

Abstract
Sickle cell disease (SCD) is an exemplar of bidirectional translational research, starting with a remarkable astute observation of the abnormally shaped red blood cells that motivated decades of bench research that have now translated into new drugs and genetic therapies. Introduction of hydroxyurea (HU) therapy, the only SCD-modifying treatment for >30 years and now standard care, was initiated through another clinical observation by a pediatrician. While the clinical efficacy of HU is primarily due to its fetal hemoglobin (HbF) induction, the exact mechanism of how it increases HbF remains not fully understood. Unraveling of the molecular mechanism of how HU increases HbF has provided insights on the development of new HbF-reactivating agents in the pipeline. HU has other salutary effects, reduction of cellular adhesion to the vascular endothelium and inflammation, and dissecting these mechanisms has informed bench—both cellular and animal—research for development of the 3 recently approved agents: endari, voxelotor, and crizanlizumab; truly, a bidirectional bench to bedside translation. Decades of research to understand the mechanisms of fetal to adult hemoglobin have also culminated in promising anti-sickling genetic therapies and the first-in-human studies of reactivating an endogenous (γ-globin) gene HBG utilizing innovative genomic approaches.
Introduction
Sickle cell disease (SCD) can trace its first description in the Western literature to a case report in 1910 by Herrick1 of a young dental male student from Grenada with severe malaise and anemia. Hallmarks of the disease were noted then: “healing ulcers” predominantly on the legs that lasted about a year; anemia with a “hemoglobin (Dare) 40 per cent” and jaundice (“tinge of yellow in the sclerae”), and a disease with “acute exacerbations.” Herrick1,2 also made a remarkable observation that the “red corpuscles varied much in size,” and that “the shape of the reds was very irregular,” but what especially attracted his attention was “the large number of thin, elongated, sickle-shaped and crescent-shaped forms.” He surmised “that some unrecognized change in the composition of the corpuscle itself may be the determining factor” (Figure 1).
Figure 1.
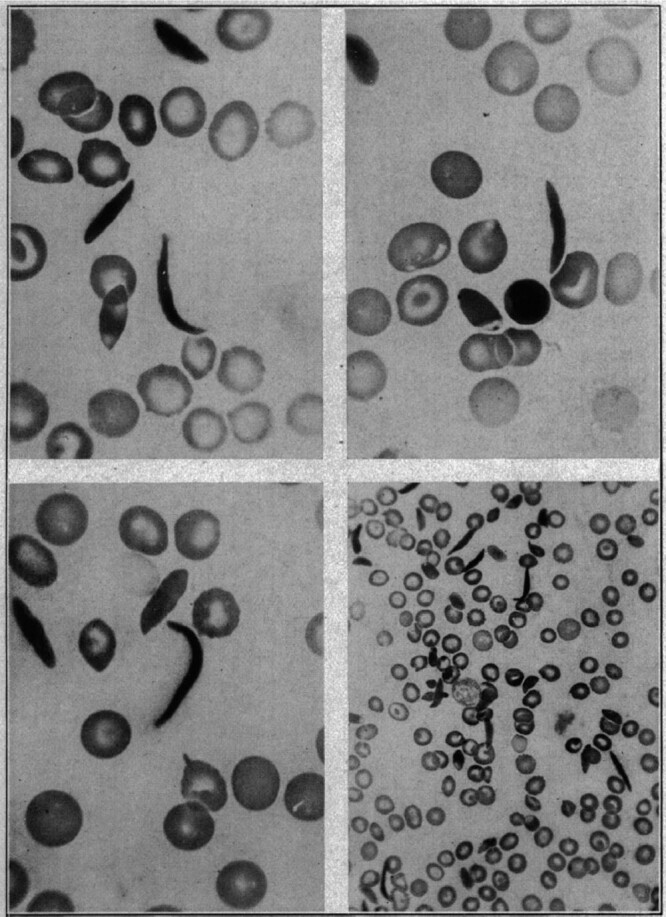
The first documented observation of sickle-shaped red blood cells. Historical figure from 1910, taken from the publication by Herrick1 with title “Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia.” (Reproduced with permission from JAMA Intern Med. 1910;6:517–521. Copyright © 1910 American Medical Association. All rights reserved.)
It was not until almost 40 years later in 1949 when Pauling and his collaborators3 discovered that the “…unrecognized change in the composition of the corpuscle” was due to an altered hemoglobin (Hb) structure, thus SCD became the first disease to be understood at a molecular level. The abnormal Hb was later shown to result from the substitution of glutamic acid by valine at position 6 of the β-globin chain of Hb4 that arose from an A>T base change (Table 1).5 Genetic simplicity of the sickle mutation in a compact gene encoding an abnormal Hb that was relatively accessible through a simple blood draw has lent SCD to many proof-of-principle and validation experiments for many years. This was facilitated by the globin genes among the first to be cloned and fully analyzed by DNA sequencing.6,7 SCD became a role model for molecular genetics, leading the way in breakthrough discoveries in areas of DNA diagnostics, population and epidemiological genetics, and more recently, genetic therapies.8,9 Certainly for the last century, studies of SCD and genetics of Hb have contributed and benefited other medical conditions more than SCD itself. In the last 10 years, however, we have gained a much better understanding of the sickle pathophysiology. We have also gained incredible insights on the switch from fetal to adult Hb10 with identification of key regulating factors such as B-cell lymphoma/leukemia 11A (BCL11A)11,12 that together, with major advances in genetic and genomic technologies,13,14 have translated into genetic-based approaches for treating SCD.
Table 1.
Terminology.
| (HbA, α2β2): consists of 2 α-globin and 2 β-globin chains and is the most common human hemoglobin tetramer, accounting for about 97% of the total red blood cell hemoglobin in adulthood |
| (HbF, α2γ2): consists of 2 α-globin and 2 γ-globin chains. This is the predominant form in the fetus and declines in the first weeks after birth |
| (HbS, α2βS2): consists of 2 α-globin and 2 mutant β-globin chains. HbS is the most common type of hemoglobin variant and the basis of sickle cell trait and sickle cell anemia |
| Sickle cell disease is caused by the presence of HbS, and includes different sickle genotypes classified according to the hemoglobin abnormality: |
| HbSS: homozygous mutation in β-globin (Glu to Val at position 6) |
| HbSC: compound heterozygotes of HbS (Glu to Val at position 6 and Glu to Lys at position 6) |
| HBS/β thal: compound heterozygotes of HbS with beta thalassemia, the latter can be either beta zero or beta plus, depending on whether beta globin is absent of present but in reduced amounts, respectively |
| Other less common sickle genotypes include compound heterozygotes of HbS with HbD Punjab (HbSD Punjab) and HbS with HbE (HbSE) |
| HbAS refers to heterozygotes or carriers of the HbS mutation: these individuals have HbS of 30%–40% and are asymptomatic. Under extreme conditions, such as physically stressful sports and severe dehydration, HbAS individuals may suffer vaso-occlusive episodes and pain. HbAS individuals are protected against falciparum malaria and can pass the mutant allele to their children |
HbA = hemoglobin A; HbD = hemoglobin D; HbE = hemoglobin E; HbF = hemoglobin F; HbS = hemoglobin S; HbSC = hemoglobin SC; HbSS = hemoglobin SS.
Here we take readers through the key discoveries, which showcases the bidirectional bench to bedside research in SCD highlighting the leaps in our understanding that have contributed to new therapeutic options in its management.
The history of SCD pathophysiology—from bench to bedside to bench
After building an electrophoresis machine, Pauling3 was able to separate normal adult hemoglobin (α2β2, HbA) from abnormal sickle hemoglobin (α2β2S, HbS) and describe SCD at a molecular level for the first time. But, many questions remained unanswered, such as how HbS lead to the formation of these “thin, elongated sickle-shaped” red cells, the key phenotype in sickle pathophysiology, motivating an enormous amount of basic science studies on the Hb polymer structure,15 thermodynamics,16,17 and kinetics18 of HbS polymerization. Since polymerization of HbS can only occur when HbS is deoxygenated,19 increasing HbS oxygen affinity as a therapeutic approach has been discussed for many years, culminating in the development of oxygen affinity modifying drugs such as voxelotor (also known as Oxbryta or GBT440). Importantly, increasing oxygen binding to HbS could also compromise oxygen delivery, as first discussed by Beutler,20 an effect that is detrimental in a disease characterized by tissue/organ damage due to oxygen deprivation.
A key bedside observation that fetal Hb (HbF) had beneficial effects was first hypothesized by the pediatrician Watson21 in 1948, who noted that African American infants with SCD were less prone to have “sickling” events in the first few months of life during which HbF gradually disappears from the blood (Table 1). Since then, multiple observational studies between 1970s and 1990s demonstrating a milder form of SCD in those patients with higher levels of HbF have been published. Clinical and population studies elucidated that the level of HbF in adults is under 2 levels of genetic control.22 Common genetic variation, historically referred to as heterocellular hereditary persistence of fetal hemoglobin (HPFH), is characterized by modest increases of HbF (1%–4% of total Hb) that are unevenly distributed among the red blood cells (RBCs). Although the HbF increases are modest in healthy adults, co-inheritance of heterocellular HPFH on a background of stress erythropoiesis, such as SCD, leads to increases in HbF levels as high as 25% with immense clinical benefits. Although familial, the inheritance pattern of heterocellular HPFH was not clear until 20 years ago, when genetic studies showed that common HbF variation behaved as a quantitative trait and the levels are predominantly genetically controlled.23 To date, 3 quantitative trait loci are known: the hemoglobin gene complex (HBB) on chromosome 11p (Xmn1-Gγ site), the BCL11A gene on chromosome 2, and the HBS1L-MYB intergenic region on chromosome 6q.24 In contrast, rare variants, historically referred to as pancellular HPFH, are inherited in a Mendelian fashion as alleles of the HBB complex. Carriers for pancellular HPFH have substantial increases in HbF levels of 15% to 30% that are homogeneously distributed among the RBCs. Pancellular HPFH is caused by substantial DNA deletions within the HBB cluster or specific single base changes in the promoters of the γ-globin genes.25 Persistence of HbF production has no clinical consequences in healthy adults, but ameliorate symptoms of SCD. Indeed, inheritance of a Mendelian form of HPFH in trans to a βS allele (HbS/HPFH) may eliminate clinical consequences of SCD, motivating enormous research on understanding how fetal HbF is repressed in adults.26
Translating clinical benefits of hydroxyurea to an improved understanding of sickle pathophysiology
The beneficial effect of HbF led to the first study of hydroxyurea (HU) in 2 patients with the HbSS form of SCD, also referred to as sickle cell anemia (see Table 1) in 1984, in which measurable and sustainable increases in HbF could be achieved with minimal toxicity, but no change in clinical course could be observed in the short period of study.27 Nonetheless, these encouraging preliminary results motivated numerous clinical trials of HU, first in adults28 and then in pediatric patients with SCD29; its overall safety profile and efficacy led to US Food and Drug Administration (FDA) approval of HU for treatment of SCD in adults in 1998 and in children in 2017.
Our understanding of sickle pathophysiology has also been greatly helped by the use of humanized sickle mouse models, which has provided new insights on adhesion, inflammation, and interactions of the sickled RBCs with their microenvironment—vasculature, neutrophils, monocytes, platelets, and the upregulation of vasculature cyto-adhesion molecules.30,31 Molecules such as P- and E-selectin, fundamental in the adhesion and activation of white blood cells, specially neutrophils, to the vasculature have been found to represent an important component of the pain crisis pathophysiology and have become therapeutic targets.32
As polymerization of deoxy-HbS is the key event that triggers the downstream consequences of SCD, several therapeutic approaches have focused on mitigation of this root cause, utilizing both genetic and pharmacological anti-sickling strategies. The best-established strategy is induction of HbF synthesis borne out not only by the plentiful clinical and epidemiological studies, but also by the kinetics and thermodynamics of the polymerization process itself. Studies of HbS polymerization kinetics posit that the delay time relative to the transit time through the microcirculation is a major determinant of whether polymerization results in irreversible sickling and hence severity in SCD. The amino acid sequence of γ-globin chain is sufficiently different from βS such that little or no γ-globin takes part in the fiber formation, so the primary effect of HbF (α2γ2) is to simply dilute the intracellular concentration of HbS.19 Because HbS polymerization is highly sensitive and dependent on intracellular HbS concentration,33 even a small decrease in HbS concentration is therapeutic because more cells can escape the small vessels before sickling occurs. Strategies that reduce HbS intracellular concentration, such as increasing HbF or the red cell volume (ie, mean corpuscular volume [MCV]), increase the delay time to sickling, while strategies that reduce adherence and shorten transit time should be therapeutic. HU inhibits ribonucleotide reductase causing reversible myelosuppression. Although the exact mechanism of HbF induction is unclear, a primary mechanism relates to the subsequent recovery or “stress erythropoiesis” and release of early erythroid progenitors that synthesize more HbF. This causes the uneven distribution of HbF among the RBCs,34 one of the reasons proposed for the variable clinical response between SCD patients.35,36 Otherwise, HU-induced HbF increase would be much more effective.
Advances in our understanding of the molecular mechanisms regulating the fetal to adult Hb switch have led to the generation of new agents that do not rely on causing “stress erythropoiesis” and they fall into 2 main groups: those that affect chromatin regulators (such as decitabine on DNA methylation and histone deacetylase [HDAC] inhibitors) and others that affect DNA-binding transcription factors. Contemporaneous genome-wide association studies11,12 identified BCL11A as the first key repressor protein for silencing of the fetal (γ) globin genes joined later by zinc finger and BTB domain-containing protein 7A (ZBTB7A), also known as leukemia related factor (LRF).37 In 2018, key studies by 2 groups showed that BCL11A and ZBTB7A each bind to a cognate recognition site within the γ-globin promoter.38,39 Besides its role as γ-globin repressor, BCL11A is also essential for B-lymphoid development.40 Identification of the key erythroid-specific enhancer elements41 was critical and important in the development of the clinical trials aimed at downregulating BCL11A using 2 different genetic approaches—lentiviral short hairpin RNA (shRNA) and clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated nuclease-9 (Cas-9) editing.42,43 Another genetic approach for reactivating endogenous γ-globin to produce high HbF is to mimic the naturally occurring HPFH variants in the γ-globin promoters by genome-editing to disable binding of BCL11A or ZBTB7A/LRF repressors.10,44 In theory, correcting the sickle mutation (rs334) is the most direct approach, as the same base change is present in all βS alleles, but homology-directed DNA repair is limited by the efficiency at which the correction is achieved and the concomitant generation of insertions/deletions and conversion of the βS gene to a β-thalassemia allele.45
New therapeutic drug targets that have evolved from molecular dissection of SCD pathophysiology
HU was originally an anti-neoplastic agent in the treatment of patients with myeloproliferative diseases, in whom it has been shown to induce variable moderate increases in HbF and MCVs,46 but HU is now probably best known as standard therapeutic agent for SCD.47,48 While the clinical efficacy of HU relates predominantly to the level of HbF increase, it also has other salutary therapeutic effects—such as reducing cellular adhesion, hemolysis, and inflammation.49 Molecular dissection of these mechanisms led to new insights on the pathophysiology of SCD (Figure 2) and new therapeutic targets on vaso-occlusion (endari), HbS polymerization (voxelotor), and vascular adhesion (crizanlizumab) that were approved by the FDA in the last 5 years (Table 2).
Figure 2.
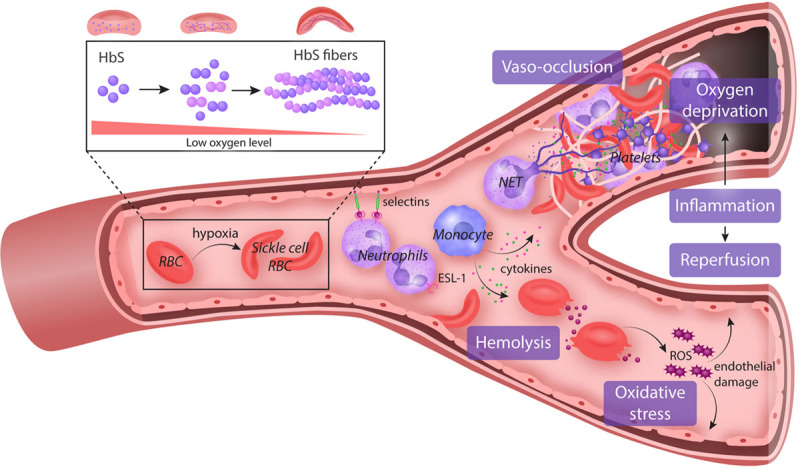
Pathophysiology of SCD. Polymerization of HbS under a state of deoxygenation is the fundamental event in the pathophysiology of SCD. These sickled RBCs become activated and interact via pro-inflammatory cytokines with the endothelium, WBCs, especially neutrophils/monocytes, and platelets. There is increased expression of pro-adhesive molecules (selectins) in the endothelial vasculature, which promote the adhesion of WBC, a key component of vaso-occlusion physiology. Platelet activation promotes further cytokine release and inflammation and also a hypercoagulable state by secreting coagulation and tissue factors. These damaged “sickle” RBCs are prone to destruction, leading to the continual release of cell-free hemoglobin which leads to depletion of hemopexin and haptoglobin. Consequently, the bioavailability of nitric oxide is reduced, leading to vascular dysfunction and end-organ damage. Both pathways triggered by the polymerization of HbS perpetuate the chronic state of inflammation seen in SCD facilitating end-organ damage. ESL-1 = E-selectin ligand-1; HbS = hemoglobin S; NET = neutrophil extracellular trap; RBC = red blood cell; ROS = reactive oxygen species; SCD = sickle cell disease; WBC = white blood cell.
Table 2.
Medications Approved and in the Pipeline for Sickle Cell Disease.
| Drug | Mechanism of Action | Phase | Others/ClinicalTrials.gov |
|---|---|---|---|
| Targeting HbS polymerization | |||
| Hydroxyurea | Ribonucleotide reductase inhibitor. The exact mechanism of HbF induction remains unknown | FDA approved | |
| Voxelotor | Binds specifically to the N-terminus of the alpha subunit of HbS and stabilizes the oxygenated state of HbS | FDA approved | |
| Panobinostat | HDAC inhibitor: increase levels of γ-globin and inducing production of HbF | Phase 1 | NCT01245179: active, not recruiting |
| Vorinostat | HDAC inhibitor: increase levels of γ-globin and inducing production of HbF | Phase 2 | NCT01000155: terminated early due to poor recruitment |
| IMR-687 | Phosphodiesterase 9 inhibitor: increasing cGMP increasing the production of HbF | Phase 2 | NCT04053803: enrolling by invitation |
| FT-4202 | PK activator: decreasing 2,3-DPG and decreasing the risk of red cell deoxygenation | Phase 2/3 | NCT04624659: recruiting |
| AG-348 (Mitapivat) | PK activator: decreasing 2,3-DPG and decreasing the risk of red cell deoxygenation | Phase 1/2 | NCT04610866: recruiting |
| Targeting pro-adhesive molecules | |||
| Crizanlizumab | Monoclonal antibody against P-selectin | FDA approved | |
| Rivipansel | Pan-selectin inhibitor with predilection for E-selectin | Phase 2 | NCT02187003: results recently published at ASH 2020 |
| Targeting inflammation | |||
| L-glutamine | Increase NADH and NAD redox potential and decrease endothelial adhesion | FDA approved in the United States | |
| Regadenoson | Adenosine A2A receptor agonist: in vitro studies show decrease iNKT activity | Phase 2 | NCT01788631: completed |
| Canakinumab | IL-1β inhibitor: targeting IL-1β which is an end product of inflammation in SCD | Phase 2 | NCT02961218: completed, results not published |
2,3-DPG= 2,3-diphosphoglycerate; ASH = American Society of Hematology; cGMP= cyclic guanosine monophosphate; FDA = Food and Drug Administration; HbF = hemoglobin F; HbS = hemoglobin S; HDAC= histone deacetylase; IL-1β = interleukin 1 beta; iNKT = invariant natural killer T cell; NAD = nicotinamide adenine dinucleotide; NADH = NAD + hydrogen (H); PK = pyruvate kinase; SCD = sickle cell disease.
Endari (L-glutamine)
L-glutamine is an essential amino acid that evolved as an anti-sickle agent through its role as a precursor for the synthesis of glutathione, nicotinamide adenine dinucleotide (NAD), and arginine, all of which protect erythrocytes from oxidative damage and indirectly maintain vascular tone.50,51 Early studies by Nihara et al52 in 7 SCD patients showed significant increases in nicotinamide adenine dinucleotide - hydrogen (NADH) and NAD redox potential, but no change in Hb concentration. In a follow-up study, erythrocytes from SCD patients who were administered L-glutamine decreased endothelial adhesion in vitro; findings interpreted as glutamine having a role in maintaining RBC membrane integrity and its interaction with the blood vessels and adhesion molecules.53 In 2017, L-glutamine became the second drug to be licensed by the FDA for patients 5 years or older with SCD (Table 2). The approval was based on a double-blind phase III trial in which 230 children and adults with either HbSS or HbS/β0 thalassemia were randomized to receive L-glutamine or placebo for 48 weeks. Compared to placebo, L-glutamine was associated with 25% reduction in the number of vaso-occlusive crisis (VOC) events (median 3.0 versus 4.0; P = 0.005), 30% lower hospitalization rates (median 2.0 versus 3.0; P = 0.005), and reduced number of episodes of acute chest syndrome, respectively. Although there were significant increases in NADH and NAD redox potential, and decreased endothelial adhesion of ex vivo treated sickle erythrocytes, there were no changes in Hb or reticulocyte counts.54 To date, however, L-glutamine has been rejected by the European Medicines Agency because of its relatively small therapeutic effects, and concerns on the high drop-out rate of 36% in the treatment arm, and 24% in the placebo arm.
L-glutamine appears to be reasonably well tolerated, but adherence is poor due to its taste and route of administration (twice daily as oral powder). As it is an amino acid, one should be cautious in its use among SCD patients in whom renal and hepatic dysfunction are not uncommon.
Voxelotor (Oxbryta/GBT440)
Voxelotor (also known as Oxbryta or GBT440) is the second anti-sickling agent that was approved by the FDA in November 2019 for the treatment of SCD in patients aged 12 years and older (Table 2). Voxelotor is anti-sickling because it stabilizes the oxygenated state of Hb through reversible binding to the amino terminus of alpha chain of Hb.55 The phase III Hemoglobin Oxygen Affinity Modulation to inhibit HbS Polymerization (HOPE) study (ClinicalTrials.gov: NCT03036813) was a randomized, placebo-control study of 274 patients of all SCD genotypes, age 12–65 years, in which voxelotor showed dose-dependent increase in Hb and decrease hemolysis markers, suggestive of decreased sickling.56 Although these findings did not correlate with a decrease in the number of pain crises in patients with SCD, the promising findings led to FDA approval in November 2019 for patients older than 12 years old with SCD. There is some concern, however, that Hb molecules with the drug bound are in a conformation that delivers very little oxygen, especially detrimental in a disease characterized by decreased oxygen delivery,57 in which case, the increase in Hb needs to be about the same as the concentration of the drug-bound, nonoxygen delivering Hb. Hopefully, these concerns are addressed in current multicenter phase III clinical studies in both adults (ClinicalTrials.gov: NCT03036813) and children (ClinicalTrials.gov: NCT02850406). In the meanwhile, it remains important to continue to monitor closely the patients while on this medication, particularly in those with prior stroke and silent cerebral infarcts. It should also be noted that HbS-voxelotor complexes, while useful in monitoring voxelotor therapy, causes interference with determination of HbS fraction in routine laboratory techniques—isoelectric-focusing gel, high-performance liquid chromatography, and capillary zone electrophoresis—of Hb fractionation.58
Crizanlizumab
Adhesion of the sickle erythrocytes and neutrophils with the vascular endothelium leads to upregulation of endothelial adhesion molecules—vascular cell adhesion molecule-1, intercellular adhesion molecule-1, and E and P selectins, facilitating vaso-occlusion. Crizanlizumab is a humanized monoclonal antibody that selectively inhibits P-selectin. The study to assess safety and impact of SelG1 with or without hydroxyurea therapy in sickle cell disease patients with pain crises (SUSTAIN) was a phase II multicenter, randomized, placebo-controlled double-blind study in which crizanlizumab was tested in 198 patients with SCD (on or not on HU) for its ability to reduce VOCs over a period of 52 weeks. Results showed a significant reduction of sickle cell-related pain crises per year in the high dose arm (5 mg/kg) as compared to the placebo (1.63 versus 2.98), and a low incidence of adverse events. Patients on the treatment arm also had an increased time-to-first VOC compared with placebo. Although side effects were relatively fewer in patients on crizanlizumab, 1 patient had an intracranial bleed. A phase III is currently ongoing to assess safety and efficacy of crizanlizumab, as this medication may alter platelet function. In November 2019, crizanlizumab (Adakveo) was FDA approved for reduction of VOCs in patients with SCD, 16 years or older (Table 2).59,60 It should be noted that crizanlizumab is a preventive therapy, administered intravenously over 30 minutes on week 0, 2, and every 4 weeks thereafter. There are recent concerns with crizanlizumab due to the increased reports of serious infusion and post-infusion reactions (https://www.crizanlizumab.info/), causing hematologists to discontinue therapy.61
Promising medications in the pipeline
Rivipansel (also known as GMI1070) is another agent targeting cell adhesion (Table 2), which was developed as a pan-selectin inhibitor, but has greatest activity against E-selectin. A phase II, randomized, placebo-controlled multicenter study in adolescents and adults showed the drug to be safe, and markedly reduced use of opioids during hospitalization (83% reduction compared to placebo) as well as a trend toward a faster resolution of VOC (41 versus 63 h).62 A phase III study of rivipansel in patients 6 years and older hospitalized for a pain crisis (ClinicalTrials.gov: NCT02187003) was recently completed, and although the drug did not reach its primary or key secondary endpoints, analyses suggested that early administration of rivipansel in vaso-occlusive events may reduce hospital stay and intravenous opioid use in pediatric and adult patients (https://doi.org/10.1182/blood-2020-134803). Although interesting, the clinical impact of rivipansel and its timely use as a preventive medication may be limited for the general SCD population.
All SCD patients have elevated pro-inflammatory cytokines (interleukin [IL]-6, tumor necrosis factor alpha [TNFα], and IL-1β), neutrophils, heme and other molecules with inflammatory potential, referred to as damage-associated molecular patterns.32 A number of anti-inflammatory agents have been investigated including corticosteroids and regadenoson, an adenosine A2A receptor agonist. Humanized sickle mouse demonstrated elevated levels of invariant natural killer T cells (iNKT) implicating their role in the pathogenesis of ischemia-reperfusion injury.63 Reduction of this subset of T cell (iNKT) activity ameliorated the inflammatory injury in the lungs in sickle mice,64 prompting studies in patients with SCD.65,66 Unfortunately, results showed that low-dose infusion of regadenoson was not sufficient to produce a statistically significant reduction in the activation of iNKT cells or in measures of clinical efficacy.66 Another study utilized the anti-iNKT cell monoclonal antibody NKTT120. High intravenous doses of NKTT120 were shown to decrease iNKT cells in adults with SCD. It should be noted, however, that the subjects in the study were in steady-state when iNKT cell activation was significantly lower compared to VOC.65 The implication is that, to be effective in VOC, much higher doses of NKTT120 (NKT Therapeutics, Inc.) may be needed.
IL-1β is a cytokine that is central in the inflammatory response and has also been shown to be elevated in subjects with SCD.67,68 Canakinumab is a humanized monoclonal antibody targeting IL-1β and has been approved by the FDA for treatment of rheumatological disorders in 2009. Its broader role as an inflammatory agent was demonstrated in subjects with previous myocardial infarcts,69 motivating an ongoing randomized double-blind placebo-controlled phase II study of subcutaneous canakinumab in patients with SCD aged 8–20 years old (ClinicalTrials.gov: NCT02961218) (Table 2). Preliminary results suggest that canakinumab improves pain scores, sleep, and school/work attendance (https://doi.org/10.1182/blood-2019-123355).
Despite high levels of HU-induced HbF, some patients continue to have sickle-related manifestations, which has been attributed to the uneven distribution of HbF among the RBCs. An alternative to increasing HbF synthesis that does not mimic stress erythropoiesis is to increase access of the transcription factors to the γ-globin genes by manipulation of the chromatin regulators (such as decitabine on DNA methylation and HDAC inhibitors). Hypermethylation of the upstream γ-globin promoter sequences is believed to be important in the Hb switch during which the γ genes are silenced by DNA methyltransferase 1 (DNMT1).70 This led to the use of 5-azacytidine, a first generation DNMT1 inhibitor, but it was quickly abandoned due to its toxicity and carcinogenicity.70 Decitabine, an analogue of 5-azacytidine, is also a potent DNMT1 inhibitor with a more favorable safety profile, but decitabine is rapidly deaminated and inactivated by cytosine deaminase if taken orally. To overcome this limitation, a clinical study combines decitabine and tetrahydrouridine (THU), a cytosine deaminase inhibitor, as a therapeutic strategy for inducing HbF (ClinicalTrials.gov: NCT01685515). A phase I study showed that decitabine-THU led to the inhibition of DNMT1 protein with induction HbF increase, and more importantly, HbF-enriched RBCs (F cells) increased to 80%. These agents did not induce cytoreduction but increased platelets count, which can be problematic in SCD patient and require further evaluation.71
HDACs are another group of regulatory molecules involved in epigenetic silencing of the γ-globin genes and have been considered as therapeutic targets for HbF induction (Table 2). Panobinostat is a pan HDAC inhibitor currently being tested in adult patients with SCD as a phase I study (ClinicalTrials.gov: NCT01245179). Increasing cellular cyclic guanosine monophosphate (cGMP) levels has also been proposed as one mechanism of HbF increase by HU.72 Phosphodiesterase 9 (PDE9) degrades cGMP, and it has been shown to be present in activated RBCs and neutrophils of patients with SCD. PDE9 inhibitors have been studied in clinical trials in patients with SCD with interesting results demonstrating elevation of HbF without deleterious effects in the bone marrow.73
Exciting drugs in the pipeline with anti-sickling properties have also been derived from a combination of bench and clinical observations. HbS polymerizes only when deoxygenated and its oxygenation is influenced by a few factors. One key factor influencing Hb oxygenation is the concentration of 2,3-diphosphoglycerate (2,3-DPG) in the RBC. Increased intracellular 2,3-DPG decreases oxygen binding and stabilizes the deoxygenated form (T form) of Hb, promoting sickling.19 It has been noted more than 50 years ago that 2,3-DPG levels in RBCs from SCD patients were significantly higher than that in healthy RBCs,74 and that adding 2,3-DPG to both healthy and SCD RBCs reduces Hb oxygen affinity.74 Decreasing 2,3-DPG as a therapeutic target has long been proposed by Poillon et al75 when they showed that considerable reduction of 2,3-DPG in sickle erythrocytes significantly reduced the sickling tendency. 2,3-DPG is an intermediate substrate in the glycolytic pathway, the only source of ATP production in RBCs. As pyruvate kinase (PK) is a key enzyme in the final step of glycolysis, enhancing its activity in red cells presents a very attractive therapeutic anti-sickling strategy as this leads to a decrease in 2,3-DPG, which increases Hb oxygenation with inhibition of the sickling process. Additionally, the concomitant increase in ATP levels restores ATP depletion in sickled RBCs and improves RBC membrane integrity. Currently, there are 3 ongoing phase I/II clinical studies of PK activation in SCD: 2 studies utilizing Mitapivat/AG-348 in HbSS patients in steady-state (ClinicalTrials.gov: NCT04000165; NCT04610866), and another (FT-4202) in healthy subjects and SCD patients (ClinicalTRials.gov: NCT03815695) (https://doi.org/10.1182/blood-2020-134269). Preliminary data showed that AG-348 data was well-tolerated and safe in subjects with SCD, and support dose-dependent changes in blood glycolytic intermediates consistent with glycolytic pathway activation accompanied by increases in Hb level and decreases in hemolytic markers (https://doi.org/10.1182/blood-2019-123113).
Mitapivat is also currently in phase II/III clinical trials in humans with PK deficiency76 (ClinicalTrials.gov: NCT02476916, NCT03548220, NCT03559699), as well as in an ongoing phase II study in subjects with nontransfusion-dependent thalassemia (ClinicalTrials.gov: NCT03692052).
Evolution of the curative approaches for SCD
Allogeneic transplantation
Hemopoietic stem cell transplantation (HSCT) had not been considered as a therapeutic option for SCD until 1984, prompted by the successful reversal of SCD in an 8-year-old SCD child who developed acute myeloid leukemia (AML).77 The patient received HSCT for the AML from a HLA-matched sister who was a heterozygous carrier for HbS (hemoglobin AS [HbAS]) (Table 1). She was cured of her leukemia and at the same time, her sickle cell complications also resolved.77,78 This successful HSCT demonstrated that reversal of SCD could be achieved without complete reversal of the hematological phenotype to normal hemoglobin genotype (HbAA), and as long as stable mixed hemopoietic chimerism after HSCT can be achieved.79
The outcomes for both children and adults who receive HLA-matched sibling donor hematopoietic stem cells (HSCs) are now excellent.80,81 Key milestones in making HLA-matched sibling donor HSCT an accepted curative option include: (1) the development of less intense conditioning regimens expanding allogeneic transplantation to adult patients who otherwise would not be able to tolerate the intense myeloablative conditioning82 and (2) that to reverse the sickle hematology, regardless of whether donors have normal hemoglobin genotype, HbAA, or are carriers for HbS (HbAS), only a minimum of myeloid chimerism of 20% is sufficient.83 Transplantation of HLA-matched sibling donor HSCs cures SCD, but to date, relatively few (~2000) patients with an average age of 10 years have benefited; the vast majority is excluded due to donor availability, toxicity related to myeloablative conditioning, and graft-versus-host disease (GvHD).81,84,85
To enable allogeneic HSCT as a therapeutic option to more patients with SCD, there is a major need to expand alternative donor sources of HSCs that include related haploidentical HSCs, matched unrelated donors, and cord blood. Of these, the most promising is related haploidentical allogeneic HSCT due to donor availability; post-transplantation cyclophosphamide has also improved safety with increased cure rates.86–88
While the overall survival was 94% in a study of unrelated cord blood transplantation for pediatric patients with SCD and thalassemia, the disease-free survival was not so good at about 50% in the SCD population.89 Compared to unrelated cord blood transplantation, related cord blood transplantation offers a better probability of success with a 2-year disease-free survival of 90% and a low risk of developing acute GvHD (11%) or chronic GvHD (6%) in pediatric patients with SCD.90
There are multiple clinical trials ongoing at this point at ClinicalTrials.gov that are assessing different techniques to improve the outcome of patients with SCD undergoing allogeneic HSCT. For more details of the different allogeneic HSCTs, we refer to a recent review.91
Autologous transplantation and genetic therapies
The genetic simplicity of the sickle mutation affecting an HSC lends itself to genetic therapies, an approach that eliminates the need to find a donor and thus, available to all patients (Table 3). Since these are the patient’s own stem cells, there is no need for immunosuppression, avoiding the risks of GvHD and immune-mediated graft rejection. Following gene modification in vitro, the patient’s own stem cells are reinfused after chemotherapy conditioning. Currently, there are 3 broad approaches: (1) Addition of lentiviral vectors (LVs) that express different versions of non- or anti-sickling genes, or a γ-globin coding sequence in a β-globin gene to increase HbF levels and decrease HbS; (2) addition of a LV that expresses erythroid-specific shRNA for BCL11A to downregulate its expression, thereby increasing γ-globin expression; and (3) editing of the BCL11A gene to delete the regulatory element controlling its expression in erythroid cells.
Table 3.
Gene Editing and Gene Therapies for Sickle Cell Disease.
| Title | ClinicalTrials.gov | Status | Mechanism | Notes |
|---|---|---|---|---|
| Gene therapies using lentiviral globin addition | ||||
| Safety and efficacy of LentiGlobin BB305 in β-thalassemia and SCD | NCT02151526 | Completed (March 10, 2020) | Lentiviral β-A-T87Q globin vector | Results published: DOI: 10.1056/NEJMoa1609677 |
| A study evaluating the safety and efficacy of the LentiGlobin BB305 drug product in severe SCD | NCT02140554 | Active, not recruitinga | BB305 lentiviral vector encoding the human β-A-T87Q globin gene | NCT03207009 and NCT02906202 related but for patients with β-thalassemia |
| Gene transfer for patients with SCD | NCT02186418 | Active, not recruiting | Autologous CD34+ hematopoietic stem cells transduced ex vivo with gamma-globin lentiviral vector | |
| Safety and feasibility of gene therapy with CSL200 | NCT04091737 | Active, not recruiting | Autologous enriched CD34+ cell fraction that contains CD34+ cells transduced with lentiviral vector encoding human γ-globinG16D and shRNA734 | |
| Stem cell gene therapy for SCD | NCT02247843 | Recruiting | βAS3 lentiviral vector-modified autologous peripheral blood stem cell transplant | |
| Safety and efficacy of gene therapy of the SCD with the lentiviral vector expressing the βAS3 globin gene in patients with SCD | NCT03964792 | Recruiting | Consists of autologous human CD34+ hematopoietic stem and progenitor cells that are enriched in CD34+ cells which have been transduced ex vivo with the lentiviral vector, expressing an βAS3 | |
| Gene transfer for SCD | NCT03282656 | Suspendedb | Lentiviral anti-BCL11A shRNA | Study paused per DSMB pending investigation of adverse event occurrence in an unrelated gene therapy study involving sickle cell patients (last update February 2021) |
| A study evaluating gene therapy with BB305 lentiviral vector in SCD | NCT04293185 | Suspendedb | CD34+ hematopoietic stem cells collected by plerixafor mobilization and apheresis, transduced with BB305 lentiviral vector encoding the human β-A-T87Q globin gene | Study suspended due to the occurrence of a suspected unexpected serious adverse reaction (last update March 2021) |
| Gene therapies using gene editing techniques | ||||
| Transplantation of CRISPR/Cas-9 corrected hematopoietic stem cells (CRISPR_SCD001) in patients with severe SCD | NCT04774536 | Not yet recruiting | Autologous CD34+ cell-enriched population that contains cells modified by the CRISPR/Cas-9 ribonucleoprotein | |
| Safety and efficacy of CRISPR/Cas-9 modified CD34+ hHSPCs | NCT03745287 | Recruiting | Autologous CD34+ hHSPCs modified with CRISPR/Cas-9 at the erythroid lineage-specific enhancer of the BCL11A gene | |
| Safety, tolerability, and efficacy of BIVV003 for autologous hematopoietic stem cell transplantation in patients with severe SCD | NCT03653247 | Recruiting | CD34+ cells transfected ex vivo with zinc finger nuclease messenger ribonucleic acid targeting the BCL11A locus | |
| Safety and efficacy of genome-edited hematopoietic stem and progenitor cells in SCD | NCT04443907 | Recruiting | Genome-edited autologous HSPC investigational drug product. Drugs: OTQ923 and HIX763 | Part C would include pediatric patients that received one of both experimental drugs |
aCurrently not recruiting due to 2 long-term follow-up patients developed myeloid malignancies.
bCurrently suspended due to findings of NCT02140554.
βAS3 = anti-sickling beta globin gene βAS3; BCL11A = B-cell lymphoma/leukemia 11A; CRISPR/Cas-9 = clustered regularly interspaced short palindromic repeats/CRISPR (C) associated nuclease-9; DSMB = Data and Safety Monitoring Board; hHSPCs = human hematopoietic stem and progenitor cells; SCD = sickle cell disease; shRNA = short hairpin RNA.
A critical component in autologous HSCT is the amount and quality of CD34+ cells that can be obtained from the patient. Historically, granulocyte colony-stimulating factor (GCS-F) had been used to obtain such cells in non-SCD patients, but the elevated white cell counts from GCS-F mobilization of CD34+ in SCD patients increases the risk of triggering acute severe pain, acute chest syndrome, and even death, and is thus contra-indicated in patients with SCD. Bone marrow harvest is another source, but CD34+ cells obtained from bone marrow harvests are suboptimal in quantity and quality, thus requiring multiple harvests, each harvesting procedure increasing the risk of triggering acute pain crisis. Development of plerixafor as an alternative approach has been crucial in optimization of CD34+ collection in patients with SCD. Plerixafor blocks the binding between chemokine CXC-receptor 4 and the stromal cell triggering mobilization of CD34+ cells into the peripheral blood stream without the uncontrolled increase of total white blood cells. Plerixafor in association with hyper-transfusion therapy has become the preferred way of mobilizing HSCs in patients with SCD.92–96
Two clinical trials (Table 3) have evolved from preclinical studies in SCD mice that showed that erythroid-specific down regulation of BCL11A is feasible and that it resulted in therapeutic elevation of HbF. One approach utilizes an shRNA embedded in a microRNA contained within a LV to limit knockdown of BCL11A to erythroid precursors.42 Of 6 patients with a median 18 months (range 7–29 mo) post-therapy, stable HbF induction of 20.4% to 41.15% was observed and the HbF was broadly distributed among the erythrocytes with F cells of 59% to 94%. Sickle complications were reduced or absent in all patients.42 The other approach utilized CRISPR-Cas editing to disrupt the key erythroid-specific enhancer in BCL11A leading to near normal Hb in 3 patients with HbF of >40% that was distributed pancellularly.43
Among the ongoing clinical trials on genetic therapy (Table 3), the most promising with the largest clinical experience relies on a lentivirus expressing a mutated β-globin βT87Q (LentiGlobin BB305) with anti-sickling properties.97 (https://ash.confex.com/ash/2020/webprogram/Paper134940.html) At the time of this review, 47 patients with SCD have been treated in 2 related clinical trials (ClinicalTrials.gov: NCT02140554 and NCT04293185).98 Unfortunately, reports of myelodysplasia and AML in 3 patients led to a temporary pause in enrolment; the clinical trial was allowed to resume when further investigation demonstrated integration of the LV to a nononcogenic gene with no disruption in expression of other genes in the vicinity. The conclusion was that the LV is unlikely to be implicated in cancer development.98,99 Exclusion of busulfan and insertional mutagenesis in these therapy-related leukemias, isolated reports of leukemias in SCD patients, with or without HU, pre-or post-transplantation,100 suggests that SCD patients may have a relatively increased risk of AML or myelodysplasia due to damage to hemopoietic stem cells related to chronic stress erythropoiesis. If so, it may be prudent to prescreen individuals with SCD for preleukemic progenitor cells as well as somatic mutations in genes involved in epigenetic regulation (DNMT3A, TET2, ASXL1), which are associated with an increased risk of developing blood cancers, referred to as clonal hematopoiesis of indeterminate potential (CHIP) origin. It has also been suggested that curative therapies should be performed in younger patients prior to acquisition of such CHIP variants or all patients should be screened for such variants prior to undergoing marrow conditioning.
Worldwide impact of SCD
SCD may have first appeared in the Western literature in 1910, but the clinical spectrum of SCD has been recognized in West Africa for centuries101 and probably existed in American slaves during the slavery period before 1910.102 Due to migration patterns, SCD is now worldwide, affecting millions globally, and the numbers are increasing.103,104 Nevertheless, SCD remains drastically more prevalent in historically malaria-endemic areas, such as sub-Saharan Africa, where carriers (HbAS) for the sickle mutation have a substantial protection against Plasmodium malariae infection. In a recent meta-analysis of SCD prevalence in subjects <5 years old, the birth prevalence of HbAS was estimated at >16,000 per 100,000 live births in Africa; much higher when compared to 800 per 100,000 live births in Europe.105–107
In 2010, an estimated 300,000 newborns were affected—projected to increase to 400,000 in 2050—of which more than 75% is in Africa. Unfortunately, 50%–80% of the infants born annually with SCD in Africa will not reach their fifth birthday. In the Republic of Congo, almost 12.5% of the pediatric patients hospitalized have SCD and the estimated annual cost of care for each of these patients is above 1000 United States dollars (USD).108 Trained personnel, access to vaccines, antibiotic prophylaxis, implementation of newborn screening, and blood products—all fundamental for the care and management of patients with SCD—are still limited resources in developing countries.109 The socioeconomic burden of SCD in Africa, and worldwide, will continue to increase with growth of the world’s population and human migration.
Although groundbreaking research is being performed in developed countries, access to the new medications—L-glutamine, voxelotor, and crizanlizumab—is limited in developing countries. In the meanwhile, studies have shown that HU is safe in malaria-endemic sub-Saharan Africa with no difference in incidence of malaria between children either on or off HU. The overall clinical benefit from HU therapy may even protect the recipients from severe effects of malaria.110–112 It should be noted, however, that prior to these studies, HU has already been demonstrated to be safe and effective as an alternative to regular blood transfusion therapy for prevention of secondary stroke in children with sickle cell anemia.113
Conclusions
SCD epitomizes the bidirectional translational research common to many other diseases. An astute observation of “elongated, sickle-shaped and crescent-shaped” RBCs has spurred the way to the uncovering of the first disease at a molecular level. Since then, SCD has been at the forefront of human genetic discovery, which has now translated into the first-in-human studies of reactivating an endogenous (γ-globin) gene utilizing innovative genomic approaches. A cure for this debilitating disease through HSCT and gene therapies is now within reach, but likely to remain available to a minority of the patients for the next few decades. A major unmet need for the vast majority now is a small molecule that targets the root cause of the disease and that can be taken orally. As new drugs and treatments are developed, it is essential that we find ways to make them accessible to all patients in both high- or low-resource countries.
Disclosures
The authors have no conflicts of interest to disclose.
Sources of funding
This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute and National Institutes of Health (SLT).
References
- 1.Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Arch Intern Med. 1910; 6:517–521. Reproduced with permission from JAMA Intern Med. 1910;6:517–521. Copyright © 1910 American Medical Association. All rights reserved. [PMC free article] [PubMed] [Google Scholar]
- 2.Herrick JB. Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. JAMA. 2014; 312:1063. [DOI] [PubMed] [Google Scholar]
- 3.Pauling L, Itano HA. Sickle cell anemia a molecular disease. Science. 1949; 110:543–548. [DOI] [PubMed] [Google Scholar]
- 4.Ingram VM. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature. 1956; 178:792–794. [DOI] [PubMed] [Google Scholar]
- 5.Goldstein J, Konigsberg W, Hill RJ. The structure of human hemoglobin. VI. The sequence of amino acids in the tryptic peptides of the beta chain. J Biol Chem. 1963; 238:2016–2027. [PubMed] [Google Scholar]
- 6.Lawn RM, Efstratiadis A, O’Connell C, et al. The nucleotide sequence of the human beta-globin gene. Cell. 1980; 21:647–651. [DOI] [PubMed] [Google Scholar]
- 7.Lauer J, Shen CK, Maniatis T. The chromosomal arrangement of human alpha-like globin genes: sequence homology and alpha-globin gene deletions. Cell. 1980; 20:119–130. [DOI] [PubMed] [Google Scholar]
- 8.Orkin SH. Molecular medicine: found in translation. Med (N Y). 2021; 2:122–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tisdale JF, Thein SL, Eaton WA. Treating sickle cell anemia. Science. 2020; 367:1198–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vinjamur DS, Bauer DE, Orkin SH. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br J Haematol. 2018; 180:630–643. [DOI] [PubMed] [Google Scholar]
- 11.Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007; 39:1197–1199. [DOI] [PubMed] [Google Scholar]
- 12.Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A. 2008; 105:1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat Biotechnol. 2020; 38:824–844. [DOI] [PubMed] [Google Scholar]
- 14.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014; 346:1258096. [DOI] [PubMed] [Google Scholar]
- 15.Cretegny I, Edelstein SJ. Double strand packing in hemoglobin S fibers. J Mol Biol. 1993; 230:733–738. [DOI] [PubMed] [Google Scholar]
- 16.Sunshine HR, Hofrichter J, Ferrone FA, et al. Oxygen binding by sickle cell hemoglobin polymers. J Mol Biol. 1982; 158:251–273. [DOI] [PubMed] [Google Scholar]
- 17.Eaton WA, Hofrichter J. Sickle cell hemoglobin polymerization. Adv Protein Chem. 1990; 40:63–279. [DOI] [PubMed] [Google Scholar]
- 18.Cellmer T, Ferrone FA, Eaton WA. Universality of supersaturation in protein-fiber formation. Nat Struct Mol Biol. 2016; 23:459–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eaton WA, Bunn HF. Treating sickle cell disease by targeting HbS polymerization. Blood. 2017; 129:2719–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beutler E. The effect of methemoglobin formation in sickle cell disease. J Clin Invest. 1961; 40:1856–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watson J. The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci. 1948; 215:419–423. [DOI] [PubMed] [Google Scholar]
- 22.Thein SL, Wood WG. The molecular basis of β thalassemia, δβ thalassemia, and hereditary persistence of fetal hemoglobin. In: Steinberg MH, Forget BG, Higgs DR, Weatherall DJ, eds. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management. 2nd ed. Cambridge, United Kingdom: Cambridge University Press; 2009:323–356. [Google Scholar]
- 23.Garner C, Tatu T, Reittie JE, et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood. 2000; 95:342–346. [PubMed] [Google Scholar]
- 24.Thein SL, Menzel S, Lathrop M, et al. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet. 2009; 18:R216–R223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Forget BG. Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci. 1998; 850:38–44. [DOI] [PubMed] [Google Scholar]
- 26.Orkin SH. Globin gene regulation and switching: circa 1990. Cell. 1990; 63:665–672. [DOI] [PubMed] [Google Scholar]
- 27.Platt OS, Orkin SH, Dover G, et al. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest. 1984; 74:652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Charache S, Terrin ML, Moore RD, et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the multicenter study of hydroxyurea in sickle cell anemia. N Engl J Med. 1995; 332:1317–1322. [DOI] [PubMed] [Google Scholar]
- 29.Wang WC, Ware RE, Miller ST, et al. Hydroxycarbamide in very young children with sickle-cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011; 377:1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang D, Xu C, Manwani D, et al. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. 2016; 127:801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaul DK, Finnegan E, Barabino GA. Sickle red cell-endothelium interactions. Microcirculation. 2009; 16:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gladwin MT, Ofori-Acquah SF. Erythroid DAMPs drive inflammation in SCD. Blood. 2014; 123:3689–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coletta M, Hofrichter J, Ferrone FA, et al. Kinetics of sickle haemoglobin polymerization in single red cells. Nature. 1982; 300:194–197. [DOI] [PubMed] [Google Scholar]
- 34.Mundee Y, Bigelow NC, Davis BH, et al. Simplified flow cytometric method for fetal hemoglobin containing red blood cells. Cytometry. 2000; 42:389–393. [DOI] [PubMed] [Google Scholar]
- 35.Steinberg MH, Lu ZH, Barton FB, et al. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Multicenter study of hydroxyurea. Blood. 1997; 89:1078–1088. [PubMed] [Google Scholar]
- 36.Steinberg MH, Chui DH, Dover GJ, et al. Fetal hemoglobin in sickle cell anemia: a glass half full? Blood. 2014; 123:481–485. [DOI] [PubMed] [Google Scholar]
- 37.Masuda T, Wang X, Maeda M, et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016; 351:285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martyn GE, Wienert B, Yang L, et al. Natural regulatory mutations elevate the fetal globin gene via disruption of BCL11A or ZBTB7A binding. Nat Genet. 2018; 50:498–503. [DOI] [PubMed] [Google Scholar]
- 39.Liu N, Hargreaves VV, Zhu Q, et al. Direct promoter repression by BCL11A controls the fetal to adult hemoglobin switch. Cell. 2018; 173:430–442.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu P, Keller JR, Ortiz M, et al. Bcl11a is essential for normal lymphoid development. Nat Immunol. 2003; 4:525–532. [DOI] [PubMed] [Google Scholar]
- 41.Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013; 342:253–257.24115442 [Google Scholar]
- 42.Esrick EB, Lehmann LE, Biffi A, et al. Post-transcriptional genetic silencing of BCL11A to treat sickle cell disease. N Engl J Med. 2021; 384:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med. 2021; 384:252–260. [DOI] [PubMed] [Google Scholar]
- 44.Orkin SH, Bauer DE. Emerging genetic therapy for sickle cell disease. Annu Rev Med. 2019; 70:257–271. [DOI] [PubMed] [Google Scholar]
- 45.Brendel C, Williams DA. Current and future gene therapies for hemoglobinopathies. Curr Opin Hematol. 2020; 27:149–154. [DOI] [PubMed] [Google Scholar]
- 46.Alter BP, Gilbert HS. The effect of hydroxyurea on hemoglobin F in patients with myeloproliferative syndromes. Blood. 1985; 66:373–379. [PubMed] [Google Scholar]
- 47.Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014; 312:1033–1048. [DOI] [PubMed] [Google Scholar]
- 48.Qureshi A, Kaya B, Pancham S, et al. Guidelines for the use of hydroxycarbamide in children and adults with sickle cell disease: a British Society for Haematology Guideline. Br J Haematol. 2018; 181:460–475. [DOI] [PubMed] [Google Scholar]
- 49.Gambero S, Canalli AA, Traina F, et al. Therapy with hydroxyurea is associated with reduced adhesion molecule gene and protein expression in sickle red cells with a concomitant reduction in adhesive properties. Eur J Haematol. 2007; 78:144–151. [DOI] [PubMed] [Google Scholar]
- 50.Quinn CT. l-Glutamine for sickle cell anemia: more questions than answers. Blood. 2018; 132:689–693. [DOI] [PubMed] [Google Scholar]
- 51.Morris CR, Hamilton-Reeves J, Martindale RG, et al. Acquired amino acid deficiencies: a focus on arginine and glutamine. Nutr Clin Pract. 2017; 32(1_suppl):30S–47S. [DOI] [PubMed] [Google Scholar]
- 52.Niihara Y, Zerez CR, Akiyama DS, et al. Oral L-glutamine therapy for sickle cell anemia: I. Subjective clinical improvement and favorable change in red cell NAD redox potential. Am J Hematol. 1998; 58:117–121. [DOI] [PubMed] [Google Scholar]
- 53.Niihara Y, Matsui NM, Shen YM, et al. L-glutamine therapy reduces endothelial adhesion of sickle red blood cells to human umbilical vein endothelial cells. BMC Blood Disord. 2005; 5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med. 2018; 379:226–235. [DOI] [PubMed] [Google Scholar]
- 55.Strader MB, Liang H, Meng F, et al. Interactions of an anti-sickling drug with hemoglobin in red blood cells from a patient with sickle cell anemia. Bioconjug Chem. 2019; 30:568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vichinsky E, Hoppe CC, Ataga KI, et al. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 2019; 381:509–519. [DOI] [PubMed] [Google Scholar]
- 57.Hebbel RP, Hedlund BE. Sickle hemoglobin oxygen affinity-shifting strategies have unequal cerebrovascular risks. Am J Hematol. 2018; 93:321–325. [DOI] [PubMed] [Google Scholar]
- 58.Rutherford NJ, Thoren KL, Shajani-Yi Z, et al. Voxelotor (GBT440) produces interference in measurements of hemoglobin S. Clin Chim Acta. 2018; 482:57–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 2017; 376:429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kutlar A, Kanter J, Liles DK, et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: a SUSTAIN study analysis. Am J Hematol. 2019; 94:55–61. [DOI] [PubMed] [Google Scholar]
- 61.Karkoska K, Quinn CT, Clapp K, et al. Severe infusion-related reaction to crizanlizumab in an adolescent with sickle cell disease. Am J Hematol. 2020; 95:E338–E339. [DOI] [PubMed] [Google Scholar]
- 62.Telen MJ, Wun T, McCavit TL, et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood. 2015; 125:2656–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wallace KL, Marshall MA, Ramos SI, et al. NKT cells mediate pulmonary inflammation and dysfunction in murine sickle cell disease through production of IFN-gamma and CXCR3 chemokines. Blood. 2009; 114:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wallace KL, Linden J. Adenosine A2A receptors induced on iNKT and NK cells reduce pulmonary inflammation and injury in mice with sickle cell disease. Blood. 2010; 116:5010–5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Field JJ, Lin G, Okam MM, et al. Sickle cell vaso-occlusion causes activation of iNKT cells that is decreased by the adenosine A2A receptor agonist regadenoson. Blood. 2013; 121:3329–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Field JJ, Majerus E, Gordeuk VR, et al. Randomized phase 2 trial of regadenoson for treatment of acute vaso-occlusive crises in sickle cell disease. Blood Adv. 2017; 1:1645–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Conran N, Belcher JD. Inflammation in sickle cell disease. Clin Hemorheol Microcirc. 2018; 68:263–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davila J, Manwani D, Vasovic L, et al. A novel inflammatory role for platelets in sickle cell disease. Platelets. 2015; 26:726–729. [DOI] [PubMed] [Google Scholar]
- 69.Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017; 377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 70.Charache S, Dover G, Smith K, et al. Treatment of sickle cell anemia with 5-azacytidine results in increased fetal hemoglobin production and is associated with nonrandom hypomethylation of DNA around the gamma-delta-beta-globin gene complex. Proc Natl Acad Sci U S A. 1983; 80:4842–4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Molokie R, Lavelle D, Gowhari M, et al. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: a randomized phase 1 study. PLoS Med. 2017; 14:e1002382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cokic VP, Andric SA, Stojilkovic SS, et al. Hydroxyurea nitrosylates and activates soluble guanylyl cyclase in human erythroid cells. Blood. 2008; 111:1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McArthur JG, Svenstrup N, Chen C, et al. A novel, highly potent and selective phosphodiesterase-9 inhibitor for the treatment of sickle cell disease. Haematologica. 2020; 105:623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Charache S, Grisolia S, Fiedler AJ, et al. Effect of 2,3-diphosphoglycerate on oxygen affinity of blood in sickle cell anemia. J Clin Invest. 1970; 49:806–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Poillon WN, Kim BC, Labotka RJ, et al. Antisickling effects of 2,3-diphosphoglycerate depletion. Blood. 1995; 85:3289–3296. [PubMed] [Google Scholar]
- 76.Grace RF, Rose C, Layton DM, et al. Safety and efficacy of mitapivat in pyruvate kinase deficiency. N Engl J Med. 2019; 381:933–944. [DOI] [PubMed] [Google Scholar]
- 77.Johnson FL, Look AT, Gockerman J, et al. Bone-marrow transplantation in a patient with sickle-cell anemia. N Engl J Med. 1984; 311:780–783. [DOI] [PubMed] [Google Scholar]
- 78.Johnson FL. Bone marrow transplantation in the treatment of sickle cell anemia. Am J Pediatr Hematol Oncol. 1985; 7:254–257. [DOI] [PubMed] [Google Scholar]
- 79.Walters MC, Patience M, Leisenring W, et al. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant. 2001; 7:665–673. [DOI] [PubMed] [Google Scholar]
- 80.Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood. 2017; 129:1548–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brodsky RA, DeBaun MR. Are genetic approaches still needed to cure sickle cell disease? J Clin Invest. 2020; 130:7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hsieh MM, Kang EM, Fitzhugh CD, et al. Allogeneic hematopoietic stem-cell transplantation for sickle cell disease. N Engl J Med. 2009; 361:2309–2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fitzhugh CD, Cordes S, Taylor T, et al. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood. 2017; 130:1946–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Joseph JJ, Abraham AA, Fitzhugh CD. When there is no match, the game is not over: alternative donor options for hematopoietic stem cell transplantation in sickle cell disease. Semin Hematol. 2018; 55:94–101. [DOI] [PubMed] [Google Scholar]
- 85.Leonard A, Tisdale JF. Stem cell transplantation in sickle cell disease: therapeutic potential and challenges faced. Expert Rev Hematol. 2018; 11:547–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bolaños-Meade J, Cooke KR, Gamper CJ, et al. Effect of increased dose of total body irradiation on graft failure associated with HLA-haploidentical transplantation in patients with severe haemoglobinopathies: a prospective clinical trial. Lancet Haematol. 2019; 6:e183–e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fitzhugh CD, Hsieh MM, Taylor T, et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical PBSCT. Blood Adv. 2017; 1:652–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de la Fuente J, Dhedin N, Koyama T, et al. Haploidentical bone marrow transplantation with post-transplantation cyclophosphamide plus thiotepa improves donor engraftment in patients with sickle cell anemia: results of an international learning collaborative. Biol Blood Marrow Transplant. 2019; 25:1197–1209. [DOI] [PubMed] [Google Scholar]
- 89.Ruggeri A, Eapen M, Scaravadou A, et al. Umbilical cord blood transplantation for children with thalassemia and sickle cell disease. Biol Blood Marrow Transplant. 2011; 17:1375–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Locatelli F, Rocha V, Reed W, et al. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood. 2003; 101:2137–2143. [DOI] [PubMed] [Google Scholar]
- 91.Salinas Cisneros G, Thein SL. Recent advances in the treatment of sickle cell disease. Front Physiol. 2020; 11:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Uchida N, Leonard A, Stroncek D, et al. Safe and efficient peripheral blood stem cell collection in patients with sickle cell disease using plerixafor. Haematologica. 2020; 105:e497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hsieh MM, Tisdale JF. Hematopoietic stem cell mobilization with plerixafor in sickle cell disease. Haematologica. 2018; 103:749–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Boulad F, Shore T, van Besien K, et al. Safety and efficacy of plerixafor dose escalation for the mobilization of CD34+ hematopoietic progenitor cells in patients with sickle cell disease: interim results. Haematologica. 2018; 103:770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Esrick EB, Manis JP, Daley H, et al. Successful hematopoietic stem cell mobilization and apheresis collection using plerixafor alone in sickle cell patients. Blood Adv. 2018; 2:2505–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lagresle-Peyrou C, Lefrère F, Magrin E, et al. Plerixafor enables safe, rapid, efficient mobilization of hematopoietic stem cells in sickle cell disease patients after exchange transfusion. Haematologica. 2018; 103:778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ribeil JA, Hacein-Bey-Abina S, Payen E, et al. Gene therapy in a patient with sickle cell disease. N Engl J Med. 2017; 376:848–855. [DOI] [PubMed] [Google Scholar]
- 98.Leonard A, Tisdale JF. A pause in gene therapy: reflecting on the unique challenges of sickle cell disease. Mol Ther. 2021; 29:1355–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hsieh MM, Bonner M, Pierciey FJ, et al. Myelodysplastic syndrome unrelated to lentiviral vector in a patient treated with gene therapy for sickle cell disease. Blood Adv. 2020; 4:2058–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ghannam JY, Xu X, Maric I, et al. Baseline TP53 mutations in adults with SCD developing myeloid malignancy following hematopoietic cell transplantation. Blood. 2020; 135:1185–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Konotey-Ahulu FI. The sickle cell diseases. Clinical manifestations including the “sickle crisis.” Arch Intern Med. 1974; 133:611–619. [PubMed] [Google Scholar]
- 102.Savitt TL. Tracking down the first recorded sickle cell patient in Western medicine. J Natl Med Assoc. 2010; 102:981–992. [DOI] [PubMed] [Google Scholar]
- 103.Piel FB, Tatem AJ, Huang Z, et al. Global migration and the changing distribution of sickle haemoglobin: a quantitative study of temporal trends between 1960 and 2000. Lancet Glob Health. 2014; 2:e80–e89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Piel FB, Patil AP, Howes RE, et al. Global epidemiology of sickle haemoglobin in neonates: a contemporary geostatistical model-based map and population estimates. Lancet. 2013; 381:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wastnedge E, Waters D, Patel S, et al. The global burden of sickle cell disease in children under five years of age: a systematic review and meta-analysis. J Glob Health. 2018; 8:021103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Archer NM, Petersen N, Clark MA, et al. Resistance to Plasmodium falciparum in sickle cell trait erythrocytes is driven by oxygen-dependent growth inhibition. Proc Natl Acad Sci U S A. 2018; 115:7350–7355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Archer NM, Petersen N, Duraisingh MT. Fetal hemoglobin does not inhibit Plasmodium falciparum growth. Blood Adv. 2019; 3:2149–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tshilolo L, Aissi LM, Lukusa D, et al. Neonatal screening for sickle cell anaemia in the Democratic Republic of the Congo: experience from a pioneer project on 31 204 newborns. J Clin Pathol. 2009; 62:35–38. [DOI] [PubMed] [Google Scholar]
- 109.Rahimy MC, Gangbo A, Ahouignan G, et al. Effect of a comprehensive clinical care program on disease course in severely ill children with sickle cell anemia in a sub-Saharan African setting. Blood. 2003; 102:834–838. [DOI] [PubMed] [Google Scholar]
- 110.Opoka RO, Ndugwa CM, Latham TS, et al. Novel use of hydroxyurea in an African Region with Malaria (NOHARM): a trial for children with sickle cell anemia. Blood. 2017; 130:2585–2593. [DOI] [PubMed] [Google Scholar]
- 111.Tshilolo L, Tomlinson G, Williams TN, et al. Hydroxyurea for children with sickle cell anemia in sub-Saharan Africa. N Engl J Med. 2019; 380:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.John CC, Opoka RO, Latham TS, et al. Hydroxyurea dose escalation for sickle cell anemia in sub-Saharan Africa. N Engl J Med. 2020; 382:2524–2533. [DOI] [PubMed] [Google Scholar]
- 113.Lagunju IA, Brown BJ, Sodeinde OO. Stroke recurrence in Nigerian children with sickle cell disease treated with hydroxyurea. Niger Postgrad Med J. 2013; 20:181–187. [PubMed] [Google Scholar]