

Abstract
Ovarian cancer is a chemoresponsive tumor with very high initial response rates to standard therapy consisting of platinum/paclitaxel. However, most women eventually develop recurrence, which rapidly evolves into chemo-resistant disease. Persistence of ovarian cancer stem cells (OCSC) at the end of therapy has been shown to contribute to resistant tumors. In this study, we demonstrate that the long non-coding RNA HOTAIR is overexpressed in HGSOC cell lines. Furthermore, HOTAIR expression was upregulated in OCSC compared to non-CSC, ectopic overexpression of HOTAIR enriched the ALDH+ cell population and HOTAIR overexpression increased spheroid formation and colony forming ability. Targeting HOTAIR using peptide nucleic acid-PNA3®, which acts by disrupting the interaction between HOTAIR and EZH2, in combination with a DNMT inhibitor inhibited OCSC spheroid formation and decreased the percentage of ALDH+ cells. Disrupting HOTAIR-EZH2 with PNA3® in combination with the DNMTi on the ability of OCSC to initiate tumors in vivo as xenografts was examined. HGSOC OVCAR3 cells were treated with PNA3® in vitro and then implanted in nude mice. Tumor growth, initiation and stem cell frequency were inhibited. Collectively, these results demonstrate that blocking HOTAIR-EZH2 interaction combined with inhibiting DNA methylation is a potential approach to eradicate OCSCs and block disease recurrence.
Keywords: ovarian cancer, epigenetics, cancer stem cells, HOTAIR, methylation
Introduction
Ovarian cancer is the fifth leading cause of death among U.S. women [1]. Currently, the standard therapy for OC consists of debulking surgery followed by chemotherapy. Although most patients are chemoresponsive in the initial stages, the majority of patients experience tumor relapse and recurrent OC rapidly evolves into chemoresistant disease, which is universally fatal[1]. The persistence of residual tumor cells, often referred to as cancer stem cells (CSCs), is now widely accepted as a critical factor contributing to OC chemoresistance[2–4]. Furthermore, their enrichment after chemotherapy in preclinical models of OC [2, 5, 6] as well as OC patient samples [7] strongly support the premise that ovarian CSCs contribute to tumor relapse and disease recurrence. Thus, therapeutically disrupting cancer stemness has the potential to eradicate tumor residuals and delay or prevent recurrence [3, 8].
CSCs share certain characteristics with normal stem cells, including the ability to self-renew, differentiate, and the heterogenous cell populations of the parental tumor [2]. Biological features of ovarian CSCs (OCSCs) such as the ability to form anchorage-independent spheroids, over-expression of ABC drug-transporters, and loss of cell-cell contact contribute to drug resistance and metastasis in these cells [9, 10]. Upregulation of EMT-related genes and stemness markers as well as downregulation of differentiation-related genes, such as homeobox (HOX) genes, and altered expression of non-coding RNAs are characteristics of OCSCs [10].
The long non-coding (lnc) RNAs HOX antisense intergenic RNA (HOTAIR) has been correlated with chemoresistant OC and poor patient outcomes [11]. Originally identified as a lncRNA located in the HOXC cluster on chromosome 12 that regulates the HOXD gene cluster on chromosome 2 in trans [12], HOTAIR has been shown to play a key role in chromatin remodeling and transcription. Interaction with HOTAIR is required for polycomb repressive complex 2 (PRC2) occupancy at specific loci, H3K27me3 by EZH2 and subsequent gene repression[13]. To therapeutically target HOTAIR, we recently designed a peptide nucleic acid (PNA3) that inhibited the HOTAIR-EZH2 interaction and showed treatment of OC cells with PNA3 decreased invasion and re-sensitized OC cells to cisplatin. Furthermore, PNA3 decreased ALDH1A1 activity in ALDH (+) OC cells [14], suggesting that targeting HOTAIR in OCSC may be a potential therapeutic approach in the disease.
DNA methylation contributes to several key characteristics of OCSC, including self-renewal ability and tumor initiation capacity [15, 16] DNA hypomethylating agents (DNMT inhibitors, DNMTi) alone or in combination with other therapeutics prevented platinum-induced enrichment of OCSC in vitro and in vivo in mouse xenografts [6, 17, 18]. Furthermore, HOTAIR and DNA methylation were both associated with platinum resistance in OC [19, 20], suggesting that dual epigenetic targeting of these epigenetic factors could be an effective approach in OC. To achieve this objective, in the current study we first demonstrate that HOTAIR is enriched in OCSCs compared to non-OCSCs and overexpression of HOTAIR promotes expansion of OCSCs. Combined targeting of HOTAIR and DNMT blocks spheroid formation ability of OCSCs, OC proliferation and clonogenic survival in vitro. Furthermore, treatment with both a HOTAIR inhibitor and a DNMTi decreases tumorigenesis and decreases tumor-initiating capacity in vivo in mouse xenografts. Our studies identify for the first time a combinatory therapeutic approach based on targeting a lncRNA and DNA methylation in OCSC.
Material and Methods
Cell culture, reagents and drug treatments:
High-grade serous ovarian cancer (HGSOC) cell lines (OVCAR3, CAOV3, OVCAR5, COV362, Kuramochi) were maintained in culture as described previously [21]. Cell lines were tested for mycoplasma contamination (Lonza, cat #LT07–318) every 6 months. Cells were authenticated by short tandem repeat (S7TR) analysis in 2017 (IDEXX BioAnalytics, Columbia, MO). Cells were treated with the DNMTi (guadecitabine, Astex Pharmaceuticals, Inc) for 72 hours, following by 24 hours of peptide nucleic acid (PNA) treatment (HOTAIR inhibitor PNA3® acts by disrupting the interaction between HOTAIR and EZH2 and the negatrive control PNA4 has no effect on the interation [20]). As a normal cell control, human ovarian surface epithelial cells (HOSE) were used (Department of Obstetrics and Gynecology, Indiana University School of Medicine ). HOSE were obtained from normal ovaries of five patients by scraping the OSE, placing in in short-term culture and expanding (two to four passages), as we have described previously [22]. The purity of the HOSE cells was confirmed by keratin and vimentin immunostaining [22]. For HOTAIR overexpression, cell lines were tranfected with lipofectamine 3000, following manufacturer’s protocol. Full-length HOTAIR was cloned into pAV5S vector containing a 98-mer aptamer sequence and as a vector control, aptamer cloned into pAV5S was used to avoid any RNA-dependent signaling effects [20].
Western blot:
Cells were treated with DNMTi (guadecitabine; 20–1000 nM) for 72 hours, and proteins were extracted from treated cells with RIPA buffer (Thermo Fisher, cat #89900). Protein concentrations were quantified with the Bradford assay (Bio-Rad, cat #5000001) following the manufacturer’s protocol. Lysates were subjected to SDS-PAGE and transferred to a PVDF membrane by standard methods [20]. Western blots were probed with primary antibodies anti-Snail (Cell Signing Technologies, cat #3879), anti-p-NF-κB (Cell Signaling Technologies, cat #3033), anti-p-β-catenin Cell Signaling Technologies, cat #4176), anti-histone H3 (Cell Signaling Technologies, cat #4499), anti-GAPDH (Cell Signaling Technologies, cat #5174), and anti-acetyl-histone H3(Cell Signaling Technologies, cat #8173). After incubation with corresponding secondary antibodies conjugated with horseradish peroxidase, ECL kit (Thermo Scientific, cat #32106) was utilized to visualize the protein bands.
qRT-PCR:
RNA was isolated from cultured cells using RNeasy mini kit (Qiagen, 74104) following manufacturer’s protocol. Concentrations were determined by using the absorbance at 260 nm, and purity was assessed based on the 260/280 nm absorbance ratio. Total RNA (2μg) was reverse transcribed with the following manufacturer’s instructions. qPCR was performed using Lightcycler 480 SYBER Green I Master kit (Roche Diagnostics, cat #04707516001) and HOTAIR (Forward primer 5’-GTGGTTTATCTTGCACCCCTCATTCTCAAGCCCCAGCCAGGGAA-3’, and reverse primer 5’-TTCCCTGGCTGGGGCTTGAGAATGAGGGGTGCAAGATAAACCAC-3’. mRNA expression levels were determined using Lightcycler software version 3.5 (Roche Applied Science) and normalized to EEF1A1: Forward primer: 5’-GCCCCAGGACACAGAGACTTTATC-3’;Reverse primer: 5’-CAACACCAGCAACAATCAG-3’).
Cell proliferation assay:
Cells were collected after drug treatments as previously described [20] and then were seeded at a concentration of 1000 cells per well in 96-well plates and 3-(4.5-dimethylthiazol-2-yl)-2.5-diphenyl tetrazolium bromide (MTT, Thermoo Scientific, cat #M6494) assay was performed at day 1–4 as previously described [19].
Spheroid and colony formation assay:
Cells were seeded at 60–70% confluency in 10cm plates and treated at indicated times as previously described. 500 cells were plated in triplicates in 24-well ultra-low adherent plates (Corning, cat #3473) with 1ml of stem cell medium (for spheroid formation assay) or 6-well plates in 2ml fresh complete media (for colony formation assay). Cells were allowed to grow for 14 days for spheroid formation or 5–7 days for colony formation. Spheroid size and morphology were assessed using a Zeiss Axiovert 40 inverted microscope with Axio-Vision software (Carl Zeiss MicroImaging). Colonies were stained with crystal violet (0.5%). Spheres or clusters smaller than 100 μm were not counted.
Combination index and synergism:
Cells were treated and plated as indicated for clonogenic survival assays. Following treatment, the percent survival subtracted from 100% was indicative of the fraction affected (FA). Subsequent combination indices, and synergism determination were calculated by the Chou-Talalay method [23].
Aldefluor assay and flow cytometry:
ALDH1 enzymatic activity was measured using the Aldefluor assay kit according to the manufacturer’s instructions (Stemcell Technologies, cat. #01700), as described previously[18, 24]. The test ALDH1-positive population was gated using control cells incubated with the ALDH inhibitor, diethylamino benzaldehyde (DEAB).
Mouse xenograft experiments:
OVCAR3 cells were treated with the DNMTi for 72 hours, following by 24 hours of PNA treatment, mixed with matrigel at 1:1 ratio and injected subcutaneously into the right flank of NSG female mice (5,000, 20,000 and 50,000 cells). Tumor size was measured once a week with a caliper, and tumor volume was determined using the formula V=½ × L × W2, where L is the longest tumor diameter and W is the perpendicular tumor diameter. Xenograft tumors were collected at the end of study and dissociated into single-cell suspension using tumor dissociation kit (Miltenyi Biotec, cat #130–095-929) in combination with the gentleMACS™ Dissociator, prior to spheroid formation assay and RNA purification. OCSC frequency and significance were calculated by Extreme Limiting Dilution Analysis (ELDA) software (http://bioinf.wehi.edu.au/software/elda/).
Statistical analysis:
All data are presented as mean values ± SD of at least three biological experiments unless otherwise indicated. The estimate variation within each group was similar therefore Student’s t-test was used to statistically analyze the significant difference among different groups. For mouse xenograft study, statistical significance was determined using a Student two-tailed t-test.
Results
HOTAIR expression drives OCSC phenotypes
The lncRNA HOTAIR is frequently overexpressed in human OC and correlates with chemoresistance and poor patient prognosis [20, 25]. To investigate HOTAIR as a therapeutic target in OCSCs, HOTAIR expression was examined in a panel of HGSOC cell lines. Increased expression of HOTAIR in OC cells compared to normal ovarian surface epithelial cells was observed (Fig. 1A). Moreover, HOTAIR expression was greater in OCSC (ALDH+) compared to non-CSC (ALDH-) (Fig. 1B). OVCAR3 cells displayed low basal HOTAIR expression but high expression of HOTAIR (Fig. 1A and B), and ectopic overexpression of HOTAIR in OVCAR3 cells (Supplementary Fig. S1A) upregulated expression of p-NF-κB and the EMT-related gene SNAIL, while p-β-catenin expression was unchanged (Supplementary Fig. S1B). Furthermore, HOTAIR overexpression significantly increased ALDH expression (Fig. 1D) and ALDH activity (Fig. 1E) compared to empty vector control. Functionally, overexpression of HOTAIR in OVCAR3 increased spheroid and colony forming ability compared to control cells (Fig. 1F). Collectively, these results indicate the potential for HOTAIR to drive non-CSCs to a more stem-like state.
Figure 1. HOTAIR promotes OC stemness phenotypes.
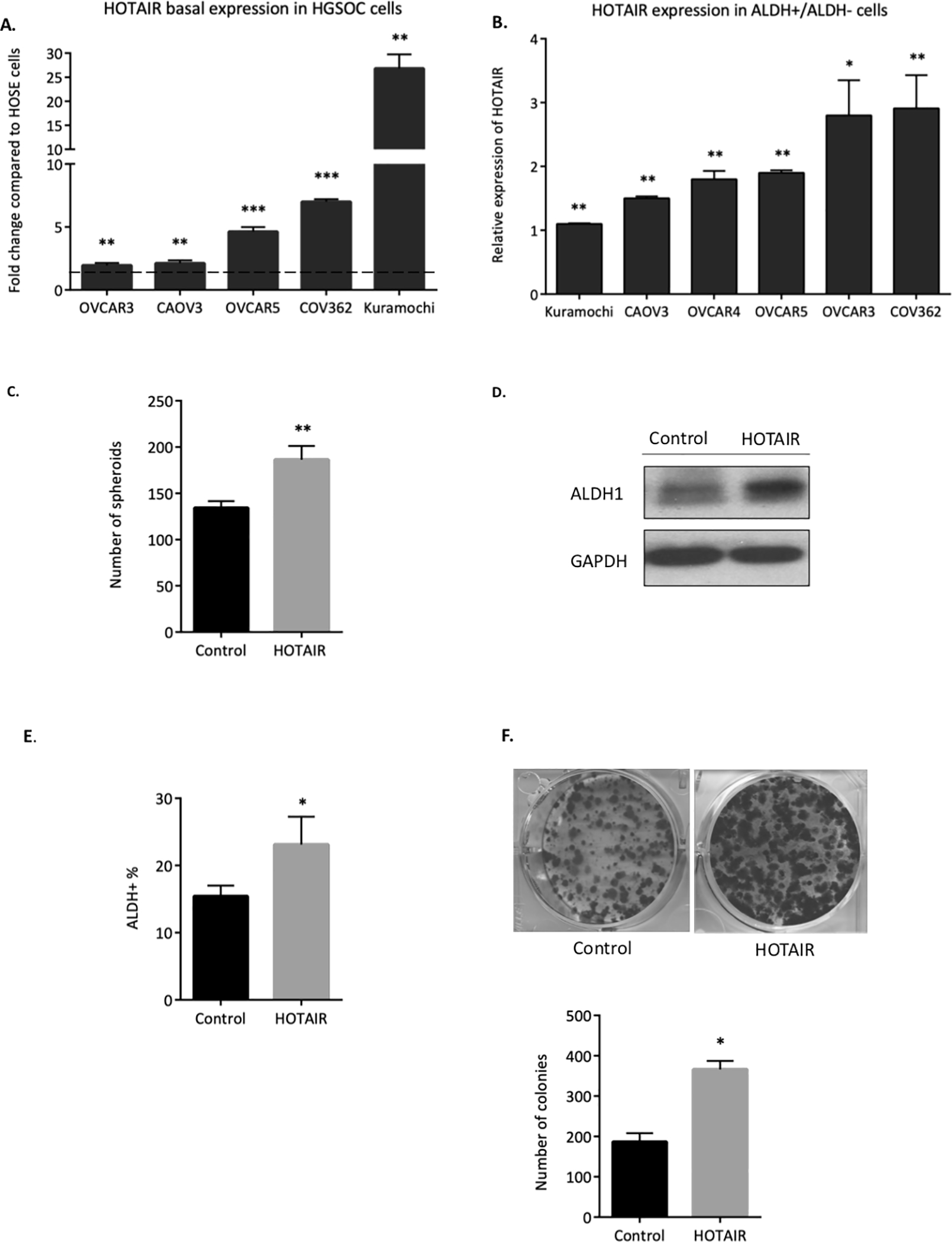
A) HOTAIR expression in a panel of HGSOC cell lines was compared to normal ovarian surface epithelial (NOSE) cells. Dashed line represents HOTAIR expression in human ovarian surface epithelial (HOSE) cells. B) HOTAIR expression in ALDH+ cells compared to ALDH- cells in a panel of HGSOC cell lines. ALDH+ vs ALDH- cells was sorted via FACS sorting. C) OVCAR3 collected after stable transfection of HOTAIR overexpression plasmid. ALDH+ population were determined by ALDEFLUOR assay using flow cytometry. D) Western blot showing higher ALDH1 in HOTAIR overexpressing cells. E) 500 control and HOTAIR overexpressing cells were plated in 24-well non-adherent conditions in stem cell medium. Number of spheroids were counted after 14 days in culture. F) Representative images of colony formation (upper panel) and quantification (lower panel).
Inhibiting both HOTAIR and DNA methylation reduces OC stemness
As an interplay between HOTAIR and DNA methylation in OC chemoresistance [26] and the OCSC phenotype have been reported [27], it was of interest to examine the effect of inhibiting both HOTAIR and DNA methylation on OCSC. First, to determine the effective dose of the DNMTi, OVCAR3 and OVCAR5 cells were treated with 20–1000nM guadecitabine and western blot analysis for DNMT1 expression was performed. Those doses reduced DNMT1 expression, with 100nM guadecitabine demonstrating similar activity in both cell lines based on DNMT1 level (Fig. 2A, B). Next, OVCAR3 and OVCAR5 cells were treated with 100nM guadecitabine for 72 hours or PNA3 for 24 hours alone, or the DNMTi for 72 hours followed by PNA3 for 24 hours and assayed for spheroid formation and ALDEFLUOR activity. PNA3 treatment alone significantly decreased the number of spheroids in both cell lines (Fig. 2C, D), and PNA3 plus guadecitabine further decreased spheroid formation in OVCAR5 cells compared to PNA3 treatment alone (Fig. 2C, D). However, a further decrease in spheroid formation was not observed in OVCAR3 cells. FACS analysis revealed that in both OVCAR3 and OVCAR5, single agent and combination treatments significantly reduced the percentage of ALDH+ cells compared to DMSO-treated cells (representative scatter plots are shown in Fig. 2E, F). Although the combination treatment reduced the %ALDH+ cells compared to either drug alone in both OC cell lines, the decreases were not statistically significant (OVCAR3: DMSO 19.0±3.14 vs. PNA3 alone 11.2±1.81 vs. guadecitabine alone 8.12±1.45 vs. PNA3 + guadecitabine 5.21±2.62; OVCAR5: DMSO control 10.9±1.71 vs. PNA3 alone 2.89±0.29 vs. guadecitabine alone 2.71±0.28 vs. PNA3 + guadecitabine 1.26±0.49). PNA4 was served as negative control for PNA3 and no significant differences in spheroid formation, and percentage of ALDH+ cells between PNA4- and DMSO-treated cells (Supplemental Fig. S2A, B).
Figure 2. Effect of inhibiting HOTAIR and DNA methylation on ovarian cancer stem cells.
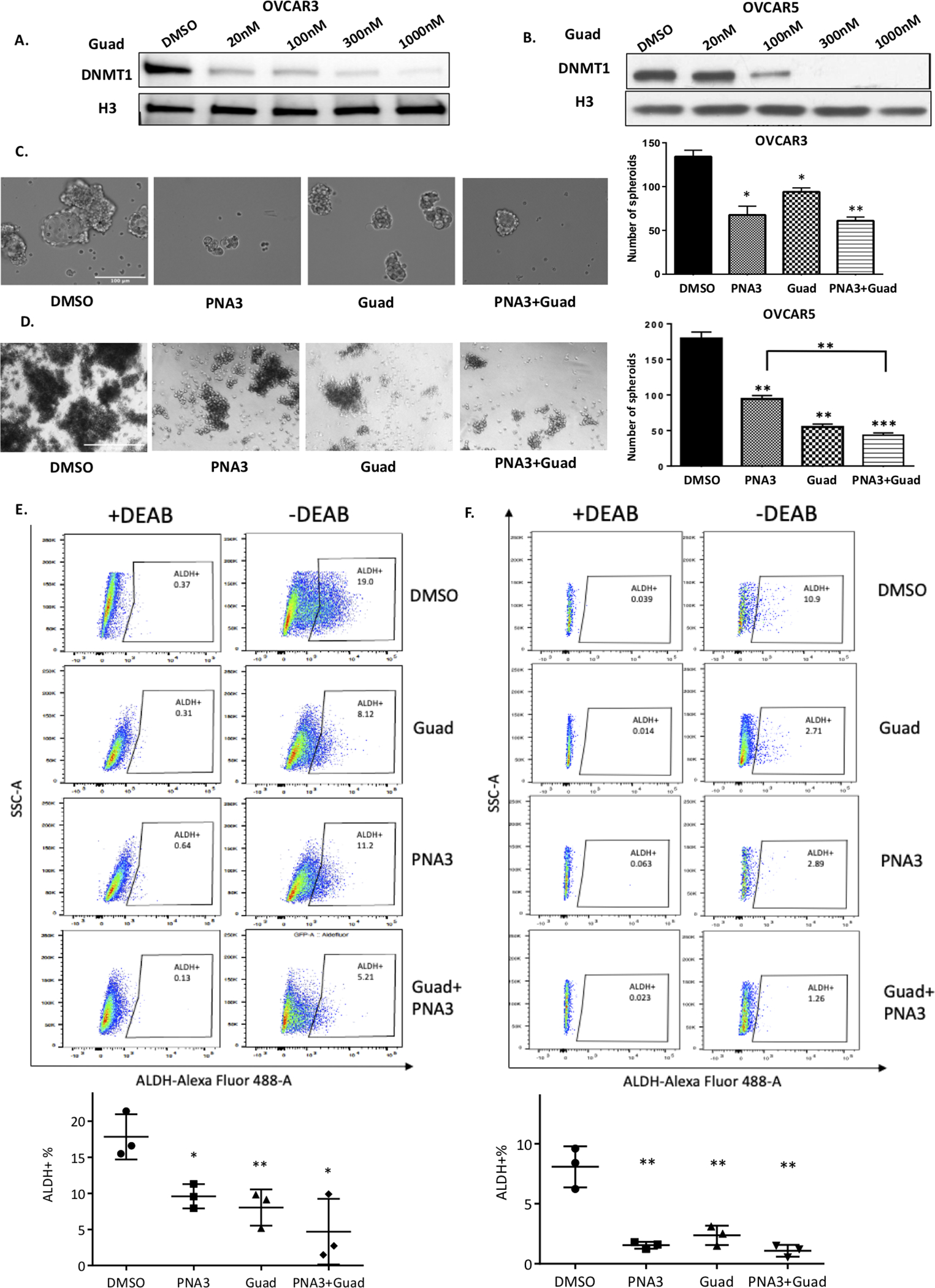
A) OVCAR3 or B) OVCAR5 were treated with hypomethylating agent guadecitabine for 72 hours (daily) at indicated dosages. Protein was isolated and immunoblots were performed to examine DNMT1 protein. H3 protein was used as the loading control. C) OVCAR3 (500 cells) and D) OVCAR5 (500 cells) were treated with HOTAIR inhibitor PNA3 (100nM for 24 hours) with guadecitabine (100nM) and or guadecitabine plus PNA3. Cells were replated in 24-well non-adherent conditions after treatment. Representative images of spheroid formation (left panel) and quantification (right panel). E) OVCAR3 and F) OVCAR5 ALDH+ populations were determined by ALDEFLUOR assay using flow cytometry (upper panel). Quantification shown below. Error bars represent SEM; n= 3 independent experiments of triplicate assays. Data are presented as mean ± SEM with p< 0.05 (*), p< 0.01 (**), and p< 0.005 (***).
To examine the effect of PNA3 in combination with DNMTi on OC cell growth, clonogenic survival assays were performed. It was first of interest to test the combination for a synergistic interaction using the Chou-Talalay method [28]. Co-administration of PNA3 (100nM) and guadecitabine (100–1000nM) showed a synergistic inhibitory effect (CI<1) on cell survival in OVCAR3 (Fig. 3A). In OVCAR5, only the higher dosages of guadecitabine (500nM, 1000nM) were synergistic with 100nM PNA3 (Fig. 3B). OVCAR3 and OVCAR5 cells were treated with the 100nM of guadecitabine and 100nM of PNA3 and re-plated for clonogenic survival assay. Compared to the single agents only, combined treatment with PNA3 plus DNMTi inhibited colony formation in both cell lines (Fig. 3C, D). In cell proliferation assays, PNA3 or guadecitabine alone decreased proliferation in both OVCAR3 and OVCAR5 cells, with the combined treatment further inhibited proliferation of both OC cell lines compared to single treatment (Fig. 3E, F). PNA4 did not alter clonogenic survival compared to DMSO treatment (Supplemental Fig. S2C,D).
Figure 3. Inhibiting HOTAIR and DNA methylation blocks ovarian tumorigenesis.
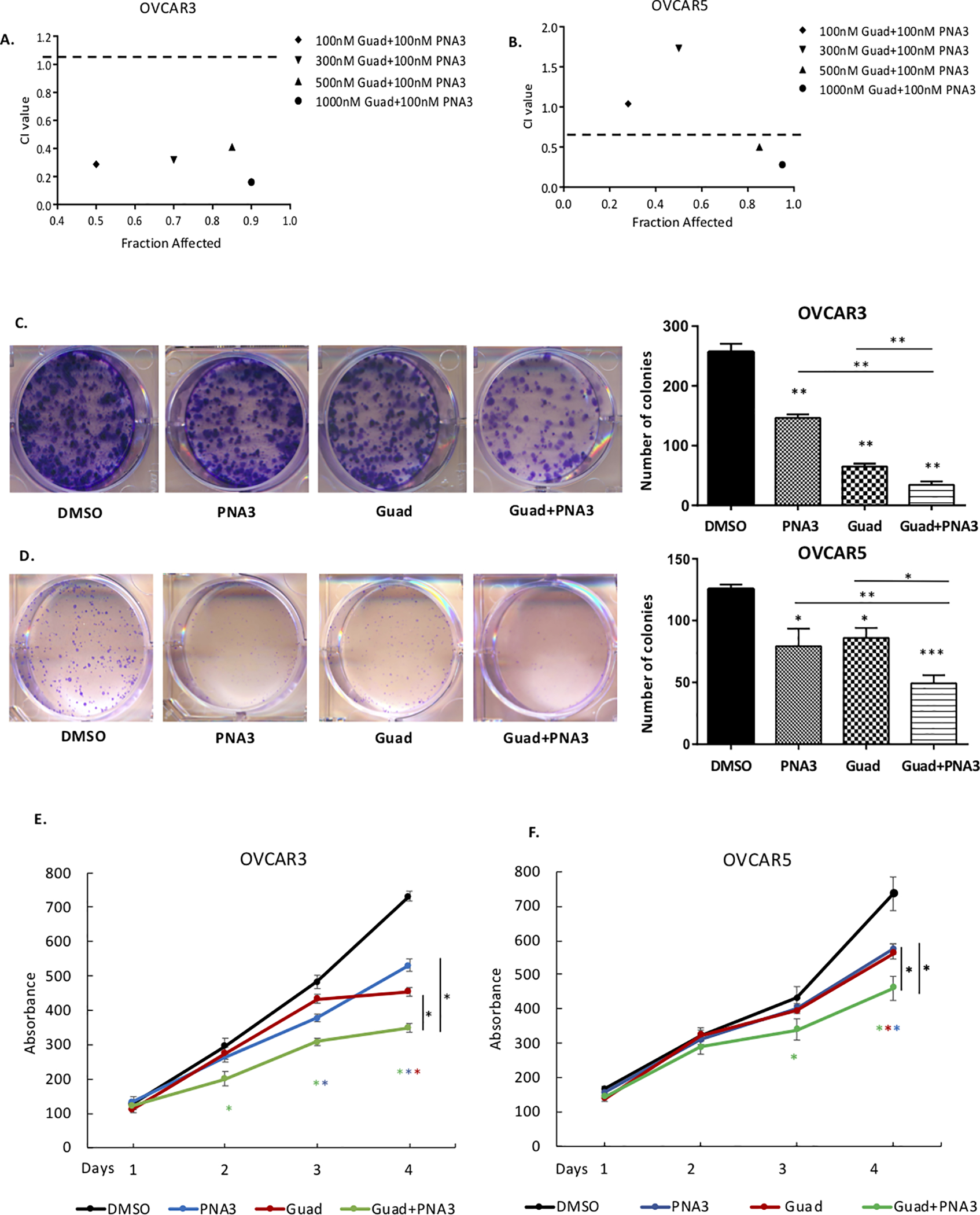
A) OVCAR3 or B) and OVCAR5 cells were treated with hypomethylating agent guadecitabine for 72 hours alone or HOTAIR inhibitor PNA3 at indicated dosages and subjected to clonogenic survival assay to determine drug efficacy; x-axis is indicative of Fraction Affected, the y-axis is indicative of the Combination Index (CI). Combinations beneath the black dashed line are synergistic. C) OVCAR3 and D) OVCAR5 cells were treated with guadecitabine (100nM daily for 72 hours), alone or following with PNA3 (100nM, 24 hours), then re-plated for clonogenic survival assay. Left panel, representative images of colonies. Right panel, quantification of the clonogenic survival assay. E) OVCAR3 and F) OVCAR5 proliferation determined by MTT assay after guadecitabine and PNA3 treatment as described above. Error bars represent SEM; n= 3 independent experiments of triplicate assays. Data are presented as mean± SEM with p< 0.05 (*), p< 0.01 (**), and p< 0.005 (***).
Inhibiting HOTAIR and DNA methylation reduces OCSC tumor initiation capacity in vivo
To further investigate the effect of the PNA3-DNMTi combination on OCSCs, a tumor initiation study was conducted in mice. FACs sorting was performed to obtain ALDH+ from the whole cell population of OVCAR3. The OVCAR3-ALDH+ cells were treated daily with 100nM guadecitabine for 72 hours or PNA3 for 24 hours alone, or guadecitabine for 72 hours followed by PNA3 for 24 hours, and 5,000, 20,000 or 50,000 ALDH+ cells from each treatment group were injected subcutaneously into mice. Tumor growth and volume were measured once a week starting on day 21 post-injection. Regardless of the cell number injected, PNA3 and DNMTi treatment alone significantly reduced tumor volume and the combination treatment further reduced tumor volume (Fig. 4A & B, 5,000-cell injection; Fig. 5A & B, 20,000-cell injection; Suppl. Fig. S3A & B, 50,000-cell injection). In addition to tumor growth, tumor initiation frequency was assessed. At weeks 5 and 6, single agent treatments reduced tumor initiation capacity of 5,000 cells, as 2/6 and 0/6 mice formed tumors in the guadecitabine and PNA3 treated mice, respectively (Fig. 4C). By week 8, tumor formation in mice initially injected with 5,000 cells was observed in 5/6 and 3/6 guadecitabine- and PNA3-treated mice, respectively (Fig. 5C). However, the combination treatment was more effective at inhibiting tumor formation throughout the study compared to either drug alone, and by week 8, tumor formation was observed in only one mouse initially injected with 5,000 ALDH+ cells (Fig. 4C). In mice injected with 20,000 ALDH+ cells, single agents were ineffective at inhibiting tumor initiation; however, tumor initiation capacity was inhibited by the PNA3-DNMTi combination treatment in 5/7 mice at week 5 and 3/7 mice at week 7 (Fig. 5C). In 50,000 cell injection group, the combination treatment was only moderately effective at inhibiting tumor, with 4/6 mice forming tumors by week 5 (Supplementary Fig. S3C).
Figure 4. Inhibiting HOTAIR and DNMT blocks ovarian tumor initiation.
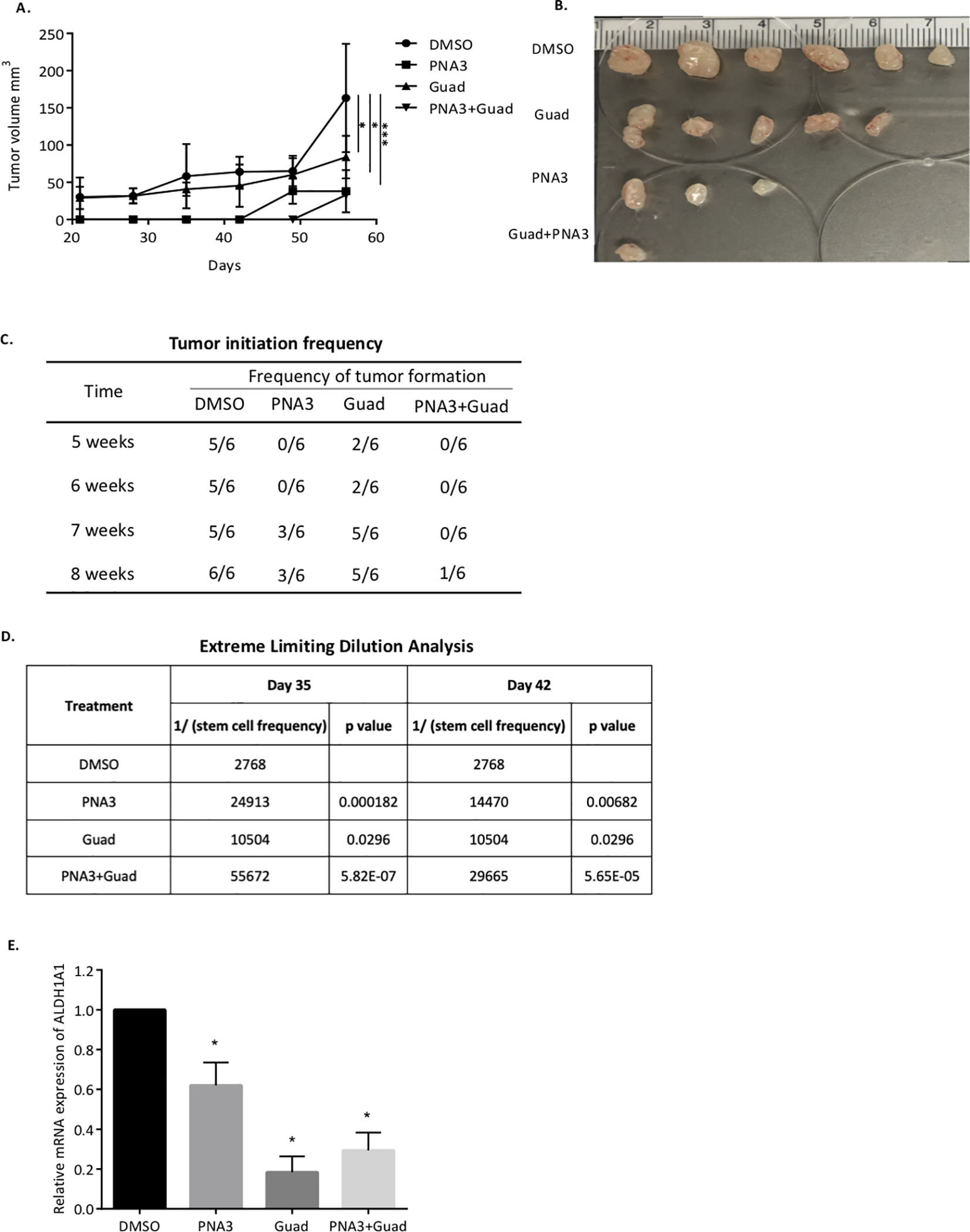
A) Xenograft tumor growth curve of different treatment groups. OVCAR3 sorted ALDH+ cells were treated with guadecitabine alone or with PNA3. 5,000 cells were injected s.c. into mice. B) Images of tumors collected at end of study. C) Quantification of tumor-formation in mice. D) Stem cell frequency estimates were calculated using Extreme Limiting Dilution Assay software.
Figure 5. Effect of inhibiting HOTAIR and DNMT on ovarian tumors initiated with 20,000 OVCAR3 cells.
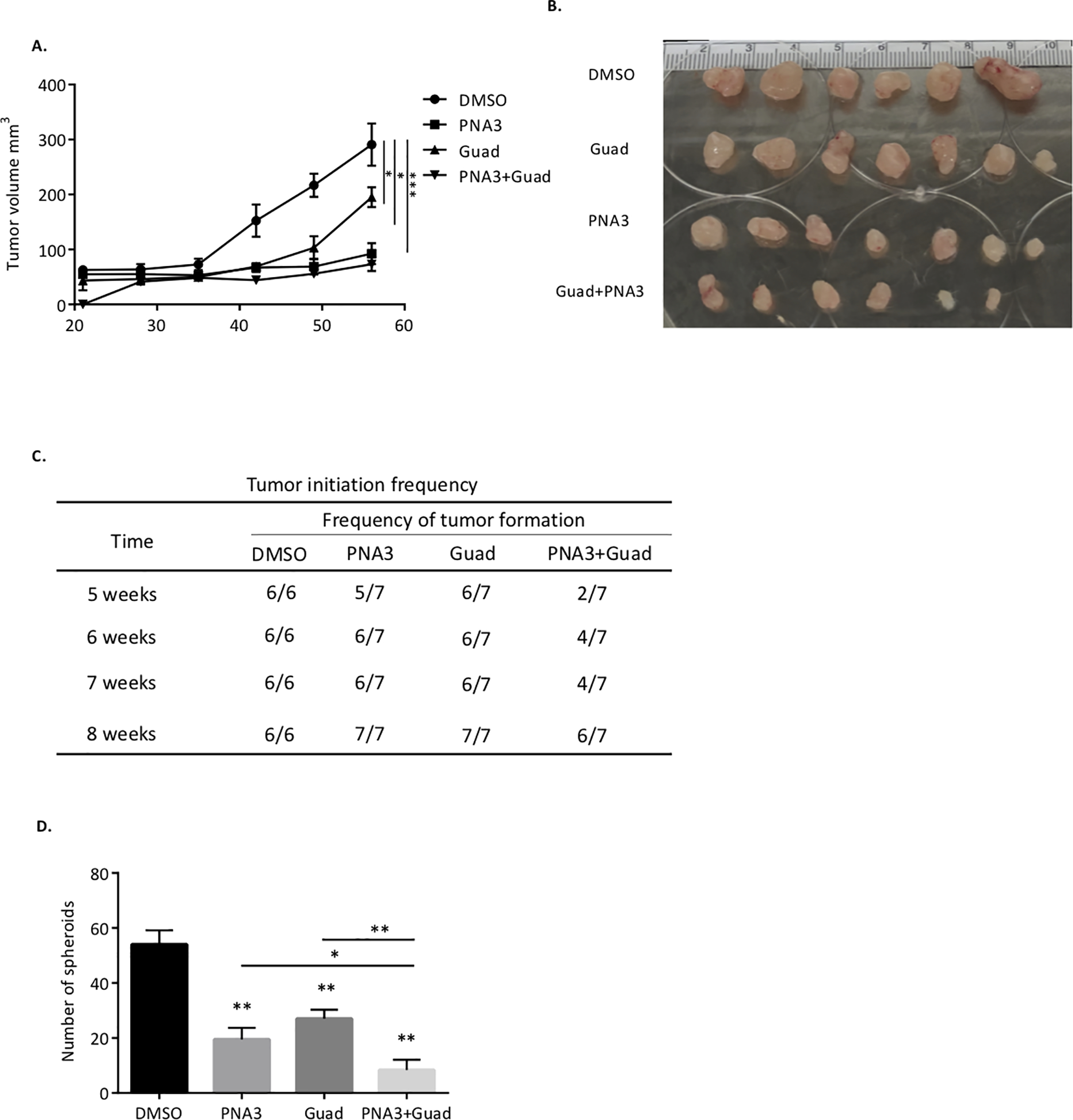
A) Xenograft tumor growth curve of different treatment groups. OVCAR3 sorted ALDH+ cells were treated with guadecitabine alone or with PNA3. 20,000 ALDH+ cells were injected s.c. into mice. B) Images of tumors collected at end of study. C) Quantification of tumor formation in mice. D) Quantification of spheroid formation by tumor cells collected from mice at end of study. Data are presented as mean ± SEM with p< 0.05 (*), p< 0.01 (**), and p< 0.005 (***).
The extreme limiting dilution analysis (ELDA) webtool was used to determine stem cell frequency on day 35 and 42 post- cancer cell injection. Stem cell frequency in the PNA3-treated group was ~10-fold less on day 35 and ~5 fold less on day 42 compared to control (Fig. 4D). In the guadecitabine-treated group, stem cell frequency was ~5-fold less compared to the control group at both days (Fig. 4D). Furthermore, based on ELDA, the PNA3 plus DNMTi combination was significantly more effective at reducing stem cell frequency than either treatment alone, resulting in 20-fold less stem cell frequency than control on day 35 and 10-fold less on day 42 in 5,000 cell injection group (Fig. 4D).
At the end of the 12-week study, tumors were harvested and dissociated into single-cell suspensions for spheroid formation assay and ALDH1A1 gene expression. In mice injected with 20,000 (Fig. 5D) or 50,000 ALDH+ cells (Supplementary Fig. S3D), spheroid formation capacity was significantly decreased by the single agent treatments. A further reduction in spheroid formation was only observed in in the 20,000 ALDH+ cell-injected mice treated with the PNA3-guadecitabine combination compared to either drug alone (Fig. 5D). However, due to a limited number of cells in the 5,000 injection group at the end of the 12 weeks, we were not able to asses spheroid formation. The single cell suspensions were also subjected to qRT-PCR analysis to determine ALDH1A1 expression. In the mice injected with 5,000 ALDH+ cells, mRNA level of ALDH1A1 was significantly reduced in both single agent and combination treated groups (Fig. 4E). Collectively, these data demonstrated that inhibiting both HOTAIR interaction and DNMT effectively reduced the OCSC population and delayed tumor initiation capacity in vivo.
Discussion
Chemotherapy decreases tumor bulk but leaves behind residual tumor cells that are capable of regenerating ovarian tumors. As these residal cells, termed OCSCs, are hypothesized to be a key source for emergence of recurrent tumors, developing therapeutic strategies that target OCSCs is a area of critical importance. Aberrant epigenetic changes in OCSCs have been reported [6], and we previously demonstrated the therapeutic potential of targeting DNMTs in OCSC [6, 18]. Here we report a new potential therapeutic approach based on combined targeting of both DNA methylation and the epigenetic regulator HOTAIR in OCSCs. We show that combining a HOTAIR inhibitor with a DNMTi is highly synergistic in reducing tumorigencity, suggesting that inhibiting both the HOTAIR-EZH2 interaction and DNA methylation effectively impaired the tumor initiation capacity of OCSCs. These novel findings have several implications.
First, we show that HOTAIR expression is upregulated in OCSCs compared to non-OCSCs and that HOTAIR overexpression significantly contributes to stemness phenotypes, including ALDH+ percentage, spheroid formation, clonogenic survival. Furthermore, expression of the mesenchymal maker SNAIL is upregulated in HOTAIR overexpressing cells, in agreement with previous reports on upregulation of Snail and Twist in OCSCs [29, 30], which are both associated with the process of epithelial-to-mesenchymal transition (EMT) [31, 32]. Furthermore, as chemotherapy-induced Snail activation has been shown to drive cancer stemness [6, 33], our results indicate a potential role for HOTAIR in regulating OCSCs via modulation of EMT. In addition, we observed that p-NF-κB expression was significantly elevated in HOTAIR overexpressing cells, in agreement with our finding that a positive feedback loop exists between HOTAIR and NF-κB and drives OC chemoresistance [20]. Furthermore, as we previously demonstrated that the HOTAIR inhibitor PNA3 effectively resensitizes tumor cells to chemotherapy both in vivo and in vitro [14], together with the current study, our data support a potential mechanism of regulation of OCSCs by HOTAIR through EMT and NF-κB pathway.
DNMTi have been utilized in combination therapies to overcome OC chemoresistance [18, 34–38]. Epigenetic priming using low doses of guadecitabine sensitized OC cells [19] and patient tumors [34–36] to platinum chemotherapy, and DNMTi sensitized BRCA-proficient HGSOC to PARP inhibitors [28, 39].The current study provides a new avenue for a epigenetic therapy strategy based on a DNMTi-HOTAIRi combination to potentially overcome chemoresistance in HGSOC. Furthermore, our pre-clinical findings suggest that the effectiveness of this strategy may be due to a direct targeting of OCSCs and the reversal of OCSC driven chemotherapy resistance. The synergistic effect of inhibiting both DNMT1-HOTAIR indicates that the drugs may be targeting the products of genes in parallel and highly connected pathways. DNMTs regulate EMT process and CSCs phenotypes in various cancers [40], and regulators of DNMT1 were involved in these process, such as miRNAs, oncogenes, and cytokines [41–43]. HOTAIR and DNMT1 interaction in several cancers has been shown [44, 45], and HOTAIR knockdown downregulates DNMT1 and decreases DNA methylation on both a global level as well as at gene promoters [44].HOTAIR-EZH2 interaction and global hypomethylation are known to induce cancer cell differentiation and stem cell features, potentially augmenting the efficacy of combining HOTAIRi and DNMT1i. However, the exact mechanism by which the PNA3-guadecitabine eradicates OCSCs requires further investigation.
We show using an in vivo limiting dilution assay that the dual inhibition approach reduces tumor initiation capacity, stem cell frequency and tumor burden, indicating that the DNMTi sensitizes OC cells to HOTAIR inhibition. Recent studies suggest that inter-conversion and dynamic equilibrium between CSCs and non-CSCs exists in many cancers [45–49]. By using a purified CSC population in breast cancer, Yang et al [50] reported that the proportion of non-CSCs increased with each generation; in addition, in the purified non-CSC population, the CSC population increased overtime [6, 51]. In a purified OCSC population implanted in mice, we showed the capacity of OCSCs forming tumor with very limited cell numbers, starting from 5,000 cells. Also, we show that in vitro epigenetic drug treatment dramatically reduces frequency and tumor initiation capacity of CSCs. Combination treatment showed significant efficacy compared to single agent alone, indicating the additive or synergistic effect of the two agents, however, the underlying mechanism is still unkown. ALDH1A1 expression and spheroid formation capacity of tumor cells collected at the end of the study, indiacating the effect of both DNMT1 inhibitor and PNA3 are quite stable. Taken together, we suggest that the combination blocked cancer cell inter-conversion, including of non-CSCs to CSCs and consequently tumor burden.
In summary, we show the efficacy of PNA3-guadecitabine combination on reducing OC stemness phenotypes and tumor initiation capacity. This anti-tumor effect is due to direct targeting of OCSCs and CSC-driven tumorigenesis. To our knowledge, this is the first report on targeting OCSCs by inhibiting both the HOTAIR-EZH2 interaction and DNA methylation. Furthermore, we show that although the combination specifically targets a small subset of cells, i.e. OCSC, the overall effect on OC tumorigenesis is impactful. The synergy between PNA3-guadecitabine indicates that the drug combination alters genes and presumably highly connected pathways in parallel. This study opens up a new avenue for combined epigenetic targeting of OCSCs in HGSOC patients.
Supplementary Material
Acknowledgements
We thank Vaishnavi Muralikrishnan (Indiana University School of Medicine) for helpful discussions and manuscript preparation. We thank Dr. Mohammad Azab and Dr. Harold N. Keer (Astex Pharmaceuticals, Inc.) for providing guadecitabine. We thank Christiane Hassel (Flow Cytometry Core Facility, Indiana University, Bloomington, IN) for technical assistance with flow cytometry. This work was funded by the Congressionally Directed Medical Research Programs, Department of Defense, Ovarian Cancer Research Program Award Number W81XWh-17–1-0076, a Collaborative Research Development grant from the Ovarian Cancer Research Alliance and Van Andel Institute through the Van Andel Institute-Stand Up To Cancer Epigenetics Dream Team. Stand Up To Cancer is a division of the Entertainment Industry Foundation, administered by AACR and IU Simon Comprehensive Cancer Center P30 CA82709–21.
Financial support: Department of Defense Ovarian Cancer Research Program Grant Number W81XWH-17–1-0076; Ovarian Cancer Research Alliance grant number 458788; Van Andel Institute through the Van Andel Institute – Stand Up To Cancer Epigenetics Dream Team. Stand Up To Cancer is a division of the Entertainment Industry Foundation, administered by AACR; IU Simon Comprehensive Cancer Center P30 CA82709–21.
Footnotes
Disclaimers: none; no conflicts of interest to disclose.
References
- 1.Torre LA, et al. , Ovarian cancer statistics, 2018. CA Cancer J Clin, 2018. 68(4): p. 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu L, McArthur C, and Jaffe RB, Ovarian cancer stem-like side-population cells are tumourigenic and chemoresistant. Br J Cancer, 2010. 102(8): p. 1276–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zong X and Nephew KP, Ovarian Cancer Stem Cells: Role in Metastasis and Opportunity for Therapeutic Targeting. Cancers (Basel), 2019. 11(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang S, et al. , Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res, 2008. 68(11): p. 4311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nacarelli T, et al. , NAMPT Inhibition Suppresses Cancer Stem-like Cells Associated with Therapy-Induced Senescence in Ovarian Cancer. Cancer Res, 2020. 80(4): p. 890–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, et al. , Epigenetic targeting of ovarian cancer stem cells. Cancer Res, 2014. 74(17): p. 4922–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown JR, et al. , Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight, 2020. 5(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saygin C, et al. , Targeting Cancer Stemness in the Clinic: From Hype to Hope. Cell Stem Cell, 2019. 24(1): p. 25–40. [DOI] [PubMed] [Google Scholar]
- 9.Bapat SA, et al. , Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res, 2005. 65(8): p. 3025–9. [DOI] [PubMed] [Google Scholar]
- 10.Steg AD, et al. , Stem Cell Pathways Contribute to Clinical Chemoresistance in Ovarian Cancer. Clinical Cancer Research, 2012. 18(3): p. 869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esteller M, Non-coding RNAs in human disease. Nature Reviews Genetics, 2011. 12(12): p. 861–874. [DOI] [PubMed] [Google Scholar]
- 12.Gupta RA, et al. , Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature, 2010. 464(7291): p. 1071–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qu X, et al. , HOX transcript antisense RNA (HOTAIR) in cancer. Cancer Lett, 2019. 454: p. 90–97. [DOI] [PubMed] [Google Scholar]
- 14.Ozes AR, et al. , Therapeutic targeting using tumor specific peptides inhibits long non-coding RNA HOTAIR activity in ovarian and breast cancer. Sci Rep, 2017. 7(1): p. 894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landen CN Jr., et al. , Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol Cancer Ther, 2010. 9(12): p. 3186–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones PA, Issa JP, and Baylin S, Targeting the cancer epigenome for therapy. Nat Rev Genet, 2016. 17(10): p. 630–41. [DOI] [PubMed] [Google Scholar]
- 17.Srivastava P, et al. , Immunomodulatory action of the DNA methyltransferase inhibitor SGI-110 in epithelial ovarian cancer cells and xenografts. Epigenetics, 2015. 10(3): p. 237–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, et al. , IL-6 mediates platinum-induced enrichment of ovarian cancer stem cells. JCI Insight, 2018. 3(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang F, et al. , The novel, small-molecule DNA methylation inhibitor SGI-110 as an ovarian cancer chemosensitizer. Clin Cancer Res, 2014. 20(24): p. 6504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ozes AR, et al. , NF-kappaB-HOTAIR axis links DNA damage response, chemoresistance and cellular senescence in ovarian cancer. Oncogene, 2016. 35(41): p. 5350–5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haley J, et al. , Functional characterization of a panel of high-grade serous ovarian cancer cell lines as representative experimental models of the disease. Oncotarget, 2016. 7(22): p. 32810–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahluwalia A, et al. , DNA methylation in ovarian cancer. II. Expression of DNA methyltransferases in ovarian cancer cell lines and normal ovarian epithelial cells. Gynecol Oncol, 2001. 82(2): p. 299–304. [DOI] [PubMed] [Google Scholar]
- 23.Chou TC and Talalay P, Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul, 1984. 22: p. 27–55. [DOI] [PubMed] [Google Scholar]
- 24.Zong X, et al. , EZH2-mediated Downregulation of the Tumor Suppressor DAB2IP Maintains Ovarian Cancer Stem Cells. Cancer Res, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, et al. , Overexpression of long non-coding RNA HOTAIR leads to chemoresistance by activating the Wnt/beta-catenin pathway in human ovarian cancer. Tumour Biol, 2016. 37(2): p. 2057–65. [DOI] [PubMed] [Google Scholar]
- 26.Teschendorff AE, et al. , HOTAIR and its surrogate DNA methylation signature indicate carboplatin resistance in ovarian cancer. Genome Med, 2015. 7: p. 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, et al. , Downregulated lincRNA HOTAIR expression in ovarian cancer stem cells decreases its tumorgeniesis and metastasis by inhibiting epithelial-mesenchymal transition. Cancer Cell Int, 2015. 15: p. 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pulliam N, et al. , An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin Cancer Res, 2018. 24(13): p. 3163–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Latifi A, et al. , Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J Cell Biochem, 2011. 112(10): p. 2850–64. [DOI] [PubMed] [Google Scholar]
- 30.Zhang R, et al. , Inhibitory effects of metformin at low concentration on epithelial-mesenchymal transition of CD44(+)CD117(+) ovarian cancer stem cells. Stem Cell Res Ther, 2015. 6: p. 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nieto MA, The snail superfamily of zinc-finger transcription factors. Nat Rev Mol Cell Biol, 2002. 3(3): p. 155–66. [DOI] [PubMed] [Google Scholar]
- 32.Yang J, et al. , Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell, 2004. 117(7): p. 927–39. [DOI] [PubMed] [Google Scholar]
- 33.Lu Z, et al. , Long non-coding RNA NKILA inhibits migration and invasion of non-small cell lung cancer via NF-kappaB/Snail pathway. J Exp Clin Cancer Res, 2017. 36(1): p. 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fang F, et al. , Decitabine reactivated pathways in platinum resistant ovarian cancer. Oncotarget, 2014. 5(11): p. 3579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang F, et al. , Genomic and Epigenomic Signatures in Ovarian Cancer Associated with Resensitization to Platinum Drugs. Cancer Res, 2018. 78(3): p. 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matei D, et al. , Epigenetic resensitization to platinum in ovarian cancer. Cancer Res, 2012. 72(9): p. 2197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeller C, et al. , Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene, 2012. 31(42): p. 4567–76. [DOI] [PubMed] [Google Scholar]
- 38.Oza AM, et al. , A Randomized Phase II Trial of Epigenetic Priming with Guadecitabine and Carboplatin in Platinum-resistant, Recurrent Ovarian Cancer. Clin Cancer Res, 2020. 26(5): p. 1009–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muvarak NE, et al. , Enhancing the Cytotoxic Effects of PARP Inhibitors with DNA Demethylating Agents - A Potential Therapy for Cancer. Cancer Cell, 2016. 30(4): p. 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee E, et al. , DNMT1 Regulates Epithelial-Mesenchymal Transition and Cancer Stem Cells, Which Promotes Prostate Cancer Metastasis. Neoplasia, 2016. 18(9): p. 553–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teng Y, et al. , DNMT1 ablation suppresses tumorigenesis by inhibiting the self-renewal of esophageal cancer stem cells. Oncotarget, 2018. 9(27): p. 18896–18907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zagorac S, et al. , DNMT1 Inhibition Reprograms Pancreatic Cancer Stem Cells via Upregulation of the miR-17–92 Cluster. Cancer Res, 2016. 76(15): p. 4546–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu R, et al. , DNMT1-microRNA126 epigenetic circuit contributes to esophageal squamous cell carcinoma growth via ADAM9-EGFR-AKT signaling. Clin Cancer Res, 2015. 21(4): p. 854–63. [DOI] [PubMed] [Google Scholar]
- 44.Li X, et al. , A novel interplay between HOTAIR and DNA methylation in osteosarcoma cells indicates a new therapeutic strategy. J Cancer Res Clin Oncol, 2017. 143(11): p. 2189–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mani SA, et al. , The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell, 2008. 133(4): p. 704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meyer MJ, et al. , Dynamic regulation of CD24 and the invasive, CD44posCD24neg phenotype in breast cancer cell lines. Breast Cancer Res, 2009. 11(6): p. R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liang Y, et al. , Stem-like cancer cells are inducible by increasing genomic instability in cancer cells. J Biol Chem, 2010. 285(7): p. 4931–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scheel C, et al. , Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell, 2011. 145(6): p. 926–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Iliopoulos D, et al. , Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A, 2011. 108(4): p. 1397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang G, et al. , Dynamic equilibrium between cancer stem cells and non-stem cancer cells in human SW620 and MCF-7 cancer cell populations. Br J Cancer, 2012. 106(9): p. 1512–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Scheel C and Weinberg RA, Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int J Cancer, 2011. 129(10): p. 2310–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.