

Abstract
Introduction:
Inositol 1,4,5-trisphosphate receptors (IP3Rs) are intracellular calcium (Ca2+) release channels located on the endoplasmic/sarcoplasmic reticulum. The availability of the structure of the ligand-binding domain of IP3Rs has enabled the design of compatible ligands, but the limiting step remains their actual effectiveness in a biological context.
Areas covered:
This article summarizes the compelling literature on both agonists and antagonists targeting IP3Rs, emphasizing their strengths and limitations. The main challenges toward the discovery and development of IP3 receptor modulators are also described.
Expert opinion:
Despite significant progress in recent years, the pharmacology of IP3R still has major drawbacks, especially concerning the availability of specific antagonists. Moreover, drugs specifically targeting the three different subtypes of IP3R are especially needed.
Keywords: Adenophostin, IP3, IP3R, ITPR, Xestospongin, 2-APB, Drug Design, Structure
1. Introduction
Inositol 1,4,5-trisphosphate receptors (IP3Rs) are intracellular calcium (Ca2+) release channels located on the sarco/endoplasmic reticulum (SR/ER), the major Ca2+ store in the cells. IP3Rs are the link between many extracellular stimuli and the initiation of intracellular Ca2+ signaling, which controls several responses, including cell division, synaptic transmission, and gene expression [1, 2].
Three different subtypes of IP3Rs are expressed in mammalian cells, namely IP3R1-2-3, which show ~70% of homology, despite originating from different genes.
Organized in a “mushroom-like” structure, IP3Rs consist of a tetrameric complex in which each monomer includes five domains: suppressor domain, IP3 binding core domain, regulatory domain, transmembrane domain, and C-terminus domain [3–9]. The intrinsic complexity of IP3R structure, with multiple regulatory sites, contemplates the potential participation of the receptor to a dense network of cellular processes [10–18]. However, the current knowledge of IP3R in human disease remain quite limited and one of the aspects contributing to the relative low number of investigations about IP3R-related disease pathogenesis is the inadequate availability of drugs effectively and selectively targeting these receptors. This limitation raises not trivial issues for studying the receptor behavior in a disease-context. The interest in developing novel ligands of IP3R has always been substantial. The first studies designed in order to develop IP3R drugs aimed to clarify the complex mechanisms of receptor activation, mapping the key residues for ligand-receptor interaction. The structure of the ligand-binding domain of IP3R has enabled the design of compatible ligands [2, 5, 6, 19], although the limiting step remains their actual effectiveness in biological context.
Here we provide an overview of the drugs currently known to target IP3R, agonists, and antagonists. A comprehensive and rational analysis of the available literature is fundamental to overcome the current limitations and to highlight the need for specific molecules targeting IP3R. Indeed, the development of effective drugs could produce important advances in the biomedical research on IP3R. On one side, it can prompt the design of new and attractive therapeutic strategies for diseases with certain pathogenic implications of IP3R. On the other side, the availability of effective drugs can be useful to better understand the biology of IP3R, exploring its putative involvement in other settings.
2. IP3R agonists
The endogenous ligand of IP3Rs is IP3 (Figure 1A), a water-soluble second messenger produced by hydrolysis of phosphatidylinositol 4,5-bisphosphate. Specifically, IP3 interacts with the inositol binding core (IBC) of IP3R, an allosteric site that, after the interaction with IP3, triggers the opening of its intrinsic Ca2+ channel [20]. The crystal structure of IP3R1 has revealed that the IBC consists of two subdomains, β and α; the interface between β and α domains creates a positively charged pocket optimal for the allocation of phosphates groups of IP3 [19]. Site-directed mutagenesis studies demonstrated that several Arg and Lys residues in this pocket are directly involved in the interaction with IP3, which is supposed to induce a conformational change of IP3R structure, leading to channel opening [21]. Of course, the development of synthetic ligands of IP3R has exploited the molecular structure of IP3 as a model to obtain effective analogs.
Figure 1.
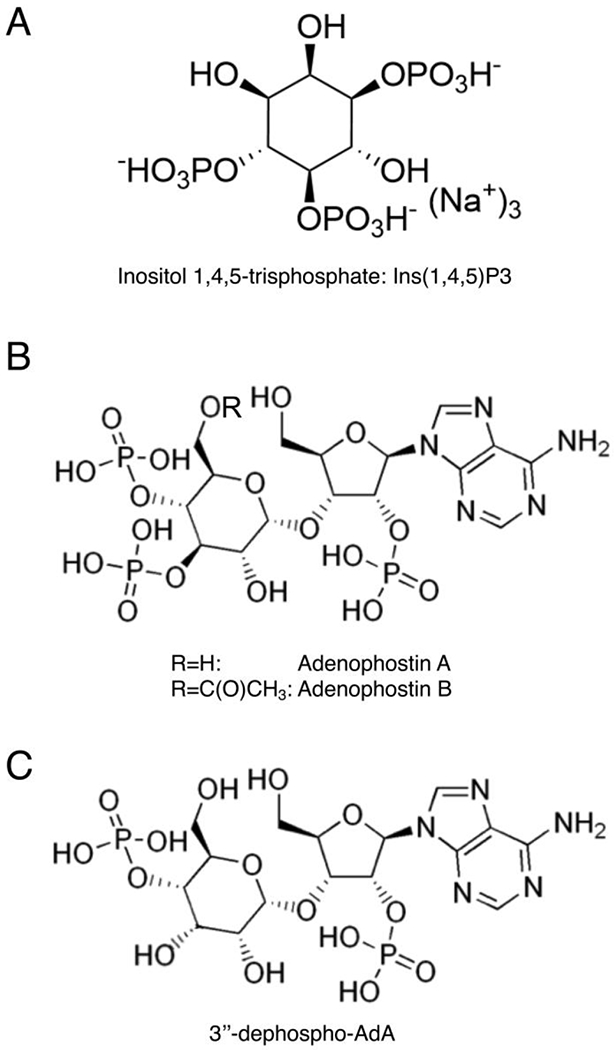
Structures of Inositol 1,4,5-trisphosphate (IP3, A) and adenophostins (B,C).
In 1993, two potent agonists of IP3Rs, adenophostins A and B, were isolated from the culture broth of Penicillium brevicompactum SANK 11991 and SANK 12177 [22]. In particular, they were more effective (nearly 10-fold) then the endogenous ligand, showing their potent action on all three IP3R subtypes. The structure of adenophostin A and B (Figure 1B) inspired many groups in synthesizing new ligands with additional modifications and properties. For instance, 3″-dephospho-AdA is an analog of AdA-A that lacks a phosphate group (5’-P), as shown in Figure 1C. This synthetic compound was tested in DT40 cells (chicken B cell line derived from an avian leukosis virus induced bursal lymphoma) devoid of native IP3R and stably expressing single subtypes of mammalian IP3Rs, showing that this analog is effective as the parental compound, and does not have a selective action for a specific IP3R subtype [23, 24]. To better understand the role of the different phosphate groups and of the adenosine-motif in the interaction and activation of IP3Rs, another group synthesized all three possible bisphosphate analogs of AdA [25]. The study provided new information on the functional role of the chemical groups in the IP3 structure. In particular, the compound harboring 4,5 bisphosphates displayed an efficacy only 4-fold less potent than IP3; the 5-dephospho (1,4 bisphosphates) conserved an optimal activity, in line with the previous study on 3″-dephospho-AdA [23]. This evidence challenges the paradigm that the two vicinal bisphosphate groups (4,5) are critical for IP3-like activity. Conversely, the compound that was lacking the 2’-AMP was 400-fold weaker than endogenous ligand [25]. This finding strongly suggests that adenosine has a key role in determining the affinity and the interaction with IP3R, while only two phosphates groups (even not adjacent) are enough to ensure the interaction. This brilliant investigation offered new and precious information for designing new compounds as potent IP3R agonists. Another potent IP3R ligand was synthesized conjugating an aromatic group at the 5’-position of AdA, and this addition was well tolerated in the receptor binding [26]. The parental structure of AdA was also modified by replacing adenine with a triazole ring, without compromising the overall compound potency [27].
Since IP3R has a tetrameric structure, the idea to produce a tetrameric ligand more effective than the AdA monomer, also emerged. As the triazole substitution did not affect the compound potency, the same research group synthesized four homodimers of triazole-AdA analogs, connecting them by oligoethylene glycol chains. However, the resultant tetrameric structure was equipotent to AdA and its triazole-derivate [28].
Several studies have explored in detail the possible modifications of AdA, producing a high number of analogs with structural differences in phosphate groups, nucleobase motif, and at both sugars [29]. These investigations have been useful to produce an accurate model of ligand-IP3R interaction, allowing the selection of structural elements necessary for the agonist efficacy. Although the essential pharmacophore is well characterized, none of the analogs reported above is more potent than the parental compound AdA. Only recently, an agonist able to produce the same effect of AdA but at a lower concentration was produced [30], harnessing a chemical modification approach never explored before. Specifically, the bisphosphorylated glucose moiety was replaced by a cyclitol bisphosphate (d-chiro-inositol), changing an element thought to be crucial in mimic IP3 pharmacophore; despite the potentially hazardous choice, the resultant d-chiro-inositol adenophostin (Figure 2) remains among the most potent known agonists of IP3Rs, nearly 2-fold more potent than the original AdA [30]. The interesting result of using d-chiro-inositol has since then inspired other research studies focused on the application of this new approach to other AdA analogs, combining different strategies.
Figure 2.
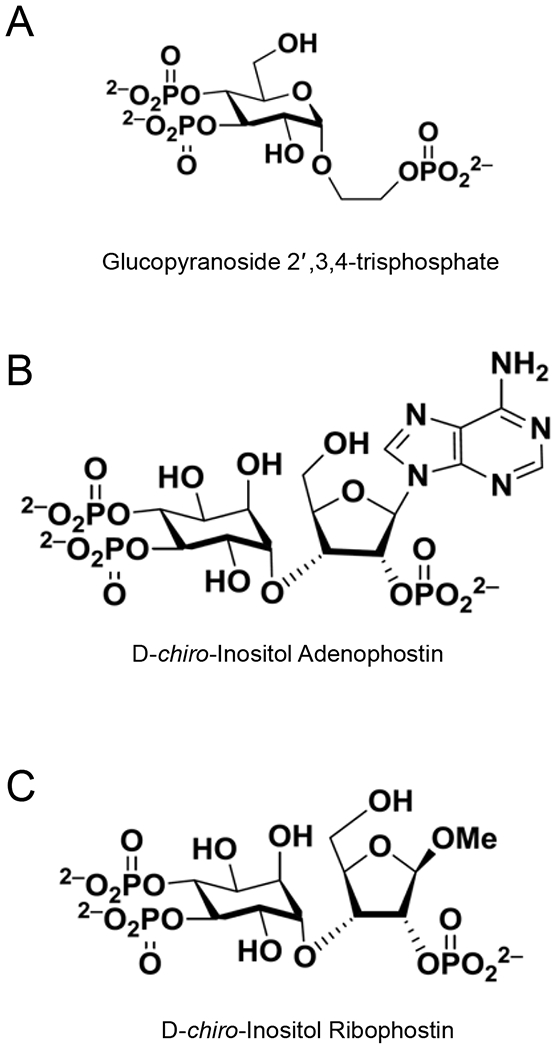
Structures of Glucopyranoside 2′,3,4-trisphosphate (A), D-chiro-Inositol Adenophostin (B) and D-chiro-Inositol Ribophostin (C).
For instance, Mills and collaborators have recently obtained a novel potent agonist [31] starting from the simplest produced AdA derivate, the glucopyranoside 2′,3,4-trisphosphate (Glc(2′,3,4)P3, Figure 2), obtained by removing adenine and part of ribose [32]. This compound is 10-fold less potent than the endogenous ligand, IP3, in inducing Ca2+ release via IP3Rs, but with the advantage of a resistance to degradation carried out by the enzymes that metabolize the endogenous IP3. To improve the potency of Glc(2′,3,4)P3 keeping the advantage of its metabolic resistance, the ribose–sugar was reintroduced, producing the ribophostin compound. Then, applying the strategy of glucose substitution with d-chiro-inositol, as previously done for AdA [30], the obtained compound, the d-chiro-inositol ribophostin (Figure 2) was more potent than parental compounds in evoking Ca2+ release, producing a new ligand with high efficacy and resistance to degradation [31]. More recently, Shipton and colleagues have generated novel active ligands for IP3R starting from d-glucose and l-glucose templates as inositol surrogates [33].
Another approach to stimulate IP3Rs is the use of caged IP3 analogs, which provide a convenient way to evaluate IP3-mediated Ca2+ liberation bypassing upstream signaling events and enabling a precise control of the timing and relative concentration of cytosolic IP3 [34–37].
Hence, the literature offers a plethora of agonists designed for IP3Rs. Nonetheless, all the available compounds derived from the endogenous IP3, resembling its structure. The IP3-like structure confers to synthetic-agonists a high specificity for IP3R, avoiding side effects on other receptors. However, the agonist-action on a specific IP3R subtype remains arduous, as IP3 is the ligand of all three receptor subtypes. This issue is also emphasized by the high degree of similarity in amino acid sequence within the IP3 binding domain of each IP3R subtype. Several studies have been conducted to find differences in recognition properties among the three-receptor subtypes, in order to use this information in designing subtype-selective ligands; these studies revealed that the three receptor subtypes have different affinity for IP3: type 2 > type 1 > type 3 [38–44]. Moreover, the 2- and 3- positions of IP3 were identified as key determinants of subtype selectivity [45, 46].
3. IP3R antagonists
If the synthesis of IP3R agonists can exploit the structure of the natural ligand as a guide, the development of receptor antagonists is more challenging. Most of the available antagonists act competing with IP3 for its binding site on IP3Rs. To achieve a good affinity in order to compete with the endogenous ligand, the antagonists should have a similar structure to IP3, too. However, their IP3-like structure increases the risk of a partial-agonist action. Few molecules have different structural nature, but often their exact mechanism of action is unknown.
3.1. Xestospongins
Xestospongin (Xe) A, B, C, and D, a group of macrocyclic bis-1-oxaquinolizidines (Figure 3) which induce relaxation of blood vessels in vivo, were isolated from the Australian marine sponge Xestospongia exigua, by Nakagawa and Endo in 1984 [47, 48]. These alkaloids share the macrocyclic 1-oxaquinolizidine structure with other vasoactive compounds including araguspongine B, C, 7S-Hydroxyxestospongin A (7-OHXeA), and demethylxestospongin B (DMXeB), which were extracted from the Xestospongia exigua and muta [49, 50]. Gafni et al. in 1997 demonstrated that XeA, C, D, dimethyl-xestospongin B (DMXeB), and araguspongine B (ArB) are able to block in a dose-dependent manner the IP3R-mediated Ca2+ release from in microsomes isolated from rabbit cerebellum [51]. Additionally, the same authors characterized the potency of these compounds to inhibit IP3R, showing that XeC exhibited the highest IP3R blockage with the half-maximal inhibitory concentration (IC50) of 358 nM, a 7-fold higher potency compared to XeA; instead, the DMXeB displayed the lowest IP3R Ca2+ release inhibition, >16-fold decreased in potency relative to XeC, with an IC50 of 5865 nM [51]. These results on IP3R inhibition were later confirmed and extended to the hydroxylated Xe derivatives 7-OH-XeA and araguspongin C (ArC), characterized by 10- to 15-fold lower potency to block IP3R-induced Ca2+ release as compared to XeC [52].
Figure 3.
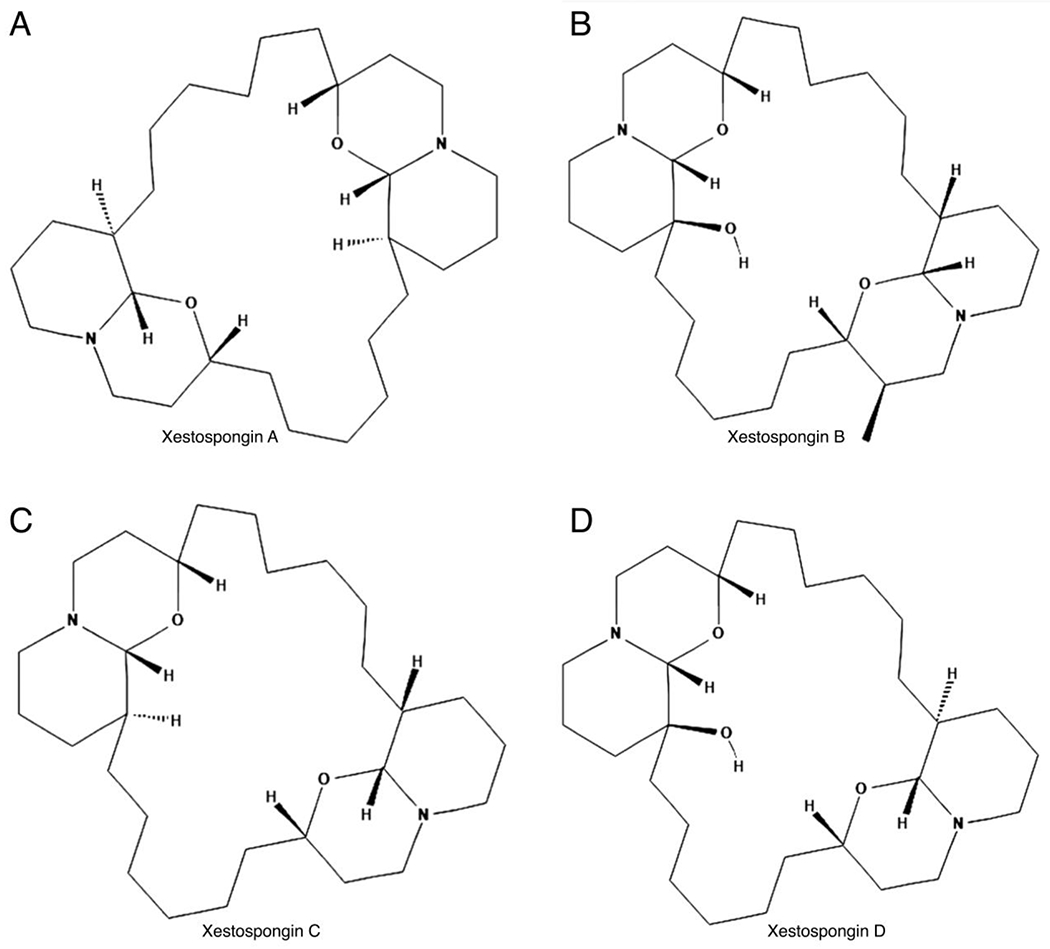
Structures of Xestospongin A (A), Xestospongin B (B), Xestospongin C (C), and Xestospongin D (D).
Xestospongins have been widely used as membrane-permeant pharmacological inhibitors of IP3R-evoked Ca2+ release, and in particular, XeC has found an extensive application for intact-cell assays, as confirmed by several authors [52–59]. Nevertheless, over the years the xestospongins inhibitory mechanism remained elusive, because Gafni et al. had reported that the blockage of IP3Rs occurs without the interaction with its IP3-binding site [51], and only a few later studies directly addressed the effects of xestospongins on IP3Rs [54, 55].
Several authors have observed that XeC is able to inhibit additional targets, including SR/ER Ca2+ ATPase (SERCA pump) [59–61], Ca2+ store and capacitative Ca2+ entry [62], L-type Ca2+ channels, and Ca2+-activated K+ channels [55]; interestingly, XeC has been shown to maintain its selectivity as IP3Rs blocker in permeabilized cells, but not in intact cells [55]. On the other hand, studies on XeC and XeB activity on SERCA pumps from isolated SR/ER membrane fractions did not evidence SERCA inhibition while they confirmed the competitive inhibition of IP3R [63, 64]. Equally important, neither XeC nor XeD, 7-OHXeA, and araguspongin C (at ≤50 μM) inhibit or interfere with SERCA1 and SERCA2 in the skeletal or cardiac SR [52]. In the same work, XeC led to weak inhibition of type 1 ryanodine receptor (RyR1), in partial agreement with a previous observation of a lack of RyR inhibition in smooth muscle SR membranes treated with XeC [55]. Instead, Xestospongins hydroxylated structures, including XeD, ArC, and 7-OH-XeA, used within the same concentration range used to observe IP3R inhibition, were found to sensitize RyR1-mediated Ca2+-induced Ca2+ release (CICR) in SR cerebellum vesicles, [52].
As discussed until here, even if the pharmacological selectivity was questionable among the different works, many researchers reported the inhibition of IP3Rs after treatment with xestospongins, with XeC having the highest potency as IP3Rs blocker. However, several other studies reported major challenges concerning the actual and effective XeC and XeD inhibition of IP3Rs.
For instance, Solovyova and colleagues observed that XeC failed to inhibit IP3R-evoked Ca2+ release in cultured dorsal root ganglion neurons, concluding that it is not possible to consider XeC as a specific inhibitor of IP3Rs [61]. Similarly, Duncan et al. reported a trend in the elevation of intracellular Ca2+ concentration, indicating an incomplete block of IP3Rs by XeD [65].
A work by Saleem and collaborators further supports the previous objections about the efficacy of XeC and XeD as IP3Rs inhibitors. The authors tested in intact (DT40) and permeabilized (HEK) cells two purified xestospongins, within a range of 5 μM (previously reported by Gafni et al. [51]) to 20 μM for XeC, and 10 μM (reported by Gafni et al. [51]) to 20 μM for XeD. Although XeC and XeD caused a statistically significant small inhibition of the maximal evoked response from IP3R1 and IP3R2, these effects were considered not sufficient to be useful as pharmacological inhibition, and only attainable at the highest concentration of XeC and XeB [66].
3.2. 2-Aminoethoxydiphenyl Borate (2-APB)
2-Aminoethoxydiphenyl Borate (2-APB, Figure 4) has been extensively used as a membrane-permeable modulator of IP3R evoked Ca2+ release [67–69]; yet, this compound has many additional effects. 2-APB inhibits the SERCA pumps [68, 69], as well as the store-operated Ca2+ entry (SOCE) and Ca2+ release-activated Ca2+ current (ICRAC) [70]. Interestingly, in an attempt to develop a more potent and selective 2-APB inhibitor, Goto and collaborators identified two structurally isomeric 2-APB analogs that were 100-fold more potent than 2-APB itself; however, the selectivity of these compounds failed since one of the 2-APB analogs activates and inhibits endogenous SOCE, depending on the concentration, while the other one inhibits SOCE [70].
Figure 4.
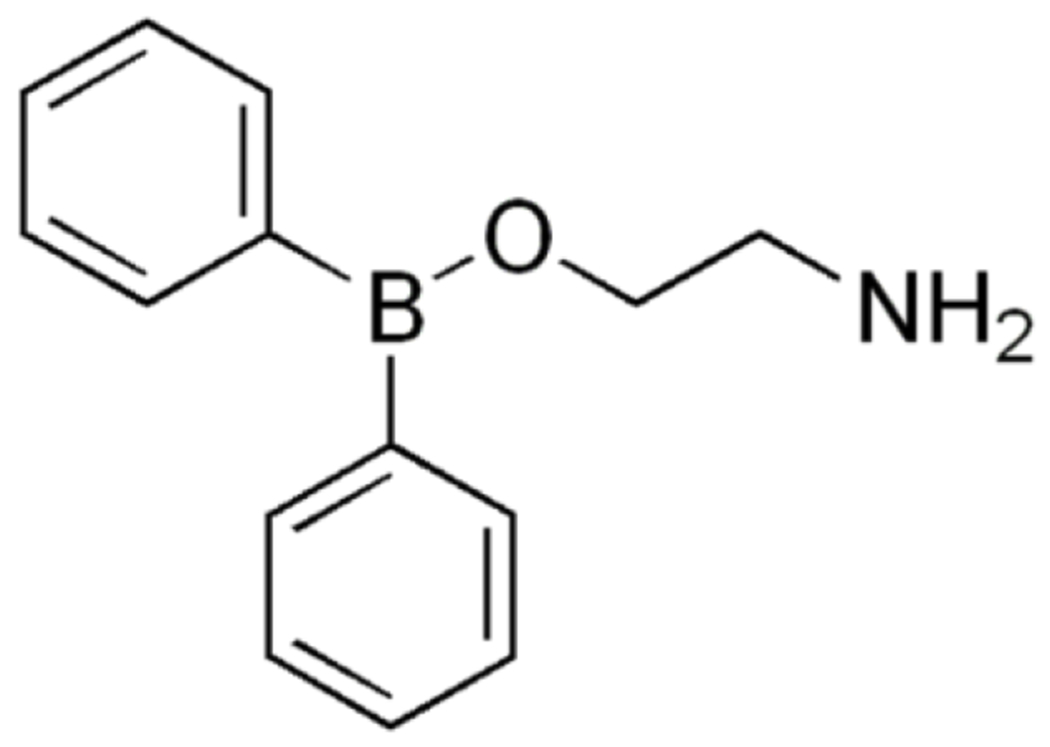
Structure of 2-Aminoethoxydiphenyl Borate (2-APB).
The inhibitory efficacy of 2-APB at different concentrations was tested in permeabilized DT40 cells, showing that 50 μM 2-APB did not affect Ca2+ uptake by the ER, in accordance with the max concentrations of 2-APB causing inhibition of SERCA; higher concentrations of 2-APB reduced the steady-state Ca2+ content [66]. In DT40-IP3R1 cells, 2-APB led to a concentration-dependent inhibition of IP3R, with a marked decreased sensitivity of IP3R1 at 50 μM 2-APB. The same concentration of 2-APB (50 μM) in permeabilized IDT40-IP3R2 and DT40-IP3R3 cells, did not show significant effects on IP3R-evoked Ca2+ release; instead, 2-APB at a concentration of 100 μM caused inhibition of IP3R3 and reduced Ca2+ uptake, but did not affect IP3-evoked Ca2+ release via IP3R2 [66].
3.3. Benzene derivatives
The class of aromatic polyphosphate derivatives has potential applicability in the field of phosphoinositides. In 1993, the compound benzene 1,2,4-trisphosphate [Bz(1,2,4)P3], harboring phosphates groups arranged in an IP3-like manner (Figure 5A), was synthesized and its ability to bind cellular proteins physiologically interacting with IP3 was tested [71]. Specifically, the Bz(1,2,4)P3 binding capacity to IP3-kinase, IP3 phosphatase, and the IP3 receptor was evaluated [71]. Despite the resistance of the compound to the phosphatase and kinase action, Bz(1,2,4)P3 was able to weakly interact with IP3R, competitively blocking the binding of 3H-IP3 in the performed assay. However, this compound retained very low action, and another study revealed that it was also able to IP3-kinase activity in vitro, emphasyzing its low specificity [72]. Yet, Bz(1,2,4)P3 was a good starting point to develop other aromatic antagonists of IP3R. In this regard, Vandeput and coworkers tested the biphenyl derivative BiPh(2,3′,4,5′,6)P5, which displayed a moderately potent capacity in inhibiting IP3R, with an IC50 value in the low micromolar range [73]. Later, the same group explored the strategy of the dimerization of benzene-derivative compounds, as an increasing binding capacity was reported for IP3–dimer [74]. In particular, two new types of dimeric benzene phosphate derivatives were generated, demonstrating their efficacy in antagonized Ca2+ release through the IP3R in saponin-permeabilized L15 fibroblasts, at a sub-micromolar dose [75]. However, the study did not evaluate the specificity of these compounds, and/or if they could affect the activity of other phosphatidylinositol partners.
Figure 5.
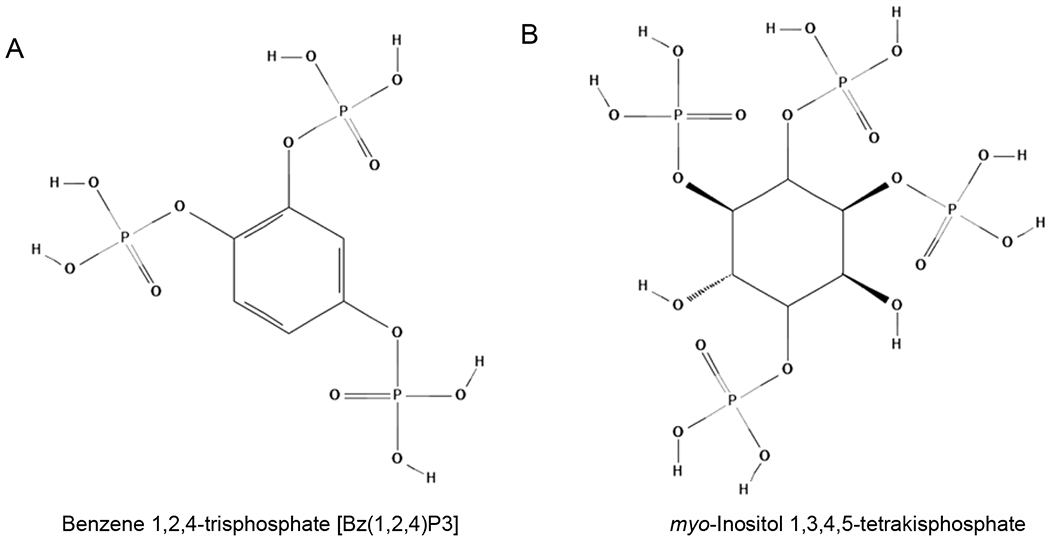
Structures of Benzene 1,2,4-trisphosphate [Bz(1,2,4)P3] (A) and myo-Inositol 1,3,4,5-tetrakisphosphate (B).
3.4. Heparin
Due to its high affinity for the IP3 active site, heparin is considered a membrane-impermeable competitive antagonist of IP3-evoked Ca2+ release [76]. However, this compound has many additional non-specific effects, including the uncoupling of receptors from G-proteins [77, 78], the activation of RyRs [79], and the inhibition of IP3-kinase [80]. The effects of heparin incubation were tested on permeabilized DT40 cells expressing each IP3R subtype, assessing the effects of IP3 on Ca2+ release from the intracellular stores [66]; In DT40-IP3R1 cells, heparin application caused parallel rightward shifts of the concentration–responses for IP3-evoked Ca2+. These results were in accord with previous works in which adenophostin A (AdA, see Figure 1), a high-affinity agonist of IP3Rs (see above: Paragraph 2), was used to evoke Ca2+ release [23, 81]. A similar analysis conducted in DT40-IP3R1 cells was replicated in DT40-IP3R2 and DT40-IP3R3 cells, assessing the effects of heparin on IP3-evoked Ca2+ release. The DT40-IP3R2 line revealed an IP3R competitive antagonism response consistent with DT40-IP3R1 [66]. An IP3R3 competitive antagonism in DT40 cells was also reported, consistent with the response from DT40-IP3R2 and DT40-IP3R3 lines, however, in this last assay it was difficult to achieve maximal IP3R3-evoked Ca2+ release, therefore, the authors used IP3 concentrations able to evoke a release of 40% Ca2+ from intracellular stores [66]. These results were consistent with previous observations showing that IP3R3 are less sensitive to IP3 than the other subtypes [23, 82].
These functional analyses contributed to define the heparin competitive antagonism with IP3 for the 3 subtypes of IP3R, founding different heparin affinities for each subtype (IP3R3 > IP3R1 ≥ IP3R2). These results were consistent with another study testing IP3 binding to mammalian IP3R expressed in Sf9 cells, showing pKD values and rank order of heparin affinity (IP3R3 > IP3R1 ~ IP3R2) [45].
3.5. Other antagonists
myo-Inositol 1,3,4,5-tetrakisphosphate (Figure 5B) and its derivatives display IP3R inhibitory action, but only at high concentrations [83]. In particular, phospho-derivatives of tetrakisphosphate are competitive antagonists of IP3R in platelets, but they are also able to act as partial agonists inducing the release of Ca2+ [84–86]. One tetrakisphosphate-derivative with methylphosphonate group was able to act as a selective competitive antagonist but at millimolar concentration [87]. Caffeine has been described as an effective antagonist of IP3R and has the advantage to be cell-permeant and to act in non-competitive manner respect to IP3 [66]. However, the main issue remains the capacity of caffeine to also act as an activator of RyR [88–91].
4. An endogenous antagonist of IP3R
In 2003, a novel antagonist was isolated from a high salt extract of crude rat brain microsomes using an affinity column with immobilized N-terminal cytoplasmic region of IP3R1 (residues 1-2217), performing the elution with IP3 [92]. Because of its isolation procedure, the compound has been named IP3R-Binding protein released with Inositol 1,4,5-Trisphosphate (IRBIT). It is composed by 530 amino acids, with an S-adenosylhomocysteine homologous domain in C-terminal extremity, while the N-terminal portion contains multiple phosphorylation sites.
The IRBIT binding site on IP3R was mapped to the IP3 binding core, suggesting that a competitive relationship occurs between the two molecules (IP3 and IRBIT). Hence, IRBIT inhibits IP3R by preventing the binding of the activator ligand (IP3). The interaction between IRBIT and IP3R has later been confirmed in a physiological context through studies of immunohistochemistry and of co-immunoprecipitation [92].
The mechanism of IP3R regulation by IRBIT emerged as unique, generating several questions and prompting other investigations. Soon, it emerged that the N-terminal domain of IRBIT was essential for the interaction with the receptor, and its phosphorylation status could modulate the inhibitory activity of IRBIT on IP3Rs [93]. Indeed, when IRBIT was exposed to alkaline phosphatase, it became no longer able to suppress receptor-IP3 binding. The concentration of IRBIT causing 50% inhibition of IP3 binding was about 0.1 mM. IRBIT can be defined as a pseudo-ligand, able to regulate the sensitivity of the receptor to its activator. This finding suggests the possibility that other cellular events can affect IP3R activity and function, for instance, by determining a different phosphorylation status of IRBIT. IRBIT released from the activated IP3R could also act as a signaling molecule downstream to the receptor, opening new research horizons [94–98]. Henceforth, the structure and the mechanism of action of IRBIT can be used to design new compounds and strategies to effectively antagonize IP3R.
5. Conclusions
Notwithstanding the great research progress in the discovery and development of effective drugs targeting IP3Rs, much more work is needed, especially in terms of specificity towards the different IP3R subtypes.
Most of the drug development studies are mainly focused on the chemistry of the ligand-receptor interaction. In numerous cases, the investigators report the receptor-kinetics evaluated in isolated-system, like microsomes. In other terms, the current studies often lack the biological approach, without testing the drug efficacy in the complexity of the entire cell or, even better, whole organism. This is the reason why the results are frequently unreliable when the developed molecules are tested in biological studies. If the combination of several strategies of chemical synthesis, including dimerization, and dephosphorylation, has been successful in producing a wide spectrum of potential IP3R ligands, the next step should be the combination with a pure biological approach, aiming to test the putative drugs in biological systems, both in physiological and pathological conditions.
6. Expert Opinion
Despite the desperate needs of IP3R targeting drugs, the pharmacology of this channel still has major drawbacks, especially in the design of specific antagonists. The available molecules exhibit low specificity or low receptor-affinity. For instance, Xestospongins which seem to be selective, exhibit weak inhibitory action on IP3-mediated Ca2+ release. 2-APB is more effective but has important off-target effects, including modulation SOCE and SERCA. When an investigator wants to know the effects of IP3R inhibition in a specific experimental setting, we suggest to use at least two different inhibitors (e.g. Xestospongins and 2-APB). Other issues are linked to membrane-impermeability of such potential effective molecules, heparin for instance. In addition to its incapacity to cross plasma-membrane, heparin also produces side effects by modulating G-proteins uncoupling.
The development of new antagonists should take into consideration the new insights emerged by the characterization of IRBIT. Albeit identified exclusively for IP3R1, IRBIT is an endogenous antagonist used physiologically by the cell, and it could be a good starting point to design effective antagonist molecules.
Another important current limitation is the lack of selectivity for the different IP3R subtypes. Most of the mammalian cells express more than one IP3R subtype. The exact functional differences among receptor subtypes remain to be established. However, different roles have emerged; for instance, IP3R1 regulates the induction of long term depression, while IP3R3 regulates apoptosis [99, 100]. In many contexts, it could be useful to target one specific receptor subtype. As some tissues or cell types exclusively express one receptor subtype, the availability of a subtype-selective drug could ensure a tissue-specific drug action. Moreover, we believe that ligands selective for the different IP3R subtypes would greatly benefit studies designed to assess the different functional role of the receptor subtypes.
HIGHLIGHTS BOX.
Inositol 1,4,5-trisphosphate receptors (IP3Rs) are intracellular calcium (Ca2+) release channels located on the endoplasmic reticulum, the major Ca2+ reservoir within the cell.
Three different subtypes of IP3Rs are expressed in mammalian cells, namely IP3R1, IP3R2, and IP3R3.
The intrinsic complexity of the recently solved structure of IP3R envisages the potential participation of the receptor to a dense network of processes.
The structure of the ligand-binding domain of IP3Rs has facilitated the design of compatible ligands, but the limiting step remains their actual effectiveness in biological context.
The availability of effective drugs targeting IP3Rs can be harnessed to better comprehend the biology of IP3R, exploring its involvement in various settings.
Acknowledgments
Funding:
The authors are funded by the National Institutes of Health via the National Heart Lung and Blood Institute (grant No. R01-HL146691) and the National Institute of Diabetes and Digestive and Kidney Diseases (via grant R01-DK123259).
Footnotes
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed
Reviewer Disclosures:
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Hamada K and Mikoshiba K, IP3 Receptor Plasticity Underlying Diverse Functions. Annu Rev Physiol, 2020. 82: p. 151–176. [DOI] [PubMed] [Google Scholar]
- 2.Lin CC, Baek K, and Lu Z, Apo and InsP(3)-bound crystal structures of the ligand-binding domain of an InsP(3) receptor. Nat Struct Mol Biol, 2011. 18(10): p. 1172–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gambardella J, Lombardi A, Morelli MB, et al. , Inositol 1,4,5-Trisphosphate Receptors in Human Disease: A Comprehensive Update. J Clin Med, 2020. 9(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Santulli G, Nakashima R, Yuan Q, et al. , Intracellular calcium release channels: an update. J Physiol, 2017. 595(10): p. 3041–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fan G, Baker ML, Wang Z, et al. , Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature, 2015. 527(7578): p. 336–41. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study shows the gating machinery of IP3R using cryogenic electron microscopy.
- 6.Serysheva II, Baker MR, and Fan G, Structural Insights into IP3R Function. Adv Exp Med Biol, 2017. 981: p. 121–147. [DOI] [PubMed] [Google Scholar]
- 7.Fan G, Baker MR, Wang Z, et al. , Cryo-EM reveals ligand induced allostery underlying InsP3R channel gating. Cell Res, 2018. 28(12): p. 1158–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamada K, Miyatake H, Terauchi A, et al. , IP3-mediated gating mechanism of the IP3 receptor revealed by mutagenesis and X-ray crystallography. Proc Natl Acad Sci U S A, 2017. 114(18): p. 4661–4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chandran A, Chee X, Prole DL, et al. , Exploration of inositol 1,4,5-trisphosphate (IP3) regulated dynamics of N-terminal domain of IP3 receptor reveals early phase molecular events during receptor activation. Sci Rep, 2019. 9(1): p. 2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartok A, Weaver D, Golenar T, et al. , IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat Commun, 2019. 10(1): p. 3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lock JT and Parker I, IP3 mediated global Ca(2+) signals arise through two temporally and spatially distinct modes of Ca(2+) release. Elife, 2020. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunt H, Tilunaite A, Bass G, et al. , Ca(2+) Release via IP3 Receptors Shapes the Cardiac Ca(2+) Transient for Hypertrophic Signaling. Biophys J, 2020. 119(6): p. 1178–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Terry LE, Alzayady KJ, Wahl AM, et al. , Disease-associated mutations in inositol 1,4,5-trisphosphate receptor subunits impair channel function. J Biol Chem, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ronkko J, Molchanova S, Revah-Politi A, et al. , Dominant mutations in ITPR3 cause Charcot-Marie-Tooth disease. Ann Clin Transl Neurol, 2020. 7(10): p. 1962–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Capel RA, Bose SJ, Collins TP, et al. , IP3-mediated Ca(2+) release regulates atrial Ca(2+)-transients and pacemaker function by stimulation of adenylyl cyclases. Am J Physiol Heart Circ Physiol, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Azumaya CM, Linton EA, Risener CJ, et al. , Cryo-EM structure of human type-3 inositol triphosphate receptor reveals the presence of a self-binding peptide that acts as an antagonist. J Biol Chem, 2020. 295(6): p. 1743–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang F, Huang L, Tso A, et al. , Inositol 1,4,5-trisphosphate receptors are essential for fetal-maternal connection and embryo viability. PLoS Genet, 2020. 16(4): p. e1008739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gambardella J, Sorriento D, Bova M, et al. , Role of Endothelial G Protein-Coupled Receptor Kinase 2 in Angioedema. Hypertension, 2020. 76(5): p. 1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bosanac I, Alattia JR, Mal TK, et al. , Structure of the inositol 1,4,5-trisphosphate receptor binding core in complex with its ligand. Nature, 2002. 420(6916): p. 696–700. [DOI] [PubMed] [Google Scholar]; •• This study revealed the crystal structure of the IP3 binding domain of IP3R, mapping the key residues for receptor-ligand interaction.
- 20.Moritsugu K, Ito T, and Kidera A, Allosteric response to ligand binding: Molecular dynamics study of the N-terminal domains in IP 3 receptor. Biophys Physicobiol, 2019. 16: p. 232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshikawa F, Morita M, Monkawa T, et al. , Mutational analysis of the ligand binding site of the inositol 1,4,5-trisphosphate receptor. J Biol Chem, 1996. 271(30): p. 18277–84. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi M, Kagasaki T, Hosoya T, et al. , Adenophostins A and B: potent agonists of inositol-1,4,5-trisphosphate receptor produced by Penicillium brevicompactum. Taxonomy, fermentation, isolation, physico-chemical and biological properties. J Antibiot, 1993. 46(11): p. 1643–7. [DOI] [PubMed] [Google Scholar]; • Seminal study reporting the discovery of adenophostins as potent agonists of IP3Rs. Most of the IP3R-ligands synthetized later are inspired by this class of molecules.
- 23.Saleem H, Tovey SC, Riley AM, et al. , Stimulation of inositol 1,4,5-trisphosphate (IP3) receptor subtypes by adenophostin A and its analogues. PLoS One, 2013. 8(2): p. e58027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rossi AM, Riley AM, Potter BV, et al. , Adenophostins: High-Affinity Agonists of IP(3) Receptors. Curr Top Membr, 2010. 66: p. 209–33. [DOI] [PubMed] [Google Scholar]
- 25.Sureshan KM, Riley AM, Thomas MP, et al. , Contribution of phosphates and adenine to the potency of adenophostins at the IP(3) receptor: synthesis of all possible bisphosphates of adenophostin A. J Med Chem, 2012. 55(4): p. 1706–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mochizuki T, Kondo Y, Abe H, et al. , Synthesis of adenophostin A analogues conjugating an aromatic group at the 5’-position as potent IP3 receptor ligands. J Med Chem, 2006. 49(19): p. 5750–8. [DOI] [PubMed] [Google Scholar]
- 27.Vibhute AM, Konieczny V, Taylor CW, et al. , Triazolophostins: a library of novel and potent agonists of IP3 receptors. Org Biomol Chem, 2015. 13(24): p. 6698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vibhute AM, Pushpanandan P, Varghese M, et al. , Synthesis of dimeric analogs of adenophostin A that potently evoke Ca(2+) release through IP3 receptors. RSC Adv, 2016. 6(89): p. 86346–86351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Correa V, Riley AM, Shuto S, et al. , Structural determinants of adenophostin A activity at inositol trisphosphate receptors. Mol Pharmacol, 2001. 59(5): p. 1206–15. [DOI] [PubMed] [Google Scholar]
- 30.Dohle W, Su X, Mills SJ, et al. , A synthetic cyclitol-nucleoside conjugate polyphosphate is a highly potent second messenger mimic. Chem Sci, 2019. 10(20): p. 5382–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mills SJ, Rossi AM, Konieczny V, et al. , d-chiro-Inositol Ribophostin: A Highly Potent Agonist of d-myo-Inositol 1,4,5-Trisphosphate Receptors: Synthesis and Biological Activities. J Med Chem, 2020. 63(6): p. 3238–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenkins DJ and Potter BV, A Ca(2+)-mobilising carbohydrate-based polyphosphate: synthesis of 2-hydroxyethyl alpha-D-glucopyranoside 2’,3,4-trisphosphate. Carbohydr Res, 1996. 287(2): p. 169–82. [DOI] [PubMed] [Google Scholar]
- 33.Shipton ML, Riley AM, Rossi AM, et al. , Both d- and l-Glucose Polyphosphates Mimic d-myo-Inositol 1,4,5-Trisphosphate: New Synthetic Agonists and Partial Agonists at the Ins(1,4,5)P3 Receptor. J Med Chem, 2020. 63(10): p. 5442–5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lock JT, Smith IF, and Parker I, Comparison of Ca(2+) puffs evoked by extracellular agonists and photoreleased IP3. Cell Calcium, 2017. 63: p. 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Decrock E, De Bock M, Wang N, et al. , Flash photolysis of caged IP3 to trigger intercellular Ca2+ waves. Cold Spring Harb Protoc, 2015. 2015(3): p. 289–92. [DOI] [PubMed] [Google Scholar]
- 36.Kantevari S, Gordon GR, MacVicar BA, et al. , A practical guide to the synthesis and use of membrane-permeant acetoxymethyl esters of caged inositol polyphosphates. Nat Protoc, 2011. 6(3): p. 327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keebler MV and Taylor CW, Endogenous signalling pathways and caged IP3 evoke Ca(2+) puffs at the same abundant immobile intracellular sites. J Cell Sci, 2017. 130(21): p. 3728–3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maranto AR, Primary structure, ligand binding, and localization of the human type 3 inositol 1,4,5-trisphosphate receptor expressed in intestinal epithelium. J Biol Chem, 1994. 269(2): p. 1222–30. [PubMed] [Google Scholar]
- 39.Wojcikiewicz RJ and Luo SG, Differences among type I, II, and III inositol-1,4,5-trisphosphate receptors in ligand-binding affinity influence the sensitivity of calcium stores to inositol-1,4,5-trisphosphate. Mol Pharmacol, 1998. 53(4): p. 656–62. [DOI] [PubMed] [Google Scholar]
- 40.Miyakawa T, Maeda A, Yamazawa T, et al. , Encoding of Ca2+ signals by differential expression of IP3 receptor subtypes. EMBO J, 1999. 18(5): p. 1303–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wagner LE 2nd and Yule DI, Differential regulation of the InsP(3) receptor type-1 and −2 single channel properties by InsP(3), Ca(2)(+) and ATP. J Physiol, 2012. 590(14): p. 3245–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gregory RB, Hughes R, Riley AM, et al. , Inositol trisphosphate analogues selective for types I and II inositol trisphosphate receptors exert differential effects on vasopressin-stimulated Ca2+ inflow and Ca2+ release from intracellular stores in rat hepatocytes. Biochem J, 2004. 381(Pt 2): p. 519–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dyer JL and Michelangeli F, Inositol 1,4,5-trisphosphate receptor isoforms show similar Ca2+ release kinetics. Cell Calcium, 2001. 30(4): p. 245–50. [DOI] [PubMed] [Google Scholar]
- 44.Mak DO, McBride S, and Foskett JK, Inositol 1,4,5-trisphosphate [correction of tris-phosphate] activation of inositol trisphosphate [correction of tris-phosphate] receptor Ca2+ channel by ligand tuning of Ca2+ inhibition. Proc Natl Acad Sci U S A, 1998. 95(26): p. 15821–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nerou EP, Riley AM, Potter BV, et al. , Selective recognition of inositol phosphates by subtypes of the inositol trisphosphate receptor. Biochem J, 2001. 355(Pt 1): p. 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mochizukia T, Tanimura A, Nezub A, et al. , Design and synthesis of indole derivatives of adenophostin A. A entry into subtype-selective IP3 receptor ligands. Tetrahedron Letters, 2010. 51: p. 977–979. [Google Scholar]
- 47.Nakagawa M EM, Structures of xestospongin A, B, C and D, novel vasodilative compounds from marine sponge Xestospongia exigua. Tetrahedron Letters, 1984. Volume 25( Issue 30): p. Pages 3227–3230. [Google Scholar]
- 48.Hoye Thomas R., J.T.N., and Yao Letitia J., A Total Synthesis of (+)-Xestospongin A/(+)-Araguspongine D. Journal of the American Chemical Society. 116: p. 2617–2618. [Google Scholar]
- 49.Kobayashi M, Kawazoe K, and Kitagawa I, Araguspongines B, C, D, E, F, G, H, and J, new vasodilative bis-1-oxaquinolizidine alkaloids from an okinawan marine sponge, Xestospongia sp. Chem Pharm Bull (Tokyo), 1989. 37(6): p. 1676–8. [DOI] [PubMed] [Google Scholar]
- 50.Althagbi HI, Alarif WM, Al-Footy KO, et al. , Marine-Derived Macrocyclic Alkaloids (MDMAs): Chemical and Biological Diversity. Mar Drugs, 2020. 18(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gafni J, Munsch JA, Lam TH, et al. , Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron, 1997. 19(3): p. 723–33. [DOI] [PubMed] [Google Scholar]
- 52.Ta TA, Feng W, Molinski TF, et al. , Hydroxylated xestospongins block inositol-1,4,5-trisphosphate-induced Ca2+ release and sensitize Ca2+-induced Ca2+ release mediated by ryanodine receptors. Mol Pharmacol, 2006. 69(2): p. 532–8. [DOI] [PubMed] [Google Scholar]
- 53.Bootman MD, Collins TJ, Mackenzie L, et al. , 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J, 2002. 16(10): p. 1145–50. [DOI] [PubMed] [Google Scholar]
- 54.Oka T, Sato K, Hori M, et al. , Xestospongin C, a novel blocker of IP3 receptor, attenuates the increase in cytosolic calcium level and degranulation that is induced by antigen in RBL-2H3 mast cells. Br J Pharmacol, 2002. 135(8): p. 1959–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ozaki H, Hori M, Kim YS, et al. , Inhibitory mechanism of xestospongin-C on contraction and ion channels in the intestinal smooth muscle. Br J Pharmacol, 2002. 137(8): p. 1207–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosado JA and Sage SO, Coupling between inositol 1,4,5-trisphosphate receptors and human transient receptor potential channel 1 when intracellular Ca2+ stores are depleted. Biochem J, 2000. 350 Pt 3: p. 631–5. [PMC free article] [PubMed] [Google Scholar]
- 57.Schafer M, Bahde D, Bosche B, et al. , Modulation of early [Ca2+]i rise in metabolically inhibited endothelial cells by xestospongin C. Am J Physiol Heart Circ Physiol, 2001. 280(3): p. H1002–10. [DOI] [PubMed] [Google Scholar]
- 58.Yuan Z, Cai T, Tian J, et al. , Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol Biol Cell, 2005. 16(9): p. 4034–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Smet P, Parys JB, Callewaert G, et al. , Xestospongin C is an equally potent inhibitor of the inositol 1,4,5-trisphosphate receptor and the endoplasmic-reticulum Ca(2+) pumps. Cell Calcium, 1999. 26(1–2): p. 9–13. [DOI] [PubMed] [Google Scholar]
- 60.Castonguay A and Robitaille R, Xestospongin C is a potent inhibitor of SERCA at a vertebrate synapse. Cell Calcium, 2002. 32(1): p. 39–47. [DOI] [PubMed] [Google Scholar]
- 61.Solovyova N, Fernyhough P, Glazner G, et al. , Xestospongin C empties the ER calcium store but does not inhibit InsP3-induced Ca2+ release in cultured dorsal root ganglia neurones. Cell Calcium, 2002. 32(1): p. 49–52. [DOI] [PubMed] [Google Scholar]
- 62.Bishara NB, Murphy TV, and Hill MA, Capacitative Ca(2+) entry in vascular endothelial cells is mediated via pathways sensitive to 2 aminoethoxydiphenyl borate and xestospongin C. Br J Pharmacol, 2002. 135(1): p. 119–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haak LL, Song LS, Molinski TF, et al. , Sparks and puffs in oligodendrocyte progenitors: cross talk between ryanodine receptors and inositol trisphosphate receptors. J Neurosci, 2001. 21(11): p. 3860–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jaimovich E, Mattei C, Liberona JL, et al. , Xestospongin B, a competitive inhibitor of IP3-mediated Ca2+ signalling in cultured rat myotubes, isolated myonuclei, and neuroblastoma (NG108–15) cells. FEBS Lett, 2005. 579(10): p. 2051–7. [DOI] [PubMed] [Google Scholar]
- 65.Duncan RS, Hwang SY, and Koulen P, Differential inositol 1,4,5-trisphosphate receptor signaling in a neuronal cell line. Int J Biochem Cell Biol, 2007. 39(10): p. 1852–62. [DOI] [PubMed] [Google Scholar]
- 66.Saleem H, Tovey SC, Molinski TF, et al. , Interactions of antagonists with subtypes of inositol 1,4,5-trisphosphate (IP3) receptor. Br J Pharmacol, 2014. 171(13): p. 3298–312. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This study offers an elegant evaluation of the efficacy and pitfalls of the several IP3R antagonists.
- 67.Maruyama T, Kanaji T, Nakade S, et al. , 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem, 1997. 122(3): p. 498–505. [DOI] [PubMed] [Google Scholar]
- 68.Missiaen L, Callewaert G, De Smedt H, et al. , 2-Aminoethoxydiphenyl borate affects the inositol 1,4,5-trisphosphate receptor, the intracellular Ca2+ pump and the non-specific Ca2+ leak from the non-mitochondrial Ca2+ stores in permeabilized A7r5 cells. Cell Calcium, 2001. 29(2): p. 111–6. [DOI] [PubMed] [Google Scholar]
- 69.Bilmen JG, Wootton LL, Godfrey RE, et al. , Inhibition of SERCA Ca2+ pumps by 2-aminoethoxydiphenyl borate (2-APB). 2-APB reduces both Ca2+ binding and phosphoryl transfer from ATP, by interfering with the pathway leading to the Ca2+-binding sites. Eur J Biochem, 2002. 269(15): p. 3678–87. [DOI] [PubMed] [Google Scholar]
- 70.Goto J, Suzuki AZ, Ozaki S, et al. , Two novel 2-aminoethyl diphenylborinate (2-APB) analogues differentially activate and inhibit store-operated Ca(2+) entry via STIM proteins. Cell Calcium, 2010. 47(1): p. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Poitras M, Bernier S, Boulay G, et al. , Interaction of benzene 1,2,4-trisphosphate with inositol 1,4,5-trisphosphate receptor and metabolizing enzymes. Eur J Pharmacol, 1993. 244(3): p. 203–10. [DOI] [PubMed] [Google Scholar]
- 72.Ward SG, Mills SJ, Liu C, et al. , D-myo-inositol 1,4,5-trisphosphate analogues modified at the 3-position inhibit phosphatidylinositol 3-kinase. J Biol Chem, 1995. 270(20): p. 12075–84. [DOI] [PubMed] [Google Scholar]
- 73.Vandeput F, Combettes L, Mills SJ, et al. , Biphenyl 2,3’,4,5’,6-pentakisphosphate, a novel inositol polyphosphate surrogate, modulates Ca2+ responses in rat hepatocytes. FASEB J, 2007. 21(7): p. 1481–91. [DOI] [PubMed] [Google Scholar]
- 74.Riley AM, Laude AJ, Taylor CW, et al. , Dimers of D-myo-inositol 1,4,5-trisphosphate: design, synthesis, and interaction with Ins(1,4,5)P3 receptors. Bioconjug Chem, 2004. 15(2): p. 278–89. [DOI] [PubMed] [Google Scholar]
- 75.Mills SJ, Luyten T, Erneux C, et al. , Multivalent benzene polyphosphate derivatives are non-Ca(2+)-mobilizing Ins(1,4,5)P3 receptor antagonists. Messenger (Los Angel), 2012. 1(2): p. 167–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ghosh TK, Eis PS, Mullaney JM, et al. , Competitive, reversible, and potent antagonism of inositol 1,4,5-trisphosphate-activated calcium release by heparin. J Biol Chem, 1988. 263(23): p. 11075–9. [PubMed] [Google Scholar]
- 77.Willuweit B and Aktories K, Heparin uncouples alpha 2-adrenoceptors from the Gi-protein in membranes of human platelets. Biochem J, 1988. 249(3): p. 857–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dasso LL and Taylor CW, Heparin and other polyanions uncouple alpha 1-adrenoceptors from G-proteins. Biochem J, 1991. 280 ( Pt 3): p. 791–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bezprozvanny IB, Ondrias K, Kaftan E, et al. , Activation of the calcium release channel (ryanodine receptor) by heparin and other polyanions is calcium dependent. Mol Biol Cell, 1993. 4(3): p. 347–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guillemette G, Lamontagne S, Boulay G, et al. , Differential effects of heparin on inositol 1,4,5-trisphosphate binding, metabolism, and calcium release activity in the bovine adrenal cortex. Mol Pharmacol, 1989. 35(3): p. 339–44. [PubMed] [Google Scholar]
- 81.Rossi AM, Sureshan KM, Riley AM, et al. , Selective determinants of inositol 1,4,5-trisphosphate and adenophostin A interactions with type 1 inositol 1,4,5-trisphosphate receptors. Br J Pharmacol, 2010. 161(5): p. 1070–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Iwai M, Michikawa T, Bosanac I, et al. , Molecular basis of the isoform-specific ligand-binding affinity of inositol 1,4,5-trisphosphate receptors. J Biol Chem, 2007. 282(17): p. 12755–64. [DOI] [PubMed] [Google Scholar]; • This study offers the molecular basis to develop isoform-specific ligands of IP3R.
- 83.Hermosura MC, Takeuchi H, Fleig A, et al. , InsP4 facilitates store-operated calcium influx by inhibition of InsP3 5-phosphatase. Nature, 2000. 408(6813): p. 735–40. [DOI] [PubMed] [Google Scholar]
- 84.Safrany ST, Wilcox RA, Liu C, et al. , Identification of partial agonists with low intrinsic activity at the inositol-1,4,5-trisphosphate receptor. Mol Pharmacol, 1993. 43(4): p. 499–503. [PubMed] [Google Scholar]
- 85.Liu C, al-Hafidh J, Westwick J, et al. , Synthesis of 1L-chiro-inositol 2,3,5-trisphosphorothioate, the first partial agonist at the platelet myo-inositol 1,4,5-trisphosphate receptor. Bioorg Med Chem, 1994. 2(4): p. 253–7. [DOI] [PubMed] [Google Scholar]
- 86.Murphy CT, Riley AM, Mills SJ, et al. , myo-inositol 1,4,6-trisphosphorothioate and myo-inositol 1,3, 6-trisphosphorothioate: partial agonists with very low intrinsic activity at the platelet myo-inositol 1,4,5-trisphosphate receptor. Mol Pharmacol, 2000. 57(3): p. 595–601. [DOI] [PubMed] [Google Scholar]
- 87.Keddie NS, Ye Y, Aslam T, et al. , Development of inositol-based antagonists for the D-myo-inositol 1,4,5-trisphosphate receptor. Chem Commun (Camb), 2011. 47(1): p. 242–4. [DOI] [PubMed] [Google Scholar]
- 88.Seo MD, Velamakanni S, Ishiyama N, et al. , Structural and functional conservation of key domains in InsP3 and ryanodine receptors. Nature, 2012. 483(7387): p. 108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Santulli G, Lewis D, des Georges A, et al. , Ryanodine Receptor Structure and Function in Health and Disease. Subcell Biochem, 2018. 87: p. 329–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Porta M, Zima AV, Nani A, et al. , Single ryanodine receptor channel basis of caffeine’s action on Ca2+ sparks. Biophys J, 2011. 100(4): p. 931–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Luo X, Li W, Kunzel K, et al. , IP3R-Mediated Compensatory Mechanism for Calcium Handling in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes With Cardiac Ryanodine Receptor Deficiency. Front Cell Dev Biol, 2020. 8: p. 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ando H, Mizutani A, Matsu-ura T, et al. , IRBIT, a novel inositol 1,4,5-trisphosphate (IP3) receptor-binding protein, is released from the IP3 receptor upon IP3 binding to the receptor. J Biol Chem, 2003. 278(12): p. 10602–12. [DOI] [PubMed] [Google Scholar]; • This study describes IRBIT, an endogenous IP3R antagonist, acting as a pseudo-ligand.
- 93.Ando H, Mizutani A, Kiefer H, et al. , IRBIT suppresses IP3 receptor activity by competing with IP3 for the common binding site on the IP3 receptor. Mol Cell, 2006. 22(6): p. 795–806. [DOI] [PubMed] [Google Scholar]
- 94.Kawaai K, Mizutani A, Shoji H, et al. , IRBIT regulates CaMKIIalpha activity and contributes to catecholamine homeostasis through tyrosine hydroxylase phosphorylation. Proc Natl Acad Sci U S A, 2015. 112(17): p. 5515–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kawaai K, Ando H, Satoh N, et al. , Splicing variation of Long-IRBIT determines the target selectivity of IRBIT family proteins. Proc Natl Acad Sci U S A, 2017. 114(15): p. 3921–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shcheynikov N, Son A, Hong JH, et al. , Intracellular Cl- as a signaling ion that potently regulates Na+/HCO3- transporters. Proc Natl Acad Sci U S A, 2015. 112(3): p. E329–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bonneau B, Ando H, Kawaai K, et al. , IRBIT controls apoptosis by interacting with the Bcl-2 homolog, Bcl2l10, and by promoting ER-mitochondria contact. Elife, 2016. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang D, Li Q, So I, et al. , IRBIT governs epithelial secretion in mice by antagonizing the WNK/SPAK kinase pathway. J Clin Invest, 2011. 121(3): p. 956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Inoue T, Kato K, Kohda K, et al. , Type 1 inositol 1,4,5-trisphosphate receptor is required for induction of long-term depression in cerebellar Purkinje neurons. J Neurosci, 1998. 18(14): p. 5366–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Khan AA, Soloski MJ, Sharp AH, et al. , Lymphocyte apoptosis: mediation by increased type 3 inositol 1,4,5-trisphosphate receptor. Science, 1996. 273(5274): p. 503–7. [DOI] [PubMed] [Google Scholar]