

Abstract
We report a systematic analysis of the DNA methylation variability in 1,595 samples of normal cell subpopulations and 14 tumor subtypes spanning the entire human B-cell lineage. Differential methylation among tumor entities relates to differences in cellular origin and to de novo epigenetic alterations, which allowed us to build an accurate machine learning-based diagnostic algorithm. We identify extensive patient-specific methylation variability in silenced chromatin associated with the proliferative history of normal and neoplastic B cells. Mitotic activity generally leaves both hyper- and hypomethylation imprints, but some B-cell neoplasms preferentially gain or lose DNA methylation. Subsequently, we construct a DNA methylation-based mitotic clock called epiCMIT, whose lapse magnitude represents a strong independent prognostic variable in B-cell tumors and is associated with particular driver genetic alterations. Our findings reveal DNA methylation as a holistic tracer of B-cell tumor developmental history, with implications in the differential diagnosis and prediction of clinical outcome.
Introduction
The process of neoplastic transformation implies a dramatic alteration of cellular identity 1. However, cancer cells partially maintain molecular imprints of the cellular lineage and maturation stage from which they originate 2. B-cell neoplasms are a paradigmatic model of this model, as the maturation stage of different B-cell neoplasms is the main principle behind the World Health Organization classification of these tumors 3. Over the last years, multiple studies analyzed the DNA methylome, a bona fide epigenetic mark related to cellular identity and gene regulation 1,4 during the entire B-cell maturation program 5 and in various B-cell neoplasms spanning the whole maturation spectrum. These include B-cell acute lymphoblastic leukemia (ALL) 6,7 derived from precursor B cells, mantle cell lymphoma (MCL) 8,9 and chronic lymphocytic leukemia 10,11 (CLL) derived from pre- and post-germinal center mature B cells, diffuse large B-cell lymphoma (DLBCL) 12 derived from germinal center B cells, and multiple myeloma (MM) 13,14 derived from terminally-differentiated plasma cells. These studies have revealed a dynamic DNA methylome during B-cell maturation as well as novel insights into the cellular origin, pathogenic mechanisms and clinical behavior of B-cell neoplasms, as reviewed in 15. However, a global analysis of the entire normal cell differentiation program and derived neoplasms is neither available for B cells nor for any other human cell lineage. Thus, we herein exploit both previously generated DNA methylation datasets as well as newly generated data to systematically decipher the sources of DNA methylation variability across B-cell neoplasms. This comprehensive approach using over 2,000 samples including training and validation series indicates that the human DNA methylome is more dynamic than previously appreciated 5,11,16 and reveals previously hidden biological insights and clinical associations. In particular, de novo disease-specific hypomethylation in active regulatory regions is associated with differential transcription factor binding and targets genes important for disease-specific pathogenesis. From the clinical perspective, we define a set of epigenetic biomarkers that can accurately classify B-cell neoplasms requiring differential clinical management and construct a DNA methylation-based mitotic clock, called epiCMIT, as a personalized predictor of clinical behavior within each B-cell neoplasm.
Results
Initial data processing and global DNA methylation dynamics in normal and neoplastic B cells
We analyzed previously published DNA methylation profiles of samples from normal and neoplastic B cells spanning the entire B-cell differentiation spectrum, all generated with the 450k microarray platform from Illumina. These included 10 normal B-cell subpopulations 5 as well as the main five categories of B-cell neoplasms, i.e. ALL 6,7, MCL 8, CLL 10,17, DLBCL (own unpublished series) and MM 13 (Fig. 1a and Supplementary Table 1). Following the guidelines of the TCGA Consortium (https://www.cancer.gov/about-nci/organization/ccg/blog/2018/bcr-tips), we selected samples containing a tumor-cell content greater than 60%. The validity of this percentage was experimentally confirmed analyzing methylation profiles of sorted and unsorted tumor cells from MCL and CLL samples (Extended Data Fig. 1a). Tumor cell content was estimated by flow cytometry 5,8,10,13,17, genetic data18 and/or lineage-specific DNA methylation patterns (Supplementary Table 2), and was highly concordant (Extended Data Fig. 1b). However, MM samples showed that DNA methylation-based estimation of tumor cell content was far lower than that estimated by flow cytometry (Extended Data Fig. 1c, d), as expected due their loss of B-cell identity 13. Interestingly, some DLBCL samples also showed a similar effect (Extended Data Fig. 1c, d), and therefore in MM and DLBCL, tumor cell content was estimated by flow cytometry and genetic data, respectively. After all filtering criteria (Methods), we generated a curated data matrix containing 1,595 high quality samples (Fig. 1a and Supplementary Table 1) with DNA methylation values for 437,182 CpGs, which was used in all downstream analyses.
Fig. 1 |. Experimental design and characterization of stably methylated regions.
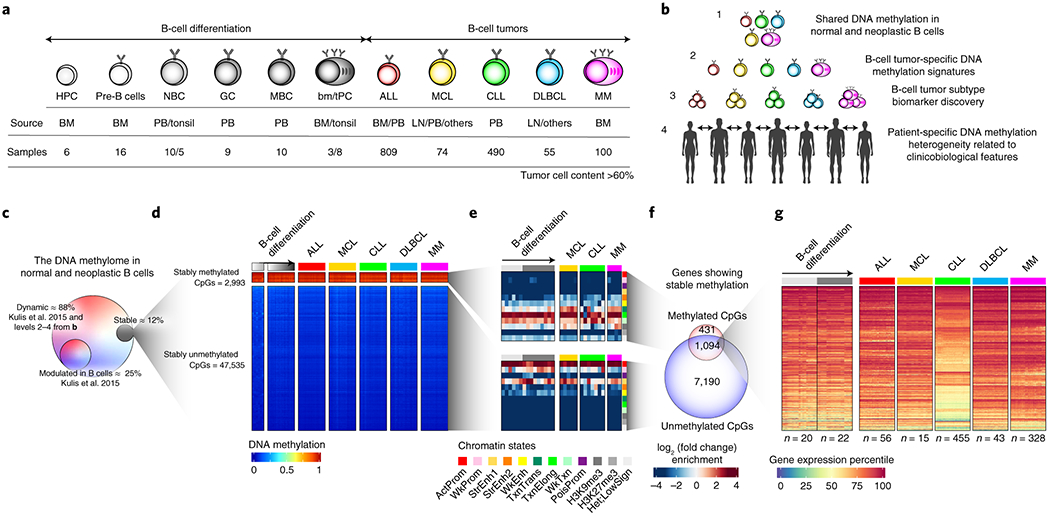
a, Experimental design, including normal B cell subpopulations, B cell tumors under study, source of the samples and number of patient samples included in the study with tumor cell content greater than 60%. HPC, hematopoietic precursor cells; pre-B, precursor B-cell and immature B cells; NBC, naïve B cells; GC, germinal center B cells; MBC, memory B cells; tPC, tonsillar plasma cells; bmPC, bone-marrow plasma cells; ALL, acute lymphoblastic leukemia; MCL, mantle cell lymphoma; CLL, chronic lymphocytic leukemia; DLBCL, Diffuse large B cell lymphoma; MM, multiple myeloma; BM, bone marrow; PB, peripheral blood; LN, lymph node.
b, Different levels of DNA methylation variability addressed in the study.
c, Percentage of CpGs whose methylation is stable in normal and neoplastic B cells, or modulated in normal B cells. Percentages are calculated over the total number of CpGs analyzed.
d, Heatmaps showing stably methylated CpGs (top) and stably demethylated CpGs (bottom) in normal and neoplastic B cell.
e, Chromatin state enrichments for stably un/methylated CpGs in normal and neoplastic B cells. All CpGs analyzed were used as background. ActProm, Active promoter; WkProm, Weak promoter; StrEnh1, Strong enhancer 1 (promoter-related); StrEnh2, Strong enhancer 2; WkEnh, Weak enhancer; TxnTrans, Transcription transition; TxnElong, Transcription elongation; WkTxn, Weak transcription; PoisProm, Poised promoter; H3K27me3, Polycomb-repressed region; H3K9me3, H3K9me3 heterochromatin; Het;LowSign, Het;LowSign heterochtomatin.
f, Overlap between the target genes of the stably methylated and unmethylated CpGs.
g, Gene expression percentiles in normal and neoplastic B cells of genes showing stable hyper- and hypomethylation.
This comprehensive dataset was used to step-wise dissect the DNA methylation variability of normal and neoplastic B cells at different levels, including cancer-specific, tumor entity-specific, tumor subtype-specific and individual-specific variability (Fig. 1b). Out of all the studied CpGs, only 12% show stable DNA methylation levels in normal and neoplastic B cells, and target expressed genes (Fig. 1c-g, Extended Data Fig. 1e-h, and Supplementary Table 3), indicating that the great majority of the DNA methylome (88%) is labile during normal B-cell development and neoplastic transformation. We could not identify any de novo epigenetic signature shared by all B-cell tumors. Therefore, the observed DNA methylation variability was related to differences among B-cell tumor entities and subtypes as well as patient-specific variability.
Disease-specific hypomethylation targeting regulatory regions is associated with transcription factor bindings and differential gene expression
An unsupervised principal component analysis showed that different B-cell neoplasms cluster separately (Fig. 2a and Extended Data Fig. 2a), with neoplasms grouped according to the maturation stage of their cellular origin, i.e. ALL together with pre-germinal center B cells and mature B-cell neoplasms together with germinal-center experienced B cells. Next, to identify DNA methylation signatures associated with malignant transformation, we focused on the 63% of genome with potential tumor-specific DNA methylation signatures (Fig. 2b). We detected varying numbers of de novo tumor-specific DNA methylation (tsDNAm) changes, ranging from 616 in CLL to 49,279 in MM (Fig. 2b, c, Extended Data Fig. 2b, c, d, Supplementary Tables 4 and 5, and Methods). Overall, hypermethylation was enriched at CpG islands and promoter-related regions, whereas hypomethylation occurred at low CpG content regions (Extended Data Fig. 2e). Remarkably, we observed that DNA methylation changes manifested differently in distinct neoplasms. ALL and DLBCL showed more tumor-specific DNA hypermethylation (tsDNAm-hyper), whereas MCL, CLL and MM acquired more tumor-specific DNA hypomethylation (tsDNAm-hypo), being this skew towards hypomethylation remarkable in MM (Fig. 2b-c). These distinct preferences among neoplasms are not apparently related to differential expression of DNA methyltransferases (DNMTs), as we could not identify any clear association between the hypermethylation/hypomethylation ratio and the DNMT1, DNMT3A or DNMT3B expression levels (Supplementary Figure 1).
Fig. 2 |. Disease-specific DNA methylation signatures.
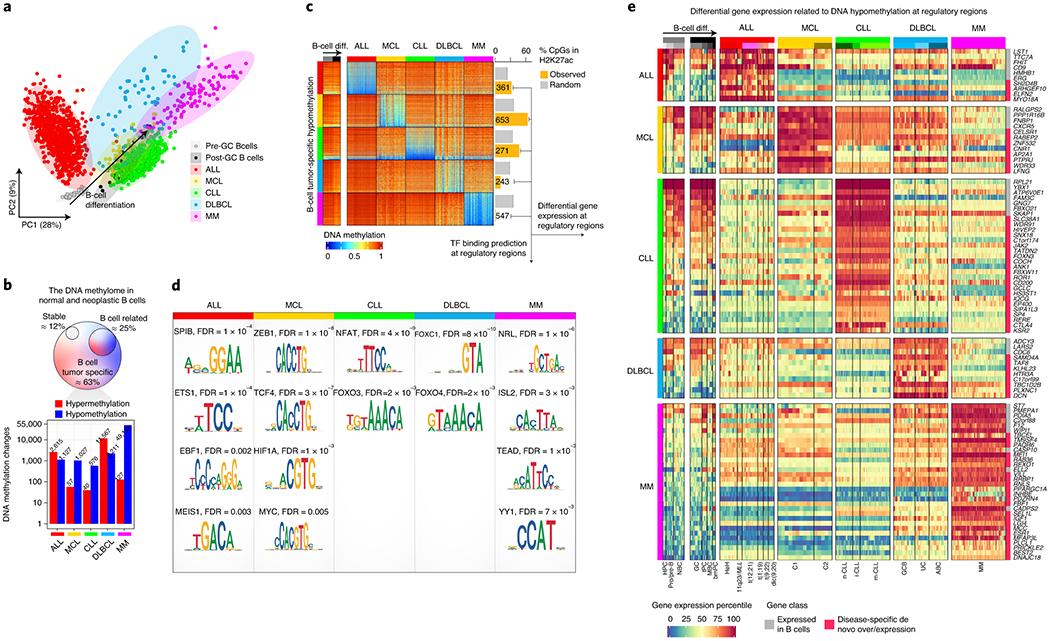
a, Principal component analysis of normal and neoplastic B-cells. Sample sizes are the same as in Fig. 1a.
b, Number of de novo DNA methylation changes in each B-cell tumor entity. Percentages are calculated over the total of 437,182 CpG analyzed. Barplots represent single data values.
c, Heatmap showing de novo B-cell tumor-specific hypomethylation and the number of CpGs falling at active regulatory regions marked by H3K27ac.
d, Enrichment of binding sites of transcription factors expressed in B-cell tumors and in regions with de novo hypomethylated CpGs located in active regulatory elements from c.
e, Differential gene expression percentiles for genes showing B-cell tumor-specific hypomethylation in active regulatory regions.
Next, we sought to identify potential upstream mediators for de novo DNA methylation signatures in each B-cell tumor. As transcription factor (TF) binding has been reported to induce hypomethylation at regulatory regions 19, we performed TFs binding site prediction analysis in active regulatory elements (i.e. marked by H3K27ac) containing tsDNAm-hypo CpG (Methods). Interestingly, the entities in which tsDNAm-hypo was predominantly located in H3K27ac regions (Fig. 2c) showed enrichments for binding sites of TFs expressed in each respective entity and with a previously reported association with their pathogenesis, such as SPI1/SPIB and EBF1 in ALL, TCF/ZEB in MCL, and NFAT in CLL (Fig. 2d, Extended Data Fig. 2f and Supplementary Table 6) 20–22. In the case of DLBCL and MM, their tsDNAm-hypo CpGs were actually depleted of active regulatory elements (Fig. 2c), suggesting that TF binding may not be a major factor leading to their tumor-specific DNA methylation signatures. However, the fraction of tsDNAm-hypo CpGs located in regulatory regions was enriched in TFs potentially involved in the respective diseases, such as FOX family in DLBCL 23, and NRL (a member of the oncogenic MAF family), ISL1, TEAD, and YY1 in MM 24–27 (Fig. 2d).
Beyond the potential role of TFs in shaping tumor-specific DNA methylation signatures, we also investigated the downstream transcriptional associations of tsDNAm-hypo signatures. An analysis of transcriptional profiles of cases from all five diseases revealed a total of 94 genes associated with tsDNAm-hypo genes expressed in a disease-specific manner (Fig. 2e). Although some of the identified genes have been shown specifically expressed in a particular disease, such as CTLA4 and KSR2 in CLL 28, this comprehensive analysis provides a rich resource of disease-specific candidate genes in which differential DNA methylation may play a role in their deregulation.
Accurate classification of 14 clinico-biological subtypes of B cell neoplasms using epigenetic biomarkers
The B-cell neoplasms shown in Fig. 1a represent broad categories which are further classified into subtypes with different clinico-biological features based on genetic, transcriptional or epigenetic features 3. These include high-hyperdiploid (HeH) ALLs, and ALLs with structural variants: rearrangements affecting 11q23/MLL, three different chromosomal translocations, i.e. t(12;21), t(1;19), and t(9;22), as well as the dicentric chromosome dic(9;20) 6; Cluster 1 (C1, DNA methylation patterns related to germinal center-inexperienced cells) and Cluster 2 (C2, DNA methylation patterns related to germinal center-experienced cells) MCLs which mostly reflect conventional and leukemic non-nodal MCLs8; naïve-like/low-programmed, intermediate/intermediate-programmed and memory-like/high-programmed CLLs 10,11, and finally DLBCLs categorized according to the cell of origin classification into germinal center B cell (GCB) and activated B cell (ABC) 29, and not according to the most recent genetic classifications30,31, whose link with epigenetic profiles deserves further investigation. In MM, a previous report did not show robust methylation differences among the distinct cytogenetic subtypes 13 and thus MM subgrouping was not included in our analyses. Here, we focused on the identification of epigenetic biomarkers that may allow a comprehensive diagnosis of B-cell tumor entities and subtypes. We built a classifier algorithm that yielded 56 CpGs as the optimal number distributed along 5 predictors (Extended Data Fig. 3a, b and Supplementary Table 7, Methods) to accurately discriminate the main B-cell tumor entities as a first step (predictor 1), and subsequently B-cell tumor subtypes as a second step (predictors 2, 3, 4 or 5) (Fig. 3a). The accuracy of the five predictors was evaluated using nested 10-fold stratified cross-validation in the training series (n=1,345) and with external validation series (n=711) (Fig. 3b). Overall, we obtained very high accuracies in the predictions in both main B-cell tumor entities (mean sensitivity was 97% for training series and 99% for validation series) and B-cell tumor subtypes (mean sensitivity was 90% for training series and 97% for validation series). This epigenetic classifier may represent the basis for a simple and accurate diagnostic tool for B-cell tumor subtypes with different clinical management (Code availability section).
Fig. 3 |. Development and validation of a DNA methylation-based diagnostic classifier of different subtypes of B cell neoplasms.
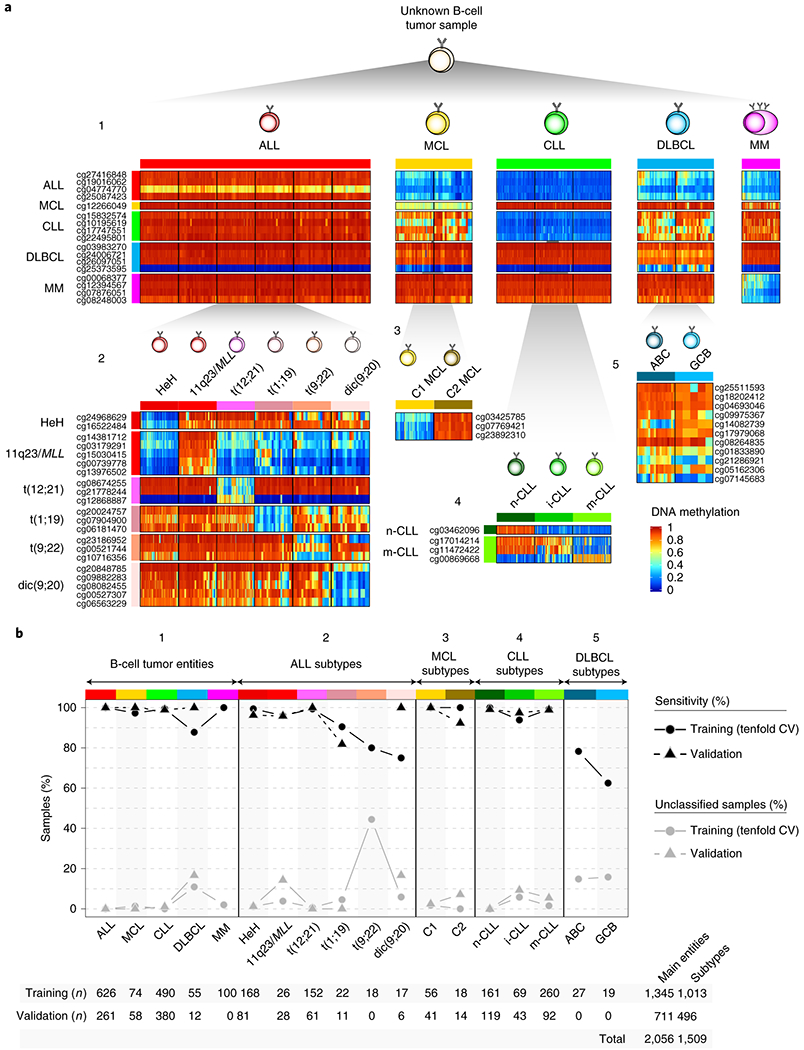
a Heatmap showing DNA methylation values of the CpGs used for the two-step pan B-cell cancer classifier. The training samples from b are represented.
b, Accuracy for the pan-B-cell cancer diagnostic classifier composed by the 5 predictors in panel a in both training and validation series. Sensitivity is represented as black circles or triangles for training or validation series, respectively. The percentage of cases without a clear prediction (unclassified) is represented in grey. The total number of samples used for both training and validation is shown at bottom.
Patient-specific DNA methylation changes are associated with silent chromatin without an impact on gene expression
To determine patient-specific changes within each tumor subtype (Fig. 1b, level 4), we computed the total number and the number of hyper- and hypomethylation changes in every single patient within each B-cell tumor subtype as compared to HPC. As each B-cell tumor entity is derived from a distinct cellular origin, this approach has the advantage of fixing a reference point for all B-cell tumors. Furthermore, each methylation change was further classified as being extensively modulated or not during normal B-cell development 5, i.e. B cell-related changes or B cell-independent changes, respectively (Fig. 4a). Overall, we found large differences in the numbers of DNA methylation changes per patient (Fig. 4a and Supplementary Table 8), and all B-cell tumors showed a similar degree of DNA methylation variability (Extended Data Fig. 4a). We also detected strikingly high correlations between the degree of B-cell related and B-cell independent DNA methylation changes (Fig. 4b, Extended Data Fig. 4b and Supplementary Table 8). This association suggests that the overall DNA methylation burden of the tumor in each individual patient may be shaped by a similar underlying phenomenon. Supporting this concept, we observed that CpGs undergoing hypomethylation both in the B cell-related and B cell-independent fractions are mainly located in low CpG-content, low-signal heterochromatin, and the associated genes are constitutively silent both in normal and neoplastic B cells (Fig. 4c-e and Extended Data Fig. 4c-f). In the case of hypermethylation, CpGs in both fractions are located mainly in promoter regions and CGIs with H3K27me3-repressed and poised-promoter chromatin states, and affect genes that remain silent across normal differentiation and neoplastic transformation of B cells (Fig. 4f-h, Extended Data Fig. 4c, g-i).
Fig. 4 |. Identification and characterization of patient-specific DNA methylation changes.
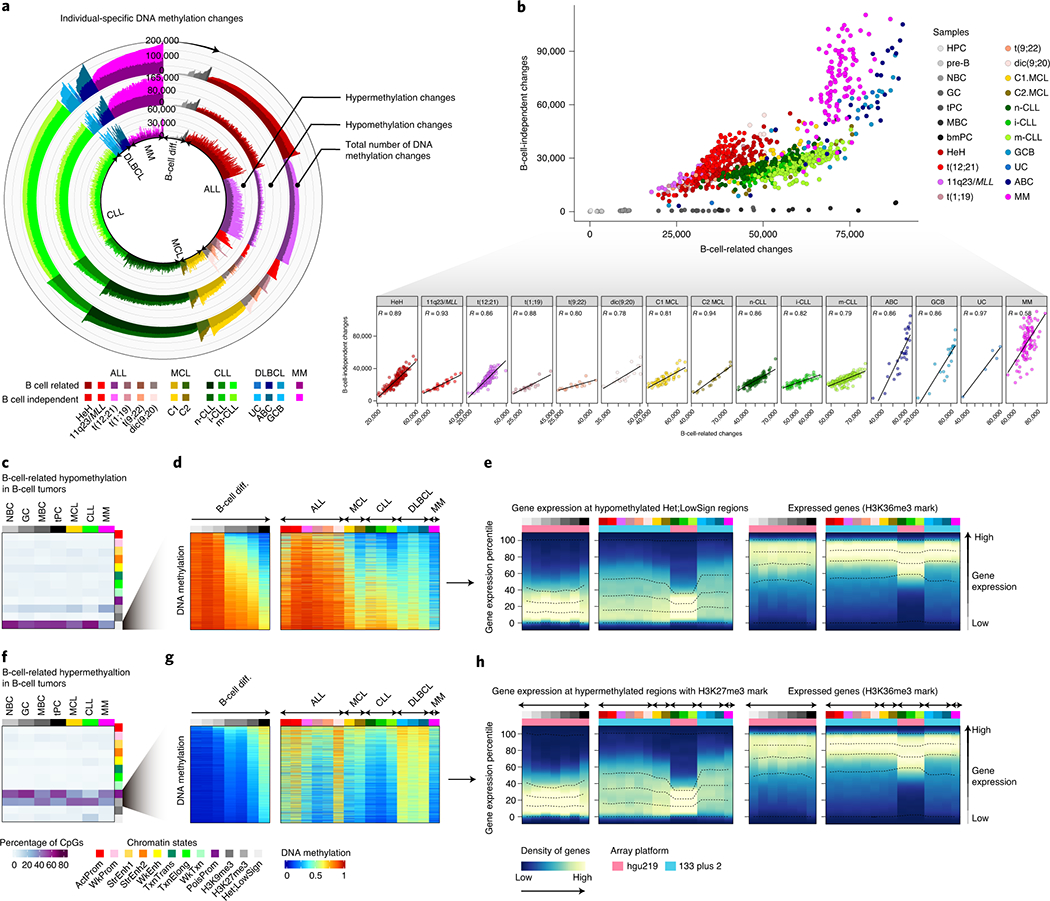
a, Number of DNA methylation changes in individual patients for normal and neoplastic B cells as compared to the hematopoietic precursor cell stage. Total number of DNA methylation changes, hypomethylation changes and hypermethylation changes are depicted at outer, middle and inner tracks, respectively. Changes are further classified and color-coded as B-cell related or B-cell independent. Sample sizes are: HPC, n=6; pre-B cells, n= 16; NBC, n=15; GC, n=9; tPC, n=8; MBC, n=10 and bmPC, n=3 donors; HeH ALL, n=168; 11q23/MLL ALL, n=26; t(12;21) ALL, n=152; t(1;19) ALL, n=22; t(9;22) ALL, n=18; dic(9;20) ALL, n=17; C1 MCL, n=56; C2 MCL, n=18; n-CLL, n=161; i-CLL, n=69; m-CLL, n=260; GCB DLBCL, n=19; ABC DLBCL, n= 27; UC DLBCL, n=5 and MM, n=100 patients. The same sample size is applied to panels b, d and g.
b, Correlation between B-cell related changes and B-cell independent changes in normal and neoplastic B-cells. R derived from linear models are shown.
c, B-cell related CpGs losing DNA methylation in B-cell tumors and the percentages in each chromatin state in normal and neoplastic B-cells. The mean of percentages per sample type is shown. Sample sizes are: NBC, n=6; GC, n=3; MBC, n=3 and tPC, n=3 donors; MCL, n=5; CLL, n=7 and MM, n=4 patients. The same sample size applies for panel f.
d, The mean of 2,000 representative CpGs per each sample subtype from panel c is represented.
e, Gene density distributed along the expression percentiles of genes associated with B-cell related CpGs losing DNA methylation in B-cell tumors at low signal heterochromatin. Expressed genes showing the H3K36me3 mark are displayed at right as a positive control. The mean for each sample type is represented. Lines represent 0, 25, 50, 75 and 100% percentiles.
f, B-cell related CpGs gaining DNA methylation in B-cell tumors and the percentages in each chromatin state in normal and neoplastic B-cells. The mean of percentages per sample type is shown.
g, The mean of 2,000 representative CpGs per each sample subtype from panel f is represented.
h, Gene density distributed along the expression percentiles of genes associated with B-cell related CpGs gaining DNA methylation in B-cell tumors in regions containing the H3K27me3 mark. Expressed genes with the H3K36me3 mark are displayed at right as a positive control. Means within each B-cell subpopulation as well as B-cell tumors are represented. Sample subtypes from panels d, e, g and h are color-coded as in panel b. Sample sizes for gene expression analyses in panels e and h are: HPC, n=3; pre-B cells, n=7; NBC, n=10; GC, n=11 tPC, n=5 donors; MBC, n=5 and bmPC, n=1 donors; HeH ALL, n=18; 11q23/MLL ALL, n=5, t(12;21) ALL, n=16, t(1;19) ALL, n=6, t(9;22) ALL, n=5, dic(9;20) ALL, n=6; C1 MCL, n=10; C2 MCL, n=5; n-CLL, n=142, i-CLL, n=64; m-CLL, n=249; GCB DLBCL, n=17, UC DLBCL, n=11, ABC DLBCL, n=15 and MM=328 patients.
Collectively, these findings indicate that most DNA methylation changes in B-cell tumor patients occur in silent chromatin regions in the absence of concurrent phenotypic changes, suggesting that a mechanism independent from gene regulation may underlie their overall DNA methylation landscape.
Development of an epigenetic mitotic clock reflecting the proliferative history of normal and neoplastic B cells
Beyond the classical role of DNA methylation as gene regulator, an accumulating body of published evidence supports the concept that hypomethylation of low CpG-content heterochromatin and hypermethylation of high CpG-content polycomb target regions accumulate during cell division in a way consistent with an epigenetic mitotic clock32–39. Here, we observe that the inter-patient methylation variability in B-cell tumors mainly affects inactive chromatin, including hypomethylation of heterochromatin and hypermethylation of regions marked with H3K27me3-containing chromatin states (Fig. 4c-h and Extended Data Fig. 4d-i). Based on this data, these DNA methylation changes most likely reflect the different tumor cell proliferative histories of individual patients. Thus, we next performed a step-wise selection of CpGs whose methylation change would reflect the cell mitotic history (Fig. 5a, Extended Data Fig. 5a and Methods). First, we selected CpGs within constitutively silenced/poised chromatin. Second, we identified CpGs methylated (≥0.9) or unmethylated (≤0.1) in HPC samples that extensively lose or gain methylation (a difference of at least 0.5) in bmPC samples. This difference was used to capture CpGs undergoing extensive methylation changes between cells with the lowest and highest proliferative histories in the B-cell lineage. Third, we obtained 184 CpGs located at constitutive H3K27me3-containing regions and 1,164 CpGs at constitutive heterochromatin which gain and lose DNA methylation upon cell division, respectively (Fig. 5a, b, Supplementary Table 9 and Methods). Fourth, we next constructed two mitotic clocks with these two sets of CpGs, one gaining DNA methylation upon cell division called epigenetically-determined Cumulative MIToses (epiCMIT)-hyper and one losing DNA methylation called epiCMIT-hypo (Fig. 5a, b and Methods). We initially evaluated both mitotic clocks in normal B cells and observed a high correlation (R=0.96, p-value<2e-16), with B-cell subpopulations distributed according to their accumulated proliferative history during B-cell differentiation and not to their current proliferation status (Fig. 5c, left panel). This association between the degree of hyper- and hypomethylation supports previous observations in colorectal cancer40 and indicates that mitotic cell division in normal B cells leaves both hyper- and hypomethylated imprints. Although this high correlation between the two mitotic clocks was also observed for MCL, CLL and DLBCL (Fig. 5c), it does not seem to be a universal phenomenon, as no correlation was observed in ALL and MM. In line with the overall trend to gain methylation in ALL and to lose methylation in MM (Fig. 2b), we observed that the epiCMIT-hyper was greater than the epiCMIT-hypo in ALL samples, and the opposite in MM. These differences do not seem to arise from differential expression of DNMTs (Supplementary Figure 1). As a final step in the epiCMIT mitotic clock development, we then selected the highest score from the epiCMIT-hyper and epiCMIT-hypo per sample to derive a unique epiCMIT value (Fig. 5a, d, Supplementary Table 9 and Methods). The epiCMIT shall then reflect the relative accumulation of mitotic cell divisions of a particular sample, including the mitotic history associated with normal cell development as well as with malignant transformation and progression. Moreover, the epiCMIT cannot be affected by a different distribution of cell cycle phases in tumor samples, since the DNA methylome remains rather stable during the whole cell cycle 41.
Fig. 5 |. Development and validation of the epiCMIT.
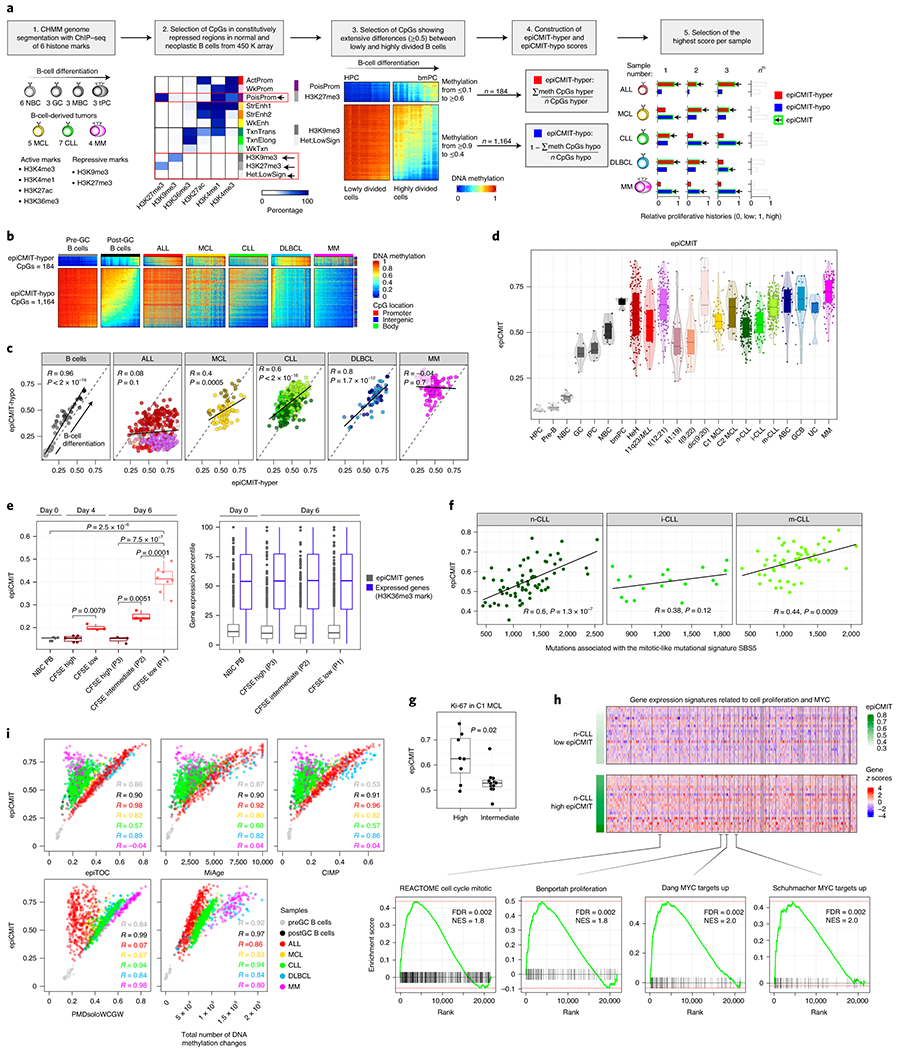
a, Steps to construct the epiCMIT-hyper, epiCMIT-hypo and epiCMIT mitotic clocks. epiCMIT, epigenetically-determined Cumulative MIToses.
b, CpGs constituting the epiCMIT-hyper (184 CpGs) and epiCMIT-hypo (1,164 CpGs) mitotic clocks.
c, Correlation between the epiCMIT-hyper and the epiCMIT-hypo in normal and neoplastic B cells. R and p-values are derived from linear models.
d, Box plot showing the distribution of epiCMIT values in normal and neoplastic B cells. Center line, box limits, whiskers and points represent the median, 25th and 75th percentiles, 1.5x interquartile range and individual samples, respectively.
e, Experimental validation of epiCMIT score with an in vitro B-cell differentiation model of primary human naïve B cells into plasma cells. The epiCMIT was calculated at day 0, day 4 and day 6 in B cells with distinct proliferative histories based on CFSE dilution. Sample sizes are: NBC-PB, n=5; CFSE-high at day 4, n=6; CFSE-low at day 4, n=3; P3 cells at day 6, n=3; P2 cells at day 6, n=3 and P1 cells at day 6, n=8. Each dot within each category is derived from a different donor, and thus represent biologically independent samples. P-values are derived from two-sided t-tests. On the right, gene expression of genes containing CpGs belonging to epiCMIT. The number of genes containing epiCMIT genes analyzed is n=1,278, and genes with CpGs mapping at H3K36me3 are n=14,598. For the box plots, center line, box limits, whiskers and points represent the median, 25th and 75th percentiles, 1.5x interquartile range and individual samples, respectively.
f, piCMIT correlates with the mitotic-like mutational signature SBS5 in CLL. R and p-values are derived from linear models. 138 CLL patients with WGS and DNA methylation data are shown. Sample sizes for CLL subtypes are: n-CLL, n=66; i-CLL, n=18 and m-CLL, n=54 patients.
g, epiCMIT is associated with high Ki67 staining in C1 MCL cases. Number of cases are n=8 and n=12 for high and intermediate Ki67 values. Two-sided t-test was used to assess statistical significance. For the box plot, center line, box limits, whiskers and points represent the median, 25th and 75th percentiles, 1.5x interquartile range and individual samples, respectively.
h, Gene set enrichment analysis (GSEA) showing that epiCMIT is associated with gene expression signatures related to cell proliferation and MYC activity in CLL. 142 n-CLL were analyzed, and 22 n-CLL samples with low and high epiCMIT are shown (15 and 85% percentiles, respectively). At top, z-score for each gene is represented. At bottom, some representative gene expression signatures enrichments are shown.
i, Correlation between the epiCMIT and previously reported mitotic clocks, including epiTOC, MiAge and PMDsoloWCGW, the pan-cancer CIMP, and the total number of DNA methylation changes accumulated since HPC stage in each patient. R’s correspond to linear regression models. The same sample for panels b, c, d and i are the same than in Fig. 4a.
Validation of the epiCMIT score as mitotic clock in normal and neoplastic B cells
The applicability of the epiCMIT as mitotic clock was validated through several perspectives. First, we used an independent in vitro B-cell differentiation model of primary NBCs into plasma cells42, in which cell divisions were controlled by carboxyfluorescein succinimidyl ester (CFSE) staining (Extended Data Fig. 5b). At days 4 and 6, different B cells were separated based on their proliferation history measured by CFSE dilution, and we observed that epiCMIT increases in cells with lower CFSE concentration, i.e. higher proliferative history (Fig. 5e, left panel). The genes related to epiCMIT-CpGs remained silenced in all these conditions regardless of the cell phenotype and proliferative history(Fig. 5e, right panel). Second, we studied the link between the epiCMIT and genetic changes using WGS data of 138 CLL patients from our cohort17. We observed that the epiCMIT was correlated with the total number of somatic mutations and with genomic complexity measured by the number of driver genetic alterations, i.e. mutations with positive selection (Extended Data Fig. 5c, d). Additionally, we measured the activity of know mutational processes through the analysis of single base substitution (SBS) signatures43 (Extended Data Fig. 5e). We detected significant correlations between our epiCMIT and signatures SBS5 and SBS1, which have been previously described as mitotic-like mutational processes (Fig. 5f and Extended Data Fig. 5f). We also identified a significant link between the epiCMIT and the non-canonical AID signature (SBS9) 17,43 in IGHV mutated CLL, possibly reflecting accumulated rounds of cell divisions in the germinal center of the ancestor B cell prior to its transformation to CLL (Extended Data Fig. 5g). Third, although the epiCMIT is aimed at capturing the proliferative history of the cell, a relationship with cell proliferation is expected in tumors (more proliferative history implies higher proliferation, although it also depends on time). Accordingly, the epiCMIT was higher in MCL cases showing high Ki-67 (a proliferation marker) than in cases with low Ki-67 expression (Fig. 5g). Furthermore, leukemic CLL cases with high epiCMIT, although not considered to be proliferative, showed higher expression of genes related with cell proliferation and MYC activity (Fig. 5h and Supplementary Table 10). Thus, these data suggest that cases with higher proliferative history also seem to have higher proliferative capacity at the time of sampling.
We next compared the epiCMIT with two previously proposed hypermethylation-based mitotic clocks called epiTOC and MiAge 37,39 (Supplementary Table 8 and Methods). In addition, we calculated a hypomethylation-based mitotic clock using a previously defined pan-cancer set of CpGs losing methylation called PMDsoloWCGW CpGs38 (Supplementary Table 8 and Methods). Focusing on hypermethylation-based mitotic clocks, the epiCMIT showed excellent correlations with epiTOC and MiAge in B-cell neoplasms acquiring polycomb-related hypermethylation (mostly ALL, but also DLBCL and MCL); a moderate correlation in the case of CLL, which acquires more hypo- than hypermethylation, and a total lack of correlation in the case of MM, which mostly loses DNA methylation (Fig. 5i upper panel and Extended Data Fig. 5h). Interestingly, identical observations were obtained comparing the epiCMIT and the widely-reported CpG island methylator phenotype (CIMP) in human cancer44, suggesting that the pan-cancer CIMP score may also represent a measure of the cell mitotic history. Interestingly, the opposite scenario was found when comparing epiCMIT with the hypomethylation-based mitotic clock PMDsoloWCGW. We showed excellent correlations between epiCMIT and PMDsoloWCGW in tumors with extensive DNA hypomethylation (mostly MM and CLL, but also MCL and DLBCL) and a null correlation in ALL (Fig. 5i, bottom panel). In spite of these striking discrepancies in ALL and MM, mitotic clocks were in general highly correlated, even though the poor overlap of their underlying CpGs, indicating that cell proliferative history can be traced with different sets of CpGs (Extended Data Fig. 5i). Additionally, we observed that epiCMIT is highly correlated with the total number of DNA methylation changes accumulated in all samples since the HPC stage, suggesting that the overall DNA methylation landscape seems to be strongly influenced by the cell proliferative history (Fig. 5i bottom panel and Extended Data Fig. 5h). Finally, epiCMIT outperformed all mitotic clocks to identify cells with different proliferative histories using the controlled setting of the in vitro B-cell differentiation model (Extended Data Fig. 5b, j), a finding that suggests its higher accuracy to trace the B-cell proliferative history. Collectively, all these analyses suggest that the epiCMIT is a more universal mitotic clock than previously reported mitotic clocks exclusively based on hyper- or hypomethylation.
A potential confusing aspect related to epiCMIT is the fact that DNA methylation changes take place during aging 45,46 and can be used to predict chronological age 47–49, as exemplified with the Horvath’s epigenetic clock 50. To study the potential relationship between mitotic activity and the aging process, we first analyzed the epiCMIT in normal B cells with low (NBC) and high (MBC) epiCMIT values in samples from infants, young adults and elderly donors (Extended Data Fig. 6a, left). This analysis did not reveal any evidence linking the epiCMIT with the chronological age of healthy donors, which indeed is accurately predicted by the Horvath’s aging clock (Extended Data Fig. 6a). In the case of B-cell tumors, we observed the same general tendency. Pediatric ALL samples show the highest epiCMIT range despite the very low age range, and thus a negligible association between epiCMIT and age. In DLBCL we observed a similar scenario, since 30 and 90-year-old patients showed similar epiCMIT levels. Only in MCL and CLL patients we observed minor correlations between epiCMIT and patient’s age (Extended Data Fig. 6a, right). We then applied the Horvath’s clock to patient samples and, as previously shown in other cancers50, we found significant epigenetic age acceleration with some pediatric ALL patients reaching an impressive predicted age over 200 years. Interestingly, we found that the epiCMIT shows a highly significant correlation with the epigenetic age predicted by Horvath’s clock in the majority of B-cell tumors subtypes (R=0.62, p-value<2el6), suggesting that epigenetic age acceleration may be related to the increased proliferation of cancer cells (Extended Data Fig. 6a, bottom). Despite this intriguing correlation that deserves further investigation, the epiCMIT and Horvath’s clocks seem to be targeting different molecular features, as their underlying CpGs show markedly distinct genomic locations, DNA methylation dynamics in normal and neoplastic B cells, chromatin enrichments and gene expression of their associated genes (Extended Data Fig. 6b-f).
The epiCMIT is a strong independent variable predicting clinical behavior in B-cell tumors
In normal B-cell maturation, the epiCMIT gradually augments as B cells proliferate, an increase that is particularly marked in highly proliferative GC B cells (Fig. 5d). In neoplastic B cells, however, the interpretation of the epiCMIT is less trivial and must be divided into two components: the epiCMIT of the cell of origin and the epiCMIT acquired in the course of the neoplastic transformation and progression (Fig. 6a). Therefore, the relative epiCMIT must be compared among patients from entities arising from the same B-cell maturation stage and should be a dynamic variable during cancer progression. Thus, we compared the epiCMIT in two paradigmatic transitions between precursor conditions and overt cancer, i.e. monoclonal gammopathy of undetermined significance (MGUS) and MM, as well as monoclonal B cell lymphocytosis (MBL) and CLL categorized according to their cellular origin. This analysis showed an overall lower epiCMIT in precursor lesions compared with overt cancer (Fig. 6b, upper panels). In line with this finding, the epiCMIT increased in paired CLL samples at diagnosis and progression before treatment as well as in sequential ALL samples at diagnosis, first relapse and second relapse (Fig. 6a, lower panels).
Fig. 6 |. Clinical impact of the epiCMIT is B-cell tumors.
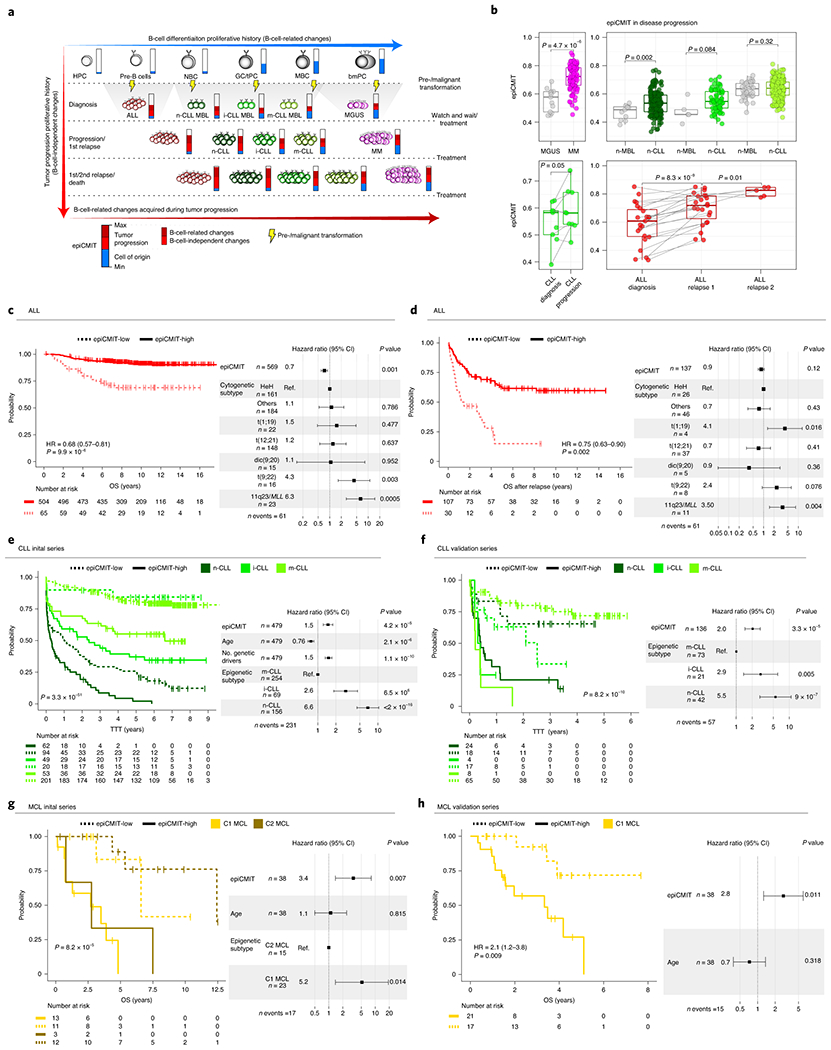
a, The epiCMIT in neoplastic B cells include the proliferative history associated with normal B-cell development and with malignant transformation and progression (blue and red components of the epiCMIT bar, respectively). B-cell tumors derive from different maturation stages, and thus they contain different normal B-cell baseline epiCMIT. Most of the B-cell related DNA methylation changes occurring in B-cell tumors relate to cell division.
b, epiCMIT evolves during disease progression. epiCMIT is lower in precursor conditions such as MGUS (n=13 patients) and MBL (n=53 patients) as compared to their respective cancer conditions CLL (n=437 patients) and MM (n=100 patients), as well as in paired CLL samples from diagnosis to progression (n=9 patients), and trios of ALL patients at diagnosis and first relapse (n=23 patients) and second relapse (n=5 patients). P-values were obtained from two-sided t-test, and paired t-test in the case of paired samples. For the box plots, center line, box limits, whiskers and points represent the median, 25th and 75th percentiles, 1.5x interquartile range and individual samples, respectively.
c, d Kaplan-Meier curves for c overall survival (OS) and d OS after relapse in ALL patients with low or high epiCMIT according to the maxstat rank statistics-based cutoff. Hazard ratio and p-value for the univariate Cox regression models are shown on the left panels. Multivariate Cox regression models with epiCMIT as continuous variable and ALL cytogenetic groups are shown on the right. Hazard ratio for epiCMIT correspond to 0.1 increments, and also in panels e, f g and h.
e,, Kaplan-Meier curves for CLL epigenetic groups based on different cellular origin divided in low and high epiCMIT according to the maxstat rank statistics-based cutoff. A multivariate Cox regression model for time to first treatment with epiCMIT as continue variable together with age, number of driver alterations and epigenetic groups based on different cellular origin is shown on the right. The results obtained with the independent validation series is shown in panel f.
g, Kaplan-Meier curves for MCL epigenetic groups based on different cellular origin divided in low and high epiCMIT according to the maxstat rank statistics-based cutoff. A multivariate Cox regression model for OS with epiCMIT as continuous variable together with epigenetic groups and age is shown on the right. Validation series for C1 MCL is shown in panel h.
Based on these observations, we next wondered whether the epiCMIT could be useful to predict the clinical behavior of B-cell neoplasms. We analyzed specific B-cell tumor subtypes based on cytogenetic subtypes (i.e. ALL) or cell of origin (i.e. MCL, CLL and DLBCL), and thus having a similar ground state proliferative history (Fig. 6a). In ALL, high epiCMIT was consistently associated with longer overall survival (OS), OS after relapse and relapse-free survival (RFS) of the patients (Fig. 6c, d and Extended Data Fig. 7a). These epiCMIT associations maintained an independent statistical significance from the well-established ALL cytogenetic groups as prognostic variable in RFS and OS, and a marginal significance in OS after relapse. In contrast to ALL, the opposite clinical scenario was observed in mature B-cell neoplasms. In each of the CLL subtypes, a high epiCMIT was strongly associated with a worse prognosis using time to first treatment (TTT) as end-point variable, both from sampling time (Fig. 6e) and in cases whose sample was obtained close to diagnosis (Extended Data Fig. 7b). Additionally, the epiCMIT as continuous variable showed a highly significant independent prognostic impact in the context of major prognostic factors in CLL, including the IGHV status and TP53 alterations (deletion and mutation) (Extended Data Fig. 7c). Overall, it seems that the epiCMIT, CLL epigenetic subgroups 10,11,51, and genomic complexity measured by the total number of driver alterations 17,52 are the most significant independent variables associated with prognosis in CLL. In addition, despite the variability of treatments in our initial CLL series, the epiCMIT also showed marginal significance in OS (Extended Data Fig. 7d). All these findings were widely confirmed in an additional series of 210 CLL patients treated mainly with chemo-immunotherapy (Fig. 6f and Extended Data Fig. 7b, d). In the case of MCL, the epiCMIT showed an independent poor prognostic impact in the two cell-of-origin subtypes (C1 and C2), an observation that was confirmed in an extended series in the more aggressive and prevalent C1 group (Fig. 6g, h). In the case of the two cell-of-origin DLBCL subtypes, our data suggest that high epiCMIT could also represent a poor prognostic variable (Extended Data Fig. 7e). Finally, our epiCMIT score showed an overall superior prognostic value compared with all the other DNA methylation-based mitotic clocks in all B-cell tumors with the largest number of patients (Extended Data Fig. 8).
epiCMIT is associated with specific genetic driver alterations in CLL
Despite the independent prognostic impact of epiCMIT and genetic alterations in CLL, we next assessed which CLL driver alterations could potentially confer a proliferative advantage to neoplastic cells, and subsequently a higher epiCMIT. To that end, we exploited 477 CLL samples in which we had DNA methylation data and whole exome sequencing (WES)17 (Fig. 7a). We initially depicted all driver genetic changes in each CLL subtype divided in high and low epiCMIT (Extended Data Fig. 9a). Next, we interrogated the levels of epiCMIT in patients with each driver genetic alteration both in the whole cohort and in each epigenetic subgroup separately (Fig. 7b, Extended Data Fig. 9b and Methods). We showed significant and positive associations of epiCMIT with 23 genetic driver alterations (Fig. 7b) 17,52. The majority of these genetic alterations have been previously linked to an adverse clinical behavior of patients, such as NOTCH1, TP53, SF3B1, ATM, BIRC3 or EGR2. Interestingly, epiCMIT showed an association with a recently identified non-coding genetic driver associated with poor prognosis in CLL, the U1 spliceosomal RNA 53. Remarkably, the presence of some genetic alterations was associated with high epiCMIT indistinctly in all patients, such as TP53, while others were particularly associated with epiCMIT within CLL subgroups, such as SF3B1 and ATM in i-CLL.
Fig. 7 |. Association between the epiCMIT and genetic driver alterations in CLL.
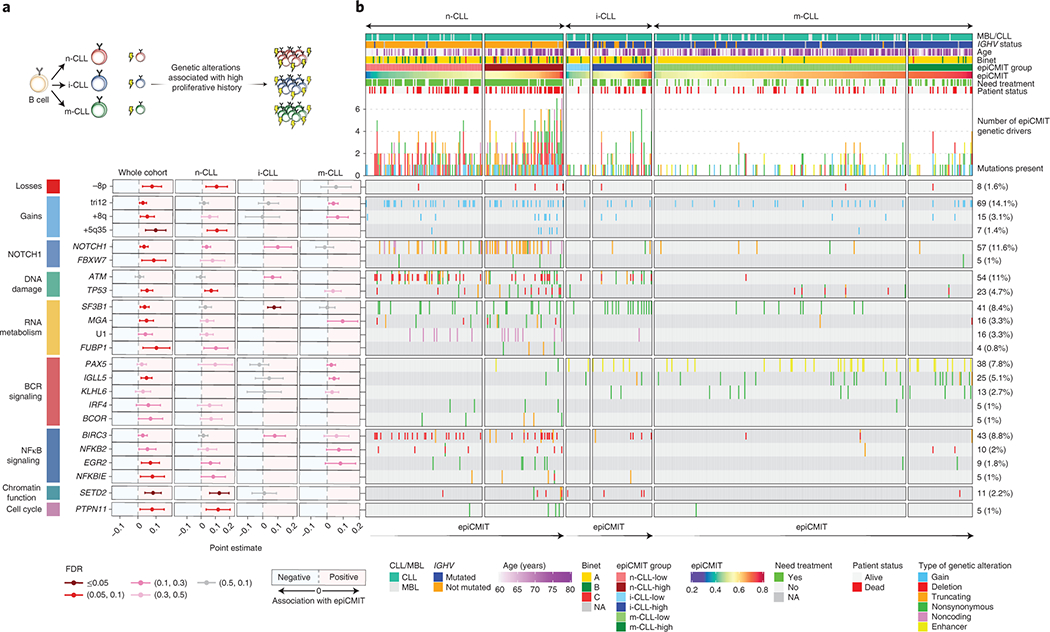
a, Illustrative scheme to represent which potential genetic driver alterations may confer a higher proliferative capacity to CLL cells.
b, Analysis of the association between the epiCMIT levels and the presence of specific driver genes grouped by signaling pathways. Point estimates with 95% confidence intervals were derived in the whole cohort using linear modelling between epiCMIT and alterations adjusted for CLL subtypes, and with two-sided t-tests within CLL subtypes. Point estimates represent the coefficient of each respective alteration in each corresponding linear model in whole cohort analysis, and the difference between the mean of CLL patients with and without each corresponding alteration for the analysis within each CLL subtypes. Point estimates are color-coded according to FDR correction. The Oncoprint shows genetic driver alterations significantly associated with higher epiCMIT with CLL epigenetic groups shown separately. Other clinicobiological features including MBL or CLL, IGHV status, Age, Binet stage, epiCMIT subgroups based on maxstat rank statistic cutoff, need for treatment and patient status are shown. Cases are ordered within each CLL subgroup from lower to higher epiCMIT values. Genetic driver alterations are depicted with different colors and shapes depending of the alteration type. Number of mutated patients as well as their percentage over the whole cohort is shown on the right. The whole CLL initial series was used for these analyses and is represented (n=490 patients).
Collectively, these results suggest that the well-established clinical impact of certain genetic alterations in CLL may be explained by their association with a high proliferative potential, being this association different for certain genetic alterations depending on the maturation state of the cellular origin.
Discussion
Here, we have followed a systematic approach to dissect the sources of DNA methylation variability of B-cell neoplasms in the context of the normal B-cell differentiation program. Overall, we found that the methylation levels of 88% of the studied CpGs are modulated in normal and/or neoplastic B cells, suggesting that the human DNA methylome is even more dynamic than previously appreciated 5,16. The extensive DNA methylation variability among different B-cell neoplasms is in part related to imprints of normal cell development, a phenomenon that has been recently used to classify not only B-cell neoplasms 8,10,11,51 but also solid tumors 2,54. In addition, each B-cell neoplasm also shows de novo disease-specific hyper- and hypomethylation, being the latter possibly related to binding of disease-specific TFs and subsequent disease-specific gene expression profiles.
In spite of the widely-reported importance of DNA methylation at regulatory regions, we identified that the majority of DNA methylation changes in B-cell neoplasms are located in inactive chromatin. These DNA methylation changes are manifested mainly in the form of hypomethylation of heterochromatin and hypermethylation of H3K27me3-containing regions, a phenomenon previously observed in colorectal cancer40. Compelling published evidences32–38 and our data support the notion that mitotic cell division leaves transcriptionally-inert epigenetic imprints onto the DNA located in repressive chromatin environments. More recently, this knowledge has led to the concept of using DNA methylation as a mitotic clock 37–39 and also has been confirmed at the single cell level 55,56. Here, we identified that using only hyper- or hypomethylation to build a mitotic clock may be insufficient to capture the mitotic history of cancer cells, as some neoplasms seem to preferentially gain or lose DNA methylation upon cell division. For instance, ALL seems to acquire broad hypermethylation upon cell division, whereas we consistently observed the opposite scenario in MM. Thus, using exclusively hyper- or hypomethylation37–39 to determine the mitotic history of MM or ALL cells would incongruently lead to the conclusion that they have not proliferated beyond their cellular origin. Therefore, to circumvent these limitations, our epiCMIT uses several filters to carefully select both hyper- and hypomethylation in CpGs. The strict filtering criteria together with the high correlation with previous cell type-independent mitotic clocks suggest the epiCMIT may represent a pan-cancer mitotic clock. Here, we showed that epiCMIT captures the entire mitotic history of B cells, including cell division associated both with normal development as well as neoplastic transformation and progression. Thus, the epiCMIT should not be compared among B-cell tumors arising from different normal counterparts but its relative magnitude must be studied in those arising from a particular maturation stage. Within each of these subgroups, the relative epiCMIT has a superior prognostic value than previous mitotic clocks and a profound independent prognostic value from other well-established clinical variables in B-cell tumors. Increased epiCMIT is associated with worse clinical outcome in CLL and MCL, suggesting that superior proliferative history before treatment seems to determine future proliferative capacity of CLL and MCL cells. Strikingly, we consistently found the opposite pattern in ALL, a finding in line with recent reports showing that the prese nee of CIMP is associated with better clinical outcome 57,58. This result may suggest that the high proliferative ALL cells of children at diagnosis (and thus having a larger proliferative history) are more efficiently killed by high intensive chemotherapy regimens59, which cannot be administrated in elderly patients such as in the case of CLL and MCL.
DNA methylation has also been used as a clock to predict the chronological age of healthy donors47–49. The epiCMIT and aging clocks such as that developed by Horvath50 seem to reflect broadly different layers of epigenetic information imprinted onto the DNA. This notion is supported by multiple perspectives, including the similar levels of epiCMIT in the same normal B-cell subpopulations regardless of donor’s age, the differential (epi)genomic and transcriptomic features between Horvath and epiCMIT clocks, and the independent prognostic value of epiCMIT and age in B-cell tumors. In spite of this overall independence of mitotic and aging clocks, we did observe a remarkable association between the epiCMIT and the epigenetic age predicted by the Horvath clock in B-cell tumors. This finding suggests that the accelerated epigenetic age reported in human cancer50 may actually reflect the mitotic activity of cancer cells. This concept is further supported by previous results indicating that the predicted age of a sample increases with in vitro cell passages50.
Finally, we found that epiCMIT is enhanced by the presence of some mutations with positive selection (i.e. driver genes) and not by random mutations, as driverless CLL patients show an overall lower epiCMIT compared with patients with abundant genetic driver alterations. We identified 23 driver genetic alterations particularly associated with higher epiCMIT levels or methylation evolution60, which may represent genetic alterations conferring a higher proliferative capacity to CLL cells. They were distributed throughout the main altered signaling pathways in CLL and were manifested differently in distinct CLL subgroups based on their cellular origin (Fig. 7b). This finding suggests that specific alterations may predispose to a higher proliferative advantage depending on the maturation stage and (epi)genetic makeup of the CLL cellular origin.
In summary, our comprehensive epigenetic evaluation of normal and neoplastic B cells spanning the entire human B-cell lineage uncovers multiple insights into the biological roles of DNA methylation in cancer, an analytic approach that may also benefit our understanding of other cancers. From a clinical perspective, DNA methylation may provide a holistic diagnostic and prognostic approach to B-cell neoplasms. Particularly, we defined an accurate and easy-to-implement pan-B-cell tumor diagnostic tool and generated a mitotic clock reflecting the proliferative history of the neoplastic cells of each patient to estimate their clinical risk, which shall represent a valuable asset in the precision medicine era.
METHODS
Quality control, normalization, filtering and annotation of DNA methylation data
We collected 450k DNA methylation array data from 913 ALL 6,7, 82 MCL 8,9, 491 CLL17, and 104 MM 13 (Supplementary Table 1). We collected also normal B cell subpopulations 5 totaling 67 samples as well as normal microenvironmental cells including 6 granulocytes, 5 CD8+ and 5 CD4+ T cells, 6 monocytes, 6 NK cells 6 whole blood samples and 6 peripheral blood mononuclear cells 61, 6 macrophages 62 and 16 endothelial cells 63. These microenvironmental cells were used to infer B-cell tumor purities through DNA methylation data. In addition, we generated genome-wide DNA methylation profiles following manufacturer’s instructions for DLBCL patients with 450k and EPIC BeadChips (Illumina) of 80 and 12 DLBCL patients, respectively, with partially available genomic data18. The analysis of these DLBCL samples was approved by the Institutional Review Board of Hospital Clinic (Barcelona, Spain), and informed consent was obtained from all patients in accordance with the Declaration of Helsinki. In total, 1,799 samples were profiled with the 450k DNA methylation microarrays. We used a custom pipeline to analyze DNA methylation data using R (version 3.6.3) packages and core Bioconductor (version 3.10) packages, with special use of minfi package (version 1.32) exclusively devoted to analyze DNA methylation data64. From the total of 485,512 probes present in the 450k array, we sequentially removed probes using the next steps: we initially removed 3,091 non-CpGs probes, 17,534 CpGs representing SNPs, 7,715 CpGs with individual-specific methylation 5, and 4,493 CpGs present in sexual chromosomes. All the remaining 452,679 CpGs had a detection p-value ≤0.01 in more than 10% of the samples. We then removed samples with bad intensity signal and/or bad probe conversions as well as those with a tumor percentage below 60% (See next section). In total, we removed 104 ALL samples, 8 MCL samples, 1 CLL sample, 25 DLBCL samples and 4 MM samples. We also removed microenvironmental cells to perform all the analyses in normal and neoplastic B cells. After all filtering criteria, we retained 1,595 samples (Supplementary Table 1 and Fig. 1a) with DNA methylation values for 452,679 CpGs, which were normalized using SWAN algorithm 65. Some CpGs showed missing values in some samples and were removed from all the subsequent analyses (with the exception of biomarker discovery, Fig. 3) and finally 437,182 CpGs were used. We used lluminaHumanMethylation450kanno.lmn12.hg19 (version 0.6) and IlluminaHumanMethylationEPICanno.ilm10b4.hg19 (version 0.6) R package to annotate all CpGs. B-cell related and B-cell independent CpGs classification was used from our previous study to separate CpGs that are significantly modulated or not during B cell differentiation, respectively5. The same pipeline was used to curate and normalize the data from the previously published 42 in vitro model of B-cell differentiation shown in Extended Data Fig. 5b and all the DNA methylation data for validation series used for the pan-B-cell tumor classifier as well as clinical associations. These include our newly generated EPIC DNA methylation data for the 12 DLBCL patients as well as other EPIC and 450K DNA methylation data previously published. In particular, we collected EPIC DNA methylation profiles for 70 MCL patients 9 and 450K and EPIC data for 380 CLL from external collaborators. Finally, to validate results in ALL, we used 183 samples included in the initial analysis (Fig. 1, 2)7 but not used to construct any classifier, and we also downloaded DNA methylation data from GSE7658566 and GSE6922967.
Inferring tumor purity through DNA methylation data
DNA methylation has been shown to represent an appropriate biological layer to infer the proportions of blood cell types in peripheral blood 68. We have previously implemented successfully this statistical framework to infer tumor purity in MCL patient samples 8. We have extended this strategy to all B-cell tumors using additional cell types to deconvolute DNA methylation data into cellular proportions including tumor cell content. We validated this approach using flow cytometry (FCM) and genetic data in MCL and CLL samples (Extended Data Fig. 1b). Briefly, we assume that B-cell tumors retain a B cell signature from its cell of origin and also have negligible proportion of normal B cells. Thus, the percentage of neoplastic B cells in a sample can be inferred by the presence of a DNA methylation signature of B cells. This B cell methylation signature was identified by two sequential steps: 1) we selected CpGs with shared methylation values during the entire B-cell maturation process (from early committed B cells to terminally-differentiated bone marrow plasma cells), and 2) from those CpGs selected above, we performed a differential DNA methylation analysis to identify CpGs whose methylation level was significantly different between B cells and the major non-neoplastic cells accompanying B cell tumors 69, namely granulocytes, T cells, monocytes, macrophages and endothelial cells. Then, with this set of CpGs representative of all major cell types present in tumor samples, we apply a linear constrained projection 68, also known as reference-based approach 70, to find the proportions of each cell type.
As a final filtering step, we retained patient samples showing at least 60% tumor cell content according to DNA methylation-based predictions in ALL, MCL and CLL samples, to FCM in MM and to genetic data in DLBCL samples.
Purity estimation from mutational and copy number variation data in DLBCL
The 80 samples included in this study were previously analyzed by whole-genome copy number (CN) arrays (Cytoscan HD, Affymetrix) and gene mutations by targeted next generation sequencing of 106 genes.18. The Allele-Specific Copy Number Analysis of Tumors (ASCAT) algorithm available at Nexus Copy Number (BioDiscovery, version 7) was used to infer the tumor purity directly from the Cytoscan HD array. The percentage of cells (or cancer cell fraction, CCF) carrying each somatic mutation found in loci not affected by a copy number alterations was calculated as CCF = 2xVAF, where VAF is the variant allele frequency of the mutation. Out of all the mutations, the highest CCF was considered as the best estimate of tumor purity of the samples based on gene mutations. As a final step, the maximum tumor purity detected by ASCAT or gene mutations was considered as the estimated tumor cell purity.
Gene expression data
Gene expression profiles using hgu219 array for normal B cells was obtained from 5 (3 hematopoietic precursor cells, 7 pre-B cells, 10 naïve B cells, 11 germinal center B cells, 5 tonsillar plasma cells, 5 memory B cells and 1 bone marrow plasma cell). Additionally, we downloaded gene expression data for 56 ALL samples profiled with 133 plus 2 array from 6, including several ALL subtypes, namely 18 HeH, 5 11q23/MLL, 16 t(12;21), 6 t(1;19), 5 t(9;22) and 6 dic(9;20). We also used 15 MCL samples profiled with 133 plus 2 arrays 71 including 10 C1 and 5 C2 MCLs. We also used previously generated gene expression data with hgu219 array for 455 CLL samples 17. For DLBCL samples, we generated gene expression data using 133 plus 2 arrays following the manufacturer’s instructions for 43 DLBCL samples, including 17 GCB, 15 ABC, and 11 unclassified. Finally, we downloaded gene expression data for 328 MM samples from 72 analyzed with the 133 plus 2 array platform. We normalized all the data using rma function available in affy (version 1.64) R package. As gene expression data come from different studies and different array platforms, we transformed all normalized gene expression values per sample to gene expression percentiles to minimize batch effects. Also, we generally used expression data to strengthen the interpretation of previous results and not for primary and discovery analyses.
Shared DNA methylation dynamics in normal and neoplastic B cells
To define CpGs whose methylation values do not change in normal and neoplastic B cells, we obtained CpGs showing differences of less than 0.25 across all normal and neoplastic B cells. Then, we classified them into hyper, partial and hypomethylated CpGs calculating the median of each CpGs for all the samples.
ChIP-seq data collection, analysis and integration
We downloaded and processed ChIP-seq data available from Blueprint73 and from a previous study in ALL74. Particularly, we used Blueprint ChIP-seq data of six histone marks, i.e.H3K4me1, H3K4me3, H3K27ac, H3K36me3, H3K27me3 and H3K9me3 available for 15 normal B cells (6 NBC, 3 GC, 3MBC and 3tPC), 5 MCLs, 7 CLLs and 4 MMs, as well as two DLBCL cell lines, i.e. KARPAS-422 and SUDHL-5 DLBCL. We next integrated these ChIP-seq data using chromHMM software75 as previously described76. Briefly, we generated a B-cell specific chromatin state model with 12 emission states using the 15 normal B cells, corrected for their corresponding input. These 12 chromatin states were ActProm (active promoter, with H3K27ac and H3K4me3 marks), WkProm (weak promoter, with H3K4me1 and H3K4me3 marks), PoisProm (poised promoter, with H3K27me3, H3K4me1 and H3K4me3 marks), StrEnh1 (strong enhancer 1, with H3K27ac, H3K4me1 and H3K4me3 marks), StrEnh2 (strong enhancer 2, with H3K27ac and H3K4me1 marks), WkEnh (weak enhancer, with H3K4me1 mark), TxnTrans (transcription transition, with H3K36me3, H3K27ac and H3K4me1 marks), TxnElong (transcription elongation, with H3K36me3 mark), WkTxn (weak transcription, with low H3K36me3 mark), H3K9me3 (H3K9me3-repressed heterochromatin), H3K27me3 (H3K27me3-repressed heterochromatin) and Het;Low;Sign (low signal heterochromatin, with the absence of all the six histone marks).Next, this model was used to assign the chromatin states in the remaining primary B-cell tumors, namely 5 MCL, 7 CLL, 5 MM, and the 2 DLBCL cell lines. In the case of ALL, we downloaded H3K27ac ChIP-seq data (generated with the ChIP-grade ab4729 from Abcam) from the NALM6 ALL cell line74. We followed the Blueprint pipeline to find H3K27ac peaks http://dcc.blueprint-epigenome.eu/#/md/chip_seq_grch37. To define regulatory regions in MCL, CLL, MM and DLBCL, we used the CHMM genome segmentation. Particularly, we used chromatin states containing H3K27ac, namely ActProm, StrEnh1, StrEnh2 and TxnTrans chromatin states. For ALL, regulatory regions were defined as regions showing H3K27ac peaks. These active regulatory regions were not merged but used in a disease-specific manner in the manuscript. To calculate CHMM enrichments of CpGs sets, we used the CpGs present in the 450k Illumina DNA methylation array as a background. To calculate CpG enrichments in regulatory regions in Fig 2c and Extended Data Fig. 2c, the number of CpGs falling in regulatory regions were compared with the same number of de novo CpGs 10,000 times randomly chosen from the DNA methylome fraction with potential tumor-specific signatures falling in regulatory regions. To select genes associated with regulatory regions (Fig. 2), we obtained gene annotation for all CpGs within regulatory regions using the lluminaHumanMethylation450kanno.lmn12.hg19 R package.
Gene Ontology Analysis
Gene ontology analyses were performed using the “gometh” function within the missMethyl R package available at Bioconductor, which takes into account the differing number of probes per gene present on the 450k array.
Tumor specific DNA methylation signatures
We performed Truncated Principal Component Analysis (PCA) using irlba package available at CRAN. Next, to find specific DNA methylation signatures in each B cell tumor, we filtered out all CpGs showing extensive modulation in B cell differentiation 5. Afterwards, we used the limma package to perform pair-wise comparisons between each B cell tumor entity. For each B cell neoplasia as compared to other B cell tumors, we retained CpGs that showed at least ≥0.25 methylation difference and FDR<0.05 in the same direction in all comparisons. We next classified the identified CpGs as hyper- or hypomethylated considering the methylation status of normal B cells.
Transcription factor binding analysis
We used the PWMEnrich package available at Bioconductor. We focused on CpGs showing specific hypomethylation in each B-cell tumor entity overlapping with regions showing H3K27ac in primary samples of MCL, CLL or MM, and cell lines in the case of ALL (NALM6) and DLBCL (KARPAS-422 and SUDHL-5) (Fig. 2c). We next extended the DNA sequence 100bps (50bps to each side) for each CpG using Bsgenome.Hsapiens.UCSC.hg19 annotation package available at Bioconductor. As a background sequences, we used 100,000 random B-cell independent CpGs. We then calculated the frequency of A, T, C and G bases in the background sequences. Next, we obtained the 537 CORE JASPAR 2018 TFs for Homo sapiens and transformed motifs to Position Weight Matrices (PWM) using previously calculated frequencies of each base to account for biases in the 450k array. We then calculated a lognormal background distribution with tiles of 100 bps to finally perform TFs binding predictions. We retrieved enrichments per group of sequences and the frequency of each TF that belongs to the Top 5% enrichment TFs, i.e. how often a TF is among the top 5% enriched TFs in all the interrogated sequences. We considered TF as relevant when being within the top 5% TFs in at least 10% of the sequences, showing an FDR ≤0.025 and consistently expressed in each respective B-cell tumor.
Construction of the classifier algorithm for B cell tumor subtypes
DNA methylation data for 1,345 samples of B-cell neoplasms was used to build a two-step classifier for the classification of the 5 main B-cell tumor entities (first step) followed by the classification B-cell tumor subtypes (second step, out of the 1,345, 1,013 samples with subtype diagnosis were available). We used the DNA methylation values of 452,679 CpGs including B-cell related and B-cell independent CpGs 5. Of note, to build the classifier we only used CpGs present in both methylation array platforms (450k and EPIC arrays).CpGs with minimal variation (interquartile range below 0.07) were removed in the training series of each one of the five predictors.
The following strategy was used to build the predictor for the main B-cell tumor entities as well as for ALL, MCL and DLBCL tumor subtypes (predictors 1, 2, 3 and 5). In the case of CLL, we used another strategy, which is subsequently described.
- For every class k,
- Rank the CpGs according to the Mann-Whitney U test p-value resulting from the comparison of samples of class k against the samples of all other classes.
- Define the signature of class k as the mean of the methylation values of the top Mk CpGs (or one minus the value for hypomethylated CpGs in class k). In case of ties in the p-value ranking, prioritize the CpG with higher mean DNA methylation change.
Train a support vector machine model with the signatures of the k classes, using a linear kernel and optimizing the cost C by cross-validation. In the case of only two classes (such as MCL or DLBCL, e.g. C1 vs C2, and ABC vs GCB subtypes), the two signatures are redundant and only one is retained.
The number of CpGs included in the signature of each class in 1) ii, vector M = {MALL, …, MGCB}, was chosen by 10-fold stratified cross-validation. Specifically, the above algorithm was repeated at each fold where all combinations of possible Mk values were tested and the values that maximized the balanced accuracy were selected. The tested values ranged from 1 to a different quantity depending on the predictor (4 for the main entities, 5 for the ALL subtypes, 20 for MCL and 20 for DLBCL).
For the classification of the three CLL subtypes (m-CLL, i-CLL, n-CLL), the described 5-CpG classifier10,51 could not be applied as one CpG (cg09637172) is not present in the EPIC array, and therefore, we reanalyzed the data to obtain a new predictor, using the following steps:
Select the 50 CpGs with the lowest Mann-Whitney U test p-value for each pairwise comparison between the three subtypes.
Apply the SVM-RFE algorithm 77 to the subset of CpGs selected in step 1.
Train a support vector machine model with the top MCLL CpGs of step 2, cost C, and a linear kernel.
A similar cross-validation strategy as the previous algorithm was used to optimize the MCLL and C parameters. The tested values were MCLL = {1, 2, …, 20} CpGs and C = 10{−3, −2, …, 3} cost. Extended Data Fig. 3d shows the balanced accuracy and sensitivities of the best performing cost for each number of CpGs.
Finally, we used two strategies to estimate the accuracy of the five predictors: (1) with nested cross-validation in the training series and (2) with a validation series. For the training series, we used 10-fold stratified cross-validation, where the optimization of the M and C parameters was independently performed at each fold using an inner stratified cross-validation step. For the validation series, we used the following data:
For ALL , we used 183 samples already included in the initial analysis (Fig. 1, 2)7 but not used to construct any classifier nor in any of the other analyses of the manuscript. Additionally, we downloaded the following DNA methylation data: GSE7658566 and GSE6922967. For MCL validations, we used DNA methylation data from 58 non-overlapping MCL cases9 (accession code EGAS00001004165). For CLL validation, we collected 450k methylation data for 109 CLL samples from a previous study 11(EGAD00010000871), and 145 CLL with 450k data and 126 CLL with EPIC data kindly provided by Dr. Thorsten Zenz and partially deposited in 78 (EGAD00010000948). Finally, for DLBCL validation we generated DNA profiles with EPIC arrays.
To more accurately represent indetermination in newly obtained samples, not all cross-validated training samples nor validation samples were assigned to an entity/subtype. Specifically, we used the svm function of the e1071 R package to obtain a probability for each entity/subtype in each one of the samples. Next, samples where the maximum probability was below 50% or multiple entities/subtypes (including the true entity) had a probability above 35% were considered unclassified.
In the case of MCL, the classification of the training series into C1 and C2 subtypes was performed using a strategy that mirrored the previously described approach8. Specifically, we first created a PCA space using all of the unfiltered methylation information in the training samples, and identified that the two first components contained most of the information related to the subtype. Then, these two components were used to fit a quadratic discriminant analysis (QDA) model that distinguished the two cell-of-origin subtypes in this new space. Finally, the validation samples were projected into the training PCA space and the fitted QDA model was applied to them. Only samples with either C1 or C2 probability ≥85% were assigned to one of the subtypes. This strategy allowed us to define a cell-of-origin subtype for the validation series using the methylation information as a whole.
Inter-patient DNA methylation heterogeneity
To analyze the variability of DNA methylation data among patients, we identify CpGs with differential methylation in each patient individually. To do this, we compared data from each single patient with the mean in HPC samples, and considered a DNA methylation change for a given CpG when a difference ≥0.25 was reached. Next, to define all the DNA methylation changes occurring in patients diagnosed with a specific B-cell tumor subtype, we selected all CpGs meeting these two criteria; 1) in at least one patient of a specific B cell tumor subtype showing an absolute methylation difference ≥0.25 as compared to HPC, and 2) all other patients in the B cell tumor subtype show the same trend, i.e. towards hypomethylation or hypermethylation.
Construction of the epiCMIT score (epigenetically-determined Cumulative MIToses)
To create the epiCMIT score, we selected all CpGs from 450k array of our entire DNA methylation matrix of normal and neoplastic B-cells (n=1,595) located in inactive regions, particularly in poised promoters (PoisProm, with H3K27me3, H3K4me1 and H3M4me3 marks), in H3K27me3 regions, in H3K9me3 regions, and in low signal heterochromatin (Het;LowSign, absence of any of the six marks analyzed). We divided this set of CpGs into two distinct sets, CpGs located in H3K27me3-repressed regions or PoisProm, and CpGs located in H3K9me3-repressed regions or Het;Low;Sign heterochromatin. We next performed differential DNA methylation analysis between normal B-cells with the lowest and the high proliferative histories, namely HPC and bmPC (step 3, Extended Data Fig. 5a) and we retained CpGs gaining DNA methylation in bmPC in H3K27me3 regions or PoisProm, and CpGs losing DNA methylation in bmPC in H3K9me3 and Het;Low;Sign heterochromatin. In addition, we imposed two key restrictions to these two sets of CpGs. First, CpGs gaining and losing methylation during cell division must respectively show a very low (<=0.1) and very high (>=0.9) methylation levels in in lowly divided cells, i.e. HPCS. Second, we retained only those CpGs showing extensive modulation between the lowly divided HPC and highly divided bmPC cells. This second condition was imposed to maximize the differences in the DNA methylation values upon cell division. With all these restrictions, we ended with 184 CpGs hypermethylated CpGs that were used to build the epiCMIT-hyper score. Conversely, we retained hypomethylated 1,164 CpGs to construct the epiCMIT-hypo mitotic score. These scores were generated using the following formulas:
Finally, to construct the epiCMIT score, we evaluated per sample both epiCMIT-hyper and epiCMIT-hypo scores, and selected the higher of the two:
As the epiCMIT score was built with 450k array data, there are 84 CpGs that are not present in the currently available EPIC array from Illumina (10 epiCMIT-hyper and 74 epiCMIT-hypo). Nonetheless, we showed high correlations between epiCMIT scores calculated with all the original CpGs with those exclusively present in both 450K and EPIC arrays (data not shown).
Determination of epiTOC, MiAge, CIMP and PMDsoloWCGW mitotic clocks and the Horvath chronological clock
To determine epiTOC37, MiAge39, CIMP79, PMDsoloWCGW38 and Horvath50 DNA methylation clocks we used their underlying CpGs overlapping with those present in our curated DNA methylation matrix. Specifically, the number of CpGs were the following: 377 out of the 385 epiTOC CpGs, 261 out of the 268 MiAge CpGs, 88 out of the 89 pan-cancer CIMP CpGs79, 5,595 out of the 6,214 PMDsoloWCGW CpGs and 351 out of the 353 Horvath CpGs. For the epiTOC and MiAge scores, we calculated them as previously indicated 37,39. For CIMP score, we used a set of previously proposed CpGs79 and used the same strategy than the epiCMIT-hyper. In the case of the PMDsoloWCGW mitotic clocks, we applied the same strategy that we used for the epiCMIT-hypo score (explained in the previous section). Finally, we used Horvath to predict age using R as previously reported 50.
Somatic mutations and mutational signature analysis in CLL
The somatic mutations found in the CLL samples used in this study were reported elsewhere 17. We considered driver alterations those reported as such in Puente et. al 2015 and Landau et. al 2015 17,52. In addition to this, a new recurrent driver mutation has been recently added to CLL, namely the U1 spliceosomal RNA 53. We obtained the U1 mutational status for 318 CLL patients already published. For the remaining 172 CLL patients from our analyses, we evaluated the U1 mutational status using rhAmp SNP Assay (Integrated DNA Technology) as previously described53. Next, the mutational signature analysis was performed following a similar framework as the one described in Alexandrov et al43,80. Briefly, de novo signature extraction was performed using a hierarchical Dirichlet process (hdp R package, https://github.com/nicolaroberts/hdp), and extracted signatures were matched to the recently described list of mutational signatures 43 based on cosine similarity and the biological knowledge of each mutational process. Signatures identified through this approach were signature SBS1, SBS5, SBS8, SBS9, SBS17b, and SBS18. Finally, the contribution of each of the previously identified signatures for each sample was measured using a fitting approach (MutationalPatterns R package). To avoid signature bleeding between samples, we iteratively removed one signature after another and the least contributing signature was censored if removal reduced the cosine similarity <0.005, with the exception of signature SBS1 and SBS5, which were always included based on their reported presence in all normal and tumor samples.
Gene Set Enrichments Analysis (GSEA)
In order to perform GSEA analysis in CLLs with different epiCMIT score, we took CLLs samples separated by their cellular origin10,51 (epigenetic groups) above 85% percentile and below 15% percentile of epiCMIT. I-CLL were excluded due to smaller sample size. We performed differential gene expression analysis using limma. We then used fgsea package to perform GSEA analyses using log FC as summary statistic to rank genes. We downloaded 5,501 curated (C2) gene signatures from Molecular Signatures Database v7.0 https://www.gsea-msigdb.org/gsea/index.jsp. We performed GSEA analysis with all these pathways filtering those with less than 5 genes and more than 5,000. We used 10,000 permutations to obtain p-values. We next selected 118 gene expression signatures related to cell proliferation and MYC in an unbiased way. These 118 expression signatures were found in R by regular expression matching with grep() R function using the following expression : grep(“CELL_CYCLE|prolifer|divi|mitotic|_CYCLING|M_PHASE|_MYC_”, names(gene_expression_signatures_names)).
epiCMIT clinical associations
We performed univariate analysis of epiCMIT score for relapse-free survival (RFS), overall survival (OS), and OS after relapse in ALL; OS and Time to First Treatment (TTT) for CLL and OS for MCL using Kaplan Meyer curves with maxstat statistics to define groups with high and low epiCMIT. The hazard ratios and their corresponding p-values are shown when epiCMIT categorization was performed. Finally, epiCMIT was assessed in OS together with ABC and GCB DLBCL transcriptomic subtypes 29. The epiCMIT prognostic value was assessed in presence of other well-established prognostic factors in all diseases with multivariate cox regression models. In ALL, this includes including Hyperdiploid ALLs (HeH), Others (including non-recurrent, undefined, <45chr,>67chr and iAMP21), t(1;19), t(12,21), dic(9;20), t(9;22) and 11q23/MLL. In MCL, we performed the multivariate Cox regression model for OS with epiCMIT together with epigenetic groups C1 and C2 and with age. Finally, in CLL we performed multivariate Cox regression models for TTT and OS with epiCMIT together with age at sampling, epigenetic groups and the total number of driver alterations considering mutations in both studies 17,52. We scaled all mitotic clocks when comparing the prognostic value among them.
Finding CLL driver alterations associated with increased epiCMIT
We analyzed the association of each genetic alteration with epiCMIT in all CLL patients, and in CLL patients belonging to each epigenetic subgroup separately. When evaluating all CLLs together, we modelled epiCMIT score with each genetic alteration using linear regression correcting by epigenetic subgroups. We used t-tests between the levels of epiCMIT in mutated and unmutated patients for each genetic alteration within each epigenetic subgroup. We derived point estimates and 95% confident intervals in both the global analysis for all CLLs and within each epigenetic subgroup for all the tests performed (p-values were corrected using FDR). We finally grouped genetic alterations most significantly associated with epiCMIT with pathways implicated in the pathogenesis of CLL. Treated and untreated patients at the time of sampling were used to perform these analyses.
Statistics and Reproducibility
Sample size and data exclusion criterion is extensively explained at section Quality control, normalization, filtering and annotation of DNA methylation data. The experiments were not randomized. The Investigators were not blinded to allocation during experiments and outcome assessment.
Data availability
DNA methylation and gene expression data that support the findings of this study have been deposited at the European Genome-phenome Archive (EGA) under accession number EGAS00001004640. Previously published DNA methylation data re-analyzed in this study can be found under accession codes: B cells, EGAS00001001196; ALL, GSE16368, GSE47051, GSE7658515, GSE6922916; MCL, EGAS00001001637, EGAS00001004165; CLL, EGAD00010000871, EGAD00010000948; MM, EGAS00001000841; In vitro B-cell differentiation model of naïve B cells from human primary samples, GSE72498. Normalized DNA methylation matrices used for all the analyses in this study are available at: http://resources.idibaps.org/paper/the-proliferative-history-shapes-the-DNA-methylome-of-B-cell-tumors-and-predicts-clinical-outcome. Published gene expression datasets can be found under the accession codes: B cells, EGAS00001001197; ALL, GSE47051; MCL, GSE36000; CLL, EGAS00000000092, EGAD00010000254; MM, GSE19784; In vitro B-cell differentiation model of naïve B cells from human primary samples, GSE72498. ChIP-seq datasets that were re-analyzed here can be found under the accession codes: GSE109377 (NALM6 ALL cell line, n=1) and EGAS00001000326 (15 normal B cells donors, and 5 MCL, 7 CLL and 4 MM patients) available from Blueprint https://www.blueprint-epigenome.eu/. Source data is available for this study. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Code availability
The source code for the DNA methylation classifier of B-cell tumors entities and subtypes and for the calculation of the epiCMIT mitotic clock can be found at https://github.com/Duran-FerrerM/Pan-B-cell-methylome. All other source code supporting the findings of this study are available from the corresponding author on reasonable request.
Extended Data
Extended Data Fig. 1. Analyses related to sample selection and annotation of stably-methylated CpGs.
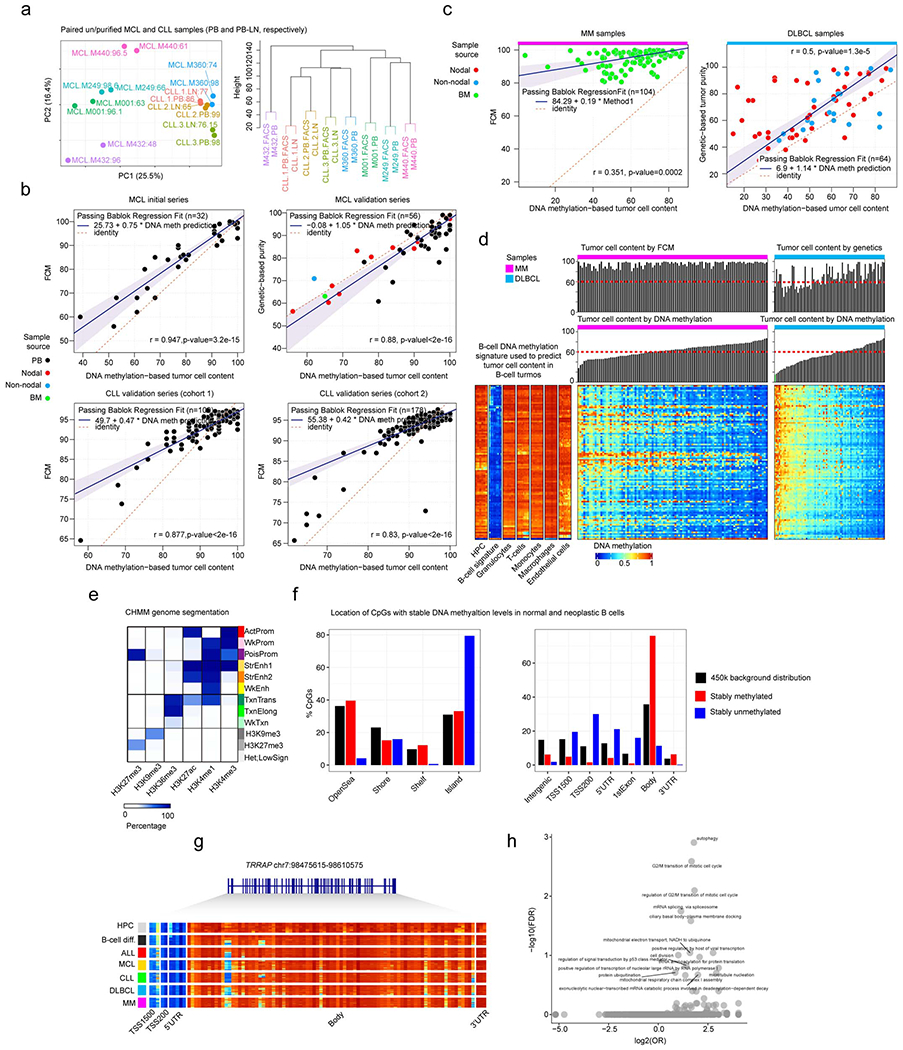
a, Principal component analysis and hierarchical clustering of synchronic unpurified/purified DNA methylation profiles obtained with EPIC array from MCL and CLL patients. Colors represent the same sample, with FCM-based purities highlighted in each sample. MCL, mantle cell lymphoma. CLL, chronic lymphocytic leukemia.
b, Correlations and Passing Bablock regression fits of gold-standard methods for tumor purity prediction (FCM and genetic-based) against DNA methylation-based tumor purity prediction for MCL and CLL patients in initial and validation series. Samples sizes are: MCL initial series, n=32; MCL validation series, n=56; CLL cohort 1, n=109 and CLL cohort 2, n=178 patients. Shaded area represents 95% confidence intervals. Pearson correlation and derived p-values are also shown.
c, Pearson correlations and Passing Bablock regression fits for gold-standard methods for tumor purity predictions (FCM and genetic-based) against DNA methylation-based tumor purity predictions in MM and DLBCL patients. Sample sizes are: MM, n=100 and DLBCL, n=55 patients and are the same as in panel d. Shaded area represents 95% confidence intervals. Pearson correlation and derived p-values are also shown.
d, Pan-B cell DNA methylation signature used to deconvolute DNA methylation data and obtain B-cell tumor purities in B-cell tumors. The DNA methylation levels for the Pan-B-cell DNA methylation signature is shown for microenvironmental cells as well as MM and DLBCL. Bar plots representing DNA-methylation based predictions as well as gold standard-based predictions for MM and DLBCL are represented on the top of the heatmaps.
e, Chromatin state genome segmentation with the CHMM software using the 6 histone marks used in the whole study for normal B cells, MCL, CLL and MM primary cases as well as for KARPAS-422 and SUDHL-5 DLBCL cells lines.
f, Genomic distribution of stably methylated and unmethylated CpGs in normal and neoplastic B cell. Barplots represent single data values.
g, Example gene showing stably unmethylated CpGs at promoters and stably methylated CpGs at gene body in normal and neoplastic B cells. A total of 98 CpGs are shown.
h, Gene ontology analysis of genes showing both stably methylated and stably unmethylated CpGs in normal and neoplastic B cells.
Extended Data Fig. 2. Characterization of tumor-specific DNA methylation signatures.

a, First 9 components of a Principal Component Analysis for normal and neoplastic B cells. Samples sizes are the same as in Fig. 1a. The same sample size applies also for panel b, c and d. Center line, box limits, whiskers and points represent the median, 25th and 75th percentiles, 1.5× interquartile range and individual samples, respectively.
b, Percentages of de novo DNA methylation signatures over the total DNA methylome. All de novo hyper- and hypomethylation from the five B-cell tumors analyzed are considered together to derive each respective percentage.
c, Heatmap showing B-cell tumor-specific hypermethylation and the number of CpGs located at active regulatory regions (marked by H3K27ac). To calculate CpG enrichments in regulatory regions, the number of CpGs falling in regulatory regions were compared with the same number of de novo CpGs 10,100 times randomly chosen from the DNA methylome fraction with potential tumor-specific signatures falling in regulatory regions.
d, Distribution of mean methylation levels of CpGs from de novo B-cell tumor-specific DNA methylation signatures across all normal and neoplastic B cell samples subtypes. The number of samples used to calculate the means is shown in Fig. 1a and the number of CpGs analyzed are those from Fig. 2b.
e, Genomic distribution for de novo DNA methylation changes in B-cell tumors. Barplots represent single data values.
f, Gene expression percentile of TFs showing the most significant p-values and frequencies for TFs binding site predictions (Methods) in de novo hypomethylation signatures in each B-cell tumor from Fig. 2d. Sample sizes for gene expression analyses in tumor samples are the same than in Fig. 4e.
Extended Data Fig. 3. DNA methylation levels and analysis of the sensitivity of the epigenetic classifier of B cell neoplasms.
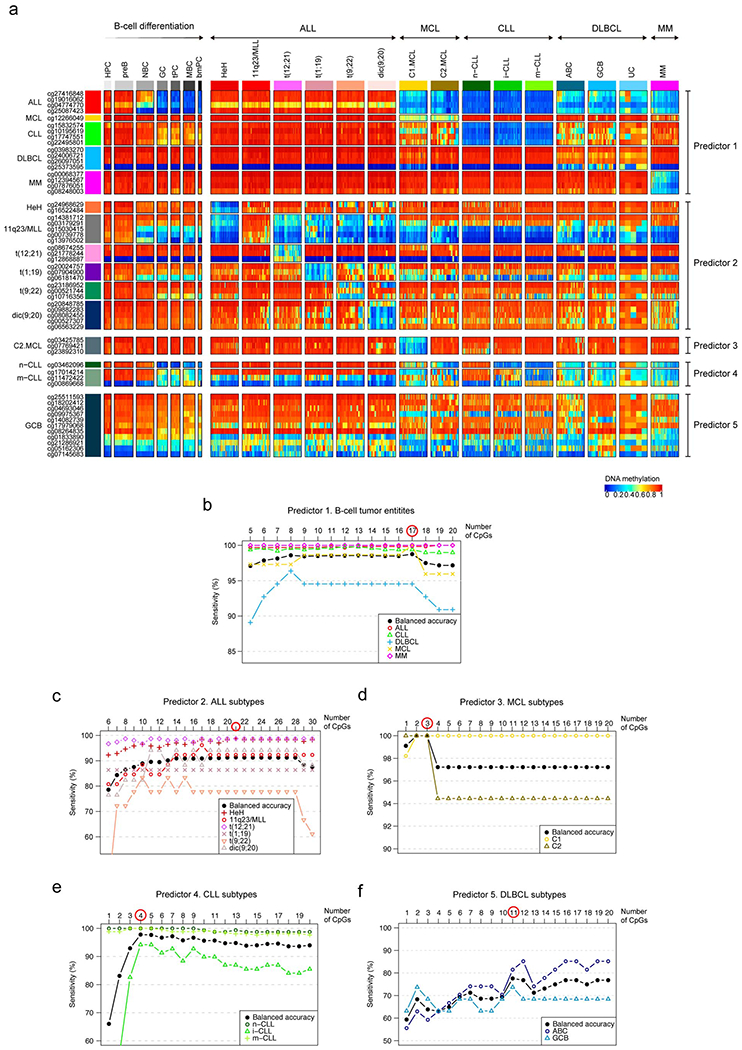
a, DNA methylation levels of all CpGs from the pan-B-cell diagnostic algorithm in normal and neoplastic B cells. Sample sizes are the training samples shown in Fig. 3b.
b, Estimated sensitivity according to the number of CpGs used in the pan-B-cell diagnostic algorithm for the classification of an unknown B-cell tumor into ALL, MCL, CLL, DLBCL or MM (first step of Fig. 3a, predictor 1). The number of CpGs selected for the predictor was chosen by maximizing the highest balanced accuracy and is indicated with a red circle. This strategy was applied also in the remaining 4 predictors to classify B-cell tumor subtypes in panels c, d, e, and f, (second step of Fig. 3a). Each B-cell tumor is represented with different shapes and colors.
c, Estimated sensitivity according to the number of CpGs used in the pan-B-cell diagnostic algorithm (predictor 2 of Fig. 3a) for the classification of ALL into the subtypes HeH, 11q23/MLL, t(12;21), t(1;19), t(9;22) and dic(9;20) while incrementing the number of CpGs (predictor 2 in Fig. 3a).
d, Estimated sensitivity according to the number of CpGs used in the pan-B-cell diagnostic algorithm (predictor 3 of Fig. 3a) for the classification of MCL into the subtypes C1 or C2 while incrementing the number of CpGs (predictor 3 in Fig. 3a).
e, Estimated sensitivity according to the number of CpGs used in the pan-B-cell diagnostic algorithm for the classification of CLL into the subtypes n-CLL, i-CLL or m-CLL while incrementing the number of CpGs (predictor 4 in Fig. 3a).
f, Estimated sensitivity according to the number of CpGs used in the pan-B-cell diagnostic algorithm for the classification of DLBCL into the subtypes ABC and GCB while incrementing the number of CpGs (predictor 5 in Fig. 3a).
Extended Data Fig. 4. Further characterization of patient-specific DNA methylation changes.
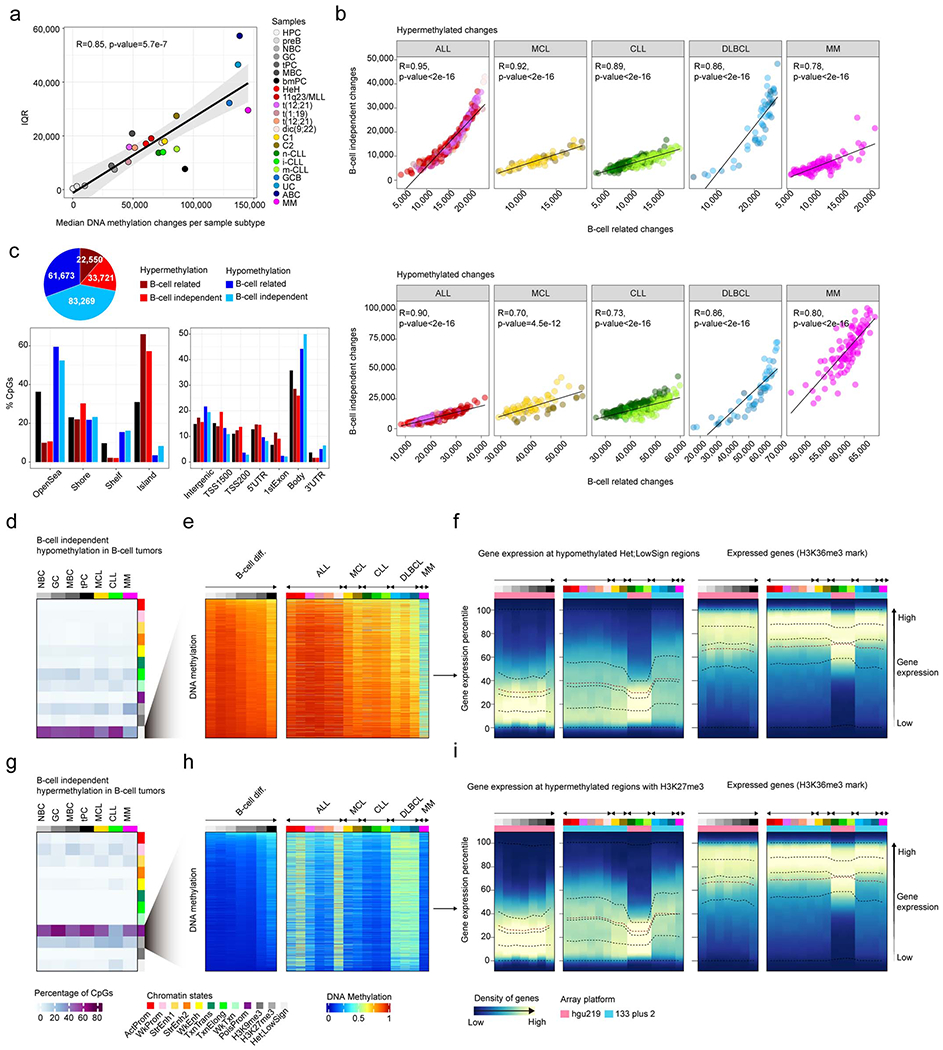
a, Variability of DNA methylation changes measured by the interquartile range (IQR) in normal and neoplastic B cells against the median number of DNA methylation changes per each subtype. R and p-values were derived from linear modelling. Shaded area represents 95% confidence interval.
b, Correlations in all B cell tumors between B-cell independent DNA methylation changes and B-cell related changes for hypermethylation (top) and hypomethylation (bottom) changes. R and p-values were derived from linear models.
c, Number of B-cell related or B-cell independent hyperor hypomethylation in B-cell tumors showing consistent patterns (Methods).
d, B-cell independent CpGs losing DNA methylation in B-cell tumors and the percentages of each chromatin state in normal and neoplastic B-cells. The mean of percentages per sample type is shown. The sample sizes are the same as in Fig. 4c and also apply for panel g.
e , The mean of 2,000 representative CpGs per each sample subtype from panel d is represented.
f, Gene density distributed along the expression percentiles of genes associated with B-cell independent CpGs losing DNA methylation at low signal heterochromatin in B-cell tumors. Expressed genes (H3K36me3) are displayed at right as control. Means within each B-cell subpopulation as well as B-cell tumors are represented.
g, B-cell independent CpGs gaining DNA methylation in B-cell tumors and the percentages in each chromatin state in normal and neoplastic B-cells.
h , The mean of 2,000 representative CpGs per each sample subtype from panel g is represented.
i, Gene density distributed along the expression percentiles of genes associated with B-cell independent CpGs gaining DNA methylation at H3K27me3 regions in B-cell tumors. Expressed genes (H3K36me3) are displayed at right as control. Means within each B-cell subpopulation as well as B-cell tumors are represented. Sample size for DNA methylation analyzes in panels a, b, c, e and h are the same as in Fig. 4a. Samples sizes for gene expression analyses in panels f and i are the same as in Fig. 4e.
Extended Data Fig. 5. Additional analyses performed to validate the epiCMIT.
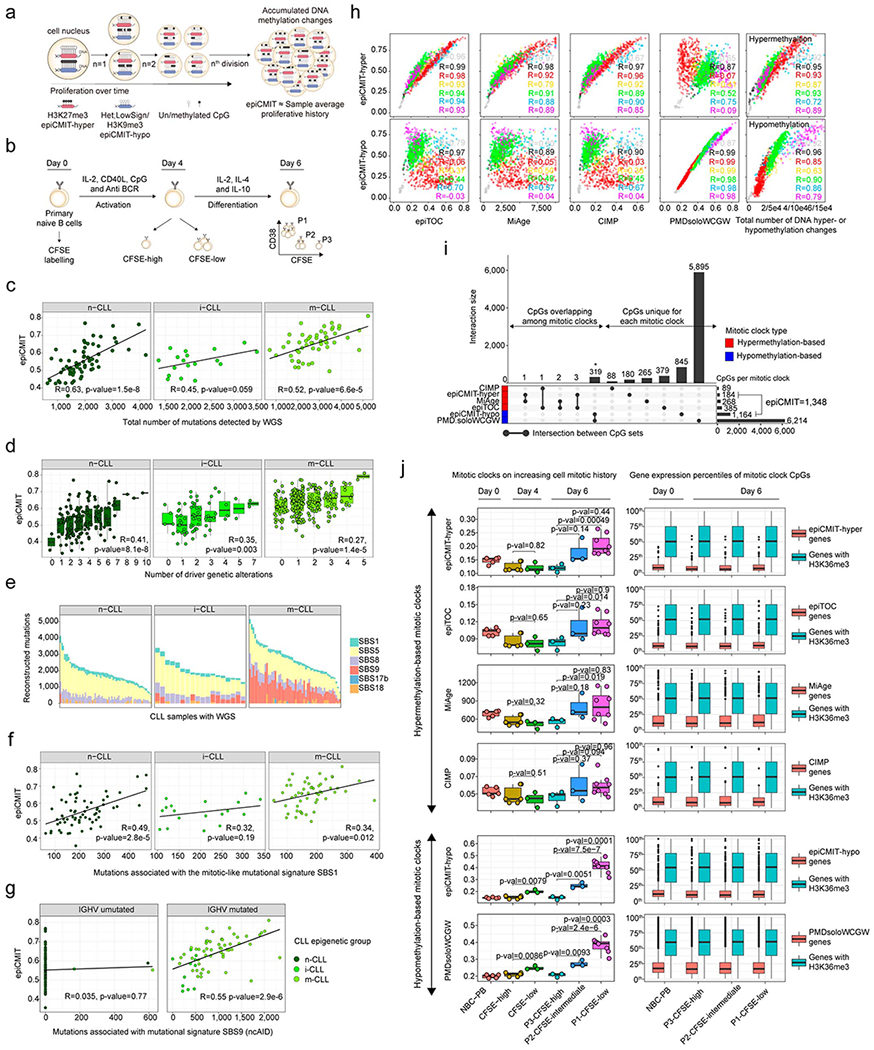
a, Illustrative scheme showing DNA methylation changes upon cell division and how they relate to epiCMIT scores.
b, In vitro B-cell differentiation model used to experimentally validate the epiCMIT score. Primary naïve B cells are differentiated into plasma cells in 6 days. At day 0, primary human B cells are incubated with Carboxyfluorescein succinimidyl ester (CFSE) and harvested with activation and proliferation cocktails necessary for plasma cell differentiation. The epiCMIT was calculated at day 0, day 4 and day 6 in B cells with different proliferative histories based on CFSE dilution.
c, The epiCMIT is correlated with total number of mutations detected by WGS in each CLL epigenetic subtype. R and p-values are derived from linear modelling. 138 CLL patient samples with WGS and DNA methylation data are shown (66 n-CLL, 18 i-CLL and 54 m-CLL). The same sample size applies for panel e, f and g.
d, The epiCMIT is correlated with CLL genomic complexity measured by the total number of driver alterations and thus with mutations with positive selection. Fitted linear regression models and derived R and p-values are shown for each group. The sample size for each number of driver alterations are: 0 drivers: n-CLL, n=2, i-CLL, n=5, m-CLL, n=44; 1 driver: n-CLL, n=14, i-CLL, n=19, m-CLL, n=119; 2 drivers: n-CLL, n=37, i-CLL, n=25, m-CLL, n= 55; 3 drivers: n-CLL, n=38, i-CLL, n= 12, m-CLL, n=28; 4 drivers: n-CLL, n=27, i-CLL, n=4, m-CLL, n=12; 5 drivers: n-CLL, n=23, i-CLL, n=2, m-CLL, n=2; 6 drivers: n-CLL, n=10, i-CLL, n=0, m-CLL, n=0; 7 drivers: n-CLL, n=7, i-CLL, n=2, m-CLL, n=0; 8 drivers: n-CLL, n=1; 9 drivers: n-CLL, n=1; 10 drivers: n-CLL, n=1. For the box plots, center line, box limits, whiskers and points represent the median, 25th and 75th percentiles, 1.5× interquartile range and individual samples, respectively.
e, Mutational signatures found in CLL with available WGS. CLL subtypes are shown separately.
f, The epiCMIT is correlated with the mitotic-like mutational signature SBS1. CLL samples are divided in CLL epigenetic subgroups. R and p-values are derived from linear models.
g, The epiCMIT is correlated with the mitotic-like mutational signatures SBS9. CLL samples are separated with the classical IGHV mutational status (98%). R and p-values are shown for each respective linear model.
h, epiCMIT-hyper CpGs and epiCMIT-hypo mitotic clocks are compared with other hyper- or hypomethylation based mitotic clocks as well as the total number of hyper- (rightmost top) or hypomethylation (rightmost bottom) changes per sample since HPC stage. R from linear models are shown. Samples sizes are the same as in Fig. 4a.
i, Overlap among the CpG used to build each mitotic clock. Barplots represent single data values.
j, Performance of all mitotic clocks in the in vitro B-cell differentiation model from panel c. The fraction of epiCMIT which gain methylation (epiCMIT-hyper) and the fraction that lose DNA methylation (epiCMIT-hypo) were analyzed together with hyper- and hypomethylation-based mitotic clocks, respectively. Biological independent sample sizes are the same as in Fig. 5e. P-values are derived from two-sided t-tests and from biological independent experiments. On the right, expression of genes containing any CpG of each respective mitotic clock as well as genes containing CpGs in H3K36me3 regions are depicted (n=14,598). The number of genes analyzed per each mitotic clock are: epiCMIT-hyper, n=155; epiTOC, n=412; MiAge, n=298; CIMP, n=102; epiCMIT-hypo, n1,123; PMDsoloWCGW, n=4053. For the box plot, center line, box limits, whiskers and points represent the median, 25th and 75th percentiles, 1.5x interquartile range and individual samples, respectively.
Extended Data Fig. 6. Comparison between the epiCMIT mitotic clock and the Horvath aging clock.
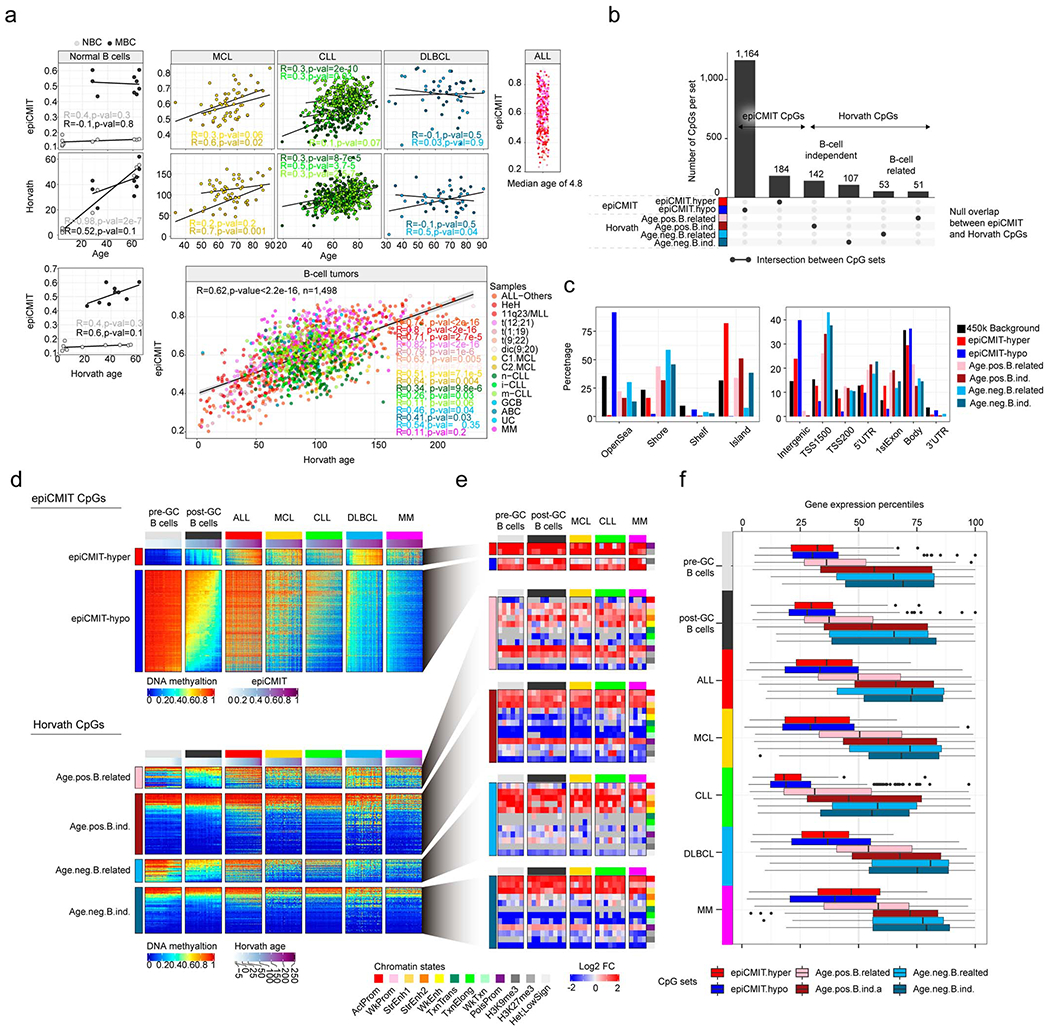
a, Correlations among epiCMIT, age and Horvath-predicted age in normal and neoplastic B cells. Samples sizes are: NBC, n=10 and MBC, n=9 donors; C1 MCL, n=40; C2 MCL, n=17; n-CLL, n=159; i-CLL, n=69; m-CLL, n=260; GCB DLBCL, n=20 and ABC DLBCL, n=28 patients. R and p-value are derived from linear models. Shaded areas represent 95% confidence intervals.
b, epiCMIT and Horvath clocks do not have any CpG in common. CpGs of the Horvath model are divided into positively associated with age (gain of methylation) and negatively associated with age (loss of methylation). In addition, they are further classified into B-cell related or B-cell independent if they are extensively modulated or not during normal B-cell differentiation. Barplots represent single data values.
c, The CpGs used to build the epiCMIT and Horvath clock show distinct genomic locations. Barplots represent single data values.
d, DNA methylation levels of the CpGs from the epiCMIT and Horvath clocks in normal and neoplastic B cells. Sample sizes are the same as in Fig. 4a.
e, The CpGs associated with the epiCMIT and Horvath clocks are located in markedly different chromatin states. Sample sizes are the same as in Fig. 4c.
f, Genes associated with epiCMIT and Horvath CpGs show distinct transcriptional states in normal and neoplastic B cells. Gene probes shared across all normalized matrices from normal and neoplastic B cells were retained and were the following: epiCMIT-hyper, n=60; epiCMIT-hypo, n=327; Age positive B-cell related, n=44; Age positive B-cell independent, n=118; Age negative B-cell related, n=49; Age negative B-cell independent, n=101. For the box plot, center line, box limits, whiskers and points represent the median, 25th and 75th percentiles, 1.5x interquartile range and individual samples, respectively. Sample size are the same as in Fig. 4e.
Extended Data Fig. 7. Additional characterization of the clinical impact of the epiCMIT in B cell tumors.
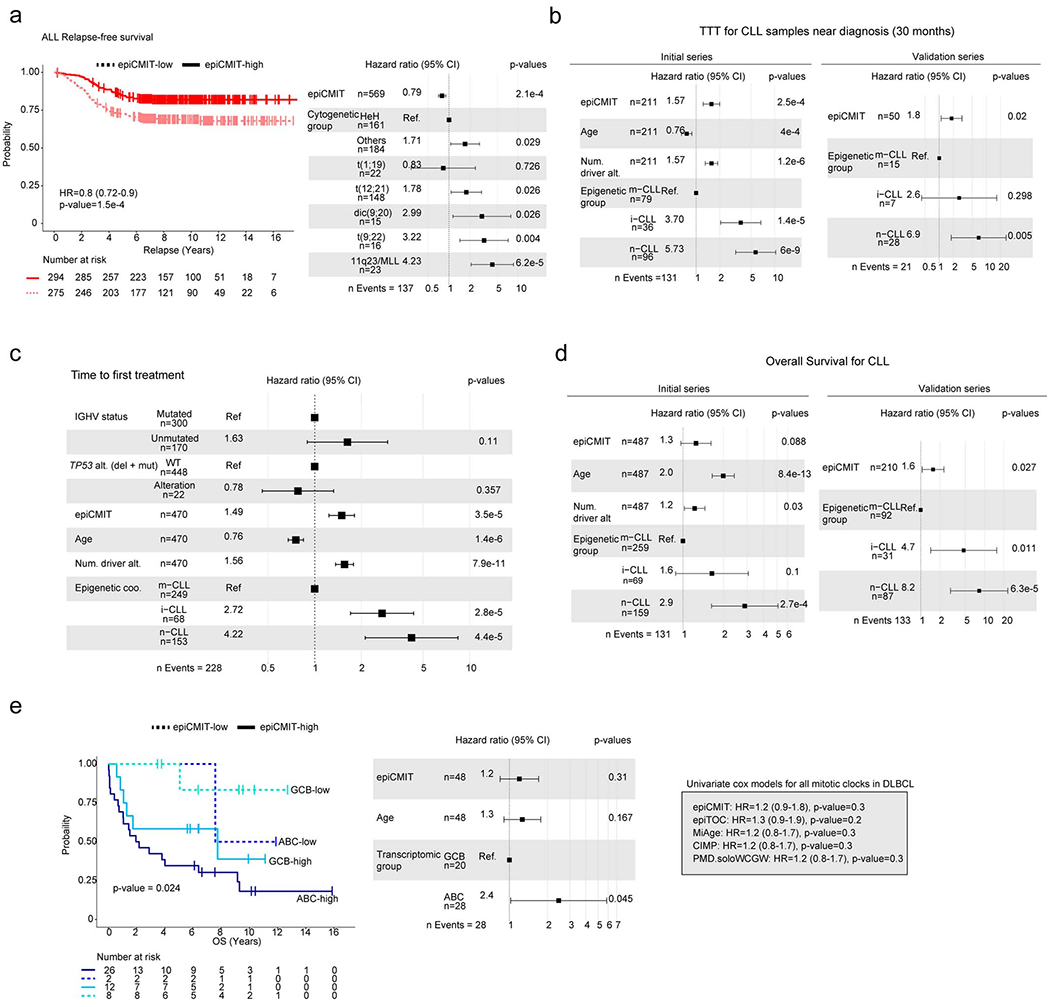
a, Kaplan-Meier curves for relapse-free survival in ALL patients with low or high epiCMIT according to the maxstat rank statistics-based cutoff. Hazard ratio and p-value for the univariate Cox regression model are shown. A multivariate Cox regression model with epiCMIT as continuous variable and ALL cytogenetic groups is shown on the right. Hazard ratio for epiCMIT correspond to 0.1 increments.
b, epiCMIT preserves its prognostic value in multivariate Cox regressions for time to first treatment in CLL patients whose samples were acquired at maximum 30 months after diagnosis both in initial and validation series.
c, epiCMIT shows independent prognostic value from major prognostic variables in CLL including IGHV mutational status and TP53 alterations (deletions and mutations) in multivariate Cox regressions for time to first treatment (TTT).
d, Multivariate cox regression models in initial and validation CLL series for overall survival with epiCMIT and important prognostic variables.
e, Kaplan-Meier curves for overall survival in GCB and ABC DLBCL patients with low or high epiCMIT according to the maxstat rank statistics-based cutoff. A multivariate Cox regression model with epiCMIT as continuous variable, the DLBCL subtype and age is shown on the right. Hazard ratio for epiCMIT correspond to 0.1 increments. On the right, univariate cox regression model for all mitotic clocks.
Extended Data Fig. 8. Clinical impact of the epiCMIT as compared to other mitotic clocks.
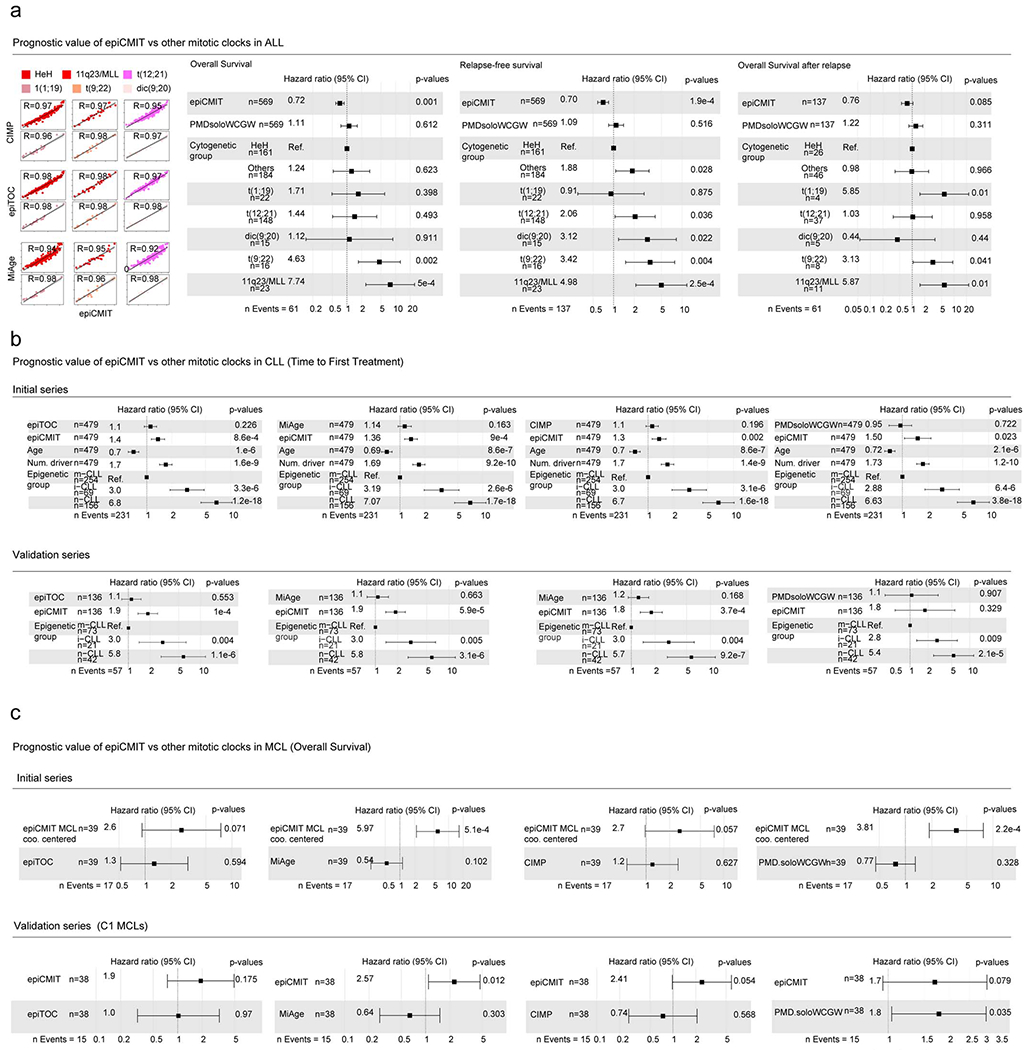
a, On the left, epiCMIT and hypermethylation-based mitotic clocks are highly correlated in ALL, creating a collinearity phenomenon in multivariate cox regression models with multiple mitotic clocks. On the right, multivariate Cox regression models with epiCMIT and PMDsoloWCGW mitotic clocks and ALL cytogenetic subgroups for overall survival, relapse-free survival and overall survival after relapse.
b, In CLL, epiCMIT shows superior prognostic value in multivariate cox models for time to first treatment than all the other mitotic clocks in both initial and validation series.
c, In MCL, epiCMIT shows an overall superior prognostic value in multivariate cox models for overall survival in both initial series (with C1 and C2 MCL subtypes) and in the validation series, which only contain C1 MCL subtypes. In the initial series, MCL subtypes with different cellular origin were not introduced in multivariate Cox regression models due to few events, and thus the epiCMIT of each MCL patient was centered according to its cellular origin (C1 or C2) to account for normal B-cell development epiCMIT (Fig. 6a).
Extended Data Fig. 9. Additional data regarding the link between the epiCMIT and genetic changes in CLL.
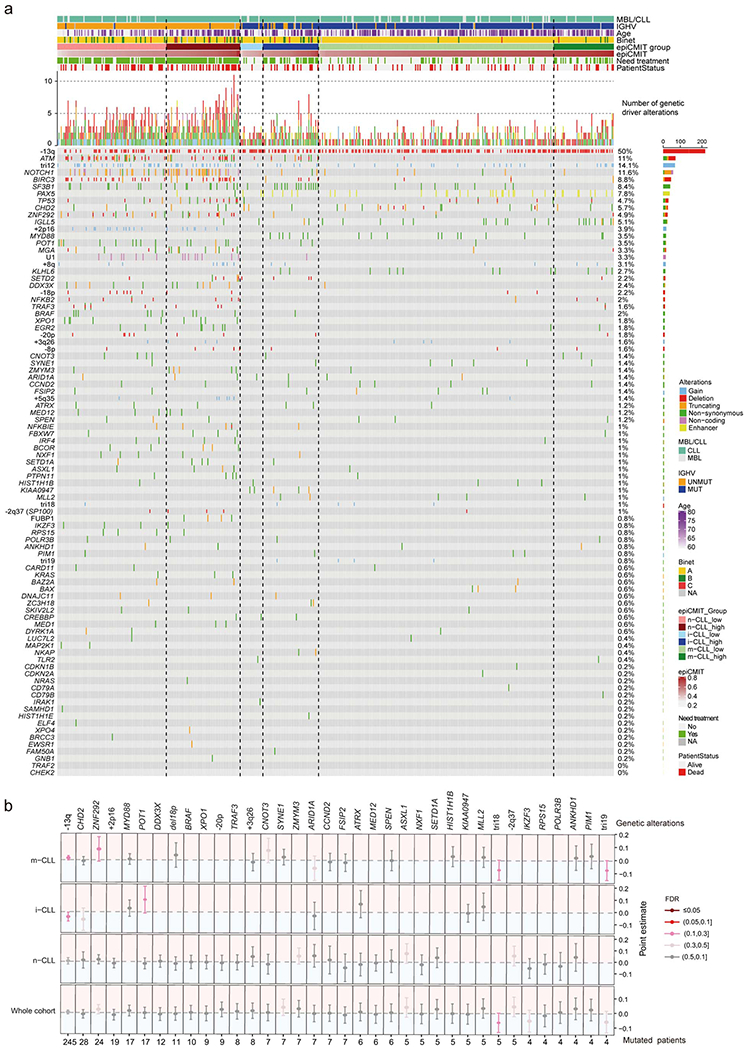
a, Oncoprint showing all genetic driver alterations considered in the whole CLL initial series composed by 490 CLL patient samples grouped by epigenetic subtypes and ordered according to increasing levels of epiCMIT (from left to right within each epigenetic subgroup). Other clinico-biological features including MBL or CLL, IGHV status, Age, Binet stage, epiCMIT subgroups based on maxstat rank statistic, need for treatment and patient status are shown. Distinct genetic driver alterations are depicted with different colors and shapes. The percentage of mutated patients and number of mutated patients for each alteration is shown at right.
b, Driver genetic alterations without clear associations with epiCMIT. Analyses were done in the whole cohort as well as within each epigenetic subgroup. Point estimates with 95% confidence intervals were derived in the whole cohort using linear modelling between epiCMIT and alterations adjusted for CLL subtypes, and with two-sided t-tests within CLL subtypes. Point estimates then represent the coefficient of each respective alteration in each corresponding linear model (whole cohort analysis) or the difference between means (CLL subtypes analysis). Point estimates are color-coded according to FDR correction. Treated and untreated patients at the moment of sampling were considered for these analyses.
Supplementary Material
ACKNOWLEDGEMENTS
This research was funded by the European Union’s Seventh Framework Programme through the Blueprint Consortium (grant agreement 282510), the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Project BCLLATLAS, grant agreement 810287), Generalitat de Catalunya Suport Grups de Recerca AGAUR 2017-SGR-1142 (to E.C.) and 2017-SGR-736 (to J.I.M.-S.), Ministerio de Ciencia, Innovación y Universidades of the Spanish Government (MCIU), Grants RTI2018-094274-B-I00 (to E.C.) and SAF2017-86126-R (to J.I.M.-S.) as well as Proyecto Medicina Personalizada PERMED (Grant PMP15/00007), which is part of Plan Nacional de I+D+I and is co-financed by the ISCIII-Sub-Directorate General for Evaluation and the European Regional Development Fund (FEDER-“Una manera de Hacer Europa”), CIBERONC (CB16/12/00225, CB16/12/00334, CB16/12/00236, and CB16/12/00489), the Accelerator award CRUK/AIRC/AECC joint funder-partnership, research funding from Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III PI17/01061 (SB), Ministerio de Ciencia, Innovación y Universidades (MCIU), RTI2018-094274-B-I00, SAF2015-64885-R (EC), the NIH grant number 1 P01CA229100 (EC), and the European Regional Development Fund “Una manera de fer Europa”, CERCA Programme/Generalitat de Catalunya. FN is supported by a pre-doctoral fellowship of the Ministerio de Economía y Competitividad (MINECO, BES-2016-076372). E.C. is an Academia Researcher of the “Institució Catalana de Recerca i Estudis Avançats” (ICREA) of the Generalitat de Catalunya. This work was partially developed at the Centro Esther Koplowitz (CEK, Barcelona, Spain). We thank Francesco Maura for his help with the analysis of mutational signatures.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
Editor summary:
Martin-Subero and colleagues analyze DNA methylation patterns in B cell tumors and developmental cells-of-origin, and develop epiCMIT, a methylation-based mitotic clock with prognostic relevance.
References
- 1.Roy N & Hebrok M Regulation of Cellular Identity in Cancer. Dev. Cell 35, 674–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoadley KA et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173, 291–304.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, J. T WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. (International Agency for Research on Cancer (IARC), 2017). [Google Scholar]
- 4.Luo C, Hajkova P & Ecker JR Dynamic DNA methylation: In the right place at the right time. Science 361, 1336–1340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kulis M et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat. Genet 47, 746–756 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nordlund J et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome biology 14, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee S-T et al. Epigenetic remodeling in B-cell acute lymphoblastic leukemia occurs in two tracks and employs embryonic stem cell-like signatures. Nucleic Acids Res. 43, 2590–602 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Queirós AC et al. Decoding the DNA Methylome of Mantle Cell Lymphoma in the Light of the Entire B Cell Lineage. Cancer Cell 30, 806–821 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nadeu F et al. Genomic and epigenomic insights into the origin, pathogenesis and clinical behavior of mantle cell lymphoma subtypes. Blood (2020). doi: 10.1182/blood.2020005289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kulis M et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet 44, 1236–1242 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Oakes CC et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat. Genet (2016). doi: 10.1038/ng.3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaknovich R et al. DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood 116, e81–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agirre X et al. Whole-epigenome analysis in multiple myeloma reveals DNA hypermethylation of B cell-specific enhancers. Genome Res. 25, 478–87 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaiser MF et al. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood 122, 219–226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oakes CC & Martin-Subero JI Insight into origins, mechanisms & utility of DNA methylation in B cell malignancies. Blood 132, blood-2018-02-692970 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Ziller MJ et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 500, 477–81 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puente XS et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature (2015). doi: 10.1038/nature14666 [DOI] [PubMed] [Google Scholar]
- 18.Karube K et al. Integrating genomic alterations in diffuse large B-cell lymphoma identifies new relevant pathways and potential therapeutic targets. Leukemia 675–684 (2017). doi: 10.1038/leu.2017.251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stadler MB et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 480, 490–495 (2011). [DOI] [PubMed] [Google Scholar]
- 20.Somasundaram R, Prasad MAJ, Ungerbäck J & Sigvardsson M Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood 126, 144–152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sánchez-Tilló E et al. The EMT activator ZEB1 promotes tumor growth and determines differential response to chemotherapy in mantle cell lymphoma. Cell Death Differ. 21, 247–257 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolf C et al. NFATC1 activation by DNA hypomethylation in chronic lymphocytic leukemia correlates with clinical staging and can be inhibited by ibrutinib. Int. J. Cancer 142, 322–333 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Blonska M et al. Jun-regulated genes promote interaction of diffuse large B-cell lymphoma with the microenvironment. Blood 125, 981–991 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huerta-Yepez S et al. Overexpression of Yin Yang 1 in bone marrow-derived human multiple myeloma and its clinical significance. Int. J. Oncol 45, 1184–1192 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Sprynski AC et al. Insulin is a potent myeloma cell growth factor through insulin/IGF-1 hybrid receptor activation. Leukemia 24, 1940–1950 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riz I & Hawley RG Increased expression of the tight junction protein TJP1/ZO-1 is associated with upregulation of TAZ-TEAD activity and an adult tissue stem cell signature in carfilzomib-resistant multiple myeloma cells and high-risk multiple myeloma patients. Oncoscience 4, 79–94 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herath NI, Rocques N, Garancher A, Eychène A & Pouponnot C GSK3-mediated MAF phosphorylation in multiple myeloma as a potential therapeutic target. Blood Cancer J. 4, e175–e175 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Navarro A et al. Improved classification of leukemic B-cell lymphoproliferative disorders using a transcriptional and genetic classifier. Haematologica 102, 360–363 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alizadeh AA et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Chapuy B et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med 24, 679–690 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitz R et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med 378, 1396–1407 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aran D, Toperoff G, Rosenberg M & Hellman A Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet 20, 670–680 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Beerman I et al. Proliferation-dependent alterations of the DNA methylation landscape underlie hematopoietic stem cell aging. Cell Stem Cell 12, 413–25 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Landan G et al. Epigenetic polymorphism and the stochastic formation of differentially methylated regions in normal and cancerous tissues. Nat. Genet 44, 1207–14 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Siegmund KD, Marjoram P, Woo Y-J, Tavaré S & Shibata D Inferring clonal expansion and cancer stem cell dynamics from DNA methylation patterns in colorectal cancers. Proc. Natl. Acad. Sci. U. S. A 106, 4828–4833 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spencer DH et al. CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell 168, 801–816.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Z et al. Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol. 17, 205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou W et al. DNA methylation loss in late-replicating domains is linked to mitotic cell division. Nat. Genet 50, 591–602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Youn A & Wang S The MiAge Calculator: a DNA methylation-based mitotic age calculator of human tissue types. Epigenetics 13, 192–206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berman BP et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet 44, 40–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vandiver AR, Idrizi A, Rizzardi L, Feinberg AP & Hansen KD DNA methylation is stable during replication and cell cycle arrest. Sci. Rep 5, 1–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caron G et al. Cell-Cycle-Dependent Reconfiguration of the DNA Methylome during Terminal Differentiation of Human B Cells into Plasma Cells. Cell Rep. 13, 1059–71 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Alexandrov LB et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Issa J CpG island methylator phenotype in cancer. Nat. Rev. Cancer 4, 988–93 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Rakyan VK et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 20, 434–439 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teschendorff AE et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 20, 440–6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bell CG et al. DNA methylation aging clocks: challenges and recommendations. Genome Biol. 20, 249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Field AE et al. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell 71, 882–895 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Horvath S & Raj K DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet (2018). doi: 10.1038/s41576-018-0004-3 [DOI] [PubMed] [Google Scholar]
- 50.Horvath S DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Queirós a C. et al. A B-cell epigenetic signature defines three biological subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia 598–605 (2015). doi: 10.1038/leu.2014.252 [DOI] [PubMed] [Google Scholar]
- 52.Landau DA et al. Mutations driving CLL and their evolution in progression and relapse. Nature 526, 525–30 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shuai S et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 574, 712–716 (2019). [DOI] [PubMed] [Google Scholar]
- 54.Rodríguez-Paredes M et al. Methylation profiling identifies two subclasses of squamous cell carcinoma related to distinct cells of origin. Nat. Commun 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gaiti F et al. Epigenetic evolution and lineage histories of chronic lymphocytic leukaemia. Nature (2019). doi: 10.1038/s41586-019-1198-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meir Z, Mukamel Z, Chomsky E, Lifshitz A & Tanay A Single-cell analysis of clonal maintenance of transcriptional and epigenetic states in cancer cells. Nat. Genet (2020). doi: 10.1038/s41588-020-0645-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Borssén M et al. DNA methylation holds prognostic information in relapsed precursor B-cell acute lymphoblastic leukemia. Clin. Epigenetics 10, 31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sandoval J et al. Genome-wide DNA methylation profiling predicts relapse in childhood B-cell acute lymphoblastic leukaemia. Br. J. Haematol 160, 406–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rhein P et al. Gene expression shift towards normal B cells, decreased proliferative capacity and distinct surface receptors characterize leukemic blasts persisting during induction therapy in childhood acute lymphoblastic leukemia. Leukemia 21, 897–905 (2007). [DOI] [PubMed] [Google Scholar]
- 60.Oakes CC et al. Evolution of DNA Methylation Is Linked to Genetic Aberrations in Chronic Lymphocytic Leukemia. Cancer Discov. 4, 348–361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
METHODS REFERENCES
- 61.Reinius LE et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One 7, e41361 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vento-Tormo R et al. IL-4 orchestrates STAT6-mediated DNA demethylation leading to dendritic cell differentiation. Genome Biol. 17, 4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brönneke S et al. DNA methylation regulates lineage-specifying genes in primary lymphatic and blood endothelial cells. Angiogenesis 15, 317–329 (2012). [DOI] [PubMed] [Google Scholar]
- 64.Aryee MJ et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30,1363–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maksimovic J, Gordon L & Oshlack A SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 13, R44 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bergmann AK et al. DNA methylation profiling of pediatric B-cell lymphoblastic leukemia with KMT2A rearrangement identifies hypomethylation at enhancer sites. Pediatr. Blood Cancer 64, 1–5 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Gabriel AS et al. Epigenetic landscape correlates with genetic subtype but does not predict outcome in childhood acute lymphoblastic leukemia. Epigenetics 10, 717–726 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Houseman EA et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 13, 86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scott DW & Gascoyne RD The tumour microenvironment in B cell lymphomas. Nat. Rev. Cancer 14, 517–534 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Teschendorff AE & Relton CL Statistical and integrative system-level analysis of DNA methylation data. Nat. Rev. Genet 19, 129–147 (2018). [DOI] [PubMed] [Google Scholar]
- 71.Navarro A et al. Molecular subsets of mantle cell lymphoma defined by the IGHV mutational status and SOX11 expression have distinct biologic and clinical features. Cancer Res. 72, 5307–5316 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Broyl A et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. October 116, 2543–2553 (2011). [DOI] [PubMed] [Google Scholar]
- 73.Stunnenberg HG, Human Epigenome Consortium & Hirst M The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery. Cell 167, 1145–1149 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Debaize L et al. Interplay between transcription regulators RUNX1 and FUBP1 activates an enhancer of the oncogene c-KIT and amplifies cell proliferation. Nucleic Acids Res. 46, 11214–11228 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ernst J & Kellis M ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beekman R et al. The reference epigenome and regulatory chromatin landscape of chronic lymphocytic leukemia. Nat. Med 24, 868–880 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Le Thi HA, Nguyen VV & Ouchani S Gene selection for cancer classification using DCA. Lect. Notes Comput. Sci. (including Subser. Lect Notes Artif. Intell. Lect. Notes Bioinformatics) 5139 LNAI, 62–72 (2008). [Google Scholar]
- 78.Dietrich S et al. Drug-perturbation-based stratification of blood cancer. J. Clin. Invest 128, 427–445 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sánchez-Vega F, Gotea V, Margolin G & Elnitski L Pan-cancer stratification of solid human epithelial tumors and cancer cell lines reveals commonalities and tissue-specific features of the CpG island methylator phenotype. Epigenetics and Chromatin 8, 1–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maura F et al. A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun (2019). doi: 10.1038/s41467-019-11037-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA methylation and gene expression data that support the findings of this study have been deposited at the European Genome-phenome Archive (EGA) under accession number EGAS00001004640. Previously published DNA methylation data re-analyzed in this study can be found under accession codes: B cells, EGAS00001001196; ALL, GSE16368, GSE47051, GSE7658515, GSE6922916; MCL, EGAS00001001637, EGAS00001004165; CLL, EGAD00010000871, EGAD00010000948; MM, EGAS00001000841; In vitro B-cell differentiation model of naïve B cells from human primary samples, GSE72498. Normalized DNA methylation matrices used for all the analyses in this study are available at: http://resources.idibaps.org/paper/the-proliferative-history-shapes-the-DNA-methylome-of-B-cell-tumors-and-predicts-clinical-outcome. Published gene expression datasets can be found under the accession codes: B cells, EGAS00001001197; ALL, GSE47051; MCL, GSE36000; CLL, EGAS00000000092, EGAD00010000254; MM, GSE19784; In vitro B-cell differentiation model of naïve B cells from human primary samples, GSE72498. ChIP-seq datasets that were re-analyzed here can be found under the accession codes: GSE109377 (NALM6 ALL cell line, n=1) and EGAS00001000326 (15 normal B cells donors, and 5 MCL, 7 CLL and 4 MM patients) available from Blueprint https://www.blueprint-epigenome.eu/. Source data is available for this study. All other data supporting the findings of this study are available from the corresponding author on reasonable request.