch-bearing microglia may be impaired in their motile abilities precluding their phagocytic function. Although microglia displayed process pouches, their processes remained motile with filopodia emanating with various degrees of extension and retraction from the pouches. Next, we studied microglial process chemotaxis in control and KA-treated mice. At 6 h of KA treatment, chemotaxis to the laser-induced injury was completed in a shorter time and thus occurred much faster following seizures than under control conditions (Figures 2E–2G), which is consistent with previous findings in slices (Avignone et al., 2008, 2015). Interestingly, pouches were maintained during chemotaxis, and new fine filopodia (Bernier et al., 2019) emanated from extant pouches. Together, these results show that MPPs increase their motility/chemotactic potential.
Seizure-induced MPPs occur and are maintained independent of microglial CX3CR1, Trem2, and P2Y12 receptors
Poor evidence of MPP engulfment may suggest impairment in the phagocytic machinery, as impaired phagocytosis of apoptotic cells following KA seizures was recently reported (Abiega et al., 2016). Furthermore, we tested Trem2 and P2Y6 receptors (Hsieh et al., 2009; Koizumi et al., 2007; Takahashi et al., 2005) in MPP formation following severe seizures. Interestingly, we found that robust MPPs formed in Trem2- and P2Y6-receptor-deficient mice (Figures 3A–3D).
Figure 3. Microglial process pouch formation and maintenance independent of various chemotactic and phagocytic proteins.
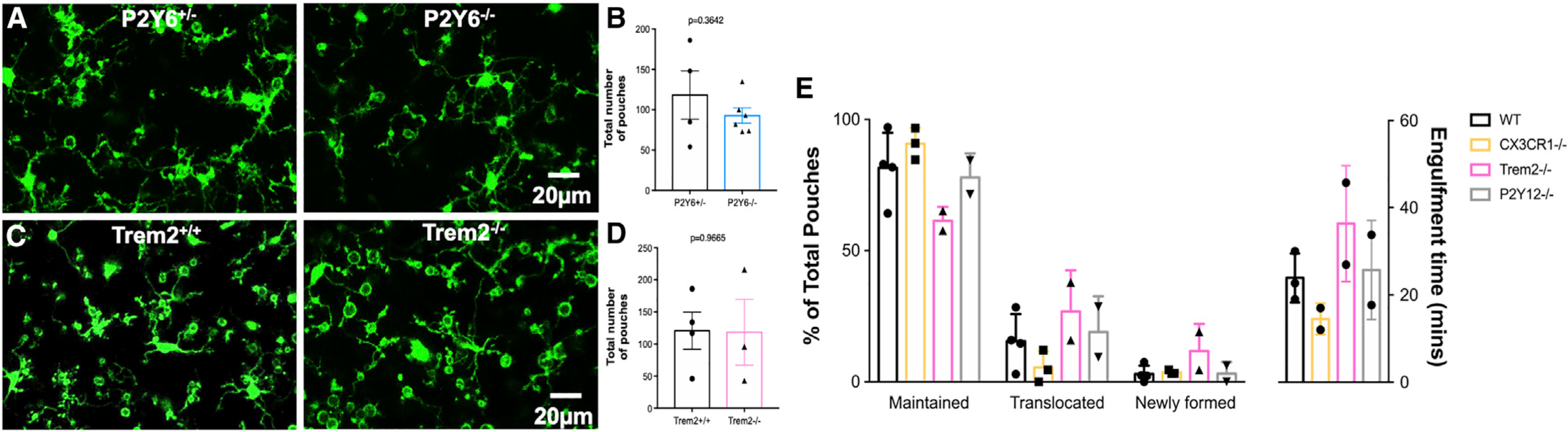
(A) Representative two-photon in vivo images from a P2Y6+/− and P2Y6−/− mouse showing microglial process pouches at 6 h of KA-induced seizures. Scale bar: 20 μm.
(B) Quantitative data of microglial process pouches per field of view in a P2Y6+/− and a P2Y6−/− mouse (n = 3 mice).
(C) Representative two-photon in vivo images from a Trem2+/+ and a Trem2−/− mouse showing microglial process pouches at 6 h of KA-induced seizures. Scale bar: 20 μm.
(D) Quantitative data of microglial process pouches per field of view in Trem2+/+ and Trem2−/− mice (n = 3 mice).
(E) Quantification of the percentage of the 3 types of process pouches during the course of 4 h of imaging categorized as either maintained, translocated toward the microglial somata, or newly formed pouches from different genotypes (left graph) as well as the time to completely engulf the wrapped material (right graph). n = 2–4 mice each. Data are presented as mean ± SEM.
Furthermore, we compared pouch maintenance in chemotactic (CX3CR1 and P2Y12R) and phagocytic (Trem2) receptor-deficient mice and report similar percentages of maintained, translocated, and newly formed pouches during 4 h of real-time imaging (Figure 3E). Therefore, the precise molecular mechanisms governing this process are not known, but our studies using a genetic approach suggest that they do not require chemotactic CX3CR1 and P2Y12R or phagocytic Trem2 and P2Y6R for their formation and/or maintenance.
Seizure-induced MPPs function without phagocytic activity
To further test the role of phagocytic activity in MPPs, we performed immunohistochemistry for CD68, a lysosomal marker active during phagocytosis, and LysoTracker, a label for active lysosomes. As expected, CD68 colocalized with LysoTracker in microglia (Figures 4A and 4B). Following KA-induced seizures, we injected LysoTracker and monitored MPPs (Figure 4C). Although microglia harbored pouches of different sizes and numbers, significant lysosomal activity was not detected in pouches (Figures 4D–4G; Video S3). Together, these results suggest that MPPs do not show robust phagocytic activity.
Figure 4. Microglial process pouches do not show phagocytic lysosomal activity.
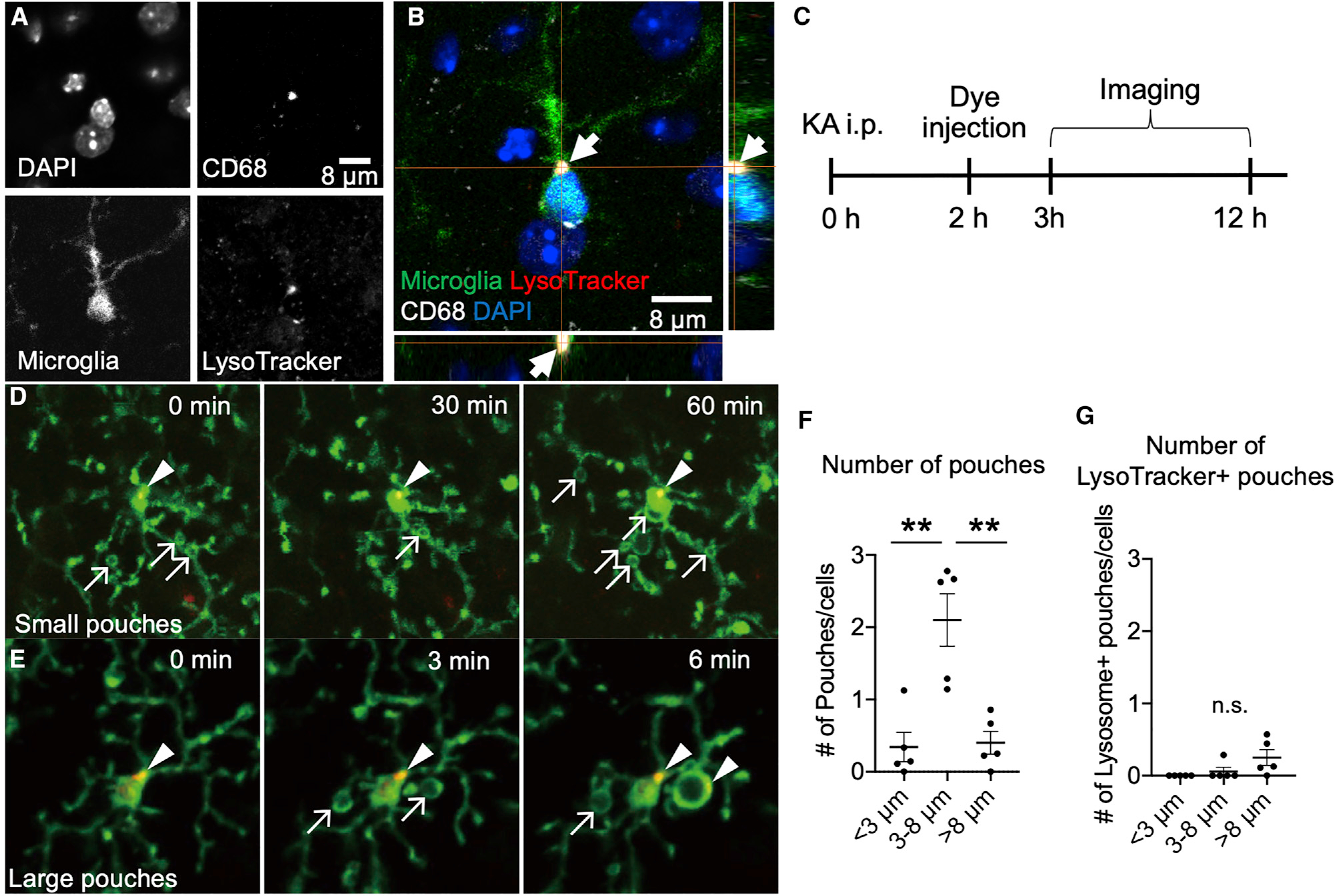
(A–C) Representative confocal image of a fixed slice stained with DAPI, CD68, CX3CR1-GFP, and LysoTracker.
(D and E) Representative two-photon in vivo images from CX3CR1GFP/+ mice after KA treatment and treated with LysoTracker. Arrows indicate pouches, and arrowheads indicate LysoTracker label. Scale bar: 10 μm.
(F) Quantification of the number of pouches of various sizes observed.
(G) Quantification of the number of LysoTracker+ pouches of various sizes observed. Data are presented as mean ± SEM.
Seizure-induced MPPs often target non-nucleic material
Next, we attempted to determine the target(s) of MPPs. MPPs were not directed toward the vasculature (Figure S3A), so we set out to determine the MPP target in fixed tissue slices. We noted an increase in pouch density in cortical and hippocampal regions at 6 h (Figures S4A–S4F). MPPs often did not contain DAPI-labeled nuclei (Figures S4A and S4G–S4J). Moreover, MPPs were more abundant in the superficial cortex (Figures 1D and 1E; Figure S1) that is not as densely populated with nuclei as deeper cortical layers. Cortical pouches varied in size ranging 2–20 μm in diameter (Figure S4K). Similar observations were made in hippocampal slices at 24 h of KA for which many pouches lacked DAPI content (Figures S4L–S4U). Although MPPs sometimes targeted DAPI-labeled nuclei in the hippocampus, MPPs did not target DAPI-labeled nuclei in superficial cortical layers (Figures S4G–S4J). These findings suggest that many non-nucleic materials are targeted by these pouches.
Seizure-induced MPPs target neuronal dendrites for resolution
Because neuronal dendrites undergo dendritic beading in response to severe seizures (Guo et al., 2016; Zeng et al., 2007) and tufts of neuronal dendrites densely populate the superficial cortex, we imaged both microglia and neurons in CX3CR1GFP/+;Thy1YFP mice (Feng et al., 2000; Jung et al., 2000) after severe seizures. Chronic imaging revealed that these dendritic beadings were observed by 6 h, of which some persisted through 72 h, although some disappeared and new ones were formed during this period (Figures 5A and 5B). Indeed, we could localize several process pouches around beaded dendrites at 6 h and 24 h after severe seizures (Figures 5C–5E; Video S4). Interestingly, only a minority of labeled beaded dendrites (~20%) were actually wrapped by microglia (Figure S5). However, this number should be considered with caution because only a few neurons were labeled in Thy1YFP mice. Real-time imaging revealed that pouches wrapped around beaded dendrites could shrink along with the bead (Figure 5F; Video S5).
Figure 5. Seizure-induced microglial process pouches target neuronal dendrites for resolution.
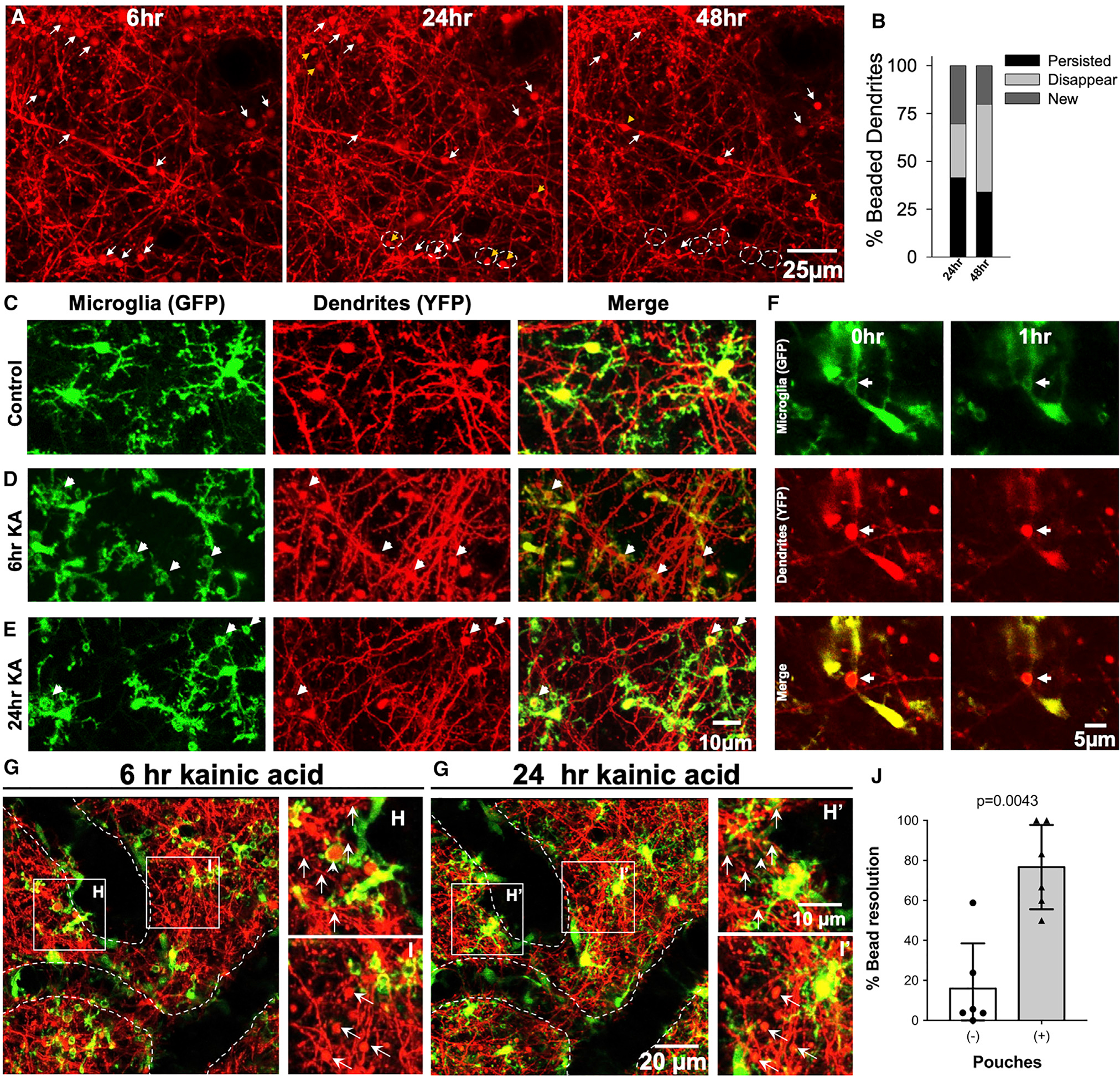
(A) Representative two-photon in vivo images through a chronically implanted cranial window from a Thy1YFP mouse from 6 to 48 h after severe (stage 5) seizures showing beaded dendrites, of which some persist (white arrows) and others are newly formed (yellow arrowhead) or disappear (dashed circles). Scale bar: 25 μm.
(B) Graphical quantification of the percentage of beaded dendrites that persist, are newly formed, or disappear between 6 and 48 h of severe (stage 5) seizures. Related to Figure 1.
(C–E) Representative two-photon in vivo images from CX3CR1GFP/+:Thy1YFP mice showing microglial pouches localized around swollen dendrites (arrows) at 6 h and 24 h after KA treatment. Scale bar: 5 μm.
(F) Representative two-photon in vivo images from a time-lapse movie collected from a CX3CR1GFP/+:Thy1YFP mouse showing a microglial pouch shrinking with the wrapped beaded dendrite that also shrinks (arrows). Scale bar: 5 μm.
(G–I’) Representative two-photon in vivo images through a chronically implanted cranial window from a CX3CR1GFP/+:Thy1YFP mouse showing dendritic beads with or without microglia at 6 h and 24 h. Data are presented as mean ± SEM. Scale bar: 20 μm in (G’) and 10 μm in (H’).
(J) Quantification of the percentage of dendritic bead resolution between 6 h and 24 h with or without microglial pouch contacts (n = 6 fields of view from 2 mice).
To assess the function of MPPs, individual dendritic beads with or without MPPs at 6 h were re-imaged at 24 h. Remarkably, beaded dendrites with microglial pouches at 6 h were 5 times more likely be resolved by 24 h (76.7% ± 7.9%; range, 50%–100% per field of view) than beaded dendrites lacking microglial pouches (16.0% ± 8.4%; range, 0%–58.8%; Figures 5G–5J). Therefore, consistent with pouch-dendritic bead shrinkage, we noted increased structural resolution of beaded dendrites with prior pouch contact when compared to beaded dendrites without pouch interactions. Our results, therefore, suggest caution in assigning phagocytic function to these structures.
DISCUSSION
In the current study, we document a microglial phenotype we have termed microglial process pouches or MPPs that (1) are robustly induced following severe seizures, (2) wrap around beaded dendrites, (3) are stably maintained for hours, (4) preserve their chemotactic abilities, (5) form independently of candidate phagocytic proteins like P2Y6 and Trem2, (6) are dynamic independent of candidate chemotactic (CX3CR1 and P2Y12) and phagocytic (P2Y6 and Trem2) mechanisms, and (7) are strongly associated with the structural resolution of beaded dendrites following severe seizures. Importantly, MPPs were widespread across brain regions including the amygdala and cerebellum, even though we mainly focused our studies on the cortex and hippocampus. These observations indicate a significant structural resolution function of microglial processes toward dendrites following severe seizures that could be harnessed for clinical therapeutics.
An interesting finding in our study is that although these seizure-induced MPPs look like previously described “phagocytic cups” (Abiega et al., 2016; Petersen and Dailey, 2004; Sierra et al., 2010; Zabel et al., 2016), they did not exhibit typical features of phagocytosis. First, they were maintained for extensive periods of time that were longer than what is typical for phagocytic processes, which range from 10 to 15 min (Zabel et al., 2016) to 0.5–2.5 h (Petersen and Dailey, 2004; Sierra et al., 2010). In our case, the majority of pouches were maintained for over 6 h.
Second, unlike other microglial phagocytic activities that target neuronal cell bodies, these MPPs often predominated in cortical regions with few cell bodies such as layer I. Rather than pouches targeting cell bodies, our results indicate that pouches targeted beaded dendritic material consistent with their localization in both the superficial cortex (Figures 1G and 1H; Figure S1) and stratum radiatum regions of the hippocampus (Figures S4G–S4J) where dendrites predominate over cell bodies. This correlation between the greater density of pouches in the superficial cortex and the abundance of dendritic tufts in the superficial cortex suggests that pouches are abundant in the cortex because of the corresponding abundance of dendrites. Therefore, we show that microglial phagocytic-like structures target dendrites in vivo. However, we failed to observe extensive phagocytosis of these structures, which is why we call them pouches.
Third, to our knowledge, the observation of shrinking microglial pouches during time-lapse imaging represents a phenotype that is not characteristic of microglia during phagocytosis when engulfment is used. The precise molecular mechanisms governing this process are not known, but our studies using a genetic approach suggest that they do not require chemotactic CX3CR1 and P2Y12R or phagocytic Trem2 and P2Y6R for their formation and/or maintenance.
Pouch shrinkage was observed with dendritic bead shrinkage in real-time. Consistent with pouch-dendritic bead shrinkage, we noted increased structural resolution of beaded dendrites with prior pouch contact when compared to beaded dendrites without pouch interactions. Dendritic beading after seizures has been previously reported in mice (Zeng et al., 2007) and patients with epilepsy (Multani et al., 1994). Moreover, they have been observed following ischemia and spreading depression (Risher et al., 2010), perhaps as a result of mitochondrial dysfunction (Greenwood et al., 2007) and cytoskeletal rearrangement (Zeng et al., 2007). Here, we show that microglia exhibit a rapid detection of these dendritic abnormalities and make contact with them by process pouches. Whether or not they perform similar functions in other pathological contexts remains to be determined.
In conclusion, although previous studies have indicated that microglia in general (Mirrione et al., 2010; Waltl et al., 2018) and microglial P2Y12 (Eyo et al., 2014) and CX3CR1 (Eyo et al., 2017) receptors in specific can be neuroprotective during seizures/epilepsy, this look into microglial-neuronal physical interactions following seizures in vivo has uncovered the formation of MPPs and that MPPs display features atypical for phagocytosis and were highly associated with a better resolution of structural dendritic injury resulting from seizures. This microglial-dependent neuroprotection is consistent with recent findings that microglial elimination exacerbates experimental seizures (Liu et al., 2020) and blocking microglia Gi, which regulates microglial process dynamics and increases network activity and spontaneous seizures (Merlini et al., 2021). Our results document a cellular process by which microglia may be facilitating such neuroprotection. Specifically, our results suggest that microglia may contribute to the structural resolution of beaded dendrites to facilitate the restoration of dendritic function. Therefore, resolution of dendritic function will need to be tested in future studies. These results highlight a neuroprotective role for microglia immediately after seizures that once fully understood could be harnessed to limit the detrimental effects of seizures in the clinic.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Long-Jun Wu (wu.longjun@mayo.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Institutional Animal Care and Use Committees at Rutgers University, Mayo Clinic and the University of Virginia approved all procedures. Adult mice of both sexes were used between 6 weeks and 6 months of age; no differences were noted between male and female mice so the data were combined. Wild-type C57/B6 mice were crossed with CX3CR1GFP/GFP mice from Jackson labs (Stock No: 005582) to generate CX3CR1GFP/+ for all experiments to visualize microglia. These mice were crossed with Thy1YFP mice from Jackson labs (Stock No: 003782) to generate CX3CR1GFP/+:Thy1YFP mice. CX3CR1GFP/GFP mice were used as CX3CR1-deficient mice. Trem2-deficient mice were provided as a gift from Dr. Marco Colonna at Washington University in St. Louis, USA. P2Y6-deficient mice were provided as a gift from Dr. Bernard Robaye from the Institut de biologie et de médecine moléculaires in Belgium. P2Y12R-deficient mice were provided as a gift from Dr. Michael Dailey at the University of Iowa. Receptor deficient mice were crossed with CX3CR1GFP/GFP mice to facilitate visualization of microglia.
METHOD DETAILS
Experimental seizure model
Six- to 16-week-old GFP reporter mice were intraperitoneally injected with kainic acid (KA) at 18 –25mg/kg (at Rutgers University and Mayo Clinic) or 24–28mg/kg (at the University of Virginia). At the University of Virginia, for unknown reasons, mice require a higher concentration of KA to elicit seizures. Seizure behavior was monitored under a modified Racine scale as follows: (1) freezing behavior; (2) rigid posture with raised tail; (3) continuous head bobbing and forepaws shaking; (4) rearing, falling, and jumping; (5) continuous level 4; and (6) loss of posture and generalized convulsion activity (Eyo et al., 2014; Racine, 1972). Mice that progressed to at least to stage 3 (“mild seizures”) were used for the study while mice that did not progress this far were discarded from the study. Mice that entered and remained in stage 5 seizures for more than 30 minutes (“severe seizures”) were used to detect the occurrence of microglial process pouches.
Cranial window surgeries
Mice were anesthetized with isoflurane (5% for induction; 1.5% for maintenance and surgery) combined with oxygen. The hair above the mouse head was shaved with a clipper (Wahl BravMini), after which the mouse was placed in a stereotactic frame (Kopf) cleaned with three alternating swabs of betadine and 75% alcohol. Mice were maintained on a heating pad during surgery and then received a local subcutaneous injection of 15–20 mL 0.25% bupivicane and lubricant eye ointment. The skin above the head was then cut to expose the skull. The skull was cleaned using a cotton swab of 3%–5% hydrogen peroxide, and a dental drill (Osada Model EXL-M40) and drill bit (Fine Science Tools, 19008–07) were used to drill open a circular > 3-mm-diameter window. During drilling, bone debris were cleared away, and the skull was frequently irrigated with sterile saline. When sufficiently thinned, the circular skull flap was carefully removed using sharp forceps. Windows were paced above the somatosensory cortex such that the skull was removed with the center at about −2.5 posterior and ± 2μm lateral to bregma. Once opened, the exposed brain surface was kept moist with sterile saline. A 3-mm glass coverslip previously sterilized in 75% ethanol was put inside the window, glued in place with cyanoacrylate glue (Krazy glue). The rest of the skull, excluding the region with the window, was then covered with IBond Total Etch glue (Heraeus) and cured with a Kerr Demi Ultra LED Curing Light (Dental Health Products). A dental cement (Tetric EvoFlow) was then applied around the glass coverslip and again cured with the curing light. Finally, a custom-made head plate was glued to the contralateral hemisphere to the window with another application of the dental cement and cured with a curing light to permanently attach the head plate to the skull.
Following this surgery, mice were either immediately transferred to the two-photon microscope for acute imaging on a heading plate or allowed to recover from anesthesia on a heating pad (~10 min) before they were returned to their home cage for chronic imaging. For chronic imaging, mice were allowed to recover from the surgery for at least 2 weeks and resting microglial morphologies were confirmed before repeated imaging commenced. Mice that showed a loss in imaging window clarity during the period of observation were discarded from the study.
Generation of brain slices
Freshly isolated hippocampal slices were prepared mice at 6 hours of KA treatment in mice that showed “severe seizures.” Briefly, mice were anesthetized and swiftly decapitated. Brains from decapitated mice were carefully removed and placed in ice-cold, oxygenated (95%O2 and5%CO2) artificial cerebrospinal fluid (ACSF) with the following composition (in mM): 124 NaCl, 25 NaHCO3, 2.5 KCl, 1 KH2PO4, 2 CaCl2, 2 MgSO4, 10 glucose, and sucrose added to make 300–320 mOsmol. Coronal slices (300μm) were prepared and transferred to a recovery chamber for 30 minutes with oxygenated ACSF with the same composition as above at room temperature before imaging.
Two-photon imaging
Experiments were conducted at room temperature with slices maintained in oxygenated ACSF with the same composition as above in a perfusion chamber at a flow rate of 2 ml/min. Microglia were typically imaged using a two-photon microscope tuned to 880–900 nm (for GFP microglia and YFP neurons or GFP microglia and Rhodamine B) with a water-immersion lens. Fluorescence was detected using two photomultiplier tubes in whole-field detection mode and a 565nmdichroic mirror with 525/50nm (green channel) and 620/60 nm (red channel) emission filters. To separate GFP and YFP in some experiments, a 509 nm dichroic mirror with 500/15 and 537/26 nm emission filters was used. The laser power was maintained at 25mW or below.
For in vivo imaging, microglia were typically imaged between the brain surface and 200μm below which corresponds to layers I-III of the cortex. For ex vivo imaging in brain slices, microglia were typically imaged between 50–150μm of the cortical surface to avoid imaging in regions where there is abundant cell death at the slice surface from the slicing procedure. For imaging, data images were collected at 1–2μm spatial and 1–5minute temporal intervals for 1–4 hours. To perform a laser injury, we focused and parked the laser 48–66x at ~250mW at 880–900nm for 0.5–3 s. All two photon images are 3D data presented in 2D as projection images.
Lysotracker Imaging
Lysosomes were visualized using LysoTracker Red DND-99 (L7528; Invitrogen). Mice that entered and remained in stage 4 or 5 seizures for more than 30 minutes (“severe seizures”) were used to observe microglial lysosomes. Mice were injected with KA and after 2 hours cranial window surgeries were performed. After opening skull, mice were injected with LysoTracker (1:200, 600 nl) into the brain parenchyma using a glass pipette and an injector (NANOJECT III; Drummond Scientific Company). Microglia and lysosomes were imaged using a two-photon microscope tuned to 920 nm with a water-immersion lens. Fluorescence was separated by a 560 nm dichroic mirror with 525/50 nm (for GFP fluorescence detection) and 630/75 nm (for LysoTracker Red fluorescence detection) emission filters. The number of pouch and their diameter were quantified using ImageJ (NIH).
Immunohistochemistry
To confirm the LysoTracker signal co-localizes with CD68 staining, we performed immunostaining. Mice were deeply anesthetized with isoflurane, and transcardially perfused with 4% paraformaldehyde (PFA). Fixed brains were post-fixed overnight in PFA and immersed in 30% sucrose for 2 days. The brains were sectioned into 30 μm slices. After a phosphate-buffered saline (PBS) wash, the slices were incubated for 1 hour in 5% bovine serum albumin (BSA) and 0.5% Triton X-100 in PBS, and then incubated at 4°C overnight with primary antibodies (anti-CD68, 1:500, Abcam, 125212) diluted in PBS. The slices were subsequently incubated with secondary antibodies (anti-rabbit Alexa 647, 1:500, Thermo Fisher Scientific) at room temperature for 3 h. Slices were then mounted on glass slides in DAPI Fluoromount-G (0100–20; Southern Biotech). Fixed tissue was imaged using a Zeiss LSM780 confocal microscope (Carl Zeiss).
QUANTIFICATION AND STATISTICAL ANALYSIS
Bead density analysis
Beads or “swellings” were identified following severe stage 5 seizures and defined as dendritic shaft swellings that were at least 3 times the size of the adjacent dendritic shaft diameter. In most cases, beads were ≥ 4–5 times the size of adjacent dendritic shafts.
Percent Pouch Depth Analysis
For microglial process pouch percent analysis at different depths (Figure 1E), the total number of pouches per 330μm x 330μm field of view was counted between 0–120μm of the brain. Then, the number of pouches were counted in bins of 20μm thickness. The percent of the total number of pouches per 20μm depth was determined by dividing the number per 20μm depth over the total number of pouches per field of view.
Pouch Density Analysis
For in vivo microglial pouch density analysis through a chronic cranial window at 6hr and 24hr (Figure 1G), specific fields of views (165μm x 165μm x 40μm thick) were followed at 6hr and then at 24hr. Pouch numbers were counted in adjacent images at the two time points. For fixed slice quantification of microglial pouch density in control and at 6hr of KA treatment (Figure S4), microglial process pouches were counted in images from 20μm thick cortical or hippocampal slices mounted in DAPI.
Comparing Pouch Density in Different Genotypes
To compare pouch density in different genotypes (Figures 3B, 3D, and 3E), the total number of pouches in a 220μm x 220μm x 120μm thick field of view was quantified and compared between genotypes.
Pouch Dynamics Analysis
To quantify pouch dynamics from time-lapse movies obtained from mice with different genotypes (Figure 3E), individual pouches were followed through 4hr movies and binned into pouches that persisted through the movie (“maintained”), showed signs of the beginnings of phagocytic activity by retracting pouches toward the cell body (“translocated”) or were formed newly during the imaging period (“newly formed”).
Engulfment Time Analysis
For engulfment analysis from time-lapse movies (Figure 3E), all phagocytic events were noted from each movie and the timing from the initiation of pouch retraction for engulfment to the time point of full engulfment in the cell body was determined.
Chemotaxis Analysis
For chemotaxis analysis from time-lapse movies, the time for microglial processes to completely encircle the laser-injury was quantified (Figure 2F) as well as the velocity of individual migrating processes was quantified (Figure 2G).
Pouch-Bead Resolution Analysis
To quantify beaded dendrite resolution (Figure 4J), dendritic beads in identical fields of view were monitored at 6hr and then 24hr and binned into possessing or lacking microglial process pouch contact. Beads present at 6hr were noted as resolved when they were absent at 24hr and the dendritic shaft was restored to lack beads.
For all experiments, data are presented as mean ± SEM. For all analyses, the analyzer was often blind to the genotype or condition except when comparing control conditions that lacked pouches and seizure conditions with an abundance of pouches or when comparing beaded dendrites with microglial pouches and beaded dendrites without microglial pouches. Student’s t test was used to establish significance.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD68, 125212 | Abcam | GR3326063-11 |
| anti-rabbit Alexa 647 | Thermo Fisher Scientific | A-21244 |
| Chemicals, peptides, and recombinant proteins | ||
| Rhodamine B | Millipore Sigma | 83689-1G |
| LysoTracker Red DND-99, L7528 | Invitrogen | 2170270 |
| Experimental models: organisms/strains | ||
| Mouse: B6.129P2(Cg)-Cx3cr1tm1Litt/J | The Jackson Laboratory | JAX: 005582 |
| Mouse: B6.Cg-Tg(Thy1-YFP)HJrs/J | The Jackson Laboratory | JAX: 003782 |
| Mouse: Trem2 knockout mouse | From Dr. Marco Colonna, Washington University in St. Louis | N/A |
| Mouse: P2Y6 knockout mouse | From Dr. Bernard Robaye, Institut de biologie et de médecine moléculaires | N/A |
| Mouse: P2Y12 knockout mouse | From Dr. Michael Dailey, University of Iowa | N/A |
Highlights.
Cortical microglia form process pouches following severe experimental seizures
Microglial process pouches target beaded dendrites following severe seizures
Microglial process pouches are long lived with little evidence for phagocytosis
Microglial process pouches correlate with the structural resolution of dendrites
ACKNOWLEDGMENTS
We thank Greg Voronin for help with discussing the project and all members of the Wu and Eyo Labs for critical discussions. We thank Dr. David Margolis at Rutgers University for help with training for the chronic imaging procedure. We also thank Dr. Justin Liu now at SUNY Buffalo who helped with collection of some initial data for this work. This work is supported by the following grants from the National Institutes of Health: R01NS088627 (L.-J.W.), R01NS112144 (L.-J.W), and K22N84392 (U.B.E.). Supplementary materials contain additional data.
Footnotes
DECLARATIONS OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109080.
REFERENCES
- Abiega O, Beccari S, Diaz-Aparicio I, Nadjar A, Layé S, Leyrolle Q, Gómez-Nicola D, Domercq M, Pérez-Samartín A, Sánchez-Zafra V, et al. (2016). Neuronal hyperactivity disturbs ATP microgradients, impairs microglial motility, and reduces phagocytic receptor expression triggering apoptosis/microglial phagocytosis uncoupling. PLoS Biol 14, e1002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Ikegaya Y, and Koyama R (2019). Microglia attenuate the kainic acid-induced death of hippocampal neurons in slice cultures. Neuropsychopharmacol. Rep 40, 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avignone E, Ulmann L, Levavasseur F, Rassendren F, and Audinat E (2008). Status epilepticus induces a particular microglial activation state characterized by enhanced purinergic signaling. J. Neurosci 28, 9133–9144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avignone E, Lepleux M, Angibaud J, and Nägerl UV (2015). Altered morphological dynamics of activated microglia after induction of status epilepticus. J. Neuroinflammation 12, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier LP, Bohlen CJ, York EM, Choi HB, Kamyabi A, Dissing-Olesen L, Hefendehl JK, Collins HY, Stevens B, Barres BA, and MacVicar BA (2019). Nanoscale surveillance of the brain by microglia via cAMP-regulated filopodia. Cell Rep 27, 2895–2908.e4. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, and Gan WB (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci 8, 752–758. [DOI] [PubMed] [Google Scholar]
- Eyo UB, and Wu LJ (2013). Bidirectional microglia-neuron communication in the healthy brain. Neural Plast 2013, 456857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, and Wu LJ (2014). Neuronal hyperactivity recruits microglial processes via neuronal NMDA receptors and microglial P2Y12 receptors after status epilepticus. J. Neurosci 34, 10528–10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Gu N, De S, Dong H, Richardson JR, and Wu LJ (2015). Modulation of microglial process convergence toward neuronal dendrites by extracellular calcium. J. Neurosci 35, 2417–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Peng J, Murugan M, Mo M, Lalani A, Xie P, Xu P, Margolis DJ, and Wu LJ (2017). Regulation of physical microglia-neuron interactions by fractalkine signaling after status epilepticus. eNeuro 3, ENEURO.0209–16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Mo M, Yi MH, Murugan M, Liu J, Yarlagadda R, Margolis DJ, Xu P, and Wu LJ (2018). P2Y12R-dependent translocation mechanisms gate the changing microglial landscape. Cell Rep 23, 959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, and Sanes JR (2000). Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28, 41–51. [DOI] [PubMed] [Google Scholar]
- Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, Engel J Jr., Forsgren L, French JA, Glynn M, et al. (2014). ILAE official report: a practical clinical definition of epilepsy. Epilepsia 55, 475–482. [DOI] [PubMed] [Google Scholar]
- Gao H, Danzi MC, Choi CS, Taherian M, Dalby-Hansen C, Ellman DG, Madsen PM, Bixby JL, Lemmon VP, Lambertsen KL, and Brambilla R (2017). Opposing functions of microglial and macrophagic TNFR2 in the pathogenesis of experimental autoimmune encephalomyelitis. Cell Rep 18, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood SM, Mizielinska SM, Frenguelli BG, Harvey J, and Connolly CN (2007). Mitochondrial dysfunction and dendritic beading during neuronal toxicity. J. Biol. Chem 282, 26235–26244. [DOI] [PubMed] [Google Scholar]
- Guo D, Arnspiger S, Rensing NR, and Wong M (2012). Brief seizures cause dendritic injury. Neurobiol. Dis 45, 348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Zeng L, Zou J, Chen L, Rensing N, and Wong M (2016). Rapamycin prevents acute dendritic injury following seizures. Ann. Clin. Transl. Neurol 3, 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser WA, and Hesdorffer DC (1990). Epilepsy: Frequency, Causes, And Consequences (Epilepsy Foundation of America; ). [Google Scholar]
- Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, and Seaman WE (2009). A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J. Neurochem 109, 1144–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Lee CL, Smith KL, and Swann JW (1998). Spine loss and other persistent alterations of hippocampal pyramidal cell dendrites in a model of early-onset epilepsy. J. Neurosci 18, 8356–8368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, and Littman DR (2000). Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol 20, 4106–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato G, Inada H, Wake H, Akiyoshi R, Miyamoto A, Eto K, Ishikawa T, Moorhouse AJ, Strassman AM, and Nabekura J (2016). Microglial Contact Prevents Excess Depolarization and Rescues Neurons from Excitotoxicity. eNeuro 3, ENEURO.0004–16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, Joshi BV, Jacobson KA, Kohsaka S, and Inoue K (2007). UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 446, 1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YU, Ying Y, Li Y, Eyo UB, Chen T, Zheng J, Umpierre AD, Zhu J, Bosco DB, Dong H, and Wu LJ (2019). Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat. Neurosci 22, 1771–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Jiang L, Wen M, Ke Y, Tong X, Huang W, and Chen R (2020). Microglia depletion exacerbates acute seizures and hippocampal neuronal degeneration in mouse models of epilepsy. Am. J. Physiol. Cell Physiol 319, C605–C610. [DOI] [PubMed] [Google Scholar]
- Merlini M, Rafalski VA, Ma K, Kim KY, Bushong EA, Rios Coronado PE, Yan Z, Mendiola AS, Sozmen EG, Ryu JK, et al. (2021). Microglial Gi-dependent dynamics regulate brain network hyperexcitability. Nat. Neurosci 24, 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirrione MM, Konomos DK, Gravanis I, Dewey SL, Aguzzi A, Heppner FL, and Tsirka SE (2010). Microglial ablation and lipopolysaccharide preconditioning affects pilocarpine-induced seizures in mice. Neurobiol. Dis 39, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Multani P, Myers RH, Blume HW, Schomer DL, and Sotrel A (1994). Neocortical dendritic pathology in human partial epilepsy: a quantitative Golgi study. Epilepsia 35, 728–736. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, and Helmchen F (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. [DOI] [PubMed] [Google Scholar]
- Petersen MA, and Dailey ME (2004). Diverse microglial motility behaviors during clearance of dead cells in hippocampal slices. Glia 46, 195–206. [DOI] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol 32, 281–294. [DOI] [PubMed] [Google Scholar]
- Risher WC, Ard D, Yuan J, and Kirov SA (2010). Recurrent spontaneous spreading depolarizations facilitate acute dendritic injury in the ischemic penumbra. J. Neurosci 30, 9859–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochefort NL, and Konnerth A (2012). Dendritic spines: from structure to in vivo function. EMBO Rep 13, 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, and Segal M (2014). Dendritic spines: the locus of structural and functional plasticity. Physiol. Rev 94, 141–188. [DOI] [PubMed] [Google Scholar]
- Sarlus H, and Heneka MT (2017). Microglia in Alzheimer’s disease. J. Clin. Invest. 127, 3240–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, and Maletic-Savatic M (2010). Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7, 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Paolicelli RC, and Kettenmann H (2019). Cien Años de Microglía: Milestones in a century of microglial research. Trends Neurosci 42, 778–792. [DOI] [PubMed] [Google Scholar]
- Swann JW, Al-Noori S, Jiang M, and Lee CL (2000). Spine loss and other dendritic abnormalities in epilepsy. Hippocampus 10, 617–625. [DOI] [PubMed] [Google Scholar]
- Szalay G, Martinecz B, Lénárt N, Kärnyei Z, Orsolits B, Judák L, Császár E, Fekete R, West BL, Katona G, et al. (2016). Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun 7, 11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Rochford CD, and Neumann H (2005). Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med 201, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe S, Saitoh S, Miyajima H, Itokazu T, and Yamashita T (2019). Microglia suppress the secondary progression of autoimmune encephalomyelitis. Glia 67, 1694–1704. [DOI] [PubMed] [Google Scholar]
- Umpierre AD, Bystrom LL, Ying Y, Liu YU, Worrell G, and Wu LJ (2020). Microglial calcium signaling is attuned to neuronal activity in awake mice. eLife 9, e56502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltl I, Käufer C, Gerhauser I, Chhatbar C, Ghita L, Kalinke U, and Löscher W (2018). Microglia have a protective role in viral encephalitis-induced seizure development and hippocampal damage. Brain Behav. Immun 74, 186–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Li Y, Wei Y, Bosco DB, Xie M, Zhao MG, Richardson JR, and Wu LJ (2020). Microglial depletion aggravates the severity of acute and chronic seizures in mice. Brain Behav. Immun 89, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabel MK, Zhao L, Zhang Y, Gonzalez SR, Ma W, Wang X, Fariss RN, and Wong WT (2016). Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 64, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng LH, Xu L, Rensing NR, Sinatra PM, Rothman SM, and Wong M (2007). Kainate seizures cause acute dendritic injury and actin depolymerization in vivo. J. Neurosci 27, 11604–11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Zabel MK, Wang X, Ma W, Shah P, Fariss RN, Qian H, Parkhurst CN, Gan WB, and Wong WT (2015). Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol. Med 7, 1179–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Liao Y, Morgan S, Mathur R, Feustel P, Mazurkiewicz J, Qian J, Chang J, Mathern GW, Adamo MA, et al. (2018). Noninflammatory changes of microglia are sufficient to cause epilepsy. Cell Rep 22, 2080–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.