

Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder etiologically linked to the loss of substantia nigra (SN) dopaminergic neurons in the mid-brain. The etiopathology of sporadic PD is still unclear; however, the interaction of extrinsic and intrinsic factors may play a critical role in the onset and progression of the disease. Studies in animal models and human post-mortem tissue have identified distinct cellular and molecular changes in the diseased brain, suggesting complex interactions between different glial cell types and various molecular pathways. Small changes in the expression of specific genes in a single pathway or cell type possibly influence others at the cellular and system levels. These molecular and cellular signatures like neuroinflammation, oxidative stress, and autophagy have been observed in PD patients’ brain tissue. While the etiopathology of PD is still poorly understood, the interplay between glial cells and molecular events may play a crucial role in disease onset and progression.
Keywords: Autophagy, Neuroinflammation, Neuron, Oxidative damage, Parkinson’s disease, Substantia nigra
Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disease, clinically defined as a movement disorder. The pathological hallmarks of PD are the degeneration of dopamine-producing neurons in the ventral midbrain substantia nigra (SN) and associated widespread intraneuronal α-synuclein aggregation in Lewy bodies (Braak et al. 2003; Maiti et al. 2017). As the disease progresses, the loss of SN dopaminergic neurons results in the typical motor symptoms, such as bradykinesia, rigidity, impaired postural balance, and a characteristic resting tremor. PD patients develop dementia as the disease progresses (Goetz 2011; Moustafa et al. 2016). In addition, prodromal symptoms have been reported by PD patients years before the onset of disease. Some of these signs include constipation, insomnia, mood disorders, depression and anxiety (Mahlknecht et al. 2015). The etiopathology of sporadic PD is still unknown; however, studies suggest that the interaction of extrinsic and intrinsic factors, such as exposure to environmental toxins and specific immune responses, may trigger disease onset (Sulzer 2007). Moreover, according to Braak’s hypothesis, the idiopathic incidence of PD may originate outside the CNS (most probably in the enteric nervous system) while “the stereotypic topographic expansion pattern of the lesions may resemble that of a falling row of dominos” (Braak et al. 2003). The α-synuclein aggregation has recently been demonstrated to begin in the enteric nervous system and propagates to the brain via the vagus nerve (Kim et al. 2019).
PD brain analysis suggests that multiple pathways may be responsible for the death of nigral neurons. An electron microscopic observation of PD brain samples suggested apoptotic, necrotic, and autophagic cell death of nigral dopaminergic neurons (Anglade et al. 1997). The presence of an autophagosomal marker, the microtubule-associated protein 1A/1B-light chain 3 (LC3), in the Lewy bodies of the PD patients supports the active role of autophagy in this disease (Tanji et al. 2013). Beclin-1, an autophagy regulator protein, is also increased in PD brains (Spencer et al. 2009). Additionally, the aggregated form of α-synuclein, which is a predominant component of Lewy bodies, may be the result of dysfunctional autophagy (Arotcarena et al. 2019). Interestingly, in the post-mortem PD brain, observed microglial activation is correlated with the presence of this aggregated α-synuclein form (Croisier et al. 2005). The active participation of inflammation in PD is seen by the presence of cytokines in PD brains (Nagatsu et al. 2000) and the presence of CD4 + T cells in PD animal models (Haque et al. 2020; Samantaray et al. 2015). Aggregated α-synuclein also induces reactive oxygen species (ROS) production in the cells (Reeve et al. 2015), and oxidative stress can exacerbate the α-synuclein aggregation (Glick et al. 2010; Scudamore and Ciossek 2018). Thus, the detection of multiple deleterious pathways in PD brains and associated animal models suggest that these pathways may interact with each other in disease initiation and progression.
Therefore, recent studies have focused on ascertaining which cellular pathways lead to the onset of pathological changes in specific brain regions. Experimental studies in animal models and human post-mortem tissues have recognized some key players in the progression of PD. Therefore, one specific event may be required to initiate cell death (primary pathway), which leads to the involvement of other secondary pathways, much like the “falling row of dominos”. Significant participants in the central nervous system (CNS) include neurons, astrocytes, microglia, oligodendrocytes, vessel-associated cells, resident innate immune cells, and immune cells from the peripheral system. A complex network of crosstalk between these cells together with cytokines and chemokines supports function of the normal brain. However, any subtle changes of extrinsic or intrinsic factors in the surrounding environment may disrupt regular interaction. Increasing numbers of studies are suggesting the critical role of neuroinflammation in neurodegenerative diseases. There is a delicate molecular balance during the neuroinflammatory process, and glial cells can help prevent or repair the damage caused by initial insults (Fig. 1). Activation of glial cells together with signals from infiltrating immune cells can contribute to pathophysiological changes in neurodegenerative diseases. Thus, it is becoming increasingly clear that a complex communication network between neurons, glia, and immune cells may play a detrimental role in the disease process.
Fig. 1.
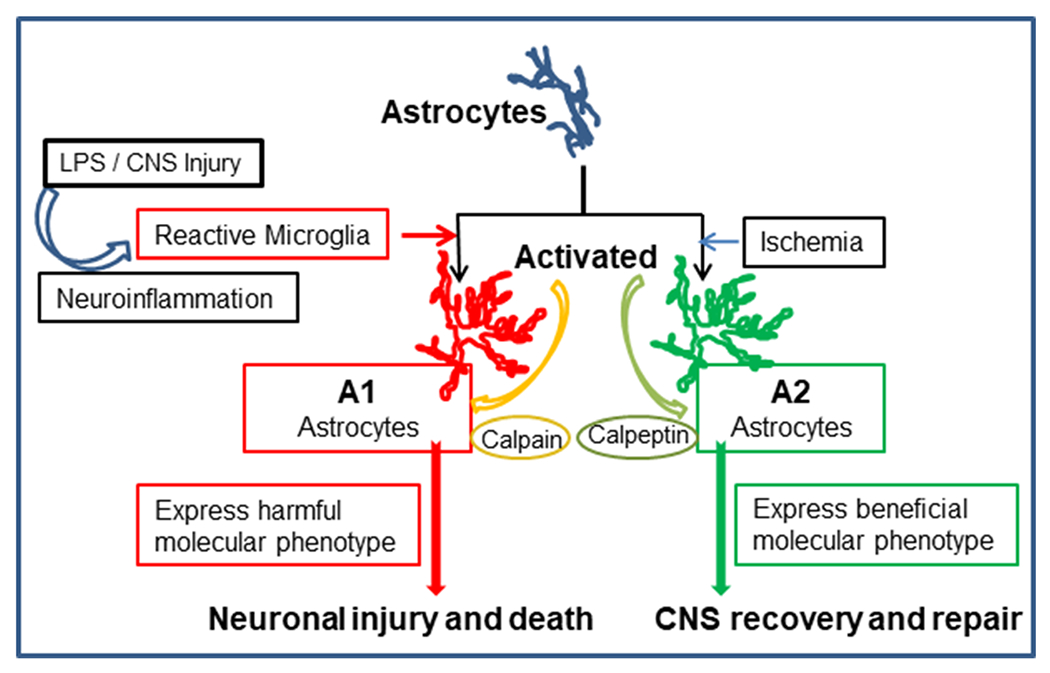
Schematic representation of astrocyte activation and differentiation following CNS injury by different types of stimuli. Microglia (triggered by different stimuli) contribute to activation of astrocytes. Depending on the micro-environment, astrocytic phenotypic differentiation may result in A1 astrocytes, which contribute to inflammation and neuronal death. However, A2 astrocytes can promote neuronal survival and recovery of function. Calpain activity may facilitate A1 astrocyte differentiation and resulting neuronal injury. However, calpain inhibition by calpeptin may contribute to neuronal survival, CNS recovery and repair through A2 astrocytic activity
Beyond sporadic PD, there are known genetic risk factors involved in initiating Parkinsonian progression (Nuytemans et al. 2010). Well defined mutations include: Leucine-Rich Repeat Kinase 2 (LRRK2), Protein DJ-1 (DJ-1), α-synuclein (SNCA), Ubiquitin C-Terminal Hydrolase L1 (UCHL1), PTEN-Induced Putative Kinase 1 (PINK1), and Parkin (PRKN) (Abeliovich and Gitler 2016; Jin and Youle 2012; Jones 2010; Liu et al. 2017; Marongiu et al. 2009; Nuytemans et al. 2010). Familial PD (autosomal dominant or recessive inheritance of the genes) patients represent only 5–10 % of PD cases. Moreover, the disease onset and progression are heterogeneous (Martinez and Peplow 2017), suggesting several subtypes of PD may exist. The critical role of mitochondria in PD is demonstrated in these known mutations, which result in mitochondrial damage and ROS formation (Klein and Westenberger 2012; Selvaraj and Piramanayagam 2019). As discussed below, animal models of PD with these mutations are valuable to aid understanding of the underlying cellular and molecular disease mechanism(s) in other non-familial cases.
Neuroinflammation
Microglia
The role of microglia in neuroinflammation has been studied in neurodegenerative diseases such as PD, Amyotrophic lateral sclerosis (ALS), and Alzheimer’s disease (AD) due to their activated presence at lesion sites in diseased patients’ brains (McGeer et al. 1988a, b). McGeer and collaborators (McGeer et al. 1988a, b) detected reactive microglia in the SN of PD and AD patients; they suggested that the presence of microglia is a sensitive indicator of pathological changes in these neurodegenerative disorders. Histological evaluation of PD patient brains also detected activated microglia and reactive astrocytes (Kam et al. 2020), suggesting neuroinflammation may be a critical factor in the disease process (Lecours et al. 2018; Long-Smith et al. 2009).
Microglia and astrocytes are known to play crucial roles in maintaining brain homeostasis (Barres 2008; Colombo and Farina 2016; Gertig and Hanisch 2014; Heithoff et al. 2021; Khakh and Sofroniew 2015; Linnerbauer et al. 2020; Paolicelli et al. 2011); alterations in glial cell activation status caused by peripheral events may eventually lead to pathological changes in the CNS. Microglia are resident innate immune cells (initial responders in the CNS) derived from the primitive hematopoietic progenitors in the yolk sac on embryonic day 8; by embryonic day 10.5, these cells colonize the developing brain (Alliot et al. 1999). Later, microglia maintain their region-specific density by self-renewal (Ajami et al. 2007; Li and Barres 2018). Microglia appear in the brain before other glial cells (astrocytes and oligodendrocytes) and neurons (Kriegstein and Alvarez-Buylla 2009), suggesting that they could play essential functions in the brain during development. They also perform immune surveillance (Nimmerjahn et al. 2005) and respond to CNS pathogens and injuries. Studies have shown that microglia actively prune synapses in the developing brain, playing a prominent role in maintaining brain homeostasis during development (Weinhard et al. 2018). A recent study by Socodato and collaborators observed synaptic pruning in adult mice following alcohol intoxication (Socodato et al. 2020), leading to anxiety-like behavior. Similarly, the regulation of neurogenesis in the adult hippocampus indicates microglial involvement in maintaining brain homeostasis later in life (De Lucia et al. 2016; Diaz-Aparicio et al. 2020; Frost and Schafer 2016; Sato 2015).
Associated with the diverse array of neurons (Lein et al. 2007), microglial populations in the brain are also heterogeneous (Li et al. 2019), and the SN (characteristically affected in PD) is densely populated by microglia (Lawson et al. 1990). Like neurons, microglial populations also demonstrate distinct region-dependent molecular identity (Grabert et al. 2016) suggesting distinct functions in different brain areas. Age and chronic stress are believed to induce morphological and functional changes (Lecours et al. 2018). Microglia and estradiol actively participate in the pre-optic area during the critical perinatal development period for sexual differentiation in male mice (Lenz et al. 2013). Transcriptome analyses in young adult male/female mice revealed the differentially expressed genes in microglia isolated from female mice compared to male mice (Villa et al. 2018). This study found that genes associated with inflammatory processes are more highly expressed in male mice. Contrary to this, females differentially expressed genes associated with inhibiting inflammatory responses and more robust repair mechanisms. The transcription factor driving the differentially expressed genes in microglia is the nuclear factor κB (NF-κB). This sex-specific difference highlights the role of microglia since PD is more prevalent in males than females by a 2:1 ratio, respectively (Cerri et al. 2019; Hirsch et al. 2016; Picillo et al. 2017). Recent studies have likewise demonstrated neuroprotective properties of low-dose estrogen treatment in rats following spinal cord injury (Cox et al. 2021). Wu and collaborators demonstrated sex- and age-specific differences in SN microglia density in mice (Wu et al. 2016). Moreover, they showed the association of activated microglia with SN dopaminergic neurons, where the high density of activated microglia leads to fewer SN neurons. Since microglia express estrogen receptors (Sierra et al. 2008; Thakkar et al. 2018; Wu et al. 2016), neuroprotective estrogen may inhibit inflammatory responses in microglia reducing the incidence of PD in women.
Regional variations in microglial phenotypes and functions indicate they are versatile cell types, regulated in part by the local microenvironment (De Biase et al. 2017; Lenz et al. 2013). The transcriptome analysis by Villa et al. (Villa et al. 2018) showed that in addition to having a neuroprotective phenotype, microglia from female mice express fewer inflammatory markers than those from male mice. In addition, De Biase et al. (De Biase et al. 2017) demonstrated a regional variation in microglial phenotypes in basal ganglia associated with local factors. A dense population of microglia in SN (Kim et al. 2000) may predispose this brain region to immune challenge. Since basal ganglia are specifically affected in PD, this variation in microglial phenotype requires more attention to further evaluate its functional role in normal and disease states.
Microglial activity is commonly categorized by proinflammatory (M1) and antiinflammatory (M2) phenotypical characteristics (Fig. 2a). The presence of neurotoxic M1 microglia at the site of neurodegeneration suggests this phenotype may have replaced the neuroprotective M2-type microglia, or transformed from M2 to M1 during the disease process (Tang and Le 2016). Since the presence of local cues plays a critical role in regulating microglial phenotypes, the presence of aggregated α-synuclein, amyloid beta (Aβ) or other unknown stimuli may promote specific microglial phenotypical expression (Ferreira and Romero-Ramos 2018; Sala Frigerio et al. 2019). Moreover, the high density of microglia in SN may produce an abundance of proinflammatory cytokines/chemokines, leading to the degeneration of nigral neurons in PD. Although incompletely understood, neuroinflammation influenced by microglia may be a significant contributing factor in PD etiology.
Fig. 2.
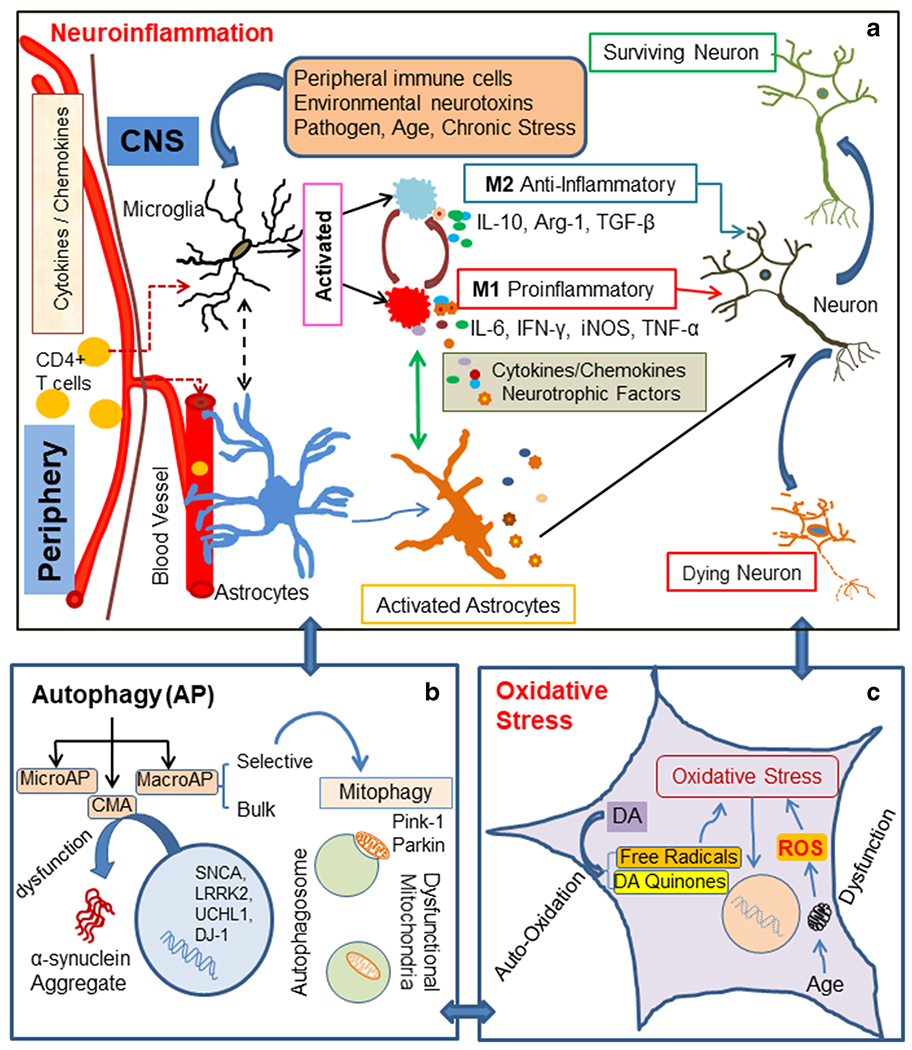
The diagram depicts several pathways involved in PD pathophysiology, a Neuroinflammation – Microglia are resident immune cells activated by various stimuli, e.g., environmental neurotoxins, pathogens, peripheral inflammation (CD4 + T cells), age, and chronic stress. These stimuli may promote divergent microglial phenotypes such as M1, a proinflammatory phenotype, which can be toxic to neurons. M1-type microglia secrete proinflammatory cytokines such as IL-1β, IL-6, IFN-γ, TNF-α, complement proteins, inducible nitric oxide synthase (iNOS), and reactive oxygen species (ROS). M2, an antiinflammatory phenotype microglia, is believed to be neuroprotective. They secrete cytokines and growth factors such as IL-10, TGF-β, brain-derived neurotrophic factor (BDNF), and arginase-1 (Arg-1). Crosstalk between microglia and astrocytes may promote a time dependent secretion of cytokines/growth factors, contributing to PD pathological conditions, b Autophagy - Neuroinflammation influences all three types of autophagy: (i) macroautophagy (MacroAP), (ii) microautophagy (MicroAP), and (iii) chaperon mediated autophagy (CMA). Genetic mutations may contribute to dysfunction of CMA, leading to α-synuclein aggregation. Mitophagy, a selective autophagy process, can clear dysfunctional mitochondria. Pink1 and Parkin maintain mitochondrial cytoarchitecture and function; they also regulate mitophagy. c Oxidative stress - Autooxidation of dopamine (DA) in dopaminergic neurons generates free radicals and DA quinone. These free radicals lead to oxidative stress. Associated with aging and specific toxins, mitochondrial dysfunction causes the generation of ROS, which ultimately results in oxidative cellular damage
A significant role of microglia in the pathogenesis of PD is also evident in PD-linked mutations. As mentioned earlier, there are well-defined genetic mutations, SNCA, LRRK2, PRKN, DJ-1, UCHL1, and PINK1, which are involved in PD. The SNCA gene encodes α-synuclein, and missense mutations and multiplications of this gene lead to the development of PD (Ibanez et al. 2009; Singleton et al. 2003). These mutations can activate microglia (Kam et al. 2020) because of the complex interplay between microglia and α-synuclein (Choi et al. 2020; Ferreira and Romero-Ramos 2018; Wang et al. 2015). Activated microglia are predominantly involved in the clearance of excess α-synuclein in the cytoplasm, α-synuclein also activates microglia, causing neuronal toxicity by production of ROS (Jin et al. 2007; Zhang et al. 2005). Furthermore, α-synuclein reduces microglial phagocytosis, leading to impaired clearance of aggregated proteins or cellular debris (Choi et al. 2015). A recent study showed that knockdown of LRRK2 inhibited microglial proinflammatory responses in cell culture model (Daher et al. 2014). SN neurons were also found to be protected in LRRK2 knockout mice following lipopolysaccharides (LPS) induced neuroinflammation as well as adeno-associated virus-mediated transduction of human α-synuclein. Interestingly, these studies also showed that under normal conditions, the LRRK2 expression was undetectable in the mid-brain of wild-type mice. However, its expression is increased in inducible nitric oxide synthase (iNOS)-positive myeloid cells in the SN neurons following overexpression of α-synuclein or exposure to LPS. These findings support the role of LRRK2 in the clearance of α-synuclein and activation of microglia. Similarly, the effect of PRKN mutation on microglia was detected in PARKIN−/−murine glial culture (Solano et al. 2008). It supported the microglial population more than the astrocytes when compared with wild-type cultures. Another cell culture study with PINK1 mutation demonstrated reduced proinflammatory and antiinflammatory cytokine production in microglia following LPS/interferon-gamma (IFN-γ) stimulation. These findings in animal models with genetic mutations and in cell culture studies strongly indicate that microglia play a critical role in the pathogenesis of PD.
Astroglia
Along with microglia, astrocytes also contribute to neurodegenerative diseases (Teismann and Schulz 2004). Glial fibrillary acidic protein (GFAP), an astrocytic marker was isolated from demyelinated multiple sclerosis (MS) plaques (Eng et al. 2000). Astrocytes are the most abundant glial cell types in the brain (Miller 2018). Based on their location and morphological features, protoplasmic and fibrous astrocytes are located in the grey and white matter, respectively (Miller 2018). The highly ramified protoplasmic astrocytes maintain integrity of the blood-brain barrier (BBB) (Alvarez et al. 2013; Cabezas et al. 2014; Heithoff et al. 2021; Obermeier et al. 2013). An increased level of proinflammatory cytokines changes BBB permeability (Wong et al. 2004). The communication changes between astrocytes and blood vessels induce astrocytes to become reactive (Alvarez et al. 2013). Moreover, studies have localized the aggregation of α-synuclein only in protoplasmic astrocytes, making these astrocytes relevant to the onset and progression of PD (Braak et al. 2006; Halliday and Stevens 2011). Astrocytes are also a critical source of specific factors required for neuronal differentiation and survival (Allen and Eroglu 2017; Barreto et al. 2011; Christopherson et al. 2005; Chung et al. 2015; Dringen 2000; Molofsky et al. 2014). Glial cell-line derived neurotrophic factor (GDNF) secreted by astrocytes promotes survival of SN neurons (Sariola and Saarma 2003; Yasuda and Mochizuki 2010). Indeed, the maintenance of SN neurons is a crucial function of astrocytes, since a decreased level of GDNF may lead to SN neuronal death and ultimately Parkinsonian symptoms (Betarbet et al. 2000). These critical functions of astrocytes indicate they could be actively involved in the pathogenesis of PD. Notably, a subpopulation of astrocytes in basal ganglia plays circuit-specific roles and could possibly regulate the striatal function (Martin et al. 2015). Further studies of this sub-population of astrocytes may provide insights into the vulnerability of basal ganglia and the resulting development of neurodegenerative diseases.
Recent genomic and transcriptomic studies suggest that astrocytes function in a highly context-dependent manner. Moreover, there are different astrocytic subpopulations, which can either support the ongoing disease process or suppress it (Bayraktar et al. 2020; Liddelow and Barres 2017; Liddelow et al. 2017; Molofsky et al. 2014; Wheeler and Quintana 2019); Lin et al. 2017; Rothhammer et al. 2016; Wheeler et al. 2020). Like microglia, two astrocyte phenotypes have been detected in the brain (Fig. 1), A1 and A2, and they are associated with neuroinflammation and ischemia, respectively (Liddelow et al. 2017). Liddelow et al. (2017) suggested that A1 astrocytes may be harmful, because they up-regulate classical complement cascade genes which damage synapses. In contrast, A2 astrocytes up-regulate many neurotrophic factors, which support neuronal growth and survival in the developing and mature brain. This study showed that activated microglia secrete proinflammatory cytokines interleukin 1 α (IL-1 α), tumor necrosis factor (TNF), and complement component 1, subcomponent q (C1q). These factors promoted A1 phenotypical astrocytes in the CNS after injury (Fig. 1). Other intrinsic and extrinsic factors also determine the fate of astrocytes following CNS insult. Some of these factors are age and sex dependent (Johnson et al. 2008). Both astrocytes and microglia are shown to release different cytokines and other inflammatory mediators during CNS inflammation (Shields et al. 2020; Tang and Le 2016; Tay et al. 2017; Yang and Zhou 2019). As depicted in Fig. 1, reactive astrocytes detected in neurodegenerative diseases are induced by activated microglia following LPS exposiue. Crosstalk between these glial cells appears to modulate inflammation (Liddelow et al. 2017). Moreover, ongoing molecular events due to oxidative stress, autophagy, or inflammation may influence each other in the neurodegenerative process (Fig. 2a–c). The genetic alterations associated with PD are also expressed in astrocytes, strongly suggesting that glial participation is crucial in the pathogenesis of the disease.
T cells
The CNS is understood to possess immune privilege. Microglia, the innate immune cells of the brain provide immune surveillance and maintenance. Unlike microglia, adaptive immune cells, such as T cells, can participate in both the inflammation and recovery processes in CNS diseases (Luckheeram et al. 2012). Activated T cells can infiltrate the brain and promote neurodegeneration as found in animal models of PD (Kustrimovic et al. 2016). In general, T cells are classified based on their use of CD4 and CD8 co-receptors to bind to major histocompatibility complex (MHC) molecules. CD8 + cytotoxic T cells interact with MHC class I molecules, whereas CD4 + helper T cells interact with and recognize the antigen presented by MHC class II molecules (Huang et al. 2009). These CD4 + T cells are further phenotypically differentiated into specific subtypes: Th1, Th2, Th17, and regulatory T cells (Tregs). Cytokine signaling pathways regulate this differentiation and activate lineage-specific transcription factors and epigenetic modifications (Chabot et al. 2001; Codarri et al. 2011). These CD4 + T cells in the CNS are observed in neurological disorders, especially in autoimmune diseases like MS (Codarri et al. 2011; Komuczki et al. 2019). CD4 + T cells are also detected in human CNS and animal models of PD (Brochard et al. 2009; Haque et al. 2020; Samantaray et al. 2015), AD (Baruch et al. 2015; Dansokho et al. 2016; Monsonego et al. 2003), and stroke (Ito et al. 2019). Their presence in neurological disorders suggests that CD4 + T cells are involved in neuroinflammation. Recent studies have shown that activation of calcium activated neutral protease (calpain) and CD4 + T cells are linked with the pathology of MS, PD, traumatic brain injury (TBI), spinal cord injury (SCI), and optic nerve crush injury, while calpain inhibition attenuates inflammatory T cells and promotes the recovery process (Haque et al. 2020; Hauben and Schwartz 2003; Kipnis et al. 2003; Moalem et al. 1999; Samantaray et al. 2015). Contrary to this, the inflammatory CD4 + T cell population present in ischemia-reperfusion injury and MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) treated mice are harmful to neurons (Brochard et al. 2009; Haque et al. 2020; Hum et al. 2007; Samantaray et al. 2015). Based on evidence in different injury models, the role of T cell sub-populations can be either detrimental or supportive at the injury site, suggesting that the presence of local cues plays a significant role in directing T cell functions (Filiano et al. 2017).
The widely-used PD animal model, 6-hydroxydopamine (6OHDA), shows an increase in microglial MHC II expression (Cicchetti et al. 2002; Fuzzati-Armentero et al. 2019), which may activate CD4 + T cells. A study by Brochard et al. (2009) demonstrated the presence of CD8 + as well as CD4 + T cells in the SN of post-mortem PD patients and mice treated with MPTP. Additionally, CD4 + T cells (not CD8 + T cells) are detrimental to SN neurons in mice treated with MPTP (Brochard et al. 2009). This study showed that CD4 + T cell toxicity was dependent on the Fas/FasL pathway rather than the IFN-γ pathway. Studies performed in our laboratory also observed a distinct subpopulation of CD4 + T cells in animal models of PD (Haque et al. 2020; Samantaray et al. 2015) following MPTP injection. Interestingly, further characterization of CD4 + T cells suggested that the inflammatory CD4 + T cells are granzyme B/perforin expressing cells in MPTP mice, while the regulatory T cell (Tregs) population decreased following MPTP injection. Moreover, the calpain inhibitor calpeptin modulated the activation of granzyme B and perforin producing splenic T cell populations after MPTP treatment, significantly inhibiting this distinct sub-population of CD4 + T cells. Treatment of MPTP mice with calpeptin restored the Treg population. Along with these changes, calpain inhibition protected SN dopaminergic neurons from degeneration (Haque et al. 2020). The mechanisms and pathways related to this CD4 + T cell function in MPTP mice are still under investigation, but suppression of inflammation by calpeptin may reduce the damage to SN neurons following MPTP treatment. Our findings (Haque et al. 2020; Podbielska et al. 2016; Samantaray et al. 2015) suggest that calpain activates microglia, astroglia, and T cells following MPTP treatment. However, calpeptin prevents calpain activation and neuroinflammation with resulting SN dopaminergic neuronal survival (Haque et al. 2020; Samantaray et al. 2015). Thus, calpain may have pathogenic potential in both neuronal cell death and immune cell activation.
Kustrimovic and collaborators (Kustrimovic et al. 2016, 2018) evaluated PD patient blood samples to evaluate the role of peripheral adaptive immunity in PD. They found CD4 + T cells expresses dopaminergic receptors (Kustrimovic et al. 2014), and immature CD4 + T cells from PD patients mostly differentiate into the Th1 lineage, a proinflammatory phenotype (Kustrimovic et al. 2018). CD4 + T cells also produced an increased amount of IFN-γ and TNF-α in PD patients, suggesting the peripheral immune system plays an important role in the disease process.
Oxidative stress
Mitochondria
Aging is one of the leading factors associated with PD and AD. Studies have demonstrated the manifestation of oxidative stress in CNS cells due to ongoing cellular activity (Fig. 2c), supporting a crucial role of the mitochondria in oxidative damage, as seen in PD (Andersen 2004; Blesa et al. 2015; Dias et al. 2013; Guo et al. 2018; Maguire-Zeiss et al. 2005). Any dysfunction of this cell organelle can be detrimental to cell function and viability. SN neurons in PD patients demonstrate specific complex I deficits (Schapira et al. 1990b) associated with mitochondrial dysfunction (Schapira et al. 1990a). MPTP and rotenone are commonly used toxins to create an animal model for PD. These toxins target complex I and damage mid-brain SN dopaminergic neurons (Betarbet et al. 2000; Hoglinger et al. 2005; Martinez and Greenamyre 2012; Testa et al. 2005) and motor neurons in the spinal cord (Samantaray et al. 2007). The extensive arborization of single nigrostriatal dopaminergic neurons (Matsuda et al. 2009) increases the susceptibility of the mitochondria to oxidative damage, due to the high energy demands of these neurons (Ge et al. 2020). Oxidative damage to mitochondria plays a crucial role in the onset and/or progression of PD. Additionally, the pathogenic form of α-synuclein inhibits complex I, causing mitochondrial degeneration (Martin et al. 2006). These studies also indicate that environmental toxins may be a significant contributing factor in sporadic PD (Betarbet et al. 2000; Johnson et al. 2019; Klingelhoefer and Reichmann 2015).
Mutations detected in PD confirm the active participation of mitochondria in PD pathogenesis (Gautier et al. 2008; Ge et al. 2020; Kumar et al. 2020; Marongiu et al. 2009; Mouton-Liger et al. 2017). PINK1 and Parkin are required to maintain mitochondrial integrity (Guo 2012); these proteins also regulate mitophagy (Deas et al. 2011; Jin and Youle 2012). ROS-induced mitochondrial dysfunction is one of the factors associated with aging (Cui et al. 2012), suggesting that mitochondrial functional impairment is detrimental for dopaminergic SN cells; however, SN neuronal loss due to mitochondrial dysfunction is not observed in all PD cases.
Reactive Oxygen Species (ROS)
Oxidative stress occurs due to the imbalance between production of ROS and the availability of antioxidants or radical scavengers (Forrester et al. 2018; Ng et al. 2013; Pizzino et al. 2017) Oxidative stress is an important factor in cell death in response to a variety of pathophysiological conditions (Fig. 2c). Different pathways generate ROS in the brain during routine cellular processes; however, sometimes either overproduction or defective clearance leads to the accumulation of these free radicals (Dias et al. 2013). Mitochondria are significant contributors to ROS production (Cui et al. 2012), but oxidative stress is also reported to result from defective lipid peroxidation (Niki 2008), DNA damage (Narciso et al. 2016), formation of insoluble Parkin aggregates, decreased E3 ligase activity (LaVoie et al. 2007), and possibly α-synuclein aggregation (Scudamore and Ciossek 2018). Dopamine metabolism also results in ROS production, probably contributing to SN dopaminergic neuron vulnerability to oxidative damage (Jenner 2003). Dopamine undergoes auto-oxidation and produces dopamine quinones along with free radicals. Monoamine oxidase (MAO) and catechol O-methyl transferase (COMT) enzymes also participate in dopamine oxidation. MAO-B, which metabolizes dopamine in the cytosol, is localized on the astrocytic outer mitochondrial membrane. Since dopaminergic neurons utilize dopamine, the dopamine quinone formation may lead to dysfunction and ultimately death of these neurons (Burbulla et al. 2017; Miyazaki and Asanuma 2009). Post-mortem analysis of brain samples from PD patients also suggests oxidative stress in SN dopaminergic neurons (Jenner 1998; Siddiqui et al. 2012). However, oxidative stress may not be the causative factor, rather induced or activated by another pathway (Jenner 1998). The prominent participation of MAO-B in PD pathogenesis is supported by increased levels of this enzyme in PD patients (Siddiqui et al. 2012), and the MAO-B inhibitor, Seleginin, is used for treatment in these patients. Calpain inhibition also reduces ROS generation and caspase – 3 activity in cultured primary rat neurons (Podbielska et al. 2016), suggesting calpain may also promote ROS production and ultimately SN neuronal death in PD. Recent studies in our laboratory observed oxidative stress and calpain activation in MPTP mice; these effects were attenuated by calpeptin, suggesting calpain inhibition is neuroprotective for SN neurons (Haque et al. 2020; Samantaray et al. 2015).
Autophagy
Autophagy is a highly conserved cellular process in which aggregate-prone proteins are targeted for cellular degradation (Fig. 2b). Autophagy dysfunction has been associated with a variety of pathologies including neurodegenerative diseases (Anglade et al. 1997; Arotcarena et al. 2019; Cerri and Blandini 2019; Glick et al. 2010; Janda et al. 2012; Lynch-Day et al. 2012; Menzies et al. 2017). The three primary types of autophagy include: (i) Macroautophagy - the primary pathway for removal of damaged cell organelles or unused proteins; (ii) Microautophagy- lysosomes directly engulf the cytoplasm by invagination, and (iii) Chaperon-mediated autophagy (CMA)- a very selective, complex pathway (Sala et al. 2016). Macroautophagy is further divided into bulk and selective modalities. Selective autophagy removes cell organelles with specific designations, e.g., mitophagy (mitochondria removal), chlorophagy (chloroplast removal); lipophagy (lipid removal), or ribophagy (ribosome removal). PINK1 and Parkin are involved in the maintenance of mitochondrial function and cytoarchitecture (Gautier et al. 2008; Palacino et al. 2004). Gautier and collaborators (Gautier et al. 2008) observed that in PINK−/− mice, the loss of mitochondrial function is specific to dopaminergic circuitry, an interesting regional specificity of PINK1 related to mitochondria function. Mitophagy as a selective form of autophagy degrades mitochondria (Fig. 2b), which is regulated by PINK1 and Parkin proteins (Deas et al. 2011; Jin and Youle 2012; Marongiu et al. 2009). In familial PD, Parkin and PINK1 mutations are the most common causes of autosomal recessive parkinsonism and possibly occur in early onset of the disease (Brooks et al. 2009). The known critical role of PINK1 and Parkin in PD highlights the significance of autophagy in the disease process. Additionally, the other known mutations involved in familial PD, including SNCA, LRRK2, UCHL1, and DJ-1, are known to alter activity of the CMA pathway (Sala et al. 2016). Since CMA also plays a role in α-synuclein clearance, the altered activity of CMA has been postulated to promote aggregation of α-synuclein in PD pathogenesis (Marongiu et al. 2009; Sala et al. 2016). In addition to functional and morphological maintenance of the mitochondrial network (Gautier et al. 2008; Palacino et al. 2004), PINK1 and Parkin also critically regulate the removal of dysfunctional mitochondria through mitophagy (Deas et al. 2011).
Anglade and collaborators (Anglade et al. 1997) observed that cell death in SN dopaminergic neurons in PD patients is due to apoptosis and autophagy (Anglade et al. 1997). The detection of more than one pathway in cell death indicates molecular heterogeneity among the SN dopaminergic neurons. Studies in PD animal models have reported dysfunction of the autophagy pathway (Lynch-Day et al. 2012). Moreover, intracellular and extracellular stress can initiate the autophagy pathway (He and Klionsky 2009), suggesting that oxidative stress due to ROS formation in PD may induce autophagy and lead to degeneration of SN neurons (Janda et al. 2012). As mentioned earlier, the neuropathological hallmark of PD is the presence of aggregated α-synuclein inclusions in the cytoplasm. The pathological accumulation of this misfolded α-synuclein in PD patients may indicate a dysfunctional protein degradation process (Arotcarena et al. 2019). Since autophagy is the only known pathway in mammalian systems to degrade damaged cellular organelles and misfolded or aggregated proteins (Glick et al. 2010), its interruption might be a critical factor in accumulation of α-synuclein protein and later onset/progression of PD.
Conclusions
Multiple regionally specific cellular and molecular pathways play active roles during the Parkinsonian disease process. Moreover, these pathways interact and influence each other under normal and pathological situations. Activated microglia and astrocytes can initiate neuroinflammation and assist in the progression of pathological damage to SN neurons. Similarly, oxidative stress may be caused by various insults, like environmental toxins or age-associated mitochondrial dysfunction, leading to ROS production. In dopaminergic neurons, auto-oxidation of dopamine is a significant source of ROS that leads to neuronal oxidative stress. The high energy demands of dopaminergic neurons during the aging process may contribute to mitochondrial dysfunction and oxidative damage. Since mitophagy clears dysfunctional mitochondria from SN neurons, mutation-related impairments in autophagy of defective proteins can allow for toxic protein accumulation in the cell. The effects of aging on these molecular pathways and cellular functions are imprecisely understood; thus, future studies of these molecular pathways and their interaction with each other during normal and pathological states are critical to developing disease-specific treatments.
Acknowledgements
This work was supported in part by funding from Veterans Administration (1I01BX002349-01, 2I01 BX001262-05, 1I01 BX004269-01), NIH-NINDS (R01 NS62327 and 1R2INS118393-01), and the South Carolina State Spinal Cord Research Fund (SCIRF-2015P-01, SCIRF-2015P-04, SCIRF-2015-I-01, SCIRF-2016 I-03, and SCIRF #2018 I-01).
Footnotes
Conflicts of interest/Competing interests The authors have no financial conflicts of interest.
Data availability Data will be available upon request after the manuscript is published.
Code availability N/A.
References
- Abeliovich A, Gitler AD (2016) Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539:207–216 [DOI] [PubMed] [Google Scholar]
- Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10:1538–1543 [DOI] [PubMed] [Google Scholar]
- Allen NJ, Eroglu C (2017) Cell Biology of Astrocyte-Synapse Interactions. Neuron 96:697–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alliot F, Godin I, Pessac B (1999) Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res 117:145–152 [DOI] [PubMed] [Google Scholar]
- Alvarez JI, Katayama T, Prat A (2013) Glial influence on the blood brain barrier. Glia 61:1939–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JK (2004) Oxidative stress in neurodegeneration: cause or consequence? Nat Med 10(Suppl):S18–25 [DOI] [PubMed] [Google Scholar]
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y (1997) Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol 12:25–31 [PubMed] [Google Scholar]
- Arotcarena ML, Teil M, Dehay B (2019) Autophagy in synucleinopathy: the overwhelmed and defective machinery. Cells 8:565–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA (2008) The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60:430–440 [DOI] [PubMed] [Google Scholar]
- Barreto GE, Gonzalez J, Torres Y, Morales L (2011) Astrocytic-neuronal crosstalk: implications for neuroprotection from brain injury. Neurosci Res 71:107–113 [DOI] [PubMed] [Google Scholar]
- Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, Tsitsou-Kampeli A, Sarel A, Cahalon L, Schwartz M (2015) Breaking immune tolerance by targeting Foxp3(+) regulatory T cells mitigates Alzheimer’s disease pathology. Nat Commun 6:7967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayraktar OA, Bartels T, Holmqvist S, Kleshchevnikov V, Martirosyan A, Polioudakis D, Ben Haim L, Young AMH, Batiuk MY, Prakash K et al. (2020) Astrocyte layers in the mammalian cerebral cortex revealed by a single-cell in situ transcriptomic map. Nat Neurosci 23:500–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 3:1301–1306 [DOI] [PubMed] [Google Scholar]
- Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR (2015) Oxidative stress and Parkinson’s disease. Front Neuroanat 9:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Rub U, Gai WP, Del Tredici K (2003) Idiopathic Parkinson’s disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (Vienna) 110:517–536 [DOI] [PubMed] [Google Scholar]
- Braak H, de Vos RA, Bohl J, Del Tredici K (2006) Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett 396:67–72 [DOI] [PubMed] [Google Scholar]
- Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, Bonduelle O, Alvarez-Fischer D, Callebert J, Launay JM et al. (2009) Infiltration of CD4 + lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Investig 119:182–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks J, Ding J, Simon-Sanchez J, Paisan-Ruiz C, Singleton AB, Scholz SW (2009) Parkin and PINK1 mutations in early-onset Parkinson’s disease: comprehensive screening in publicly available cases and control. J Med Genet 46:375–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obemraier CD, Strojny C et al. (2017) Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357:1255–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas R, Avila M, Gonzalez J, El-Bacha RS, Baez E, Garcia-Segura LM, Jurado Coronel JC, Capani F, Cardona-Gomez GP, Barreto GE (2014) Astrocytic modulation of blood brain barrier: perspectives on Parkinson’s disease. Front Cell Neurosci 8:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerri S, Blandini F (2019) Role of autophagy in Parkinson’s disease. Curr Med Chem 26:3702–3718 [DOI] [PubMed] [Google Scholar]
- Cerri S, Mus L, Blandini F (2019) Parkinson’s disease in women and men: what’s the difference? J Parkinsons Dis 9:501–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabot S, Charlet D, Wilson TL, Yong VW (2001) Cytokine production consequent to T cell-microglia interaction: the PMA/IFN gamma-treated U937 cells display similarities to human microglia. J Neurosci Methods 105:111–120 [DOI] [PubMed] [Google Scholar]
- Choi YR, Kang SJ, Kim JM, Lee SJ, Jou I, Joe EH, Park SM (2015) FcgammaRIIB mediates the inhibitory effect of aggregated alpha-synuclein on microglial phagocytosis. Neurobiol Dis 83:90–99 [DOI] [PubMed] [Google Scholar]
- Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, Zhang B, Yue Z (2020) Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun 11:1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA (2005) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120:421–433 [DOI] [PubMed] [Google Scholar]
- Chung WS, Welsh CA, Barres BA, Stevens B (2015) Do glia drive synaptic and cognitive impairment in disease? Nat Neurosci 18: 1539–1545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O (2002) Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. Eur J Neurosci 15:991–998 [DOI] [PubMed] [Google Scholar]
- Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B (2011) RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 12:560–567 [DOI] [PubMed] [Google Scholar]
- Colombo E, Farina C (2016) Astrocytes: key regulators of neuroinflammation. Trends Immunol 37:608–620 [DOI] [PubMed] [Google Scholar]
- Cox A, Capone M, Matzelle D, Vertegel A, Bredikhin M, Vamra A, Haque A, Shields DC, Banik NL (2021) Nanoparticle-based estrogen delivery to spinal cord injury site reduces local parenchymal destruction and improves functional recovery. J Neurotrauma 38: 342–352 [DOI] [PubMed] [Google Scholar]
- Croisier E, Moran LB, Dexter DT, Pearce RK, Graeber MB (2005) Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflammation 2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Kong Y, Zhang H (2012) Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct 2012:646354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher JP, Volpicelli-Daley LA, Blackburn JP, Moehle MS, West AB (2014) Abrogation of alpha-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc Natl Acad Sci USA 111:9289–9294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dansokho C, Ait Ahmed D, Aid S, Toly-Ndour C, Chaigneau T, Calle V, Cagnard N, Holzenberger M, Piaggio E, Aucouturier P et al. (2016) Regulatory T cells delay disease progression in Alzheimer-like pathology. Brain 139:1237–1251 [DOI] [PubMed] [Google Scholar]
- De Biase LM, Schuebel KE, Fusfeld ZH, Jair K, Hawes IA, Cimbro R, Zhang HY, Liu QR, Shen H, Xi ZX et al. (2017) Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron 95:341–356 e346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lucia C, Rinchon A, Olmos-Alonso A, Riecken K, Fehse B, Boche D, Perry VH, Gomez-Nicola D (2016) Microglia regulate hippocampal neurogenesis during chronic neurodegeneration. Brain Behav Immun 55:179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas E, Wood NW, Plun-Favreau H (2011) Mitophagy and Parkinson’s disease: the PINK1-parkin link. Biochim Biophys Acta 1813:623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias V, Junn E, Mouradian MM (2013) The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis 3:461–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Aparicio I, Paris I, Sierra-Torre V, Plaza-Zabala A, Rodriguez-Iglesias N, Marquez-Ropero M, Beccari S, Huguet P, Abiega O, Alberdi E et al. (2020) Microglia Actively Remodel Adult Hippocampal Neurogenesis through the Phagocytosis Secretome. J Neurosci 40:1453–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R (2000) Metabolism and functions of glutathione in brain. Prog Neurobiol 62:649–671 [DOI] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS, Lee YL (2000) Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000). Neurochem Res 25:1439–1451 [DOI] [PubMed] [Google Scholar]
- Ferreira SA, Romero-Ramos M (2018) Microglia response during Parkinson’s disease: alpha-synuclein intervention. Front Cell Neurosci 12:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filiano AJ, Gadani SP, Kipnis J (2017) How and why do T cells and their derived cytokines affect the injured and healthy brain? Nat Rev Neurosci 18:375–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK (2018) Reactive oxygen species in metabolic and inflammatory signaling. Circ Res 122–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost JL, Schafer DP (2016) Microglia: architects of the developing nervous system. Trends Cell Biol 26:587–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuzzati-Armentero MT, Cerri S, Blandini F (2019) Peripheral-central neuroimmune crosstalk in Parkinson’s disease: what do patients and animal models tell. Us? Front Neurol 10:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier CA, Kitada T, Shen J (2008) Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 105:11364–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge P, Dawson VL, Dawson TM (2020) PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson’s disease. Mol Neurodegener 15:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertig U, Hanisch UK (2014) Microglial diversity by responses and responders. Front Cell Neurosci 8:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glick D, Barth S (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221:3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz CG (2011) The history of Parkinson’s disease: early clinical descriptions and neurological therapies. Cold Spring Harb Perspect Med 1:a008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM, McColl BW (2016) Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci 19:504–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M (2012) Drosophila as a model to study mitochondrial dysfunction in Parkinson’s disease. Cold Spring Harb Perspect Med 2:a009944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JD, Zhao X, Li Y, Li GR, Liu XL (2018) Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (Review). Int J Mol Med 41:1817–1825 [DOI] [PubMed] [Google Scholar]
- Halliday GM, Stevens CH (2011) Glia: initiators and progressors of pathology in Parkinson’s disease. Mov Disord 26:6–17 [DOI] [PubMed] [Google Scholar]
- Haque A, Samantaray S, Knaryan VH, Capone M, Hossain A, Matzelle D, Chandran R, Shields DC, Farrand AQ, Boger HA et al. (2020) Calpain mediated expansion of CD4 + cytotoxic T cells in rodent models of Parkinson’s disease. Exp Neurol 330:113315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauben E, Schwartz M (2003) Therapeutic vaccination for spinal cord injury: helping the body to cure itself. Trends Pharmacol Sci 24:7–12 [DOI] [PubMed] [Google Scholar]
- He C, Klionsky DJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annual review of genetics 43:67–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heithoff BP, George KK, Phares AN, Zuidhoek IA, Munoz-Ballester C, Robel S (2021) Astrocytes are necessary for blood-brain barrier maintenance in the adult mouse brain. 69:436–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch L, Jette N, Frolkis A, Steeves T, Pringsheim T (2016) The incidence of parkinson’s disease: a systematic review and meta-analysis. Neuroepidemiology 46:292–300 [DOI] [PubMed] [Google Scholar]
- Hoglinger GU, Lannuzel A, Khondiker ME, Michel PP, Duyckaerts C, Feger J, Champy P, Prigent A, Medja F, Lombes A et al. (2005) The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J Neurochem 95:930–939 [DOI] [PubMed] [Google Scholar]
- Huang X, Reynolds AD, Mosley RL, Gendelman HE (2009) CD 4 + T cells in the pathobiology of neurodegenerative disorders. J Neuroimmunol 211:3–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, Offner H (2007) T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab 27:1798–1805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez P, Lesage S, Janin S, Lohmann E, Durif F, Destee A, Bonnet AM, Brefel-Courbon C, Heath S, Zelenika D et al. (2009) Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch Neurol 66:102–108 [DOI] [PubMed] [Google Scholar]
- Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, Sakai R, Matsuo K, Nakayama T, Yoshie O et al. (2019) Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 565:246–250 [DOI] [PubMed] [Google Scholar]
- Janda E, Isidoro C, Carresi C, Mollace V (2012) Defective autophagy in Parkinson’s disease: role of oxidative stress. Mol Neurobiol 46:639–661 [DOI] [PubMed] [Google Scholar]
- Jenner P (1998) Oxidative mechanisms in nigral cell death in Parkinson’s disease. Mov Disord 13(Suppl 1 ):24–34 [PubMed] [Google Scholar]
- Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53(Suppl 3):S26–36 (discussion S36-28) [DOI] [PubMed] [Google Scholar]
- Jin SM, Youle RJ (2012) PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci 125:795–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Shie FS, Liu J, Wang Y, Davis J, Schantz AM, Montine KS, Montine TJ, Zhang J (2007) Prostaglandin E2 receptor subtype 2 (EP2) regulates microglial activation and associated neurotoxicity induced by aggregated alpha-synuclein. J Neuroinflammation 4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RT, Breedlove SM, Jordan CL (2008) Sex differences and laterality in astrocyte number and complexity in the adult rat medial amygdala. J Comp Neurol 511:599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ME, Stecher B, Labrie V, Brundin L, Brundin P (2019) Triggers, facilitators, and aggravators: redefining Parkinson’s disease pathogenesis. Trends Neurosci 42:4–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones R (2010) The roles of PINK1 and Parkin in Parkinson’s disease. PLoS Biol 8:e1000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam TI, Hinkle JT, Dawson TM, Dawson VL (2020) Microglia and astrocyte dysfunction in parkinson’s disease. Neurobiol Dis 144:105028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Sofroniew MV (2015) Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci 18:942–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS (2000) Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. J Neurosci 20:6309–6316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kwon SH, Kam TI, Panicker N, Karuppagounder SS, Lee S, Lee JH, Kim WR, Kook M, Foss CA et al. (2019) Transneuronal propagation of pathologic alpha-Synuclein from the gut to the brain models Parkinson’s disease. Neuron 103(627–641):e627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipnis J, Nevo U, Panikashvili D, Alexandrovich A, Yoles E, Akselrod S, Shohami E, Schwartz M (2003) Therapeutic vaccination for closed head injury. J Neurotrauma 20:559–569 [DOI] [PubMed] [Google Scholar]
- Klein C, Westenberger A (2012) Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2:a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingelhoefer L, Reichmann H (2015) Pathogenesis of Parkinson disease—the gut-brain axis and environmental factors. Nat Rev Neurol 11:625–636 [DOI] [PubMed] [Google Scholar]
- Komuczki J, Tuzlak S, Friebel E, Hartwig T, Spath S, Rosenstiel P, Waisman A, Opitz L, Oukka M, Schreiner B et al. (2019) Fate-mapping of GM-CSF expression identifies a discrete subset of inflammation-driving T helper cells regulated by cytokines IL-23 and IL-lbeta. Immunity 50:1289–1304 e1286 [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A (2009) The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci 32:149–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, Acevedo-Cintron J, Jhaldiyal A, Wang H, Andrabi SA, Eacker S, Karuppagounder SS, Brahmachari S, Chen R, Kim H et al. (2020) Defects in mitochondrial biogenesis drive mitochondrial alterations in PARKIN-deficient human dopamine neurons. Stem Cell Rep 15: 629–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustrimrovic N, Rasini E, Legnaro M, Marino F, Cosentino M (2014) Expression of dopaminergic receptors on human CD4 + T lymphocytes: flow cytometric analysis of naive and memory subsets and relevance for the neuroimmunology of neurodegenerative disease. J Neuroimmune Pharmacol 9:302–312 [DOI] [PubMed] [Google Scholar]
- Kustrimovic N, Rasini E, Legnaro M, Bombelli R, Aleksic I, Blandini F, Comi C, Mauri M, Minafra B, Riboldazzi G et al. (2016) Dopaminergic receptors on CD4 + T naive and memory lymphocytes correlate with motor impairment in patients with Parkinson’s disease. Sci Rep 6:33738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustrimovic N, Comi C, Magistrelli L, Rasini E, Legnaro M, Bombelli R, Aleksic I, Blandini F, Minafra B, Riboldazzi G et al. (2018) Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: cross-sectional studies of CD4 + Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J Neuroinflammation 15:205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie MJ, Cortese GP, Ostaszewski BL, Schlossmacher MG (2007) The effects of oxidative stress on parkin and other E3 ligases. J Neurochem 103:2354–2368 [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S (1990) Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39:151–170 [DOI] [PubMed] [Google Scholar]
- Lecours C, Bordeleau M, Cantin L, Parent M, Paolo TD, Tremblay ME (2018) Microglial implication in Parkinson’s disease: loss of beneficial physiological roles or gain of inflammatory functions? Front Cell Neurosci 12:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ et al. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445: 168–176 [DOI] [PubMed] [Google Scholar]
- Lenz KM, Nugent BM, Haliyur R, McCarthy MM (2013) Microglia are essential to masculinization of brain and behavior. J Neurosci 33: 2761–2772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Barres BA (2018) Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol 18:225–242 [DOI] [PubMed] [Google Scholar]
- Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, Gulati G, Bennett ML, Sun LO, Clarke LE et al. (2019) Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 101:207–223 e210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Barres BA (2017) Reactive astrocytes: production, function, and therapeutic potential. Immunity 46:957–967 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CCJ, Yu K, Hatcher A, Huang TW, Lee HK, Carlson J, Weston MC, Chen F, Zhang Y, Zhu W, Mohila CA, Ahmed N, Patel AJ, Arenkiel BR, Noebels JL, Creighton CJ, Deneen B (2017) Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci 20:396–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnerbauer M, Wheeler MA, Quintana FJ (2020) Astrocyte crosstalk in CNS inflammation. Neuron 108:608–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HF, Ho PW, Leung GC, Lam CS, Pang SY, Li L, Kung MH, Ramsden DB, Ho SL (2017) Combined LRRK2 mutation, aging and chronic low dose oral rotenone as a model of Parkinson’s disease. Sci Rep 7:40887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long-Smith CM, Sullivan AM, Nolan YM (2009) The influence of microglia on the pathogenesis of Parkinson’s disease. Prog Neurobiol 89:277–287 [DOI] [PubMed] [Google Scholar]
- Luckheeram RV, Zhou R, Verma AD, Xia B (2012) CD4(+)T cells: differentiation and functions. Clin Dev Immunol 2012:925135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ (2012) The role of autophagy in Parkinson’s disease. Cold Spring Harb Perspect Med 2:a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire-Zeiss KA, Short DW, Federoff HJ (2005) Synuclein, dopamine and oxidative stress: co-conspirators in Parkinson’s disease? Brain Res Mol Brain Res 134:18–23 [DOI] [PubMed] [Google Scholar]
- Mahlknecht P, Seppi K, Poewe W (2015) The concept of prodromal Parkinson’s disease. J Parkinsons Dis 5:681–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti P, Manna J, Dunbar GL (2017) Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl Neurodegener 6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marongiu R, Spencer B, Crews L, Adame A, Patrick C, Trejo M, Dallapiccola B, Valente EM, Masliah E (2009) Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J Neurochem 108: 1561–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK (2006) Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci 26:41–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R, Bajo-Graneras R, Moratalla R, Perea G, Araque A (2015) Circuit-specific signaling in astrocyte-neuron networks in basal ganglia pathways. Science 349:730–734 [DOI] [PubMed] [Google Scholar]
- Martinez TN, Greenamyre JT (2012) Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid Redox Signal 16: 920–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez B, Peplow PV (2017) MicroRNAs in Parkinson’s disease and emerging therapeutic targets. Neural Regen Res 12:1945–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, Kaneko T (2009) Single nigrostriatal dopaminergic neurons fomr widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci 29:444–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Akiyama H, McGeer EG (1988a) Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol 24:574–576 [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG (1988b) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38:1285–1291 [DOI] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Sanchez J, Karabiyik M et al. (2017) Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93:1015–1034 [DOI] [PubMed] [Google Scholar]
- Miller SJ (2018) Astrocyte heterogeneity in the adult central nervous system. Front Cell Neurosci 12:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki I, Asanuma M (2009) Approaches to prevent dopamine quinone-induced neurotoxicity. Neurochem Res 34:698–706 [DOI] [PubMed] [Google Scholar]
- Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M (1999) Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med 5:49–55 [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Kelley KW, Tsai HH, Redmond SA, Chang SM, Madireddy L, Chan JR, Baranzini SE, Ullian EM, Rowitch DH (2014) Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature 509:189–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, Budson AE, Sperling R, Selkoe DJ, Weiner HL (2003) Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest 112:415–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustafa AA, Chakravarthy S, Phillips JR, Gupta A, Keri S, Polner B, Frank MJ, Jahanshahi M (2016) Motor symptoms in Parkinson’s disease: A unified framework. Neurosci Biobehav Rev 68:727–740 [DOI] [PubMed] [Google Scholar]
- Mouton-Liger F, Jacoupy M, Corvol JC, Corti O (2017) PINK1/Parkin-dependent mitochondrial surveillance: from pleiotropy to Parkinson’s disease. Front Mol Neurosci 10:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatsu T, Mogi M, Ichinose H, Togari A (2000) Cytokines in Parkinson’s disease. J Neural Transm Suppl 58:143–151 [PubMed] [Google Scholar]
- Narciso L, Parlanti E, Racaniello M, Simonelli V, Cardinale A, Merlo D, Dogliotti E (2016) The response to oxidative DNA damage in neurons: mechanisms and disease. Neural Plast 2016:3619274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng TH, Britton GJ, Hill EV, Verhagen J, Burton BR, Wraith DC (2013) Regulation of adaptive immunity; the role of interleukin-10. Front Immunol 4:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niki E (2008) Lipid peroxidation products as oxidative stress biomarkers. Biofactors 34:171–180 [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318 [DOI] [PubMed] [Google Scholar]
- Nuytemans K, Theuns J, Cmts M, Van Broeckhoven C (2010) Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31:763–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeier B, Daneman R, Ransohoff RM (2013) Development, maintenance and disruption of the blood-brain barrier. Nat Med 19:1584—1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J (2004) Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 279:18614–18622 [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L et al. (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456–1458 [DOI] [PubMed] [Google Scholar]
- Picillo M, Nicoletti A, Fetoni V, Garavaglia B, Barone P, Pellecchia MT (2017) The relevance of gender in Parkinson’s disease: a review. J Neurol 264:1583–1607 [DOI] [PubMed] [Google Scholar]
- Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F, Arcoraci V, Squadrito F, Altavilla D, Bitto A (2017) Oxidative Stress: Harms and Benefits for Human Health. Oxid Med Cell Longev 2017: 8416763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podbielska M, Das A, Smith AW, Chauhan A, Ray SK, Inoue J, Azuma M, Nozaki K, Hogan EL, Banik NL (2016) Neuron-microglia interaction induced bi-directional cytotoxicity associated with calpain activation. J Neurochem 139:440–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve AK, Ludtmann MH, Angelova PR, Simcox EM, Horrocks MH, Klenerman D, Gandhi S, Turnbull DM, Abramov AY (2015) Aggregated alpha-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis 6:e1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, Chao CC, Patel B, Yan R, Blain M, Alvarez JI, Kébir H, Anandasabapathy N, Izquierdo G, Jung S, Obholzer N, Pochet N, Clish CB, Prinz M, Pra A, Ante 1J, Quintana FJ (2016) Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med 22:586–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala G, Marinig D, Arosio A, Ferrarese C (2016) Role of chaperone-mediated autophagy dysfunctions in the pathogenesis of Parkinson’s disease. Front Mol Neurosci 9:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, Mancuso R, Chen WT, Woodbury ME, Srivastava G et al. (2019) The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Abeta plaques. Cell Rep 27: 1293–1306 e1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samantaray S, Knaryan VH, Guyton MK, Matzelle DD, Ray SK, Banik NL (2007) The parkinsonian neurotoxin rotenone activates calpain and caspase-3 leading to motoneuron degeneration in spinal cord of Lewis rats. Neuroscience 146:741–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samantaray S, Knaryan VH, Shields DC, Cox AA, Haque A, Banik NL (2015) Inhibition of calpain activation protects MPTP-induced nigral and spinal cord neurodegeneration, reduces inflammation, and improves gait dynamics in mice. Mol Neurobiol 52:1054–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sariola H, Saarmra M (2003) Novel functions and signalling pathways for GDNF. J Cell Sci 116:3855–3862 [DOI] [PubMed] [Google Scholar]
- Sato K (2015) Effects of microglia on neurogenesis. Glia 63:1394–1405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH, Holt IJ, Sweeney M, Harding AE, Jenner P, Marsden CD (1990) Mitochondrial DNA analysis in Parkinson’s disease. Mov Disord 5:294–297 [DOI] [PubMed] [Google Scholar]
- Schapira AH, Mann VM, Cooper JM, Dexter D, Daniel SE, Jenner P, Clark JB, Marsden CD (1990b) Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J Neurochem 55:2142–2145 [DOI] [PubMed] [Google Scholar]
- Scudamore O, Ciossek T (2018) Increased oxidative stress exacerbates alpha-synuclein aggregation in vivo. J Neuropathol Exp Neurol 77:443–453 [DOI] [PubMed] [Google Scholar]
- Selvaraj S, Piramanayagam S (2019) Impact of gene mutation in the development of Parkinson’s disease. Genes Dis 6:120–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields DC, Haque A, Banik NL (2020) Neuroinflammatory responses of microglia in central nervous system trauma. J Cereb Blood Flow Metab 40:S25–S33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui A, Hanson I, Andersen JK (2012) Mao-B elevation decreases parkin’s ability to efficiently clear damaged mitochondria: protective effects of rapamycin. Free Radic Res 46:1011–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Gottfried-Blackmore A, Milner TA, McEwen BS, Bulloch K (2008) Steroid hormone receptor expression and function in microglia. Glia 56:659–674 [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R et al. (2003) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. [DOI] [PubMed] [Google Scholar]
- Socodato R, Portugal CC, Canedo T, Rodrigues A, Almeida TO, Henriques JF, Vaz SH, Magalhaes J, Silva CM, Baptista FI et al. (2020) Microglia dysfunction caused by the loss of rhoa disrupts neuronal physiology and leads to neurodegeneration. Cell Rep 31: 107796. [DOI] [PubMed] [Google Scholar]
- Solano RM, Casarejos MJ, Menendez-Cuervo J, Rodriguez-Navarro JA, Garcia de Yebenes J, Mena MA (2008) Glial dysfunction in parkin null mice: effects of aging. J Neurosci 28:598–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E (2009) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci 29:13578–13588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D (2007) Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci 30:244–250 [DOI] [PubMed] [Google Scholar]
- Tang Y, Le W (2016) Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol 53:1181–1194 [DOI] [PubMed] [Google Scholar]
- Tanji K, Odagiri S, Maruyama A, Mori F, Kakita A, Takahashi H, Wakabayashi K (2013) Alteration of autophagosomal proteins in the brain of multiple system atrophy. Neurobiol Dis 49:190–198 [DOI] [PubMed] [Google Scholar]
- Tay TL, Savage JC, Hui CW, Bisht K, Tremblay ME (2017) Microglia across the lifespan: from origin to function in brain development, plasticity and cognition. J Physiol 595:1929–1945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teismann P, Schulz JB (2004) Cellular pathology of Parkinson’s disease: astrocytes, microglia and inflammation. Cell Tissue Res 318:149–161 [DOI] [PubMed] [Google Scholar]
- Testa CM, Sherer TB, Greenamyre JT (2005) Rotenone induces oxidative stress and dopaminergic neuron damage in organotypic substantia nigra cultures. Brain Res Mol Brain Res 134:109–118 [DOI] [PubMed] [Google Scholar]
- Thakkar R, Wang R, Wang J, Vadlamudi RK, Brann DW (2018) 17beta-Estradiol Regulates Microglia Activation and Polarization in the Hippocampus Following Global Cerebral Ischemia. Oxid Med Cell Longev 2018:4248526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa A, Gelosa P, Castiglioni L, Cimino M, Rizzi N, Pepe G, Lolli F, Marcello E, Sironi L, Vegeto E et al. (2018) Sex-specific features of microglia from adult mice. Cell Rep 23:3501–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Chu CH, Stewart T, Ginghina C, Wang Y, Nie H, Guo M, Wilson B, Hong JS, Zhang J (2015) alpha-Synuclein, a chemoattractant directs microglial migration via H202-dependent Lyn phosphorylation. Proc Natl Acad Sci U S A 112:E1926–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U, Exiga M, Vadisiute A, Raggioli A, Schertel A et al. (2018) Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun 9:1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler MA, Quintana FJ (2019) Regulation of astrocyte functions in multiple sclerosis. Cold Spring Harb Perspect Med 9:a029009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler MA, Clark IC, Tjon EC, Li Z, Zandee SEJ, Couturier CP, Watson BR, Scalisi G, Alkwai S, Rothhammer V, Rotem A, Heyman JA, Thaploo S, Sanmarco LM, Ragoussis J, Weitz DA, Petrecca K, Moffitt JR, Becher B, Antel JP, Prat A, Quintana FJ (2020) MAFGdriven astrocytes promote CNS inflammation. Nature 578:593–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong D, Dorovini-Zis K, Vincent SR (2004) Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol 190:446–455 [DOI] [PubMed] [Google Scholar]
- Wu SY, Chen YW, Tsai SF, Wu SN, Shih YH, Jiang-Shieh YF, Yang TT, Kuo YM (2016) Estrogen ameliorates microglial activation by inhibiting the Kir2.1 inward-rectifier K(+) channel. Sci Rep 6:22864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang QQ, Zhou JW (2019) Neuroinflammation in the central nervous system: Symphony of glial cells. Glia 67:1017–1035 [DOI] [PubMed] [Google Scholar]
- Yasuda T, Mochizuki H (2010) Use of growth factors for the treatment of Parkinson’s disease. Expert Rev Neurother 10:915–924 [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhang W, Zhou Y, Hong JS et al. (2005) Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson’s disease. FASEB J 19:533–542 [DOI] [PubMed] [Google Scholar]