

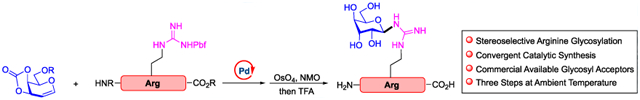
Abstract
A stereospecific convergent peptide arginine glycosylation method is reported for the first time. A recently discovered arginine glycosylation invigorated the interests of arginine modification, which has been challenging, because of the inertness of the guanidino side chain. The approach renders the arginine glycoside construction convergently. Catalyzed by palladium complex, glycals modify arginine guanidino groups in one step with high functional group tolerance at ambient temperature. The glycosylated products may be converted to glycopeptide analogues in few steps.
Graphical Abstract
Arginine is structurally unique and a critical role in biological systems.1 Its guanidino side chain is protonated at physiological pH, which makes arginine a charged species. Functionalization of the arginine side chain during the protein post-translational modification (PTM) is a commonly observed occurrence, such as citrullination2 and methylation.3 Recently, a novel PTM of arginine was reported, where guanidinium side chain was glycosylated by N-acetylglucosamine4 and rhamnose5 in pathogenic bacteria. Meanwhile, other arginine modifications are extensively investigated and well-understood.6 The investigations of arginine glycosylation have been limited and establishing a chemical methodology that could rapidly construct arginine glycosides motifs could certainly accelerate the biomedical studies of arginine PTM.
Glycosylation is one of the most opulent and critical protein PTMs. Commonly observed saccharides attachments occur at Ser/Thr/Tyr (O-Link) and Asn (N-Link) residues,7 whereas the arginine glycosylation has been a much less known process and under-utilized. Perhaps such rare occurrences are attributed to the inertness of the guanidino group.8 From a synthetic standpoint, the inability to functionalize guanidinium motifs became more compelling. Transformations involving arginine side chains often employed strong electrophiles to compensate for the relatively weak nucleophilicity of ketimine.9 The challenge of glycosylation is elevated because of the steric and electronic properties of glycosyl donors. As a case in point, in the reports of the chemical synthesis of arginine Glc N-acylation by Shao, Liu and Hu,10 the strategy for installing carbohydrates was illustrated as the union of thiourea glycosides and ornithine-bearing peptides (Figure 1a). Such maneuvers elegantly circumvent the difficulties in connecting canonical glycosyl donors and acceptors; however, the introduction of nonproteinogenic amino acid ornithine and elaborated multistep preparation of glycosyl thiourea hampers its general application. Ideally, direct construction of glycopeptides using a native arginine bearing peptide with a commonly used glycosyl donor at the reducing end would significantly improve the efficiency of arginine glycoside chemical preparation. Herein, we report a catalytic method that provides stereoselective arginine glycosylation in a convergent fashion for the first time, using glycals and commercially available arginine precursors (Figure 1b).
Figure 1.

Convergent arginine glycosylation.
In the past decades, the palladium-catalyzed glycosylation reaction has been extensively developed and successfully applied in the synthesis.11 Our previous studies have revealed the extraordinary ability of glycals toward glycosyl acceptors.12 Under palladium-mediated conditions, external nucleophiles such as Ts-NR moieties could achieve exclusive stereoselectivity and excellent yields.12b In order to fine-tune the N-glycosyl acceptor’s nucleophilicity, the Ts sulfonyl group is required as an activator, which is extremely difficult to remove13 without degrading the construct of the carbohydrates (Figure 2). We speculate that the 2,2,4,6,7-pentamethyldihy-drobenzofuran-5-sulfonyl (pbf) group could replace the Ts functional group, and the protocol for pbf removal is well-documented and mild under acidic conditions14 (cf. Figure 2). If successful, arginine glycosylation could be achieved via routinely utilized, commercially available Fmoc-Arg(pbf) and Boc-Arg(pbf) building blocks, which could significantly improve the efficiency for preparing arginine glycopeptides.
Figure 2.

Removal of sulfonyl activating groups.
We believed that glycal 1a would be an ideal glycosyl donor for initial evaluation, which was prepared via a quick two-step sequence from commercially available saccharide (Scheme 1). Glycosylation of pbf protected arginine methyl ester 2a with glycal 1a under palladium catalyzed conditions was evaluated. The combination of Pd(OAc)2 and xantphos as a ligand was first investigated, and the reaction produced desired product 3a with exclusive β-stereochemistry and 29% yield after 24 h at ambient temperature (Scheme 1, entry 1). The structure of 3a was identified by extensive one-dimensional (1D) and two-dimensional (2D) NMR experiments (see the SI for stereochemistry and regiochemistry assignments). When divalent palladium catalyst Pd(PPh3)2Cl2 was employed, however, we did not observe glycosylation product (Scheme 1, entry 2). To our surprise, zerovalent Pd(PPh3)4, or Pd2dba3·CHCl3, improved the reaction yields significantly (Scheme 1, entries 3 and 4). Next, several phosphine bidentate ligands, such as dppb, dppe, dppp, and dppf were screened (Scheme 1, entries 5–8), yet the yields were inferior (0%–21%). Chiral biaryl BINAP was evaluated, and no product was found (Scheme 1, entry 9). DPEPhos, N-xantphos, and monodentate phosphine ligand were less effective than xantphos (Scheme 1, entries 10–12). When 3 equiv of glycal 1a was used, a higher yield of 3a was obtained (Scheme 1, entry 13). Switching to Pd(PPh3)4 afforded a comparable yield but much faster reaction time (2 h). Although Pd(PPh3)4 rendered a slightly higher yield (73%) and shorter reaction time, the formation the trace amount of byproduct [(O)PPh3] from the ligand complicates the silica gel purification (same rf with certain products). Our further studies use both conditions when appropriate. Without palladium catalyst or ligand, the reaction could not occur (Scheme 1, entries 15 and 16).
Scheme 1. Reaction Optimization for Arginine Glycosylation.

a0.1 mmol 1a, 0.05 mmol 2a, 10 mol % [Pd] and 10 mol % ligand, 4 mL of CH2Cl2 were used. bIsolated yield. cNot detected. d20 mol % ligand was used. e0.15 mmol 1a and 0.05 mmol 2a were used.
With the optimized conditions in hand, we subsequently investigated the arginine glycosylation in a more-complicated system (Scheme 2). The commercially available Fmoc-Arg(pbf)-OMe successfully provided product in comparable yield (71%). We then explored a dipeptide system. Arginine–valine produced 3c smoothly with 71% yield, indicating that dipeptides could be glycosylated. Other amino acid residues with hydrophobic side chains were well-tolerated, such as arginine–alanine, in which the carboxyl was protected by Bn and produced 3d with 75% yield. For arginine–leucine with tBu, 3e was generated with 64% yield. Even for compounds with a rigid proline substrate, glycosylation had no issue and generated 3f with 70% yield. Arginine–phenylalanine afforded corresponding product 3g with moderate 53% yield. Next, we implemented glycosylations using different dipeptides involving hydrophilic side chains. Arginine–aspartic acid with a methyl-protected carboxyl group, arginine–tyrosine with a tBu-protected phenol, and arginine–lysine with a Boc-protected amine were demonstrated to be tolerated and gave good yield. Among them, arginine–lysine dipeptide generated 77% of 3j under similar conditions. In addition, we found that placing Arg(pbf) at the C-terminus of the peptides did not alter the reaction outcomes. Valine–arginine showed no difference in yield, when compared with arginine–valine. Under the reaction conditions, glycopeptide 3k was furnished with 72% yield. N-methyl-alanine-arginine and phenylalanine as glycosyl acceptors provided good results. The yield of 3l was 72% and 3m was 69%. Glycine–arginine generated product 3n with 41% yield. Dipeptide aspartic acid–arginine with a hydrophilic side chain gave a good result. Cysteine-derived dithio substrate delivered product 3p. Lastly, we found glycosylation, using tripeptide as an acceptor, produced glycoside 3q with 44% isolation yield.
Scheme 2. Reaction Scope Evaluations.

aReaction conditions: 0.15 mmol 1a, 0.05 mmol 2, 10 mol % Pd(PPh3)4, 10 mol % xantphos, and 4 mL of CH2Cl2 were used, rt, 2 h, isolated yield. b5 mol % Pd2dba3·CHCl3, 24 h, isolated yield.
Subsequently, we examined the scope of glycosyl donors (Scheme 3). Under reaction conditions, glycals 1 with dipeptide 2f furnished a variety of glycosides 3. The substituent groups such as TIPS, TBDPS, Bn, Bz, tBu and adamantyl were well-tolerated, the dipeptide glycosides 3r–3x were produced in comparable yields (41%–73%). Lipidic and fluorescein side chains could also be managed and afforded 3y and 3z. The excellent stereoselectivity of arginine glycosylation was illustrated again by introducing an exclusive α-glycosidic bond in glycopeptide 3aa, utilizing d-allal carbohydrate as the donor, albeit wth a lower yield (35%).
Scheme 3. Glycosyl Donor Evaluationa.

a0.15 mmol glycal 1, 0.05 mmol 2f and 4 mL of CH2Cl2 were used, rt, 24 h, isolated yield. bReaction time: 2 h. cReaction time: 1 h.
The practicality of the arginine glycosylation method was illustrated in Scheme 4. Glycopeptide 3a was reduced via Pd/C and H2 to generate a deoxy-sugar 4 in excellent yield. Furthermore, transformation of 3a with catalytic OsO4 and N-methylmorpholine-N-oxide afforded the corresponding glycopeptide 5a in 87% yield as a single diastereomer, which offered a chiral scaffold that could be mimicking N-acetylglucosamine or rhamnose. We expected that dihydroxylation occurred at the less sterically hindered α-face. Finally, a one-pot, two-step protocol successfully converts Arg glycoside 3g to a protecting-group-free dipeptide 6 in high yield as a single diastereomer.15 During the transformation, the pbf moiety was removed along with other common peptidyl protecting groups under acidic conditions. This protocol underscores the efficiency of our convergent arginine glycosylation methodology.
Scheme 4. Functionalization of Arginine Glycosides.

Based on the experimental data and our previously reported result,11 the arginine glycosylation should undergo a classic Tsuji–Trost reaction mechanism.16 (see Figure 3) The less sterically demanding face of glycal 1 forms a π-allyl Pd(0) species II, which should be the reaction intermediate and governs the stereoselectivity. The N-glycoside 3 can be produced upon attacking of arginine(pbf) 2 toward π-allyl Pd complex II. We do not fully understand why the xantphos attains superior reactivities, compared with other bidentate phosphine ligands. According to literature reports, we speculated that a larger cone angle between xantphos and palladium is attributed to the superior reactivity during allylation. It is well-documented that the Xantphos has a cone angle of 247° with palladium, compared to that obtained with dppe (225°), dppf (230°), and other bidentate ligands.17 The studies from van Leeuwen et al.18 suggested that a larger ligand cone angle not only promotes a faster oxidative addition, but also enhances the reaction rate of nucleophilic addition to π-allyl species and subsequent product dissociation.
Figure 3.

Proposed arginine glycosylation mechanism.
In summary, we have established a convergent method that renders challenging peptide arginine glycosylation for the first time. This practical approach could establish the glycosidic bonds of arginine with exclusive regioselectivities, and stereoselectivities. The mild reaction processes were catalyzed by a palladium complex and enjoy high amino acid residue tolerance. Both glycosyl donors and acceptors are either easily obtained or commercially available. The dual functionality of the pbf construct was highlighted as a protecting as well as a specific activating group, which could be smoothly removed along with other commonly used amino acid protecting moieties. Compared with extant methods, our convergent approach provides a facile alternative strategy for highly efficient glycosylation, which potentially could assist the biological studies toward arginine glycosides. Further investigations of the glycosylation toward other protein modifications will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
Support for this work was provided by the National Science Foundation (No. CHE 1710174), and the National Institute of Health (No. 1R35 GM138336-01) to Q.Z. National Science Foundation is acknowledged (No. CHE 17267240) for the purchase of NMR services. We thank Prof. David Crich (University of Georgia) for his valuable suggestions.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.1c01218.
Complete experimental procedures and characterization data for all new compounds (PDF)
REFERENCES
- (1).Fuhrmann J; Clancy KW; Thompson PR Chemical biology of protein arginine modifications in epigenetic regulation. Chem. Rev 2015, 115, 5413–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).van Venrooij WJ; Pruijn GJ Citrullination: a small change for a protein with great consequences for rheumatoid arthritis. Arthritis Res. 2000, 2, 249–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wu Q; Schapira M; Arrowsmith CH; Barsyte-Lovejoy D Protein arginine methylation: from enigmatic functions to therapeutic targeting. Nat. Rev. Drug Discovery 2021, DOI: 10.1038/s41573-021-00159-8. [DOI] [PubMed] [Google Scholar]
- (4).Xu C; Liu X; Zha H; Fan S; Zhang D; Li S; Xiao W A pathogen-derived effector modulates host glucose metabolism by arginine GlcNAcylation of HIF-1α protein. PLoS Pathog. 2018, 14, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lassak J; Keilhauer EC; Furst M; Wuichet K; Godeke J; Starosta AL; Chen J-M; Søgaard-Andersen L; Rohr J; et al. Arginine-rhamnosylation as new strategy to activate translation elongation factor P. Nat. Chem. Biol 2015, 11, 266–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Pan X; Luo J; Li S Bacteria-Catalyzed Arginine Glycosylation in Pathogens and Host. Front. Cell. Infect. Microbiol 2020, 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Varki A; Cummings RD; Esko JD; Freeze HH; Stanley P; Bertozzi CR; Hart GW; Etzler ME Essentials of Glycobiology, 2nd Edition; Cold Spring Harbor Laboratories Press, 2009. [PubMed] [Google Scholar]
- (8).Rao Kovvuri VR; Xue H; Romo D Generation and Reactivity of 2-Amido-1,3-diaminoallyl Cations: Cyclic Guanidine Annulations via Net (3 + 2) and (4 + 3) Cycloadditions V. Org. Lett 2020, 22, 1407–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Grundler V; Gademann K Direct Arginine Modification in Native Peptides and Application to Chemical Probe Development. ACS Med. Chem. Lett 2014, 5, 1290–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Pan M; Li S; Li X; Shao F; Liu L; Hu HG Synthesis of and specific antibody generation for glycopeptides with arginine N-GlcNAcylation. Angew. Chem. Int. Ed 2014, 53, 14517–14521. [DOI] [PubMed] [Google Scholar]; (b) Wang S; Corcilius L; Sharp PP; Rajkovic A; Ibba M; Parker BL; Payne RJ Synthesis of rhamnosylated arginine glycopeptides and determination of the glycosidic linkage in bacterial elongation factor P. Chem. Sci 2017, 8, 2296–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).For selected examples of palladium-catalyzed glycosylation, see: (a) Babu RS; O’Doherty GA A palladium-catalyzed glycosylation reaction: the de novo synthesis of natural and unnatural glycosides. J. Am. Chem. Soc 2003, 125, 12406–12407. [DOI] [PubMed] [Google Scholar]; (b) Babu RS; Zhou M; O’Doherty GA De Novo Synthesis of Oligosaccharides Using a Palladium-Catalyzed Glycosylation Reaction. J. Am. Chem. Soc 2004, 126, 3428–3429. [DOI] [PubMed] [Google Scholar]; (c) Comely AC; Eelkema R; Minnaard AJ; Feringa BL De Novo Asymmetric Bio- and Chemocatalytic Synthesis of Saccharides – Stereoselective Formal O-Glycoside Bond Formation Using Palladium Catalysis. J. Am. Chem. Soc 2003, 125, 8714–8175. [DOI] [PubMed] [Google Scholar]; (d) Sherry BD; Loy RN; Toste FD Rhenium(V)-Catalyzed Synthesis of 2-Deoxy-α-glycosides. J. Am. Chem. Soc 2004, 126, 4510–4511. [DOI] [PubMed] [Google Scholar]; (e) Kim H; Men H; Lee C Stereoselective Palladium-Catalyzed O-Glycosylation Using Glycals. J. Am. Chem. Soc 2004, 126, 1336–1337. [DOI] [PubMed] [Google Scholar]; (f) Schuff BP; Mercer GJ; Nguyen HM Palladium-Catalyzed Stereoselective Formation of α-O-Glycosides. Org. Lett 2007, 9, 3173–3176. [DOI] [PubMed] [Google Scholar]; (g) Zeng J; Ma J; Xiang S; Cai S; Liu X-W Stereoselective β-C-Glycosylation by a Palladium-Catalyzed Decarboxylative Allylation: Formal Synthesis of Aspergil-lide A. Angew. Chem., Int. Ed 2013, 52, 5134–5137. [DOI] [PubMed] [Google Scholar]; (h) Xiang S; Hoang KLM; He J; Tan Y-J; Liu X-W Reversing the Stereoselectivity of a Palladium-Catalyzed O-Glycosylation through an Inner-Sphere or Outer-Sphere Pathway. Angew. Chem. Int. Ed 2014, 54, 604–607. [DOI] [PubMed] [Google Scholar]; (i) Yao H; Zhang S-S; Leng W-L; Leow M-L; Xiang S-H; He J-X; Liao H-Z; Hoang KLM; Liu X-W Catalyst-Controlled Stereoselective O-Glycosylation: Pd(0) vs Pd(II). ACS Catal. 2017, 7, 5456–5460. [Google Scholar]; (j) Sau A; Williams R; Palo-Nieto C; Franconetti A; Medina S; Galan MC Palladium-Catalyzed Direct Stereoselective Synthesis of Deoxyglycosides from Glycals. Angew. Chem., Int. Ed 2017, 56, 3640–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Sau A; Galan MC Palladium-Catalyzed α-Stereoselective O-Glycosylation of O(3)-Acylated Glycals. Org. Lett 2017, 19, 2857–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Crich D Mechanism of a Chemical Glycosylation Reaction. Acc. Chem. Res 2010, 43, 1144–1153. [DOI] [PubMed] [Google Scholar]; (m) McKay MJ; Nguyen HM Recent Advances in Transition Metal-Catalyzed Glycosylation. ACS Catal. 2012, 2, 1563–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Li XH; Zhu JL Glycosylation via Transition-Metal Catalysis: Challenges and Opportunities. Eur. J. Org. Chem 2016, 2016, 4724–4767. [Google Scholar]; (o) Yu B Gold(I)-Catalyzed Glycosylation with Glycosyl o-Alkynylbenzoates as Donors. Acc. Chem. Res 2018, 51, 507–516. [DOI] [PubMed] [Google Scholar]; (p) Leng W-L; Yao H; He J-X; Liu X-W Venturing beyond Donor-Controlled Glycosylation: New Perspectives toward Anomeric Selectivity. Acc. Chem. Res 2018, 51, 628–639. [DOI] [PubMed] [Google Scholar]
- (12).(a) Dai Y; Tian B; Chen H; Zhang Q Palladium-Catalyzed Stereospecific C-Glycosylation of Glycals with Vinylogous Acceptors . ACS Catal. 2019, 9, 2909–2915. [Google Scholar]; (b) Dai Y; Zheng J; Zhang Q General Strategy for Stereoselective Synthesis of β-N-Glycosyl Sulfonamides via Palladium-Catalyzed Glycosylation. Org. Lett 2018, 20, 3923–3927. [DOI] [PubMed] [Google Scholar]
- (13).Javorskis T; Orentas E Chemoselective Deprotection of Sulfonamides Under Acidic Conditions: Scope, Sulfonyl Group Migration, and Synthetic Applications. J. Org. Chem 2017, 82, 13423–13439. [DOI] [PubMed] [Google Scholar]
- (14).Carpino LA; Shroff H; Triolo SA; Mansour E–SME; Wenschuh H; Albericio F The 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl group (Pbf) as arginine side chain protectant. Tetrahedron Lett. 1993, 34, 7829–7832. [Google Scholar]
- (15).The relative stereochemistry of C2 and C3 hydroxyl groups are determined by coupling constant analysis. The larger coupling constant between C1 and C2 hydrogen (J = 9.0 Hz) indicates an anti-configuration, which reveal the OsO4 approaches from the convex face of the glycal 3g. For coupling constant analysis of a similar system, please see: Altona C; Haasnoot CAG Prediction of anti and gauche vicinal proton-proton coupling constants in carbohydrates: A simple additivity rule for pyranose rings. Org. Magn. Reson 1980, 13, 417. [Google Scholar]
- (16).(a) Tsuji J Catalytic Reactions via π-AllylPalladium complexes. Pure Appl Chem. 1982, 54, 197–206. [Google Scholar]; (b) Trost BM; Van Vranken DL Asymmetric Transition Metal-Catalyzed Allylic Alkylations. Chem. Rev 1996, 96, 395–422. [DOI] [PubMed] [Google Scholar]; (c) Trost BM; Crawley ML Asymmetric Transition-Metal-Catalyzed Allylic Alkylations: Applications in Total Synthesis. Chem. Rev 2003, 103, 2921–2944. [DOI] [PubMed] [Google Scholar]; (d) Trost BM; Thaisrivongs DA Strategy for Employing Unstabilized Nucleophiles in Palladium-Catalyzed Asymmetric Allylic Alkylations. J. Am. Chem. Soc 2008, 130, 14092–14093. [DOI] [PubMed] [Google Scholar]; (e) Trost BM; Zhang T; Sieber JD Catalytic asymmetric allylic alkylation employing heteroatom nucleophiles: a powerful method for C–X bond formation. Chem. Sci 2010, 1, 427–440. [Google Scholar]; (f) Trost BM; Krische MJ; Berl V; Grenzer EM Chemo-, Regio-, and Enantioselective Pd-Catalyzed Allylic Alkylation of Indolocarbazole Pro-aglycons. Org. Lett 2002, 4, 2005–2008. [DOI] [PubMed] [Google Scholar]; (g) Guppi SR; Zhou M-Q; O’Doherty GA De Novo Asymmetric Synthesis of Homoadenosine via a Palladium-Catalyzed N-Glycosylation. Org. Lett 2006, 8, 293–296. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Yang J; Mercer GJ; Nguyen HM Palladium-Catalyzed Glycal Imidate Rearrangement: Formation of α- and β-N-Glycosyl Trichloroacetamides. Org. Lett 2007, 9, 4231–4234. [DOI] [PubMed] [Google Scholar]; (i) Mercer GJ; Yang J; McKay MJ; Nguyen HM Palladium(II)-Catalyzed Rearrangement of Glycal Trichloroacetimi-dates: Application to the Stereoselective Synthesis of Glycosyl Ureas. J. Am. Chem. Soc 2008, 130, 11210–11218. [DOI] [PubMed] [Google Scholar]; (j) Zhang Q; Sun J; Zhu Y; Zhang F; Yu B An Efficient Approach to the Synthesis of Nucleosides: Gold(I)-Catalyzed N-Glycosylation of Pyrimidines and Purines with Glycosyl ortho-Alkynyl Benzoates. Angew. Chem., Int. Ed 2011, 50, 4933–4936. [DOI] [PubMed] [Google Scholar]; (k) Xiang S-H; He JX; Ma J-M; Liu X-W One-pot synthesis of β-N-glycosyl imidazole analogues via a palladium-catalysed decarboxylative allylatio. Chem. Commun 2014, 50, 4222–4224. [DOI] [PubMed] [Google Scholar]; (l) Ji L; Xiang S-H; Leng W-L; Le Mai Hoang K; Liu X-W Palladium-Catalyzed Glycosylation: Novel Synthetic Approach to Diverse N-Heterocyclic Glycosides. Org. Lett 2015, 17, 1357–1360. [DOI] [PubMed] [Google Scholar]
- (17).Martinelli J; Watson D; Freckmann D; Barder T; Buchwald SJ Palladium-Catalyzed Carbonylation Reactions of Aryl Bromides at Atmospheric Pressure: A General System Based on Xantphos. J. Org. Chem 2008, 73, 7102–7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).van Haaren R; Goubitz K; Fraanje J; van Strijdonck G; Oevering H; Coussens B; Reek J; Kamer P; van Leeuwen P An X-ray Study of the Effect of the Bite Angle of Chelating Ligands on the Geometry of Palladium(allyl) Complexes: Implications for the Regioselectivity in the Allylic Alkylation. Inorg. Chem 2001, 40, 3363–3372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.