

Abstract
Cardiac injury remains a major cause of morbidity and mortality worldwide. Despite significant advances, a full understanding of why the heart fails to fully recover function after acute injury, and why progressive heart failure frequently ensues, remains elusive. No therapeutics, short of heart transplantation, have emerged to reliably halt or reverse the inexorable progression of heart failure in the majority of patients once it has become clinically evident. To date, most pharmacologic interventions have focused on modifying hemodynamics (reducing afterload, controlling blood pressure and blood volume) or on modifying cardiac myocyte function. However, important contributions of the immune system to normal cardiac function and the response to injury have recently emerged as exciting areas of investigation. Therapeutic interventions aimed at harnessing the power of immune cells hold promise for new treatment avenues for cardiac disease. Here, we review the immune response to heart injury, its contribution to cardiac fibrosis, and the potential of immune modifying therapies to affect cardiac repair.
Keywords: Heart disease, fibrosis, immunology, immunotherapy, CAR T
Introduction
The body responds to various cardiac injuries via complex acute and chronic adaptive processes to maintain pump function. Central to this process is inflammation and immune cell signaling. For example, in the setting of acute ischemic injury, immune-mediated activation of cardiac fibrosis is necessary to avoid catastrophic myocardial rupture. Activation of resident interstitial fibroblasts and recruitment of those derived from the epicardium result in scar formation that maintains chamber integrity. However, in many cases, the initially beneficial fibroblast response becomes mal-adaptive, resulting in excess extracellular matrix (ECM). This over-accumulation of ECM both stiffens the myocardium and negatively alters the cardiomyocyte niche resulting in progressive deterioration of cardiac function. While fibroblast activation has been well studied, the multifaceted role of immune signaling and its effects on acute and chronic fibrosis have only recently gained widespread traction in the cardiovascular research community. The emerging understanding is poised to revolutionize the treatment of myocardial diseases through targeted immune modulators. Furthermore, advances in T cell engineering offer exciting future directions for development of novel therapeutics.
Immune response to cardiac injury
The immune response to cardiac injury is complex (see figure 1) and remains only partially understood. Differences in the types of injury, host and environmental factors all modulate the immune response. The potent sterile inflammation of the injured heart depends on a multitude of signals. Damage associated molecular patterns (DAMPs) released by dying cells, the cytosolic DNA sensing pathway (cGAS-STING), and cardiomyocyte NLRP3 inflammasome (activated through CaMKIIδ) are each responsible for initiating the pro-inflammatory environment during myocardial injury.1–5 Furthermore, the many different cell types that make up the immune system interact with one another and with resident cardiac fibroblasts, endothelial and myocardial cells. Some examples of the beneficial and injurious immune cell sub-type responses to injury are summarized below.
Figure 1. Immune cell involvement after cardiac injury.
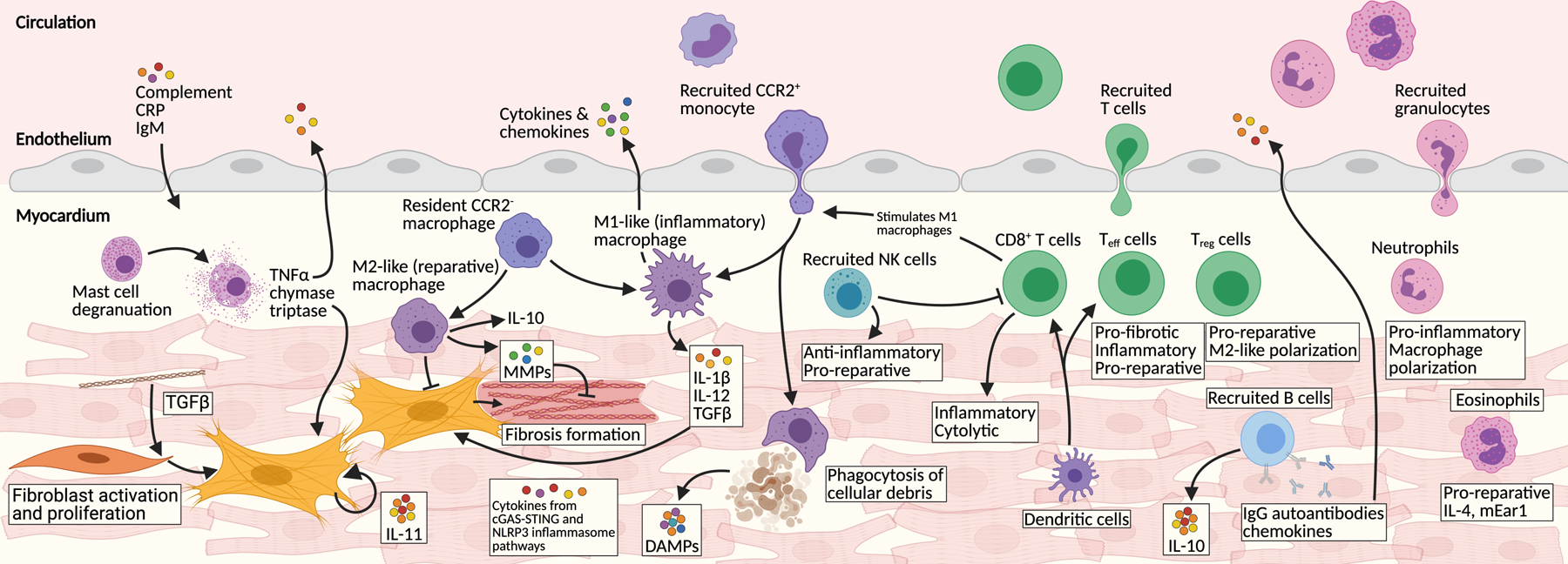
Numerous immune components are involved in the injured myocardium. The main components are represented here. Stressed cells initiate inflammatory secretory programs through the cytosolic DNA sensing cGAS-STING pathway. Dying cells release damage associated patterns (DAMPs) which act as potent signaling molecules. C-reactive protein (CRP), IgM, and other circulating factors permeate the injured heart. Tissue-resident macrophages proliferate and additional CCR2+ monocytes are recruited from circulation. Macrophages polarize between M1-like (inflammatory) and M2-like (reparative) phenotypes. Dendritic cells are potent activators of recruited T cells, which are major immune infiltrates. CD8+ cytotoxic and several effector T (Teff) cells signal with M1 macrophages and many other pro-inflammatory pathways. In contrast some Teff and regulatory T cells (Treg) infiltrate the heart and stimulate M2-like macrophages, ECM remodeling and overall heart repair. B cells produce many chemokines, cytokines, and autoantibodies. Natural killer (NK) cells are recruited and play a mainly reparative role. Many neutrophils are recruited where they stimulate M1-like macrophages and secrete pro-inflammatory cytokines. Eosinophils also infiltrate the heart and help stimulate tissue recovery. One of the major downstream targets of this inflammatory milieu are cardiac fibroblasts. Tissue-resident fibroblasts proliferate and activate and produce detrimental fibrosis.
One of the first immune cell types to respond to injury is tissue-resident macrophages. These self-renewing cells are established early in the embryonic myocardium from the yolk sac and fetal liver.6 Macrophages are critical for myocardial development, including the proper formation of the cardiac lymphatic system and they facilitate proper electrical conduction.7–9 Recently, a fascinating role for macrophages has been described in the maintenance of cardiac energy homeostasis, mediated by disposal of spent/defective mitochondria that are extruded by cardiac myocytes and engulfed by resident macrophages.10 This process utilizes components of the autophagy pathway and is enhanced in times of myocardial stress. Depletion of cardiac macrophages disrupts this process and results in activation of the inflammasome and cardiac dysfunction.10 This observation may help to explain the long-appreciated role of autophagy in metabolic dysfunction in heart failure.11
In the setting of injury, activated fibroblasts (discussed below), cardiomyocytes, and various other immune cells release cytokines to polarize existing macrophages and chemokines to recruit additional monocytes from the circulation.8,12,13 Tissue-resident CCR2− macrophages are activated and proliferate.14 These cells seem to serve several key protective/reparative roles in the post-injury heart, including promoting neo-vascularization.6,8,12–14 In contrast, the potent inflammatory signaling environment (including IL-1β) setup by numerous cell types recruits many infiltrating immune cells from the circulation.15 Infiltrating CCR2+ monocytes mature into macrophages and may outnumber those arising from tissue-resident cells.8 Pro-inflammatory Ly6chigh macrophages produce a potent cocktail of cytokines including (but not limited to) IL-1β, IL-6, and TNFα.8,16 Historically, the polarization of macrophages has been referred to as M1-like (detrimental) and M2-like (reparative) phenotypes without consideration of their tissue source. Using this terminology, the incoming CCR2+ macrophages are thought to mainly polarize into the M1-like phenotype, especially in the early phases of injury-response. Once there, pro-inflammatory macrophages secrete many cytokines and play apoptotic, cytolytic and proteolytic roles.8,13 The timing and ratio between these two phenotypes of macrophages is critical.17 Tipping the balance towards M2-like macrophages is sufficient to stimulate heart repair and improve function following injury in animal models.8,18,19 Further breakdown of the individual contributions of subgroups of macrophages in cardiac disease is the subject of intense research.20 Similarly, the mechanism underlying atherosclerotic disease is heavily dependent on infiltration of clonal monocytes.21 Clonal hematopoiesis of indeterminant potential (CHIP) has also been implicated in ischemic heart failure.22 Further refinement of our understanding of macrophage subtypes responding to various myocardial injuries, along with the development of interventions capable of targeting specific subtypes, may provide attractive therapeutic avenues for development.
Alongside the recruitment of monocytes, the inflammatory environment of the injured heart also attracts significant neutrophil infiltrates. Neutrophils participate in various aspects of myocardial injury response. These include amplifying damaging pro-inflammatory signals, production of reactive-oxygen species, and secretion of proteolytic enzymes to remodel the ECM.23–25 However, experimental depletion of all neutrophils before cardiac injury negatively impacts the ability of the heart to recover.26 Mechanistically, neutrophils promote cardiac recovery by polarizing macrophages towards a reparative phenotype.26 Neutrophil recruitment and participation in the post-infarct heart have recently been comprehensively reviewed by Daseke, et al.27 Targeting subsets of pro-inflammatory neutrophils might therefore offer opportunities for therapy.
Eosinophils are also recruited to the injured heart. Increased levels of circulating eosinophils have been associated with risk of coronary artery disease and, in rare examples, excessive myocardial infiltration can result in eosinophilic myocarditis.28 However, recent research also suggests a protective role for eosinophils in the heart following acute cardiac injury. Depletion of eosinophils exacerbates cardiac dysfunction and fibrosis after infarction.29,30 Eosinophil-specific IL-4 and mEar1 expression has been demonstrated to block H2O2 and hypoxia induced cardiomyocyte death and dampen fibroblast activation.30 The full role of eosinophils in the injured myocardium remains to be elucidated.
Tissue-resident mast cells proliferate in response to numerous cardiac injuries. Degranulated mast cells release many pro-inflammatory signaling molecules (including TNF-α and histamines) and proteases (including chymase, renin, and tryptase) involved in fibroblast activation.31,32 Mast cells are mainly pro-fibrotic in the injured heart and interestingly, this negative impact is somewhat abrogated in the presence of estrogen.33 In contrast, natural killer cells appear to play multiple, complex roles to protect the injured myocardium. Natural killer cells limit damaging innate immune cell infiltration and activity through restraining chemokine production as well as secreting INF-γ, perforin and other anti-inflammatory chemokines.34 Additionally, natural killer cells have been shown to directly inhibit activated fibroblasts from over-producing collagen, limit cardiomyocyte apoptosis and stimulate neovascularization.35,36
The adaptive immune system is also integral to the pro-inflammatory environment of the injured heart. It plays multifaceted roles following acute myocardial infarction that have only recently begun to be fully elucidated.37 In chronic pressure-overload, dendritic cells (professional antigen-presenting cells) accumulate potent γ-ketoaldyhydes, which activate a pro-inflammatory program of reactive oxygen species, IL-1β, IL-6, and IL-23 secretion.38 Dendritic cells also increase expression of T cell co-stimulatory proteins. Ultimately, dendritic cells in the hypertensive heart serve to promote T cell proliferation (especially CD8+ subsets) and promote polarization into a pro-inflammatory phenotype.38–40 Isolated dendritic cells from hypertensive mice transplanted into naïve mice prime the recipient towards the ill-effects of high blood pressure.38 Post-myocardial infarction, cross-priming dendritic cells are partially responsible for sustained myocardial injury and decreased left ventricular function.39 On the other hand, there is some evidence that injection of dendritic cells primed in injured hearts helps to coordinate a beneficial, pro-reparative myocardium through coordination of regulatory T cells and shifting macrophage polarization to the M2-like phenotype.41 More work is required to dissect and understand the potentially beneficial and deleterious functions of dendritic cells in the injured heart.
Both B and T lymphocytes also play important roles in cardiac homeostasis and response to injury. For example, mice lacking PD-1 (essential for B cell differentiation) spontaneously develop dilated cardiomyopathy with increased levels of cardiomyocyte-specific IgG autoantibodies.42 Interestingly, patients with end-stage heart failure can often harbor multiple anti-cardiomyocyte antibodies as recently reviewed in detail by García-Rivas, et al.43 In the setting of acute decompensation and ischemia-reperfusion, B cells are responsible for production of potent apoptotic signals and autoantibody-activation of the complement cascade.43,44 B cell depletion in hypertensive mice results in lower fibrotic burden.45 Furthermore, B cells in the injured heart signal to CD4+ T cells through MHC-II and to monocytes.43,46 This interplay is dependent on the IL-1 pathway and MyD88, with MyD88 knockout mice lacking the robust fibrosis seen in induced myocarditis in wild-type animals.47 IL-10, produced primarily by B cells, has emerged as a critical anti-inflammatory (protective) cytokine that targets the potent NF-κB/STAT3 signaling pathway, which can be augmented through blockade of T cell costimulation.48,49
Certain infiltrating T cells are protective in the injured myocardium. Subsets of CD4+ non-specific effector T (Teff) cells have been shown to protect against post-inflammatory fibrosis in an experimental model of myocarditis.50 The authors suggest multiple potential mechanisms for protective effects of non-specific Teff cells. Essentially, if the balance shifts away from antigen-specific Teff cells to heart non-specific Teff cells, the levels of pro-inflammatory cytokines such as IL-17A are reduced and fibrosis is limited.50 Separately, our lab previously elucidated an immune-based cardioprotective mechanism through which the epicardium recruits CD4+ T regulatory (Treg) cells in a Hippo/INF-γ –dependent process.51 Furthermore, Foxp3+ Treg cells have been shown to stimulate recovery of both injured skeletal and heart muscle.52–54 Accumulating Treg and certain MYHCA-specific T helper cells have multiple roles, driving pro-reparative phenotypes.52
Many classes of pro-inflammatory T cells infiltrate the injured myocardium in response to dendritic cell activation and chemotactic signals. Activated fibroblasts, macrophages, endothelial cells, and cardiomyocytes produce additional chemotactic signals attracting T cells (through CXCR3 and endothelial ICAM-1), including the NLRP3 inflammasome, CCL2 ligand, GM-CSF, IL-6 and CXCL1/9/10, amongst others.55–57 Once in the myocardium, activated macrophages also contribute to the stimulation of T cells.8 Pro-inflammatory CD8+ T cells, activated by dendritic cells, produce potent cytokines including IL-17, IFN-γ, and TNFα.38,39,58 CD8+ T cells are required for M1-like macrophage infiltration and pro-inflammatory cytokine and chemokine secretion.58 In addition to cytotoxic T cells, certain CD4+ T cells also exhibit damaging effects in the injured heart and are required for the progression of pressure-overload injury into heart failure.56,59–61 Adoptive transfer of splenic T cells from heart failure mice was sufficient to induce heart failure in naïve recipient mice.59 T cell infiltrates have pleotropic effects on virtually every cell type in the injured myocardium. One main role is the direct activation of cardiac fibroblasts and the induction of fibrosis.62 The rapid, pro-inflammatory response of CD4+ and CD8+ T cells may be beneficial in the acute phase of injury to enhance scar formation but may be maladaptive in the long term.
The development of myocardial fibrosis
The multi-faceted immune response to injury impacts many aspects of cardiac function. Among these, regulation of cardiac fibrosis is particularly important in both acute and chronic settings.63,64 Tissue-resident fibroblasts differentiate from multiple sources early during cardiogenesis and are a self-sustaining population.65 At homeostasis, these cardiac fibroblasts are primarily responsible for the secretion and maintenance of the extracellular matrix. After an injury, resident fibroblasts activate, alter gene expression programs (including pro-inflammatory cytokine production), proliferate and accelerate matrix production.66–70 Additionally, in the setting of myocardial infarction, activated fibroblasts are recruited from the epicardium through a process of epithelial-mesenchymal transformation and migration into underlying regions of damaged myocardium.51,71,72 In the setting of pressure-overload, fibroblasts are also activated in the vessel wall leading to peri-arteriolar fibrosis that can impair vascular function.73 Fibroblast activation occurs in response to multiple cytokines such as transforming growth factor beta (TGFβ), which is produced by a variety of cells including immune cells and active when liberated from its latent form in the matrix.74–77 TGFβ signaling is mainly propagated within fibroblasts through phosphorylation of SMAD2/3 which complexes with SMAD4 to directly regulate transcription.75,78–81 Additional signaling cues are received from the local inflammatory environment and spread through multiple signaling pathways including WNT, MEK/ERK, and JNK.82 Recently, autocrine interleukin-11 has also been implicated in fibroblast activation.83 The extracellular matrix (ECM) produced by activated fibroblasts maintains tissue integrity, limiting cardiac dilatation and rupture.84,85 However, in chronic injury the resulting fibrosis contributes negatively to myocardial health.
Myocardial fibrosis has been associated with poor clinical outcomes and negatively impacts heart physiology both directly and indirectly.86–89 First, the excess ECM directly alters the compliance of the heart muscle. The stiffness associated with myocardial fibrosis places increase workload on the cardiomyocytes, fueling their dysfunction and decreasing both diastolic and systolic function.63,64,89,90 Furthermore, excess collagen networks have been linked to electrical conduction defects and arrhythmias.91 Perivascular fibrosis has been associated with impaired coronary blood flow and microvessel rarefication contributing to latent myocardial hypoxia.73 Finally, both activated fibroblasts and the composition of the ECM signal to infiltrating immune cells.87,92 For example, the extracellular matrix protein tenascin-c has been shown to accelerate macrophage recruitment and to activate a pro -inflammatory/-fibrotic (M1-like) transcriptome.93 Interestingly, another neonatal extracellular matrix protein, agrin, can stimulate murine cardiac regeneration.94
Fibroblast activation is less homogenous than was originally appreciated. Revolutions in single cell and single nuclei RNA sequencing have uncovered many seemingly unique fibroblast populations before and after injury with distinct transcriptome signatures.20,95–98 The lineage relationship between these subsets and their physiologic importance remains to be fully elucidated. Further evidence for different subsets of fibroblasts contributing to disease pathology comes from recent research in rheumatoid arthritis. Croft et al., identified two subpopulations of fibroblasts in injured joints.99 The first group of fibroblasts (marked by FAP+/Thy1− expression) were responsible primarily for bone and cartilage-degradation, while a second population of FAP+/Thy1+ fibroblasts contributed to joint inflammation.99 This example suggests that further refinement in our understanding of distinct sub-classes of activated fibroblasts in the heart may reveal distinct roles in cardiac pathology and inform targeted therapeutics.
Immune modulators as cardiac therapies
The potential for immune modulation to impact the heart has been appreciated for decades, and clinicians have long recognized that acute infection and associated immune activation can negatively impact cardiac function.100–102 Recently, the role of immune modulation in atherosclerosis has been clearly demonstrated, as reviewed elsewhere.21,103 Broad immunosuppression in acute ischemic heart failure through the administration of corticosteroids or nonsteroidal anti-inflammatory drugs (NSAIDS) is contraindicated and can lead to an increased incidence of cardiac rupture, and their use in chronic forms of heart failure has generally produced either no or a negative effect.104–106 Similarly, broad immune suppression through colchicine administration has also failed to demonstrate any benefit in multiple heart diseases and is often contraindicated.107–109 The challenge is to identify targeted immune-modulators that can specifically affect components of the immune response to favor recovery after injury and/or diminish adverse remodeling.
Evidence to support the potential for immune modulation in heart failure has emerged, surprisingly, from studies focused on the mechanism of potential benefit observed in some cardiac stem cell studies. Despite decades of work, cardiac stem cells remain elusive, yet improvement in cardiac function is sometimes observed after delivery of various cell types to the injured heart. The delivered cells generally do not survive, engraft, or differentiate into functional myocytes. Instead, they seem to stimulate an inflammatory response through activated macrophages that can result in enhanced cardiac function, at least temporarily.110 In seminal work, Vagnozzi, et al. showed that following ischemia-reperfusion injury, a myocardial injection of freeze-thaw killed cells resulted in equal improvement in function to injection of live putative stem cells.110 More importantly, a local injection of zymosan (a potent stimulator of the innate immune system) was also sufficient to produce functional improvements.110 This effect was due to recruitment of CCR2+ and CX3CR1+ (M2-like) macrophages which are beneficial following injury.110 The authors described endothelial cell, but not cardiomyocyte, proliferation and beneficial extracellular matrix remodeling. Thus, agents that augment M2-like macrophages may prove beneficial.
Targeted disruption of the complement cascade following myocardial infarction was hypothesized to tip the balance towards myocardial recovery. Early evidence of monoclonal antibodies blocking C5 cleavage were encouraging in animal models.111,112 However, clinically similar antibodies (pexelizumab) have yet to demonstrate beneficial effect above standard percutaneous coronary reperfusion therapy following reperfusion injury associated with myocardial infarction.113 While clinical trials failed to show clear benefit of targeted interference of the complement cascade, the approach may still hold promise.114
One of the first cytokines targeted in heart failure was TNFα.115 This potent inflammatory cytokine was identified as a circulating biomarker, clinically correlated with heart failure severity.116 Research laboratories were quick to follow up this observation with animal model experimentation. It was found that administration of exogenous TNFα was sufficient to induce progressive left ventricular dysfunction.117 Further, animals co-infused with TNFα and its antagonist partially reversed the induced left ventricular dysfunction.117 Based on these observations and others, several clinical trials were conducted to test safety and efficacy in humans of anti-TNFα therapy. Unfortunately, while intravenous etanercept infusions were well tolerated, RENEWAL analysis of the RECOVER and RENAISSANCE clinical trials revealed that TNFα blockade did not yield beneficial reductions in morbidity and mortality.118 Despite these initial unsuccessful clinical trials, the original concept of therapeutically modulating the post-injury immune system remains attractive.119
Another cytokine that has been tested in heart failure is C-X-C motif chemokine ligand 12 (CXCL12; also known as stromal cell-derived factor 1 or SDF-1). Studies in animal models suggested potential benefit, perhaps via recruitment of monocytes or other cells through a CXCR4 dependent mechanism.120 Unfortunately, a phase II clinical trial in patients with heart failure with reduced ejection fraction (EF<40%) determined that transcatheter endomyocardial delivery of CXCL12 provided no cardiac functional improvements detectable at four months.121 Further refinement in dosing, delivery and timing of cytokine-based therapeutics may beget clinical benefit.
Targeted disruption of the fibroblast activation induced by immune cells after injury has also been considered as a potential therapeutic approach. In this regard, inhibition of the TGFβ receptor kinase pathway has received particular attention.122,123 Experiments in animals have yielded conflicting results, with some finding cardiac benefits and others worrisome increases to mortality and left ventricular dysfunction.122,124–127 In systemic sclerosis, disruption of the TGFβ pathway was not successful at controlling fibrosis and may have even been responsible for adverse events (although sample size was small).128 In contrast, TGFβ blockade with perfenidone has demonstrated benefits in fibrosis-based interstitial lung diseases.129,130 At least one clinical trial of anti-TGFβ blockade in heart failure patients has been initiated.131 While we await these results, developing more targeted approaches may yield beneficial results.65 Partial evidence for this comes from studies in mice with cardiomyocyte-specific TGFβ receptor inactivation, which favors cardiac recovery through an immune-modulatory mechanism.132
Targeted or engineered immune-based therapeutics
Recent efforts have begun to focus on the use of biomaterials, biologics and targeted cell and gene therapy to modify specific components of the immune response to cardiac injury (see Figure 2).133 For example, targeted inhibition of the coagulation cascade (factor Xa, but not IIa) has demonstrated beneficial cardiac remodeling following injury by dampening harmful inflammation.134 Clever chemical engineering has resulting in graphene oxide complexes functionalized with IL-4, that appear to coax cardiac macrophages from M1-like to M2-like pro-reparative phenotype and limit damaging inflammation when injected into post-myocardial infarction hearts.135 Several other macrophage modulating molecules are in various stages of development in non-cardiovascular injuries that may be worth evaluating in heart injury models.136 These include lipoxin A4 and resolvin D1, which were shown to improve resolution following implanted biomaterial scaffolds through an M2-like macrophage mechanism.137 Many other naturally derived biomaterials have been shown to scavenge radical oxygen species and can be loaded with other anti-inflammatory molecules to further alter the microenvironment.136 It may be possible to target lipid nanoparticles loaded with mRNA or other agents to specific immune cell types, taking advantage of therapeutic platforms developed and validated as part of the COVID-19 vaccination efforts.
Figure 2. Potential immune-modulating targets to promote cardiac repair.
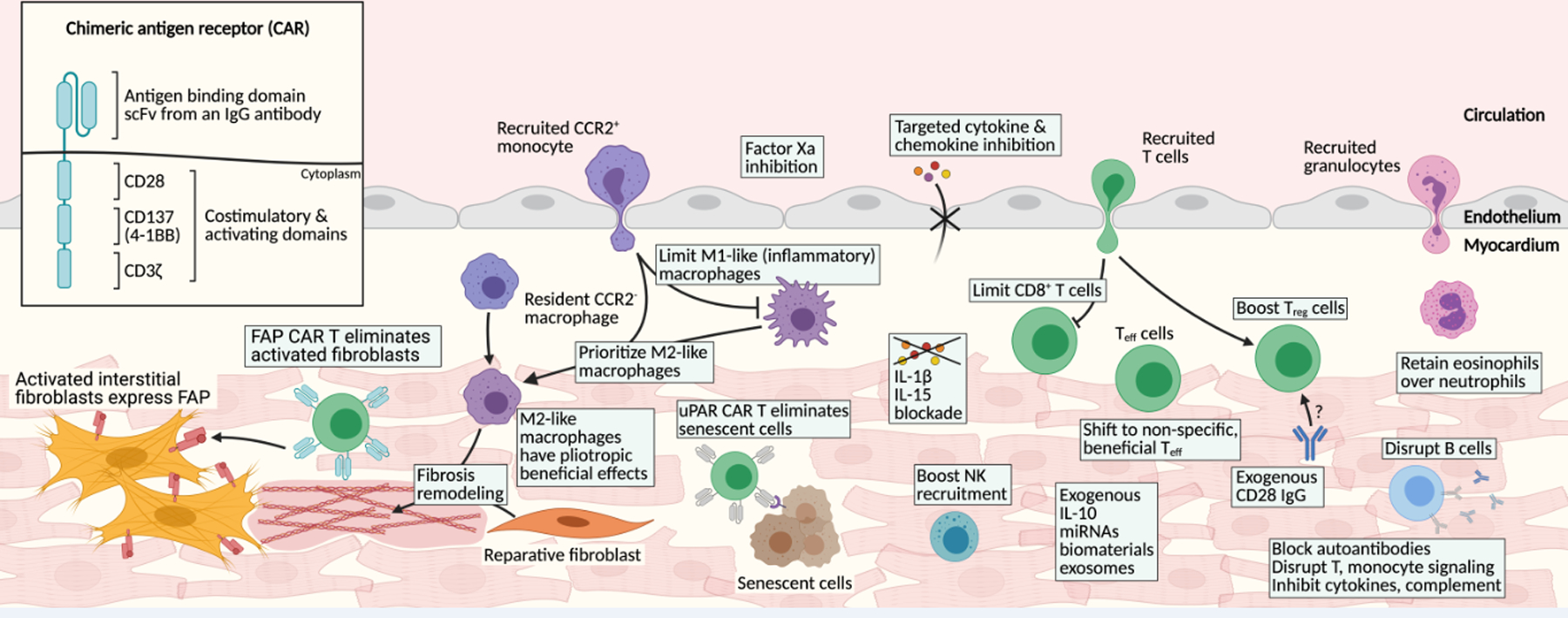
Many immune modulatory targets exist within the complex signaling network in the injured myocardium. Chimeric antigen receptor (CAR) constructs include antigen recognition domains and intracellular signaling domains, which are engineered into cytotoxic T cells. CAR T cells designed against activated fibroblasts and senescent cells both offer exciting directions to improve cardiac repair in the failing heart. Shifting the balance of macrophage polarization to favor the M2-like, reparative phenotype, may produce beneficial effects. Limiting the cytotoxic CD8+ and heart-specific CD4+ T cell infiltrate in favor of boosting regulatory (Treg) cells, beneficial subsets of effector T (Teff) cells and natural killer (NK) cells have each been shown to benefit cardiac repair. Similarly, shifting the balance to eosinophils over neutrophils may benefit recovery. Disrupting B cell pathways offers potential benefit. Targeted interleukin blockade (IL-1, IL-11, IL-15, TGFβ, etc.) or reparative cytokine (IL-10) administration may limit damage of excessive (or improperly timed) inflammation. Lastly, the development of novel chemical materials and biological interventions (including miRNAs and exosomes) offer exciting targeted immune modulators.
Immune-modulating exosomes and miRNAs may hold potential for enhancing repair following cardiac injury. Exosomes are naturally-occurring small extracellular vesicles filled with a variety of biological molecules, which may offer avenues for immune modulation in cardiac injury.138 Advances in engineered exosomes offers the potential for unique cardiac delivery vehicles of immune-modulating therapeutics such as miRNAs. Multiple miRNAs including miR-155, miR-146a, miR-181b, miR-208, miR-29b and others are under investigation for promoting heart repair through immune modulation, with several in current clinical trials.139
Blocking specific cytokines offers a potential avenue for therapy. Interleukin 1β (IL-1β) is a pleotropic cytokine with various inflammatory functions in the injured heart, mainly augmenting injury. Systemic blockade of IL-1 signaling is typically achieved with either a competitive inhibitor of the receptor (canakinumab) or blocking antibodies against IL-1β (anakinra).105 This approach has shown modest beneficial effect in animal models of myocardial infarction.140,141 In clinical trials canakinumab has been well tolerated and resulted in avoidance of post-myocardial infarction heart failure and reduction in mortality in heart failure patients.142,143 Furthermore, canakinumab has demonstrated beneficial effect in atherosclerotic disease.144 The beneficial outcomes observed following IL-1 pathway disruption are exciting with the caveat that less targeted IL-1 interference (low-dose methotrexate) did not recapitulate the results obtained with canakinumab in atherosclerotic events and resulted in increased incidence of negative side effects.145 This trial re-enforces that targeted immunotherapeutics are likely to offer more potential benefits than broad immune modulators. Interestingly, SGLT2 inhibitors, which have demonstrated benefits in heart failure patients, may be operating through IL-1β and/or NLRP3 inflammasome mechanisms, supporting the notion of careful inflammation modulation.146–148 Similarly, both interleukin 15 and interleukin 11 blockade are under investigation as a potential cytokine targets for cardiac disease.83,149 Interleukin 11 appears to be a central player in the immune-fibrosis axis in multiple organs, reinforcing its attractiveness as a potential therapeutic target.83,150,151
Certain cytokines have been shown to stimulate repair and may be attractive therapeutics. In the injured heart, IL-10 is typically thought of as pro-reparative; however, in other contexts it can be pro-inflammatory.152 A recent structural study has elucidated a biased signaling mechanism for IL-10, analogous to GPCR/β-arrestin signaling.153 Furthermore, the authors demonstrate how engineered variants of IL-10 can strongly skew the intracellular signaling response.153 This paradigm of fine-tuning biased receptor signaling through ligand optimization, well accepted in the field of GPCR signaling, offers exciting opportunities for anti-inflammatory therapeutics.
A recent study identified cardioprotective T regulatory-like cells in the post-myocardial infarcted heart. These CD4+ T cells react with myosin heavy chain alpha and accumulate in the injured myocardium of both mice and human patients, and appear to be beneficial in mice when delivered exogenously before induction of myocardial infarction.52 Related Foxp3+ Treg cells have been shown to be beneficial in the injured mouse and rat heart, either with an infusion of autologous Treg or a CD28 antibody (which augments the natural recruitment of Treg cells).51,54,154 Boosting the recruitment and accumulation of Treg in the injured heart as demonstrated in animal models, may be an attractive future direction for human therapeutics.
CAR T cells for heart disease
Genetically engineered cytotoxic T cells that have been redirected to recognize a specific antigen were developed in the late twentieth century following the success of adoptive transfer of T cells to control viral infection.155–160 This was accomplished by expressing a chimeric antigen receptor (CAR) on activated CD8+ T cells. The chimeric antigen receptor is a combination of a single-chain variable fragment (scFv) against a specific antigen linked to intracellular T cell signaling and co-stimulatory molecules (see figure 2 inset). Typically, these include at least a CD3-zeta tyrosine phosphorylation activation motif, as well as 4–1BB and CD28 co-stimulatory signaling domains, though this design has many iterations. This all-in-one protein design results in one-step cytotoxicity, induced through antigen binding alone, as opposed to T cell receptor (TCR) mediated cytotoxicity which requires MHC class I antigen presentation, CD3 recruitment to the TCR, and costimulatory signaling.161 CAR T cells were quickly adapted to target cancerous cells and have revolutionized the treatment of refractory acute lymphoblastic leukemia and diffuse large B-cell lymphoma.161,162
In the setting of heart failure, it is attractive to consider targeting activated cardiac fibroblasts with engineered T cells. This is made possible because activated cardiac fibroblasts are sufficiently different from quiescent, homeostatic fibroblasts such that they express unique cell surface markers and can therefore be specifically targeted. An early indication that this approach might be effective to reverse fibrosis and improve function after injury came from mouse studies in which activated fibroblasts were eliminated by genetic ablation using diphtheria toxin in animals engineered to express the diphtheria toxin receptor on activated fibroblasts.163 In the setting of pressure-overload injury, heart function was improved and fibrotic burden was decreased by removal of activated fibroblasts.163
In order to provide a mechanism to translate these findings to humans, we sought to target activated fibroblasts with an engineered T cell. To find a unique molecular marker of activated fibroblasts, we utilized transcriptomics to compare healthy human hearts to dilated and hypertrophic hearts. Consistent with the literature, we found that both fibroblast activation protein alpha (FAP) and periostin were highly upregulated in both disease states when compared to healthy hearts.164 In the adult, FAP protein expression is largely restricted to activated fibroblasts in various disease states and after injury. It is also expressed by proliferative mesenchyme in many forms of cancer.165 Although a secreted form of FAP can be identified in the circulation of healthy humans, it does not correlate with stromal cell expression or serve as a reliable biomarker.166,167 Importantly, this protein is virtually absent in healthy adult tissues.165 FAP CAR T cells have been demonstrated to effectively target and reduce FAP+ cancer associated fibroblasts.168 Using a mouse model of cardiac fibrosis and heart failure produced by the constant infusion of angiotensin II and phenylephrine, we showed that FAP CAR T cells were effective at restoring cardiac function and significantly lowering the burden of fibrosis even when the CAR T cells were delivered after fibrosis has already accumulated.164 Resorption of previously accumulated ECM is likely the result of macrophage, non-targeted fibroblasts and neutrophil activity which predominates once the cells producing the excessive matrix are destroyed by the CAR T therapy.
One of the advantages of FAP as an antigen target is the ability to visualize expression in vivo using imaging probes. Initial FAP-PET (Positron Emission Tomography) probes were developed using antibodies and tested extensively in mice.169 Loktev, et al. extended these findings by attaching gallium-68 to a synthetic FAP inhibitor and showed that cancer-associated fibroblasts, which resemble activated cardiac fibroblasts, can be directly imaged in both mice and humans.170 These powerful imaging tools have been used successfully to image cardiac fibrosis in humans and represent an exciting screening tool for clinical evaluation of heart failure patients prior to FAP CAR T intervention and for monitoring of therapeutic efficacy.171,172
The use of CAR T cells to target senescent cells is also in development. Senescent cells are characterized by permanent cell cycle arrest and secretion of high levels of inflammatory molecules producing a senescence-associated secretory phenotype (SASP). Cellular senescence has been associated with many age-related disorders including cardiac pathologies and arrhythmias.173,174 Clearance of senescent cells may ameliorate the decrease in cardiac performance associated with aging and improve recovery following injury.175,176 Recently, Amor, et al. described an anti-senescence CAR T cell directed against urokinase-type plasminogen activator receptor, a marker of at least some senescent cells.177 Following adoptive transfer of anti-senescence CAR T cells, mice with induced liver fibrosis or lung adenocarcinoma showed improvement in organ function.177 To our knowledge, this approach has not yet been studied in animal models of cardiac disease.
CAR T cell therapy is associated with important acute toxicities. Infusion of CAR T products, including the FDA approved anti-CD19 CAR T cells (tisagenlecleucel and axicabtagene ciloleucel) can induce potentially lethal cytokine release syndrome.178,179 Fortunately this syndrome is typically transient and can be managed with IL-6 blockade (tocilizumab) and supportive care.179–181 Anti-CD19 CAR T therapy is also rarely associated with neurotoxicity, potentially through unintended, on-target elimination of CD19+ mural cells.182 Especially pertinent for this discussion are cases of cardiotoxicity following engineered T cell administration potentially due to unforeseen cross-reactivity.183,184 The binding strength of the individual CAR to the target antigen is also important. In animal trials of preliminary FAP CAR constructs, some toxicity was observed, although it is mitigated by changing the antibody upon which the CAR was based.168,185 These examples highlight the need for restricted target antigen expression limited to the pathologic cells of interest, and for further safety studies for potential cardiac CAR T products. In support of FAP as a potential target, a recent study found that genetic ablation of FAP does not impair cardiac function in healthy animals or result in cardiotoxicity following myocardial infarction.186 Furthermore, target-cell specificity can be greatly increased through the use of dual-specific engineered T cells. In this approach, two antigens are chosen and the intracellular signaling domains are split between the two separate chimeric antigen receptors.187 Therefore, the CAR T must engage both antigens before cytolysis occurs. An alternative approach is using a bispecific T cell engager (BiTE) to increase specificity and reduce unwanted off-target effects.188,189 By refining the target antigen beyond FAP, anti-fibrotic CAR T cells hold great promise.
Unlike in cancer where curative therapy requires elimination of every cancerous cell, CAR T therapy for fibrosis may be effective at restoring function even if only a fraction of the disease-causing fibroblasts are destroyed. Dosing can therefore be titrated, and cells can be engineered to have only transient activity, eliminating potential long-term toxicities. It is critical to tune the quantity, potency, and longevity of the CAR to ensure that the beneficial, homeostatic fibroblasts remain within the myocardium to maintain the healthy ECM after the excessive fibrosis is reduced. Furthermore, subsequent acute myocardial ischemic events require robust fibroblast activation. A transient anti-fibrotic CAR T is therefore required. Various mechanisms to produce transient CAR T cells have been described, including the use of kill switches or required co-activator small molecules.190 Alternatively, CARs can be engineered using mRNA rather than lentiviral vectors such that the CAR is no longer expressed after the mRNA decays (over the course of several days) or after dilution due to cell division.191 The ability to target specific subsets of pathologic fibroblasts in various disease states, in the heart and in other organs, may emerge as additional unique cell surface markers are identified. Cell therapy for cardiac diseases is an exciting field, still in its infancy.
Conclusion
The pathology of myocardial injury and repair is coordinated by a complex web of intersecting inflammatory pathways and immune cell types. Our understanding of the beneficial and harmful contributions of specific immune cells and cytokines is an exciting and emerging field of investigation. Targeted modulation of the immune system as a mechanism to boost myocardial recovery and repair is an important goal of cardio-immunology. M2-like macrophages, natural killer cells, and regulatory T cells represent especially promising targets for therapeutic intervention. Furthermore, the prospect of engineering the immune system to reduce both fibrosis and senescence in aging and failing hearts offers potential future therapeutic avenues. Other engineered immune cells, such as CAR macrophages may also be on the horizon.192 Altogether, targeted manipulation of the immune system to benefit the injured heart is an exciting and promising field of active study.
Acknowledgements
We thank Cheryl Smith, the members of the Epstein Lab, and the immune-fibrosis axis in ischemic heart failure Leducq transatlantic network for discussion. Figures were created using BioRender.com.
Sources of Funding
This work was supported by NIH R35 HL140018, T32 HL007843–23, the Cotswold Foundation, the WW Smith Endowed Chair and the Leducq Foundation.
Nonstandard Abbreviations and Acronyms:
- CAR
Chimeric antigen receptor
- cGAS-STING
cyclic GMP-AMP synthase stimulator of interferon genes
- CHIP
Clonal hematopoiesis of indeterminate potential
- COVID-19
Coronavirus disease 2019
- CXCL12
C-X-C motif chemokine ligand 12 (previously known as stromal cell-derived factor 1 or SDF-1)
- DAMPs
Damage associated molecular patterns
- ECM
Extracellular matrix
- EF
Ejection fraction (%)
- FAP
Fibroblast activation protein
- IL-1
Interleukin 1
- M1-like
Pro-inflammatory macrophage
- M2-like
Pro-reparative macrophage
- MHC-II
Major histocompatibility complex class II
- NK
Natural killer cell
- NLRP3
NLR family pyrin domain containing 3
- PET
Positron emission tomography
- scFv
Single-chain variable fragment
- SGLT2
Sodium/glucose cotransporter 2
- TCR
T cell receptor
- Teff
Effector T cell
- TGFβ
Transforming growth factor β
- TNFα
Tumor necrosis factor
- Treg
Regulatory T cell
Footnotes
Disclosures
JAE and HA are scientific founders, but not financially vested in Teefib Bio, a biotech company devoted to evaluating the use of anti-fibrotic engineered immune cells in non-oncologic diseases. JAE reports research funding from Calico Life Sciences to develop novel anti-senescent CAR T cells.
References
- 1.Jaén RI, Val-Blasco A, Prieto P, Gil-Fernández M, Smani T, López-Sendón JL, Delgado C, Boscá L, Fernández-Velasco M. Innate Immune Receptors, Key Actors in Cardiovascular Diseases. JACC Basic to Transl Sci. 2020;5:735–749. Available from: 10.1016/j.jacbts.2020.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS–STING signalling. Nat Rev Mol Cell Biol. 2020;21:501–521. Available from: 10.1038/s41580-020-0244-x [DOI] [PubMed] [Google Scholar]
- 3.Cao DJ, Schiattarella GG, Villalobos E, Jiang N, May HI, Li T, Chen ZJ, Gillette TG, Hill JA. Cytosolic DNA Sensing Promotes Macrophage Transformation and Governs Myocardial Ischemic Injury. Circulation. 2018;137:2613–2634. Available from: 10.1161/CIRCULATIONAHA.117.031046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu D, Cui Y-X, Wu M-Y, Li L, Su L-N, Lian Z, Chen H. Cytosolic DNA sensor cGAS plays an essential pathogenetic role in pressure overload-induced heart failure. Am J Physiol Circ Physiol. 2020;318:H1525–H1537. [DOI] [PubMed] [Google Scholar]
- 5.Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, Brown JH. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca2+/Calmodulin-Dependent Protein Kinase II δ Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation. 2018;138:2530–2544. Available from: 10.1161/CIRCULATIONAHA.118.034621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. Available from: 10.1016/j.immuni.2013.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cahill TJ, Sun X, Ravaud C, Villa del Campo C, Klaourakis K, Lupu I-E, Lord AM, Browne C, Jacobsen SEW, Greaves DR. Tissue-resident macrophages regulate lymphatic vessel growth and patterning in the developing heart. Development. 2021;148:dev194563. Available from: 10.1242/dev.194563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lavine KJ, Pinto AR, Epelman S, Kopecky BJ, Clemente-Casares X, Godwin J, Rosenthal N, Kovacic JC. The Macrophage in Cardiac Homeostasis and Disease: JACC Macrophage in CVD Series (Part 4). J Am Coll Cardiol. 2018;72:2213–2230. Available from: 10.1016/j.jacc.2018.08.2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wülfers EM, Seemann G, Courties G, et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell. 2017;169:510–522.e20. Available from: 10.1016/j.cell.2017.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicolás-Ávila JA, Lechuga-vieco AV, Esteban-Martínez L, Sánchez-Díaz M, Díaz-García E, Santiago DJ, Rubio-Ponce A, Li JLY, Balachander A, Quintana JA, et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell. 2020;183:94–109.e23. Available from: 10.1016/j.cell.2020.08.031 [DOI] [PubMed] [Google Scholar]
- 11.Abdullah CS, Alam S, Aishwarya R, Miriyala S, Bhuiyan MAN, Panchatcharam M, Pattillo CB, Orr AW, Sadoshima J, Hill JA, Bhuiyan MS. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respiration. Sci Rep. 2019;9:1–20. Available from: 10.1038/s41598-018-37862-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ, Mann DL. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci. 2014;111:16029–16034. Available from: 10.1073/pnas.1406508111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma Y, Mouton AJ, Lindsey ML. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl Res. 2018;191:15–28. Available from: 10.1016/j.trsl.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao HM, Weinheimer C, et al. Tissue Resident CCR2- and CCR2+ Cardiac Macrophages Differentially Orchestrate Monocyte Recruitment and Fate Specification Following Myocardial Injury. Circ Res. 2019;124:263–278. Available from: 10.1161/CIRCRESAHA.118.314028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, Wojtkiewicz GR, Tricot B, Iwamoto Y, Sun Y, et al. Targeting Interleukin-1β Reduces Leukocyte Production After Acute Myocardial Infarction. Circulation. 2015;132:1880–1890. Available from: 10.1161/CIRCULATIONAHA.115.016160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gombozhapova A, Rogovskaya Y, Shurupov V, Rebenkova M, Kzhyshkowska J, Popov SV, Karpov RS, Ryabov V. Macrophage activation and polarization in post-infarction cardiac remodeling. J Biomed Sci. 2017;24:13. Available from: 10.1186/s12929-017-0322-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilgendorf I, Gerhardt LMS, Tan TC, Winter C, Holderried TAW, Chousterman BG, Iwamoto Y, Liao R, Zirlik A, Scherer-Crosbie M, et al. Ly-6C high Monocytes Depend on Nr4a1 to Balance Both Inflammatory and Reparative Phases in the Infarcted Myocardium. Circ Res. 2014;114:1611–1622. Available from: 10.1161/CIRCRESAHA.114.303204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res. 2020;116:1101–1112. Available from: 10.1093/cvr/cvz336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shiraishi M, Shintani Y, Shintani Y, Ishida H, Saba R, Yamaguchi A, Adachi H, Yashiro K, Suzuki K. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest. 2016;126:2151–2166. Available from: 10.1172/JCI85782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McLellan MA, Skelly DA, Dona MSI, Squiers GT, Farrugia GE, Gaynor TL, Cohen CD, Pandey R, Diep H, Vinh A, et al. High-Resolution Transcriptomic Profiling of the Heart During Chronic Stress Reveals Cellular Drivers of Cardiac Fibrosis and Hypertrophy. Circulation. 2020;142:1448–1463. Available from: 10.1161/CIRCULATIONAHA.119.045115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jaiswal S, Libby P. Clonal haematopoiesis: connecting ageing and inflammation in cardiovascular disease. Nat Rev Cardiol. 2020;17:137–144. Available from: 10.1038/s41569-019-0247-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorsheimer L, Assmus B, Rasper T, Ortmann CA, Ecke A, Abou-El-Ardat K, Schmid T, Brüne B, Wagner S, Serve H, et al. Association of Mutations Contributing to Clonal Hematopoiesis with Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019;4:25–33. Available from: 10.1001/jamacardio.2018.3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daseke MJ, Valerio FM, Kalusche WJ, Ma Y, DeLeon-Pennell KY, Lindsey ML. Neutrophil proteome shifts over the myocardial infarction time continuum. Basic Res Cardiol. 2019;114:1–13. Available from: 10.1007/s00395-019-0746-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, Quaife-Ryan GA, Al-Sharea A, Pernes G, Dragoljevic D, et al. Neutrophil-derived S100A8/A9 amplify granulopoiesis after myocardial infarction. Circulation. 2020;1080–1094. Available from: 10.1161/CIRCULATIONAHA.119.043833 [DOI] [PMC free article] [PubMed]
- 25.Vinten-Johansen J Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc Res. 2004;61:481–497. Available from: 10.1016/j.cardiores.2003.10.011 [DOI] [PubMed] [Google Scholar]
- 26.Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S. Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J. 2017;38:187–197. Available from: 10.1093/eurheartj/ehw002 [DOI] [PubMed] [Google Scholar]
- 27.Daseke MJ, Chalise U, Becirovic-Agic M, Salomon JD, Cook LM, Case AJ, Lindsey ML. Neutrophil signaling during myocardial infarction wound repair. Cell Signal. 2021;77:109816. Available from: 10.1016/j.cellsig.2020.109816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheung CC, Constantine M, Ahmadi A, Shiau C, Chen LYC. Eosinophilic Myocarditis. Am J Med Sci. 2017;354:486–492. Available from: 10.1016/j.amjms.2017.04.002 [DOI] [PubMed] [Google Scholar]
- 29.Toor IS, Rückerl D, Mair I, Ainsworth R, Meloni M, Spiroski AM, Benezech C, Felton JM, Thomson A, Caporali A, et al. Eosinophil Deficiency Promotes Aberrant Repair and Adverse Remodeling Following Acute Myocardial Infarction. JACC Basic to Transl Sci. 2020;5:665–681. Available from: 10.1016/j.jacbts.2020.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Yang C, Liu T, Deng Z, Fang W, Zhang X, Li J, Huang Q, Liu C, Wang Y, et al. Eosinophils improve cardiac function after myocardial infarction. Nat Commun. 2020;11:1–15. Available from: 10.1038/s41467-020-19297-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W, Chancey AL, Tzeng H, Zhou Z, Lavine KJ, Gao F, Sivasubramanian N, Barger PM, Mann DL. The Development of Myocardial Fibrosis in Transgenic Mice With Targeted Overexpression of Tumor Necrosis Factor Requires Mast Cell – Fibroblast Interactions. Circulation. 2011;124:2106–2116. Available from: 10.1161/CIRCULATIONAHA.111.052399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janicki JS, Brower GL, Gardner JD, Forman MF, Stewart JAJ, Murray DB, Chancey AL. Cardiac mast cell regulation of matrix metalloproteinase-related ventricular remodeling in chronic pressure or volume overload. Cardiovasc Res. 2006;69:657–665. Available from: 10.1016/j.cardiores.2005.10.020 [DOI] [PubMed] [Google Scholar]
- 33.Levick SP, Melndez GC, Plante E, McLarty JL, Brower GL, Janicki JS. Cardiac mast cells: The centrepiece in adverse myocardial remodelling. Cardiovasc Res. 2011;89:12–19. Available from: 10.1093/cvr/cvq272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ong S, Ligons DL, Barin JG, Wu L, Talor MV, Diny N, Fontes JA, Gebremariam E, Kass DA, Rose NR, Čiháková D. Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am J Pathol. 2015;185:847–861. Available from: 10.1016/j.ajpath.2014.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ayach BB, Yoshimitsu M, Dawood F, Sun M, Arab S, Chen M, Higuchi K, Siatskas C, Lee P, Lim H, et al. Stem cell factor receptor induces progenitor and natural killer cell-mediated cardiac survival and repair after myocardial infarction. Proc Natl Acad Sci U S A. 2006;103:2304–2309. Available from: 10.1073/pnas.0510997103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ong SF, Rose NR, Čiháková D. Natural killer cells in inflammatory heart disease. Clin Immunol. 2017;175:26–33. Available from: 10.1016/j.clim.2016.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forte E, Panahi M, Baxan N, Ng FS, Boyle JJ, Branca J, Bedard O, Hasham MG, Benson L, Harding SE, et al. Type 2 MI induced by a single high dose of isoproterenol in C57BL/6J mice triggers a persistent adaptive immune response against the heart. J Cell Mol Med. 2021;25:229–243. Available from: 10.1111/jcmm.15937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirabo A, Fontana V, De Faria APC, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. 2014;124:4642–4656. Available from: 10.1172/JCI74084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forte E, Perkins B, Sintou A, Kalkat HS, Papanikolaou A, Jenkins C, Alsubaie M, Chowdhury RA, Duffy TM, Skelly DA, et al. Cross-Priming Dendritic Cells Exacerbate Immunopathology After Ischemic Tissue Damage in the Heart. Circulation. 2021;143:821–836. Available from: 10.1161/CIRCULATIONAHA.120.044581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ngwenyama N, Kirabo A, Aronovitz M, Velázquez F, Carrillo-Salinas F, Salvador AM, Nevers T, Amarnath V, Tai A, Blanton RM, et al. Isolevuglandin-Modified Cardiac Proteins Drive CD4+ T-Cell Activation in the Heart and Promote Cardiac Dysfunction. Circulation. 2021;143:1242–1255. Available from: 10.1161/CIRCULATIONAHA.120.051889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choo EH, Lee JH, Park EH, Park HE, Jung NC, Kim TH, Koh YS, Kim E, Seung KB, Park C, et al. Infarcted Myocardium-Primed Dendritic Cells Improve Remodeling and Cardiac Function after Myocardial Infarction by Modulating the Regulatory T Cell and Macrophage Polarization. Circulation. 2017;135:1444–1457. Available from: 10.1161/CIRCULATIONAHA.116.023106 [DOI] [PubMed] [Google Scholar]
- 42.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. Available from: 10.1126/science.291.5502.319 [DOI] [PubMed] [Google Scholar]
- 43.García-Rivas G, Castillo EC, Gonzalez-Gil AM, Maravillas-Montero JL, Brunck M, Torres-Quintanilla A, Elizondo-Montemayor L, Torre-Amione G. The role of B cells in heart failure and implications for future immunomodulatory treatment strategies. ESC Hear Fail. 2020;7:1387–1399. Available from: 10.1002/ehf2.12744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yasuda M, Takeuchi K, Hiruma M, Iida H, Tahara A, Itagane H, Toda I, Akioka K, Teragaki M, Oku H, et al. The complement system in ischemic heart disease. Circulation. 1990;81:156–163. Available from: 10.1161/01.cir.81.1.156 [DOI] [PubMed] [Google Scholar]
- 45.Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, Kim HA, Krishnan SM, Lewis CV, Salimova E, et al. Obligatory role for B cells in the development of angiotensin II-dependent hypertension. Hypertension. 2015;66:1023–1033. Available from: 10.1161/HYPERTENSIONAHA.115.05779 [DOI] [PubMed] [Google Scholar]
- 46.Smith SC, Allen PM. Expression of myosin-class II major histocompatibility complexes in the normal myocardium occurs before induction of autoimmune myocarditis. Proc Natl Acad Sci U S A. 1992;89:9131–9135. Available from: 10.1073/pnas.89.19.9131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blyszczuk P, Kania G, Dieterle T, Marty RR, Valaperti A, Berthonneche C, Pedrazzini T, Berger CT, Dirnhofer S, Matter CM, et al. Myeloid differentiation factor-88/interleukin-1 signaling controls cardiac fibrosis and heart failure progression in inflammatory dilated cardiomyopathy. Circ Res. 2009;105:912–920. Available from: 10.1161/CIRCRESAHA.109.199802 [DOI] [PubMed] [Google Scholar]
- 48.Verma SK, Krishnamurthy P, Barefield D, Singh N, Gupta R, Lambers E, Thal M, MacKie A, Hoxha E, Ramirez V, et al. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-κB. Circulation. 2012;126:418–429. Available from: 10.1161/CIRCULATIONAHA.112.112185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kallikourdis M, Martini E, Carullo P, Sardi C, Roselli G, Greco CM, Vignali D, Riva F, Ormbostad Berre AM, Stølen TO, et al. T cell costimulation blockade blunts pressure overload-induced heart failure. Nat Commun. 2017;8:1–14. Available from: 10.1038/ncomms14680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zarak-Crnkovic M, Kania G, Jaźwa-Kusior A, Czepiel M, Wijnen WJ, Czyż J, Müller-Edenborn B, Vdovenko D, Lindner D, Gil-Cruz C, et al. Heart non-specific effector CD4+ T cells protect from postinflammatory fibrosis and cardiac dysfunction in experimental autoimmune myocarditis. Basic Res Cardiol. 2020;115:1–15. Available from: 10.1007/s00395-019-0766-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramjee V, Li D, Manderfield LJ, Liu F, Engleka KA, Aghajanian H, Rodell CB, Lu W, Ho V, Wang T, et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J Clin Invest. 2017;127:899–911. Available from: 10.1172/JCI88759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rieckmann M, Delgobo M, Gaal C, Büchner L, Steinau P, Reshef D, Gil-Cruz C, ter Horst EN, Kircher M, Reiter T, et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J Clin Invest. 2019;129:4922–4936. Available from: 10.1172/JCI123859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, Mathis D. A Special Population of regulatory T Cells Potentiates muscle repair. Cell. 2013;155:1282–1295. Available from: 10.1016/j.cell.2013.10.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weirather J, Hofmann UDW, Beyersdorf N, Ramos GC, Vogel B, Frey A, Ertl G, Kerkau T, Frantz S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ Res. 2014;115:55–67. Available from: 10.1161/CIRCRESAHA.115.303895 [DOI] [PubMed] [Google Scholar]
- 55.Nehra S, Gumina RJ, Bansal SS. Immune cell dilemma in ischemic cardiomyopathy: to heal or not to heal. Curr Opin Physiol. 2021;19:39–46. Available from: 10.1016/j.cophys.2020.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. 2018;18:733–744. Available from: 10.1038/s41577-018-0065-8 [DOI] [PubMed] [Google Scholar]
- 57.Ngwenyama N, Salvador AM, Velázquez F, Nevers T, Levy A, Aronovitz M, Luster AD, Huggins GS, Alcaide P. CXCR3 regulates CD4+ T cell cardiotropism in pressure overload–induced cardiac dysfunction. JCI Insight. 2019;4:1–18. Available from: 10.1172/jci.insight.125527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma F, Feng J, Zhang C, Li Y, Qi G, Li H, Wu Y, Fu Y, Zhao Y, Chen H, Du J, Tang H. The Requirement of CD8+ T Cells To Initiate and Augment Acute Cardiac Inflammatory Response to High Blood Pressure. J Immunol. 2014;192:3365–3373. Available from: 10.4049/jimmunol.1301522 [DOI] [PubMed] [Google Scholar]
- 59.Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, Prabhu SD. Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ Hear Fail. 2017;10:1–12. Available from: 10.1161/CIRCHEARTFAILURE.116.003688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, Delage C, Calise D, Dutaur M, Parini A, Pizzinat N. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation. 2014;129:2111–2124. Available from: 10.1161/CIRCULATIONAHA.113.007101 [DOI] [PubMed] [Google Scholar]
- 61.Nevers T, Salvador AM, Grodecki-Pena A, Knapp A, Velázquez F, Aronovitz M, Kapur NK, Karas RH, Blanton RM, Alcaide P. Left ventricular t-cell recruitment contributes to the pathogenesis of heart failure. Circ Hear Fail. 2015;8:776–787. Available from: 10.1161/CIRCHEARTFAILURE.115.002225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nevers T, Salvador AM, Velazquez F, Ngwenyama N, Carrillo-Salinas FJ, Aronovitz M, Blanton RM, Alcaide P. Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J Exp Med. 2017;214:3311–3329. Available from: 10.1084/jem.20161791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Díez J, González A, Kovacic JC. Myocardial Interstitial Fibrosis in Nonischemic Heart Disease, Part 3/4. J Am Coll Cardiol. 2020;75:2204–2218. Available from: 10.1016/j.jacc.2020.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frangogiannis NG, Kovacic JC. Extracellular Matrix in Ischemic Heart Disease, Part 4/4: JACC Focus Seminar. J Am Coll Cardiol. 2020;75:2219–2235. Available from: 10.1016/j.jacc.2020.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Furtado MB, Nim HT, Boyd SE, Rosenthal NA. View from the heart: cardiac fibroblasts in development, scarring and regeneration. Development. 2016;143:387–397. Available from: 10.1242/dev.120576 [DOI] [PubMed] [Google Scholar]
- 66.Moore-Morris T, Guimarães-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, et al. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J Clin Invest. 2014;124:2921–2934. Available from: 10.1172/JCI74783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ivey MJ, Kuwabara JT, Pai JT, Moore RE, Sun Z, Tallquist MD. Resident fibroblast expansion during cardiac growth and remodeling. J Mol Cell Cardiol. 2018;114:161–174. Available from: 10.1016/j.yjmcc.2017.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, Kamran P, Müller AMS, Volz KS, Tang Z, et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ Res. 2014;115:625–635. Available from: 10.1161/CIRCRESAHA.115.303794 [DOI] [PubMed] [Google Scholar]
- 69.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, J Lin S-C, Aronow BJ, Tallquist MD, Molkentin JD. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. Available from: 10.1038/ncomms12260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mouton AJ, Ma Y, Rivera Gonzalez OJ, Daseke MJ, Flynn ER, Freeman TC, Garrett MR, DeLeon-Pennell KY, Lindsey ML. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res Cardiol. 2019;114:1–16. Available from: 10.1007/s00395-019-0715-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhou B, Honor LB, He H, Qing M, Oh JH, Butterfield C, Lin RZ, Melero-Martin JM, Dolmatova E, Duffy HS, Von Gise A, Zhou P, Hu YW, Wang G, Zhang B, Wang L, Hall JL, Moses MA, McGowan FX, Pu WT. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J Clin Invest. 2011;121:1894–1904. Available from: 10.1172/JCI45529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.González-Rosa JM, Peralta M, Mercader N. Pan-epicardial lineage tracing reveals that epicardium derived cells give rise to myofibroblasts and perivascular cells during zebrafish heart regeneration. Dev Biol. 2012;370:173–186. Available from: 10.1016/j.ydbio.2012.07.007 [DOI] [PubMed] [Google Scholar]
- 73.Dai Z, Aoki T, Fukumoto Y, Shimokawa H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J Cardiol. 2012;60:416–421. Available from: 10.1016/j.jjcc.2012.06.009 [DOI] [PubMed] [Google Scholar]
- 74.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. Available from: 10.1038/359693a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee S-JJ, et al. Fibroblast-specific TGF-β–Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127:3770–3783. Available from: 10.1172/JCI94753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J Clin Invest. 2010;120:3520–3529. Available from: 10.1172/JCI42028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brooks WW, Conrad CH. Myocardial fibrosis in transforming growth factor β1 heterozygous mice. J Mol Cell Cardiol. 2000;32:187–195. Available from: 10.1006/jmcc.1999.1065 [DOI] [PubMed] [Google Scholar]
- 78.Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nat Rev Drug Discov. 2012;11:790–811. Available from: 10.1038/nrd3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Russo I, Cavalera M, Huang S, Su Y, Hanna A, Chen B, Shinde A V., Conway SJ, Graff J, Frangogiannis NG. Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program. Circ Res. 2019;124:1214–1227. Available from: 10.1161/CIRCRESAHA.118.314438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Y, Feng X, Wu R, Derynck R. Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature. 1996;383:168–172. Available from: 10.1038/383168a0 [DOI] [PubMed] [Google Scholar]
- 81.Bujak M, Ren G, Kweon HJ, Dobaczewski M, Reddy A, Taffet G, Wang XF, Frangogiannis NG. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–2138. Available from: 10.1161/CIRCULATIONAHA.107.704197 [DOI] [PubMed] [Google Scholar]
- 82.Działo E, Tkacz K, Błyszczuk P. Crosstalk between the TGF-β and WNT signalling pathways during cardiac fibrogenesis. Acta Biochim Pol. 2018;65:341–349. Available from: 10.18388/abp.2018_2635 [DOI] [PubMed] [Google Scholar]
- 83.Schafer S, Viswanathan S, Widjaja AA, Lim W-W, Moreno-Moral A, DeLaughter DM, Ng B, Patone G, Chow K, Khin E, et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552:110–115. Available from: 10.1038/nature24676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ichihara S, Senbonmatsu T, Price E, Ichiki T, Gaffney FA, Inagami T. Targeted deletion of angiotensin II type 2 receptor caused cardiac rupture after acute myocardial infarction. Circulation. 2002;106:2244–2249. Available from: 10.1161/01.CIR.0000033826.52681.37 [DOI] [PubMed] [Google Scholar]
- 85.Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, Saito M, Fukuda K, Nishiyama T, Kitajima S, et al. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med. 2008;205:295–303. Available from: 10.1084/jem.20071297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Frangogiannis NG. The extracellular matrix in ischemic and nonischemic heart failure. Circ Res. 2019;125:117–146. Available from: 10.1161/CIRCRESAHA.119.311148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Frangogiannis NG. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. 2019;65:70–99. Available from: 10.1016/j.mam.2018.07.001 [DOI] [PubMed] [Google Scholar]
- 88.Flynn JE, Mann FD. The presence and pathogenesis of endocardial and subendocardial degeneration, mural thrombi, and thromboses of the thebesian veins in cardiac failure from causes other than myocardial infarction. Am Heart J. 1946;31:757–768. Available from: 10.1016/0002-8703(46)90502-9 [DOI] [PubMed] [Google Scholar]
- 89.Assomull RG, Prasad SK, Lyne J, Smith G, Burman ED, Khan M, Sheppard MN, Poole-Wilson PA, Pennell DJ. Cardiovascular Magnetic Resonance, Fibrosis, and Prognosis in Dilated Cardiomyopathy. J Am Coll Cardiol. 2006;48:1977–1985. Available from: 10.1016/j.jacc.2006.07.049 [DOI] [PubMed] [Google Scholar]
- 90.Villari B, Campbell SE, Hess OM, Mall G, Vassalli G, Weber KT, Krayenbuehl HP. Influence of collagen network on left ventricular systolic and diastolic function in aortic valve disease. J Am Coll Cardiol. 1993;22:1477–1484. Available from: 10.1016/0735-1097(93)90560-N [DOI] [PubMed] [Google Scholar]
- 91.Nattel S. Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin Electrophysiol. 2017;3:425–435. Available from: 10.1016/j.jacep.2017.03.002 [DOI] [PubMed] [Google Scholar]
- 92.Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010;48:504–511. Available from: 10.1016/j.yjmcc.2009.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shimojo N, Hashizume R, Kanayama K, Hara M, Suzuki Y, Nishioka T, Hiroe M, Yoshida T, Imanaka-Yoshida K. Tenascin-C may accelerate cardiac fibrosis by activating macrophages via the integrin αVβ3/Nuclear Factor-κB/Interleukin-6 Axis. Hypertension. 2015;66:757–766. Available from: 10.1161/HYPERTENSIONAHA.115.06004 [DOI] [PubMed] [Google Scholar]
- 94.Bassat E, Mutlak YE, Genzelinakh A, Shadrin IY, Baruch Umansky K, Yifa O, Kain D, Rajchman D, Leach J, Riabov Bassat D, et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature. 2017;547:179–184. Available from: 10.1038/nature22978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi KC, Akkad A-D, Herndon CN, Arduini A, Papangeli I, et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation. 2020;142:466–482. Available from: 10.1161/CIRCULATIONAHA.119.045401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, Rosenthal NA, Pinto AR. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018;22:600–610. Available from: 10.1016/j.celrep.2017.12.072 [DOI] [PubMed] [Google Scholar]
- 97.Litviňuková M, Talavera-lópez C, Maatz H, Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M, Lee M, et al. Cells of the adult human heart. Nature. 2020;588:466–472. Available from: 10.1038/s41586-020-2797-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hu P, Liu J, Zhao J, Wilkins BJ, Lupino K, Wu H, Pei L. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018;32:1344–1357. Available from: 10.1101/gad.316802.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, Savary L, Wehmeyer C, Naylor AJ, Kemble S, et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. 2019;570:246–251. Available from: 10.1038/s41586-019-1263-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Corrales-Medina VF, Alvarez KN, Weissfeld LA, Angus DC, Chirinos JA, Chang C-CH, Newman A, Loehr L, Folsom AR, Elkind MS, et al. Association Between Hospitalization for Pneumonia and Subsequent Risk of Cardiovascular Disease. JAMA. 2015;313:264. Available from: 10.1001/jama.2014.18229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kadoglou NPE, Bracke F, Simmers T, Tsiodras S, Parissis J. Influenza infection and heart failure—vaccination may change heart failure prognosis? Heart Fail Rev. 2017;22:329–336. Available from: 10.1007/s10741-017-9614-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Knowlton KU, Badorff C. The Immune System in Viral Myocarditis. Circ Res. 1999;85:559–561. Available from: 10.1161/01.RES.85.6.559 [DOI] [PubMed] [Google Scholar]
- 103.Lawler PR, Bhatt DL, Godoy LC, Lüscher TF, Bonow RO, Verma S, Ridker PM. Targeting cardiovascular inflammation: Next steps in clinical translation. Eur Heart J. 2021;42:113–131. Available from: 10.1093/eurheartj/ehaa099 [DOI] [PubMed] [Google Scholar]
- 104.Stubbs DF. Positive Synergism between Diuretics and Methylprednisolone Following Acute Myocardial Infarction. J Int Med Res. 1986;14:21–24. Available from: 10.1177/03000605860140S104 [DOI] [PubMed] [Google Scholar]
- 105.Huang S, Frangogiannis NG. Anti-inflammatory therapies in myocardial infarction: failures, hopes and challenges. Br J Pharmacol. 2018;175:1377–1400. Available from: 10.1111/bph.14155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schmidt M, Lamberts M, Olsen A-MS, Fosbøll E, Niessner A, Tamargo J, Rosano G, Agewall S, Kaski JC, Kjeldsen K, et al. Cardiovascular safety of non-aspirin non-steroidal anti-inflammatory drugs: review and position paper by the working group for Cardiovascular Pharmacotherapy of the European Society of Cardiology. Eur Hear J - Cardiovasc Pharmacother. 2016;2:108–118. Available from: 10.1093/ehjcvp/pvv054 [DOI] [PubMed] [Google Scholar]
- 107.Verma S, Eikelboom JW, Nidorf SM, Al-Omran M, Gupta N, Teoh H, Friedrich JO. Colchicine in cardiac disease: A systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc Disord. 2015;15:1–15. Available from: 10.1186/s12872-015-0068-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Deftereos S, Giannopoulos G, Panagopoulou V, Bouras G, Raisakis K, Kossyvakis C, Karageorgiou S, Papadimitriou C, Vastaki M, Kaoukis A, et al. Anti-inflammatory treatment with colchicine instable chronic heart failure. A prospective, randomized study. JACC Hear Fail. 2014;2:131–137. Available from: 10.1016/j.jchf.2013.11.006 [DOI] [PubMed] [Google Scholar]
- 109.D’Amario D, Cappetta D, Cappannoli L, Princi G, Migliaro S, Diana G, Chouchane K, Borovac JA, Restivo A, Arcudi A, et al. Colchicine in ischemic heart disease: the good, the bad and the ugly. Clin Res Cardiol. 2021; Available from: 10.1007/s00392-021-01828-9 [DOI] [PMC free article] [PubMed]
- 110.Vagnozzi RJ, Maillet M, Sargent MA, Khalil H, Johansen AKZ, Schwanekamp JA, York AJ, Huang V, Nahrendorf M, Sadayappan S, Molkentin JD. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature. 2020;577:405–409. Available from: 10.1038/s41586-019-1802-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vakeva AP, Agah A, Rollins SA, Matis LA, Li L, Stahl GL. Myocardial infarction and apoptosis after myocardial ischemia and reperfusion: Role of the terminal complement components and inhibition by anti-C5 therapy. Circulation. 1998;97:2259–2267. Available from: 10.1161/01.CIR.97.22.2259 [DOI] [PubMed] [Google Scholar]
- 112.Thomas TC, Rollins SA, Rother RP, Giannoni MA, Hartman SL, Elliott EA, Nye SH, Matis LA, Squinto SP, Evans MJ. Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Mol Immunol. 1996;33:1389–1401. Available from: 10.1016/S0161-5890(96)00078-8 [DOI] [PubMed] [Google Scholar]
- 113.Armstrong PW, APEX-AMI Investigators. Pexelizumab for Acute ST-Elevation Myocardial Infarction in Patients Undergoing Primary Percutaneous Coronary Intervention. JAMA. 2007;297:43. Available from: 10.1001/jama.297.1.43 [DOI] [PubMed] [Google Scholar]
- 114.Emmens RW, Wouters D, Zeerleder S, van Ham SM, Niessen HWM, Krijnen PAJ. On the value of therapeutic interventions targeting the complement system in acute myocardial infarction. Transl Res. 2017;182:103–122. Available from: 10.1016/j.trsl.2016.10.005 [DOI] [PubMed] [Google Scholar]
- 115.Baumgarten G, Knuefermann P, Mann DL. Cytokines as Emerging Targets in the Treatment of Heart Failure. Trends Cardiovasc Med. 2000;10:216–223. Available from: 10.1016/S1050-1738(00)00063-3 [DOI] [PubMed] [Google Scholar]
- 116.Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated Circulating Levels of Tumor Necrosis Factor in Severe Chronic Heart Failure. N Engl J Med. 1990;323:236–241. Available from: 10.1056/NEJM199007263230405 [DOI] [PubMed] [Google Scholar]
- 117.Bozkurt B, Kribbs SB, Clubb FJ, Michael LH, Didenko VV, Hornsby PJ, Seta Y, Oral H, Spinale FG, Mann DL. Pathophysiologically relevant concentrations of tumor necrosis factor- α promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–1391. Available from: 10.1161/01.CIR.97.14.1382 [DOI] [PubMed] [Google Scholar]
- 118.Mann DL, McMurray JJV, Packer M, Swedberg K, Borer JS, Colucci WS, Djian J, Drexler H, Feldman A, Kober L, et al. Targeted Anticytokine Therapy in Patients with Chronic Heart Failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation. 2004;109:1594–1602. Available from: 10.1161/01.CIR.0000124490.27666.B2 [DOI] [PubMed] [Google Scholar]
- 119.Mann DL. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ Res. 2015;116:1254–1268. Available from: 10.1161/CIRCRESAHA.116.302317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Askari AT, Unzek S, Popovic ZB, Goldman CK, Forudi F, Kiedrowski M, Rovner A, Ellis SG, Thomas JD, DiCorleto PE, et al. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet. 2003;362:697–703. Available from: 10.1016/S0140-6736(03)14232-8 [DOI] [PubMed] [Google Scholar]
- 121.Chung ES, Miller L, Patel AN, Anderson RD, Mendelsohn FO, Traverse J, Silver KH, Shin J, Ewald G, Farr MJ, et al. Changes in ventricular remodelling and clinical status during the year following a single administration of stromal cell-derived factor-1 non-viral gene therapy in chronic ischaemic heart failure patients: The STOP-HF randomized Phase II trial. Eur Heart J. 2015;36:2228–2238. Available from: 10.1093/eurheartj/ehv254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Okada H, Takemura G, Kosai KI, Li Y, Takahashi T, Esaki M, Yuge K, Miyata S, Maruyama R, Mikami A, et al. Postinfarction gene therapy against transforming growth factor-β signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005;111:2430–2437. Available from: 10.1161/01.CIR.0000165066.71481.8E [DOI] [PubMed] [Google Scholar]
- 123.Nagaraju CK, Robinson EL, Abdesselem M, Trenson S, Dries E, Gilbert G, Janssens S, Van Cleemput J, Rega F, Meyns B, et al. Myofibroblast Phenotype and Reversibility of Fibrosis in Patients With End-Stage Heart Failure. J Am Coll Cardiol. 2019;73:2267–2282. Available from: 10.1016/j.jacc.2019.02.049 [DOI] [PubMed] [Google Scholar]
- 124.Frantz S, Hu K, Adamek A, Wolf J, Sallam A, Maier SKG, Lonning S, Ling H, Ertl G, Bauersachs J. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol. 2008;103:485–492. Available from: 10.1007/s00395-008-0739-7 [DOI] [PubMed] [Google Scholar]
- 125.Van Erp C, Irwin NG, Hoey AJ. Long-term administration of pirfenidone improves cardiac function in mdx mice. Muscle and Nerve. 2006;34:327–334. Available from: 10.1002/mus.20590 [DOI] [PubMed] [Google Scholar]
- 126.Yamazaki T, Yamashita N, Izumi Y, Nakamura Y, Shiota M, Hanatani A, Shimada K, Muro T, Iwao H, Yoshiyama M. The antifibrotic agent pirfenidone inhibits angiotensin II-induced cardiac hypertrophy in mice. Hypertens Res. 2012;35:34–40. Available from: 10.1038/hr.2011.139 [DOI] [PubMed] [Google Scholar]
- 127.Mirkovic S, Seymour AML, Fenning A, Strachan A, Margolin SB, Taylor SM, Brown L. Attenuation of cardiac fibrosis by pirfenidone and amiloride in DOCA-salt hypertensive rats. Br J Pharmacol. 2002;135:961–968. Available from: 10.1038/sj.bjp.0704539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Denton CP, Merkel PA, Furst DE, Khanna D, Emery P, Hsu VM, Silliman N, Streisand J, Powell J, Åkesson A, et al. Recombinant human anti-transforming growth factor β1 antibody therapy in systemic sclerosis: A multicenter, randomized, placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum. 2007;56:323–333. Available from: 10.1002/art.22289 [DOI] [PubMed] [Google Scholar]
- 129.King TE, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, Gorina E, Hopkins PM, Kardatzke D, Lancaster L, et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N Engl J Med. 2014;370:2083–2092. Available from: 10.1056/nejmoa1402582 [DOI] [PubMed] [Google Scholar]
- 130.Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, Axmann J, Kirchgaessler KU, Samara K, Gilberg F, Cottin V. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. 2020;8:147–157. Available from: 10.1016/S2213-2600(19)30341-8 [DOI] [PubMed] [Google Scholar]
- 131.Lewis GA, Schelbert EB, Naish JH, Bedson E, Dodd S, Eccleson H, Clayton D, Jimenez BD, McDonagh T, Williams SG, et al. Pirfenidone in Heart Failure with Preserved Ejection Fraction—Rationale and Design of the PIROUETTE Trial. Cardiovasc Drugs Ther. 2019;33:461–470. Available from: 10.1007/s10557-019-06876-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rainer PP, Hao S, Vanhoutte D, Lee DI, Koitabashi N, Molkentin JD, Kass DA. Cardiomyocyte-Specific Transforming Growth Factor β Suppression Blocks Neutrophil Infiltration, Augments Multiple Cytoprotective Cascades, and Reduces Early Mortality After Myocardial Infarction. Circ Res. 2014;114:1246–1257. Available from: 10.1161/CIRCRESAHA.114.302653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Andreadou I, Cabrera-Fuentes HA, Devaux Y, Frangogiannis NG, Frantz S, Guzik T, Liehn EA, Gomes CPC, Schulz R, Hausenloy DJ. Immune cells as targets for cardioprotection: new players and novel therapeutic opportunities. Cardiovasc Res. 2019;115:1117–1130. Available from: 10.1093/cvr/cvz050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gadi I, Fatima S, Elwakiel A, Nazir S, Al-Dabet MM, Rana R, Bock F, Manoharan J, Gupta D, Biemann R, et al. Different DOACs Control Inflammation in Cardiac Ischemia-Reperfusion Differently. Circ Res. 2020;513–529. Available from: 10.1161/circresaha.120.317219 [DOI] [PMC free article] [PubMed]
- 135.Han J, Kim YS, Lim MY, Kim HY, Kong S, Kang M, Choo YW, Jun JH, Ryu S, Jeong HY, et al. Dual Roles of Graphene Oxide to Attenuate Inflammation and Elicit Timely Polarization of Macrophage Phenotypes for Cardiac Repair. ACS Nano. 2018;12:1959–1977. Available from: 10.1021/acsnano.7b09107 [DOI] [PubMed] [Google Scholar]
- 136.Julier Z, Park AJ, Briquez PS, Martino MM. Promoting tissue regeneration by modulating the immune system. Acta Biomater. 2017;53:13–28. Available from: 10.1016/j.actbio.2017.01.056 [DOI] [PubMed] [Google Scholar]
- 137.Vasconcelos DP, Costa M, Amaral IF, Barbosa MA, Águas AP, Barbosa JN. Modulation of the inflammatory response to chitosan through M2 macrophage polarization using pro-resolution mediators. Biomaterials. 2015;37:116–123. Available from: 10.1016/j.biomaterials.2014.10.035 [DOI] [PubMed] [Google Scholar]
- 138.Barani B, Rajasingh S, Rajasingh J. Exosomes: Outlook for Future Cell-Free Cardiovascular Disease Therapy. In: Xiao J, Cretoiu S, eds. Advances in experimental medicine and biology. Singapore: Springer Nature; 2017. p. 285–307. Available from: 10.1007/978-981-10-4397-0_19 [DOI] [PubMed] [Google Scholar]
- 139.Piotto C, Julier Z, Martino MM. Immune Regulation of Tissue Repair and Regeneration via miRNAs—New Therapeutic Target. Front Bioeng Biotechnol. 2018;6:1–10. Available from: 10.3389/fbioe.2018.00098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, Giuseppe GL, Biondi-Zoccai, Houser JE, Qureshi IZ, et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation. 2008;117:2670–2683. Available from: 10.1161/CIRCULATIONAHA.107.740233 [DOI] [PubMed] [Google Scholar]
- 141.Toldo S, Mezzaroma E, Van Tassell BW, Farkas D, Marchetti C, Voelkel NF, Abbate A. Interleukin-1β blockade improves cardiac remodelling after myocardial infarction without interrupting the inflammasome in the mouse. Exp Physiol. 2013;98:734–745. Available from: 10.1113/expphysiol.2012.069831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GGL, Van Tassell BW, Robati R, Roach LM, Arena RA, Roberts CS, Varma A, et al. Interleukin-1 Blockade With Anakinra to Prevent Adverse Cardiac Remodeling After Acute Myocardial Infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot Study). Am J Cardiol. 2010;105:1371–1377.e1. Available from: 10.1016/j.amjcard.2009.12.059 [DOI] [PubMed] [Google Scholar]
- 143.Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, Libby P, Glynn RJ, Ridker PM. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. 2019;139:1289–1299. Available from: 10.1161/CIRCULATIONAHA.118.038010 [DOI] [PubMed] [Google Scholar]
- 144.Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119–1131. Available from: 10.1056/nejmoa1707914 [DOI] [PubMed] [Google Scholar]
- 145.Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, Mam V, et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N Engl J Med. 2019;380:752–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zannad F, Ferreira JP, Pocock SJ, Anker SD, Butler J, Filippatos G, Brueckmann M, Ofstad AP, Pfarr E, Jamal W, Packer M. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: a meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet. 2020;396:819–829. Available from: 10.1016/S0140-6736(20)31824-9 [DOI] [PubMed] [Google Scholar]
- 147.Prattichizzo F, De Nigris V, Micheloni S, La Sala L, Ceriello A. Increases in circulating levels of ketone bodies and cardiovascular protection with SGLT2 inhibitors: Is low-grade inflammation the neglected component? Diabetes, Obes Metab. 2018;20:2515–2522. Available from: 10.1111/dom.13488 [DOI] [PubMed] [Google Scholar]
- 148.Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, Rim JH, Hwang I, Lee CJ, Lee M, et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun. 2020;11:1–11. Available from: 10.1038/s41467-020-15983-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Guo L, Liu M, Huang J, Li J, Jiang J, Wang J. Role of interleukin‐15 in cardiovascular diseases. J Cell Mol Med. 2020;24:7094–7101. Available from: 10.1111/jcmm.15296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ng B, Dong J, D’Agostino G, Viswanathan S, Widjaja AA, Lim W-W, Ko NSJ, Tan J, Chothani SP, Huang B, et al. Interleukin-11 is a therapeutic target in idiopathic pulmonary fibrosis. Sci Transl Med. 2019;11:eaaw1237. Available from: 10.1126/scitranslmed.aaw1237 [DOI] [PubMed] [Google Scholar]
- 151.Widjaja AA, Singh BK, Adami E, Viswanathan S, Dong J, D’Agostino GA, Ng B, Lim WW, Tan J, Paleja BS, et al. Inhibiting Interleukin 11 Signaling Reduces Hepatocyte Death and Liver Fibrosis, Inflammation, and Steatosis in Mouse Models of Nonalcoholic Steatohepatitis. Gastroenterology. 2019;157:777–792.e14. Available from: 10.1053/j.gastro.2019.05.002 [DOI] [PubMed] [Google Scholar]
- 152.Saraiva M, Vieira P, O’Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med. 2020;217:1–19. Available from: 10.1084/jem.20190418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Saxton RA, Tsutsumi N, Su LL, Abhiraman GC, Mohan K, Henneberg LT, Aduri NG, Gati C, Garcia KC. Structure-based decoupling of the pro- and anti-inflammatory functions of interleukin-10. Science. 2021;371:eabc8433. Available from: 10.1126/science.abc8433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tang TT, Yuan J, Zhu ZF, Zhang WC, Xiao H, Xia N, Yan XX, Nie SF, Liu J, Zhou SF, et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res Cardiol. 2012;107:1–17. Available from: 10.1007/s00395-011-0232-6 [DOI] [PubMed] [Google Scholar]
- 155.Kuwana Y, Asakura Y, Utsunomiya N, Nakanishi M, Arata Y, Itoh S, Nagase F, Kurosawa Y. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochem Biophys Res Commun. 1987;149:960–968. Available from: 10.1016/0006-291X(87)90502-X [DOI] [PubMed] [Google Scholar]
- 156.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the γ or ζ subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90:720–724. Available from: 10.1073/pnas.90.2.720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Full F, Lehner M, Thonn V, Goetz G, Scholz B, Kaufmann KB, Mach M, Abken H, Holter W, Ensser A. T Cells Engineered with a Cytomegalovirus-Specific Chimeric Immunoreceptor. J Virol. 2010;84:4083–4088. Available from: 10.1128/jvi.02117-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bohne F, Chmielewski M, Ebert G, Wiegmann K, Kürschner T, Schulze A, Urban S, Krönke M, Abken H, Protzer U. T Cells Redirected Against Hepatitis B Virus Surface Proteins Eliminate Infected Hepatocytes. Gastroenterology. 2008;134:239–247. Available from: 10.1053/j.gastro.2007.11.002 [DOI] [PubMed] [Google Scholar]
- 159.Rooney CM, Smith CA, Ng CY, Loftin S, Li C, Krance RA, Brenner MK, Heslop HE. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet. 1995;345:9–13. Available from: 10.1016/S0140-6736(95)91150-2 [DOI] [PubMed] [Google Scholar]
- 160.Singh AK, McGuirk JP. CAR T cells: continuation in a revolution of immunotherapy. Lancet Oncol. 2020;21:e168–e178. Available from: 10.1016/S1470-2045(19)30823-X [DOI] [PubMed] [Google Scholar]
- 161.Feins S, Kong W, Williams EF, Milone MC, Fraietta JA. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol. 2019;94:S3–S9. Available from: 10.1002/ajh.25418 [DOI] [PubMed] [Google Scholar]
- 162.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Sci Transl Med. 2011;3:95ra73–95ra73. Available from: 10.1126/scitranslmed.3002842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Möllmann H, et al. Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ Res. 2016;118:1906–1917. Available from: 10.1161/CIRCRESAHA.116.308643 [DOI] [PubMed] [Google Scholar]
- 164.Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573:430–433. Available from: 10.1038/s41586-019-1546-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Fitzgerald AA, Weiner LM. The role of fibroblast activation protein in health and malignancy. Cancer Metastasis Rev. 2020;39:783–803. Available from: 10.1007/s10555-020-09909-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Hamson EJ, Keane FM, Tholen S, Schilling O, Gorrell MD. Understanding fibroblast activation protein (FAP): Substrates, activities, expression and targeting for cancer therapy. Proteomics - Clin Appl. 2014;8:454–463. Available from: 10.1002/prca.201300095 [DOI] [PubMed] [Google Scholar]
- 167.Puré E, Blomberg R. Pro-tumorigenic roles of fibroblast activation protein in cancer: back to the basics. Oncogene. 2018;37:4343–4357. Available from: 10.1038/s41388-018-0275-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wang LCS, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, Antzis M, Cotner CE, Johnson LA, Durham AC, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. 2014;2:154–166. Available from: 10.1158/2326-6066.CIR-13-0027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Laverman P, van der Geest T, Terry SYAA, Gerrits D, Walgreen B, Helsen MM, Nayak TK, Freimoser-Grundschober A, Waldhauer I, Hosse RJ, et al. Immuno-PET and Immuno-SPECT of Rheumatoid Arthritis with Radiolabeled Anti-Fibroblast Activation Protein Antibody Correlates with Severity of Arthritis. J Nucl Med. 2015;56:778–783. Available from: 10.2967/jnumed.114.152959 [DOI] [PubMed] [Google Scholar]
- 170.Loktev A, Lindner T, Mier W, Debus J, Altmann A, Jäger D, Giesel F, Kratochwil C, Barthe P, Roumestand C, Haberkorn U. A Tumor-Imaging Method Targeting Cancer-Associated Fibroblasts. J Nucl Med. 2018;59:1423–1429. Available from: 10.2967/jnumed.118.210435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Windisch P, Zwahlen DR, Giesel FL, Scholz E, Lugenbiel P, Debus J, Haberkorn U, Adeberg S. Clinical results of fibroblast activation protein (FAP) specific PET for non-malignant indications: systematic review. EJNMMI Res. 2021;11:18. Available from: 10.1186/s13550-021-00761-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Siebermair J, Köhler MI, Kupusovic J, Nekolla SG, Kessler L, Ferdinandus J, Guberina N, Stuschke M, Grafe H, Siveke JT, et al. Cardiac fibroblast activation detected by Ga-68 FAPI PET imaging as a potential novel biomarker of cardiac injury/remodeling. J Nucl Cardiol. 2020; Available from: 10.1007/s12350-020-02307-w [DOI] [PMC free article] [PubMed]
- 173.Swynghedauw B Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–62. Available from: 10.1152/physrev.1999.79.1.215 [DOI] [PubMed] [Google Scholar]
- 174.Song P, Zhao Q, Zou MH. Targeting senescent cells to attenuate cardiovascular disease progression. Ageing Res Rev. 2020;60:101072. Available from: 10.1016/j.arr.2020.101072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jia K, Dai Y, Liu A, Li X, Wu L, Lu L, Bao Y, Jin Q. Senolytic Agent Navitoclax Inhibits Angiotensin II-Induced Heart Failure in Mice. J Cardiovasc Pharmacol. 2020;76:452–460. Available from: 10.1097/FJC.0000000000000878 [DOI] [PubMed] [Google Scholar]
- 176.Dookun E, Walaszczyk A, Redgrave R, Palmowski P, Tual‐Chalot S, Suwana A, Chapman J, Jirkovsky E, Donastorg Sosa L, et al. Clearance of senescent cells during cardiac ischemia–reperfusion injury improves recovery. Aging Cell. 2020;19:1–15. Available from: 10.1111/acel.13249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Amor C, Feucht J, Leibold J, Ho Y-J, Zhu C, Alonso-Curbelo D, Mansilla-Soto J, Boyer JA, Li X, Giavridis T, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. 2020;583:127–132. Available from: 10.1038/s41586-020-2403-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378:439–448. Available from: 10.1056/NEJMoa1709866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. 2020;383:2255–2273. Available from: 10.1056/NEJMra2026131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, Maus MV, Park JH, Mead E, Pavletic S, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol Blood Marrow Transplant. 2019;25:625–638. Available from: 10.1016/j.bbmt.2018.12.758 [DOI] [PubMed] [Google Scholar]
- 181.Ruella M, June CH. Predicting Dangerous Rides in CAR T Cells: Bridging the Gap between Mice and Humans. Mol Ther. 2018;26:1401–1403. Available from: 10.1016/j.ymthe.2018.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Parker KR, Migliorini D, Perkey E, Yost KE, Bhaduri A, Bagga P, Haris M, Wilson NE, Liu F, Gabunia K, et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell. 2020;183:126–142.e17. Available from: 10.1016/j.cell.2020.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. Available from: 10.1182/blood-2013-03-490565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jamal FA, Khaled SK. The Cardiovascular Complications of Chimeric Antigen Receptor T Cell Therapy. Curr Hematol Malig Rep. 2020;15:130–132. Available from: 10.1007/s11899-020-00567-4 [DOI] [PubMed] [Google Scholar]
- 185.Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CCR, Restifo NP, Rosenberg SA. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. 2013;210:1065–1068. Available from: 10.1084/jem.20130110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hoffmann DB, Fraccarollo D, Galuppo P, Frantz S, Bauersachs J, Tillmanns J. Genetic ablation of fibroblast activation protein alpha attenuates left ventricular dilation after myocardial infarction. PLoS One. 2021;16:e0248196. Available from: 10.1371/journal.pone.0248196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. Available from: 10.1038/nbt.2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Goebeler ME, Bargou RC. T cell-engaging therapies — BiTEs and beyond. Nat Rev Clin Oncol. 2020;17:418–434. Available from: 10.1038/s41571-020-0347-5 [DOI] [PubMed] [Google Scholar]
- 189.De Sostoa J, Fajardo CA, Moreno R, Ramos MD, Farrera-Sal M, Alemany R. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J Immunother Cancer. 2019;7:1–15. Available from: 10.1186/s40425-019-0505-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hong M, Clubb JD, Chen YY. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell. 2020;38:473–488. Available from: 10.1016/j.ccell.2020.07.005 [DOI] [PubMed] [Google Scholar]
- 191.Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, Carroll RG, June CH, Grupp SA. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther. 2011;22:1575–1586. Available from: 10.1089/hum.2011.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, Schmierer M, Gabrusiewicz K, Anderson NR, Petty NE, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38:947–953. Available from: 10.1038/s41587-020-0462-y [DOI] [PMC free article] [PubMed] [Google Scholar]