

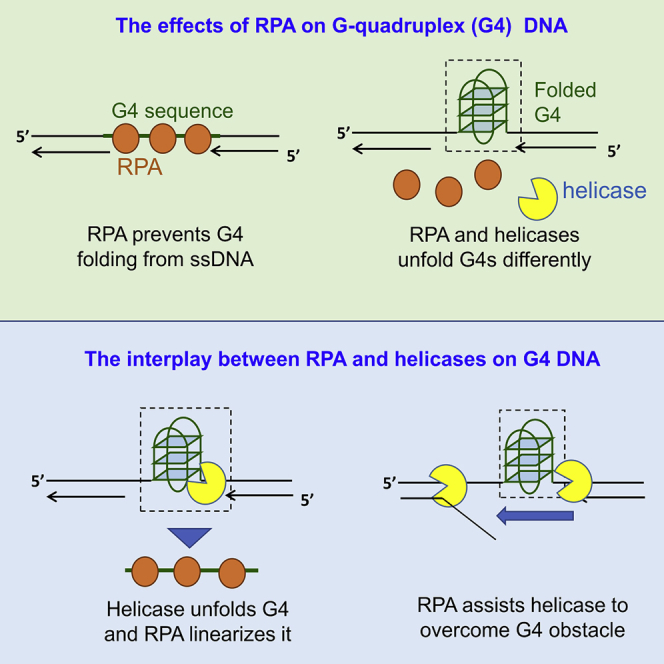
Summary
G-quadruplexes (G4s) are non-canonical DNA structures with critical roles in DNA metabolisms. To resolve those structures that can cause replication fork stalling and genomic instability, single-stranded DNA-binding proteins and helicases are required. Here, we characterized the interplay between RPA and helicases on G4s using single-molecule FRET. We first discovered that human RPA efficiently prevents G4 formation by preempting ssDNA before its folding. RPA also differentially interacts with the folded G4s. However, helicases such as human BLM and yeast Pif1 have different G4 preferences from RPA mainly based on loop lengths. More importantly, both RPA and these helicases are required for the stable G4 unfolding, as RPA promotes helicase-mediated repetitive unfolding into durative linear state. Furthermore, BLM can traverse G4 obstacles temporarily disrupted by RPA and continue to unwind downstream duplex. We finally proposed the mechanisms underlying above functions of RPA in preventing, resolving, and assisting helicases to eliminate G4s.
Subject areas: Molecular structure, Molecular biology
Graphical abstract
Highlights
-
•
RPA efficiently prevents G4 formation by preempting ssDNA before its folding
-
•
Loop length may direct folded G4s to different unfolding way by RPA and helicases
-
•
RPA promotes helicase-mediated repetitive G4 unfolding into durative linear state
-
•
RPA assists BLM to overcome G4 obstacle and continue to unwind downstream duplex
Molecular structure; Molecular biology
Introduction
G-rich DNA sequences may fold into G-quadruplexes (G4s), which are the four-stranded nucleic acid structures held together by Hoogsteen bonds and further stabilized by monovalent cations (Bochman et al., 2012; Varshney et al., 2020). About 716,310 G4 sequences have been identified in the human genome (Chambers et al., 2015), which are particularly enriched in gene promoters, introns, replication origins, and telomeres (Maizels and Gray, 2013; Lombardi and Londono-Vallejo, 2020). Due to their essential biological functions in critical cellular processes, including initiation of DNA replication, regulation of DNA transcription, telomere maintenance, and anti-cancer drug targeting (Cimino-Reale et al., 2016), G4s have attracted great attention in recent years (Lerner and Sale, 2019). It is worth noting that, in living cells, G4s dynamically fluctuate between the folded and unfolded states (Di Antonio et al., 2020). The folding/unfolding of G4s is controlled by specialized interacting proteins, thus the biological functions of G4s can be elaborately regulated (Lerner and Sale, 2019; Mendoza et al., 2016). Importantly, the clear connections between G4s and a number of human diseases have been established by recent studies (Maizels, 2015). Therefore, understanding the G4 regulation mechanism, i.e., how G4s are prevented or unwound by proteins, becomes very significant, as these stable secondary structures can lead to the stalling of replication forks and cause genomic instability.
It is known that single-stranded DNA binding proteins (SSB) interact with G4s (Lerner and Sale, 2019). As the most abundant SSB (∼1 μM in vivo) and the first responder of ssDNA in eukaryotes, replication protein A (RPA) is a highly conserved protein with critical roles in DNA replication, recombination, and repair (Chen and Wold, 2014). RPA has three subunits of 70-kDa (RPA1), 32-kDa (RPA2), and 14-kDa (RPA3), and a trimerization core is formed through the interaction between each subunit (Chen and Wold, 2014; Fan and Pavletich, 2012). Recently, RPA was reported to prevent the formation of G-rich structures suggested to be G4s at the lagging-strand telomeres to allow the maintenance of chromosome ends (Audry et al., 2015). In contrast, RPA1 mutation provokes the accumulation of those G-rich structures and the recruitment of homologous recombination factors (Audry et al., 2015). Besides preventing G4 formation, RPA was also reported to disrupt the folded G4 structures in the 5′–3′ polarity (Salas et al., 2006; Safa et al., 2014, 2016; Lancrey et al., 2018). RPA trimerization core preferentially recognizes G4 or G-rich DNA, likely responsible for unfolding G4s (Prakash et al., 2011a, 2011b). Interestingly, Qureshi et al. recently discovered that the decreases in G4 thermal stability (the energy difference between the folded and unfolded G4) did not necessarily lead to the increases in the G4 unfolding efficiency by RPA (Qureshi et al., 2012), leaving the major determinant of RPA-mediated G4 unfolding still unknown. Given that RPA is ubiquitous in the cell nucleus, it is thus capital to understand its impact on G4 thermodynamics and G4 resolution at the molecular level.
A large number of proteins are required to remove the folded G4 barriers and maintain the processive DNA replication and transcription, among which specialized DNA helicases have been extensively studied (Mendoza et al., 2016; Sauer and Paeschke, 2017; Lansdorp and van Wietmarschen, 2019). It is worth noting that all G4 helicases including BLM, WRN, DHX36, etc. shared a common mechanism of inducing the repetitive unfolding/refolding of G4 structures (Tippana et al., 2016; Hou et al., 2015; Wu et al., 2015, 2017). Although RPA can stimulate the helicase activity of WRN and BLM on duplex DNA by direct interaction between those proteins (Lee et al., 2018; Qin et al., 2020), the interplay between RPA and helicases in G4 DNA unfolding were still poorly understood. Recently, the interaction of BLM and WRN with RPA was detected to suppress G4s in human cancer cell lines, facilitated by the HERC2 protein (Wu et al., 2018). RPA and Pif1 were also reported to remove G4s coordinately at both leading and lagging strands in DNA replication in yeast cells (Maestroni et al., 2020). These evidences then raise the important questions that helicases may require RPA to fulfill their function of resolving G4s. How RPA and helicases are coordinated with each other in G4 unfolding at the molecular level remains elusive.
In this report, we studied the interaction between G4 DNA, RPA, and helicases mainly using single-molecule fluorescence resonance energy transfer (smFRET), which allows us to accurately monitor the folding/unfolding of G4 DNA in real time at the molecular level (Lee et al., 2019). We first considered the prevention effect of RPA on G4 structure formation starting from an unfolded structure by rapidly applying RPA and K+ to the G4 strand. Then, we addressed the unfolding effect of RPA on the well-folded G4s, which were prepared by slowly annealing in K+. Next, we looked at the unwinding effect of helicases on the folded G4s and finally to the same G4s together with RPA. As G4 structures may exist in different DNA environments during DNA replication, repair, etc., we also varied the G4 DNA constructs by using different linker lengths between G4 and duplex, as well as G4s with different polarity related to the duplex. Our results revealed that RPA significantly prevents G4 formation by preempting ssDNA before the folding. More importantly, RPA plays multifaceted roles complementary to helicases in processing the folded G4 DNA. First, the folded G4s are potentially directed into the RPA- or helicases-mediated unfolding pathways differentially by their loop lengths. For instance, the short-looped G4s were repetitively resolved by RPA but poorly unfolded by BLM and Pif1. Second, RPA promotes helicase-mediated repetitive G4 unfolding into the durative linear state, i.e., both helicases and RPA are necessary in the case of stable G4 unfolding. Third, RPA temporarily disrupts those short-looped G4s that can be poorly unfolded by helicase, allowing helicase to traverse the G4 obstacles and then efficiently unwind the downstream duplex. Overall, our results highlight the pivotal roles of RPA in eliminating G4s in DNA metabolisms such as replication, transcription, and repair.
Results
RPA significantly prevents the formation of G4 structures by preempting ssDNA before the folding
In DNA replication or transcription, the transient exposure of ssDNA could lead to the formation of G4 structures (Lerner and Sale, 2019). As the first responder to ssDNA in the eukaryotic cell, RPA may interfere with G4 formation. Here, at the single-molecule level, we mimicked the dynamic G4-folding process from ssDNA with the coexistence of two opposing factors K+ and RPA (Figure 1A). A 27-bp partial duplex with the 3′-ssDNA harboring G4 sequence was used. Cy3 and Cy5 were labeled at the end of the ssDNA and the sixth nucleotide inside the duplex, respectively. A 4-nt linker was placed between the duplex and G4 motif to avoid the steric hindrance for G4 folding.
Figure 1.

RPA significantly suppresses the folding of G-rich DNA sequences into G4 structures
(A) Schematic diagram of the Cy3- and Cy5-labeled DNA harboring G4 motif. K+ will drive G4 sequence folding into G4 structure; however, on the opposite, the tight occupation by RPA (a trimer composed of 70A, 70B, and 70C domains in the 70 kD subunit; the 32 kD subunit; and the 14 kD subunit) may maintain G4 motif in the linearized state.
(B–G) The upper panels are the FRET distributions of G4 motifs in 25 mM Tris-HCl before and 4 min after the addition of 200 mM KCl with 5 mM MgCl2. The lower panels are the FRET distributions after the addition of RPA in 200 mM KCl and 5 mM MgCl2 to the G4 DNA substrates originally prepared in 25 mM Tris-HCl.
(H) The fractions of linearized G4s were obtained from the FRET peaks at ∼E0.2. The error bars were obtained from at least three repetitive experiments. Data are presented as mean ± SEM.
First, we examined the formation of 3-layered G4s with three 1-nt to 5-nt loops (named as 3G4LN in Table S1, N denotes the loop lengths). The 3-layered G4 with 1-, 2-, and 1-nt loops was referred to as 3G4L121. All substrates were prepared in 25 mM Tris-HCl free of monovalent cations to avoid G4 folding. Large variations in FRET efficiencies exist among the various oligos in the absence of cations (Figures 1B–1G), possibly due to the different DNA lengths, different DNA flexibility, or formation of some partial structures. Although the physiologically relevant K+ is ∼100 mM, to promote G4 folding from ssDNA (Figure 1A), a high concentration of 200 mM KCl was used together with 5 mM MgCl2, which can further stabilize G4s (Hou et al., 2017). Upon the addition of ions (Figures 1B–1G, upper panels), the FRET distributions show the significant shift to the right, reflecting the formation of G4 structures (Figure S1). The different peak widths may suggest the different structural homogeneity possibly including the G4 intermediate states. According to the literature (Chen and Wold, 2014), the physiological concentration of RPA might be as high as 1 μM. To observe the interaction between single-molecule RPA and DNA, a low RPA concentration at nanomolar level (10 nM) was used. Besides, to mimic the effects of the possible concentration variations of RPA inside the cell, an RPA concentration gradient was also applied. Interestingly, when 10–500 nM RPA in the folding buffer was introduced to the G4 sequences that were originally placed in 25 mM Tris-HCl, different results from above were observed (Figures 1B–1G, lower panels). Figure 1H demonstrated the fractions of G4s at the linearized state, i.e., the leftmost peak at ∼ E0.2 in FRET distributions. Besides 3G4L1, the formation of G4 structures in other substrates was all remarkably prevented. Particularly, G4s with long loops such as 4-nt or 5-nt can be thoroughly suppressed by 50 nM RPA. Even the high stabilized G4s such as 3G4L121 can be essentially prohibited by 100 nM RPA. Similar phenomena were associated with G4s consisting of different G-tetrad layers (Figures S2A–S2E).
To further elucidate the mechanism underlying the prevention of G4 formation by RPA, the dynamic FRET traces were analyzed. Upon the addition of the G4 folding buffer, the FRET levels of all G4 sequences immediately rise to higher levels in ∼0.1–0.2 s (Figures 2A–2F, panel 1). However, in the presence of RPA, different types of traces were observed. First, the FRET level immediately goes up and stays at higher values, indicating the folding of G4s. Second, G4s with very short loops (3G4L121) display quick fluctuations, reflecting the interaction of RPA with the rapidly folded G4s (Figure 2A). The FRET traces of 3G4L1 in the presence of RPA are similar to that of 3G4L121 (not shown). Those G4s might be temporarily and partially unfolded instead of being stably linearized, as the ∼E0.2 state cannot be observed. Third, the FRET level immediately drops to and maintains at ∼E0.2, reflecting the stable linearization of the G4 sequence possibly by multiple RPA, as each RPA has an 8-nt binding mode (Wang et al., 2019). Fourth, the FRET level rises to higher values at first, indicating the formation of higher-order structures, and then drops to ∼ E0.2, reflecting the subsequent linearization by RPA.
Figure 2.

RPA prevents the formation of G4 structures by preempting ssDNA before its folding
(A–F) The representative FRET traces of G4 DNA substrates prepared in 25 mM Tris-HCl, pH 7.5 upon the addition of 200 mM KCl and 5 mM MgCl2 (panel 1) and RPA in 200 mM KCl with 5 mM MgCl2 (panels 2–4). The red arrow indicates the addition of ions or proteins, i.e., the start of the flow to the ∼25 μL flow cell.
(G) The fractions of FRET traces showing different types of changes upon the addition of RPA. All the single-molecule traces in each experiment were examined and classified into different types.
Figure 2G further reveals that the fractions of the above four types differ significantly in different substrates. Importantly, the fractions of traces showing constant FRET decreases (Type 3) rise clearly with the increases in RPA concentration, i.e., DNA is quickly linearized by RPA before it can be folded. Therefore, the appearance of the above different types of traces may be caused by the competition between the cation-driven G4 folding (determined by G4 folding energy barrier) and RPA-binding-mediated DNA linearization (sensitive to RPA concentration and G4 sequence length) as shown in Figure S2F. Interestingly, no traces with initial FRET decreases were observed to rise to higher levels later, suggesting that the stable linearization by RPA thoroughly prevents its folding. In consideration of the lower KCl concentration ∼100 mM inside the cells, we speculated that the prevention effect of RPA on G4 folding from ssDNA may be more significant than that in 200 mM KCl (Table S2). Altogether, the above evidences suggest that the exposed ssDNA sequences harboring G4 motifs are not necessarily folded into G4 structures owning to the high abundance of RPA inside the cell (Chen and Wold, 2014).
Long loops and high thermal stability can protect the preformed G4 structures against RPA
According to the above results, G4 structures may emerge once RPA has not effectively prohibited their folding from ssDNA. Besides, there are proteins inside the cell such as nucleolin and LARK that promote G4 structure folding (Gonzalez et al., 2009; Niu et al., 2019). Therefore, we further investigated the dynamic interaction between RPA and the pre-folded G4 structures (Figure 3A). For this purpose, G4 substrates were prepared by incubating at 95°C for 5 min and then slowly cooling down to room temperature to allow the well folding of G4 structures. However, in the above section, we aimed to address the influences of RPA on the folding of G4 structures from ssDNA; therefore, RPA and ions were applied simultaneously to the unfolded G4 strands without low annealing in advance. Indeed, G4s generated by slow annealing procedure display stronger CD signals than that just by quick mixing (Figures S1B, S1D, and S1E), implying the more pronounced G4 formation. It is worth noting that a high concentration of 200 mM KCl was used above to promote the G4 folding from ssDNA against RPA; however, in the following assays on the interactions between the proteins and the preformed G4s, 100 mM or lower concentration of ions were adopted to be consistent with the physiological conditions and the previous G4 unwinding studies (Wu et al., 2015; Voter et al., 2018; Hou et al., 2017; Salas et al., 2006; Zhou et al., 2014; Tippana et al., 2016).
Figure 3.

The interaction between RPA and different preformed G4 structures
(A) The schematic diagram of the experimental design. Preformed G4 substrates were anchored at the coverslip surface, and then different concentrations of RPA were added to unfold the G4 structures. For G4 substrates prepared in 100 mM KCl (or 100 mM NaCl), the reaction buffer of RPA is composed of 100 mM KCl (or 100 mM NaCl) and 5 mM MgCl2.
(B and C) The left panels are the FRET distributions of 2G4 in 100 mM KCl and 3G4L3 in 100 mM NaCl before and 4 min after the addition of 10 nM RPA. Washing the channels with buffer leads to little difference in the FRET distributions. The right panels are the representative traces. The red arrow indicates the addition of RPA.
(D–F) The left panels are the FRET distributions of 3G4L3, 3G4L4, and 3G4L5 in 100 mM KCl before and 4 min after the addition of 100 nM RPA. The right panels are the representative traces.
The 2-layered G4 substrate with 2-, 3-, and 2-nt loops prepared in 100 mM KCl was examined first (Figure 3B). Upon the addition of 10 nM RPA, the FRET levels of most molecules decreased immediately from ∼E0.8 to ∼E0.2, reflecting the complete G4 disruption. 3G4L3 prepared in 100 mM NaCl was also efficiently resolved into the linearized state (Figure 3C). Both two-step and one-step decreases can be observed in the traces of these two substrates. Interestingly, flushing buffers to the chamber results in no significant change in the FRET distributions, suggesting that the G4 sequences were stably bound by RPA. In sharp contrast, 3G4L3, 3G4L4, and 3G4L5 prepared in 100 mM KCl were poorly disrupted (Figures 3D–3F) because the addition of 100 nM RPA only leads to the slight FRET decreases from ∼E0.8 to ∼E0.7 (the 3G4L4 and 3G4L5 were not tested in NaCl due to the low thermal stability as shown in Table S2). Using the FRET-melting analysis, Figure S3 further corroborated that RPA significantly destabilizes 2G4 in 100 mM KCl but has a much poor unfolding effect on the long-looped 3G4L4, consistent with the observations in Figures 3B and 3E. Besides, the preformed G4s with more than 3 layers such as 4G4 (Figure S4A) and 5G4 (Figure S4B) were poorly unfolded by RPA as well.
Interestingly, about 30% of 3G4L3 traces show an increase of FRET then undergo FRET decrease to ∼E0.2 in Figure 2G at 100 nM RPA; however, in Figure 3D, most traces display a slight FRET decrease and the small peak at ∼E0.2 is only ∼10%. We speculated that this discrepancy could be caused by the different G4-forming procedures that may lead to different G4 folding status and stabilities. Based on the pink curve in the upper panel of Figure 1E that shows a broad FRET distribution, 3G4L3 were not homogeneously folded (likely with folding intermediates) after the addition of KCl. However, 3G4L3 forms uniform structures by the slow annealing as shown by the single peak in black color in Figure 3D. This is also consistent with the CD signal differences in Figure S1D between the slow annealing and rapid mixing procedures. All the above evidences suggest that G4 folds more completely in the heating and slow annealing process. Therefore, the differences between Figures 2G and 3D may be caused by the existence of some partially folded G4s in the rapid mixing that can be easily unfolded by RPA (Figure 1E, upper panel, pink curve). However, RPA can potently disrupt the 3G4L3 in 100 mM NaCl prepared by the slowly annealing in Figure 3C. The major reason should be the much lower stability of 3G4L3 in 100 mM NaCl than in 100 mM KCl (49.6 versus 66.8°C).
To further compare the different interacting modes between RPA and different G4 structures, gel filtration was performed with two selective G4s with and without unfolding by RPA based on the above smFRET assays. Figure S4C–D suggests that two or more RPA molecules may associate with 3G4L3 in 100 mM NaCl, leading to stable G4 unfolding as shown in Figure 3C; however, RPA only forms one kind of complex with 4G4, possibly by interacting with the linker. To verify this speculation, a partial duplex with a 4-nt 3′-ssDNA was designed (Figure S4E). Indeed, the addition of RPA leads to the slight decreases of FRET level from ∼E0.9 to ∼E0.7, and the traces clearly show the dynamic binding between them. Therefore, RPA may only dynamically interact with the linker shown in Figures 3D–3F, S4A, and S4B without disrupting the G-tetrads.
Short-looped G4 structures are rapidly and repetitively resolved by RPA
After addressing the interaction between RPA and long-looped G4s, we next examined the RPA-mediated unfolding of short-looped G4s including 3G4L1 and 3G4L121. In fact, the binding affinities between RPA and 3G4L1 or 3G4121 are almost indistinguishable from that between RPA and 3G4L3 (KD 6.3–8.6 nM, Figure S5). Besides, the KD values are slightly higher than that between RPA and ssDNA (∼4 nM) in similar experiment conditions (Wang et al., 2019), reflecting the differences between G4 and ssDNA in RPA binding. Thereafter, the dynamic interactions between RPA and those two G4s were carefully studied by smFRET (Figure 4A and S6).
Figure 4.

RPA can rapidly and repetitively disrupt the G4 structures with very short loops
(A) Schematic diagram of the experimental design. The G4 substrate has three loops of 1-, 2-, and 1-nt, respectively.
(B) The FRET distributions of 3G4L121 before and 4 min after the addition of varying concentrations of RPA.
(C) The representative FRET traces. tG4 denotes the duration time of G4 folding in the presence of RPA. tnG4 denotes the duration time of G4 unfolding.
(D) The interaction of RPA with G4 structure being placed at the 5′-end of the duplex DNA.
(E) The FRET distributions of 3G4L121∗ before and 4 min after the addition of RPA.
(F) The FRET traces in 50–100 nM RPA.
Panels 1 in Figures 4B and 4C confirmed that stable G4 structures have been predominantly formed. The G4 folding state at ∼E0.9 is slightly higher than the ∼E0.8 of other long-looped G4s in Figure 3 likely due to the different G4-folding topologies (Figure S1). Surprisingly, the addition of 10 nM RPA induces the abrupt decrease/increase of FRET levels between ∼E0.9 and ∼E0.35 (tnG4 ∼0.9 s, tG4 ∼15.5 s, Figure S6A), reflecting the quick G4 unfolding/refolding. As the complete and stable G4 unfolding generates an FRET at ∼E0.2 (Figures 1B and 1C), the E0.35 state here may reflect the partially unfolded G4. The non-specific interaction of RPA with the substrate without G4 unfolding can be ruled out, as the dynamic binding of RPA to the 4-nt linker only decreases the FRET signal slightly from ∼E0.9 to ∼E0.7 (Figure S4E). With the increases in protein concentrations, the FRET oscillations of both 3G4L121 and 3G4L1 become much more frequent in most molecules with the FRET distributions significantly shifting to the left (Figures 4B, 4C, and S6B). These evidences strongly suggest that RPA can rapidly and repetitively unfold the short-looped G4s. To further corroborate the above observations, we also placed G4 structures at the 5′-end of the duplex DNA (Figure 4D). The frequent FRET decrease/increase was observed in ∼25% short-looped G4 at 100 nM RPA (Figures 4E and 4F), reflecting the repetitive G4 unfolding, and the tnG4 was ∼1 s, very close to that in 3G4L121. We speculated that the differences between Figures 4B and 4E could be the results of different space lengths between the duplex and G4 rather than the different polarity. However, the change of FRET in 3G4L3∗ can hardly be observed (Figure S6C), consistent with that at the 3′-end of the duplex (Figure 3D). Hence one can see that whether a G4 can be resolved by RPA should be dependent on its intrinsic properties rather than the orientation relative to duplex DNA. As increasing or decreasing the linker significantly increases or decreases the frequency of FRET oscillations (Figure S7), we speculate that RPA may load onto the ssDNA linker by 70A or 70B domains in 5′–3′ polarity (Safa et al., 2014), and then the trimerization core destabilizes G4 by attracting the guanines in G-tetrads (Prakash et al., 2011b).
To understand why G4s undergo quick unfolding/refolding in 100 mM KCl when RPA exists, we then decreased G4 thermal stability by preparing 3G4L121 in 100 mM NaCl. It shows the same parallel topology (Figure S1B); however, the Tm value significantly decreased from above 88°C–57.4°C (Table S2). Upon the addition of RPA, a minor peak emerged at ∼E0.2 (Figure S6D), and stable G4 unfolding can be observed in about 15% traces. Although the repetitive unfolding still exists, the duration of the FRET state at ∼E0.35 increases compared with that in 100 mM KCl (0.9 s versus 1.6 s). This evidence suggests that the quick FRET fluctuation behavior in 100 mM KCl should be caused by the stronger tendency of G4 refolding.
BLM and Pif1 helicases display different G4 substrate preferences from RPA mainly based on loop length
As the majority of G4s in humans and Saccharomyces cerevisiae possesses three loops with each longer than 3-nt (Chambers et al., 2015; Capra et al., 2010; Wang et al., 2018), to resolve those G4s, specialized helicases may be required based on our above results. To verify this speculation, we further characterized the G4 substrate preference of the typical G4 helicase BLM from the RecQ family with the 3′–5′ polarity (Wu et al., 2015). We constructed a DNA substrate with a 14-nt 3′-ssDNA tail for BLM helicase to associate (referred to as 3G4L3S, Figure 5A). The linker between G4 and duplex was decreased to 2-nt to avoid the helicases to unwind duplex directly from the linker. Figure 5B shows that the G4 structure is uniformly folded at ∼E0.9, and no significant change can be observed when only 40 nM BLM was added, consistent with our previous study (Wu et al., 2015). Upon the addition of 40 nM BLM and 25 μM ATP, the FRET distribution shifts to the left and the trace displays frequent fluctuations between ∼E0.9 and ∼E0.3 (Figure 5C), reflecting the transient and possibly partial G4 unfolding/refolding in the ATP-dependent mode (Wu et al., 2015). However, to resolve G4s with longer or shorter loops (Figure S8), much higher concentrations of protein and ATP are required. Those evidences suggest that BLM is quite efficient at resolving human telomere G4 (3G4L3) but poorer at other G4s, possibly related to the peculiar parallel/antiparallel hybrid topology (Figure S1D).
Figure 5.

The helicases-mediated unfolding of G4 DNA structures with different loop lengths
(A) The 3-layered G4 substrate with three 3-nt loops for the 3′–5′ helicase. The ssDNA tail and stem duplex are 14-nt and 29-bp, respectively.
(B) FRET distributions before and after the addition of 40 nM BLM and 25 μM ATP in 25 mM Tris-HCl, 50 mM KCl, and 5 mM MgCl2. Adding only 40 nM BLM without ATP has no detectable effect on the FRET distribution of G4.
(C) The representative trace.
(D) The 3-layered G4 substrate with three 4-nt loops for the 5′–3′ helicase. The ssDNA tail and stem duplex are 26-nt and 15-bp, respectively.
(E) The remaining fractions of 3G4L4S∗ and 15bp2nt on the coverslip surface before and 4 min after the addition of 40 nM Pif1 and 0.5 mM ATP in 25 mM Tris-HCl, 50 mM KCl, and 5 mM MgCl2. Pif1 cannot unwind the duplex directly from the 2-nt ssDNA linker. The error bars were obtained from at least three repetitive experiments. Data are presented as mean ± SEM.
(F) FRET distributions of the remaining 3G4L4S∗ molecules on coverslip before and after the addition of Pif1. Adding only 40 nM Pif1 without ATP has no obvious effect on the FRET distribution of G4.
(G) The representative traces.
(H) The 3-layered G4 substrate with three loops of 1-, 2-, and 1-nt for Pif1 helicase.
(I) FRET distributions of 3G4L121S∗ before and 4 min after the addition of 80 nM Pif1 and 1 mM ATP. Even after 10 min, there is no obvious G4 unfolding.
(J) The representative trace. No G4 unfolding can be observed.
We next examined Pif1 helicase, which is conserved from bacteria to humans (Byrd and Raney, 2017). S. cerevisiae Pif1, which stands out among various G4 helicases due to its potent activity, was used (Hou et al., 2015). The DNA substrate includes a 26-nt ssDNA tail at the 5′-end of a 3-layered G4 with 4-nt loops (Figure 5D). According to our previous studies (Hou et al., 2015; Wang et al., 2018; Zhang et al., 2016), a stem duplex of 15-bp that cannot be spontaneously unwound by Pif1 without ATP was used here. Figure 5E further rules out the possibility that Pif1 may unwind the duplex DNA directly from the linker region using the substrate 15bp2nt (Table S1). Indeed, Pif1 is able to transiently unfold the G4s with relatively long loops in the ATP-dependent mode and then continue to unwind the downstream duplex (Figures 5E–5G). However, Pif1 cannot resolve G4s with very short loops at all (Figures 5H–5J), consistent with our previous reports (Wang et al., 2018), and the absence of unfolding was still true at ∼10 min incubation time (Figure 5I).
The above findings suggest that the short-looped G4s were poorly processed by BLM and Pif1 helicases, possibly because those G4s usually display high thermal stability (Table S2). However, those G4s were repetitively and transiently unfolded by RPA even at 10 nM. It is worth noting that DHX36 can only disrupt c-Myc Pu22 G4 (3G4L121 herein) very slightly by pulling 1-nt out (Chen et al., 2018). Besides, although DDX5 has been reported to resolve c-Myc G4 recently (Wu et al., 2019), it is still unknown to what extent the G4 structure can be disrupted. Therefore, we speculate that RPA may have potential roles in processing these types of short-looped G4 DNA.
RPA promotes helicases-mediated repetitive G4 unfolding into the durative linearized state
Although it has been well established that specialized DNA helicases such as BLM and Pif1 are responsible for resolving G4 structures during DNA replication and repair, RPA was also reported to participate in the helicases-mediated G4 unfolding from in vivo studies (Wu et al., 2018; Maestroni et al., 2020; Dahan et al., 2018; Sparks et al., 2019). Therefore, we next examined the function of RPA in helicases-mediated G4 unfolding at the single-molecule level.
We first chose BLM helicase, and the 3-layered G4 with 3-nt loops, which can be transiently unfolded by BLM but poorly unfolded by RPA, was used (Figure 6A). We incubated 3G4L3S with RPA for 10 min and then introduced BLM with ATP. Indeed, the addition of RPA caused very limited damage to the G4 structure; however, a prominent peak occurred at ∼E0.2 after the subsequent addition of BLM and ATP (Figure 6B). The FRET traces were also maintained at ∼E0.2 after BLM addition (Figure 6C). Those evidences indicate that the coating of ssDNA adjacent to G4 by RPA leads to the stable G4 unfolding by BLM, in sharp contrast with the repetitive G4 unfolding/refolding induced by only BLM (Figure 5C). We also carried out experiments by adding BLM and ATP together with RPA (Figure 6D). As a result, over 40% of molecules show the stable G4 unfolding (Figures 6E and 6F). However, no significant unfolding can be observed if we incubated 3G4L3S with BLM first and then introduced ATP with RPA. The possible reason might be that the association between BLM and substrate is not very strong; therefore, BLM may fall off and be washed away during the addition of RPA.
Figure 6.

RPA promotes BLM-mediated G4 unfolding into the durative linearized state
(A) RPA was incubated with the substrate 3G4L3S first, and then BLM and ATP were introduced in the reaction buffer containing 50 mM KCl and 5 mM MgCl2.
(B) The FRET distributions of 3G4L3S in different concentrations of RPA and after the addition of BLM to the RPA-bound 3G4L3S.
(C) Representative traces after the addition of BLM and ATP to the RPA-bound 3G4L3S.
(D) RPA and BLM were added simultaneously to the 3G4L3S.
(E) The fractions of different types of traces at different concentrations of RPA and BLM with 25 μM ATP.
(F) Representative traces of 3G4L3S in the mixture of 20 nM RPA, 40 nM BLM, and 25 μM ATP.
We further investigated the interplay between RPA and Pif1 in G4 unfolding. As RPA is a highly conserved trimer between human and S. cerevisiae yeast (Chen and Wold, 2014), human RPA was used here instead. The DNA substrate with a 26-nt ssDNA tail at the 5′-end of a 5-layered G4 was employed (Figure S9A), as the G4s over 3 layers were expected to be poorly unfolded by RPA. On one hand, 5–100 nM RPA indeed has negligible effects on 5G4S∗ (Figure S9B); on the other hand, 10 nM Pif1 transiently unfolds 5G4S in a highly repetitive manner (Figure S9C, upper panel). However, with the coexistence of Pif1 and RPA, G4 structures can be stably unfolded (Figures S9B and S9C). Due to the very low ATP concentration of 25 μM, no significant duplex DNA unwinding can be observed. We also changed the order of protein addition by incubating Pif1 (or RPA) with 5G4S∗ for 10 min and then introducing RPA (or Pif1) together with ATP. In both conditions, the FRET distributions shift to the lower band at ∼E0.2, with the FRET traces showing stable G4 unfolding (Figures S9D–S9I). These evidences further suggest that RPA significantly promotes the helicases-mediated transient and repetitive G4 DNA unfolding into the linearized state, no matter which one binds to the substrate first.
Helicase can traverse the G4 obstacle disrupted by RPA and efficiently unwind the downstream duplex
According to our results in Figures 5 and S8, some G4 structures, particularly the short-looped G4s, were poorly unfolded by BLM and Pif1 helicases. It is possible that they may create obstacles to those helicases in DNA metabolism (Lerner and Sale, 2019). Therefore, we further investigated the interplay between RPA and BLM on those types of G4 substrate. To this end, the 3-layered G4 with 1-, 2-, and 1-nt loops, which can be repetitively unfolded by RPA but poorly unfolded by BLM, was chosen; 60 nM BLM and 0.5 mM ATP were used, as 3G4L121 can barely be unfolded in this condition (Figure S8C). Based on the above results, a 4-nt segment was placed between G4 and duplex for RPA to well associate and then efficiently disrupt G4 (Figure 7A).
Figure 7.

RPA assists BLM to overcome the G4 structure and continue to unwind the downstream duplex DNA
(A) The schematic diagram of the substrate. The ssDNA tail and duplex stem are 14-nt and 27-bp, respectively. There is a 4-nt linker between G4 and duplex.
(B) The remaining fractions of duplexes on coverslip before and 4 min after the addition of 100 nM RPA, 60 nM BLM, and 0.5 mM ATP and the mixture of 100 nM RPA, 60 nM BLM, and 0.5 mM ATP. The error bars were obtained from at least three repetitive experiments. Data are presented as mean ± SEM.
(C) The FRET distributions of DNA molecules on coverslips before and after the addition of proteins.
(D) Selected trace after the addition of 100 nM RPA.
(E and F) Selected traces after the addition of 100 nM RPA, 60 nM BLM, and 0.5 mM ATP.
Upon the addition of 100 nM RPA together with 60 nM BLM and 0.5 mM ATP, ∼60% of DNA molecules disappeared in 4 min, reflecting the efficient unwinding of the 27-bp downstream duplex (Figure 7B). In contrast, very little duplex unwinding can be observed with only RPA or only BLM. Besides, the FRET distributions of remaining molecules on the surface show the significant shift to the left, indicating the unfolding of G4 structures (Figure 7C). There is a small peak at ∼E0.2, corresponding to the stable linearized state. The FRET traces display similar oscillations with the coexistence of RPA and BLM to that with only RPA (Figures 7D, 7E, S10A, and S10B), and tnG4 and tG4 are ∼0.9 s and ∼1.3 s in both conditions (Figure S10C). More importantly, the simultaneous signal loss of the two fluorophores after FRET oscillations can be observed within 2 min in ∼40% traces (Figure 7F), indicating the abrupt unwinding of the downstream duplex DNA during the repetitive G4 unfolding/refolding process.
Figures S10D and S10E further rules out the possibility that BLM may associate with the 4-nt ssDNA linker and further unwind the 27-bp duplex. Therefore, it is most likely that RPA partially or completely unfolded the G4 in 3G4L121S, then BLM got the chance to traverse the obstacle. As a control, the linker was then decreased to 2-nt, which would decrease the G4-unfolding efficiency of RPA. Indeed, there is no significant downstream duplex unwinding in the same experimental condition, although a similar FRET decrease/increase can be occasionally observed (Figures S10F and S10G).
Discussion
In this work, we systematically investigated the interactions between G4 DNA, RPA, and helicases at the single-molecule level. Our study highlights the functions of RPA in preventing, resolving, and assisting helicases to eliminate G4s during DNA metabolisms (DNA replication, for example, Figure 8). The in-depth mechanisms will be discussed below.
Figure 8.

The proposed mechanisms of RPA in eliminating G4 structures
DNA replication was taken for example. G4 structures can be formed in both the leading and lagging strands. Here the G4 in the lagging strand was shown. In scheme 1, RPA quickly and directly binds to the exposed G4 sequence, directly inhibiting its folding. Once the G4 sequence escapes RPA, it may well fold into G4 structures. In scheme 2, RPA is able to constantly disrupt the low stabilized G4s or repetitively unfold the short-looped G4s. However, RPA is poor at resolving G4s with long loops and high stability. Then in scheme 3, RPA assists the specialized helicases to remove those G4 obstacles no matter RPA coats the adjacent ssDNA at first or the helicase binds to the G4 substrate at first.
RPA may significantly suppress G4 formation
Recently, a high-resolution sequencing-based method has identified 716,310 G4 sequences in the human genome, including a large amount of G4s with noncanonical long loops (G4s with loop 1–3 nt are only 28%; loop 4–5 nt ∼25%; loop 6–7 nt ∼22%; longer loops ∼15%) (Chambers et al., 2015). As RPA is a highly abundant protein (∼1 μM) and the first responder to ssDNA inside the cell (Chen and Wold, 2014), it is necessary to reconsider the folding of G4s from ssDNA in vivo with the presence of RPA. In this study, we characterized the dynamic G4 formation process at different concentrations of RPA in the folding buffer of 200 mM KCl and 5 mM MgCl2. Our results indicate that direct RPA binding onto ssDNA significantly prevents the folding of G4 structures (Figure 8, scheme 1), particularly as ssDNA can be stably bound by multiple RPA molecules (Wang et al., 2019). Whether a specific G4 structure can be formed is indeed competition between two opposing factors. The first one is the rate of ion-driven G4 folding, which may mainly depend on the energy barrier. The second one is the rate of RPA binding, which is very sensitive to both the RPA concentration and the length of the G4 sequence.
It is noteworthy that G4s with loops over 3-nt such as 3G4L4 can even be significantly inhibited by 10 nM RPA. G4s with strong thermal stability such as c-Myc Pu22 G4 (3G4L121) and insulin-linked polymorphic region G4 (4G4) can also be suppressed to ∼45% by 100 nM RPA, implying that those G4s may not assuredly be formed in vivo due to the quick coating by RPA. Altogether, although a large amount of G4 motifs have been identified in the human genome, over 60% possess loops over 3-nt. Our results possibly suggest that those long-looped G4s may not be formed in the presence of RPA inside the cell. It is also possible that there are additional factors inside the cell such as nucleolin and LARK to promote the folding of G4 structures (Gonzalez et al., 2009; Niu et al., 2019). Different from those G4s, the folding of 3G4L1 was barely influenced, consistent with its uniqueness in causing genome instability (Piazza et al., 2015), possibly due to the very high folding rate.
The proposed mechanism of RPA-mediated unfolding of preformed G4s
If the G4 sequences successfully fold into G4 structures, we speculated that some of those preformed G4s may be directly overcome by RPA as shown in Figure 8, scheme 2. In fact, RPA was known to unfold preformed G4 with the 5′–3′ polarity from the studies of the Saintomé group (Salas et al., 2006; Safa et al., 2014, 2016; Lancrey et al., 2018). Further studies revealed that the trimerization core of RPA is responsible for disrupting G4s by interacting with Guanines (Prakash et al., 2011a, 2011b). However, the efficiency of RPA-mediated G4 unfolding is not consistent with the thermal stability of G4 (Qureshi et al., 2012), and the reason is not clear.
Here, we systematically addressed the dynamic interaction of RPA with a variety of preformed G4s. In general, the types of G4s that can be disrupted by RPA are limited, in sharp contrast with its potent inhibitory functions in G4s formation. Interestingly, we discovered that the loop lengths and thermal stability altogether determined the G4 unfolding efficiency by RPA, with the proposed mechanisms shown in Figure 9A. The footprint of each domain in RPA is about 3-nt (Fan and Pavletich, 2012). Due to the high affinity of 70A-B domains to ssDNA, RPA may first bind to the ssDNA linker by 70A or 70B. For G4s with long loops (≥3-nt), the CDE-core may prefer to interact with the loops instead of the G-tetrads (Prakash et al., 2011b). Therefore, RPA can only disrupt G4s with low thermal stability such as human telomeric G4 in 100 mM NaCl (Tm ∼ 50°C). However, RPA poorly disrupts long-looped G4s with relatively higher stability such as 3G4L3, 3G4L4, and 3G4L5 in 100 mM KCl. Interestingly, although G4s with short loops (1- or 2-nt) usually display very strong thermal stability, RPA can induce very quick unfolding/refolding in those G4s. We speculate that, instead of binding the single-stranded loops, the trimerization core of RPA may attract the Guanines in G-tetrad layers with high affinity (Prakash et al., 2011b) and then disrupt the short-looped G4s possibly with Guanines flipping out (Voter et al., 2018).
Figure 9.

The possible interacting modes of RPA with G4 structures in the absence or presence of helicases
(A) The proposed mechanisms of the interaction between RPA and different types of G4 DNA. First, for G4s with long loops (≥3-nt) and low thermal stability (Tm approximately or lower than ∼50°C), RPA may first load onto the substrate at the 4-nt linker by 70A or 70B domains. Then the trimerization core dynamically interacts with the loops, leading to the destabilization of the G4 structure. In addition, the G4 structures can be finally stably linearized by multiple RPA molecules. Second, for G4s with long loops (≥3-nt) and high thermal stability (Tm obviously higher than ∼50°C), the trimerization core predominantly interacts with the loops and the G4 structures are still intact. Third, for G4s with short loops (<3-nt), the trimerization core interacts with the Guanines in the G-tetrads, resulting in the complete or partial G4 unfolding. However, the unfolded G4s have a very strong tendency to refold back. Therefore, quick and repetitive unfolding/refolding can be observed.
(B) The proposed functions of RPA in helicases-mediated G4 unfolding. Once the G4 structure was unfolded by helicases into intermediate or ssDNA states, RPA may rapidly invade into the exposed strand by one-dimensional diffusion on the ssDNA and finally, maintain the G4 strand at the linearized state.
(C) Once the G4 was unfolded by RPA, helicase was able to traverse the G4 obstacle and further unwind the downstream duplex DNA.
It is worth noting that, in the recent paper of Sparks et al., RPA was observed to assist polymerase δ to synthesize DNA over G4 obstacles (Sparks et al., 2019). Interestingly, although G4 with (G3T7)3G3 sequence has the lowest thermal stability than other G4s, no assisting effect can be observed when RPA was present. This may be explained by our results that long loops can protect G4s against RPA. In the study of Ray et al., a linear increase was observed in the steady-state stability of G4s against RPA-mediated unfolding with increased G-tetrad layers or decreasing loop lengths (Ray et al., 2013). Besides, Ray et al. showed that RPA can efficiently resolve long-looped G4s. This inconsistency from ours may be mainly caused by the different experimental strategies, as G4 substrates were annealed in 50 mM NaCl and then incubated with RPA in 150 mM KCl for 15 min in their study (Ray et al., 2013). It is possible that the long-looped G4 sequences fold insufficiently in 50 mM NaCl (Tm∼40°C or even lower), and then in 150 mM KCl there will be competition between ion-driven G4 folding and RPA-binding mediated linearization as shown in our Figure 1. However, in the present study, we allowed the G4 structures to be adequately folded in 100 mM KCl by slow annealing and then recorded the dynamic interaction between the preformed G4 structures and RPA upon protein addition in 100 mM KCl and 5 mM MgCl2. As a result, the long-looped G4s show the different results from Ray et al.
The relationship between RPA and helicases in resolving G4s
RPA is able to stimulate the helicase activity of BLM and WRN on duplex DNA, significantly promoting the unidirectional unwinding and increasing processivity by the direct interaction with the helicases (Qin et al., 2020; Lee et al., 2018). It is well known that G4 structures undergo repetitive unfolding/refolding in the presence of helicases (Varshney et al., 2020). Therefore, RPA and helicases may also interact with each other in G4 unfolding. Indeed, RPA and Pif1 were reported to cooperatively remove G4 structures at both leading and lagging stands in vivo (Maestroni et al., 2020). The complementary roles of Pif1 and RPA in stimulating DNA replication through G4s were also demonstrated (Sparks et al., 2019; Dahan et al., 2018). In addition, HERC2 was reported to facilitate BLM and WRN helicases to suppress G4s by releasing RPA onto the ssDNA (Wu et al., 2018). All these evidences suggest that the interaction or cooperation between G4 helicases and RPA may be very crucial for smooth G4 replication in the cell.
Our study indicated that RPA plays multifaceted roles complementary to helicases in G4 DNA unfolding. First, RPA is good at disrupting short-looped G4s and poor at resolving G4s with long loops. Meanwhile, helicases are good at processing a variety of G4s except that with very short loops. Therefore, G4 loop lengths may potentially direct the G4 structures toward the different RPA- or helicases-mediated unfolding pathways. Second, RPA substantially changed the highly repetitive G4 unfolding mode of helicases and promotes stable and complete unfolding, which is initiated by helicases (Figure 8, scheme 3). Helicases alone cannot stably open G4s but rather unfold partially and transiently (repetitive unfolding); however, in the presence of RPA, G4s are stably unfolded. Thus, both helicases and RPA are necessary in the case of stable G4 unfolding. As RPA is the first ssDNA responder in DNA replication and repair, we speculated that RPA may initially coat on the ssDNA adjacent to G4. Interestingly, helicases may directly load onto the RPA-bound ssDNA according to the previous report (Crickard et al., 2019) and then unfold the adjacent G4s catalyzed by ATP. Once the structures are unfolded by helicases even a little, RPA may further invade into the strand and completely disrupt the G4s into the linearized state by one-dimensional diffusion (Nguyen et al., 2014) and impede the refolding of G4 (Figure 9B). If the helicase is recruited to G4 sites at first, RPA can still cause the complete and stable unfolding of G4s by transiently capturing the exposed ssDNA. It is also possible that in scheme 3, helicases (present at low concentration compared to RPA) may assist RPA to unfold the G4s.
In addition to the above interactions, for the G4s that were poorly unfolded by helicase but efficiently unfolded by RPA, helicase was able to traverse the obstacle that was disrupted by RPA and further unwind the downstream duplex efficiently (Figure 9C). Therefore, it is possible that RPA may also play potential roles in assisting the replicative helicases to bypass the G4 obstacles in DNA replication. The interplay between RPA and helicases in G4 unfolding should be different from that in duplex DNA unwinding. In the latter case, there are direct and specific interactions between RPA and helicases, which significantly stimulate the unwinding activity of helicases with increased processivity (Qin et al., 2020; Lee et al., 2018). However, in the former case, there should be no specific interactions between them, as the truncated BLM-core was used.
Limitations of the study
Although here we provided the evidences for the cooperation of RPA and BLM in overcoming the short-looped G4 structures, the further study would be necessary to address whether RPA is able to help other helicases to bypass the G4 barriers.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli strain BL21 (DE3) | NEB | Cat#C2527H |
| E. coli strain Rosetta (DE3) | Boule and Zakian, 2007 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| D-glucose | Sigma-Aldrich | Cat#G8270-100g |
| glucose oxidase | Sigma-Aldrich | Cat#G2133-50KU |
| Catalase | Sigma-Aldrich | Cat#C9322-5g |
| Trolox | Sigma-Aldrich | Cat#238813-1g |
| Streptavidin | Invitrogen | Cat#43-4301 |
| mPEG-SC | Laysan bio | Cat#mPEG-SC-5000 |
| Biotin-PEG-SC | Laysan bio | Cat#Biotin-mPEG-SC-5000 |
| Oligonucleotides | ||
| GGTTGGTGTGGTTGG; see Table S1 for sequences used in smFRET, CD, and FRET-melting assay. | This paper | N/A |
| GGGTGGGTGGGTGGG; see Table S1 | This paper | N/A |
| GGGTGGGTAGGGTGGG; see Table S1 | This paper | N/A |
| GGGTTGGGTTGGGTTGGG; see Table S1 | This paper | N/A |
| GGGTTAGGGTTAGGGTTAGGG; see Table S1 | This paper | N/A |
| GGGTTTTGGGTTTTGGGTTTTGGG; see Table S1 | This paper | N/A |
| GGGTTTTTGGGTTTTTGGGTTTTTGGG; see Table S1 | This paper | N/A |
| GGGGTTAGGGGTTAGGGGTTAGGGG; see Table S1 | This paper | N/A |
| GGGGTGTGGGGACAGGGGTGTGGGG; see Table S1 | This paper | N/A |
| GGGGGTTAGGGGGTTAGGGGGTTAGGGGG; see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid: p11d-tRPA | Henricksen et al., 1994 | N/A |
| Plasmid: PET-15b-SUMO | Wu et al., 2015 | N/A |
| Plasmid: PET-28a | Boule and Zakian, 2007 | N/A |
| Software and algorithms | ||
| SmCamera | Lee at al., 2019 | http://ha.med.jhmi.edu/resources/#1464200861600-0fad9996-bfd4 |
| Matlab2014 | MathWorks | https://www.mathworks.com/ |
| Origin 2019 | OriginLab | https://www.originlab.com/ |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Xi-Miao Hou (houximiao@nwsuaf.edu.cn).
Materials availability
Materials and the information used for the experiments are available upon reasonable request.
Data and code availability
No new resources have been generated. Further information about data supporting the conclusions of this manuscript will be made available by the lead contact to any qualified researchers.
Experimental model and subject details
E. coli strains for in vitro studies
The E. coli strain BL21 (DE3) was used to express RPA and BLM proteins. The E. coli strain Rosetta (DE3) was used to express Pif1 protein. These e. coli cells were incubated at 37oC in LB medium for 4 h, and then the protein expression was induced by IPTG at 18oC for 16 h.
Method details
DNA constructs
All oligonucleotides were purchased from Sangon Biotech (Shanghai, China). The sequences and labeling positions of the oligonucleotides were listed in Table S1. For the DNA constructs used in single-molecule measurements, DNA was annealed with the 1:2 mixture of the stem and G4 strands by incubating the mixture at 95°C for 5 min and then slowly cooling down to room temperature in about 7 h. The strand without biotin was used in excess to reduce the possibility of having the non-annealed strand anchored at the coverslip surface. The concentration of the stem strand was 100 nM. All annealing procedures were carried out in 25 mM Tris-HCl, pH 7.5 with or without 100 mM monovalent cations.
Protein purification
The expression and purification of RPA were carried out essentially according to Henricksen et al (Henricksen et al., 1994). Escherichia coli strain BL21 (DE3) was transformed with a plasmid p11d-tRPA for recombinant human RPA that permits the co-expression of RPA1, RPA2, and RPA3 at 18°C for 16 h. Then, RPA was purified with Affi-Gel blue, hydroxyapatite (BioRad), and Q-sepharose chromatography columns (GE healthcare). The purified protein was finally eluted in phosphate buffer containing ∼300 mM KCl (pH 7.5) and stored at −80°C. The RPA reaction buffer contained 50–200 mM KCl or NaCl, 5 mM MgCl2 in 25 mM Tris-HCl, pH 7.5. The human BLM-core (truncated BLM642-1290) was expressed and purified as previously described (Wu et al., 2015). The protein was expressed in the E. coli strain BL21 (DE3) at 18°C for 16 h and then purified by FPLC with sequential chromatography on Ni-NTA (GE Healthcare, Chicago, IL, USA) and Superdex200 10/300 GL column (GE Healthcare). S. cerevisiae Pif1 was expressed and purified essentially according to Boule and Zakian (Boule and Zakian, 2007). The protein was expressed in the E. coli strain Rosetta (DE3) at 18°C for 16 h and then purified by FPLC with sequential chromatography on Ni-NTA (GE Healthcare, Chicago, IL, USA) and cationic exchange column Hi-Trap SP (GE Healthcare). These proteins were usually tested by the ssDNA-binding assay as quality control and displayed good activity in our previous studies (Wang et al., 2018, 2019; Wu et al., 2015; Hou et al., 2015).
Single-molecule fluorescence data acquisition
Single-molecule fluorescence experiments were performed as described previously (Hou et al., 2015). The coverslips (Fisher Scientific) and slides were cleaned thoroughly by a mixture of sulfuric acid and hydrogen peroxide, acetone, and sodium ethoxide, then the surfaces of coverslip were coated with a mixture of 99% methoxy poly (ethylene glycol) (m-PEG-5000, Laysan Bio) and 1% of biotin-PEG (biotin- PEG-5000, Laysan Bio, Inc.). Streptavidin (10 μg/mL) was added to the microfluidic chamber made of the PEG-coated coverslip and incubated for 10 min. After washing, 50 pM DNA was added to the chamber where it was immobilized for 10 min. Then, the free DNA was removed by washing with the reaction buffer. Afterward, the chamber was filled with the reaction buffer with an oxygen-scavenging system (0.8% D-glucose, 1 mg/mL glucose oxidase, 0.4 mg/mL catalase, and 4 mM Trolox) to prevent photobleaching and photoblinking. Imaging was initiated before the protein flowed into the chamber. We used an exposure time of 100 ms for all single-molecule measurements at a constant temperature of 22°C.
FRET data analysis
The FRET efficiency was calculated using IA/(ID + IA), where ID and IA represent the donor and acceptor intensities, respectively. Basic data analysis was carried out by scripts written in MATLAB, and all data fitting was performed by Origin 8.0. All fluorescent spots in each movie were selected unless the trace showed a poor signal: noise ratio lower than 3:1 or the intensity changes of the donor and acceptor did not match well (the changes of the two fluorescent intensities were not strictly inverse correlated). The FRET histograms were constructed from more than 300 traces in most cases (Table S3). The fluorescence of all molecules in each FRET distribution was recorded during the same amount of time. The number of Cy5 spots was counted to represent the number of remaining DNA molecules on the surface.
Circular dichroism (CD) spectropolarimetry
CD experiments were performed with a Bio-Logic MOS450/AF-CD optical system (Bio-Logic Science Instruments, France). A 5 μM solution of G4 sequences was incubated at 95°C for 5 min and then slowly cooled down to room temperature in about 7 h. CD spectra were recorded in the UV (220–320 nm) regions in 0.75 nm increments with an averaging time of 2 s at 25°C.
FRET-melting assay
FRET-melting experiments were conducted with the FAM-TAMRA dual-labeled oligomers using a Rotor-Gene Q real-time PCR machine (Qiagen) as described in a previous study (De Cian et al., 2007). The oligonucleotides were generally measured at 0.5 μM strand concentration in 25 mM Tris-HCl, pH 7.5 containing different concentrations of NaCl or KCl, and 5 mM MgCl2. The emission of the FAM fluorophore was normalized between 0 and 1, and the melting temperature Tm was determined accordingly. To investigate the effects of protein on G4 structures, the G4s were prepared by slow cooling from 95°C to room temperature in about 7 h. Then the protein and G4s were mixed and incubated for 5 min. Next, the sample was heated from 25°C to 95°C and the FAM intensity was recorded.
Equilibrium DNA-binding assay with RPA
The binding of RPA to DNA was analyzed by a fluorescence polarization assay using Infinite F200 PRO (Tecan group, Switzerland) at 25°C. DNA was annealed with the 1:1 mixture of the stem and G4 strands by incubating the mixture at 95°C for 5 min and then slowly cooling down to room temperature in about 7 h. Various amounts of protein were added to a 150-μL aliquot of binding buffer (25 mM Tris-HCl, pH 7.5; 100 mM KCl, 5 mM MgCl2) containing 5 nM DNA. Each sample was allowed to equilibrate in the solution for 5 min, after which the fluorescence polarization was measured.
Gel filtration
DNA substrates were slowly annealed in 25 mM Tris-HCl, pH 7.5, and 100 mM KCl or NaCl with a 1:1 mixture of the stem and ssDNA strands (Table S1). Afterward, 2 μM DNA was incubated with varying amounts of RPA for 5 min. The gel filtration was performed at 25°C using an FPLC system (GE Healthcare) with a Superdex-200 column (analytical grade) equilibrated with binding buffer (25 mM Tris-HCl, pH 7.5; 100 mM KCl or NaCl) (Wang et al., 2019). A 100-μL aliquot of the RPA-DNA complex was loaded onto the column and eluted at a flow rate of 0.3 mL/min with the buffer. The absorbances were monitored at 280 and 260 nm ultraviolet rays.
Quantification and statistical analysis
FRET distributions
The numbers of the FRET traces used in each FRET distribution was shown in Table S3. All those traces from the same experiment were combined together. Then the FRET histograms were constructed with Origin 8.0. See the details in the Method.
Time distributions
The duration time of G4 at the folded state or the unfolded state were measured manually in each trace (see the details in the legends of Figures 4 and S6). Then those data were combined together and the time distribution were constructed. The time of G4 at folded state was fitted by the single-exponential decay, and the time of G4 at unfolded state was fitted by the Gaussian distribution with Origin 8.0.
DNA unwinding fractions
The number of Cy5 spots in each movie was counted to represent the number of remaining DNA molecules on the surface. Then the remaining fractions was obtained by normalization with the initial molecule numbers. At least three repeated experiments were performed, and the error bar can be obtained. See the details in the legends of Figures 5 and 7.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (32071225, 31870788, and 32071291) and the Chinese Universities Scientific Fund (Z109021718). The research was conducted within the context of the International Associated Laboratory “Helicase-mediated G-quadruplex DNA unwinding and Genome Stability.” We thank Dr. Wen-Qiang Wu at Henan University and Dr. Xing-Hua Zhang at Wuhan University for insightful discussions.
Author contributions
X.H., B.S., and X.X. designed and supervised the study. Y.W., T.G., Y.Z., and C.L. conducted the experiments and analyzed the data. X.H. wrote the manuscript with input from all authors.
Declaration of interests
The authors declare no competing interests.
Published: May 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102493.
Supplemental information
References
- Audry J., Maestroni L., Delagoutte E., Gauthier T., Nakamura T.M., Gachet Y., Saintome C., Geli V., Coulon S. RPA prevents G-rich structure formation at lagging-strand telomeres to allow maintenance of chromosome ends. EMBO J. 2015;34:1942–1958. doi: 10.15252/embj.201490773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochman M.L., Paeschke K., Zakian V.A. DNA secondary structures: stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012;13:770–780. doi: 10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boule J.B., Zakian V.A. The yeast Pif1p DNA helicase preferentially unwinds RNA DNA substrates. Nucleic Acids Res. 2007;35:5809–5818. doi: 10.1093/nar/gkm613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd A.K., Raney K.D. Structure and function of Pif1 helicase. Biochem. Soc. Trans. 2017;45:1159–1171. doi: 10.1042/BST20170096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra J.A., Paeschke K., Singh M., Zakian V.A. G-quadruplex DNA sequences are evolutionarily conserved and associated with distinct genomic features in Saccharomyces cerevisiae. PLoS Comput. Biol. 2010;6:e1000861. doi: 10.1371/journal.pcbi.1000861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers V.S., Marsico G., Boutell J.M., Di Antonio M., Smith G.P., Balasubramanian S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015;33:877–881. doi: 10.1038/nbt.3295. [DOI] [PubMed] [Google Scholar]
- Chen M.C., Tippana R., Demeshkina N.A., Murat P., Balasubramanian S., Myong S., Ferre-D'amare A.R. Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36. Nature. 2018;558:465–469. doi: 10.1038/s41586-018-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R., Wold M.S. Replication protein A: single-stranded DNA's first responder Dynamic DNA-interactions allow replication protein A to direct single-strand DNA intermediates into different pathways for synthesis or repair. Bioessays. 2014;36:1156–1161. doi: 10.1002/bies.201400107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimino-Reale G., Zaffaroni N., Folini M. Emerging role of G-quadruplex DNA as target in anticancer therapy. Curr. Pharm. Des. 2016;22:6612–6624. doi: 10.2174/1381612822666160831101031. [DOI] [PubMed] [Google Scholar]
- Crickard J.B., Xue C., Wang W., Kwon Y., Sung P., Greene E.C. The RecQ helicase Sgs1 drives ATP-dependent disruption of Rad51 filaments. Nucleic Acids Res. 2019;47:4694–4706. doi: 10.1093/nar/gkz186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahan D., Tsirkas I., Dovrat D., Sparks M.A., Singh S.P., Galletto R., Aharoni A. Pif1 is essential for efficient replisome progression through lagging strand G-quadruplex DNA secondary structures. Nucleic Acids Res. 2018;46:11847–11857. doi: 10.1093/nar/gky1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cian A., Guittat L., Kaiser M., Sacca B., Amrane S., Bourdoncle A., Alberti P., Teulade-Fichou M.P., Lacroix L., Mergny J.L. Fluorescence-based melting assays for studying quadruplex ligands. Methods. 2007;42:183–195. doi: 10.1016/j.ymeth.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Di Antonio M., Ponjavic A., Radzevicius A., Ranasinghe R.T., Catalano M., Zhang X., Shen J., Needham L.M., Lee S.F., Klenerman D., Balasubramanian S. Single-molecule visualization of DNA G-quadruplex formation in live cells. Nat. Chem. 2020;12:832–837. doi: 10.1038/s41557-020-0506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J., Pavletich N.P. Structure and conformational change of a replication protein A heterotrimer bound to ssDNA. Genes Dev. 2012;26:2337–2347. doi: 10.1101/gad.194787.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez V., Guo K.X., Hurley L., Sun D. Identification and characterization of nucleolin as a c-myc G-quadruplex-binding protein. J. Biol. Chem. 2009;284:23622–23635. doi: 10.1074/jbc.M109.018028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henricksen L.A., Umbricht C.B., Wold M.S. Recombinant replication protein A: expression, complex formation, and functional characterization. J. Biol. Chem. 1994;269:11121–11132. [PubMed] [Google Scholar]
- Hou X.M., Fu Y.B., Wu W.Q., Wang L., Teng F.Y., Xie P., Wang P.Y., Xi X.G. Involvement of G-triplex and G-hairpin in the multi-pathway folding of human telomeric G-quadruplex. Nucleic Acids Res. 2017;45:11401–11412. doi: 10.1093/nar/gkx766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou X.M., Wu W.Q., Duan X.L., Liu N.N., Li H.H., Fu J., Dou S.X., Li M., Xi X.G. Molecular mechanism of G-quadruplex unwinding helicase: sequential and repetitive unfolding of G-quadruplex by Pif1 helicase. Biochem. J. 2015;466:189–199. doi: 10.1042/BJ20140997. [DOI] [PubMed] [Google Scholar]
- Lancrey A., Safa L., Chatain J., Delagoutte E., Riou J.F., Alberti P., Saintome C. The binding efficiency of RPA to telomeric G-strands folded into contiguous G-quadruplexes is independent of the number of G4 units. Biochimie. 2018;146:68–72. doi: 10.1016/j.biochi.2017.11.017. [DOI] [PubMed] [Google Scholar]
- Lansdorp P., van Wietmarschen N. Helicases FANCJ, RTEL1 and BLM act on guanine quadruplex DNA in vivo. Genes (Basel) 2019;10:870. doi: 10.3390/genes10110870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.Y., Mcnerney C., Myong S. G-quadruplex and protein binding by single-molecule FRET microscopy. Methods Mol. Biol. 2019;2035:309–322. doi: 10.1007/978-1-4939-9666-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M., Shin S., Uhm H., Hong H., Kirk J., Hyun K., Kulikowicz T., Kim J., Ahn B., Bohr V.A., Hohng S. Multiple RPAs make WRN syndrome protein a superhelicase. Nucleic Acids Res. 2018;46:4689–4698. doi: 10.1093/nar/gky272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner L.K., Sale J.E. Replication of G Quadruplex DNA. Genes (Basel) 2019;10:95. doi: 10.3390/genes10020095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi E.P., Londono-Vallejo A. A guide to computational methods for G-quadruplex prediction. Nucleic Acids Res. 2020;48:1603. doi: 10.1093/nar/gkaa033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestroni L., Audry J., Luciano P., Coulon S., Geli V., Corda Y. RPA and Pif1 cooperate to remove G-rich structures at both leading and lagging strand. Cell Stress. 2020;4:48–63. doi: 10.15698/cst2020.03.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maizels N. G4-associated human diseases. EMBO Rep. 2015;16:910–922. doi: 10.15252/embr.201540607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maizels N., Gray L.T. The G4 genome. PLoS Genet. 2013;9:e1003468. doi: 10.1371/journal.pgen.1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza O., Bourdoncle A., Boule J.B., Brosh R.M., Mergny J.L. G-quadruplexes and helicases. Nucleic Acids Res. 2016;44:1989–2006. doi: 10.1093/nar/gkw079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen B., Sokoloski J., Galletto R., Elson E.L., Wold M.S., Lohman T.M. Diffusion of human replication protein A along single-stranded DNA. J. Mol. Biol. 2014;426:3246–3261. doi: 10.1016/j.jmb.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu K.K., Xiang L.J., Jin Y., Peng Y.L., Wu F., Tang W.H., Zhang X.J., Deng H.M., Xiang H., Li S. Identification of LARK as a novel and conserved G-quadruplex binding protein in invertebrates and vertebrates. Nucleic Acids Res. 2019;47:7306–7320. doi: 10.1093/nar/gkz484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza A., Adrian M., Samazan F., Heddi B., Hamon F., Serero A., Lopes J., Teulade-Fichou M.P., Phan A.T., Nicolas A. Short loop length and high thermal stability determine genomic instability induced by G-quadruplex-forming minisatellites. EMBO J. 2015;34:1718–1734. doi: 10.15252/embj.201490702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash A., Kieken F., Marky L.A., Borgstahl G.E. Stabilization of a G-quadruplex from unfolding by replication protein A using potassium and the porphyrin TMPyP4. J. Nucleic Acids. 2011;2011:529828. doi: 10.4061/2011/529828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash A., Natarajan A., Marky L.A., Ouellette M.M., Borgstahl G.E. Identification of the DNA-binding domains of human replication protein A that recognize G-quadruplex DNA. J. Nucleic Acids. 2011;2011:896947. doi: 10.4061/2011/896947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Z., Bi L., Hou X.M., Zhang S., Zhang X., Lu Y., Li M., Modesti M., Xi X.G., Sun B. Human RPA activates BLM's bidirectional DNA unwinding from a nick. Elife. 2020;9:e54098. doi: 10.7554/eLife.54098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi M.H., Ray S., Sewell A.L., Basu S., Balci H. Replication protein A unfolds G-quadruplex structures with varying degrees of efficiency. J. Phys. Chem. B. 2012;116:5588–5594. doi: 10.1021/jp300546u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S., Qureshi M.H., Malcolm D.W., Budhathoki J.B., Celik U., Balci H. RPA-mediated unfolding of systematically varying G-quadruplex structures. Biophys. J. 2013;104:2235–2245. doi: 10.1016/j.bpj.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safa L., Delagoutte E., Petruseva I., Alberti P., Lavrik O., Riou J.F., Saintome C. Binding polarity of RPA to telomeric sequences and influence of G-quadruplex stability. Biochimie. 2014;103:80–88. doi: 10.1016/j.biochi.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Safa L., Gueddouda N.M., Thiebaut F., Delagoutte E., Petruseva I., Lavrik O., Mendoza O., Bourdoncle A., Alberti P., Riou J.F., Saintome C. 5' to 3' unfolding directionality of DNA secondary structures by replication protein A: g-quadruplexes and duplexes. J. Biol. Chem. 2016;291:21246–21256. doi: 10.1074/jbc.M115.709667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas T.R., Petruseva I., Lavrik O., Bourdoncle A., Mergny J.L., Favre A., Saintome C. Human replication protein a unfolds telomeric G-quadruplexes. Nucleic Acids Res. 2006;34:4857–4865. doi: 10.1093/nar/gkl564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer M., Paeschke K. G-quadruplex unwinding helicases and their function in vivo. Biochem. Soc. Trans. 2017;45:1173–1182. doi: 10.1042/BST20170097. [DOI] [PubMed] [Google Scholar]
- Sparks M.A., Singh S.P., Burgers P.M., Galletto R. Complementary roles of Pif1 helicase and single stranded DNA binding proteins in stimulating DNA replication through G-quadruplexes. Nucleic Acids Res. 2019;47:8595–8605. doi: 10.1093/nar/gkz608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tippana R., Hwang H., Opresko P.L., Bohr V.A., Myong S. Single-molecule imaging reveals a common mechanism shared by G-quadruplex-resolving helicases. Proc. Natl. Acad. Sci. U S A. 2016;113:8448–8453. doi: 10.1073/pnas.1603724113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney D., Spiegel J., Zyner K., Tannahill D., Balasubramanian S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020;21:459–474. doi: 10.1038/s41580-020-0236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voter A.F., Qiu Y., Tippana R., Myong S., Keck J.L. A guanine-flipping and sequestration mechanism for G-quadruplex unwinding by RecQ helicases. Nat. Commun. 2018;9:4201. doi: 10.1038/s41467-018-06751-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Wang Q.M., Wang Y.R., Xi X.G., Hou X.M. DNA-unwinding activity of Saccharomyces cerevisiae Pif1 is modulated by thermal stability, folding conformation, and loop lengths of G-quadruplex DNA. J. Biol. Chem. 2018;293:18504–18513. doi: 10.1074/jbc.RA118.005071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.M., Yang Y.T., Wang Y.R., Gao B., Xi X.G., Hou X.M. Human replication protein A induces dynamic changes in single-stranded DNA and RNA structures. J. Biol. Chem. 2019;294:13915–13927. doi: 10.1074/jbc.RA119.009737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., Xing Z., Tran E.J., Yang D. DDX5 helicase resolves G-quadruplex and is involved in MYC gene transcriptional activation. Proc. Natl. Acad. Sci. U S A. 2019;116:20453–20461. doi: 10.1073/pnas.1909047116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W., Rokutanda N., Takeuchi J., Lai Y., Maruyama R., Togashi Y., Nishikawa H., Arai N., Miyoshi Y., Suzuki N. HERC2 facilitates BLM and WRN helicase complex interaction with RPA to suppress G-quadruplex DNA. Cancer Res. 2018;78:6371–6385. doi: 10.1158/0008-5472.CAN-18-1877. [DOI] [PubMed] [Google Scholar]
- Wu W.Q., Hou X.M., Li M., Dou S.X., Xi X.G. BLM unfolds G-quadruplexes in different structural environments through different mechanisms. Nucleic Acids Res. 2015;43:4614–4626. doi: 10.1093/nar/gkv361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.Q., Hou X.M., Zhang B., Fosse P., Rene B., Mauffret O., Li M., Dou S.X., Xi X.G. Single-molecule studies reveal reciprocating of WRN helicase core along ssDNA during DNA unwinding. Sci. Rep. 2017;7:43954. doi: 10.1038/srep43954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Wu W.Q., Liu N.N., Duan X.L., Li M., Dou S.X., Hou X.M., Xi X.G. G-quadruplex and G-rich sequence stimulate Pif1p-catalyzed downstream duplex DNA unwinding through reducing waiting time at ss/dsDNA junction. Nucleic Acids Res. 2016;44:8385–8394. doi: 10.1093/nar/gkw669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R.B., Zhang J.C., Bochman M.L., Zakian V.A., Ha T. Periodic DNA patrolling underlies diverse functions of Pif1 on R-loops and G-rich DNA. Elife. 2014;3:e02190. doi: 10.7554/eLife.02190. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new resources have been generated. Further information about data supporting the conclusions of this manuscript will be made available by the lead contact to any qualified researchers.