

Abstract
Objective
Sensory loss with normal nerve conduction studies (NCS) from focal sensory root inflammatory demyelination is characteristic of chronic immune sensory polyradiculopathy (CISP). However, nonpure cases involving motor and distal sensory nerves exist (CISP-plus). We hypothesize that CISP-plus and CISP are fundamentally part of the same syndrome through comparison of clinical, neurophysiologic, and pathologic features.
Methods
CISP-plus (primary dorsal root with lesser motor and sensory nerve involvement) and CISP cases were retrospectively analyzed (1986–2019).
Results
We identified 44 CISP-plus and 28 CISP cases (n = 72) with 86% (38/44) of patients with CISP-plus and 79% (22/28) of patients with CISP experiencing imbalance. On examination, large fiber sensory loss was present in 98% (43/44) of patients with CISP-plus and 96% (27/28) of patients with CISP. Gait ataxia was evident in 93% (41/44) of patients with CISP-plus and 79% (22/28) of patients with CISP. Mild distal weakness was common in CISP-plus (75%, 33/44). NCS showed mild abnormalities in all patients with CISP-plus and were normal (by definition) in all patients with CISP. Elevated CSF protein, slowing of somatosensory evoked potentials, and MRI root enhancement occurred in most CISP-plus and CISP cases. Eleven CISP-plus nerve biopsies showed loss of large myelinated fibers and onion-bulb formations, most prominent in rootlet biopsies. Immunotherapy resulted in marked improvement of gait ataxia in 84% (27/32) of patients with CISP-plus and 93% (13/14) of patients with CISP with return to normal neurologic examination in half (25/46).
Conclusion
The recognition of CISP-plus expands the spectrum of CIDP by combining CISP-plus (predominant sensory polyradiculopathy with mild motor and sensory nerve involvement) with pure CISP (focal sensory polyradiculopathy) together as proximal sensory CIDP.
Although chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) usually presents with weakness,1,2 sensory-predominant forms have been described,3,4 including the recently recognized paranodal neuropathies (contactin-1–associated CIDP).5,6 In 2004, our group described a restricted form of sensory CIDP, chronic immune sensory polyradiculopathy (CISP), characterized by selective involvement of sensory nerve roots.7 A similar acute form of immune-mediated pure sensory polyradiculopathy has been described recently.8 CISP presents with sensory loss, gait ataxia, falls, large fiber sensory deficits, reduced reflexes, and preserved muscle strength. Because the pathology is confined to sensory roots (preganglionic) and motor nerves are spared, nerve conduction studies and EMG (NCS/EMG) are normal.7 The presence of slowed somatosensory evoked potentials (SSEP), CSF protein, and enlarged dorsal roots on MRI support the diagnosis of CISP.9-12 Dorsal lumbar rootlet biopsies confirm inflammatory demyelination.7 CISP has been recognized as atypical CIDP and is included in the European Federation of Neurologic Societies/Peripheral Nerve Society (EFNS/PNS) CIDP criteria.13 Since CISP's description, we have noted patients with a similar clinical syndrome but with mild distal weakness and mild abnormalities on NCS, implying the pathology is not restricted to sensory nerve roots (figure 1A, right panel). We call this nonpure form CISP-plus.
Figure 1. Anatomical and Somatosensory Localization of Pathophysiology in Chronic Immune Sensory Polyradiculopathy (CISP) and CISP-Plus.

(A) Anatomical pictorial representation of the nerves affected in (A.a) CISP (isolated sensory root involvement) and in (A.b) CISP-plus (primarily but not solely sensory root involvement). The dark shaded area represents the dorsal root where the primary pathology occurs in both conditions and the dotted area represents lesser involvement of other nerves (shown are ventral root, dorsal root ganglion, and spinal nerve) that occurs in CISP-plus but not in CISP. (B) Median nerve somatosensory evoked potential (SSEP) tracings from patients with CISP (B.a) and patients with CISP-plus (B.b) show examples where slowing occurs before the cervical spine site (N13) and within the segment between the clavicular and cervical spine sites (N9–N13, the segment containing the sensory root). The absolute and interpeak latencies values (in milliseconds) from the median SSEP studies as well as the normal values are listed at the bottom. These SSEP findings show that the slowing in both conditions primarily occurs at the sensory root level.
The purpose of this study is to introduce and define the entity of CISP-plus by presenting a cohort of CISP-plus cases and comparing their clinical, neurophysiologic, pathologic, and radiologic characteristics and treatment response with a cohort of CISP cases.
Methods
Standard Protocol Approvals, Registrations, and Patient Consents
The Mayo Clinic Institutional Review Board approved human specimen acquisition and review of patients' histories. We retrospectively reviewed the Mayo Clinic (Rochester, Minnesota) database for coded diagnosis of CIDP, polyganglionopathy, sensory neuropathy, sensory ataxia, and CISP from 1986 to 2019. Medical records of 16,542 potential patients were identified and reviewed; only patients who fulfilled our inclusion criteria and were evaluated by a neurologist were included. We reviewed NCS/EMG, SSEP, quantitative sensory testing (QST), autonomic reflex screen, nerve biopsies, MRIs, CSF testing, and serology for paraneoplastic or autoimmune causes of sensory neuropathy.
Clinical Criteria
Patients with CISP were defined using the previously published criteria7: (1) a sensory syndrome without weakness; (2) normal NCS/EMG studies; (3) imaging studies (usually MRI) that exclude brain, cerebellum, spinal cord, or compressive nerve root lesions that could explain the clinical syndrome; and (4) SSEP or imaging abnormalities (usually MRI) consistent with nerve root involvement. Patients with CISP-plus were defined using the following criteria: (1) a predominant sensory syndrome with only mild distal or no weakness; (2) mild abnormalities on NCS/EMG (motor or sensory) that do not fully explain the clinical syndrome (including reduced compound muscle action potentials and sensory nerve action potentials [taking normal age values into account]); (3) imaging studies (usually MRI) that exclude brain, cerebellum, spinal cord, or compressive nerve root lesions that could explain the clinical syndrome; and (4) SSEP or imaging abnormalities (usually MRI) consistent with nerve root involvement. Sensory gait ataxia was defined as impaired Romberg test, abnormal tandem walking, or marked difficulty walking without assistance with normal muscle strength and cerebellar function.
Patients in whom the pathologic process was predominantly localized to the peripheral nerves were excluded. Excluded diagnoses included typical sensory CIDP,3 sensory neuronopathy (Sjögren and paraneoplastic syndromes), or known causes of neuropathy including uremia, alcoholism, vitamin deficiency, heavy metal intoxication, collagen vascular diseases, thyroid disease, and hereditary neuropathies. Patients with diabetes were excluded unless the neurologic syndrome predated the diabetes.
Physiologic Testing and Imaging
NCS/EMG and SSEP studies were performed using Mayo Clinic laboratory normal values14 and Nicolet Viking EDX (Natus Neurology, Madison, WI) and Cadwell Sierra Summit (Cadwell Industries, Inc., Kennewick, WA). QST was performed on the dorsal foot with the CASE IV system (WR Medical Electronics, Maplewood, MN).15 Autonomic testing was performed using autonomic reflex screen (WR Medical Electronics).16
MRIs from patients with CISP-plus and patients with CISP including spine, plexus, and peripheral nerves were reviewed blinded to the final diagnosis by 2 authors (S.S. and K.K.A.). All studies were screened for increased T2 signal, contrast enhancement within nerves or roots, and clumping or thickening of roots.
Nerve Histology
Nerve biopsies were performed and processed in our peripheral nerve laboratory using standard histologic and immunohistochemistry stains (CD45, CD68) prepared on paraffin sections.17 Semithin epoxy sections were stained with methylene blue. Teased fibers were prepared and graded by previously defined pathologic criteria.17,18 Morphometric analysis was performed on epoxy sections using our Imaging System for Nerve Morphometry.19
Data Availability
De-identified data are available upon reasonable request.
Results
Summary of Demographic Data
We identified 44 CISP-plus and 28 CISP (total = 72) cases. A total of 15/28 of the CISP cases have previously been reported.7 The median time from first neurologic documented visit to final follow-up was 19 months (range 1–287) for all cases: 25 months for CISP-plus and 18 months for CISP (p = 0.002). The median age at onset overall was 58 years (range 18–80): 60 years (range 40–81) for CISP-plus and 57 years (range 18–80) for CISP. There were 44 male patients (61%) overall, with 28 male patients (64%) in the CISP-plus cohort and 16 male patients (57%) in the CISP cohort. The median duration of symptoms overall was 36 months (range 1–360): 48 months (range 1–360) in CISP-plus and 24 months (range 1–216) in CISP. The overall median time from symptom onset to diagnosis for the combined groups was 3 years (range 1–30): 4 years for CISP-plus (range 1–30) and 2 years for CISP (range 1–18) (p = 0.06) with 12 patients having very delayed diagnosis of more than 10 years after symptom onset (4 CISP and 8 CISP-plus; figure 2).
Figure 2. Diagnosis Delay in Patients With Chronic Immune Sensory Polyradiculopathy (CISP) and CISP-Plus.
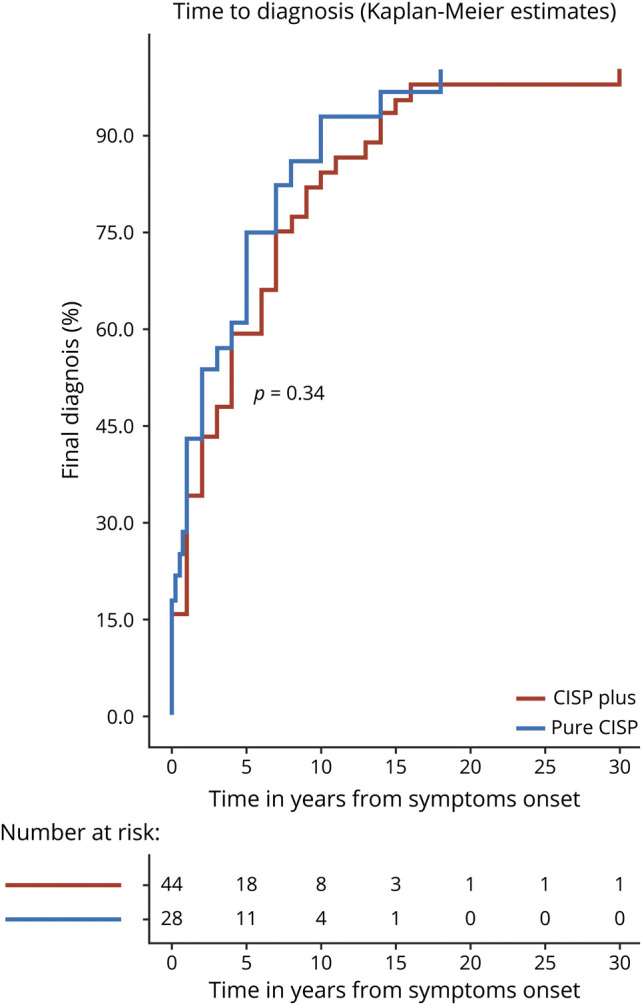
Time to diagnosis plots comparing patients with CISP to patients with CISP-plus showing that both groups have delayed diagnoses and that 12 patients from both groups were diagnosed >10 years after symptom onset (to the right of the dotted line). The trend to a more delayed diagnosis in CISP-plus compared to CISP (p = 0.06) may be due to the nerve conduction findings being more in keeping with axonal and not demyelinating pathology in CISP-plus. Such findings may wrongly point physicians away from an inflammatory demyelinating neuropathy in CISP-plus.
Clinical Manifestations With Sensory Predominant Presentations
All patients with CISP-plus and patients with CISP (n = 72) had progressive sensory predominant presentations. Patients presented with symptoms of gait imbalance (often debilitating) and sensory loss in 85% (61/72) overall: 86% (38/44) in CISP-plus and 79% (22/28) in CISP. Falls were common and reported in 57% (41/72) of cases overall: 57% (25/44) in CISP-plus and 57% (16/28) in CISP. Use of gait aids was necessary for almost half of cases; 45% (29/64) patients overall required gait aids: 46% (19/41) of patients with CISP-plus and 43% (10/23) of patients with CISP. More specifically, 27% (11/41) of patients with CISP-plus and 26% (6/23) of patients with CISP used canes (p = nonsignificant [NS]); 10% (4/41) of patients with CISP-plus and no patient with CISP used walkers; 12% (5/41) of patients with CISP-plus and 18% (4/22) of patients with CISP used wheelchairs (p = NS). Prickling paresthesia was commonly reported: in 77% (34/44) of patients with CISP-plus and in 68% (19/28) of patients with CISP (p = NS). In contrast, troublesome neuropathic pain at presentation was uncommon, reported by only 5% (2/44) of patients with CISP-plus and no patient with CISP (p = NS). Overall, symptoms of CISP-plus were very similar to those of CISP.
Proprioception or vibration loss (large fiber modalities) were the most common findings on neurologic examination, present in 97% (70/72) of cases overall: 98% (43/44) in CISP-plus and 96% (27/28) in CISP (p = NS). Reduced or absent deep tendon reflexes (knees or ankle) were evident in 90% (65/72) of cases overall: 89% (39/44) in CISP-plus and 93% (26/28) in CISP. Sensory gait ataxia was noted on examination in 88% (63/72) overall: 93% (41/44) in CISP-plus and in 79% (22/28) in CISP. In CISP-plus, 66% (25/38) of patients had positive Romberg testing and 70% (19/27) had a pathologic tandem walk, whereas in CISP, 65% (13/20) of patients had positive Romberg testing (n = NS) and 60% (12/20) had a pathologic tandem walk (p = NS). By definition, no patients with CISP had weakness on neurologic examination (0/28), but 75% (33/44) of patients with CISP-plus had mild weakness involving distal lower limb segments (limited to mild weakness of toe flexors or extensors; p < 0.001). Neurologic examination findings were similar except for mild distal weakness in CISP-plus.
Neurophysiologic Evaluation: NCS/EMG, SSEP, QST, and Autonomic Testing
All 72 patients had NCS/EMG performed at our EMG laboratory within a year of the time of diagnosis. By definition, all patients with CISP (28/28) had normal NCS/EMG in terms of the sensory syndrome (2 had separate, coexisting chronic L5 radiculopathy on EMG findings). Minimal NCS abnormalities were present in all CISP-plus cases (44/44), characterized by mildly prolonged F-wave latencies in 73% (32/44); reduced motor and sensory amplitudes in 57% (25/44) and 57% (25/44), respectively; mild slowing of motor conduction velocities in 41% (18/44); and mild slowing of sensory conduction velocities in 68% (30/44). None of the patients with CISP-plus met the EFNS/PNS electrophysiologic criteria for CIDP13 and in no cases were the electrophysiologic abnormalities significant enough to explain the severe clinical sensory symptoms and signs (especially sensory ataxia). All 72 patients had preserved or normal for age sural sensory nerve action potentials (5 patients with CISP-plus and 0 patients with CISP older than 60 years had absent sural potentials, which is normal for age in our laboratory). Needle EMG was normal in the majority (38/44) of patients with CISP-plus, with 6/44 patients demonstrating a mild motor polyradiculopathy with sparse fibrillation potentials confined to distal muscles (in feet more than hands) on needle examination that was interpreted as having minimal clinical relevance.
SSEPs were performed in 40 patients with CISP-plus and in 23 patients with pure CISP. Overall, 76% (48/63) of patients showed SSEP abnormality: 78% (31/40) of patients with CISP-plus and 74% (17/23) of patients with CISP had slowing of SSEP (p = NS). In median CISP-plus studies, 56% (18/32) were abnormal, and in tibial studies, 91% (30/33) were abnormal. In comparison, 50% (8/16) and 94% (15/16) of median and tibial CISP studies, respectively, were abnormal (p = NS). More specifically, in abnormal median studies, 61% (11/18) of patients with CISP-plus and 88% (7/8) of patients with CISP had prolonged N13 (cervical) latencies or prolonged N9-N13 interpeak latencies (segments that include cervical roots; figure 1B). In addition, 33% (6/18) of patients with CISP-plus and 13% (1/8) of patients with CISP had nonlocalizable median slowing. One CISP-plus median study showed mild prolongation of the N9 latency (clavicular response) in keeping with slowing of peripheral nerves. In the tibial studies, 27% (8/30) patients with CISP-plus and no patient with CISP had prolonged N22 (lumbar spine) potentials (segment containing lumbar roots) and 73% (22/30) of patients with CISP-plus and 100% (15/15) of patients with CISP had poorly localizable slowing.
QST was performed in 33/44 patients with CISP-plus and in 14/28 patients with CISP. In patients with CISP-plus, 88% (29/33) had abnormalities with large fiber modalities (reduced vibration in 64% [n = 21/33] and light touch in 54% [n = 18/33]), being somewhat more affected than small myelinated (reduced cooling in 48% [n = 16/33]) or unmyelinated fiber modalities (reduced heat pain in 24% [hypoalgesia, n = 8/33] and increased heat pain in 9% [hyperalgesia, n = 3/33]). Similarly, in patients with CISP, 93% (13/14) had abnormalities with large fiber modalities (reduced vibration in 86% [n = 12/14] and touch in 50% [n = 7/14]), being more affected than small myelinated (reduced cooling in 36% [n = 5/14]) or unmyelinated fiber modalities (reduced heat pain in 7% [hypoalgesia, n = 1/14] or increased heat pain in 14% [hyperalgesia, n = 2/14]). These results indicate that in both groups large myelinated sensory fibers are more involved than small myelinated or unmyelinated sensory fibers.
Autonomic reflex screen was performed in 29 patients with CISP-plus and 10 patients with pure CISP. The median composite autonomic scoring scale score for CISP-plus was 2 (range 0–7) and for CISP was 0 (range 0–4).20 This indicates mild autonomic involvement for both groups, similar to the minimal autonomic involvement seen in CIDP.21
Nerve Pathologic Findings
Nerve biopsies from 11 patients with CISP-plus were regraded by 2 authors (K.S. and P.J.B.D.). No additional CISP nerve biopsies than previously reported were performed.7 The biopsied CISP-plus nerves included 3 lumbar dorsal rootlet, 2 fascicular sciatic, and 6 distal cutaneous nerve biopsies (1 superficial radial and 5 sural). Electron microscopy was performed in 3 rootlets, 1 sciatic, 1 superficial radial, and 1 sural nerve. Teased fiber preparations were performed in all and showed increased rates of segmental demyelination highest in rootlet biopsies (mean 21%; range 9%–33%), next highest in sciatic biopsies (mean 15%; range 14%–16%), and lowest in distal cutaneous biopsies (mean 3%; range 0%–12%, figure 3). Onion-bulbs were seen on teased preparations from rootlet biopsies (figure 3, A and B).
Figure 3. Segmental Demyelination on Teased Fiber Preparations in Chronic Immune Sensory Polyradiculopathy (CISP)–Plus.
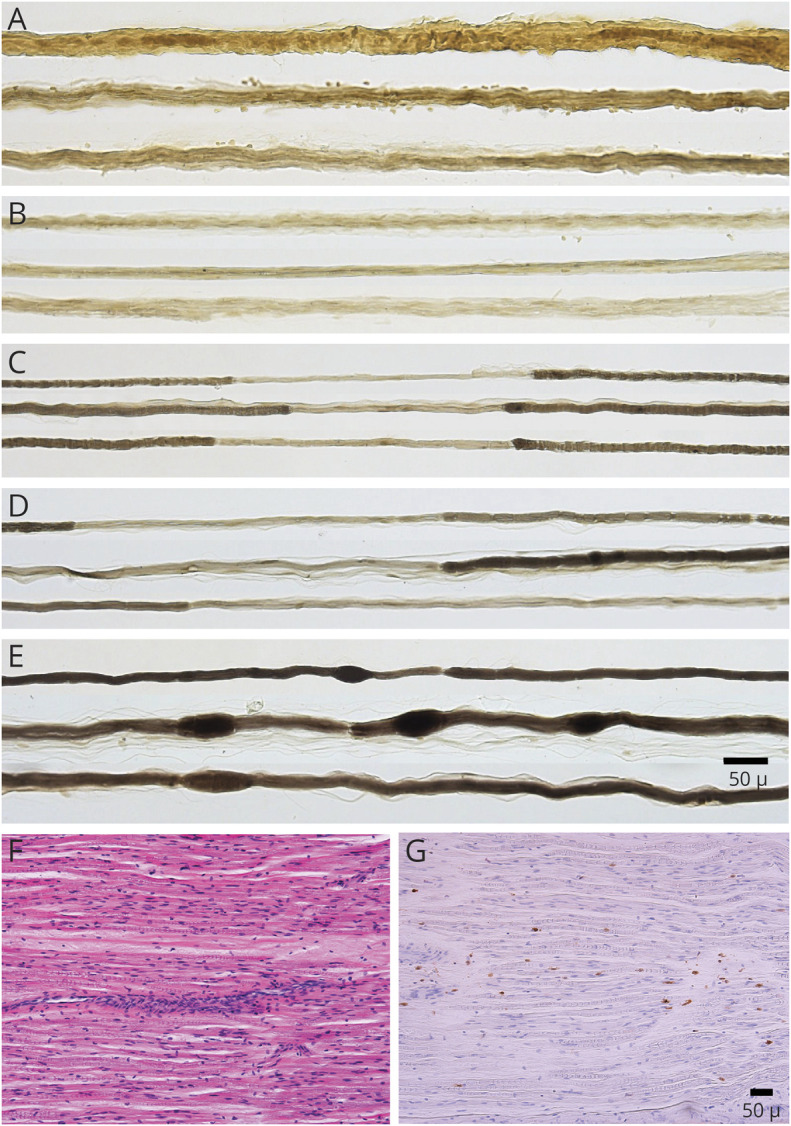
Teased myelinated nerve fiber preparations from patients with CISP-plus taken from proximal and distal nerves show chronic demyelination and remyelination at different stages and severities. (A, B) Dorsal rootlet biopsies show onion-bulb formations (thickened strands of multiple Schwann cell processes surrounding nerve fibers). (C) Fascicular sciatic biopsy and (D) sural biopsy show areas of ongoing demyelination and remyelination. (E) Sural biopsy demonstrates myelin reduplication (small tomaculae; thickened areas of myelin along the lengths of nerve fibers). (F, G) Serial longitudinal paraffin sections of a CISP-plus biopsy show abnormal amounts of scattered endoneurial inflammation on hematoxylin & eosin (F) and CD45 (G) preparations. The teased fiber changes show that in CISP-plus ongoing chronic demyelination and remyelination occurs at multiple nerve levels but are the most severe in the sensory roots.
Morphometric analysis was performed on all 11 CISP-plus biopsies and the overall density of myelinated fibers was normal or mildly reduced (figures 4 and 5). The main pathologic finding was reduced density of large myelinated nerve fibers, more pronounced in proximal biopsies and less evident in distal biopsies, confirmed by fiber size histograms. Hence, all rootlet biopsies had unimodal small fiber peaks (figure 4, B and D). Of 2 sciatic biopsies, one had a unimodal small fiber peak (figure 5B), whereas the other had an abnormal bimodal distribution with the large fiber peak occurring at a smaller fiber size than normal. In the 6 distal cutaneous nerve biopsies, 2 biopsies (1 superficial radial and 1 sural) had unimodal small fiber peaks (figure 5, C and D), another had the large fiber peak occurring at smaller fiber size than normal, and the other 3 had normal fiber size distributions.
Figure 4. Loss of Large Myelinated Fibers and Onion-Bulbs in Dorsal Lumbar Rootlets of Chronic Immune Sensory Polyradiculopathy (CISP)–Plus.

Dorsal lumbar rootlet biopsies (A.a, B.a, C.a, D.a) and their corresponding myelinated fiber histograms (A.b, B.b, C.b, D.b) from a normal control (A) and from 3 patients with CISP-plus (B–D). The sections are methylene blue–stained epoxy preparations. The control biopsy (A) and histogram shows a normal bimodal size distribution. The CISP-plus biopsies are arranged in order from least (B) to most severely (D) affected. Note that in all 3 CISP-plus rootlet biopsies, the density of myelinated fibers is preserved but the size distribution is altered with loss of large myelinated fibers and a relative increase in the number of small myelinated fibers. These findings are confirmed in the histograms with loss of large myelinated fiber peaks (at 8.5 μm on the control) and larger amplitudes of the small myelinated fiber peak (at 3.0 μm). Note the frequent onion-bulbs (arrowheads) as well as the demyelinated axons surrounded by onion-bulbs (arrows) in (C) and (D). The loss of large myelinated sensory fibers demonstrated here correlates well with the clinical syndrome of gait ataxia. The severe pathologic findings from dorsal roots in CISP-plus are similar to those found in CISP rootlet biopsies. Morphometric analysis showed the mean myelinated fiber density (MF/mm2) of 3 dorsal rootlets in CISP-plus was 16,556/mm2 (SD ± 5,917/mm2) and was similar to the densities of CISP and controls dorsal root biopsies. Individual densities were as follows: (B) 21,218, (C) 18,552, and (D) 9,899/mm2. The 3 normal dorsal rootlet control biopsies had a mean MF density of 16,700/mm2 (SD +/1,200/mm2) and 3 CISP dorsal rootlet biopsies had a mean MF density of 14,300/mm2 (SD ± 1,850/mm2).
Figure 5. Loss of Large Myelinated Fibers and Onion-Bulbs in Limb Nerves of Chronic Immune Sensory Polyradiculopathy (CISP)–Plus.

Nerve biopsies (A.a, B.a, C.a, D.a) taken from proximal and distal limb nerves from a normal control (A) and from 3 patients with CISP-plus (B–D) and their corresponding myelinated fiber histograms (A.b, B.b, C.b, D.b). The biopsies are methylene blue–stained epoxy sections. All CISP-plus biopsies have reduced numbers of large myelinated fibers and a unimodal size distribution with a small fiber peak at about 3.5 μm on the histograms. (A) Normal control sural nerve shows normal myelinated fiber density (MF) and bimodal size distribution (with peaks at 3.5 μm and 9 μm). CISP-plus biopsies of sural (B) and superficial radial nerves (C) show increased regenerating nerve clusters in (C) (arrowheads). (D) CISP-plus fascicular sciatic nerve biopsy shows occasional onion-bulb formations (arrows). The CISP-plus nerve biopsies demonstrate that large myelinated fiber loss and onion-bulb formation are not restricted to the sensory root level (as in CISP) but extend to more distal nerves to a lesser degree. Morphologic analysis for 2 fascicular sciatic biopsies showed the mean MF was 6,656/mm2 (6,742 and 6,570/mm2), for the superficial radial biopsy the MF was 9,153/mm2, and for 5 sural nerve biopsies the mean MF was 5,724/mm2 (SD ± 1,864/mm2). Individual sural densities were 4,107, 4,413, 5,114, 6,280, and 8,706/mm2; 4 of the sural nerves were mildly reduced (4,000–6,500/mm2) and 1 was normal (6,500–13,000/mm2) per our laboratory MF density values.
Onion-bulb formations were found frequently in proximal biopsies. Two of the 3 lumbar rootlet biopsies had dense large onion-bulb formations (figure 4, C and D; and figure 6, A and B); 1 sciatic biopsy had frequent moderate-sized onion-bulb formations (figure 5D; and figure 6, C and D), while no distal cutaneous biopsies had frequent onion-bulb formations (although rare small onion-bulbs were seen) (figure 6, G and H). Some fibers had myelin that was too thin for the axonal size, especially in proximal biopsies, suggesting remyelination. Abnormal degrees of scattered endoneurial inflammation were seen in all but one sural biopsy (figure 3, F and G). The CISP-plus biopsies were compared to 3 previously published lumbar rootlet CISP biopsies and 3 postmortem lumbar rootlet controls.7 The rootlet biopsies from CISP-plus and CISP were very similar, with loss of large fibers, unimodal small myelinated fiber size distribution, and onion-bulb formations, whereas the controls all had normal bimodal distribution (small and large myelinated fiber peaks) and no onion-bulb formations (figure 4A).
Figure 6. Onion-Bulb Formation on Electron Microscopy in Chronic Immune Sensory Polyradiculopathy (CISP)–Plus.

Electron micrographs taken from different nerves of patients with chronic immune sensory polyradiculopathy plus (CISP-plus) show evidence of chronic demyelination and abortive repair. The left column is at low magnification and the right column is at high magnification. (A, B) Dorsal lumbar rootlet biopsy shows frequent large onion-bulbs. (C, D) Fascicular sciatic nerve biopsy shows frequent but smaller onion-bulbs than were found in the dorsal roots. (E, F) Superficial radial nerve biopsy shows frequent regenerating nerve clusters. (G, H) Sural nerve biopsy shows rare small onion-bulbs. The chronic demyelinating changes occur at all levels but are most frequent and severe in proximal nerves (especially at the sensory root level).
CSF and MRI Radiology Evaluations
CSF studies were performed in all 44 patients with CISP-plus and in 26 patients with CISP. The median CSF protein was 67 g/dL (range 19–455) in CISP-plus and 71.5 g/dL (range 31–161) in CISP (p = NS). Overall, 95% (42/44) of patients with CISP-plus and 92% (24/26) of patients with CISP had elevated CSF proteins. Six patients (5 CISP-plus and 1 CISP) had a mild lymphocytic pleocytosis (median 12 cells/mm3; range 4–44).
In CISP-plus, lumbar spine MRI was available in 36 patients, 33 with contrast. The most common abnormality was nerve root enhancement (axial images) in 73% (24/33) followed by thickening or clumping of nerve roots in 55% (20/36) and cauda equina enhancement (sagittal images) in 24% (8/33). Among patients with CISP, 21 had imaging available, 14 with contrast. The most common abnormality was thickening or clumping of nerve roots (axial images) in 57% (12/21), followed by nerve root enhancement (axial images) in 50% (7/14), and cauda equina enhancement (sagittal images) in 42% (6/14). In the CISP-plus cohort, 10 patients had MRI of lumbar (n = 8) or brachial plexus (n = 2). Abnormal plexus contrast enhancement was seen in 90% (9/10), with thickening of femoral or sciatic nerves, and of the divisions of the brachial or lumbosacral plexus (figure 7).
Figure 7. MRI Abnormalities in Chronic Immune Sensory Polyradiculopathy (CISP)–Plus.
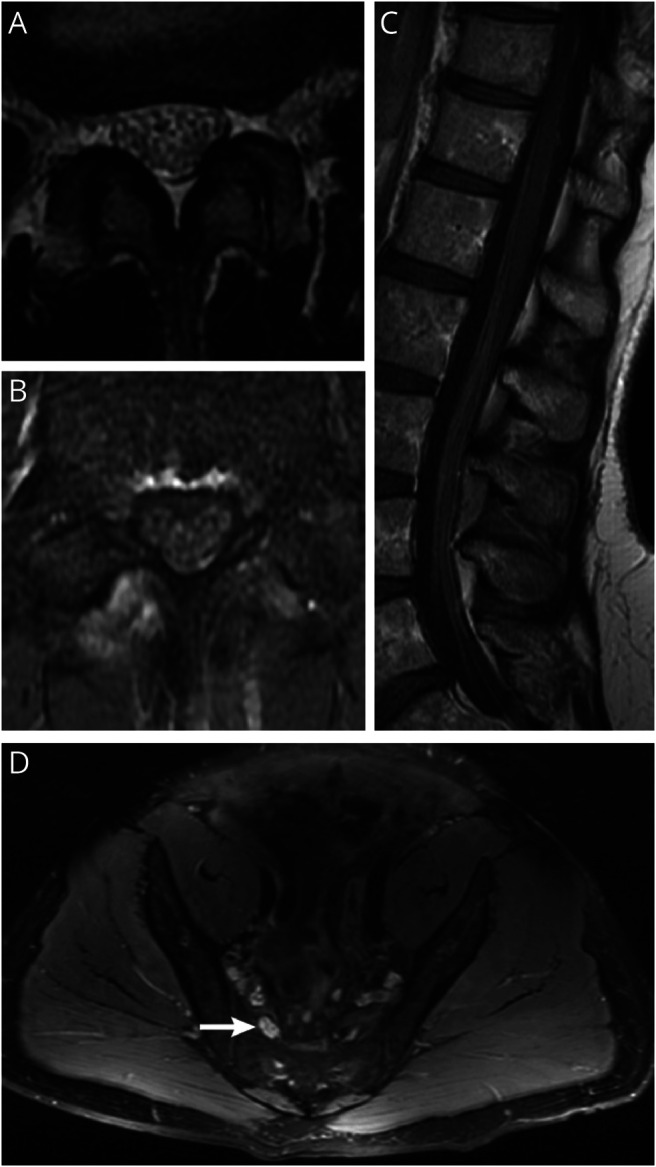
(A) T2 axial lumbar MRI from a patient with CISP-plus shows clumping and thickening of lumbar nerve roots. (B) The same patient and lumbar level as in (A) with T1 postcontrast imaging shows contrast enhancement of the roots. (C) Sagittal T1 postcontrast imaging of the cauda equina from a patient with CISP-plus shows enlargement and enhancement of the lumbar roots. (D) T2 axial MRI of the lumbosacral plexus from a patient with CISP-plus shows enlarged nerves with increased T2 signal of the lumbosacral plexus on the right (arrow) compared to the left.
Central Demyelination and Other Associations
Six of our patients had concurrent central demyelinating disease (diagnosed as coexisting multiple sclerosis, 3 with CISP-plus and 3 with CISP) with 3 previously reported.22 One patient with CISP-plus had longstanding HIV infection and was being treated with antiretroviral therapy. Two patients (1 of 4 CISP-plus and 1 of 6 CISP tested) were positive for contactin-1/CASPR1 antibody confirmed by cell-based assay and immunofluorescence.
Immunotherapy and Clinical Outcomes
Forty patients with CISP-plus and 17 patients with CISP were treated with immunotherapy. The initial immunotherapy was IV immunoglobulins (IVIg) in 80% (32/40) of CISP-plus and 82% (14/17) of CISP cases. IV methylprednisolone (IVMP) was used initially in 15% (6/40) of CISP-plus and 18% (3/17) in CISP cases. Plasmapheresis (PLEX) and rituximab were each used initially in 1 CISP-plus case (3%, 1/40). A second immunotherapy was used in 11 CISP-plus cases (IVMP in 8; IVIg, PLEX, and azathioprine, each in 1) and in 2 CISP cases (PLEX and IVIg, each in 1). Two patients with CISP-plus needed a third immunotherapy: rituximab (1) or PLEX (1).
Treatment response and outcome data were available in 32 CISP-plus and 14 CISP cases and showed clear improvement for the group as a whole. At baseline, all patients with CISP-plus had abnormal neurologic examinations: 87.5% (28/32) had gait ataxia and 19 used ambulatory aids (11 canes, 5 walkers, and 3 wheelchairs). At baseline, all patients with CISP had abnormal neurologic examinations and 10 used ambulatory aids (6 canes and 4 walker/wheelchair combination). At posttreatment follow-up, 84% (27/32) of patients with CISP-plus and 93% (13/14) of patients with CISP (p = NS) showed marked improvement of gait ataxia and had reduced numbness and paresthesia. Three of 32 patients with CISP-plus were stable and 2/32 worsened. One patient with CISP was stable and none worsened. Neurologic functional outcomes (assessed at final neurologic visit) including needing assistance in daily tasks were markedly better after treatment (p < 0.001, odds ratio [OR] 40.25, confidence interval [CI] 6.5–249.1). Overall, 54% (25/46) and 59% (19/32) of patients with CISP-plus and 43% (6/14) of patients with CISP returned to normal neurologic examinations. Nine patients with CISP-plus (cane 7, wheelchair 2) and 6 patients with CISP (cane 5, scooter 1) still required ambulatory aids; the need for ambulatory aid was unknown in the rest. Overall for patients with CISP and patients with CISP-plus combined, use of ambulatory aids went from 29 at baseline to 15 posttreatment, with OR 30 (p < 0.001, CI 6.02–156.08). Two nonrelated deaths occurred from cancer (1) and liver failure (1) in the CISP-plus cohort.
Discussion
Since the original description of CISP, we have become aware of patients presenting with a clinical syndrome similar to CISP but in whom the disease extends beyond dorsal roots to also involve motor and postganglionic sensory nerve fibers, resulting in mild distal weakness and mild abnormalities on NCS/EMG (figure 1A). Knowing how to best classify such patients has been problematic. They cannot correctly be classified as CISP because their disease is not isolated to the dorsal root and many do not exclusively have sensory symptoms. However, they also do not meet the diagnostic criteria for classical CIDP or sensory CIDP, which require demyelinating abnormalities on NCS.
In our study, we introduce the concept and terminology of CISP-plus by identifying patients with nonpure, sensory predominant, inflammatory polyradiculopathy and comparing them to patients with pure CISP. Our intent is to show that patients with CISP-plus and patients with CISP are alike. We found striking similarities in the clinical, SSEP, QST, CSF, MRI, and pathologic features as well as in the treatment response between the CISP-plus and CISP cohorts, supporting our belief that they should be classified together. Both groups present with large fiber sensory loss, imbalance, frequent falls, and little pain. Use of gait aids is necessary in almost half of the patients in both groups. On examination, both groups have reduced light touch, vibration and proprioception, hyporeflexia or areflexia, and gait ataxia. One important difference (which explains why they have been thought of as different conditions) is the presence of weakness, which is distal and lower limb predominant (toe flexor and extensor) in CISP-plus. This degree of weakness is mild and nondisabling, and its clinical relevance in CISP-plus is small; severe gait impairment and profound sensory loss are the main sources of disability in both CISP-plus and CISP.
Neurophysiologic tests are similar between groups. SSEP is a useful diagnostic tool and should be considered in all suspected CISP-plus or CISP cases.7,12,23 Often SSEP slowing is nonlocalizable (as evidenced by delayed cortical responses) but this confirms that pathology exists somewhere along the somatosensory pathways. The tibial studies are often nonlocalizable due to the frequent absence of the lumbar spine potential (N22) (location of sensory roots). However, in one-quarter of CISP-plus SSEP studies, the tibial N22 response was preserved and prolonged, confirming lumbar root slowing. Furthermore, for many, the median (upper limb) SSEP in both CISP-plus and CISP localizes the slowing to the cervical sensory root level (prolonged N13 or N9-N13 interpeak latencies) indicative of focal root demyelination (figure 1B). QST results mirror the clinical syndrome and provide evidence that large fiber abnormalities (reduced light touch and vibration) predominate for both CISP-plus and CISP cohorts. As expected, symptoms referable to small myelinated and unmyelinated fibers (neuropathic pain and autonomic dysfunction) and corresponding abnormalities on QST (reduced cooling and heat pain sensation) and autonomic testing are less common. CSF proteins are at least moderately elevated in the majority of patients with CISP-plus and patients with CISP and support an inflammatory root process. MRI of the lumbar spine shows nerve root thickening, clumping, and enhancement in more than half of patients with CISP-plus and patients with CISP (figure 7). In addition, there is radiographic evidence of peripheral nerve involvement beyond the root level in patients with CISP-plus, with most plexus MRI studies being abnormal. The main neurophysiologic difference between CISP-plus and CISP is found on NCS/EMG, because CISP by definition excludes NCS/EMG abnormalities whereas CISP-plus allows them. The NCS abnormalities in CISP-plus are mild, do not meet electrophysiologic criteria for CIDP in any cases, and incorrectly imply that the pathology is axonal predominant. The majority of needle EMG studies in CISP-plus are normal and only rarely show a mild motor polyradiculopathy with distal denervation.
The similarities in nerve pathology observed in the CISP-plus and CISP cohorts are also noteworthy and dorsal lumbar rootlet biopsies are essentially indistinguishable. Both show loss of large myelinated nerve fibers, a resultant unimodal small myelinated fiber size distribution (figure 4), onion-bulb formations (figures 4 and 6), increased rates of segmental demyelination (figure 3), and scattered endoneurial inflammation (figure 3, F and G). Nerve biopsies distal to the dorsal root were not performed in patients with CISP at our institution because the disease is restricted to the dorsal root. In contrast, targeted fascicular sciatic and distal cutaneous nerve biopsies were performed in CISP-plus and showed similar findings as were found in the rootlet biopsies but of lesser severity (figures 3, 5, and 6). These are the same pathologic findings typically seen in classical CIDP.1,2
Response to immunotherapy in CISP-plus and CISP was also similar with marked improvement of neurologic deficits, especially the sensory ataxia. Approximately half of patients with CISP-plus and patients with CISP returned to normal neurologic examinations including resolution of weakness (in CISP-plus) with treatment (25/46 overall). For all patients, the most widely used immunotherapies were IVIg and IVMP. Many patients with falls and severe imbalance for years improved with immunotherapy, suggesting that treatment is effective even after many years. The likely explanation is that ongoing inflammatory demyelination is amenable to delayed treatment whereas axonal degeneration is not.
Recently, some authors have described chronic immune sensorimotor polyradiculopathy (CISMP), a syndrome that shares some features with CISP, including having the pathologic process localized to proximal nerve segments, lack of demyelination on NCS, thickened nerve roots on MRI, and elevated CSF protein.24,25 The question could be asked whether patients with CISP-plus should be classified as CISMP. For the cases described here, CISP-plus is a more correct term. Most cases of CISMP have a somewhat different clinical presentation—usually in CISMP there is prominent weakness (especially lower limbs) and less frequent sensory ataxia, whereas weakness is minimal in CISP-plus and sensory ataxia is almost universally present. Consequently, CISP-plus cases should not be classified as CISMP. However, we acknowledge that these conditions may lie on a continuum and that borderline cases between the 2 entities exist, making classification difficult. CISP-plus is a more suitable term to describe our patients as it highlights their sensory and dorsal root predominant presentation and connects them to CISP.
There is a strong justification to classify CISP-plus and CISP together as sensory variants of CIDP but maintaining the distinction from CISMP and typical sensory CIDP. The evidence is overwhelming that these are forms of inflammatory demyelinating neuropathy primarily localized to dorsal nerve roots. It is likely that CISP-plus and CISP are part of one disease that in some cases is restricted to the sensory nerve root (CISP) while in others extends beyond to involve motor and sensory fibers to a lesser degree (CISP-plus). Furthermore, CISP-plus may be even more important to recognize than CISP, as we found more cases of CISP-plus than CISP during the same time period (although we acknowledge that this is not an epidemiologic study).
CISP-plus and CISP are both difficult to recognize and diagnose. The diagnosis of CISP is challenging because despite patients having profound sensory deficits, NCS/EMG are normal, leading to some patients being labeled as having a conversion disorder.7 Similarly, CISP-plus is difficult to diagnose because patients’ NCS/EMG abnormalities are mild and not severe enough to explain their clinical deficits, they are not in the demyelinating range, and they do not meet the EFNS/PNS criteria for CIDP.13 In fact, the NCS/EMG findings likely falsely point physicians away from considering an inflammatory demyelinating disorder and towards an axonal pathology. Consequently, the possibility of CIDP is probably not considered by many physicians evaluating patients with CISP-plus, which may explain the trend for more delay in diagnosis of CISP-plus vs CISP (figure 2). It is important for physicians, especially neurologists, to be aware of both conditions so they can recognize and treat them promptly. Both are responsive to immunotherapy, especially to IVIg, even years after disease onset, and with treatment neurologic examinations often return to normal.
In our study, it was important to include patients with CISP-plus with coexisting infectious or immune-mediated diseases as their clinical features mimicked the rest of the cohort and were not explained by their concomitant disorders. One patient with CISP-plus with longstanding HIV infection and 6 with concurrent central demyelinating disease (3 CISP-plus and 3 CISP) had improvement of ataxia with IVIg. Two patients (1 with CISP and 1 with CISP-plus) were positive for contactin-1 antibodies, which are known to be associated with inflammatory sensory-predominant neuropathies.5,6 Finding co-occurring autoimmune diseases supports our hypothesis that CISP-plus and CISP are immune-mediated neuropathies.
The limitations of our study include being retrospective and not population-based, therefore the frequency of CISP-plus vs CISP cases may be skewed. Because the study was retrospective, patients were evaluated in a nonstandardized manner, including the choice of immunotherapy and the treatment follow-up. However, many patients received comprehensive and uniform evaluations because they were personally seen by 2 of the authors (P.J.B.D. or C.J.K.).
Herein we introduce the syndrome of CISP-plus. CISP-plus is similar to CISP. Both present with sensory loss and sensory ataxia; they differ in that CISP-plus is not isolated to the dorsal nerve root and slight distal weakness and mild NCS/EMG abnormalities often exist. We propose that CISP-plus and CISP are 2 similar subtypes of the same disorder and that the diagnostic criteria used to diagnose CISP cases have incorrectly excluded CISP-plus cases. CISP-plus and CISP are both atypical subtypes of CIDP and are responsive to immunotherapy, and CISP-plus should be included in the spectrum of sensory predominant CIDP.
Glossary
- CI
confidence interval
- CIDP
chronic inflammatory demyelinating polyradiculoneuropathy
- CISMP
chronic immune sensorimotor polyradiculopathy
- CISP
chronic immune sensory polyradiculopathy
- EFNS
European Federation of Neurological Societies
- IVIg
IV immunoglobulin
- IVMP
IV methylprednisolone
- MF
myelinated fibers
- NCS
nerve conduction studies
- NS
nonsignificant
- OR
odds ratio
- PLEX
plasmapheresis
- PNS
Peripheral Nerve Society
- QST
quantitative sensory testing
- SSEP
somatosensory evoked potential
Appendix. Authors

Footnotes
Podcast: NPub.org/isbulv
Study Funding
No targeted funding reported.
Disclosure
S. Shelly, K. Shouman, P. Paul, J. Engelstad, K.K. Amrami, and R.J. Spinner report no disclosures. D. Dubey reports receiving research support from the Center of Multiple Sclerosis and Autoimmune Neurology, Translational Research Innovation and Test Development Office, and Grifols Pharmaceuticals; has consulted for UCB Pharmaceuticals (all compensation for consulting activities is paid directly to Mayo Clinic); has a patent pending for KLHL11 as marker of neurologic autoimmunity; and is on the editorial board of Journal of Clinical Medicine. R. Vazquez Do Campo and P.J. Dyck report no disclosures. C.J. Klein reports he is on the therapeutic CMTA advisory board. P.J.B. Dyck reports no disclosures. Go to Neurology.org/Nhttps://n.neurology.org/lookup/doi/10.1212/WNL.0000000000011792 for full disclosures.
References
- 1.Dyck PJ, Lais AC, Ohta M, Bastron JA, Okazaki H, Groover RV. Chronic inflammatory polyradiculoneuropathy. Mayo Clin Proc 1975;50:621–637. [PubMed] [Google Scholar]
- 2.Stino AM, Naddaf E, Dyck PJ, Dyck PJB. Chronic inflammatory demyelinating polyradiculoneuropathy: diagnostic pitfalls and treatment approach. Muscle Nerve 2021;63:157–169. [DOI] [PubMed] [Google Scholar]
- 3.Oh SJ, Joy JL, Kuruoglu R. Chronic sensory demyelinating neuropathy: chronic inflammatory demyelinating polyneuropathy presenting as a pure sensory neuropathy. J Neurol Neurosurg Psychiatry 1992;55:677–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ayrignac X, Viala K, Koutlidis RM, et al. . Sensory chronic inflammatory demyelinating polyneuropathy: an under-recognized entity? Muscle Nerve 2013;48:727–732. [DOI] [PubMed] [Google Scholar]
- 5.Querol L, Nogales-Gadea G, Rojas-Garcia R, et al. . Antibodies to contactin-1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol 2013;73:370–380. [DOI] [PubMed] [Google Scholar]
- 6.Dubey D, Honorat JA, Shelly S, et al. . Contactin-1 autoimmunity: serologic, neurologic, and pathologic correlates. Neurol Neuroimmunol Neuroinflamm 2020;7:e771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinnreich M, Klein CJ, Daube JR, Engelstad J, Spinner RJ, Dyck PJB. Chronic immune sensory polyradiculopathy: a possibly treatable sensory ataxia. Neurology 2004;63:1662–1669. [DOI] [PubMed] [Google Scholar]
- 8.Vazquez do Campo R, Dyck PJB, Boon AJ, Tracy JA. Acute immune sensory polyradiculopathy: a new variant of Guillain‐Barré syndrome [letter to the editor]. Muscle Nerve 2021;63:E28–E30. [DOI] [PubMed] [Google Scholar]
- 9.Trip SA, Saifee T, Honan W, et al. . Chronic immune sensory polyradiculopathy with cranial and peripheral nerve involvement. J Neurol 2012;259:1238–1240. [DOI] [PubMed] [Google Scholar]
- 10.Rajabally YA, Uncini A. Outcome and its predictors in Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry 2012;83:711–718. [DOI] [PubMed] [Google Scholar]
- 11.Zuberbuhler P, Minarro D, Binaghi D, Marchesoni C, Pardal A, Reisin R. Chronic inflammatory sensory polyradiculopathy [in Paris]. Medicina 2013;73:259–262. [PubMed] [Google Scholar]
- 12.Salhi H, Corcia P, Remer S, Praline J. Somatosensory evoked potentials in chronic inflammatory demyelinating polyradiculoneuropathy. J Clin Neurophysiol 2014;31:241–245. [DOI] [PubMed] [Google Scholar]
- 13.Joint Task Force of the EFNS and the PNS. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of neurological Societies and the Peripheral Nerve Society: first revision. J Peripher Nerv Syst 2010;15:1–9. [DOI] [PubMed] [Google Scholar]
- 14.Daube JR, Rubin DI. Needle electromyography. Muscle Nerve 2009;39:244–270. [DOI] [PubMed] [Google Scholar]
- 15.Dyck PJ, Zimmerman I, Gillen DA, Johnson D, Karnes JL, O'Brien PC. Cool, warm, and heat-pain detection thresholds: testing methods and inferences about anatomic distribution of receptors. Neurology 1993;43:1500–1508. [DOI] [PubMed] [Google Scholar]
- 16.Low PA, Tomalia VA, Park KJ. Autonomic function tests: some clinical applications. J Clin Neurol 2013;9:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dyck PJ, Dyck PJB, Engelstad J. Pathologic alteration in nerve. In: Dyck PJ, Thomas PK, eds. Peripheral Neuropathy, 4th ed. Elsevier; 2005:733–829. [Google Scholar]
- 18.Xu M, Pinto M, Sun C, et al. . Expanded teased nerve fibre pathological conditions in disease association. J Neurol Neurosurg Psychiatry 2019;90:138–140. [DOI] [PubMed] [Google Scholar]
- 19.Zimmerman IR, Karnes JL, O'Brien PC, Dyck PJ. Imaging system for nerve and fiber tract morphometry: components, approaches, performance, and results. J Neuropathol Exp Neurol 1980;39:409–419. [DOI] [PubMed] [Google Scholar]
- 20.Low PA. Composite autonomic scoring scale for laboratory quantification of generalized autonomic failure. Mayo Clin Proc 1993;68:748–752. [DOI] [PubMed] [Google Scholar]
- 21.Figueroa JJ, Dyck PJB, Laughlin RS, et al. . Autonomic dysfunction in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology 2012;78:702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suanprasert N, Taylor BV, Klein CJ, et al. . Polyneuropathies and chronic inflammatory demyelinating polyradiculoneuropathy in multiple sclerosis. Mult Scler Relat Disord 2019;30:284–290. [DOI] [PubMed] [Google Scholar]
- 23.Clerici AM, Nobile-Orazio E, Mauri M, Squellati FS, Bono GG. Utility of somatosensory evoked potentials in the assessment of response to IVIg in a long-lasting case of chronic immune sensory polyradiculopathy. BMC Neurol 2017;17:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thammongkolchai T, Suhaib O, Termsarasab P, Li Y, Katirji B. Chronic immune sensorimotor polyradiculopathy: report of a case series. Muscle Nerve 2019;59:658–664. [DOI] [PubMed] [Google Scholar]
- 25.Khadilkar S, Patel B, Mansukhani KA, Jaggi S. Two cases of chronic immune sensorimotor polyradiculopathy: expanding the spectrum of chronic immune polyradiculopathies. Muscle Nerve 2017;55:135–137. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
De-identified data are available upon reasonable request.