

Summary
Targeting the signaling pathway of growth differentiation factor 8 (GDF8), also known as myostatin, has been regarded as a promising strategy to increase muscle mass in the elderly and in patients. Accumulating evidence in animal models and clinical trials has indicated that a rational approach is to inhibit a limited number of transforming growth factor β (TGF-β) family ligands, including GDF8 and activin A, without affecting other members. Here, we focused on one of the endogenous antagonists against TGF-β family ligands, follistatin-like 3 (FSTL3), which mainly binds and neutralizes activins, GDF8, and GDF11. Although bivalent human FSTL3 Fc-fusion protein was rapidly cleared from mouse circulation similar to follistatin (FST)-Fc, monovalent FSTL3-Fc (mono-FSTL3-Fc) generated with the knobs-into-holes technology exhibited longer serum half-life. Systemic administration of mono-FSTL3-Fc in mice induced muscle fiber hypertrophy and increased muscle mass in vivo. Our results indicate that the monovalent FSTL3-based therapy overcomes the difficulties of current anti-GDF8 therapies.
Subject areas: Musculoskeletal medicine, Physiology, Human metabolism
Graphical abstract
Highlights
-
•
FSTL3-Fc has a more specific binding profile for TGF-β family ligands than ActRIIB-Fc.
-
•
Bivalent two-armed FSTL3-Fc is rapidly cleared from mouse circulation.
-
•
Monovalent FSTL3-Fc has longer serum half-life and causes systemic muscle hypertrophy.
-
•
ActRIIB-Fc-related side effects are not detected in monovalent FSTL3-Fc-treated mice.
Musculoskeletal medicine; Physiology; Human metabolism
Introduction
Skeletal muscle, a component of the locomotor system, has been highlighted as a key regulator of systemic energy homeostasis. Skeletal muscle secretes several cytokines and proteins named myokines, and it also functions as a primary site for glucose uptake and metabolism (Cruz-Jentoft and Sayer, 2019; Johnson et al., 2013; Lee and Jun, 2019). In addition, skeletal muscle is reported to induce a microenvironment with less inflammation, sheltering a population of T cells from systemic inflammation and preventing their exhaustion (Wu et al., 2020). Aging and various systemic diseases are associated with a decrease in the mass and function of skeletal muscle, which not only reduces the quality of life of patients but also leads to higher morbidity and mortality (Cruz-Jentoft and Sayer, 2019; Johnson et al., 2013). Therefore, several factors involved in “muscle wasting diseases” are regarded as novel therapeutic targets (Furrer and Handschin, 2019).
Growth differentiation factor 8 (GDF8), also known as myostatin, is a member of the transforming growth factor β (TGF-β) family, which acts as an inhibitor of normal muscle growth (Cohen et al., 2015; Morikawa et al., 2016). Several genetic and biological studies indicate that inhibition of GDF8 promotes skeletal muscle hypertrophy across many species, including humans (Cohen et al., 2015; McPherron et al., 1997; Schuelke et al., 2004). Conversely, nude mice xenografted with GDF8-producing cells exhibit cachectic phenotype, with muscle wasting and fat loss (Zimmers et al., 2002). In addition, closely related members of the TGF-β family, i.e., GDF11 and activins, redundantly function as negative regulators of muscle growth (Egerman et al., 2015; Lee et al., 2005). Therefore, targeting the GDF8 signaling pathway has been regarded as a promising strategy to increase muscle mass (Argilés et al., 2018; Cohen et al., 2015; Marty et al., 2017). GDF8 signals mainly through the type II receptor ActRIIB (encoded by the ACVR2B gene) and the type I receptors ALK4 (ACVR1B) and ALK7 (ACVR1C) of the TGF-β family, which activate and phosphorylate transcription factors SMAD2 and SMAD3 (Morikawa et al., 2016). ActRIIB binds to numerous ligands including, activins (activin A, B, C, and E), GDF8, GDF11, bone morphogenetic protein 9 (BMP9), and BMP10.
Several molecules have been developed to block the GDF8 pathway, and some of them are/were in clinical trials (Furrer and Handschin, 2019; Lee and Jun, 2019; Marty et al., 2017): (i) monoclonal antibodies targeting GDF8, (ii) monoclonal antibodies targeting its receptor, ActRIIB, (iii) extracellular ligand traps, such as ActRIIB decoys, and (iv) endogenous antagonists, such as follistatin (FST). However, anti-GDF8 antibodies, including MYO-029 (stamulumab), LY2495655 (landogrozumab), PF-06252616 (domagrozumab), and REGN1033 (trevogrumab), did not show statistically significant improvements in muscle strength as expected. This might be explained by the functional redundancy of the ligands, although more work is needed to understand the connection of muscle mass increases with functional muscle mass during the inhibition of GDF8 and other TGF-β ligands. Consistently, combination of neutralizing antibodies against activin A and GDF8 was sufficient to regulate muscle wasting phenotype in mice and monkeys (Latres et al., 2017). On the other hand, targeting a broad spectrum of TGF-β family members caused adverse effects. The clinical trial of ACE-031, a recombinant fusion protein of the extracellular domain of ActRIIB and the Fc domain of IgG (ActRIIB-Fc) that functions as an extracellular ligand trap, was discontinued due to a statistically significant risk of bleeding, and this can be explained by undesired inhibitions of BMP9, BMP10, and other angiogenic TGF-β family members (Campbell et al., 2017). A rational approach to tackle muscle wasting diseases is therefore to inhibit a limited number of TGF-β family ligands, including GDF8 and activin A, without affecting the others, such as BMP9/10. One of the promising candidate inhibitors for this purpose is FST. Recently, FST-derived biological products were shown to effectively rescue muscle wasting phenotype (Castonguay et al., 2019; Datta-Mannan et al., 2013; Glasser et al., 2018; Iskenderian et al., 2018; Kota et al., 2009; Lodberg et al., 2019; Pearsall et al., 2019; Yaden et al., 2014), one of which was shown to significantly increase muscle volume with local administration in phase 2 clinical trials (Acceleron Pharma ACE-083: ClinicalTrials.gov NCT02927080, NCT03124459). For systemic administration, however, FST-Fc in circulation can be a target of rapid proteolysis (Castonguay et al., 2019). Although protein engineering strategies, such as deletion or amino acid substitution of the heparin-binding site (HBS) of FST, prolonged its serum half-life in mice (Datta-Mannan et al., 2013; Iskenderian et al., 2018; Lodberg et al., 2019), it is still unclear whether the modifications affect its ligand selectivity and immunogenicity in humans.
Here, we have focused on another endogenous antagonist follistatin-like 3 (FSTL3, also known as follistatin-related gene or FLRG) (Chang, 2016). Unlike FST, which binds to several BMPs, FSTL3 has a more restricted binding profile for TGF-β family ligands; its main targets are activins, GDF8, and GDF11 (Le et al., 2018; Sidis et al., 2006). We found that conventional or bivalent human FSTL3 Fc-fusion protein (bi-FSTL3-Fc) efficiently antagonized the ligands in vitro and in vivo, but it was rapidly cleared from mouse circulation. However, monovalent human FSTL3 Fc-fusion protein (mono-FSTL3-Fc), generated with the knobs-into-holes technology (Atwell et al., 1997; Ridgway et al., 1996), exhibited longer serum half-life. mono-FSTL3-Fc protein was applicable for systemic administration and increased muscle mass in mice. Thus, our results demonstrate the potential of monovalent FSTL3-Fc as a therapeutic agent to treat muscle wasting disease, without affecting other ligands of the TGF-β family.
Results
FSTL3 has a more restricted binding profile for TGF-β family ligands
To develop a single molecule applicable for systemic administration without large amino acid substitutions, we focused on endogenous antagonists for TGF-β family ligands, including FST family members (reviewed in Chang, 2016). Among them, FSTL3 binds and potently neutralizes activins, GDF8, and GDF11, while the other FST family members, such as FSTL1, mainly antagonize BMPs (Chang, 2016). FSTL3 was isolated from human and mouse serum as a component of circulating GDF8-containing protein complex (Hill et al., 2002). FSTL3 differs from FST in several aspects (Figure 1A); FSTL3 lacks an HBS that is essential for binding to cell surface proteoglycans (Datta-Mannan et al., 2013; Iskenderian et al., 2018; Lodberg et al., 2019); it also lacks a domain equivalent to the third FST domain (FSD3) of FST, which plays critical roles when FST binds and neutralizes BMPs (Stamler et al., 2008). As a result, FSTL3 is less localized to the cell membrane, existing as a circulating protein. In addition, FSTL3 lacks inhibitory activity against BMPs, while it has slightly weaker inhibitory activity against GDF8 (IC50 is 1.2–7.9 nM) and comparable inhibitory activity against activin A compared to FST (Le et al., 2018; Sidis et al., 2006; Walker et al., 2017). We thus generated human FSTL3 Fc-fusion protein (bivalent, hereafter bi-FSTL3-Fc) using HEK293T cells stably expressing it (Figure 1B).
Figure 1.

Bivalent FSTL3-Fc is a potent inhibitor that specifically binds to activin A, GDF8, and GDF11
(A) Schematic presentation of the three isoforms of FST and FSTL3. NTD, N-terminal domain; FSD, follistatin domain; C, C-terminal domain; HBS, heparin-binding site.
(B) Schematic presentation of bivalent FSTL3-Fc protein used in this study. See also Supplemental information for cDNA sequence.
(C–G) In vitro binding assays between bi-FSTL3-Fc protein immobilized by anti-human Fc antibody and biotinylated ligands. The data represent mean ± SD from n = 3 independent experiments. Ligand-binding parameters for bi-FSTL3-Fc as determined by in vitro binding assays (G). ALK1-Fc is a positive control for BMP9.
First, we evaluated binding affinity between recombinant bi-FSTL3-Fc and ligands in vitro. bi-FSTL3-Fc protein was immobilized by anti-Fc antibody, and binding of biotinylated ligands was examined. As reported previously, bi-FSTL3-Fc bound to recombinant activin A, GDF8, and GDF11 proteins in vitro, while the binding affinity to BMP9 was low (Figures 1C–1G). It is of note that biotinylation might slightly disrupt FSTL3 ligand binding. However, concerning BMP9, targets of biotinylation were the N-terminus and 10 lysine residues in BMP9 monomers. Because of the low biotinylation ratio per molecule (2.76 biotin/BMP9 homodimer, Figure 1G), it is unlikely that the biotinylated ligands lost binding affinity because of the biotinylation.
We then assessed ligand-neutralizing activities of bi-FSTL3-Fc using cultured reporter cells. Hs578T human breast cancer cells with the 9×CAGA-Luc reporter for SMAD2/3 (Harada et al., 2019) were used as reporter cells to monitor the activation of activin/TGF-β, while HepG2 hepatocellular carcinoma cells or HMEC-1, an immortalized human microvascular endothelial cell line with the BRE-Luc reporter for SMAD1/5/8 (Morikawa et al., 2011), were used for BMP signaling. bi-FSTL3-Fc potently inhibited SMAD2/3 signal transduction activated by activin A, activin B, GDF8, and GDF11 (Figures 2A and 2B). On the other hand, bi-FSTL3-Fc did not affect SMAD2/3 signaling activated by TGF-β3 nor SMAD1/5/8 signaling activated by BMP6 or BMP9 (Figures 2A, 2C and 2D). Consistent with in vitro binding assays, reporter cell-based ligand-neutralizing assays showed that bi-FSTL3-Fc efficiently bound and neutralized activin A, activin B, GDF8, and GDF11 (Figure 2E).
Figure 2.

Bivalent FSTL3-Fc neutralizes activin A, activin B, GDF8, and GDF11
(A and B) Ligand neutralization by bi-FSTL3-Fc, measured in Hs578T reporter cells with 9×CAGA-Luc for SMAD2/3. The data represent mean ± SD from n = 6 (activin A, GDF8, GDF11, and TGF-β3) or n = 3 (activin B) independent experiments.
(C) Ligand neutralization by bi-FSTL3-Fc, measured in HepG2 reporter cells with BRE-Luc for SMAD1/5/8. The data represent mean ± SD from n = 3 independent experiments.
(D) Ligand neutralization by bi-FSTL3-Fc, measured in HMEC-1-reporter cells with BRE-Luc for SMAD1/5/8. The data represent mean ± SD from n = 3 independent experiments.
(E) Ligand-binding parameters for bi-FSTL3-Fc as determined by reporter cell-based assay. (Right) Validation of activation of SMAD signaling pathway after treatment with indicated ligands.
Bivalent FSTL3-Fc is rapidly cleared from mouse circulation after systemic administration
We conducted a series of experiments in wild-type (WT) mice to determine whether systemic administration of bi-FSTL3-Fc exerts systemic effects. Mice were injected with 10 mg/kg of bi-FSTL3-Fc using the intravenous (i.v.), subcutaneous (s.c.), or intraperitoneal (i.p.) routes, and serum from the injected mice was analyzed (Figures 3A–3C and Figures S1A–S1D). Control Fc was detected at least 48 hr after administration (Figure S1D). In contrast, bi-FSTL3-Fc was detected at 1 hr after administration, and then, it was rapidly cleared from serum within 6 hr irrespective of the route of administration (Figures 3B, 3C, S1B, and S1C). Immunohistochemistry using anti-human Fc antibody showed that bi-FSTL3-Fc was detected in the liver, spleen, kidney, and pancreas, especially on the surface of liver sinusoidal endothelial cells (Figures 3D and S2–S4). The data suggest that bi-FSTL3-Fc in the present form is unlikely to sufficiently exhibit its effects by systemic administration.
Figure 3.

Bivalent FSTL3-Fc is efficiently cleared from mouse circulation
(A) Schematic presentation of the protocol. bi-FSTL3-Fc or control Fc was injected into male mice systemically, and blood was temporally collected from the tail at 0‒48 hr after injection (n = 2 independent experiments).
(B and C) Immunoblot analysis for FSTL3 in reduced serum taken from mice intravenously (i.v.) (B) or intraperitoneally (i.p.) (C) injected with bi-FSTL3-Fc (10 mg/kg) at the indicated time points (n = 2 independent experiments; the identifying number represents each mouse). See also Figure S1, which contains results of subcutaneous (s.c.) injection (Figure S1B), immunoblot analysis for human IgG Fc of the same sera (Figure S1C), and results of control Fc (10 mg/kg) (Figure S1D).
(D) Immunohistochemistry for human IgG Fc in mouse tissues at 5 hr after intravenous injection of bi-FSTL3-Fc. Images are representative of different experiments (n = 2 independent samples), scale bar: 100 μm.
To rule out the possibility that bi-FSTL3-Fc lost its bioactivity in vivo, we assessed whether intramuscular administration of bi-FSTL3-Fc increases muscle mass of WT mice. To confirm the distribution after intramuscular administration, dye (trypan blue) was injected into the right calf, and the staining was observed mainly in the right posterior hindlimb (Figures 4A and 4B). We then injected bi-FSTL3-Fc protein (100 μg), twice weekly for 2 weeks in one side only (Figure 4C), following the local administration regimen of FST-Fc (Castonguay et al., 2019; Pearsall et al., 2019). bi-FSTL3-Fc treatment increased the weight of the gastrocnemius muscle (GC) and hamstring muscle (Ham) in the injected side by approximately 25% compared to that of the un-injected side or that of control-Fc-treated mice (Figures 4D and 4E). Detailed histological analysis showed that muscle fibers exhibited a hypertrophic change, rather than a hyperplastic change (Figures 4F and 4G). The distribution of myofiber size was shifted toward larger sizes, and the means of the fiber sizes were statistically significantly increased in the bi-FSTL3-Fc-treated group, indicating that b-iFSTL3-Fc was bioactive in vivo. These local effects were also observed after a 4-week treatment (Figures S5A–S5C). However, the body weight of the mice was not affected even after the 4-week treatment (Figures S5D and S5E). This suggests that intramuscular administration of bi-FSTL3-Fc does not cause systemic effects.
Figure 4.

Local administration of bivalent FSTL3-Fc increases muscle mass in healthy mice
(A) Schematic presentation of intramuscular injection (i.m.) of bi-FSTL3-Fc into the right calf.
(B) Distribution of trypan blue 10 min after i.m. injection into the right calf.
(C) Schematic presentation of the protocol. bi-FSTL3-Fc, ActRIIB-Fc, or control Fc was injected intramuscularly into the right calf (hindlimb) of 6-week-old male mice, twice weekly for 2 weeks. All mice were sacrificed 2 days after final injection (n = 10 for each group).
(D) Representative macroscopic images of GC muscles excised from mice with 2-week local administration of bi-FSTL3-Fc, ActRIIB-Fc, or control Fc. Muscles on the right hindlimb were injected, while muscles on the left side were used as contralateral counterparts. Images are representative of different experiments (n = 10 independent samples). Scale bar: 1 cm.
(E) Normalized weight of muscles excised from mice with 2-week local administration of bi-FSTL3-Fc, ActRIIB-Fc, or control Fc. Muscle weight was normalized to the body weight. Data are means ± SD from n = 10 independent experiments. Differences between the conditions were analyzed by analysis of variance (ANOVA) with Tukey-Kramer post hoc test for multiple comparison; n.s., not significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. GC, gastrocnemius; QF, quadriceps femoris; TA, tibialis anterior; and Ham, hamstrings; uninjected, left hindlimb; injected, right hindlimb.
(F) Representative cross-sectional images of myofibers in the GC muscle excised from mice with 2-week local administration of bi-FSTL3-Fc, ActRIIB-Fc, or control Fc. Top: immunohistochemistry (IHC) for laminin, middle: quantification using Hybrid Cell Count software, bottom: hematoxylin and eosin staining (H&E). Images are representative of different experiments (n = 5 independent samples). Scale bar: 100 μm.
(G) Quantification and distribution of muscle fiber cross-sectional area of (F). The differences in muscle fiber cross-sectional area were analyzed using Wilcoxon rank-sum test, and p values were adjusted with Benjamini-Hochberg correction for multiple comparisons; ∗∗∗∗p < 0.0001.
(H) Normalized weight of the pancreas of mice after a 2-week treatment. Weight of the pancreas was normalized to the body weight. Data are means ± SD from n = 10 independent experiments. Differences between the conditions were analyzed by ANOVA with Tukey-Kramer post hoc test; n.s., not significant, ∗∗p < 0.01.
(I) The blood glucose concentration of mice after a 2-week-treatment. Mice were fasted for 2.5 hr. Data are means ± SD from n = 10 independent experiments. Differences between the conditions were analyzed by ANOVA with Tukey-Kramer post hoc test; n.s., not significant, ∗p < 0.05.
Ligand trap with ActRIIB-Fc is shown to cause undesirable systemic effects, as well as systemic muscle hypertrophy. ActRIIB-Fc stimulates extramedullary hematopoiesis in the spleen but not in the bone marrow (Latres et al., 2017; Yamawaki et al., 2013). In addition, ActRIIB-Fc increases pancreas weight with elevated blood glucose and impaired glucose tolerance (Latres et al., 2017). Similar to the results of systemic administration of ActRIIB-Fc, local administration of ActRIIB-Fc tended to increase pancreas weight and elevate blood glucose concentration (Figures 4H and 4I) but did not cause macroscopic and histological changes in the spleen (Figures S6A and S6B). On the other hand, blood glucose concentration was not affected in the bi-FSTL3-Fc-treated mice. We could not detect any macroscopic and histological changes in the spleen and pancreas either (Figures 4H, 4I, S6A, and S6B).
Local bivalent FSTL3-Fc administration increases muscle mass in a mouse model of Duchenne muscular dystrophy
These data prompted us to evaluate whether bi-FSTL3-Fc can be a potential treatment option for targeted local therapy in a mouse model of Duchenne muscular dystrophy, C57BL/10ScSn-Dmdmdx/J mice (mdx mice). Intramuscular administration of bi-FSTL3-Fc (100 μg, right calf, twice weekly for 2 weeks) increased the weight of GC and Ham muscles in the injected side by approximately 30% compared to that of the un-injected side or that of control-Fc-treated mice (Figures 5A and 5B). Histological analysis confirmed that muscle fibers exhibited a hypertrophic change (Figures 5C and 5D). These data showed that intramuscular administration of bi-FSTL3-Fc exerts local effects without evidence of systemic effects, similar to the case of FST-Fc/ACE-083 (Pearsall et al., 2019).
Figure 5.

Local administration of bivalent FSTL3-Fc increases muscle mass in a mouse model of Duchenne muscular dystrophy
(A) Representative macroscopic images of GC muscles excised from mice with local administration of bi-FSTL3-Fc or control Fc. Muscles on the right side were injected, while muscles on the left side were used as contralateral counterparts. bi-FSTL3-Fc or control Fc was injected intramuscularly into the right calf (hindlimb) of 6-week-old mdx mice, twice weekly for 2 weeks. All mice were sacrificed 2 days after final dose (n = 6 for each group). Scale bar: 1 cm.
(B) Normalized weight of muscles excised from mice with local administration of bi-FSTL3-Fc or control Fc. Muscle weight was normalized to the body weight. Data are means ± SD from n = 6 independent experiments. Differences between the conditions were analyzed by ANOVA with Tukey-Kramer post hoc test for multiple comparison; n.s., not significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001.
(C) Representative cross-sectional images of myofibers in the GC muscle excised from mice with local administration of bi-FSTL3-Fc or control Fc (IHC for laminin). Scale bar: 100 μm.
(D) Quantification and distribution of muscle fiber cross-sectional area of (C). The differences in muscle fiber cross-sectional area were analyzed using Wilcoxon rank-sum test, and p values were adjusted with Benjamini-Hochberg correction for multiple comparisons; ∗∗∗∗p < 0.0001.
Monovalent FSTL3-Fc protein has longer serum half-life and increases muscle mass after systemic administration
Since FSTL3 is a circulating protein (Hill et al., 2002; Roh et al., 2019), we hypothesized that fusion with Fc was responsible for the rapid clearance from mouse circulation. It is of note that control Fc had long serum half-life (Figure S1D), suggesting that the rapid clearance is not caused by the specific amino acid sequences of the Fc domain of human IgG1. Instead, the dimerization of Fc fragments and the resultant bivalent form of the bi-FSTL3-Fc protein (Figure 1B) may negatively affect its serum half-life. Therefore, we developed mono-FSTL3-Fc using the knobs-into-holes technology (Atwell et al., 1997; Ridgway et al., 1996) (Figures 6A and S7A–S7C). In vitro pull-down assays showed that mono-FSTL3-Fc did not interact with BMP9 (Figure S7D). mono-FSTL3-Fc had slightly weaker inhibitory activity against TGF-β family ligands in the reporter cell-based ligand-neutralizing assays compared to bi-FSTL3-Fc, which could be explained by the difference in the valency of the molecule (Figures 6B–6D and S8A). It is of note that recombinant FSTL3 and mono-FSTL3-Fc had relatively high IC50 values for GDF8 compared to those of activin A as reported previously (Figures 6B–6D and S8A) (Le et al., 2018; Sidis et al., 2006; Walker et al., 2017). On the other hand, ActRIIB-Fc and FST288 had comparable inhibitory activity against activin A, GDF8, and GDF11, although the recombinant FST288 protein we used in this study was less potent compared to that of the previous studies (Le et al., 2018; Sidis et al., 2006; Walker et al., 2017). We then evaluated whether mono-FSTL3-Fc was applicable for systemic administration. After i.p. or i.v injection into WT mice (10 mg/kg), mono-FSTL3-Fc was detected in the serum of the injected mice, even at 48 hr after injection (Figures 6E and S8B), and its concentration was more than 20 μg/mL at 48 hr after i.p. injection (Figures 6F and S8C). Immunohistochemistry using anti-human Fc antibody showed that mono-FSTL3-Fc was detected in the liver, spleen, kidney, and pancreas, especially on the surface of endothelial cells of blood vessels (Figures S9A and S9B). The staining pattern resembled that of control Fc and was different from that of bi-FSTL3-Fc (Figures 3D and S2–S4).
Figure 6.

Monovalent FSTL3-Fc protein has longer serum half-life and increases muscle mass after systemic administration in mice
(A) Schematic presentation of monovalent FSTL3-Fc protein used in this study. See also Supplemental information for cDNA sequence.
(B) Ligand neutralization by mono-FSTL3-Fc, measured in Hs578T-reporter cells with 9×CAGA-Luc for SMAD2/3. The data represent mean ± SD from n = 3 independent experiments.
(C) Ligand neutralization by mono-FSTL3-Fc, measured in HMEC-1-reporter cells with BRE-Luc for SMAD1/5/8. The data represent mean ± SD from n = 3 independent experiments.
(D) Ligand-binding parameters for mono-FSTL3-Fc as determined by reporter cell-based assay. ∗ indicates data from Figure 2C, ∗∗ indicates data from Figure S8A, and ∗∗∗ indicates data from Figure 2D.
(E) Immunoblot analysis for FSTL3 in reduced serum taken from mice at indicated time points after i.p. or i.v. injection with mono-FSTL3-Fc (10 mg/kg) (n = 2 independent experiments. #, ψ, Δ, or ▲ represents each mouse).
(F) Time course of serum concentration of FSTL3-Fc or control Fc measured by anti-human IgG Fc ELISA. Serum was taken at the indicated time points after injection with monovalent- or bivalent-FSTL3-Fc or control Fc (10 mg/kg). The data represent mean ± SD from n = 3 independent experiments, except for bi-FSTL3-Fc, at 96 hr (n = 2, because of anesthesia-related death). Differences between the conditions were analyzed using analysis of variance (ANOVA) followed by Tukey-Kramer post hoc test for multiple comparison; n.s., not significant, ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001.
We then evaluated the systemic effects of mono-FSTL3-Fc in WT mice. Intraperitoneal injection of mono-FSTL3-Fc (10 mg/kg, twice weekly for 2 weeks) was performed, following the regimen of ActRIIB-Fc (Cadena et al., 2010; Pistilli et al., 2011). mono-FSTL3-Fc treatment increased the body weight of the mice in a dose-dependent manner compared to that of control-Fc-treated mice (Figure 7A). Consistently, it dose dependently increased the weight of hindlimb muscles on both sides by approximately 20% compared to that of control-Fc-treated mice (Figures 7B and 7C). Histological analysis confirmed that muscle fibers exhibited a hypertrophic change (Figures 7D and 7E). These data indicated that mono-FSTL3-Fc caused systemic muscle hypertrophy, similar to ActRIIB-Fc/ACE-031. Furthermore, we were not able to find any macroscopic and histological changes in organs and blood glucose concentration in mono-FSTL3-Fc-treated mice and control-Fc-treated mice, even with the highest dose 30 mg/kg (Figures 7F and 7G).
Figure 7.

Systemic administration of monovalent FSTL3-Fc increases muscle mass in healthy mice
(A) Body weight of mice injected with mono-FSTL3-Fc or control Fc before and after the start of the injection. Data are means ± SD from n = 7 (control Fc and 10 mg/kg mono-FSTL3-Fc) or n = 6 (3 mg/kg and 30 mg/kg mono-FSTL3-Fc) independent experiments. Differences between the conditions were analyzed by ANOVA with Tukey-Kramer post hoc test for multiple comparison; n.s., not significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(B) Representative macroscopic images of hindlimb muscles and QF and GC muscles excised from mice with i.p. injection of mono-FSTL3-Fc or control Fc (10 mg/kg). Scale bar: 1 cm.
(C) Normalized weight of muscles excised from mice with i.p. of mono-FSTL3-Fc or control Fc at the indicated protein concentration. Muscle weight was normalized to the body weight. Data are means ± SD from n = 7 (control Fc and 10 mg/kg mono-FSTL3-Fc) or n = 6 (3 mg/kg and 30 mg/kg mono-FSTL3-Fc) independent experiments. Differences between the conditions were analyzed by ANOVA with Tukey-Kramer post hoc test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(D) Representative cross-sectional images of myofibers in the QF excised from mice with local administration of mono-FSTL3-Fc or control Fc (IHC for laminin). Scale bar: 100 μm.
(E) Quantification and distribution of muscle fiber cross-sectional area of (D). The differences in muscle fiber cross-sectional area were analyzed using Wilcoxon rank-sum test; ∗∗∗∗p < 0.0001.
(F) Normalized weight of tissue of mice after a 2-week-treatment. Tissue weight was normalized to the body weight. Data are means ± SD from n = 7 (control Fc and 10 mg/kg mono-FSTL3-Fc) or n = 6 (3 mg/kg and 30 mg/kg mono-FSTL3-Fc) independent experiments. Differences between the conditions were analyzed by ANOVA with Tukey-Kramer post hoc test; n.s., not significant.
(G) The blood glucose concentration of mice after a 2-week-treatment. Mice were fasted for 2.5 hr. Data are means ± SD from n = 7 (control Fc and 10 mg/kg mono-FSTL3-Fc) or n = 6 (3 mg/kg and 30 mg/kg mono-FSTL3-Fc) independent experiments. Differences between the conditions were analyzed by ANOVA with Tukey-Kramer post hoc test; n.s., not significant.
Systemic administration of monovalent FSTL3-Fc increases muscle mass in a mouse model of Duchenne muscular dystrophy
Finally, we evaluated whether mono-FSTL3-Fc has comparable therapeutic effects to RAP-031/ActRIIB-Fc, a mouse version of ACE-031, in the disease model mice. Intraperitoneal injection of mono-FSTL3-Fc (10 mg/kg, twice weekly for 2 weeks) tended to increase body weight of the mice after a 2-week treatment (Figure 8A). Consistently, mono-FSTL3-Fc and RAP-031/ActRIIB-Fc increased the weight of hindlimb muscles except for tibialis anterior (TA) on both sides by approximately 25% compared to that of control-Fc-treated mice (Figures 8B and 8C). RAP-031/ActRIIB-Fc (estimated molecular weight [MW]: 78 kDa) is bivalent and can bind and neutralize more ligands than mono-FSTL3-Fc (estimated MW: 76 kDa) per unit weight. Nevertheless, the difference between mono-FSTL3-Fc and RAP-031 was not significant in terms of their effects on the weight of the quadriceps femoris (QF), GC, and Ham muscles (Figures 8B and 8C). Muscle mass of TA of mdx mice was not affected by either mono-FSTL3-Fc or ActRIIB-Fc (Figure 8C), suggesting that TA muscle may be less responsive to anti-GDF8 therapy. Histological analysis confirmed that muscle fibers exhibited a hypertrophic change (Figures 8D and 8E). Moreover, mono-FSTL3-Fc and RAP-031/ActRIIB-Fc-treated mice had greater forelimb grip strength than did control-Fc-treated mice (Figure 8F). These data indicated that systemic administration of mono-FSTL3-Fc exerted systemic effects, similar to ActRIIB-Fc/ACE-031. On the other hand, it did not affect blood glucose concentration (Figure 8G). mono-FSTL3-Fc did not cause macroscopic nor histological changes in other organs we examined (Figure 8H). In conclusion, we have developed mono-FSTL3-Fc, which overcomes the difficulties of current anti-GDF8 therapies; it does not bind to BMP9 and it is applicable for systemic administration with longer serum half-life (Figure 8I).
Figure 8.

Systemic administration of monovalent FSTL3-Fc increases muscle mass in a mouse model of Duchenne muscular dystrophy
(A) Body weight of mice injected with mono-FSTL3-Fc, control Fc, or ActRIIB-Fc before and after the start of the injection. Data are means ± SD from n = 12 (mono-FSTL3-Fc and RAP-031/ActRIIB-Fc on day 0 and day 3) or n = 11 (control Fc and RAP-031/ActRIIB-Fc after day 7) independent experiments. Because the body weight of one mouse treated with RAP-031/ActRIIB-Fc decreased more than the criteria on day 3, the mouse was excluded from the analysis thereafter. Differences between the conditions were analyzed by ANOVA with Tukey-Kramer post hoc test for multiple comparison; n.s., not significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
(B) Representative macroscopic images of hindlimb muscles and QF and GC muscles excised from mice with i.p. injection of mono-FSTL3-Fc, RAP-031/ActRIIB-Fc, or control Fc. Scale bar: 2 cm (upper) and 1 cm (lower).
(C) Normalized weight of muscles excised from mice with i.p. of mono-FSTL3-Fc, control Fc, or ActRIIB-Fc. Muscle weight was normalized to the body weight. Data are means ± SD from n = 8 (mono-FSTL3-Fc) or n = 7 (control Fc and RAP-031/ActRIIB-Fc) individuals. Differences between the conditions were analyzed by ANOVA with Tukey-Kramer post hoc test; n.s., not significant, ∗∗∗p < 0.001.
(D) Representative cross-sectional images of myofibers in the QF muscle excised from mice with local administration of mono-FSTL3-Fc, RAP-031/ActRIIB-Fc, or control Fc (IHC for laminin). Images are representative of different experiments (n = 5 independent samples). Scale bar: 100 μm.
(E) Quantification and distribution of muscle fiber cross-sectional area of (D). The differences in muscle fiber cross-sectional area were analyzed using Wilcoxon rank-sum test, and p values were adjusted with Benjamini-Hochberg correction for multiple comparisons; n.s., not significant, ∗∗∗∗p < 0.0001.
(F) Grip strength of the forelimb treated with mono-FSTL3-Fc, RAP-031/ActRIIB-Fc, or control Fc for 2 weeks. Data are means ± SD from n = 7 individuals. Differences between the conditions were analyzed by Welch's t test ANOVA with Tukey-Kramer post hoc test; n.s., not significant, ∗p < 0.05.
(G) The blood glucose concentration of mice after a 2-week treatment. Mice were fasted for 2.5 hr. Data are means ± SD from n = 8 (mono-FSTL3-Fc) or n = 7 (control Fc and RAP-031/ActRIIB-Fc) independent experiments. Differences between the conditions were analyzed by ANOVA with Tukey-Kramer post hoc test.
(H) Normalized weight of tissue of mice after a 2-week treatment. Tissue weight was normalized to the body weight. Data are means ± SD from n = 8 (mono-FSTL3-Fc) or n = 7 (control Fc and RAP-031/ActRIIB-Fc) independent experiments. Differences between the conditions were analyzed by ANOVA with Tukey-Kramer post hoc test; n.s., not significant, ∗p < 0.05, ∗∗p < 0.01∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
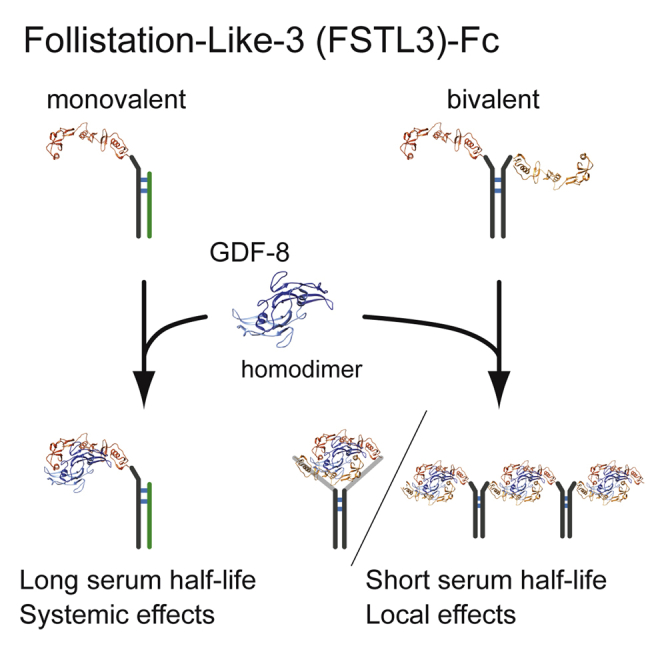
(I) Schematic presentation of the characteristics of mono- and bi-valent FSTL3-Fc. Two FSTL3 and GDF-8 homodimers form a complex (PDB code: 3SEK (Cash et al., 2012)). Although conventional or bivalent FSTL3-Fc has shorter serum half-life, monovalent FSTL3-Fc has longer serum half-life and increases muscle mass under systemic administration.
Discussion
FST-derived biological products, including FST-Fc, are thought to be promising therapeutic options for the treatment of muscle wasting diseases and cancer cachexia since FST is able to inhibit multiple TGF-β family members that negatively regulate muscle growth. ActRIIB-Fc (ACE-031) was discontinued because of undesirable inhibition of BMP9, BMP10, and other angiogenic TGF-β family members (Campbell et al., 2017). Although the newly developed FST-Fc protein (ACE-083) did not bind to BMP9 and BMP10 (Pearsall et al., 2019), it still inhibited other TGF-β family ligands in addition to the negative regulators of muscle mass. FST also antagonized BMP6 and BMP7 (Pearsall et al., 2019; Sidis et al., 2006), inhibition of which is implicated in iron overload (Andriopoulos et al., 2009; Meynard et al., 2009) and kidney disease (Zeisberg et al., 2003), respectively. Another limitation of FST-Fc is that it is only available for local administration (Castonguay et al., 2019; Pearsall et al., 2019).
Fusion to the IgG Fc domain is an established way to prolong serum half-life in vivo by neonatal Fc receptor (FcRn)-mediated recycling and transcytosis, which protects IgG and Fc-fusion proteins from lysosomal degradation (Rath et al., 2015). Actually, the half-life of ActRIIB-Fc was reported to be 4 days or longer (Chiu et al., 2013; Zhou et al., 2010). Fusion to Fc also enables us to use efficient methods of protein purification. Since FSTL3 is a circulating protein (Hill et al., 2002; Roh et al., 2019), we first assumed that bi-FSTL3-Fc can be used systemically. However, bi-FSTL3-Fc protein was rapidly cleared from serum, and, at least to some extent, it was deposited into the liver, spleen, kidney, and pancreas (Figures 3D and S1B–S1D). We thus hypothesized that fusion to the Fc fragment to obtain a bivalent protein with two arms (Figure 1B) caused the rapid clearance of the recombinant protein. Indeed, mono-FSTL3-Fc had prolonged serum half-life (Figure 6). Although the bi-FSTL3-Fc protein is “bivalent and monospecific”, the target ligands make (homo-)dimers with two epitopes on one dimer (Figure 8I). In this case, the bi-FSTL3-Fc protein resembles a biparatopic bispecific antibody. As shown in the case of a heterodimeric fusion protein ActRIIB:ALK4-Fc (Li et al., 2021), one molecule of Fc-protein with two arms can pinch one GDF8 homodimer (Figure 8I). However, our data suggest that this is not a major binding form between bi-FSTL3-Fc and GDF8, possibly because of steric hindrance. Rather, bi-FSTL3-Fc can make protein complex containing multiple bi-FSTL3-Fc through two epitopes on the GDF8 homodimers (Figure 8I). It is of note that a protein complex with multiple Fc regions is reported to cross-link multiple FcRn molecules, which results in lysosomal degradation rather than recycling or transcytosis (Weflen et al., 2013). Similarly, a bispecific antibody (Datta-Mannan et al., 2016) and a bispecific/biparatopic antibody that targets two different epitopes of IL-6 (Kasturirangan et al., 2017) are rapidly cleared from serum and detected in the liver in mice. Thus, monovalent Fc, or one-armed Fc, is a rational solution to overcome the limitation. We generated mono-FSTL3-Fc using human Fc with the knobs-into-holes mutations (Atwell et al., 1997; Ridgway et al., 1996). Although it might cause anti-drug antibodies in mouse even in a 2-week regimen (Bautista et al., 2012), therapeutic effects of mono-FSTL3-Fc were not inferior to those of RAP-031 (the murine homolog of ACE-031/ActRIIB-Fc) in our mouse experiments (Figure 8).
There are several reported side effects of ActRIIB-Fc in addition to the phenotype in the vasculature (Campbell et al., 2017): extramedullary hematopoiesis in the spleen (Latres et al., 2017; Yamawaki et al., 2013) and increased pancreas weight with impaired glucose tolerance (Latres et al., 2017). Although these can be explained by off-target inhibition of other TGF-β family members not involved in the regulation of muscle mass, it is also possible that these are on-target effects caused by inhibition of GDF11. It is of note that the phenotype observed in hematopoiesis and pancreatic β cells in mice treated with ActRIIB-Fc is not reported under combination of anti-activin A and anti-GDF8 antibodies (Latres et al., 2017).
On the other hand, the phenotype related to GDF11 inhibition is still under debate. One possible reason is that amino acid residues are highly conserved between GDF8 and GDF11, which makes it difficult to generate specific antibodies against each protein (Egerman et al., 2015). It is of note that therapeutic anti-GDF8 antibodies in clinical trials may, at least to some extent, inhibit GDF11 in addition to GDF8. However, the phenotype of hematopoiesis and pancreatic β cells has not been reported in these trials (Furrer and Handschin, 2019; Lee and Jun, 2019; Marty et al., 2017). Consistently, luspatercept (also known ACE-536; ActRIIB-Fc with modification in the extracellular domain of ActRIIB that targets mainly GDF11) enhances erythroid maturation and increases hemoglobin concentration in animal models and patients with low-risk myelodysplastic syndromes (Fenaux et al., 2020; Suragani et al., 2014); in this case, inhibition of GDF11 stimulates hematopoiesis in the bone marrow and not in the spleen. Concerning the phenotype in the pancreas, genetic ablation of Gdf11 in the pancreas attenuated maturation of pancreatic β cells (Harmon et al., 2004), while Fstl3 knockout mice had increased pancreatic islet number and size and β-cell hyperplasia (Mukherjee et al., 2007). In contrast, Fstl3-transgenic mice displayed improved insulin sensitivity and muscle insulin signaling (Brandt et al., 2015). The discrepancy between gain of function and loss of function of FSTL3 in vivo can be explained by the fact that the anti-GDF8 therapy increases muscle mass and improves glucose intolerance (Guo et al., 2009). Although the “GDF11-related phenotype of anti-GDF8 therapy” was not detected in our mouse model after administration of mono-FSTL3-Fc (Figures 7 and 8), it should be carefully evaluated when using mono-FSTL3-Fc in clinical settings.
In summary, our data provide evidence that monovalent FSTL3-based therapy overcomes the difficulties of current anti-GDF8 therapies. FSTL3 has a more restricted binding profile for TGF-β family ligands, and mono-FSTL3-Fc can be administered systemically. We thus believe that FSTL3-based therapy could be a promising therapeutic option for the treatment of muscle wasting diseases. In addition, mono-FSTL3-Fc can be an alternative approach for treating several diseases, such as lung adenocarcinoma (Marini et al., 2018) and osteoarthritis (Tang et al., 2020), in which exogenous FST has therapeutic effects.
Limitations of the study
Our current study provides in vitro and in vivo evidence that a monovalent form of FSTL3-Fc overcomes issues with current anti-GDF8/myostatin therapies and can be a promising treatment option to increase muscle mass in muscle wasting and neuromuscular diseases. However, additional studies are needed to optimize dose and administration regimen. It is also important to evaluate therapeutic effects in other mouse cohorts, such as female mice and aged mice and other disease model mice, although we used young WT male mice and a mouse model of Duchenne muscular dystrophy to evaluate biological effects of mono-FSTL3-Fc. In addition, the side effects of mono-FSTL3-Fc should be carefully evaluated in future pre-clinical studies in different animal models.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-human IgG Fc | Bethyl Laboratories | Cat#A80-105A; RRID: AB_67482 |
| Rabbit polyclonal anti-laminin | Abcam | Cat# ab11575; RRID: AB_298179 |
| Rabbit polyclonal anti-human FSTL3 | Abcam | ab170680 |
| Mouse monoclonal anti-BMP9 | R&D | Cat# MAB3209; RRID: AB_2247656 |
| Goat polyclonal anti-rabbit IgG with Alexa Fluor594 | Thermo Fisher SCIENTIFIC | Cat# A11012; RRID: AB_141359 |
| Goat polyclonal anti-mouse IgG light chain specific with HRP | Jackson ImmunoResearch Labs | Cat# 115-035-174; RRID: AB_2338512 |
| Goat polyclonal anti-rabbit IgG with HRP | Cell Signaling Technology | Cat# 7074S; RRID: AB_2099233 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human IgG1 Fc | This paper | N/A |
| Bivalent Human FSTL3-Fc | This paper | N/A |
| Monovalent Human FSTL3-Fc | This paper | N/A |
| Human Activin A | R&D | Cat# 338-AC |
| Human Activin B | R&D | Cat# 659-AB |
| Human TGF-β3 | R&D | Cat# 243-B3 |
| Human BMP6 | R&D | Cat# 507-BP |
| Human BMP9 | R&D | Cat# 3209-BP |
| Human GDF8 | Pepro Tech | Cat# 120-00 |
| Human GDF11 | Pepro Tech | Cat# 120-11 |
| Human ALK1-Fc | R&D | Cat# 370-AL |
| Murine ActRIIB-murine Fc | R&D | Cat# 3725-RB |
| murine ActRIIB-murine Fc/RAP-031 | Acceleron Pharma | N/A |
| Human Follistatin 288 | R&D | Cat# 5836-FS-025 |
| Human FSTL3 | BioLegend | Cat# 789604 |
| trypan blue | BIO-RAD | Cat# 1450021 |
| Human Insulin | Gibco/Thermo Fisher Scientific | Cat# 12585014 |
| Puromycin | InvivoGen | Cat# ant-pr |
| Hygromycin | Invitrogen/Thermo Fisher Scientific | Cat# 10687010 |
| Lipofectamine 2000 | Invitrogen/Thermo Fisher Scientific | Cat# 11668019 |
| HiTrap Protein A HP antibody purification columns | GE Healthcare Life Sciences/Cytiva | Cat# 17040201 |
| HRP conjugated streptavidin | BioLegend | Cat# 405210 |
| TMB One Component Substrate | Bethyl Laboratories | Cat# E102 |
| FreeStyle 293 Expression Medium | Thermo Fisher Scientific | Cat# 12338026 |
| Critical Commercial Assays | ||
| Biotin Labeling Kit-NH2 | Dojindo Laboratories | Cat# LK03 |
| Pierce Biotin Quantitation Kit | Thermo Fisher Scientific | Cat# 28005 |
| Anti-human IgG ELISA | Bethyl Laboratories | Cat# E80-114 |
| Dual-luciferase Reporter Assay System | Promega | Cat# E1910 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T cells | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| Human: HepG2 cells | ATCC | Cat# HB-8065; RRID: CVCL_0027 |
| Human: Lenti-X 293T cells | TaKaRa Bio | Cat# 632180; RRID: CVCL_4401 |
| Human: HMEC-1 cells | Dr. T. Lawley Xu et al. (1994) |
RRID: CVCL_0307 |
| Human: Hs578T-9×CAGA-Luc | Harada et al. (2019) | N/A |
| Human: HepG2-BRE-Luc | This paper | N/A |
| Human: HMEC-1-BRE-Luc | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: CD2F1/slc | Sankyo Labo Service Corporation | Slc:CDF1 |
| Mouse: C57BL/10ScSn-Dmdmdx/J | CLEA Japan | C57BL/10-mdx |
| Recombinant DNA | ||
| CSII-EF-RfA | Dr. H. Miyoshi Riken BRC |
Cat# RDB04387 |
| CSII-CAG-MCS-IRES-Puro | Harada et al. (2019) | N/A |
| CSII-CAG-MCS-IRES-Hyg | Harada et al. (2019) | N/A |
| pcDNA3-Fc | This paper | N/A |
| pcDNA3-FSTL3-Fc | This paper | N/A |
| pcDNA3-Fc(T366S/L368A/Y407V) | This paper | N/A |
| pcDNA3-FSTL3-Fc(T366W) | This paper | N/A |
| CSII-CAG-Fc-IRES-Puro | This paper | N/A |
| CSII-CAG-FSTL3-Fc-IRES-Puro | This paper | N/A |
| CSII-CAG-FSTL3-Fc(T366S/L368A/Y407V)-IRES-Puro | This paper | N/A |
| CSII-CAG-Fc(T366W)-IRES-Hyg | This paper | N/A |
| CSII-BRE-Luc-SV40-Hyg | Morikawa et al. (2011) | N/A |
| Software and Algorithms | ||
| R software | R-project.org | https://www.R-project.org |
| GraphPad Prism 6 | GraphPad Software | version 6 |
| Hybrid Cell Count software | Keyence | Cat# BZ-H3C |
| Other | ||
| Amicon Ultra-15 Ultracel-30 kDa | Millipore, Merck | Cat# C7715 |
| VivaSpin 20 MW | Sartorius | Cat# VS2041 |
| Dynabeads Protein A | Thermo Fisher Scientific | Cat# 10001D |
| TC 20 Automated Cell Counter | BIO-RAD | Cat# 1450102 |
| ImageQuant LAS 4000 | Fujifilm | Cat# LAS-4000 |
| BZ-X700 microscope | Keyence | Cat# BZ-X 700 |
| digital force indicator | Imada | Cat# DST-20N |
| Lab Gluco | ForaCare Suisse AG | Cat# 4239R1006 |
| Model 680 Microplate Reader | BIO-RAD | Cat# 1681101 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Masato Morikawa (morikawa-tky@umin.ac.jp).
Materials availability
Materials used or generated in this study will be available upon reasonable request, and a material transfer agreement may be required.
Data and code availability
This study did not generate or analyze datasets or code.
Experimental model and subject details
Mouse experiments
All animal experiments were reviewed and approved by the Animal Ethics Committee of the University of Tokyo, and performed in accordance with the institutional guidelines. Briefly, 5∼6-week-old male CD2F1 mice were purchased from Sankyo Labo Service Corporation (Tokyo, Japan) and maintained to acclimate to the animal facility for 1 week. C57BL/10ScSn-Dmdmdx/J mice (mdx mice) were purchased from CLEA Japan (Tokyo, Japan). Mice were injected with 100 μg (50 μL) of hFc, bi-FSTL3-hFc, or ActRIIB-Fc intramuscularly (i.m.) into the right calf twice weekly for 2 or 4 weeks. Systemic administration of mono-FSTL3-Fc and hFc was performed at 10 mg/kg twice weekly for 2 weeks via i.p. injection. Two days after all treatments, mice were sacrificed by inhalation anesthesia with isoflurane, and tissues were extirpated, weighed, and used to make tissue blocks for histological analysis. All mice were fasted for 2.5 h, and blood was taken from the tail vein. The blood glucose concentrations in the tail vein samples were measured by Lab Gluco (ForaCare Suisse AG, St. Gallen, Switzerland).
Skeletal muscle histology
Histological analysis of skeletal muscle was performed essentially as described previously (Ojima et al., 2020). The QF, Ham, TA, and GC muscles were isolated from CD2F1 and mdx mice. The mid belly region of the GC and QF muscle was embedded in OCT and snap-frozen in isopentane and acetone mixture (1:2) cooled with a cooling apparatus, UT2000F (Leica Microsystems, Wetzlar, Germany). Cryostat transverse sections were 5 μm in thickness. Frozen sections were fixed with 4% PFA/PBS, and then stained with anti-laminin antibody (ab11575; Abcam). Images of the section were obtained using a BZ-X700 microscope (Keyence, Osaka, Japan) using n=5 mice for each group/condition, and at least 1 slide for each mouse. Quantification was performed using Keyence software. The average area of GC or QF muscle fibers was calculated from at least 350 individual muscle fibers, which were automatically quantified by a Hybrid Cell Count software (Keyence).
Grip strength measurement
Grip strength of the forelimb of mdx mice after a 2-week treatment was measured by digital force indicator (DST-20N; Imada, Aichi, Japan). Mice were acclimated to the grip bar for 5 days. On day 13 after first injection, mice were tested in five independent trials and the maximum force (N) was recorded.
Cell culture
HEK293T (female) and HepG2 (male) cells were obtained from the American Type Culture Collection (ATCC). Lenti-X 293T (female) cells were purchased from Clontech (TaKaRa Bio, Shiga, Japan). HMEC-1 (male), an immortalized human dermal microvascular endothelial cell line, was obtained from Dr. T. Lawley, and was cultured as described previously (Suzuki et al., 2008; Xu et al., 1994). Hs578T (female)-reporter cells with 9×CAGA-Luc construct were described previously (Harada et al., 2019). HepG2-reporter cells and HMEC-1-reporter cells with BRE-Luc were generated as described previously (Morikawa et al., 2011).
HEK293T, Lenti-X 293T, and HepG2-reporter cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), supplemented with 10% (v/v) fetal bovine serum (FBS) (HyClone, GE Healthcare Life Sciences/Cytiva, Marlborough, MA, USA) and 100 U/ml penicillin-streptomycin (Gibco). Hs578T-reporter cells were maintained in DMEM (Gibco) supplemented with 10% FBS (HyClone) and 10 μg/ml insulin (Gibco).
Cultured cells were routinely tested for mycoplasma contamination using e-Myco VALid-Q mycoplasma qPCR detection kit (iNtRON Biotechnology, Korea).
Method details
Reagents and antibodies
Recombinant human Activin A, Activin B, TGF-β3, BMP6, BMP9, Follistatin 288, and ALK1-Fc were purchased from R&D Systems (Minneapolis, MN, USA). Recombinant human FSTL3 was purchased from BioLegend (San Diego, CA, USA). GDF8 and GDF11 were purchased from Pepro Tech (Rocky Hill, NJ, USA). Murine ActRIIB-murine Fc was purchased from R&D Systems, and murine ActRIIB-murine Fc/RAP-031 was kindly provided by Acceleron Pharma (Cambridge, MA, USA).
The following antibodies were used: anti-human IgG Fc (Bethyl Laboratories, Montgomery, TX, USA), anti-human FSTL3 (ab170680; Abcam, Cambridge, UK), anti-Laminin (ab11575; Abcam), and anti-BMP9 (MAB3209; R&D).
Plasmid construction
The coding region of the human FSTL3 (GenBank accession NM_005860.3) gene was amplified by PCR and subcloned between BamHI and XhoI sites of pcDNA3-Fc, which contains Fc constant region (Asp 6-Lys 232) of human IgG1 (GenBank: AEV43323.1).
For knobs-into-holes using T366W, T366S/L368A/Y407V (Atwell et al., 1997; Ridgway et al., 1996) was generated by site-directed mutagenesis using PCR with specific primers. The FSTL3-Fc fragment was transferred into the CSII-CAG-MCS-IRES-Puro or CSII-CAG-MCS-IRES-Hyg lentiviral vector (Harada et al., 2019), which was derived from the CSII-EF-RfA lentiviral vector. Control Fc fragment was prepared by replacing FSTL3 with an efficient signal peptide. All the constructs for lentivirus production were kindly provided by Dr. H. Miyoshi (deceased, formerly RIKEN, Japan).
Lentivirus production
Recombinant lentiviruses were produced by transient transfection using Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific) in HEK293T or Lenti-X 293T cells, and were concentrated using the Lenti-X Concentrator (Clontech).
hFSTL3-Fc protein sequence
The amino acid sequence of monovalent hFSTL3-Fc protein is as follows:
([signal peptide]-hFSTL3-hFc(T366W))
[MRPGAPGPLWPLPWGALAWAVGFVSS]
MGSGNPAPGGVCWLQQGQEATCSLVLQTDVTRAECCASGNIDTAWSNLTHPGNKINLLGFLGLVHCLPCKDSCDGVECGPGKACRMLGGRPRCECAPDCSGLPARLQVCGSDGATYRDECELRAARCRGHPDLSVMYRGRCRKSCEHVVCPRPQSCVVDQTGSAHCVVCRAAPCPVPSSPGQELCGNNNVTYISSCHMRQATCFLGRSIGVRHAGSCAGTPEEPPGGESAEEEENFV
ISTGGGTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLWCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
([signal peptide]-hFc(T366S/L368A/Y407V))
[MRPGAPGPLWPLPWGALAWAVGFVSS]
MGTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLSCAVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLVSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
The amino acid sequence of bivalent hFSTL3-Fc protein ([signal peptide]-hFSTL3-hFc):
[MRPGAPGPLWPLPWGALAWAVGFVSS] MGSGNPAPGGVCWLQQGQEATCSLVLQTDVTRAECCASGNIDTAWSNLTHPGNKINLLGFLGLVHCLPCKDSCDGVECGPGKACRMLGGRPRCECAPDCSGLPARLQVCGSDGATYRDECELRAARCRGHPDLSVMYRGRCRKSCEHVVCPRPQSCVVDQTGSAHCVVCRAAPCPVPSSPGQELCGNNNVTYISSCHMRQATCFLGRSIGVRHAGSCAGTPEEPPGGESAEEEENFV ISSTMVRSDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
Protein purification
Recombinant FSTL3-Fc or control Fc was purified from HEK293T cells stably expressing them. Cells were cultured in FreeStyle 293 Expression Medium (Thermo Fisher Scientific) without serum. After 3 days of cell culture, the supernatant was used for purification. The protein was captured on a protein A column (HiTrap Protein A HP, GE Healthcare Life Sciences/Cytiva) and eluted with 0.1 M sodium citrate (pH 3.5) after over 20 times column volume wash with 20 mM sodium phosphate (pH 6.8). The protein solution was neutralized with 1 M Tris-HCl (pH 9.0) to adjust the pH to 7.0‒7.4 immediately after elution. Pooled fractions from the elution were concentrated and the vehicle was changed into PBS by ultrafiltration (Amicon Ultra-15 Ultracel-30 kDa, Millipore, Merck; VivaSpin 20 MW cut off 50 kDa and 100 kDa, Sartorius, Göttingen, Germany). The quality of the purified proteins was assessed by SDS-PAGE. The concentration of the proteins was determined by anti-human IgG ELISA (Human IgG ELISA Quantitation Set, Bethyl Laboratories).
In vitro binding assay with biotinylated ligands
Binding assays between recombinant ligands and recombinant bi-FSTL3-Fc were performed using biotinylated ligands. Activin A, GDF8, and GDF11 were biotinylated using Biotin Labeling Kit-NH2 (LK03; Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's recommendations. A part of the ligands was used for quantitation of biotinylation (Pierce Biotin Quantitation Kit; Thermo Fisher Scientific). Briefly, 96-well plates were coated with rabbit anti-human IgG Fc (A80-105A; Bethyl Laboratories) for 1 h at room temperature. Biotinylated ligands and 2 nM hFSTL3-Fc or control Fc were incubated with 0.5% BSA/PBS for 30 min at 37°C. After blocking with 1% BSA/50 mM Tris-HCl, 140 mM NaCl (pH 8.0), the ligand and protein mixtures were incubated in the well for 1 h at room temperature. HRP conjugated streptavidin (405210; BioLegend) was used to detect ligands for 1 h at room temperature. After TMB (E102; Bethyl Laboratories) and HRP reaction for 10 min in the dark, 0.18 M H2SO4 was added to each well to stop the reaction. The absorbance of the well at 450 nm was measured on a plate reader.
Protein pulldown assay
Protein pulldown assay was performed using Dynabeads Protein A (Invitrogen, Thermo Fisher Scientific), according to the manufacturer’s instructions. Thirty μl of beads were washed once in PBS with 0.02% Tween 20 (PBST), resuspended in 200 μl of PBST, and preincubated with 1 μg of recombinant Fc protein at room temperature for 10 min. The beads were washed once in 200 μl of PBST, and then incubated with 500 ng of recombinant BMP9 in 200 μl of PBST for 10 min. The beads were then washed three times in 200 μl of PBST and eluted with SDS-PAGE sample buffer without DTT, and subjected to SDS-PAGE. Since anti-BMP9 (MAB3209; R&D) recognizes non-reduced BMP9 dimer, SDS-PAGE was performed under non-reduced conditions. Western blot analysis was performed using the indicated antibodies.
Western blot analysis
Western blot analysis was performed essentially as described previously (Morikawa et al., 2019). Cells were lysed in lysis buffer containing 20 mM HEPES, pH 7.4, 100 mM NaCl, 0.5 mM EDTA, 10% glycerol, and 0.2% NP-40 with cOmplete EDTA-free protease inhibitor cocktail (Roche Life Science, Basel, Switzerland). Samples were separated by SDS-PAGE, blotted onto a polyvinylidene difluoride (PVDF) membrane (Pall, New York, NY, USA), and the chemiluminescence signal was detected by a luminescence image analyzer (LAS-4000; Fujifilm, Tokyo, Japan).
Luciferase assays
Hs578T-reporter cells with 9×CAGA-Luc construct (Harada et al., 2019) and HepG2-reporter cells or HMEC1-reporter cells with BRE-Luc (Morikawa et al., 2011) were used for luciferase reporter assay. The reporter cells were pre-treated with bi- or mono-FSTL3-Fc for 1 h in DMEM with 2% or 10% FBS (Hs578T- and HepG2-reporter cells) or EGM-2 and Opti-MEM (Gibco) with 0.5% serum (HMEC1-reporter cells). Cells were stimulated with indicated amounts of ligands for 24 h and were harvested and assayed for luciferase activity. Firefly luciferase activities of the cell lysates were determined using the Dual-luciferase Reporter Assay System (Promega, Madison, WI, USA).
Quantification and statistical analysis
The statistical analyses were performed using GraphPad Prism 6 (San Diego, CA, USA) and R software (https://www.R-project.org). The differences between two experimental groups were analyzed using Welch's ttest or analysis of variance (ANOVA) followed by Tukey-Kramer post hoc test for multiple comparison. The differences in muscle fiber cross-section area, which did not follow a normal distribution, were analyzed using Wilcoxon rank sum (Mann-Whitney) tests, and p values were adjusted with Benjamini-Hochberg correction for multiple comparisons. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 and ∗∗∗∗p < 0.0001.
Acknowledgments
We thank Dr. H. Miyoshi (deceased, formerly RIKEN, Japan) for the lentivirus vector system; Dr. R. Kumar and Acceleron Pharma for a kind gift of RAP-031/ActRIIB-Fc; Drs. K. Miyazawa, C.-H. Heldin, and P.I. Arvidsson for discussion; and Dr. M. Harada and Ms. M. Kobayashi for technical help. We also thank the Science for Life Laboratory Drug Discovery and Development Platform (SciLifelab DDD) for technical advice. This work was supported by KAKENHI [Grants-in-Aid for Scientific Research (C) (grant number 19K07683 to M.M.)], Grant-in-Aid for Scientific Research (S) from Japan Society for the Promotion of Science (JSPS) (15H05774 to K.M.), and Grant-in-Aid for Scientific Research on Innovative Area on Integrated Analysis and Regulation of Cellular Diversity (17H06326 to K.M. and M.M.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. T.O. was supported by JSPS Research Fellowship for Young Scientists (DC2).
Author contributions
M.M. conceived the project. T.O. and M.M. designed the study and performed experiments. Y.M., K.O., and D.K. helped with the experiments. F.I. and P.-Å.N. provided expertise and feedback. M.M. and K.M. supervised the study. T.O. and M.M. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 21, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102488.
Contributor Information
Masato Morikawa, Email: morikawa-tky@umin.ac.jp.
Kohei Miyazono, Email: miyazono@m.u-tokyo.ac.jp.
Supplemental information
References
- Andriopoulos B., Jr., Corradini E., Xia Y., Faasse S.A., Chen S., Grgurevic L., Knutson M.D., Pietrangelo A., Vukicevic S., Lin H.Y. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat. Genet. 2009;41:482–487. doi: 10.1038/ng.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argilés J.M., Stemmler B., López-Soriano F.J., Busquets S. Inter-tissue communication in cancer cachexia. Nat. Rev. Endocrinol. 2018;15:9–20. doi: 10.1038/s41574-018-0123-0. [DOI] [PubMed] [Google Scholar]
- Atwell S., Ridgway J.B., Wells J.A., Carter P. Stable heterodimers from remodeling the domain interface of a homodimer using a phage display library. J. Mol. Biol. 1997;270:26–35. doi: 10.1006/jmbi.1997.1116. [DOI] [PubMed] [Google Scholar]
- Bautista A.C., Salimi-Moosavi H., Jawa V. Universal immunoassay applied during early development of large molecules to understand impact of immunogenicity on biotherapeutic exposure. AAPS J. 2012;14:843–849. doi: 10.1208/s12248-012-9403-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt C., Hansen R.H., Hansen J.B., Olsen C.H., Galle P., Mandrup-Poulsen T., Gehl J., Pedersen B.K., Hojman P. Over-expression of Follistatin-like 3 attenuates fat accumulation and improves insulin sensitivity in mice. Metabolism. 2015;64:283–295. doi: 10.1016/j.metabol.2014.10.007. [DOI] [PubMed] [Google Scholar]
- Cadena S.M., Tomkinson K.N., Monnell T.E., Spaits M.S., Kumar R., Underwood K.W., Pearsall R.S., Lachey J.L. Administration of a soluble activin type IIB receptor promotes skeletal muscle growth independent of fiber type. J. Appl. Physiol. 2010;109:635–642. doi: 10.1152/japplphysiol.00866.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C., McMillan H.J., Mah J.K., Tarnopolsky M., Selby K., McClure T., Wilson D.M., Sherman M.L., Escolar D., Attie K.M. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: results of a randomized, placebo-controlled clinical trial. Muscle Nerve. 2017;55:458–464. doi: 10.1002/mus.25268. [DOI] [PubMed] [Google Scholar]
- Cash J.N., Angerman E.B., Kattamuri C., Nolan K., Zhao H., Sidis Y., Keutmann H.T., Thompson T.B. Structure of myostatin.follistatin-like 3: N-terminal domains of follistatin-type molecules exhibit alternate modes of binding. J. Biol. Chem. 2012;287:1043–1053. doi: 10.1074/jbc.M111.270801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castonguay R., Lachey J., Wallner S., Strand J., Liharska K., Watanabe A.E., Cannell M., Davies M.V., Sako D., Troy M.E. Follistatin-288-Fc fusion protein promotes localized growth of skeletal muscle. J. Pharmacol. Exp. Ther. 2019;368:435–445. doi: 10.1124/jpet.118.252304. [DOI] [PubMed] [Google Scholar]
- Chang C. Agonists and antagonists of TGF-β family ligands. Cold Spring Harb. Perspect. Biol. 2016;8:a021923. doi: 10.1101/cshperspect.a021923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C.S., Peekhaus N., Weber H., Adamski S., Murray E.M., Zhang H.Z., Zhao J.Z., Ernst R., Lineberger J., Huang L. Increased muscle force production and bone mineral density in ActRIIB-Fc-treated mature rodents. J. Gerontol. A. Biol. Sci. Med. Sci. 2013;68:1181–1192. doi: 10.1093/gerona/glt030. [DOI] [PubMed] [Google Scholar]
- Cohen S., Nathan J.A., Goldberg A.L. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015;14:58–74. doi: 10.1038/nrd4467. [DOI] [PubMed] [Google Scholar]
- Cruz-Jentoft A.J., Sayer A.A. Sarcopenia. Lancet. 2019;393:2636–2646. doi: 10.1016/S0140-6736(19)31138-9. [DOI] [PubMed] [Google Scholar]
- Datta-Mannan A., Croy J.E., Schirtzinger L., Torgerson S., Breyer M., Wroblewski V.J. Aberrant bispecific antibody pharmacokinetics linked to liver sinusoidal endothelium clearance mechanism in cynomolgus monkeys. MAbs. 2016;8:969–982. doi: 10.1080/19420862.2016.1178435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta-Mannan A., Yaden B., Krishnan V., Jones B.E., Croy J.E. An engineered human follistatin variant: insights into the pharmacokinetic and pharmocodynamic relationships of a novel molecule with broad therapeutic potential. J. Pharmacol. Exp. Ther. 2013;344:616–623. doi: 10.1124/jpet.112.201491. [DOI] [PubMed] [Google Scholar]
- Egerman M.A., Cadena S.M., Gilbert J.A., Meyer A., Nelson H.N., Swalley S.E., Mallozzi C., Jacobi C., Jennings L.L., Clay I. GDF11 increases with age and inhibits skeletal muscle regeneration. Cell Metab. 2015;22:164–174. doi: 10.1016/j.cmet.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenaux P., Platzbecker U., Mufti G.J., Garcia-Manero G., Buckstein R., Santini V., Díez-Campelo M., Finelli C., Cazzola M., Ilhan O. Luspatercept in patients with lower-risk myelodysplastic syndromes. N. Engl. J. Med. 2020;382:140–151. doi: 10.1056/NEJMoa1908892. [DOI] [PubMed] [Google Scholar]
- Furrer R., Handschin C. Muscle wasting diseases: novel targets and treatments. Annu. Rev. Pharmacol. Toxicol. 2019;59:315–339. doi: 10.1146/annurev-pharmtox-010818-021041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasser C.E., Gartner M.R., Wilson D., Miller B., Sherman M.L., Attie K.M. Locally acting ACE-083 increases muscle volume in healthy volunteers. Muscle Nerve. 2018;57:921–926. doi: 10.1002/mus.26113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo T., Jou W., Chanturiya T., Portas J., Gavrilova O., McPherron A.C. Myostatin inhibition in muscle, but not adipose tissue, decreases fat mass and improves insulin sensitivity. PLoS One. 2009;4:e4937. doi: 10.1371/journal.pone.0004937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada M., Morikawa M., Ozawa T., Kobayashi M., Tamura Y., Takahashi K., Tanabe M., Tada K., Seto Y., Miyazono K. Palbociclib enhances activin-SMAD-induced cytostasis in estrogen receptor-positive breast cancer. Cancer Sci. 2019;110:209–220. doi: 10.1111/cas.13841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon E.B., Apelqvist Å.A., Smart N.G., Gu X., Osborne D.H., Kim S.K. GDF11 modulates NGN3+ islet progenitor cell number and promotes β-cell differentiation in pancreas development. Development. 2004;131:6163–6174. doi: 10.1242/dev.01535. [DOI] [PubMed] [Google Scholar]
- Hill J.J., Davies M.V., Pearson A.A., Wang J.H., Hewick R.M., Wolfman N.M., Qiu Y. The myostatin propeptide and the follistatin-related gene are inhibitory binding proteins of myostatin in normal serum. J. Biol. Chem. 2002;277:40735–40741. doi: 10.1074/jbc.M206379200. [DOI] [PubMed] [Google Scholar]
- Iskenderian A., Liu N., Deng Q., Huang Y., Shen C., Palmieri K., Crooker R., Lundberg D., Kastrapeli N., Pescatore B. Myostatin and activin blockade by engineered follistatin results in hypertrophy and improves dystrophic pathology in mdx mouse more than myostatin blockade alone. Skelet. Muscle. 2018;8:34. doi: 10.1186/s13395-018-0180-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson M.L., Robinson M.M., Nair K.S. Skeletal muscle aging and the mitochondrion. Trends Endocrinol. Metab. 2013;24:247–256. doi: 10.1016/j.tem.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasturirangan S., Rainey G.J., Xu L., Wang X., Portnoff A., Chen T., Fazenbaker C., Zhong H., Bee J., Zeng Z. Targeted Fcγ receptor (FcγR)-mediated clearance by a biparatopic bispecific antibody. J. Biol. Chem. 2017;292:4361–4370. doi: 10.1074/jbc.M116.770628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota J., Handy C.R., Haidet A.M., Montgomery C.L., Eagle A., Rodino-Klapac L.R., Tucker D., Shilling C.J., Therlfall W.R., Walker C.M. Follistatin gene delivery enhances muscle growth and strength in nonhuman primates. Sci. Transl. Med. 2009;1:6ra15. doi: 10.1126/scitranslmed.3000112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latres E., Mastaitis J., Fury W., Miloscio L., Trejos J., Pangilinan J., Okamoto H., Cavino K., Na E., Papatheodorou A. Activin A more prominently regulates muscle mass in primates than does GDF8. Nat. Commun. 2017;8:15153. doi: 10.1038/ncomms15153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le V.Q., Iacob R.E., Tian Y., McConaughy W., Jackson J., Su Y., Zhao B., Engen J.R., Pirruccello-Straub M., Springer T.A. Tolloid cleavage activates latent GDF8 by priming the pro-complex for dissociation. EMBO J. 2018;37:384–397. doi: 10.15252/embj.201797931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.H., Jun H.S. Role of myokines in regulating skeletal muscle mass and function. Front. Physiol. 2019;10:42. doi: 10.3389/fphys.2019.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.J., Reed L.A., Davies M.V., Girgenrath S., Goad M.E., Tomkinson K.N., Wright J.F., Barker C., Ehrmantraut G., Holmstrom J. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc. Natl. Acad. Sci. U S A. 2005;102:18117–18122. doi: 10.1073/pnas.0505996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Fredericks M., Cannell M., Wang K., Sako D., Maguire M.C., Grenha R., Liharska K., Krishnan L., Bloom T. ActRIIB:ALK4-Fc alleviates muscle dysfunction and comorbidities in murine models of neuromuscular disorders. J. Clin. Invest. 2021;131:e138634. doi: 10.1172/JCI138634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodberg A., van der Eerden B.C.J., Boers-Sijmons B., Thomsen J.S., Bruel A., van Leeuwen J., Eijken M. A follistatin-based molecule increases muscle and bone mass without affecting the red blood cell count in mice. FASEB J. 2019;33:6001–6010. doi: 10.1096/fj.201801969RR. [DOI] [PubMed] [Google Scholar]
- Marini K.D., Croucher D.R., McCloy R.A., Vaghjiani V., Gonzalez-Rajal A., Hastings J.F., Chin V., Szczepny A., Kostyrko K., Marquez C. Inhibition of activin signaling in lung adenocarcinoma increases the therapeutic index of platinum chemotherapy. Sci. Transl. Med. 2018;10:aat3504. doi: 10.1126/scitranslmed.aat3504. [DOI] [PubMed] [Google Scholar]
- Marty E., Liu Y., Samuel A., Or O., Lane J. A review of sarcopenia: enhancing awareness of an increasingly prevalent disease. Bone. 2017;105:276–286. doi: 10.1016/j.bone.2017.09.008. [DOI] [PubMed] [Google Scholar]
- McPherron A.C., Lawler A.M., Lee S.J. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Meynard D., Kautz L., Darnaud V., Canonne-Hergaux F., Coppin H., Roth M.P. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat. Genet. 2009;41:478–481. doi: 10.1038/ng.320. [DOI] [PubMed] [Google Scholar]
- Morikawa M., Derynck R., Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016;8:a021873. doi: 10.1101/cshperspect.a021873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa M., Koinuma D., Tsutsumi S., Vasilaki E., Kanki Y., Heldin C.H., Aburatani H., Miyazono K. ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Res. 2011;39:8712–8727. doi: 10.1093/nar/gkr572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa M., Mitani Y., Holmborn K., Kato T., Koinuma D., Maruyama J., Vasilaki E., Sawada H., Kobayashi M., Ozawa T. The ALK-1/SMAD/ATOH8 axis attenuates hypoxic responses and protects against the development of pulmonary arterial hypertension. Sci. Signal. 2019;12:aay4430. doi: 10.1126/scisignal.aay4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A., Sidis Y., Mahan A., Raher M.J., Xia Y., Rosen E.D., Bloch K.D., Thomas M.K., Schneyer A.L. FSTL3 deletion reveals roles for TGF-β family ligands in glucose and fat homeostasis in adults. Proc. Natl. Acad. Sci. U S A. 2007;104:1348–1353. doi: 10.1073/pnas.0607966104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojima C., Noguchi Y., Miyamoto T., Saito Y., Orihashi H., Yoshimatsu Y., Watabe T., Takayama K., Hayashi Y., Itoh F. Peptide-2 from mouse myostatin precursor protein alleviates muscle wasting in cancer-associated cachexia. Cancer Sci. 2020;111:2954–2964. doi: 10.1111/cas.14520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearsall R.S., Davies M.V., Cannell M., Li J., Widrick J., Mulivor A.W., Wallner S., Troy M.E., Spaits M., Liharska K. Follistatin-based ligand trap ACE-083 induces localized hypertrophy of skeletal muscle with functional improvement in models of neuromuscular disease. Sci. Rep. 2019;9:11392. doi: 10.1038/s41598-019-47818-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistilli E.E., Bogdanovich S., Goncalves M.D., Ahima R.S., Lachey J., Seehra J., Khurana T. Targeting the activin type IIB receptor to improve muscle mass and function in the mdx mouse model of Duchenne muscular dystrophy. Am. J. Pathol. 2011;178:1287–1297. doi: 10.1016/j.ajpath.2010.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath T., Baker K., Dumont J.A., Peters R.T., Jiang H., Qiao S.W., Lencer W.I., Pierce G.F., Blumberg R.S. Fc-fusion proteins and FcRn: structural insights for longer-lasting and more effective therapeutics. Crit. Rev. Biotechnol. 2015;35:235–254. doi: 10.3109/07388551.2013.834293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridgway J.B., Presta L.G., Carter P. Knobs-into-holes' engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9:617–621. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- Roh J.D., Hobson R., Chaudhari V., Quintero P., Yeri A., Benson M., Xiao C., Zlotoff D., Bezzerides V., Houstis N. Activin type II receptor signaling in cardiac aging and heart failure. Sci. Transl. Med. 2019;11:aau8680. doi: 10.1126/scitranslmed.aau8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuelke M., Wagner K.R., Stolz L.E., Hubner C., Riebel T., Komen W., Braun T., Tobin J.F., Lee S.J. Myostatin mutation associated with gross muscle hypertrophy in a child. N. Engl. J. Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- Sidis Y., Mukherjee A., Keutmann H., Delbaere A., Sadatsuki M., Schneyer A. Biological activity of follistatin isoforms and follistatin-like-3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology. 2006;147:3586–3597. doi: 10.1210/en.2006-0089. [DOI] [PubMed] [Google Scholar]
- Stamler R., Keutmann H.T., Sidis Y., Kattamuri C., Schneyer A., Thompson T.B. The structure of FSTL3.activin A complex. Differential binding of N-terminal domains influences follistatin-type antagonist specificity. J. Biol. Chem. 2008;283:32831–32838. doi: 10.1074/jbc.M801266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suragani R.N., Cadena S.M., Cawley S.M., Sako D., Mitchell D., Li R., Davies M.V., Alexander M.J., Devine M., Loveday K.S. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat. Med. 2014;20:408–414. doi: 10.1038/nm.3512. [DOI] [PubMed] [Google Scholar]
- Suzuki Y., Montagne K., Nishihara A., Watabe T., Miyazono K. BMPs promote proliferation and migration of endothelial cells via stimulation of VEGF-A/VEGFR2 and angiopoietin-1/Tie2 signalling. J. Biochem. 2008;143:199–206. doi: 10.1093/jb/mvm215. [DOI] [PubMed] [Google Scholar]
- Tang R., Harasymowicz N.S., Wu C.L., Collins K.H., Choi Y.R., Oswald S.J., Guilak F. Gene therapy for follistatin mitigates systemic metabolic inflammation and post-traumatic arthritis in high-fat diet-induced obesity. Sci. Adv. 2020;6:eaaz7492. doi: 10.1126/sciadv.aaz7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker R.G., Czepnik M., Goebel E.J., McCoy J.C., Vujic A., Cho M., Oh J., Aykul S., Walton K.L., Schang G. Structural basis for potency differences between GDF8 and GDF11. BMC Biol. 2017;15:19. doi: 10.1186/s12915-017-0350-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weflen A.W., Baier N., Tang Q.J., Van den Hof M., Blumberg R.S., Lencer W.I., Massol R.H. Multivalent immune complexes divert FcRn to lysosomes by exclusion from recycling sorting tubules. Mol. Biol. Cell. 2013;24:2398–2405. doi: 10.1091/mbc.E13-04-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Weisshaar N., Hotz-Wagenblatt A., Madi A., Ma S., Mieg A., Hering M., Mohr K., Schlimbach T., Borgers H. Skeletal muscle antagonizes antiviral CD8+ T cell exhaustion. Sci. Adv. 2020;6:eaba3458. doi: 10.1126/sciadv.aba3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Swerlick R.A., Sepp N., Bosse D., Ades E.W., Lawley T.J. Characterization of expression and modulation of cell adhesion molecules on an immortalized human dermal microvascular endothelial cell line (HMEC-1) J. Invest. Dermatol. 1994;102:833–837. doi: 10.1111/1523-1747.ep12382086. [DOI] [PubMed] [Google Scholar]
- Yaden B.C., Croy J.E., Wang Y., Wilson J.M., Datta-Mannan A., Shetler P., Milner A., Bryant H.U., Andrews J., Dai G. Follistatin: a novel therapeutic for the improvement of muscle regeneration. J. Pharmacol. Exp. Ther. 2014;349:355–371. doi: 10.1124/jpet.113.211169. [DOI] [PubMed] [Google Scholar]
- Yamawaki K., Ueda S., Okada T., Oshima T., Kakitani M., Kato T., Tomizuka K. Adult-specific systemic over-expression reveals novel in vivo effects of the soluble forms of ActRIIA, ActRIIB and BMPRII. PLoS One. 2013;8:e78076. doi: 10.1371/journal.pone.0078076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisberg M., Bottiglio C., Kumar N., Maeshima Y., Strutz F., Muller G.A., Kalluri R. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am. J. Physiol. Ren. Physiol. 2003;285:F1060–F1067. doi: 10.1152/ajprenal.00191.2002. [DOI] [PubMed] [Google Scholar]
- Zhou X., Wang J.L., Lu J., Song Y., Kwak K.S., Jiao Q., Rosenfeld R., Chen Q., Boone T., Simonet W.S. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531–543. doi: 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- Zimmers T.A., Davies M.V., Koniaris L.G., Haynes P., Esquela A.F., Tomkinson K.N., McPherron A.C., Wolfman N.M., Lee S.J. Induction of cachexia in mice by systemically administered myostatin. Science. 2002;296:1486–1488. doi: 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze datasets or code.