

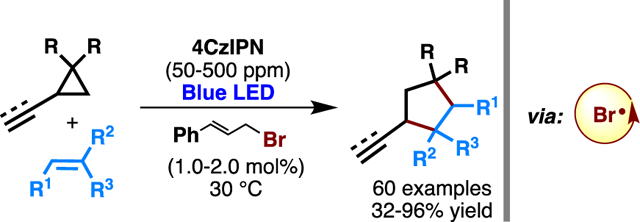
Abstract
We report here a mild, safe, and user-friendly bromine radical catalysis system that enables efficient [3 + 2] cycloaddition of diversely substituted vinyl- and ethynylcyclopropanes with a broad range of alkenes, including drug-like molecules and pharmaceuticals. Key to the success is the use of photosensitizing triplet-state β-fragmentation of a judiciously selected precatalyst, cinnamyl bromide, to generate bromine radicals in a controlled manner using parts per million-level photocatalyst (i.e., 4CzIPN) loading.
Keywords: catalysis, bromine radical, photocatalysis, energy transfer, cycloaddition
Graphical Abstract
Excited-state molecules, compared to those of the ground state, possess distinct electronic configurations, which alter the spin-multiplicity and bond strength, thus exhibiting unique chemical properties and reactivity.1 Although direct photoexcitation is straightforward to access excited-state reactivity, the majority of organic molecules require ultraviolet (UV) light irradiation and quartz reaction vessels that challenge practicality, selectivity, and functional group tolerance.2 Recently, visible-light photocatalysis has emerged as a sustainable and powerful tool to access excited-state reactivity under mild conditions, enabling numerous bond-forming reactions.3 Most examples have utilized single electron-transfer (ET) mechanisms, that is, photoredox catalysis,4 while transformations proceeding through energy transfer (EnT) catalysis, that is, photosensitization,5 are relatively underdeveloped. EnT catalysis is unique in that it largely relies on the relative triplet energies of the photocatalyst (PC) and the substrates. Upon photosensitization, the partial diradical character of a substrate may significantly lower the kinetic barriers toward otherwise disfavored transformations, such as [2 + 2] photocycloadditions,6 photoisomerization of alkenes,7 and others.8
For billions of years nature has utilized EnT to convert solar energy into chemical energy (Figure 1a).9 This synergy has inspired synthetic chemists to develop excited-state organometallic catalysis via photosensitization as a novel activation pathway.10,11 A paradigm of such catalysis is triplet excited-state NiII complex-catalyzed C–C and C–X (X = O, N) cross coupling reactions, (Figure 1b).11 Another elegant example employed two-center photocatalysis for the generation and trapping of (hetero)aryl radicals.12 Specifically, Ru(bpy)2Cl2 was used to absorb visible light and transfer the energy to pyrene, enabling the efficient single-electron reduction of (hetero)aryl halides and triflates (Figure 1c). Although the concept of photosensitization-initiated catalysis has been established as a robust strategy with great synthetic potential, the excited-state activation mode and further application in discovering valuable and unique bond-forming reactions are still in their infancy.
Figure 1.
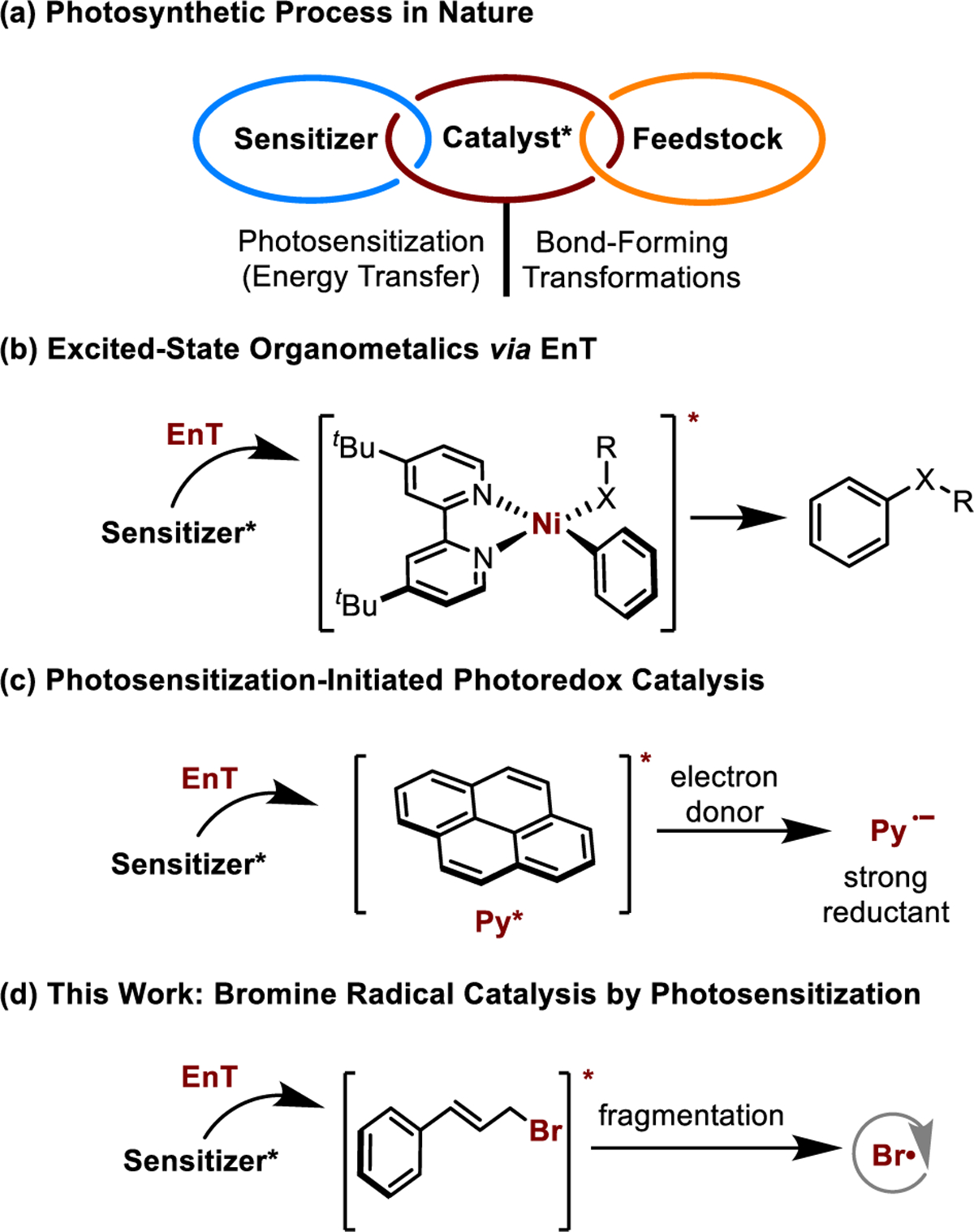
Photosensitization-Initiated Excited-State Catalysis
Heteroatom-centered radicals, for example, bromine ity.13 One of the intriguing features of the bromine radical is that it can reversibly add to carbon–carbon π-bonds (via addition and elimination).14 Exploiting this reversibility, we envisioned a bromine radical catalysis system for the activation of alkenes or alkynes. To the best of our knowledge, such catalysis still remains unknown. The immediate challenges to realize this bromine radical catalysis is the need for mild and controlled generation of bromine radicals. Two classical strategies to deliver a bromine radical includes (i) photolysis of molecular bromine (Br2), a highly toxic, corrosive, and volatile liquid,15 and (ii) thermolysis of N-bromosuccinimide (NBS), mean while generating reactive succinimide radicals that can abstract hydrogen atoms from weak C–H bonds (e.g., allylic C–H bonds).16 The resulting carbon radical, after hydrogen atom abstraction (HAT), may terminate the bromine radical. In light of the [2 + 2] photocycloaddition of styrene derivatives,8a we hypothesized that, upon photosensitization, the partial diradical character of an excited-state styrene derivative, for example, cinnamyl bromide, would allow β-fragmentation to liberate a bromine radical that may be intercepted for catalysis (Figure 1d).17
[3 + 2] cycloaddition of vinyl cyclopropanes and alkenes is a typical reaction to evaluate the addition/elimination radical replay reactivity. It is important to note that such a cycloaddition has been well-established through classical thiyl18 and tin19 radical catalysis. However, several limitations still remain. First, commonly used precursors of tin or thiyl radicals are highly toxic and smelly, including tin hydride, thiols, or disulfides. Second, harsh conditions for radical initiation, such as high temperature or UV light irradiation, negatively impacts practicality. Here, we report a method for mildly controlling bromine radicals through parts per million (ppm) level EnT photocatalysis, which enables efficient cycloaddition of vinyl- and ethynylcyclopropane and alkenes with excellent functional group tolerance.
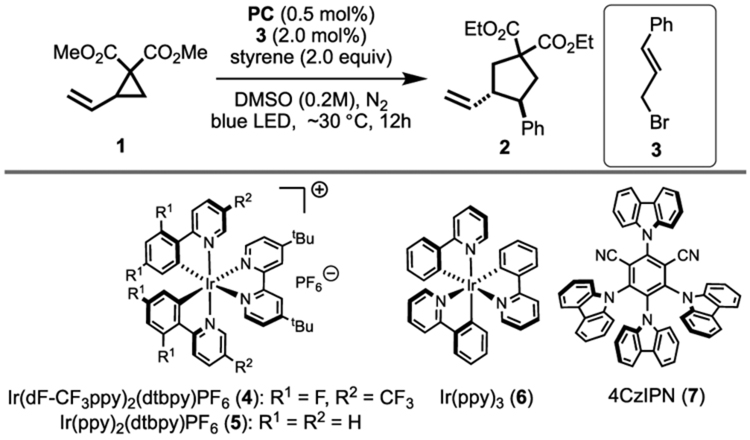
Initial hypothesis validation began with investigation of the reaction between 1,1-dimethoxycarboxyl-2-vinylcyclopropane (1) and styrene as the substrates in dimethyl sulfoxide (DMSO), and 34 W blue light-emitting diode (LED) as the light source (Table 1). Inspired by MacMillan’s photoredox strategy to form bromine radicals,20 catalyst combinations of 4 with various bromide salts in different solvents were evaluated (Table S1). Unfortunately, none of these experiments resulted in any conversion after 12 h. We then turned our attention to the strategy of photosensitization to generate bromine radicals. Because of the possibility of recombination of the bromine and carbon radicals, the use of sub-stoichiometric quantitites of 3 should favor high conversion. Gratifyingly, the [3 + 2] cycloaddition of 1 (1.0 equiv) and styrene (2.0 equiv) proceeded in the presence of 4 (0.5 mol %) and 3 (2.0 mmol %) to yield the cyclopentane product 2 in 95% NMR yield (Table 1, entry 1). The subsequent examination of popular photocatalysts revealed a correlation between reaction efficiency and the PCs triplet energy (ET). More specifically, reactions utilizing PCs with higher ET resulted in higher yields, which is consistent with the photoinduced EnT mechanism (entries 1, 2, and 4). The donor–acceptor fluorophore 4CzIPN (7)21 provides comparable yield to the precious iridium-based photocatalyst 4. We chose 7 for further optimization. Importantly, no conversion was observed in the absence of light, PC, or 3 (entries 6–8). Moreover, attempts on lower catalyst loadings were successful such that 0.005 mol % (50 ppm) of 7 and 1.0 mol % of 3 enabled the formation of 2, as a 70:30 (trans/cis) mixture of diastereomers, in 88% isolated yield (entry 10), highlighting the robustness of this photocatalytic system.
Table 1.
Optimization of Conditions for Visible Light-Gated Bromine Radical-Catalyzed [3 + 2] of Vinylcyclopropane 1 and Styrenea
 | ||||
|---|---|---|---|---|
| entry | PC | ET (kcal/mol) | [Br] | yieldb (%) |
| 1 | 4 | 61 | 3 | 95 |
| 2 | 5 | 49 | 3 | 11 |
| 3 | 6 | 55 | 3 | 0 |
| 4 | 7 | 58 | 3 | 93 |
| 5c | 7 | 58 | 3 | 0 |
| 6 | – | – | 3 | 0 |
| 7 | 7 | 58 | – | 0 |
| 8d,e | 7 | 58 | 3 | 93 |
| 9d,f | 7 | 58 | 3 | 92(88)g |
| 10d,h | 7 | 58 | 3 | 30 |
All reactions were Cycloaddition performed on 0.2 mmol scale. Regardless of reaction conditions, dr was 70:30.
Determined by 1H NMR using trimethoxylbenzene as an internal standard.
No light.
1.0 mol % 3 was used.
0.1 mol % 4CzIPN was used.
50 ppm 4CzIPN was used.
Isolated yield.
10 ppm 4CzIPN was used.
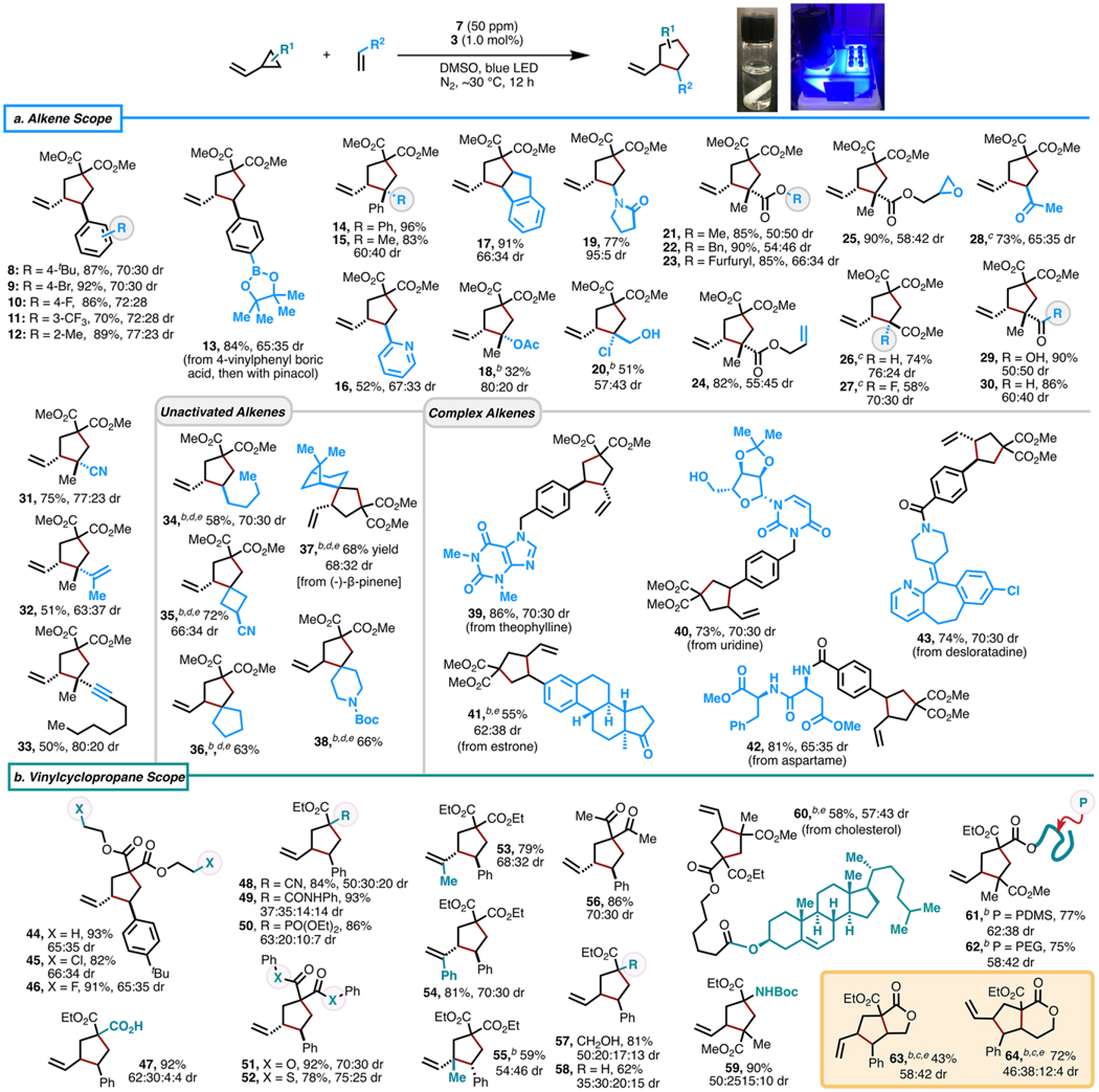
With optimal conditions in hand, we investigated the substrate scope (Table 2). Generally, a broad range of vinylcyclopropanes and alkenes is amenable to this protocol, affording vinylcyclopentane products in moderate to excellent yields and with distereomeric ratios (dr) ranging from 50:50 to 95:5. Reactions of 1 with styrenes bearing substituents on the phenyl ring or at α-position, including alkyl and aryl groups, proceed well to give products 8–15 in good to excellent yields (70–96% yield). Notably, boric acid is tolerated (13, 84% yield after a subsequent reaction with pinacol), providing opportunities for late-stage functionalization through cross-coupling reactions. 2-Vinylpyridine and 1H-indene readily participate in this transformation (16, 48% yield; 17, 91% yield). Electron-rich alkenes also engage in reactivity to afford products (18–20, 32–77% yield). It is noteworthy that the cycloaddition of 1 with N-vinylpyrrolidone exhibited excellent diastereoselectivity (dr = 95:5). With a diverse set of electron deficient alkenes, we demonstrate excellent functional group tolerance of fluorine, epoxide, ketone, carboxylic acid, aldehyde, and nitrile (21–31, 58–90% yield). Cycloadditions of diene and enyne substrates afford corresponding products in moderate yields (32, 51% yield; 33, 50% yield). Although a higher loading of 7 (500 ppm) was used, good yields were maintained across a range of unactivated alkenes (34–38, 58–72% yield). In the reaction of (−)-β-pinene, the bridged bicyclic motif is retained (37), suggesting that the ring-forming step in this cycloaddition is fast.
Table 2.
Scope of Visible Light-Gated Bromine Radical-Catalyzed [3 + 2] Cycloaddition of Vinylcyclopropanes and Alkenesa
|
Conditions: vinylcyclopropane (0.20 mmol), alkene (0.40 mmol), 7 (50 ppm), 3 (1.0 mol %), DMSO (0.2 M), 30 °C, 34 W blue LED, 12 h.
2.0 mol % 3 was used.
0.1 M in DMSO.
0.4 M in DMSO.
500 ppm of 7 was used.
To demonstrate the utility of this new protocol, we selected several complex molecules of diverse biological activities for late-stage functionalization. 4-Vinylbenzylated theophylline, a phosphodiesterase inhibitor,22 and uridine, one of the standard nucleosides, were efficient substrates (39–40, 73–86% yield). Because of a solubility issue in DMSO, an estrone derivative affords product 41 in only moderate yield (55%). Aspartame, a dipeptide sweetener, is converted into 42 in 81% yield. Gratifyingly, this [3 + 2] cycloaddition is effective for 4-vinyl benzoylated desloratadine (43, 74% yield), a popular tricyclic antihistamine.
We next explored the vinylcyclopropane scope (Table 2b). Diverse substituents on either the malonate motif (44–46, 82–93% yield), vinyl group (53–54, 79–81% yield), or cyclopropane scaffold (55, 59% yield) are tolerated. This cycloaddition is also amenable to a range of electron-deficient patterns bearing various functionalities, such as carboxylic acid (47, 92% yield), amide (49, 93% yield), phosphonate (50, 86% yield), alcohol (57, 86% yield), and carbamate (59, 90% yield), delivering corresponding products in good to excellent yield (47–52, 56–59, 62–93% yield). Reaction of a substrate containing a hydrophobic cholesterol motif is challenging in DMSO but provides 60 in 58% yield. Vinylcyclopropanes bearing tethered poly(dimethylsiloxane) (PDMS) or poly-(ethylene glycol) (PEG) efficiently react with methyl methacrylate (MMA) to afford products 61 and 62 in 77% and 75% yield, respectively. Moreover, well-designed substrates with pendant alkenyl functionality possibly engage in intramolecular reactivity to access advanced bicyclic scaffolds. This concept is demonstrated in the successful synthesis of γ- and δ-lactones 63 (43% yield) and 64 (72% yield).
For the reaction mechanism, a Stern–Volmer quenching experiment reveals the feasible interaction between 7 and 3 (Figure 2d). However, the single-electron oxidation pathway to generate bromine radicals is thermodynamically unfavorable ([E1/2red(7*/7•−)] = +1.37 vs standard calomel electrode (SCE), [E 1/2OX (3/3•+)] = 2.01 V vs SCE, see Figure S5), suggesting that previously proposed photosensitization via EnT is likely the dominant pathway. Several control experiments were performed to gain insight into this reaction mechanism. First, radical trapping using (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) supports the formation of cinnamyl carbon radical (Figure 2a). However, the bromine radical-TEMPO adduct was not detected, presumably due to stability issues. Second, reactivity of other precatalysts, such as NBS and Br2, was investigated (Figure 2b). Photosensitization of NBS or direct photolysis of Br2 provides similar catalytic reactivity, but in much lower yields (41% and 16% yield, respectively), evidencing the key role of bromine radical in this transformation. Third, epimerization of 66 under optimal conditions strongly supports the radical mechanism (Figure 2c). Furthermore, temporal on/off control on the reaction (Figure 2e) showcases the light-controlled feature of this bromine radical catalysis. Accordingly, we proposed a catalytic cycle as shown in Figure 2f. Upon visible light irradiation, PC 7 is capable of sensitizing triplet-state fragmentation of 3 to release a bromine radical. Addition of the bromine radical to the vinyl group of 1 gives a transient radical species I, which undergoes a fast but reversible ring-opening process to furnish intermediate II. A subsequent radical addition of II to alkenes, followed by a ring-closure reaction, affords intermediate IV. In this step, the spin center shifts to the position for β-elimination, giving the final product, meanwhile regenerating the bromine radical to enter into further catalytic cycles. Efficient radical–radical coupling of bromine radical and cinnamyl carbon radical leads to the dormant precatalyst (3), which is evidenced by the high-yielding recovery of 3 in TEMPO trapping experiment (81%, Figure 2b). Possibly, Br2, formed through recombination of two bromine radicals, may be another dormant species within this photosystem.
Figure 2.
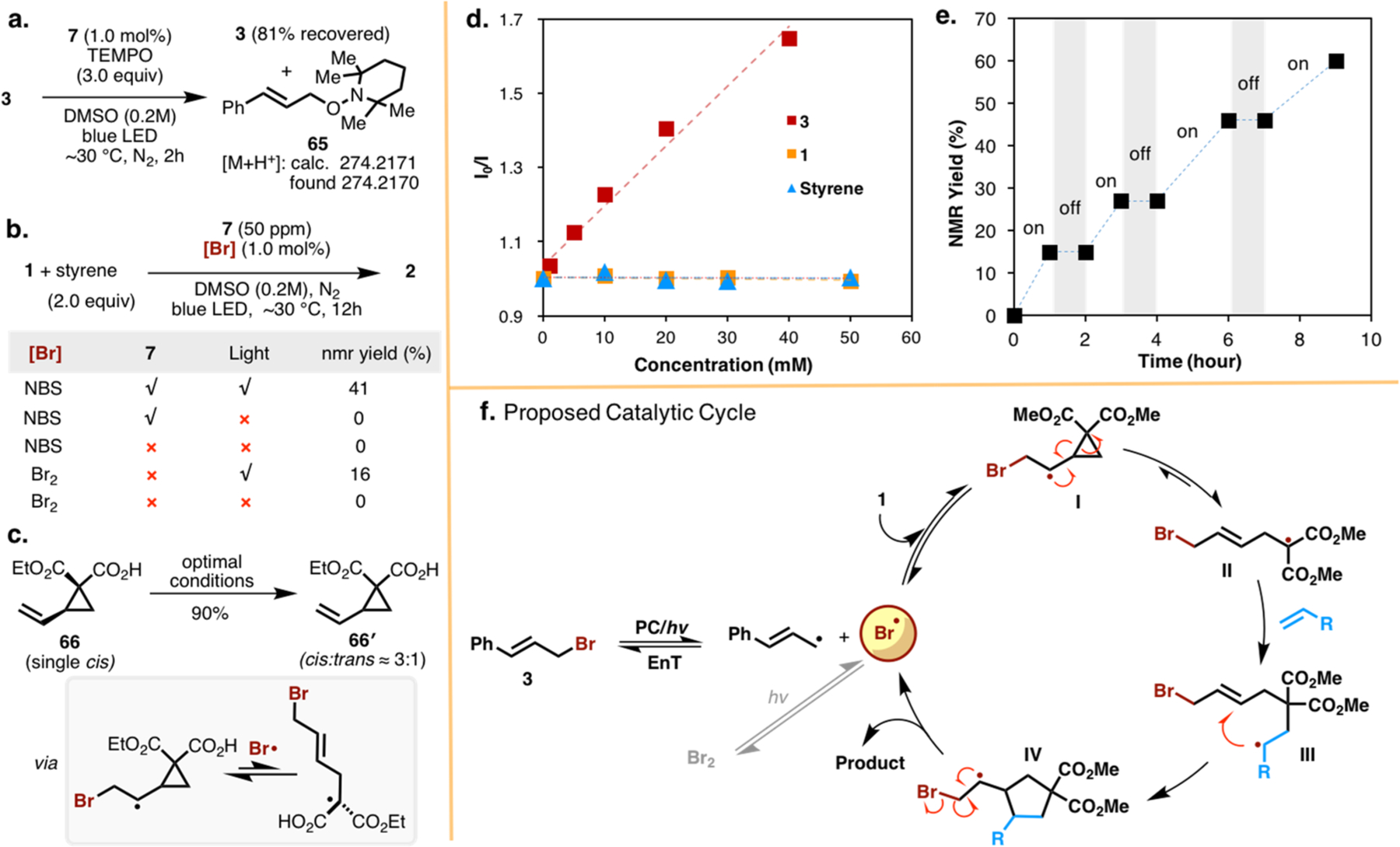
Mechanistic studies. (a) TEMPO trapping experiment. (b) Examination of alternative precatalysts. (c) Radical epimerization experiment. (d) Stern–Volmer quenching experiment. (e) “On/off” experiment and (f) proposed catalytic cycle.
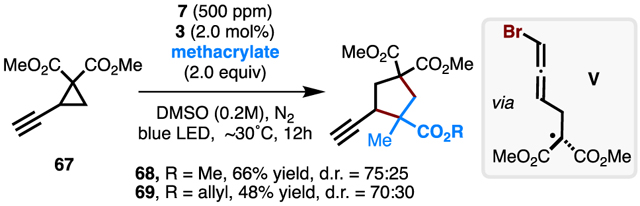
Lastly, we examined the performance of bromine radicals in the activation of C–C triple bonds. Under optimal conditions, in situ-generated bromine radicals add to alkyne group of ethynylcyclopropane 67, followed by a similar ring-opening reaction, to give the key allenyl intermediate V (eq 1). Trapping of V with methacrylates successfully affords ethynylcyclopentanes 68 and 69 in 66% and 48% yield, respectively. Continued efforts to functionalize more challenging molecules (such as alkanes via HAT) utilizing this mild bromine radical catalysis are underway.
 |
(1) |
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by Colorado State University and the National Institute of General Medical Sciences of the National Institutes of Health under Award No. R35GM119702. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c00281.
Experimental procedures, analytical data, and NMR spectra (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Hammond GS; Turro NJ Organic Photochemistry. Science 1963, 142, 1541–1553. [DOI] [PubMed] [Google Scholar]
- (2).Hoffmann N Photochemical Reactions as Key Steps in Organic Synthesis. Chem. Rev 2008, 108, 1052–1103. [DOI] [PubMed] [Google Scholar]
- (3).Schultz DM; Yoon TP Solar Synthesis: Prospects in Visible Light Photocatalysis. Science 2014, 343, 1239176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Prier CK; Rankic DA; MacMillan DWC Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Strieth-Kalthoff F; James MJ; Teders M; Pitzer L; Glorius F Energy Transfer Catalysis Mediated by Visible Light: Principles, Applications, Directions. Chem. Soc. Rev 2018, 47, 7190–7202. [DOI] [PubMed] [Google Scholar]; (b) Zhou Q-Q; Zou Y-Q; Lu L-Q; Xiao W-J Visible-Light-Induced Organic Photochemical Reactions Through Energy-Transfer Pathways. Angew. Chem., Int. Ed 2019, 58, 1586–1604. [DOI] [PubMed] [Google Scholar]
- (6).For selected examples:; (a) Ikezawa H; Kutal C; Yasufuku K; Yamazaki H Direct and Sensitized Valence Photoisomerization of a Substituted Norbornadiene. Examination of the Disparity between Singlet- and Triplet-state Reactivities. J. Am. Chem. Soc 1986, 108, 1589–1594. [Google Scholar]; (b) Zou Y-Q; Duan S-W; Meng X-G; Hu X-Q; Gao S; Chen J-R; Xiao W-J Visible Light Induced Intermolecular [2 + 2]-Cycloaddition Reactions of 3-Ylideneoxindoles through Energy Transfer Pathway. Tetrahedron 2012, 68, 6914–6919. [Google Scholar]; (c) Lu Z; Yoon TP Visible Light Photocatalysis of [2 + 2] Styrene Cycloadditions by Energy Transfer. Angew. Chem., Int. Ed 2012, 51, 10329–10332. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lei T; Zhou C; Huang M-Y; Zhao L-M; Yang B; Ye C; Xiao H; Meng Q-Y; Ramamurthy V; Tung CH; Wu L-Z General and Efficient Intermolecular [2 + 2] Photodimerization of Chalcones and Cinnamic Acid Derivatives in Solution through Visible-Light Catalysis. Angew. Chem., Int. Ed 2017, 56, 15407–15410. [DOI] [PubMed] [Google Scholar]; (e) Zhao J; Brosmer JL; Tang Q; Yang Z; Houk KN; Diaconescu PL; Kwon O Intramolecular Crossed [2 + 2] Photocycloaddition through Visible Light-Induced Energy Transfer. J. Am. Chem. Soc 2017, 139, 9807–9810. [DOI] [PubMed] [Google Scholar]; (f) Hörmann FM; Chung TS; Rodriguez E; Jakob M; Bach T Evidence for Triplet Sensitization in the Visible-Light-Induced [2 + 2] Photocycloaddition of Eniminium Ions. Angew. Chem., Int. Ed 2018, 57, 827–831. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhu M; Zheng C; Zhang X; You S-L Synthesis of Cyclobutane-Fused Angular Tetracyclic Spiroindolines via Visible-Light-Promoted Intramolecular Dearomatization of Indole Derivatives. J. Am. Chem. Soc 2019, 141, 2636–2644. [DOI] [PubMed] [Google Scholar]; For selected examples of enantioselective [2 + 2] photocycloaddition, see:; (h) Alonso R; Bach T A Chiral Thioxanthone as an Organocatalyst for Enantioselective [2 + 2] Photocycloaddition Reactions Induced by Visible Light. Angew. Chem., Int. Ed 2014, 53, 4368–4371. [DOI] [PubMed] [Google Scholar]; (i) Blum TR; Miller ZD; Bates DM; Guzei IA; Yoon TP Enantioselective Photochemistry through Lewis Acid-Catalyzed Triplet Energy Transfer. Science 2016, 354, 1391–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Tröster A; Alonso R; Bauer A; Bach T Enantioselective Intermolecular [2 + 2] Photocycloaddition Reactions of 2(1H)-Quinolones Induced by Visible Light Irradiation. J. Am. Chem. Soc 2016, 138, 7808–7811. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Skubi KL; Kidd JB; Jung H; Guzei IA; Baik M-H; Yoon TP Enantioselective Excited-State Photoreactions Controlled by a Chiral Hydrogen-Bonding Iridium Sensitizer. J. Am. Chem. Soc 2017, 139, 17186–17192. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Daub ME; Jung H; Lee BJ; Won J; Baik M-H; Yoon TP Enantioselective [2 + 2] Cycloadditions of Cinnamate Esters: Generalizing Lewis Acid Catalysis of Triplet Energy Transfer. J. Am. Chem. Soc 2019, 141, 9543–9547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For selected examples, see:; (a) Hammond GS; Turro NJ; Leermakers PA The Mechanisms of Photoreactions in Solution. IX. Energy Transfer from the Triplet States of Aldehydes and Ketones to Unsaturated Compounds. J. Phys. Chem 1962, 66, 1144–1147. [Google Scholar]; (b) Singh K; Staig SJ; Weaver JD Facile Synthesis of Z-Alkenes via Uphill Catalysis. J. Am. Chem. Soc 2014, 136, 5275–5278. [DOI] [PubMed] [Google Scholar]; (c) Metternich JB; Gilmour R A Bio-Inspired, Catalytic E → Z Isomerization of Activated Olefins. J. Am. Chem. Soc 2015, 137, 11254–11257. [DOI] [PubMed] [Google Scholar]; (d) Metternich JB; Gilmour R One Photocatalyst, n Activation Modes Strategy for Cascade Catalysis: Emulating Coumarin Biosynthesis with (−)-Riboflavin. J. Am. Chem. Soc 2016, 138, 1040–1045. [DOI] [PubMed] [Google Scholar]; (e) Singh K; Fennell CJ; Coutsias EA; Latifi R; Hartson S; Weaver JD Light Harvesting for Rapid and Selective Reactions: Click Chemistry with Strain-Loadable Alkenes. Chem. 2018, 4, 124–137. [Google Scholar]
- (8).For selected examples, see:; (a) Farney EP; Yoon TP Visible-Light Sensitization of Vinyl Azides by Transition-Metal Photocatalysis. Angew. Chem., Int. Ed 2014, 53, 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brachet E; Ghosh T; Ghosh I; König B Visible Light C–H Amidation of Heteroarenes with Benzoyl Azides. Chem. Sci 2015, 6, 987–992. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Scholz SO; Farney EP; Kim S; Bates DM; Yoon TP Spin-Selective Generation of Triplet Nitrenes: Olefin Aziridination through Visible-Light Photosensitization of Azidoformates. Angew. Chem., Int. Ed 2016, 55, 2239–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Münster N; Parker NA; van Dijk L; Paton RS; Smith MD Visible Light Photocatalysis of 6π Heterocyclization. Angew. Chem., Int. Ed 2017, 56, 9468–9472. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Teders M; Henkel C; Anhauser L; Strieth-Kalthoff F; Goméz-Suárez A; Kleinmans R; Kahnt A; Rentmeister A; Guldi DM; Glorius F The Energy-Transfer-Enabled Biocompatible Disulfide-Ene Reaction. Nat. Chem 2018, 10, 981–988. [DOI] [PubMed] [Google Scholar]
- (9).Mirkovic T; Ostroumov EE; Anna JM; VanGrondelle R; Govindjee; Scholes, G. D. Light Absorption and Energy Transfer in the Antenna Complexes of Photosynthetic Organisms. Chem. Rev 2017, 117, 249–293. [DOI] [PubMed] [Google Scholar]
- (10).Yoo W-J; Tsukamoto T; Kobayashi S Visible Light-Mediated Ullmann-Type C–N Coupling Reactions of Carbazole Derivatives and Aryl Iodides. Org. Lett 2015, 17, 3640–3642. [DOI] [PubMed] [Google Scholar]
- (11).(a) Heitz DR; Tellis JC; Molander GA Photochemical Nickel-Catalyzed C–H Arylation: Synthetic Scope and Mechanistic Investigations. J. Am. Chem. Soc 2016, 138, 12715–12718. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Welin ER; Le C; Arias-Rotondo DM; McCusker JK; MacMillan DWC Photosensitized, Energy Transfer-Mediated Organometallic Catalysis through Electronically Excited Nickel(II). Science 2017, 355, 380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kim T; McCarver SJ; Lee C; MacMillan DWC Sulfonamidation of Aryl and Heteroaryl Halides through Photo-sensitized Nickel Catalysis. Angew. Chem., Int. Ed 2018, 57, 3488–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kudisch M; Lim CH; Thordarson P; Miyake GM Energy Transfer to Ni-Amine Complexes in Dual Catalytic, Light- Driven C-N Cross-Coupling Reactions. J. Am. Chem. Soc 2019, 141, 19479–19486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For original report, see:; (a) Ghosh I; Shaikh RS; König B Sensitization-Initiated Electron Transfer for Photoredox Catalysis. Angew. Chem., Int. Ed 2017, 56, 8544–8549. [DOI] [PubMed] [Google Scholar]; For further discussion of the mechanism, see:; (b) Marchini M; Bergamini G; Cozzi PG; Ceroni P; Balzani V Photoredox Catalysis: The Need to Elucidate the Photochemical Mechanism. Angew. Chem., Int. Ed 2017, 56, 12820–12821. [DOI] [PubMed] [Google Scholar]; (c) Ghosh I; Bardagi JI; König B Reply to “Photoredox Catalysis: The Need to Elucidate the Photochemical Mechanism”. Angew. Chem., Int. Ed 2017, 56, 12822–12824. [DOI] [PubMed] [Google Scholar]
- (13).Saikia I; Borah AJ; Phukan B Use of Bromine and Bromo- Organic Compounds in Organic Synthesis. Chem. Rev 2016, 116, 6837–7042. [DOI] [PubMed] [Google Scholar]
- (14).Studer A; Curran DP Catalysis of radical reactions: a radical chemistry perspective. Angew. Chem., Int. Ed 2016, 55, 58–102. [DOI] [PubMed] [Google Scholar]
- (15).Goehring RR Bromine. e-EROS Encyclopedia of Reagents for Organic Synthesis; Wiley, 2001; DOI: 10.1002/047084289X.rb256. [DOI] [Google Scholar]
- (16).Virgil SC; Jenkins PR; Wilson AJ; Romero MD G. N-Bromosuccinimide. e-EROS Encyclopedia of Reagents for Organic Synthesis; Wiley, 2006; DOI: 10.1002/047084289X.rb318.pub2. [DOI] [Google Scholar]
- (17).The photoisomerization of 2-phenylallyl chloride was previously observed, which arose from the generation and recombination of a radical pair. See:; Xu B; Tambar UK Remote Allylation of Unactivated C(sp3)−H Bonds Triggered by Photogenerated Amidyl Radicals. ACS Catal. 2019, 9, 4627–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) Miura K; Fugami K; Oshima K; Utimoto K Synthesis of Vinylcyclopentanes from Vinylcyclopropanes and Alkenes Promoted by Benzenethiyl Radical. Tetrahedron Lett. 1988, 29, 5135–5138. [Google Scholar]; (b) Feldman KS; Berven HM; Weinreb PH 2,2-Dihalovinylcyclopropanes as Highly Diastereoselective Three-Atom Addends in Phenylthio Radical Mediated Vinylcyclopentane Synthesis. J. Am. Chem. Soc 1993, 115, 11364–11365. [Google Scholar]; (c) Hashimoto T; Kawamata Y; Maruoka K An Organic Thiyl Radical Catalyst for Enantioselective Cyclization. Nat. Chem 2014, 6, 702–705. [DOI] [PubMed] [Google Scholar]; (d) Hashimoto T; Takino K; Hato K; Maruoka K A Bulky Thiyl-Radical Catalyst for the [3 + 2] Cyclization of N-Tosyl Vinylaziridines and Alkenes. Angew. Chem., Int. Ed 2016, 55, 8081–8035. [DOI] [PubMed] [Google Scholar]
- (19).Zhang H; Curran DP A Short Total Synthesis of (±)-Epimeloscine and (±)-Meloscine Enabled by a Cascade Radical Annulation of a Divinylcyclopropane. J. Am. Chem. Soc 2011, 133, 10376–10378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zhang P; Le C; MacMillan DWC Silyl radical activation of alkyl halides in metallaphotoredox catalysis: a unique pathway for cross-electrophile coupling. J. Am. Chem. Soc 2016, 138, 8084–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Luo J; Zhang J Donor-acceptor fluorophores for visible-light promoted organic synthesis: photoredox/Ni dual catalytic C(sp3)-C(sp2) cross-coupling. ACS Catal. 2016, 6, 873–877. [Google Scholar]
- (22).Boswell-Smith V; Cazzola M; Page CP Are phosphodiesterase 4 inhibitors just more theophylline? J. Allergy Clin. Immunol 2006, 117, 1237–1243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.