

Abstract
Bis(phosphine) copper hydride complexes are uniquely able to catalyze direct dearomatization of unactivated pyridines with carbon nucleophiles, but the mechanistic basis for this result has been unclear. Here we show that, contrary to our initial hypotheses, the catalytic mechanism is monometallic and proceeds via dearomative rearrangement of the phenethylcopper nucleophile to a Cpara-metalated form prior to reaction at heterocycle C4. Our studies support an unexpected heterocycle-promoted pathway for this net 1,5-Cu-migration beginning with a doubly dearomative imidoyl-Cu-ene reaction. Kinetics, substituent effects, computational modeling, and spectroscopic studies support the involvement of this unusual process. In this pathway, the CuL2 fragment subsequently mediates a stepwise Cope rearrangement of the doubly dearomatized intermediate to the give the C4-functionalized 1,4-dihydropyridine, lowering a second barrier that would otherwise prohibit efficient asymmetric catalysis.
Graphical Abstract
Dearomative addition reactions of carbon nucleophiles (Figure 1, A–D)1 build up complexity starting from relatively abundant heteroaromatic substrates and give access to products spanning several oxidation states, including pyridines and piperidines, the two most common heterocycles in FDA-approved drugs.2 However, harsh reagents or reaction conditions are typically required for direct organometallic addition to pyridine (Figure 1, A),1c,3 and catalysis of such reactions (Figure 1, C) was undocumented,4 to our knowledge, until 2018.5–7 Broadly useful C–C-bond-forming dearomatization methods have instead relied on stoichiometric preactivation of the heterocycle (Figure 1, B) to achieve efficient reaction under mild conditions.1 This powerful tactic has been the subject of hundreds of methodological contributions from numerous research groups,1c and there now exist many options for effecting dearomatization with a specific sense of regioselectivity (i.e., 1,4-addition or 1,2-addition, Figure 1, B). However, these methods often require installation or removal of activating groups or preparation of non-commercial nucleophiles in separate operations, and thus commit significant effort to non-strategic transformations. Certain forms of stereocontrol have also been difficult to achieve within this reactivity paradigm. In particular, diastereofacial discrimination by nucleophiles approaching pyridinium ions at C4 is challenging,8 and asymmetric catalysis of 1,4-dearomatization9 remains rare1c despite numerous successes realizing 1,2-selective variants.10
Figure 1.
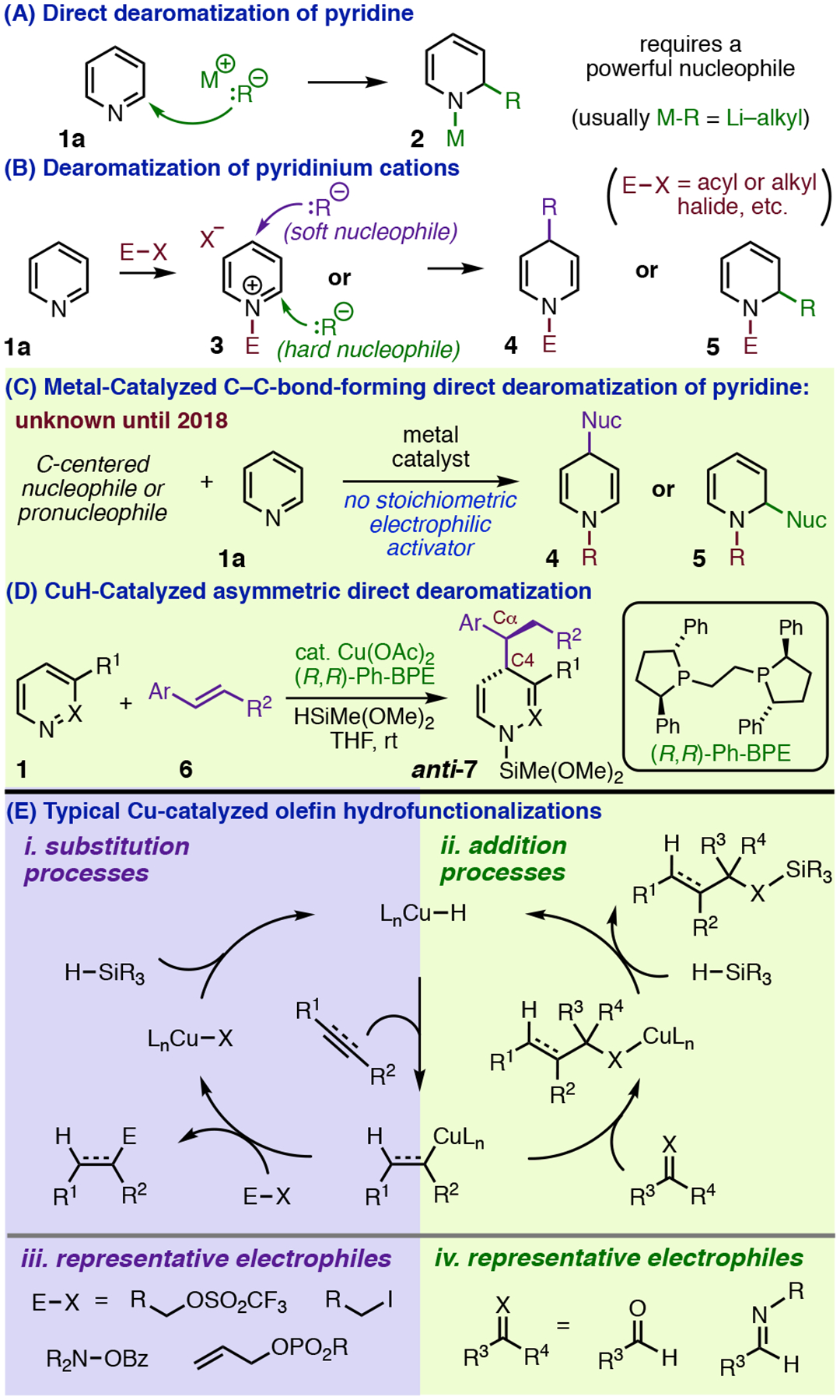
C–C-Bond-forming dearomatization of pyridine.
We recently described a new type of dearomative transformation (Figure 1, C) that operates directly on pyridine and generates the nucleophiles catalytically from olefin precursors (6, Figure 1, D),4 making it even more efficient than direct organometallic addition. The labile nature of the N-silyl group11 incorporated into the product also obviates separate protecting group manipulations. The reaction was notable for being both highly enantio- and 1,4-selective, and it gave 1,4-dihydropyridines (1,4-DHPs, i.e., anti-7, Figure 1, D) with very high diastereoselectivity in all but a few cases.4 However, we did not have a clear mechanistic rationale for these selectivity outcomes. The fundamental result that pyridine reacts so readily under these conditions was also surprising given that it is a traditionally problematic dearomatization substrate and far less intrinsically reactive than the kinds of electrophiles typically used in CuH-catalyzed hydrofunctionalization reactions (Figure 1, E).12
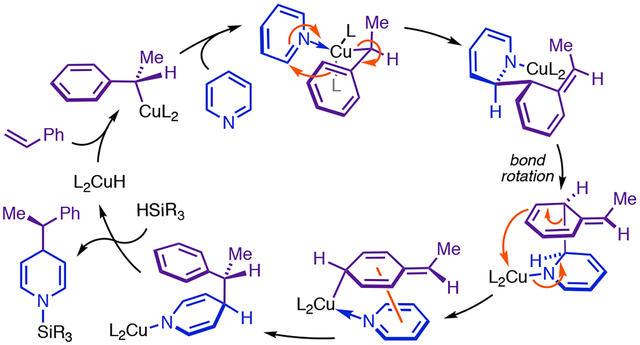
Our early observations suggested that two CuL2 fragments were likely involved in the dearomative addition step (Figure 2, A).4 However, the mechanistic investigations presented in this article show that the major pathway exclusively involves monometallic intermediates and entails dearomatization of the phenethylcopper nucleophile (9a, Figure 2, A and B) prior to its ultimate delivery to heterocycle C4 (see Figure 2, B, ii for labeling conventions). Based on pioneering work by Schomaker,13 we initially thought that 9a might undergo stepwise dearomative 1,5-Cu-migration (steps ii + iii, Figure 2, B, i) to give Cpara-metalated intermediate 13aa (after coordination, step iv, Figure 2, B, i), which we hypothesized would generate 10aa through a polar isomerization topologically equivalent to a 5,5-sigmatropic rearrangement (step v, Figure 2, B, i). While our experiments support the involvement of both 13aa and the 5,5-rearrangement in the major pathway, they also indicate that the heterocycle must be functioning as an electrophile in the most difficult step of the phenethyl isomerization. In our revised pathway, doubly dearomative imidoyl-Cu-ene reaction14 of 14aa (step iii, Figure 2, B, ii) provides an unexpectedly facile pathway for net 1,5-Cu-migration of the phenethyl group, with 13aa forming via retro-ene reaction (step v, Figure 2, B, ii) of transient 1,2-DHP 15aa. Notably, the retro-ene and 5,5-rearrangement (steps v + vi, Figure 2, B, ii) can be viewed as a stepwise Cope reaction in which 13aa is a fleeting intermediate. Cu(I)-mediation of this Cope rearrangement in a two-step manner is critical for successful asymmetric catalysis and results in the reaction’s dramatic intolerance of para-substituted styrenes.
Figure 2.
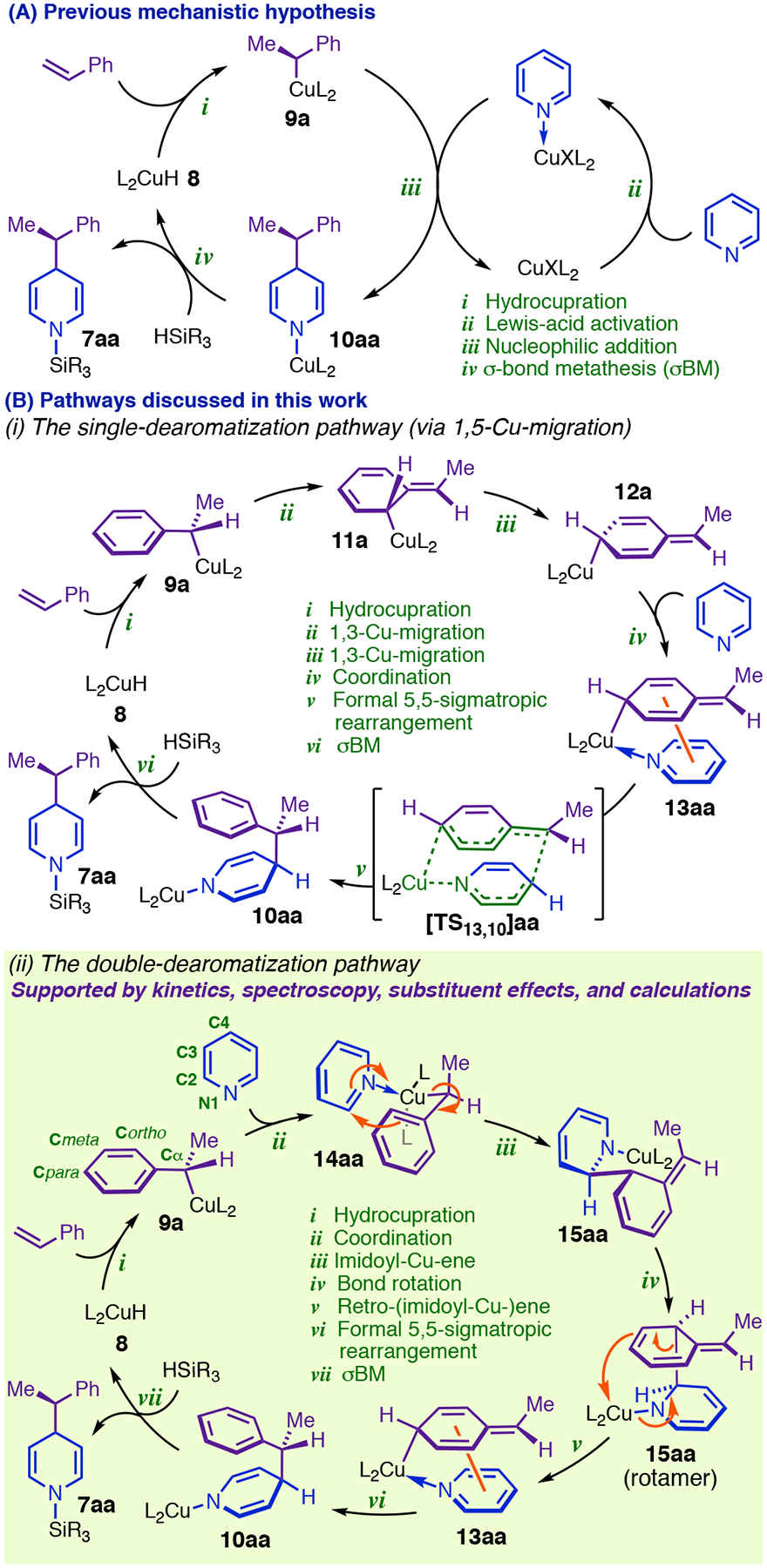
Mechanistic proposals for Cu-catalyzed direct dearomatization.
Background
At the outset, we imagined that Cu(I) might catalyze a new dearomative transformation analogous to the recently reported 1,2-α-phenethylation of imines (Figure 3, A).15 However we expected that dearomative addition of an organocopper nucleophile would require activation of the heterocycle by an N-bound Lewis acid, and we conjectured that the hydrosilane reductants used in CuH catalysis would be insufficiently electrophilic for this purpose (we have subsequently proven than the silane is not responsible for activation, vide infra). Lewis-acid activation by Cu(I) was on the other hand precedented,16 although addition via a monometallic intermediate seemed unlikely given that it would involve a highly distorted metalacyclobutane-like transition state (TS14,2, Figure 3, B). Bimetallic carbonyl-addition mechanisms (Figure 3, C–D)17 suggested that a more favorable transition state (TS18,2, Figure 3, E) might be attainable with a second CuL2 fragment bridging the nucleophile and heterocycle. However, when we exposed pyridine to DMMS, styrene, and precursors to [(S,S)-Ph-BPE]CuH, we indeed saw facile catalytic dearomatization but obtained 1,4-DHP 7aa almost exclusively (Figure 3, F).4 Although this regioselectivity was incompatible with our mechanistic hypothesis, a study of heterocycle reactivity trends nevertheless suggested that activation by Cu(I) was important as expected; in particular, C2-substitution on the heterocycle profoundly slowed the reaction, even with groups (e.g., CF3)4,18 that increase the intrinsic electrophilicity of the heterocycle but interfere with coordination to bulky Lewis acids.
Figure 3.
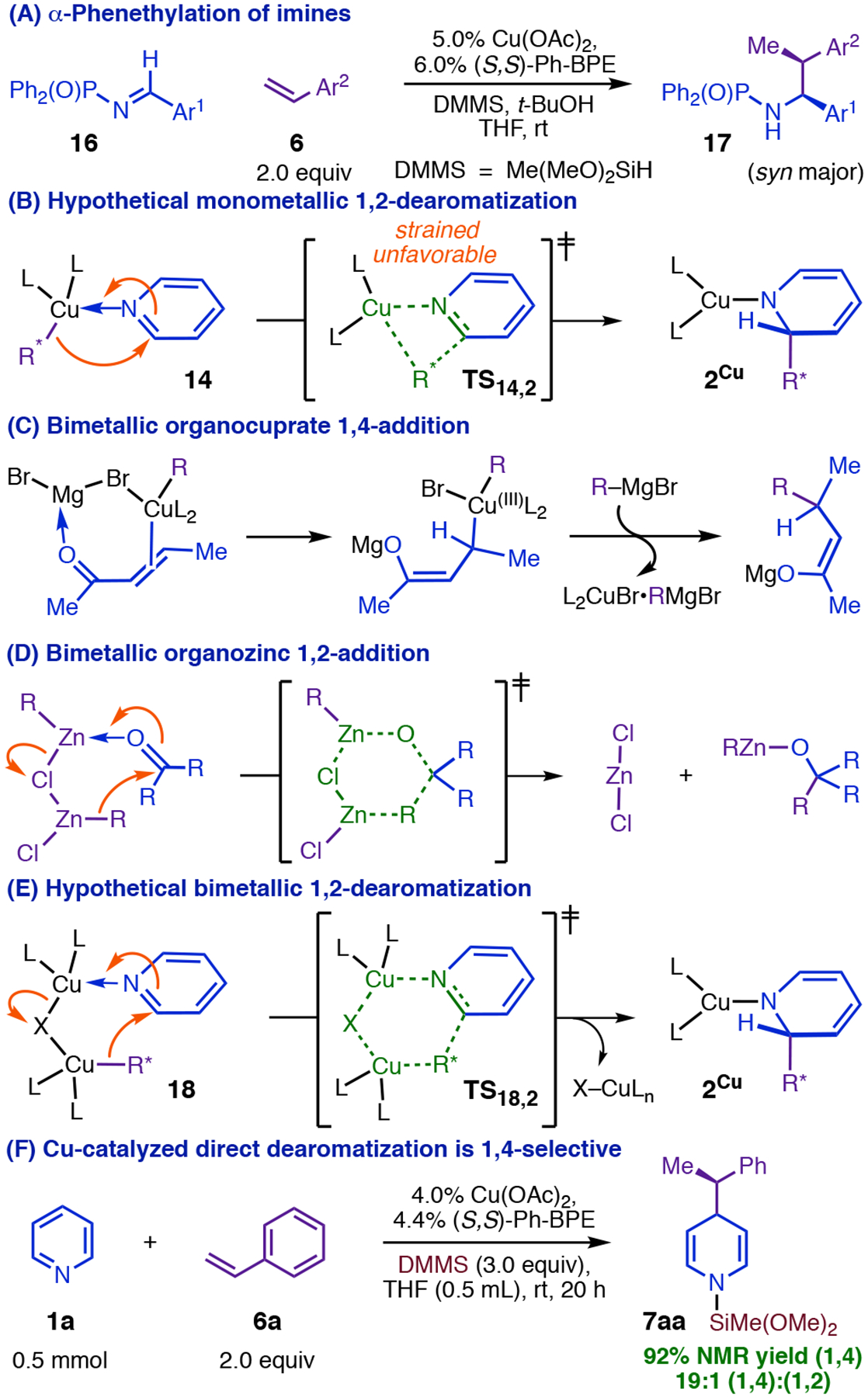
Cu-catalyzed direct dearomatization occurs with regioselectivity opposite that predicted.
C4-selective addition with Cu(I)-activation of both reaction partners was difficult to reconcile with monometallic (Figure 4, A) or closed bimetallic transition states (Figure, 3, E). However, open bimetallic transition states had structural precedent. For example, Aggarwal’s 1,4-dearomatization of pyridiniums with phenethylboronates8 occurs through an open transition state (TS19,7, Figure 4, B) with invertive addition by 19a at C4, and our group has proposed a very similar mode of attack (Figure 4, C) for stereoinvertive addition of phenethylcopper nucleophiles to imines.15 Planarization of Cα in Aggarwal’s reaction creates the potential for favorable π-stacking19 between the phenyl and pyridinium rings (Figure 4, B), leading to a preference for anti-selective addition, which avoids an otherwise disruptive clash between the Cα methyl and the out-of-plane carbonyl group at C3.8 We realized that we could explain one of the most surprising properties of the Cu-catalyzed dearomatization by invoking a similar π-stacking interaction (see TS9,10, Figure 4, D). With the exception of those bearing strong π-acceptor groups, styrenes with substituents at Cpara have generally been among the best substrates for the asymmetric olefin hydrofunctionalization reactions our group had previously reported,20 including all that use the catalyst system employed here. However, the presence of para-F or para-OMe groups completely suppressed catalytic dearomatization,4 while para-Me slowed the reaction twentyfold and caused profound erosion of the 1,4-selectively (Table 1). These findings cannot be attributed to simple electronic perturbation of the arene given that the corresponding ortho-substituted styrenes are all excellent dearomatization substrates (7ab, 7ad, 22ab, and 22ag, Table 1). Rather there must be a specific interaction, unique to the dearomatization mechanism, affecting Cpara substituents and one or more of the other reacting fragments. TS9,10 (Figure 4, D) exhibits such an interaction: formation of a parallel-displacement ππ-complex between 9 and the heterocycle would project a para-group toward the bulky coordination sphere of the CuL2 fragment bound at N1.
Figure 4.
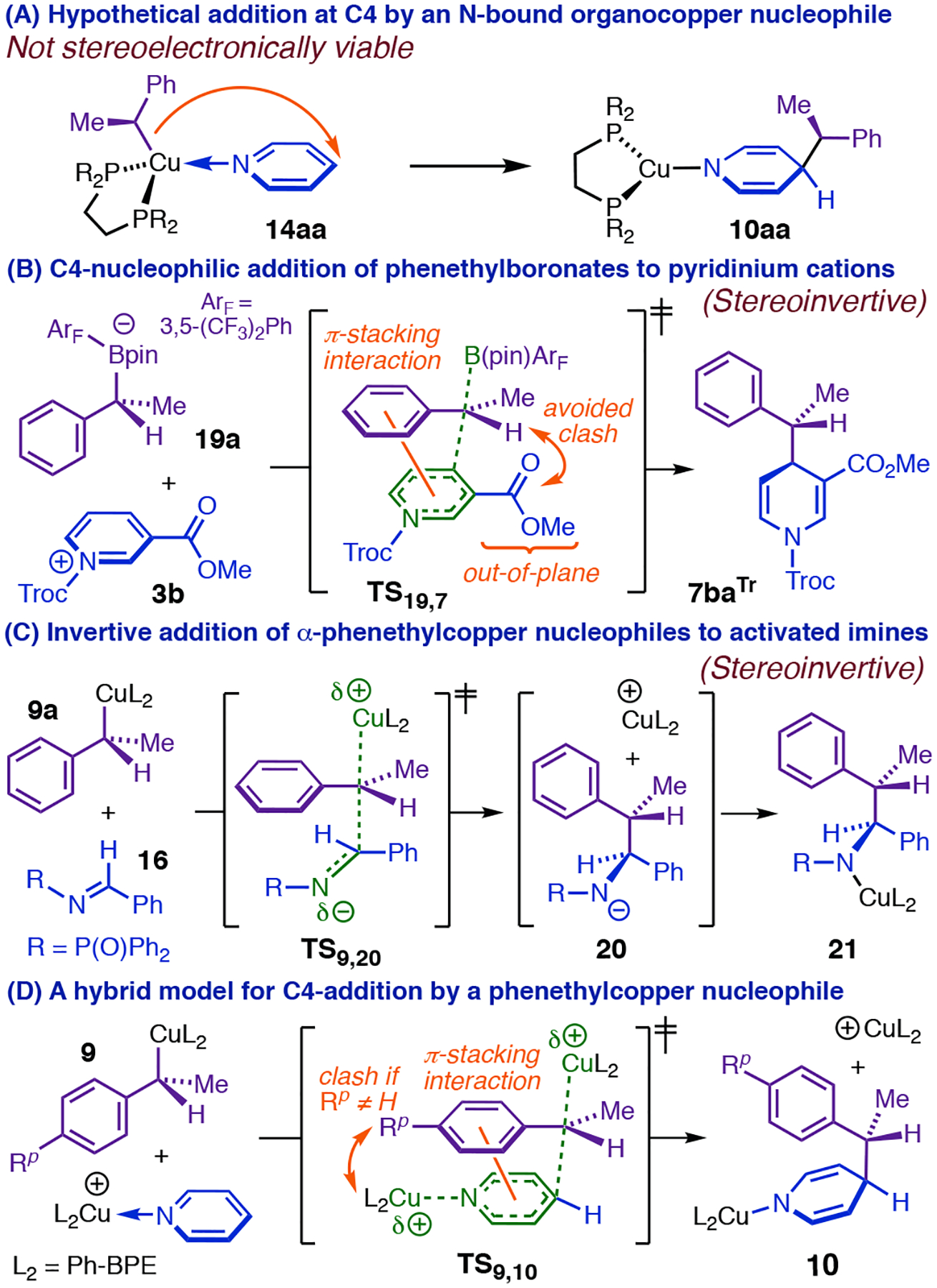
Development of an open bimetallic transition state model.
Table 1.
The Styrene-Para-Group Effect.
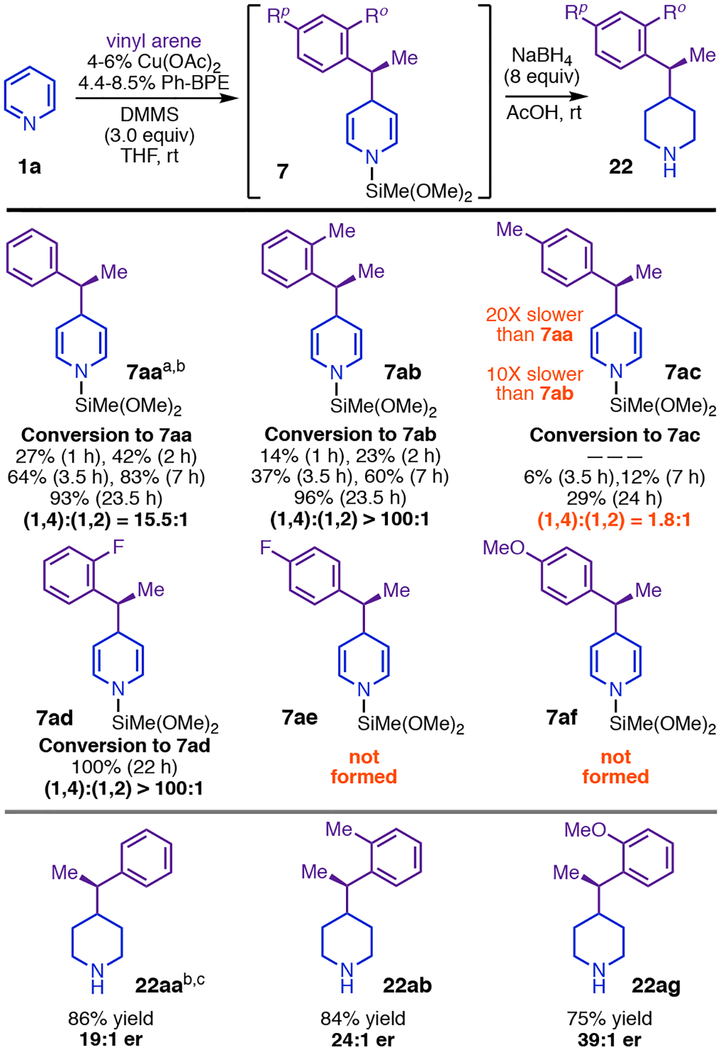
|
Reactions used (R,R)-Ph-BPE,
see supporting information for details,
reactions used (S,S)-Ph-BPE.
Despite its explanatory power, we had difficulty adopting the bimetallic transition-state model upon learning that the dearomatization is in fact highly selective for net retention of the Cα stereocenter in (S,S)S-9 (Table 2).4 We and others have shown that hydrocupration is enantiodetermining in various CuH-catalyzed reactions,21–23 which implies that the majority of product is generated by stereospecific reaction of the major phenethylcopper diastereomer with the electrophile. Thus, major product Cα-(R)-anti-7 (Table 2) cannot be formed via invertive attack by the nucleophile unless stereodetermination occurs in a fundamentally different manner in the dearomatization (i.e., achieves net retention through invertive preferential reaction of the minor phenethylcopper diastereomer). At the same time, we were unaware of precedent for models similar to TS9,10 (Figure 4, D) that might yield stereoretention, which would require the CuL2 fragment of the nucleophile to depart from the same face that the electrophile approaches. Such models would not entail planarization of Cα, and thus it seemed less assured that they would permit π-stacking or incur a para-group clash. Further, although the (anti):(syn) ratios we observed with substituted heterocycles (Table 2) did not exhibit easily intuited correlations with substituent properties, the selectivity trends were clearly difficult to rationalize by analogy to the stereochemical model for dearomatization with phenethylboronates. Highly diastereoselective phenethylboronate addition requires a sterically demanding out-of-plane carbonyl group at C3 (Figure 4, B);8 in contrast, the Cu-catalyzed dearomatization occurred with excellent selectivity with small C3 substituents like Me and MeO (12:1–30:1 dr; e.g., 7ca, Table 2)18 and gave much lower dr’s with nicotinates and nicotinamides (e.g., 7ba, Table 2). In fact, pyridazine, which lacks a C3 substituent altogether, gave one of the highest dr’s we have observed (≥ 28:1; 7ea Table 2).
Table 2.
Representative Stereochemical Outcomes.
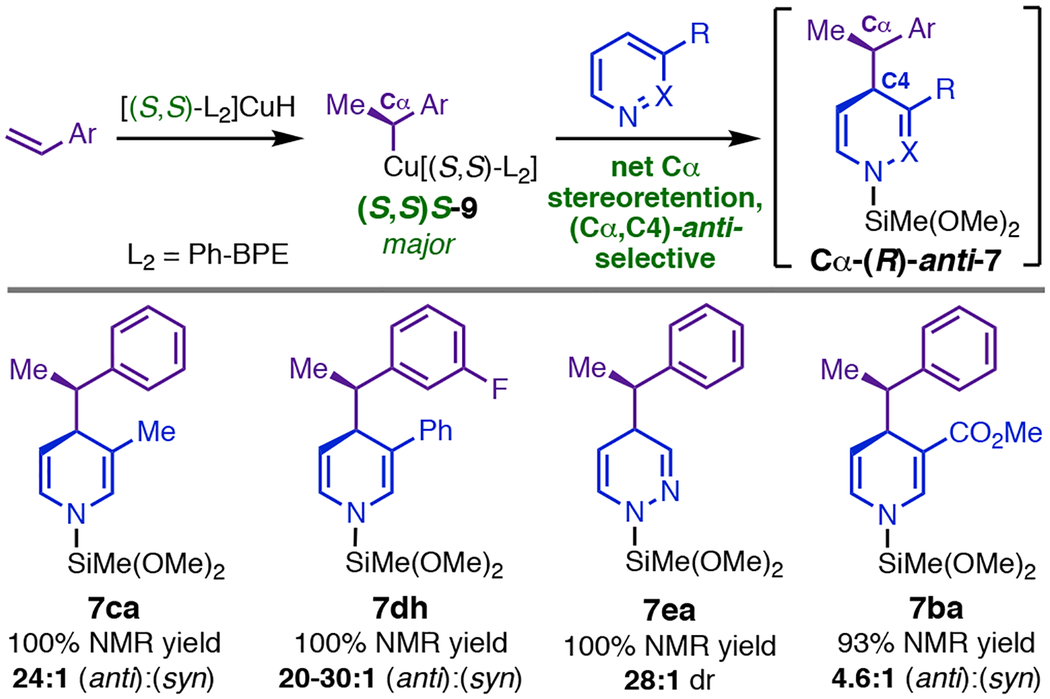
|
Results and Discussion
1. Identification of the catalyst resting state and observation of kinetically controlled phenethylcopper dr during catalysis.
Reaction kinetics could confirm or invalidate bimetallic pathways, provided that we knew the catalyst resting state. We expected that the molecular formula for the turnover-limiting transition state (TLTS) might be evident from kinetic orders if the resting state contained one catalyst species of known composition in large excess over all others (i.e., contained a MACS, most abundant catalyst species). In that case, the rate law would be determined by the linear sequence of steps starting from the MACS and ending with the turnover-limiting step (TLS).24 The dearomative step that consumes 9 (Table 2) was a likely TLS given its expected difficulty, and the formation of 9 in in a facile, exergonic step (calculated ΔG‡ = 12.2 kcal/mol; calculated ΔG° = −11.2 kcal/mol for styrene) thus suggested that the phenethylcopper nucleophile should be a major component of the resting state. Hartwig’s mechanistic studies of Cu-catalyzed hydroboration entailed characterizing phenethylcopper complexes derived from DTBM-SEGPHOS by 19F and 31P NMR spectroscopy and, in the case of minor diastereomer (S)S-9cSEG (Figure 5, A), by X-ray crystallographic analysis, showing these species to be monometallic, Y-shaped complexes.22 We find that analogous (S,S)-Ph-BPE complexes form as major hydrocupration products under conditions relevant to catalytic dearomatization. Successively exposing THF-d8 solutions of Cu(OAc)2 and (S,S)-Ph-BPE to DMMS and styrene-α-13C (Figure 5, B) gave mixtures containing three principal phosphorus-containing species, as shown by 31P NMR spectroscopy. One was free Ph-BPE, which resonated as a sharp singlet with the expected shift at −60 °C; this signal broadened at higher temperatures due to chemical exchange with the other two components. These latter species were clearly products of reaction with styrene, appearing as doublets (Figure 5, C) having similar coupling constants (2JP,C(S,S)S-9a = 22.5 Hz, 2JP,C(S,S)R-9a = 22.8 Hz). Suggestively of the epimerization process described by Hartwig for 9cSEG (Figure 5, A),22 the distribution of the hydrocupration products greatly favored the major species initially but approached an apparent equilibrium ratio < 1.8:1 over the course of four days. Confirming the minimal connectivity in 9a (Figure 5, B), the benzylic 13C resonances for the products were a pair of triplets (Figure 5, D) that occurred in the same ratio as the 31P doublets and exhibited triplet couplings virtually identical to the 2JP,C values of the respective corresponding 31P signals. We corroborated that the species assigned as (S,S)S-9a and (S,S)R-9a (Figure 5, B) were stereoisomers by introducing a much faster pathway for attaining the apparent diastereomeric equilibrium. When the Ph-BPE is enantiomerically pure, phenethylcopper diastereomers only interconvert by epimerization at Cα, via relatively slow β-hydride-elimination/reinsertion. However, we have found that the species assigned as (S,S)S-9a and (S,S)R-9a (Figure 5, B) undergo ligand exchange with free Ph-BPE on the NMR timescale.18 The result of a heterochiral ligand exchange reaction, e.g., (S,S)S-9a + (R,R)-Ph-BPE ⇌ (R,R)S-9a + (S,S)-Ph-BPE, is rapid interconversion of diastereomers by changing the configuration of the ancillary ligand. Thus, conducting hydrocupration with racemic Ph-BPE must immediately generate 9a (Figure 5, E) in its equilibrium diastereomer ratio. In the event, hydrocupration of styrene (6a) with racemic Ph-BPE gave [(R,R)R-9a + (S,S)S-9a] and [(R,R)S-9a + (S,S)R-9a] in a static ratio of 1.43:1 (Figure 5, E) by the time of the first analysis, from which we have calculated equilibrium constants of 0.699 and 0.489 for the respective processes in Figure 5, B and E.25 This rapid equilibration method enabled us to study the relative reactivity and selectivity of the individual phenethylcopper diastereomers toward catalytic dearomatization (vide infra).
Figure 5.
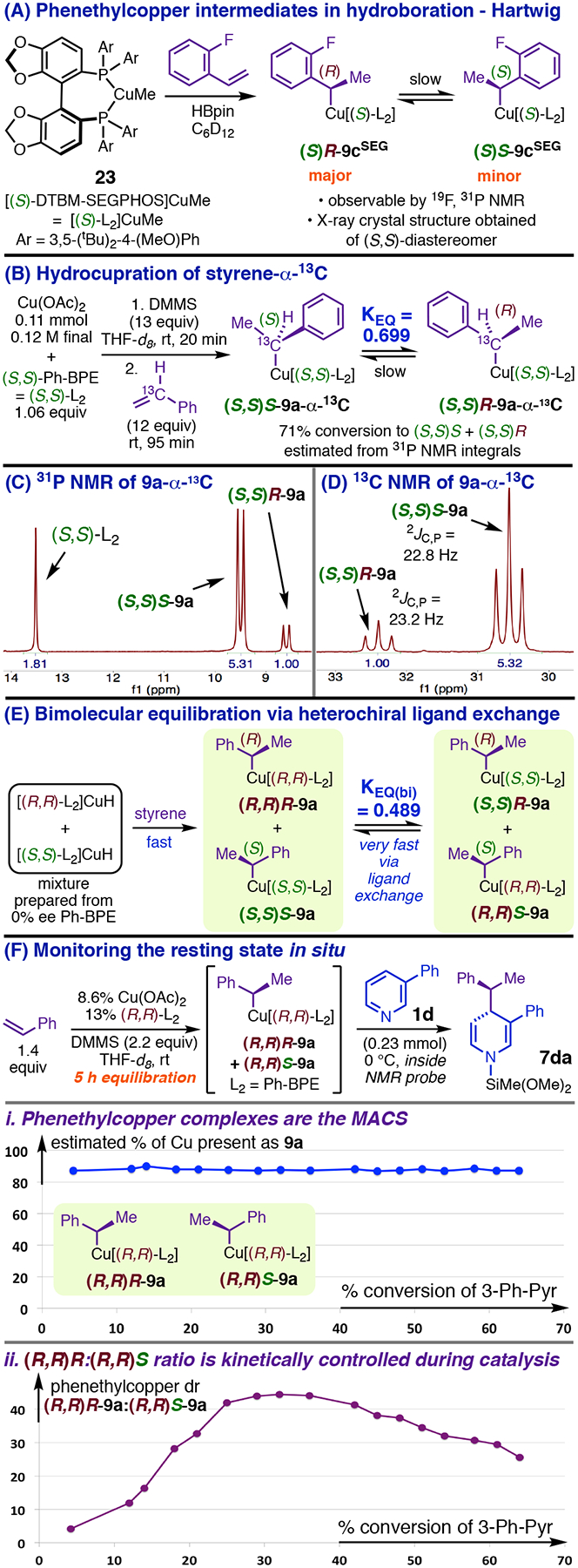
Phenethylcopper complexes are the MACS during dearomatization and exhibit mechanistically significant stereochemical dynamics.
The phenethylcopper complexes (particularly the major diastereomer) were indeed the MACS during dearomatization of pyridine or 3-Ph-pyridine with styrene or 3-fluorostyrene. Depending on the exact conditions, we estimated that total phenethylcopper made up ca. 70–88% of all copper-containing species (based on integration of the 31P NMR signals) after hydrocupration conducted with excesses of styrene and DMMS, and 75% (based on 19F integrals) to 90% (based on 31P integrals) of all copper-containing species after similar hydrocupration of kinetics model substrate 3-fluorostyrene.26 Apart from a distribution of low-level species, some of which formed in variable amounts and appeared to be decomposition products of the phenethylcopper complexes, no other observable copper compounds were present. Addition of 3-Ph-pyridine to a hydrocupration mixture led to rapid catalytic dearomatization (Figure 5, F) but did not generate any new observable catalyst species or markedly alter the approximate proportion of metal present as 9a (plotted using 31P-based estimates in Figure 5, F, i). However, we did observe mechanistically significant variation in the dr of 9a as the catalytic reaction proceeded (figure 5, F, ii). This experiment used 9a that had been aged in the absence of heterocycle for >5 h (Figure 5, F), during which time extensive epimerization occurred, giving an (R,R)R:(R,R)S ratio of 4:1 at the start of the catalytic reaction. Over the first 30% conversion of 1d (Figure 5, F), the (R,R)R:(R,R)S ratio increased to a maximum of 45:1 before gradually declining again at higher conversion (Figure 5, F, ii). Both phenethylcopper diastereomers present when the heterocycle is added are consumed by the dearomative addition step, leading to regeneration of [(R,R)-Ph-BPE]CuH. Hydrocupration then regenerates (R,R)R-9a with high kinetic selectivity in the presence of 1d (Figure 5, F), which leads to subsequent catalytic recycling occurring much faster than (R,R)R-9a epimerizes.27 This causes the initial dramatic increase in the (R,R)R:(R,R)S ratio. The catalyst dr gradually decreases at higher conversions because epimerization of (R,R)R-9a (which has rate ~ k[(R,R)R-9a]28 under these conditions) becomes increasingly competitive with turnover as the reactant required for dearomatization is depleted. It is implied that the rate expression for the TLS of the dearomatization has a nonzero kinetic order in at least one of the substrates.
2. The dearomative addition step is stereoretentive.
Mechanistic ramifications of the kinetically controlled catalyst dr enabled us to demonstrate that the invertive nucleophilic approach trajectory in Figure 4, B and C, cannot be operative in Cu-catalyzed dearomatization, invalidating the transition state model in Figure 4, D. For catalytic reactions that continuously regenerate the major diastereomer of 9a ((R,R)R-9a + (S,S)S-9a if both Ph-BPE enantiomers are present; Figure 5, E) with high kinetic selectivity, a straightforward equilibrium calculation implies that heterochiral ligand exchange (Figure 5, E) should generate resting states containing 9a in a steady-state diastereomer ratio governed by
| (eq 1), |
where ϕ is the mol fraction of Ph-BPE supplied as the major enantiomer, is the equilibrium constant (0.489) for heterochiral ligand exchange, and 2ψ is the steady-state mol fraction of 9a present as (R,R)S-9a + (S,S)R-9a (Figure 5, E).29 Thus one can generate catalyst mixtures having very different diastereomeric composition by varying the er of the ligand. This enabled us to correlate the rate and diastereoselectivity of the catalytic dearomatization with the proportion of 9a (Figure 5, F) present as the minor diastereomer.30,31 It became clear from those studies that (R,R)S-9a undergoes dearomative addition to 3-phenylpyridine at a comparable rate to (R,R)R-9a (Figure 5, F), but with severely eroded anti-selectivity.18 Given that catalytic dearomatization is highly anti-selective under typical conditions, this implies that reaction via (R,R)S-9a is not normally a major contributor to total product formation. It also signals that simple substrate-controlled interactions are not capable of explaining how diastereodetermination occurs in the dearomative addition. Having established that both diastereomers of 9 (Table 2) directly undergo dearomative addition, it became possible to show that (R,R)S-9 is not selective for the major 1,4-DHP enantiomer. Conducting dearomatization with 100% (R,R)-Ph-BPE under conditions implied by the observed catalyst stereochemical dynamics to increase the proportion of total product formed via (R,R)S-932,33 significantly erodes the enantioselectivity and diastereoselectivity of the catalytic reaction.18 Thus, only (R,R)R-9 selectively generates the major 1,4-DHP product enantiomer, and because these two species have the same absolute configuration at Cα, the dearomative addition step must itself be stereoretentive.
3. Dearomative addition involves a single CuL2 fragment: Empirical determination of the molecular formula of the TLTS for dearomatization of 3-phenylpyridine.
We began kinetics studies with the model reaction in Figure 6, A, which had ideal properties for calorimetric reaction-progress kinetics analysis.34 The near-perfect overlay of rate/[Cu] (Figure 6, B) for two reactions differing only in catalyst loading shows that the reaction is first-order in copper. This implies that no additional equivalents of Cu are incorporated into the catalyst during the sequence leading from the MACS to the TLTS, and because the MACS is monometallic, the TLTS can only contain one copper atom. The general bimetallic mechanism in Figure 2, A cannot be correct. Further, the linearity of the plots in Figure 6, C shows that the reaction is first-order in one reactant and zero-order in the others, and in that case, the invariance of the reaction rate when [DMMS] and [3-F-styrene] are varied simultaneously (Figure 6, C) implies that both have kinetic orders of zero.35 This means that no DMMS36 is incorporated into the TLTS, and that no 3-fluorostyrene is incorporated beyond that already present in 9h (Figure 6, A). It also means that the reaction is first-order in heterocycle. With regard to stoichiometry, the TLTS must be composed of one equivalent of the phenethylcopper complex, and one equivalent of 3-phenylpyridine (Figure 6, D).
Figure 6.
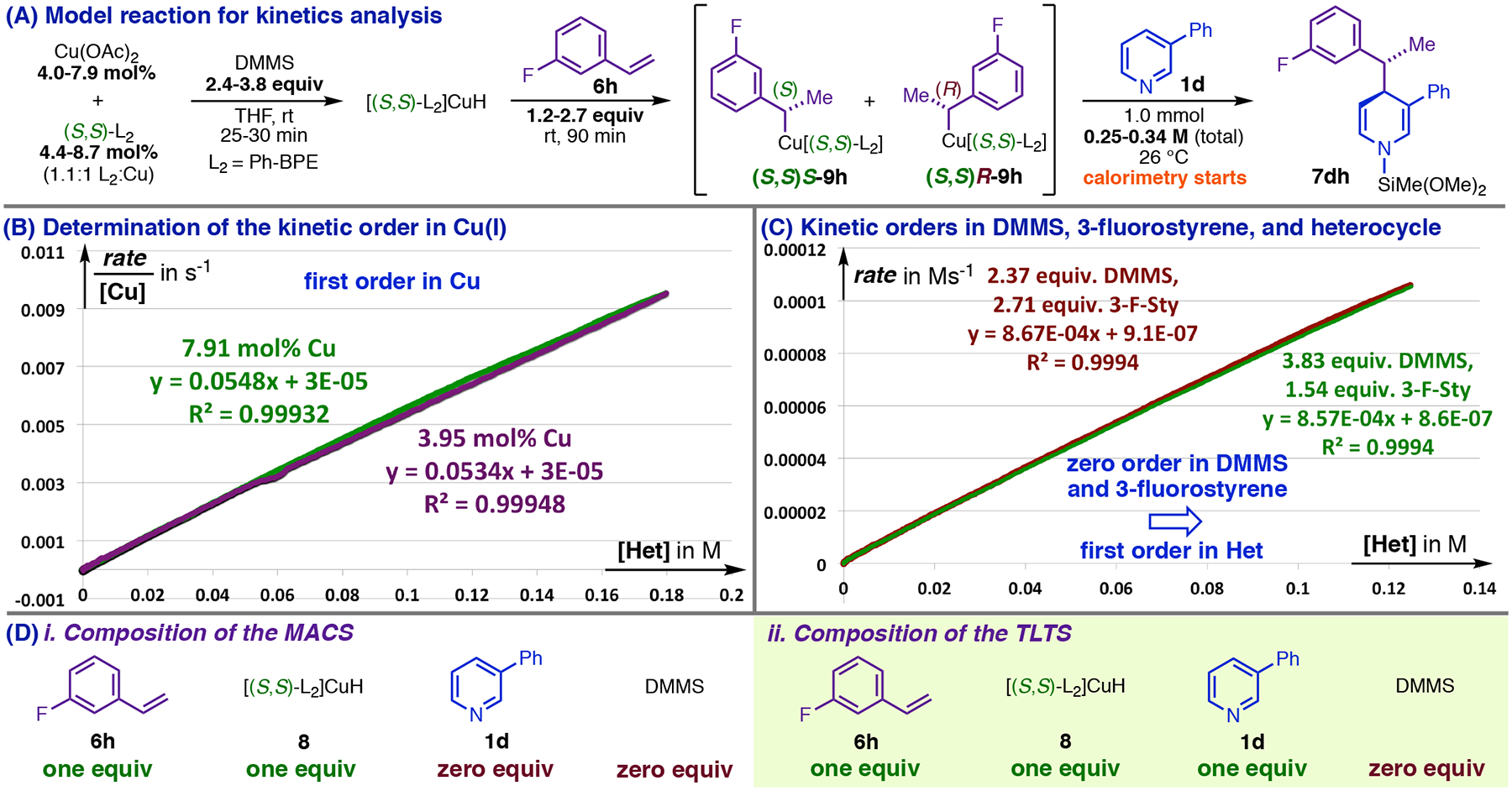
Determination of the molecular formula for the TLTS of dearomatization of 1d using reaction kinetics.
4. The single-dearomatization pathway: Rearrangement of 9 to a Cpara-metalated isomer and 5,5-rearrangement.
The reaction kinetics necessitated a major reappraisal of the likely requirements for dearomative reactivity. Specifically, we needed to formulate a stereoelectronically feasible mechanism by which a single CuL2 fragment simultaneously activates the electrophile and nucleophile while mediating transfer of the latter to a remote position on the heterocycle. We additionally knew that this pathway needed to retain the phenethylcopper Cα stereocenter and generate 1,4-DHP’s with robust anti-selectivity. It occurred to us that these conditions could be fulfilled simultaneously only if the C4-Cα bond-forming event occurred from an isomerized form of the phenethylcopper nucleophile. Schomaker has shown that Cortho-halogenated phenethylcopper complexes (e.g., 9i, Figure 7, A) undergo dearomative 1,3-Cu-migration en route to halogen-rearrangement products (24, Figure 7, A).13 Successive 1,3-migrations starting from 9a (Figure 7, B) would furnish 12a, whose dative complex (13aa), we reasoned, might generate N-Cu-1,4-DHP 10aa through a polar isomerization topologically equivalent to a 5,5-sigmatropic rearrangement (henceforth ‘5,5-rearrangement’; Figure 7, B). Modeling at the CPCM(THF)/BP86-D3(BJ)/6–311+G(d,p)–SDD(Cu)//B3LYP/6–31G(d)–SDD(Cu)37 level of theory located [TS13,10]aa 3.7 kcal/mol higher in energy than 13aa and situated 13aa only 8.5 kcal/mol higher than the MACS (Figure 7, C). It is noteworthy that 13aa is destabilized relative to 14aa (Figure 7, C) by far less (+6.6 kcal/mol) than the resonance energy of a phenyl ring (30–36 kcal/mol by various estimates).38 A serious penalty for dearomatization of the phenethyl group only becomes evident if the heterocycle is removed to give free 12a, which lies 17.3 kcal/mol above isomeric 9a (Figure 7, C). Given that the Cu–N dative bonds in 13aa and 14aa should be comparably stabilizing, the fact that heterocycle dissociation by 13aa is 10.7 kcal/mol more endergonic than dissociation by 14aa (Figure 7, C) suggests that secondary interactions between the 2,5-cyclohexadienyl ring and the heterocycle are important to stabilizing 13aa. The optimized structure for 13aa (Figure 8, A) supports this interpretation: the two rings are arranged in the characteristic geometry of a parallel-displacement π-stacking complex (distance between centroids = 3.58 Å for 13aa versus 3.5 Å for the parallel-displacement benzene dimer),39 permitting Coulombic interactions between rings over a large area (alignment of complementary charges is depicted with colored circles in Figure 8, A). The structures of 13aa and 5,5-rearrangement transition state [TS13,10]aa (Figure 8, B) closely emulate the π-stacking interaction conjectured to exist within bimetallic TS9,10 (Figure 8, B), although we note that dearomatization via 13aa would result in the opposite stereochemical outcome: In [TS13,10]aa (Figure 8, B), the CuL2 fragment of the nucleophile is present on the same face that the electrophile approaches, leading to retention of configuration at Cα.
Figure 7.
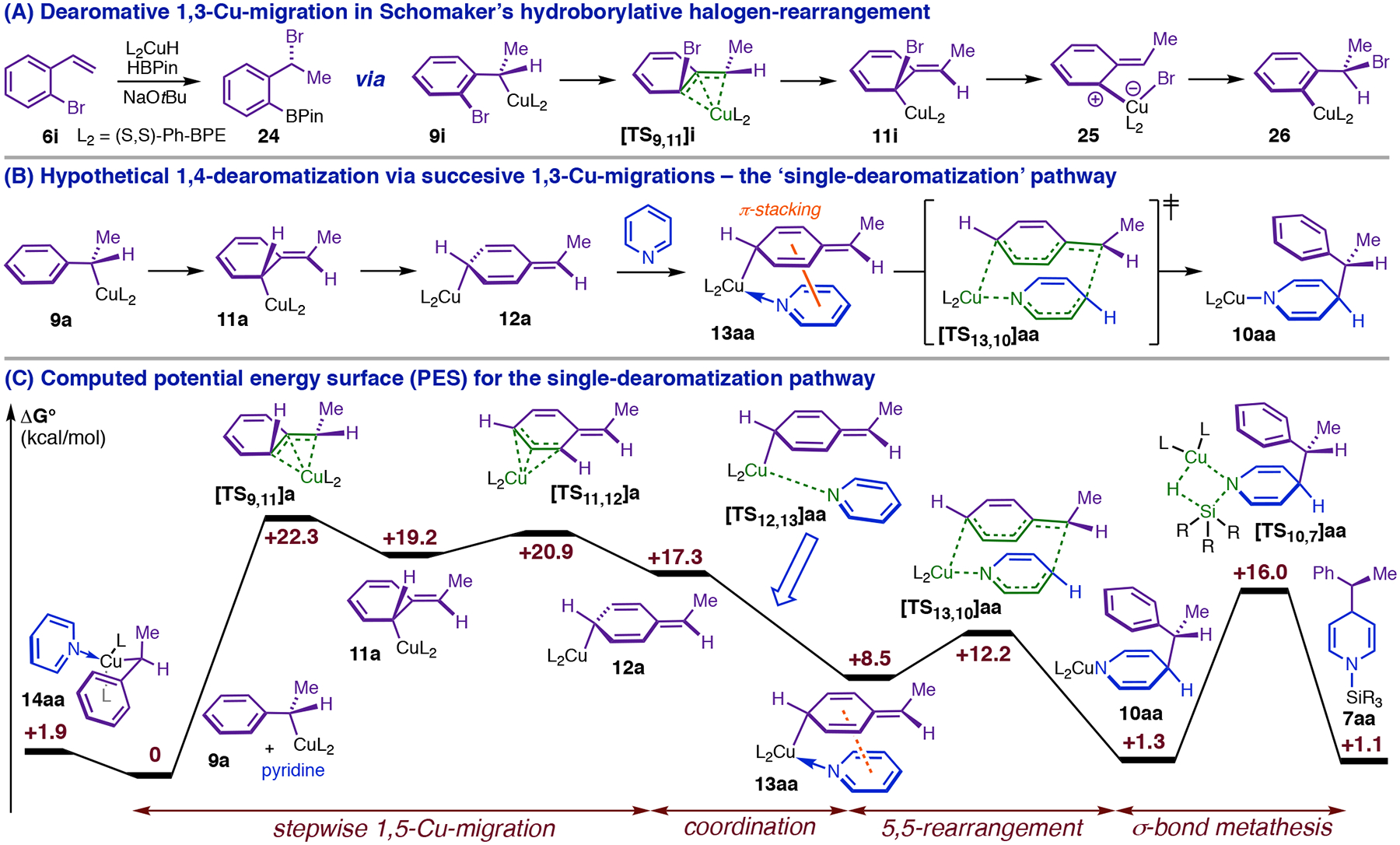
The single-dearomatization pathway: Precedent and computed energy surface.
Figure 8:
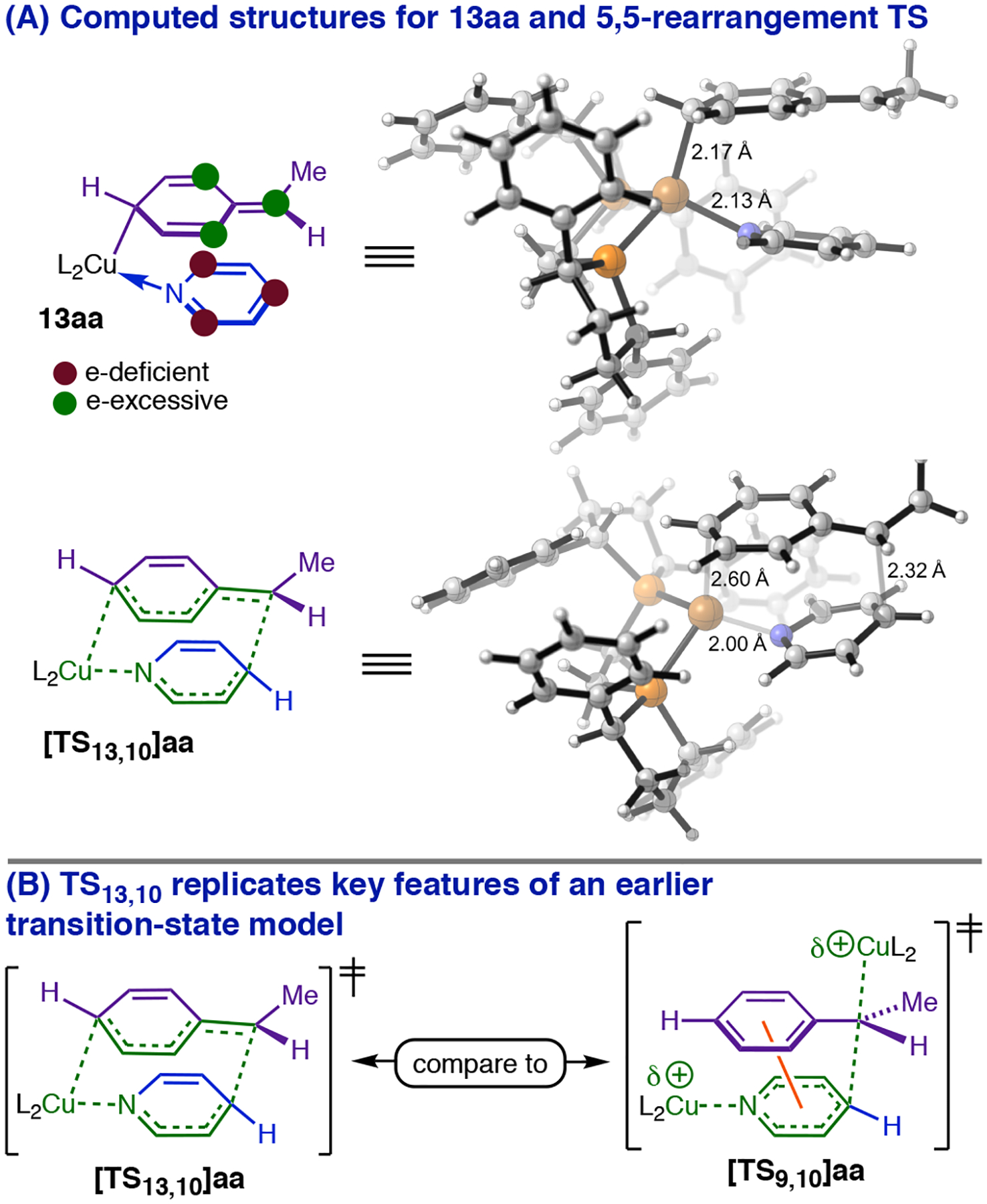
Models for Cpara-metalated intermediate 13aa and the 5,5-rearrangement transition state.
5. An imidoyl-Cu-ene pathway for dearomatizing the phenethylcopper nucleophile: Formulation and precedent.
Strong stabilization and a low barrier to [TS13,10]aa make 13aa a viable catalytic intermediate (Figure 7, C), but they also make the computed PES for the single-dearomatization mechanism inconsistent with the reaction kinetics. The most challenging step in the pathway (Figure 7, C) is the initial dearomative 1,3-Cu-migration, yielding predicted kinetic orders of zero for all reaction components except Cu(I). Further, heterocycle substituent effects on the rate of dearomatization (Table 3) imply that the heterocycle is not only present, but also behaving as an electrophile in the TLS: electron-donor C3-substituents slow the reaction (compare 1f to 1d and 1c to 1a, Table 3), whereas groups that stabilize negative charge markedly accelerate it (compare 1a to 1d and 1d to 1g, Table 3).40 The single-dearomatization mechanism would only predict the correct rate law and substituent effects if the transition state for 5,5-rearrangement (corresponding to [TS13,10]aa in Figure 7, C) were in general the true energy maximum of the dearomatization PES, and this would require dramatic changes to the energies in Figure 7. Further, the (para-MsPh) group in 1g (Table 3) is sufficiently activating to change the rate law, resulting in kinetic dependence on DMMS in addition to 1g (vide infra), which implies that the 5,5-rearrangement barrier (relative to the phenethylcopper) and the σBM barrier would both need to significantly surpass the 1,3-Cu-migration barrier for this substrate. As it is not easily reconciled with empirical rate data, we consider the single-dearomatization mechanism a problematic candidate for being the general 1,4-selective pathway. However, our modeling indicates that the intermediacy of 1,3-Cu-migration product 11a en route to 13aa (Figure 7, B) should enable formation of the 1,2-DHP regioisomer via an imidoyl-Cu-ene reaction with pyridine (Figure 9. A and B). Although 1,2-DHPs were formed in trace or undetectable quantities in the majority of the dearomatization reactions we have investigated, the single-dearomatization pathway is a plausible source of the minor 1,2-addition products we have observed with certain substrates.
Table 3.
Heterocycle Substituent Effects on the Rate of Dearomatization.
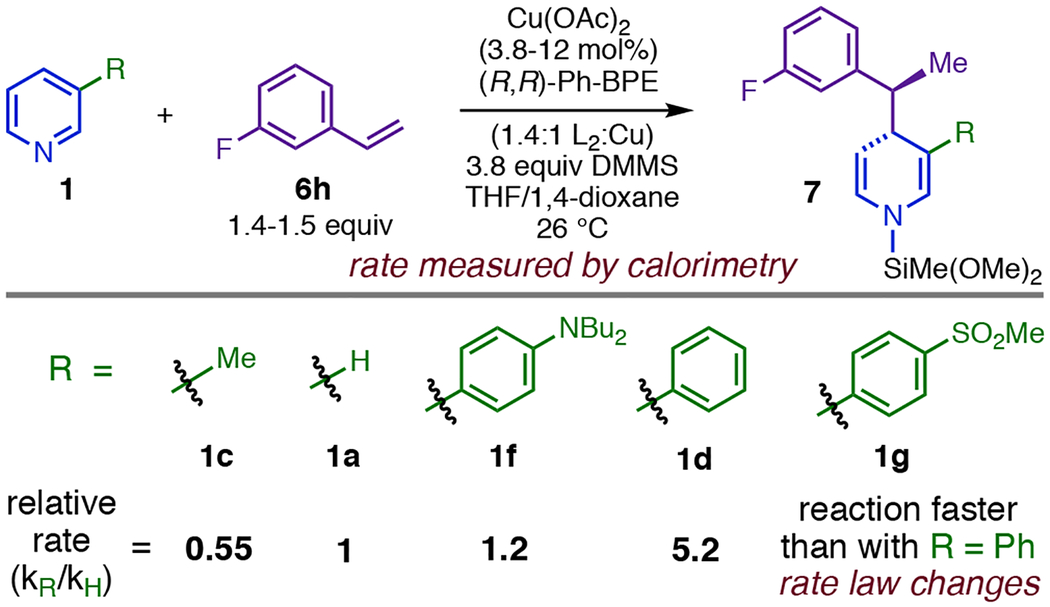
|
Figure 9.
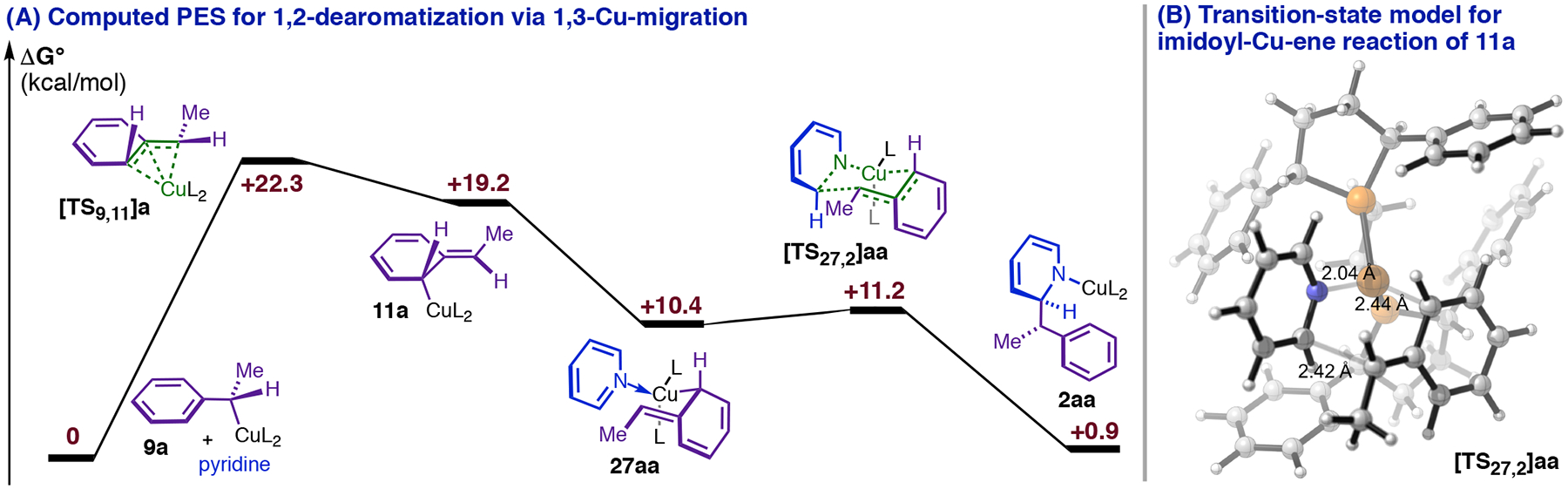
The 1,3-Cu-migration product 11a in the single-dearomatization pathway is predicted to undergo facile 1,2- addition.
Rearrangement of free 9a (Figure 7, B) is not the only way to generate Cpara-metalated intermediates. The inconsistencies described above are eliminated if coordination of heterocycle occurs before the 1,5-Cu-migration process. Brown’s C2-borylation of 3-haloheterocycles,14 which involves a chair imidoyl-Cu-ene reaction (28 → 29, Figure 10, A) suggests an alternative pathway to 13aa (Figure 8, A) involving a turnover-limiting polar reaction of the phenethylcopper nucleophile with the heterocycle. That an imidoyl-Cu-ene reaction might form the basis for a 1,4-selective process is evident from the reaction of diallylcalcium with pyridine,41 in which ene product 33 undergoes Cope rearrangement to C4-functionalized 1,4-DHP 34 (Figure 10, B). Yang has provided evidence for electrophilic dearomatization of phenethylcopper nucleophiles via an imidoyl-Cu-ene reaction in a Cortho-cyanation with tosylcyanamides42 (Figure 10, C). Thus we investigated whether phenethylcopper nucleophiles could engage in an analogous imidoyl-Cu-ene reaction with pyridine
Figure 10.
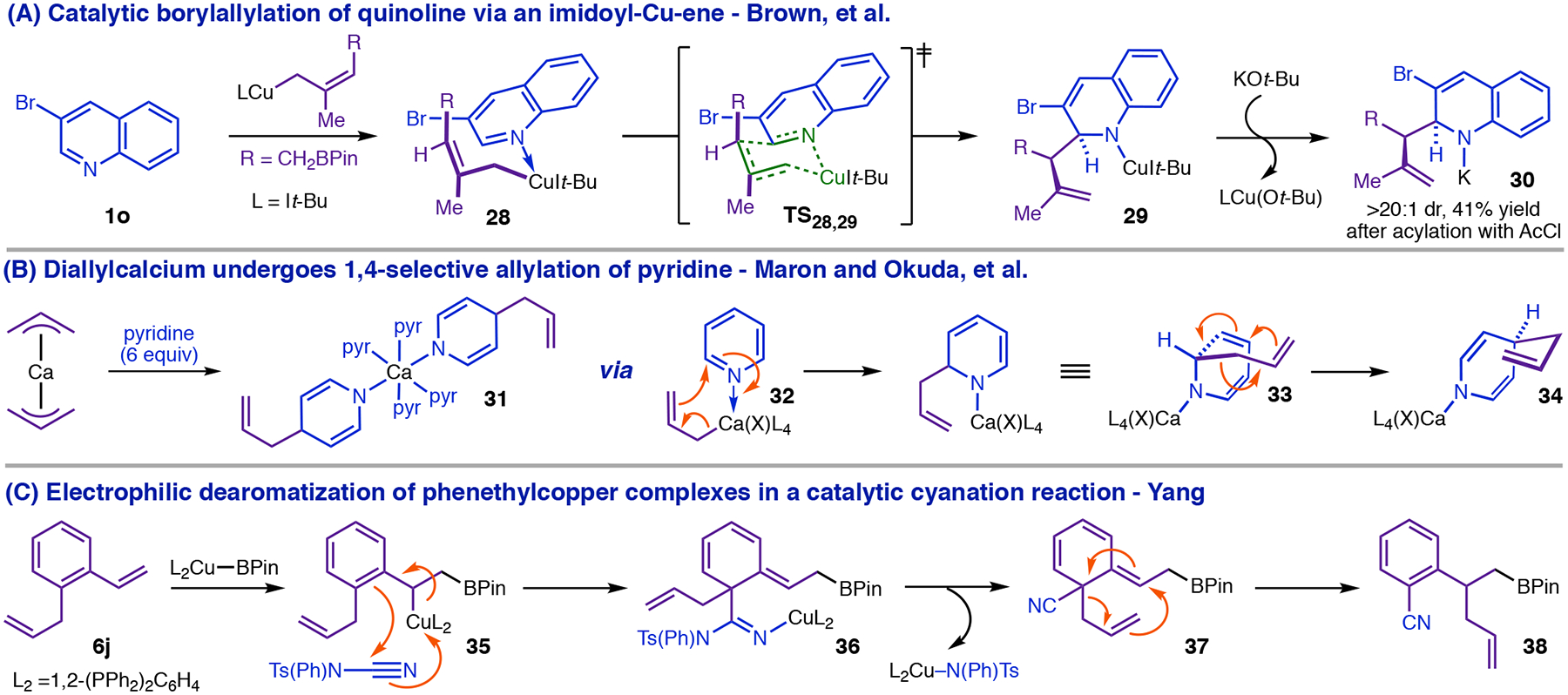
Precedent for 1,4-dearomatization via an imidoyl-Cu-ene reaction of the phenethylcopper intermediate.
6. Computational investigation of the doubly dearomative ene: PES, energy-decomposition analysis, and aromaticity as a function of the reaction coordinate.
The hypothesized ene would have the remarkable property of simultaneously dearomatizing two aromatic rings. Despite this, our modeling identified a transition state ([TS14,15]aa, Figure 11, A and B) for the ene only 25.8 kcal/mol above 9a (Figure 11, A), which corresponds to a reaction rate significant on the timescale of days at 298K. It is unintuitive that a doubly dearomative transformation would occur at ambient temperature in the absence of highly energetic reactants, but our investigations provide some insight into why this reaction should be feasible. We calculate a −28 kcal/mol free-energy change for a hypothetical ene (eq 2, Figure 11, C) involving similar changes in formally localized bonds but no dearomatization. The amount of energy released in this step, presumably due to replacement of C–N π- and Cu–C σ-bonds with stronger C–N and C–C σ-bonds, is close to current estimates for the aromatic stabilization energy of pyridine (30–31 kcal/mol).38,43 Thus, coupling these bonding changes to destruction of aromaticity could largely offset or overcompensate the dearomative penalty for the heterocycle, leaving a residual penalty for the doubly dearomative ene (eq 3, Figure 11, C) closer in magnitude to the aromatic stabilization energy of just the phenyl ring. Accordingly, the predicted barrier to 1,3-Cu-migration by 9a (+22.3 kcal/mol, Figure 7, C), which dearomatizes only one ring but fails to generate more favorable localized bonds, is only modestly lower than the barrier to the doubly dearomative ene. Taking the energy change of eq 2 as an estimate for the total energy change not due to dearomatization in eq 3 (Figure 11, C) enables estimation of the resonance energy lost in eq 3 as simply ΔG°(eq 3) − ΔG°(eq 2) = 48 kcal/mol, a value much larger than the overall thermodynamic penalty for the reaction (+20 kcal/mol in the gas phase).18 Consideration of how computational indices of aromaticity change as the ene reaction progresses affords a unique perspective on the nature of the double-dearomatization event. Figure 11, D, plots the NICS(0) indices44 (defined as the negative of the magnetic shielding value calculated for a virtual proton at the center of the ring in question) as functions of the ene reaction coordinate for the pyridine ring (blue line), phenyl ring (purple line), and pericyclic ring (i.e., the ring defined by the bonds that are changing; green line). NICS(0) values for the pyridine and phenyl rings initially indicate strong aromaticity and decrease monotonically throughout the reaction, entering a phase of particularly rapid dearomatization near 40% extent-of-reaction. Both rings attain their maximum rates of dearomatization at the transition state, showing that the transformation as modeled is truly a simultaneous double-dearomatization. The apparent aromaticity of the pericyclic ring reaches its maximum value immediately after the transition state and decreases thereafter. While the NICS(0) value for this ring is not a pure measure of its aromatic stabilization,44b the manner which it evolves nevertheless suggests that the concerted double-dearomatization is kinetically facilitated by transient formation of a new aromatic orbital array that confers additional stabilization to the transition state.
Figure 11.
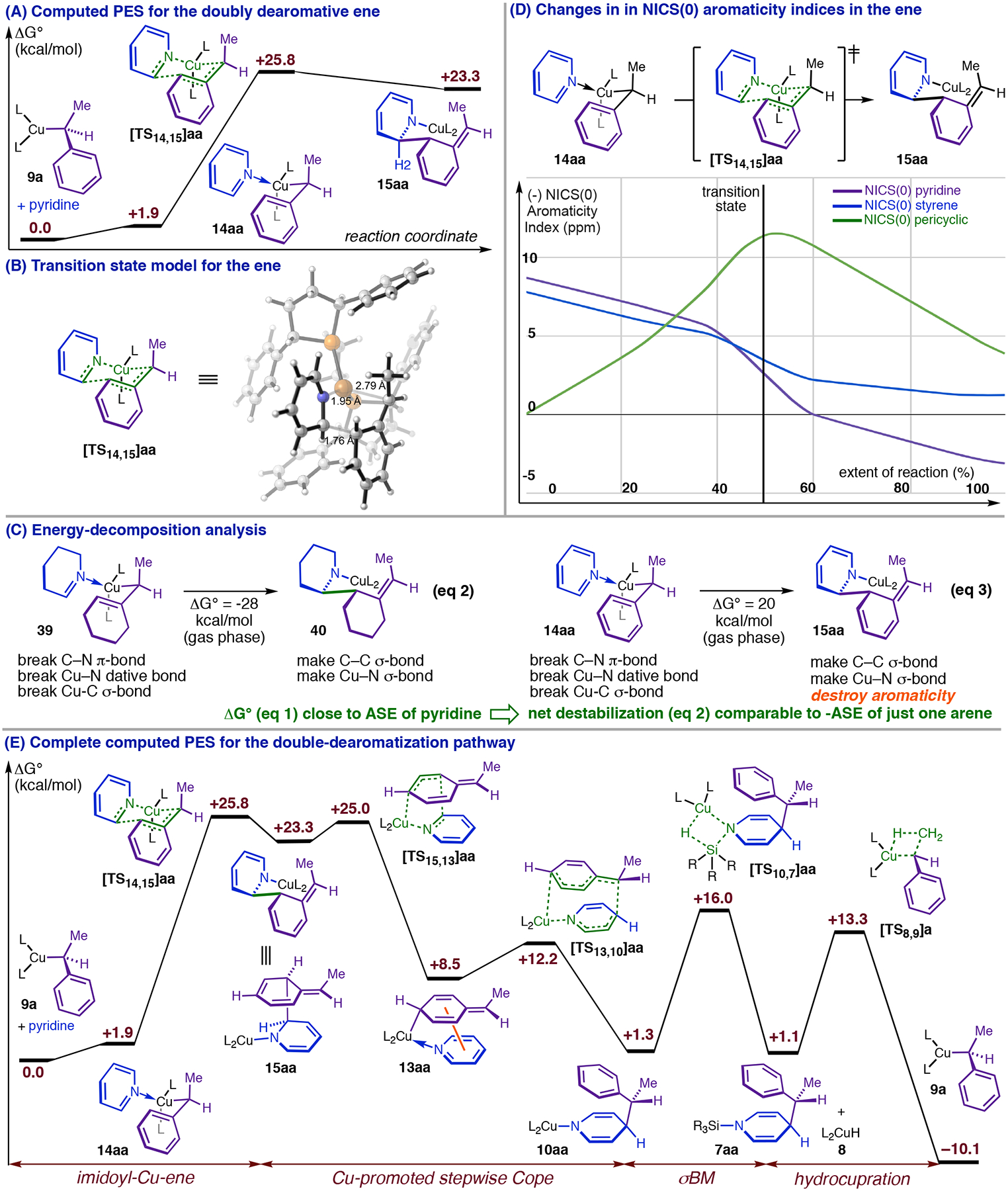
The double-dearomatization pathway.
7. Retro-ene of the doubly dearomatized intermediate gives 13: Heterocycle promotion of the 1,5-Cu-migration.
We attempted to model a concerted Cope rearrangement of 15aa, which would give 10aa (Figure 11, E) analogously to the presumptive mechanism of the rearrangements in Figure 10, B and C. However, the transition state we found ([TS15,13]aa, Figure 11, E) exhibits C2–Cortho cleavage, but without inception of bonding between C4 and Cα. Instead, the two dearomatized rings begin to reorient themselves parallel to one another, and Cpara puckers out of the plane while engaging Cu(I) in a new interaction, forming previously identified 13aa (Figure 11, E). This step is thus just the reverse of the imidoyl-Cu-ene reaction that would generate doubly dearomatized intermediate 15aa from the Cpara-metalated isomer of 14aa (Figure 11, E; the step is henceforth referred to as the retro-ene). Viewed within the complete dearomatization PES (Figure 11, E), 13aa appears as an intermediate in a stepwise Cope rearrangement (15aa → 13aa → 10aa, Figure 11, E). while 15aa is a fleeting intermediate in a heterocycle-mediated net 1,5-Cu-migration (14aa → 15aa → 13aa, Figure 11, E). Although the predicted total barrier to the double-dearomatization mechanism (+25.8 kcal/mol) is slightly greater than the predicted barrier to 1,3-Cu-migration by 9a (Figure 7, C), the rate law and substituent effects support double-dearomatization being the major reaction pathway. Given that heterocycle coordination to 9a is not intrinsically favorable, the preference for generating 13aa this way implies that the sequence 9a → 14aa → 15aa → 13aa (Figure 2, B, ii) is faster than 9a → 11a → 12a → 13aa (Figure 2, B, i), which makes the ene/retro-ene sequence a heterocycle-promoted pathway for net 1,5-Cu-migration. The ability of the heterocycle to couple dearomatization to favorable changes in localized bonding and stabilize dearomatized intermediates provides a plausible chemical basis for this result.
8. The double-dearomatization mechanism correctly predicts the rate law for dearomatization of 1d.
The double-dearomatization PES (Figure 11, E) straightforwardly predicts the correct rate law and observed resting state for dearomatization of 1d (Figure 6, A).45 The stability of 9a (−1.9 kcal/mol) relative to dative complex 14aa correctly implies that the latter should not be a significant resting state component, and the relatively low barrier to regeneration of 9a after the TLS implies that no 8 or 10aa should be present during catalysis; i.e., 9a is required to be the MACS. The simplified catalytic cycle in Scheme 1 can be used for theoretical analysis of a generic reaction via the double-dearomatization pathway because the ene/retro-ene/5,5-rearrangement sequence is kinetically indistinguishable from a unimolecular elementary step (modeled as 14 → 10 in Scheme 1 and referred to as dearomative isomerization). Heterocycle binding to 9 is expected to be much more facile than the other steps, making 9 + 1 ⇌ 14 a pseudoequilibrium (Scheme 1; refer to this scheme for definition of all kinetic parameters). The PES implies that dearomative isomerization should be the TLS, and because 9 is the MACS, the rate law for the catalytic reaction can be determined from the sequence 9 ⇄ 14 → 10 (Scheme 1), thus (eq 4), in agreement with experiment.46
Scheme 1.
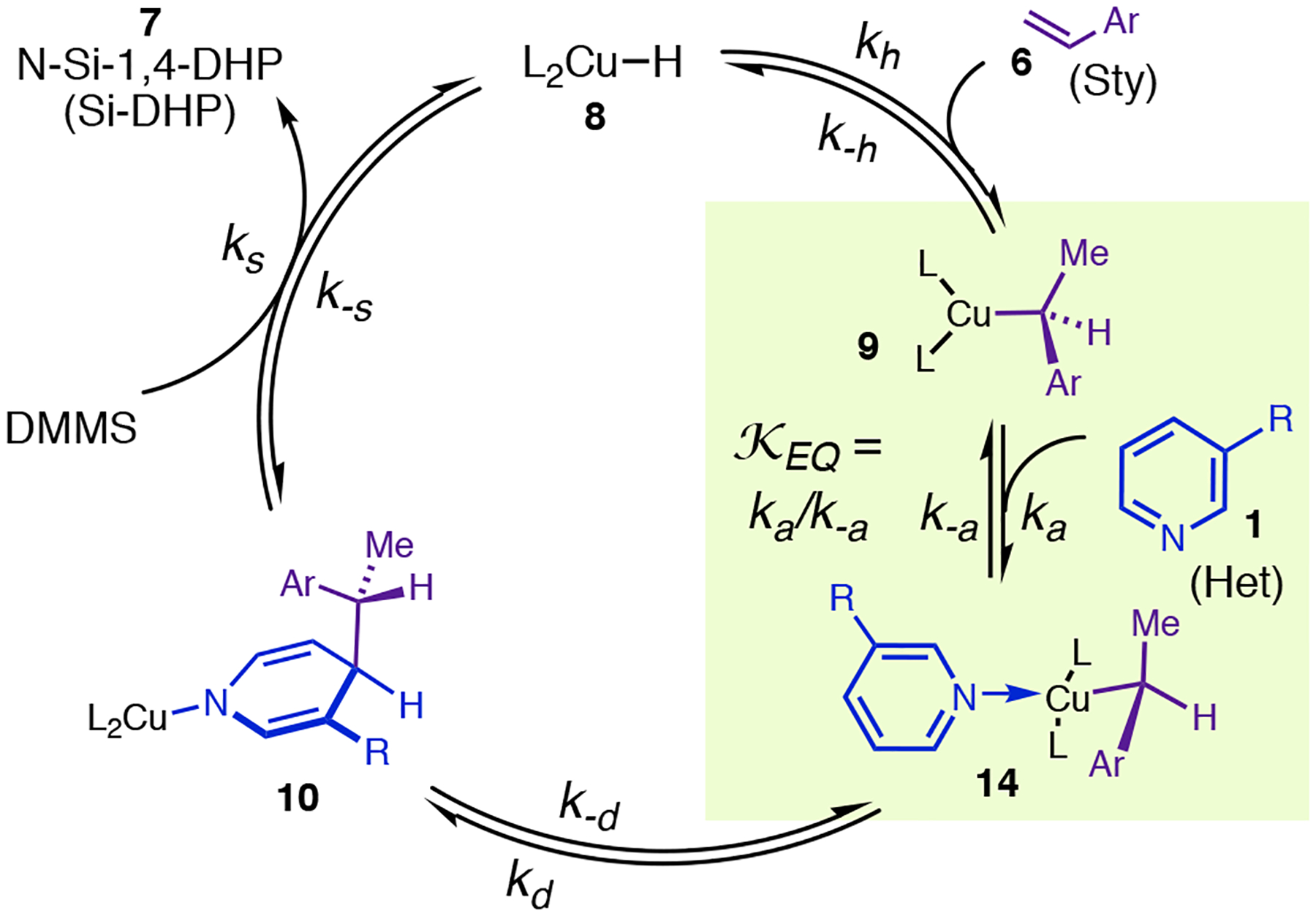
Simplified Catalytic Cycle for Rate-Law Derivations.
Figure 13:
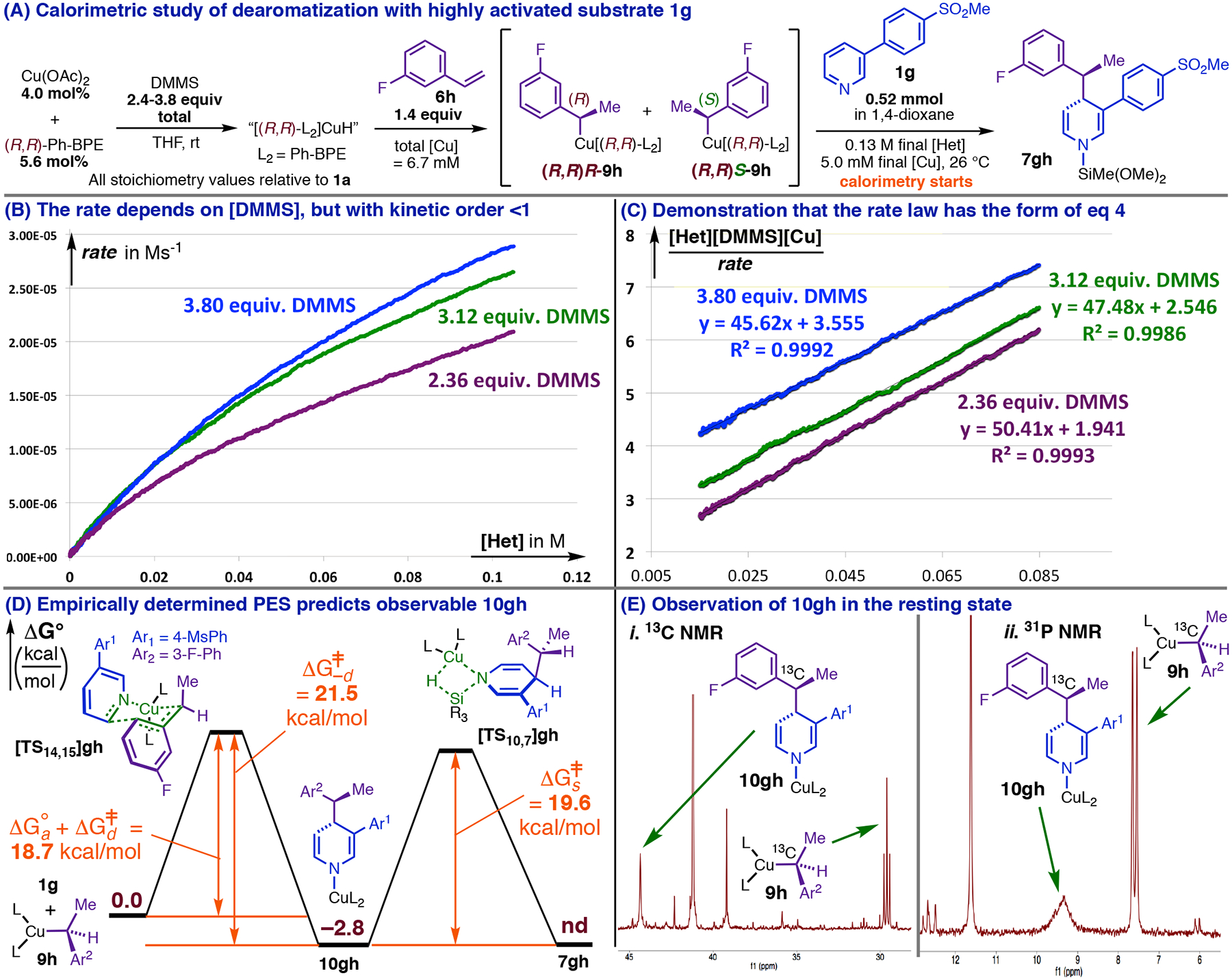
Saturation kinetics in the dearomatization of 1g imply an energy surface like that predicted for the double-dearomatization and similarly imply that 10gh should be observable.
9. Predicted saturation kinetics and altered resting state for very electron-deficient heterocycles.
Highly reactive pyridines are among the best substrates for asymmetric dearomatization and thus form an intrinsically important heterocycle class. They also provide opportunities for empirical study of proposed steps and intermediates that occur after the TLS for 1d (Figure 6, A) and consequently cannot be probed via kinetics or spectroscopy using that substrate. Figure 12 shows how strongly anion-stabilizing C3 substituents are expected to affect the energies of key structures in the double-dearomatization PES. Because the doubly dearomative ene (14 → 15, Figure 12) and the ene leading from 13 to 15 (i.e., the reverse of the retro-ene, Figure 12) both have nucleophilic addition character, anion-stabilizing C3 groups must lower the barriers to both (i.e., decrease the energies of TS14,15 and TS15,13 relative to 9a; Figure 12). Activating C3 groups also lower the energy of doubly dearomatized intermediate 15 (Figure 12), which contains a fully developed metalloenamine functional group. These changes lower the rate-controlling barrier to dearomative isomerization (i.e., increase kd). Our group’s earlier study of substituent effects on the rate of σBM by Cu-carboxylate complexes21 revealed marked slowing of that reaction by groups that attenuate the nucleophilicity of the carboxylate ligand, and we expect similar deceleration here with substituents that attenuate the nucleophilicity of 10 (Figure 12). This implies an increase in the energy of σBM transition state TS10,7 relative to 10, whereas 10 itself should be stabilized by the substituent much like 15 (Figure 12). If the opposing effects on the dearomative isomerization and σBM steps are large enough, their barriers will become comparable (i.e., kd and ks will have similar values) Because 10aa is predicted to be very close in energy to 9a in the absence of a substituent (Figure 11, E), the double-dearomatization PES indicates that even modest additional stabilization should render 10 isoenergetic with or potentially more stable than 9 (Figure 12). Consequently, 10 (Figure 12) must become a major component of the resting state if the barriers to dearomative isomerization and σBM are similar. Hydrocupration is expected to remain kinetically irrelevant, however: this step cannot exhibit a (heterocycle) substituent effect, and its barrier is already significantly below that of σBM (−2.7 kcal/mol) in the absence of a substituent (Figure 11, E).
Figure 12.
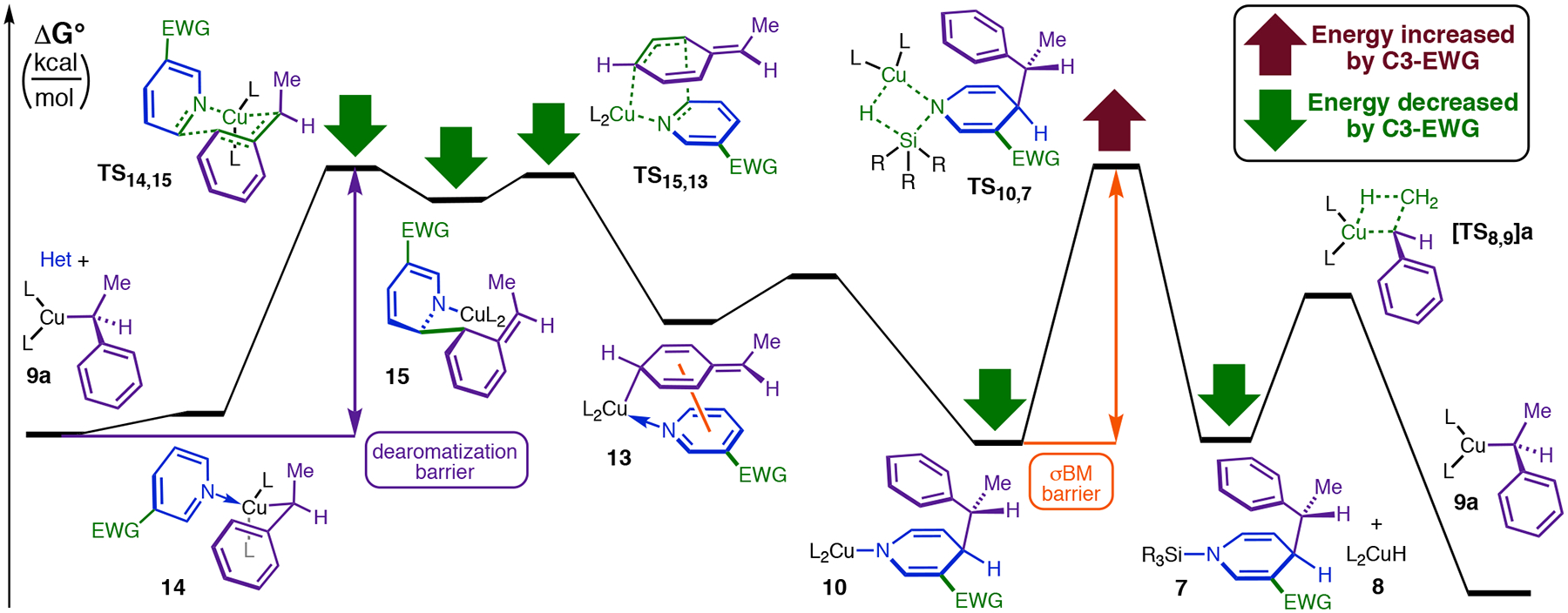
Predicted qualitative double-dearomatization PES for heterocycles with very electron-withdrawing C3 substituents.
Dearomatization of 1g (Figure 13, A) is faster than that of any other pyridine we have studied (Table 3) and clearly obeys a different rate law, making it an excellent model reaction for testing these predictions. We have confirmed that this reaction exhibits no kinetic dependence on 3-fluorostyrene. If this result is stipulated, and no assumptions are made about the relative values of kd, ks, and k−d, then the catalytic cycle in Scheme 1 must obey saturation rate law eq 5 (see the SI for derivation), in which kD is defined as and [Cu]A is the total concentration of all active catalyst species (note that the rate law for 3-phenylpyridine is just an instance of eq 5 for which kD, k−d≪ ks).
| (eq 5) |
The discussion of substituent effects implies that kd and ks should be comparable for 1g (Figure 13, A), and thus its rate law must correspond to an instance of eq 5 in which [Het] and [DMMS] both appear with non-negligible coefficients in the denominator. Taking advantage of the fact that [DMMS] is linearly related to [Het] over the course of the reaction as [DMMS] = [Het] + (χ − 1)[Het]0, where χ is the number of equivalents of DMMS used and [Het]0 is the initial heterocycle concentration, one can write
| (eq 6). |
Eq 6 provides a convenient graphical way to diagnose whether the kinetics of a given dearomative reaction obey eq 5, and, if so, to estimate rate constants for the kinetically relevant steps. If the rate does obey eq 5, then plots of ([Het][DMMS][Cu]/rate) using empirical rate and concentration data must be linear functions of [Het] having slope N ≅ (kD + s)/kDks.46 The Y-intercepts Y (χ, [Het]0) of those plots must in turn constitute a linear function of (χ − 1) [Het]0 having slope n ≅ 1/kD:46
| (eq 7) |
Implication also goes in the other direction: given respective kinetic orders of zero and one for [Sty] and [Cu], if plots of ([Het][DMMS][Cu]/rate) using actual rate and concentration data are found to be linear functions of [Het], then the true rate law must conform to an instance of eq 5. This is because, with the exception of trivial examples like rate = k[Het][Cu]A and rate = k[DMMS][Cu]A (which are themselves limiting cases of eq 5), multiplying (1/rate) by [Het][DMMS][Cu]A will only yield a linear function of [Het] if the denominator of rate is a linear function of [Het], and if [Het] and [DMMS] both appear with exponents of one in the numerator of rate. Graphically determined linear parameters then indicate which limiting case of eq 5 is applicable. In particular, straightforward algebraic manipulations (see the SI) show that [DMMS] and [Het] must both appear with non-negligible coefficients in the denominator of rate if Y(χ, [Het]0) is found to be a (non-constant) linear function of (χ − 1)[Het]0 for which n ≠ N.
Plots of ([Het][DMMS][Cu]/rate) for three reactions differing only in χ (Figure 13, C; the untransformed rate curves are in Figure 13, B) were indeed linear except at very high conversion, where the heatflow data became erratic. In agreement with eq 6, the slopes of these lines are similar; treating all slope estimates as resulting from identical measurements would yield an average (kD + ks/kDks) ≅ 47.8 Ms with standard deviation 1.88 (five measurements). Further, the Y-intercepts of the lines in Figure 13, C (obtained by extrapolation) are monotonically increasing with (χ − 1)[Het]0 as required by eq 7, and fit well enough to a line (R2 = 0.97) to enable estimation of n ≅ 1/kD = 8.58 Ms and (k−d/kDks) ≅ 0.345 M2s from its slope and intercept, respectively.18 The values of N and n are indeed very different, which demonstrates that dearomatization of 1g (Figure 13, A) obeys the saturation rate law predicted by the double-dearomatization mechanism. This supports the catalytic cycle having the form in Scheme 1. Equations for the linear parameters were solvable for estimates for kD (0.117 M–1s–1), ks (0.0255 M−1s−1) and k−d (1.02•10−3s−1),48 which correspond by way of the Eyring relationship to the approximate energy landscape in Figure 13, D. These energies are in excellent qualitative agreement with how the double-dearomatization PES is expected to change under the influence of an anion-stabilizing C3 substituent (Figure 12). They also mirror the prediction of Fig. 12 that 10gh should be a major resting state component during dearomatization of 1g (Figure 13, D).
10. Observation of N-Cu-1,4-DHP intermediate 10gh.
Monitoring of the catalytic dearomatization of 1g (Figure 13, A) with 3-fluorostyrene-α-13C by 13C, 31P, and 19F NMR spectroscopy confirmed that a new major catalyst species was present having the diagnostic 13C, 19F, and 31P NMR resonances (Figure 13, E) expected for 10gh: in particular, its 13C (Figure 13, E, i) and 19F signals indicate the presence of a fluorophenethyl group bound at Cα to C rather than Cu. These signals were associated with a new 31P resonance (Figure 13, E, ii), indicating the presence of a CuL2 fragment (NB, neither these nor any analogous signals were evident for a control reaction mixture prepared from 3-phenylpyridine). The 13C, 19F, and 31P resonances assigned to 10gh (Figure 13, E) were all relatively large near the start of the catalytic reaction and simultaneously diminished as conversion of 1g (Figure 13, A) increased. Phenethylcopper intermediate 9h, initially present in a much smaller amount than usual, gradually increased in abundance at the expense of 10gh (Figure 13, E), resuming its typical role as the MACS only at very high conversion of 1g (Figure 13, A; the proportion of observable phosphorus present as 9h + 10gh was conserved throughout the catalytic reaction). This is precisely how the distribution of catalyst species is expected to change throughout the reaction if dearomative isomerization and σBM have similar barriers, as in Fig 12: the dearomative isomerization becomes more nearly turnover-limiting as the reaction proceeds because the concentration of 1g (Figure 13, A) becomes very low, while the concentration of DMMS, used in fivefold excess, remains relatively high. The predictive power of the double-dearomatization mechanism thus enabled us to confirm the involvement of 10 (Figure 12), show that 10 must be close in energy to 9 (Figure 12) as implied by our modeling (Figure 11, E), calculate a rate constant and approximate activation barrier for σBM (Figure 13, D), and corroborate our computational prediction that σBM should be significantly more challenging than hydrocupration.
11. The double-dearomatization mechanism predicts the observed stereochemical outcomes.
The double-dearomatization model also predicts the correct stereo- and regio-chemical outcomes. Unlike 11a (Figure 9, A), 15aa (Figure 11, A) lacks a viable pathway to 1,2-DHP 2aa (Figure 9, A) and only generates stable species upon rearomatizing the phenyl ring via reversion to 14aa or forward reaction to 1,4-DHP 10aa (Figure 11, E). The PES in Figure 11, A and E, pertains to dearomatization via ene chair-conformer ax-[TS14,15]aa (Figure 14, A) because our calculations indicate that it is more favorable than its equatorial-Me counterpart (ΔΔG‡ = −2.4 kcal/mol). Attaining eq-[TS14,15]aa requires a serious clash between the Me group and Ph-BPE (Figure 14, B), whereas ax-[TS14,15]aa projects the methyl away from the phosphine. Two additional diastereomeric chairs are possible for C3-substituted heterocycles, but one (pro-syn-[TS14,15], Figure 14, C) causes a clash between the equatorial C3 group and the approaching phenyl ring, while the other (pro-anti-[TS14,15], Figure 14, D) incurs no additional penalties. The only chair retro-ene reaction available to pro-anti-15 generates 13 in the conformer (pro-anti-13) leading to N-cuprated DHP anti-10 (Figure 14, D). At this point, stereorandomization prior to formation of anti N-Si-1,4-DHP would require either dissociation of heterocycle from 13 (+8.8 kcal/mol above 13aa, Figure 7, C) or 180° N–Cu rotation of 13; however, both of these processes disrupt the π-stacking interaction critical for stabilizing 13, making them unfavorable relative to regeneration of the phenethylcopper resting state via 5,5- rearrangement/σBM/hydrocupration (which occurs with a total barrier of 7.5 kcal/mol for 13aa → 10aa → 7aa + 8 → 9aa, Figure 11 E). As a result, the stepwise Cope/σBM sequence should be stereospecific. Predicted energy differences for ax-[TS14,15] and eq-[TS14,15] (analogues of the structures in Figure 14A and B) for C6-attack on various C3-substituted heterocycles known to give high diastereoselectivity were in the range of 1.3−3.0 kcal/mol, which indicates that the double- dearomatization pathway should have a robust preference for the observed anti-1,4-DHP. Further, because the ene, retro-ene and 5,5-rearrangements are all suprafacial, the Cα configuration of the phenethylcopper nucleophile is retained throughout the remainder of the cycle.
Figure 14:
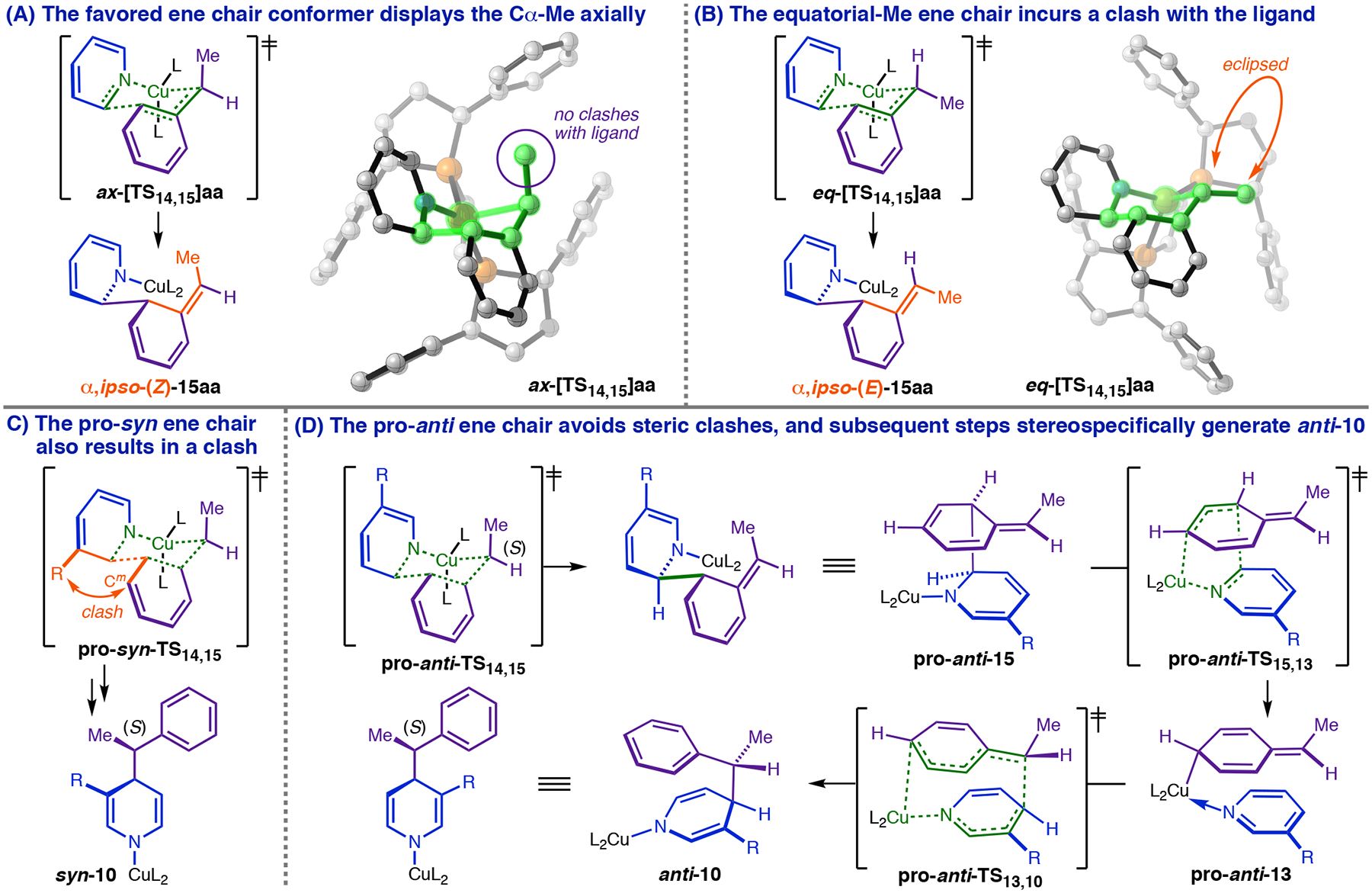
The double-dearomatization mechanism predicts the correct diastereo- and enantio-selectivity.
12. Mechanistic basis for the styrene-para-group effect.
The double-dearomatization mechanism explains the severe deterioration or failure of the asymmetric dearomatization observed with para-substituted styrenes. The barrier to the retro-ene step is low relative to intermediate 15aa (Figure 11, E) but still very near the energy maximum of the PES. Structural changes that destabilize the retro-ene transition state (exemplified by [TS15,13]aa in Figure 11, E) are expected to make it the TLTS, and if they do not commensurately stabilize 15, this will increase the total barrier to 1,4-dearomatization. While the doubly dearomatized intermediate derived from styrene permits close approach between Cu and Cpara (Figure 15, A), the presence of a para-Me or para-Ph would lead to formation of a very sterically unfavorable Cu-Ctert bond in the retro-ene step. In fact, the transition state we located for rearrangement for 15ak (Figure 15, B) does not correspond to a retro-ene reaction, but rather to a concerted Cope rearrangement. The Cu-Cpara interaction is lost entirely in [TS15,10]ak (Figure 15, B); it exhibits a Cu-Cpara distance much longer (+1.2 Å) than that in [TS15,13]aa (Figure 15, A), as is required to avoid clashes with Ph-BPE. We calculate that the concerted Cope rearrangement of 15ak is 4.0 kcal/mol more difficult than the retro-ene rearrangement of 15aa (Figure 15, A), which is indeed enough to make the overall 1,4-dearomatization more challenging. The expected outcome for a modest increase in the 1,4-dearomatization barrier is erosion of enantioselectivity due to increased lifetime and epimerization of the phenethylcopper (NB 4-phenylstyrene gives virtually racemic 1,4-DHP product). Larger increases are expected to cause 1,4-dearomatization to fail or cease to be competitive with other processes. Thus, the ability of Cu to provide a low-barrier stepwise alternative to the concerted Cope appears to be critical for successful enantioselective catalysis. For para-heteroatom-substituted styrenes, which result in compete failure of the catalytic dearomatization, additional factors may further affect the viability of the Cpara-metalated intermediate (e.g., decomposition via elimination or radical reactions).
Figure 15.
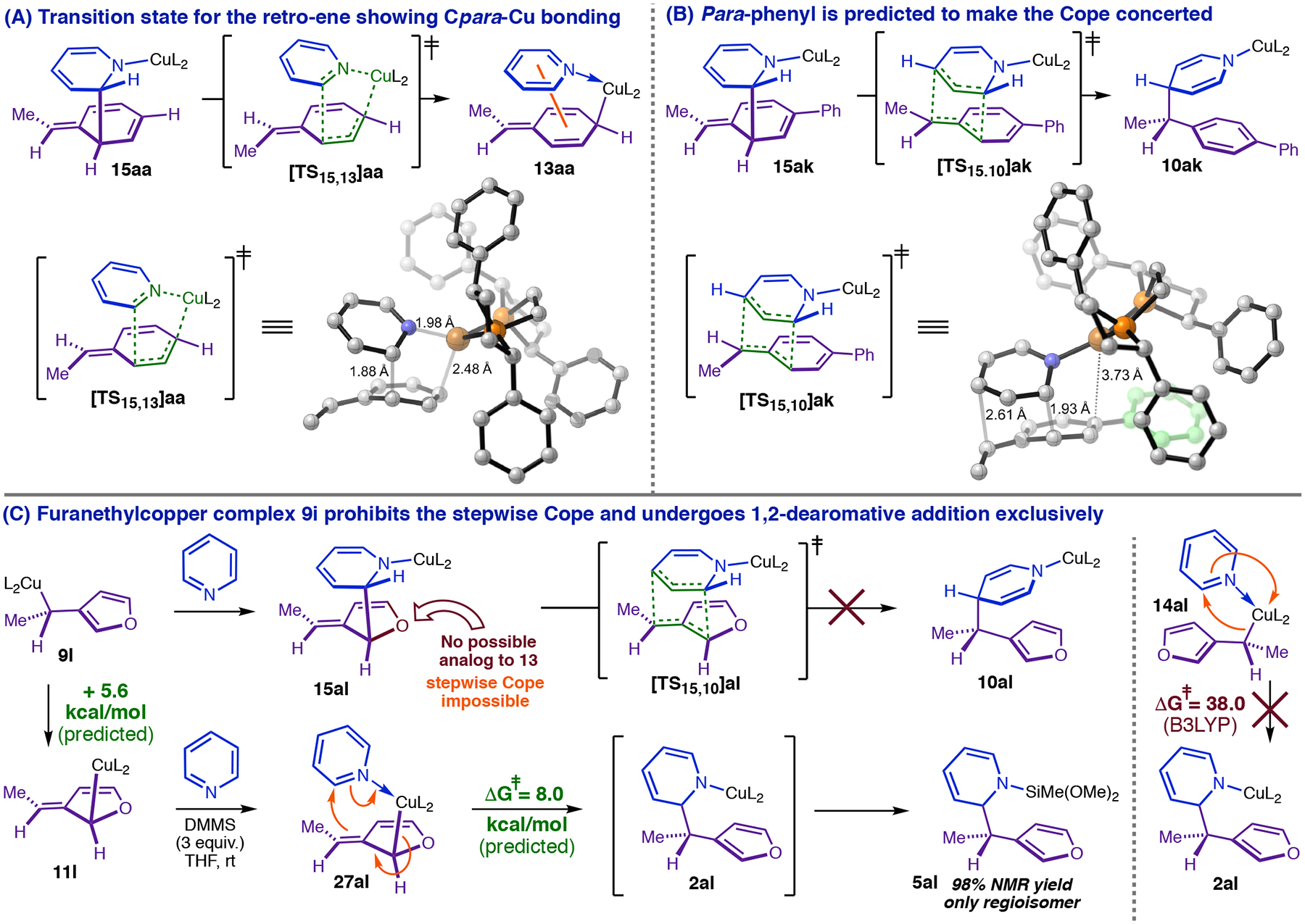
Mechanistic basis for the styrene-para-group effect.
13. Organocopper species that cannot generate Cpara-metalated intermediates undergo 1,2-selective dearomatization: the case of 3-vinylfuran.
The double-dearomatization model predicts that conjugated nucleophiles lacking the structural features required for generating Cpara-metalated isomers should still undergo dearomatization, albeit with complete reversal in regioselectivity. This result is partially anticipated by the work of Brown, et al. showing that allylcopper complexes react with halopyridines and haloquinolines with exclusive 1,2-selelctivity (see e.g., Figure 10, A).14 The dearomatization of α-furanethylcopper complex 9l with pyridine is particularly illuminating (Figure 15, C). Prior to formulating the new mechanistic models described in this article, we expected 9l to undergo 1,4-selective dearomatization analogously to α-phenethylcopper nucleophiles. However, dearomatization of 9l via the ene/retro-ene pathway is impossible because the furan ring lacks a para-carbon; in order to give 1,4-DHP, this substrate would have to rearrange via the unfavorable concerted Cope (Figure 15, C). Instead, 3-vinylfuran (6l), undergoes catalytic dearomatization to give 1,2-DHP 5al exclusively and in nearly quantitative yield (Figure 15, C). Furan has a much lower aromatic stabilization energy (ca. 15 kcal/mol)43a than benzene; consistent with this, our calculations indicate that dearomative 1,3-Cu-migration should be much more facile for 9l (+5.6 kcal/mol, Figure 15, C) than 9a (+22.3 kcal/mol, Figure 9, A). Further, we find that 27al should have a low barrier (+8.0 kcal/mol relative to 9l) to forming 1,2-DHP 2al (Figure 15, C) via imidoyl-Cu-ene reaction with pyridine (note: this is the same pathway that would generate 1,2-DHP’s from phenethylcopper nucleophiles in the single-dearomatization pathway, Figure 9). Corroborating our preliminary expectations (Figure 3, B), monometallic direct 1,2-addition of 9l (Figure 15, C, ΔG‡ = +38.0 kcal/mol) does not appear to be a viable pathway for generating the observed product.
Conclusion
Our theoretical investigations have elucidated two possible pathways for Cu-catalyzed asymmetric dearomatization of unactivated heterocycles, both of which involve an unusual 5,5-rearrangement occurring within the dative complex of a Cpara-metalated isomer of the phenethylcopper nucleophile. The first pathway generates this intermediate via successive 1,3-Cu-migration steps, making it a plausible source of the 1,2-DHP products we observe in certain cases where 1,4-dearomatization is particularly inefficient. However, our theoretical analysis of this single-dearomatization pathway yields an erroneous catalytic rate law and is inconsistent with substituent effects and other reactivity trends indicating that the heterocycle functions as an electrophile in the TLS of the 1,4-selective reaction. The double-dearomatization pathway generates the Cpara-metalated intermediate through a turnover-limiting polar reaction of the phenethylcopper with the heterocycle and is consequently consistent with all known experimental properties of the catalytic reaction. This pathway is also notable for correctly predicting the regio-, diastereo-, and enantio-selectivity of the catalytic reaction. It is seemingly unintuitive that simultaneous dearomatization of two arenes would be facile under such mild conditions. However, this result is readily understood in terms of very thermodynamically favorable bonding changes associated with a generic imidoyl-Cu-ene reaction and plausible stabilization of the transition state via transient formation of a new aromatic orbital array. The new modes of reactivity documented in this work illustrate how chiral Cu(I) complexes can facilitate fundamental new catalytic transformations of a sort long considered exceptionally challenging.
Supplementary Material
ACKNOWLEDGMENT
The National Institutes of Health under award number R35- GM122483 supported research reported in this publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Dr. Walt Massefski and Dr. Bruce Adams for assistance with design of NMR experiments and Dr. Sheng Guo (MIT) and Ms. Claudia Keller (Spirochem) for sharing observations that ultimately proved to be mechanistically significant; e.g., the regiochemical outcome of dearomatization with 3-vinylfuran (SG). We thank Dr. Veronika Kottisch (MIT) and Dr. Alex Schuppe (MIT) for assistance with preparation of this manuscript, and Professor Donna Blackmond (TSRI) for making us aware of reference 31b. We thank the National Institutes of Health for a supplemental grant for the purchase of supercritical fluid chromatography (SFC) equipment (GM058160-17S1).
Footnotes
Supporting Information
Procedural information and characterization data (PDF)
Complete calorimetry data from a representative kinetics experiment (xlsx)
The authors declare no competing financial interests.
REFERENCES
- (1).For reviews on pyridine dearomatization and dihydropyridine chemistry, see:; (a) Lavilla R Recent Developments in the Chemistry of Dihydropyridines. J. Chem. Soc., Perkin Trans 1, 2002, 1141–1156. [Google Scholar]; (b) Ahamed M; Todd MH Catalytic Asymmetric Additions of Carbon-Centered Nucleophiles to Nitrogen-Containing Aromatic Heterocycles. Eur. J. Org. Chem 2010, 2010, 5935–5942. [Google Scholar]; (c) Bull JA; Mousseau JJ; Pelletier G; Charette AB Synthesis of Pyridine and Dihydropyridine Derivatives by Regio- and Stereoselective Addition to N-Activated Pyridines. Chem. Rev 2012, 112, 2642–2713. [DOI] [PubMed] [Google Scholar]; (d) Zhuo C-X; Zhang W; You S-L Catalytic Asymmetric Dearomatization Reactions. Angew. Chem. Int. Ed 2012, 51, 12662–12686. [DOI] [PubMed] [Google Scholar]; (e) Ding Q; Zhou X; Fan R Recent Advances in Dearomatization of Heteroaromatic Compounds. Org. Biomol. Chem 2014, 12, 4807–4815. [DOI] [PubMed] [Google Scholar]; (f) Bertuzzi G; Bernardi L; Fochi M Nucleophilic Dearomatization of Activated Pyridines. Catalysts 2018, 8, 632–665. [Google Scholar]
- (2).Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (3).Comins DL; Abdullah AH Regioselective Addition of Grignard Reagents to 1-Acylpyridinium Salts. A Convenient Method for the Synthesis of 4-Alkyl(aryl)pyridines. J. Org. Chem 1982, 47, 4315–4319. [Google Scholar]
- (4).Gribble MW Jr.; Guo S; Buchwald SL Asymmetric Cu-Catalyzed 1,4-Dearomatization of Pyridines and Pyridazines Without Preactivation of the Heterocycle or Nucleophile. J. Am. Chem. Soc 2018, 140, 5057–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Reports on catalyzed direct addition of hydride or silyl nucleophiles to pyridine had appeared in preceding years and some are conceptually related to the Cu-catalyzed asymmetric dearomatization; the reaction with styrenes in Figure 1, D, can be viewed formally as a diverted 1,4-hydrosilylation of pyridine in which the hydride is intercepted by an olefin to generate a carbon-centered nucleophile rather than undergoing attack at heterocycle C4. See; (a) Oshima K; Ohmura T; Suginome M Palladium-Catalyzed Regioselective Silaboration of Pyridines Leading to the Synthesis of Silylated Dihydropyridines. J. Am. Chem. Soc 2011, 133, 7324–7327. [DOI] [PubMed] [Google Scholar]; (b) Gutsulyak DV; van der Est A; Nikonov GI Facile Catalytic Hydrosilylation of Pyridines. Angew. Chem. Int. Ed 2011, 50, 1384–1387. [DOI] [PubMed] [Google Scholar]; (c) Hill MS; Kociok-Köhn G; MacDougall DJ; Mahon MF; Weetman C Magnesium Hydrides and the Dearomatisation of Pyridine and Quinoline derivatives. Dalton Trans 2011, 40, 12500–12509. [DOI] [PubMed] [Google Scholar]; (d) Königs CDF; Klare HFT; Oestreich M Catalytic 1,4-Selective Hydrosilylation of Pyridines and Benzannulated Congeners. Angew. Chem. Int. Ed 2013, 52, 10076–10079. [DOI] [PubMed] [Google Scholar]; (e) Dudnik AS; Weidner VL; Motta A; Delferro M; Marks TJ Nat. Chem 2014, 6, 1100–1107. [DOI] [PubMed] [Google Scholar]; (f) Gandhamsetty N; Park S; Chang S Atom-Efficient Regioselective 1,2-Dearomatization of Functionalized Pyridines by an Earth-Abundant Organolanthanide Catalyst. J. Am. Chem. Soc 2015, 137, 15176–15184.26580152 [Google Scholar]; (g) Intemann J; Bauer H; Pahl J; Maron L; Harder S Calcium Hydride Catalyzed Highly 1,2-Selective Pyridine Hydrosilylation. Chem. Eur. J 2015, 21, 11452–11461. [DOI] [PubMed] [Google Scholar]; (h) Fan X; Zheng J; Li ZH; Wang H Organoborane Catalyzed Regioselective 1,4-Hydroboration of Pyridines. J. Am. Chem. Soc 2015, 137, 4916–4919. [DOI] [PubMed] [Google Scholar]; (i) Kaithal A; Chatterjee B; Gunanathan C Ruthenium-Catalyzed Regioselective 1,4-Hydroboration of Pyridines. Org. Lett 2016, 18, 3402–3405. [DOI] [PubMed] [Google Scholar]; (j) Zhang F; Song H; Zhuang X; Tung C-H; Wang W Iron-Catalyzed 1,2-Selective Hydroboration of N-Heteroarenes. J. Am. Chem. Soc, 2017, 139, 17775–17778. [DOI] [PubMed] [Google Scholar]; (k) Yu H-C; Islam SM; Mankad NP Cooperative Heterobimetallic Substrate Activation Enhances Catalytic Activity and Amplifies Regioselectivity in 1,4-Hydroboration of Pyridines. ACS Catal 2020, 10, 3670–3675. [Google Scholar]
- (6).You’s elegant work on hydrogenative C–C-bond-forming dearomatization also addresses the challenges associated with stoichiometric electrophilic preactivation, but these reactions are distinct from catalyzed direct addition of carbon nucleophiles (Figure 1, C) in that they involve C–C-bond-forming nucleophilic addition after dearomative semireduction of the heterocycle. See; (a) Wang S-G; Zhang W; You S-L Construction of Spiro-tetrahydroquinolines via Intramolecular Dearomatization of Quinolines: Free of a Preinstalled Activation Group. Org. Lett 2013, 15, 1488–1491. [DOI] [PubMed] [Google Scholar]; (b) Wang S-G; You S-L Hydrogenative Dearomatization of Pyridine and an Asymmetric aza-Friedel-Crafts Alkylation Sequence. Angew. Chem. Int. Ed 2014, 53, 2194–2197. [DOI] [PubMed] [Google Scholar]
- (7).An orthogonal approach to catalytic dearomatization without stoichiometric activation involves intramolecular substitution reactions in which the heterocycle is the nucleophilic component. See; (a) Yang Z-P; Wu Q-F; You S-L Direct Asymmetric Dearomatization of Pyridines and Pyrazines by Iridium-Catalyzed Allylic Amination Reactions. Angew. Chem. Int. Ed 2014, 53, 6986–6989. [DOI] [PubMed] [Google Scholar]; (b) Yang Z-P; Wu Q-F; Shao W; You S-L Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization Reaction of Pyridines, Pyrazines, Quinolines, and Isoquinolines. J. Am. Chem. Soc 2015, 137, 15899–15906. [DOI] [PubMed] [Google Scholar]
- (8).Mohiti M; Rampalakos C; Feeney K; Leonori D; Aggarwal VK Asymmetric Addition of Chiral Boron-Ate Complexes to Cyclic Iminium Ions. Chem. Sci 2014, 5, 602–607. [Google Scholar]
- (9).(a) For examples, seeMancheño OG; Asmus S; Zurro M; Fischer T Highly Enantioselective Nucleophilic Dearomatization of Pyridines by Anion-Binding Catalysis. Angew. Chem., Int. Ed 2015, 54, 8823–8827. [DOI] [PubMed] [Google Scholar]; (b) Bertuzzi G; Sinisi A; Caruana L; Mazzanti A; Fochi M; Bernardi L Catalytic Enantioselective Addition of Indoles to Activated N-Benzylpyridinium Salts: Nucleophilic Dearomatization of Pyridines with Unusual C-4 Regioselectivity. ACS Catal 2016, 6, 6473–6477. [Google Scholar]; (c) Bertuzzi G; Sinisi A; Pecorari D; Caruana L; Mazzanti A; Bernardi L; Fochi M Nucleophilic Dearomatization of Pyridines under Enamine Catalysis: Regio-, Diastereo-, and Enantioselective Addition of Aldehydes to Activated N-Alkylpyridinium Salts. Org. Lett 2017, 19, 834–837. [DOI] [PubMed] [Google Scholar]; (d) Flanigan DM; Rovis T Enantioselective N-Heterocyclic Carbene-Catalyzed Nucleophilic Dearomatization of Alkyl Pyridiniums. Chem. Sci 2017, 8, 6566–6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For examples of catalytic asymmetric 1,2-selective dearomatization of activated substrates, see, e.g.:; (a) Sun Z; Yu S; Ding Z; Ma D Enantioselective Addition of Activated Terminal Alkynes to 1-Acylpyridinium Salts Catalyzed by Cu−Bis(oxazoline) Complexes. J. Am. Chem. Soc 2007, 129, 9300–9301. [DOI] [PubMed] [Google Scholar]; (b) Black DA; Beveridge RE; Arndtsen BA Copper-Catalyzed Coupling of Pyridines and Quinolines with Alkynes: A One-Step, Asymmetric Route to Functionalized Heterocycles. J. Org. Chem 2008, 73, 1906–1910. [DOI] [PubMed] [Google Scholar]; (c) Fernández-Ibáñez MA; Maciá B; Pizzuti MG; Minnaard AJ; Feringa BL Catalytic Enantioselective Addition of Dialkylzinc Reagents to N-Acylpyridinium Salts. Angew. Chem. Int. Ed 2009, 48, 9339–9341. [DOI] [PubMed] [Google Scholar]; (d) Pappoppula M; Cardoso FSP; Garrett BO; Aponick A Enantioselective Copper-Catalyzed Quinoline Alkynylation. Angew. Chem. Int. Ed 2015, 54, 15202–15206. [DOI] [PubMed] [Google Scholar]; (e) Lutz JP; Chau ST; Doyle AG Nickel-Catalyzed Enantioselective Arylation of Pyridine. Chem. Sci 2016, 7, 4105–4109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yu S; Sang HL; Ge S Enantioselective Copper-Catalyzed Alkylation of Quinoline N-Oxides with Vinylarenes. Angew. Chem. Int. Ed 2017, 56, 15896–15900. [DOI] [PubMed] [Google Scholar]; (g) Robinson DJ; Spurlin SP; Gorden JD; Karimov RR Enantioselective Synthesis of Dihydropyridines Containing Quaternary Stereocenters Through Dearomatization of Pyridinium Salts. ACS Catal 2020, 10, 51–55. [Google Scholar]
- (11).For recent work on nucleophilic dearomatization using traceless activation with BF3 see; (a) Wang D; Wang Z; Liu Z; Huang M; Hu J; Yu P Strategic C–C Bond-Forming Dearomatization of Pyridines and Quinolines. Org. Lett 2019, 21, 4459–4463. [DOI] [PubMed] [Google Scholar]; (b) Wang D; Jiang Y; Dong L; Li G; Sun B; Désaubry L; Yu P One-Pot Selective Saturation and Functionalization of Heteroaromatics Leading to Dihydropyridines and Dihydroquinolines. J. Org. Chem [online early access]. DOI: 10.1021/acs.joc.0c00314. Published Online: Mar 18, 2020. https://pubs.acs.org/doi/abs/10.1021/acs.joc.0c00314 (accessed Mar 28, 2020). [DOI] [PubMed] [Google Scholar]
- (12).(a) For examples, seePirnot MT; Wang Y-M; Buchwald SL Copper Hydride Catalyzed Hydroamination of Alkenes and Alkynes Angew. Chem. Int. Ed 2016, 55, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang H; Buchwald SL Copper-Catalyzed, Enantioselective Hydrofunctionalization of Alkenes. In Organic Reactions; Denmark SE, Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2019; pp 121–205. and refs. therein. [Google Scholar]
- (13).(a) Grigg RD; Van Hoveln R; Schomaker JM Copper-Catalyzed Recycling of Halogen Activating Groups via 1,3-Halogen Migration. J. Am. Chem. Soc 2012, 39, 16131–16134. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Van Hoveln R; Hudson BM; Wedler HB; Bates DM; Le Gros G; Tantillo DJ; Schomaker JM Mechanistic Studies of Copper(I)-Catalyzed 1,3-Halogen Migration. J. Am. Chem. Soc 2015, 137, 5346–5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dearomative allylation of quinoline by an imidoyl-Cu-ene can be found in Smith KB; Huang Y; Brown MK Copper-Catalyzed Heteroarylboration of 1,3-Dienes with 3-Bromopyridines: A cine Substitution. Angew. Chem. Int. Ed 2018, 57, 6146–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yang Y; Perry IB; Buchwald SL Copper-Catalyzed Enantioselective Addition of Styrene-Derived Nucleophiles to Imines Enabled by Ligand-Controlled Chemoselective Hydrocupration. J. Am. Chem. Soc 2016, 138, 9787–9790. [DOI] [PMC free article] [PubMed] [Google Scholar]; The transition state model shown in Figure 3, D is provided in the SI.
- (16).For example, see Liu RY; Yang Y; Buchwald SL Regiodivergent and Diastereoselective CuH-Catalyzed Allylation of Imines with Terminal Allenes. Angew. Chem. Int. Ed, 2016, 55, 14077–14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).(a) Harutyunyan SR; López F; Browne WR; Correa A; Peña D; Badorrey R; Meetsma A; Minnaard AJ; Feringa BL On the Mechanism of the Copper-Catalyzed Enantioselective 1,4-Addition of Grignard Reagents to α,β-Unsaturated Carbonyl Compounds. J. Am. Chem. Soc 2006, 128, 9103–9118. [DOI] [PubMed] [Google Scholar]; (b) Metzger A; Bernhardt S; Manolikakes G; Knochel P MgCl2-Accelerated Addition of Functionalized Organozinc Reagents to Aldehydes, Ketones, and Carbon Dioxide. Angew. Chem. Int. Ed 2010, 49, 4665–4668. [DOI] [PubMed] [Google Scholar]
- (18).See the SI for details.
- (19).π-stacking interactions have been invoked as critical stereocontrol elements in several other diastereoselective pyridinium additions. See ref. 1b and examples therein.
- (20).(a) See, e.g.,Wang Y-M; Buchwald SL Enantioselective CuH-Catalyzed Hydroallylation of Vinylarenes. J. Am. Chem. Soc 2016, 138, 5024–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gribble MW Jr.; Pirnot MT; Bandar JS; Liu RY; Buchwald SL Asymmetric Copper Hydride-Catalyzed Markovnikov Hydrosilylation of Vinylarenes and Vinyl Heterocycles. J. Am. Chem. Soc, 2017, 139, 2192–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhou Y; Bandar JS; Buchwald SL Enantioselective CuH-Catalyzed Hydroacylation Employing Unsaturated Carboxylic Acids as Aldehyde Surrogates. J. Am. Chem. Soc 2017, 139, 8126–8129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Bandar JS; Pirnot MT; Buchwald SL Mechanistic Studies Lead to Dramatically Improved Reaction Conditions for the Cu-Catalyzed Asymmetric Hydroamination of Olefins. J. Am. Chem. Soc 2015, 137, 14812–14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Xi Y; Hartwig JF Mechanistic Studies of Copper-Catalyzed Asymmetric Hydroboration of Alkenes. J. Am. Chem. Soc 2017, 139, 12758–12772. [DOI] [PubMed] [Google Scholar]
- (23).Although not primarily concerned with hydrofunctionalization, Hoveyda’s study on the stereoselectivity of Cu-catalyzed olefin carboborylation is highly pertinent to this discussion:; Lee J; Radomkit S; Torker S; del Pozo J; Hoveyda AH Mechanism-Based Enhancement of Scope and Enantioselectivity for Reactions Involving a Copper-Substituted Stereogenic Carbon Centre. Nat. Chem 2018, 10, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).For a more rigorous treatment of the derivation of catalytic rate laws for single cycles under steady-state conditions when there is a MACS and a TLS, see; Helfferich FG Kinetics of Multistep Reactions, 2nd Edition; Green NJB, Ed.; Comprehensive Chemical Kinetics 40; Elsevier: Amsterdam, 2004, pp 227–239. [Google Scholar]
- (25).Note that these pathways must have different equilibrium constants in order to give the same equilibrium dr because they have different molecularity. See the SI.
- (26).These estimates were made using parameters sets that had been optimized for quantitative accuracy. See the SI.
- (27).For a more detailed discussion of the mechanistic basis for this phenomenon, see the SI.
- (28).The rate of epimerization is actually a first order function of the fractional distance from equilibrium, but when the fractional distance from equilibrium is very large (i.e., the dr is very high), the rate is well approximated by the equation given.
- (29).This equation is derived in the SI and specifically pertains to an idealized reaction in which the major phenethylcopper diastereomer is continuously regenerated with perfect kinetic selectivity. This scenario well approximates the reactions under study because the hydrocupration step has very high kinetic selectivity.
- (30).In section 3, we demonstrate that a single CuL2 fragment is present in the dearomative addition transition state. If the addition mechanism were bimetallic, as initially thought, then the differences in rate and selectivity observed when both enantiomers of Ph-BPE are present could be attributed to differences in reactivity or selectivity for competing homo- and hetero-chiral bimetallic pathways: the latter are not possible when the Ph-BPE is enantiomerically pure. However, because only one CuL2 fragment is present in the addition step, the differences in rate and diastereoselectivity we observe can only be due to differences in the intrinsic reactivity of the major and minor phenethylcopper diastereomers.
- (31).The phenomenon of a ligand’s enantiomeric composition affecting the dr of the catalyst and consequently the diastereoselectivity of the catalytic reaction is closely related to both the Kagan model for nonlinear effects with catalysts bearing multiple chiral spectator ligands and the ‘formal enantioselectivity refinement described by Jacobsen.; (a) Girard C; Kagan HB Nonlinear Effects in Asymmetric Synthesis and Stereoselective Reactions: Ten Years of Investigation. Angew. Chem. Int. Ed 1998, 37, 2922–2959. [DOI] [PubMed] [Google Scholar]; (b) Zhang W; Lee NH; Jacobsen EN Nonstereospecific Mechanisms in Asymmetric Addition to Alkenes Result in Enantiodifferentiation after the First Irreversible Step. J. Am. Chem. Soc 1994, 116, 425–426. [Google Scholar]
- (32).For example, if dearomatization is conducted with very low catalyst loading, then formation of a resting state with high phenethylcopper dr via catalytic recycling occurs while the reaction is still at extremely low conversion. In that case, premature termination of the reaction strictly lowers the amount of product formed via the minor phenethylcopper by reducing the amount of product formed during the phase of substantially eroded phenethylcopper dr that occurs at high conversion. We find that this increases the ee and the dr of the product. NB, reduced ee of product obtained upon complete reaction cannot be attributed to configurational instability. See the SI for more detail.
- (33).Given that the TLS of the dearomatization evidently had higher molecularity than the epimerization step (which we show in section 3 is due to a kinetic order in 1d for dearomatization), running reactions at higher concentration necessarily increases the kinetic preference for dearomatization of (R,R)R-9 over epimerization of (R,R)R-9, and we find that this markedly improves the enantioselectivity of the reaction. See the SI for details. This result in in agreement with a mechanistic postulate of ours in ref. 20b and conclusions discussed ref. 23.
- (34).For discussion of the fundamentals of the reaction-progress kinetics analysis methodology employed here, see; (a) Blackmond DG Angew. Chem. Int. Ed. Reaction Progress Kinetic Analysis 2005, 44, 4302–4320. [DOI] [PubMed] [Google Scholar]; (b) Blackmond DG Kinetic Profiling of Catalytic Organic Reactions as a Mechanistic Tool. J. Am. Chem. Soc 2015, 137, 10852–10866. [DOI] [PubMed] [Google Scholar]
- (35).If, e.g., the kinetic order were one in DMMS, it would be zero in styrene, and then the effect of varying [DMMS] and [3-F-styrene] would be the same as if only [DMMS] were changed. The same argument would apply, mutatis mutandis, if it were instead 3-F-styrene that had a kinetic order of one. Consequently, simultaneous variation of [DMMS] and [3-F-styrene] can only fail to affect the rate if the kinetic orders are zero for both.
- (36).DMMS could be incorporated into the TLS without there being a kinetic dependence on [DMMS] if the heterocycle were effectively saturated with DMMS at all relevant DMMS concentrations, but we have confirmed spectroscopically that there is negligible association between DMMS and pyridine in THF-d8, making this situation untenable.
- (37).For a description of the computational methodology employed in this article and relevant references, see the SI.
- (38).Schleyer P. v. R.; Puhlhofer F Recommendations for the Evaluation of Aromatic Stabilization Energies. Org. Lett 2002, 4, 2873–2876. [DOI] [PubMed] [Google Scholar]
- (39).Janowski T; Pulay P High Accuracy Benchmark Calculations on the Benzene Dimer Potential Energy Surface. Chem. Phys. Lett 2007, 447, 27–32. [Google Scholar]
- (40).For a more detailed discussion of the interpretation of the substituent effects in Table 3, see Section 2.4 of the SI.
- (41).Jochmann P; Dols TS; Spaniol TP; Perrin L; Maron L; Okuda J Insertion of Pyridine into the Calcium Allyl Bond. Angew. Chem. Int. Ed 2010, 49, 7795–7798. [DOI] [PubMed] [Google Scholar]
- (42).(a) Yang Y; Liu P Mechanism and Origins of Selectivities in the Copper-Catalyzed Dearomatization-Induced ortho C–H Cyanation of Vinylarenes. ACS Catal 2015, 5, 2944–2951. [Google Scholar]; (b) Yang Y Regio- and Stereospecific 1,3-Allyl Group Transfer Triggered by a Copper-Catalyzed Borylation/ortho-Cyanation Cascade. Angew. Chem. Int. Ed 2016, 55, 345–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).(a) Cyrański MK Energetic Aspects of Cyclic Pi-Electron Delocalization: Evaluation of the Methods of Estimating Aromatic Stabilization Energies. Chem. Rev 2005, 105, 3773–3811. [DOI] [PubMed] [Google Scholar]; (b) Fishtik I; Grimme S Accurate Evaluation of the Resonance Energies of Benzene and Pyridine via Cyclic Reference State. Phys. Chem. Chem. Phys 2012, 14, 15888–15896. [DOI] [PubMed] [Google Scholar]
- (44).(a) Schleyer P. v. R.; Maerker C; Dransfeld A; Jiao H; Hommes N. J. R. v. E. Nucleus-Independent Chemical Shifts. J. Am. Chem. Soc 1996, 118, 6317–6318. [DOI] [PubMed] [Google Scholar]; (b) Chen Z; Wannere CS; Corminboeuf C; Puchta R; Schleyer P. v. R. Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion. Chem. Rev 2005, 105, 3842–3888. [DOI] [PubMed] [Google Scholar]
- (45).Thorough derivations of the general rate law for the cycle in Scheme 1 and kinetics equations 4–7 are provided in the SI.
- (46).Interestingly, one can derive the correct rate law for dearomatization of 1d without knowledge of the abundance of 9a relative to 8 or 10aa using instead the fact that the rate constant for β-hydride elimination must be comparable to or smaller than kd for 3-phenylpyridine, which follows from the observation that the dr of 9a is kinetically controlled during catalysis. See the SI.
- (47).Relating linear parameters (N and n) determined graphically from plots of ([DMMS][Het][Cu])/rate to expressions in terms of individual rates constants by way of eq 6 requires information about the relationship between [Cu] and [Cu]A. The equations for N and n provided use the ‘approximately equals’ sign (≅) because we are employing the approximation that [Cu] ≅ [Cu]A. Quantifying the latter value with high precision would be logistically prohibitive and unnecessary for obtaining estimates of kD, kS, and k−d of sufficiently high quality for our purposes. Our kinetics and spectroscopic studies indicate that active catalyst species make up the large majority of copper, with [Cu]A = Q[Cu], where Q is constant over the course of a reaction and invariant with respect to modest changes in reaction conditions. It is therefore justifiable to set [Cu] ≅ [Cu]A, although it can be expected that this will result in each of the individual rate constants being slightly underestimated.
- (48).Details of these calculations are provided in the SI.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.