

Abstract
Purpose:
As hypoxia can mediate resistance to immunotherapy, we investigated the safety, tolerability, and efficacy of combining evofosfamide, a prodrug that alleviates hypoxia, with ipilimumab, an immune checkpoint inhibitor, in immunologically “cold” cancers, which are intrinsically insensitive to immunotherapy, as well as in “hot/warm” metastatic cancers that are, atypical of such cancers, resistant to immunotherapy.
Patients and Methods:
In a Phase I, 3+3 dose-escalation trial (NCT03098160), evofosfamide (400–640 mg/m2) and ipilimumab (3 mg/kg) were administered in four 3-week cycles. The former was administered on days 1 and 8 of cycles 1–2, while the latter was administered on day 8 of cycles 1–4. Response was assessed using irRECIST, and retreatment was allowed, if deemed beneficial, after completion of cycle 4 or at progression.
Results:
Twenty-two patients were enrolled, of whom 21 were evaluable, encompassing castration-resistant prostate cancer (n=11), pancreatic cancer (n=7), immunotherapy-resistant melanoma (n=2), and HPV-negative head and neck cancer (n=1). Drug-related hematologic toxicities, rash, fever, nausea, vomiting, and elevation of liver enzymes were observed in > 10% of patients. The most common drug-related grade 3 adverse event was ALT elevation (33.3%). Two patients discontinued ipilimumab and four required evofosfamide de-escalation due to toxicity. Of 18 patients with measurable disease at baseline, three (16.7%) achieved partial response and 12 (66.7%) achieved stable disease. The best responses were observed at 560 mg/m2 evofosfamide. Pre-existing immune gene signatures predicted response to therapy, while hypermetabolic tumors predicted progression. Responders also showed improved peripheral T cell proliferation and increased intra-tumoral T cell infiltration into hypoxia.
Conclusions:
No new or unexpected safety signals were observed from combining evofosfamide and ipilimumab, and evidence of therapeutic activity was noted.
Keywords: CTLA-4, hypoxia, pancreatic cancer, prostate cancer, head and neck cancer
Introduction
Tumor hypoxia predicts poor outcomes across all cancers, and has long been recognized as a critical driver of resistance to both chemo- and radiotherapy (1) and, more recently, of immune evasion (2,3). Indeed, we recently showed that T cells fail to accumulate in hypoxic zones of prostate cancers, and that hypoxia suppressess key elements of the tumor stroma to establish and maintain an immune “cold” and immunotherapy-resistant state (4). Accordingly, alleviation of hypoxia has been demonstrated to reverse T cell exclusion and restore therapeutic sensitivity to immunotherapy in pre-clinical models of prostate cancer, head and neck cancer, melanoma, and lung cancer (3–6).
Immune checkpoint blockade, particularly by antibodies targeting the cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and/or the programmed death 1 (PD-1) T cell co-inhibitory receptors, has revolutionized therapy for numerous cancers. These antibodies contributed significantly to a record drop in US cancer deaths in the last year of reporting, although efficacy remains limited outside of immunologically “hot” or “warm” cancers that are already infiltrated with immune cells and/or that generate more mutational neoantigens (7). In these “best case” settings, response rates to checkpoint blockade range from lows of 15% (in HPV-negative head and neck, bladder, and gastric cancer) to as high as 87% (in Hodgkin’s lymphoma). Most patients, however, present with “cold” tumors that are almost completely resistant to checkpoint blockade (7,8), leaving the overall percentage of patients achieving objective clinical response below 15%, even within approved indications (9). Thus, developing therapies that sensitize “cold” cancers to checkpoint blockade has become a preeminent goal in cancer immunotherapy.
While the role of hypoxia in mediating immune privilege has been previously recognized in “cold” tumors (10), recent data also show a pivotal role in driving checkpoint resistance in “hot” tumors like melanoma (11). We previously showed that evofosfamide (TH-302) significantly reduces hypoxia across murine cancer models via a tissue remodeling process that restores normal, healthy vasculature capable of maintaining oxygenated blood supply (4,5). Evofosfamide is a hypoxia-activated, 2-nitroimidazole prodrug of bromo-isophosphoramide mustard, a cytotoxin that was clinically successful in numerous trials as an adjuvant to chemotherapy until failing a pivotal Phase III trial with gemcitabine in pancreatic cancer (12). Follow up studies showed, however, that this failure was due to poor bioavailability from a new formulation of the drug, resulting in sub-optimal dosing (13). Evofosfamide-mediated remodeling of hypoxic zones and the associated immunosuppressive microenvironment may enable ipilimumab, an antibody that blocks CTLA-4, to trigger immune rejection of otherwise resistant solid tumors (14). Additionally, evofosfamide-induced immunogenic cell death may enhance presentation of tumor antigens, leading to clonal expansion of tumor-specific T cells, especially in combination with ipilimumab (15,16). Therefore, we sought to evaluate the safety and tolerability of combining evofosfamide and ipilimumab in patients with advanced solid tumors (NCT03098160).
Patients and Methods
Study Design and Participants
This Phase I study assessed the safety, preliminary efficacy, and pharmacokinetics of evofosfamide in combination with ipilimumab. Adult patients with metastatic or locally advanced pancreatic cancer, castration-resistant prostate cancer, melanoma refractory to immune checkpoint blockade, or HPV- head and neck squamous cell carcinoma were eligible. Other eligibility criteria included an Eastern Cooperative Oncology Group (ECOG) score of ≤ 2, adequate organ function within 7 days of day 1 of therapy and, measurable disease as defined by the Immune Related Response Evaluation Criteria in Solid Tumors (irRECIST) in pancreatic cancer, melanoma, or HPV- head and neck cancer (17,18). As conventional RECIST 1.1 does not account for the response pattern typically seen with immune checkpoint inhibitors, i.e., extended time to response and tumor response after initial disease progression, we chose to use irRECIST, which requires confirmation of disease progression on two occasions to ensure that true disease progression is observed rather than pseudoprogression due to early, transient tumor enlargement following infiltration by immune cells. On the other hand, patients with castration-resistant prostate cancer could have measurable or evaluable disease defined by any of the following: PSA progression, as indicated by a minimum of two rising values (three measurements) obtained a minimum of 7 days apart with the last result being ≥ 1.0 ng/mL; new or increasing non-bone disease (RECIST 1.1 criteria); positive bone scan with two or more new lesions. Major exclusion criteria were history of grade 3 or 4 adverse events from prior ipilimumab or ipilimumab combination therapy, known risk factors for bowel perforation, long term systemic steroid use (>10 mg daily prednisone equivalent initiated > 2 weeks before start of study treatment), and presence of active or uncontrolled autoimmune disease.
The protocol was a single-center open-label trial approved by the institutional review board at MD Anderson Cancer Center, and conducted in compliance with the Declaration of Helsinki. Written informed consent was obtained from all patients or their legal representatives.
Procedures
A standard 3+3 design was utilized to combine 3 mg/kg ipilimumab with escalating doses of evofosfamide. In particular, 3–6 patients were enrolled sequentially at 400 mg/m2 evofosfamide, 480 mg/m2, 560 mg/m2, and 640 mg/m2 (Supplementary Table S1). Evofosfamide was administered as a 60-minute intravenous infusion on days 1 and 8 of cycles 1 and 2 of four 3-week cycles. Ipilimumab was administered as a 90-minute intravenous infusion on day 8 of each cycle for a maximum of four doses. Patients who achieve complete response, partial response, or stable disease after completion of cycle 4, as assessed by irRECIST, were eligible for re-treatment, as were patients who experience disease progression at any time after start of treatment, if the investigator determined that such re-treatment had positive benefit-risk ratio, in consultation with the sponsor’s medical monitor. In patients eligible for re-treatment, the same dose and schedule for evofosfamide and ipilimumab were used for cycles 5 to 8.
Study Assessments
Investigators used irRECIST at baseline and every two cycles to determine tumor response. Safety was evaluated by history at baseline and at regular intervals, along with physical and laboratory assessments. Severity of adverse events was assessed according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.03 (CTCAE 4.03), beginning after the first dose and up to 30 days after the last dose.
For pharmacokinetic analysis, blood samples were drawn predose and at 0.5, 1, 2, and 4 hours after evofosfamide infusion. Cmax, AUClast, and Tmax for evofosfamide and bromo-isophosphoramide mustard (Br-IPM) were derived from plasma concentrations using non-compartmental analysis. For analysis of circulating biomarkers, blood samples were drawn before the first dose and after each 3-week cycle. Following whole-blood collection, peripheral blood mononuclear cells were separated at room temperature along a Ficoll gradient (Histopaque-1077, Sigma), as previously described (19). At the conclusion of the trial, peripheral blood mononuclear cell samples were analyzed by 18-parameter flow cytometry on a BD LSR II instrument. For pharmacodynamic analysis, pimonidazole, a weak base that allows quantification of tumor hypoxia by immunohistochemistry, was administered with patient consent 5–24 hours prior to tumor biopsy, at an oral dose of 0.5 g/m2 (20). Paired tumor biopsies (at baseline and at week 7) were obtained and were analyzed by flow cytometry for quantitative and qualitative changes in hypoxia and in the immune microenvironment as previously described (4). DNA and RNA were also isolated using AllPrep DNA/RNA Mini Kit (Qiagen). RNA sequencing was performed by Avera in 2 × 100 bp paired-end runs.
RNA sequencing data were analyzed in collaboration with Baylor College of Medicine Multi-Omics Data Analysis Core. Briefly, sequencing reads were trimmed using Trim Galore (The Babraham Institute), mapped using HISAT (21) against the human genome build UCSC hg38, and quantified using StringTie (22) against the Gencode model (23). Gene expression (FPKM) was quantile-normalized in R, and genes differentially expressed between tumor and normal samples were identified by parametric t-test at p value < 0.05 and fold change 1.25. Pathway enrichment was evaluated in GSEA (http://software.broadinstitute.org/gsea/index.jsp) (24); significance was achieved for adjusted q-values (q < 0.25). Heat maps were generated using Matplotlib, NumPy, and SciPy libraries in Python. Sequencing data have been deposited in GEO under accession number GSE159237.
Outcomes
The safety profile of combining evofosfamide and ipilimumab, as well as the recommended Phase II dose of evofosfamide in this context, were the primary endpoints, with objective response rate and pharmacokinetics as secondary endpoints. Baseline and change from baseline in peripheral blood and tumor tissue immune and hypoxia parameters were exploratory endpoints.
Dose-limiting toxicity was defined as any of the following treatment-emergent adverse events: grade 3 or 4 non-hematologic toxicity; grade 3 or 4 nausea, vomiting, or diarrhea that cannot be managed medically; grade 3 or 4 AST or ALT elevation lasting > 7 days, grade 4 neutropenia lasting > 5 days, or febrile neutropenia. The surveillance period for dose-limiting toxicity was 21 days (cycle 1).
Statistical Analysis
Descriptive statistics were used to summarize safety, tumor response, and biomarker data. Safety data are reported using preferred terms defined in the Medical Dictionary for Regulatory Activities. Enrollment of up to 30 patients was anticipated in order to determine recommended Phase II dose. Safety was assessed in all patients who received one or more doses of evofosfamide or ipilimumab. A safety review committee reviewed safety and pharmacokinetic data prior to each dose escalation. Tumor response was assessed in all patients, and objective response rate was calculated as the proportion of patients with measurable disease at baseline who subsequently experienced complete or partial response. For prostate cancer patients with evaluable biomarkers, a decline in prostate specific antigen (PSA) of > 50% from baseline, measured twice at least 4 weeks apart, was considered a response. For subjects with an observed decline in PSA while on study, subsequent PSA progression was defined as ≥ 25% increase and ≥ 2 ng/mL above nadir, confirmed by a second measurement at least 3 weeks later. For patients with no observed decline in PSA while on study, progression was defined as ≥ 25% increase and ≥ 2 ng/mL above baseline after at least 12 weeks on study. Similarly, a decline in CA 19–9 of > 50% from baseline was considered a response; such response was considered confirmed if the decline is observed twice at least 4 weeks apart.
Results
Twenty-two patients were enrolled at MD Anderson’s Department of Investigational Cancer Therapeutics between June 2017 and March 2019. Three patients were enrolled at 400 mg/m2 evofosfamide, six at 480 mg/m2, six at 560 mg/m2, and six at 640 mg/m2. All patients received 3 mg/kg ipilimumab as described in Patients and Methods. One patient enrolled at 560 mg/m2 withdrew consent before initiating treatment. Thus, 21 patients were included in safety analysis.
Baseline patient characteristics are shown in Table 1. Median age was 67 years (range 47–78), with patients being 81.0% male (17/21) and 19.0% (4/21) female. ECOG performance status was 1 in 81.0% (17/21) and 0 in 19.0% (4/21). Eleven patients had metastatic castration-resistant prostate cancer, seven had pancreatic cancer, two had checkpoint blockade-refractory melanoma, and one had checkpoint blockade-refractory HPV-negative head and neck cancer. Fifteen patients (71.4%) had received ≥ 3 prior lines of systemic therapy, and four (19.0%) had received prior immune checkpoint inhibitor therapy.
Table 1.
Baseline patient characteristics
| Characteristics | N (%) N = 21 |
|---|---|
| Median Age (range) | 66 years (47–78) |
| Gender | |
| Female | 4 (19.0) |
| Male | 17 (81.0) |
| ECOG* performance status | |
| 0 | 4 (19.0) |
| 1 | 17 (81.0) |
| Diagnosis | |
| castration-resistant prostate cancer | 11 (52.4) |
| Pancreatic cancer | 7 (33.3) |
| Melanoma | 2 (9.5) |
| HPV- head and neck squamous cell carcinoma | 1 (4.8) |
| Median number of prior systemic therapies (range) | 4 (1–9) |
| Prior immune checkpoint inhibitor | 4 (19.0) |
| HPV- head and neck squamous cell carcinoma | 1 (4.8) |
| Melanoma | 2 (9.5) |
| Castration-resistant prostate cancer | 1 (4.8) |
| Median number of evofosfamide doses (range) | 4 (2–8) |
| Median number of ipilimumab doses (range) | 4 (1–8) |
ECOG Eastern Cooperative Oncology Group
Treatment was discontinued for all three patients enrolled at 400 mg/m2 evofosfamide due to disease progression. Similarly, treatment was discontinued for four patients at 480 mg/m2 due to disease progression (66.7%), while the other two withdrew from the study (33.3%). At 560 mg/m2 (n=6), treatment was discontinued for five patients due to disease progression (83.3%), while ipilimumab was discontinued for one patient due to toxicity (16.7%). At 640 mg/m2 (n=6), treatment was discontinued for four patients (66.7%) due to disease progression and ipilimumab was discontinued for one patient due to toxicity (16.7%). One patient remained on treatment at 640 mg/m2 (16.7%) at the time of analysis. As no dose-limiting toxicity was observed, 640 mg/m2 was considered the maximum tolerated dose, although 560 mg/m2 is the recommended Phase II dose based on response as detailed below.
Safety
A total of 193 treatment-related adverse events occurred in 21 patients, of which 162 (83.9%) were grade 1 or 2 events and 31 (16.1%) were grade ≥ 3 events (Table 2). There were 29 serious adverse events. The most common any grade treatment related adverse events were hematological (anemia 100%, thrombocytopenia 71.4%, leukopenia 61.9%, lymphopenia 57.1%, neutropenia 33.3%), rash (81.0%), elevated liver enzymes (ALT 90.5%, AST 71.4%, alkaline phosphatase 23.8%), fever (47.6%), nausea (23.8%), vomiting (38.1%), diarrhea (19.0%), constipation (19.0%), and colitis (14.3%). There were six treatment-related grade ≥ 3 adverse events at 400 mg/m2, eight at 480 mg/m2, ten at 560 mg/m2, and seven at 640 mg/m2. The most frequent treatment related grade 3 adverse events included elevated ALT (33.3%), elevated AST (19.0%), anemia, thrombocytopenia, lymphopenia (14.3% each), rash, hypotension, and neutropenia (9.5% each). Three patients required transfusions of packed red blood cells, but none required platelet transfusions. One patient at 640 mg/m2 had delayed grade 3 thrombocytopenia following five cycles of treatment, an episode that lasted more than 3 weeks. One patient had grade 3 ipilimumab-related lymphocytic myocarditis and grade 3 ipilimumab-related transaminitis. The former was treated with intravenous immunoglobulin and the latter required treatment with mycophenolate mofetil, resulting in complete resolution of both. One patient experienced grade 3 ipilimumab-related transaminitis and grade 3 ipilimumab- or evofosfamide-related pneumonitis, both of which resolved with steroids. One patient had grade 3 ipilimumab-related colitis that resolved with infliximab. No grade 5 adverse events were reported. Overall, four patients (19.0%) (one at 480 mg/m2, two at 560 mg/m2, and one at 640 mg/m2) required evofosfamide de-escalation due to toxicity. One patient at 640 mg/m2 discontinued ipilimumab due to toxicity. None of the adverse events met the criteria for dose-limiting toxicity.
Table 2.
Treatment-related adverse events occurring in >10% of patients or are ≥ grade 3
| Adverse event | Any Grade | Grade 1–2 | Grade ≥ 3 |
|---|---|---|---|
| Anemia | 21 (100.0) | 18 (85.7) | 3 (14.3) |
| Rash | 17 (81.0) | 15 (71.4) | 2 (9.5) |
| Thrombocytopenia | 15 (71.4) | 12 (57.1) | 3 (14.3) |
| Elevated alanine aminotransferase | 15 (71.4) | 11 (52.4) | 4 (19.0) |
| Leukopenia | 13 (61.9) | 13 (61.9) | 0 (0.0) |
| Lymphopenia | 12 (57.1) | 9 (42.9) | 3 (14.3) |
| Elevated aspartate aminotransferase | 11 (52.4) | 10 (47.6) | 1 (4.8) |
| Fever | 10 (47.6) | 10 (47.6) | 0 (0.0) |
| Vomiting | 8 (38.1) | 8 (38.1) | 0 (0.0) |
| Neutropenia | 7 (33.3) | 5 (23.8) | 2 (9.5) |
| Nausea | 5 (23.8) | 5 (23.8) | 0 (0.0) |
| Elevated alkaline phosphatase | 5 (23.8) | 4 (19.0) | 1 (4.8) |
| Transaminitis | 4 (19.0) | 1 (4.8) | 3 (14.3) |
| Fatigue | 4 (19.0) | 3 (14.3) | 1 (4.8) |
| Diarrhea | 4 (19.0) | 4 (19.0) | 0 (0.0) |
| Constipation | 4 (19.0) | 4 (19.0) | 0 (0.0) |
| Hypomagnesemia | 3 (14.3) | 3 (14.3) | 0 (0.0) |
| Colitis | 3 (14.3) | 0 (0.0) | 3 (14.3) |
| Myalgia | 3 (14.3) | 3 (14.3) | 0 (0.0) |
| Hypotension | 2 (9.5) | 0 (0.0) | 2 (9.5) |
| Sinus tachycardia | 1 (4.8) | 0 (0.0) | 1 (4.8) |
| Lymphocytic myocarditis | 1 (4.8) | 0 (0.0) | 1 (4.8) |
| Pneumonitis | 1 (4.8) | 0 (0.0) | 1 (4.8) |
Response
All 21 patients were evaluable at the time of analysis, although only 18 had measurable disease at baseline. Two of these 18 patients achieved confirmed partial response (11.1%), one of whom had HPV-negative head and neck cancer and the other had metastatic castration-resistant prostate cancer (Fig. 1A). One additional patient with metastatic castration-resistant prostate cancer had an unconfirmed partial response (5.6%); thus, the objective response rate was 16.7%. Twelve patients (66.7%) had stable disease (five with metastatic castration-resistant prostate cancer, five with pancreatic cancer, and two with checkpoint blockade-refractory melanoma). Overall disease control rate (complete response + partial response + stable disease) was 83.3% (15/18). Five patients (two with pancreatic cancer, two with metastatic castration-resistant prostate cancer, and one with HPV-negative head and neck cancer) were re-treated. Two patients who achieved partial response in the first course of treatment, had a repeat partial response on re-treatment (one with metastatic castration-resistant prostate cancer and one with HPV-negative head and neck cancer), while all others continued to present stable disease (one with metastatic castration-resistant prostate cancer and two with pancreatic cancer). The median duration of response for patients with partial response was 85 days (range 78–183 days), while the median duration of disease control (complete response + partial response + stable disease) was 64 days (range 2–244 days) (Fig. 1B and C).
Figure 1. Responses to evofosfamide and ipilimumab combination therapy.
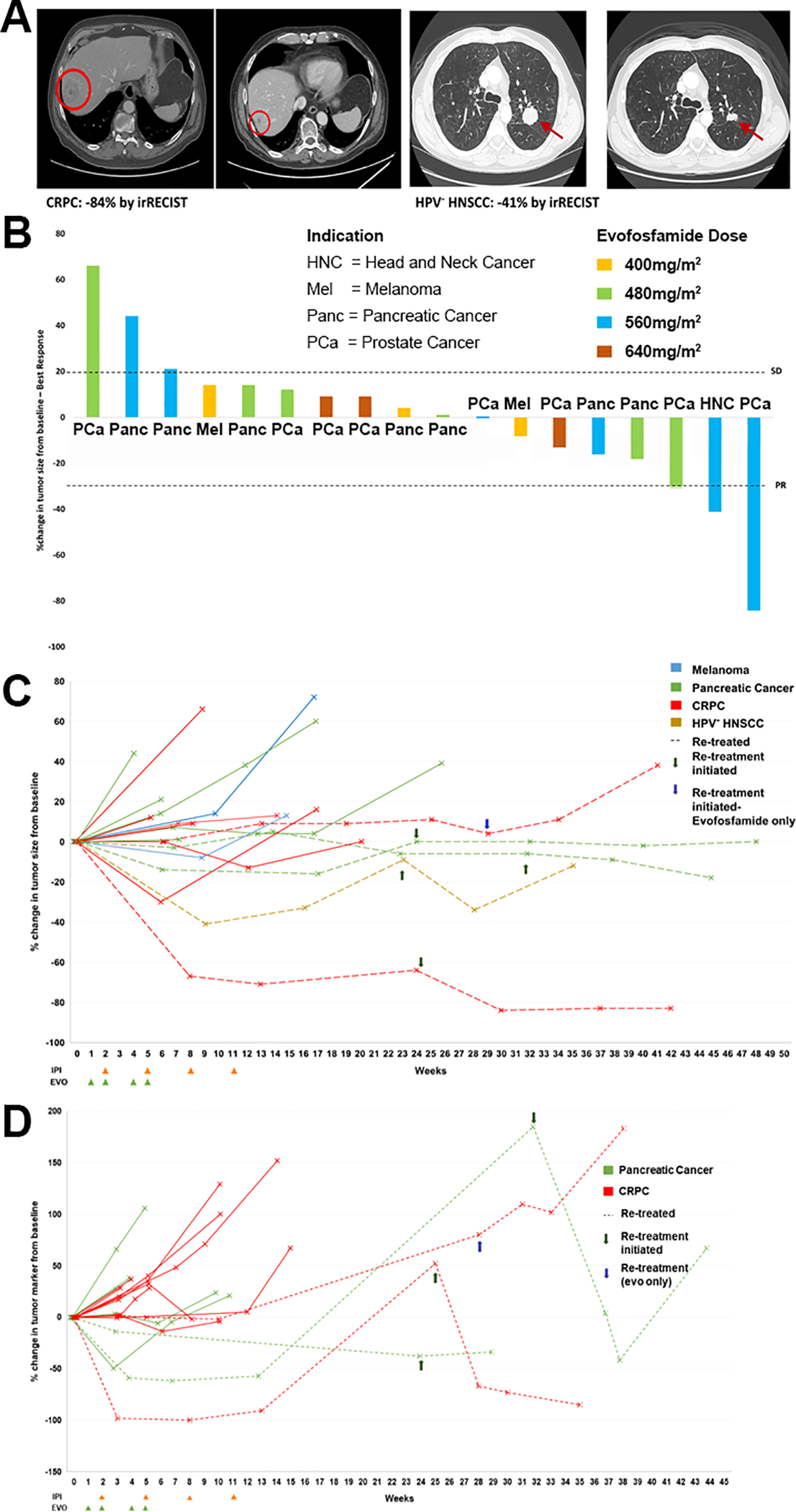
A) Best response scans for a prostate cancer and HPV- head and neck cancer patient who achieved partial response at 560 mg/m2 evofosfamide. B) Waterfall plot showing best percent change in tumor size across all four doses of evofosfamide (color coded) with tumors indicated on the X axis. C) Spider plot showing changes in tumor size over time for all patients with measurable lesions. D) CA 19–9 or PSA changes from baseline were tracked over time for indicated patients. Green arrows indicate retreatment with ipilimumab and evofosfamide. The blue arrow indicates re-treatment with evofosfamide only.
Serial tumor markers were also assessed for 18 of 21 evaluable patients (11 with metastatic castration-resistant prostate cancer and seven with pancreatic cancer). One patient with pancreatic cancer had a confirmed CA 19–9 response (62% decrease from baseline) as best response. One patient with metastatic castration-resistant prostate cancer had a confirmed PSA response (100% decrease from baseline) as best response. The three patients with non-measurable disease at baseline had metastatic castration-resistant prostate cancer, of whom one had confirmed PSA progression (48% increase from baseline) as best response, and two had stable PSA as best response (Fig. 1D).
At 400 mg/m2 (n=3), all three patients achieved stable disease as best response. At 480 mg/m2 (n=6), one patient with prostate cancer achieved an unconfirmed partial response (16.7%) as best response, four achieved stable disease (66.7%), and one progressed (16.7%). Best outcomes were achieved at 560 mg/m2 (n=6), with two patients (one with prostate cancer and one with HPV-negative head and neck cancer) achieving confirmed partial response (33.3%), two achieving stable disease (33.3%), and two experiencing progressive disease (33.3%) as best response. At 640 mg/m2 (n=6), three patients achieved stable disease (50.0%), two achieved stable PSA (33.3%), and one experienced PSA progression (16.7%) as best response.
Pharmacokinetics
Pharmacokinetic studies were performed at 560 mg/m2 and 640 mg/m2 evofosfamide. No differences in mean plasma concentration, Tmax, Cmax, and AUClast were observed for evofosfamide and its active metabolite Br-IPM (Supplemental Fig. S1). These studies demonstrated a plateau of active evofosfamide exposure at 560 mg/m2 (Cmax = 0.18 ug/mL) that was higher than the active range defined in the successful Phase II Maestro study (Cmax = 0.13 ug/mL), and which was never achieved in non-Japanese patients at Phase III (Cmax = 0.08 ug/mL) (13).
Pharmacodynamics
For all pharmacodynamic analyses, patients were classified into four groups (partial response, stable disease ≥ 60 days, SD < 60 days, and progressive disease), of which the first two groups were considered “responders” and the latter two “non-responders” based on molecular clustering. In addition, we note that stable disease lasting less than 60 days reflects a lack of durable response to therapy and can include patients who were slowly but steadily progressing. In contrast, stable disease lasting more than 60 days indicates continuing response to therapy beyond the treatment period. These response classifications were all based on irRECIST excepting two prostate cancer patients whose progressive disease was diagnosed based on PSA progression. Both patients are included in the analysis of peripheral blood mononuclear cells (PBMC) and RNA, and one is included in the flow cytometry analysis of paired biopsies.
Peripheral blood mononuclear cells (PBMC) collected pre-treatment, on-treatment (generally after four doses of evofosfamide [range 2–6] and two doses of ipilimumab [range 1–3]), and post-treatment were available for five non-responding patients (three progressive disease and two stable disease < 60 days) and seven responding patients (three partial response and four stable disease ≥ 60). Statistically significant accumulation of proliferative CD8+ and CD4+ effector T cells, as measured by Ki67 expression, was observed in responders relative to non-responders (Fig. 2A and Supplemental Fig. S2). Non-responders also tended to accumulate exhausted peripheral CD8+ T cells (PD-1+ LAG3+) (Fig. 2B). These peripheral T cell responses are plotted across therapy for individual patients in Supplemental Fig. S3. Levels of the immunosuppressive enzyme arginase also trended higher in circulating myeloid-derived suppressor cells at every time point examined (Fig. 2C). Similarly, we found significantly higher pre-treatment levels of arginase in circulating dendritic cells (CD11c+b-) in non-responders compared to responders (Fig. 2D). A similar trend was observed in monocytic dendritic cells (CD11c+b+). Mirroring arginase, we found a higher fraction of PD-L1 expression pre-treatment in dendritic cells from non-responders versus responders (Fig. 2E). Taken together, the data suggest the potential of a suppressive phenotype in the circulating myeloid repertoire as a potential pre-treatment biomarker, and of peripheral effector T cell proliferation as a pharmacodynamic biomarker of response to co-treatment with evofosfamide and ipilimumab.
Figure 2. Improved peripheral anti-tumor immunity signature in responding versus non-responding patients.
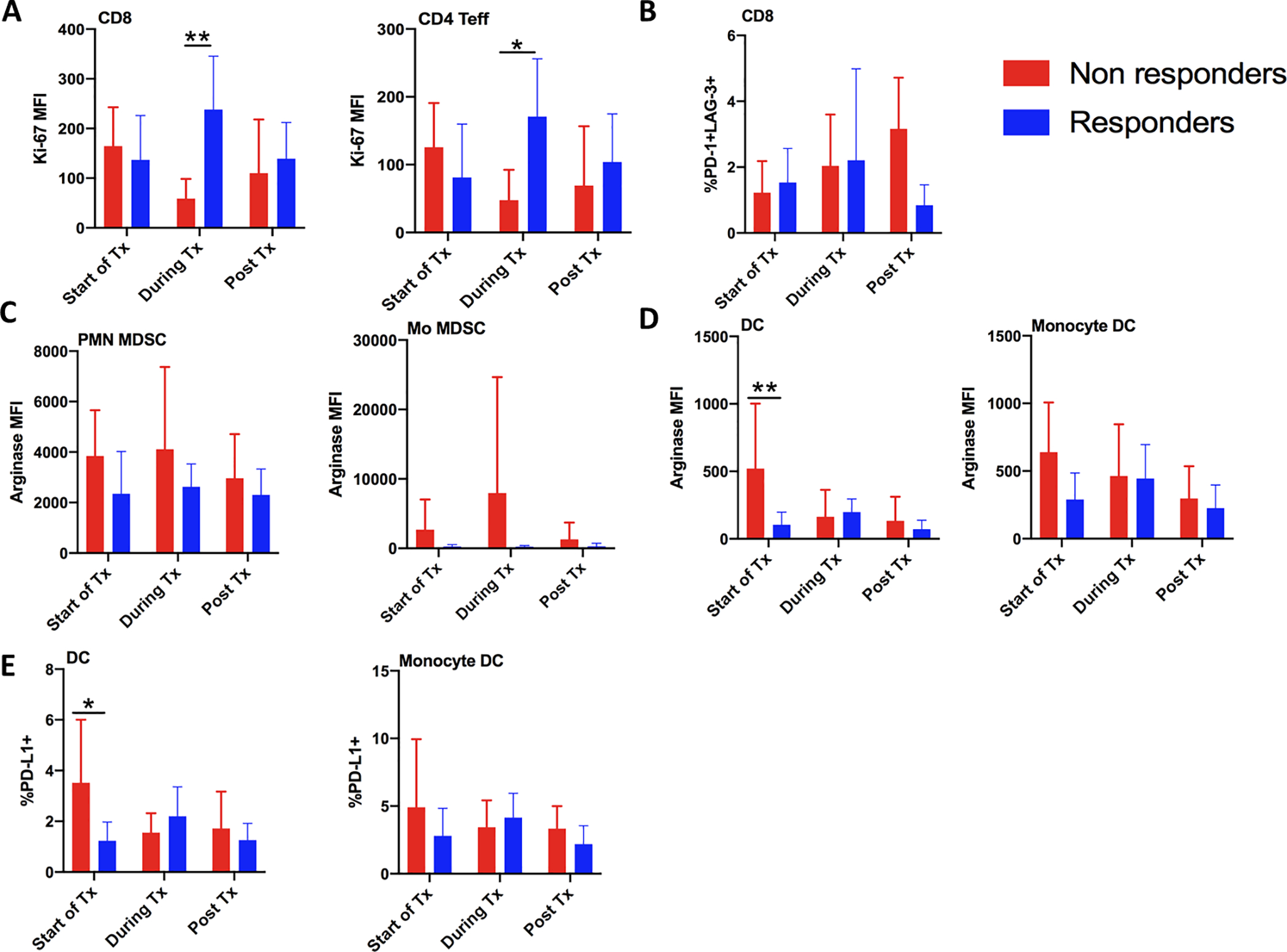
Peripheral blood mononuclear cells were isolated from patients prior to, during, and post treatment, and assessed for immune composition and function by 20-color flow cytometry. A) Proliferation of proliferating CD8 and CD4 T effector cells was analyzed by Ki-67 expression B) Percentage of exhausted PD-1+ LAG-3+ CD8 T cells. C,D) Arginase expression was measured in C) immune suppressive PMN and Mo myeloid-derived suppressor cells, and in D) dendritic cells and monocyte-derived dendritic cells. E) Percent of dendritic cells and monocyte-derived dendritic cells expressing PD-L1 was also measured. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 by two-way ANOVA (Sidak’s multiple comparisons test)
We obtained pre-treatment tumor biopsies from 16 patients, nine of whom also consented to on-treatment biopsies at week 7. These biopsies were analyzed by 20-parameter flow cytometry to evaluate the effect of evofosfamide and ipilimumab co-treatment on hypoxia and on the immune composition of the tumor microenvironment (Supplemental Fig. S4). Hypoxia was assessed using Hypoxyprobe, an antibody to the pimonidazole adduct and conjugated to the fluorescent dye ATTO 594. Consistent with restoration of anti-tumor T cell immunity, CD8+ cytotoxic T cells and CD4+ effector T cells tended to accumulate in responders (Fig. 3A). Greater densities of T cells and antigen-presenting dendritic cells in hypoxic regions also correlated with response; statistically significant differences in CD4+ effector T cells were observed even with the small number of samples available for analysis (Fig. 3B). In addition, responders tended to have reduced proliferation of immune-suppressive CD33+CD14-CD15- tumor-associated macrophages (Fig. 3C). Responses for all of these populations are plotted for individual patients in Supplemental Fig. S5A. Both CD4 and CD8 hypoxia-exposed T cells tended to exhibit higher PD-1 expression compared to their non-hypoxia resident counterparts; however, this difference only reached statistical significance for pre-treatment CD8 T cells (Fig. 3D and Supplemental Fig. S5B). We also observed a non-significant trend toward lower PD-1 expression by hypoxia-exposed CD8 T cells (PD-1 mean MFI of 6565 versus 3139) following treatment with evofosfamide and ipilimumab. We hope to investigate this phenomenon further in a larger study. Increased PD-L1 expression correlated with better response to therapy, consistent with the generation of an inflammatory tumor microenvironment, as shown for a representative patient who achieved partial response (Fig. 3E).
Figure 3. Evofosfamide and ipilimumab therapy increases immune activation of the tumor microenvironment in responders.
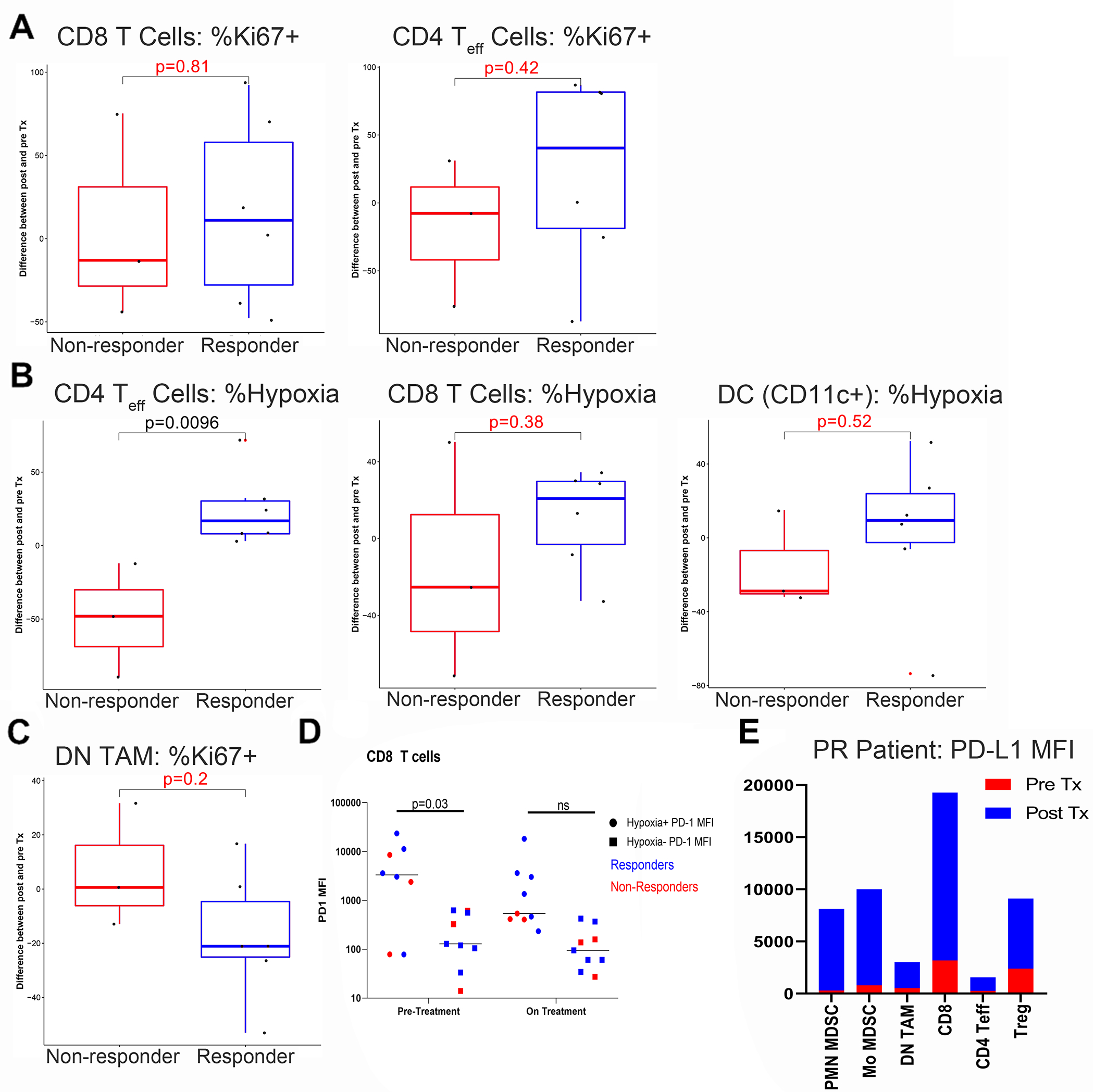
Patient biopsies were obtained at baseline and at week 7, and assessed for hypoxia and immune composition and function by 20-color flow cytometry. A) Proliferation of tumor-infiltrating CD8 and CD4 T effector cells was analyzed based on Ki-67 expression. B) Patients were given oral pimonidazole 5–24 hours prior to biopsy to evaluate tumor hypoxia using anti-pimonidazole antibody conjugated to the fluorescent dye ATT0 594 (Hypoxyprobe), and densities of T cells and dendritic cells in hypoxic areas was assessed. C) Proliferation of immune-suppressive CD33+CD14-CD15- tumor-associated macrophages was evaluated by Ki-67 expression. D) Hypoxia-exposed (pimonidazole+, circles) vs non-hypoxia resident (pimonidazole-, squares) CD8 T cells were analyzed for PD-1 expression pre- and on-treatment (cycle 3 day 8). E) PD-L1 expression was assessed in tumor-infiltrating T cells and myeloid cells in a representative responding patient. P values are indicated for Student’s t-test comparing responders and non-responders, with significant values listed in black and insignificant values listed in red or marked “ns” for the purpose of assessing trends.
RNA sequencing data showed that patients who eventually achieved partial response have distinct tumor gene expression profiles pre-treatment from patients who eventually experienced progressive disease and stable disease < 60 days (Fig. 4A). These differences were also apparent in comparing responders overall to non-responders (Fig. 4B and Supplemental Table 2). Significant differences were also observed between responders and non-responders on treatment (Fig. 4C and Supplemental Table 2), although gene expression was altered in response to therapy in both responders and non-responders (Supplemental Fig. S6). Gene set enrichment analysis against the Hallmark gene set collection revealed that patients who eventually responded to treatment show evidence of pre-existing adaptive and innate anti-tumor immunity (Fig. 4D). Hence, restoring T cell access to and survival in formerly hypoxic areas of the tumor may be sufficient to sensitize cancers in this setting to T cell checkpoint blockade. On the other hand, patients who progress on treatment show a hypermetabolic phenotype at baseline. In this environment, relieving hypoxia still leaves T cells at a tremendous metabolic disadvantage, likely explaining the lack of response to CTLA-4 blockade. Finally, enrichment analysis against the transcription factor targets collection showed that targets of HIF-1α, the cardinal hypoxia-responsive transcription factor, accumulated in non-responders on treatment, but diminished in responders (Fig. 4E).
Figure 4. Changes in gene expression in response to evofosfamide + ipilimumab therapy.
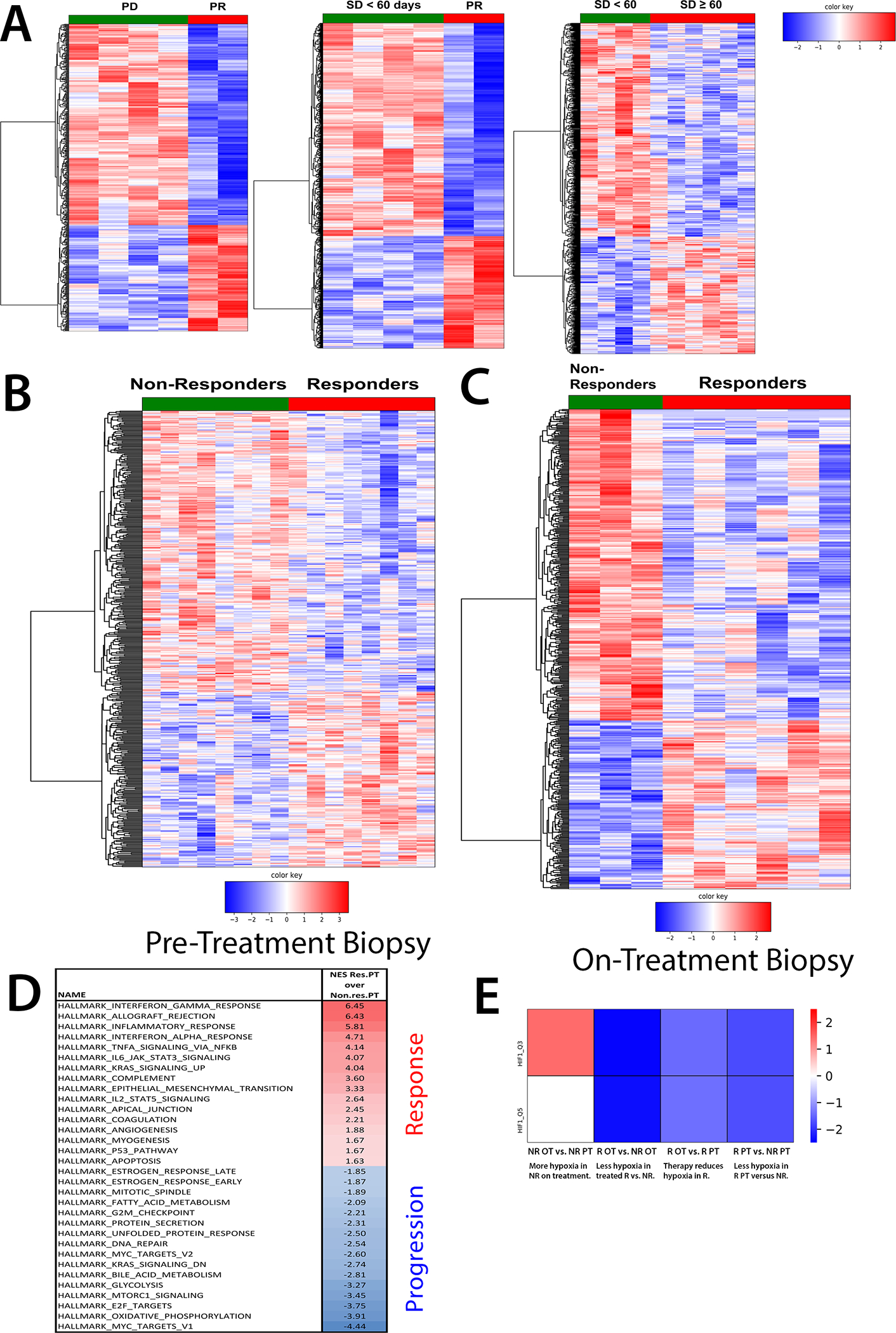
RNA was isolated using All-prep (Qiagen) from tumor biopsies collected pre- and on-treatment (cycle 3, day 8). RNA sequencing was performed by Avera and bioinformatic analysis was performed by Baylor College of Medicine Multi-omics core. A) Heat map of genes significantly differentially expressed prior to treatment between patients who achieve partial response (PR) and who experience progressive disease (PD), between patients who achieve partial response and stable disease < 60 days, and between patients who achieved stable disease ≥ 60 and < 60 days. B,C) Heat map of genes significantly differentially expressed B) prior to and C) on treatment between responders (partial response and stable disease ≥ 60 days) versus non-responders (progressive disease and stable disease < 60 days). (D) Enrichment scores of select gene sets (FDR < 0.25) enriched in responding over non-responding patients, as mapped against the Hallmark gene set collection. E) A heat map of changes in HIF-1α targets (NES shown, FDR < 0.25 except where indicated), as mapped against the transcription factor targets collection.
Discussion
Co-treatment with evofosfamide and ipilimumab appears to be both safe and well tolerated, with most drug-related adverse events being grade 1–2. Common grade ≥ 3 adverse events included hematologic toxicities that were not dose-limiting, that were comparable across evofosfamide doses, and that were similar to those observed with single-agent evofosfamide (25). Grade 3 ALT/AST elevation and grade ≥ 3 rash were observed more frequently than previously reported with either agent alone, suggesting a possible additive effect (25,26). Other immune-related grade 3 adverse events were comparable to that previously reported with single-agent ipilimumab (26). On the other hand, mucositis was significantly less frequent (9.5%) compared to single-agent evofosfamide and was exclusively all grade 1–2. Of note, adverse events were well-managed with supportive measures and dose interruption, except in one patient with immune hepatitis who required immunosuppression with mycophenolate mofetil. Collectively, safety data indicate that toxicities from combining evofosfamide and ipilimumab were comparable to that of either agent as monotherapy with the exception of milder mucositis, elevated but not dose-limiting hepatotoxicity, more frequent skin rashes, and, in one instance, delayed/prolonged thrombocytopenia. The safety profile is also comparable to that of the combination of evofosfamide with gemcitabine, as assesswed in a randomized Phase III study (MAESTRO) of 693 patients with advanced pancreatic cancer (12).
We observed multiple partial responses in hormone- and chemotherapy-refractory prostate and HPV-negative head and neck cancer patients, as well as stable disease in pancreatic cancer and checkpoint-blockade refractory melanoma, for whom treatment options are typically scarce. While ipilimumab was previously demonstrated to be of limited efficacy against metastatic castration-resistant prostate cancer (29,30), it is important to note that patients in these prior studies were chemotherapy-naïve and, in most cases, were treated with 10 mg/kg, rather than 3 mg/kg, ipilimumab. Given these differences, the lack of similarity between the transcriptomic response we observed and the classic transcriptomic response to ipilimumab in melanoma (31), and the transcriptomic separation between responders and non-responders and between pre- and on-treatment biopsies, we assert that these responses are the result of therapeutic cooperativity between evofosfamide and ipilimumab. Of note, we determined the recommended Phase II evofosfamide dose when combined with 3 mg/kg ipilimumab to be 560 mg/m2, at which two confirmed partial responses were observed, even though the maximum tolerated dose appeared to be 640 mg/m2, at which neither response nor dose-limiting toxicity was observed.
Further, the observation of excellent disease control among patients who were re-treated indicates that treatment with a limited number of cycles, followed by re-treatment if necessary, may be feasible with this combination, thereby avoiding prolonged and undue toxicities. However, the duration of response and duration of disease control were relatively short for many patients, highlighting the need for correlative studies and better combinatorial strategies, including, potentially, regimens thah block PD-1, as evofosfamide synergized with anti-CTLA-4 and anti-PD-1 antibodies in multiple pre-clinical models of murine prostate cancer (4), and as PD-L1 accumulated in responders. Indeed, a follow-up study to combine evofosfamide with anti-CTLA-4 and anti-PD-1 is planned.
Although prior trials demonstrated that levels of circulating myeloid-derived suppressor cells can be predictive of subsequent responses to ipilimumab, the phenotypic traits of these cells (e.g., arginase expression) were not examined (27). We have now observed that CD8+ and CD4+ effector T cells significantly accumulate in responders versus non-responders while on treatment, even in a small cohort. This not only suggests the value of monitoring peripheral T cell phenotypes as a biomarker of response to the combination of evofosfamide and ipilimumab, but also shows that this regimen elicits a peripherally measurable mobilization of circulating effector T cells in responding patients.
Comprehensive flow cytometry of pre- and on-treatment tumor biopsies also revealed a T cell inflamed milieu in responders. T cell proliferation, activation, and survival under hypoxia also improved or trended toward improvement, accompanied by concomitant reduction in myeloid-derived suppressor cells and tumor-associated macrophages. These observations suggest that evofosfamide cooperates with ipilimumab to remodel the tumor microenvironment of traditionally immune-refractory cancers to one that is conductive to T cell persistence and function. The data also reinforce the notion that specific biomarkers can be used to assess the efficacy of the combination.
Gene expression analysis indicated that a hypermetabolic phenotype at baseline often precludes response. Similarly, we found that a similar hypermetabolic phenotype featuring elevated oxidative phosphorylation and heightened glycolysis was highly enriched when a checkpoint blockade-sensitive melanoma cell line was multiply passaged in vivo in mice receiving immune checkpoint inhibitors until complete immunotherapy resistance was acquired (28). Thus, we speculate that hypoxia reduction alone may be insufficient to relieve T cell inhibition due metabolic starvation, as happens in the context of a hypermetabolic tumor.
In summary, the combination of evofosfamide and ipilimumab was well tolerated, with no new or unexpected safety signals. Anti-tumor activity was observed not only in heavily pretreated patients, but also in checkpoint blockade-refractory tumors, although the response duration was limited. Nevertheless, biomarker-driven patient selection and/or addition of PD-1 blockade may further augment and extend the clinical response.
Supplementary Material
Translational Relevance.
Tumor hypoxia can drive resistance to immunotherapy, and is pervasive in immunologically “cold” tumors such as pancreatic and prostate cancer. Thus, such tumors are typically resistant to T cell checkpoint blockade. This Phase I study evaluated the safety and efficacy of combining hypoxia reduction (through evofosfamide) with blockade of the immune checkpoint (through ipilimumab), in patients with advanced metastatic or locally advanced pancreatic cancer, HPV-negative head and neck cancer, melanoma refractory to immune checkpoint inhibitors, and castration-resistant prostate cancer. Data indicate that the combination regimen is not only well tolerated, but also elicits both objective response and prolonged disease stabilization in cancers for which treatment options are exceptionally limited. Correlative studies identified gene signatures associated with response versus resistance to treatment, as well as patterns of immune activation associated with response.
Acknowledgements
We thank Molecular Templates, Inc. and Threshold, Inc., particularly Charles Hart, Tillman Pearce, and Barry Selick, for supporting this trial. We thank Amadeo Biter for editorial support.
Financial Support:
This trial and associated correlative studies were supported by Molecular Templates, Inc.
Footnotes
Conflict of Interest: M.A.C. has ownership in, receives honoraria from, is founder of, and advisor to ImmunoGenesis, Inc. M.A.C. receives royalties from the Patent “Methods and Composition for Localized Secretion of Anti-CTLA-4 Antibodies”. J.P.H., E.T.W., and T.F.W. are employees of Molecular Templates, Inc.
References
- 1.Saggar JK, Yu M, Tan Q, Tannock IF. The tumor microenvironment and strategies to improve drug distribution. Front Oncol 2013;3:154 doi 10.3389/fonc.2013.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hatfield SM, Kjaergaard J, Lukashev D, Belikoff B, Schreiber TH, Sethumadhavan S, et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1alpha-dependent and extracellular adenosine-mediated tumor protection. J Mol Med (Berl) 2014;92(12):1283–92 doi 10.1007/s00109-014-1189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med 2015;7(277):277ra30 doi 10.1126/scitranslmed.aaa1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest 2018. doi 10.1172/JCI96268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jamieson SM, Tsai P, Kondratyev MK, Budhani P, Liu A, Senzer NN, et al. Evofosfamide for the treatment of human papillomavirus-negative head and neck squamous cell carcinoma. JCI Insight 2018;3(16) doi 10.1172/jci.insight.122204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res 2017;5(1):9–16 doi 10.1158/2326-6066.CIR-16-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018;362(6411) doi 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science 2018;359(6382):1350–5 doi 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haslam A, Prasad V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw Open 2019;2(5):e192535 doi 10.1001/jamanetworkopen.2019.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zemek RM, Chin WL, Nowak AK, Millward MJ, Lake RA, Lesterhuis WJ. Sensitizing the Tumor Microenvironment to Immune Checkpoint Therapy. Front Immunol 2020;11:223 doi 10.3389/fimmu.2020.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016;165(1):35–44 doi 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cutsem EV, Lenz H-J, Furuse J, Tabernero J, Heinemann V, Ioka T, et al. MAESTRO: A randomized, double-blind phase III study of evofosfamide (Evo) in combination with gemcitabine (Gem) in previously untreated patients (pts) with metastatic or locally advanced unresectable pancreatic ductal adenocarcinoma (PDAC). Journal of Clinical Oncology 2016;34(15_suppl):4007– doi 10.1200/JCO.2016.34.15_suppl.4007. [DOI] [Google Scholar]
- 13.Higgins JP, Sarapa N, Kim J, Poma E. Unexpected pharmacokinetics of evofosfamide observed in phase III MAESTRO study. Journal of Clinical Oncology 2018;36(15_suppl):2568– doi 10.1200/JCO.2018.36.15_suppl.2568. [DOI] [Google Scholar]
- 14.Chouaib S, Noman MZ, Kosmatopoulos K, Curran MA. Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017;36(4):439–45 doi 10.1038/onc.2016.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ai M, Budhani P, Sheng J, Balasubramanyam S, Bartkowiak T, Jaiswal AR, et al. Tumor hypoxia drives immune suppression and immunotherapy resistance. J Immunother Cancer 2015;3(Suppl 2):P392-P doi 10.1186/2051-1426-3-S2-P392. [DOI] [Google Scholar]
- 16.Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. The Journal of Clinical Investigation 2018;128(11):5137–49 doi 10.1172/JCI96268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bohnsack O, Hoos A, Ludajic K. Adaptation and modification of the immune related response criteria (IRRC): IrRECIST. Journal of Clinical Oncology 2014;32(15_suppl):e22121–e doi 10.1200/jco.2014.32.15_suppl.e22121. [DOI] [Google Scholar]
- 18.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. New England Journal of Medicine 2010;363(8):711–23 doi 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carthon BC, Wolchok JD, Yuan J, Kamat A, Ng Tang DS, Sun J, et al. Preoperative CTLA-4 blockade: tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin Cancer Res 2010;16(10):2861–71 doi 10.1158/1078-0432.CCR-10-0569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gross MW, Karbach U, Groebe K, Franko AJ, Mueller-Klieser W. Calibration of misonidazole labeling by simultaneous measurement of oxygen tension and labeling density in multicellular spheroids. Int J Cancer 1995;61(4):567–73 doi 10.1002/ijc.2910610422. [DOI] [PubMed] [Google Scholar]
- 21.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015;12(4):357–60 doi 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 2015;33(3):290–5 doi 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 2019;47(D1):D766–D73 doi 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiss GJ, Infante JR, Chiorean EG, Borad MJ, Bendell JC, Molina JR, et al. Phase 1 study of the safety, tolerability, and pharmacokinetics of TH-302, a hypoxia-activated prodrug, in patients with advanced solid malignancies. Clin Cancer Res 2011;17(9):2997–3004 doi 10.1158/1078-0432.CCR-10-3425. [DOI] [PubMed] [Google Scholar]
- 26.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010;363(8):711–23 doi 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer immunology, immunotherapy : CII 2014;63(3):247–57 doi 10.1007/s00262-013-1508-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaiswal AR, Liu AJ, Pudakalakatti S, Dutta P, Jayaprakash P, Bartkowiak T, et al. Melanoma Evolves Complete Immunotherapy Resistance through the Acquisition of a Hypermetabolic Phenotype. Cancer Immunol Res 2020;8(11):1365–80 doi 10.1158/2326-6066.CIR-19-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients With Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J Clin Oncol 2017;35(1):40–7 doi 10.1200/JCO.2016.69.1584. [DOI] [PubMed] [Google Scholar]
- 30.Slovin SF, Higano CS, Hamid O, Tejwani S, Harzstark A, Alumkal JJ, et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: results from an open-label, multicenter phase I/II study. Ann Oncol 2013;24(7):1813–21 doi 10.1093/annonc/mdt107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350(6257):207–11 doi 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.