

Abstract
Over the last three decades, polymeric micelles have emerged as a highly promising drug delivery platform for therapeutic compounds. Particularly, poorly soluble small molecules with high potency and significant toxicity were encapsulated in polymeric micelles. Polymeric micelles have shown improved pharmacokinetic profiles in preclinical animal models and enhanced efficacy with a superior safety profile for therapeutic drugs. Several polymeric micelle formulations have reached the clinical stage and are either in clinical trials or are approved for human use. This furthers interest in this field and underscores the need for additional learning of how to best design and apply these micellar carriers to improve the clinical outcomes of many drugs. In this review, we provide detailed information on polymeric micelles for the solubilization of poorly soluble small molecules in topics such as the design of block copolymers, experimental and theoretical analysis of drug encapsulation in polymeric micelles, pharmacokinetics of drugs in polymeric micelles, regulatory approval pathways of nanomedicines, and current outcomes from micelle formulations in clinical trials. We aim to describe the latest information on advanced analytical approaches for elucidating molecular interactions within the core of polymeric micelles for effective solubilization as well as for analyzing nanomedicine’s pharmacokinetic profiles. Taking into account the considerations described within, academic and industrial researchers can continue to elucidate novel interactions in polymeric micelles and capitalize on their potential as drug delivery vehicles to help improve therapeutic outcomes in systemic delivery.
Keywords: active pharmaceutical ingredient, block copolymer, clinical trials, drug, drug delivery, polymeric micelle, solubilization
Graphical Abstract

1. Introduction
Biocompatible polymers have been extensively employed in pharmaceutical science as excipients for traditional pharmaceutical formulations and more recently in nanomedicines for enhancing therapeutic outcomes of potent drugs [1, 2]. About three decades ago, micelles formed by amphiphilic block copolymers in aqueous solution were conceived as carriers for poorly soluble therapeutic compounds that were either covalently attached to polymer chains [3] or non-covalently incorporated in the micelles [4]. Since then, the applications of amphiphilic block copolymers in the design of polymeric micelles as therapeutics have been extensively studied [5–8]. A variety of novel block copolymers have been proposed to develop micelle-based delivery systems as potential nanomedicines for humans [9–12]. Many significant advances in polymeric micelles have been made to optimize the delivery of therapeutic molecules. Such advances have driven an increasing number of polymeric micelle drug formulations to enter clinical trials for regulatory approval [12, 13].
The design of block copolymers is intended to effectively encapsulate therapeutic compounds into polymeric micelles by various molecular interactions which results in the protection of the cargo from the external environment and improvements of the pharmacokinetic (PK) profile [14]. Ideal polymeric micelle formulations are expected to improve therapeutic outcomes of the encapsulated drug due to the functionalities of the formulation conferred by the polymer. The PK profile of the therapeutic compounds encapsulated in polymeric micelles differ from native compounds, because micelles are capable of releasing the cargo in a controlled manner from the core during systemic circulation. Structural features of polymeric micelles, such as the hydrophilic shell, help to avoid both unexpected drug loss from serum components and prevent opsonization by the complement system which typically result in the rapid clearance of drugs from systemic circulation [15, 16]. Based on these functionalities derived from the polymer formulations, the overall PK profile of the therapeutic compounds such as maximum systemic concentration, area under the curve (AUC), clearance (CL), volume of distribution (Vd), and biodistribution can be improved [13]. Furthermore, ideal polymeric micelles are expected to reduce the toxicity of the therapeutic compounds. The safety profile of compounds within polymeric micelles could improve therapeutic outcomes by expanding the therapeutic window. Side effects could be largely mitigated in both preclinical and clinical studies by using polymeric micelle formulations. This could also greatly affect the quality of life of the patients [11, 17].
Polymeric micelle systems exploit block copolymers for the delivery of therapeutic compounds such as small molecule drugs, proteins, and nucleic acids [2]. The desired physicochemical properties of block copolymers will vary based on the physicochemical properties of each therapeutic compound. Generally speaking, hydrophobic small molecules could be encapsulated in amphiphilic block copolymers which have both hydrophilic and hydrophobic blocks [4, 18, 19]. The amphiphilic block copolymers can spontaneously self-assemble into a core-shell polymeric micelle even in the absence of the therapeutic molecules. The drug molecules can be physically entrapped (“solubilized”) in the hydrophobic core of such micelles. Small, water insoluble drugs can also be chemically conjugated to the core-forming block of a block copolymer and the resulting amphiphilic block copolymeric conjugates then self-assemble into a polymeric micelle containing the conjugated drug in the core. Meanwhile, biopolymers usually require charged blocks to be encapsulated in polymeric micelles by electrostatic interactions [20, 21].
In this review we focus on the applications of the polymeric micelle technology for the delivery of poorly soluble small molecules. Among these applications, the exploitation of amphiphilic block copolymers as carriers for poorly soluble small molecules in polymeric micelles, has shown the potential to improve therapeutic outcomes, and several polymeric micelle drugs have reached the clinical stage of evaluation and regulatory approval for cancer treatment. For example, paclitaxel has been physically encapsulated in polymeric micelles to improve the systemic PK profile and alleviate drug-induced side effects, such as neurotoxicity, in both preclinical studies and clinical trials [6, 7, 9, 22]. One such formulation, Genexol® PM, has received regulatory approval in South Korea and other countries as a cancer therapeutic.
Many comprehensive reviews and collections on polymeric micelles have been published over last two decades which describe the general aspects of formulation and applications for the delivery of small drugs and biopolymers [2, 23], or that highlight specific delivery strategies, such oral drug delivery [24], biological response modifying effects of block copolymers [25] or field-responsive micelles [26, 27]. We refer the readers to these and other publications for additional insights and historical perspectives. In this review, we focus on the basic principles and current advances in polymeric micelle systems for the delivery of poorly soluble small molecules with a particular emphasis on the systemic drug delivery (Figure 1). The current state of the literature reflects that the physical entrapment of water insoluble small molecules into the micelle core is the most feasible path to clinical translation for polymeric micelles, so this is where we will focus our efforts.
Figure 1.

Schematic illustration of polymeric micelles for delivery of poorly soluble drugs
In the subsequent sections, we consider the current state of the polymeric micelle field from four different principal angles. In Section 2, we focus on the polymeric materials used for the manufacturing of polymeric micelles with a particular emphasis on the materials which have or could be developed for clinical use. The various possible blocks in block copolymers such as hydrophilic shell forming blocks with anti-fouling properties, hydrophobic blocks, and blocks with alternative interaction mechanisms are described along with their roles in the solubilization of poorly soluble small molecules.
In Section 3, we consider the pivotal problem of drug loading in polymeric micelles with the aim to maximize the payload of the drug and decrease relative amounts of a polymeric excipient used. To this end, we analyze the drug-polymer interactions within polymeric micelles focusing on the theory, modeling and experiment. Here, multidisciplinary approaches for investigating detailed molecular interactions between drugs and block copolymers are introduced to improve the understanding of the solubilization processes and to aid in the informed development of polymeric micelles for effective drug delivery. We also discuss in this section key innovations in the analysis of high loaded micelles, such as drug partitioning measurements, nuclear magnetic resonance (NMR) spectroscopy, small angle neutron scattering (SANS), and fluorescence analysis of host-guest interactions.
In Section 4, we focus on the highly complex problems of polymeric micelle drug PK and biodistribution that remains a constant subject for active research. Here we focus on key metrics for PK studies of nanoformulations, in particular those which relate to the tumor distribution of the polymeric micelle drugs. We discuss advanced experimental methods of PK analysis of polymeric micelle drugs as well as theoretical and modeling approaches. The role of the drug release characteristics along with the hydrodynamic size and morphology of polymeric micelles in the drug distribution to the disease site is thoroughly discussed. We also discuss the potential for the combination therapy with multiple drugs co-loaded in polymeric micelles from the standpoint of both improved PK and delivery as well as improved therapeutic efficacy. Last but not the least, this section focuses on the relationship between the drug loading and excipient derived toxicity in polymeric micelles as a foundation for successful clinical translation of the polymeric micelle drugs.
Finally, in Section 5 we focus on the preclinical and clinical translation of polymeric micelle products. Here we discuss various methods for polymeric micelle preparation from the standpoint of their scalability and translational potential. The discussion of the regulatory approval of nanomedicines, including polymeric micelles, is provided with the objective to assist academic and industrial scientists in considering regulatory approval challenges and opportunities throughout the formulation discovery and development process. Lastly, we discuss several key examples of polymeric micelles which have been translated successfully to the clinic. The concluding Section 6 focuses on future directions.
We would like to emphasize that polymeric micelle systems are special and present unique advantages over many other nanosized drug carrier systems. One issue with many nanoparticle carriers is that if they penetrate into tumors, or other sites of action, the drug release is slow, uncontrolled, or inefficient. Polymeric micelles are dynamic systems. Because of this, they release the drug to their target much easier than many more rigid, “solid” nanoparticle systems. On the other hand, due to their dynamic character, the micelles can lose drug on the way to the target. In light of this, the pharmaceutical development of these formulations must balance both drug loading and release to improve drug therapeutic indices by polymeric micelle delivery. These properties are governed by drug-polymer interactions as well as structural parameters of the block copolymers, which could be finely optimized. Therefore, this review has a particular emphasis on such parameters which have maximal influence over these properties as well as recent advances in the analysis of the complex interactions between drugs and block copolymers in these uniquely dynamic drug delivery systems.
2. Functionalities of polymeric micelles as a delivery platform for poorly soluble small molecules
Amphiphilic block copolymers self-assemble in aqueous media to form micelles that have hydrophilic shells and hydrophobic cores. The shell prevents aggregation and precipitation of the micelles while also protecting the therapeutic cargo. The core holds the micelle together and solubilizes poorly soluble small molecules. In general, diblock copolymers (A–B) or triblock copolymers (A–B–A) of hydrophilic (A) and hydrophobic (B) blocks are most often employed for the preparation of polymeric micelle formulations. However, B-blocks exhibiting interactions other than purely hydrophobic have recently been developed for the encapsulation of poorly soluble compounds [28]. These block copolymers exhibit very complex and interdependent interactions. For example, the work of Kozlov et al. showed that both hydrophobic and hydrophilic blocks participate in formation of the microenvironment for poorly soluble compounds in the micelles [29]. The Luxenhofer group has shown that hydrophilic blocks can play substantial role in drug polymer interaction especially in highly drug loaded polymeric micelles [30]. Not only does the structural composition of each block play a role, but the length of each block, and thus polymer molecular weight, are important as well. Additionally, the amount of drug loaded into the micelles can affect stability, morphology, and the size of the micelles in aqueous solution. The complex interdependency of block structure and block lengths make for highly tunable properties with unique capacities for drug solubilization. However, it also makes understanding these interactions and the intelligent design of block copolymers more challenging.
When considering biological interactions, the hydrophilic shell plays a critical role in the polymeric micelles. Utilizing hydrophilic blocks which have “anti-fouling” properties reduces the binding of serum components (serum proteins and complement system) and protects the encapsulated drug, thus avoiding the unexpected loss of the cargo during systemic circulation. To this end, polymeric micelles should be designed to minimize these interactions. Otherwise, polymeric micelles could be readily cleared from the body by plasma protein adsorption and/or complement activation leading to the removal of the entire micelle along with the drug within its core by the reticuloendothelial system (RES) [31, 32]. The RES removes immune complexes in healthy people and consists of phagocytic cells in circulation and tissues. To avoid this system, several hydrophilic blocks have been introduced into the structure of block copolymers to endow anti-fouling properties to the polymeric micelles (Table 1) [33, 34]. The functionalities of the hydrophilic shells were extensively studied and according to those studies, physicochemical properties of hydrophilic polymers such as molecular weight and surface density were closely related to the stability, systemic circulation time, and biodistribution of polymeric micelles in vivo [16, 35].
Table 1.
Hydrophilic polymers commonly used for constructing amphiphilic block copolymers.
| Polymer | Chemical structure | Synthesis | Properties and comments | Ref |
|---|---|---|---|---|
| PEG1 |
 
|
Living anionic ring-opening polymerization (ROP) of ethylene oxide | Most often used hydrophilic polymer with stealth property. Used in clinically approved nanoformulations including polymer micelle (Genexol®PM). Potential immunogenicity and accelerated blood clearance (ABC) phenomenon. The only shell-forming polymer that is used in clinically approved products as of today. | [40–43] |
| Poly(2-oxazoline)s |
 
|
Living cationic ring-opening polymerization (LCRP) of 2-oxazoline monomers | Both polymers are evaluated as PEG replacement PMeOx is more hydrophilic than PEG. | [22, 44–46] |
| Poly(sarcosine) |  |
Living polymerization of α-amino acid-N carboxyanhydrides | Evaluated as PEG replacement. Biodegradable. | [47–49] |
| Polysaccharides |
  
|
Enzymatic synthesis | Used as a component in block and graft copolymers. Highly variable molecular weight. Dextran has been used as excipient in clinically approved injectable products (FERAHEME®). Biodegradable. | [50–53] |
| Miscellaneous |
   
|
Atom transfer radical polymerization; reversible addition fragmentation chain transfer | Potential immunogenicity noted (PVP). Nonbiodegradable. | [57–64] |
polyethyleneglycol
methoxy-PEG
hydroxy-PEG
poly(2-methyl-2-oxazoline)
poly(2-ethyl-2-oxazoline)
poly(vinylpyrrolidone)
poly(N,N-dimethylacrylamide)
poly[N-(2-hydroxypropyl) methacrylamide]
poly(methyl methacrylate)
The hydrophobic block of block copolymers is intended to solubilize poorly soluble drugs in the core and control the release of the drug from the polymeric micelles [36–38]. Hydrophobic interactions between drugs and hydrophobic block of amphiphilic block copolymers are very well recognized as one principal factor in solubilizing the drugs in polymeric micelles. Such interactions help to retain the drug in the core and may retard the release rate of the drug to the external solution. Additional molecular interactions existing in the core, such as hydrogen bonding and pi-pi interactions, are no less significant as they can strengthen the molecular interactions between the polymer and the drug in the core [28, 39]. Many hydrophobic polymers have been synthesized and evaluated as core-forming blocks in polymeric micelles and show the capacity to solubilize poorly soluble drugs (Table 2).
Table 2.
Hydrophobic polymers commonly used for constructing amphiphilic block copolymers.
| Polymer | Chemical structure | Synthesis | Ref | |
|---|---|---|---|---|
| Polyethers |
 
|
Anionic ROP of respective alkylene oxides | PPO as a component of poloxamers – PEO-PPO-PEO triblock copolymers that are widely used in pharmaceutical formulations including SP1049C polymeric micelles in clinical trials. Commercially available (poloxamers). | [43, 54, 55] |
| Polyesters |
  
|
ROP of cyclic monomers | PLA is used clinically approved polymeric micelles drugs (Genexol® PM, Nanoxel® M). PLGA has been used as biodegradable surgical suture in clinic (Vicryl®). Biodegradable. | [40, 56–58] |
| Poly(amino acid)s |
 
|
Living polymerization of α-amino acid-N carboxyanhydrides | Biodegradable. Increased hydrophobicity by benzyl pendant group. | [59–61] |
| Poly(2-oxazoline)s |
|
LCRP of 2-oxazoline monomers | Versatile library of polymer structures, ultra-high loading capacity for several poorly-soluble drugs (ex. paclitaxel, etoposide) | [22, 45, 62] |
| Poly(2-oxazine)s |
 
|
LCRP of 2-oxazine monomers | Ultra-high loading capacity several poorly-soluble drugs (ex. curcumin) | [30, 46, 63–65] |
poly(propylene oxide)
poly(D,L-lactide)
poly(D,L-lactide-co-glycolide)
poly(ε-caprolactone)
poly(β-benzyl-l-aspartate)
poly(γ-benzyl-α, l-glutamate)
poly(2-isopropyl-2-oxazoline)
poly(2-n-propyl-2-oxazoline)
poly(2-n-butyl-2-oxazoline)
poly(2-n-propyl-oxazine)
poly(2-n-butyl-2-oxazine)
In this section, frequently employed blocks of block copolymers will be identified and their functionality in delivery platforms will be discussed.
2.1. Hydrophilic blocks and anti-fouling polymers in block copolymers
2.1.1. Polyethylene glycol
Polyethyleneglycol (PEG) (also known as poly(ethylene oxide) (PEO)) has been the most frequently employed hydrophilic, shell-forming block in polymeric micelles thus far due to its safety profile in humans and classification as “Generally Regarded as Safe” (GRAS) by the US Food and Drug Administration (USFDA). Low molecular weight PEG and PEG-conjugates of 20 kDa or less, have a low incidence of toxicity [66–68]. PEG has been the gold standard for anti-fouling polymers throughout nanomedicine. When PEG forms the hydrophilic shell of the polymeric micelles, its hydrophilicity and flexibility help the micelle avoid the adsorption of plasma proteins and opsonization processes which may cause the clearance of the cargo and polymeric micelles by the RES [35]. PEG with molecular weights ranging from 1 to 6 kDa are an ideal molecular weight for endowing nanoparticles with efficient anti-fouling properties and are frequently being employed to prepare block copolymers for drug delivery [34, 69, 70]. The anti-fouling mechanism of PEG has been comprehensively investigated in many studies. They reveal that primarily steric repulsion by PEG minimizes the adsorption of plasma components on polymeric micelles and the physical properties of PEG, such as sufficient flexibility and aqueous solubility, play a significant role on the anti-fouling properties as well [38]. Both the surface density and the MW of PEG are critical parameters when forming the shell. These both influence the conformation of PEG on the surface of the polymeric micelle where a “brush-like” conformation is preferred to sterically repel complement and plasma proteins [35, 71]. It was also reported that PEG conformation ultimately affects the circulation time and clearance of polymeric micelles in vivo [35]. That is, micelles with a higher PEG density and a brush-like conformation had increased AUC in vivo which is essential for improved efficacy of polymeric micelle formulations. PEG polymers have also been used in delivery involving mucosal barriers, because PEG can confer mucus penetrating properties. These mucus penetrating properties are conferred by the polarity of the molecule and overall net neutral charge which has been shown to enhance the penetration of nanomedicines through mucosal barriers [72, 73].
Synthesis of PEG is usually done by anionic ring opening polymerization (ROP) of ethylene oxide and this synthetic process generates well-defined PEG with a narrow molecular weight distribution [43, 74, 75]. The modification on the end group of PEG by appropriate chemical reagents (end-capping moiety) can expand the structural versatility of PEG [76]. The chemical versatility of the end group of PEG includes additional reactive moieties for ligand labeling which enables further conjugation with other species of polymers to prepare target-specific block copolymers.
Recently, the phenomenon of accelerated blood clearance (ABC) of PEG has gained a lot of attention due to its detrimental effects on the nanoparticle therapeutics which utilize PEG shielding [77]. It is well studied that systemic exposure to PEG may cause ABC in humans [78]. This phenomenon mainly arises from development of anti-PEG antibodies. ABC primarily occurs in human patients treated with PEGylated proteins as well as liposomal formulations coated with PEG [79]. In a Phase I study of refractory gout, patients were treated with a PEG-uricase therapy. About one third of the treatment group, who had previously been treated with pegylated therapies at some point, had previously developed anti-PEG antibodies in the body. This resulted in lower AUC’s and poor efficacy of the PEG-uricase treatment [80]. Another study by Sherman et al. reported that end-group of PEG contributed to the immune response to PEG-protein conjugates [81]. They compared the immunogenicity of mPEG-protein conjugates and HO-PEG-protein conjugates using enzyme-linked immunosorbent assays. It was found that the methoxy group of the mPEG-protein contributed to a significantly higher immune response than that of hydroxy group of HO-PEG. This study indicates that the end-group of PEG may affect the ABC phenomenon due to differential affinity to anti-PEG-antibodies.
The clinically approved doxorubicin liposome formulation, DOXIL®, has also shown the ability to induce the ABC phenomenon in human patients mainly due to the development of anti-PEG antibodies in the body after the initial treatment of DOXIL® [82]. The effect of hydrophilic chains of liposomal formulation on the genesis of ABC phenomenon was extensively studied by Dr. Szoka’s group [83]. They found that both PEG hydrophilic shells and PMeOx hydrophilic shells on liposomes induced the ABC phenomenon in rats after the initial dose of the same liposomes. Other hydrophilic polymers such as poly[N-(2-hydroxypropyl) methacrylamide] (HPMA), poly(vinylpyrrolidone) (PVP), poly(N,N-dimethylacrylamide) (PDMA) and poly(N-acryloyl morpholine) did not induce ABC, indicating these polymers may have superior anti-fouling properties. However, these polymers did not have as long of circulation times during the initial dose. In our view, the conclusion that these polymers may have superior anti-fouling properties requires more extensive verification.
Interestingly, in contrast to ABC phenomenon induced by liposomes with PEG shielding and PEGylated proteins, previous studies revealed that polymeric micelle formulations with PEG shielding did not induce significant ABC phenomenon in preclinical animal models as determined by the reduced anti-PEG antibody production. According to Shiraishi et al., anti-PEG antibodies did not affect the PK of PEG-b-poly(b-benzyl L-aspartate) (PEG-b-PBLA) polymeric micelles, while the PK profile of PEG-liposomes was marked by significantly decreased circulation times after repeated dosing [84]. Another study revealed that the hydrophobic block of PEG-conjugates was closely related to the binding of anti-PEG antibodies [85]. That is, proximal hydrophobic blocks are another key factor for the binding of PEG-specific anti-PEG antibodies to PEG moieties. Thus, polymeric micelle formulations with optimal PEG length and density on the surface may be less of a concern in promoting the ABC phenomenon than their liposomal counterparts. Nevertheless, the ABC phenomenon remains a concern for the use of PEGs and the field is actively searching for suitable replacements.
2.1.2. Hydrophilic Poly(2-oxazoline)s
Poly(2-oxazoline) (POx)-based block copolymers recently gained a lot of interest as novel biomaterials due to their biocompatibility and chemical versatility [44, 45]. Hydrophilic POx such as poly(2-methyl-2-oxazoline) (PMeOx) and poly(2-ethyl-2-oxazoline) (PEtOx) have shown the anti-fouling properties to avoid rapid clearance by the RES in vivo. These studies demonstrated the potential of these hydrophilic POx as stealth polymers [86]. POx can be readily synthesized via living cationic ring opening polymerization (LCRP) and recently block copolymers composed of POx have demonstrated scalable synthesis and chemical versatility [45].
As for the anti-fouling properties of POx, Zhang et al. reported that both PMeOx and PEtOx had extremely low protein adsorption and cell adhesion that is comparable to that of PEG-coating [87]. Interestingly, the modification on the end group of those polymers had minimal effect on the protein adsorption, unlike with PEG. However, the length of the polymer was significantly related to the anti-fouling properties with longer block lengths exhibiting better anti-fouling up until a certain point where the effect of additional block length was negligible. Another study done by Pidhatika et al. clarified long-term anti-fouling properties of PMeOx coatings [88]. They found that PMeOx had excellent anti-fouling properties comparable to PEG for short term protein exposures. However, for a long-term exposure to media, it was found that only PMeOx, but not PEG, could maintain the anti-fouling properties. This superiority of PMeOx was due to the lack of degradation of PMeOx in biological fluids. In the case of PEG, though it had anti-fouling properties at the early time points, it gradually degraded in biological fluids resulting in the loss of anti-fouling properties. This study, along with others, indicate that PMeOx may actually have superior anti-fouling properties to that of PEG [89]. Currently, only PEtOx is approved as food additives by the USFDA [90] and the safety profile of POx in humans, such as the biodegradation of POx, needs to be investigated for the further clinical development of POx-based micelle formulations. POx hydrophilic blocks have also demonstrated improved mucus penetrating properties which could be useful in the oral delivery of polymer micelles [91]. They showed that PMeOx had superior muco-penetrating properties, as measured by the diffusion coefficient in gastric mucus, compared to silica nanoparticles. PEtOx also showed some muco-penetrating enhancement, but less so than PMeOx. Overall, hydrophilic POx polymers, and especially PMeOx, have emerged as highly attractive anti-fouling stealth polymers which have the potential to replace PEG in these applications.
2.1.3. Other reported anti-fouling polymers
Several other hydrophilic polymers have been identified which show anti-fouling properties in preclinical models, suggesting their potential to be applied as shielding agents in polymeric micelles. Hydrophilic poly(amino acid)s were employed in amphiphilic block copolymers as anti-fouling agents to form the outer shell of the polymeric micelles. The biodegradability of poly(amino acid)s by endogenous proteases in vivo potentially confers the safety of these materials in the body [92]. However, this could mean that anti-fouling properties are not sustained for long durations like those seen with POx systems. The synthesis of hydrophilic poly(amino acid)s can be done via anionic ROP using N-carboxyanhydride of amino acids to generate poly(aspartic acid) (P(Asp)), poly(glutamic acid) (P(Glu)), and poly(sarcosine) [93]. Among hydrophilic poly(amino acid)s, poly(sarcosine) has shown effective anti-fouling properties in recent studies [49, 94, 95].
Polysaccharides such as dextran, heparin, chitosan, hyaluronic acid, and chondroitin sulfate have also shown anti-fouling properties and inhibited protein adsorption on the particle surface in biological fluids. Interestingly, some studies revealed that dextran as a shielding agent for nanoparticles displayed anti-fouling effects and prolonged circulation in animal models [96, 97]. A comprehensive and concise review on polysaccharides as anti-fouling agents was reported by Doh et al. and this review may provide useful information for researchers in selecting suitable polysaccharides with anti-fouling properties [50].
Several studies have investigated the anti-fouling properties of PVP. PVP can be synthesized via radical polymerization, and it has traditionally been used as an excipient in formulation design [98]. Allegedly, both the pyrrolidone moiety and amide groups in the side chain are closely related to the anti-fouling properties of PVP, but comprehensive mechanisms of these properties are still unknown [99].
Several other hydrophilic polymers such as PDMA, HPMA, and other zwitterionic polymers have been reported as anti-fouling macromolecules [100–102]. Those polymers are expected to be suitable for the development as block copolymers for the efficient delivery of poorly soluble small molecules in polymeric micelles formulations.
2.2. Hydrophobic polymers in block copolymers
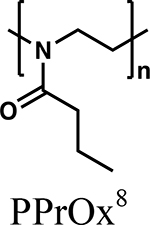
Hydrophobic segments of block copolymers play an essential role in solubilizing and encapsulating poorly soluble drugs in the core of polymeric micelles. The core of the polymeric micelles features a hydrophobic environment which allows for the entrapment of poorly soluble drugs via hydrophobic and potentially other types of interactions. This allows encapsulated drug to stably reside in the core during systemic circulation and gradually be released to the external environment. Hydrophobic segments of block copolymers can vary widely in their structure in order to effectively encapsulate poorly soluble drugs (Table 2). Commonly employed hydrophobic polymers are polyethers and polyesters. More recently, a variety of POx and poly(2-oxazine) (POzi) based polymers such as poly(2-n-butyl-2-oxazoline) (PBuOx) and poly(2-n-butyl-2-oxazine) (PBuOzi) have gained much attention due to their high loading for physically encapsulating drugs [45, 63, 65].
2.2.1. Polyethers
Polyethers have been used as the core-forming segment for encapsulating hydrophobic drugs. Generally, polyethers are synthesized via ring-opening anionic polymerization of alkenes to produce well-defined polymers with low polydispersity index (PDI) and molecular weight (MW) distributions [43, 54]. PPO and poly(butylene oxide) have shown hydrophobic properties and, when incorporated in block copolymers, have the capacity to solubilize hydrophobic drugs [4, 103]. PEO-PPO-PEO copolymers, which are called poloxamer (also known under the trademark of BASF formerly as Pluronic® and currently Kolliphor® P grade), are often exploited as block copolymers for solubilizing hydrophobic drugs and preparing polymeric micelle formulations [23]. In fact these block copolymers were the first used for the delivery of non-covalently incorporated drug in polymeric micelles. This concept was introduced by our group in the late 1980s and was initially termed “micellar microcontainer”, but is now widely known as a “micellar nanocontainer” [4]. In that study Pluronic® block copolymer micelles were used to solubilize a neuroleptic drug, haloperidol, and the micelles were conjugated with insulin or antibody to neurospecific antigens to deliver this neuroleptic to the brain. Subsequently Pluronic® block copolymers were extensively studied by our group and many others as materials for design of polymeric micelles for drug delivery. A notable property of select Pluronic® block copolymers to act as biological response modifiers to sensitize multidrug resistant (MDR) and cancer stem cells with respect to anticancer chemotherapeutics was widely reported and reviewed from mechanistic and translational points of view [25, 104]. Pluronic® block copolymers were also used in the first polymeric micelle drug formulation for cancer chemotherapy that entered clinical evaluation in early 2000s. Particularly, Pluronic® block copolymers were employed to manufacture the SP1049C formulation which is composed of an anticancer drug, doxorubicin, solubilized in the mixture of Pluronic® F127 and Pluronic® L61 [23]. This polymeric micelle drug is discussed in further detail in Section 5.3. Extensive review of the properties of poloxamers as micellar carriers for small molecule drugs as well as biological response modifiers can be found elsewhere [23, 25].
2.2.2. Polyesters
Polyesters are other exemplary hydrophobic polymer candidates which are frequently used in the formulation design of polymeric micelles. Synthesis of polyesters is commonly done by ring-opening polymerization of cyclic esters and this synthetic strategy is known to produce high molecular weight polyesters with narrow polydispersity [105]. One major advantage of using polyesters is their biodegradability [60]. The in vivo degradation process of the polyester backbone prevents the undesired accumulation of the polymer in the body, thus reducing the risk of chronic toxicity [60]. Examples of polyesters for solubilizing hydrophobic drugs are PCL, PDLLA and PLGA [106–108]. The block copolymers composed of the hydrophobic polyester block and hydrophilic block, such as PEG, were often utilized to formulate micelle systems. For example, micelle formulations prepared using PCL-b-PEG-b-PCL showed high loading up to 28 % of paclitaxel [109]. The clinically approved polymeric micelle formulation of paclitaxel Genexol® PM formulation exploits mPEG-b-PDLLA to solubilize paclitaxel and is discussed in greater detail in Section 5.3 [110].
2.2.3. Hydrophobic Poly(amino acid)s
Poly(amino acid) have often been used as hydrophobic core-forming blocks in amphiphilic block copolymers for solubilizing poorly-soluble drugs. Synthesis of poly-amino acids is usually done via living polymerization of α-amino acid N-carboxyanhydrides [61]. Commonly used hydrophobic poly(amino acid)s are poly(β-benzyl-l-aspartate) (PBLA) and poly(γ-benzyl-α, l-glutamate) (PBLG). According to Thambi et al., PEG-b-PBLG bearing the disulfide bond (PEG-SS-PBLG) could solubilize poorly soluble camptothecin and form micelles in solution [59]. The micelles displayed 20–125 nm size and the drug loading capacity was up to 12 %. PEG-b-PBLA block copolymer was employed to form polymeric micelles for the physical encapsulation of doxorubicin [2]. The micelle formulation exhibited 15–20% of doxorubicin loading and a 57–70 nm of size distribution.
2.2.3. Polyoxazolines and polyoxazines
POx and POzi block copolymers were recently introduced for drug delivery applications and have shown high potential as materials for polymeric micelle drug carriers [45, 63, 111]. The synthesis of POx and POzi can be achieved via LCRP process which results in strictly linear polymers of low molar mass distribution (PDI = Mw/Mn from 1.01–1.3) and defined degrees of polymerization [45, 86]. POx and POzi represent a versatile library of polymer structures. Depending on the 2-substitution of the 2-oxazoline or 2-oxazine monomers, the water-solubility of the resulting polymers range from highly hydrophilic MeOx or EtOx described above to highly hydrophobic, e.g. 2-nonyl-2-oxazoline (NOx) [112]. Such structural variability makes easily accessible an expanded library of POx- and POzi-based block copolymers that can be used to produce polymeric micelle formulations of structurally diverse, poorly soluble drugs [64].
Triblock A-B-A copolymers of POx consisting of hydrophobic PBuOx block with two flanking hydrophilic PMeOx blocks, PMeOx-b-PBuOx-b-PMeOx, have shown unprecedentedly high loading for many poorly soluble drugs [22, 45, 113, 114]. Our group, and others, have reported several polymeric micelle systems composed of POx-based block copolymers [22, 28, 63, 113–115]. We have screened potential hydrophobic drug candidates and found that many hydrophobic drugs can be efficiently solubilized in these POx systems with extremely high drug loading (sometimes approaching or even exceeding 50% by weight drug loading) [116]. For example, paclitaxel was extremely well-solubilized in POx up to a paclitaxel concentration of 40 mg/mL in aqueous solution to form well-defined spherical micelles with a size of less than 50 nm [22]. The maximum loading of paclitaxel in POx was up to 50 % which potentially minimizes the amounts of excipients in formulation design (Figure 2). Stability studies confirmed that the POx-paclitaxel polymeric micelles were stable in aqueous media for a month without any loss of paclitaxel. A number of other hydrophobic drugs such as etoposide, 3rd generation of taxanes, and vismodegib as well as multiple drug combinations were shown to be solubilized in the POx micelle system with high loading [113, 117, 118]. Due to its high drug loading capacity and safety profile, the POx system has drawn a lot of interest for use as a polymer carrier for drug delivery.
Figure 2.

Comparison of various paclitaxel (PTX) formulations that are either clinically approved (Abraxane and Taxol by USFDA, Genexol-PM by South Korea’s Ministry of Food and Drug Safety) or undergone clinical trials (NK105) with the POx/PTX polymeric micelle formulation. Taxol contains only about 1% wt. of active ingredient (m(PTX)max/m(total)), while Genexol-PM and NK105 have much higher drug loadings. The maximal paclitaxel concentration in solution (PTXmax) achieved with all four formulations is below 10 g/L, while POx/PTX can reach almost 50 g/L. Compared to Abraxane and POx-PTX, NK105 and Genexol-PM formulations are significantly diluted down for injection, so that final PTX concentrations ([PTX]inj) are well below 1 g/L [6, 7]. Of all compared PTX formulations, the novel POx/PTX polymeric micelle formulation exhibits the highest maximum tolerated dose (MTD) in mice. Reprinted with permission from [22] Copyright 2016, Elsevier.
Interestingly, it was recently reported that such high loading capacity of poorly soluble drugs in POx micelle was due to the structure of both the hydrophilic and hydrophobic blocks in POx triblock copolymers [119]. For example, PMeOx (which is more hydrophilic than PEG) was well-hydrated in the shell of highly drug-loaded POx micelles and had less interaction with loaded drug compared to PEtOx (which is similarly hydrophilic to PEG). With highly hydrophilic properties, PMeOx in the shell could support super high drug loading micelle formulation and stabilize the micelle structure in solution. Also, a variety of hydrophobic POx blocks have been used to produce micelles with different cores which show differential solubilization profiles with respect to various poorly soluble drugs [63]. For example, one study varied the side chain structure in the core forming blocks of POx and POzi A-B-A triblock polymers to determine the effect on drug loading [63]. The triblock copolymers composed of PMeOx-b-PBuOx-b-PMeOx and PMeOx-b-PBuOzi-b-PMeOx were exploited to prepare these polymeric micelle formulations. In particular, the solubility of the drug curcumin was compared between the two triblock polymers. This POzi system has an extra carbon in the hydrophobic block backbone compared to the PMeOx-b-PBuOx-b-PMeOx system, and this confers differential solubilizing capacity – the same drug, curcumin, is much less soluble in the POx only system than the POzi polymer. Meanwhile, paclitaxel was highly soluble in PMeOx-b-PBuOx-b-PMeOx but is less soluble its POzi analog [63].
Also, we have recently reported a POx based diblock copolymer which has a hydrophobic block consisting of a triazine ring and exhibits both hydrophobic interaction and likely pi-pi stacking capabilities [28]. In this work, first PMeOx-b-poly(2-methoxycarboxyethyl-2-oxazoline) is synthesized followed by the conversion of the methyl ester group to a triazine ring structure via the condensation of N,N-dimethylbiguanide. Prior to adding the ring structure, the polymer was unable to form micelles in aqueous solution. This polymer structure conferred some unique solubilization characteristics likely due increased modalities of drug-polymer interaction, such as hydrogen bonding and pi-pi stacking. These examples illustrate the breadth of chemistry and functionalization that can readily be performed with POx and POzi based systems for the rational design of polymeric micelle systems.
POx-based polymers have shown little degradation in biological fluids in the short-term, but it was reported that POx can be degraded in the long-term via oxidative degradation [45]. Although extensive body of work in preclinical models is available about the safety profile of PMeOx-b-PBuOx-b-PMeOx (see for example [22]), the safety profile analysis of other amphiphilic block copolymers containing various hydrophobic POx or POzi blocks in humans has not been conducted. This evaluation is needed before proceeding to clinical use in the future.
2.3. Stimuli-responsive block copolymers in polymeric micelle formulations
Polymers with stimuli-responsive properties are of interest as materials for polymeric micelle design due to their ability to modulate drug delivery by internal chemical or external physical stimuli. Upon exposure to either chemical stimulus (pH, hypoxia, redox, enzyme activity) or physical stimulus (light, temperature), the physicochemical properties of polymers, e.g solubility, within the micelles can be altered. This can be tailored to modulate the delivery functionality of polymeric micelles, such as their accumulation at disease sites or release rate of the drug cargo. This section introduces some stimuli-responsive polymers as components of amphiphilic block copolymers for delivery of poorly soluble drugs and discusses their principal functions in drug delivery.
Temperature-responsive hydrophilic polymers are often incorporated in block copolymers used for drug delivery. Such polymers are characterized by lower critical solution temperature (LCST) and/or upper critical solution temperature (UCST) which define the mode of modulation of the polymer solubility by temperature. Thus far, polymers with LCST have been preferably employed in temperature-responsive polymeric carriers for drug delivery. At temperatures below LCST such polymers are hydrophilic and water-soluble, but above LCST they dehydrate and transform to hydrophobic polymers that phase-separate from a solution. If such a polymer comprises the shell of a polymeric micelle that is stable in dispersion below the LCST, once the temperature exceeds the LCST (such as upon injection or trafficking to a hyperthermic site) the micelle precipitates and the drug could be rapidly released to the environment [120]. Due to this temperature-responsive property, polymeric micelles with shell-forming constituents exhibiting LCST behavior could be employed as carriers for therapeutic drugs in conjunction with local hyperthermia [121]. Commonly used polymers with LCST in the physiologically meaningful range include poly(N-isopropylacrylamide) [122], poly(N,N-dimethylacrylamide) [123] and their copolymers. The LCST values of such polymers are well-defined and can be fine-tuned by changing the copolymer composition to enable temperature response of the resulting polymeric micelles within a desired temperature range [124, 125].
For example, Sun et al. reported that hydrophilic poly(N-isopropylacrylamide) in amphiphilic block copolymers exhibited temperature-sensitive behavior that facilitated the release of a loaded drug (doxorubicin) from the polymeric micelles [126]. An in vitro cytotoxicity assay confirmed the temperature-sensitive micelle formulation showed enhanced cytotoxicity in MCF-7 cells above LCST (37°C) compared to that below LCST (20°C). Wang et al reported on a thermo-sensitive amphiphilic diblock copolymer, poly(N,N-isopropylacrylamide-co-N-hydroxymethylacrylamide)-b-PCL with an LCST of ~38°C [127]. The polymeric micelles of this copolymer loaded with doxorubicin released the drug in vitro in a temperature-dependent fashion. At 14 °C only 20% of doxorubicin was released during 200 h, while at 43 °C over 80% of the drug was released during the same time.
Additional useful temperature-responsive polymers with LCST behavior are poly(2-isopropyl-2-oxazoline) [128], elastin-like polypeptides (ELPs) [129], and some substituted polysaccharides [130]. ELPs are polypeptides composed of amino acids which exhibit elastin like properties that potentially induce LCST phase transition behavior. The LCST behavior of ELPs is dependent on the additional amino acid moiety among amino acid sequence as repeating unit. Frequently employed ELPs has polypeptide structure with valine (Val), proline (Pro), glycine (Gly), and additional amino acid which may determine the physicochemical properties of ELPs such as LCST and their assembly properties in solution.
Another important class of stimuli-responsive polymers are pH-sensitive polymers that were employed for targeted drug delivery to acidic compartments in the body. Physicochemical properties of such pH-sensitive polymers can be modulated by changes in the environmental pH enabling the release of the cargos at target sites. Endosomes and the tumor microenvironment where the local pH is slightly acidic are the targeted spaces for such pH-sensitive polymers. Several pH-sensitive polymers have been employed in polymeric micelle formulations including polycations such as poly(histidine) [131], poly(4-vinylpyridine) [132], poly(N,N-dimethylaminoethylmethacrylate) [133], poly(β-amino ester) (PBAE) [134], and polyanions, such as poly(acrylic acid) [135], poly(methacrylic acid) (PMAA) [136], and poly(sulfonamides) [137].
For example, the carboxylic groups in PMAA are ionized and charged at the physiologic pH 7.4 which renders the polymer soluble, while at acidic pH these groups protonate and the polymer becomes insoluble due to the presence of methyl groups in the backbone of the main chain [136]. Therefore, PMAA can be used as pH-sensitive shell-forming block in polymeric micelles that can enable precipitation of the micelle and release of the drug in the acidic environment. On the other hand, the imidazole groups in poly(histidine) are uncharged at the physiologic pH 7.4 and become protonated at acidic pH. Therefore, poly(histidine) can be used as a core-forming block in the polymeric micelles that is hydrophobic at the extracellular pH but becomes positively charged and soluble upon acidification, which can result in the local release of an incorporated drug [131].
Another class of pH-responsive polymers are degradable polymers such as poly(β-amino ester)s (PBAE) synthesized by Michael step-growth polymerization using diacrylates and amines [138]. Due to the tertiary amine groups in the polymer structure, PBAEs exhibit pH-sensitive behavior with the polymer being insoluble at neutral pH but degrading to soluble fragments in acidic and alkali environments. Such properties allow to employ PBAE as a core-forming segment for solubilizing poorly soluble drugs in PEG-b-PBAE polymeric micelles at physiological pH 7.4 [139]. In weakly acidic environments the PBAE protonates, the micelle core swells, and the drug is rapidly released. Due to the presence of the cationic charge, PBAE-based copolymers were also employed as carriers for nucleic acids [140]. Molecular diversity of diacrylates and amines greatly expands the library of PBAE-based copolymers available for delivery of drugs and nucleic acids [140–142].
2.4. Drug conjugates and complexes with block copolymers
The drug-polymer conjugates were introduced in the early work by Helmut Ringsdorf in 1970s to improve drug solubility, toxicity, and body biodistribution, which was followed by extensive studies on the development of drug-polymer conjugates [143, 144]. One problem often encountered with hydrophobic drugs conjugated to water-soluble polymers is that as the amount of drug conjugated increases the hydrophobicity of the conjugate also increased, resulting in its aggregation. This was the rationale for the proposal by Helmut Ringsdorf to use block copolymers in which one block is used as solubility enhancer – e.g. hydrophilic PEG and another as drug attachment scaffold that can be highly modified with drug [145]. In early 80-ies, Ringsdorf used PEG-polypeptide block copolymers with hydrophobic and cyclophosphamide-containing side groups, where upon conjugation of the drug, the drug conjugate block becomes hydrophobic and the entire block copolymer becomes amphiphilic resulting in drug-conjugate self-assembly into the polymeric micelles [143]. In these micelles, the core was formed by the conjugated block and shell from the PEG block. According to their approach, a drug is chemically conjugated to the core-forming block of the copolymer via a carefully designed pH- or enzyme-sensitive linker, that can be cleaved to release a drug in its active form within a cell. The appropriate choice of conjugating bond depends on specific applications.
This same concept was used by Kataoka and colleagues in a series of works leading to development of polymeric micelle NK911, which was evaluated clinically [146, 147]. The original approach developed by this group used doxorubicin conjugated to the poly(Asp) chain of PEG-b-P(Asp) block copolymer through an amide bond [3, 148]. However, the conjugated drug was not easily released from the micelle core, resulting in negligible drug activity; albeit, the toxicity to the body was also decreased and circulation time greatly increased [149]. Therefore, in the subsequent development, free doxorubicin was added in the formulation and solubilized in the hydrophobic core formed by PEG-b-poly(Asp)-doxorubicin conjugate utilizing the “like-dissolves-like” principle [150].
In subsequent studies, a different type of “conjugates” was developed using PEG-b-P(Asp) and PEG-b-P(Glu) – which can form a coordination complex with drugs containing transition metal complexes such as cis-dichlorodiammineplatinum(II) (cisplatin, CDDP), dichloro(1,2-diaminocyclohexane)platinum(II) (DACHPt) and cis-oxalato-(trans-l)-1,2-diaminocyclohexane-platinum(II) (oxaliplatin) [151]. In this case the micelle formation of the block copolymer is driven by complexation among the carboxylic acid groups on the polyacid block and the metal of the drug molecule. One example is CDDP-containing micelles formed by reacting PEG-b-poly(Asp) and CDDP [152]. The complex of PEG-b-P(Asp) and CDDP spontaneously self-assembled into polymeric micelles with a very narrow size distribution. The drug was released from the micelles via ligand exchange with chloride ions in biological milieu. A similar polymeric micelle formulation where CDDP was coordinated with PEG-b-P(Glu) [153] evolved into a clinically evaluated polymeric micelle drug, NC-6004 which is described below in Section 5.3. Kataoka’s group also reported on PEG-b-P(Glu) polymeric micelles containing another platinum drug DACHPt [154]. Similarly to CDDP, DACHPt was attached via coordination bonding of the platinum to the carboxylic groups of P(Glu) block and the active drug was released via ligand exchange of DACHPt with chloride ions in environment. The polymeric micelle drug NC-4016 based on the PEG-b-P(Glu) and DACHPt complex is undergoing clinical trials and is described in Section 5.3.
2.5. Unimolecular micelles and cross-linked micelles
The performance of polymeric micelles is intimately related to micelle stability, drug loading, release kinetics, circulation time, and biodistribution. The formation of polymeric micelles from amphiphilic block copolymers in solution is thermodynamically favorable when above the critical micelle concentration (CMC) of the amphiphilic macromolecules. Below the CMC, micelles in solution tend to dissociate and the loaded drug may be unexpectedly dispersed in the solution. Since polymeric micelles are significantly diluted upon administration into the body, classical amphiphilic block copolymers may disassemble after injection, which results in the loss of micelle functionalities. For these reasons, structurally or chemically modified polymeric carrier systems have been introduced aiming for the optimal drug release from the micelles. Such approaches are unimolecular micelles, core-crosslinked micelles, and shell-crosslinked micelles.
Unimolecular micelles are topologically similar to self-assembled micelles, but consist of single polymer molecules with covalently linked amphiphile block copolymer chains [155]. Dendrimers are commonly used as building blocks to prepare unimolecular micelles, because of their well-defined globular architecture, high-branching, and controlled surface functionality [156, 157]. To increase the loading of poorly soluble drugs the dendrimer core can be modified with a hydrophobic block, followed by the attachment of the hydrophilic chains. For example, Wang et al. reported on an amphiphilic 16-arm star block copolymer consisting of inner lipophilic PCL and outer PEG blocks [158]. The core of the polymer was a polyamidoamine (PAMAM) dendrimer of generation 2 with 16 terminal OH groups. These OH groups were used to initiate polymerization of ε-caprolactone to form PCL blocks and then the free ends of PCL were coupled with PEG chains. The micelle formulation from the resulting 16-arm star-block copolymer, stPCL-PEG16 exhibited high loading of a hydrophobic drug, etoposide up to 22% w/w and did not show toxicity on porcine kidney epithelial cells. However, it was pointed out that despite the star-block architecture the drug loaded micelles still represented aggregates of several unimolecular micelles assembled together due to relatively loose 16 PEG chain outer shell [158]. To increase the density of the PEG chains in the shell a similar design was employed to manufacture a 32-arm star-block copolymer using PAMAM dendrimer of generation 3 coupled with either PCL or poly(L-lactide) (PLLA) and then with PEG [159]. In this case, however, the loading with respect to the same drug, etoposide was much lower - 7.8 w/w for stPCL-PEG32 and 4.3 w/w % for stPLLA-PEG32. Generally speaking, loading of unimolecular star-block copolymer micelles with hydrophobic drugs is more challenging than that of linear amphiphilic block copolymers, probably, due to lack of conformational flexibility of polymeric chains covalently attached to the same structural node.
Chemical crosslinking approaches were also employed to improve the stability and circulation time of self-assembled polymeric micelles. Either the shell or core could be crosslinked, and the drug release could be modulated by cleavage of the crosslinks [160–163]. The resulting cross-linked micelles are, in essence, single molecules of nanoscale size that are stabile upon dilution, shear forces and environmental variations (e.g. changes in pH, ionic strength, solvents etc.) [155]. Various shell-crosslinked polymeric micelles were reported by several groups, showing the effect of shell-crosslinking on the drug release profile from the micelle formulations [164–167]. For example, Chang et al. reported a shell-crosslinkable poly(methyl methacrylate)-b-poly(N-isopropylacrylamide-co-N-acryloxysuccinimide) block copolymer. Shell-crosslinking of the polymeric micelle was done by the addition of ethylenediamine in the micelle solution. An in vitro release study showed a more sustained release of prednisolone acetate from the shell-crosslinked micelle compared to uncrosslinked micelles [168].
Bronich et al. reported polymeric micelles with cross-linked ionic cores prepared by using complexes of PEG-b-PMAA copolymer and divalent metal cations [169]. These complexes self-assemble into small spherical micelles with PEG shell and PMAA -cation complex core. Crosslinking of the core followed by removal of the divalent metal cations by dialysis in the presence of a chelating agent results in formation of a nanogel-like structure with covalently linked PMAA core and outer PEG shell. The core can be further loaded with various drugs that can interact with the carboxylic groups, such as CDDP or doxorubicin. As an example in a study by Kim et al. the cross-linking was done by using 1-(3-dimethylaminopropyl)-3-ethyl-carbodiimide hydrochloride and 1,2-ethylenediamine [170]. After removal of the divalent cations doxorubicin was incorporated into the core of such micelles via electrostatic and hydrophobic interactions, and the micelle exhibited high loading of the drug up to 50% wt/wt. In acidic solution, the protonation of carboxylic acid moieties on PMAA accelerated the release of doxorubicin. This design was used extensively in subsequent studies with a number of crosslinked core micelles prepared using either PEG-b-PMAA or PEG-b-poly(Glu) or other similar copolymers which in some cases were additionally modified with hydrophobic groups in the core-forming blocks to facilitate their self-assembly before cross-linking [171–175]. Various cross-linkers including biodegradable ones were used to facilitate loading and release of the drugs in such micelles. Notable examples of these studies include loading of the core of the core-crosslinked micelles with CDDP, DACHPt and other anti-cancer drugs and multiple drug combinations that then were used to treat cancer in animal models [176–178]. Increased blood circulation time and tumor distribution of drugs incorporated in such core-crosslinked micelles along with decreased toxicity and improved anti-tumor effects of the micellar drugs were reported. Successful targeted delivery of these drug-loaded micelles to the tumors using tumor-specific ligands attached to the micelle outer PEG chains were also described [179–181]. However, the clinical translation of the cross-linked micelle designs has not occurred yet, with one of the greatest challenges in our view being consistency of the cross-linking chemistry and characterization of the chemical composition including the number and spatial distribution of the crosslinks in the resulting crosslinked micelles.
3. Drug-polymer interactions within polymeric micelles: theory, modeling and experiment
The self-assembly of amphiphilic block copolymers composed of immiscible blocks elicits the formation of core-shell micelle architecture in aqueous media when the block copolymer concentration is above CMC [182]. Several factors may affect the size and morphology of micelles in solution. These factors include 1) structural parameters of the block copolymer, such as chemical structure of the repeating units in each block, molecular mass of the blocks, and their mass ratio, 2) block copolymer concentration, as well as 3) the environmental parameters, such as the temperature, ionic strength and/or pH for blocks containing ionizable groups (e.g. polyelectrolytes) [182]. The assembled micelles feature highly ordered macromolecular structure having segregated hydrophobic compartment in the core surrounded by a hydrophilic shell on the outer surface of the micelle which confines the overall micelle architecture. From a thermodynamic perspective, the self-assembly process is driven by the minimization of the interfacial free energy [14]. As a first approximation, 1) the hydrophobic segment collapses and aggregates, which decreases the contact area of this segment with the aqueous environment, and 2) the hydrophilic segment becomes hydrated and forms a shell, which further masks the core surface and reduces the interaction between the hydrophobic segment and water. In addition to the interfacial free energy, the micelle thermodynamics and the resulting micelle shapes are critically dependent on the steric repulsion of the hydrophilic chains in the shell and the stretching of the hydrophobic chains in the core [183].
Hydrophobic small molecules can be physically encapsulated (solubilized) in the core of the micelle during the self-assembly of amphiphilic block copolymers, which brings about the formation of polymeric micelles in aqueous solution. A traditional view is that the solubilization process primarily driven by hydrophobic interactions between the incorporated molecules and the hydrophobic domains of the micelles formed by segregated copolymer blocks in the micelle core. The molecular interactions between encapsulated drugs and hydrophobic blocks not only assist the formation of the micelle, but also further stabilize micelle structure in solution. Additional cohesive forces such as van der Waals forces, driven by the proximity of hydrophobic drug and hydrophobic segment of the polymer in the core, could lower the CMC of the micelle, resulting in further stabilization of the micelle structure upon dilution [184, 185]. Recent studies on the experimental analysis of polymeric micelles have shown that the drug-polymer compatibility was achieved via more complex molecular mechanisms of interaction than simple hydrophobic interactions [63, 65, 115]. Other molecular interactions such as hydrogen bonding or pi-pi interactions are known to affect CMC values of drug-loaded polymeric micelles and facilitate enhanced stability of the micelles [186]. In some cases, these interactions can involve not only the core-forming hydrophobic blocks but also the shell-forming hydrophilic blocks and selected drugs can be at least partially incorporated in the shell of the micelle [46, 65]. In other cases, the hydrophilic blocks were reported to interpenetrate into the core of the micelle and affect the micelle CMC as well as the partitioning of the drug between the micelle and the external milieu [29]. For the drug containing micelles, the molecular interactions between the solubilized drug and the micelle are also important parameters that can affect not only the stability of the micelle but also its size and morphology, thereby strongly influencing the biological performance of the polymeric micelles as drug delivery vehicles [114, 115].
The CMC and the partitioning of the drug between the micelle and the external milieu has long been considered a thermodynamic measure of the stability of the micelle as a drug carrier in equilibrium conditions [29, 187]. The extent of the partitioning is defined by the value of the partitioning coefficient, which depends on the drug solubility in the aqueous solution and drug-polymer interactions in the micelle core. Upon micellar drug dilution in water, the drug partitions itself in the external solution. Once the block copolymer concentration drops below the CMC the micelle disintegrates into single block copolymer molecules (“unimers”) releasing the remaining drug. In more complex biological environments, the various biological molecules present in these environments, such as serum proteins in blood, can bind the drug, thereby shifting the equilibrium towards drug release. Kinetic stability of the assembled micelles reveals the dynamic character of the micelles in aqueous media and its stability in solution over time [23, 182]. Upon dilution or external environmental changes, the dynamics among individual micelles, such as exchange of polymer chains and the merging/disruption of the micelle structure, determine the stability of the micelle structure over time [23, 182]. The dynamic distribution of the drugs between the polymeric micelles and various body compartments after administration of drug-containing micelles in the organism plays a pivotal role in the PK of the drug [188].
A theoretical understanding of the solubilization processes of poorly soluble drugs by amphiphilic block copolymers can be helpful to inform the design of novel drug delivery carriers. Computational approaches can facilitate formulation discovery and design by sparing loss of time and cost for experiments based on trial-and-error learning for drug solubilization in polymeric micelles. Various computational approaches have been proposed to predict the compatibility between the drug and polymer during the encapsulation process, such as using the solubility parameters (SPs) [189], Flory-Huggins interaction parameters [190], Molecular Dynamics (MD) [191], and quantitative structure property relationship (QSPR) [116] (Table 3). Experimental validation of the prediction data from the aforementioned approaches has been performed. In-depth physicochemical analysis of drug-loaded polymeric micelles by experimental approaches have revealed detailed molecular interactions between drug and polymer systems which form micelles. These interactions play important roles both during the self-assembly process and in the dynamic behavior of polymeric micelles in solution, such as drug release to the external milieu. Investigation of the molecular interactions provides explanations for the role of structural factors of both components in polymeric micelles. The interpretation from those investigations gives us comprehensive insight into the plausible molecular interactions during micelle formation and guidance in the development and intelligent design of polymeric micelle-based delivery systems. In this section, we will describe computational and experimental approaches which explain the drug solubilization process by polymeric micelles as well as showcase the recent progress in the characterization of polymeric micelles for the efficient design of polymeric micelle-based delivery systems.
Table 3:
Theoretical approaches for solubilization of poorly soluble drugs in polymeric micelles
| Approach | Advantage | Limitations | Examples and result |
|---|---|---|---|
| Prediction of miscibility of a drug and core-forming block based on similarity of their Hansen’s SPs estimated using GCMs | Allows for simple and rapid estimate of drug-polymer miscibility | Does not account for excluded volume, concentration of solutes, configuration and conformation of molecules and drug interactions with shell | Ellipticine with PEG-b-PCL and PEG-b-PDLLA. Hansen’s SPs predictions correlate with experimental drug loading and release [192] Sagopilone with PEG-b-PCL, PEG-b-PDLLA and PEG-b-PLLA. Hansen’s SPs are not predictive. Drug solubilization accompanied by supersaturation [193] Five drugs with eighteen POx and POzi-based triblock copolymers. Hansen SPs predicted solubilizing trends for a given drug among different copolymers. The prediction would not allow comparing different drugs with each other. [64] |
| Uses a classic Flory-Huggins solution theory for a binary mixture. Predicts miscibility of a drug and a block copolymer based on the Flory-Huggins interaction parameter χFH which is estimated using Hansen’s SPs. | Prediction miscibility of binary mixtures with accurate estimation of enthalpy changes. In many cases allows correct ranking of solubilization of different drugs in one polymer, or compares solubilization of one drug in different polymers | Does not account for polymer-solvent interactions, excluded volume effects, and geometry of molecules. Cannot distinguish between isomers that have identical chemical structures but different constitution and configuration. Underestimates polar and Coulomb interactions. | Eleven drugs with PEG-b-PCL. χFH ranks drug solubilization consistent with experimental data for a large drug set. [194] Five drugs with mPEG-b-PCL. χFH ranks drug solubilization for any one copolymer; not the dependence on PCL length [195] Eight drugs in PEG-b-poly(ε-caprolactone-co-trimethylene carbonate) micelles. χFH predicts trend in drug solubilization and effect of core forming block composition. Uses interaction parameters for core and shell blocks. [196] Doxorubicin with di-block copolymers of mPEG and modified PCL. χFH successfully predicts experimental drug solubilization depending on core block composition. [197] Cucurbitacin I and di-block copolymers of PEG and modified PCL. χFH successfully predicts experimental drug solubilization depending on core block composition. [198] Bicalutamide and di-block copolymers of mPEG and PLLA-based blocks. χFH successfully predicts experimental drug solubilization depending on core block composition. [199] Indomethacin with mPEG-b-poly(ε-decalactone) and mPEG-b-PCL. χFH gives opposite results for drug solubilization in poly(e-decalactone) vs PCL core, presumably, due to difference in core crystallinity. [200] Five drugs in eighteen POx and POzi-based triblock copolymer. χFH determined by different methods are not predictive of drugs solubilization. [64] |
| Computer simulation method for analyzing the physical movements of atoms and molecules. | Allows computing Hansen’s SPs, Flory-Huggins interaction parameters and free energy of mixing. Can successfully predict the solubility of the drugs in the micelles, localization of the drug in the micelle, size and morphology of the drug-loaded micelles, drug localization within the micelle. | Limitation on time span and the system size | Nimodipine, fenofibrate Cucurbitacin B and Cucurbitacin I in PEG-b-PCL diblock copolymer and branched multi-block copolymer. χFH values are consistent with experimental drug solubilization. MD simulations account for drug interaction with both PEG and PCL blocks and correctly predict binding of drugs with linear and multi-block copolymers. [201–203] Pyrene, nile red, and indomethacin with mPEG-b-PDLLA. χFH and free energy of mixing from MD simulations correctly predict the trend in the solubilization of drugs in the micelles. MD simulations correctly account for effects of block length and ratio. [204] Itraconazole with PEG-b-PLGA. MD simulation reveals that the drug localizes primarily at the interface, while the core of the micelle remains empty; explains relatively low loading of this drug. [205] Curcumin, paclitaxel and vitamin D3 with PEG-b-oligo(desaminotyrosyl-tyrosine octyl ester suberate)-b-PEG. χFH and free energy of mixing from MD simulations correctly predict a trend in drug solubilization. [206] Doxorubicin with poly{γ-2-[2-(2-methoxyethoxy)-ethoxy]-ethoxy-ε-caprolactone}-b-poly(γ-alkoxy-ε-caprolactone). MD simulations predict drug solubility for polymers differing in side chains structures. [207] Camptothecin with mPEG-b-PBAE. Predicts drug solubility_and provide information about the shape, size and morphological transitions in drug loaded micelles. Provides insight on in the drug release mechanism. [208] |
| Powerful model with sufficient amount of dataset for statistics can predict property such as drug loading in the micelle | Requires large dataset for model development | Doxorubicin with 15 star polymers of different architecture containing PCL, poly[(2-diethylamino)ethyl methacrylate] and poly(poly(ethyleneglycol)methacrylate) blocks. The QSPR approach was able to establish A quantitative relationship between the polymer architecture and drug loading established. [209, 210] Many drugs in several POx-based amphiphilic copolymers. Based on large data set, predicted loading of eight drugs in the micelles with 75% accuracy. [116] |
3.1. Theoretical and computational approaches
3.1.1. Hildebrand and Hansen solubility parameters
Two substances are mutually soluble when the free energy of their mixing (ΔGmix) is negative. The free energy is defined by the following Equation 1:
| (Equation 1) |
where ΔHmix is the enthalpy of mixing, ΔSmix is the entropy of mixing, and T is the absolute temperature.
According to the modern interpretation of the Hildebrand approach (for historical reference, please, see [211, 212]), the mixing enthalpy can be estimated from the Equation 2:
| (Equation 2) |
where ϕ1 and ϕ2 are the volume fractions of the drug and the polymer and δ1 and δ2 are the SPs of the drug and the polymer, respectively.
Hildebrand’s SPs are determined by the square root of the cohesive energy density (CED) that corresponds to the molecular self-interaction energies and is expressed in MPa1/2. This parameter is determined by Equation 3:
| (Equation 3) |
where δHIL is the Hildebrand SP, Hcoh is the cohesive enthalpy needed to infinitely separate a unit volume of molecules from each other, R is the universal gas constant, and V is the molar volume [213].
Equation 1 The entropy term −TΔSmix in this equation is negative and therefore the substances are miscible when the enthalpy term ΔHmix is negligible. Based on this the two components with similar SP values are predicted to be miscible. According to the Hildebrand’s SPs, two components are predicted to be miscible when the difference in SPs is less than 2 MPa1/2 [214, 215].
In order to predict the solubility of polymers in solvents and account for a broader range of molecular interactions, such as dissimilar patterns of polar and hydrogen-bonding interactions, Hansen has proposed to use a multi-dimensional SP expressed as the square root of a sum of dispersion, polar, and hydrogen-bonding components as shown in Equation 4:
| (Equation 4) |
where δT, is the Hansen’s SP, δd, δp, and δh are the partial dispersion, dipole-dipole and hydrogen-bonding components, each corresponding to their respective partial energies of cohesion [211].
In the case of Hansen’s SP, two components are predicted to be miscible when the difference in SPs is less than (or equal) to the interaction sphere radius defined by Equation 5:
| (Equation 5) |
where δd, δp, and δh are the partial SP of the components 1 and 2, and Ro is radius of interaction sphere in Hansen space.
The Hansen’s SPs of various substances can be determined experimentally or estimated using the group contribution method (GCM), which estimates CED as the sum of partial contributions from all functional and structural groups of a molecule (see [213] for further review). Several studies on drug and macromolecule compatibilities revealed that the enthalpies of mixing derived from the calculation of Hansen’s SP could predict the solubilization of hydrophobic drugs by amphiphilic block copolymers. Although, in some cases the predictions were not satisfactory (Table 3).
An exemplary study from the Allen group showcased the application of SPs as an indicator of polymer-drug compatibility in order to formulate the anticancer drug, ellipticine [192]. The physicochemical analysis of polymer–drug pairs was performed and the difference in total and partial SPs, as well as enthalpies of mixing, were compared using a range of biodegradable polymers. The partial and total SPs of polymer candidates and ellipticine were calculated using the GCM. Interestingly, the efficiency of drug loading in micelle formulations for PEG-b-PCL and PEG-b-PDLLA block copolymers was in good agreement with the prediction of drug compatibility with the core-forming polymer blocks using SPs. Also, along with the compatibility prediction, the release profile of ellipticine from each formulation was closely related to the SPs. Specifically, a compatible polymer, such as PCL, sustained the release of ellipticine for over 6 days, while a less compatible polymer, such as PDLLA, showed a faster release of ellipticine which was complete within 3 days. These results indicate that Hansen’s SPs could predict polymer–drug compatibility in the context of both drug solubilization and drug release in polymeric micelles.
A subsequent study by the Kissel group, using similar PEG-b-PCL, PEG-b-PDLLA and PEG-b-PLLA block copolymers and an anti-cancer drug, sagopilone, concluded to the contrary-that Hansen’s SPs were not in good agreement with the experimental data such as solubilization capacity and micelle stability [193]. In this case the drug loading was high and the micelles were supersaturated with the drug, which could have contributed to the lack of correlation with the SPs. Interestingly, there was a drastic difference in the drug solubilization in the micelles with PDLLA and PLLA core forming blocks. This was not reflected in the SPs which do not distinguish between stereoisomers. The PDLLA and PLLA also differed in their aggregation behavior and degree of crystallinity that most likely affected the placement of the drug in the core.
Recently Luxenhofer’s group published a comprehensive study involving five different drugs and eighteen A-B-A type amphiphilic block copolymers with hydrophilic PMeOx block (A) and hydrophobic POx- or POzi-based blocks (B) [64]. The Hansen’s SPs were calculated using GCMs as well as estimated experimentally by determining solubility of polymers in solvents with different polarity. The experimental approach was proven to be more complex and difficult to interpret because of the possibility of self-assembly of the block copolymers. The experimentally derived Hansen’s SPs poorly correlated with the solubility data. However, the SPs obtained using Hoftyzer−van Krevelen method, and to a lesser extent the computer-aided Yamamoto Molecule Break method, were able to classify the drug-polymer compatibility fairly well. However, the computational methods in some cases were misleading, especially in the case of non-solubilizable drugs. They also could not account for small changes in the chemical structure of the side chains of the polymers that had tremendous impact on polymer−drug compatibility. No differentiation between two different drugs would have been possible using these SPs only. Overall, the study suggested limited applicability of the Hansen’s SP approach for predicting drug solubilization in POx- and POzi-based triblock copolymer systems.
3.1.2. Flory-Huggins solution theory
The classic Flory-Huggins solution theory is based on the lattice model to describe the thermodynamic behavior of polymer solutions. According to this theory, the free energy of mixing of a solvent and a polymer at a constant temperature and pressure is expressed by Equation 6:
| (Equation 6) |
where ϕ1 and ϕ2 are the volume fractions of the solvent and the polymer, n1 and n2 are the number of moles of the solvent and the polymer, and χFH is the Flory-Huggins interaction parameter.
In the classic Flory-Huggins theory the interaction parameter χFH is a scalar quantity that accounts for the enthalpy of mixing of a solvent and a polymer and is expressed by Equation 7:
| (Equation 7) |
where δ1 and δ2 are the SPs of the solvent and the polymer, and V1 is the molar volume of the solvent [216, 217].
According to the Flory-Huggins theory, two components are predicted to be miscible if χFH is less than 0.5, or phase separated if χFH > 0.5. The χFH values can be determined by estimating SP values for each component as described above in 3.1.1.
Several studies were reported that related experimental results and theoretical estimates using classic Flory-Huggins theory for drug solubilization in polymeric micelles (Table 3). These publications suggest that the interaction parameters in some cases were able to correctly predict the trends in solubilization of different drugs in a block copolymer micelle or rank the solubility of a drug in different micelles. In most of these studies the χFH values were estimated using the partial Hansen’s SP calculated by GCM.
One of the earliest works in this area by Glen Kwon’s group reported on eleven drugs in polymeric micelles of PEG-b-PCL [194]. They estimated the Flory-Huggins parameter for drugPCL interactions and concluded that as the value of this parameter decreased the solubility of the drug in the micelles increased. Then they prepared a series of prodrug derivatives of geldanamycin with varying alkyl chain lengths in an attempt to lower χFH and thereby improve the solubilization in the micelle. This approach was shown to be successful as the prodrugs with the lower interaction parameters indeed demonstrated enhanced solubilization. Similar observations were reported by Helen Burt’s group who examined five poorly soluble drugs (etoposide, paclitaxel, plumbagin, curcumin, and indomethacin) in polymeric micelles of mPEG-b-PCL [195]. They also described inverse correlations between the drug solubilization and χFH for any single copolymer. However, this study also revealed a limitation of the classic Flory-Huggins approach for the polymeric micelles. That is, for each single drug the experimentally measured partitioning between the micelle and external solution depended on the PCL block length, which was not reflected in the estimated χFH.
Nevertheless, several other studies compared the solubilization of model drugs in polymeric micelles formed by various PCL- and PLA-based copolymers using experimental and theoretical approaches. Latere Dwan’Isa et al. reported on eight poorly soluble drugs (carbamazepine, cimetidine, furosemide, hydrocortisone, indomethacin, ketoconazole, ketoprofen, risperidone) in PEG-b-poly(ε-caprolactone-co-trimethylene carbonate) micelles [196]. An interesting aspect of this work is that to predict the drug-polymer compatibility they took into account interaction parameters for both hydrophilic and hydrophobic blocks, de facto suggesting that the drug could interact not only with the core but also with the shell. The resulting rankings agreed reasonably well with the experimentally measured drug solubilities in the micelles. For the block copolymers with varying composition of the core-forming block the solubility of a single drug (furosemide) positively correlated with the content of the trimethylene carbonate co-monomer in this block. This also was consistent with the drug-polymer miscibility prediction based on χFH. Yan et al. estimated the drug core compatibility for doxorubicin in the polymeric micelles of mPEG-b-PCL and mPEG-b-poly[(ε-caprolactone-co-γ-(carbamic acid benzylester)-ε-caprolactone] [197]. They reported that the Flory-Huggins parameters could predict differences in drug solubilization and controlled release between these micelle systems. Mahmud et al. examined solubilization and release of cucurbitacin I in PEG-b-PCL, PEG-b-poly(R-benzylcarboxylate-ε-caprolactone) and PEG-b-poly(R-cholesteryl carboxylate-ε-caprolactone) micelles [198]. They also found that the interaction parameter correctly predicted the trend of the drug loading. However, in this case the release profiles of the drug from the micelle were not predictable. Rather, the release of the drug depended on the core viscosity which likely controls the drug diffusion from the micelle. Danquah et al. compared the solubilization of bicalutamide in polymeric micelles formed by PLLA-based copolymers [199]. The drug solubilization in mPEG-b-poly(carbonate-co-lactide), containing additional carbonate groups in the core-forming block, exceeded that in mPEG-b-PLLA, which was again consistent with the prediction based on the Flory-Huggins interaction parameter. However, both Mahmud et al. [198] and Danquah et al. [199] noted the that the numerical values of estimated χFH substantially exceeded 0.5 even for the drug-polymer pairs that displayed good solubilization. The lack of consistency with the classic Flory-Huggins theory which uses, χFH < 0.5, for polymer-solute miscibility was accounted for by non-random chain distribution within the micelle and specific molecular interactions formed or destroyed upon incorporation of the drug in the polymeric micelle [199]. Likewise, it was pointed out that the use of the 0.5 as a miscibility cutoff is not suitable in the cases when Coulombic or hydrogen bonding interactions are involved [198]. As a result, in most cases the classic theory χFH can only be used for polymer-drug interactions ranking to reveal possible trends in drug solubilization and in some cases it can be false.
Indeed, several authors suggested limitations of the Flory-Huggins approach for predicting the solubility of the drugs in polymeric micelles. For example, Kakde et al. pointed out the discrepancy of the prediction using χFH estimates and experimental data for indomethacin in mPEG-b-poly(ε-decalactone) and mPEG-b-PCL micelles [200]. The χFH values estimated in this work predicted that the drug solubility in poly(ε-decalactone) core was less than that in PCL core, which contradicted the experimental data. The erroneous prediction was attributed to the decreased crystallinity of the poly(ε-decalactone) core compared to PCL that was not accounted for in the classic Flory-Huggins approach. An extensive study using five drugs and eighteen various POx- and POzi-based block copolymers by Lubtow et al. reported that the Flory−Huggins interaction parameters estimated using various GCMs did not correctly estimate the experimental drug solubilization [64]. As discussed in the previous section, they also concluded that the Hansen SP values could better predict some trends in identifying good and poor solubilizers among the block copolymers.
Overall, further advancement in the application of the Flory-Huggins theory in polymeric micelle field is needed to correctly predict the solubilization of diverse drugs in a wider variety of block copolymers. The classic method fails to take into consideration the geometry of the molecules involved, the excluded volume interactions that are especially prevalent for long-chain copolymers, and cannot distinguish between isomers that have identical chemical structures but different constitution and configuration [201]. It also tends to underestimate polar and coulomb interactions, and does not provide a straightforward approach for accounting for the drug interactions with both core- and shell-forming block.
3.1.3. Molecular dynamics
Computational approaches, such as MD, have been extensively utilized to investigate the molecular interactions in polymeric micelles and obtain microscopic insights into the solubilization of small drug molecules in the polymeric micelles [218]. MD is based on application of Newton’s second law for the computation of the interactions among molecules to trace the successive molecular motions or conformational changes of the components in the solution [219]. In the case of polymeric micelles, MD simulations of the interactions between hydrophobic drug and block copolymers in a given model are computed to generate the next successive conformations. This can simulate the process of self-assembly during drug loading in the micelle core [218]. The time scale required for these simulations is usually on the order of microseconds or longer, which often makes fully atomistic simulations impossible, or at the very least extremely expensive and impractical with the currently available computational power. Therefore, coarse-grained MD simulations are more frequently exploited, in which the number of the degrees of freedom is reduced in order to simplify and expedite the simulations [218]. For example, in the dissipative particle dynamics (DPD), a coarse-grained methodology commonly used for simulating the dynamic and rheological properties of fluids, several atoms or repeat units are grouped together and presented by a single bead [208]. Similar to MD simulation, time evolution in DPD is governed by Newton’s equation of motion.
Several studies have investigated the drug loading and molecular interactions within polymeric micelles using the MD modeling approach (Table 3). In contrast to the SP approach, the MD simulations are able to account for specific factors, such as hydrogen bonding or spatial distribution of hydrophobic drugs in the micelle core [203]. Therefore, the combination of Flory-Huggins theory and MD could produce more reliable predictions of the drug-polymer compatibility than the interaction parameter calculated from SP alone. Moreover, the MD simulations can also account for drug interactions with both shell and core forming blocks as well for the interactions of the blocks with each other. For a mixture of a drug D containing volume fractions of a drug D (ϕD) and a diblock copolymer A-B (ϕAB=ϕA + ϕB) the free energy of mixing can be determined by Equation 8:
| (Equation 8) |
where rAB is the sum of the ratios of the molar volumes of each A and B to D, and χeff is the effective interaction parameter [204]. This effective interaction parameter is defined using the volume fractions of blocks A (fA) and B (fB) and the binary interaction parameters χAD, χBD and χAB related by Equation 9:
| (Equation 9) |
Patel et al. successfully performed a series of MD modeling studies to elucidate the essential factors in the solubilization of hydrophobic drugs (fenofibrate, nimodipine, cucurbitacin B and cucurbitacin I) in mPEG-b-PCL micelles [201–203]. They found that Flory-Huggins interaction parameters computed by MD simulations could successfully predict the experimental solubility of the drugs in the micelles, whereas those calculated by the GCM deviate from the experimental observations. Their MD simulations accounted for the drug interactions with both PEG and PCL blocks. They predicted that the increase in the PCL block length should enhance solubilization of the drugs due to additional polar interactions and hydrogen bond formation between a drug and PCL. Moreover, the simulations suggested differential localization of drugs within a micelle depending on the drug and block copolymer ratio. For example, nimodipine at high drug loading has higher solubility in PCL and localizes deeper within the core, whereas fenofibrate clusters around both PCL and PEG. The MD simulations also predicted the effects of matching molecular architecture of the drug molecule and polymer chain. A branched multi-block copolymer with three PCL blocks attached to the same PEG chain was predicted to be a better solubilizer for cucurbitacin [203]. Due to an even distribution of the hydrogen bond donors and acceptors, the molecules could form a greater number of hydrogen bonds with branching PCL chains compared to a linear PEG-b-PCL. An opposite trend in solubilization was predicted for nimodipine and fenofibrate that have the hydrogen bond acceptors only and could not bind effectively with the branched multiblock copolymer, although no experimental evidence was provided to validate these predictions in this study.
More recently Erlebach et al. applied MD simulation to elucidate the polymer–drug compatibility in mPEG-b-PDLLA micelles [204]. To facilitate the computation, they obtained Flory−Huggins interaction parameters from the MD simulations that avoided explicit consideration of the actual copolymer chains. The predictions from those Flory−Huggins parameters correctly accounted for the effects of the block length and the PEG:PDLLA ratio and were in reasonable agreement with the experimental data for the encapsulation of several hydrophobic molecules (pyrene, Nile red, and indomethacin). MD simulation was also used to understand a relatively low loading of itraconazole in the PEG-b-PLGA polymeric micelles [205]. The results demonstrated that the drug loading primarily occurred at the water-polymer interfaces, while the core of the micelle tends to remain empty. MD simulation could also rank order drugs by their loading in polymeric micelles in a good agreement with experimental data. For example, Costache et al. used a combination of MD and docking calculations which predict preferred orientation of a drug to a polymer for ranking three drugs (curcumin, paclitaxel and vitamin D3) by the energies of binding of with PEG-b-oligo(desaminotyrosyl-tyrosine octyl ester suberate)-b-PEG micelles [206]. Furthermore, MD simulations can predict drug solubility for polymers differing in side chain structure. Hao et al. successfully used MD simulations to predict particle size and drug (doxorubicin) loading in the micelles of various substituted poly{γ−2-[2-(2-methoxyethoxy)ethoxy]ethoxy-ε-caprolactone}-b-poly(γ-alkoxy-ε-caprolactone) [207].
In addition to the drug loading characteristics the MD simulations could provide information about the shape, size and morphological transitions in drug loaded polymeric micelles. For example, the MD and DPD simulations were integrated to investigate the micellization of the pH-sensitive amphiphilic block copolymer, mPEG-b-PBAE [208]. This multi-scale simulation suggests that the PBAE block upon protonation undergoes the transition from hydrophobic to hydrophilic. This is accompanied by a transition of the shape of the polymeric aggregates from spherical to disk-like micelles and finally to vesicles, as dictated by the counterbalance of free energies for the formation of shell, interface, and core. The loading of a drug (camptothecin) in such aggregates predicted by the Flory-Huggins parameter and MD simulation was in good agreement with the experimental values. The drug was loaded into both hydrophobic core and core interface with hydrophilic shell loading. Similar to literature reports, high loading could induce a morphology transition from micelles to vesicles. The simulations also provided insight in the drug release and suggested that camptothecin was released from the micelles and/or vesicles upon protonation of the PBAE block.
Overall, these studies elucidated the molecular interactions occurring in the core of the drug-containing micelles and suggested that MD simulation can be useful for analyzing some important trends of drug solubilization in polymeric micelle systems.
3.1.4. Quantitative structure property relationship
MD approaches are often inappropriate for large data sets commonly encountered in polymeric micelles drug formulation research where the MD simulations could require enormous time, cost, and computational power [218]. Instead, statistical approaches such as QSPR can be exploited for the prediction of polymer–drug compatibility. Several promising studies were published recently that conducted QSPR modeling for the prediction of drug solubilization in polymeric micelles. QSPR modeling is based on the statistical analysis of the data sets and has been frequently used in the field of medicinal chemistry and chemical toxicology for the prediction of efficacy and/or toxicity of small molecules [220]. Wu et al. reported the development of a series of QSPR models to assess the loading of doxorubicin in polymeric micelles using the genetic function approximation algorithm [209, 210]. In this study the polymeric micelles were formed by either four-arm or six-arm star polymers consisting of PCL, poly[(2-diethylamino)ethyl methacrylate], and poly(poly(ethyleneglycol)methacrylate) blocks. The QSPR approach was able to establish a quantitative relationship between the polymer structure and drug loading.
To maximize the quality of prediction by QSPR, a large data set is required. Recently, Alves et al. have shown possibility of generalizing the use of QSPR for cheminformatics-driven discovery of polymeric micelle formulations across multiple drugs to predict their solubilization in PMeOx-b-PBuOx-b-PMeOx tri-block copolymer micelles (Figure 3) [116]. A total of 41 hydrophobic compounds were tested at various drug concentrations either individually or in combination with each other, which produced over four hundred data points reporting various drug solubilities. The study demonstrated that the computational QSPR modeling could predict the solubility of water-insoluble drugs with a high accuracy of up to 75 %. These models were employed for virtual screening of drug libraries, and eight drugs predicted to have either good or poor solubilization in these polymeric micelles were selected. Three putative positives, as well as three putative negative hits, were confirmed experimentally. The success of this computer-aided strategy suggests its broad utility for predicting drug compatibility in a given micelle system. Further work in this QSPR modeling involving adding more polymers to the database could allow for the informed selection of a given polymer to solubilize a desired drug molecule.
Figure 3.

Study design of cheminformatics-driven discovery of polymeric micelle formulations for poorly soluble drugs. From [116]. Reprinted with permission from AAAS.
3.2. Experimental approaches
Many experimental approaches have been employed for the physicochemical characterization of block copolymer micellization, structure, and morphology [221]. Generally, the polymeric micelle stability, size and size distribution, along with the drug loading and drug release profiles are considered key properties. These properties of the polymeric micelle-based drug delivery systems affect their performance in vivo. The combination of several physicochemical techniques is normally used to determine the relevant parameters of drug-loaded polymeric micelles. Typically, (a) the zeta potential, z-average size, and polydispersity of polymeric micelle particle over time is determined by dynamic light scattering; (b) the particle number-average size, size distribution, and concentration by nanoparticles tracking analysis; and (c) the particle morphology by transmission electron microscopy (TEM), cryo-TEM, and/or atomic force microscopy. Size exclusion chromatography and gel permeation chromatography are also often used to determine MW and PDI of the polymers. NMR spectroscopy can be used to confirm polymer structure as well as monitor reaction progress and determine block lengths of block copolymers. The drug-polymer interactions and partitioning are sometimes characterized by fluorescence- or ultraviolet spectroscopy [29, 30]. The drug concentration present in polymeric micelle dispersion is determined by high performance liquid chromatography, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry, or similar analytical techniques. The polymeric micelle stability after preparation and storage in aqueous dispersions are commonly assessed for over two weeks by measuring the particle size and polydispersity index, drug concentration, and particle concentration [22]. Also, the drug release profiles in vitro are studied by dialysis under “sink conditions”, i.e. against external solutions containing excess of drug-binding molecules, such as serum albumins. The retention of the drug in polymeric micelles in the presence of the serum proteins in vitro is characterized by the adsorption column chromatography [22].
In this section, we would like to focus mainly on those approaches that are applied to drug-polymer interactions in polymeric micelle formulations. We will not cover the more traditional physicochemical characterization mentioned in the previous paragraph which is commonplace in the field. A more-fine, molecular-level characterization of the drug and polymer interactions, as well as the short- and long-range order of the drug and polymer chain distribution within the micelle are also needed to understand the size, morphology, and stability of drug-containing polymeric micelles in aqueous dispersions. Such studies can update the simplistic representation of polymeric micelles as a simple core-shell structure with strictly segregated polymer chains and drug being exclusively localized in the micelle core. It is necessary to explore the microstructures within the drug-containing polymeric micelles that potentially govern the solubilization capacity as well as the drug distribution in vivo. Several such studies describing these intricacies are described below.
3.2.1. Interactions with core and shell-forming blocks in drug partitioning
Studying of drug partitioning between the polymeric micelles and external solution has been one approach to probe drug-polymer interactions and determine the thermodynamic characteristics of such interactions. Generally, the partitioning of the drug between the micelle and external solution is determined by the partition coefficient, P determined by Equation 10:
| (Equation 10) |
where Cm and Cw are the concentrations of the drug in the micelle and in the water, respectively.
The P value is a general thermodynamic characteristic depending on all types of interactions existing between the drug and polymer in the micelles and drug and solvent in the external solution. It represents a measure of the free energy of transfer from the solution to the micelle, ΔGmw, as presented by Equation 11:
| (Equation 11) |
The partitioning coefficients have been determined for many drugs in polymeric micelles (for example, poloxamers) [29]. By comparing the partitioning coefficients between different polymeric micelles for a given drug, one can probe differences in the thermodynamic contributions of drug interactions with these micelles. The work by Kozlov et al. determined the incremental contributions of the free energy of the transfer of methylene group from an aqueous environment into the micellar hydrophobic core for a significant number of poloxamers (triblock copolymers consisting of PEO-b-PPO-b-PEO) that differ in the PEO and PPO block lengths (Figure 4). This was accomplished by measuring the partitioning coefficients for a homologous series of alkylated fluorescein probes with different lengths of alkyl substituents and determining the partial contribution of the free energy per one methylene group. Typically, the free energy of transfer from aqueous to a water-immiscible organic solvent of a methylene group is −3.1 kJ/mol. Surprisingly, for the poloxamer polymeric micelles this value was less suggesting that the core environment was less hydrophobic than that of organic solvent and the free energy of transfer strongly depended on both hydrophobic PPO and hydrophilic PEO length. This implies that both types of blocks contributed to the microenvironment of the methylene group in the polymeric micelle cores. In other words, the probe molecule in the micelle was coming in contact with both hydrophilic and hydrophobic blocks in the core. The more hydrophobic copolymers, P103 and P123, having long PPO and short PEO blocks had values similar to that of an immiscible organic solvent. As the length of PEO chain increased the apparent penetration of PEO chain in the core also increased and the core environment became less hydrophobic. The authors detailed that an increase in the PEO length allowed increased distribution of ethylene oxide units and water into the micellar core, decreasing its hydrophobicity and lowering the partitioning coefficient [29]. In contrast, as the length of the PPO chain increased the core was becoming more hydrophobic due to a greater share of propylene oxide units in the core. Using alkylated fluorescein probes with varying lengths of alkyl substituents the “size” of the core was also probed as the break point in the incremental dependence of the partitioning coefficient (−ΔGmw) on the alkyl substituent length [29].
Figure 4.

(A) Partitioning coefficients of CH3−(CH2)N−Flu probes vs the number of methylene groups in the alkyl radical obtained for P103, P105, and F108. (B) Schematic presentation of Pluronic micelle using spherical lattice model described in [222–225]. Hydrophobic PO units (black filled circles) localize in the central part of the micelle, while hydrophilic EO units (black empty circles) and water molecules (empty cells) fill external layers. Parts a−c show incorporation of a solute (probe) having alkyl radicals of varying length (gray filled circles) and a polar fluorescent group (gray empty circle). Note unfavorable contacts between hydrophobic groups of the solute and water molecules or EO units in the case of the probes with shorter radicals. Reprinted with permission from [29] Copyright 2009, American Chemical Society.
The study also pointed out that the log(P) of the drug in a given polymer was strongly correlated to log(CMC) suggesting that similar types of interactions are guiding the partitioning of the hydrophobic drug and self-assembly of poloxamer block copolymers into a micelle. The limitation of this approach was that the partition coefficient probe measurement was done at very low drug loading when the molecules incorporated in the micelles did not perturb strongly the polymer chains arrangement within the micelle structure. For the highly drug-loaded polymeric micelles, different techniques are needed to characterize the drug-polymer interactions.
3.2.2. Drug-polymer interactions in polymeric micelles by NMR spectroscopy
Solid-state NMR (ssNMR) spectroscopy can be a valuable experimental approach to study drug-containing polymeric micelles since the chemical shifts and the respective peak widths depend on the local environment and interactions between the drug and polymer molecules. Advanced NMR techniques have been employed for the characterization of drug and polymer interactions within the polymeric micelles. Callari et al. showcased the utility of 1D and 2D ssNMR to investigate the correlation between drug loading and size of polymeric micelles [226]. They used a diblock copolymer composed of polymerized fructose methacrylate as a hydrophilic shell-forming block and PMAA as a core-forming block which was conjugated with a platinum drug (dichloroplatinum;1,10-phenanthroline) via coordination bonds. They compared micelles with varied drug loading and from ssNMR analysis concluded that as the drug loading increased the chain mobility and swelling in the core and shell of the micelle decreased. They further related these observations to the cytotoxicity of the higher and lower loaded micelles in the cells [226]. The lower loaded micelle had greater uptake and in vitro cytotoxicity, presumably due to the softer micellar structure facilitating interactions between the micelles and the cells.
Another study using ssNMR by Pöppler et al. investigated polymeric micelles with various drug loading by physical entrapment (Figure 5) [65]. They used amphiphilic A-B-A triblock copolymer PMeOx-b-poly(2-n-propyl-2-oxazine)-b-PMeOx (PMeOx-b-PPrOzi-b-PMeOx) and curcumin as a model hydrophobic drug. Interestingly, ssNMR analysis of the changes in the chemical shifts and cross-peaks in 2D correlation revealed that the degree of curcumin loading in polymeric micelle affected the localization of the drug in the micelle. At a low loading, the drug primarily accumulated in the core of the micelle where it interacted with the hydrophobic PPrOzi block via hydrogen bonding between phenolic OH group of curcumin and the amide group in the polymer backbone. At higher loading, there was an increased interaction of the drug with the carbonyl-carbon of the hydrophilic PMeOx blocks.
Figure 5.

Schematic model of the structural changes of the polymeric micelles upon loading with curcumin based on the solid-state NMR data and complementary insights. For each loading stage, the additionally occurring interaction site is depicted. Reproduced with permission from [65].
Haider et al. followed up on this previous work and examined drug-polymer interactions in polymeric micelles formed by several A-B-A triblock copolymers, that had the same core forming block but different hydrophilic blocks - PMeOx-b-PPrOzi-b-PMeOx, PMeOx-b-PPrOzi-b-PEtOx and PEtOx-b-PPrOzi-b-PEtOx [46]. The drug (curcumin) loading was extremely high when more hydrophilic PMeOx was used as the shell forming block in the polymeric micelles. Such micelles were also highly stable in solution. On the contrary, PEtOx was much less efficient as a hydrophilic shell and the respective micelles did not solubilize as much curcumin and were less stable at higher loading. The micelles with the mixed PEtOx and PMeOx shell displayed an intermediate result. The authors further used several physicochemical methods to demonstrate the interactions of the drug with the polymers. They confirmed their previous observation that at low drug loading the drug is predominantly localized within the hydrophobic core. As the drug loading in the polymeric micelle increased the interactions of the hydrophilic blocks with the drug became evident. Notably, they observed drastic differences in drug interaction with PMeOx and PEtOx. 1H-NMR spectroscopy revealed that the latter displayed much greater propensity for interacting with the drug at the lower loadings, which is possibly reducing the colloidal stability of the drug loaded micelles by shrinking the shell. This study suggests a significant role for drug interactions with the hydrophilic block in achieving extremely-high drug loadings. Overall, 1D ssNMR is able to probe the polymer-drug interactions of super high loaded polymeric micelles.
However, not all intermolecular interactions with these amorphous polymer-drug micelles can be easily probed using 1D ssNMR. The inclusion of highly complex molecules, such as paclitaxel, which has 51 individual protons, can significantly complicate the analysis. To this end, Grune et al. have recently utilized 14N-1H Heteronuclear Multiple Quantum Coherence (HMQC) ssNMR to show the interactions of the PMeOx-b-PBuOx-b-PMeOx triblock copolymer and paclitaxel [227]. Following the work of Callari et al. and Pöppler et al., they found additional ways in which the loading of drugs in the polymeric micelle affect the intermolecular interactions. In the HMQC ssNMR they can visualize the cross-peak interactions between nitrogens and hydrogens [227]. At a low 10:2 (weight polymer:weight paclitaxel) loading, all cross peaks occur in the aliphatic region of the 1H-NMR spectrum indicating that primarily interactions between aliphatic hydrogens on the polymer and the nitrogens on the polymer backbone are occurring. Due to the micellar structure, these interactions are most likely intermolecular in nature (i.e. not within the same unimer). At a higher loading of 10:4, additional peaks on the HMQC can be seen which are in the 4.5–8.0 ppm range on the 1H-NMR spectrum. These peaks indicate that as paclitaxel loading increases, there is an increased interaction between polymer nitrogens and hydrogens involved in the aromatic ring systems and hydrogen bonding of paclitaxel [227]. The signal at the negative ppm shift on the nitrogen NMR spectrum continued to increase with increased paclitaxel loading indicating increased interactions with the polymer amides. Therefore, 2D HMQC ssNMR provides additional structural insights, and in combination with 1D NMR, SANS, MD, and other advanced characterization techniques, yields a better understanding of drug-polymer interactions.
3.2.3. Microstructure of polymeric micelles by small angle neutron scattering
SANS is an experimental method to probe materials structure at the nanometer to micrometer scale using elastic neutron scattering at small angle of scattering. This method has been previously used to reveal the microstructure of the core and shell of drug-free polymeric micelles [228] and more recently extended to the thorough characterization of drug-loaded micelles. Schultz et al. reported on the morphological change of micelle structure upon the incorporation of paclitaxel in PMeOx-b-PBuOx-b-PMeOx polymeric micelles [115]. In the absence of the drug, this particular block copolymer formed worm-like micelles as shown in atomic force microscopy and cryo-TEM with an effective hydrodynamic diameter of about 200 nm. As the drug was loaded in the micelles at some critical point (>8% paclitaxel loading w/w), the micelles underwent a structural transition from worm-like micelles to small and uniform spherical micelles of about 45–50 nm. Further physicochemical analysis of these paclitaxel loaded polymeric micelles by SANS revealed that the micelle core represents a raspberry-like sphere morphology with a diameter of 4 to 6 nm. This was interpreted as the presence in the core of multiple paclitaxel-rich domains (raspberry nodules) which increased in size from about 2.4 to 3.4 nm as the drug loading increased. This was the first study to show the microstructures in polymeric micelles, which could possibly assist ultra-high drug-loading of POx-based polymeric micelles.
The Luxenhofer group recently followed with a similar analysis using the SANS technique which was extended to polymeric micelles containing a different drug, curcumin [46]. In this case a broader set of POx- and POzi-based triblock copolymers was used. Consistent with the NMR study discussed in the previous section, this drug revealed differential localization depending on its loading amount in the micelles. As the drug loading amount increased, the SANS data suggests that curcumin initially located in the micelle core shifted to the periphery forming an inner “shell” within the micelles. These data clearly show that a better understanding of the inner morphology of the drug-loaded micelles can be achieved using the SANS technique.
3.2.4. Host-guest interactions by fluorescence analysis
Fluorescence spectroscopy analysis can yield valuable insights into drug-polymer interactions in polymeric micelles in the case of drugs with fluorophore groups which are sensitive to the microenvironment. For example, Luxenhofer and colleagues have utilized curcumin as a means for investigating host-guest interactions in polymeric micelles. Curcumin is a very hydrophobic drug with fluorescent properties, such as peak positions and quantum yield, which are highly dependent on the local microenvironment [30]. Major changes in curcumin peak absorption position were observed when this drug was encapsulated into POx- and POzi-based micelle systems. The absorption band at 345 nm completely disappeared when encapsulated in both polymers which may be due to hydrogen bonding between the keto and enol groups of curcumin and the carbonyl groups on the polymer, which indicates that hydrogen bonding plays a key role in curcumin solubilization. Additionally, a shift in the absorbance maximum from 432 nm to 414 nm was observed as the concentration of curcumin in the POzi-based polymeric micelle increased indicating drug localization in a less polar microenvironment and the further exclusion of water from the core with increased curcumin loading.
Interestingly, as the curcumin loading increased, the micelle size as measured by dynamic light scattering first decreased and then increased [30]. Possibly, at low loading both curcumin and water are present in the micelle core. As curcumin accumulates the size decreases as water is excluded from the core, and then the size proceeds to increase due to the continuing accumulation of the curcumin in the core.
Additionally, the quantum yield of curcumin was able to elucidate additional properties of the polymer-drug interactions [30]. At the same level of curcumin absorbance (same curcumin concentration), a higher fluorescence intensity was observed in the POx polymer compared to the POzi polymer. This must mean that the fluorescence quantum yield is higher in the POx polymer due to a decrease in non-radiative decay pathways. They hypothesize that this is due to the decreased mobility of the POx polymer when compared to the POzi. The POzi has an additional methylene unit in the hydrophobic block background which confers an added degree of flexibility and enables alternative non-radiative decay pathways for excited state curcumin. This indicates that the added degree of flexibility may play a critical role in the differential solubilization capacity of POzi with regard to curcumin.
Additionally, they used fluorescence to probe the dynamic behavior of these micelles [30]. The addition of powder polymer to the polymeric micelle formulations altered the Polymer/Drug ratio without changing the concentration of the curcumin. After this addition, in both the POx and POzi polymers, the curcumin fluorescence increased indicating the mobility and dynamic exchange/behavior of these polymer systems. The work of the Luxenhofer lab in this regard shows the powerful nature of using curcumin and other microenvironment sensitive fluorophores for probing host-guest interactions in polymeric micelles.
3.2.5. In vitro drug release analysis
One essential step in the physicochemical characterization of polymeric micelle systems is the in vitro drug release studies. Utilizing these studies, one can visualize general release kinetics from a particular formulation or gain insight into the role of different drug-polymer interaction mechanisms occurring within the polymeric micelle formulation. These experiments are often performed using dialysis under sink conditions. Sink conditions ensure that complete dissolution of the drug from the polymeric micelle system is feasible. Literature varies on the definition of sink conditions, but in general the value is around 5 times the volume necessary for saturation of the drug in the system [229]. That is, the drug must be released in a volume of liquid which is 5 times greater than that which is needed to solubilize the total amount of drug in the system. Performing these experiments under sink conditions is essential for reliable data.
There are two issues with drug dissolution and sink conditions only addresses one of them. Sink conditions address the potential for saturation of the drug in the system, but not for the potential of saturation of drug in the dosage form. Water must penetrate into the polymeric micelle system and solubilize the drug for it to be released into the surrounding media. Thus, the drug dissolution into the water solubilized phase in the micelle can be a rate limiting step for the overall release. However, this same kind of limitation exists during in vivo circulation, so the use of sink conditions for in vitro release is still a good initial assessment of drug release behavior [229].
Additionally, these in vitro release experiments can be performed in either buffers, such as PBS, or in a serum solution which better mimics the in vivo environment. Studies performed in serum will often show quicker release profiles as hydrophobic drugs can directly transfer from the micelle to the serum protein molecules and the latter can serve as carrier for the drug to the external solution. Experiments in both conditions are important and give a more complete picture of polymeric micelle drug release behavior. To establish sink conditions for very poorly soluble drugs in the dialysis experiment serum albumin is added sometimes to the external solution to avoid the use of excessively large volumes.
A study by He et al. demonstrated how one could probe the distribution of hydrophobic drugs between the micellar fraction and serum bound fraction [22]. They incubated POx block copolymers with serum solutions for 1 and 4 hours to mimic what the polymeric micelles might experience in vivo. This was followed by the separation of the samples on a reversed phase solid phase extraction columns. The column could effectively bind the serum albumin (and associated drug) as well what little free, unbound drug was in the system. Micelle bound drug was quickly eluted from the column. An acid/methanol wash could then release the rest of the drug from the column. Using this technique, they were able to show that about 83% of paclitaxel was in the micellar fraction of the solution. The commercial Taxol formulation only showed 20% of paclitaxel being eluted in this fraction. This technique can be used to effectively probe how micelles alter drug distribution in the body.
As discussed above (Section 3.1.1) Allen group used Hansen’s SPs to predict whether or not a given drug, ellipticine, was compatible with varying block copolymer systems including PEG-b-PCL and PEG-b-PDLLA [192]. They then performed in vitro drug release studies. The polymer with the lowest enthalpy of mixing (most negative and thermodynamically favorable), PEG-b-PCL showed the strongest retention of the drug when compared to the PEG-b-PDLLA system. The release rate was also dependent on the drug : polymer loading ratio. The higher loading ratios of 1:4 showed slower release rates than the 1:10 ratio. This was attributed to the “like dissolves like” principle as more drug present in the micelle core creates a more suitable and stable environment for the drug to exist in. In another study, Hwang et. al. synthesized an A-B diblock copolymer with a triazine ring based hydrophobic B block and PMeOx A block [28]. They compared the drug release of paclitaxel and bruceantin from the micelles of this copolymer to their release from a PMeOx-b-PBuOx-b-PMeOx triblock copolymer micelles. While the rate of release of bruceantin from both types of the micelles was approximately the same, the rate of release of paclitaxel was drastically different. Their work showed that over 24 hours paclitaxel was not completely released from the PBuOx polymer while it was completely released from the triazine ring-based polymer. While limited in scope, this study shows that in vitro release can give some insights into the differential affinity between drugs and various polymeric micelle core structures.
A study by the Kataoka group showed the development of pH sensitive drug release from PEG-b-P(aspartate hydrazone adriamycin) [147]. This kind of technology can improve drug targeting abilities by only allowing drug release from the micelles under certain external stimuli. Other groups have explored this concept as well [230, 231]. These studies highlight the need to explore the in vitro drug release of formulations in different pH conditions. For example, the tumor microenvironment is relatively acidic compared to that of circulating blood. Analysis of in vitro drug release in lower pH’s can aid in the optimization of pH-sensitive formulations and limit the number of costly in vivo studies by ruling out certain formulations prior to in vivo analysis. Other common stimuli which are used to trigger drug release are temperature and local redox environment [27, 231]. We refer the readers to the following reviews for details on the design and implementation of stimuli sensitive polymeric micelles [232–234].
4. Drug loading, pharmacokinetics and distribution of polymeric micelles
Polymeric micelles have shown the unique potential to deliver critically important drugs with high drug loadings thereby minimizing excipient use and improving overall drug PK and therapeutic indices. This has led to many formulations going into clinical trial. There are many intertwined factors which influence polymeric micelle behavior in vivo and the pharmacodynamic response. This complex interplay between polymeric micelle physicochemical properties and PK behavior is not well understood. As a whole, in nanomedicine PK behavior is complex, and it is even more complex for polymeric micelles due to their unique, dynamic nature. Many factors influence this PK behavior such as drug loading, particle size, morphology, and the presence of multiple therapeutic agents. Each of these can uniquely affect polymeric micelle PK and add additional layers and considerations to PK modeling. In this section, we will not consider polymeric micelle-cell interactions which effect internalization. Rather, we will cover fundamental considerations for approaching and understanding the PK of polymeric micelles. With this information in-hand, we hope the reader can better design and execute PK experiments to expedite the progress of promising formulations in the pre-clinical stage.
4.1. Drug loading and excipient derived toxicity in polymeric micelles
The physicochemical properties of polymeric micelle products are closely related to the success of novel polymeric micelles in clinical trials. These properties must be well analyzed to validate the characteristics of the polymeric micelle products in the form of a certificate of the analysis to identify unexpected inconsistencies during the manufacturing process [221, 235].
One critical parameter is the loading efficiency (LE) which represents the portion of the drug entrapped in the micelles during solubilization process:
| (Equation 12) |
It is important for formulation to maintain a high LE to ensure that the formulation process is economical and the drug loss is minimized.
Another parameter is the loading capacity (LC) of the drug within the polymeric micelle which is defined as:
| (Equation 13) |
The ratio of drug to excipient can be one of the significant factors for successful polymeric micelle products as therapeutics. High LC of polymeric micelles is desirable as it reduces the amount of excipient being used in the formulation, thus minimizing undesired toxicity derived from the excipients.
Excipient derived toxicity has often occurred in patients and was a dose limiting factor in clinical treatments. For example, the Taxol® formulation of paclitaxel consists of Cremophor EL and anhydrous ethanol [236]. Though Taxol® has shown anti-cancer efficacy in human patients and is still approved for humans, these excipients cause toxicity and limit the ability to dose Taxol® [237]. Patients must often be pre-treated with a corticosteroid before being administered Taxol® to prevent these hypersensitivity reactions. Due to the biocompatible nature of some block copolymers, polymeric micelles that encapsulate paclitaxel have shown reduced excipient-derived toxicity. For instance, it was shown that the Cremophor EL-free Genexol® PM formulation of paclitaxel (approved in South Korea and several other countries) was much safer than Taxol® in cancer patients (390 and 200 mg/m2 doses approved, respectively) and there were much less concern of hypersensitivity reactions compared to those that can be observed during Taxol® treatment [11]. Some other paclitaxel polymeric micelle formulations, such NK105, were also reported to have lower toxicity in clinical trials compared to Taxol® [238].
Our group has reported on the ultra-high LC of paclitaxel in POx micelles which resulted in superior safety of the drug in preclinical animal models. POx formulation could encapsulate paclitaxel up to 4:5 weight ratio of drug and excipient, resulting in LC of ~45%. Maximum tolerated dose (MTD) of paclitaxel in mouse was about 7.5-fold higher in POx micelle (150 mg/kg) compared to that of Taxol (20 mg/kg) [22]. Also, according to Alves et al., POx micelles could solubilize many poorly soluble small molecules with high LC (Table 4) [116]. Another paper out of our lab showed decreased liver and kidney toxicity in animals treated at the MTD dose of POx-Paclitaxel versus the clinically approved Abraxane and Taxol formulations. The POx formulation showed no indications for organ toxicity. Additionally, the activation of the complement system, which could lead to hypersensitivity reactions and premature clearance of the formulation by macrophages, was evaluated. The Taxol formulation showed a significantly higher activation of the complement pathway when compared to the POx formulation. This is partly due to the significantly decreased amount of excipient necessary to deliver the given PTX dose. This could also be due to an increased biocompatibility and/or decreased immunoactivation in response to POx polymer [118]. This study indicates that both the biocompatibility of excipients and high LC are required to minimize excipient-derived toxicity. A high LC also increases the amount of drug per micelle, which can increase the amount of drug internalized into cells [114]. This is especially true if the internalization processes become saturated so only a given number of micelles can be taken up per unit time. It has also been noted that as polymer concentrations increase, this can inhibit the caveolae mediated endocytosis at least in the case of certain poloxamers with sufficiently long PPO blocks [239]. For these reasons, it is critical to maximize LC in polymeric micelle formulations.
Table 4.
Examples of active pharmaceutical ingredients with improved solubility in PMeOx-b-PBuOx-b-PMeOx micelles ranked by LE and LC. (Modified from Ref.[116]. Reprinted with permission from AAAS).
| Compound | Aqueous solubility (mg/mL)a | logPa | Solubility in POx micelle (10 mg/mL POx) | Fold increased | LE % (mean) | LC % (mean) |
|---|---|---|---|---|---|---|
| ABT-263 | 0.000212 | 7.77 | 8.00 | 37736 | 100.0 | 44.4 |
| Podophyllotoxin | 0.114 | 1.5 | 7.62 | 67 | 95.2 | 43.2 |
| Etoposide | 0.1b | 0.60c | 7.34 | 73 | 91.8 | 42.3 |
| Simvastatin | 0.0122 | 4.68c | 6.98 | 572 | 87.2 | 41.1 |
| Efavirenz | 0.00855 | 4.6c | 6.90 | 807 | 86.2 | 40.8 |
| Cisplatin prodrug (C6) | insoluble | - | 6.78 | - | 84.8 | 40.4 |
| VE-822 | 0.0401 | 3.1 | 6.42 | 160 | 80.2 | 26.7 |
| Paclitaxel | 0.00556 | 3c | 5.05 | 908 | 63.1 | 30.4a |
| AZD5363 | insoluble | 1.31 | 4.98 | - | 62.3 | 33.3 |
| Cisplatin prodrug (C4) | insoluble | - | 4.68 | - | 58.5 | 31.9 |
| Teniposide | 0.0598 | 1.24c | 4.58 | 77 | 57.2 | 31.4 |
| Cisplatin prodrug (C10) | insoluble | - | 4.30 | - | 53.7 | 23.9 |
| AZD8055 | 0.241 | 2.87 | 4.06 | 17 | 50.8 | 28.9 |
| Docetaxel | 0.0127 | 2.4 | 3.71 | 292 | 46.4 | 19.0 |
| Rutin | 0.125 | 0.15 | 3.61 | 29 | 45.1 | 26.5 |
Data obtained from drugbank (https://www.drugbank.ca/) and predicted by either ALOGPS (http://www.vcclab.org/lab/alogps/) or ChemAxon (https://chemaxon.com/) when available, in other cases we refer to compounds “insoluble” if their solubility is less than 0.1 mg/ml.
US patent (US4772589A).
Experimental data obtained from drugbank (https://www.drugbank.ca/), The experimental value is from [22] and is different from that listed in [116], which was a mean of several values obtained for different conditions of micelle preparation.
High LC is also important for combination polymeric micelle formulations that contain two or more drugs in a single micelle for combination therapy. For the effective and safe delivery of drug combinations, the combination drugs should be well-solubilized in the polymeric micelle so that the amount of the excipient and injection volume can be minimized for parenteral injection [114, 188, 240]. The effects of the co-loading of drug combinations in the same polymeric micelle carriers are further considered below.
4.2. Pharmacokinetic analysis of polymeric micelle drugs
Knowing the PK profile for all medicines, traditional and nano-based, is extremely important. The primary goals of clinical PK is to enhance the efficacy and decrease the toxicity of a patient’s drug therapy. Researchers look for strong correlations between a drug’s concentration in various compartments (plasma, tumor, liver, kidney, etc.) and pharmacologic responses. Insights into these relationships and mechanistic PK differences between formulations can elucidate mechanisms of pharmacodynamic activity and improve patient outcomes. However, the application of basic PK principles to nano-based pharmaceuticals is not trivial and requires some additional considerations. The advent of nanomedicines introduces additional complexities to traditional PK studies, modeling, and analysis. This is reflected in the increasing number of studies focusing on the delivery of the nanoparticles to tumors, which is the most extensively studied facet of nanoparticle drug PK.
Despite a significant body of literature on the use of various nanoparticles for cancer drug delivery, the complex interactions between solid tumor physiology and nano-sized drugs are not fully understood. One notable example is an analysis by Wilhelm et al. that has questioned the utility of nanoparticles for the treatment of solid tumors due to presumed low tumor delivery efficiency and extent of tumor penetration [241]. However, they used a non-standard metric for tumor delivery defined as percent injected dose (%ID) in tumor = (AUCtumor/tend)*tumor mass. This metric reduces the time-concentration data to a single average value, neglects overall exposure time and does not relate the tumor and systemic exposure [242, 243]. A recent reanalysis by Price et al., based on the same dataset, has cast major doubt on the validity of Wilhelm et al. conclusions [244]. The studies included in the Wilhelm et al. analysis which reported matched tumor and blood concentration versus time data were re-evaluated by Pierce et al. using classical PK endpoints. These classical PK endpoints were compared to the unestablished %ID in tumor metric used in the Wilhelm et al. study. The conclusion was that the %ID in tumor was poorly correlated with the standard PK metrics which describe nanoparticle tumor delivery (AUCtumor/AUCblood ratio). The relative tumor delivery of nanoparticles was ~100-fold greater as assessed by the standard AUCtumor/AUCblood ratio than by %ID in tumor. Therefore, the flaw of the Wilhelm et al. analysis is that %ID in tumor does not relate tumor exposure to systemic exposure, as AUC ratio does, and is therefore not a true measure of the tumor delivery efficiency. Moreover, a rigorous nanoparticle PK analysis of tumor drug delivery should have accounted for Cmax in the tumor, nanoparticle drug release, tumor free drug exposure and overall exposure time.
Another important issue is that most studies have only measured total drug (i.e., encapsulated plus released), and not the released drug which is pharmacodynamically active. In many cases, the nanoparticle encapsulated drug dominates the total drug profile for nanoparticle formulations. Thus, the nanoparticle-encapsulated drug uptake into the tumor can often be inferred from the total drug profile. However, in many cases, a considerable portion of the drug can be released by nanoparticles before they reach their final destination. This is definitely the case of such dynamic systems as polymeric micelles that can partition the drug between the micelle and its external environment. Therefore, it is always best to measure these distinct fractions individually as it is the released drug fraction that correlates with toxicity and efficacy [245]. The PK analysis of traditional, excipient-based formulations usually treats the drug in two forms: that which is protein bound and that which is free and unbound. The unbound drug is the fraction which is considered pharmacodynamically active form. The protein bound drug is not pharmacologically active, and not available for metabolism. However, a lot of PK analysis of nanoparticle formulated agents measures the total drug in the blood or plasma, and does not consider the difference between protein bound and free drug. The introduction of nanocarriers adds an additional layer of complexity to PK analysis, as there is another drug fraction to consider that is a nanoparticle encapsulated drug. The existence of three drug fractions, protein bound, nanoparticle bound/encapsulated, and free unbound drug makes it difficult to interpret PK profiles. Both unbound and nanoparticle bound drug can enter tumor microenvironments, but only the unbound drug is pharmacological activity, thus the nanoparticles must release their encapsulated drug to the tumor microenvironment after local uptake. Additionally, the protein bound drug fraction in target tissues can be different than when in the blood. Thus, one must measure all three fractions in all tissues in order to obtain the full PK picture. However, to the best of our knowledge there are no methods at this time to measure encapsulated/protein bound/unbound drug fractions in tissue and this is an important area of research.
The Nanotechnology Characterization Laboratory (NCL) has recently developed the Stable Isotope Tracer Ultrafiltration Assay (SITUA) to probe these complex nanomedicine PK profiles [246]. The SITUA concept is based on an assumption that a tracer amount of isotopically labeled drug in plasma will behave identically to drug that is released from the nanomedicine with regard to protein binding. The isotopically labeled tracer added to nanomedicine-containing plasma becomes a measure of the free drug fraction in the system, which can then be used to calculate nanomedicine encapsulated, unencapsulated protein bound, and unbound drug fractions simultaneously. The system is spiked with the isotopically labeled tracer which rapidly achieves binding equilibrium with plasma proteins. The plasma sample is then transferred to an ultrafiltration device and the sample is separated by centrifugation. One first determines the % bound isotopically labeled drug (D*) using Equation 14 below. A known amount of D* is spiked into the plasma sample, and measurement of the D* in the reservoir and filtrate are then used to determine % Bound D*, which the NCL claims behaves similarly to the non-isotopically labeled drug, D. From there, Equation 15 can be used to determine the unencapsulated drug concentration. With these two values determined, the nanomedicine encapsulated drug can be determined using Equation 16. These equations yield values for nanomedicine encapsulated drug, unencapsulated protein bound drug, and unencapsulated free drug.
| (Equation 14) |
| (Equation 15) |
| (Equation 16) |
This approach enables the analysis of the aforementioned subpopulations of drugs derived from nanomedicines in the systemic circulation in PK studies and can also be used clinically [247]. In a recent study, the authors applied the SITUA method to analyze the PK profiles of several clinically approved nanomedicines and their generic or nanosimilar formulations, some of which lack bioequivalence testing. This study showcases the utility of pragmatic methodology for the measurement of drug subpopulations in plasma [247]. They reported extensive bioequivalence studies on several nanomedicines, to compare PK profiles of follow-on formulations to reference products (Janssen’s DOXIL® vs. Sun Pharma’s doxorubicin hydrochloride liposome formulation and Celgene’s Abraxane® vs. Samyang’s Genexol® PM) [247]. SITUA was employed in the analysis of plasma samples from animal PK studies in order to quantify the subpopulation of each product. They demonstrated that both doxorubicin (DOXIL® and Sun Pharma’s formulation) and paclitaxel (Abraxane® and Samyang’s Genexol® PM) nanoformulations had comparable encapsulated/unencapsulated/unbound PK parameters. Bioequivalence analysis by statistical analysis (two one-sided t-test) revealed that the Abraxane® and Genexol® PM formulations were bioequivalent in total drug PK parameters. However, the generic Taxol formulation did show some marked differences in key PK parameters when compared to Abraxane® and Genexol® PM. This study could have been improved by using a higher animal number and a crossover study design to increase data variability. Nonetheless, this novel methodology showcased advanced analysis of drug subpopulations of nanoformulations, which are closely related to therapeutic outcomes, as well as a pragmatic analytical approach to determine the bioequivalence of follow-on formulations and reference products, in order to facilitate 505(b)(2) regulatory review. Therefore, SITUA method fills a major gap in nano-based pharmaceutical research-that is, the analysis of distinct drug fractions in PK. It is of paramount importance that we can measure all three fractions over time, and SITUA is one of the first steps towards this goal to elucidate how PK of various fractions influences pharmacodynamic activity.
While this SITUA assay is an invaluable tool for nanomedicine PK analysis, its application to polymeric micelles which encapsulate hydrophobic drug by physical entrapment may have additional levels of complexity. It is important to consider that, in contrast to other common nanoformulations, polymeric micelles formulations exist as dynamic structures in which drugs are loaded via non-covalent interaction resulting in gradual drug release to the external environment and can be reabsorbed by the micelles [246]. The SITUA assay assumes the isotopically labeled drug, D*, only equilibrates with the plasma protein bound drug and not with the nanomedicine bound drug (polymeric micelle encapsulated in our case) during the timescale of this assay. Thus, the assay assumes that the polymeric micelles are not behaving dynamically under the timespan of the assay: that is, there is no exchange between free D* and drug which is encapsulated in the polymeric micelles. If exchange between unbound drug and micelle-bound drug is rapid, similar to protein binding, then the SITUA identifies the micelle binding as an increase in protein binding. This was the case for the SITUA when it was applied to the Taxol formulation in which Cremophor EL micelles bind in rapid equilibrium with unbound drug; this Cremophor EL micelle binding was observed as an increase in protein binding that correlated with formulation concentration, and influenced paclitaxel PK by decreasing both clearance and volume of distribution [247]. While micelle drug exchange may occur at a slower rate than protein drug exchange, this was not the case for Taxol, and there is a significant need for the evaluation of this phenomena. It is possible that this dynamic exchange of D* into the micelles occurs on a longer timescale than the assay takes to run (~10 min, dependent upon the time for the tracer to reach equilibrium with protein). If so, then the assay should be straightforward to identify the polymeric micelle encapsulated drug fractions. However, this could be different for all polymeric micelle systems and should be individually evaluated. Until then, the SITUA assay should be cautiously applied to polymeric micelle drug systems and additional considerations such as dependence of the bound D* fraction on the formulation concentration should be taken in account. This is contrary to polymeric micelle formulations prepared via stable covalent conjugation which may only react upon certain external stimuli and release the cargo via the cleavage of the covalent bond.
Overall, polymeric micelle formulations that physically encapsulate hydrophobic drugs are gradually releasing drug via both diffusion of the drug from the core in the external solution and drug binding to plasma proteins. Thus, polymeric micelle formulations may exist in systemic circulation as a dynamic system, comprised of the multiple forms of hydrophobic drug as discussed above. This dynamic nature must be accounted for in the development of PK models. A recent study by our lab explored the complex, dynamic nature of POx polymeric micelles and the PK activity of these formulations [188]. This has also been studied earlier by Bulitta et al., who came to develop similar models to represent the regular micelle and emulsions systems [248]. They compared the PK in cancer patients of Taxol and a Cremophor-free D-α-Tocopherol PEG succinate (TPGS)-based nanoemulsion formulation of paclitaxel. They sought to develop a mechanistic model in humans to model both the free and total paclitaxel in the system. Although the Bulitta et al. model was more comprehensive, there were some key similarities between these two studies and we will consider the Wan et al. model [188] as an example. Rather than treating the drug as being injected into a single central compartment, the system must be modeled as “polymeric micelle” compartment and a “plasma bound” compartment. In the Wan et al. study, this modeling approach was applied using a POx-paclitaxel polymeric micelle formulation. They also used a tumor “effector” compartment as seen in Figure 6. There was no measurement of the free, unbound drug which is the shortcoming of this model as the unbound drug is pharmacologically active and contributes to the transfer of drug between compartments. However, the unbound drug fraction is minimal in systemic circulation and neglecting its measurement in a model is inconsequential as this fraction is generally, and thus mimics the total unencapsulated drug profile. The work showed that the critical parameter was the micelle encapsulated drug permeability into the tumor effector compartment. When the micellar drug permeability into the tumor is much less than the plasma bound drug permeability (Scenario A), then changes in the micelle drug retention have little effect on the tumor AUC. However, when the micelle permeability is similar to or better than that of plasma bound drug (Scenario B), then the micelle retention has a major effect of the tumor AUC. That is, stronger micelle retention leads to significantly increased tumor AUC (Figure 7). This is because for both scenarios the clearance of plasma bound drug is assumed, based on experimental evidence, to be higher than that of micelle bound drug. Scenario B, in our opinion, is the more realistic and biologically relevant scenario. Thus, it is important for formulation designers to maximize the drug retention in the micelles to improve overall tumor drug exposure profiles.
Figure 6. A three-compartmental model describing the PM drug delivery to a tumor.

The drug is administered as bolus in the form of PM (1) and is subsequently distributed between the plasma (2) and tumor (3) compartments. The PK constants correspond to: k12 - rate of drug transfer from PM to plasma; k21 – rate of drug re-capture from plasma to PM; k13 - rate of transfer (permeability) of the micellar drug to tumor; k23 - rate of transfer of the plasma bound drug to tumor; k31 and k32 – rates of drug reabsorption from tumor to PM and plasma, respectively; k10 and k20 - micellar and plasma bound drug clearances, respectively. The model assumes that the drug solubility in blood is very low and the free drug form in the blood is therefore neglected. Reprinted with permission from [188].
Figure 7.

The drug amount-time profiles for the tumor obtained using a three-compartmental model. Simulations for basic scenarios A (solid lines) and B (dashed lines) are shown. The three-compartmental model is presented in Figure 6 and the values of the PK parameters used in simulation were varied. In the A scenario, the penetration of micellar bound drug into the tumor is much less than that of plasma bound drug. In scenario B, the micellar bound drug penetration is comparable to that of plasma bound drug. As you move through A0 and B0 up to A5 and B5, this shows the effect of increased drug retention in the micelle. All units are arbitrary. Reprinted with permission from [188].
The Wan et al. study did not discriminate between the micelle bound and released drug within the tumor assuming that all drug will be pharmacologically active if delivered to the tumor, which may be a fair assumption for the micelles used in the study. However, as general case both drug subpopulations should be accounted for as the Cmax of the unbound form achieved in the tumor is strongly correlated to pharmacodynamic activity. Measuring drug concentrations in tumors can be a bit more complex than measuring concentrations in plasma, as there is some in the tumor interstitial area as well as intracellularly. The lysis of cells and digestion of tumors is often necessary for these measurements. Microdialysis can help separate the two subpopulations and in some cases noninvasive imaging techniques such as NMR, quantitative autoradiography, position emission tomography can be used to visualize drug concentrations and localization in the tumors [249].
It should be pointed out that both Wan et al. [188] and Bulitta et al. [248] models relied on stability of the micelles and did not account for a situation where formulation could influence the unbound drug concentration, like in the case of equilibrium micelle binding. Such a case would involve dependence of the PK on the micelle concentration and would need to be considered separately. An additional layer of complexity for the modeling of the PK of polymeric micelle drugs is that in systemic circulation, just as in vitro, the CMC of polymeric micelle materials can play a central role in the PK. Upon dilution of the polymeric micelles, should the polymer concentration decrease below the CMC, the micelles will disassemble over time altering the drug populations in circulation. In this case, there will eventually be no “nanoparticle encapsulated” fraction thus increasing the protein bound and free, unbound concentrations in circulation. The CMC of polymeric micelles should be low enough to endure dilution upon infusion, thus the micelle structure would be intact during the systemic circulation and avoid unexpected drug loss [23]. While concerns over CMC are valid, it is also important to note that this is not an instantaneous process, rather it could be kinetically dominated. While the micelle disassembly upon dilution below the CMC is thermodynamically favored, the disassembly process can take hours, especially in micelles with “glassy cores” [250, 251]. It is also important to note that the CMC of a micelle system could depend on the drug loading in the micelle. A higher drug loading could improve micelle stability in circulation. A previous study by Batrakova et al. showed that the clearance of the polymer (Pluronic P85) was determined by the release of the single polymer chains (unimers) and was unaffected by delivery of a dose with the polymer concentration being above or below the CMC [252]. This indicates that unimers were cleared by the kidneys while micelles bypassed glomerular filtration. However, this does not apply to drug which would be contained in the micelles. When polymer concentration drops below the CMC, the redistribution of drug into the unbound fractions can then increase the observed clearance (but not necessarily depending on rate limiting metabolic processes). Only unbound drug can be metabolized, so as polymer concentration is decreased below the CMC, the equilibrium shifts as drug is slowly removed from the “micellar compartment” and the unbound concentration can increase making more drug available for metabolism.
While nanomedicine PK is highly complex, it is clear that the dynamic nature of polymeric micelles presents many additional considerations and opportunities in analytical processing of samples and the PK modeling of drug systems. This is still an area where there is an immense need for further work and characterization of these fundamental PK processes. Additional analytical techniques may need to be developed to probe polymeric micelle systems which can account for their dynamic nature. For now, we must be sure to utilize proper PK metrics, such as the tumor/plasma AUC ratio, and validate novel metrics against traditional analyses. A proper understanding of this complex PK and design of polymeric micelle PK analysis will ensure that literature moving forward is reliable and truly represents the field.
4.3. Hydrodynamic size and morphology of polymeric micelles and drug distribution
Hydrodynamic size of polymeric micelles is another important factor to consider for clinical applications. It is well-established that the size distribution of nano-sized particles may affect their biodistribution when administered, resulting in either extended systemic circulation or faster clearance from the body. Particles with size ranging over 200 nm could be caught by the liver, while smaller nanoparticles less than 10 nm would be easily cleared by the kidney [239]. In some previous studies, it was shown that sizes from 50 nm–100 nm are effective in preclinical models, suggesting ideal particle size distribution is necessary for efficacy of polymeric micelles [253, 254]. The size of polymeric micelles currently in clinical trials are less than 100 nm [6, 7]. Thus, one may speculate that size range from 20 nm to 100 nm can be efficacious as polymeric micelle formulations. In fact, the study by Kataoka’s group has shown that tumor accumulation of the polymeric micelles depends both on the particle size and tumor desmoplasticity with smaller polymeric micelles (size) displaying better accumulation on more desmoplastic tumors [255]. To increase penetration into such tumors the particle size of the drug carriers must be minimized. Kataoka and colleagues have shown that when the drug is encapsulated in small polymeric micelles (~30 nm), both the penetration of the micelles into the desmoplastic tumors and anti-tumor effect of the drug can be improved [255]. An alternative approach discussed in the next section, is the co-delivery of two drugs where one drug is used to modify the tumor microenvironment and reduce desmoplasticity while the other drug acts as a traditional chemotherapy. Recently, a study from our lab showed the improved PK profile of the drug vismodegib when encapsulated in a POx micelle for treatment of a pediatric brain tumor – medulloblastoma [113]. The POx-vismodegib formulation showed a 1.6-times increase in tumor AUC and overall improved delivery to the brain. The micelle formulation also resulted in a lower Vd which could lead fewer off-target affects than the conventional formulation’s less than ideal distribution to peripheral tissues [113].
Not only hydrodynamic size, but also micelle morphology can play a role in PK and distribution. For example, the Discher group was to our knowledge the first to report on drastic PK differences between spherical and worm-like polymeric micelles made from the same block copolymer [256]. The Discher Lab used PEO-b-PCL block copolymers and have reported on the increased systemic circulation times of worm-like polymeric micelles compared to spherical micelles. The Discher lab did not use drug loaded micelles for their study so only monitored the polymer distribution. In contrast, Wan et al. have recently reported on the highly loaded worm-like POx-based micelles with therapeutically relevant concentrations of the drugs [114]. This study suggested that the worm-shaped POx micelles co-loaded with two drugs (etoposide and hydrophobic cisplatin analog) have increased accumulation of the drugs into tumor compared to spherical micelles loaded with just one drug. They reported that the overall AUC of the drugs in the tumor from worm-like micelles were higher than from spherical micelles. However, other factors, in particular better retention of co-loaded drugs compared to single-drug micelles, rather than the shape alone could have also contributed to this phenomenon as discussed below. In addition, although worm-like micelles circulate longer and could eventually accumulate more in the tumor, they can release the drug before they reach the tumor. Therefore, as drug is released from the micelles over time this does not necessarily mean that drug exposure in the tumors is higher from worm-like micelles. The further comparison of highly loaded spherical and worm-like polymeric micelles is needed to consider morphology of micelles and how this may affect circulation, biodistribution, accumulation, and targeting.
Other factors must be considered as well which may affect the hydrodynamic size and shape of polymeric micelles in systemic circulation. One such factor is the effect of dilution of polymeric micelles by plasma. This dilution as discussed above could possibly result in a concentration below the CMC causing the disassembly of the polymeric micelles altogether but could also lead to changes size and morphology of the micelles above the CMC. Additionally, surface charge of polymeric micelles could affect their biodistribution. Lastly, the release profile of drug from polymeric micelles can subsequently affect the hydrodynamic size of polymeric micelles over time. Some studies have shown that the release of drug, and lower Drug/Polymer ratios allow the incorporation of solvent (water) into the micelle core which increases the hydrodynamic size [30]. If this size increase is large enough, this could affect key PK parameters such as clearance and volume of distribution as well as penetration of micelles in the tumors.
4.4. Polymeric micelle combination therapies and effects on drug retention and pharmacokinetics
Combination therapy has been largely exploited for the treatment of various types of cancer based on related pathways of oncogenesis or utilizing a combination of agents which affect the tumor microenvironment [257, 258]. Frequently employed combinations are 1) a combination of several chemotherapies for killing cancer cells, 2) a combination of chemotherapy with additional agents, such as tumor microenvironment modifiers, or more recently 3) a combination of immunotherapy with anticancer agents [259]. The latest study highlighted that even the best combinations of the chemotherapeutic agents determined in vitro as having the most potent and highly synergistic effect against non-small cell lung cancer (NSCLC) cells may not be effective against the in vivo tumor even at the MTD of those combinations [259]. In contrast, resiquimod, an imidazoquinoline TLR 7/8 agonist, solubilized in POx block copolymer (PMeOx-b-PBuOx-b-PMeOx) micelles had a superior tumor inhibitory effect in a metastatic model of lung adenocarcinoma, relative to anti-PD1 immune checkpoint blockade therapy as well as platinum-based chemotherapy, which is the mainstay of treatment for NSCLC. Investigation of the in vivo immune status following resiquimod in polymeric micelles treatment showed that resiquimod-based stimulation of antigen-presenting cells in the tumor microenvironment resulted in the mobilization of anti-tumor CD8+ immune response [259]. This study demonstrates the promise of optimally delivered and nanoformulated immunomodulating therapeutic agents and possibly their combinations with chemotherapy for treatments of metastatic NSCLC. Ideal combination therapy requires precise drug exposure to induce synergistic therapeutic effects. Thus, the administration and subsequent disposition of combinations to target tissue with the desired ratio of the delivered agents is highly warranted.
Hydrophobic small molecule combination therapy is hindered due to poor PK profiles of the molecules, similar to that of single drug therapies. However, these combination therapies are complicated even further by the inclusion of additional active pharmaceutical ingredient (API). One of the main factors that determine PK profiles of small molecule drugs is the aqueous solubility which hinders administration of such molecules via the parenteral route. For this reason, the effective solubilization of hydrophobic small molecules and co-encapsulation of drug combinations in a single micelle is important for successful combination therapy. The ability to solubilize multiple drugs in the same micelle can yield more predictable PK profiles and allow for the solubility of new molecules and additional combination options. Although, as of today no polymeric micelle formulation for combination therapy is USFDA approved, but the design of pharmacologically effective drug combinations in polymeric micelles is highly warranted.
Kwon and colleagues conducted a series of studies on a drug combination therapy for cancer therapy using polymeric micelles [240, 260, 261]. In 2009, they reported that paclitaxel, etoposide, docetaxel, and 17-AAG were solubilized in PEG-b-PDLLA block copolymers as either single drug or combination therapy. Polymeric micelles of 2- and 3-drug combinations such as paclitaxel/17-AAG, etoposide/17-AAG, docetaxel/17-AAG and paclitaxel/etoposide/17-AAG were small (~30–40 nm) and displayed enhanced drug solubility up to 3–4 mg/mL in aqueous media [260]. In a follow-up study, 3-in-1 PEG-b–PDLLA micelles were prepared for poorly soluble multidrug systems such as 17-AAG, paclitaxel, and rapamycin [240]. The 3-in-1 micelle featured 40 nm hydrodynamic size and improved drug solubility (approximately up to 3 mg/mL in aqueous solution). In vitro cytotoxicity studies and combination index (CI) analysis displayed a synergistic effect of drug components in 3-in-1 micelles for MCF-7 and 4T1 breast cancer cell lines. This group also reported that 3-in-1 PEG-b-PDLLA micelle delivering high doses of paclitaxel, 17-AAG, and rapamycin significantly increased the exposures of paclitaxel and rapamycin in mice compared to single drug micelles [261]. In contrast, at a lower dose of 3-in-1 micelle, PK differences of individual drugs were marginal. These data indicate that the PK profile of drug combinations in polymeric micelle formulations can be improved by the functionalities of polymeric micelles such as the solubilization capacity and increased dose tolerability.
Often times, the inclusion of a drug which is highly soluble in a micelle system with a drug which is not typically highly soluble in the system, can synergistically increase the solubility of the non-soluble drug in the system. In POx micelles, the inclusion of poorly compatible drugs with well-compatible, such as paclitaxel, allows for their encapsulation and increased aqueous solubility. This alters the PK of both paclitaxel and the other drug. Han et al. reported the synergistic solubilization of multiple drugs in a single micelle system with high LC resulting in formation of ultra-high loaded polymeric micelle drug formulations. A variety of poorly-soluble drugs (paclitaxel, docetaxel, etoposide, 17-allylamino-17-demethoxygeldanamycin (17-AAG), bortezomib) were solubilized in POx-based block copolymer (PMeOx-b-PBuOx-b-PMeOx) to prepare binary and ternary drug combinations (Figure 8) [117]. POx-based polymeric micelles were able to solubilize multi-drug combinations with an extremely high LC of up to 50 % (48.7 % LC for 1:1:1 ratio of paclitaxel:17-AAG:etoposide and 48.4 % LC for 1:1:1 ratio of paclitaxel:17-AAG:bortezomib). Also, POx micelles co-loaded with three drugs (paclitaxel, 17-AAG, and etoposide) displayed improved formulation stability in aqueous media. Presumably, multiple drug components in the core of polymeric micelle could further stabilize the polymeric micelle systems. Alternatively, drugs which form highly stable single drug systems (like paclitaxel), could increase the stability of those drugs which do not form stable single drug micelles. This stabilization can allow for unique therapeutic combinations which are not available in single drug micelle systems. An in vitro cytotoxicity assay confirmed synergistic cytotoxicity of micellar etoposide/17-AAG and micellar bortezomib/17-AAG in cancer cells.
Figure 8.

Multiple chemotherapeutic agents in high capacity poly(2-oxazoline) micelles. (Drug designations: BTZ – bortezomib, DTX – docetaxel, ETO – etoposide, PTX – paclitaxel) Reprinted with permission from [117] Copyright 2012, American Chemical Society.
Wan et al. reported POx-based combination therapy of etoposide and an alkylated cisplatin prodrug for the treatment of lung cancers [114]. The POx block copolymer (PMeOx-b-PBuOx-b-PMeOx) was employed to solubilize drug combinations in a single high-capacity vehicle (over 50 % wt. drug in dispersed phase). The combination polymeric micelle featured nanosized and worm-like morphology which was rarely reported previously for drug-loaded polymeric micelles. Interestingly, drug release from combination polymeric micelle was slower, presumably due to the formation of stable micelle formulations with the co-loading of drugs. PK analysis of combination polymeric micelles exhibited enhanced drug exposure to the target site compared to single micelle, mixture of single micelle, and free drugs. Along with the strong synergistic effect of the combination, a superior anti-tumor activity of combination polymeric micelles was observed in preclinical lung cancer models.
Wan et al. also reported the co-delivery of paclitaxel and an alkylated cisplatin prodrug as polymeric micelles for the treatment of ovarian and breast cancer [188]. The drug combination was effectively solubilized in a POx-based block copolymer with high LC (over 50 %) and stable polymeric micelles for two-drug combinations were formed. Drug-coloaded micelles had slower release of the drug components to plasma and improved drug exposure of both drugs to the tumor site (Table 5). Superior anti-tumor activity of combination therapy was confirmed in ovarian and breast cancer preclinical models. Interestingly, PK simulations of the polymeric micelles in a three-compartment model as previously discussed revealed that a decreased release rate of drug components from the micelle could be related to the improved tumor delivery of the drug combination (Figure 7). This study indicates to us that co-loading of drugs in a single micelle would be beneficial for the delivery of the combination therapy. Also, an in-depth analysis of the PK profile/simulation and its correlation with physicochemical properties of polymeric micelles would be helpful for designing ideal polymeric micelle formulations.
Table 5.
PK parameters of C6CP and paclitaxel in plasma and tumor after administering polymeric micelle in A2780/CisR tumor bearing mice. Reprinted with permission from [188] Copyright 2018, Elsevier.
| Parametersa | C6CPb, 20 mg/kg | paclitaxel, 20 mg/kg | |||||
|---|---|---|---|---|---|---|---|
| paclitaxel/C6CP polymeric micelle | C6CP polymeric micelle | Parameter ratioc | paclitaxel/C6CP polymeric micelle | paclitaxel polymeric micelle | Parameter ratiod | ||
| Plasma | t1/2, α (h) | 7.67 | 6.20 | 1.24 | 5.12 | 3.67 | 1.40 |
| Cmax (μg/mL) | 9.40 | 4.71 | 1.99 | 6.51 | 4.62 | 1.41 | |
| AUClast (h*μg/mL) | 34.80 | 30.00 | 1.16 | 18.41 | 9.78 | 1.88 | |
| Clobs (mL/h/kg) | 527.40 | 541.00 | 0.97 | 984.06 | 1434.66 | 0.69 | |
| Vdobs (mL/kg) | 5077.11 | 7963.56 | 0.64 | 10112.89 | 7689.6 | 1.32 | |
| Tumor | AUClast (h*μg/g) | 110.92 | 38.68 | 2.87 | 111.14 | 87.28 | 1.27 |
| Cmax (μg/g) | 9.86 | 4.34 | 2.27 | 8.34 | 5.38 | 1.55 | |
| Tmax (h) | 1 | 1 | 1 | 1 | |||
t1/2, α, half-life at the biodistribution phase; Cmax, maximum plasma concentration; AUClast, area under the curve from time 0–15 h; Clobs, observed total body clearance; Vdobs, total volume of distribution observed; Tmax, time of maximum concentration
Hydrophobic derivatives of cisplatin with aliphatic chains of 6 carbon atoms at the axial positions
paclitaxel/C6CP polymeric micelle: C6CP polymeric micelle
paclitaxel/C6CP polymeric micelle: paclitaxel polymeric micelle
5. Polymeric micelles in clinical trials and regulatory approval for human use
5.1. Polymeric micelle manufacturing considerations
The manufacturing process of polymeric micelles should be considered throughout formulation development in order to produce micelle formulations with consistent physicochemical properties and achieve the desired scale of production. The selection of applicable manufacturing processes may impact characteristics of the final polymeric micelle formulation such as drug loading, size distribution, and stability in aqueous media, which are critical factors for translation [262]. Several processes are available to produce polymeric micelles with uniform size distributions and stability. However, not all processes are created equal. That is, what works for one polymer-drug system may not be feasible in another, and not all processes are conducive to a manufacturing scale.
One of the most common drug-loaded polymeric micelle preparation techniques is the thin film hydration method, which is fairly conducive to scale-up in a batch-wise manner. In this method, polymers and drugs are solubilized in a common organic solvent and the solution is evaporated under air flow or at a reduced pressure to form a polymer-drug thin film. This film is then hydrated with an aqueous solution and the polymer and drug are dispersed into the solution as drug loaded micelles [22, 263–265]. In this technique, it is often critical that a dry thin film is achieved with no residual solvent. Often times after thin film formation, the film will be placed under vacuum for several hours to ensure solvent removal is complete. Additionally, solvent selection is a key parameter which is explored in several studies [116, 193, 264]. First of all, both the drug and polymer must be soluble in the selected organic solvent. However, this does not necessarily mean that all good solvents for a polymer-drug system will produce stable, uniform polymeric micelles. It is important to explore several solvent systems when possible. There must be intimate mixing to avoid crystallization of the drug during the thin film formation, and this cannot be achieved in every solvent. Alternatively, if melting temperature allows, the co-melting of the polymer and drug can be performed without a solvent in order to prepare the micelles.
One study compared the use of thin film hydration and sonication alone. In the sonication process an anticancer drug, sagopilone, and PEG-b-PLLA, PEG-b-PDLLA or PEG-b-PCL were added to an aqueous solution and sonicated to form micelles [193]. In that study, when comparing the sonication method and thin film hydration technique, they determined that the process which forms more stable micelles is polymer dependent. Some key factors are the block copolymer chemical composition, block lengths, and the polymer/drug ratio. However, in our experience, the sonication process alone yields poor results, because the drug tends to precipitate faster than it solubilizes into the micelle core.
One way to control this process is by using flash precipitation, which was in part pioneered by the Prud’Homme lab. In flash precipitation, an aqueous and organic phase are mixed rapidly with engineered geometries in a special “jet mixer” instrument which allows for the synthesis of uniform nanoparticles [266, 267]. This process has been brought to scale by BASF for the generation of β-carotene nanoparticles [268]. It can be optimized by increasing mixing times to obtain drug-containing polymeric micelles and block copolymer coated drug nanoparticles with desired size, coating, and other characteristics [269]. The mixing performance can be maximized by using microfluidic mixer systems, leading to the highest mixing efficiency and homogeneous reaction environment of the mixed solutions under continuous flow condition. However, the process is not universal and has challenges especially for drugs with relatively low logP values that tend to dissolve faster than they incorporate into nanoparticles. Moreover, the organic solvents must still be removed by dialysis or freeze drying, which is discussed below. Overall the use of rapid mixing for preparation of drug containing micelles is in our view an area for further research and development, potentially at industrial scale.
One technique that is commonly used in laboratory micelle preparation which is not conducive to industrial scale up is dialysis. Pure organic solutions of micelle and drug, with appropriate dialysis cut-off membranes, are dialyzed against pure water for a few days while the water is changed out frequently. As organic solvent in the dialysis bag is replaced with aqueous media, drug loaded polymeric micelles begin to form [270–273]. With dialysis, the processing times are long and the complete removal of organic solvent and free, unloaded drug from the solution is difficult. This is often paired with freeze drying for a more complete solvent removal, but scale up of the dialysis method remains a challenge. In one study, the use of just dialysis did not produce a uniform particle population or nanosized polymeric micelles [274]. They compared this pure dialysis approach with a hybrid approach. In the hybrid approach, polymer (PEO-b-PCL) and drug are dissolved in an organic phase which is added dropwise to an excess of aqueous media. As the organic phase enters aqueous solution, polymeric micelles spontaneously form which encapsulate the drug. These solutions can then be dialyzed against aqueous media to remove solvent and free drug. This approach yielded smaller (20–50 nm) and more uniform micelle sizes. In both methods, the particle size was stable after dilution. The selection of organic solvent used also had an effect on polydispersity once again highlighting the need to evaluate multiple solvent systems in order to optimize formulation stability.
An alternative to dialysis is using cosolvent evaporation. Drug and polymer are dissolved in organic solvent and are added dropwise, or sometimes rapidly, to an aqueous phase. The solution can then be heated and/or placed under vacuum to remove the volatile organic solvents and produce stable aqueous micellar solutions [275, 276]. If there is any residual drug precipitated, then a brief centrifugation step can remove drug precipitates (this is applicable to any of the discussed methods). It is also important to note that any combination of the aforementioned methods could potentially be used to prepare stable micelles. In these studies, organic solutions were added to an aqueous media, dialyzed, sonicated, and then freeze dried to produce stable formulations [273, 277].
Another technique, oil in water emulsions, is common in pharmaceutical development and can be applied to polymer micelle preparation. Polymer and drug are dissolved in a water immiscible organic solvent which is then added to an aqueous solution to prepare an emulsion [278, 279]. The mixture is then stirred and/or heated to remove the organic solvent. Oil in water emulsions are commonly produced at manufacturing scales, but the complete removal of organic solvent from them can be difficult. Thus, the use of common emulsion solvents such as tetrahydrofuran, chloroform, and acetone could be problematic for the development of injectable polymeric micelle formulations. However, the addition of a freeze-drying step could help completely remove undesired excipients from the formulation.
Freeze drying, when done under particular conditions, can be used to remove small amounts of residual organic solvents. Moreover, freeze drying alone has been used to prepare stable micelle formulations. In one study, a tert-butanol solution of drug was added to an aqueous solution of a polymer (PVP-b-PDLLA), and then the mixture was freeze dried to form stable micelles upon resuspension in aqueous media [280, 281]. The tert-butanol is highly compatible with the one-step freeze drying process. However, the use of tert-butanol in this process was unique to the system of PVP-b-PDLLA block copolymer and could not be applied, for example, to PEG-b-PDLLA since PEG is practically insoluble in tert-butanol.
The freeze drying process is highly conducive to scale up. Freeze drying after preparation of micelles can also be used to stabilize the formulation for longer periods of time [264]. Sometimes, the aqueous stability of polymeric micelles can be limited, so long term storage of powdered polymeric micelle solutions could be more feasible. To ensure the rapid dissolution of the powdered polymeric micelles, their mixing with excipients such as lactose makes this composition instantly dissolvable in water allowing the product to be dried to a solid. This approach was used, for example, for preparation of the dry from of SP1049C polymeric micelle formulation of doxorubicin [282].
Lastly, a more recent and complex method which has been developed involves the use of supercritical fluids [283, 284]. A full review on the theory of using supercritical fluids in drug delivery systems can be found here [285]. Using supercritical fluids in polymeric micelle preparation allows for faster processing times, easy solvent removal, and no long freeze drying or dialysis steps. Common solvents for this are trifluoromethane and carbon dioxide. A solution is brought up to pressure to dissolve the polymer and drug as a supercritical fluid and then pressure is quickly removed allowing for micelles to form and the solvent to evaporate. It can then be washed with water to form stable aqueous solutions. We refer the readers to this more comprehensive review for additional information on supercritical fluid use [285].
As we have explained, several factors in these processes should be considered critical such as the selection of organic solvent, number of overall steps, yield of the polymeric micelle from the pure drug and excipient (de-facto LE), sterilization process for endotoxin-free formulation, and final formulation design (solution or lyophilized powder for reconstitution). Minimizing the steps in the manufacturing process to increase LE and mitigate the detrimental side effects of manufacturing processes (e.g. formulation instability) is highly desirable. For this purpose, the thin film hydration method could be the most plausible option since 1) there are the fewest steps 2) organic solvent removal is easy during film formation 3) it avoids dialysis and potential contamination from water. As the final formulation, the lyophilized powder form is the better option since it may be helpful to avoid contamination or drug release/degradation of micelle formulations in aqueous media.
At a larger scale of production, the manufacturing of polymeric micelle products with reproducible physicochemical properties is of importance. For this purpose, proper establishment of the good manufacturing process (GMP) is necessary to follow industrial standards and ensure the quality of the final nanomedicine products. Also, adapting a systemic management system based on the principle of Quality by Design for the manufacturing is useful in order to develop efficient quality control system and define key parameters for mass production of polymeric micelle formulation. The key parameters such as amount of API and excipients, purity of solvents, presence of impurities and processing variables (time, temperature and pressure) can be useful factors to define the cause of unexpected quality issues or non-reproducibility of the products during the production. International Conference on Harmonisation (ICH) guidelines Q8 Pharmaceutical Development of the European Medicines Agency (EMA) and Q11 Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities) of the USFDA provide such guidelines to provides knowledge on the application of scientific approaches and quality risk management for the development of a product and its manufacturing process [286, 287].
5.2. Regulatory approval of nanomedicines
No new medical product is without risks, and these must be assessed throughout the drug development pipeline, and will ultimately be evaluated to either approve or reject the use in human or veterinary patients by regulatory agencies worldwide, based on evaluation of quality, safety and efficacy, as well as a final risk-benefit assessment. Examples of such Agencies include the USFDA, the UK Medicines and Healthcare Products Regulatory Agency (MHRA), the EMA, Brazilian Health Surveillance Agency (abbreviated in the home country as ANVISA), Health Canada, Australian Therapeutic Goods Administration (TGA), Japanese Pharmaceuticals and Medical Devices Agency (PMDA) and the Ministry of Health, Labor, and Welfare (MHLW), the National Medical Products Administration (NMPA) of China, and the Ministry of Health of Russian Federation to name a few. The purpose of these agencies is to protect public health through the evaluation and review of the quality, efficacy, and safety for innovative medical products at the moment of marketing approval and to provide a framework for the continued evaluation of technologies and therapeutic options. Below, we will focus on the approval process used in the US and Europe. The move towards nano-based pharmaceutical platforms are expected to improve one or several aspects related to drug dissolution, bioavailability, metabolism, clearance, and distribution profiles, improving the therapeutic index, often by reduction of safety risks in clinical use [288–290]. While improving many facets of drug delivery, the use of nanocarriers changes the frame of traditional regulatory procedures. Encapsulation in nanoparticles changes the size, surface properties, and other characteristics which affect in vivo drug behavior when compared to traditional formulations composed of low molecular weight API and excipients. Regulatory appraisal has to deal with its impact on safety and efficacy profiles. This calls for increased scrutiny and rigor in the review process [288, 291, 292].
In the US, all new pharmaceuticals are reviewed and approved based on the well-established regulatory framework which is based on a weight-of-evidence approach. Such an approach considers each new drug on a case-by-case basis. Since each new formulation has unique properties, the USFDA recommends sponsors engaging the Agency early in the development and pre-submission process so as to obtain timely and adequate guidance for studies critical for the given drug product. The available guidance for industry documents containing specific recommendations for different categories of drug products are accessible on the USFDA website (https://www.fda.gov/drugs/guidance-compliance-regulatory-information/guidances-drugs). Among them are specific recommendations for nano-based products (https://www.fda.gov/science-research/nanotechnology-programs-fda/nanotechnology-guidance-documents, https://www.fda.gov/science-research/nanotechnology-programs-fda/nanotechnology-guidance-documents) [291]
The traditional application process in the US for New Drug Application (NDA) of novel human pharmaceuticals, including those containing nanomaterials, occurs through the 505(b)(1) regulatory pathway. In addition, the 505(b)(2) pathway exists and applies to products closely related to the innovators. If an API has been reformulated into a new ‘nanocarrier’, then it may proceed along either the 505(b)(1) with non-similar PK profile to the innovator or the 505(b)(2) route if the PK profile is similar. Utilizing the 505(b)(2) pathway then allows for the use of clinical data from the Reference Listed Drug (RLD). However, by following the 505(b)(2) pathway, one must still demonstrate some kind of improved outcome whether therapeutic, improved shelf-life/stability, increased convenience, reduced metabolic burden, or some other quality which distinguishes it from the innovator product. The 505(j) pathway for the approval of generics requires the demonstration of “sameness” with only small changes to formulation allowed. This pathway can be more challenging for nano-based pharmaceuticals than traditional small molecule formulations due to the added complexity inherent when working with nanomaterials. These pathways are authorized by the Food, Drugs, and Cosmetics Act and apply to non-biologic drugs. Biologics, on the other hand, are licensed through the Public Health Service Act. Innovators follow a 351(a) pathway whereas ‘biosimilars’ are approved through the 351(k) pathway [293]. Nano-based pharmaceuticals and biologics represent the category of complex drug formulations which are often heterogeneous in nature. This heterogeneous nature makes it impossible to produce identical follow-on drugs and demands a more adequate route to regulatory approval. That is, there is not a distinct or definitive set of critical quality attributes (CQAs), which can accurately predict the in vivo similarity of two distinct nano-based pharmaceuticals or biologic products. Thus, a wide variety of evidence is needed to show equivalence which includes long term clinical monitoring and extensive PK data [288, 293–295]. For example, the SITUA method (See section 4.2) developed by the NCL has shown the potential to probe the complex PK behaviors of nano-based pharmaceuticals [247]. This kind of analysis can provide strong evidence for the similarity of two nanomedicines.
A robust and consistent manufacturing process for nano-based pharmaceuticals is of paramount importance to regulatory approval. While drug nanocrystal, liposomal, iron nanoparticle, and a few others have been approved by the USFDA, there is not yet a clinically approved polymeric micelle formulation in the United States [296, 297]. In fact, the USFDA has released a specific guidance for Liposomal formulations as they have become more common in the marketplace [298]. Two polymeric micelle formulations, Genexol® PM and Nanoxel® M, have been approved by regulatory agencies as discussed in Section 5.3. The relationship between nano-based pharmaceutical physicochemical properties and in vivo behavior is an active area of investigation. Unlike in traditional drug formulations, similar physicochemical properties of nano-based pharmaceuticals are not often a predictor of similar in vivo behavior [288, 293, 295] Once thoroughly assessed, our understanding of these relationships can help to improve how we design and evaluate nano-based pharmaceuticals and, in particular, block copolymer micelle systems. Nano-based pharmaceuticals are usually synthetic in origin and do not fall under the ‘biologics’ class of drugs. However, there are some biologics which are nanosized and exhibit unique properties due to these dimensions. This creates a highly complex classification of nanoscale drugs. This high degree of complexity can make complete physicochemical characterization a significant challenge, especially for follow-on or generic formulations, which has also been addressed at different levels both by the NCL (USA) and European Pharmacopoeia [293, 299]
Similar to the USFDA’s 351(k) route for biosimilars, the EU regulatory bodies have developed a ‘nanosimilar’ route for nano-based pharmaceuticals. For the approval of nanosimilars, the EMA follows a stepwise approach [293, 299] which includes in vitro quality assessments, pre-clinical biodistribution, clinical PK, and then therapeutic equivalence. The nanosimilar formulation must show comparable character at each step of the process and cannot rely on in vitro or preclinical animal model data alone, making a case for having some level of clinical evaluation to be necessary for nanosimilars. On the other hand, the USFDA does not rely on pre-clinical animal data and instead focuses on extensive physicochemical characterization followed by clinical PK data [291, 293]. These data sets provide necessary information to predict “sameness” of two nanoformulations. However, there is still a high attrition rate for new drugs, and new nano-based pharmaceuticals are subject to this as well. The development of new clinical models which better replicate patient pathophysiology are highly warranted, and this is a key place where physician-scientists can help to evaluate these formulations and push the field forward by lowering the attrition rate of new medicines. That is, the responsibility does not fall solely on the USFDA, but also on researchers (both in Academia and Industry) to develop better pre-clinical models that can more accurately assess the potential for clinical efficacy similarity.
The nanosimilarity approach should not be confused with the fact that a nano-formulation of an already existing API, is to be considered by default as a new medicinal product, that needs to go through an almost full dossier for marketing authorization application (MAA). That was the case for Caelyx (DOXIL® in USA) in 1996 (https://www.ema.europa.eu/en/medicines/human/EPAR/caelyx-pegylated-liposomal) or later on for Abraxane® in 2005 (https://www.ema.europa.eu/en/documents/assessment-report/abraxane-epar-public-assessment-report_en.pdf), or more recently in 2018 to Vyxeos® (https://www.ema.europa.eu/en/documents/assessment-report/vyxeos-epar-public-assessment-report_en.pdf) just to give three distinct, well-known examples.
The EMA has developed since 1995 a Scientific Advise procedure that is very useful for companies to use, when assessing the relevance of the evidence gathered at the moment of designing their clinical studies. This procedure is well supported for small enterprises and larger companies dealing with orphan drugs, which makes EMA an attractive pole for translational research in highly entrepreneurial ecosystems around academia involving new company incubators and translational accelerators that foster the launch of active and innovative Start-Ups. The system is expected to have a high impact in the future of medicines innovation in Europe (https://www.ema.europa.eu/en/human-regulatory/overview/supporting-smes).
The application of the complexity issue to block copolymers is apparent. Block copolymers and polymeric micelles do not consist of one single molecular weight or sized species. Rather, they consist as a distribution range and thus should be described by their averaged molecular masses and mass distribution for block copolymers and mean size and PDI for micelles as well as. Both of these must be under tight control for optimal consideration in the regulatory process. Micelle morphology and zeta potential could also play critical roles in the interactions of polymeric micelles with cells and proteins. Performing in vitro drug release studies, in the most biologically relevant matrix possible, is essential to predicting in vivo stability. The ideal matrix for these studies would be blood plasma, but a buffer containing albumin can be helpful as well, when blood serum is unavailable [221, 235].
The in vivo stability of polymeric micelles in particular can be difficult to assess. The dynamic nature of polymeric micelles means the polymeric micelle dissociates over time often leading to changing measurements of free, unbound drug and that which is physically encapsulated in the micelle. It is important to measure all “species” of drug present in the blood and determine which of these is most critical to therapeutic efficacy [291]. Otherwise, extensive characterization using the methods discussed in Section 3 above are useful to support approval. In general, for the nano-based pharmaceuticals the physicochemical sameness is more difficult to relate to the same therapeutic performance because the physiochemical characteristics that may affect performance of a delivered drug are more complex and could affect therapeutic performance in different ways. However, as the understanding of this relationship improves one should expect that a more robust and well-defined regulatory process develops. In the meantime, the researchers in the field of translational nanomedicine could utilize resources available from the government sponsored programs (e.g., the U.S. National Cancer Institute Sponsored NCL, https://ncl.cancer.gov/; the USFDA Learning Portal, https://www.fda.gov/training-and-continuing-education/fda-learning-portal-students-academia-and-industry; European Union Nanomedicine Characterization Lab, http://www.euncl.eu/; EMA, https://www.ema.europa.eu/en/partners-networks/international-activities/training-opportunities-non-eu-regulators). Some of these resources including the USFDA and EMA, contain trainings and workshops for students and pharmaceutical scientists to help better understand the drug approval process. Moreover, since 2012 the USFDA implemented Generic Drug User Fee Amendment (GDUFA) that provides additional resources for developers of generic medicines (https://www.fda.gov/industry/fda-user-fee-programs/generic-drug-user-fee-amendments). Other available resources, such as the NCL provide a standardized assay cascade protocols (https://ncl.cancer.gov/resources/assay-cascade-protocols) and assist nanomedicine community with conducting IND-enabling preclinical studies (https://ncl.cancer.gov/working-ncl/ncl-assaycascade-application-process). Consolidated efforts among researchers in regulatory agencies, government, industry and academia, and other nanomedicine stakeholders (e.g., educators, patients, physicians) are required to advance the science and make regulatory approval for nanomedicines more straight forward [235].
5.3. Clinical status of polymeric micelle formulations
A number of polymeric micelle formulations utilizing the physical entrapment of poorly soluble small molecules have been reported in literature. Since then, many polymeric micelle formulations have reached clinical trials in several countries [9, 10, 300]. Examples of polymeric micelle drugs which have obtained regulatory approval or clinical evaluation are presented in Table 6. These drugs have all been for cancer indications. The first polymeric micelle which was clinically approved (in South Korea) was Genexol® PM. Of the listed drugs, two have been approved (Genexol® PM and Nanoxel® M in South Korea) and two have completed phase 3 (NK105 and NC-6004). Some selected examples are considered below in greater detail.
Table 6.
Examples of polymeric micelle-based drug products in clinical trials or approved.
| Product code name | API | Polymer | Physicochemical properties | Development Stage | Comments | Trial code and/or Ref. |
|---|---|---|---|---|---|---|
| Genexol® PM1 | Paclitaxel | mPEG-b-PDLLA | Size: 20–50 nm, LC: 16.7% | Approved in South Korea, Philippines, India, and Vietnam | Enabled higher doses of paclitaxel with decreased toxicity in phase I study, Improved overall response rate (58.5%) compared to that of Taxol® (21–54%) in patients with metastatic breast cancer in phase II study. | [9, 11] NCT00876486 NCT02064829 |
| Nanoxel® M1 | Docetaxel | mPEG-b-PDLLA | Size: 25.4 nm | Approved in South Korea | Comparable efficacy and superior safety profile compared to that of conventional docetaxel formulated in polysorbate 80 (Taxotere) [13] | NCT01336582 NCT02639858 |
| NK1052 | Paclitaxel | mPEG-b-modified P(Asp) | Size: 85 nm LC: 23% | Phase 3 (completed) | Improved plasma AUC of paclitaxel and reduced hypersensitivity reaction compared to conventional paclitaxel [238]. Failed to improve efficacy of paclitaxel in phase III study [307]. | NCT01644890 |
| NC-60043 | Cisplatin | PEG-b-P(Glu) coordination complex | Size: 28 nm LC: 39 % | Phase 3 (completed) | Improved safety profile and patient’s quality of life, while showed modest efficacy in phase II study [309]. | NCT02043288 |
| SP1049C4 | Doxorubicin | Pluronic® L61 and F127 | Size: < 30 nm LC: 8.2 % | Phase 2 (completed) | Shown efficacy of doxorubicin as a single agent in phase II study | [10, 17] |
| NK0125 | SN-38 | PEG-b-P(Glu) covalent drug-copolymer conjugate | Size: 20 nm LC: 20 % | Phase 2 (completed) | Showed efficacy in patients with sensitive relapsed small cell lung cancer and toxicity was manageable. | NCT00951054 NCT00951613 |
| CPC6346 (CriPec®) | Docetaxel | PEG-b-P(HPMAm-Lacn) covalent drug-copolymer conjugate | Size: 66 nm LC: 12 % | Phase 2 (recruiting) | Possible improved safety profile, skin toxicity was seen at high dose in phase I study | NCT02442531 NCT03742713 |
| NK9117 | Doxorubicin | PEG-b-P(Asp) covalent drug-copolymer conjugate | Size: 40 nm | Phase 1 (completed) | Comparable toxicity profile of doxorubicin to free doxorucin, No infusion-related reaction in phase I study | [146] |
| NC-40168 | Oxaliplatin | PEG-b-P(Glu) coordination complex | Size: 40 nm LC: 32 % | Phase 1 (completed) | No results available yet | NCT03168035 |
Developed by Samyang Biopharmaceuticals Corp.
originally developed by NanoCarrier Co., Ltd. and licensed to Nippon Kayaku Co., Ltd.
developed by NanoCarrier Co., Ltd. in collaboration with Orient Europharma Co., Ltd. (drug is bound to the P(Glu) block of the block copolymer via coordination bonds)
originally developed by Supratek Pharma Inc. and acquired by SoftKemo Pharma Corp. (now termed SKC1049)
developed jointly by NanoCarrier Co., Ltd. and Nippon Kayaku Co., Ltd. (SN-38 is chemically conjugated to the P(Glu) block of the copolymer)
developed by Cristal Therapeutics (cdocetaxel is covalently conjugated m-PEG-b-poly[N-(2-hydroxypropyl)methacrylamide lactate] (mPEG-b-p(HPMAm-Lacn) copolymer)
developed by Nippon Kayaku Co., Ltd. (doxorubicin is chemically conjugated to the P(Asp) block of the copolymer, and the free drug is solubilized in the micelles of the resulting conjugate)
developed by NanoCarrier Co., Ltd. (drug is bound to the P(Glu) block of the block copolymer via coordination bonds).
5.3.1. Genexol® PM
Genexol® PM is a Cremophor EL-free, polymeric micelle formulation incorporating paclitaxel as the API. Originally developed by Samyang Biopharmaceuticals Corp., this is the first polymeric micelle formulation approved for human use in South Korea and several other countries for patients with metastatic breast cancer (MBC), NSCLC, and ovarian cancer. Genexol® PM consists of paclitaxel in polymeric micelles of mPEG-b-PDLLA (mPEG: 2,000 g/mol, PDLLA: 1,750 g/mol, PDI: 1.0–1.2). The micelles are manufactured by the solid dispersion method using the thin film hydration approach and are nano-sized particles with well-defined spherical structure in aqueous media (20–50 nm in diameter and 16.7% LC of paclitaxel) [6]. Under sink conditions, paclitaxel was slowly released from the micelle with 65% released at 24 hours and 95% released at 48 hours [110].
In a phase I clinical trial in South Korea, twenty-one patients entered into a dose-escalation study and were treated with Genexol® PM ranging from 135 mg/m2 to 390 mg/m2 without premedication of hydrocortisone and histamine blocker [11]. No acute hypersensitivity reactions were observed, while neuropathy, myalgia, and neutropenia were observed which limited the highest dose. The MTD was determined to be 390 mg/m2 for Genexol® PM, which is higher than that of the commercially available paclitaxel formulations Taxol® (200 mg/m2) and Abraxane® (300 mg/m2) [11].
Based on the established toxicity profile, a multi-centered phase II study of Genexol® PM with cisplatin was performed on patients with advanced NSCLC in South Korea [301]. In this study, the patients were administered Genexol® PM 230 mg/m2 and cisplatin 60 mg/m2. The overall response rate was 37.7% and the median survival period was 21.7 months, which indicated significant improvements compared to previous clinical trials conducted with Taxol® 175–200 mg/m2 and cisplatin 75–80 mg/m2 [301]. Another phase II study in South Korea reported the clinical benefit of Genexol® PM in patients with metastatic breast cancer [9]. Forty-one patients were enrolled in the study and administered with a Genexol® PM infusion (300 mg/m2) every 3 weeks. The overall response rate was 58.5%, which is superior to that of Abraxane® (47.6%) and Taxol® (21–54%). With improved therapeutic outcomes in clinical trials conducted in South Korea, Genexol® PM received regulatory approval in South Korea and was marketed there since 2007 for the treatment of MBC, NSCLC, and ovarian cancers. In addition to South Korea it received marketing approval in several other countries including India, Serbia, Philippines, and Vietnam. A completed phase III study in South Korea further evaluated the efficacy and safety of Genexol® PM (260 mg/m2) compared to paclitaxel (175–200 mg/m2) in recurrent or MBC (ClinicalTrials.gov: NCT00876486) [302], but the results of this trial have not been reported yet. Other clinical trials of Genexol® PM were conducted for patients with advanced biliary tract cancer (Genexol® PM 100 mg/m2 with gemcitabine 1,000 mg/m2) [303], with unresectable thymic epithelial tumors (Genexol® PM 230 mg/m2 with cisplatin 70 mg/m2) [304], with ovarian cancer (as first-line treatment) (Genexol® PM 260 mg/m2) [305], which posted positive results on therapeutic outcomes from treatment with Genexol® PM. In conclusion, Genexol® PM could improve the dosing of paclitaxel and the safety of the drug in patients. In clinical trials Genexol® PM demonstrated improved therapeutic outcomes as a formulation which limited hypersensitivity reactions in patients through the elimination of toxic excipients. Although Genexol® PM has shown some hypersensitivity reaction in the clinic, probably due to the PEG, this is not nearly as much of a concern as for Taxol® which contains Cremophor EL [301]. The tolerable dose of paclitaxel was increased by virtue of polymeric micelle formulation, resulting in an increased MTD. From the results of a bioequivalence study, it is expected that it could gain regulatory approval in the United States via the 505(b)(2) pathway.
The clinical development of Genexol® PM in the USA (under the trade name of Cynviloq™) was initiated by Sorrento Therapeutics after the exclusive distribution rights to Genexol® PM were acquired by Sorrento Therapeutics in 2013. In 2014, bioequivalence studies of Cynviloq™ versus Abraxane® were conducted in patients with metastatic or locally recurrent breast cancer and patients with NSCLC (ClinicalTrials.gov: NCT02064829). Preliminary positive data in eight patients was reported in 2014 which potentially supported the bioequivalence of the two products (https://sorrentotherapeutics.com/sorrento-announces-first-patient-dosed-in-registration-trial-to-evaluate-bioequivalence-between-cynviloq-and-abraxane/). The bioequivalence of Cynviloq™ to Abraxane® could grant 505(b)(2) pathway approval by the USFDA and expedite the regulatory process by avoiding extensive clinical trials to validate efficacy versus the standard of care [13]. No updates are available on the bioequivalence of these formulations since Cynviloq™ was acquired in 2015 by NantWorks, which was founded by Dr. Patrick Soon-Shiong who developed Abraxane®.
5.3.2. Nanoxel® M
Nanoxel® M is a docetaxel-loaded polymeric micelle formulation which was developed by Samyang Biopharmaceuticals Corp. and received regulatory approval in South Korea in 2012 (according to announcement from Samyang Biopharm (https://samyangbiopharm.com/eng/ProductIntroduce/injection) and Korea Pharmaceutical Information Center (http://www.health.kr/searchDrug/result_drug.asp?drug_cd=2012122700008). Also, Nanoxel® M is currently under clinical evaluation in the USA. The formulation is composed of docetaxel in mPEG-b-PDLLA (mPEG: 2,000 g/mol and PDLLA: 1,765 g/mol) polymeric micelle [306]. It is manufactured by the thin-film hydration method, which produces uniform micelles with a hydrodynamic size of 25.4 nm [306]. However, Nanoxel® M has limited micelle stability in solution after reconstitution (up to 6 h in saline) [306]. A phase I clinical trial was conducted in South Korea (NCT01336582) and it was reported that Nanoxel® M (70 mg/m2) exhibited an improved drug safety profile compared to the conventional docetaxel formulation in patients with advanced solid tumors [13]. This is believed to be due to the removal of toxicity of Polysorbate 80 that is contained in a conventional docetaxel formulation, Taxotere. Currently, additional clinical trials are recruiting participants to evaluate the efficacy and safety of Nanoxel® M in recurrent or metastatic head and neck squamous cell carcinoma (Nanoxel® M 75 mg/m2, ClinicalTrials.gov: NCT02639858), Nanoxel® M and oxaliplatin for patients with metastatic esophageal squamous cell carcinoma (Nanoxel® M 75 mg/m2 and oxaliplatin 120 mg/m2, ClinicalTrials.gov: NCT03585673), and the safety of Nanoxel® M in patients with other various types of cancers (ClinicalTrials.gov: NCT04066335).
5.3.3. NK105
NK105 is a paclitaxel-loaded polymeric micelle formulation which was originally developed by Kataoka’s group and NanoCarrier Co., Ltd. in the early 1990s and advanced to clinical trials (phase III completed, ClinicalTrials.gov: NCT01644890). NK105 is composed of paclitaxel and modified mPEG-b-P(Asp) block copolymer (mPEG = 12,000 g/mol and P(Asp) = 8,000 g/mol) where half of the carboxylic groups P(Asp) block is modified with hydrophobic 4-phenyl-1-butanol to increase its hydrophobicity and improve drug incorporation [7]. The modified P(Asp) block, which forms the hydrophobic core of the micelle, enhances the drug loading in the micelle core via physical entrapment [7]. NK105 exhibited 23% of drug loading with a hydrodynamic size of approx. 90 nm [7].
In a phase I trial of NK105, nineteen patients with various type of cancers (pancreatic, bile duct, gastric, and colonic) were recruited to examine the safety and PK of NK105. NK105 doses ranging from 10 mg/m2 to 180 mg/m2 was administered to patients without premedication. The MTD of NK105 was determined to be 180 mg/m2 due to dose limiting hematological toxicity (neutropenia). The PK profile of NK105 showed that the plasma AUC of paclitaxel from NK 105 (at 150 mg/m2) was approximately 15-fold higher than that of paclitaxel from Taxol® (210 mg/m2) [238]. Also, NK105 was well tolerated exhibiting reduced hypersensitivity reactions in patients.
A phase II trial of NK105 recruited 57 patients with advanced gastric cancer after the failure of first-line chemotherapy [300]. The patients were administered NK105 (at 150 mg/m2 paclitaxel) without anti-allergic premedication. The results of the phase II study revealed that NK105 showed modest activity and tolerability for paclitaxel. The overall response rate was 25% with median progress free survival of 3.0 months and median overall survival of 14.4 months [300]. In this study, conventional drug was not administered to patients since there was no standard of care for advanced gastric cancer. Thus, the interpretation of the results from the phase II study of NK105 is difficult. In July 2016, an open-label phase III non-inferiority trial of NK105 in patients with metastatic or recurrent breast cancer was completed (ClinicalTrials.gov: NCT01644890) [307]. Four hundred thirty-six patients were enrolled in the study and administered either NK105 (65 mg/m2) or conventional paclitaxel (80 mg/m2). The results of the phase III trial revealed that the primary endpoint (statistical non-inferiority of progression-free survival) was not met (the median progression free survival (PFS) of 8.4 and 8.5 months for NK105 and paclitaxel, respectively, and the median overall survival of 31.2 and 36.2 months, and overall response rates of 31.6% and 39.0%, respectively) [307]. However, the incidence of peripheral sensory neuropathy among patients treated with NK105 was decreased compared to that of paclitaxel, indicating an improved toxicity profile of the drug in NK105 [307]. Overall, NK105 successfully improved the PK and safety profiles of paclitaxel by incorporating the drug into a polymeric micelle formulation. However, NK105 failed to improve the efficacy of paclitaxel in clinical trials.
NC-6004
Originally developed by Kataoka’s group and NanoCarrier Co., Ltd., NC-6004 is a cisplatin-containing polymeric micelle formed via coordination of platinum drug with the P(Glu) segment of the PEG-b-P(Glu) block copolymer (PEG: 12,000 g/mol and P(Glu): 6,000 g/mol) [153]. The complex was formed between the platinum metal and the carboxylic acid group in the P(Glu) segment of the block copolymer resulting in a cisplatin-loaded core in NC-6004. NC-6004 exhibited stable micelles in solution and slowly released cisplatin from the micelle for over 150 h in physiological saline. NC-6004 exhibited a narrow size distribution in solution with a size of 28 nm and loading of cisplatin in NC-6004 was up to 39% LC [153].
In a phase I clinical trial, a total of 17 patients with various type of advanced solid tumors were recruited to evaluate the efficacy and safety of NC-6004 [308]. NC-6004 was administered in doses ranging from 10 mg/m2 to 120 mg/m2. Drug toxicities were observed at the dose of 90 mg/m2 of NC-6004, and 120 mg/m2 of NC-6004 was determined as the MTD due to renal impairment and hypersensitivity reactions in patients [308]. Plasma samples were processed via gel-filtration and ultrafiltration methods to analyze the subpopulations of cisplatin in plasma such as total platinum, platinum in NC-6004, and extra-micellar platinum [308]. In the PK profile, the amounts of low-molecular mass platinum, including cisplatin released from NC-6004, were marginal compared to gel-filterable platinum (micelle encapsulated) and total platinum, indicating sustained release of cisplatin from NC-6004 in systemic circulation. Also, NC-6004 exhibited extended half-life and increased AUC compared to cisplatin infused as an aqueous solution due to the prolonged blood circulation of NC-6004.
A Phase Ib/II trial of NC-6004 with gemcitabine was conducted in patients with advanced solid tumors to evaluate the safety and tolerability (ClinicalTrials.gov: NCT02240238) [309]. NC-6004 was administered to patients at 60 mg/m2 to 180 mg/m2 (on day 1) with gemcitabine (1,250 mg/m2, on days 1 and 8) every 3 weeks. A dose of 135 mg/m2 of NC-6004 was determined as the MTD and common hematologic adverse events were leukopenia and thrombocytopenia. Tumor shrinkage, partial responses, and stable disease were observed in 55%, 15%, and 70% of total patients, respectively [309]. A Phase III clinical trial of NC-6004 with gemcitabine in patients with locally advanced or metastatic pancreatic cancer has been completed, but no results reported yet (ClinicalTrials.gov: NCT02043288).
Overall, NC-6004 formed a stable polymeric micelle with a sustained release of cisplatin. Clinical trials of NC-6004 demonstrated long circulation and sustained drug release of NC-6004 in patients. Also, NC-6004 could improve the drug toxicity profile, such as reducing nephrotoxicity. Additional clinical trials are ongoing on NC-6004 in patient with head and neck cancer and pancreatic cancers.
SP1049C
SP1049C is a doxorubicin-loaded Pluronic®-based formulation which was developed by Supratek Pharma Inc. and was the first polymeric micelle drug formulation to enter clinical trials in 1999 [17]. It was then evaluated in Phase II clinical trials and has shown positive results [10]. The formulation was prepared by the blending of two Pluronic® block copolymers (L61 and F127) which consist of PEO and PPO at a 1:8 weight ratio of L61:F127 [23] and compounding this mixture with doxorubicin. Initially the micellar formulation was prepared before injection by dissolving doxorubicin in the sterile aqueous solution of the block copolymer mixture. Subsequently the dry form of the SP1049C was dev eloped that is reconstituted by adding saline. Physicochemical analysis of SP1049C revealed that the micelles had well-defined spherical morphology with a size of less than 30 nm, and the doxorubicin LC was 8.2 %. According to Batrakova et al., Pluronic® L61 exhibited sensitization of multidrug-resistant cancer cells, thereby enhancing the cytotoxicity of doxorubicin while F127 showed stabilization of the doxorubicin-loaded micelle formulation in aqueous solution [23]. In drug-sensitive tumors, SP1049C has the same efficacy as doxorubicin but, at the same time, is highly active against multidrug-resistant tumors and cancer stem cells [310, 311].
In the phase I clinical trial, SP1049C exhibited a similar PK profile of doxorubicin to the conventional doxorubicin formulation and a similar MTD of 70 mg/m2 in the patients with metastatic or recurrent solid tumors [17]. Interestingly, doxorubicin-related toxicity, such as hand–foot syndrome was less prevalent in the SP1049C treated patients compared to those treated with conventional doxorubicin. Later, a Phase II trial demonstrated that SP1049C has a notable single-agent activity as well as an acceptable safety profile in patients with advanced carcinoma of the esophagus and gastroesophageal junction [10]. Patients treated with SP1049C at a dose 75 mg/m2 (doxorubicin equivalents) had an objective response rate of 47% in the evaluable patient population, and 43% in the intent-to-treat population along with the median overall survival of 10 months and PFS of 6.6 months. The principal toxicity concern, neutropenia, was manageable and reversible, and in line with that expected from doxorubicin 75 mg/m2 in the standard formulation. Notably, doxorubicin (API of SP1049C) is considered to be inactive (response rates less than 20%) in advanced adenocarcinoma of the esophagus and is not used in this indication. In 2008, the SP1049C obtained a special protocol assessment on a single approvable Phase 3 trial in refractory upper gastrointestinal adenocarcinoma and has obtained an orphan drug designation in adenocarcinoma of the esophagus from USFDA. In addition, SP1049C has obtained two USFDA orphan drug designations for the carcinoma of the esophagus and gastric cancer. However, no clinical data has been reported since then. The development of SP1049C was suspended as a result of the economic crisis of 2008. In late 2016 patent rights to SP1049C were acquired by SoftKemo Pharma Corp. to complete the final development of the novel anticancer therapeutic now code named SKC1049. Overall, SP1049C may have several applications: as a new agent with novel mechanism of action for combination therapy; salvage therapy; and as the first line single agent in doxorubicin indications.
6. Conclusions and future directions
Polymeric micelle formulations for poorly soluble small molecules have been extensively studied over three decades as a versatile platform for drug delivery. Their ability to be customized and tailored to specific needs is a distinct advantage over other drug delivery systems. While simple at first pass, it is clear that polymeric micelles represent a much more complex system than early understanding suggests. With some successful preclinical results, several polymeric micelle formulations have entered clinical trials, but only a few have received regulatory approval for human use. There have been many challenges affecting their ability to navigate the regulatory pathway, which we have worked to address in this review.
In Section 2, we have focused on the types of polymeric materials that are used for the manufacturing of polymeric micelles. Block copolymers segments required for forming amphiphilic block copolymers were described in order to aid in the proper selection of block copolymer components for the efficient solubilization of target molecules. Although many polymeric materials were studied during the previous three decades, there are only a few such materials used for the manufacturing of micelles which have reached the clinical stage of development. These include several hydrophobic polymers that are used to design the core-forming blocks of the polymeric micelles and just one hydrophilic polymer, PEG, used to manufacture the shell of the micelles, which is applied in several nanoformulations in clinical studies. There are limitations to the use of the current materials including relatively poor drug incorporation in some cases, toxicity, and, in the case of PEG, unfavorable immunological interactions such as antibody responses. Therefore, the development of novel materials that are safe, enable high drug loading, and are “immunologically inert” is needed. In our opinion POx- and POzi- based block copolymers satisfy these requirements and deserve future research, and more useful materials are likely to emerge in the future.
In Section 3, we have extensively described polymeric micelle formulations for the delivery of poorly soluble small drug molecules. While initially thought to solubilize based on simple hydrophobic interactions, recent advances in analytical techniques have revealed new insights into these drug delivery systems. For example, drug-polymer interactions are not simply limited to the hydrophobic blocks. In fact, the hydrophilic, shell-forming blocks also play an intimate roll in solubilization. Hansen’s solubility parameters and Flory-Huggins theory are successful predictors of polymer-drug compatibility in certain cases, but in others a more complex analysis is needed to account for all the interactions taking place between polymers and drugs. To improve our understanding of drug solubilization, multi-disciplinary approaches for investigating detailed molecular interactions between hydrophobic segments of block copolymers and encapsulated drugs were described. Recent advances in ssNMR, fluorescence analysis, MD, and SANS techniques have proven effective in probing the intimate interactions taking place in the micelle core and beyond. Computational methods, like QSPR, have shown promise for predicting polymer-drug compatibility and will surely be at the forefront of this field moving forward. Section 3 provides the formulation chemist with a key set of theoretical and practical tools for characterizing novel formulations to better prepare for pre-clinical formulation analysis.
In Section 4, we discuss many of the intricacies of analyzing polymeric micelle formulations. Due to their unique dynamic nature, we must place additional considerations on their PK analysis. We layout many of the considerations such as protein binding changes, concerns with CMC, and morphology and size considerations amongst many others. A limited understanding of the complex dynamics of polymeric micelle formulations has hindered their translation into clinics. In this review, we have highlighted some of the key problems still facing the field today which, when addressed early in formulation development, may increase the translatability of these dynamic formulations. Section 4 can be considered a basic guide to the design of and analysis of polymeric micelle formulations to help the researcher expedite their preclinical formulation and PK analysis.
Clinical investigations of polymeric micelle formulations have revealed promising therapeutic outcomes for human use, while we have also witnessed a number of clinical trial failures from other polymeric micelle formulations. Section 5 provided the reader with an introduction to the regulatory pathway and challenges facing the approval of polymeric micelles and nanomedicines in general. We hope that our section, in concert with the provided references, can help the reader better prepare for regulatory approval early and often throughout the formulation development process. Previous sections provide the scientist with some of the tools they need to gather CQAs in preparation for regulatory approval, which can hopefully expedite translation and improve clinical outcomes.
It is also important to point out important areas of research and future directions that were deliberately left outside of the focused consideration of the current review. One very important area is the use of ionic block copolymers for the drug delivery of biopolymers. The introduction of cationic block copolymers that contain polycation blocks to bind negatively charged nucleic acids and water soluble anionic blocks to ensure micelle stability in solution [312–314] have resulted in a myriad of studies focusing on the use of polymeric micelles for the delivery of a variety of therapeutic molecules including plasmid DNA (pDNA), antisense oligonucleotides, messenger RNA (mRNA), small interfering RNA (siRNA) as well as negatively charged drug molecules such as nucleoside triphosphates [314–317]. Proteins, being polyampholytes, can also be formulated into polymeric micelles with either cationic or anionic block copolymers [318, 319]. This technology can also be expanded for the delivery of supramolecular biopolymer complexes including oligomeric enzymes, multienzyme complexes [320], or protein and nucleic acid complexes, such as Cas9 and guide RNA [321, 322]. In all these cases, the polyelectrolyte blocks of the block copolymers bind electrostatically with the oppositely charged molecules forming a polyion complex, which is usually insoluble and becomes segregated within the core of polymeric micelles. The hydrophilic blocks of these block copolymers form a shell around the core that stabilizes the micelles in aqueous dispersion. These structures sometimes are called “polyion complex micelles” or “block ionomer complexes” [323–325]. In selected cases, in addition to the electrostatic interactions, the hydrophobic interactions of the reacting molecules with each other, or the formation of hydrogen bonds between block copolymer and therapeutic molecules, can play an essential role in the self-assembly and stabilization of such polymeric micelles [326]. Despite great advancements, none of these technologies have reached the clinical stage at this time and therefore we left them outside of the current consideration. These important technologies have some key features that are common with amphiphilic block copolymer micelles discussed in this review but also are dissimilar in certain fundamental aspects, such as mechanisms of formation, stability, interactions with the components of body fluids, and cell entry which therefore require a separate update and review.
We also would like to point out that most, but not all, of the studies discussed in this paper focus on the delivery of small molecules using polymeric micelles to treat cancer. All examples of clinically approved polymeric micelle drugs or polymeric micelle drugs in clinical development discussed here belong to the area of cancer therapy. In addition, numerous studies have been reported in preclinical animal models using small molecule drugs in polymeric micelles of amphiphilic block copolymers for the treatment of various other diseases and conditions, such as autoimmune diseases, cardiovascular disease, dementia, germ infection, ocular disease, pain management, pulmonary arterial hypertension, skin disease, spinal cord injury, and wound healing. In these studies, the polymeric micelles were shown to greatly improve drug solubility and formulation stability, improve PK and bioavailability of the drug at the target site (e.g., longer systemic circulation after IV injection, increased brain exposure, enhanced drug disposition in skin or corneal permeability), decrease drug toxicity and unwanted side effects of treatments, increase specific pharmacological activity of the drug, and in some cases show bioequivalence to the clinically approved formats of the drug. We summarized examples of these studies in Table 7 to provide the reader with the insight about the breadth of potential future applications and possible clinical developments of the polymeric micelle drug formulations beyond cancer.
Table 7.
Examples of formulations of poorly soluble small molecule drugs in polymeric micelles of amphiphilic block copolymers evaluated for treatment of various diseases and other medical use.
| Therapeutic agent | Drug class / mechanism | Block copolymers | Main study results |
|---|---|---|---|
| Dexamethasone | Corticosteroids /Anti-inflammatory | PEG-b-PCL | The polymeric micelles exhibited longer systemic circulation compared to conventional drug format and accumulated preferentially in inflamed joints. Reduced joint swelling, bone erosion, and inflammatory cytokine expression were observed in joint tissue and serum for the micelle formulation treatments in a rat model [327]. |
| Betamethasone phosphate | Corticosteroids /Anti-inflammatory | PLA and PEG-b-PLGA | In in vivo studies, a 35% decrease in paw inflammation was observed in the first day of treatment and the efficacy of the formulation maintained for 9 days with a single injection. In AbIA mice, a single injection of the micelle formulation resulted in complete remission of the inflammatory response after 1 week [328]. |
| Andrographolide | Labdane diterpenoids / Anti-inflammatory and anti-platelet aggregation | PEG-b-poly(propylene sulphide) | Due to the reactive oxygen species (ROS)-responsive nature of the polymer, the micelle not only serves as a stimuli-responsive drug carrier to quickly release andrographolide, but also consumes ROS at the pathologic sites, resulting in synchronical alleviation of inflammation and oxidative stress [329]. |
| Rivastigmine | Acetylcholinesterase inhibitor | PEG-b-PCL | The PK study in rats indicated that the brain uptake of rivastigmine-loaded micelle was significantly higher than that of the free drug. Pharmacodynamic studies using the Morris water maze test confirmed that faster regain of memory loss with micellar formulation when compared to the free drug solution [330]. |
| Amphotericin B | Aminoglycosides / Antifungal | Pluronic F127 | Amphotericin B-containing Poloxamer P407-based polymeric micelles were the most efficient among the polymeric micelle, amphotericin B and Ambisome® in a mouse model of Leishmania amazonensis. Low parasitism, Th1 immunity and no significant toxicity were seen in the treated animals [331]. |
| Protoporphyrin IX | Porphyrin / Photosensitizer | PEG-b -PCL and PCL-b- PBAE | Bacterial killing of multidrug resistant Staphylococcus aureus biofilms by protoporphyrin IX-loaded micelles was superior compared to the control formulation. Staphylococcus aureus infection was eradicated by daily intravenous injection of the micelle formulation combined with its light-activation at the infected site in the mouse models of the bacteria infection [332]. |
| Naphthoquinone derivative | Naphthoquinones / Antileishmanial | Poloxamer 407 | The polymeric micelle formulation was more effective to treat Leishmania amazonensis infected mice in comparison to amphotericin B and its liposomal formulation, Ambisome®. No severe toxicity was identified in the treated animal model [333]. |
| 8-Hydroxyquinoline | Quinolones / Antifungal | Poloxamer 407 | The 8-hydroxyquinoline loaded poloxamer 407 micelle showed significant reduction in the average lesion diameter of the infected tissue and in the parasite burden in the skin and all infected organs (spleen, liver and draining lymph nodes) compared to controls in the mouse model of Leishmania amazonensis infection [334]. |
| Itraconazole | Azoles /Antifungal | Linear-dendritic PEG-b-PCL | Itraconazole loaded micelle had similar efficacy against Candida albicans to that of the free drug in in vitro. In vivo PK study in healthy mice demonstrated that the micelle formulation could improve tissue distribution of itraconazole [335]. |
| PDLLA and PEG-b-PDLLA | Single or repeated doses of itraconazole in rats and dogs equivalent to the drug clinical doses, were well tolerated. The plasma levels of the micellar itraconazole and its major metabolite were comparable to those of the control formulations (Sporanox® Injection and Oral Solution) [336]. | ||
| Efavirenz | Benzoxazines / Antiretrovirals | Pluronic F127 and Tetronic® T904 | The polymeric micelle formulation exhibited a dramatic drug solubility increase (over 8400-fold) and superior stability in comparison with the control formulations [337]. |
| Ocular disease | |||
| α-Lipoic acid | Heterocyclic thia fatty acids / Antioxidant | Polyvinyl caprolactam-b-poly(vinyl acetate)-b-PEG | The polymeric micelle formulation improved solubility and accumulation of enhanced α-lipoic acid into the bovine cornea compared to regular eye drops format of this agent. Drug containing polymeric micelles tolerated dilution in lachrymal fluid and freeze-drying [338]. |
| Diclofenac | Phenylacetic acid derivatives /Anti-inflammatory | PEG-b-PCL | The polymeric micelles increased diclofenac permeability up to 17-fold compared to that of conventional diclofenac eye drops in the rabbit cornea. In vivo PK study revealed that the diclofenac in the polymeric micelles has 2-fold greater drug exposure than the eye drops [339]. |
| Pain management | |||
| Propofol | Cumenes / Anesthetics | PVP-b-PDLLA | Propofol loaded polymeric micelles induced anesthesia effect in healthy rats. PK profile of the drug in the micelles was not significantly different from that of an emulsion formulation (Diprivan®) [340]. |
| Rapamycin | Macrolides / mTOR inhibitor | PEG-b-PCL | In a rat model of pulmonary arterial hypertension of mTOR inhibitor Rapamycin in polymeric micelles administered intravenously displayed lower toxicity, increased accumulation in lungs, and similar efficacy as the free drug in DMSO administered intraperitoneally [341]. |
| Tacrolimus | Macrolides / Immunosuppressant | mPEG-b-dihexyl substituted polylactide | Tacrolimus in polymeric micelle formulation showed improved drug solubility (by 400-fold), and drug disposition in porcine and human skin compared to tacrolimus ointment formulation (Protopic®) [342]. |
| Retinoic acid | Vitamers of vitamin A / Anti-acne | mPEG-b-dihexyl substituted polylactide | Retinoic acid in polymeric micelles showed improved drug disposition in porcine and human skin compared to commercially available gel formulation (Retin-A micro) [343]. |
| Spinal cord injury | |||
| Methylprednisolone | Steroids / Anti-inflammatory | PEG-b-PPO-b-PEG | Polymeric micelles increased the methylprednisolone level in plasma and spinal cord compared to the free drug solution in rabbit model. Increased expression of anti-apoptotic proteins was seen in animal group treated with micellar [344]. |
| Dexamethasone acetate | Corticosteroids /Anti-inflammatory | mPEG-b-PCL | Polymeric micelles improved dexamethasone solubility and the drug neuroprotective effect in the hemisection spinal cord injury model of rats [345]. |
| Zonisamide | Benzisoxazoles /Anticonvulsant | mPEG-b-PLLA-poly(trimethylene carbonate) | In the hemisection spinal cord injury rat model IV injection of zonisamide in polymeric micelles improved the motor function and neuron density compared to the effect of the drug in dimethyl sulfoxide [346]. |
| Wound healing | |||
| Curcumin | Curcuminoids / Anti-inflammatory and antioxidant activities | PEG-b-PCL | Curcumin loaded polymeric micelle exhibited good tissue adhesion and could release curcumin for an extended period in vitro. In animal excision model and histopathologic examination in rats, the formulation exhibited enhancement of cutaneous wound repair [347]. |
Polymeric micelle formulations hold a clinical importance as a delivery platform for poorly soluble small molecules, and for this purpose current polymeric micelle formulation systems have to further evolve to serve as efficient drug carriers. We believe that the comprehensive analysis of drug encapsulation and subsequent drug release profile in systemic circulation will provide insight for the future design of novel polymeric micelle systems for human use. We hope that this review aids in the further development and intelligent design of polymers for specific, drug-tailored applications in the future. In particular, we believe strongly in the role of computational methods and AI for the for the drug-oriented design of polymers. This drug-oriented design can improve the efficiency of translation from lab, to pre-clinical, to clinical applications. Altogether, the educated, informed design and innovative analysis of polymeric micelle formulations should capitalize on their potential and capabilities as essential drug carriers allowing us to increase their ability to improve clinical outcomes in systemic delivery.
Acknowledgement:
We gratefully acknowledge kind assistance of Rogério Gaspar of University of Lisbon, as well as Marina A. Dobrovolskaia and Stephan T. Stern, both of the Nanotechnology Characterization Laboratory, US Frederick National Laboratory for Cancer Research, who read select sections of this paper and provided valuable insight. This work was supported by the National Cancer Institute (NCI) Alliance for Nanotechnology in Cancer and U54CA198999 (Carolina Center of Cancer Nanotechnology Excellence), U01CA198910. Partial support of DH and JDR work on polymeric micelles came from R01NS102627 awarded to Dr. Tim Gershon at UNC-Chapel Hill.
Abbreviations
- %ID
percent injected dose
- 17-AAG
17-allylamino-17-demethoxygeldanamycin
- ABC
accelerated blood clearance
- ANVISA
Brazilian Health Surveillance Agency
- API
active pharmaceutical ingredient
- AUC
area under the curve
- CDDP, cisplatin
cis-dichlorodiaminepatinum(II)
- CED
cohesive energy density
- CI
combination index
- CL
clearance
- CMC
critical micelle concentration
- CQA
critical quality attribute
- DACHPt
dichloro(1,2-diaminocyclohexane)platinum(II)
- DPD
dissipative particle dynamics
- ELP
elastin-like polypeptide
- EMA
European Medicines Agency
- GCM
group contribution method
- GMP
good manufacturing process
- GRAS
generally regarded as safe
- HPMA
poly[N-(2-hydroxypropyl)methacrylamide]
- ICH
International Conference on Harmonisation
- LC
loading capacity
- LCRP
living cationic ring-opening polymerization
- LCST
lower critical solution temperature
- LE
loading efficiency
- MAA
marketing authorization application
- MBC
metastatic breast cancer
- MD
molecular dynamics
- MDR
multidrug resistant
- MHLW
the Ministry of Health, Labor, and Welfare
- MHRA
Medicines and Healthcare Products Regulatory Agency
- mPEG
methoxy-PEG
- mRNA
messenger RNA
- MTD
maximum tolerated dose
- MW
molecular weight
- NCL
Nanotechnology Characterization Laboratory
- NDA
New Drug Application
- NMPA
the National Medical Products Administration
- NMR
nuclear magnetic resonance
- NSCLC
non-small cell lung cancer
- OH-PEG
hydroxy-PEG
- Oxaliplatin
cis-oxalato-(trans-l)-1,2-diaminocyclohexane-platinum(II)
- P(Asp)
poly(aspartic acid)
- P(Glu)
poly(glutamic acid)
- PAMAM
polyamidoamine
- PBAE
poly(β-amino ester)
- PBLA
poly(β-benzyl-l-aspartate)
- PBLG
poly(γ-benzyl-α, l-glutamate)
- PBuOx
poly(2-n-butyl-2-oxazoline)
- PBuOzi
poly(2-n-butyl-2-oxazine)
- PCL
poly(Ɛ-caprolactone)
- PDI
polydispersity index
- PDLLA
poly(D,L-lactide)
- PDMA
Pharmaceutical and Medical Devices Agency
- PDMA
poly(N,N-dimethylacrylamide)
- pDNA
plasmid DNA
- PEG
polyethyleneglycol
- PEO
poly(ethylene oxide)
- PEtOx
poly(2-ethyl-2-oxazoline)
- PFS
progression free survival
- PiPrOx
poly(2-isopropyl-2-oxazoline)
- PK
pharmacokinetic
- PLGA
poly(D,L-lactide-co-glycolide)
- PLLA
poly(L-lactide)
- PMeOx
poly(2-methyl-2-oxazoline)
- PMMA
poly(methacrylic acid)
- PMMA
poly(methyl methacrylate)
- POx
poly(2-oxazoline)
- POzi
poly(2-oxazine)
- PPO
poly(propylene oxide)
- PPrOx
poly(2-n-propyl-2-oxazoline)
- PPrOzi
poly(2-n-propyl-oxazine)
- PTX
paclitaxel
- PVP
poly(vinylpyrrolidone)
- QSPR
quantitative structure property relationship
- RES
reticuloendothelial system
- RLD
reference listed drug
- ROP
ring-opening polymerization
- ROS
reactive oxygen species
- SANS
small angle neutron scattering
- siRNA
small interfering RNA
- SITUA
stable isotope tracer ultrafiltration assay
- SP
solubility parameter
- ssNMR
solid-state NMR
- TEM
transmission electron microscopy
- TGA
Therapeutic Goods Administration
- UCST
upper critical solution temperature
- USFDA
US Food and Drug Administration
- Vd
volume of distribution
Footnotes
Conflict of Interest: Kabanov is the co-developer of SP1049C and has interest in SoftKemo. He is also a co-founder and interested in the commercial success of DelAqua Pharmaceuticals Inc. which has the intent of developing of polymeric micelle drug formulations. Kabanov is co-inventor on US Patent 9.402,908B2 pertinent to the subject matter. The other authors have no competing interests to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bharate SS, Bharate SB, Bajaj A, Interactions and incompatibilities of pharmaceutical excipients with active pharmaceutical ingredients: a comprehensive review, J. excip. food chem, 1 (2016) 1131. [Google Scholar]
- [2].Cabral H, Miyata K, Osada K, Kataoka K, Block Copolymer Micelles in Nanomedicine Applications, Chem. Rev, 118 (2018) 6844–6892. [DOI] [PubMed] [Google Scholar]
- [3].Yokoyama M, Miyauchi M, Yamada N, Okano T, Sakurai Y, Kataoka K, Inoue S, Characterization and anticancer activity of the micelle-forming polymeric anticancer drug adriamycin-conjugated poly (ethylene glycol)-poly (aspartic acid) block copolymer, Cancer Res, 50 (1990) 1693–1700. [PubMed] [Google Scholar]
- [4].Kabanov AV, Chekhonin V, Alakhov VY, Batrakova E, Lebedev A, Melik-Nubarov N, Arzhakov S, Levashov A, Morozov G, Severin E, The neuroleptic activity of haloperidol increases after its solubilization in surfactant micelles: micelles as microcontainers for drug targeting, FEBS Lett, 258 (1989) 343–345. [DOI] [PubMed] [Google Scholar]
- [5].Kwon GS, Kataoka K, Block copolymer micelles as long-circulating drug vehicles, Adv. Drug Deliv. Rev, 16 (1995) 295–309. [Google Scholar]
- [6].Kim SC, Kim DW, Shim YH, Bang JS, Oh HS, Wan Kim S, Seo MH, In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy, J. Control. Release, 72 (2001) 191–202. [DOI] [PubMed] [Google Scholar]
- [7].Hamaguchi T, Matsumura Y, Suzuki M, Shimizu K, Goda R, Nakamura I, Nakatomi I, Yokoyama M, Kataoka K, Kakizoe T, NK105, a paclitaxel-incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel, Br. J. Cancer, 92 (2005) 1240–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Matsumura Y, Poly (amino acid) micelle nanocarriers in preclinical and clinical studies, Adv. Drug Deliv. Rev, 60 (2008) 899–914. [DOI] [PubMed] [Google Scholar]
- [9].Lee KS, Chung HC, Im SA, Park YH, Kim CS, Kim SB, Rha SY, Lee MY, Ro J, Multicenter phase II trial of Genexol-PM, a Cremophor-free, polymeric micelle formulation of paclitaxel, in patients with metastatic breast cancer, Breast Cancer Res. Treat, 108 (2008) 241–250. [DOI] [PubMed] [Google Scholar]
- [10].Valle JW, Armstrong A, Newman C, Alakhov V, Pietrzynski G, Brewer J, Campbell S, Corrie P, Rowinsky EK, Ranson M, A phase 2 study of SP1049C, doxorubicin in P-glycoprotein-targeting pluronics, in patients with advanced adenocarcinoma of the esophagus and gastroesophageal junction, Invest New Drugs, 29 (2011) 1029–1037. [DOI] [PubMed] [Google Scholar]
- [11].Kim TY, Kim DW, Chung JY, Shin SG, Kim SC, Heo DS, Kim NK, Bang YJ, Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies, Clin. Cancer Res, 10 (2004) 3708–3716. [DOI] [PubMed] [Google Scholar]
- [12].Cabral H, Kataoka K, Progress of drug-loaded polymeric micelles into clinical studies, J. Control. Release, 190 (2014) 465–476. [DOI] [PubMed] [Google Scholar]
- [13].Houdaihed L, Evans JC, Allen C, Overcoming the Road Blocks: Advancement of Block Copolymer Micelles for Cancer Therapy in the Clinic, Mol. Pharm, 14 (2017) 2503–2517. [DOI] [PubMed] [Google Scholar]
- [14].Kataoka K, Harada A, Nagasaki Y, Block copolymer micelles for drug delivery: design, characterization and biological significance, Adv. Drug Deliv. Rev, 47 (2001) 113–131. [DOI] [PubMed] [Google Scholar]
- [15].Owens DE 3rd, Peppas NA, Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles, Int. J. Pharm, 307 (2006) 93–102. [DOI] [PubMed] [Google Scholar]
- [16].Photos PJ, Bacakova L, Discher B, Bates FS, Discher DE, Polymer vesicles in vivo: correlations with PEG molecular weight, J. Control. Release, 90 (2003) 323–334. [DOI] [PubMed] [Google Scholar]
- [17].Danson S, Ferry D, Alakhov V, Margison J, Kerr D, Jowle D, Brampton M, Halbert G, Ranson M, Phase I dose escalation and pharmacokinetic study of pluronic polymer-bound doxorubicin (SP1049C) in patients with advanced cancer, Br. J. Cancer, 90 (2004) 2085–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Savić R, Eisenberg A, Maysinger D, Block copolymer micelles as delivery vehicles of hydrophobic drugs: micelle–cell interactions, J. Drug Target, 14 (2006) 343–355. [DOI] [PubMed] [Google Scholar]
- [19].Lee H, Zeng F, Dunne M, Allen C, Methoxy poly(ethylene glycol)-block-poly(delta-valerolactone) copolymer micelles for formulation of hydrophobic drugs, Biomacromolecules, 6 (2005) 3119–3128. [DOI] [PubMed] [Google Scholar]
- [20].Kakizawa Y, Kataoka K, Block copolymer micelles for delivery of gene and related compounds, Adv. Drug Deliv. Rev, 54 (2002) 203–222. [DOI] [PubMed] [Google Scholar]
- [21].Kabanov VA, Kabanov AV, Interpolyelectrolyte and block ionomer complexes for gene delivery: physico-chemical aspects, Adv. Drug Deliv. Rev, 30 (1998) 49–60. [DOI] [PubMed] [Google Scholar]
- [22].He Z, Wan X, Schulz A, Bludau H, Dobrovolskaia MA, Stern ST, Montgomery SA, Yuan H, Li Z, Alakhova D, Sokolsky M, Darr DB, Perou CM, Jordan R, Luxenhofer R, Kabanov AV, A high capacity polymeric micelle of paclitaxel: Implication of high dose drug therapy to safety and in vivo anti-cancer activity, Biomaterials, 101 (2016) 296–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kabanov AV, Batrakova EV, Alakhov VY, Pluronic® block copolymers as novel polymer therapeutics for drug and gene delivery, J. Control. Release, 82 (2002) 189–212. [DOI] [PubMed] [Google Scholar]
- [24].Gaucher G, Satturwar P, Jones MC, Furtos A, Leroux JC, Polymeric micelles for oral drug delivery, Eur. J. Pharm. Biopharm, 76 (2010) 147–158. [DOI] [PubMed] [Google Scholar]
- [25].Batrakova EV, Kabanov AV, Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers, J. Control. Release, 130 (2008) 98–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lee ES, Oh KT, Kim D, Youn YS, Bae YH, Tumor pH-responsive flower-like micelles of poly(L-lactic acid)-b-poly(ethylene glycol)-b-poly(L-histidine), J. Control. Release, 123 (2007) 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Neradovic D, Van Nostrum CF, Hennink WE, Thermoresponsive polymeric micelles with controlled instability based on hydrolytically sensitive N-isopropylacrylamide copolymers, Macromolecules, 34 (2001) 7589–7591. [Google Scholar]
- [28].Hwang D, Ramsey JD, Makita N, Sachse C, Jordan R, Sokolsky-Papkov M, Kabanov AV, Novel poly (2-oxazoline) block copolymer with aromatic heterocyclic side chains as a drug delivery platform, J. Control. Release, 307 (2019) 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kozlov MY, Melik-Nubarov NS, Batrakova EV, Kabanov AV, Relationship between pluronic block copolymer structure, critical micellization concentration and partitioning coefficients of low molecular mass solutes, Macromolecules, 33 (2000) 3305–3313. [Google Scholar]
- [30].Lubtow MM, Marciniak H, Schmiedel A, Roos M, Lambert C, Luxenhofer R, Ultra-High to Ultra-Low Drug-Loaded Micelles: Probing Host-Guest Interactions by Fluorescence Spectroscopy, Chemistry, 25 (2019) 12601–12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].He G, Ma LL, Pan J, Venkatraman S, ABA and BAB type triblock copolymers of PEG and PLA: a comparative study of drug release properties and “stealth” particle characteristics, Int. J. Pharm, 334 (2007) 48–55. [DOI] [PubMed] [Google Scholar]
- [32].Xiao K, Li Y, Luo J, Lee JS, Xiao W, Gonik AM, Agarwal RG, Lam KS, The effect of surface charge on in vivo biodistribution of PEG-oligocholic acid based micellar nanoparticles, Biomaterials, 32 (2011) 3435–3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Shan X, Yuan Y, Liu C, Tao X, Sheng Y, Xu F, Influence of PEG chain on the complement activation suppression and longevity in vivo prolongation of the PCL biomedical nanoparticles, Biomed. Microdevices, 11 (2009) 1187–1194. [DOI] [PubMed] [Google Scholar]
- [34].Logie J, Owen SC, McLaughlin CK, Shoichet MS, PEG-graft density controls polymeric nanoparticle micelle stability, Chem. Mater, 26 (2014) 2847–2855. [Google Scholar]
- [35].Shiraishi K, Sanada Y, Mochizuki S, Kawano K, Maitani Y, Sakurai K, Yokoyama M, Determination of polymeric micelles’ structural characteristics, and effect of the characteristics on pharmacokinetic behaviors, J. Control. Release, 203 (2015) 77–84. [DOI] [PubMed] [Google Scholar]
- [36].Velluto D, Demurtas D, Hubbell JA, PEG-b-PPS diblock copolymer aggregates for hydrophobic drug solubilization and release: cyclosporin A as an example, Mol. Pharm, 5 (2008) 632–642. [DOI] [PubMed] [Google Scholar]
- [37].Lin WJ, Juang LW, Lin CC, Stability and release performance of a series of pegylated copolymeric micelles, Pharm. Res, 20 (2003) 668–673. [DOI] [PubMed] [Google Scholar]
- [38].Zhang Y, Zhuo RX, Synthesis and in vitro drug release behavior of amphiphilic triblock copolymer nanoparticles based on poly (ethylene glycol) and polycaprolactone, Biomaterials, 26 (2005) 6736–6742. [DOI] [PubMed] [Google Scholar]
- [39].Shi Y, Lammers T, Storm G, Hennink WE, Physico-Chemical Strategies to Enhance Stability and Drug Retention of Polymeric Micelles for Tumor-Targeted Drug Delivery, Macromol. Biosci, 17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Qiao M, Chen D, Ma X, Liu Y, Injectable biodegradable temperature-responsive PLGA–PEG–PLGA copolymers: synthesis and effect of copolymer composition on the drug release from the copolymer-based hydrogels, Int. J. Pharm, 294 (2005) 103–112. [DOI] [PubMed] [Google Scholar]
- [41].Li F, Li S, El Ghzaoui A, Nouailhas H, Zhuo R, Synthesis and gelation properties of PEG− PLA− PEG triblock copolymers obtained by coupling monohydroxylated PEG− PLA with adipoyl chloride, Langmuir, 23 (2007) 2778–2783. [DOI] [PubMed] [Google Scholar]
- [42].Bogdanov B, Vidts A, Van Den Buicke A, Verbeeck R, Schacht E, Synthesis and thermal properties of poly (ethylene glycol)-poly (ϵ-caprolactone) copolymers, Polymer, 39 (1998) 1631–1636. [Google Scholar]
- [43].Herzberger J, Niederer K, Pohlit H, Seiwert J, Worm M, Wurm FR, Frey H, Polymerization of ethylene oxide, propylene oxide, and other alkylene oxides: synthesis, novel polymer architectures, and bioconjugation, Chem. Rev, 116 (2016) 2170–2243. [DOI] [PubMed] [Google Scholar]
- [44].Luxenhofer R, Han Y, Schulz A, Tong J, He Z, Kabanov AV, Jordan R, Poly (2-oxazoline) s as Polymer Therapeutics, Macromol. Rapid Commun, 33 (2012) 1613–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lorson T, Lubtow MM, Wegener E, Haider MS, Borova S, Nahm D, Jordan R, Sokolski-Papkov M, Kabanov AV, Luxenhofer R, Poly(2-oxazoline)s based biomaterials: A comprehensive and critical update, Biomaterials, 178 (2018) 204–280. [DOI] [PubMed] [Google Scholar]
- [46].Haider MS, Lübtow MM, Endres S, Aseyev V, Pöppler A-C, Luxenhofer R, Think Beyond the Core: The Impact of the Hydrophilic Corona on the Drug Solubilization Using Polymer Micelles, (2019). [DOI] [PubMed] [Google Scholar]
- [47].Cui S, Pan X, Gebru H, Wang X, Liu J, Liu J, Li Z, Guo K, Amphiphilic star-shaped poly (sarcosine)-block-poly (ε-caprolactone) diblock copolymers: one-pot synthesis, characterization, and solution properties, J. Mater. Chem. B, 5 (2017) 679–690. [DOI] [PubMed] [Google Scholar]
- [48].Deng Y, Zou T, Tao X, Semetey V, Trepout S, Marco S, Ling J, Li M-H, Poly (ε-caprolactone)-block-polysarcosine by ring-opening polymerization of sarcosine N-thiocarboxyanhydride: synthesis and thermoresponsive self-assembly, Biomacromolecules, 16 (2015) 3265–3274. [DOI] [PubMed] [Google Scholar]
- [49].Birke A, Ling J, Barz M, Polysarcosine-containing copolymers: Synthesis, characterization, self-assembly, and applications, Prog. Polym. Sci, 81 (2018) 163–208. [Google Scholar]
- [50].Doh KO, Yeo Y, Application of polysaccharides for surface modification of nanomedicines, Ther Deliv, 3 (2012) 1447–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Jeong Y-I, Do Hyung Kim C-WC, Yoo J-J, Choi KH, Kim CH, Ha SH, Kang DH, Doxorubicin-incorporated polymeric micelles composed of dextran-b-poly (DL-lactide-co-glycolide) copolymer, Int. J. Nanomedicine, 6 (2011) 1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kim DH, Kim M-D, Choi C-W, Chung C-W, Ha SH, Kim CH, Shim Y-H, Jeong Y-I, Kang DH, Antitumor activity of sorafenib-incorporated nanoparticles of dextran/poly (dl-lactide-co-glycolide) block copolymer, Nanoscale Res. Lett, 7 (2012) 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Verma MS, Liu S, Chen YY, Meerasa A, Gu FX, Size-tunable nanoparticles composed of dextran-b-poly (D, L-lactide) for drug delivery applications, Nano Res, 5 (2012) 49–61. [Google Scholar]
- [54].Wilms D, Stiriba SE, Frey H, Hyperbranched polyglycerols: from the controlled synthesis of biocompatible polyether polyols to multipurpose applications, Acc. Chem. Res, 43 (2010) 129–141. [DOI] [PubMed] [Google Scholar]
- [55].Choi YK, Bae YH, Kim SW, Star-shaped poly (ether− ester) block copolymers: Synthesis, characterization, and their physical properties, Macromolecules, 31 (1998) 8766–8774. [Google Scholar]
- [56].Ghasemi R, Abdollahi M, Zadeh EE, Khodabakhshi K, Badeli A, Bagheri H, Hosseinkhani S, mPEG-PLA and PLA-PEG-PLA nanoparticles as new carriers for delivery of recombinant human Growth Hormone (rhGH), Sci. Rep, 8 (2018) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zheng X, Kan B, Gou M, Fu S, Zhang J, Men K, Chen L, Luo F, Zhao Y, Zhao X, Preparation of MPEG–PLA nanoparticle for honokiol delivery in vitro, Int. J. Pharm, 386 (2010) 262–267. [DOI] [PubMed] [Google Scholar]
- [58].Rainbolt EA, Washington KE, Biewer MC, Stefan MC, Recent developments in micellar drug carriers featuring substituted poly (ε-caprolactone) s, Polym. Chem, 6 (2015) 2369–2381. [Google Scholar]
- [59].Thambi T, Yoon HY, Kim K, Kwon IC, Yoo CK, Park JH, Bioreducible block copolymers based on poly (ethylene glycol) and poly (γ-benzyl L-glutamate) for intracellular delivery of camptothecin, Bioconjug. Chem, 22 (2011) 1924–1931. [DOI] [PubMed] [Google Scholar]
- [60].Kamaly N, Yameen B, Wu J, Farokhzad OC, Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release, Chem. Rev, 116 (2016) 2602–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].González-Henríquez CM, Sarabia-Vallejos MA, Rodríguez-Hernández J, Strategies to fabricate polypeptide-based structures via ring-opening polymerization of N-carboxyanhydrides, Polymers, 9 (2017) 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Luxenhofer R, Schulz A, Roques C, Li S, Bronich TK, Batrakova EV, Jordan R, Kabanov AV, Doubly amphiphilic poly(2-oxazoline)s as high-capacity delivery systems for hydrophobic drugs, Biomaterials, 31 (2010) 4972–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].L btow MM, Hahn L, Haider MS, Luxenhofer R, Drug specificity, synergy and antagonism in ultrahigh capacity poly (2-oxazoline)/poly (2-oxazine) based formulations, J. Am. Chem. Soc, 139 (2017) 10980–10983. [DOI] [PubMed] [Google Scholar]
- [64].Lubtow MM, Haider MS, Kirsch M, Klisch S, Luxenhofer R, Like Dissolves Like? A Comprehensive Evaluation of Partial Solubility Parameters to Predict Polymer-Drug Compatibility in Ultrahigh Drug-Loaded Polymer Micelles, Biomacromolecules, 20 (2019) 3041–3056. [DOI] [PubMed] [Google Scholar]
- [65].Poppler AC, Lubtow MM, Schlauersbach J, Wiest J, Meinel L, Luxenhofer R, Loading-Dependent Structural Model of Polymeric Micelles Encapsulating Curcumin by Solid-State NMR Spectroscopy, Angew Chem. Int. Ed. Engl, 58 (2019) 18540–18546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Rudmann DG, Alston JT, Hanson JC, Heidel S, High molecular weight polyethylene glycol cellular distribution and PEG-associated cytoplasmic vacuolation is molecular weight dependent and does not require conjugation to proteins, Toxicol. Pathol, 41 (2013) 970–983. [DOI] [PubMed] [Google Scholar]
- [67].Webster R, Elliott V, Park BK, Walker D, Hankin M, Taupin P, PEG and PEG conjugates toxicity: towards an understanding of the toxicity of PEG and its relevance to PEGylated biologicals, PEGylated protein drugs: Basic science and clinical applications, Springer; 2009, pp. 127–146. [Google Scholar]
- [68].Veronese FM, PEGylated protein drugs: basic science and clinical applications, Springer Science & Business Media; 2009. [Google Scholar]
- [69].Miller T, Rachel R, Besheer A, Uezguen S, Weigandt M, Goepferich A, Comparative investigations on in vitro serum stability of polymeric micelle formulations, Pharm. Res, 29 (2012) 448–459. [DOI] [PubMed] [Google Scholar]
- [70].Emami J, Rezazadeh M, Hasanzadeh F, Sadeghi H, Mostafavi A, Minaiyan M, Rostami M, Davies N, Development and in vitro/in vivo evaluation of a novel targeted polymeric micelle for delivery of paclitaxel, Int. J. Biol. Macromol, 80 (2015) 29–40. [DOI] [PubMed] [Google Scholar]
- [71].Song X, Zhao S, Fang S, Ma Y, Duan M, Mesoscopic Simulations of Adsorption and Association of PEO-PPO-PEO Triblock Copolymers on a Hydrophobic Surface: From Mushroom Hemisphere to Rectangle Brush, Langmuir, 32 (2016) 11375–11385. [DOI] [PubMed] [Google Scholar]
- [72].Xu Q, Ensign LM, Boylan NJ, Schon A, Gong X, Yang JC, Lamb NW, Cai S, Yu T, Freire E, Hanes J, Impact of Surface Polyethylene Glycol (PEG) Density on Biodegradable Nanoparticle Transport in Mucus ex Vivo and Distribution in Vivo, ACS Nano, 9 (2015) 9217–9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Maisel K, Reddy M, Xu Q, Chattopadhyay S, Cone R, Ensign LM, Hanes J, Nanoparticles coated with high molecular weight PEG penetrate mucus and provide uniform vaginal and colorectal distribution in vivo, Nanomedicine (Lond), 11 (2016) 1337–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Gong C, Shi S, Dong P, Kan B, Gou M, Wang X, Li X, Luo F, Zhao X, Wei Y, Qian Z, Synthesis and characterization of PEG-PCL-PEG thermosensitive hydrogel, Int. J. Pharm, 365 (2009) 89–99. [DOI] [PubMed] [Google Scholar]
- [75].Xiong M-H, Wu J, Wang Y-C, Li L-S, Liu X-B, Zhang G-Z, Yan L-F, Wang J, Synthesis of PEG-armed and polyphosphoester core-cross-linked nanogel by one-step ring-opening polymerization, Macromolecules, 42 (2009) 893–896. [Google Scholar]
- [76].Akiyama Y, Harada A, Nagasaki Y, Kataoka K, Synthesis of poly (ethylene glycol)-block-poly (ethylenimine) possessing an acetal group at the PEG end, Macromolecules, 33 (2000) 5841–5845. [Google Scholar]
- [77].Lila ASA, Kiwada H, Ishida T, The accelerated blood clearance (ABC) phenomenon: clinical challenge and approaches to manage, J. Control. Release, 172 (2013) 38–47. [DOI] [PubMed] [Google Scholar]
- [78].Yang Q, Lai SK, Anti-PEG immunity: emergence, characteristics, and unaddressed questions, Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol, 7 (2015) 655–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Armstrong JK, Hempel G, Koling S, Chan LS, Fisher T, Meiselman HJ, Garratty G, Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients, Cancer, 110 (2007) 103–111. [DOI] [PubMed] [Google Scholar]
- [80].Ganson NJ, Kelly SJ, Scarlett E, Sundy JS, Hershfield MS, Control of hyperuricemia in subjects with refractory gout, and induction of antibody against poly(ethylene glycol) (PEG), in a phase I trial of subcutaneous PEGylated urate oxidase, Arthritis Res. Ther, 8 (2006) R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Sherman MR, Williams LD, Sobczyk MA, Michaels SJ, Saifer MG, Role of the methoxy group in immune responses to mPEG-protein conjugates, Bioconjug. Chem, 23 (2012) 485–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chanan-Khan A, Szebeni J, Savay S, Liebes L, Rafique NM, Alving CR, Muggia FM, Complement activation following first exposure to pegylated liposomal doxorubicin (Doxil®): possible role in hypersensitivity reactions, Ann. Oncol, 14 (2003) 1430–1437. [DOI] [PubMed] [Google Scholar]
- [83].Kierstead PH, Okochi H, Venditto VJ, Chuong TC, Kivimae S, Frechet JMJ, Szoka FC, The effect of polymer backbone chemistry on the induction of the accelerated blood clearance in polymer modified liposomes, J. Control. Release, 213 (2015) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Shiraishi K, Hamano M, Ma H, Kawano K, Maitani Y, Aoshi T, Ishii KJ, Yokoyama M, Hydrophobic blocks of PEG-conjugates play a significant role in the accelerated blood clearance (ABC) phenomenon, J. Control. Release, 165 (2013) 183–190. [DOI] [PubMed] [Google Scholar]
- [85].Shiraishi K, Kawano K, Maitani Y, Aoshi T, Ishii KJ, Sanada Y, Mochizuki S, Sakurai K, Yokoyama M, Exploring the relationship between anti-PEG IgM behaviors and PEGylated nanoparticles and its significance for accelerated blood clearance, J. Control. Release, 234 (2016) 59–67. [DOI] [PubMed] [Google Scholar]
- [86].Gaertner FC, Luxenhofer R, Blechert B, Jordan R, Essler M, Synthesis, biodistribution and excretion of radiolabeled poly(2-alkyl-2-oxazoline)s, J. Control. Release, 119 (2007) 291–300. [DOI] [PubMed] [Google Scholar]
- [87].Zhang N, Pompe T, Amin I, Luxenhofer R, Werner C, Jordan R, Tailored poly(2-oxazoline) polymer brushes to control protein adsorption and cell adhesion, Macromol. Biosci, 12 (2012) 926–936. [DOI] [PubMed] [Google Scholar]
- [88].Pidhatika B, Rodenstein M, Chen Y, Rakhmatullina E, Muhlebach A, Acikgoz C, Textor M, Konradi R, Comparative stability studies of poly(2-methyl-2-oxazoline) and poly(ethylene glycol) brush coatings, Biointerphases, 7 (2012) 1. [DOI] [PubMed] [Google Scholar]
- [89].Bludau H, Czapar AE, Pitek AS, Shukla S, Jordan R, Steinmetz N, POxylation as an alternative stealth coating for biomedical applications, Eur. Polym. J, 88 (2017) 679–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wang X, Li X, Li Y, Zhou Y, Fan C, Li W, Ma S, Fan Y, Huang Y, Li N, Liu Y, Synthesis, characterization and biocompatibility of poly(2-ethyl-2-oxazoline)-poly(D,L-lactide)-poly(2-ethyl-2-oxazoline) hydrogels, Acta Biomater, 7 (2011) 4149–4159. [DOI] [PubMed] [Google Scholar]
- [91].Mansfield ED, Victor R, Kowalczyk RM, Grillo I, Hoogenboom R, Sillence K, Hole P, Williams AC, Khutoryanskiy VV, Side chain variations radically alter the diffusion of poly (2-alkyl-2-oxazoline) functionalised nanoparticles through a mucosal barrier, Biomater. Sci, 4 (2016) 1318–1327. [DOI] [PubMed] [Google Scholar]
- [92].Itaka K, Ishii T, Hasegawa Y, Kataoka K, Biodegradable polyamino acid-based polycations as safe and effective gene carrier minimizing cumulative toxicity, Biomaterials, 31 (2010) 3707–3714. [DOI] [PubMed] [Google Scholar]
- [93].Roweton S, Huang S, Swift G, Poly (aspartic acid): synthesis, biodegradation, and current applications, J. Environ. Polym. Degrad, 5 (1997) 175–181. [Google Scholar]
- [94].Huesmann D, Sevenich A, Weber B, Barz M, A head-to-head comparison of poly (sarcosine) and poly (ethylene glycol) in peptidic, amphiphilic block copolymers, Polymer, 67 (2015) 240–248. [Google Scholar]
- [95].Lau KHA, Ren C, Sileika TS, Park SH, Szleifer I, Messersmith PB, Surface-grafted polysarcosine as a peptoid antifouling polymer brush, Langmuir, 28 (2012) 16099–16107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Karmali PP, Chao Y, Park JH, Sailor MJ, Ruoslahti E, Esener SC, Simberg D, Different effect of hydrogelation on antifouling and circulation properties of dextran-iron oxide nanoparticles, Mol. Pharm, 9 (2012) 539–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Cao B, Li L, Wu H, Tang Q, Sun B, Dong H, Zhe J, Cheng G, Zwitteration of dextran: a facile route to integrate antifouling, switchability and optical transparency into natural polymers, ChemComm, 50 (2014) 3234–3237. [DOI] [PubMed] [Google Scholar]
- [98].Torchilin VP, Levchenko TS, Whiteman KR, Yaroslavov AA, Tsatsakis AM, Rizos AK, Michailova EV, Shtilman MI, Amphiphilic poly-N-vinylpyrrolidones: synthesis, properties and liposome surface modification, Biomaterials, 22 (2001) 3035–3044. [DOI] [PubMed] [Google Scholar]
- [99].Jiang J, Zhu L, Zhu L, Zhang H, Zhu B, Xu Y, Antifouling and antimicrobial polymer membranes based on bioinspired polydopamine and strong hydrogen-bonded poly(N-vinyl pyrrolidone), ACS Appl. Mater. Interfaces, 5 (2013) 12895–12904. [DOI] [PubMed] [Google Scholar]
- [100].Zhao C, Li L, Zheng J, Achieving highly effective nonfouling performance for surface-grafted poly(HPMA) via atom-transfer radical polymerization, Langmuir, 26 (2010) 17375–17382. [DOI] [PubMed] [Google Scholar]
- [101].Muppalla R, Rana HH, Devi S, Jewrajka SK, Adsorption of pH-responsive amphiphilic copolymer micelles and gel on membrane surface as an approach for antifouling coating, Appl. Surf. Sci, 268 (2013) 355–367. [Google Scholar]
- [102].Zhao J, Shi Q, Luan S, Song L, Yang H, Shi H, Jin J, Li X, Yin J, Stagnaro P, Improved biocompatibility and antifouling property of polypropylene non-woven fabric membrane by surface grafting zwitterionic polymer, J. Membr. Sci, 369 (2011) 5–12. [Google Scholar]
- [103].Elsabahy M, Perron ME, Bertrand N, Yu GE, Leroux JC, Solubilization of docetaxel in poly(ethylene oxide)-block-poly(butylene/styrene oxide) micelles, Biomacromolecules, 8 (2007) 2250–2257. [DOI] [PubMed] [Google Scholar]
- [104].Alakhova DY, Kabanov AV, Pluronics and MDR reversal: an update, Mol. Pharm, 11 (2014) 2566–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Cameron DJ, Shaver MP, Aliphatic polyester polymer stars: synthesis, properties and applications in biomedicine and nanotechnology, Chem. Soc. Rev, 40 (2011) 1761–1776. [DOI] [PubMed] [Google Scholar]
- [106].Makadia HK, Siegel S, Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier, Polymers, 3 (2011) 1377–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Allen C, Yu Y, Maysinger D, Eisenberg A, Polycaprolactone-b-poly (ethylene oxide) block copolymer micelles as a novel drug delivery vehicle for neurotrophic agents FK506 and L-685,818, Bioconjug. Chem, 9 (1998) 564–572. [DOI] [PubMed] [Google Scholar]
- [108].Xiao RZ, Zeng ZW, Zhou GL, Wang JJ, Li FZ, Wang AM, Recent advances in PEG-PLA block copolymer nanoparticles, Int. J. Nanomedicine, 5 (2010) 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Zhang L, He Y, Yu M, Song C, Paclitaxel-loaded polymeric nanoparticles based on PCL-PEG-PCL: preparation, in vitro and in vivo evaluation, J. Control. Release, 152 Suppl 1 (2011) e114–116. [DOI] [PubMed] [Google Scholar]
- [110].Werner ME, Cummings ND, Sethi M, Wang EC, Sukumar R, Moore DT, Wang AZ, Preclinical evaluation of Genexol-PM, a nanoparticle formulation of paclitaxel, as a novel radiosensitizer for the treatment of non-small cell lung cancer, Int. J. Radiat, 86 (2013) 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].He Z, Schulz A, Wan X, Seitz J, Bludau H, Alakhova DY, Darr DB, Perou CM, Jordan R, Ojima I, Kabanov AV, Poly(2-oxazoline) based micelles with high capacity for 3rd generation taxoids: preparation, in vitro and in vivo evaluation, J Control Release, 208 (2015) 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Seo Y, Schulz A, Han Y, He Z, Bludau H, Wan X, Tong J, Bronich TK, Sokolsky M, Luxenhofer R, Poly (2-oxazoline) block copolymer based formulations of taxanes: effect of copolymer and drug structure, concentration, and environmental factors, Polym. Adv. Technol, 26 (2015) 837–850. [Google Scholar]
- [113].Hwang D, Dismuke T, Tikunov A, Rosen EP, Kagel JR, Ramsey JD, Lim C, Zamboni W, Kabanov AV, Gershon TR, Sokolsky-Papkov M, Poly(2-oxazoline) nanoparticle delivery enhances the therapeutic potential of vismodegib for medulloblastoma by improving CNS pharmacokinetics and reducing systemic toxicity, bioRxiv, (2020) 2020.04.30.068726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Wan X, Min Y, Bludau H, Keith A, Sheiko SS, Jordan R, Wang AZ, Sokolsky-Papkov M, Kabanov AV, Drug Combination Synergy in Worm-like Polymeric Micelles Improves Treatment Outcome for Small Cell and Non-Small Cell Lung Cancer, ACS Nano, 12 (2018) 2426–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Schulz A, Jaksch S, Schubel R, Wegener E, Di Z, Han Y, Meister A, Kressler J, Kabanov AV, Luxenhofer R, Papadakis CM, Jordan R, Drug-induced morphology switch in drug delivery systems based on poly(2-oxazoline)s, ACS Nano, 8 (2014) 2686–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Alves VM, Hwang D, Muratov E, Sokolsky-Papkov M, Varlamova E, Vinod N, Lim C, Andrade CH, Tropsha A, Kabanov A, Cheminformatics-driven discovery of polymeric micelle formulations for poorly soluble drugs, Sci. Adv, 5 (2019) eaav9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Han Y, He Z, Schulz A, Bronich TK, Jordan R, Luxenhofer R, Kabanov AV, Synergistic combinations of multiple chemotherapeutic agents in high capacity poly(2-oxazoline) micelles, Mol. Pharm, 9 (2012) 2302–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].He Z, Schulz A, Wan X, Seitz J, Bludau H, Alakhova DY, Darr DB, Perou CM, Jordan R, Ojima I, Kabanov AV, Luxenhofer R, Poly(2-oxazoline) based micelles with high capacity for 3rd generation taxoids: preparation, in vitro and in vivo evaluation, J. Control. Release, 208 (2015) 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Haider MS, Lübtow MM, Endres S, Forster S, Flegler VJ, Böttcher B, Aseyev VO, Pöppler A-C, Luxenhofer R, Think beyond the core: The impact of the hydrophilic corona on the drug solubilization using polymer micelles, ACS Appl. Mater. Interfaces, (2020). [DOI] [PubMed] [Google Scholar]
- [120].Karimi M, Sahandi Zangabad P, Ghasemi A, Amiri M, Bahrami M, Malekzad H, Ghahramanzadeh Asl H, Mahdieh Z, Bozorgomid M, Ghasemi A, Rahmani Taji MRB, Hamblin MR, Temperature-responsive smart nanocarriers for delivery of therapeutic agents: applications and recent advances, ACS Appl. Mater. Interfaces, 8 (2016) 21107–21133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Chung J, Yokoyama M, Yamato M, Aoyagi T, Sakurai Y, Okano T, Thermo-responsive drug delivery from polymeric micelles constructed using block copolymers of poly (N-isopropylacrylamide) and poly (butylmethacrylate), J. Control. Release, 62 (1999) 115–127. [DOI] [PubMed] [Google Scholar]
- [122].Wei H, Cheng S-X, Zhang X-Z, Zhuo R-X, Thermo-sensitive polymeric micelles based on poly (N-isopropylacrylamide) as drug carriers, Prog. Polym. Sci, 34 (2009) 893–910. [Google Scholar]
- [123].Liu S, Tong Y, Yang Y, Thermally sensitive micelles self-assembled from poly (N-isopropylacrylamide-co-N, N-dimethylacrylamide)-b-poly (D, L-lactide-co-glycolide) for controlled delivery of paclitaxel, Mol. Biosyst, 1 (2005) 158–165. [DOI] [PubMed] [Google Scholar]
- [124].Cammas S, Suzuki K, Sone C, Sakurai Y, Kataoka K, Okano T, Thermo-responsive polymer nanoparticles with a core-shell micelle structure as site-specific drug carriers, J. Control. Release, 48 (1997) 157–164. [Google Scholar]
- [125].Kohori F, Sakai K, Aoyagi T, Yokoyama M, Yamato M, Sakurai Y, Okano T, Control of adriamycin cytotoxic activity using thermally responsive polymeric micelles composed of poly (N-isopropylacrylamide-co-N, N-dimethylacrylamide)-b-poly (D, L-lactide), Colloids Surf. B Biointerfaces, 16 (1999) 195–205. [Google Scholar]
- [126].Sun X-L, Tsai P-C, Bhat R, Bonder E, Michniak-Kohn B, Pietrangelo A, Thermoresponsive block copolymer micelles with tunable pyrrolidone-based polymer cores: structure/property correlations and application as drug carriers, J. Mater. Chem. B, 3 (2015) 814–823. [DOI] [PubMed] [Google Scholar]
- [127].Wang X, Li S, Wan Z, Quan Z, Tan Q, Investigation of thermo-sensitive amphiphilic micelles as drug carriers for chemotherapy in cholangiocarcinoma in vitro and in vivo, Int. J. Pharm, 463 (2014) 81–88. [DOI] [PubMed] [Google Scholar]
- [128].Diehl C, Schlaad H, Thermo-responsive polyoxazolines with widely tuneable LCST, Macromol. Biosci, 9 (2009) 157–161. [DOI] [PubMed] [Google Scholar]
- [129].Meyer DE, Shin B, Kong G, Dewhirst M, Chilkoti A, Drug targeting using thermally responsive polymers and local hyperthermia, J. Control. Release, 74 (2001) 213–224. [DOI] [PubMed] [Google Scholar]
- [130].Prabaharan M, Mano JF, Stimuli-responsive hydrogels based on polysaccharides incorporated with thermo-responsive polymers as novel biomaterials, Macromol. Biosci, 6 (2006) 991–1008. [DOI] [PubMed] [Google Scholar]
- [131].Wu H, Zhu L, Torchilin VP, pH-sensitive poly (histidine)-PEG/DSPE-PEG co-polymer micelles for cytosolic drug delivery, Biomaterials, 34 (2013) 1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Zhang W, Shi L, Ma R, An Y, Xu Y, Wu K, Micellization of thermo-and Ph-responsive triblock copolymer of poly (ethylene glycol)-b-poly (4-vinylpyridine)-b-poly (N-isopropylacrylamide), Macromolecules, 38 (2005) 8850–8852. [Google Scholar]
- [133].Car A, Baumann P, Duskey JT, Chami M, Bruns N, Meier W, pH-responsive PDMS-b-PDMAEMA micelles for intracellular anticancer drug delivery, Biomacromolecules, 15 (2014) 3235–3245. [DOI] [PubMed] [Google Scholar]
- [134].Kim MS, Hwang SJ, Han JK, Choi EK, Park HJ, Kim JS, Lee DS, pH-Responsive PEG-Poly (β-amino ester) Block Copolymer Micelles with a Sharp Transition, Macromol. Rapid Commun, 27 (2006) 447–451. [Google Scholar]
- [135].Liu H, Chen H, Cao F, Peng D, Chen W, Zhang C, Amphiphilic block copolymer poly (acrylic acid)-b-polycaprolactone as a novel pH-sensitive nanocarrier for anti-cancer drugs delivery: In-Vitro and in-vivo evaluation, Polymers, 11 (2019) 820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Luo Y-L, Yang X-L, Xu F, Chen Y-S, Zhao X, pH-triggered PMAA-b-HTPB-b-PMAA copolymer micelles: physicochemical characterization and camptothecin release, Colloid Polym. Sci, 292 (2014) 1061–1072. [Google Scholar]
- [137].Chen S, Tan Z, Li N, Wang R, He L, Shi Y, Jiang L, Li P, Zhu X, Highly efficient intracellular drug delivery with a negatively charged hyperbranched polysulfonamine, Macromol. Biosci, 11 (2011) 828–838. [DOI] [PubMed] [Google Scholar]
- [138].Lynn DM, Langer R, Degradable poly (β-amino esters): synthesis, characterization, and self-assembly with plasmid DNA, J. Am. Chem. Soc, 122 (2000) 10761–10768. [Google Scholar]
- [139].Li W, Sun J, Zhang X, Jia L, Qiao M, Zhao X, Hu H, Chen D, Wang Y, Synthesis and Characterization of pH-Responsive PEG-Poly (β-Amino Ester) Block Copolymer Micelles as Drug Carriers to Eliminate Cancer Stem Cells, Pharmaceutics, 12 (2020) 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Green JJ, Langer R, Anderson DG, A combinatorial polymer library approach yields insight into nonviral gene delivery, Acc. Chem. Res, 41 (2008) 749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Kaczmarek JC, Patel AK, Kauffman KJ, Fenton OS, Webber MJ, Heartlein MW, DeRosa F, Anderson DG, Polymer–lipid nanoparticles for systemic delivery of mRNA to the lungs, Angew. Chem, 128 (2016) 14012–14016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Kaczmarek JC, Kauffman KJ, Fenton OS, Sadtler K, Patel AK, Heartlein MW, DeRosa F, Anderson DG, Optimization of a degradable polymer–lipid nanoparticle for potent systemic delivery of mRNA to the lung endothelium and immune cells, Nano Lett, 18 (2018) 6449–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Bader H, Ringsdorf H, Schmidt B, Watersoluble polymers in medicine, Angew. Makromolek. Chem, 123 (1984) 457–485. [Google Scholar]
- [144].Ringsdorf H, Structure and properties of pharmacologically active polymers, J. Polym. Sci., Polym. Symp, Wiley Online Library, 1975, pp. 135–153. [Google Scholar]
- [145].Emmelius M, Hörpel G, Ringsdorf H, Schmidt B, Polymeric Micelles and Liposomes as Potential Drug Carriers, Polymers in Medicine II, Springer; 1986, pp. 313–331. [Google Scholar]
- [146].Matsumura Y, Hamaguchi T, Ura T, Muro K, Yamada Y, Shimada Y, Shirao K, Okusaka T, Ueno H, Ikeda M, Phase I clinical trial and pharmacokinetic evaluation of NK911, a micelle-encapsulated doxorubicin, Br. J. Cancer, 91 (2004) 1775–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K, Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy, Bioconjug. Chem, 16 (2005) 122–130. [DOI] [PubMed] [Google Scholar]
- [148].Yokoyama M, Block copolymers as drug carriers, Crit. Rev. Ther. Drug, 9 (1992) 213–248. [PubMed] [Google Scholar]
- [149].Yokoyama M, Okano T, Sakurai Y, Ekimoto H, Shibazaki C, Kataoka K, Toxicity and antitumor activity against solid tumors of micelle-forming polymeric anticancer drug and its extremely long circulation in blood, Cancer Res, 51 (1991) 3229–3236. [PubMed] [Google Scholar]
- [150].Yokoyama M, Fukushima S, Uehara R, Okamoto K, Kataoka K, Sakurai Y, Okano T, Characterization of physical entrapment and chemical conjugation of adriamycin in polymeric micelles and their design for in vivo delivery to a solid tumor, J. Control. Release, 50 (1998) 79–92. [DOI] [PubMed] [Google Scholar]
- [151].Osada K, Christie RJ, Kataoka K, Polymeric micelles from poly (ethylene glycol)–poly (amino acid) block copolymer for drug and gene delivery, J. R. Soc. Interface, 6 (2009) S325–S339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Yokoyama M, Okano T, Sakurai Y, Suwa S, Kataoka K, Introduction of cisplatin into polymeric micelle, J. Control. Release, 39 (1996) 351–356. [Google Scholar]
- [153].Uchino H, Matsumura Y, Negishi T, Koizumi F, Hayashi T, Honda T, Nishiyama N, Kataoka K, Naito S, Kakizoe T, Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats, Br. J. Cancer, 93 (2005) 678–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Cabral H, Nishiyama N, Okazaki S, Koyama H, Kataoka K, Preparation and biological properties of dichloro (1, 2-diaminocyclohexane) platinum (II)(DACHPt)-loaded polymeric micelles, J. Control. Release, 101 (2005) 223–232. [DOI] [PubMed] [Google Scholar]
- [155].Batrakova EV, Bronich TK, Vetro JA, Kabanov AV, Polymer micelles as drug carriers, Nanoparticulates as drug carriers, (2006) 57–93. [Google Scholar]
- [156].Liu M, Kono K, Fréchet JM, Water-soluble dendritic unimolecular micelles:: Their potential as drug delivery agents, J. Control. Release, 65 (2000) 121–131. [DOI] [PubMed] [Google Scholar]
- [157].Kojima C, Kono K, Maruyama K, Takagishi T, Synthesis of polyamidoamine dendrimers having poly (ethylene glycol) grafts and their ability to encapsulate anticancer drugs, Bioconjug. Chem, 11 (2000) 910–917. [DOI] [PubMed] [Google Scholar]
- [158].Wang F, Bronich TK, Kabanov AV, Rauh RD, Roovers J, Synthesis and evaluation of a star amphiphilic block copolymer from poly (ε-caprolactone) and poly (ethylene glycol) as a potential drug delivery carrier, Bioconjug. Chem, 16 (2005) 397–405. [DOI] [PubMed] [Google Scholar]
- [159].Wang F, Bronich TK, Kabanov AV, Rauh RD, Roovers J, Synthesis and Characterization of Star Poly (ε-caprolactone)-b-Poly (ethylene glycol) and Poly (l-lactide)-b-Poly (ethylene glycol) Copolymers: Evaluation as Drug Delivery Carriers, Bioconjug. Chem, 19 (2008) 1423–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Shuai X, Merdan T, Schaper AK, Xi F, Kissel T, Core-cross-linked polymeric micelles as paclitaxel carriers, Bioconjug. Chem, 15 (2004) 441–448. [DOI] [PubMed] [Google Scholar]
- [161].Cajot S, Lautram N, Passirani C, Jérôme C, Design of reversibly core cross-linked micelles sensitive to reductive environment, J. Control. Release, 152 (2011) 30–36. [DOI] [PubMed] [Google Scholar]
- [162].Yang Z, Zheng S, Harrison WJ, Harder J, Wen X, Gelovani JG, Qiao A, Li C, Long-circulating near-infrared fluorescence core-cross-linked polymeric micelles: synthesis, characterization, and dual nuclear/optical imaging, Biomacromolecules, 8 (2007) 3422–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Jiménez-Pardo I, González-Pastor R, Lancelot A, Claveria-Gimeno R, Velázquez-Campoy A, Abian O, Ros MB, Sierra T, Shell Cross-Linked Polymeric Micelles as Camptothecin Nanocarriers for Anti-HCV Therapy, Macromol. Biosci, 15 (2015) 1381–1391. [DOI] [PubMed] [Google Scholar]
- [164].Kim Y, Pourgholami MH, Morris DL, Lu H, Stenzel MH, Effect of shell-crosslinking of micelles on endocytosis and exocytosis: acceleration of exocytosis by crosslinking, Biomater. Sci, 1 (2013) 265–275. [DOI] [PubMed] [Google Scholar]
- [165].Mocanu G, Nichifor M, Stanciu MC, New shell crosslinked micelles from dextran with hydrophobic end groups and their interaction with bioactive molecules, Carbohydr. Polym, 119 (2015) 228–235. [DOI] [PubMed] [Google Scholar]
- [166].Mocanu G, Nichifor M, Sacarescu L, Dextran based polymeric micelles as carriers for delivery of hydrophobic drugs, Curr. Drug Deliv, 14 (2017) 406–415. [DOI] [PubMed] [Google Scholar]
- [167].Gaber M, Elhasany KA, Sabra S, Helmy MW, Fang J-Y, Khattab SN, Bekhit AA, Teleb M, Elkodairy KA, Elzoghby AO, Co-Administration of Tretinoin Enhances the Anti-Cancer Efficacy of Etoposide via Tumor-Targeted Green Nano-Micelles, Colloids Surf. B Biointerfaces, (2020) 110997. [DOI] [PubMed] [Google Scholar]
- [168].Chang C, Wei H, Wu D-Q, Yang B, Chen N, Cheng S-X, Zhang X-Z, Zhuo R-X, Thermo-responsive shell cross-linked PMMA-bP (NIPAAm-co-NAS) micelles for drug delivery, Int. J. Pharm, 420 (2011) 333–340. [DOI] [PubMed] [Google Scholar]
- [169].Bronich TK, Keifer PA, Shlyakhtenko LS, Kabanov AV, Polymer micelle with cross-linked ionic core, J. Am. Chem. Soc, 127 (2005) 8236–8237. [DOI] [PubMed] [Google Scholar]
- [170].Kim JO, Kabanov AV, Bronich TK, Polymer micelles with cross-linked polyanion core for delivery of a cationic drug doxorubicin, J. Control. Release, 138 (2009) 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Kim JO, Nukolova NV, Oberoi HS, Kabanov AV, Bronich TK, Block ionomer complex micelles with cross-linked cores for drug delivery, Polym. Sci. Ser. A, 51 (2009) 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Kim JO, Oberoi HS, Desale S, Kabanov AV, Bronich TK, Polypeptide nanogels with hydrophobic moieties in the cross-linked ionic cores: synthesis, characterization and implications for anticancer drug delivery, J. Drug Target, 21 (2013) 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Desale SS, Raja SM, Kim JO, Mohapatra B, Soni KS, Luan H, Williams SH, Bielecki TA, Feng D, Storck M, Band V, Cohen SM, Band H, Bronich TK, Polypeptide-based nanogels co-encapsulating a synergistic combination of doxorubicin with 17-AAG show potent anti-tumor activity in ErbB2-driven breast cancer models, J. Control. Release, 208 (2015) 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Kim JO, Sahay G, Kabanov AV, Bronich TK, Polymeric micelles with ionic cores containing biodegradable cross-links for delivery of chemotherapeutic agents, Biomacromolecules, 11 (2010) 919–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Oberoi HS, Nukolova NV, Bronich TK, Dichloro (1, 2-Diaminocyclohexane) Platinum (II)(DACHPt) Loaded Polymer Micelles with Cross-Linked Core: Preparation and Characterization, PMSE prepr., NIH Public Access, 2011, pp. 630. [PMC free article] [PubMed] [Google Scholar]
- [176].Oberoi HS, Nukolova NV, Zhao Y, Cohen SM, Kabanov AV, Bronich TK, Preparation and in vivo evaluation of dichloro (1, 2-diaminocyclohexane) platinum (II)-loaded core cross-linked polymer micelles, Chemother. Res. Pract, 2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Desale SS, Cohen SM, Zhao Y, Kabanov AV, Bronich TK, Biodegradable hybrid polymer micelles for combination drug therapy in ovarian cancer, J. Control. Release, 171 (2013) 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Desale SS, Soni KS, Romanova S, Cohen SM, Bronich TK, Targeted delivery of platinum-taxane combination therapy in ovarian cancer, J. Control. Release, 220 (2015) 651–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Nukolova NV, Oberoi HS, Cohen SM, Kabanov AV, Bronich TK, Folate-decorated nanogels for targeted therapy of ovarian cancer, Biomaterials, 32 (2011) 5417–5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Nukolova NV, Oberoi HS, Zhao Y, Chekhonin VP, Kabanov AV, Bronich TK, LHRH-targeted nanogels as a delivery system for cisplatin to ovarian cancer, Mol. Pharm, 10 (2013) 3913–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Liu T, Romanova S, Wang S, Hyun MA, Zhang C, Cohen SM, Singh RK, Bronich TK, Alendronate-Modified Polymeric Micelles for the Treatment of Breast Cancer Bone Metastasis, Mol. Pharm, 16 (2019) 2872–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Owen SC, Chan DP, Shoichet MS, Polymeric micelle stability, Nano today, 7 (2012) 53–65. [Google Scholar]
- [183].Allen C, Maysinger D, Eisenberg A, Nano-engineering block copolymer aggregates for drug delivery, Colloids Surf. B Biointerfaces, 16 (1999) 3–27. [Google Scholar]
- [184].Lee J, Cho EC, Cho K, Incorporation and release behavior of hydrophobic drug in functionalized poly(D,L-lactide)-block-poly(ethylene oxide) micelles, J. Control. Release, 94 (2004) 323–335. [DOI] [PubMed] [Google Scholar]
- [185].Kwon G, Naito M, Yokoyama M, Okano T, Sakurai Y, Kataoka K, Micelles based on AB block copolymers of poly (ethylene oxide) and poly (. beta.-benzyl L-aspartate), Langmuir, 9 (1993) 945–949. [Google Scholar]
- [186].Shi Y, van Steenbergen MJ, Teunissen EA, Novo L, Gradmann S, Baldus M, van Nostrum CF, Hennink WE, Pi-Pi stacking increases the stability and loading capacity of thermosensitive polymeric micelles for chemotherapeutic drugs, Biomacromolecules, 14 (2013) 1826–1837. [DOI] [PubMed] [Google Scholar]
- [187].Kabanov AV, Nazarova IR, Astafieva IV, Batrakova EV, Alakhov VY, Yaroslavov AA, Kabanov VA, Micelle formation and solubilization of fluorescent probes in poly (oxyethylene-b-oxypropylene-b-oxyethylene) solutions, Macromolecules, 28 (1995) 2303–2314. [Google Scholar]
- [188].Wan X, Beaudoin JJ, Vinod N, Min Y, Makita N, Bludau H, Jordan R, Wang A, Sokolsky M, Kabanov AV, Co-delivery of paclitaxel and cisplatin in poly (2-oxazoline) polymeric micelles: Implications for drug loading, release, pharmacokinetics and outcome of ovarian and breast cancer treatments, Biomaterials, 192 (2019) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Thakral S, Thakral NK, Prediction of drug–polymer miscibility through the use of solubility parameter based Flory–Huggins interaction parameter and the experimental validation: PEG as model polymer, J. Pharm. Sci, 102 (2013) 2254–2263. [DOI] [PubMed] [Google Scholar]
- [190].Chun BJ, Lu J, Weck M, Jang SS, Characterization of molecular association of poly (2-oxazoline) s-based micelles with various epoxides and diols via the Flory–Huggins theory: a molecular dynamics simulation approach, Phys. Chem. Chem. Phys, 17 (2015) 29161–29170. [DOI] [PubMed] [Google Scholar]
- [191].Loverde SM, Klein ML, Discher DE, Nanoparticle shape improves delivery: rational coarse grain molecular dynamics (rCG-MD) of taxol in worm-like PEG-PCL micelles, Adv. Mater, 24 (2012) 3823–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Liu J, Xiao Y, Allen C, Polymer-drug compatibility: a guide to the development of delivery systems for the anticancer agent, ellipticine, J. Pharm. Sci, 93 (2004) 132–143. [DOI] [PubMed] [Google Scholar]
- [193].Richter A, Olbrich C, Krause M, Kissel T, Solubilization of sagopilone, a poorly water-soluble anticancer drug, using polymeric micelles for parenteral delivery, Int. J. Pharm, 389 (2010) 244–253. [DOI] [PubMed] [Google Scholar]
- [194].Forrest ML, Zhao A, Won C-Y, Malick AW, Kwon GS, Lipophilic prodrugs of Hsp90 inhibitor geldanamycin for nanoencapsulation in poly (ethylene glycol)-b-poly (ε-caprolactone) micelles, J. Control. Release, 116 (2006) 139–149. [DOI] [PubMed] [Google Scholar]
- [195].Letchford K, Liggins R, Burt H, Solubilization of hydrophobic drugs by methoxy poly(ethylene glycol)-block-polycaprolactone diblock copolymer micelles: theoretical and experimental data and correlations, J. Pharm. Sci, 97 (2008) 1179–1190. [DOI] [PubMed] [Google Scholar]
- [196].Latere Dwan’Isa J-P, Rouxhet L, Preat V, Brewster M, Arien A, Prediction of drug solubility in amphiphilic di-block copolymer micelles: the role of polymer-drug compatibility, Pharmazie, 62 (2007) 499–504. [PubMed] [Google Scholar]
- [197].Yan J, Ye Z, Chen M, Liu Z, Xiao Y, Zhang Y, Zhou Y, Tan W, Lang M, Fine tuning micellar core-forming block of poly (ethylene glycol)-block-poly (ε-caprolactone) amphiphilic copolymers based on chemical modification for the solubilization and delivery of doxorubicin, Biomacromolecules, 12 (2011) 2562–2572. [DOI] [PubMed] [Google Scholar]
- [198].Mahmud A, Patel S, Molavi O, Choi P, Samuel J, Lavasanifar A, Self-associating poly(ethylene oxide)-b-poly(alpha-cholesteryl carboxylate-epsilon-caprolactone) block copolymer for the solubilization of STAT-3 inhibitor cucurbitacin I, Biomacromolecules, 10 (2009) 471–478. [DOI] [PubMed] [Google Scholar]
- [199].Danquah M, Fujiwara T, Mahato RI, Self-assembling methoxypoly(ethylene glycol)-b-poly(carbonate-co-L-lactide) block copolymers for drug delivery, Biomaterials, 31 (2010) 2358–2370. [DOI] [PubMed] [Google Scholar]
- [200].Kakde D, Taresco V, Bansal KK, Magennis EP, Howdle SM, Mantovani G, Irvine DJ, Alexander C, Amphiphilic block copolymers from a renewable ε-decalactone monomer: prediction and characterization of micellar core effects on drug encapsulation and release, J. Mater. Chem. B, 4 (2016) 7119–7129. [DOI] [PubMed] [Google Scholar]
- [201].Patel S, Lavasanifar A, Choi P, Application of molecular dynamics simulation to predict the compatability between water-insoluble drugs and self-associating poly (ethylene oxide)-b-poly (ε-caprolactone) block copolymers, Biomacromolecules, 9 (2008) 3014–3023. [DOI] [PubMed] [Google Scholar]
- [202].Patel SK, Lavasanifar A, Choi P, Roles of nonpolar and polar intermolecular interactions in the improvement of the drug loading capacity of PEO-b-PCL with increasing PCL content for two hydrophobic Cucurbitacin drugs, Biomacromolecules, 10 (2009) 2584–2591. [DOI] [PubMed] [Google Scholar]
- [203].Patel SK, Lavasanifar A, Choi P, Molecular dynamics study of the encapsulation capability of a PCL-PEO based block copolymer for hydrophobic drugs with different spatial distributions of hydrogen bond donors and acceptors, Biomaterials, 31 (2010) 1780–1786. [DOI] [PubMed] [Google Scholar]
- [204].Erlebach A, Ott T, Otzen C, Schubert S, Czaplewska J, Schubert US, Sierka M, Thermodynamic compatibility of actives encapsulated into PEG-PLA nanoparticles: In Silico predictions and experimental verification, J. Comput. Chem, 37 (2016) 2220–2227. [DOI] [PubMed] [Google Scholar]
- [205].Wilkosz N, Łazarski G, Kovacik L, Gargas P, Nowakowska M, Jamróz D, Kepczynski M, Molecular insight into drug-loading capacity of PEG–PLGA nanoparticles for itraconazole, J. Phys. Chem. B, 122 (2018) 7080–7090. [DOI] [PubMed] [Google Scholar]
- [206].Costache AD, Sheihet L, Zaveri K, Knight DD, Kohn J, Polymer− drug interactions in tyrosine-derived triblock copolymer nanospheres: a computational modeling approach, Mol. Pharm, 6 (2009) 1620–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Hao J, Cheng Y, Ranatunga RU, Senevirathne S, Biewer MC, Nielsen SO, Wang Q, Stefan MC, A combined experimental and computational study of the substituent effect on micellar behavior of γ-substituted thermoresponsive amphiphilic poly (ε-caprolactone) s, Macromolecules, 46 (2013) 4829–4838. [Google Scholar]
- [208].Luo Z, Jiang J, pH-sensitive drug loading/releasing in amphiphilic copolymer PAE–PEG: Integrating molecular dynamics and dissipative particle dynamics simulations, J. Control. Release, 162 (2012) 185–193. [DOI] [PubMed] [Google Scholar]
- [209].Wu W, Zhang C, Lin W, Chen Q, Guo X, Qian Y, Zhang L, Quantitative structure-property relationship (QSPR) modeling of drug-loaded polymeric micelles via genetic function approximation, PLoS One, 10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Wu W, Zhang R, Peng S, Li X, Zhang L, QSPR between molecular structures of polymers and micellar properties based on block unit autocorrelation (BUA) descriptors, Chemom. Intell. Lab. Syst, 157 (2016) 7–15. [Google Scholar]
- [211].Hansen CM, The universality of the solubility parameter, Ind. Eng. Chem. Prod. Res. Dev, 8 (1969) 2–11. [Google Scholar]
- [212].Hildebrand JH, SOLUBILITY J Am. Chem. Soc, 38 (1916) 1452–1473. [Google Scholar]
- [213].Hansen CM, Hansen solubility parameters: a user’s handbook, CRC press; 2007. [Google Scholar]
- [214].Forster A, Hempenstall J, Tucker I, Rades T, Selection of excipients for melt extrusion with two poorly water-soluble drugs by solubility parameter calculation and thermal analysis, Int. J. Pharm, 226 (2001) 147–161. [DOI] [PubMed] [Google Scholar]
- [215].Gupta J, Nunes C, Vyas S, Jonnalagadda S, Prediction of solubility parameters and miscibility of pharmaceutical compounds by molecular dynamics simulations, J. Phys. Chem. B, 115 (2011) 2014–2023. [DOI] [PubMed] [Google Scholar]
- [216].Flory PJ, Thermodynamics of high polymer solutions, J. Chem. Phys, 10 (1942) 51–61. [Google Scholar]
- [217].Huggins ML, Some properties of solutions of long-chain compounds, J. Phys. Chem, 46 (1942) 151–158. [Google Scholar]
- [218].Thota N, Jiang J, Computational amphiphilic materials for drug delivery, Front. Mater, 2 (2015) 64. [Google Scholar]
- [219].Frenkel D, Smit B, Understanding molecular simulation: from algorithms to applications, Elsevier; 2001. [Google Scholar]
- [220].McKim JM, Bradbury SP, Niemi GJ, Fish acute toxicity syndromes and their use in the QSAR approach to hazard assessment, Environ. Health Perspect, 71 (1987) 171–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Sakai-Kato K, Nishiyama N, Kozaki M, Nakanishi T, Matsuda Y, Hirano M, Hanada H, Hisada S, Onodera H, Harashima H, General considerations regarding the in vitro and in vivo properties of block copolymer micelle products and their evaluation, J. Control. Release, 210 (2015) 76–83. [DOI] [PubMed] [Google Scholar]
- [222].Hurter PN, Scheutjens JM, Hatton TA, Molecular modeling of micelle formation and solubilization in block copolymer micelles. 1. A self-consistent mean-field lattice theory, Macromolecules, 26 (1993) 5592–5601. [Google Scholar]
- [223].Hurter PN, Scheutjens JM, Hatton TA, Molecular modeling of micelle formation and solubilization in block copolymer micelles. 2. Lattice theory for monomers with internal degrees of freedom, Macromolecules, 26 (1993) 5030–5040. [Google Scholar]
- [224].Linse P, Micellization of poly (ethylene oxide)-poly (propylene oxide) block copolymers in aqueous solution, Macromolecules, 26 (1993) 4437–4449. [Google Scholar]
- [225].Linse P, Micellization of poly (ethylene oxide)-poly (propylene oxide) block copolymer in aqueous solution: effect of polymer impurities, Macromolecules, 27 (1994) 2685–2693. [Google Scholar]
- [226].Callari M, De Souza PL, Rawal A, Stenzel MH, The Effect of Drug Loading on Micelle Properties: Solid-State NMR as a Tool to Gain Structural Insight, Angew Chem. Int. Ed. Engl, 56 (2017) 8441–8445. [DOI] [PubMed] [Google Scholar]
- [227].Grüne M, Luxenhofer R, Iuga D, Brown SP, Poppler A-C, 14 N-1 H HMQC solid-state NMR as a powerful tool to study amorphous formulations–an exemplary study of paclitaxel loaded polymer micelles, J. Mater. Chem. B, (2020). [DOI] [PubMed] [Google Scholar]
- [228].Wu G, Chu B, Schneider DK, SANS study of the micellar structure of PEO/PPO/PEO aqueous solution, J. Phys. Chem, 99 (1995) 5094–5101. [Google Scholar]
- [229].Siepmann J, Siepmann F, Sink conditions do not guarantee the absence of saturation effects, Int. J. Pharm, 577 (2020) 119009. [DOI] [PubMed] [Google Scholar]
- [230].Ma B, Zhuang W, Liu G, Wang Y, A biomimetic and pH-sensitive polymeric micelle as carrier for paclitaxel delivery, Regen. biomater, 5 (2018) 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Chen W, Zhong P, Meng F, Cheng R, Deng C, Feijen J, Zhong Z, Redox and pH-responsive degradable micelles for dually activated intracellular anticancer drug release, J. Control. Release, 169 (2013) 171–179. [DOI] [PubMed] [Google Scholar]
- [232].Li Y, Gao GH, Lee DS, Stimulus-sensitive polymeric nanoparticles and their applications as drug and gene carriers, Adv. Healthc. Mater, 2 (2013) 388–417. [DOI] [PubMed] [Google Scholar]
- [233].Felber AE, Dufresne M-H, Leroux J-C, pH-sensitive vesicles, polymeric micelles, and nanospheres prepared with polycarboxylates, Adv. Drug Deliv. Rev, 64 (2012) 979–992. [DOI] [PubMed] [Google Scholar]
- [234].Bae Y, Kataoka K, Intelligent polymeric micelles from functional poly (ethylene glycol)-poly (amino acid) block copolymers, Adv. Drug Deliv. Rev, 61 (2009) 768–784. [DOI] [PubMed] [Google Scholar]
- [235].Gaspar R, Duncan R, Polymeric carriers: preclinical safety and the regulatory implications for design and development of polymer therapeutics, Adv. Drug Deliv. Rev, 61 (2009) 1220–1231. [DOI] [PubMed] [Google Scholar]
- [236].Campos FC, Victorino VJ, Martins-Pinge MC, Cecchini AL, Panis C, Cecchini R, Systemic toxicity induced by paclitaxel in vivo is associated with the solvent cremophor EL through oxidative stress-driven mechanisms, Food Chem. Toxicol, 68 (2014) 78–86. [DOI] [PubMed] [Google Scholar]
- [237].Sparreboom A, van Zuylen L, Brouwer E, Loos WJ, de Bruijn P, Gelderblom H, Pillay M, Nooter K, Stoter G, Verweij J, Cremophor EL-mediated alteration of paclitaxel distribution in human blood: clinical pharmacokinetic implications, Cancer Res, 59 (1999) 1454–1457. [PubMed] [Google Scholar]
- [238].Hamaguchi T, Kato K, Yasui H, Morizane C, Ikeda M, Ueno H, Muro K, Yamada Y, Okusaka T, Shirao K, Shimada Y, Nakahama H, Matsumura Y, A phase I and pharmacokinetic study of NK105, a paclitaxel-incorporating micellar nanoparticle formulation, Br. J. Cancer, 97 (2007) 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Sahay G, Alakhova DY, Kabanov AV, Endocytosis of nanomedicines, J. Control. Release, 145 (2010) 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Shin HC, Alani AW, Cho H, Bae Y, Kolesar JM, Kwon GS, A 3-in-1 polymeric micelle nanocontainer for poorly water-soluble drugs, Mol. Pharm, 8 (2011) 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WC, Analysis of nanoparticle delivery to tumours, Nat. Rev. Mater, 1 (2016) 1–12. [Google Scholar]
- [242].Siegel RA, Kirtane AR, Panyam J, Assessing the benefits of drug delivery by nanocarriers: A partico/pharmacokinetic framework, IEEE Trans. Biomed. Eng, 64 (2016) 2176–2185. [DOI] [PubMed] [Google Scholar]
- [243].McNeil SE, Evaluation of nanomedicines: stick to the basics, Nat. Rev. Mater, 1 (2016) 1–2. [Google Scholar]
- [244].Price LS, Stern ST, Deal AM, Kabanov AV, Zamboni WC, A reanalysis of nanoparticle tumor delivery using classical pharmacokinetic metrics, Sci. Adv, 6 (2020) eaay9249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Ambardekar VV, Stern ST, NBCD pharmacokinetics and bioanalytical methods to measure drug release, Non-biological complex drugs, Springer; 2015, pp. 261–287. [Google Scholar]
- [246].Skoczen S, McNeil SE, Stern ST, Stable isotope method to measure drug release from nanomedicines, J. Control. Release, 220 (2015) 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Skoczen SL, Snapp KS, Crist RM, Kozak D, Jiang X, Liu H, Stern ST, Distinguishing Pharmacokinetics of Marketed Nanomedicine Formulations Using a Stable Isotope Tracer Assay, ACS Pharmacology Translational Science, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Bulitta JB, Zhao P, Arnold RD, Kessler DR, Daifuku R, Pratt J, Luciano G, Hanauske A-R, Gelderblom H, Awada A, Jusko WJ, Mechanistic population pharmacokinetics of total and unbound paclitaxel for a new nanodroplet formulation versus Taxol in cancer patients, Cancer Chemother. Pharmacol, 63 (2009) 1049–1063. [DOI] [PubMed] [Google Scholar]
- [249].Chu J, Gallo JM, Application of microdialysis to characterize drug disposition in tumors, Adv. Drug Deliv. Rev, 45 (2000) 243–253. [DOI] [PubMed] [Google Scholar]
- [250].Nishiyama N, Kataoka K, Nanostructured devices based on block copolymer assemblies for drug delivery: designing structures for enhanced drug function, Polymer Therapeutics II, Springer; 2006, pp. 67–101. [Google Scholar]
- [251].Kabanov AV, Alakhov VY, Pluronic® block copolymers in drug delivery: From micellar nanocontainers to biological response modifiers, Crit. Rev. Ther. Drug, 19 (2002). [DOI] [PubMed] [Google Scholar]
- [252].Batrakova EV, Li S, Li Y, Alakhov VY, Elmquist WF, Kabanov AV, Distribution kinetics of a micelle-forming block copolymer Pluronic P85, J. Control. Release, 100 (2004) 389–397. [DOI] [PubMed] [Google Scholar]
- [253].Caster JM, Yu SK, Patel AN, Newman NJ, Lee ZJ, Warner SB, Wagner KT, Roche KC, Tian X, Min Y, Wang AZ, Effect of particle size on the biodistribution, toxicity, and efficacy of drug-loaded polymeric nanoparticles in chemoradiotherapy, Nanomedicine, 13 (2017) 1673–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Lee H, Fonge H, Hoang B, Reilly RM, Allen C, The effects of particle size and molecular targeting on the intratumoral and subcellular distribution of polymeric nanoparticles, Mol. Pharm, 7 (2010) 1195–1208. [DOI] [PubMed] [Google Scholar]
- [255].Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, Terada Y, Kano MR, Miyazono K, Uesaka M, Nishiyama N, Kataoka K, Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size, Nat. Nanotechnol, 6 (2011) 815–823. [DOI] [PubMed] [Google Scholar]
- [256].Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE, Shape effects of filaments versus spherical particles in flow and drug delivery, Nat. Nanotechnol, 2 (2007) 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Miao L, Guo S, Lin CM, Liu Q, Huang L, Nanoformulations for combination or cascade anticancer therapy, Adv. Drug Deliv. Rev, 115 (2017) 3–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Hu Q, Sun W, Wang C, Gu Z, Recent advances of cocktail chemotherapy by combination drug delivery systems, Adv. Drug Deliv. Rev, 98 (2016) 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [259].Vinod N, Hwang D, Azam SH, Van Swearingen AE, Wayne E, Fussell SC, Sokolsky-Papkov M, Pecot CV, Kabanov AV, High-capacity poly (2-oxazoline) formulation of TLR 7/8 agonist extends survival in a chemo-insensitive, metastatic model of lung adenocarcinoma, Sci. Adv, 6 (2020) eaba5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Shin HC, Alani AW, Rao DA, Rockich NC, Kwon GS, Multi-drug loaded polymeric micelles for simultaneous delivery of poorly soluble anticancer drugs, J. Control. Release, 140 (2009) 294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Shin HC, Cho H, Lai TC, Kozak KR, Kolesar JM, Kwon GS, Pharmacokinetic study of 3-in-1 poly(ethylene glycol)-block-poly(D, L-lactic acid) micelles carrying paclitaxel, 17-allylamino-17-demethoxygeldanamycin, and rapamycin, J. Control. Release, 163 (2012) 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Hussein YH, Youssry M, Polymeric micelles of biodegradable diblock copolymers: Enhanced encapsulation of hydrophobic drugs, Materials, 11 (2018) 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [263].Lee H, Zeng F, Dunne M, Allen C, Methoxy poly (ethylene glycol)-block-poly (δ-valerolactone) copolymer micelles for formulation of hydrophobic drugs, Biomacromolecules, 6 (2005) 3119–3128. [DOI] [PubMed] [Google Scholar]
- [264].Liggins R, Burt H, Polyether–polyester diblock copolymers for the preparation of paclitaxel loaded polymeric micelle formulations, Adv. Drug Deliv. Rev, 54 (2002) 191–202. [DOI] [PubMed] [Google Scholar]
- [265].Salimi A, Zadeh BSM, Kazemi M, Preparation and optimization of polymeric micelles as an oral drug delivery system for deferoxamine mesylate: in vitro and ex vivo studies, Res. Pharm. Sci, 14 (2019) 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [266].Markwalter CE, Pagels RF, Wilson BK, Ristroph KD, Prud’homme RK, Flash nanoprecipitation for the encapsulation of hydrophobic and hydrophilic compounds in polymeric nanoparticles, J. Vis. Exp, (2019) e58757. [DOI] [PubMed] [Google Scholar]
- [267].Shen H, Hong S, Prud’homme RK, Liu Y, Self-assembling process of flash nanoprecipitation in a multi-inlet vortex mixer to produce drug-loaded polymeric nanoparticles, J. Nanoparticle Res, 13 (2011) 4109–4120. [Google Scholar]
- [268].Gindy ME, Prud’homme RK, Multifunctional nanoparticles for imaging, delivery and targeting in cancer therapy, Expert Opin. Drug Deliv, 6 (2009) 865–878. [DOI] [PubMed] [Google Scholar]
- [269].Tao J, Chow SF, Zheng Y, Application of flash nanoprecipitation to fabricate poorly water-soluble drug nanoparticles, Acta Pharm. Sin. B, 9 (2019) 4–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [270].Kedar U, Phutane P, Shidhaye S, Kadam V, Advances in polymeric micelles for drug delivery and tumor targeting, Nanomedicine, 6 (2010) 714–729. [DOI] [PubMed] [Google Scholar]
- [271].Lavasanifar A, Samuel J, Kwon GS, Micelles of poly (ethylene oxide)-block-poly (N-alkyl stearate L-aspartamide): synthetic analogues of lipoproteins for drug delivery, J. Biomed. Mater. Res, 52 (2000) 831–835. [DOI] [PubMed] [Google Scholar]
- [272].Aliabadi HM, Lavasanifar A, Polymeric micelles for drug delivery, Expert Opin. Drug Deliv, 3 (2006) 139–162. [DOI] [PubMed] [Google Scholar]
- [273].Kim SY, Shin IG, Lee YM, Cho CS, Sung YK, Methoxy poly (ethylene glycol) and ϵ-caprolactone amphiphilic block copolymeric micelle containing indomethacin.: II. Micelle formation and drug release behaviours, J. Control. Release, 51 (1998) 13–22. [DOI] [PubMed] [Google Scholar]
- [274].Vangeyte P, Gautier S, Jérôme R, About the methods of preparation of poly (ethylene oxide)-b-poly (ε-caprolactone) nanoparticles in water: Analysis by dynamic light scattering, Colloids Surfaces A, 242 (2004) 203–211. [Google Scholar]
- [275].Aliabadi HM, Brocks DR, Lavasanifar A, Polymeric micelles for the solubilization and delivery of cyclosporine A: pharmacokinetics and biodistribution, Biomaterials, 26 (2005) 7251–7259. [DOI] [PubMed] [Google Scholar]
- [276].Xie X, Ma Y, Huang L, Cai M, Chen Y, Luo X, Effect factors of micelle preparation for a pH-sensitive copolymer containing zwitterionic sulfobetaines, Colloids Surfaces A, 468 (2015) 31–39. [Google Scholar]
- [277].Taillefer J, Jones MC, Brasseur N, Van Lier J, Leroux JC, Preparation and characterization of pH-responsive polymeric micelles for the delivery of photosensitizing anticancer drugs, J. Pharm. Sci, 89 (2000) 52–62. [DOI] [PubMed] [Google Scholar]
- [278].Patil YB, Toti US, Khdair A, Ma L, Panyam J, Single-step surface functionalization of polymeric nanoparticles for targeted drug delivery, Biomaterials, 30 (2009) 859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Zhang J, Jiang W, Zhao X, Wang Y, Preparation and characterization of polymeric micelles from poly (d, l-lactide) and methoxypolyethylene glycol block copolymers as potential drug carriers, Tsinghua Sci. Technol, 12 (2007) 493–496. [Google Scholar]
- [280].Le Garrec D, Gori S, Luo L, Lessard D, Smith D, Yessine M-A, Ranger M, Leroux J-C, Poly (N-vinylpyrrolidone)-block-poly (D, L-lactide) as a new polymeric solubilizer for hydrophobic anticancer drugs: in vitro and in vivo evaluation, J. Control. Release, 99 (2004) 83–101. [DOI] [PubMed] [Google Scholar]
- [281].Fournier E, Dufresne M-H, Smith DC, Ranger M, Leroux J-C, A novel one-step drug-loading procedure for water-soluble amphiphilic nanocarriers, Pharm. Res, 21 (2004) 962–968. [DOI] [PubMed] [Google Scholar]
- [282].Klinski E, Patel K, Pietrzynski G, Alakhov V, Doxorubicin formulations for anti-cancer use, United States Patent Application 2007. 0196493.
- [283].Tyrrell ZL, Shen Y, Radosz M, Near-critical fluid micellization for high and efficient drug loading: encapsulation of paclitaxel into PEG-b-PCL micelles, J. Phys. Chem. C, 115 (2011) 11951–11956. [Google Scholar]
- [284].Tyrrell Z, Winoto W, Shen Y, Radosz M, Block Copolymer Micelles Formed in Supercritical Fluid Can Become Water-Dispensable Nanoparticles: Poly (ethylene glycol)−block-Poly (ϵ-caprolactone) in Trifluoromethane, Ind. Eng. Chem. Res, 48 (2009) 1928–1932. [Google Scholar]
- [285].Chakravarty P, Famili A, Nagapudi K, Al-Sayah MA, Using supercritical fluid technology as a green alternative during the preparation of drug delivery systems, Pharmaceutics, 11 (2019) 629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Guideline IHT, Pharmaceutical development, Q8 (R2) Current Step, 4 (2009). [Google Scholar]
- [287].McDonald K, Ho K, ICH Q11: development and manufacture of drug substances–chemical and biotechnological/biological entities, GaBI J, 1 (2012) 142–144. [Google Scholar]
- [288].Tyner KM, Zheng N, Choi S, Xu X, Zou P, Jiang W, Guo C, Cruz CN, How has CDER prepared for the nano revolution? A review of risk assessment, regulatory research, and guidance activities, AAPS J, 19 (2017) 1071–1083. [DOI] [PubMed] [Google Scholar]
- [289].D’Mello SR, Cruz CN, Chen M-L, Kapoor M, Lee SL, Tyner KM, The evolving landscape of drug products containing nanomaterials in the United States, Nat. Nanotechnol, 12 (2017) 523–529. [DOI] [PubMed] [Google Scholar]
- [290].Chen M-L, John M, Lee SL, Tyner KM, Development considerations for nanocrystal drug products, AAPS J, 19 (2017) 642–651. [DOI] [PubMed] [Google Scholar]
- [291].Drug Products, Including Biological Products, that Contain Nanomaterials–Guidance for Industry, US Food Drug Administration, 2017. [Google Scholar]
- [292].de Vlieger JS, Crommelin DJ, Tyner K, Drummond DC, Jiang W, McNeil SE, Neervannan S, Crist RM, Shah VP, Report of the AAPS Guidance Forum on the FDA draft guidance for industry:”drug products, including biological products, that contain nanomaterials”, AAPS J, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Mühlebach S, Regulatory challenges of nanomedicines and their follow-on versions: A generic or similar approach?, Adv. Drug Deliv. Rev, 131 (2018) 122–131. [DOI] [PubMed] [Google Scholar]
- [294].Kapoor M, Lee SL, Tyner KM, Liposomal drug product development and quality: current US experience and perspective, AAPS J, 19 (2017) 632–641. [DOI] [PubMed] [Google Scholar]
- [295].Lawrence XY, Akseli I, Allen B, Amidon G, Bizjak TG, Boam A, Caulk M, Doleski D, Famulare J, Fisher AC, Advancing product quality: a summary of the second FDA/PQRI conference, AAPS J, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [296].Anselmo AC, Mitragotri S, Nanoparticles in the clinic: An update, Bioengineering & Translational Medicine, 4 (2019) e10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [297].Jarvis M, Krishnan V, Mitragotri S, Nanocrystals: A perspective on translational research and clinical studies, Bioengineering & translational medicine, 4 (2019) 5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Liposome Drug Products: Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation. Guidance for Industry, US Food Drug Administration, 2018. [Google Scholar]
- [299].Bartlett JA, Brewster M, Brown P, Cabral-Lilly D, Cruz CN, David R, Eickhoff WM, Haubenreisser S, Jacobs A, Malinoski F, Summary report of PQRI workshop on nanomaterial in drug products: current experience and management of potential risks, AAPS J, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Kato K, Chin K, Yoshikawa T, Yamaguchi K, Tsuji Y, Esaki T, Sakai K, Kimura M, Hamaguchi T, Shimada Y, Matsumura Y, Ikeda R, Phase II study of NK105, a paclitaxel-incorporating micellar nanoparticle, for previously treated advanced or recurrent gastric cancer, Invest. New Drugs, 30 (2012) 1621–1627. [DOI] [PubMed] [Google Scholar]
- [301].Kim D-W, Kim S-Y, Kim H-K, Kim S-W, Shin SW, Kim J, Park K, Lee M, Heo DS, Multicenter phase II trial of Genexol-PM, a novel Cremophor-free, polymeric micelle formulation of paclitaxel, with cisplatin in patients with advanced non-small-cell lung cancer, Ann. Oncol, 18 (2007) 2009–2014. [DOI] [PubMed] [Google Scholar]
- [302].Park IH, Sohn JH, Kim SB, Lee KS, Chung JS, Lee SH, Kim TY, Jung KH, Cho EK, Kim YS, An open-label, randomized, parallel, phase III trial evaluating the efficacy and safety of polymeric micelle-formulated paclitaxel compared to conventional cremophor EL-based paclitaxel for recurrent or metastatic HER2-negative breast cancer, Cancer Res. Treat, 49 (2017) 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [303].Kim JY, Do YR, Song HS, Cho YY, Ryoo HM, Bae SH, Kim JG, Chae YS, Kang BW, Baek JH, Multicenter phase II clinical trial of Genexol-PM® with gemcitabine in advanced biliary tract cancer, Anticancer Res, 37 (2017) 1467–1473. [DOI] [PubMed] [Google Scholar]
- [304].Kim HS, Lee JY, Lim SH, Sun J-M, Lee SH, Ahn JS, Park K, Moon SH, Ahn M-J, A prospective phase II study of cisplatin and Cremophor EL-free paclitaxel (Genexol-PM) in patients with unresectable thymic epithelial tumors, J. Thorac. Oncol, 10 (2015) 1800–1806. [DOI] [PubMed] [Google Scholar]
- [305].Lee S-W, Kim Y-M, Cho CH, Kim YT, Kim SM, Hur SY, Kim J-H, Kim B-G, Kim S-C, Ryu H-S, An open-label, randomized, parallel, phase II trial to evaluate the efficacy and safety of a cremophor-free polymeric micelle formulation of paclitaxel as first-line treatment for ovarian cancer: a Korean Gynecologic Oncology Group study (KGOG-3021), Cancer Res. Treat, 50 (2018) 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [306].Lee S-W, Yun M-H, Jeong SW, In C-H, Kim J-Y, Seo M-H, Pai C-M, Kim S-O, Development of docetaxel-loaded intravenous formulation, Nanoxel-PM™ using polymer-based delivery system, J. Control. Release, 155 (2011) 262–271. [DOI] [PubMed] [Google Scholar]
- [307].Fujiwara Y, Mukai H, Saeki T, Ro J, Lin YC, Nagai SE, Lee KS, Watanabe J, Ohtani S, Kim SB, Kuroi K, Tsugawa K, Tokuda Y, Iwata H, Park YH, Yang Y, Nambu Y, A multi-national, randomised, open-label, parallel, phase III non-inferiority study comparing NK105 and paclitaxel in metastatic or recurrent breast cancer patients, Br. J. Cancer, 120 (2019) 475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Plummer R, Wilson R, Calvert H, Boddy A, Griffin M, Sludden J, Tilby M, Eatock M, Pearson D, Ottley C, A Phase I clinical study of cisplatin-incorporated polymeric micelles (NC-6004) in patients with solid tumours, Br. J. Cancer, 104 (2011) 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Subbiah V, Grilley-Olson JE, Combest AJ, Sharma N, Tran RH, Bobe I, Osada A, Takahashi K, Balkissoon J, Camp A, Phase Ib/II Trial of NC-6004 (Nanoparticle Cisplatin) plus gemcitabine in patients with advanced solid tumors, Clin. Cancer Res, 24 (2018) 43–51. [DOI] [PubMed] [Google Scholar]
- [310].Batrakova EV, Li S, Brynskikh AM, Sharma AK, Li Y, Boska M, Gong N, Mosley RL, Alakhov VY, Gendelman HE, Effects of pluronic and doxorubicin on drug uptake, cellular metabolism, apoptosis and tumor inhibition in animal models of MDR cancers, J. Control. Release, 143 (2010) 290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [311].Zhao Y, Alakhova DY, Zhao X, Band V, Batrakova EV, Kabanov AV, Eradication of cancer stem cells in triple negative breast cancer using doxorubicin/pluronic polymeric micelles, Nanomedicine, 24 (2020) 102124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [312].Kabanov AV, Kabanov VA, DNA complexes with polycations for the delivery of genetic material into cells, Bioconjug. Chem, 6 (1995) 7–20. [DOI] [PubMed] [Google Scholar]
- [313].Harada A, Kataoka K, Formation of polyion complex micelles in an aqueous milieu from a pair of oppositely-charged block copolymers with poly (ethylene glycol) segments, Macromolecules, 28 (1995) 5294–5299. [Google Scholar]
- [314].Kataoka K, Togawa H, Harada A, Yasugi K, Matsumoto T, Katayose S, Spontaneous formation of polyion complex micelles with narrow distribution from antisense oligonucleotide and cationic block copolymer in physiological saline, Macromolecules, 29 (1996) 8556–8557. [Google Scholar]
- [315].Vinogradov SV, Kohli E, Zeman AD, Cross-Linked Polymeric Nanogel Formulations of 5 ‘-Triphosphates of Nucleoside Analogues: Role of the Cellular Membrane in Drug Release, Mol. Pharm, 2 (2005) 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Lai TC, Bae Y, Yoshida T, Kataoka K, Kwon GS, pH-sensitive multi-PEGylated block copolymer as a bioresponsive pDNA delivery vector, Pharm. Res, 27 (2010) 2260–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Baba M, Itaka K, Kondo K, Yamasoba T, Kataoka K, Treatment of neurological disorders by introducing mRNA in vivo using polyplex nanomicelles, J. Control. Release, 201 (2015) 41–48. [DOI] [PubMed] [Google Scholar]
- [318].Efremenko EN, Lyagin IV, Klyachko NL, Bronich T, Zavyalova NV, Jiang Y, Kabanov AV, A simple and highly effective catalytic nanozyme scavenger for organophosphorus neurotoxins, J. Control. Release, 247 (2017) 175–181. [DOI] [PubMed] [Google Scholar]
- [319].Manickam DS, Brynskikh AM, Kopanic JL, Sorgen PL, Klyachko NL, Batrakova EV, Bronich TK, Kabanov AV, Well-defined cross-linked antioxidant nanozymes for treatment of ischemic brain injury, J. Control. Release, 162 (2012) 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Klyachko NL, Manickam DS, Brynskikh AM, Uglanova SV, Li S, Higginbotham SM, Bronich TK, Batrakova EV, Kabanov AV, Cross-linked antioxidant nanozymes for improved delivery to CNS, Nanomedicine, 8 (2012) 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Rui Y, Wilson DR, Choi J, Varanasi M, Sanders K, Karlsson J, Lim M, Green JJ, Carboxylated branched poly (β-amino ester) nanoparticles enable robust cytosolic protein delivery and CRISPR-Cas9 gene editing, Sci. Adv, 5 (2019) eaay3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, Mackley VA, Chang K, Rao A, Skinner C, Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair, Nat. Biomed. Eng, 1 (2017) 889–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [323].Li Y, Bronich TK, Chelushkin PS, Kabanov AV, Dynamic properties of block ionomer complexes with polyion complex cores, Macromolecules, 41 (2008) 5863–5868. [Google Scholar]
- [324].Harada A, Kataoka K, Effect of charged segment length on physicochemical properties of core− shell type polyion complex micelles from block ionomers, Macromolecules, 36 (2003) 4995–5001. [Google Scholar]
- [325].Kabanov AV, Kabanov VA, Interpolyelectrolyte and block ionomer complexes for gene delivery: physico-chemical aspects, Adv. Drug Deliv. Rev, 30 (1998) 49–60. [DOI] [PubMed] [Google Scholar]
- [326].Jiang Y, Fay JM, Poon CD, Vinod N, Zhao Y, Bullock K, Qin S, Manickam DS, Yi X, Banks WA, Kabanov AV, Nanoformulation of Brain-Derived Neurotrophic Factor with Target Receptor-Triggered-Release in the Central Nervous System, Adv. Funct. Mater, 28 (2018) 1703982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [327].Wang Q, Jiang J, Chen W, Jiang H, Zhang Z, Sun X, Targeted delivery of low-dose dexamethasone using PCL–PEG micelles for effective treatment of rheumatoid arthritis, J. Control. Release, 230 (2016) 64–72. [DOI] [PubMed] [Google Scholar]
- [328].Ishihara T, Kubota T, Choi T, Higaki M, Treatment of experimental arthritis with stealth-type polymeric nanoparticles encapsulating betamethasone phosphate, J. Pharmacol. Exp. Ther, 329 (2009) 412–417. [DOI] [PubMed] [Google Scholar]
- [329].Wu T, Chen X, Wang Y, Xiao H, Peng Y, Lin L, Xia W, Long M, Tao J, Shuai X, Aortic plaque-targeted andrographolide delivery with oxidation-sensitive micelle effectively treats atherosclerosis via simultaneous ROS capture and anti-inflammation, Nanomedicine, 14 (2018) 2215–2226. [DOI] [PubMed] [Google Scholar]
- [330].Mohamadpour H, Azadi A, Rostamizadeh K, Andalib S, Saghatchi Zanjani MR, Hamidi M, Preparation, Optimization, and Evaluation of Methoxy Poly (ethylene glycol)-co-Poly (ε-caprolactone) Nanoparticles Loaded by Rivastigmine for Brain Delivery, ACS Chem. Neurosci, 11 (2020) 783–795. [DOI] [PubMed] [Google Scholar]
- [331].Tavares GS, Mendonça DV, Miyazaki CK, Lage DP, Soyer TG, Carvalho LM, Ottoni FM, Dias DS, Ribeiro PA, Antinarelli LM, A Pluronic® F127-based polymeric micelle system containing an antileishmanial molecule is immunotherapeutic and effective in the treatment against Leishmania amazonensis infection, Parasitol. Int, 68 (2019) 63–72. [DOI] [PubMed] [Google Scholar]
- [332].Liu Y, van der Mei HC, Zhao B, Zhai Y, Cheng T, Li Y, Zhang Z, Busscher HJ, Ren Y, Shi L, Eradication of Multidrug-Resistant Staphylococcal Infections by Light-Activatable Micellar Nanocarriers in a Murine Model, Adv. Funct. Mater, 27 (2017) 1701974. [Google Scholar]
- [333].Mendonça DV, Tavares GS, Lage DP, Soyer TG, Carvalho LM, Dias DS, Ribeiro PA, Ottoni FM, Antinarelli LM, Vale DL, In vivo antileishmanial efficacy of a naphthoquinone derivate incorporated into a Pluronic® F127-based polymeric micelle system against Leishmania amazonensis infection, Biomed. Pharmacother, 109 (2019) 779–787. [DOI] [PubMed] [Google Scholar]
- [334].dos Reis Lage LM, Barichello JM, Lage DP, Mendonça DVC, Carvalho AMRS, Rodrigues MR, Menezes-Souza D, Roatt BM, Alves RJ, Tavares CAP, An 8-hydroxyquinoline-containing polymeric micelle system is effective for the treatment of murine tegumentary leishmaniasis, Parasitol. Res, 115 (2016) 4083–4095. [DOI] [PubMed] [Google Scholar]
- [335].Li S, Wei W, Jia W, Zhao L, Xu H, Zhou F, Zhu L, Song Z, Feng S, Feng R, Itraconazole-loaded micelles based on linear-dendritic poly (ethylene glycol)-b-poly (ε-caprolactone), J. Biomater. Sci. Polym. Ed, 29 (2018) 2299–2311. [DOI] [PubMed] [Google Scholar]
- [336].Yi Y, Yoon HJ, Kim BO, Shim M, Kim S-O, Hwang S-J, Seo MH, A mixed polymeric micellar formulation of itraconazole: Characteristics, toxicity and pharmacokinetics, J. Control. Release, 117 (2007) 59–67. [DOI] [PubMed] [Google Scholar]
- [337].Chiappetta DA, Facorro G, de Celis ER, Sosnik A, Synergistic encapsulation of the anti-HIV agent efavirenz within mixed poloxamine/poloxamer polymeric micelles, Nanomedicine, 7 (2011) 624–637. [DOI] [PubMed] [Google Scholar]
- [338].Alvarez-Rivera F, Fernández-Villanueva D, Concheiro A, Alvarez-Lorenzo C, α-Lipoic acid in Soluplus® polymeric nanomicelles for ocular treatment of diabetes-associated corneal diseases, J. Pharm. Sci, 105 (2016) 2855–2863. [DOI] [PubMed] [Google Scholar]
- [339].Li X, Zhang Z, Li J, Sun S, Weng Y, Chen H, Diclofenac/biodegradable polymer micelles for ocular applications, Nanoscale, 4 (2012) 4667–4673. [DOI] [PubMed] [Google Scholar]
- [340].Ravenelle F, Vachon P, Rigby-Jones A, Sneyd J, Le Garrec D, Gori S, Lessard D, Smith D, Anaesthetic effects of propofol polymeric micelle: a novel water soluble propofol formulation, Br. J. Anaesth, 101 (2008) 186–193. [DOI] [PubMed] [Google Scholar]
- [341].Segura-Ibarra V, Amione-Guerra J, Cruz-Solbes AS, Cara FE, Iruegas-Nunez DA, Wu S, Youker KA, Bhimaraj A, Torre-Amione G, Ferrari M, Rapamycin nanoparticles localize in diseased lung vasculature and prevent pulmonary arterial hypertension, Int. J. Pharm, 524 (2017) 257–267. [DOI] [PubMed] [Google Scholar]
- [342].Lapteva M, Mondon K, M ller M, Gurny R, Kalia YN, Polymeric micelle nanocarriers for the cutaneous delivery of tacrolimus: a targeted approach for the treatment of psoriasis, Mol. Pharm, 11 (2014) 2989–3001. [DOI] [PubMed] [Google Scholar]
- [343].Lapteva M, Möller M, Gurny R, Kalia YN, Self-assembled polymeric nanocarriers for the targeted delivery of retinoic acid to the hair follicle, Nanoscale, 7 (2015) 18651–18662. [DOI] [PubMed] [Google Scholar]
- [344].Chen C-L, Chang S-F, Lee D, Yang L-Y, Lee Y-H, Hsu CY, Lin S-J, Liaw J, Bioavailability effect of methylprednisolone by polymeric micelles, Pharm. Res, 25 (2008) 39–47. [DOI] [PubMed] [Google Scholar]
- [345].Wang Y, Wu M, Gu L, Li X, He J, Zhou L, Tong A, Shi J, Zhu H, Xu J, Effective improvement of the neuroprotective activity after spinal cord injury by synergistic effect of glucocorticoid with biodegradable amphipathic nanomicelles, Drug Deliv, 24 (2017) 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [346].Li J, Deng J, Yuan J, Fu J, Li X, Tong A, Wang Y, Chen Y, Guo G, Zonisamide-loaded triblock copolymer nanomicelles as a novel drug delivery system for the treatment of acute spinal cord injury, Int. J. Nanomedicine, 12 (2017) 2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [347].Gong C, Wu Q, Wang Y, Zhang D, Luo F, Zhao X, Wei Y, Qian Z, A biodegradable hydrogel system containing curcumin encapsulated in micelles for cutaneous wound healing, Biomaterials, 34 (2013) 6377–6387. [DOI] [PubMed] [Google Scholar]